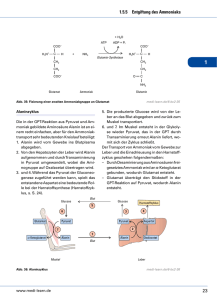
Rolle der Aminosäuren im Stoffwechsel
Werbung

Rolle der Aminosäuren im Stoffwechsel freie AS aus zugeführtem Protein und hydrolysiertem Körperprotein = 0.05 % 70 g Im Gegensatz zu Fettsäuren und Glucose sind freie AS NICHT speicherbar 70-80 % davon im Muskel! Resorption und Verteilung der Aminosäuren im Organismus • Darm: Verlust essentieller AS, besonders Thr (Bestandteil proteolyse-resistenter Mucine in endothelialen Schleimschichten!) • Mucosa: Glu, Gln, Asp Æ Energie Arg Æ Citrullin • Niere: Arg-Synthese • zentrale Rolle der Leber: Leber Proteinbiosynthese und -abbau Ketosäuren Æ Fettsäuren, Glucose Ammoniak Æ Harnstoff genaue Menge nicht resorbierter AS unbekannt • Muskulatur: nimmt v.a. verzweigtkettige AS auf: Val, Leu, Ile Aminosäuren - Hauptabbauwege Aminosäuren Transaminierung α-Ketosäuren dehydrierende Decarboxylierung oxidative Desaminierung (Glutamat-DH) biogene Amine NH3 Harnstoffzyklus Niere - Urin Fettsäure-Thioester (Acetyl-CoA) Decarboxylierung Oxidation (Aminoxidasen) Fettsäuren Citrat-Zyklus: je nach Stoffwechsellage Æ Abbau zu CO2 + H20 oder Æ Aufbau von Kohlenhydraten, Fetten... AS-Abbau: 1. Stickstoff (N2) - Abspaltung Generelles: nicht benötigte AS werden abgebaut (normal: < 50 %) bei Säugern: AS-Abbau hauptsächlich in der Leber aber auch periphere Organe können AS zum Energiegewinn (ATP-Bildung) zu CO2, H2O und NH3 abbauen in Energieübertragungswegen kommen keine StickstoffEnergieübertragung Stickstoff haltigen Verbindungen vor Æ Aminogruppe entfernen durch Transaminierung und oxidative Desaminierung Ammoniak (NH3) ist cytotoxisch Æ muss entgiftet werden Die zentralen Aminoäuren und ihre zugehörigen α-Ketosäuren (α-KS) • sind am häufigsten an Umbaureaktionen (Transaminierungen) beteiligt • hohe intrazelluläre Konzentration dieser AS: 10- bis 50-fach erhöht gegenüber Plasma Transaminierung Prinzip der Reaktion: AS α-KG α-KS Glu • Aminogruppe (-NH2) wird auf α-Ketoglutarat (α-KG; = 2-Oxo-glutarat) übertragen: AS Æ α-KS • abhängig von Pyridoxalphosphat (PALP) • reversibel Æ AS-Synthese aus α-KS ABER: ABER:für fürLysin Lysin(Lys, (Lys,K) K)und undThreonin Threonin(Thr, (Thr,T): T): keine keineAminotransferasen Aminotransferasen Die beiden wichtigsten Aminotransferasen AspartatAminotransferase (AST, ASAT, GOT = Glutamat-OxalacetatTransaminase) AlaninAminotransferase (ALT, ALAT, GPT = Glutamat-PyruvatTransaminase) ••hohe hoheKonzentration Konzentrationdieser dieserEnzyme Enzymein inLeber, Leber,Myocard, Myocard,Hirn Hirn ••Isoformen Isoformenin inCytosol Cytosolund undMitochondrien Mitochondrien Æ ÆDiagnostik Diagnostik Pyridoxalphosphat (PLP, PALP) Vitamin B6 = Pyridoxal leicht sauer leicht basisch substituiertes Pyridin Aldehydgruppe bildet kovalente Schiff-Basen: prosthetische Gruppe (Coenzym) z.B. in Aminotransferasen Mechanismus der PALP-abhängigen Transaminierung an der Aldehydgruppe des PALP: protoniertes PALP wirkt als Elektronenfalle Amino-Stickstoff Amino-Stickstoffwird wirdnicht nichtfrei! frei! PALP-abhängige Reaktionen Spaltung der Bindung a), b) oder c) c) Aldolasen: Aldolspaltung (Glycin vom Rest der AS abgespalten) a) Aminotransferasen: Transaminierung (AS + α-KG Æ α-KS + Glu) b) Decarboxylasen: Decarboxylierung (AS Æ Amin + CO2) weitere Reaktionen: Dehydratasen = Desaminasen: α-,ß-Eliminierung (AS Æ α-KS + NH3) PALP-abhängige Reaktionen: im AS-Stoffwechsel, in der HämoglobinBiosynthese, bei der GABA-Biosynthese, im Glycogen-Abbau PALP-abhängige Decarboxylierung Spaltung Spaltungzwischen zwischenα-C α-Cund und Carboxylgruppe Carboxylgruppe Æ ÆFreisetzung Freisetzungdes desAmins Amins und undCO CO2 Ox 2 DAO Säure Aminosäure Amin Vorkommen Cystein (Cys, C) Cysteamin Coenzym A Asparaginsäure (Asp, D) ß-Alanin Coenzym A Glutaminsäure (Glu, E) γ-Aminobuttersäure (GABA) Neurotransmitter Histidin (His, H) Histamin Mediator, Hormon Dihydroxy-Phenylalanin (DOPA) Dopamin Neurotransmitter Tryptophan (Trp, W) Tryptamin Hormon 5-Hydroxy-Tryptophan Serotonin Neurotransmitter α-,ß-Eliminierung und Spaltung Spaltungder derBindungen Bindungenzwischen zwischen α-C α-Cund undseinem seinemHHsowie sowie ß-C und seinem Substituenten ß-C und seinem Substituenten ↓↓ Eliminierung Eliminierungvon vonWasser Wasser(bzw. (bzw.HH22S) S) Æ ÆIminosäure IminosäureÆ Æspontane spontaneHydrolyse Hydrolyse zu zuα-KS α-KS++NH NH3 Aldolspaltung Spaltung Spaltungder derBindung Bindungzwischen zwischenα-C α-C und undß-C ß-C ↓↓ Glycin Glycinvom vom‚Rest‘ ‚Rest‘der derAminosäure Aminosäure abgetrennt abgetrennt 3 [Aminoacrylat] Abbau von Ser, Cys zu Pyruvat Thr zu α-Ketobutyrat auch Met, Gly, His ß-, γ-Eliminierung bei Homoserin, Homocystein Folat Æ Tetrahydrofolat (FH4) (sind Coenzyme der Purin- und Pyrimidinsynthese und des Met-Stoffwechsels) Oxidative Desaminierung wichtigste Reaktion: Glutamat-Dehydrogenase (GLDH): • Gleichgewichtsreaktion; allosterische Aktivierung (ADP, GDP) • Aminogruppe als NH3 freigesetzt (Æ) oder NH3 wird fixiert (Å) • Zwischenprodukt Schiff-Base (Iminosäure) – wie bei Transaminierung • mitochondriales Enzym hohe hoheKonzentration Konzentrationder derGLDH GLDHin inder derLeber Leber(Æ (ÆDiagnostik) Diagnostik) Hyperaktive GLDH führt zu Hypoglykämischer Hyperinsulinämie Mutationen im GLDH-Gen: „gain-of-function“: erhöhter Umsatz von Glu keine Hemmung durch ATP/GTP ↓ a) ATP-Synthese = ATP/ADP-Quotient steigt ↓ Insulin-Ausschüttung ↓ Blutglucosespiegel sinkt b) erhöhter NH3-Spiegel (Hyperammonämie) Die wichtigsten NH3 liefernden Reaktionen • oxidative Desaminierung (GLDH) • andere Aminosäure-Desaminierungen z.B. basische AS, α-,ß-Eliminierung, Lyasen • Aminoxidasen (Mono-, Diaminoxidasen = MAO, DAO) • Transaminierung von Glutamin Q: Q + α-KS + H20 Æ α-KG + AS + NH4+ • Purinnucleotid-Desaminasen • Pyrimidinabbau: Pyrimidinabbau (d)CMP-Desaminase, Ringspaltungen U, T molekulare molekulareUrsache Ursachefür fürNeurotoxizität Neurotoxizitätvon vonNH NH33:: Hemmung Hemmungdes desCitratcyclus Citratcyclusdurch durchα-KG-Mangel α-KG-Mangelim imGehirn. Gehirn. Ammoniak NH3 • ~ 25% des im Körper gebildeten Ammoniaks entsteht im Darm. • bei normalem Blut-pH (7.4): 98% NH4+ und 2% NH3: NH3 + H20 Æ NH4+ + OH• NH3 toxisch: freie Diffusion durch Nervenzellmembran • in Muskulatur wird NH3 z.T. aufgenommen Î nur die arterielle Ammoniakkonzentration korreliert mit der im ZNS, nicht die venöse! • Entgiftung & Ausscheidung als Harnstoff Ammoniak-Entgiftung I • Vermeidung der NH3 -Freisetzung: = Übertragung von Aminogruppen auf Ketoverbindungen durch: Aspartatzyklus Transaminierungen: Transaminierungen - Aminogruppen auf α-KG übertragen Aspartatzyklus: Aspartatzyklus - ist verknüpft mit Harnstoff-, AMP- und Purinbiosynthese - Aminogruppen auf Citrullin bzw. Nukleotide (X) übertragen Ammoniak-Entgiftung II • kovalente Fixierung von NH3 durch: • GLDH (mitochondrial) Æ Rückreaktion; zentrale Rolle des Glutamats ABER: Glutamat kann BlutHirn-Schranke nicht passieren! • Glutaminsynthetase (ebenfalls mitochondrial) • Harnstoffzyklus Æ Harnstoff aus der Leber in die Niere Æ Ausscheidung mit Urin Periphere Gewebe reichen Stickstoff an Leber extrahepatische Zellen: Zellen Transaminierungen, GLDH Æ Glutamat Æ Glutamin-Synthetase (besonders Gehirn): Gehirn Glutamin Glutaminist istDER DER Transporter Transportervon von Amino-Stickstoff Amino-Stickstoff zu zuLeber, Leber,Niere Niere Muskelzellen nutzen außerdem die Alanin-Aminotransferase Rückreaktion, um Alanin aus Pyruvat herzustellen: Alanin Alaninist istdie die AS ASmit mitder der zweithöchsten zweithöchsten Plasmakonz. Plasmakonz. Glukose-Alanin-Zyklus: Zusammenspiel von Muskel und Leber • Glukose-Alanin-Zyklus hat besondere Bedeutung im arbeitenden Muskel • Gln hauptsächlich aus Asp und Glu erzeugt = Transporter für Kohlenstoffgerüste aus der Proteolyse • Niere: Glutaminase I, II Æ NH4+ im Urin Entgiftung des NH3 im Harnstoffzyklus • 0.5–1.5 mol (30-90 g) Harnstoff / Tag • Fixierung: NH3 + CO2 • CO2 diffundiert ins Mito Æ Carbo-Anhydrase (CA) Æ HCO3• Ornithin/Citrullin Antiport KrebsHenseleitZyklus • N-Acetylglutamat = allosterischer Aktivator (essentiell; Schnellregulation) • Bilanz: NH4+ + HCO3- + 3 ATP + Asp + 2 H2O Harnstoff + 2 ADP + AMP + Pi + Fumarat Zusammenspiel des Harnstoffzyklus mit anderen Stoffwechselwegen GLDH E Æ α-KG + NH3 (Glutaminase QÆE + NH3) = NADH-liefernde Reaktionen Störungen des Harnstoffzyklus Symptome Hyperammonämie Enzephalopathie respiratorische Alkalose Ursachen • genetische Enzymdefekte: Inzidenz ca. 1:25.000, früher: Stickstoffreduktionskost, Zufuhr von α-Ketosäuren • erworbene Lebererkrankungen wie Leberzirrhose • akutes Leberversagen z.B. durch Knollenblätterpilz- oder Paracetamolvergiftung. Enzymdefekte im Harnstoffzyklus OTC-Mangel: Mangel Carbamoyl-P diffundiert ins Cytoplasma → Pyrimidinsynthese ↑ → Orotacidurie (Diagnose) Krankheit Hyperammonämie I (kongenitale Ammoniak-Intox.) Hyperammonämie II Citrullinämie Argininbernsteinsäurek. (Argininosuccinaturie) Hyperargininämie N-AcetylglutamatSynthasemangel Defektes Enzym CPS I OTC ArgininosuccinatSynthetase (AsS) ArgininosuccinatLyase (AsL) Arginase (R-ase) AsL R-ase AsS OTC CPS I Folge StickstoffAnhäufung als Glycin und Glutamin StickstoffAnhäufung als Citrullin Ornithin fehlt (ArgininosuccinatAusscheidung) Arginin angehäuft, Orn fehlt N-Acetyl-glutamat- CPS I inaktiv Synthetase (allosterisch) Behandlung proteinarme Diät mit Benzoat + Phenylacetat Æ Konjugation von Glycin und Glutamin Æ Ausscheidung proteinarme Diät mit viel Arg Æ OrnSynthese steigern Diät ohne Arginin siehe Hyperammonämie I Einschränkung des Harnstoffzyklus durch Leberinsuffizienz gesund z.B. durch Infektion, Alkohol, Medikamente Æ Leberfunktion auf 10 – 20 % eingeschränkt häufig Fibrosen Æ Bluthochdruck der Pfortader (portale Hypertens.) Bildung von Umgehungskreisläufen (Anastomosen) Ammoniakspiegel steigt hepatische Enzephalopathie (Ammoniakvergiftung des Gehirns) Coma hepaticum Insuffizienz ! Diagnostik von Lebererkrankungen Enzyme des Aminosäurestoffwechsels: Messung der Plasmakonzentrationen Störungen des Gallengangsystems: • γ-Glutamyl-Transpeptidase (γ-GT) – epitheliales Enzym erhöhte Plasmakonzentration Leberschäden: • Aminotransferasen (Transaminasen): Isoenzyme in Cytosol und Mitochondrien normale höchste Aktivitäten: Alanin-Aminotransferase (ALT) im Cytosol, Aspartat-Aminotransferase (AST) v.a. im Mito • Glutamat-Dehydrogenase (GLDH) im Mitochondrium • Diagnostik: AST/ALT < 1: normal, aber auch bei Entzündung leichte Leberschäden: Anstieg ALT schwere Leberschäden: AST und GLDH Harnstoff, NH3 und Säure-Basen-Haushalt • Fixierung von NH4+ und HCO3- im Harnstoffzyklus gekoppelt • ACIDOSE: Protonenüberschuss im Blut (pH < 7.37) Leber: weniger Hydrogencarbonat HCO3- Æ Harnstoffsynthese ↓; NH4+ : in Glutamin (Gln) fixiert Niere: aus Glutamin Æ NH4+ freigesetzt Æ Ausscheidung • ALKALOSE: Protonenmangel im Blut (pH > 7.44) Leber: mehr HCO3- Æ NH4+ benötigt: Hydrolyse von Gln Æ Harnstoffsynthese↑; Niere: Glukoneogenese gestoppt (keine Gln-Desaminierung) Æ keine Kopplung von H+ an NH3 Æ Protoneneinsparung Niere Zusammenfassung Stationen der Ammoniakentsorgung: Gewebe: Fixierung von NH3 in Glu und Gln Leber: Harnstoffzyklus Niere: Freisetzung von NH3 aus Gln, Ausscheidung von Harnstoff (Urea) und NH4+ Aminosäureabbau: 2. Kohlenstoffgerüste Die 20 Standard-AS werden über 7 Moleküle in den CC eingespeist: CitratCyclus Hämsynthese Fettsäuren Abbau essentieller und bedingt essentieller AS nach Entfernung der Amino (NH2-)-Gruppe Æ dehydrierende Decarboxylierung Amino- glucogenes ketogenes Bemerkungen säure Abbauprodukt Abbauprodukt Lysin 2x Acetyl-CoA irreversible Transaminierung Methio- Succinyl-CoA abhängig von Vitamin B6, B12 nin Cys Æ Pyruvat und Folsäure ThreoSuccinyl-CoA Acetaldehyd Æ 2 Wege: α, ß-Eliminierung nin Gly Æ Æ Pyruvat Acetyl-CoA oder Aldolspaltung Isoleucin Succinyl-CoA Acetyl-CoA Valin Succinyl-CoA Leucin Acetyl-CoA, über ß-Hydroxy-ß-methylAcetoacetat glutaryl-CoA (HMG-CoA) Æ Cholesterolsynthese Phenyl- Tyrosin ÆFumarat Acetoacetat Tetrahydrobiopterin-, alanin Ascorbinsäure-abhängig Tyrosin Fumarat Acetoacetat Trp Alanin Æ Pyruvat 2x Acetyl-CoA PLP-abhängig Histidin α-Ketoglutarat PLP-abhängig Cystein Pyruvat - * * *historisch: historisch:Bildung Bildungvon vonGlukose Glukosebzw. bzw.Acetoacetat Acetoacetatim imdiabetischen diabetischenTier Tier Methionin: abhängig von 3 Vitaminen Remethylierung (Vit. B12) Æ Mangel durch N20 (Narkose): Synthase inaktiv neurologische Schäden ==SAM; SAM; AdoMet; AdoMet; aktives aktives Methionin Methionin Transmethylierung (Folsäure) 5 α-Ketobutyrat + Cystein PropionylPropionylCoA CoA Pyruvat Pyruvat 5 = Transsulfurierung mit PALP (aus Vit. B6) Enzymdefekte Æ Homocystinurie bzw. Cystathionurie 2 Wege für den Abbau von Threonin Aldolspaltung α-,ß-Eliminierung α-Ketobutyrat PropionylCoA Essentielle AS der Aspartat- und Pyruvatfamilie Bedeutung • essentielle AS: Aspartatfam. Æ Lys, Met, Thr; Pyruvatfam. Æ Val, Ile, Leu • Lysin: mögliche Vorstufe von Carnitin (mito. Fettsäure-Carrier) Hydroxy-Lysin: Bestandteil von Kollagen • Methionin: S-Adenosylmet = ‚aktives Methyl‘: wichtigster Methylgruppendonator für N- oder O-Methylierung (C1Übertragung; z.B. Bildung von Kreatin) • Pathobiochemie: Pathobiochemie Homocystinurie (beim Met-Abbau): Endothelschädigung, Arteriosklerose Acidose durch gestörten Abbau von Propionyl-CoA (Abbau von Met, Thr, Val, Ile) Abbau verzweigtkettiger Aminosäuren (Pyruvat-Familie) • verzweigtkettige AS (Val, Ile, Leu) v.a. in Muskel (Skelett, Herz) und Niere, auch im Gehirn abgebaut • die anderen essentiellen AS in der Leber • dehydrierende Decarboxylierung zu Fettsäure-CoA-Thioestern Threonine Propionyl-CoA-Carboxylase Biotin-abhängig Enzymdefekt/Biotinmangel: Propionylacidämie L-Methylmalonyl-CoA-Isomerase Vit. B12 (Cobalamin)-abhängig Enzymdefekt/Vit. B12-Mangel: Methylmalonacidämie Verzweigtketten- (Ahornsirup-) krankheit Transaminierung dehydrierende Decarboxylierung Enzymdefekt Enzymdefektinindehydrierender dehydrierenderDecarboxylierung Decarboxylierung zentralnervöse zentralnervöseStörungen, Störungen,Azidose Azidose Körperflüssigkeiten Körperflüssigkeitenund undAtemluft: Atemluft:würzig-süßlicher würzig-süßlicherGeruch Geruch sofortige sofortigeDiät: Diät:arm arman anVal, Val,Ile, Ile,Leu Leu(sind (sindessentiell!) essentiell!) Aromatenfamilie I: Abbau von Phenylalanin Phenylalanin-Hydroxylase Hydroxylierung Transaminierung 1. Hydroxylierungen, Biopterin-abhängig (Tyrosin = Abbauprodukt von Phenylalanin) 2. oxidative Ringspaltung Hydroxylierung Decarboxylierung (Vit. C abhängig) ox. Ringspaltung Isomerisierung Aromatenfamilie II: Bedeutung v. Phe und Tyr ••Vorstufen Vorstufenvon vonPigmenten Pigmenten(Melanin; (Melanin;Synthesestörung: Synthesestörung: Albinismus), Albinismus),Neurotransmittern Neurotransmittern(Noradrenalin) (Noradrenalin) und und Hormonen Hormonen(Adrenalin, (Adrenalin,Thyroxin) Thyroxin) Phenylketonurie (PKU): • häufigste Störung des AS-Stoffwechsels • 1:10.000 (heterozygot: 1:50!) • > 240 Mutationen der Phenylalanin-Hydroxylase • Phe-Abbau auf alternativen Wegen Æ (neuro)tox. Produkte • frühe Diagnose! • Phe-arme Diät bis mindestens 10 Jahre Aromatenfamilie III: Tryptophan Tryptophan = Indolyl-Alanin Abbau: Biopterin-abhängig 1. oxidative Spaltung der Ringe 2. Abtrennung Ala-Seitenkette Æ Alanin + Acetoacetat Tryptophan Tryptophan •• verstärkt verstärktProteinbiosynthese Proteinbiosyntheseininder derLeber Leber(ist (istdort dortnormalerweise normalerweisedie dieam am geringsten geringstenkonzentrierte konzentrierteAS) AS) •• Provitamin Provitaminfür fürNicotinsäure-Synthese Nicotinsäure-Synthese(Æ (ÆNAD(P)) NAD(P))Æ Æ Pellagra Pellagra==kombinierter kombinierterTryptophanTryptophan-und undVitaminmangel Vitaminmangel(Niacin) (Niacin) •• Vorstufe Vorstufebiogener biogenerAmine: Amine:Serotonin Serotonin(5-Hydroxytryptamin), (5-Hydroxytryptamin),Melatonin Melatonin (Epiphysenhormon), (Epiphysenhormon),Tryptamin Tryptamin Histidin Stickstoff #1 als NH3 abgespalten (Histidase) Stickstoff #2 als Formiminogruppe auf Tetrahydrofolat übertragen PALP-abhängige Decarboxylierung Æ biogenes Amin Histamin: Æ Freisetzung NO in Gefäßendothel Æ Relaxation glatter Gefäßmuskeln allergische Reaktion! 3-Methyl-Histidin 3-Methyl-HistidinininActin, Actin,Myosin Myosin Æ Æim imUrin: Urin:Indikator Indikatorfür fürMuskelprotein-Abbau Muskelprotein-Abbau Cystein (Serinfamilie) im Extrazellularraum als Cystin (oxidierende Umgebung) : Abbau durch α-,ß-Eliminierung: Disulfid [Aminoacrylat] • Vorstufe von Taurin (Aminoethylsulfonsäure) Æ Konjugation von Gallensäuren (Taurocholsäure) • Cystinurie Æ AS-Transporterstörung (rBAT-Gen), betrifft auch Lys, Arg, Orn (Diaminocarbonsäuren), ebenfalls vermehrte Ausscheidung von Homocystein Abbau nicht- und semiessentieller AS Aminosäure Arginin glucogenes Abbauprodukt Ornithin Æ Glu Æ α-Ketoglutarat Prolin Glu Æ α-Ketoglutarat Glutamin Glu Æ α-Ketoglutarat ketogenes Produkt - Glutamat - α-Ketoglutarat - Asparagin Asp Æ Oxalacetat Aspartat Glycin Alanin Serin Oxalacetat, Fumarat Pyruvat Pyruvat (Glycin) Æ Pyruvat - Bemerkungen über Glutamat-Semialdehyd über Glutamat-semialdehyd Aminogruppendonator; Glutaminase Zentralsubstanz im ASStoffwechsel, Neurotransmitter! Aminogruppendonator; Asparaginase wichtigste glucogene AS •• liefern liefernletztlich letztlichalle alleOxalacetat Oxalacetat(C4-Gerüst) (C4-Gerüst)Æ Æ glucogen glucogen •• aber: PyruvatÆ ÆAcetylCoA AcetylCoAÆ ÆÆ ÆFettsäuren Fettsäuren aber:Pyruvat •• Abbau Abbauin infast fastallen allenGeweben Geweben Bedeutung nichtessentieller Aminosäuren • Aspartat/Asparagin, Glutamat/Glutamin Æ Transaminierungen... • Prolin: Prolin als Hydroxyprolin (Hyp) in Kollagen (ca. 25 %) Hyp im Urin = Maß für Kollagen-Stoffwechsel • Arginin: Arginin - Freisetzung von NO (+ Citrullin) durch NO-Synthase Å siehe auch Histamin! - Vorstufe für Polyamine (Spermin, Spermidin) Æ stabilisieren DNA - Ausgangsstoff für Kreatinsynthese (Kreatinphosphat = Energiereserve im Muskel) • Glycin: Glycin Biosynthese von Häm (+ Succinyl-CoA) und Purinen • Serin: Serin - Vorstufe für Purinbasen, Phospholipide, Cystein - Hydroxymethylgruppe (C1-Verbindung) kann auf Tetrahydrofolsäure übertragen werden • Alanin: Alanin D- und L-Form in Murein-Zellwand der Bakterien C1-Verbindungen im AS-Stoffwechsel Biotin = Vit. H Harnstoffderivat bindet CO2 an Position N1, überträgt es auf Pyruvat, Acetyl-CoA, Propionyl-CoA C1-Einheiten C1-Einheitenwerden werdengeliefert geliefertvon: von: Ado-Met) Methionin Met Æ Æaktiviert aktiviert((Ado-Met) Æüberträgt überträgtMethyl Methyl(-CH (-CH33))auf aufHistidin, Histidin,DNA; DNA; MethioninÆ bei beiSynthese Synthesevon vonKreatin, Kreatin,Adrenalin, Adrenalin,Cholin Cholin Glycin Æ Methylen Methylen(-CH (-CH22-)-) GlycinÆ Übertragung Serin Æ Methyl Methyl Übertragungauf aufTetrahydrofolat Tetrahydrofolat SerinÆ Histidin Æ Formimino Formimino HistidinÆ (-CH=NH) Synthese (-CH=NH) Synthesevon vonMethionin, Methionin,Thyminnucleotiden Thyminnucleotiden Der vollständige Abbau von AS ... ...zu zuCO CO22++HH2200++NH NH33liefert liefertEnergie Energie(ca. (ca.17 17kJ/g) kJ/g) essentielle AS nichtessentielle AS Zahlenwerte = ATP-Äquivalente Glucose: Stearinsäure (C18): Ethanol: 38 ATP-Äquivalente (17 kJ/g) 148 ATP-Äquivalente (39 kJ/g) 114 ATP-Äquivalente (30 kJ/g) Zusammenfassung: AS im Stoffwechsel ¾Synthese ¾Synthesevon vonKörperprotein: Körperprotein:Verfügbarkeit Verfügbarkeitessentieller essentieller AS! AS! ¾Energie ¾Energieliefernde lieferndeSubstrate: Substrate: tgl. tgl.Umsatz Umsatz4.4g/kg 4.4g/kgbzw bzw37 37mmol/kg mmol/kgübertrifft übertrifft FettFett-bzw. bzw.Kohlenhydratumsatz; Kohlenhydratumsatz; AS ASam amEnergiegewinn Energiegewinnmit mit15-20% 15-20%beteiligt beteiligt ¾glucogene ¾glucogeneAS ASÆ ÆGlucosehomöostase Glucosehomöostase ¾Quelle ¾Quellefür fürStickstoff Stickstoffund undorganischen organischenSchwefel Schwefel für fürBiosynthesen Biosynthesen ¾zellulärer ¾zellulärerAS-Pool AS-Poolgespeist gespeistaus ausendogenem endogenemProteinabbau Proteinabbau und undAS ASaus ausNahrung Nahrung Aminosäure-Stoffwechsel unter verschiedenen äußeren Bedingungen • myotrop (anabol): AS von Leber → Muskel; positive N2-Bilanz durch: Insulin (AS-Transportsysteme!), Testosteron, Somatotropin; Æ mTOR bewirkt Anschalten der Ribosomenbiogenese und Translation • hepatotrop (negative Stickstoff-Bilanz): Proteinabbau zuerst in Gastrointestinaltrakt, Leber, Niere, Pankreas Muskel (größtes Organ) verliert höchste Absolutmenge an Protein durch Aktivierung von Calpainen und Proteasom ↓ Nahrungskarenz = Glukose fehlt; Acetyl-CoA Æ glucogene Substanz; Lactat, Pyruvat Æ keine Netto-Neusynthese von Glukose Î Proteinabbau: Alanin Transport Muskel Æ Leber: Glukoneogenese Glycin in die Niere Æ Guconeogenese ÆÆ Hypoalaninämie (Muskelabbau reduziert) Verlust von > 50% der Muskelmasse Æ Tod • Stress, Infektionen, Verletzungen, Tumoren, AIDS Æ höhere Stickstoffausscheidung (Verlust von Körperprotein) Æ Æ Kachexie Proteinqualität – Biologische Wertigkeit • Fehlen einer essentiellen AS Æ Proteinbiosynthese • unterschiedliche Verdaubarkeit (%) Muttermilch 95 Kartoffel 71 Ei Fisch Rindfleisch Kuhmilch Gelatine 87-97 83 76-93 81-90 25 Soja Hefe Hafer Reis Mais Weizen 64 63 62 57-63 36-54 30-52 pflanzliche pflanzlicheProteine Proteine––im immenschlichen menschlichenOrganismus Organismusoft oftunvollständig unvollständig hydrolysiert hydrolysiert Æ Ægemischte gemischteMahlzeit: Mahlzeit:essentielle essentielleAS ASergänzen: ergänzen: Getreide Getreide(arm (arman anLys, Lys,ausreichend ausreichendTrp) Trp) ++Bohnen Bohnen(arm (arman anTrp, Trp,ausreichend ausreichendLys) Lys) Protein-Energie-Malnutrition (PEM) Kwashiorkor: „Entwöhnungskrankheit“ zu wenig Protein: essentielle AS fehlen Hypoalbuminämie („Wasserbauch“) Marasmus (grch.: verwelken) zu wenig Protein Hypoalbuminämie (Ödeme) generell zu wenig Energiezufuhr