Bedeutung von Tumorantigenen bei Hauttumorpatienten
Werbung

Bedeutung von Tumorantigenen bei Hauttumorpatienten Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften (Dr. rer. nat.) Fakultät Naturwissenschaften Universität Hohenheim Institut für Ernährungsmedizin, Fachgebiet Ernährungsphysiologie und Genderforschung Klinische Kooperationseinheit für Dermato-Onkologie, Deutsches Krebsforschungszentrum vorgelegt von Nicole Wiedemann geb. Gottstein aus Pforzheim 2008 Dekan: Prof. Dr. rer. nat. Heinz Breer 1. berichtende Person: Prof. Dr. rer. nat. Stefan Eichmüller 2. berichtende Person: Prof. Dr. rer. nat. Christiane Bode Eingereicht am: 25.06.2008 Mündliche Prüfung am: 27.11.2008 Die vorliegende Arbeit wurde am 23.09.2008 von der Fakultät Naturwissenschaften der Universität Hohenheim als „Dissertation zur Erlangung des Doktorgrades der Naturwissenschaften“ angenommen. Was lange währt, wird endlich gut Die vorliegende Arbeit wurde am Deutschen Krebsforschungszentrum (DKFZ) Heidelberg in der Abteilung „Klinische Kooperationseinheit für Dermato-Onkologie“ von Prof. Dr. med. Dirk Schadendorf in der Arbeitsgruppe Tumorantigene unter der Betreuung von Prof. Dr. rer. nat. Stefan Eichmüller angefertigt. DANKSAGUNG An dieser Stelle möchte ich mich von Herzen bei allen bedanken, die zum Gelingen dieser Arbeit beigetragen haben: Herrn Prof. Dr. Stefan Eichmüller möchte ich sehr für die Überlassung des interessanten Themas, die hervorragende Betreuung dieser Arbeit, die vielen hilfreichen Diskussionen und die optimalen Arbeitsbedingungen danken. Ich habe mich in seiner Arbeitsgruppe sehr wohl gefühlt und viele hilfreiche Anregungen erhalten. Im Besonderen schätze ich seinen Einsatz für seine Mitarbeiter, der mir die Teilnahme an einem internationalen Immuntherapie Workshop sowie an verschiedenen nationalen und internationalen Kongressen ermöglichte. Bei Frau Prof. Dr. Christiane Bode möchte ich mich herzlich für die Betreuung dieser Arbeit von Seiten der Universität Hohenheim bedanken sowie für ihr immerwährendes Interesse an diesem Thema. Ihre jährlichen Besuche am DKFZ in Verbindung mit unseren Treffen sowie die Möglichkeit, in ihrer Arbeitsgruppe meine Fortschritte vorstellen zu können, stellten wichtige Elemente der Entstehung dieser Arbeit dar. Herrn Prof. Dr. Dirk Schadendorf danke ich für die freundliche Aufnahme in die Abteilung sowie die hilfreichen Diskussionen und die Möglichkeit der Vorstellung der Ergebnisse meiner Arbeit. Mein besonderer Dank gilt weiterhin Dr. Friederike Fellenberg für die Einführung in die Methoden zu Beginn der Arbeit sowie die herzliche Aufnahme in die Arbeitsgruppe und Dr. Sascha Bazhin für die Weitergabe seines immensen Wissens hinsichtlich Proteinaufreinigung, seiner kontinuierlichen fachlichen Unterstützung und der Durchsicht des Manuskripts. Für die freundliche und kompetente Unterstützung hinsichtlich Multiplex-Serologie und Auswertung derselben danke ich Dr. Tim Waterboer, Dr. Peter Sehr und Dr. Michael Pawlita. Des Weiteren gilt mein Dank Fr. Dr. Kopp-Schneider für die kompetente Beratung bei der statistischen Auswertung der Ergebnisse. Für die Bereitstellung der HPLC-Anlage und Anleitung bei der Proteinaufreinigung danke ich Jürgen Kretschmer und Prof. Dr. Ponstingl. Ein großer Dank gebührt ebenfalls allen Blut- und Gewebespendern. Dem gesamten Dermato-Onkologie-Team danke ich für eine sehr schöne und lustige Zeit sowie die angenehme und hilfsbereite Atmosphäre. Während dieser abwechslungsreichen Zeit durfte ich viele motivierende Kaffeepausen und Kochabende erleben. Elke Dickes, Anita Dahlke und Judith Bartels möchte ich für die tatkräftige Unterstützung im Labor und viel Humor danken. Dr. Tanja Hartmann, Dr. Claudia Kistler und Dr. Mieun Lee danke ich für hilfreiche Diskussionen und Anregungen sowie kurzweilige Arbeitstage. Bei Dr. Hanno Ehlken, Eva Mattern, Dr. Dirk Usener, Nadine Willner, Andreas Lange und Brigitte Geschwendt bedanke ich mich für eine schöne gemeinsame Zeit. Nicht vergessen werde ich die liebe- und verständnisvolle Unterstützung meiner Eltern, meiner gesamten Familie und all meiner Freunde während der Anfertigung dieser Arbeit. Ohne die Ermöglichung des Studiums der Ernähungswissenschaften durch meine Eltern und ihren Rückhalt wäre diese Arbeit nicht möglich gewesen. Mein innigster Dank geht an meinen Mann Frank, der während dieser schönen und stürmischen Zeit immer mit viel Geduld, Motivation und Liebe an meiner Seite stand und mich stets ertragen und unterstützt hat. PUBLIKATIONEN Artikel Hartmann TB, Mattern E, Wiedemann N, van Doorn R, Willemze R, Niikura T, Hildenbrand R, Schadendorf D, Eichmüller SB. 2008. Identification of selectively expressed genes and antigens in CTCL. Exp Dermatol, 17(4):324-34. Bazhin A, Wiedemann N, Schnölzer M, Schadendorf D, Eichmüller SB. 2006. Expression of GAGE family proteins in malignant melanoma. Cancer Letters 251(2):258-67. Devitt G, Meyer C, Wiedemann N, Eichmüller S, Kopp-Schneider A, Haferkamp A, Hautmann R, Zöller M. 2005. Serological analysis of human renal cell carcinoma. Int J Cancer, 118(9):2210-9. Gottstein N, Ewins BA, Eccleston C, Hubbard GP, Kavanagh IC, Minihane AM, Weinberg PD, Rimbach G. 2003. Effect of genistein and daidzein on platelet aggregation and monocyte and endothelial function. Br J Nutr 89:607-615. Bingham M, Gibson G, Gottstein N, de Pascual-Teresa S, Minihane AM, Rimbach GH. 2003. Gut metabolism and cardioprotective effects of dietary isoflavones. Curr Top Nutraceut Res 1(1):31-48. Tagungsbeiträge Wiedemann N, Bazhin AV, Schadendorf D, Eichmüller SB. 2005. GAGE protein expression in malignant melanoma. The 6th World Congress on Melanoma 2005:200. Wiedemann N, Bazhin AV, Schadendorf D, Eichmüller SB. 2005. A new polyclonal antibody reveals GAGE protein expression in malignant melanoma CIMT 2005. Wiedemann N, van Doorn R, Willemze R, Schadendorf D, Eichmüller SB. 2005. SADA - a serum antibody detection array for analyzing the humoral immune response against tumor antigens. International Symposium on the Biology and Immunology of Cutaneous Lymphoma 2005. Gottstein N, van Doorn R, Willemze R, Schadendorf D, Eichmüller S. 2004. Analyse der humoralen Immunantwort gegen Tumorantigene mittels Serum-Antikörper Detektions Arrays. Akt Dermatol 08/09:368. Gottstein N, Schadendorf D, Eichmüller S. 2004. The humoral immune response against tumor antigens analyzed by serum antibody detection arrays. DKFZ Graduate Forum 2004; unpublished. VERZEICHNISSE i INHALTSVERZEICHNIS INHALTSVERZEICHNIS........................................................................................................... I ABBILDUNGSVERZEICHNIS ............................................................................................... VI TABELLENVERZEICHNIS ................................................................................................. VIII ABKÜRZUNGSVERZEICHNIS.............................................................................................. IX 1 EINLEITUNG ..........................................................................................................1 1.1 IMMUNÜBERWACHUNG IN DER HAUT ..........................................................................1 1.2 IMMUNÜBERWACHUNG VON TUMOREN .......................................................................2 1.3 DAS KUTANE T-ZELL LYMPHOM .................................................................................4 1.3.1 Klassifikation...............................................................................................................5 Mycosis fungoides ...................................................................................................................5 Sézary Syndrom .......................................................................................................................6 1.3.2 Stadieneinteilung.........................................................................................................6 1.3.3 Pathogenese ................................................................................................................7 1.3.4 Epidermotropismus .....................................................................................................8 1.3.5 Diagnose und Prognose ..............................................................................................9 1.3.6 Therapieformen .........................................................................................................10 1.4 DAS MALIGNE MELANOM ..........................................................................................12 1.4.1 Stadieneinteilung.......................................................................................................13 1.4.2 Pathogenese ..............................................................................................................14 1.4.3 Diagnose und Prognose ............................................................................................16 1.4.4 Therapie ....................................................................................................................18 1.5 TUMORANTIGENE ......................................................................................................19 1.5.1 Klassen von Tumorantigenen ....................................................................................20 1.5.2 Identifizierung von Tumorantigenen .........................................................................24 1.6 2 ZIEL DIESER ARBEIT ..................................................................................................26 MATERIAL UND METHODEN .........................................................................27 MATERIAL ..............................................................................................................................27 2.1 CHEMIKALIEN ...........................................................................................................27 2.2 GERÄTE, VERBRAUCHSMATERIALIEN UND FERTIGE KITS..........................................30 2.3 PUFFER UND LÖSUNGEN ............................................................................................33 2.3.1 Bakterienstämme .......................................................................................................35 2.3.2 Agar und Medien .......................................................................................................35 VERZEICHNISSE ii 2.3.3 Vektoren ....................................................................................................................36 2.3.4 Plasmide ....................................................................................................................36 2.3.5 Seren..........................................................................................................................36 2.3.6 Antikörper..................................................................................................................37 2.3.7 Zelllinien....................................................................................................................38 2.4 SOFTWARE ................................................................................................................38 METHODEN ............................................................................................................................39 2.5 SADA .......................................................................................................................39 2.5.1 Auswahl der Antigene................................................................................................39 2.5.2 Präabsorption der Patientenseren ............................................................................40 2.5.3 Transduktion..............................................................................................................43 2.5.4 Blotting und immunologischer Screen ......................................................................44 2.5.5 In vivo Exzision .........................................................................................................46 2.5.6 Plasmid-DNA-Isolierung...........................................................................................47 2.5.7 Spaltung der Phagemide ...........................................................................................47 2.5.8 Agarose-Gelelektrophorese.......................................................................................47 2.6 ELISA UND MULTIPLEX-SEROLOGIE ........................................................................48 2.6.1 DNA-Klonierungen....................................................................................................48 2.6.2 Proteinexpression der Antigene in E. coli.................................................................53 2.6.3 SDS-PAGE und Western Blot....................................................................................54 2.6.4 Antigen-Titration im GST-X-tag-ELISA....................................................................56 2.6.5 Methodik der Multiplex-Serologie ............................................................................57 2.7 EXPRESSIONSANALYSE VON GAGE-7B AUF RNA- UND PROTEINEBENE ..................60 2.7.1 Isolierung von RNA ...................................................................................................60 2.7.2 RNA-Gelelektrophorese ............................................................................................60 2.7.3 Herstellung von cDNA ..............................................................................................61 2.7.4 RT-PCR .....................................................................................................................62 2.7.5 RNA-Proben ..............................................................................................................63 2.7.6 Herstellung von Dialyseschläuchen ..........................................................................63 2.7.7 Rekombinante Expression von GAGE-7b als GST-Fusionsprotein ..........................63 2.7.8 Aufschluss der Bakterien ...........................................................................................63 2.7.9 Aufreinigung eines GST-Fusionsproteins an Glutathion Sepharose-Beads .............64 2.7.10 Abspaltung des GST vom Fusionsprotein .................................................................65 2.7.11 Ionenaustauschchromatographie ..............................................................................65 2.7.12 Umpufferung von Proteinlösungen ...........................................................................66 VERZEICHNISSE iii 2.7.13 Matrix Assisted Laser Desorption/Ionisation-Time of Flight (MALDI-TOF) ..........67 2.7.14 Immunisierung von Kaninchen..................................................................................67 2.7.15 ELISA ........................................................................................................................68 2.7.16 Aufreinigung von Antikörpern aus Kaninchenantiserum..........................................69 2.7.17 Bestimmung von Antikörpern in Serum mittels Western Blot ...................................70 2.7.18 Western Blot ..............................................................................................................71 2.7.19 Immunhistologie ........................................................................................................71 2.7.20 Zellkultur ...................................................................................................................72 3 ERGEBNISSE ........................................................................................................74 3.1 SADA .......................................................................................................................75 3.1.1 Aufbau des SADA ......................................................................................................75 3.1.2 Eigenschaften der ausgewählten Antigene................................................................75 3.1.3 Validierung des SADA: Phagenmobilität und Monoklonalität .................................81 3.1.4 Validierung des SADA: Sensitivität...........................................................................82 3.1.5 Validierung des SADA: Reproduzierbarkeit .............................................................83 3.1.6 Seroreaktivitäten aller SADA-Antigene gegen CTCL-, MM-Patienten und Kontrollpersonen......................................................................................................84 3.2 ELISA UND MULTIPLEX-SEROLOGIE ........................................................................88 3.2.1 Die Grundlage der Multiplex-Serologie ...................................................................88 3.2.2 Verwendete Antigene in der Multiplex-Serologie .....................................................90 3.2.3 Qualitätskontrolle der Antigene und der Multiplex-Analyse ....................................91 3.2.4 Festlegung der Cut-off-Werte und statistische Auswertung......................................93 3.2.5 Untersuchung der Serumantikörperantwort gegen die verwendeten Antigene in einem MM-Kollektiv.................................................................................................95 3.2.6 Untersuchung der Serumantikörperantwort gegen die verwendeten Antigene in einem CTCL-Kollektiv............................................................................................107 3.2.7 3.3 Monitoring der Serumantikörperantwort in immunbehandelten CTCL-Patienten.119 EXPRESSIONSANALYSE DES CANCER TESTIS ANTIGENS GAGE-7B ........................125 3.3.1 Identifizierung von GAGE-7b und Homologie der GAGE-Familie ........................125 3.3.2 Expressionsanalyse von GAGE-7b mRNA ..............................................................125 3.3.3 Antikörper-Herstellung mittels GST-GAGE-7b-tag-Fusionsprotein ......................127 3.3.4 Evaluierung der optimalen Expressions- und Aufreinigungsbedingungen für das rekombinante GST-GAGE-7b-tag-Protein.............................................................128 VERZEICHNISSE 3.3.5 iv Aufreinigung des GST-GAGE-7b-tag-Proteins an Glutathion Sepharose und Abspaltung des GST-Fusionspartners mittels Thrombin .......................................130 3.3.6 Zusätzliche Aufreinigung des GAGE-7b-tag-Proteins mittels Anionenaustauschchromatographie (MonoQ) und Identifizierung von GAGE-7b mittels MALDI-TOF...........................................................................................................131 3.3.7 Immunisierung eines Kaninchens mit aufgereinigtem GAGE-7b-tag-Protein .......134 3.3.8 Isolierung der spezifischen GAGE-7b-Antikörper aus dem Antiserum ..................135 3.3.9 Spezifität des GAGE-7b-Antikörpers ......................................................................137 Western Blot ........................................................................................................................137 Immunzytologie ...................................................................................................................138 Multiplex-Serologie.............................................................................................................139 3.3.10 Expressionsanalyse des Proteins GAGE-7b ...........................................................140 Expression in Kontrollgewebe ............................................................................................140 Expression in Zelllinien und Tumorgewebe ........................................................................141 Zelluläre Lokalisation von GAGE-7b .................................................................................143 3.3.11 Serumantikörperantworten gegen GAGE-7b ..........................................................146 4 DISKUSSION .......................................................................................................147 4.1 VERWENDUNG VON TUMORANTIGENEN ..................................................................147 4.1.1 Prognose und Diagnose ..........................................................................................148 4.1.2 Immuntherapie ........................................................................................................148 4.2 SADA .....................................................................................................................151 4.2.1 Validierung des SADA.............................................................................................152 4.2.2 Untersuchung von humoralen Immunantworten mittels SADA ..............................153 Serumantikörperantworten in CTCL-Patienten ..................................................................155 HD-MM-48..........................................................................................................................155 HD-CL-02............................................................................................................................156 Serumantikörperantworten in MM-Patienten .....................................................................156 GBP-5a und GBP-5ta..........................................................................................................157 HD-MM-19..........................................................................................................................157 4.2.3 SADA als Methode zur Bestimmung der Antikörperantwort ..................................158 4.2.4 Vergleich von SADA und SEREX ............................................................................159 4.2.5 SADA - Fazit und Ausblick......................................................................................160 4.3 4.3.1 MULTIPLEX-SEROLOGIE ..........................................................................................161 Qualitätskontrolle und statistische Auswertung......................................................162 VERZEICHNISSE v 4.3.2 Humorale Immunantworten von MM-Patienten .....................................................164 4.3.3 Humorale Immunantworten von CTCL-Patienten ..................................................168 4.3.4 Humorale Immunantworten von immunbehandelten CTCL-Patienten...................172 4.3.5 Vergleich von Multiplex-Serologie und SADA/SEREX...........................................174 4.3.6 Multiplex-Serologie im Vergleich zu ELISA in der Krebsdiagnostik......................178 4.3.7 Multiplex-Serologie – Fazit und Ausblick...............................................................180 4.4 DAS CANCER TESTIS ANTIGEN GAGE-7B ..............................................................181 4.4.1 Spezifität des Antikörpers........................................................................................182 4.4.2 Expressionsanalyse von GAGE-7b..........................................................................183 4.4.3 Serologische Untersuchung von GAGE-7b.............................................................187 4.4.4 GAGE-7b als Ziel einer Immuntherapie .................................................................188 4.4.5 GAGE-7b – Fazit und Ausblick...............................................................................189 4.5 CTA UND TAA IN DER KREBSTHERAPIE .................................................................190 4.6 CTA UND TAA IN DER KREBSDIAGNOSTIK UND -PROGNOSTIK ...............................192 5 ZUSAMMENFASSUNG .....................................................................................195 6 SUMMARY...........................................................................................................196 7 LITERATURVERZEICHNIS ............................................................................197 8 ANHANG ..............................................................................................................229 LEBENSLAUF ..........................................................................................................................232 EIDESTATTLICHE ERKLÄRUNG ......................................................................................233 VERZEICHNISSE vi ABBILDUNGSVERZEICHNIS Abbildung 1-1: Die Schichten der Haut mit der Angabe der Schichtenfolge (Gratzl 2002)....1 Abbildung 1-2: Integrierte Immunantwort gegen Tumorantigene (modifiziert nach Sahin et al. 1999 und Banchereau et al. 2000) .............................................................3 Abbildung 1-3: Lokalisation von GAGE-7b (http://www.ncbi.nlm.nih.gov/entrez/) ............23 Abbildung 1-4: SEREX-Methode...........................................................................................25 Abbildung 2-1: Schema der SADA-Methode.........................................................................46 Abbildung 2-2: Programm der Klonierungs-PCR ..................................................................50 Abbildung 2-3: Programm der Sequenzier-PCR ....................................................................52 Abbildung 2-4: Programm der cDNA-Synthese.....................................................................61 Abbildung 2-5: Programm der GAPDH-PCR ........................................................................62 Abbildung 3-1: Gelelektrophoretische Auftrennung der mit Restriktionsenzymen gespaltenen Plasmide von vier SADA-Klonen nach einer in vivo Exzision ...82 Abbildung 3-2: Entwickelte SADA-Membranen verschiedener Serumverdünnungen..........83 Abbildung 3-3: Repräsentative SADA-Membranen verschiedener Seren .............................85 Abbildung 3-4: Schematische Darstellung von „GST-capture-ELISA“ (A) und MultiplexSerologie (B) ................................................................................................89 Abbildung 3-5: Schematische Darstellung des Luminex-Analysegeräts ...............................90 Abbildung 3-6: Titration der Antigene für die Multiplex-Serologie im GST-X-tag-ELISA .92 Abbildung 3-7: Darstellung der kumulativen Distribution des Kontroll- und MM-Kollektivs für MAGE-A1, A3 und A9 ..........................................................................97 Abbildung 3-8: Darstellung der kumulativen Distribution des Kontroll- und MM-Kollektivs für LAGE-1a, GAGE-7b und CAMEL ........................................................98 Abbildung 3-9: Darstellung der kumulativen Distribution des Kontroll- und MM-Kollektivs für se70-2, se57-2 und GBP-5ta...................................................................99 Abbildung 3-10: Darstellung der kumulativen Distribution des Kontroll- und MM-Kollektivs für cTAGE-5a N- und C-terminal sowie p53.............................................100 Abbildung 3-11: Darstellung der kumulativen Distribution des Kontroll- und MM-Kollektivs für p16 ........................................................................................................101 Abbildung 3-12: Vergleich der prozentualen Seroreaktivität der Kontrollen mit allen MMPatienten (A) und den verschiedenen MM-Stadiengruppen (B)................103 Abbildung 3-13: Monitoring der Serumantikörperantwort von acht MM-Patienten .............106 Abbildung 3-14: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für MAGE-A1, A3 und A9.......................................................110 Abbildung 3-15: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für LAGE-1a, GAGE-7b und CAMEL ....................................111 Abbildung 3-16: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für se70-2, se57-1 und GBP-5ta ...............................................112 Abbildung 3-17: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für cTAGE-5a N- und C-terminal sowie p53 ...........................113 VERZEICHNISSE vii Abbildung 3-18: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für p16.......................................................................................114 Abbildung 3-19: Vergleich der prozentualen Seroreaktivität der Kontrollen mit allen CTCLPatienten (A) und den verschiedenen CTCL-Stadiengruppen (B).............115 Abbildung 3-20: Monitoring der Serumantikörperantwort von zwölf CTCL-Patienten ........117 Abbildung 3-21: Darstellung der Serumantikörperantwort der 19 immunbehandelten CTCLPatienten im Verlauf der Therapie .............................................................121 Abbildung 3-22: Darstellung der Serumantikörperantwort der fünf mit einem Masernimpfstoff behandelten CTCL-Patienten im Verlauf der Therapie .............................123 Abbildung 3-23: Multiples Alignment der GAGE-Proteine...................................................126 Abbildung 3-24: Repräsentative RT-PCR mit beiden GAGE-Primerpaaren .........................126 Abbildung 3-25: Analyse der Antikörper K5101 und K5102 im Western Blot .....................128 Abbildung 3-26: Coomassie-Gel zur Bestimmung der optimalen Expressionsbedingungen für GST-GAGE-7b-tag ....................................................................................129 Abbildung 3-27: Coomassie-Gel zum Vergleich der Lyse von GST-tag-Protein in der French Press gegenüber Sonifizierung und Homogenisieren (A) und zur Bestimmung des optimalen pH-Werts während der Bindung von GSTGAGE-7b-tag-Protein an Glutathion Sepharose (B)..................................130 Abbildung 3-28: Coomassie-Gel zur Aufreinigung von GST-GAGE-7b-tag und Abspaltung des GST-Fusionspartners ...........................................................................131 Abbildung 3-29: Chromatogramm der Ionenaustauschchromatographie für GAGE-7b-tag .132 Abbildung 3-30: Coomassie-Gele der Proben und Fraktionen der Ionenaustauschchromatographie.........................................................................................133 Abbildung 3-31: Titration der Kaninchenseren vor und nach Immunisierung im ELISA .....135 Abbildung 3-32: Titration des Antiserums und der isolierten Antikörper nach den unterschiedlichen Aufreinigungsschritten..................................................136 Abbildung 3-33: Western Blot an verschiedenen GST-Fusionsproteinen unter Verwendung des monospezifischen GAGE-7b-Antikörpers...........................................138 Abbildung 3-34: Überprüfung der Spezifität des GAGE-7b-Antikörpers mittels Immunzytologie .........................................................................................139 Abbildung 3-35: Western Blot an Kontrollgewebe mit GAGE-7b-Antikörper .....................141 Abbildung 3-36: Western Blot an verschiedenen Zelllinien mit GAGE-7b-Antikörper........142 Abbildung 3-37: Western Blot an MM-Tumorgewebe mit GAGE-7b-Antikörper................143 Abbildung 3-38: Immunzytologische und –histologische Untersuchungen mit GAGE-7bAntikörper ..................................................................................................145 Abbildung 3-39: Serumantikörperantworten gegen GAGE-7b ermittelt mit Western Blot und DAB (A) sowie Chemilumineszenz (B).....................................................146 VERZEICHNISSE viii TABELLENVERZEICHNIS Tabelle 1-1: TNM-Stadieneinteilung für kutane Lymphome......................................................7 Tabelle 1-2: Stadiengruppierung der TNM-Einteilung ...............................................................7 Tabelle 1-3: Melanom TNM-Klassifikation..............................................................................13 Tabelle 1-4: Überarbeitete „American Joint Committee on Cancer“ (AJCC) klinische Stadieneinteilung des Melanoms...........................................................................13 Tabelle 2-1: Ausgewählte Antigene für die SADA-Methode ...................................................39 Tabelle 2-2: E. coli-Lysis-Ansatz zur Herstellung der lytischen Säulen...................................42 Tabelle 2-3: Zusammensetzung des Transduktionsansatzes .....................................................43 Tabelle 2-4: Anordnung der Phagen auf den SADA-Membranen 1 und 2 ...............................45 Tabelle 2-5: Primer für die Klonierung in den pGEX-4T-3tag-Vektor.....................................49 Tabelle 2-6: Klonierungs-PCR-Materialien und Pipettierschema.............................................50 Tabelle 2-7: Verwendete Primer in der Sequenzier-PCR..........................................................52 Tabelle 2-8: Zusammensetzung von Trenn- und Sammelgel für die SDS-PAGE ....................55 Tabelle 2-9: Pipettierschema für den cDNA-Synthese-Ansatz .................................................61 Tabelle 2-10: Reaktionsansatz für die RT-PCR ..........................................................................62 Tabelle 2-11: In der RT-PCR verwendete Primer .......................................................................63 Tabelle 2-12: Immunisierungsschema der Firma Biogenes ........................................................67 Tabelle 2-13: Im Zentralen Tierlabor des DKZF verwendetes Immunisierungsschema ............68 Tabelle 3-1: mRNA Expression und Eigenschaften der SADA-Antigene ...............................76 Tabelle 3-2: Seroreaktivitäten aller SADA-Antigene, statistische Auswertung und Ergebnisse im sekundären SEREX..........................................................................................86 Tabelle 3-3: Übersicht über die verwendeten humanen Tumorantigene...................................91 Tabelle 3-4: Cut-off-Werte für alle Antigene ...........................................................................94 Tabelle 3-5: Veränderung der Serumantikörperantwort gegen alle Antigene in 19 immunbehandelten CTCL-Patienten im Verlauf der Therapie ...........................120 Tabelle 3-6: Veränderung der Serumantikörperantwort gegen alle Antigene in fünf immunbehandelten CTCL-Patienten im Verlauf der Therapie ...........................122 Tabelle 3-7: Expressionsanalyse der beiden GAGE-Gruppen in Zelllinien und Geweben.....127 Tabelle 3-8: Verwendung des GAGE-7b-Antikörpers in der Multiplex-Serologie ................140 Tabelle 3-9: Zusammenfassung der Ergebnisse der Expressionsanalyse von GAGE-7b .......144 Tabelle 4-1 Vergleich der Seroreaktivitäten von Ctrl, MM- und CTCL-Patienten in der Multiplex-Serologie, im SADA und SEREX......................................................176 VERZEICHNISSE ix ABKÜRZUNGSVERZEICHNIS ABCDE-Regel ADCC ADP AFP AG AJCC ALM Amp AP AP-1 APC APS ART AS ATCC ATP mAU BAX BCIP BF-1 bp BPB BRCA1 BSA CaCl2 CBCL CBS-K CD CDA CDK4 CDKN2A CEA CGH CIAP CKAP2 CMV CRP CTA CTCL CTL CTLA Ctrl D Da DAB DC DDRT-PCR DKFZ Asymmetrie, Begrenzung, Colorit, Durchmesser, Erhabenheit-Regel Antikörperabhängige zellvermittelte Zytotoxizität (“antibody-dependent cell – mediated cytotoxicity“) Adenosindiphosphat Alpha-1-Fetoprotein Arbeitsgruppe „American Joint Committee on Cancer“ Akrolentiginöses Melanom Ampicillin Alkalische Phosphatase Aktivierungsprotein-1 („activator protein-1“) Antigen-präsentierende Zellen („antigen presenting cells“) Ammoniumperoxidsulfat Adenosindiphosphat-Ribosyltransferase Aminosäure „American Tissue and Culture Collection“ Adenosintriphosphat Milli Absorptionseinheiten („milli absorption units“) B-Zell Leukämie/Lymphom 2 (BCL2)-assoziiertes X Protein 5-Brom-4-chlor-3-indolyl-phosphat „Brain factor 1“ Basenpaare Bromphenolblau Brustkrebs 1 („Breast Cancer 1“) Rinderserumalbumin (“bovine serum albumin”) Calciumchlorid Kutanes B-Zell Lymphom Siperchemiclock Differenzierungscluster (“clusters of differentiation”) Zellteilung Autoantigen (“cell division autoantigen”) Zyklin-abhängige Kinase 4 („cyclin-dependent kinase 4“) Zyklin-abhängiger Kinaseinhibitor 2A (“cyclin-dependent kinase inhibitor 2A”) Karzinoembryonales Antigen („carcinoembryonic antigen“) “Comparative Genomic Hybridization“ “Calf Intestine Alkaline Phosphatase” “Cytoskeletton-associated protein 2” Zytomegalo Virus (“cytomegalo virus”) C-reaktives Protein Cancer Testis Antigen Kutanes T-Zell Lymphom („cutaneous T cell lymphoma“) Zytotoxische T-Zellen („cytotoxic T cells“) Kutanes T-Lymphozyten Antigen („cutaneous T lymphocyte antigen“) Kontrolle(n) Differentiell Dalton 3,3`-Diaminobenzidin Dendritische Zellen („dendritic cells“) „Differential Display RT-PCR“ Deutsches Krebsforschungszentrum VERZEICHNISSE DMF DMPC DMSO DNA cDNA DNEL2 DPK DTT EBV EDTA EEF1A1 ELISA EORTC ERK EST EtOH FCS bFGF FHL2 GAPDH GBP GC GCC2 GDP GMP GOLGA GRIP GST GTP h HCl HDM2 HERV-K10 HEXIM-1 HHV-8 HIV-1 HLA HMB-45 H2O2 ddH2O HPLC HPV HRP H2SO4 Hsp HTLV-1 HYREX IFN-α IgE sIL-2R IL-10 N,N-Dimethylformamid Dimethylpyrocarbonat Dimethylsulfoxid Desoxyribonukleinsäure (“deoxyribonucleic acid”) “Copy DNA” “Dynein heavy chain” “Dendritic cell derived protein kinase” Dithiothreitol Epstein Barr Virus Ethylendiamintetraessigsäure Eukaryotischer Translations-Elongationsfaktor 1 alpha 1 “Enzyme-linked immunosorbent assay” „European Organization for Research and Treatment of Cancer“ Extrazelluläre regulatorische Proteinkinase „Expressed sequence tag“ Ethanol Fötales Kälberserum („fetal calf serum“) “Basic fibroblast growth factor” Vier-und-ein-halb LIM Protein 2 Glycerin-Aldehyd-3-Phosphat-Dehydrogenase Guanylat-bindendes Protein Glutathion-Casein “GRIP and coiled-coil domain-containing 2” Guanosindiphosphat Guanosinmonophosphat Golgi Autoantigen Glutamat-Rezeptor-interagierendes Protein Glutathion-S-Transferase Guanosintriphosphat Stunde Salzsäure „Human double minute 2“ Humanes endogenes Retrovirus Antigen K10 Hexamethylen-bis-acetamid-induzierbar 1 Humanes Herpesvirus-8 Humanes Immundefizienz Virus Typ 1 Humanes Leukozyten Antigen (“human leukocyte antigen”) „Human melanoma black-45“ Wasserstoffperoxid bidestilliertes Wasser Hochleistungsflüssigkeits-Chromatographie (“High pressure liquid chromatography”) Humanes Papillomavirus Meerrettich Peroxidase (“horseradish peroxidase”) Schwefelsäure Hitze Schock Protein Humanes T-Zell-lymphotropes Virus Typ 1 „DNA hybridization analysis of recombinantly expressed cDNA libraries“ Interferon-α Immunglobulin E Löslicher IL-2 Rezeptor („soluble IL-2 receptor“) Interleukin-10 x VERZEICHNISSE IMP1 IP-10 IPTG JNK k KCl KH2PO4 KRAB LAR LB Lck LDH LMM M m MAGE MALDI-TOF MAP2 MAPK6 MART-1 MC1R MF MFI MgCl2 MgSO4 MHC MHD MIA min MM 8-MOP MOPS α-MSH MUM-1 n NaAc NaCl Na2CO3 NaHCO3 Na2HPO4 Na3N NaOH NBT NFATC3 NFκB NHL NHS (NH4)2SO4 NK-Zellen NMM NP Insulin-ähnlicher Wachstumsfaktor 2 mRNA Bindungsprotein 1 IFN-γ-induziertes Protein-10 Isopropyl-β-D-thiogalactosid c-Jun N-terminale Kinase kilo Kaliumchlorid Kaliumdihydrogenphosphat Krüppel-assoziierte Box „Leukocyte antigen-related“ Luria Bertani Lymphozytenprotein Tyrosinkinase Laktatdehydrogenase Lentigo-maligna-Melanom Marker Männlich Melanom Antigen „Matrix assisted laser desorption/ionisation-time of flight“ Mikrotubuli-assoziiertes Protein 2 Mitogen-aktivierte Proteinkinase 6 „Melanoma antigen recognized by T cells 1“ Melanocortin-1 Rezeptorgen Mycosis fungoides Median Fluoreszenz-Intensität („mean fluorescence intensity“) Magnesiumchlorid Magnesiumsulfat Haupthistokompatibilitätskomplex (“major histocompatibility complex”) MAGE Homologie Domäne „Melanoma inhibitory activity“ Minuten Malignes Melanom 8-Methoxypsoralen „3-[N-morpholino]2-hydroxypropansulfonic acid“ α-Melanozyten-stimulierendes Hormon Melanom-assoziiertes Antigen (mutiert) („melanoma ubiquitous mutated“) Anzahl Natriumacetat Natriumchlorid Natriumcarbonat Natriumhydrogencarbonat Dinatriumhydrogenphosphat Natriumazid Natriumhydroxid Nitrobluetetrazolium Nukleärer Faktor von aktivierten T-Zellen 3 Nukleärer Faktor kappa B Non-Hodgkin Lymphome N-hydroxysuccinimid Ammoniumsulfat Natürliche Killerzellen Noduläres malignes Melanom Nukleäres Protein xi VERZEICHNISSE dNTP NUDC OPA-Puffer ORF PAMP PARD3ta PARP PAX9 PBL PBMC PBS PBS-T PCR Pfu PMNC PRR PSID P-TEPb PTP4A2 PUVA RAP140 RAYS pRB Rbbp2h1a RCC RDA RIF1 RNA mRNA siRNA RSV Rt RT-PCR SADA SAF SAGE SAPK SART3 SCP-1 retSDR2 SDS-PAGE sek SEREX SKIP SMARTA SS SSM STAT5 STK17b SV desoxy-Nukleosidtriphosphat “Nuclear distribution gene C homolog” “One Phor All”-Puffer Offener Leserahmen Pathogen-assoziierte molekulare Muster („pathogen-associated molecular patterns”) “Partitioning defective 3 homolog” Poly-ADP-Ribose-Polymerase “Paired Box 9” Periphere Blut Lymphozyten Periphere Blut mononukleäre Zellen Phosphatgepufferte Salzlösung PBS-Tween Polymerasekettenreaktion „Plaque forming units“ Periphere mononukleäre Zellen Mustererkennungsrezeptoren („pattern recognition receptors“) „Putative S1 RNA-binding domain protein“ Positiver transkriptioneller Elongationsfaktor b Protein Tyrosin Phosphatase IVA2 Psoralen plus ultraviolettes Licht Retinoblastom-assoziiertes Protein 140 Rekombinant exprimierte Antigene auf Hefeoberfläche Retinoblastom Retinoblastom-bindendes Protein 2 Homolog 1a Nierenzellkarzinom („renal cell carcinoma“) „Representational Difference Analysis“ Rezeptor-interagierender Faktor 1 Ribonukleinsäure Boten-RNA („messenger“ RNA) „small interfering“ RNA „Respiratory syncytial virus“ Raumtemperatur Reverse Transkriptase-Polymerasekettenreaktion Serumantikörper-Detektionsarray T-Zell-Aktivierungsfaktor „Serial Analysis of Gene Expression“ Stress-aktivierte Proteinkinase „Squamous cell carcinoma antigen recognized by T-cells 3“ „Synaptonemal complex protein 1“ Retinale kurzkettige Dehydrogenase/Reduktase Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese (“sodium dodecyl sulphate polyacrylamide gel electrophoresis”) Sekunde „Serological analysis of recombinantly expressed cDNA clones“ SKI-interagierendes Protein „Serological mini-arrays of recombinant tumor antigens“ Sézary Syndrom Superfiziell spreitendes Melanom „Signal Transducer and Activator of Transcription 5“ Serin/Threonin Kinase 17b Säulenvolumen xii VERZEICHNISSE cTAGE-1 Strep-PE TAA TBS-T TCR TEMED TH-Zellen TIL TMB TNF-α TRP-1 TRIP4 TSEB U Ub UACA UBE3A ÜN UICC UPM UTR UVA WHO w wo X-Gal ZNF ZNS Kutanes T-Zell Lymphom Antigen 1 („Cutaneous T-cell lymphoma antigen 1“) Streptavidin-R-Phycoerythrin Tumor-assoziiertes Antigen TBS-Tween T-Zell-Rezeptor („T cell receptor“) N,N,N`,N`-Tetramethylethylendiamin T-Helferzellen Tumor-infiltrierende Lymphozyten Tetramethylbenzidin Tumor Nekrosis Faktor-α „Tyrosin Related Protein 1“ Thyroidhormon Rezeptor-interagierendes Protein 4 „Total skin electron beam radiation“ Unit Ubiquitär UVEAL Autoantigen Ubiquitin Protein Ligase E3A Über Nacht Internationale Union gegen den Krebs („Unio internationalis contra cancrum“) Umdrehungen pro Minute “Untranslated regions” Ultraviolettes Licht A Weltgesundheitsorganisation („World Health Organisation“) Weiblich Woche 5-Brom-4-chlor-3-indolyl-β-D-galaktopyranosid Zink Finger Protein Zentrales Nervensytem xiii EINLEITUNG 1 1 EINLEITUNG Die Haut ist das größte Organ des menschlichen Körpers. Sie begrenzt das Körperinnere gegen die Umwelt und ist aus 3 Schichten aufgebaut: der Epidermis (Oberhaut), dem Korium (Dermis, Lederhaut) und der Subcutis (Unterhaut). Ein gefäßloses, mehrschichtiges, verhornendes Plattenepithel bildet die Epidermis, die zum größten Teil aus Keratinozyten aufgebaut ist. Zudem sind dort unter anderem die pigmentbildenden Zellen der Haut (Melanozyten) und Zellen des Immunsystems (Antigen-präsentierende Langerhans-Zellen) lokalisiert. Die Dermis enthält als festes, unterstützendes Gewebe Gefäße, Schweißdrüsen, Haarfollikel, Kollagen und ElastinFasern. Die Subcutis schließlich besteht im wesentlichen aus Binde- und Fettgewebe (Abbildung 1-1) (Thews et al. 1999). Sowohl beim kutanen T-Zell Lymphom (CTCL) als auch beim malignen Melanom (MM) ist die Haut das primär betroffene Organ. Stratum corneum Stratum lucidum Stratum granulosum Stratum spinosum Stratum basale Stratum papillare Dermis Stratum reticulare Knäuel(Schweiß)drüse Subcutis Abbildung 1-1: Die Schichten der Haut mit der Angabe der Schichtenfolge (Gratzl 2002) Die Haut ist aus drei Schichten aufgebaut: der Epidermis, der Dermis und der Subcutis. 1.1 Immunüberwachung in der Haut Die Haut stellt die primäre Grenzfläche zwischen dem Körper und der Umwelt dar und ist deshalb nicht nur eine physikalische Barriere, sondern enthält ebenfalls eine Reihe aktiver Verteidigungsmechanismen gegen biologische, chemische und physikalische Angriffe von außen. Um diese Angriffe abzuwehren, wird sowohl das angeborene als auch das adaptive Immunsystem der Haut aktiviert (Kupper and Fuhlbrigge 2004). Bei einer Epithelverletzung oder dem Eindringen von Pathogenen werden zunächst Komponenten des angeborenen EINLEITUNG 2 Immunsystems, die Hautzellen (Keratinozyten und Fibroblasten) sowie Immunzellen (Langerhans-Zellen, dermale dendritische Zellen (DC) und Mastzellen), aktiviert, die antimikrobielle Peptide, chemotaktische Proteine und Zytokine freisetzen (Yang et al. 2001). Dies resultiert unter anderem in der Rekrutierung von Immunzellen aus dem Blut. Die Mechanismen der angeborenen Immunantwort führen zudem dazu, dass das adaptive Immunsystem, das auf T- und B-Zellen basiert, die Antigen-spezifische Rezeptoren exprimieren, aktiviert wird. Dieses System stellt sicher, dass jede T-Zelle ihr passendes Antigen zur richtigen Zeit am richtigen Ort findet. Im ersten Schritt treffen Antigen-präsentierende Zellen (APC), die das in der Haut lokalisierte Antigen aufgenommen haben, in Lymphknoten der Haut mit naiven T-Zellen zusammen. T-Zellen mit passendem Rezeptor binden das von APC präsentierte Antigen und werden zusätzlich über kostimulatorische Signale der DC aktiviert. Das B7-Protein der APC bindet an das Differenzierungscluster 28 (CD28)-Protein der T-Zelle. Zumeist trägt nur ein T-Zellklon den passenden T-Zell-Rezeptor (TCR) für ein Antigen. Nach der Antigenerkennung erfolgt die selektive Proliferation (klonale Selektion) des spezifischen TZellklons, um die Erkennung des Antigens durch zahlreiche aktivierte T-Zellen sicherzustellen. Im zweiten Schritt werden Antigen-spezifische Effektor-Gedächtnis T-Zellen produziert, die „Homing“ Rezeptoren exprimieren, mit deren Hilfe sie an den Ort der Antigenlokalisation, der Haut, gelangen können. Im letzten Schritt werden zentrale Gedächtnis- und Effektorzellen gebildet, die andere Gewebe des Körpers als die Haut auf die Antigenexpression hin untersuchen (Kupper and Fuhlbrigge 2004). Dies gewährleistet die Ausbildung einer langlebigen, erworbenen Immunantwort. 1.2 Immunüberwachung von Tumoren Krebs ist die gängige Bezeichnung für einen bösartigen Tumor, der alle Organe des Körpers, Epithelgewebe, das blutbildende oder das Immunsystem befallen kann. Zu Grunde liegt eine Funktionsstörung in den Regulationsmechanismen der Zelle, die zu unkontrolliertem Wachstum und Replikation der Zelle führt. Körpereigenes Gewebe wächst autonom und irreversibel, so dass es zu ausgeprägtem Verlust verschiedener spezifischer Zell- und Gewebefunktionen kommen kann (Pschyrembel 1998). In Deutschland sind Tumorerkrankungen (25%) die zweithäufigste Todesursache nach den Herzkreislauferkrankungen (46%) (Statistisches Bundesamt Deutschland 2003). Das Immunsystem besitzt das Potenzial, neoplastische Zellen zu töten. Für verschiedene Tumorerkrankungen (vor allem MM und Nierenzellkarzinom (RCC)) wurden bereits spontane Regressionen berichtet (Bastian, 2003; Hammad et al. 2003). Hinweise für sowohl T-Zell- als EINLEITUNG 3 auch humorale Immunogenität vieler Tumorerkrankungen wurden beschrieben (Pfreundschuh et al. 1978; Sahin et al. 1995). Bei der Erkennung von Tumoren aktiviert das Immunsystem zwei verschiedene Komponenten: das angeborene und das erworbene Immunsystem. Tumorimmunität wird durch die Effektormechanismen des angeborenen Immunsystems initiiert und durch die Zellen des erworbenen Immunsystems weitergeführt, wobei DC eine zentrale regulatorische Rolle spielen (Abbildung 1-2) (Banchereau et al. 2000). Antikörper-mediierte Tumorzelllyse PAMP Tumorzelle Tumorantigen Erkennung PRR Vorläufer DC Tumorzellkörper B-Zelle Hilfe durch Zytokinfreisetzung CD4 Freisetzung von IFN-α Aufnahme von Tumorantigen und Tumorzellkörper TCR CD4+ TH Tumorzelllyse Direkte Tumorzelllyse MHC II TCR CD4 CD8 MHC II MHC I Reife DC APC CD8+ CTL NKMakro- Zelle phage NK-TZelle TCR Kostimulatorische Signale Erworbene Immunität Angeborene Immunität Unreife DC Abbildung 1-2: Integrierte Immunantwort gegen Tumorantigene (modifiziert nach Sahin et al. 1999 und Banchereau et al. 2000) Vorläufer dendritische Zellen (DC) erkennen Pathogen-assoziierte molekulare Muster (PAMP) der Tumorzelle durch Mustererkennungsrezeptoren (PRR). Dies führt zu einer Interferon-α (IFN-α)Freisetzung durch die DC, welche Makrophagen, Natürliche Killer (NK)-T-Zellen und NK-Zellen aktiviert. Die aktivierten Zellen töten die Tumorzellen ab, und Tumorzellkörper werden freigesetzt. Die Zellkörper und freigesetzte Tumorantigene werden von unreifen DC aufgenommen, die zu reifen DC differenzieren und Tumorantigene zur Selektion von tumorspezifischen Lymphozyten präsentieren. Differenzierungscluster 8+ (CD8+) T-Zellen lysieren Tumorzellen unmittelbar, wohingegen ausgewählte CD4+ T-Helferzellen (TH) Zytokine freisetzen, um CD8+ T-Zellen und B-Zellen zu stimulieren. Von Tumoren sezernierte oder durch Tumorzelllyse freigesetzte Tumorantigene werden durch spezifische BZellen über deren membrangebundenes Immunglobulin (Ig) aufgenommen. B-Zellen, die Tumorantigene EINLEITUNG 4 aufgenommen haben, werden durch TH–Zellen stimuliert, um signifikante Titer spezifischer Immunglobuline der IgG-Klasse sezernieren zu können. APC: Antigen-präsentierende Zelle, MHC: Haupthistokompatibilitätskomplex, TCR: T-Zell-Rezeptor, CTL: Zytotoxische T-Zelle. Zunächst werden Tumormoleküle („pathogen-associated molecular patterns“, PAMP) von Vorläufer-DC erkannt. Die DC setzen Interferon-α (IFN-α) frei, welches Natürliche Killer (NK)-T-Zellen, NK-Zellen und Makrophagen aktiviert. Dies führt zu einer direkten oder durch IFN-γ-vermittelten Abtötung der Tumorzelle. Um die erworbene Immunität zu aktivieren, nehmen unreife DC Tumorzellkörper und Tumorproteine von Tumorzellen auf, die zuvor von Zellen der angeborenen Immunantwort abgetötet wurden. Die Tumorproteine werden prozessiert und cross-präsentiert. Das heißt, dass die gebildeten Peptide sowohl von CD8+ T-Zellen über den Haupthistokompatibilitätskomplex (MHC) Klasse I Weg als auch von CD4+ T-Zellen über den MHC Klasse II Weg erkannt werden können. Im Anschluss differenzieren CD8+ T-Zellen zu zytotoxischen T-Zellen (CTL). Diese lysieren Tumorzellen, die das entsprechende antigene Protein über MHC Klasse I Moleküle präsentieren, unmittelbar. CD4+ T-Helferzellen (THZellen) stimulieren CD8+ T-Zellen und naive B-Zellen durch die Freisetzung von Zytokinen. Spezifische B-Zellen können Tumorproteine über membranständiges Immunglobulin (Ig) aufnehmen und werden von CD4+ T-Zellen zur antigenspezifischen Differenzierung angeregt. Die ausdifferenzierten, reifen Plasma-B-Zellen sezernieren spezifische Antikörper. Damit findet neben der Zell-vermittelten Lyse durch CD8+ T-Zellen die Antikörper-vermittelte Lyse der Tumorzellen durch Plasma-B-Zellen statt (Abbildung 1-2) (Sahin et al. 1999; Banchereau et al. 2000). Diese antikörperabhängige zellvermittelte Zytotoxizität (ADCC) besteht in der Zerstörung von Tumorzellen, die durch von B-Zellen sezernierte Antikörper bedeckt sind, durch NK-Zellen. Somit wird deutlich, dass Tumorproteine essentiell für die Ausbildung einer erworbenen Immunität gegen den Tumor sind. Im folgenden sollen nun zunächst die beiden Tumorerkrankungen, das CTCL und das MM, und anschließend die Tumorantigene dargestellt werden. 1.3 Das kutane T-Zell Lymphom Kutane Lymphome stellen Neoplasien mit dermatotropem Ursprung dar, die durch die autonome Proliferation einzelner T- oder B-Lymphozyten entstehen. Sie unterscheiden sich stark in ihrer Morphologie sowie klinischen Ausprägung und Prognose (Burg et al. 1984; Sterry et al. 1997). Sowohl das CTCL als auch das kutane B-Zell Lymphom (CBCL) zählen zu den primär kutanen Lymphomen, die klinisch und biologisch eine heterogene Gruppe der Non-Hodgkin Lymphome (NHL) bilden (Querfeld et al. 2005). Ein Viertel aller NHL werden jährlich als primär kutane Lymphome diagnostiziert. Die Inzidenz dieser sehr seltenen Erkrankung ist 0,5-1/100.000 EINLEITUNG 5 (Willemze et al. 1997; Groves et al. 2000). Ungefähr 75% der primär kutanen Lymphome sind CTCL, wohingegen ca. 25% der Fälle CBCL darstellen (Willemze et al. 2005). Kutane Lymphome betreffen zumeist Patienten im höheren Lebensalter, und treten bei Männern zweimal häufiger auf als bei Frauen (Kim et al. 2003). Eine Metastasierung in Lymphknoten und innere Organe wird für fortgeschrittene Stadien berichtet. Im Weiteren werden die in ihrem klinischen Verlauf zumeist als aggressive beschriebenen CTCL besprochen. 1.3.1 Klassifikation Das CTCL beschreibt eine klinisch und histologisch sehr heterogene Gruppe von Neoplasien, deren Gemeinsamkeit zumeist in der malignen Proliferation von CD4+ TH2-Zellen liegt (Dummer et al. 1996a). Zu dieser Gruppe zählen laut WHO-EORTC-Klassifikation (Willemze et al. 2005), die sich auf klinisch, histo- und zytomorphologische, phäno- und genotypische Kriterien stützt, die Mycosis fungoides (MF) und ihre Varianten (follikulotropische MF, pagetoide Retikulose, elastolytisches Lymphom („granulomatous slack skin“)), das Sézary Syndrom (SS), das adulte CTCL, die primär kutanen CD30+ lymphoproliferativen Erkrankungen (anaplastisches großzelliges CTCL, lymphomatoide Papulose), das subkutane Pannikulitis-artige CTCL, die extranodalen NK/T-Zell Lymphome und die primär kutanen peripheren CTCL (aggressives epidermotrophes CD8+ CTCL, kutanes γδ CTCL, kutanes CD4+ klein- und mittelzelliges pleomorphes CTCL). Hierbei gelten das SS und die primär kutanen peripheren CTCL als besonders aggressiv mit Überlebenszeiten unter 5 Jahren. Im folgenden sollen nun die beiden häufigsten Formen, die MF und das SS, genauer beschrieben werden. Mycosis fungoides Die MF ist mit 50% aller primär kutanen Lymphome die häufigste Entität und macht ca. 1% aller NHL aus (Kim et al. 2003; Willemze et al. 2005). Aufgrund ihres indolenten Verlaufs stellt sie eine niedrig maligne Erkrankung dar, die eine langsame Progression über Jahre und teilweise Jahrzehnte aufweist (Weinstock and Horm 1988). Für Patienten mit begrenzter Erkrankung (<10% der Körperoberfläche) wurden ähnliche Überlebenszeiten wie für Kontrollpersonen nachgewiesen (Kim et al. 2003). Die Überlebenszeiten sinken mit zunehmendem Stadium, und bei 10% aller Patienten schreitet die Krankheit in höhere Stadien fort (Kim et al. 2003). Der Phänotyp der MF-Tumorzellen kann meist als CD4+, CDw29+, CD2+, CD3+, CD45RO+ und CD30- beschrieben werden, der demjenigen reifer T-Gedächtnis-Zellen entspricht. Die Zellen besitzen eine charakteristische zerebriforme Morphologie (Slater, 1991; Burg et al., 1997; Sterry et al., 1997). Ein charakteristisches Merkmal der MF sind Pautrier’sche Mikroabszesse, die eine Ansammlung von atypischen Lymphozyten in der Epidermis darstellen (Nickoloff 1988). EINLEITUNG 6 Sézary Syndrom Das SS wird als leukämische Variante der MF verstanden, ist eine seltene, nur bei Erwachsenen auftretende Erkrankung und hat eine deutlich schlechtere Prognose als die MF. Die mittlere Überlebenszeit beträgt 2-4 Jahre (Willemze et al. 1997; Scarisbrick et al. 2001). Nach zunächst mildem Verlauf kommt es schnell zu Knotenbildung mit Übergang in ein hochmalignes, immunoblastisches T-Zell Lymphom. Das SS ist durch Erythrodermie, typischerweise mit Befall der Handflächen und Fußsohlen, und generalisierter Lymphadenopathie gekennzeichnet. Oft findet man im Blut, den Lymphknoten und der Haut die sogenannten Sézary- oder LutznerZellen. Diese malignen T-Lymphozyten exprimieren den IFN-γ-Rezeptor AP-1. Immunhistochemisch sind Sézary-Zellen CD3+, CD4+, CD45RO-, CD8- und CD30-. Zirkulierende Zellen sind durch einen Verlust der Oberflächenmarker CD7 und CD26 charakterisiert (Willemze et al. 1997; Willemze et al. 2005). 1.3.2 Stadieneinteilung Die Stadieneinteilung der CTCL erfolgt mittels TNMB-Klassifikation. Diese wurde jedoch vor allem für die MF und das SS entwickelt, und ist somit nur bedingt für andere CTCL-Formen gültig (Kerl H 1987; UICC 1993). T steht für Primärtumor, N für Lymphknoten, M für Fernmetastasen und B für Blut (Tabelle 1-1). In Anlehnung an die TNM-Einteilung in Tabelle 1-1 kann eine Stadiengruppierung der Patienten vorgenommen werden. Das klinische Bild eines CTCL-Patienten in Stadium IIIB weist beispielsweise eine generalisierte Erythrodermie (T4), anormale regionäre Lymphknoten ohne histologisch erkennbaren Befall (N1) und keine Fernmetastasen (M0) auf. Patienten in frühen Stadien (IA-IIA) haben in der Regel eine sehr gute Prognose mit Überlebenszeiten von 10-20 Jahren, wohingegen in späten Stadien (III, SS) ungünstige Prognosen mit Überlebenszeiten von unter 5 Jahren vorherrschen (Tabelle 1-2). EINLEITUNG Tabelle 1-1: 7 TNM-Stadieneinteilung für kutane Lymphome In dieser Tabelle ist die TNMB-Stadieneinteilung für kutane Lymphome nach (Kim et al. 2005) wiedergegeben. Die Abkürzung TNMB steht für Primärtumor (T), Lymphknoten (N), Fernmetastasen (M) und Blut (B). Kategorie Definition T: Haut (Primärtumor) T1 Begrenzte Plaques, Papeln oder ekzematöse Herde, <10% Körperoberfläche T2 Disseminierte Plaques, Papeln oder erythematöse Herde, ≥10% Körperoberfläche T3 Tumoren (einer oder mehrere) T4 Generalisierte Erythrodermie N: Lymphknoten N0 Regionäre Lymphknoten klinisch nicht befallen N1 Regionäre Lymphknoten klinisch anormal, histologisch nicht befallen N2 Regionäre Lymphknoten klinisch nicht befallen, histologisch befallen N3 Regionäre Lymphknoten klinisch anormal, histologisch befallen M: Nicht-regionärer extrakutaner Befall („Fernmetastasen“) M0 Kein nicht-regionärer extrakutaner Befall M1 Nicht-regionärer extrakutaner Befall B: Blut B0 Keine zirkulierende atypische (Sézary) Zellen B1 Zirkulierende atypische (Sézary) Zellen vorhanden Tabelle 1-2: Stadiengruppierung der TNM-Einteilung In dieser Tabelle ist die Einteilung der Stadien des CTCL nach Kim et al. 2005 dargestellt. Stadium IA IB IIA IIB IIIA IIIB IVA IVB T 1 2 1,2 3 4 4 1,2,3,4 1,2,3,4 N 0 0 1 0,1 0 1 2,3 0,1,2,3 M 0 0 0 0 0 0 0 1 5-Jahres-Überlebensrate [Jahre] 96 73 73 44 44 44 27 27 1.3.3 Pathogenese Die Ätiologie der Entstehung von CTCL ist unbekannt. Viele verschiedene Gründe wurden bislang diskutiert, die von Infektionen über Onkogene, Zytokine und Rauchen bis hin zu Umweltfaktoren (z.B. ultraviolettes Licht) reichen. Hintergrund scheint eine chronische Antigenstimulation zu sein, die durch chromosomale Alteration (Mao et al. 2002), bakterielle (Abrams et al. 1999) oder virale Infektion (Herne et al. 2003) hervorgerufen wird. Viele Chromosomen können klonale Alterationen aufweisen (Thangavelu et al. 1997), die Onkogene, Tumorsuppressorgene und Zellzyklus-regulierende Gene verändern. Insbesondere EINLEITUNG 8 das Chromosom 1 ist häufig betroffen, so dass die Region zwischen 1p22 und 1p36 ein Gen enthalten könnte, das entweder in der malignen Transformation oder Progression von MF und SS eine wichtige Rolle spielt (Thangavelu et al. 1997). Zudem treten Alterationen häufig mit Genverlusten auf 1p, 17p, 10q und 19 sowie mit Gengewinnen auf 4q, 18 und 17q auf (Mao et al. 2002). In frühen Stadien werden nur wenige proliferierende Zellen in malignen Läsionen gefunden, die durch Überexpression von bcl-2 der Apoptose entgehen (Dummer et al. 1995). In späten Stadien jedoch können Mutationen in Genen wie ras, c-myc, tal-1, p53 und p16 auftreten, die die onkogene Transformation vermitteln (Kanavaros et al. 1994; Neri et al. 1995; McGregor et al. 1999; Navas et al. 2002). Ein stimulierender Effekt des Chlamydia Pneumoniae-assoziierten Sézary T-Zell-Aktivierungsfaktor (SAF) wurde bei SS-Zellen entdeckt (Abrams et al. 1999). In einigen malignen Lymphomgeweben konnten virale Sequenzen nachgewiesen werden: das Retrovirus humanes TZell-lymphotropes Virus Typ 1 (HTLV-1), welches in der Ätiologie der adulten T-Zell Leukämie eine große Rolle spielt (Hall et al. 1991; Pancake et al. 1995; Kikuchi et al. 1997), das humane Herpesvirus-8 (HHV-8) (Pawson et al. 1996; Sander et al. 1996) und das Epstein Barr Virus (EBV) (Shimakage et al. 2001). Frühe Stadien der MF besitzen häufig ein TH1 Zytokinprofil, wohingegen es in späten Stadien zu einer Verschiebung des Profils nach TH2 kommt. Hier ist die Interleukin-10 (IL-10) Produktion erhöht und IFN-γ erniedrigt (Asadullah et al. 1996; Dummer et al. 1996b). SS-Zellen proliferieren nach einer Induktion mit IL-7, IL-2 und IL-15 und sezernieren IL-7 und IL-15 (Foss et al. 1994; Dobbeling et al. 1998). Dies impliziert einen autokrinen Wachstumsmechanismus in der Pathogenese von SS. Zudem regulieren IL-7 und IL-15 die Expression des Protoonkogens cmyb und des Transkriptionsfaktors „Signal Transducer and Activator of Transcription“ 5 (STAT5), die wiederum die Expression von bcl-2 erhöhen und damit die Apoptose der SSZelllinie SeAx verhindern (Qin et al. 2001). 1.3.4 Epidermotropismus Die Wanderung von zytologisch atypischen T-Lymphozyten in die Epidermis wird als Epidermotropismus bezeichnet. Der Epidermotropismus unterscheidet kutane Lymphome (CTCL und CBCL) von allen anderen NHL. Der molekulare Mechanismus für dieses Phänomen ist äußerst komplex. Zahlreiche Faktoren sind vermutlich beteiligt. Das kutane Lymphozyten Antigen (CTLA), ein Haut-Homing-Rezeptor, wird im allgemeinen nicht auf T-Lymphozyten der entzündlich veränderten Haut exprimiert, jedoch auf allen CTCL-Zellen (Rook und Heald 1995). CTLA ermöglicht CTCL-Zellen durch die Interaktion mit E-Selektin, einem EINLEITUNG 9 Adhäsionsmolekül auf Endothelzellen der kutanen Gefäße, das Einwandern in die Haut. Das IFN-γ-induzierte Protein-10 (IP-10), welches in MF- und SS-Läsionen überexprimiert ist und chemotaktisch auf CD4+ T-Zellen wirkt, spielt vermutlich ebenfalls eine Rolle bei der Entstehung des Epidermotropismus (Sarris et al. 1995; Tensen et al. 1998). Darüber hinaus wurden Veränderungen des Immunsystems in MF und SS nachgewiesen: die Abnahme an NKZellen und Eosinophilen, die verringerte T-Zell-Antwort auf Antigene und die Zunahme von IgE- und IgA-Titern (Rook und Heald 1995). 1.3.5 Diagnose und Prognose Die Diagnosestellung erfolgt primär nach klinischen und histo- sowie immunhistologischen Kriterien, wobei die Abgrenzung zu anderen chronisch entzündlichen Dermatosen schwierig ist. Auch Routine-Laborparameter werden überprüft wie Laktatdehydrogenase (LDH) (Whittaker et al. 2003), da LDH und das T-Stadium nach klinischen Gesichtspunkten wichtige prognostische Faktoren darstellen (Marti et al. 1991). Histopathologisch wird ein bandartiges Infiltrat von kleinen bis mittelgroßen hyperchromatischen, zerebriformen Zellen in der papillären Dermis nachgewiesen. Spezifisch sind Pautrier‘sche Mikroabszesse, die in fortgeschrittenen Krankheitsstadien auftreten. Im Blut können stadienunabhängig maligne Zellen auftreten, und besonders beim SS sind Sézary-Zellen im Blut ein entscheidendes Kriterium (Muche et al. 1997). Die Menge an zirkulierenden malignen Zellen korreliert invers mit der Prognose des Patienten (Muche et al. 1997), und ein Verlust des Oberflächenmarkers CD7 korreliert häufig mit einer schlechten Prognose (Bernengo et al. 1998). Der Verlust der pan-T-Zell-Marker CD2, CD3 und CD5 wird häufig, gerade in fortgeschrittenen Stadien, berichtet und ist in der Diagnose von MF hilfreich (Ralfkiaer 1994). Mittlerweile umfasst die Routinediagnostik auch molekularbiologische Analysen wie die TCR-Genumstellung („TCR gene rearrangement“), um die klonale Population an malignen Zellen zu definieren. Hierfür werden der Southern Blot, mit einer Sensitivität von 90-95%, und die Polymerasekettenreaktion (PCR) mit einer Sensitivität von 100% verwendet (Guitart und Kaul 1999; Russell-Jones und Whittaker 1999). Die Sensitivität der Detektion kann durch die Verwendung von Mikrodissektion erhöht werden (Yazdi et al. 2003). Eine hohe Konzentration an löslichem IL-2 Rezeptor (sIL-2R) ist mit einer schlechten Prognose und eine hohe Expression an p53 ist mit fortgeschrittenem CTCL-Stadium assoziiert (Lauritzen et al. 1995; Wasik et al. 1996). Hohe Mengen an CD8+ T-Zellen in Hautbiopsien korrelieren mit einer besseren Überlebenschance (Hoppe et al. 1995). Serumkonzentrationen von Neopterin, β2-Mikroglobulin und sIL-2R sind in SS-Patienten signifikant erhöht. Zudem stellt Neopterin einen prognostischen Parameter für nicht-leukämische EINLEITUNG 10 CTCL dar (Hassel et al. 2004). Schließlich ist die TNM-Klassifikation ein wichtiges Kriterium neben Patientenalter, LDH-Konzentration und T-Zellklonalität, da der Befall der Haut (TStadium) und die extrakutane Erkrankung (N-Stadium, Lymphknoten oder viszerale Organe) mit der Prognose des Patienten korreliert (Querfeld et al. 2005). 1.3.6 Therapieformen Das Ziel der derzeit angewandten Therapieformen besteht darin, Symptome, kosmetisches Erscheinungsbild und regressionsfreie Überlebenszeit zu verbessern, da CTCL im fortgeschrittenen Stadium kaum Heilungschancen besitzen. Das Stadium der Erkrankung bedingt die Behandlung, so dass topische (örtliche) Therapien vor allem in frühen Stadien und systemische Therapien in späten Stadien Anwendung finden (Dummer et al. 1996a). Die Psoralen plus ultraviolettes Licht (PUVA)-Therapie basiert auf der Hemmung der Desoxyribonukleinsäure (DNA)- und Ribonukleinsäure (RNA)-Synthese durch die Aufnahme von 8-Methoxypsoralen (8-MOP), welches photosensibilisierend wirkt. Nach UV-Bestrahlung (330 – 430nm) interkaliert 8-MOP mit der DNA und bildet Vernetzungen zwischen verschiedenen DNA-Strängen aus. Es entstehen mono- oder bifunktionale Thymine, Genmutationen oder Schwesterchromatiden-Austausche. Die DNA-Synthese wird gehemmt. Vor allem in frühen Stadien angewandt erzielt die PUVA-Therapie Remissionsraten von ca. 60% (Molin et al. 1981; Herrmann et al. 1995). Insbesondere für die Therapie des SS ist die extrakorporelle Photophorese geeignet. Die peripheren mononukleären Zellen (PMNC) werden aus dem Blut isoliert, in Gegenwart von 8-MOP mit UVA bestrahlt und reinjiziert. Ansprechraten von 73% werden berichtet (Edelson et al. 1991). Strahlentherapie in Form von UVB wird erfolgreich in frühen Stadien der Erkrankung eingesetzt und erzielt Remissionsraten von 83% (Ramsay et al. 1992). Da CTCL-Zellen radiosensitiv sind, wird die Radiotherapie vor allem unter Verwendung von Elektronenstrahlung („total skin electron beam radiation“) mit begrenzter Eindringtiefe für die gesamte Haut eingesetzt. Eine hohe Effizienz insbesondere in frühen Stadien mit begrenzter Plaquebildung wird berichtet (Hoppe 1991; Quiros et al. 1997). Die Anwendung der topischen Therapie, vor allem in frühen Stadien, erfolgt lokal auf die Hautoberfläche. Mechlorethamin, ein Stickstoff-Lost-Zytostatikum, besitzt alkylierende Wirkung in der Mitose-Phase der Zelle. Ansprechraten von bis zu 70% werden je nach Stadium erzielt (Ramsay et al. 1988; Vonderheid et al. 1989). Carmustin, ein Nitrosoharnstoffderivat, verwendet denselben Wirkmechanismus und führt zu stadienabhängigen Ansprechraten von 60- EINLEITUNG 11 90% (Zackheim 1994). Kortikosteroide stellen eine effiziente Therapie im Ekzemstadium der MF dar (Zackheim et al., 1998). Die systemische Chemotherapie dient vor allem der palliativen Behandlung von Patienten mit wiederauftretender oder fortgeschrittener Erkrankung, jedoch ohne anhaltenden Erfolg. Unter anderem werden Methotrexat, Stickstoff-Lost-Derivate, Cisplatin, Doxorubicin und Vindesin verwendet. Patienten im fortgeschrittenen Stadium können ebenso mit einer Kombinationstherapie aus alkylierenden Substanzen und Doxorubicin oder Vinca Alkaloiden behandelt werden (Rosen und Foss 1995). Retinoide, Derivate des Vitamins A, wirken antiproliferativ und werden systemisch in frühen Tumorstadien eingesetzt. Auch Kombinationstherapien von niedrigen Dosen mit IFN-α, PUVA oder Chemotherapie finden Anwendung (Kessler et al. 1987; Molin et al. 1987; Dreno et al. 1991). Das neue Retinoid Bexaroten erzielte Therapieerfolge in der Hälfte der behandelten Patienten in fortgeschrittenen Stadien (Duvic et al. 2001). Interferone, insbesondere IFN-α, werden erfolgreich in der CTCL-Therapie eingesetzt. Ihr Effektormechanismus ist komplex. IFN-α wirkt proliferativ auf Keratinozyten und immunregulatorisch auf Makrophagen und NK-Zellen (Nickoloff et al. 1984; Saiki et al. 1986). In vorbehandelten Patienten wurden Ansprechraten von 45-64% mit Remissionen von 2-jähriger Dauer erreicht (Bunn et al. 1986; Olsen et al. 1989). Der Einsatz von Interleukinen in der CTCL-Therapie ist möglich. IL-2 induziert die T-ZellAktivierung und Proliferation sowie die CD8+- und NK-Zell-Toxizität. IL-12 stimuliert die IFNγ Produktion (Rook et al. 2001). Durch die Aktivierung von TH1-Zellen haben IL-2 und IL-12 einen günstigen Effekt auf das CTCL als Neoplasie der TH2-Zellen. Für IL-2 werden Ansprechraten von 67% berichtet, und für IL-12 ergeben sich in frühen Stadien Ansprechraten von 50% (Marolleau et al. 1995; Rook et al. 1999). Denileukin diftitox (DAB389IL-2, Ontak) ist ein rekombinantes Fusionsprotein, das die membrantranslozierenden und katalytischen Domänen des Diphtherietoxins und humanes IL-2 kombiniert. Da bei Lymphompatienten eine hohe Expression an IL-2-Rezeptoren vorherrscht (59%), kann durch Denileukin diftitox die Proteinsynthese maligner Zellen gehemmt und Ansprechraten um 40% erzielt werden (Saleh et al. 1998). Monoklonale Antikörper zählen mittlerweile zum Behandlungsspektrum von CTCL. Sieben von acht MF-Patienten sprachen auf den chimären CD4-Antikörper an (Knox et al. 1996). Die Hälfte aller MF-Patienten sprachen auf CAMPATH-1H an, einen CD52-Antikörper (Lundin et al. 1998). Neue Therapieansätze wie Vakzinierungen mit DC, die mit Tumorlysat, Peptiden, DNA EINLEITUNG 12 oder RNA beladen sind (Maier et al. 2003), und Histondeacetylase Inhibitoren, die die Expression von Tumorsuppressorgenen wiederherstellen können, werden angewendet (Duvic et al. 2003; Piekarz et al. 2004). 1.4 Das maligne Melanom Das MM ist ein bösartiger Tumor, der von den pigmentbildenden Zellen, den Melanozyten, ausgeht. Dieser stark immunogene, neuroektodermale Tumor tritt vor allem an der Haut, aber auch an den Schleimhäuten, der Aderhaut oder den Hirnhäuten auf. Das MM ist für etwa 90% der Mortalität an Hautkrebs verantwortlich, da eine frühe Tendenz zur aggressiven, lymphogenen und hämatogenen Metastasierung besteht (Pschyrembel 1998). Die MM-Inzidenz in der weißen Bevölkerung nimmt weltweit seit 25 Jahren zu und korreliert mit der Helle der Hautfarbe und der Nähe zum Äquator. In Mitteleuropa treten 10-12 Fälle pro 100.000 Einwohner und Jahr auf, wohingegen die höchste Inzidenz (50-60/100.000) in Australien auftritt (Garbe und Blum 2001; Stang und Jockel 2003). In stärker pigmentierten Bevölkerungsgruppen ist das hier seltene MM vor allem auf die Schleimhaut, Handflächen und Fußsohlen beschränkt (Garbe 1997a). MM treten zu 5-10% familiär (Greene et al. 1985), bei Frauen häufiger an den unteren Extremitäten (35%) und bei Männern häufiger am Stamm (57%) auf (Volkenandt et al. 1999). Klinisch und histologisch lassen sich vier MM-Haupttypen unterscheiden (Clark et al. 1969): Das superfiziell spreitende Melanom (SSM) ist mit 60% der häufigste MM-Typ. Es ist häufig am Rumpf lokalisiert. Das noduläre maligne Melanom (NMM) betrifft 20% der Patienten. Da dieser MM-Typ primär ein vertikales Wachstum besitzt und oft amelanotisch ist, ergibt sich häufig eine schlechte Prognose. Am Lentigo-maligna-Melanom (LMM) erkranken etwa 10%. Besonders bei älteren Menschen tritt dieser Tumor auf dem Boden einer Lentigo maligna (Pigmentläsion) in lichtexponierten Körperregionen (Gesicht, Kopf) auf. Das akrolentiginöse Melanom (ALM) wird in Deutschland nur in etwa 5% diagnostiziert. Diese in Afrika und Asien häufigste Form ist an Hand-, Fußflächen und im Nagelbereich lokalisiert. 5% aller Melanomfälle sind seltene MM-Varianten, zu denen unter anderem MM der Schleimhäute, amelanotische MM und desmoplastische MM zählen. EINLEITUNG 13 1.4.1 Stadieneinteilung Die Stadieneinteilung des MM erfolgt nach der TNM-Klassifikation in 4 Stadien. Die entscheidenden prognostischen Kriterien sind die Tumordicke des Primärmelanoms, der Lymphknotenbefall und die Fernmetastasierung (Tabelle 1-3) (Balch et al. 2001). Tabelle 1-3: Melanom TNM-Klassifikation Die Tabelle verdeutlicht die TNM-Klassifikation des MM nach Balch et al. 2001. LDH: Laktatdehydrogenase. T-Kriterium T1 T2 T3 T4 N-Kriterium N1 N2 N3 M-Kriterium M1a M1b M1c Dicke ≤ 1,0mm 1,01-2,0mm 2,01-4,0mm > 4,0mm Anzahl metastatischer Lymphknoten 1 2-3 4 und mehr Lokalisation Subkutan, nodulär Lunge Alle anderen viszeralen Metastasen Alle Fernmetastasen Ulzeration (U) a: ohne U, Clark level II/III; b: mit U, Clark level IV/V a: ohne U; b: mit U a: ohne U; b: mit U a: ohne U; b: mit U Art a: Mikrometastasen; b: Makrometastasen a: Mikrometastasen; b: Makrometastasen; c: In-transit Metastasen/Satelliten-Metastasen ohne metastatische Lymphknoten Serum LDH Normal Normal Normal Erhöht Das Stadium I besitzt eine sehr gute Prognose (90-95% 5-Jahres-Überlebensrate). Diese verschlechtert sich mit dem Auftreten von Satelliten-Metastasen (50-70%), regionären Lymphknotenmetastasen (20-50%) und Fernmetastasen (10-20%) kontinuierlich (Tabelle 1-4) (Rogers und Braun 2002). Tabelle 1-4: Überarbeitete „American Joint Committee on Cancer“ (AJCC) klinische Stadieneinteilung des Melanoms Dieses System der Stadieneinteilung entspricht der aktuellen Klassifikation durch das „American Joint Committee on Cancer“ von 2002 und wurde verändert nach Rogers und Braun 2002. Ein Melanoma in situ (Tis) ist definitionsgemäß nur auf die Epidermis oberhalb der Basalmembran ausgedehnt und damit nicht in der Lage zu metastasieren. Stadium 0 IA IB IIA IIB IIC III IV T Tis 1a 1b oder 2a 2b oder 3a 3b oder 4a 4b jedes jedes N 0 0 0 0 0 0 1-3 jedes M 0 0 0 0 0 0 0 1a-c 5-Jahres-Überlebensrate [Jahre] 100 95,3 89,0-90,9 77,4-78,7 63,0-67,4 45,1 26,7-69,5 6,7-18,8 EINLEITUNG 14 1.4.2 Pathogenese In der Pathogenese des MM spielt eine Kombination von verschiedenen genetischen (somatische und hereditäre) und epigenetischen Veränderungen wie Promotor-Methylierung sowie Umweltfaktoren, die somatische Mutationen auslösen können, eine wichtige Rolle. Im folgenden werden die häufigsten pathogenetischen Faktoren dargestellt. Signifikante Risikofaktoren für die Entstehung eines MM sind insbesondere die Anzahl gewöhnlicher melanozytärer Nävi, das Vorhandensein atypischer, dysplastischer melanozytärer Nävi, die Anzahl aktinischer Lentigines, die Haarfarbe und der Hauttyp (Garbe 1997b). Der Nävuszellnävus, das Muttermal, ist eine gutartige Ansammlung von Nävuszellen, die seit der Geburt vorhanden oder im Lauf des Lebens neu aufgetreten sind. Ihre Gesamtanzahl sowie ihre Art erhöhen das MM-Risiko (Bauer und Garbe 2003). Die UV-Exposition korreliert bei allen Altersgruppen mit der Nävusanzahl. Sonnenschutz führt bei Kindern und Jugendlichen zu verminderter Nävusneubildung (Darlington et al. 2002; Milne et al. 2002). Die Mehrheit epidemiologischer Studien identifiziert Hellhäutigkeit (Hautfarbe Typ I) als Risikofaktor für die MM-Entstehung (Tucker und Goldstein 2003). In Bezug auf die Pigmentierung von Individuen spielt ebenfalls die Haarfarbe eine entscheidende Rolle. Rothaarige weisen ein doppeltes MMRisiko auf im Vergleich zu Schwarzhaarigen (Bliss et al. 1995). Verschiedene Allele des Melanocortin-1 Rezeptorgens (MC1R) werden in Zusammenhang mit dem Phänotyp helle Haut und rote Haare gebracht (Valverde et al. 1995). MC1R kodiert für den G-Protein gekoppelten Rezeptor des α-Melanozyten-stimulierenden Hormons (α-MSH). Bei der Bindung von α-MSH an MC1R wird die Produktion von rot/gelbem Pheomelanin-Pigment auf braun/schwarzes Eumelanin umgestellt (Sturm et al. 1998). MC1R-Varianten weisen eine geringere Kapazität auf, diesen Wechsel in der Melanin-Produktion zu bewirken. Das MC1R-Gen ist mit dem Auftreten von Sommersprossen und einem erhöhten Risiko der MM-Entstehung assoziiert (Valverde et al. 1996; Palmer et al. 2000). Schließlich stellen familiäre MM, die zwei oder mehr MM-Fälle bei Familienmitgliedern ersten Grades voraussetzen, einen bedeutenden pathogenetischen Faktor dar. Diese Patientengruppe entwickelt MM häufig in jüngeren Lebensjahren. Es treten dünnere und multiple Primärmelanome auf (Barnhill et al. 1992). Durch ihre DNA-schädigende Wirkung gilt die UV-Strahlung als bedeutendster umweltbedingter Risikofaktor in der MM-Pathogenese. Sowohl UVA als auch UVB können an der malignen Transformation von Melanozyten beteiligt sein (Ley 1997; Atillasoy et al. 1998). Vor allem hohe UV-Exposition mit Entstehung von Sonnenbrand in der Kindheit und intensive periodische UVExposition (Freizeit und Urlaub) in jungen Jahren stellen ein großes Risiko dar (Autier und Dore 1998; Whiteman et al. 2001). Sonnenbrand ist jedoch auch im Erwachsenenalter ein Risikofaktor EINLEITUNG (Elwood und Jopson 1997). Studien über 15 Einwanderer in Regionen mit hoher Sonneneinstrahlung belegen die Bedeutung der UV-Strahlung in Zusammenhang mit der MMEntstehung (Armstrong und Kricker 2001). Keimbahnmutationen als Risikofaktoren für das MM wurden vor allem bei Tumorsuppressorgenen und Onkogenen im familiären MM identifiziert. Die entsprechenden Proteine spielen eine zentrale Rolle im Zellwachstum, der Zellzyklusregulation und der DNAReparatur. Mutationen werden am häufigsten für das Tumorsuppressorgen „Cyclin-dependent kinase inhibitor 2A“ (CDKN2A) (Kamb et al. 1994) und die Onkogene Zyklin-abhängige Kinase 4 (CDK4) (Zuo et al. 1996) und N-ras (Demunter et al. 2001) nachgewiesen. CDKN2A kodiert für 2 Proteine, deren inaktivierte Signalwege stark mit der MM-Entstehung assoziiert sind. p16INK4A kontrolliert CDK4 und CDK6, blockiert die Retinoblastom (pRB)-Phosphorylierung und arretiert somit Zellen in der G1 Phase (Serrano et al. 1993). p14ARF interagiert mit „Human double minute 2“ (HDM2) und stabilisiert p53 durch die Verhinderung der HDM2-vermittelten p53-Degradation (Stott et al. 1998). 25-50% aller familiärer MM besitzen eine Keimbahnmutation in CDKN2A (Hussussian et al. 1994; Kamb et al. 1994). HDM2 ist im MM häufig überexprimiert, jedoch vermutlich kein Onkogen, da die Überexpression mit verbessertem klinischem Ergebnis korreliert (Polsky et al. 2001; Polsky et al. 2002). Der Cyclin D1/CDK4Komplex vermittelt die pRB-Phosphorylierung, die durch die Bindung des Komplexes an p16INK4A verhindert wird. Tritt eine Punktmutation im CDK4-Gen wie bei drei Familien mit familiärem MM auf, stört dies die Bindung von CDK4 an p16INK4A, nicht aber an Cyclin D1 (Zuo et al. 1996). Cyclin D1 ist im ALM durch eine Gen-Amplifizierung überexprimiert. Eine onkogene Rolle dieses Gens wurde in Tierversuchen mit Antisense-Behandlung nachgewiesen, die zu Apoptose und Tumorverkleinerung führen (Sauter et al. 2002). p53-Mutationen sind eher selten im MM. Mutationshäufigkeiten liegen bei 10-25% (Sparrow et al. 1995; Zerp et al. 1999). Eine Korrelation zwischen Mutation und schlechter Prognose wird vermutet (Daniotti et al. 2004). Ras-Proteine sind Guanosintriphosphat (GTP)-bindende Proteine, die durch MMassoziierte Mutationen ihre GTPase Aktivität verlieren und stets in aktiver, GTP-gebundener Form in der unkontrolliert proliferierenden Zelle vorliegen (Macaluso et al. 2002). Aktivierende N-ras-Mutationen kommen im MM mit bis zu 30% vor (Demunter et al. 2001). 70% der MM besitzen eine mutierte Serin/Threonin Kinase BRAF (Davies et al. 2002). Die häufigste Mutation führt zu einem ständig aktiven Protein, das eine erhöhte extrazelluläre regulatorische Proteinkinase (ERK) 1/2-Aktivierung bedingt. Da BRAF-Mutationen selten in mukosalen MM aber häufig in Nävi detektiert werden, stellen sie vermutlich ein frühes neoplastisches Ereignis in der MM-Entwicklung dar, das allerdings noch von zusätzlichen Faktoren abhängig ist (Pollock et EINLEITUNG 16 al. 2003; Edwards et al. 2004). Das erste, die Apoptose hemmende Onkogen ist bcl-2. Es zählt zu einer Familie intrazellulärer Proteine, die im Zuge der Regulierung der Caspase-Aktivierung DNA-Fragmentierung und Apoptose einleiten (Cory und Adams 2002). In primären und metastatischen MM ist bcl-2 überexprimiert und mit Tumorprogression assoziiert (Leiter et al. 2000). Der wichtigste autokrine Wachstumsfaktor für die maligne Transformation von Melanozyten ist der „basic fibroblast growth factor“ (bFGF). Um zu überleben, müssen Melanozyten über den bFGF-Rezeptor kommunizieren. In normalen Melanozyten wird bFGF nicht exprimiert. Die Expression ist jedoch mäßig bis hoch in dysplastischen Naevi und im MM (Halaban 1991). 1.4.3 Diagnose und Prognose Zur Abgrenzung einer Pigmentläsion von einem MM wird zunächst die ABCDE-Regel angewandt, die sich auf 5 Kriterien stützt: A für Asymmetrie, B für unregelmäßige Begrenzung, C für uneinheitliche Farbe (Colorit), D für einen Durchmesser größer als 5mm und E für Erhabenheit (AWMF 2005). Zusätzlich wird zur differentialdiagnostischen Abklärung die Dermatoskopie eingesetzt (Kittler et al. 2002). Nach der Exzision eines Primärmelanoms ist eine histologische Beurteilung unerlässlich, die insbesondere den Tumortyp, die Tumordicke nach Breslow und die Eindringtiefe nach Clark erfassen muss. Zudem kann eine immunhistologische Charakterisierung der Läsion unter Verwendung von Antikörpern gegen Differenzierungsantigene wie S100, „human melanoma black-45“ (HMB-45), MelanA und Tyrosinase durchgeführt werden (Busam und Jungbluth 1999; Orchard 2000). Allerdings ist bis heute kein histologischer Marker bekannt, der spezifisch und zugleich sensitiv ist (de Wit et al. 2004). S100 zeichnet sich durch eine hohe Sensitivität aus, erreicht jedoch nur eine geringe Spezifität. S100 wird auch in nicht malignen Zellen exprimiert. HMB-45 weist eine hohe Spezifität auf, wird allerdings nur in 80% der Melanomzellen detektiert (Cochran et al. 2001). MelanA und Tyrosinase haben ebenfalls eine hohe Spezifität aber eine geringe Sensitivität. Deshalb wird S100 häufig in Kombination mit einem oder mehreren Differenzierungsantigenen für die Immunhistologie verwendet. „Melanoma ubiquitous mutated“ (MUM-1) wurde kürzlich als neuer Marker für primäre und metastasierende MM identifiziert und ist sensitiver als MelanA und spezifischer als S100 (Sundram et al. 2003). Zum Nachweis der exprimierten Gene wird neben der Immunhistologie auch die Reverse Transkriptase (RT)-PCR herangezogen. Diese kann zum Nachweis von Tumorzell-RNA in Lymphknoten, Blut oder anderen Körperflüssigkeiten angewandt werden. Da bei gesunden Menschen keine Melanozyten im Blut zirkulieren, ist der Nachweis von MelanA und Tyrosinase im Blut mittels RT-PCR relativ sensitiv (Georgieva et al. EINLEITUNG 17 2002). Andere angebrachte Marker, die in der RT-PCR seit kurzem Anwendung finden, sind „Cancer Testis Antigene“ (CTA), insbesondere der Melanom Antigen (MAGE)-A-Familie (Miyashiro et al. 2001; Reynolds et al. 2003). Serologische Marker sind für das MM bereits bekannt, allerdings fehlt auch hier ein Marker, der sowohl in der Diagnostik, Prognostik als auch der Verlaufskontrolle gleichermaßen einsetzbar ist. S100β ist gegenwärtig der am besten geeignete Serummarker des MM, dessen Einsatz in der Diagnose und Früherkennung limitiert ist, aber einen hohen prognostischen Wert im fortgeschrittenen Krankheitsstadium besitzt (Bottoni et al. 2003). In 55% der Patienten, die auf eine Chemotherapie ansprachen, normalisierte sich der S100β Wert. In nur 5% der Patienten, die eine Progression aufwiesen, normalisierte sich der S100β Wert (Hamberg et al. 2003). Obwohl ein erhöhter LDH-Serumspiegel als Klassifikationsparameter in die aktuelle Stadieneinteilung der AJCC aufgenommen wurde, konnte der Zusammenhang eines erhöhten LDH-Serumspiegels mit einer schlechten Prognose für Stadium IV Patienten in einigen Studien nicht bestätigt werden (Hauschild et al. 1999; Hamberg et al. 2003). Desweiteren stellte sich die Serumkonzentration an C-reaktivem Protein (CRP) als spezifischer als LDH in der Erkennung einer Progression von StadiumI/II nach Stadium IV dar (Deichmann et al. 2004). Im klinischen Alltag wird neben S100β und LDH „melanoma inhibitory activity“ (MIA) als Serummarker verwendet. MIA ist deutlich stabiler als S100β (Djukanovic et al. 2001). Die MIA-Serumkonzentration korreliert eindeutig mit dem Erkrankungsstadium (Bosserhoff und Buettner 2002). Allerdings ist S100β als prognostischer Indikator MIA überlegen (Deichmann et al. 1999; Stahlecker et al. 2000). Wie erläutert werden heute einige serologische Marker für die Prognose und Verlaufskontrolle angewandt. Jedoch fehlt es an diagnostischen Markern zur Früherkennung des MM Der wohl wichtigste prognostische Faktor ist die Tumordicke. Weitere wichtige Kriterien sind die Tumorlokalisation, das Alter und das Geschlecht des Patienten. Bei einer gegebenen Eindringtiefe des Tumors haben Patienten mit Tumoren an den Extremitäten, jünger als 60 Jahre und Frauen eine bessere Prognose (Schuchter et al. 1996). In primären MM ist HDM2 ein unabhängiger Überlebensmarker und seine Überexpression ist mit verbessertem klinischen Ergebnis korreliert (Polsky et al. 2001). Laut Healy et al. 1998 ist p53 Überexpression signifikant mit verbesserten Überlebenschancen assoziiert. Metallothionein scheint ein weiterer guter prognostischer Marker zu sein, da seine Überexpression mit einem erhöhten Progressionsrisiko einhergeht (Weinlich et al. 2003). In der vertikalen Wachstumsphase ist CD40 Expression mit einer signifikant kürzeren, tumorfreien Überlebenszeit assoziiert (van den Oord et al. 1996). Zudem ist der Verlust an Melastatin „messenger“ RNA (mRNA) Expression in MM mit einem erhöhten Metastasierungsrisiko assoziiert. Der Melastatin Status korreliert mit der tumorfreien EINLEITUNG 18 Überlebenszeit unabhängig von der Tumordicke (Duncan et al. 1998; Duncan et al. 2001). Schließlich wird den CTA eine Rolle als prognostische Marker zugeschrieben, die jedoch bisher nur durch eine Assoziation zwischen MAGE Expression und tumor-infiltrierenden Lymphozyten unterstützt wird (Busam et al. 2000). Hierbei besitzen tumor-infiltrierende Lymphozyten einen prognostischen Wert in primären MM (Clemente et al. 1996). 1.4.4 Therapie Während MM-Patienten im Stadium I und II relativ gute Chancen auf vollständige Heilung haben, ist die Therapie im Stadium III und IV häufig palliativer Natur, da sich das MM hier als sehr strahlen- und äußerst chemoresistent erweist. MM-Patienten im Stadium I können durch chirurgische Entfernung des Primärtumors mit ausreichendem Sicherheitsabstand geheilt werden (Balch et al. 1993). Neben der Exzision des Primärtumors wird bei Stadium IB und II zusätzlich eine Wächter („Sentinel“)Lymphadenektomie durchgeführt (Morton et al. 1993). Durch Resektion der betroffenen Lymphknoten und adjuvanter (vorbeugender) Behandlung können Patienten mit Melanomen der Stadien II und III behandelt werden. Adjuvante Therapien haben nach der Exzision des Primärtumors das Ziel, die Chancen einer anhaltenden Tumorfreiheit unter Verwendung von Chemotherapeutika und Immunmodulatoren wie IL-2 und IFN-α-2a oder –2b zu erhöhen. Eine Meta-Analyse identifizierte IFN-α als wirksamste Substanz bei der Senkung des Sterbe- und Rezidivrisikos (Wheatley et al. 2002). Die Strahlentherapie und teilweise auch die Thermoradiotherapie werden bei der Bekämpfung von vor allem Knochen- und Hirnmetastasen eingesetzt, die sich der chirurgischen Therapie entziehen (AWMF 2005). Für Stadium IV Patienten ist die Prognose mit einer Überlebenszeit von 6-9 Monaten infaust (AWMF 2005). Obwohl der Nutzen einer systemischen Chemotherapie hinsichtlich Verlängerung des Überlebens und Verbesserung der Lebensqualität im Allgemeinen limitiert ist, kann sie im fortgeschrittenen MM-Stadium unter palliativen Gesichtspunkten eingesetzt werden. Zumeist wird heute Dacarbazin als Monotherapeutikum oder in einer Polychemotherapie verwendet. Bei Metastasen im zentralen Nervensystem (ZNS) wird häufig das ZNS-gängige Temozolamid benutzt. Da Chemotherapien im metastasierten Stadium insgesamt wenig erfolgreich sind (Ansprechraten 15-20%) (Middleton et al. 2000; Young et al. 2001), finden experimentelle Immuntherapien Anwendung und erzielten erste Erfolge. Verschiedene Formen der Immunisierung unter anderem mit Peptiden von Differenzierungsantigenen (Wang et al. 1999a) mit und ohne Adjuvantien, mit DC beladen mit Tumorantigenen oder Tumorzellen (Nestle et al. 1998), mit adoptivem Transfer EINLEITUNG 19 von T-Zellen (Rosenberg et al. 1988) und mit viralen Transfektionssystemen (Overwijk et al. 1999) wurden verwendet. In einer ersten Phase III Studie wurden MM-Patienten im Stadium IV mit autologen Peptid-beladenen DC immunisiert oder mit Dacarbazin Chemotherapie behandelt (Schadendorf et al. 2006). Peptide verschiedener, bekannter Tumorantigene (z.B. MAGE-A1, Tyrosinase) wurden zur Beladung der DC ausgewählt. Obgleich kein statistisch signifikanter Unterschied in der Effektivität der Immuntherapie gegenüber der Chemotherapie nachgewiesen wurde, konnten vergleichbare Ansprechraten (niedrig, < 6%) in beiden Studienarmen berechnet werden (Schadendorf et al. 2006). In Zusammenhang mit Immuntherapien wurden bereits viele Tumorantigene, vor allem aus der Gruppe der CTA, eingesetzt. Aufgrund des Nachweises von spontanen jedoch seltenen Regressionen wird vermutet, dass Krebszellen Autoantigene exprimieren, die vom Immunsystem erkannt werden. Die Immunantwort, die gegen die Krebszellen gebildet wird, ist allerdings zu schwach, um das Tumorwachstum und die Metastasierung zu verhindern. Deshalb versuchen viele Therapien mittlerweile die vorhandene Immunantwort zu verstärken (Overwijk und Restifo 2000). 1.5 Tumorantigene In Tumorzellen werden wesentliche biologische Unterschiede hinsichtlich der Proteinexpression im Vergleich zu den nicht-malignen Ursprungszellen berichtet. Zum einen können Gene von molekularen Alterationen betroffen sein, die nur in Tumoren und einzelnen Geweben oder in einer bestimmten Entwicklungsphase exprimiert werden. Zum anderen können Veränderungen der molekularen Struktur auftreten, die durch Punktmutationen, Spleißvarianten, chromosomale Translokationen, posttranslationale Modifikationen oder Translation alternativer Leserahmen bedingt sind (Gilboa 1999). Zusätzlich werden Tumor-spezifische Antigene, die nur auf Tumorzellen lokalisiert sind, und die häufigeren Tumor-assoziierten Antigene unterschieden, die sowohl auf Tumorzellen als auch auf normalen Zellen exprimiert werden. Bei Erkennung dieser Tumor-assoziierten Strukturen durch das spezifische Immunsystem des Tumorpatienten spricht man von Tumorantigenen (Sahin et al. 1999). Die Erkennung von Antigenen auf neoplastischen Zellen durch das Immunsystem kann zur Initiierung von zytotoxischen Effektormechanismen führen und die Zerstörung der Krebszellen bewirken (Shankaran et al. 2001). Insbesondere Fremdantigene, die durch bakterielle und virale Infektionen in den Körper gelangen, können eine starke Immunantwort induzieren (Cooper et al. 1999; Gilboa 1999). Im Gegensatz dazu ist die Immunantwort gegen körpereigene Antigene eher schwach, da häufig eine immunologische Toleranz aufgrund von klonaler Deletion (Herndon et al. 2005) oder Anergie (Pape et al. 1998; Lee et al. 1999) der T-Zellen vorliegt. Um eine Immunabwehr gegen den Tumor aufzubauen, muss die immunologische Toleranzschwelle gegenüber den körpereigenen Antigenen EINLEITUNG 20 überwunden werden wie beispielsweise durch die Aktivierung oder Erhöhung von spezifischen CTL oder reaktiven Antikörpern (van der Bruggen et al. 1991; Sahin et al. 1995). Heute finden Tumorantigene Anwendung in der Diagnose sowie dem Monitoring von Tumorerkrankungen und der Therapie von Tumorpatienten in Form von Vakzinierungstherapien. 1.5.1 Klassen von Tumorantigenen Tumorantigene können aufgrund ihrer Immunogenität, ihres Expressionsprofils und ihrer molekularen Strukturveränderungen in fünf unterschiedliche Gruppen eingeteilt werden. Onkofetale Antigene Im Embryo exprimierte Proteine, die in gesunden adulten Zellen nicht, jedoch in malignen Zellen re-exprimiert werden, werden als onkofetale Antigene bezeichnet. Die beiden bedeutensten Vertreter dieser Antigengruppe sind das karzinoembryonale Antigen (CEA) und Alpha-1-Fetoprotein (AFP). CEA ist ein Tumormarker für alle gastrointestinalen Tumore sowie zahlreiche andere Tumorarten (Mulcahy und Benson 1999). AFP wird zur Diagnose von Leberkarzinomen verwendet und tritt bei Testis- und Gebärmuttertumoren auf (Lamerz 1997; Yuen und Lai 2005). Virale Antigene Einige Erkrankungen sind mit einer definierten viralen Infektion assoziiert. Zelluläre Transformationen werden berichtet. Dies deutet auf Virus-assoziierte-Antigene in der Tumorentstehung hin. Das EBV scheint eine Rolle im Burkitt Lymphom zu spielen (Murray et al. 1992). In Krebspatienten wurden bereits reaktive Antikörper gegen die EBVMembranproteine LMP2A und 2B nachgewiesen (Lennette et al. 1995). Im Zusammenhang mit Leukämie wurde das humane endogene Retrovirus Antigen (HERV-K10) beschrieben (Depil et al. 2002). Die Onkoproteine E6 und E7 werden von humanen Papillomaviren (HPV) kodiert (Steinwaerder et al. 2001). Durch die Bindung von E7 an pRB und E6 an p53 werden beide Tumorsuppressorgene inaktiviert (Munger et al. 1992). Insbesondere die HPV-Subtypen HPV-16 und –18 sind ursächlich an der Zervixkarzinom-Entstehung beteiligt (Meschede et al. 1998). Schließlich spielen das Hepatitis B Virus und das Hepatitis C Virus eine entscheidende Rolle in der Entstehung von Leberzellkarzinomen (Gentilini et al. 1997). Antigene durch molekulare Veränderungen Eine durch Punktmutation oder strukturelle Alteration veränderte Gensequenz kann zu einer veränderten Aminosäure (AS)-sequenz des kodierten Proteins und somit zu einem einzigartigen Protein führen. Dies kann die Entstehung neuer antigener Epitope und einer Immunantwort EINLEITUNG 21 gegen zuvor „normale“, immuntolerante Proteine zur Folge haben (Lurquin et al. 1989). Im p53 Gen werden häufig Mutationen berichtet, die unter anderem im Ovarialkarzinom zu Antikörperantworten führen (Hogdall et al. 2002). Das ras-Onkogen weist ebenfalls häufig Mutationen auf zum Beispiel bei 90% aller Pankreaskarzinome (Aguirre et al. 2003). Mutationen von CDK4 und des β-Catenin Gens treten geringfügig häufiger auf (Wolfel et al. 1995; Rubinfeld et al. 1997). Durch Translokation können neue Fusionsproteine entstehen, die Antigene kodieren wie beispielsweise das sogenannte Philadelphia Chromosom. Das Fusionsprotein wird durch die Translokation des Abl-Gens von Chromosom 9 auf das bcr-Gen von Chromosom 22 gebildet. Dieses stark immunogene Antigen ist charakteristisch für die chronisch myeloische Leukämie (Bocchia et al. 1996; Berke et al. 2000). Nach der Identifizierung von „human leukocyte antigen“ (HLA)-abhängigen Epitopen des Bcr/Abl-Gens konnten CTLs generiert werden (Berke et al. 2000). Kürzlich konnte ebenfalls nachgewiesen werden, dass das Immunsystem alternative Spleißprodukte, neue offene Leserahmen (ORF), Pseudogene und sogar Produkte des antisense Strangs der DNA erkennen kann (Van Den Eynde et al. 1999; Moreau-Aubry et al. 2000). Beispielsweise sind reaktive Antikörper gegen das Spleißprodukt des Restins in Kontrollseren und Seren von Krebspatienten zu finden (Bilbe et al. 1992). Überexprimierte Tumorantigene Die Überexpression eines ubiquitär vorhandenen Antigens durch verstärkte Transkription oder erhöhte Proteinstabilität kann zu einer Überschreitung der Toleranzschwelle und somit zu einer Immunantwort führen. Dieses Phänomen konnte unter anderem für die Carbon-Anhydrase XII bei RCC (Tureci et al. 1998) und das EphA3 Gen bei MM, Lungenkarzinomen und Sarkomen (Chiari et al. 2000) bestätigt werden. Bekanntester Vertreter dieser Tumorantigengruppe ist das Her-2/neu Gen (Disis et al. 1994), welches sich als membranständiges Antigen für eine Therapie mit monoklonalen Antikörpern (Herceptin) eignet. Herceptin ist bereits seit 2000 in Deutschland für die Therapie bei Brustkrebs zugelassen (Hehl 2001). Differenzierungsantigene Differenzierungsantigene stellen eine Gruppe von Zelltyp-spezifischen Proteinen dar, die sowohl in Tumorzellen (Melanomzellen) als auch im differenzierten Normalgewebe (ausschließlich in Melanozyten) vor der Tumorentstehung exprimiert werden. Hierzu zählen die am Melaninstoffwechsel beteiligte Tyrosinase (Brichard et al. 1993), gp100 (Bakker et al. 1994), die „Tyrosinase Related Proteins“ 1 und 2 (TRP1/gp75; TRP2) (Wang et al. 1995; Wang et al. 1996) EINLEITUNG 22 und das „Melanoma antigen recognized by T-cells 1“ (MART-1)/MelanA (Coulie et al. 1994). Ein Zeichen für eine Immunantwort gegen diese Antigene ist eine Vitiligo (Overwijk und Restifo 2000). Bei Progression des MM tendieren Melanomzellen zur Dedifferenzierung und zum Verlust dieser Antigene (Sahin et al. 1999). Differentiell exprimierte Antigene treten ebenfalls bei anderen Tumorerkrankungen auf wie beispielsweise Galektin 4 beim Kolonkarzinom (Scanlan et al. 1998) und „Renal cell carcinoma associated antigen“ (RAGE) beim RCC (Gaugler et al. 1996). Cancer Testis Antigene Die CTA sind durch ihr charakteristisches Expressionsmuster gekennzeichnet. Neben ihrer Expression in zahlreichen humanen Tumorentitäten werden sie lediglich in den Spermatogonien der Testis, im Trophoblast, im Ovar und in der Plazenta exprimiert (Scanlan et al. 2004). Dementsprechend sind CTA tumorspezifische Antigene, da die genannten Keimbahngewebe durch ihre fehlende HLA-Expression immunprivilegierte Gewebe darstellen (Fiszer und Kurpisz 1998). Dies führte zu der Theorie, dass anomale Expression von CTA in Krebsgeweben die (Re) Transkription von einem Zellprogramm, das nach der Ausdifferenzierung der Keimbahn- zur somatischen Zelle stillgelegt wurde, reflektiert (Old 2001). Der wichtigste bisher bekannte Mechanismus, der zur Entstehung der ektopischen Expression der CTA in Tumoren und Keimbahnzellen führt, ist die genomweite Demethylierung der CpG-Inseln. Sie ist für die Aktivierung von in tumorfreien Geweben nicht exprimierten Genen verantwortlich. Diese Promoter-Demethylierung konnte für einige CTA in direktem Zusammenhang mit der Aktivierung von Tumorzellen (De Smet et al. 1996) sowie in Testisgewebe (Choi und Chae 1993) beschrieben werden. Auch wenn es sich bei dieser Gruppe von Antigenen um SelbstAntigene handelt, besitzen einzelne CTA ein hohes immunogenes Potential. Viele CTA sind in Multigenfamilien organisiert und häufig auf dem X-Chromosom, das oft ein Ort für Störungen in der Methylierung ist, lokalisiert. Ausnahmen sind beispielsweise das „Synaptonemal complex protein 1“ (SCP-1) (Kondoh et al. 1997) und „cutaneous T-cell lymphoma antigen 1“ (cTAGE-1) (Usener et al. 2003b). Anfang der 90er Jahre wurde das erste CTA mittels der genetischen Methode identifiziert: MAGE-1, welches später in MAGE-A1 umbenannt wurde (van der Bruggen et al. 1991). Bisher konnten 44 CTA-Genfamilien identifiziert und näher charakterisiert werden (Scanlan et al. 2004). Charakteristisch für CTA ist, dass nur wenige Tumorzellen eine Expression aufweisen. Dies weist auf eine inhomogene Verteilung der CTA im Tumor hin. Häufig werden mehrere CTA in einem Tumor exprimiert. In EINLEITUNG 23 40% Brusttumoren und 65% MM wurde die Expression von drei oder mehr CTA detektiert (Sahin et al. 1998). Heute sind 12 Mitglieder der MAGE-A-Familie bekannt. Daneben wurden die MAGE-B- und C-Familien identifiziert (Chomez et al. 2001). Andere MAGE-Familien (D und L) sind ubiquitär exprimiert und zählen nicht zu den CTA. Alle Gene der MAGE-A Familie sind auf dem langen Arm des X Chromosoms, der Region Xq28, lokalisiert. Zumeist wurde bisher die Expression der CTA MAGE-A1 bis -A4, -A6 und A-12 analysiert. Seit kurzem wird die Expression der anderen MAGE-A Mitglieder untersucht. Für das Pseudogen MAGE-A7 konnte keine Expression nachgewiesen werden. MAGE-A9 wurde in knapp 30% der CTCL Tumorgewebe detektiert (Eichmuller et al. 2003). Der vollständige ORF all dieser Gene ist im letzten Exon kodiert. Die MAGE Homologie Domäne (MHD) ist eine Konsensusregion von 170 AS mit zentraler Lage im abgeleiteten Protein jedes Familienmitgliedes. Aufgrund der zentralen Anordnung der MHD wird eine konservierte Funktion vermutet. Eine weitere Multigenfamilie stellt GAGE mit vormals 9 Mitgliedern (GAGE-1 bis –7, -7b und – 8) (Van den Eynde et al. 1995; De Backer et al. 1999) dar. Zwischenzeitlich wurden insgesamt 16 Familienmitglieder auf dem Chromosom Xp11.23 mittels BLAST-Untersuchung identifziert und eine neue Nomenklatur festgelegt (Gjerstorff und Ditzel, 2008). In dieser Arbeit wird jedoch die alte Nomenklatur, GAGE-7b, für GAGE-12I verwendet. Mit Ausnahme von GAGE-1 unterscheiden sich alle Familienmitglieder der GAGE-Familie in einzelnen Nukleotidsubstitutionen. GAGE-1 weist eine Insertion von 143 Basenpaaren auf. In Abbildung 1-3 ist die Lokalisation der Familienmitglieder auf Xp11.2 – p11.4 dargestellt. Abbildung 1-3: Lokalisation von GAGE-7b (http://www.ncbi.nlm.nih.gov/entrez/) GAGE-7b (GAGE-12I) ist auf dem Chromosom Xp11.23 lokalisiert und teilt ein Gen mit GAGE-4, -5 und –7. Die verschiedenen GAGE-Familienmitglieder unterscheiden sich nur in einzelnen Nukleotidsubstitutionen. Bisher wurde die Expression verschiedener GAGE-Gene auf mRNA-Ebene in über 30% humaner Tumore, die auch MM und Lungenzellkarzinom beinhalten, beschrieben. Desweiteren war die GAGE-Expression mit einer schlechten Prognose in MM und metastasiertem Neuroblastom korreliert (Cheung et al. 1999; Cheung et al. 2000). Jedoch wurde eine geringe EINLEITUNG 24 GAGE-Expression auch in peripheren Blutproben von gesunden Kontrollen gemessen (Oltra et al. 2004). Die Aufklärung der biologischen Funktion von CTA sowohl in der Gametogenese als auch in Tumoren steht bisher am Anfang. Das SCP-1 Protein spielt eine Rolle bei der korrekten Paarung der homologen Chromosomen während der Meiose (Schmekel et al. 1996). MAGE-A1 wurde als autonomer Hemmstoff der Transkription durch seine Bindung mit dem SKI-interagierenden Protein (SKIP) beschrieben (Laduron et al. 2004). Im NOTCH1 Signaltransduktionsweg bindet MAGE-A1 an SKIP und rekrutiert Histondeacetylase, wodurch der NOTCH1 Weg gestört wird. Der C-Terminus von MAGE-A4, der ebenfalls an SKIP binden kann, wurde als ein Bindungspartner des Onkoproteins Gankyrin identifiziert und unterdrückt durch diese Bindung die onkogene Aktivität von Gankyrin (Nagao et al. 2003). Da zudem Necdin, ein weiteres Mitglied der MAGE-Familie, die Zellproliferation unterbinden kann (Taniura et al. 1999), könnten die MAGE Gene multifunktionelle Regulatormoleküle darstellen. 1.5.2 Identifizierung von Tumorantigenen Zwei klassische Strategien zur Identifizierung von Tumorantigenen unter Verwendung von Tumor-reaktiven CTL sind bekannt: die „genetische“ (De Plaen et al. 1997) und die „biochemische“ Methode (Falk et al. 1991). Für die genetische Methode wird eine Tumor „copy“ DNA (cDNA)-Bank in Zielzellen exprimiert und mit CTL, die aus Tumor-infiltrierenden Lymphozyten (TILs) oder peripheren Blut Lymphozyten (PBLs) von Krebspatienten generiert wurden, durchsucht (Rosenberg 1996). Diese Methode diente bislang der Charakterisierung einer Vielzahl von Tumorantigenen, die sowohl von CD8+ (van der Bruggen et al. 1991; Bakker et al. 1994) als auch CD4+ T-Zellen (Wang et al. 1999b; Wang et al. 1999c) erkannt werden. Die biochemische Methode nutzt von MHC-Molekülen eluierte Peptide, die zunächst mittels Hochleistungsflüssigkeits-Chromatographie (HPLC) und anschließender Massenspektrometrie identifiziert und auf ihre Reaktivität mit CTL getestet werden (Hunt et al. 1992; Cox et al. 1994). Neben der zellulären Immunantwort wird auch der humorale Arm des Immunsystems zur Detektion von Tumorantigenen herangezogen. „Serological analysis of recombinantly expressed cDNA clones“ (SEREX) stellt eine systematische molekularbiologische Methode zur Identifizierung von Tumorantigenen dar, die das Antikörperrepertoire von Tumorpatienten zur Durchsuchung von Tumor-cDNA-Banken nach antigenen Strukturen verwendet (Abbildung 1-4) (Sahin et al. 1995; Eichmuller et al. 2001). EINLEITUNG Testis Blotten der Proteine 5’ cap Inkubation mit Patientenserum Präabsorbtion gegen Phagen- und E. coliProteine Patientenserum Nitrozellulosemembran Tumorgewebe 25 mRNA 3’ AAAA Farbreaktion Transduktion von E. coli Alkalische Phosphatasegekoppelter antihumanes IgG Antikörper Isolierung des positiven Klons Antikörper aus Patientenserum Phagenbank Molekularbiologische Analysen (cDNA-Bank) Abbildung 1-4: Antigen SEREX-Methode Aus „messenger“ RNA (mRNA) von Testis- oder Tumor-Gewebe wird eine Phagenbank hergestellt. Die Elemente einer Lambda-ZAP Phagenbank (sogenannte Phagemide) beinhalten einen Phagenanteil und ein vollständiges Plasmid (pBluescript) mit „copy“ DNA (cDNA) Insert. Mit Phagemiden transduzierte Bakterien exprimieren Phagenproteine und im Insert kodiertes Protein und werden anschließend von Phagen lysiert. Exprimierte Proteine werden geblottet und auf Reaktionen mit Patientenserum überprüft. Dieses ist durch Präabsorption gegen E. coli- und Phagenproteine gereinigt und final 1:100 verdünnt. Sichtbar wird die Protein (Antigen)-Antikörper-Bindung durch eine Farbreaktion mittels eines zweiten anti-humanen Immunglobulin G (IgG) spezifischen Antikörpers, an den eine alkalische Phosphatase gekoppelt ist. Ein positiver Klon wird schließlich von der Agarplatte isoliert, das Plasmid aus dem Phagemiden ausgeschnitten und dessen Insert sequenziert und somit identifiziert. Bisher konnten weit über 2000 körpereigene Antigene bei verschiedenen Tumorarten beschrieben werden (Old und Chen 1998; Scanlan et al. 1998). Zudem konnten bereits bekannte T-Zell-Tumorantigene mittels SEREX als B-Zell-Tumorantigene bestätigt werden und umgekehrt (Sahin et al. 1995; Jager et al. 2000b). Allerdings werden vor allem Antigene, die hochtitrige IgG-Immunantworten hervorrufen und zytoplasmatisch lokalisiert sind, identifiziert. Eine Weiterentwicklung der SEREX-Methode ist die „DNA hybridization analysis of recombinantly expressed cDNA libraries“ (HYREX)-Methode, die der Identifizierung und Isolierung homologer cDNAs in einer Phagen-Plasmidbank dient. Dies ermöglicht die Suche nach verwandten Sequenzen und Familienmitgliedern eines Gens. Die systematische Genanalyse unter Verwendung von DNA-Datenbanken zählt ebenfalls zum Methodenspektrum in der Tumorantigen-Identifizierung. Sie identifiziert potentielle Zielstrukturen, deren immunogene Wirkung gegenüber CTL oder IgG-Antikörper von Krebspatienten im Nachhinein überprüft wird. Wichtige Techniken im Zusammenhang mit differentiellem Screening und subtraktiver Hybridisierung stellen die „Comparative Genomic Hybridization“ (CGH) (Forus et al. 1998), die „Differential Display RT-PCR“ (DDRT-PCR) EINLEITUNG 26 (Musholt et al. 1997), die „Representational Difference Analysis“ (RDA) (Gure et al. 2000), die „Suppression Subtractive Hybridization“ (SSH) (Hufton et al. 1999) und die „Serial Analysis of Gene Expression“ (SAGE) (Matsuzaki et al. 2005) dar. Inzwischen werden häufig Gen Chips (Microarrays) für die Analyse der Genexpression herangezogen. 1.6 Ziel dieser Arbeit Das Ziel dieser Arbeit ist es, Antikörperantworten gegen ausgewählte Tumorantigene des CTCL und des MM, die mittels SEREX und HYREX an verschiedenen Phagenbanken identifiziert wurden, auf ihren Wert in Diagnose, Monitoring und Prognose des CTCL und MM zu untersuchen. Hierbei werden Häufigkeiten von Antikörperantworten von CTCL- und MMPatienten im Vergleich zu Kontrollpersonen evaluiert. Dies soll mittels zweier serologischer Methoden erfolgen: dem Serumantikörper-Detektionsarray (SADA), einer abgewandelten Form des SEREX, die etabliert wird, und der Multiplex-Serologie. Das CTA GAGE-7b soll auf RNA- und Proteinebene auf seine Expression im CTCL und MM analysiert werden, um eine Aussage über eine mögliche Verwendung dieses CTA in der Therapie der beiden Krebserkrankungen treffen zu können. Hierfür wird zunächst rekombinantes Glutathion-S-Transferase (GST)-Fusionsprotein hergestellt, das entsprechende Protein von seinem GST-Partner abgespalten und aufgereinigt. Das somit erhaltene Protein wird zur Generierung von polyklonalen Antikörpern verwendet, die im Western Blot und in der Immunzytologie und -histologie eingesetzt werden. MATERIAL UND METHODEN 27 2 MATERIAL UND METHODEN In diesem Kapitel sind alle verwendeten Materialien und Methoden beschrieben. Die Zusammensetzung der Lösungen, die Materialien, die benutzten Geräte und deren Hersteller sind nachfolgend aufgeführt. Die in dieser Arbeit untersuchten Antigene wurden mit der „serological analysis of recombinantly expressed copy deoxyribonucleic acid (cDNA) clones“ (SEREX)- oder „hybridization analysis of recombinantly expressed cDNA clones“ (HYREX)-Methode an einer Testis-, Lymphom-, malignes Melanom (MM)- und einer Zelllinien (MyLa)-Bank identifiziert (Eichmuller et al. 2001; Usener et al. 2003a; Usener et al. 2003b; Ehlken et al. 2004; Fellenberg et al. 2004; Gerhardt et al. 2004; Hartmann et al. 2004) oder lagen als IMAG-Klon vor. MATERIAL Materialien werden entweder unter der Rubrik „Allgemein“ bei einer Verwendung in mehreren Methoden aufgeführt oder in Verbindung mit der jeweiligen Methode. 2.1 Chemikalien Allgemein • • • • • • • • • • • • • • • • • • • • • • Agar Agarose Antibiotika: Ampicillin (Amp), Kanamycin, Tetrazyklin Aqua ad iniectabilia Braun Bromphenolblau Natriumsalz (BPB) Dimethylsulfoxid (DMSO) 1kbp DNA-Leiter Essigsäure, 100% Ethanol (EtOH), 98% Ethidiumbromid Ethylendiamintetraessigsäure (EDTA) Formaldehyd (37%) Glycerol Glycin Hefeextrakt Isopropanol Isopropyl-β-D-thiogalactosid (IPTG) Magermilchpulver Magnesiumsulfat (MgSO4) Natriumacetat (NaAc) Natriumazid (Na3N) Natriumchlorid (NaCl) Sigma-Aldrich, Steinheim, D Sigma-Aldrich, Steinheim, D Sigma-Aldrich, Steinheim, D B.Braun Melsungen AG, Melsungen, D Merck, Darmstadt, D Merck, Darmstadt, D GeneCraft, Lüdinghausen, D J.T. Baker, Devener, NL Riedel-de-Haen, Seelze, D Roth, Karlsruhe, D Acros Organics, Geel, B Roth, Karlsruhe, D J.T. Baker, Deventer, NL Gerbu, Gaiberg, D Gerbu, Gaiberg, D Riedel-de-Haen, Seelze, D AppliChem, Darmstadt, D Roth, Karlsruhe, D Neolab, Heidelberg, D Roth, Karlsruhe, D AppliChem, Darmstadt, D J.T. Baker, Deventer, NL MATERIAL UND METHODEN • • • • • • • • • • Natriumhydrogencarbonat (NaHCO3) Natriumhydroxid (NaOH) Nukleotid (dNTP)-Mix “long range” One Phor All Buffer Plus (OPA-Puffer) Phosphatgepufferte Salzlösung (PBS) w/o Ca2+, Mg2+ Protease Inhibitor Tabletten, EDTA-frei Salzsäure (HCl) Trizma Base Trypton Tween 20 28 AppliChem, Darmstadt, D J.T. Baker, Deventer, NL Peqlab, Erlangen, D Amersham Pharmacia, Piscataway, USA Biochrom KG, Berlin, D Roche, Mannheim, D J.T. Baker, Deventer, NL Roth, Karlsruhe, D AppliChem, Darmstadt, D Sigma-Aldrich, Steinheim, D Serumantikörper-Detektionsarray (SADA) • • • • • • • • • • Affinitätsadsorbens (aktivierte Glutardialdehyd-Matrix) 5-Brom-4-chlor-3-indolyl-β-d-galaktopyranosid (X-Gal) 5-Brom-4-chlor-3-indolyl-phosphat (BCIP) N,N-Dimethylformamid (DMF) Gelatine Magnesiumchlorid (MgCl2 x 6H2O) Maltose Nitrobluetetrazolium (NBT) NZ-Amin A Restriktionsenzyme: KpnI, SmaI Roche, Mannheim, D Sigma-Aldrich, Steinheim, D Sigma-Aldrich, Steinheim, D Merck, Darmstadt, D Sigma-Aldrich, Steinheim, D Merck, Darmstadt, D AppliChem, Darmstadt, D Sigma-Aldrich, Steinheim, D Sigma-Aldrich, Steinheim, D Amersham Biosciences, Braunschweig, D Klonierung • • • • • • • • • • Calciumchlorid (CaCl2) Calf Intestine Alkaline Phosphatase (CIAP) 10x CIAP Reaktionspuffer T4-DNA-Ligase 5x DNA-Ligase-Puffer High Dye Formamid Lambda-Marker PWO-Polymerase PWO-Polymerase Reaktionspuffer komplett Restriktionsenzyme: BamHI, EcoRI, SalI XmaI Merck, Darmstadt, D Amersham Biosciences, Braunschweig, D Amersham Biosciences, Braunschweig, D Amersham Biosciences, Braunschweig, D Amersham Biosciences, Braunschweig, D Applied Biosystems, Darmstadt, D MBI Fermentas, St. Leon-Rot, D Peqlab, Erlangen, D Peqlab, Erlangen, D Amersham Biosciences, Braunschweig, D New England BioLabs, Frankfurt a. Main, D Proteinexpression • • • Antibiotikum: Chloramphenicol BioRad Protein Assay (Bradford Reagenz) Rinderserumalabumin (BSA, Pentax Fr. V) Roche, Mannheim, D BioRad, München, D Serva, Heidelberg, D Natriumdodecylsulfat (SDS)-Polyacrylamid-Gelelektrophorese (PAGE) und Western Blot • • • Ammoniumperoxidsulfat (APS) Coomassie Brillant Blau G-250 Coomassie Brillant Blue R Roth, Karlsruhe, D Roth, Karlsruhe, D Gerbu, Gaiberg, D MATERIAL UND METHODEN • • • • • • • • • 29 ECL Plus Amersham Biosciences, Little Chalfont, UK β-Mercaptoethanol Sigma-Aldrich, Steinheim, D Methanol Fluka, Deisenhofen, D Natriumdodecylsulfat (sodium dodecyls., SDS) Gerbu, Gaiberg, D 37,5% Polyacrylamid BioRad, München, D Ponceau S AppliChem, Darmstadt. D Precision Plus Protein Standard Dual Color BioRad, München, D Saccharose J.T. Baker, Deventer, NL N,N,N’,N’-Tetramethylethylendiamin p.A. Roth, Karlsruhe, D (TEMED) „Enzyme-linked immunosorbent assay“ (ELISA) und Multiplex-Serologie • • • • • • • • • • • • • Casein Dinatriumhydrogenphosphat (Na2HPO4) Glutathion-Casein (GC) Kaliumchlorid (KCl) p.A. Kaliumdihydrogenphosphat (KH2PO4) Natriumcarbonat (Na2CO3) Polyvinylalkohol (mittleres Molekulargewicht 30-70 kilo Dalton (kDa)) Polyvinylpyrrolidon (mittleres Molekulargewicht 360kDa) Schwefelsäure (H2SO4) Streptavidin-R-Phycoerythrin (Strep-PE) Superchemiclock (CBS-K) Tetramethylbenzidin (TMB) Wasserstoffperoxid (H2O2) Ribonukleinsäure (RNA), kettenreaktion (PCR) • • • • • • • cDNA und 10x Biotherm Reaktionspuffer Biotherm Taq Polymerase Dimethylpyrocarbonat (DMPC) DNase I, RNase-frei Formamid Ionenaustauscherharz, AG 501-X8 Resin 3-[N-morpholino]2-hydroxypropansulfonic acid (MOPS) Sigma-Aldrich, Steinheim, D Sigma-Aldrich, Steinheim, D P. Sehr, DKFZ, Heidelberg, D Merck, Darmstadt, D Merck, Darmstadt, D Sigma-Aldrich, Steinheim, D Sigma-Aldrich, Schnelldorf, D Sigma-Aldrich, Schnelldorf, D Neolab, Heidelberg, D Molecular Probes, Eugene, USA Chemicon, Temecula, USA Sigma-Aldrich, Steinheim, D J.T. Baker, Deventer, NL Reverse Transkriptase (RT)-Polymerase- NatuTec, Frankfurt am Main, D NatuTec, Frankfurt am Main, D Sigma-Aldrich, Steinheim, D Roche, Mannheim, D Roth, Karlsruhe, D BioRad, München, D Sigma-Aldrich, Steinheim, D Proteinexpression und Aufreinigung • • • • • • • 1,4-Dithiotreitol (DTT) L-Glutathion, reduziert Glutathion Sepharose 4B Guanidiumhydrochlorid Lysozym Thrombin Triton-X-100 Roth, Karlsruhe, D Sigma-Aldrich, Steinheim, D Amersham Biosciences, Uppsala, SE Roth, Karlsruhe, D Sigma-Aldrich, Steinheim, D Amersham Biosciences, Piscataway, USA Sigma-Aldrich, Steinheim, D MATERIAL UND METHODEN Immunisierung von Kaninchen, ELISA und Aufreinigung von Antikörpern • • • Ethanolamin Freund’sches Adjuvans inkomplett Freund’sches Adjuvans komplett Sigma-Aldrich, Steinheim, D Sigma-Aldrich, Steinheim, D Sigma-Aldrich, Steinheim, D Western Blot und Immunhistologie • • • • • • • • Aceton 3,3’-Diaminobenzidin (DAB) Eindeckmedium: VectaShield Mounting Med. Levamisol Mayer’s Hämalaun Lösung Natriumcitrat-Dihydrat Tris-HCl, pH 8,5 Ziegenserum, normal J.T. Baker, Deventer, NL Biomol, Hamburg, D Vector Laboratories, Burlingame, USA Vector Laboratories, Burlingame, USA AppliChem, Darmstadt, D Roth, Karlsruhe, D Linaris, Wertheim, D Vector Laboratories, Burlingame, USA 2.2 Geräte, Verbrauchsmaterialien und fertige Kits Allgemein • • • • • • • • • • • • • • • • Biophotometer Brutschränke Bunsenbrenner und Vierfuß Dewa-Stickstoffbehälter Feinwaage AG245 Geldokumentationssystem E.A.S.Y. 429 K Gelelektrophoresekammer (Wide mini subcell GT), Schlitten, Kämme Inkubationsschüttler G25 Inkubator/Trockenschrank Kühlschränke (4°C, -20°C, -80°C) Magnetrührer MR 3002 Mikrowelle Micromat Millipore-Anlage pH-Meter 220 PCR-Maschine Pipetten • • • • • • • • Pipettierhilfe accu-jet PowerPac 300 Power Supply Sonifikator Stickstofftank Chronos Thermoblock Vortexer Reax 2000 Waage Wasserbäder • • • Zentrifuge Avanti-J27 Zentrifuge Biofuge fresco/pico Zentrifuge Minifuge GL Eppendorf, Hamburg, D Kendro, Langenselbold, D Buddeberg, Mannheim, D KGW Isotherm, Karlsruhe, D Mettler-Toledo, Greifensee, CH Herolab, Wiesloch, D BioRad, München, D New Brunswick Scientific, Nürtingen, D Heraeus, Hanau, D Liebherr, Ochsenhausen, D Heidolph, Schwabach, D AEG, Nürnberg, D Millipore, Schwalbach, D Mettler-Toledo, Schwerzenbach, CH MJ Research/BioRad, München, D Eppendorf, Hamburg, D Gilson, Middleton, USA BRAND, Wertheim, D BioRad, München, D Hielscher, Teltow, D Messer Griesheim, Krefeld, D Eppendorf, Harmburg, D Heidolph, Schwabach, D Sartorius, Göttingen, D Köttermann Labortechnik, Uetze, D Julabo Labortechnik, Seelbach, D Beckman Coulter, Krefeld, D Hereaus, Hanau, D Heraeus, Hanau, D 30 MATERIAL UND METHODEN • • • • • • • • • • • • • • • • • • Bechergläser (50,100,250,500,1000mL) Fisher Scientific, Schwerte, D Eppendorf Tubes (0,2;0,5;1,5;2mL) Eppendorf, Hamburg, D Erlenmeyerkolben mit/ohne Schikanen Schott, Mainz, D (250mL, 1L) Falconröhrchen (15,50mL) Becton Dickinson, Heidelberg, D Faltenfilter Filtrak, Bärenstein, D Filter (0,2µm) Sartorius, Göttingen, D Glaspipetten (1,2,5,10,20mL) mit Pipettierhilfe BioRad, München, D Küvetten (UVette ultraviolett (UV)/VIS) Eppendorf, Hamburg, D Messzylinder (25,50,100,250,1000mL) Hirschmann, Eberstadt, D Pasteur Capillary Pipettes WU, Mainz, D Pipettenspitzen (1000er, 100er, 10er) Gilson, Middleton, USA Eppendorf, Hamburg, D Plastik-Petrischalen (80/150mm) Greiner, Kremsmünster, A Plastikpipetten Sarsted, Nürnbrecht, D Schlauchklemme (1cm) Roth, Karlsruhe, D Schraubflaschen (250,500,1000mL) Schott, Mainz, D Serum-Monovetten Sarstedt, Nürnbrecht, D Skalpell Feather, Osaka, JP Spritzen (1,5,10,20,50mL) Henke Sass Wolf, Tuttlingen, D SADA • • • • • • • • • • • Rotationsschüttler Unimax 2010 Schüttler Promax 2020 Überkopfrotor Überkopfrotor Reax 2 Glas-Petrischalen Nitrozellulose Blotting Membran Nunc 9mL Tubes mit Schraubkappe Nunclon 96 Lochplatte (steril) OmniTray Protran Nitrozellulose Transfer Membran TSP Screening Stempel Heidolph, Schwabach, D Heidolph, Schwabach, D Snijders, Tilburg, NL Heidolph, Schwabach, D Greiner, Frickenhausen, D Sartorius, Göttingen, D Nunc, Wiesbaden, D Nunc, Wiesbaden, D Nunc, Wiesbaden, D Schleicher & Schüll, Dassel, D Nunc, Wiesbaden, D Klonierung • • ABI PRISM TM 310 Genetic Analyzer UV-Tisch Applied Biosystems, Darmstadt, D Konrad Benda Laborgeräte, Wiesloch, D Proteinexpression • Hochdruckhomogenisator, Emulsiflex C-5 Avestin, Ottawa, CA SDS-PAGE und Western Blot • • • • • Entwicklergerät Agfa Curix 60 Folienschweißgerät Geltrocknungsrahmen (Gel drying frames) Mini-Protean II/III Electrophoresis Cell Mini Trans Blot Electrophoretic Transfer Cell Agfa, Köln, D Serverin, Sundern, D Sigma-Aldrich, Steinheim, D BioRad, München, D BioRad, München, D 31 MATERIAL UND METHODEN • • • • • Röntgenfilmexpositionskassette Celluphanfolien HyperfilmTM ECL Standardprospekthülle Whatman Filterpapier 3MM Dr. Goos-Suprema, Heidelberg, D Sigma-Aldrich, Steinheim, D Amersham Biosciences, Little Chalfont, UK Esselte Leitz, Stuttgart, D Whatman, Maidstone, UK ELISA und Multiplex-Serologie • • • • • • • • • • ELISA-Reader µ-Quant Luminex 100 (Analyzer) Luminex SD (Puffer-Reservoir) Luminex XYP (xy-Rastertisch für Waschpl.) Mehrkanalpipette Precision, elektrisch Ultraschallbad Vakuum-Absaug-Einrichtung f. Waschplatten 96 Lochplatten, polysorb MultiScreen BV Filtrationsplatten (Waschpl.) SeroMap Microspheres Deelux Labortechnik, Gödenstorf, D Luminex Corp., Austin, USA Luminex Corp., Austin, USA Luminex Corp., Austin, USA Biozym, Hamburg, D Bandelin Sonorex, Berlin, D Millipore, Eschborn, D Nunc, Wiesbaden, D Millipore, Eschborn, D Luminex Corp., Austin, USA RNA, cDNA und RT-PCR • • • • Mahlbecher aus Teflon Mahlkugeln aus Wolfram/Stahl Rotationsschüttler Unimax 2010 Schwingmühle MM301 Retsch, Haan, D Retsch, Haan, D Heidolph, Schwabach, D Retsch, Haan, D Proteinexpression und Aufreinigung • • • • • • • • • Äkta-Purifier 10/100 mit Box 900, Monitor pH/C 900, Monitor UV-900, Pump-900, Fraction Collector-900 Dounce-Homogenisator Kühlkabine Minicoldlab 2023 Vakuumzentrifuge: Speed Vac Concentrator Dialysierschlauch MonoQ HR 5/5 NAPTM 5 Säule Plastiksäule, leer, aus QIAprep Midi Spin Kit Rotilab-Spritzenfilter 0,8µm Amersham Pharmacia, San Francisco, USA Wheaton Science Products, Millville, USA LKB Bromma, Stockholm, SE Savant/Thermoquest, Egelsbach, D Neolab, Heidelberg, D Amersham Pharmacia, Uppsala, SE Amersham Biosciences, Uppsala, SE Qiagen, Hilden, D Roth, Karlsruhe, D Immunisierung von Kaninchen, ELISA und Aufreinigung von Antikörpern • • Gammacell 1000 D (Cäsium-137) Chinchilla Bastard Kaninchen • • Kanüle MicrolanceTM 3 N-hydroxysuccinimid (NHS)-aktivierte Sepharose 4 Fast Flow Atomic energy of Canada lim., Ottawa, CA Ivanovas, Kisslegg, Biogenes, Berlin, D Becton Dickinson, Drogheda, IR Amersham Biosciences, Uppsala, SE Western Blot und Immunhistologie • • DakoCytomation Stift (Fettstift) Digitalkamera 32 DakoCytomation, Glostrup, DK MATERIAL UND METHODEN • • • • • Mikroskop DMLS Zytospin-Zentrifuge Cytospin 2 COSTAR 1,2mL Cluster Tubes COSTAR, Cluster Tube, 8-cap strip Objektträger und Deckgläser 33 Leica, Bensheim, D Shandon/Thermo, Dreieich, D Corning, Wiesbaden, D Corning, Wiesbaden, D R. Langenbrinck, Emmendingen, D Zellkultur • • • • • • • Brutschrank Function Line/Hera cell Casy 1 Modell TT Mikroskop DMIL Wasserbad Zentrifuge RT7 Gewebekulturflaschen Kryoröhrchen Heraeus, Hanau, D Schärfe System, Reutlingen, D Leica, Bensheim, D Köttermann Labortechnik, Uetze, D Kendro/Sorvall, Langenselbold, D Greiner, Frickenhausen, D Greiner, Frickenhausen, D Fertige Kits • • • • • • • • ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit Alkaline Phosphatase Rabbit IgG ABC Kit AP Substrat Kit Avidin/Biotin Blocking Kit iScript cDNA Synthesis Kit QIAprep Spin Mini Prep Kit QIAquick Gel Extraction Kit Rneasy Mini Kit Applied Biosystems, Darmstadt, D Linaris, Wertheim, D Linaris, Wertheim, D Linaris, Wertheim, D BioRad, München, D Qiagen, Hilden, D Qiagen, Hilden, D Qiagen, Hilden, D 2.3 Puffer und Lösungen Alle Puffer wurden in bidestilliertem Wasser (ddH2O) angesetzt, wenn nicht anders vermerkt. Alle Prozentangaben sind in Gewichtsprozent (w/v) angegeben. Allgemein 5x DNA-Ladepuffer: IPTG-Stammlösung: 50x TAE-Puffer: 0,1M EDTA pH 8,0, 0,1% BPB, 1% SDS, 50% Glycerin 0,5M IPTG, sterilfiltrieren, in Aliquots bei –20°C lagern 40mM Trizma Base pH 7,8 (pH mit Essigsäure einstellen), 10mM NaAc, 1mM EDTA SADA BCIP-Lösung: Blockierungslösung: 10x CDS-Puffer: X-Gal-Stammlösung: Entwicklungslösung: Maltose-Stammlösung: MgSO4-Stammlösung: NBT-Lösung: 10x Serumverdünnungslösung: 50mg BCIP/mL DMF, bei –20°C lagern 5% Milchpulver in 1x TBS 0,1M Trizma Base pH 9,5, 5mM MgCl2 x 6H2O, 0,1M NaCl 250mg X-Gal/mL DMF, in Aliquots bei –20°C lagern 0,2% NBT-Lösung, 0,1% BCIP-Lösung in 1x CDS, frisch ansetzen 10% Maltose, autoklavieren 0,2M MgSO4 x 7H2O, autoklavieren 50mg NBT/mL 70% DMF, bei 4°C lagern 2% Magermilchpulver, 0,25% Na3N MATERIAL UND METHODEN SM-Puffer: 10x TBS-Puffer: 10x TBS-Tween (TBS-T): 34 0,58% NaCl, 0,2% MgSO4 x 7H2O, 0,05M Tris-HCl, 0,01% Gelatine, autoklavieren 0,5M Trizma Base pH7,5, 8,78% NaCl 10x TBS, 0,5% Tween SDS-PAGE und Western Blot Blockierungslösung: 5% Milchpulver in PBS-T Coomassie Brillant Blue-Lösung: 4 Coomassie Brillant Blue R Tabletten (= 200mg Coomassie), 10% Essigsäure, 40% Methanol Entfärber-Lösung: 10% Methanol, 5% Essigsäure Färbelösung (koll. Coomassie): 80% Färbestammlösung, 20% Methanol, frisch ansetzen Färbestammlösung (kolloidales Lösung A: 2% Phosphorsäure, 10% (NH4)2SO4 add 1L Coomassie): ddH2O; Lösung B: 1g Coomassie Brillant Blue G-250 in 20mL ddH2O; Lösung A in B lösen Fixierungslösung: 40% Methanol, 10% Essigsäure 4x Lower Buffer: 1,5M Trizma Base pH 8,8, 0,4% SDS PBS-Tween (PBS-T): 1x PBS pH 7,4, 0,05% Tween Ponceau S-Lösung: 0,2% Ponceau S, 1% Essigsäure 10x Running Buffer: 0,25M Trizma Base pH 8,3, 1% SDS, 1,92M Glycin 5x SDS-PAGE-Probenpuffer: 0,0625M Tris-HCl pH 6,8, 2% SDS, 10% Saccharose, 0,001% BPB, 5% β-Mercaptoethanol, in Aliquots bei -20°C lagern Stripping-Lösung: 0,0625M Tris-HCl pH 6,8, 2% SDS, 0,7% βMercaptoethanol 1x Transferpuffer: 10% 10x Transferpuffer, 20% Methanol 10x Transferpuffer: 25mM Trizma Base pH 8,8, 0,19M Glycin, 0,037% SDS 4x Upper Buffer: 0,5M Trizma Base pH 6,8, 0,4% SDS ELISA und Multiplex-Serologie Casein-Blockpuffer: Casein-Blockpuffer (Luminex): Coating-Puffer: Lagerungspuffer: PBS (für ELISA): Serumvorinkubationspuffer: Substratpuffer: TMB-Stammlösung: Waschpuffer (PBS-T): 0,2% Casein in PBS-T, frisch ansetzen 0,1% Casein in 1xPBS, frisch ansetzen 50mM Carbonatpuffer pH 9,6, 1 Teil 50mM Na2CO3 mit 4 Teilen 50mM NaHCO3 mischen, pH einstellen, bei 4°C lagern Casein-Blockpuffer (Luminex), 0,05% NaN3 137mM NaCl, 3mM KCl, 1,5mM KH2PO4, 8mM Na2HPO4, Herstellung als 10x Lösung, zum Gebrauch mit ddH2O auf 1x verdünnt 1x PBS, 0,1% Casein, 2mg/mL Lysat von Glutathion-STransferase (GST)-tag exprimierenden Bakterien, 0,5% Polyvinylalkohol, 0,8% Polyvinylpyrrolidon, 2,5% CBS-K 100mM NaAc pH 6,0 (pH mit Essigsäure einstellen), bei 4°C lagern 10mg TMB/mL DMSO, bei –20°C lagern, lichtempfindlich 1x PBS (für ELISA), 0,05% Tween 20 RNA, cDNA und RT-PCR DMPC-H2O: 10x DNase-Testpuffer: 0,1% DMPC, 30min bei Rt inkubieren, DMPC durch Autoklavieren inaktivieren 1M NaAc pH 5,0, 50M MgSO4, autoklavieren MATERIAL UND METHODEN Formamid, deionisiert: 10x MOPS-Puffer: RNA-Ladepuffer: RNA-Laufpuffer: 35 500mL Formamid, 50g Ionenaustauscherharz, 30min rühren, Harz mit Faltenfilter entfernen 400mM MOPS, 100mM NaAc, 10mM EDTA pH 8,0, pH 7,0 einstellen, autoklavieren, lichtgeschützt aufbewahren 50% Glycerol, 1mM EDTA, 0,4% BPB, 0,0004% Ethidiumbromid 50% deionisiertes Formamid, 7,4% Formaldehyd, 1x MOPS in DMPC- H2O Proteinexpression und Aufreinigung Buffer high: Buffer low: Elutionspuffer: Puffer A: Puffer B: 0,1M Tris-HCl pH 8,5, 0,5M NaCl 0,1M NaAc pH 4,5, 0,5M NaCl 50mM Tris-HCl, 10mM reduziertes Glutathion, pH 8,0 50mM Tris-HCl pH 7,5 50mM Tris-HCl pH 7,5, 1M NaCl Immunisierung von Kaninchen, ELISA und Aufreinigung von Antikörpern Bindungspuffer: Kopplungspuffer: NHS-Elutionspuffer: NHS-Puffer A: NHS-Puffer B: NHS-Waschpuffer: 2.3.1 Bakterienstämme E. coli BL21 E. coli BL21 Rosetta D3 E. coli SOLR E. coli TOP10 OneShotTM E. coli XL1 Blue (MRF) 2.3.2 0,02M Tris-HCl, pH 8,0 0,2M NaHCO3, 0,15M NaCl, pH 8,2 0,2M Glycin, 0,15M NaCl, pH 2,5 0,5M Ethanolamin, 0,5M NaCl, pH 8,3 0,1M NaAc, 0,5M NaCl, pH 4,0 0,3M NaCl Novagen/Merck Biosciences, Bad Soden, D Novagen/Merck Biosciences, Bad Soden, D, freundlicherweise von Dr. Pawlita zur Verfügung gestellt Stratagene, La Jolla, USA Invitrogen, Karlsruhe, D Stratagene, La Jolla, USA Agar und Medien Allgemein LB-Medium: Luria Bertani (LB)-Agar: 1% Trypton, 0,5% Hefeextrakt, 1% NaCl, pH 7,4, autoklavieren LB-Medium, 1,5% Agar, autoklavieren, Zugabe von Antibiotikum nach dem Abkühlen des Mediums auf ca. 55°C, Gießen der Platten SADA LB-Komplettmedium: NZY-Agar: NZY-Medium: NZY-Top-Agar: LB-Medium, 0,01M MgSO4, 0,2% Maltose NZY-Medium, 1,5% Agar, autoklavieren, Gießen der Platten 0,5% NaCl, 0,5% Hefeextrakt, 0,2% MgSO4, 1% NZ-Amin, pH 7,5, autoklavieren NZY-Medium, 0,7% Agarose, autoklavieren Zellkultur Dulbecco’s modified Eagle Medium (DMEM) Fötales Kälberserum (FCS) PAA Laboratories, Cölbe, D Pan Biotecs, Aidenbach, D MATERIAL UND METHODEN L-Glutamin Melanocyte Growth Medium PBS w/o Ca2+, Mg2+ (1x) Penicillin/Streptomycin RPMI 1640 SupplementMix Trypsin/EDTA (1x) 2.3.3 Stratagene (Sehr et al. 2001), freundlicherweise von Dr. Pawlita zur Verfügung gestellt Plasmide IMAGp998M145158.1 2.3.5 Biochrom, Berlin, D Promocell, Heidelberg, D PAA Laboratories, Pasching, A PAA Laboratories, Pasching, A Biochrom, Berlin, D Promocell, Heidelberg, D PAA Laboratories, Pasching, A Vektoren pBluescript SK pGEX-4T3-tag 2.3.4 36 Deutsches Resourcenzentrum für Genomforschung, Berlin, D Seren Seren Tumorart Typ Anzahl Seren Insgesamt 209 SADA: 18 MM Alter w: 7 m: 11 - w: 88 m: 112 21-92 - - Dummer, Zürich, CH m: 5 53-64 Dummer, Zürich, CH Stadium I-IV CTCL Adenovirus CTCL Masernimpfstoff CTCL MF CTCL MF, SS CTCL MF, SS Ctrl Geschlecht Luminex: 191 (je Stadium ca. 47-49 Patienten) 67 (20 Patienten mit je 1-10 Seren) 45 (5 Patienten mit je 5-12 Seren) 65 (52 Patienten mit je 1-4 Seren) 26 43 (41 Patienten mit 1-2 Seren) Insgesamt 200 100 w: 17 m:35 w: 10 m: 17 w: 18 m:22 20-93 - 19-66 w: 42 m:58 21-67 35-92 35-92 100 Herkunft Klinikum Mannheim, klinische Kooperationseinheit für DermatoOnkologie, Mannheim, D Willemze, Leiden, NL Dippel, Mannheim, D Dummer, Zürich, CH Blutbank, Mannheim, D MM: Malignes Melanom, CTCL: Kutanes T-Zell Lymphom, MF: Mycosis fungoides, SS: Sézary Syndrom, Ctrl: Kontrollen, w: Weiblich, m: Männlich. MATERIAL UND METHODEN 2.3.6 Antikörper Art Primärantikörper 37 Spezifität Goat anti-GST (polyklonal) Mouse anti-Aktin (monoklonal) Mouse anti-tag (monoklonal) Verdünnung Hersteller 1:50.000 Amersham Biosciences, 1:5000 Freiburg, D Anwendung WB, ELISA 1:5000 ICN, Ohio, USA WB 1:10.000 1:1000 M. Pawlita, DKFZ, Heidelberg, D, MacArthur, 1984 WB, ELISA Strep-PE-markiert T. Waterboer, DKFZ, 1:100 Lu mouse anti-tag Heidelberg, D (monoklonal) Biotinyliert goat anti1:1000 Dianova, Hamburg, D Lu human IgA, IgG, IgM Cy3-markierter goat 1:1000 Dianova, Hamburg, D Lu anti-rabbit IgG Donkey anti-goat IgG1:10.000 Santa Cruz, Heidelberg, D WB, ELISA HRP SekundärDonkey anti-human 1:10.000 Dianova, Hamburg, D WB, ELISA antikörper IgG-HRP Goat anti-human IgG1:2500 Dianova, Hamburg, D SADA AP Goat anti-mouse IgG1:10.000 Santa Cruz, Heidelberg, D WB, ELISA HRP Goat anti-rabbit IgG1:10.000 Santa Cruz, Heidelberg, D WB, ELISA HRP cTAGE-5a 1:50 BioGenes, Berlin, D Lu (Peptidantikörper) Selbst K5 (MAGE-A9, 1:1000 Tierstall des DKFZ, WB, ELISA, Lu polyklonal) 1:1600 Heidelberg, D generierte K80 (GAGE-7b, 1:200 Tierstall des DKFZ, WB, ELISA, Lu, Antipolyklonal) 1:100 Heidelberg, D IC, IH K5101 (GAGE-7b, körper 1:5000 Biogenes, Berlin, D Unspezifisch polyklonal) K5102 (GAGE-7b, 1:5000 Biogenes, Berlin, D Unspezifisch polyklonal) GST: Glutathion-S-Transferase, Strep-PE: Streptavidin-R-Phycoerythrin, DKFZ: Deutsches Krebsforschungszentrum, HRP: „Horseradish peroxidase“, AP: Alkalische Phosphatase, Ig: Immunglobulin, cTAGE: CTCL Antigen, MAGE: Melanom Antigen, SADA: SerumantikörperDetektionsarray, ELISA: „Enzyme-linked immunosorbent assay“, WB: Western Blot, Lu: Luminex/Multiplex-Serologie, IC: Immunzytologie, IH: Immunhistologie. MATERIAL UND METHODEN 2.3.7 38 Zelllinien Zelllinie Cou-L3 HH HuT78 MyLa SeAx MeWo Ma-Mel-04 Ma-Mel-05 Ma-Mel-11 Ma-Mel-12 Ma-Mel-16 Ma-Mel-21 Ma-Mel-37b Ma-Mel-47 Ma-Mel-52 SK-Mel-023 UKRV-Mel-02 UKRV-Mel-06a UKRV-Mel-14a UKRV-Mel-21a UKRV-Mel-27 UKRV-Mel-31 HaCat Melanozyten Tumorart CTCL MM Lokalisation/Typ Herkunft Mycosis Fungoides M. Bagot, Créteil, F Lymphomatoide Papulosis ATCC, Manassas, USA Sézary Syndrom ATCC, Manassas, USA Mycosis Fungoides K. Kaltoft, Aahrus, DK Sézary Syndrom K. Kaltoft, Aahrus, DK Metastase: Lymphknoten Old, New York, USA Metastase: Harnblase Metastase: Bauch Metastase: Mediastinum Klinische Metastase: Bauch, kutan Kooperationseinheit für Dermato-Onkologie, Prof. Metastase: Dünndarm Dr. Dirk Schadendorf, Metastase: kutan DKFZ, Heidelberg, D Metastase: Lymphknoten Metastase: Lymphknoten Metastase: Haut Oberarm rechts Old, New York, USA Metastase: Pleura-Exudat Klinische Metastase: kutan Kooperationseinheit Metastase: kutan Dermato-Onkologie, Prof. Metastase: Lymphknoten Dr. Dirk Schadendorf, Metastase: kutan DKFZ, Heidelberg, D Metastase: kutan Immortalisierte humane Haut Keratinozyten - - Norbert Fusenig, DKFZ, Heidelberg, D - PromoCell, Heidelberg, D ATCC: “American Tissue and Culture Collection” 2.4 Software HUSAR / GCG Package Luminex 100 IS 2.2 SP1 Software Microsoft Windows / Office 2003/XP DKFZ, Heidelberg, D Luminex Corp., Austin, USA Microsoft Corp., Unterschleißheim, D MATERIAL UND METHODEN 39 METHODEN 2.5 SADA Die SADA-Methode bildete die Grundlage zur Bestimmung der Tumorspezifität zahlreicher Antigene auf serologischer Ebene. Diese Methode stellt eine Variante der SEREX-Methode dar, mit welcher viele Antigene gleichzeitig auf ihre Seroreaktivität mit einem Serum getestet werden können. Die verwendeten Antigene wurden im Verlauf von fünf Jahren in verschiedenen Projekten für Doktorarbeiten in der Arbeitsgruppe identifiziert. 2.5.1 Auswahl der Antigene Die Auswahl der Antigene wurde anhand ihrer zuvor ermittelten serologischen Reaktivität in sekundären SEREX-Experimenten, der Ergebnisse im virtuellen Northern und ihres Zusammenhangs mit Krebserkrankungen anhand der in Pubmed (http://www.ncbi.nlm. nih.gov/entrez/query.fcgi?db=PubMed) vorhandenen Literatur getroffen. Im sekundären SEREX wird die Reaktivität eines bestimmten Klons mit einer gewissen Anzahl an Seren, zumeist bis zu 20 Seren, ermittelt. Der virtuelle Northern ist über den Link genes/genefinder auf der CGAPHomepage (http://cgap.nci.nih.gov) zu erreichen und simuliert einen virtuellen Northern. Dem Programm liegen Veröffentlichungen von „expressed sequence tags“ (ESTs) in Genbank/EMBL zu Grunde, von denen bekannt ist, aus welchem Gewebe sie isoliert wurden. Ein Unterprogramm legt bekannte ESTs aufgrund ihrer Überlappung übereinander und erstellt daraufhin Gen-Cluster. Das Ergebnis ist eine Zahl, die das Verhältnis von der Anzahl der ESTs, die in den Gen-Cluster des zu untersuchenden Klons fallen, zu der Gesamtzahl aller ESTs in diesem Gewebe darstellt. Je größer der Zahlenwert ist, desto mehr wird dieses Gen abgelesen. Die Analyse wird sowohl für Normal- als auch Tumorgewebe durchgeführt. Die Grenzen des virtuellen Northern liegen bei geringen Gesamtzahlen für ESTs, unbekannten Genen und differentiell gespleißten Genprodukten, die durch ein Standard-Gen-Clustering nicht erfasst werden können. Die ausgewählten Antigene lassen sich Tabelle 2-1 entnehmen. Tabelle 2-1: Ausgewählte Antigene für die SADA-Methode In dieser Tabelle sind der Name, der Kontrolltyp, das homologe Gen und die Zugangsnummer aller im SADA verwendeter Antigene aufgeführt. Name PINCH Uveal Autoantigen (UACA) „Leerer“ Phage (ohne Insert) Kontrolltyp Positivkontrolle Positivkontrolle Negativkontrolle Zugangsnummer U09284 NM_018003 - MATERIAL UND METHODEN 40 Name GBP-5a GBP-5ta pGAGEt Genname oder Homologes Gen laut GenBank Zugangsnummer Guanylat-bindendes Protein (GBP)-5a AF430643 Guanylat-bindendes Protein-5ta AF328727 GAGE-3 NM_001473 Glutamat-Rezeptor-interagierendes Protein (GRIP) and coiled-coil se1-1 AF273042 domain-containing 2 (GCC2) se2-1 Synaptonemal complex protein 1 (SCP1) AF273043 se2-5 Partitioning defective 3 homolog (PARD3ta) AF177228 HD-CL-12 Zink Finger Protein 195 (ZNF195) AF003540 pMAGE-A9ag MAGE-A9 NM_005365 cTAGE-1 Kutanes T-Zell Lymphom Antigen 1 AF177229 cTAGE-5a Kutanes T-Zell Lymphom Antigen 5a AF338233 se20-7 Golgi Autoantigen, Golgin Subfamilie a, 4 (GOLGA4) AF273047 du20-10L4 Zytoskelett assoziiertes Protein 2 (CKAP2) AF177227 HD-CL-04 Jak und Mikrotubuli interagierendes Protein 2 (KIAA0555) AF273057 HD-CL-03 Retinale kurzkettige Dehydrogenase/Reduktase (retSDR2) AF273056 HD-CL-09 Serin/Threonin Kinase “dendritic cell derived protein kinase” (DPK) AF328731 HD-MM-01 Dynein heavy chain (DNEL-2) AJ000522 HD-MM-02 PLU-1 AJ132440 HD-MM-03 Retinoblastom-bindendes Protein 2 Homolog 1a (Rbbp2h1a) AJ243706 se57-1 CTCL Tumorantigen se57-1 AF273051 se89-1 Retinoblastom-assoziiertes Protein 140 (RAP140) AF273053 HD-MM-48 Rezeptor-interagierender Faktor 1 (RIF1 Variante 2) NM_001006945 HD-MM-08 Hexamethylen-bis-acetamid-induzierbar 1 (HEXIM-1) AB021179 HD-MM-16 Humanin AY029066 HD-MM-19 Zink Finger Protein 133 (ZNF133) BC001887 HD-MM-23 „Leukocyte antigen-related“ (LAR)-interagierendes Protein 1a/b U22815/6 HD-CL-02 Inositol-Polyphosphat-5-Phosphatase AF273055 HD-CL-08 Eukaryotischer Translations-Elongationsfaktor 1 alpha 1 (EEF1A1) AF328730 Nukleärer Faktor von aktivierten T-Zellen, zytoplasmatisch, HD-CL-14 NM_004555 Calcineurin-abhängig (NFATC3) HD-CL-17 Hefe ribosomales Protein S28 Homolog D14530 Schuppenförmiges Zell Karzinom Antigen erkannt von T-Zellen 3 HD-CL-24 AB020880 (SART3) HD-CL-30 Chromosom 8 EST Sequenz, Klon RP11-14I17 AC011726 se20-4 Zellteilung Autoantigen (CDA-1) AF273046 se33-1 Nukleäres Protein 220 (NP220) AF273049 se37-2 Ubiquitin Protein Ligase E3A (UBE3A) AF273050 pMAGE-A3ag MAGE-A3 NM_05362 HD-CL-41 Serin/Threonin Kinase 17b (STK17b) BC016040 HD-CL-49 Nukleäre Verteilung Gen C Homolog (NUDC) BC015153 HD-CL-50 Mitogen-aktivierte Proteinkinase 6 (MAPK6) BC035492 HD-CL-51 Putative S1 RNA-bindende Domäne Protein (PS1D) BC007446 HD-CL-52 Hypothetisches Protein BM-009 BF691904 HD-CL-53 Protein Tyrosin Phosphatase IVA2 (PTP4A2) U14603 HD-CL-54 Chromosom 8, Klon RP11-621B3 AC046174 2.5.2 Präabsorption der Patientenseren Die SADA-Methode bedient sich der Antikörper aus Seren von Tumorpatienten und Kontrollpersonen, um die humorale Immunantwort gegen zahlreiche Antigene zu untersuchen. Hierzu wurde Vollblut nach Koagulation in Serum-Monovetten 10 Minuten (min) bei 2500 MATERIAL UND METHODEN 41 Umdrehungen pro Minute (UPM), Rotor 2150, zentrifugiert und das gewonnene Serum bei -20°C gelagert. Seren von 100 CTCL-, 18 MM-Patienten und 100 Kontrollen (Ctrl) wurden verwendet. Die Präabsorption erfolgte, um reaktive Antikörper gegen die Antigene des VektorSystems, E. coli und lysierter E. coli-Fragmente zu entfernen, und damit unspezifische Reaktionen zu vermeiden. Isolierung eines „leeren“ Phagen Das Plasmid pBluescript trägt neben einer Amp-Resistenz ein lacZ Gen inklusive eines Polylinkers. Eine Insertion eines Gens führt somit zu einer Inaktivierung des lacZ Gens und kann im lacZ deletierten E. coli Stamm MRF für eine Blau-Weiß Selektion verwendet werden. Ein blauer Plaque auf einer X-gal-Amp-Nähragarplatte weist auf Phagen ohne Insert, ein weißer Plaque dagegen auf einen Phagen mit Insert hin. Ohne Insert kann das LacZ Gen abgelesen werden, so dass mit der im Bakterienstamm MRF gebildeten Permease (lac-Y Gen) und Transacetylase (lac-A Gen) ein funktionsfähiges Lactose-Operon synthetisiert wird. Die Galaktosidase kann den Farbstoff X-Gal in einen blauen Azoe-Farbstoff umwandeln. Mit Insert wird keine Galaktosidase gebildet, somit auch kein blauer Farbstoff. Mehrere „leere“ Phagen wurden isoliert und jeweils 1 Stunde (h) in 500µL SM-Puffer unter Schütteln aus dem Agar gelöst. Nach der Inkubation wurden die Agarreste mittels Zentrifugation pelletiert (13.000UPM, 4min). Der Überstand, die Phagenlösung, wurde abgenommen und bei 4°C aufbewahrt. Verwendet wurde dieser „leere“ Phage als Negativkontrolle im Array und um Antikörper gegen das Vektor-System und lysierte E. coli-Fragmente zu entfernen. Präabsorptionsschritt I, mechanisch zerkleinerte E. coli In diesem ersten Präabsorptionsschritt werden zunächst E. coli-Fragmente und Proteine an das Adsorbens der Säulenmatrix gebunden. Die „mechanische Säule“ wurde hergestellt, indem eine überwachsene, über Nacht (ÜN) 50mL E. coli MRF-Kultur pellettiert (2000UPM, 15min), das Pellet in 5mL 1x PBS gelöst und die Zellen mittels Sonifizierung auf Eis (10x 20 Sekunden (sek)) aufgebrochen wurden. Das gewonnene Lysat wurde gleichmäßig auf 10 Schraubdeckelröhrchen, die jeweils 0,2g Affinitätsadsorbens (aktivierte Glutardialdehyd-Matrix) enthielten, verteilt, mit 1x PBS auf 4mL aufgefüllt und ÜN bei 4°C im Überkopfrotor inkubiert. Die Säulen wurden anschließend 3-mal mit 4mL 1x TBS inklusive 0,1% NaN3 1h bei 4°C gewaschen (Waschschritt). Das 1:10 mit Serumverdünnungslösung vorverdünnte Serum wurde mit der beladenen Matrix über Nacht bei 4°C im Überkopfrotor inkubiert. Das präabsorbierte Serum wurde bei 4°C aufbewahrt und die Säule wieder hergestellt (3-mal mit 4mL 0,05M Tris, MATERIAL UND METHODEN 42 pH 3,0). Die Säule konnte nach dem Äquilibrieren (Waschschritt) bei 4°C aufbewahrt werden und war für maximal 0,6mL unverdünntes Serum zu verwenden. Präabsorptionsschritt II: lysierte E. coli Die „lytische Säule“ dient dazu, dem Serum mittels lysierter E. coli-Fragmente und Phagenelemente unspezifische Antikörper zu entziehen. E. coli MRF wurde in 50mL LBKomplettmedium bis zu einer optischen Dichte bei 600nm (OD600) von 0,5 schüttelnd inkubiert. Die Kultur wurde pelletiert (1500UPM, 15min) und in 2mL 0,01M MgSO4 aufgenommen. Anschließend wurde die E. coli MRF-Kultur mit einem „leeren“ Phagen in LB-Komplettmedium für 4h bei 37°C schüttelnd transduziert. In Tabelle 2-2 ist die Zusammensetzung des E. coli Lysis-Ansatzes für die lytischen Säulen aufgelistet. Im Anschluss wurde die restliche E. coliKultur zugegeben und weitere 2h bei 37°C schüttelnd inkubiert. Die lysierten Bakterien wurden zusätzlich mittels Sonifizierung auf Eis 10x 20sec zerkleinert. Die mit 0,2g Matrix beladene Säule wurde mit diesem Lysat ÜN bei 4°C im Überkopfrotor inkubiert und wie die mechanische Säule äquilibriert. Das bereits mit der mechanischen Säule präinkubierte Serum wurde mit der hergestellten lytischen Säule bei 4°C ÜN auf dem Überkopfrotor weiter präabsorbiert, dekantiert und bei 4°C aufbewahrt. Die Säulen wurden anschließend wieder hergestellt und konnten zur Präabsorption von bis zu 0,6mL unverdünntem Serum verwendet werden. Tabelle 2-2: E. coli-Lysis-Ansatz zur Herstellung der lytischen Säulen Der E. coli-Lysis-Ansatz zusammen. setzt sich aus LB-Medium, Maltose, MRF und dem „leeren“ Phagen Reagenzien LB-Medium Maltose, 10mM E. coli XL1 MRF „Leerer“ Phage Volumen 5mL 100µL 200µL 500µL Präabsorptionsschritt III: Blot der lysierten E. coli-Proteine Um nach der mechanischen und lytischen Säule noch vorhandene unspezifische Antikörper zu entfernen, verwendet die lytische Folie ebenfalls lysierte E. coli-Fragmente. Der Unterschied zur lytischen Säule besteht darin, dass hier lysierte Bakterien auf Nitrozellulose-Membran geblottet werden. Es wurden 2 verschieden große lytische Folien hergestellt: kleine und große. Für die kleine lytische Folie (Durchmesser 8,5cm) wurden 1-2µL isolierter „leerer“ Phage mit 200µL MRF (OD600=0,5) gemischt und 15min inkubiert. 600µL MRF und 2-4µL Phagenlösung wurden für die große lytische Folie (Durchmesser 14cm) verwendet. Die Suspension wurde mit heißem Top-Agar (ca. 50°C) auf eine NZY-Agarplatte ausplattiert und ÜN bei 37°C inkubiert MATERIAL UND METHODEN 43 (Konfluenz der Bakterien). Anschließend wurde jeweils eine Nitrozellulose-Membran 4h und eine zweite ÜN bei 37°C aufgelegt und geblottet. Nach erfolgtem Blot wurden die Membranen an der Luft getrocknet und bei Raumtemperatur (Rt) aufbewahrt. Das Serum aus den Präabsorptionsschritten I und II wurde 1/33 verdünnt, auf eine mit 1x TBS angefeuchtete, kleine lytische Folie gegeben und ÜN oder tagsüber bei 4°C inkubiert. Pro Serum wurde dieser Schritt 5-mal mit jeweils einer neuen lytischen Folie wiederholt. Zuletzt wurde das präabsorbierte Serum auf eine finale Konzentration von 1/100 verdünnt und auf unspezifische Reaktionen an einer Phagenbank getestet. Falls Reaktionen gegenüber E. coli MRF und/oder Phagen bestanden, wurde mit großen lytischen Folien weiter präabsorbiert. 2.5.3 Transduktion Über Nacht wurde zunächst eine 50mL LB-Komplettmedium-Kultur des MRF E. coli-Stamms mittels Picken einer Einzelkolonie von einer MRF-LB-Tetrazyklin-Kulturplatte hergestellt. Die Platte konnte eine Woche lang verwendet werden. Am Wochenbeginn wurde mit 200µL dieser ÜN-Kultur eine 50mL LB-Komplettmedium-Kultur angeimpft und bis zu einer OD600 von 0,5 schüttelnd inkubiert. Bei dieser OD befinden sich die Bakterien in der exponentiellen Wachstumsphase und sind somit am stoffwechselaktivsten. Titration der Phagenlösungen Alle verwendeten Phagen waren als monoklonale Stocklösung, die bei –80°C gelagert wurde, vorhanden. Ein µL dieser Lösungen wurde pro Phage für einen Transduktionsansatz in einer kleinen Petrischale verwendet (Tabelle 2-3). Tabelle 2-3: Zusammensetzung des Transduktionsansatzes Die Zusammensetzung des Transduktionsansatzes aus MRF, NZY-Top-Agar und IPTG für den jeweiligen Petrischalentyp kann dieser Tabelle entnommen werden. Anzahl Platten / Material MRF (OD=0,5) NZY-Top-Agar IPTG 1 kleine Petrischale 200µL 4 mL 8µL 1 große Petrischale 600µL 7-8 mL 15µL 5 große Petrischalen/ 5 OmniTrays 3mL 40 mL 75µL Hierfür wurden 200µL E. coli MRF mit der Phagenlösung 15min bei 37°C inkubiert, anschließend mit IPTG und NZY-Top-Agar vermischt und auf einer NZY-Agarplatte ausplattiert. Das IPTG dient der Induktion des Galaktosidase-Gens auf dem Plasmid. Nachdem der Agar sich verfestigt hatte, wurden die Platten kopfüber bei 37°C ÜN inkubiert. Mehrere Phagenplaques eines Klons wurden isoliert (siehe Isolierung eines „leeren“ Phagen), damit ein MATERIAL UND METHODEN 44 hoher Phagentiter erreicht werden konnte, und sterilfiltriert (Porengröße 0,2µm), um Pilzwachstum zu verhindern. Nach angemessener Verdünnung erfolgte ein erneuter Transduktionsansatz, um die Phagenkonzentration zu bestimmen. Die Phagenlösung wurde bei 4°C gelagert, wobei die Phagenkonzentration monatlich neu bestimmt wurde. Transduktion Laut Tabelle 2-3 wurden MRF, NZY-Top-Agar und IPTG auf eine NZY-Agar-OmniTray-Platte gegeben und 2h bei 37°C kopfüber inkubiert. Mittels eines 96 Lochstempels wurden ca. 0,5µL Phagenlösung aus einer 96 Lochplatte (150µL Phagenlösung pro Loch) in Duplikaten auf den wachsenden MRF-Rasen übertragen. Die Phagenlösungen waren zuvor für jeden Klon auf 2000 „Plaque forming units“ (Pfu) pro µL eingestellt worden. Somit konnten pro OmniTray-PlattenAnsatz bis zu 22 verschiedene Antigene plus eine Negativ- („leerer“ Phage) und eine Positivkontrolle (PINCH oder „UVEAL Autoantigen“ (UACA)) aufgespottet werden. Die Anordnung der Phagen auf den Membranen ist Tabelle 2-4 zu entnehmen. Nach dem Einsickern der Phagenlösung in den Agar wurden die Platten kopfüber ÜN bei 37°C inkubiert. 2.5.4 Blotting und immunologischer Screen Am darauf folgenden Tag waren die gebildeten Phagenplaques im Bakterienrasen sichtbar. Die gebildeten rekombinanten Proteine wurden pro OmniTray-Platte auf eine NitrozelluloseMembran für 4h bei 37°C geblottet. Die abgezogenen Blots wurden vorsichtig von Agarresten befreit und 3-mal 10min, schüttelnd in 1x TBS-T gewaschen. Im Anschluss wurden alle Membranen bis zu ihrer Verwendung bei –20°C eingefroren. Nach dem Auftauen wurden die Membranen nach einem zweimaligen Waschschritt für 10min in 1x TBS-T 1h bei Rt schüttelnd mit Blockierungslösung inkubiert, um unspezifische Bindungen abzusättigen. Die Membranen wurden 3-mal für 10min in 1x TBS gewaschen (Waschschritt) und halbiert. Die Bindung der Antikörper aus dem präabsorbierten Patienten- bzw. Kontrollserum, das gesammelt und wieder verwendet wurde, erfolgte ÜN bei 4°C. Am folgenden Tag wurde der sekundäre Antikörper (1:2500 in 0,5% Milchpulver (w/v) in 1x TBS) nach einem Waschschritt 1h lang mit den Membranen inkubiert. Als sekundärer Antikörper wurde ein gegen das Fc-Fragment humaner IgG gerichteter, in einer Ziege hergestellter, spezifischer Antikörper verwendet, der an eine alkalische Phosphatase (AP) gekoppelt ist. Nach einem Waschschritt und einer Äquilibrierung in 1x CDS, pH 9,5 wurde der Blot in frisch angesetzter Entwicklungslösung für 2h bei 37°C schüttelnd entwickelt. MATERIAL UND METHODEN Tabelle 2-4: 45 Anordnung der Phagen auf den SADA-Membranen 1 und 2 Dargestellt ist die Anordnung der SADA-Phagenklone auf den Membranen 1 und 2. Ph. = Phage. SADA-Membran 1 „Leerer“ Ph. „Leerer” Ph. GBP-5ta se2-5 se2-5 se2-1 cTAGE-5a cTAGE-5a cTAGE-1 HD-CL-03 HD-CL-03 HD-CL-04 HD-MM-03 HD-MM-03 HD-MM-02 PINCH PINCH se89-1 GBP-5ta se2-1 cTAGE-1 HD-CL-04 HD-MM-02 se89-1 „Leerer“ Ph. „Leerer” Ph. se57-1 se57-1 HD-CL-09 HD-CL-09 HD-MM-01 HD-MM-01 se20-7 se20-7 du20-10L4 du20-10L4 HD-CL-12 HC-CL-12 pMAGE-A9 pMAGE-A9 pGAGE-t PGAGE-t se1-1 se1-1 PINCH PINCH GBP-5a GBP-5a GBP-5a GBP-5a PINCH PINCH se1-1 se1-1 pGAGE-t pGAGE-t pMAGE-A9 pMAGE-A9 HD-CL-12 HC-CL-12 du20-10L4 du20-10L4 se20-7 se20-7 HD-MM-01 HD-MM-01 HD-CL-09 HD-CL-09 se57-1 se57-1 „Leerer“ Ph. „Leerer” Ph. se89-1 HD-MM-02 HD-CL-04 cTAGE-1 se2-1 GBP-5ta se89-1 PINCH PINCH HD-MM-02 HD-MM-03 HD-MM-03 HD-CL-04 HD-CL-03 HD-CL-03 cTAGE-1 cTAGE-5a cTAGE-5a se2-1 se2-5 se2-5 GBP-5ta „Leerer“ Ph. „Leerer” Ph. SADA-Membran 2 „Leerer“ Ph. „Leerer” Ph. HD-MM-08 HD-CL-02 HD-CL-02 HD-MM-23 HD-CL-24 HD-CL-24 HD-CL-17 se37-2 se37-2 se33-1 HD-CL-50 HD-CL-50 HD-CL-49 HD-CL-54 HD-CL-54 HD-CL-53 HD-MM-08 HD-MM-23 HC-CL-17 se33-1 HD-CL-49 HD-CL-53 HD-MM-48 HD-MM-19 HD-CL-14 se20-4 HD-CL-41 HD-CL-52 HD-MM-48 UACA UACA HD-MM-19 HD-MM-16 HD-MM-16 HC-CL-14 HD-CL-08 HC-CL-08 se20-4 HD-CL-30 HD-CL-30 HD-CL-41 pMAGE-A3 pMAGE-A3 HD-CL-52 HD-CL-51 HD-CL-51 HD-CL-51 HD-CL-51 pMAGE-A3 pMAGE-A3 HD-CL-30 HD-CL-30 HD-CL-08 HC-CL-08 HD-MM-16 HD-MM-16 UACA UACA HD-CL-52 HD-CL-41 se20-4 HC-CL-14 HD-MM-19 HD-MM-48 HD-CL-53 HD-CL-49 se33-1 HD-CL-17 HD-MM-23 HD-MM-08 HD-CL-53 HD-CL-54 HD-CL-54 HD-CL-49 HD-CL-50 HD-CL-50 se33-1 se37-2 se37-2 HC-CL-17 HD-CL-24 HD-CL-24 HD-MM-23 HD-CL-02 HD-CL-02 HD-MM-08 „Leerer“ Ph. „Leerer” Ph. HD-CL-52 HD-CL-41 se20-4 HD-CL-14 HD-MM-19 HD-MM-48 Der Nachweis erfolgte mittels einer Farbumsetzung des NBT/BCIP in der Entwicklungslösung durch die AP. Ein positiver Klon erscheint blau-violett auf der geblotteten Membran. Die Bindung der Serumantikörper gegen die geblotteten Proteine eines Klones und damit die Reaktion des Serums gegen das jeweilige Antigen wurde im Vergleich mit der Negativkontrolle auf demselben Blot bestimmt. In Abbildung 2-1 ist das Schema der SADA-Methode dargestellt. MATERIAL UND METHODEN 46 Stempel Substratreaktion 96 well Platte 150µL Phagenlösung pro well Phagenlösung: ca. 2000 Phagen/µL Phagen in Duplikaten mittels Stempel auf Agar spotten Inkubation mit sekundärem Antikörper 1:2500 in 0.5% Milch in 1xTBS Inkubation mit präabsorbiertem Serum 1:100 ÜN bei 4°C NZY-Agar mit MRF (E. coliStamm) + IPTG OmniTray (Nunc) Blockieren mit 5% Milch in 1xTBS Phagen ÜN bei 37°C inkubieren Blotten der Proteine 4h bei 37°C auf Nitrozellulose Membran Phagenplaque Abbildung 2-1: Membranen bis zur Verwendung bei -20°C lagern Schema der SADA-Methode Die Phagen wurden mittels eines Stempels auf eine Agarplatte übertragen, die exprimierten Phagenklonproteine auf Nitrozellulosemembranen geblottet und eingefroren. Nach dem Auftauen wurden die Membranen blockiert und mit präabsorbiertem Patienten- oder Kontrollserum inkubiert. Die Detektion einer positiven Serumreaktion erfolgte durch eine Inkubation mit sekundärem Antikörper und einer abschließenden Substratreaktion. IPTG: Isopropyl-β-D-thiogalactosid, ÜN: Über Nacht, h: Stunde. 2.5.5 In vivo Exzision Bei der in vivo Exzision wird der pBluescript Phagemid aus dem Uni-Zap XR Vektor unter Verwendung des Helfer-Phagen ExAssist, der für F1 Proteine kodiert, isoliert. Die HelferPhagen werden verwendet, um das Wirtsgenom aus dem Lambda Phagen am λ Ursprung auszuschneiden. Das Plasmid wird mit dem Insert isoliert, da der λ Ursprung an das pBluescript Phagemid grenzt. Da stets mehrere Phagenplaques für die Stocklösungen gepickt wurden, diente diese Methode der Bestätigung der Monoklonalität der verwendeten Phagen. Um das Plasmid aus dem entsprechenden Phagenklon zu gewinnen, wurden Kulturplatten der E. coli-Stämme MRF (Tetrazyklin-Resistenz) und SOLR (Kanamycin-Resistenz) angelegt. Beide Stämme wurden in 50mL LB-Komplettmedium bis zu einer OD600 von 1,0 kultiviert, pelletiert und in 50mL 10mM MgSO4 resuspendiert. Nach einer 15-minütigen Inkubation bei 37°C von 200µL MRF-Lösung mit 250µL Phagenlösung und 1µL Helfer-Phage, wurde die Phagen- MATERIAL UND METHODEN 47 Bakterien-Lösung nach der Zugabe von 3mL LB-Medium für 3h geschüttelt. Die Bakterienzellen wurden 20min bei 65-70°C lysiert und anschließend abzentrifugiert. 50µL des phagenhaltigen Überstands wurden mit 200µL kompetenten E. coli SOLR-Bakterien für 15min inkubiert und auf LB-Amp-Platten ausplattiert. Da das Plasmid eine Amp-Resistenz trägt, wurden die erfolgreich transduzierten Bakterien ÜN bei 37°C selektioniert. Von jeder LB-AmpPlatte wurden je Klon 3 Kolonien gepickt und in LB-Amp ÜN bei 37°C hochgezogen. Sowohl der phagenhaltige Überstand als auch die LB-Amp-Platten konnten bei 4°C mehrere Wochen gelagert werden. 2.5.6 Plasmid-DNA-Isolierung Das Qiaprep Spin Miniprep Kit wurde gemäß des Protokolls des Herstellers zur Isolierung der Plasmide aus den Bakterien verwendet. Eine 4mL ÜN-LB-Amp-Kultur (100µg/mL Amp) des entsprechenden Klons wurde pelletiert und in 250µL kaltem Puffer P1 inklusive RNase resuspendiert. Nach der Zugabe von 250µL Puffer P2 und 350µL Puffer N3 wurden Zelltrümmer und Proteine abzentrifugiert und der Überstand auf die mitgelieferte Säule gegeben. Die Säule enthält ein Glasfaservlies, an das die DNA bindet, die in mehrmaligen Schritten zunächst mit Puffer und anschließend mit ethanolhaltigem Puffer PE gewaschen wurde. Die Plasmid-DNA wurde schließlich in Aqua ad iniectabilia Braun oder mitgeliefertem TE-Puffer eluiert. 2.5.7 Spaltung der Phagemide Die Restriktionsspaltung dient dazu, die cDNA aus dem Plasmid herauszuschneiden und somit die Größe des Inserts zu bestimmen. Das Plasmid enthält einen Polylinker, welcher zahlreiche Schnittstellen für Restriktionsenzyme besitzt. Da die Schnittstellen SmaI und KpnI das Insert flankieren, wurden sie für die Restriktion herangezogen. Für eine Probespaltung wurden für 0,5µg Plasmid 1 Unit (U) Restriktionsenzym eingesetzt. Die Spaltung erfolgte 2h bei 37°C unter Verwendung des OPA-Puffers in einer Endkonzentration von 1x oder 2x in Anlehnung an die Erfordernisse des Enzyms. Das gespaltene Plasmid wurde in einem DNA-Agarose-Gel im Vergleich zu einer DNA-Leiter elektrophoretisch aufgetrennt. 2.5.8 Agarose-Gelelektrophorese Die Agarose (1-2%) wurde mit 1x TAE-Puffer aufgekocht, in eine Gelkammer gegossen und polymerisierte beim Abkühlen aus. Nach einer gelelektrophoretischen Auftrennung der Proben von 40-50min bei 80V wurde das Gel für 5-10min in ein Ethidiumbromidbad gelegt, um die DNA anzufärben. Da Ethidiumbromid unter UV-Licht fluoresziert, konnte das Ergebnis mit einem Geldokumentationssystem festgehalten werden. MATERIAL UND METHODEN 48 2.6 ELISA und Multiplex-Serologie Die ELISA-Methode sowie die davon abgeleitete Multiplex-Serologie ermöglichen die quantitative oder zumindest semiquantitative Bestimmung von Antigen oder Antikörper. Engvall und Perlman beschrieben als erste Wissenschaftler die ELISA-Methode (Engvall und Perlman 1971). In dieser Arbeit wurden ELISA und Multiplex-Serologie für 16 unterschiedliche Antigene in einer neuen Form des GST-X-tag-ELISA (capture ELISA) und der Multiplex-Serologie als serologische Tests zum Nachweis von Antikörpern gegen Tumorantigene des CTCL und MM verwendet. Der GST-X-tag-ELISA wurde von Dr. C. Gänzler, Dr. P. Sehr und Dr. M. Pawlita am Deutschen Krebsforschungszentrum entwickelt und in den USA zum Patent angemeldet (US 60/305259). Die Multiplex-Serologie im Sinne eines Suspensions-Arrays als Antikörperscreeningsystem für komplexe Antikörperantworten (Luminex-Methode) wurde in derselben Arbeitsgruppe von Dr. T. Waterboer etabliert (Waterboer 2005; Waterboer et al. 2005). Für beide Methoden liegen die in E. coli überexprimierten Proteinantigene als GSTFusionsproteine vor. Durch Bindung des GST-Teils an immobilisiertes Glutathion, das sich entweder auf einer ELISA-Platte oder aber an Beads gekoppelt befindet, wird das lösliche Antigen durch einen in situ-Affinitätsaufreinigungsschritt aus dem bakteriellen Lysat immobilisiert und aufgereinigt. Die Herstellung des GC und die Kopplung desselben an die Beads wurde von Dr. P. Sehr und Dr. T. Waterboer übernommen. 2.6.1 DNA-Klonierungen 2.6.1.1 Plasmide Alle in dieser Arbeit verwendeten Tumorantigene lagen im Amp-resistenten pBluescript-Vektor der Firma Stratagene vor und konnten im Weiteren für Umklonierungen verwendet werden. Das Originalplasmid für GAGE-7 wurde als IMAG-Klon IMAGp998M145158.1 vom Deutschen Ressourcenzentrum für Genomforschung erworben. Die Antigene MAGE-A1, -A3, -A9, LAGE1a, CAMEL, GAGE-7b, cTAGE-5a N- und C-terminal wurden selbst umkloniert, wobei mittels des Bioinformatik-Systems HUSAR des DKFZ der offene Leserahmen („open reading frame“, ORF) des jeweiligen Gens ausgewählt wurde, der für das längste Protein kodiert. Die Antigene se57-1, se70-2 und GBP-5ta wurden von Dr. F. Fellenberg umkloniert. Die Humane Papillomavirus (HPV)- und Papavovirus-Antigene BK, JC, LPV und HPV1 sowie die Tumorsuppressorgene p16 und p53 wurden von Dr. P. Sehr und Dr. K. Michael kloniert. Ausgehend von den umklonierten Plasmiden wurden alle verwendeten Proteine der Arbeitsgruppe von S. Eichmüller selbst exprimiert. Die viralen GST-Fusionsproteine sowie die Tumorsuppessorgene wurden von der Arbeitsgruppe von Dr. M. Pawlita zur Verfügung gestellt. MATERIAL UND METHODEN 49 Der verwendete pGEX-Vektor (pGEX-4T-3tag) ist ein IPTG-induzierbarer Expressionsvektor mit tac-Promoter, der rekombinante Proteine als Doppelfusionsproteine mit N-terminaler GST (26kDa) und einem C-terminalen Undecapeptid des großen T-Antigens des simian virus 40 (SV40, KPPTPP PEPET) exprimiert. Die Amp-Resistenz des Vektors dient der Selektion bei Klonierungen. 2.6.1.2 Auswahl der Primer und Klonierungs-PCR Um ein PCR-Amplifikat für die Klonierung zu erhalten, wurden zunächst Forward Primer entworfen, die eine eingebaute Restriktionsschnittstelle, vier bis sechs überhängende Basen des Vektors und die ersten 19-21 Basen des ORF des jeweiligen Antigens enthielten. Die Reverse Primer waren ähnlich aufgebaut, nur wurden hier die letzten 19-21 Basen des ORF ohne Stopcodon verwendet (Tabelle 2-5). Mit Hilfe der PCR konnte eine ausreichende Menge an Amplifikat zur anschließenden Klonierung erhalten werden. Ein Klonierungs-PCR-Ansatz ist in Tabelle 2-6 dargestellt. Aus Abbildung 2-2 ist das verwendete PCR-Programm ersichtlich, das für jedes Primerpaar entsprechend modifiziert wurde. Tabelle 2-5: Primer für die Klonierung in den pGEX-4T-3tag-Vektor Die unterstrichenen Basen kennzeichnen die eingebauten Restriktionsschnittstellen (1BamHI, 2SalI, 3 XmaI, 4EcoRI,) für die Klonierungs-PCR in den pGEX-4T-3tag-Vektor. Alle Primer wurden von Wolfgang Weinig, Abteilung Oligonukleotidsynthese/DNA-Sequenzierung, DKFZ, hergestellt. ORF: Offener Leserahmen, bp: Basenpaar, kDa: kilo Dalton. Tumorantigen MAGEA1 MAGEA3 MAGEA9 LAGE-1a CAMEL long GAGE-7b cTAGE5a Nterminal cTAGE5a Cterminal Primer for Primer rev ccg cgt gga tcc1 atg tct ctt gag cag agg agt ctg cga att ccc ggg3 tgc ctc ttg agc aga ccg cgt gga tcc1 atg tct ctc gag cag agg a tc ccc gaa ttc4 tat gca ggc cga agg cca g tc ccc gaa ttc4 tgc aca ggg ggt tcg acg ggc g ccg cgt gga tcc4 tat gag ttg gcg agg aag at agg ttt gtc gac2 gac tcc ctc ttc ctc ctc tct c agg ttt gtc gac2 ctc ttc ccc ctc tct caa a agg ttt ccc ggg3 aga ctc cct ctt gct cct ct agg ttt gtc gac2 gcg cct ctg ccc tga ggg a agg ttt gtc gac2 gaa ccg ccc ctg gtc ggg gga ga agg ttt gtc gac2 aca ctg tga ttg ctt ttc acc ccg cgt gga tcc1 atg aga cca gat tct aat cg cgt gga tcc1 cac agg aaa tta aca gta gag gaa a ORF AnlagerungsProteingröße [bp] temperatur 930 60°C 70kDa 942 60°C 75kDa 945 45°C 72kDa 540 58,4°C 47kDa 351 60°C 40kDa 351 58,4°C 45kDa g ttt gtc gac2 ctc cag ctc ttc agt ggc at 1224 55°C 75kDa agg ttt gtc gac2 ggt ttc ttg ctg tgg t 1107 55°C 75kDa MATERIAL UND METHODEN Tabelle 2-6: 50 Klonierungs-PCR-Materialien und Pipettierschema Die Tabelle führt die Reagenzien, ihre Endkonzentration und das pipettierte Volumen in einer Klonierungs-PCR auf. PCR: Polymerasekettenreaktion, NTP: Nukleotid-Triphosphat, U: Unit. Reagenzien Plasmid (1:100 verdünnt) PWO-Polymerase 10x PWO Reaktionspuffer komplett Forward Primer Reverse Primer dNTP-Mix (Nukleotide) Aqua ad iniectabilia Braun 95°C 95°C 5min 1min Denaturierung 45-60°C 1min Anlagerung 72°C 2min Elongation Volumen 1,5µL 2,5µL 2,5µL 1,0µL 1,0µL 0,5µL 16,0µL Endkonzentration 60ng/µL 2,5U 1x 1,0µM 1,0µM 0,8µM - 72°C 10min finale Elongation 4°C Aufbewahrung 35 Zyklen Abbildung 2-2: Programm der Klonierungs-PCR Die Anlagerungstemperatur der Primer wurde für jedes Primerpaar entsprechend gewählt, wobei alle anderen Parameter unverändert blieben. min: Minute. 2.6.1.3 Restriktionsverdau Restriktionsendonukleasen, die keine Schnittstelle im jeweiligen Gen besaßen, wurden zur Restriktionsspaltung eingesetzt. Für die Spaltung eines PCR-Amplifikats wurden die jeweiligen Enzyme wie folgt verwendet: 24U EcoRI, 60U BamHI, 10U XmaI und 75U SalI. Die verwendete Enzymmenge wurde für den Spaltungsansatz des Vektors verdoppelt. Um die Spaltung sicherzustellen wurde mit einem Enzymüberschuss gearbeitet. OPA-Puffer wurde entsprechend des Enzyms in Endkonzentrationen von 1x oder 2x eingesetzt. Für Klonierungen erfolgte die Spaltung des Vektors und des PCR-Produkts ÜN bei 37°C. 2.6.1.4 CIAP-Behandlung Zur Verhinderung einer Religation der Vektor-DNA nach dem Restriktionsverdau wurde eine Dephosphorylierung der gespaltenen Enden unter Verwendung von CIAP durchgeführt. CIAP katalysiert die Hydrolyse von 5'-Phosphatenden der DNA. Zunächst wurden die Restriktionsendonukleasen 10min bei 65°C hitzeinaktiviert. Anschließend wurde die VektorDNA in Anwesenheit von 10x CIAP Reaktionspuffer (500mM Tris-HCl pH 9,0, 10mM MgCl2) und 2 U/µL CIAP 30min bei 37°C dephosphoryliert. Nach einer gelelektrophoretischen Auftrennung der Vektor-DNA und des PCR-Amplifikats wurde eine Gelextraktion durchgeführt. MATERIAL UND METHODEN 51 2.6.1.5 Gelextraktion Vektor- und Insert-DNA wurden mittels langwelligem UV-Licht detektiert, aus dem AgaroseGel ausgeschnitten und nach dem Protokoll des Herstellers mit dem Gelextraktionskit der Firma Qiagen aus dem Gel aufgereinigt. Zu Beginn wurde das Gelstück in entsprechendem Volumen des Puffers QG aufgenommen, 10min bei 50°C gelöst und nach Zugabe von Isopropanol auf die mitgelieferte Säule gegeben. Die durch Zentrifugation der Säule im Glasfaservlies gebundene DNA, wurde mit Puffer QG und ethanolhaltigem Puffer PE gewaschen. Nach dem Entfernen der letzten Reste an EtOH durch Zentrifugation, wurde die DNA mit Aqua ad iniectabilia Braun eluiert. Die Konzentration an gelextrahierter Vektor- und Insert-DNA wurde für die weitere Verarbeitung anhand eines Agarose-Gels im Vergleich zu der Konzentration eines λ-Markers abgeschätzt. 2.6.1.6 Ligation Im Ligationsansatz wurden 0,067 U/µL T4-DNA-Ligase und 1x Ligase-Puffer (50mM Tris-HCl pH 7,6, 10mM MgCl2, 1mM Adenosintriphosphat (ATP), 1mM DTT, 5% Polyethylenglycol) verwendet. Das molare Verhältnis von Vektor zu Insert wurde wie folgt berechnet: MolaresVerhältnis = Vektor[bp ] Insert[bp ] Dementsprechend wurde das Verhältnis von Vektor zu Insert zwischen 5:1 und 15:1 gewählt. Ligiert wurde für 5min bei Rt. 2.6.1.7 Herstellung kompetenter Bakterien Nach der Anzucht einer LB-Agarkulturplatte ÜN bei 37°C von Bakterien (TOP10, BL21, Rosetta D3) aus einem Glycerolstock, wurden mit einer gewachsenen Kolonie 100mL LBMedium angeimpft. Beim Erreichen einer OD600=0,5 wurden die Zellen abzentrifugiert (4000UPM, 10min) und das Pellet in 10mL kaltem 50mM CaCl2 aufgenommen. Es folgte eine Inkubation der Bakterien für 30min auf Eis. Die pelletierten Bakterien (4000UPM, 5min) wurden in 4mL 50mM CaCl2 gelöst. Durch Zugabe von 50% Glycerol (autoklaviert) in gleichen Volumina konnten die Bakterien in Aliquots von 200µL in flüssigem Stickstoff eingefroren und bei –80°C gelagert werden. Die kompetenten Zellen wurden durch Transformation eines bekannten Plasmids überprüft. MATERIAL UND METHODEN 52 2.6.1.8 Transformation Um die ligierte Vektor- und Insert-DNA in kompetente Bakterien zu transformieren, wurden die Bakterien auf Eis aufgetaut und der komplette Ligationsansatz zugegeben. Auf eine Inkubation des Transformationsansatzes für 30min auf Eis folgte ein Hitzeschock von 42°C für 45sek und eine Abkühlung für 2min auf Eis. Bevor die transformierten Bakterien 1h bei 37°C inkubierten, wurden 250µL warmes LB-Medium zugefügt. Der Transformationsansatz wurde anschließend auf einer LB-Amp-Platte ausplattiert und über Nacht bei 37°C herangezogen. Zur Überprüfung der Klonierung wurden mehrere Einzelkolonien von den Platten gepickt und in 3mL LB-AmpKulturen über Nacht bei 37°C schüttelnd inkubiert. Die Plasmid-DNA der Kulturen wurde wie unter 2.5.6 beschrieben isoliert und mittels Probespaltung unter Verwendung geeigneter Restriktionsenzyme analysiert. Zur Proteinexpression wurden kompetente BL21 oder Rosetta D3 Bakterien mit den Plasmiden der Expressionsvektoren transformiert. 2.6.1.9 Sequenzierung Umklonierte cDNA Abschnitte oder PCR-Produkte wurden mit dem ABI PRISM TM 310 „Genetic Analyser“ und dem „Big Dye Terminator Cycle Sequencing Ready Reaction Kit“ sequenziert. Dazu wurden in der Regel Standard-Primer aus dem Vektor verwendet, die das Insert oder die PCR-Primer flankierten (Tabelle 2-7). Tabelle 2-7: Verwendete Primer in der Sequenzier-PCR Jeweils einer der aufgeführten Primer wurde in einer Sequenzier-PCR verwendet. Primer T3 T7 pGEX.for pGEX.rev Sequenz aat taa ccc tca cta aag gg gta ata cga ctc act ata ggg c ggg ctg gca agc cac gtt tgg tg ccg gga gct gca tgt gtc aga gg Vektor pBluescript pBluescript pGEX-4T-3tag pGEX-4T-3tag In der Sequenzier-PCR wurden 400ng Plasmid eingesetzt, welche mit 12,5µM Primer und 4µL BigDye-Mix im Thermocycler mittels des nachfolgenden Programms markiert wurden (Abbildung 2-3). 95°C 15sek Denaturierung 50°C 15sek Anlagerung 60°C 4min Elongation 4°C Aufbewahrung 25 Zyklen Abbildung 2-3: Programm der Sequenzier-PCR Die Sequenzier-PCR wurde stets nach diesem Schema durchgeführt. min: Minute, sek: Sekunde. MATERIAL UND METHODEN 53 Nach der Sequenzier-PCR musste die DNA für den Einsatz in der Sequenzierung aufgereinigt werden. Hierzu wurde eine EtOH-Fällung durchgeführt. Zu einem PCR-Reaktionsansatz wurden 2µL 3M NaAc pH 5,0 und 50µL EtOH (absolut) gegeben, 10min auf Eis gefällt und 20min bei 13.000UPM, Rotor PP1/97, abzentrifugiert. Das DNA-Pellet wurde mit 70% EtOH gewaschen, 10min bei 13.000UPM, Rotor PP1/97, zentrifugiert und bei Rt getrocknet. Die so erhaltene DNA wurde in 20µL Formamid aufgenommen und im Sequenziergerät ABI PRISM TM 310 Genetic Analyzer nach der Ketten-Abbruch-Methode sequenziert. Die erhaltenen Sequenzen wurden mittels der GCG/HUSAR Programme ausgewertet. 2.6.1.10 Computer-Programme Sequenzanalysen, Primerauswahl, die Bestimmung der offenen Leserahmen, Datenbank-Suchen und Sequenzvergleiche wurden unter Verwendung verschiedener GCG-Programme (HUSAR, DKFZ) durchgeführt. 2.6.2 Proteinexpression der Antigene in E. coli Die Proteinexpression der verwendeten Tumorantigene erfolgte entweder im proteasedefizienten E. coli-Stamm BL21 (MAGE-A1, -A3, -A9, GAGE-7b, CAMEL, GBP-5ta, cTAGE-5a N- und C-terminal, BK, JC, LPV1, p16, p53) oder in E. coli BL21 Rosetta D3 (LAGE-1a, se57-1, se702, HPV1). Die letztere BL21-Subspezies enthält ein zusätzliches Plasmid, das für sechs in E. coli selten gebrauchte tRNAs codiert und zur Selektion ein Chloramphenicol-Resistenzgen trägt. In großen Proteinen kommen statistisch gehäuft diese in E. coli selten gebrauchte Codons vor. Deshalb werden große Proteine häufig besser in BL21 Rosetta D3 in voller Länge exprimiert. Proteine in E. coli BL21 wurden in LB-Medium mit 100µg/mL Amp, Proteine in BL21 Rosetta D3 in LB-Medium mit 100µg/mL Amp und 20µg/mL Chloramphenicol exprimiert. 2.6.2.1 Kulturbedingungen und Ernte der Bakterien Von allen zu exprimierenden GST-Fusionsproteinen wurden entsprechende LB-AmpAgarkulturplatten von Glycerolstocks der Bakterienkulturen angelegt. Zur Expression der GSTFusionsproteine wurde eine 50mL Vorkultur des jeweiligen BL21 Bakterienstamms mit einer Kolonie von der Kulturplatte in LB-Amp-Medium angeimpft und bei 37°C ÜN auf einem Rotationsschüttler (200UPM) inkubiert. Je 5mL der dicht gewachsenen Vorkultur wurden in sechs Erlenmeyerkolben mit je 250mL LB-Amp-Medium überführt und bei 37°C schüttelnd bis zu einer OD600=0,7-0,9 kultiviert. Nach dem Test der optimalen Expressionsbedingungen für jedes Antigen wurden MAGE-A1, -A3, -A9, GAGE-7b, CAMEL und GBP-5ta bei Rt 6h und LAGE-1a, se57-1, se70-2 sowie cTAGE-5a N- und C-terminal bei 16°C ÜN exprimiert. Durch MATERIAL UND METHODEN 54 Zugabe von IPTG in einer Endkonzentration von 0,25mM wurde die Expression der Proteine induziert. Am Ende der Inkubationszeit wurden die Bakterien mittels Zentrifugation 10min bei 5000UPM und 4°C in einer Sorvall Zentrifuge mit JA14-Rotor geerntet und die Überstände verworfen. Jedes Bakterien-Pellet wurde in 20mL 1x PBS resuspendiert und bei -20°C gelagert. 2.6.2.2 Aufschluss der Bakterien mittels French Press Nach dem Auftauen des Bakterienlysats bei Rt im Wasserbad wurde pro 1,5L Kultur eine halbe Tablette „Protease Inhibitor Cocktail CompleteTM“ zugegeben und das Lysat auf Eis gelagert, um Degradation der Proteine zu vermeiden. Die Lyse der Bakterien wurde mittels Hochdruckhomogenisator Emulsiflex-C5 (French Press) durchgeführt. Die French Press wurde ÜN bei 4°C vorgekühlt und wurde während des Gebrauchs ständig von außen mit Eis gekühlt. Jede Bakteriensuspension wurde dreimal aufgeschlossen. Anschließend erfolgte die Trennung löslicher Proteine von Zelltrümmern und unlöslichen Proteinen mittels Zentrifugation 30min bei 4°C und 14.000UPM in einer Sorvall-Zentrifuge mit JA17-Rotor. 2.6.2.3 Proteinbestimmung nach Bradford und Lagerung der Proteine Im geklärten Lysat (Lysat nach Zentrifugation) wurde die Proteinkonzentration mittels Bradford bestimmt. Hierzu wurden 10µL des Lysats mit 790µL ddH2O und 200µL Bradford Reagenz (BioRad Protein Assay Reagenz) versetzt und die Extinktion bei 595nm gegen einen Blindwert (ohne Protein) gemessen. Anhand einer Verdünnungsreihe von BSA konnte die genaue Proteinkonzentration berechnet werden. Im Anschluss wurde das geklärte Lysat mit wasserfreiem Glycerin in einer Endkonzentration von 50% gemischt und konnte ohne einzufrieren bei -20 °C gelagert werden. 2.6.3 SDS-PAGE und Western Blot Neben der Konzentrationsbestimmung der Fusionsproteine wurden diese als weitere Qualitätskontrolle im Western Blot und Coomassie-Gel nach SDS-PAGE auf ihre Größe und Expression hin überprüft. Hierfür wurde das BioRad Protean II und III Gelsystem verwendet. Die Lysate wurden in 5x SDS-PAGE-Probenpuffer (Endkonzentration 1x) aufgenommen und 5min bei 99°C erhitzt. Nach dem Säubern der Glasplatten mit 70% EtOH und dem Einsetzen derselben in die Apparatur konnte das Gelsystem verwendet werden. Für die SDS-Mini-Gele wurde zunächst das Trenngel (12% Acrylamid) gegossen, mit Isopropanol überschichtet und auspolymerisiert. Anschließend wurde das Gel mit dem Gießen des Sammelgels und Einsetzen der Kämme fertiggestellt. Die Zusammensetzung des Trenn- und Sammelgels ist Tabelle 2-8 zu entnehmen. MATERIAL UND METHODEN Tabelle 2-8: 55 Zusammensetzung von Trenn- und Sammelgel für die SDS-PAGE Die folgende Zusammensetzung des Trenn- und Sammelgels für die „sodium dodecyl sulphate polyacrylamide gel electrophoresis“ (SDS-PAGE) wurde verwendet. APS: Ammoniumpersulfat, TEMED: N,N,N`,N`-Tetramethylethylendiamin, ddH2O: bidestilliertes Wasser. Reagenz ddH2O 4x Lower Buffer 4x Upper Buffer Polyacrylamid 10% APS TEMED Trenngel (15% Polyacrylamid) 6,0mL 4,3mL 7,0mL 42,2µL 19,1µL Sammelgel (6% Polyacrylamid) 7,4mL 3,3mL 2,1mL 128,1µL 17,1µL Als Marker wurden 3µL des „Precision Plus Protein Standard Dual Color“ aufgetragen. Die Proteine wurden bei 130V für 90-100min in 1x Running Buffer aufgetrennt. Im Anschluss konnten die Polyacrylamid-Gele entsprechend der Anforderungen mit Coomassie gefärbt oder geblottet werden. 2.6.3.1 Coomassie-Färbung der SDS-PAGE-Gele Bei der Verwendung der weniger sensitiven Methode der Coomassie-Färbung wird ein Polyacrylamid-Gel für 30min in Coomassie Brillant Blue-Lösung bei Rt schüttelnd gefärbt und ÜN bei Rt mit Entfärber-Lösung entfärbt. Die sensitive Methode verwendet hingegen kolloidales Coomassie zur Färbung. Hier wird das Gel zunächst in Fixierungslösung fixiert. Eine Inkubation mit der frisch angesetzten Färbelösung erfolgt ÜN bei Rt. Erscheint das Gel am folgenden Tag ausreichend gefärbt, wird in 25% Methanol schüttelnd bei Rt kurz entfärbt. Um Gele haltbar zu machen, wurden sie in Wasser gewaschen, in einem Geltrocknungssystem zwischen zwei Celluphanfolien eingespannt und bei Rt mehrere Tage getrocknet. 2.6.3.2 Blot der Proteine Zur Detektion der Proteine mit Antikörpern wurden die Polyacrylamid-Gele nach der Auftrennung in einer BioRad Nassblotkammer zwischen vier Whatman-Papieren auf eine Nitrozellulosemembran transferiert. Der Transfer erfolgte mit 1x Transferpuffer 1h bei 100V und wurde mit einer Ponceau-Rot-Färbung überprüft (einminütige Inkubation in Ponceau S-Lösung und Entfärben mit PBS-T). 2.6.3.3 Antikörper-Inkubation und Detektion von Proteinen Um unspezifische Antikörper- und Proteinbindungsstellen der Membran zu sättigen, wurden die Membranen für 2h bei Rt mit Blockierungslösung inkubiert. Nach dreimaligem Waschen der Membran in PBS-T für 10min (Waschschritt) wurde die Inkubation der Erstantikörper in 0,5% MATERIAL UND METHODEN 56 Milchpulver (w/v) in PBS-T mit der Membran 1h bei Rt oder ÜN bei 4°C durchgeführt. Nach einem Waschschritt wurden die mit Peroxidase gekoppelten Zweitantikörper (1:10.000 in 0,5% Milchpulver (w/v) in PBS-T) 45min bei Rt auf die Membran gegeben. Der letzte Waschschritt wurde mit der Inkubation der Membran mit ECL plus-Reagenz nach Herstellerprotokoll für 1min abgeschlossen. Die in Klarsichtfolie luftblasenfrei eingeschweißte Membran wurde in der Dunkelkammer Röntgenfilmen mit Expositionszeiten von 10 sek bis 5min ausgesetzt, um bei der stattfindenden Entwicklung die gebundenen Antikörper zu visualisieren. Um eine Membran mit mehreren Antikörpern zu untersuchen, wurde sie nach der Detektion der Proteine mit einem Antikörper gestrippt. Hierfür wurde die Membran nach der Inkubation mit ECL-Reagenz in PBS-T gewaschen und anschließend 30min bei 57°C mit Stripping-Lösung inkubiert. Nach einem dreimaligen Waschschritt für 10min in PBS-T konnte die Membran für den nächsten Antikörper verwendet werden. Eine Membran konnte mindestens 2-mal gestrippt werden. Um die Qualität von eukaryotischen Proteinproben zu überprüfen und deren Quantität abzuschätzen, wurde ein Anti-β-Aktin Antikörper an derselben Membran eingesetzt und mit dem zuvor mit einem anderen Antikörper erhaltenen Ergebnis verglichen. 2.6.4 Antigen-Titration im GST-X-tag-ELISA Die Antigen-Titration der selbst hergestellten Proteine war notwendig, um die Proteine auf ihre Verwendbarkeit in der Luminex-Methode zu testen. Bei jeder Antigen-Titration betrug das Standardvolumen 100µL pro Loch der 96 Loch ELISA-Platte, falls nicht anders angegeben. ÜN wurde die 96 Lochplatte mit 2ng/µL GC in Coating-Puffer bei 4°C beschichtet. Am folgenden Morgen wurde die Platte ausgeschüttet und auf einem Papierhandtuchstapel ausgeklopft. Freie Proteinbindungsstellen der Plattenoberfläche wurden mit 180µL CaseinBlockpuffer pro Loch 1h bei Rt blockiert. Die zusätzlichen 80µL Blockierungspuffer pro Loch stellten sicher, dass über dem Standardvolumen von 100µL keine unspezifische Anlagerung von Antikörpern und anschließende Detektion im ELISA-System stattfand. Nach dem Ausschütten und Ausklopfen der Platte wurden die Antigene in einer zwölfstufigen Verdünnungsreihe in Casein-Blockpuffer mit einer Anfangskonzentration von 100µg Antigen pro Loch auf die Platte gegeben und für 1h bei Rt inkubiert. Pro Antigen wurden Doppelwerte der Verdünnungsreihe gewählt, da jedes Antigen mit zwei Antikörpern (anti-GST und anti-tag) untersucht wurde. Im Anschluss wurde die Platte durch dreimaliges Eintauchen in eine mit PBS-T (Waschpuffer) gefüllte Plastikwanne gewaschen (Waschschritt). Anschließend wurde die Platte mit dem jeweiligen Erstantikörper (Maus anti-tag 1:1000 oder Ziege anti-GST 1:10.000) in Casein- MATERIAL UND METHODEN 57 Blockpuffer für 1h bei Rt beschickt. Nach einem Waschschritt wurde ein mit Meerrettich Peroxidase (HRP) gekoppelter Zweitantikörper Ziege Anti-Maus oder Ziege Anti-Ziege (1:10.000) in Casein-Blockpuffer 1h bei Rt in den entsprechenden Löchern inkubiert. Nach einem Waschschritt fand die Farbreaktion mit 10µg/mL TMB und 0,003% (v/v) H2O2 als Substrat in Substratpuffer statt. Die Farbreaktion wurde bei entsprechender Färbung nach 5 bis 8min durch Zugabe von 50µL 1M Schwefelsäure pro Loch gestoppt. Die Extinktion der gesamten Platte wurde am ELISA-Lesegerät bei einer Wellenlänge von 450nm gemessen. Die Messwerte wurden gegen die Titrationsstufen linear/linear oder linear/logarithmisch aufgetragen. Der linear/linear Liniengraph diente zur Bestimmung der Sättigungsmenge des Antigens. Der linear/logarithmische Graph beschrieb den Titrationsverlauf. Entsprachen die Ergebnisse des jeweiligen Antigens im Western Blot und bei der AntigenTitration im ELISA den nachfolgend aufgeführten Anforderungen, konnte das Antigen in der Multiplex-Serologie eingesetzt werden. Im Western Blot musste eine prominente Bande mit dem entsprechenden Molekulargewicht des jeweiligen Antigens unter Verwendung beider Antikörper (anti-GST und anti-tag) detektiert werden. Im ELISA bestätigte ein Erreichen der Sättigung der Titrationskurve bei 25µg Protein pro Loch eine ausreichende Menge an Volle-LängeAntigenprotein. 2.6.5 Methodik der Multiplex-Serologie Im Unterschied zum ELISA werden die GST-Fusionsproteine bei der Multiplex-Serologie an GC-Beads gebunden. Diese Tatsache und die Möglichkeit der Vermessung der unterschiedlichen Beadsorten in einem Luminex-Gerät stellen sicher, dass bis zu 1000 Seren parallel mit zahlreichen Antigenen gemessen werden können. Deshalb wurde die Reaktivität aller Seren nur mittels der Multiplex-Serologie ermittelt. 2.6.5.1 Beladung der GC-Beads mit Antigenen Alle Bakterienlysate wurden auf 1mg Protein in 1mL Casein-Blockpuffer verdünnt. Pro Serum wurden 3000 Beads einer Beadsorte mit dem jeweiligen Antigen des vorbereiteten Bakterienlysats 1h bei Rt im Dunkeln zum Schutz der Farbmoleküle jeder Beadsorte schüttelnd (250UPM) beladen. Bis zu 3 Millionen Beads für die Analyse von 1000 Seren konnten mit 1mL Bakterienlysat beladen werden. Danach wurden die Beads 2min bei 13.000UPM pelletiert und dreimal mit 1mL Casein-Blockpuffer gewaschen. Schließlich wurden die Beads in einem angemessenen Volumen an Casein-Blockpuffer oder Lagerungspuffer aufgenommen. MATERIAL UND METHODEN 58 2.6.5.2 Vorinkubation von Seren Die Seren wurden in einer Verdünnung von 1:50 für 1h bei Rt auf einem Schüttler mit dem Serumvorinkubationspuffer in einer ELISA-Platte präabsorbiert. Diese Vorinkubation diente dazu, das Entstehen von Hintergrundreaktionen mit bakteriellen Proteinen und den GST- und tag-Anteilen der Antigen-Fusionsproteine zu verhindern. 2.6.5.3 Durchführung der Multiplex-Serologie-Methode Zunächst wurde jede Beadsorte 6x 30sek im Ultraschallbad behandelt, um die Beads zu vereinzeln. Danach wurde eine Beadmischung hergestellt, indem verschiedene Beadsorten beladen mit unterschiedlichen Antigenen gemischt wurden. Je 50µL präabsorbiertes Serum und Beadmix wurden in einer mit Casein-Blockpuffer äquilibrierten 96 Lochfilterplatte gemischt und für 1h bei Rt im Dunkeln auf einem Schüttler inkubiert (Serumendverdünnung 1:100). Wenn nicht anders beschrieben, wurde mit 100µL Lösung pro Loch gearbeitet. Danach wurden die Beads dreimal mit Blockpuffer auf einer Vakuum-Absaugstation gewaschen. Nach Zugabe des biotinylierten Sekundärantikörpers (1:1000 in Casein-Blockpuffer) wurde der Ansatz erneut wie oben inkubiert. Auf einen weiteren Waschschritt folgend wurden die Beads für 30min mit StrepPE (1:1000 in Casein-Blockpuffer) inkubiert. Schließlich wurden die Beads gewaschen und zur Messung in Casein-Blockpuffer aufgenommen. Während der Messung wurde neben der Autofluoreszenz die Reporter-Fluoreszenz der Beads einzeln gemessen. 2.6.5.4 Analyse der Beads Die Beadsuspension wird im Luminex-Gerät in einer Mikrokapillare vereinzelt und in einer Durchfluss-Küvette mit einer Flussrate von 1µL/sek gemessen. Dabei regt der rote (Klassifizierungs-) Laser die Beads zur Identifizierung der Beadsorte bei einer Wellenlänge von 635nm an. Hingegen regt der grüne (Reporter-)Laser die Beads bei einer Wellenlänge von 532nm an und ermittelt die Reaktivität des jeweiligen Serums mit dem entsprechenden Antigen. Da eine Photodiode die Reporter-Fluoreszenz der Beads zwischen 565 und 585nm detektiert, können unterschiedlicher Fluoreszenz-Farbstoffe (z.B. Cy3, PE, Alexa® 532) verwendet werden. Die Analyse von Beads ist auch in einer Lösung mit überschüssigem, nicht gebundenem StrepPE möglich, weil die Hintergrund-Fluoreszenz der Lösung bei 575nm automatisch von der Reporter-Fluoreszenz der Beads abgezogen wird. Ein Analog/Digital-Wandler speichert die Signalintensität anschließend im 15bit-Format, so dass die Skala der Signalintensitäten von 0 bis 32768 (215) relative Einheiten als Median Fluoreszenz-Intensität („median fluorescence intensity“, MFI) reicht. Pro Antigen wurden 100 Beads gemessen und deren MFI bestimmt. MATERIAL UND METHODEN 59 2.6.5.5 Verarbeitung und Analyse der gemessenen Daten Zusätzlich zu den zu analysierenden Antigenen wurde grundsätzlich GST als Antigen mitgeführt, um aus den Rohdaten („Brutto“-Werte) durch Subtraktion des GST-Wertes Hintergrund-korrigierte „Netto“-Werte errechnen zu können. Negative Netto-Werte wurden auf 0 gesetzt solange keine Werte unter –100 MFI erreicht wurden. War dies der Fall, wurde das betreffende Serum von der Analyse ausgeschlossen. Ob Serum in das entsprechende Loch pipettiert worden war, wurde unter Verwendung der durchschnittlichen Reaktivität des Serums gegen alle verwendeten Antigene ermittelt. Lag dieser berechnete Wert unter der durchschnittlichen Autofluoreszenz der verwendeten Beadsorten, wurde das Serum nachgetestet. 2.6.5.6 Bestimmung des Cut-off-Wertes für jedes Antigen Um eine Aussage treffen zu können, ob ein Serum positiv oder negativ mit dem jeweiligen Antigen reagierte, wurde für jedes Antigen ein spezifischer Cut-off-Wert aus den Messwerten von 100 Kontrollseren der Blutbank berechnet und unter Berücksichtigung der kumulativen Distribution aller gemessener Seren festgelegt. Von der Normalverteilung der Reaktivitäten ausgehend, wurde iterativ der Mittelwert plus zweifacher Standardabweichung für alle humanen Antigene und für alle viralen Antigene der Mittelwert plus dreifacher Standardabweichung zusätzlich unter Wegfall der außerhalb dieser Grenzen liegenden Messwerte berechnet. 2.6.5.7 Evaluierung der serologischen Daten Die Evaluierung der serologischen Daten erfolgte unter Verwendung von Microsoft Office Excel. Es wurden Säulendiagramme und kumulative Distributionen erstellt. Bei der kumulativen Distribution werden zunächst die Messwerte nach ihrer Größe sortiert und anschließend jedem Messwert ein Platz auf einer Skala von 1 bis 100% zugewiesen. Damit ergibt sich eine Verteilung der Seren einer Kohorte, die im Vergleich zu den Seren einer anderen Kohorte z.B. Ctrl beurteilt werden kann. Zur Bewertung von Serumverläufen wurde festgelegt, dass ein signifikanter Anstieg einer Seroreaktivität auftritt, wenn eine der nachfolgenden Seroreaktivitäten eine doppelte Reaktivität aufweist, deren Wert zudem über dem Cut-off-Wert des jeweiligen Antigens liegt. Ein signifikanter Abfall wurde definiert als Halbierung der Seroreaktivität bei einer nachfolgenden Seroreaktivität, solange die erste Seroreaktivität über dem Cut-off-Wert des jeweiligen Antigens lag. MATERIAL UND METHODEN 60 2.6.5.8 Statistische Auswertung der serologischen Daten – „Fisher’s Exact Test“ Der „Fisher’s Exact Test“ ist ein Unabhängigkeitstest, der häufig in der medizinischen Forschung angewendet wird. Dieser Test wird bei einer 2 x 2 Matrix verwendet, wenn die Ereignisse zweier unabhängiger Gruppen in jeweils nur eine sich gegenseitig ausschließende Kategorie fallen. Festgestellt werden soll, ob ein Unterschied zwischen den beiden Gruppen besteht hinsichtlich der Verteilung der Ereignisse in die beiden unterschiedlichen Kategorien. In dieser Arbeit wurden die MFI-Werte der einzelnen Seren anhand des Cut-off-Wertes jedes Antigens in die beiden Gruppen negative und positive Reaktivität entsprechend der Lage des Serum MFI-Wertes unter oder über dem Cut-off-Wert eingeteilt. Sobald der berechnete p-Wert unter dem Signifikanzniveau von p = 0,05 lag, wurde der Test als statistisch signifikant gewertet. 2.7 Expressionsanalyse von GAGE-7b auf RNA- und Proteinebene Um die Expression des Antigens sowohl auf RNA- als auch auf Proteinebene in MM- und CTCL-Zelllinien und Patienten zu untersuchen, wurden RNA und Protein isoliert sowie ein polyklonaler Antikörper in Kaninchen hergestellt. 2.7.1 Isolierung von RNA Die Gesamt-RNA wurde aus in flüssigem Stickstoff eingefrorenen Zellpellets von Zelllinien und Gewebe unter Verwendung des RNeasy Kit und nach Herstellerprotokoll isoliert. Die Zellpellets von 1x 107 Zellen oder 100mg Gewebe wurden in 1mL RNeasy-Lysis-Puffer und 1% β-Mercaptoethanol aufgenommen und in einer Schwingmühle homogenisiert. Für Gewebe wurde das Lysat nach der Homogenisierung zusätzlich über eine Qiagen Mini Shredder Säule gegeben, um unlösliche Zellbestandteile zu entfernen. Der Durchfluss oder das Lysat der Zellpellets wurden nach der Homogenisierung im Qiagen Mini RNeasy Kit weiterverarbeitet. Zusätzlich zum Protokoll des Herstellers wurde nach dem ersten Waschen der RNA-Säule mit RW1 Puffer ein DNase-Verdau auf der Säule durchgeführt. Zur DNase-Behandlung wurden 80µL DNase (20U) in 1x DNase-Testpuffer mit der Säule für 15min bei Rt inkubiert. Die RNA wurde mit DMPC-Wasser eluiert und nachfolgend mit kaltem 96% EtOH und 3M NaAc gefällt. Die RNA-Konzentration wurde anschließend photometrisch bestimmt. 2.7.2 RNA-Gelelektrophorese Um die Qualität der isolierten RNA zu überprüfen, wurde eine Agarose–Gelelektrophorese mit MOPS-Puffer durchgeführt. Hierfür wurden 1-2µg RNA mit 2 Volumenteilen RNA-Puffer gemischt und 10min bei 65°C denaturiert, damit die RNA einzelsträngig vorlag. Anschließend MATERIAL UND METHODEN 61 wurde die RNA-Lösung mit 2µL RNA-Ladepuffer versetzt und in einem 1% Agarose 1x MOPSGel bei 80V elektrophoretisch aufgetrennt. Die Integrität der RNA wurde bestätigt, wenn bei der Begutachtung des RNA-Gels im Geldokumentationssystem die Banden der 28S und 18S ribosomalen RNA deutlich erkennbar waren. 2.7.3 Herstellung von cDNA cDNA wurde aus der isolierten Gesamt-RNA hergestellt. Hierfür wurde der iScript cDNA Synthesis Kit verwendet, der mit Hilfe der RT den komplementären Strang zu der Gesamt-RNA polymerisiert. 1µg RNA wurde für die cDNA-Synthese eingesetzt. Während der cDNA-Synthese stellt der RNase-Inhibitor sicher, dass keine RNA-Degradation auftritt. Sowohl Oligo (dT)- als auch random Hexamer-Primer, die sich an den PolyA-Schwanz bzw. zufällig an die RNA anlagern, wurden in der Synthesereaktion verwendet. Tabelle 2-9 ist der Ansatz für die cDNA-Synthese dargestellt. Um die Primer an die RNA anzulagern, wurde der Reaktionsansatz 5min auf 25°C erwärmt. 30min lang polymerisierte die RT bei 42°C in 5’-Richtung von den Primern ausgehend den komplementären Strang. Eine Inaktivierung der RT erfolgte für 5min bei 85°C (Abbildung 2-4). Tabelle 2-9: Pipettierschema für den cDNA-Synthese-Ansatz * 1µg der in wässriger Lösung vorliegenden RNA ergibt zusammen mit Wasser 15µL. Der komplette Reaktionsansatz verwendet insgesamt 20µL. RNA: Ribonukleinsäure, DMPC: Dimethylpyrocarbonat. Reagenz 5x iScript Reaction Mix iScript Reverse Transkriptase RNA DMPC-H2O Volumen pro Ansatz 4µL 1µL Gemäß Konzentration* Ad 20µL Endkonzentration 1x Keine Angaben 1µg - Inaktivierung der RT Annealing 25 °C 5min Transkription 42°C 30min 85°C 5min 4°C Aufbewahrung Abbildung 2-4: Programm der cDNA-Synthese Diese schematische Darstellung der cDNA-Synthese stellt das verwendete Programm dar. min: Minute. Die cDNAs wurden bei -20°C gelagert und ihre Qualität in einer RT-PCR mit einem HaushaltsGen, Glycerin-Aldehyd-3-Phosphat-Dehydrogenase (GAPDH), überprüft (Abbildung 2-5). MATERIAL UND METHODEN 95°C 95°C 5min 30 sec 57°C 30 sec 72°C 72°C 30 sec 10 min 62 4°C Aufbewahrung 35 Zyklen Abbildung 2-5: Programm der GAPDH-PCR Die Qualitätsbestimmung der cDNA-Synthese erfolgte anhand einer PCR mit GAPDH-Primern. min: Minute, sec: Sekunde. 2.7.4 RT-PCR Die Expression des jeweiligen Antigens wurde unter Verwendung der RT-PCR in MM- und CTCL-Zelllinien und Geweben untersucht. Mittels der PCR ist es möglich, eine geringe Kopienzahl eines DNA-Abschnittes zu amplifizieren. Hierzu werden spezifische OligoNukleotide/Primer, die im gewünschten Fragment liegen, verwendet. Ein PCR-Reaktionsansatz enthielt die in Tabelle 2-10 aufgeführten Reagenzien. Tabelle 2-10: Reaktionsansatz für die RT-PCR Der 10x Reaktionspuffer enthält 160mM (NH4)2SO4, 670mM Tris HCl (pH 8,0), 15mM MgCl2 und 0,1% Tween 20. Das verwendete DMSO simuliert eine „Hot-Start“ PCR durch Einzelstrangstabilisierung. NTP: Nukleotidtriphosphat, DMSO: Dimethylsulfoxid, cDNA: „copy“ DNA. Reagenz 10x Biotherm Reaktionspuffer Biotherm Taq Polymerase dNTP-Mix Forward Primer Reverse Primer DMSO cDNA Aqua ad iniectabilia Braun Volumen pro Ansatz 2,5µL 0,5µL 0,5µL 1µL 1µL 2,5µL Gemäß Konzentration Ad 25µL Endkonzentration 1x 2,5U 0,8µM 1µM 1µM 10% 0,1 - 1ng - In Tabelle 2-11 sind alle Primer, die für die Expressionsanalyse verwendet wurden, aufgeführt. Für jedes Primer-Paar wurde die Annealing-Temperatur mittels Gradienten-PCR optimiert. Bei jedem PCR-Programm blieben die Denaturierung der doppelsträngigen cDNA bei 95°C, der Syntheseschritt der Polymerase bei 72°C, die Zyklenanzahl (35) und der Salzgehalt (MgCl2) unverändert (siehe Abbildung 2-2). Als Positivkontrolle diente entweder Testis cDNA oder cDNA einer positiven Zelllinie, als Negativkontrolle wurde Aqua ad iniectabilia Braun verwendet. Die PCR-Produkte wurden anschließend gelelektrophoretisch aufgetrennt, mit dem Geldokumentationssystem visualisiert und im Vergleich zur DNA-Leiter analysiert. MATERIAL UND METHODEN Tabelle 2-11: 63 In der RT-PCR verwendete Primer Jede RT-PCR wurde mit einer Positivkontrolle (cDNA aus Testis oder einer positiven Zelllinie) und einer Negativkontrolle (Aqua ad iniectabilia Braun) in mindestens zwei unabhängigen Experimenten durchgeführt. bp: Basenpaare, GAPDH: Glycerin-Aldehyd-3-Phosphat-Dehydrogenase. Untersuchtes Antigen GAPDH GAGE-1,-2,-8 GAGE-3 bis 7b Forward Primer Reverse Primer ccg ttt aca tgt tcc aat atg att cca c gac caa gac gct acg tag (vde44) gac caa ggc gct atg tac (vv-1) tca tat ttg gca ggt ttt tct aga c cca tca gga cca tct tca (vde24) cca tca gga cca tct tca (vde24) Produkt- Anlagerungslänge Temperatur 638bp 57°C 244bp 56°C 244bp 58°C Referenz (Eichmuller et al. 2001) (De Backer et al. 1999) (De Backer et al. 1999) 2.7.5 RNA-Proben Die Untersuchung der Expression erfolgte in umgeschriebener cDNA aus RNA-Proben von Gewebe von 7 verschiedenen MM-Patienten, 5 CTCL-Zelllinien (Cou-L3, HH, HuT78, MyLa und SeAx), 17 MM-Zelllinien (MeWo, MA-Mel-04, -05, -11, -12, -16, -21, -37b, -47, -52, SKMel-023, UKRV-Mel-02, -06a, -14a, -21a, -27, -31), Keratinozyten (HaCat-Zelllinie) und Melanozyten. 2.7.6 Herstellung von Dialyseschläuchen 20cm lange Stücke von Dialyseschläuchen mit einer Porengröße von 12-14kDa wurden 10min in 2% NaHCO3, 1mM EDTA pH 8,0 gekocht und anschließend in ddH2O gewaschen. Nach einem weiteren Kochschritt für 10min in 1mM EDTA pH 8,0 konnten die in ddH2O gewaschenen Schläuche in 20% EtOH bei 4°C bis zu ihrer weiteren Verwendung gelagert werden. 2.7.7 Rekombinante Expression von GAGE-7b als GST-Fusionsprotein Für die Proteinexpression und Aufreinigung ausreichender Mengen rekombinanten Proteins zur Herstellung von Antikörpern wurde der kodierende Abschnitt der cDNA von GAGE-7b in den pGEX-4T-3tag-Vektor kloniert (siehe 2.6.1) und in BL21-Bakterien exprimiert (siehe 2.6.2). Die somit erhaltenen 20mL Bakteriensuspensionen in 1x PBS konnten monatelang bis zu ihrer weiteren Aufarbeitung bei –20°C gelagert werden. 2.7.8 Aufschluss der Bakterien Die aufgetauten Bakteriensuspensionen wurden mittels French Press (siehe 2.6.2.2) oder mittels Sonifizierung aufgeschlossen. Neben der Zugabe einer Protease Inhibitor Tablette pro 20mL Suspension wurde dem Ansatz für die Sonifizierung Lysozym (50µg/mL), Triton-X-100 (0,002%), EDTA (0,1mM, pH 8,0) und DTT (1mM) zugegeben. Zunächst wurde die Suspension MATERIAL UND METHODEN 64 in einen Dounce-Homogenisator überführt und mit 15 Hüben homogenisiert. Nach 15-maliger Sonifizierung für 10sek wurde das Homogenisat einer 30-minütigen Zentrifugation bei 14.000UPM und 4°C, Rotor JA-17, unterzogen (geklärtes Lysat). Diese Zentrifugation folgte ebenfalls auf den Aufschluss der Bakterienzellen in der French Press. Anschließend wurde das geklärte Lysat durch einen Filter mit 0,8µm Porengröße von größeren Verunreinigungen geklärt. 2.7.9 Aufreinigung eines GST-Fusionsproteins an Glutathion Sepharose-Beads Das rekombinant exprimierte GST-Fusionsprotein wurde über die Affinität der N-terminalen Glutathion-S-Transferase zu Glutathion aufgereinigt und mittels reduziertem Glutathion eluiert oder das Fusionsprotein abgespalten. Dabei wurde das Protein vom Großteil der E. coli-Proteine befreit. Für eine Aufreinigung wurde Protein aus 3L Bakterienkultur verwendet, wobei ein Pellet von 1,5L Bakterienkultur in 20mL 1x PBS aufgenommen wurde. Die Aufreinigung erfolgte sowohl im Batch- als auch im Säulenverfahren. Für das Batch-Verfahren wurden zunächst 3mL Glutathion Sepharose 4B (50% Beads in 20% EtOH) bei 5000UPM für 10min, Rotor 2150, in einem 50mL Falconröhrchen zentrifugiert und anschließend 2-mal mit 5 Säulenvolumina (SV) 1x PBS gewaschen. Die gewaschenen Beads wurden in 2SV 1x PBS aufgenommen und ÜN bei 4°C auf einem Überkopfrotor mit dem geklärten Lysat inkubiert. Am folgenden Morgen wurden die Glutathion Sepharose-Beads abzentrifugiert (5000UPM, 10min, 4°C) und dreimal mit 7SV 1x PBS gewaschen, um unspezifisch gebundene Proteine zu entfernen. Anschließend wurde das GST-Fusionsprotein mit 1mL Elutionspuffer pro 1SV Glutathion Sepharose für 10min bei Rt, für 1h bei Rt und ÜN bei 4°C eluiert. Um reduziertes Glutathion des Elutionspuffers aus der Proteinlösung zu entfernen, wurde sie ÜN bei 4°C gegen 1x PBS dialysiert. Das rekombinante Protein wurde in den Eluaten mit dem anti-GST- und dem anti-tag-Antikörper im Western Blot detektiert. Für das Säulenverfahren wurde eine Plastiksäule mit Glutathion Sepharose-Beads bis zu einem Bettvolumen von 3mL gefüllt und mit 10-20SV 1x PBS bei einer Fließgeschwindigkeit von 2mL pro min gewaschen, wobei der pH zwischen 6,5 und 7,4 lag. Das geklärte Lysat des GSTFusionsproteins wurde bei Rt dreimal mit einer Fließgeschwindigkeit von 0,5mL pro min über die Säule gegeben. Um unspezifische Proteine zu entfernen, wurde die Säule mit 20 – 50SV 1x PBS gewaschen, bis die OD bei 280nm im Photometer unverändert blieb und einen Wert knapp über 0 ergab. Das gebundene GST-Fusionsprotein konnte anschließend unter Verwendung von 3SV Elutionspuffer eluiert werden oder der GST-Teil mittels Thrombin abgespalten werden. Nach der Elution oder Abspaltung des jeweiligen Proteins wurden Reste an gebundenem GSTProtein mit Elutionspuffer entfernt (sofern zuvor keine Elution durchgeführt wurde) und die MATERIAL UND METHODEN 65 Glutathion Sepharose-Säule mittels 3SV 6M Guanidin-HCl, Buffer high und Buffer low von ausgefallenen, verunreinigenden Proteinen befreit. Zwischen den jeweiligen Anwendungen wurde intensiv mit 1x PBS gewaschen, um die Integrität der Beads zu gewährleisten. Die wiederhergestellte Säule wurde in 20% EtOH bei 4°C gelagert und konnte für dasselbe Protein wieder verwendet werden. 2.7.10 Abspaltung des GST vom Fusionsprotein Die Abspaltung des GST-Moleküls vom Fusionsprotein erfolgte im Säulenverfahren. Nach der Bindung des GST-Proteins an die Glutathion Sepharose-Beads und nachfolgenden Waschschritten wurde die Säule, falls nötig, mit 3SV 1x PBS pH 7,4 äquilibriert bevor 1SV 1x PBS pH 7,4 und 100U Thrombin auf die Säule gegeben wurden. Während der 2-stündigen Inkubation bei Rt wurde die Säule alle 15min mit einer Pasteurpipette umgerührt, um zu gewährleisten, dass das zugegebene Thrombin alle Thrombinschnittstellen zwischen dem GSTMolekül und dem Fusionspartner erreicht. Am Ende der Inkubationszeit wurde das abgespaltene Protein mit 3SV 1x PBS pH 7,4 in jeweils 3mL Aliquots eluiert, die bei –80°C gelagert wurden. Das GST-Molekül sollte laut Hersteller an den Beads gebunden bleiben. Die Proteinkonzentration der Aliquots wurde im Photometer bestimmt, da die Bradford-Methode für diese Proteinkonzentrationsbestimmung zu ungenau war. Die Proteinkonzentration wurde nach folgender Formel von Warburg-Christian unter Berücksichtigung der Absorption bei 280nm und 260nm berechnet: Proteinkonzentration [mg/mL] = 1,45 x OD280nm – 0,74 x OD260nm Anschließend wurde das abgespaltene Fusionsprotein auf seine Reinheit hin im Coomassie-Gel und mittels Western Blot unter Verwendung der anti-GST- und anti-tag-Antikörper untersucht. 2.7.11 Ionenaustauschchromatographie Da nach der Abspaltung des GST-Moleküls noch verunreinigende Proteine im Coomassie-Gel sichtbar waren, war es nötig, die Proteinlösung weiter aufzureinigen. Dies war mittels einer Ionenaustauschchromatographiesäule (MonoQ) möglich, weil der isoelektrische Punkt des Zielproteins und des GST-Moleküls ausreichend weit auseinander lagen. Zunächst wurde die Proteinlösung an das Säulenmaterial gebunden, um anschließend die mittels eines Salzgradienten aufgetrennten Proteine nacheinander zu eluieren. Die Ionenaustauschchromatographie wurde in der Arbeitsgruppe von Prof. Dr. Herwig Ponstingl unter der Anleitung von Jürgen Kretschmer durchgeführt. MATERIAL UND METHODEN 66 Zum Schutz der Proteine wurde die Chromatographie bei 4°C durchgeführt. Alle verwendeten Puffer wurden mittels eines Filters von 0,45µm Porengröße filtriert, um die Funktionsfähigkeit der HPLC-Anlage zu gewährleisten. Mit Puffer A wurde die Äkta-HPLC-Anlage gründlich gespült, um Reste des Lagerungspuffers (20% EtOH) vollständig zu entfernen. Puffer B diente der Elution der gebundenen Proteine mit einem Salzgradienten. Nach dem Spülen der Anlage wurde die MonoQ HR 5/5-Säule luftblasenfrei bei einer Flussrate von 0,5mL/min angeschlossen. Die Höhe der Säulenmatrix betrug 5cm, der Durchmesser 0,5cm. Die Säule wurde vor dem Probenauftrag zweimal mit einem Salzgradienten über 15min eluiert, damit sich keine Proteinreste mehr auf der Säule befanden. Anschließend wurde die Säule mit Puffer A äquilibriert bis die Absorption bei 280nm konstant blieb. Zumeist wurden die ersten beiden Elutionen nach der Abspaltung des Fusionsproteins von seinem GST-Partner für die Ionenaustauschchromatographie herangezogen. Hierfür wurden diese Fraktionen 1:10 in Puffer A verdünnt, um die Salzkonzentration auf ungefähr 15mM NaCl zu verringern. Die Probenaufgabe erfolgte mit einer Flussrate von 0,5mL/min. Vor Beginn des Salzgradienten wurden alle ungebunden Proteine in einem Waschgang mit Puffer A von der Säule entfernt. Sobald kein ungebundenes Protein mehr von der Säule gewaschen wurde, begann die Elution der an die Säule gebundenen Proteine durch einen Salzgradienten in 45min von 0,15M auf 1M NaCl. Die Flussrate während des Salzgradienten betrug 1mL/min. Während des gesamten Laufs wurde das Eluat in 1mL Fraktionen gesammelt. Der Gesamtproteingehalt wurde als Absorption bei 280nm gemessen und im Chromatogramm aufgezeichnet. In Coomassie-Gelen konnte die Reinheit der Fraktionen bestimmt und über ihre Verwendung zur Immunisierung von Kaninchen entschieden werden. Nach der Elution der Proteine wurde die Säule über 15min mit einer Flussrate von 1mL/min von 1M auf 0M NaCl auf einen Waschgang mit Puffer A vorbereitet und schließlich in Lagerungspuffer bei Rt gelagert. 2.7.12 Umpufferung von Proteinlösungen Da die Proteinlösungen zumeist nicht in 1x PBS (nötig für die Immunisierung der Kaninchen) oder aber zu gering konzentriert vorlagen, war ein Umpufferungsschritt nötig. Dieser wurde mit NAPTM 5 Säulen (Sephadex G-25 DNA Grade) durchgeführt. Die 1mL Säulchen wurden zunächst mit 10SV 1x PBS äquilibriert, um Reste des Lagerungspuffers zu entfernen und das richtige Milieu zur Umpufferung zu schaffen. Im Anschluss wurde die in der SpeedVac auf ca. 500µL eingeengte Proteinlösung zur Proteinbindung auf die Säule gegeben. Der Durchfluss, der keine Proteine enthalten sollte, wurde aufgefangen.. Schließlich wurden die Proteine mit 1mL 1x PBS eluiert, die Säule mit 1x PBS gewaschen und unter 20% EtOH bei 4°C gelagert. Die Säule konnte für dasselbe Protein wieder verwendet werden. MATERIAL UND METHODEN 67 2.7.13 Matrix Assisted Laser Desorption/Ionisation-Time of Flight (MALDI-TOF) Nach der weitergehenden Aufreinigung des Proteins mittels Chromatographie und entsprechender Umpufferung wurden die in Coomassie-Gelen sichtbaren Proteinbanden ausgeschnitten und zur Bestätigung der Proteinidentität in der Arbeitsgruppe von Frau Dr. Martina Schnölzer (Zentrale Proteinanalytik, DKFZ, Heidelberg) unter Verwendung der MALDI-TOF Fingerprint-Technik analysiert. Die ausgeschnittenen Banden wurden mit Trypsin verdaut und mit massenspektrometrischen Methoden identifiziert (Hofmann et al. 2002). Nach einer „Post source decay fragmentation“ Analyse und anschließender Datenbanksuche konnten die Proteine identifiziert werden. 2.7.14 Immunisierung von Kaninchen Biogenes Um die Expression von GAGE-7b auf Proteinebene zu untersuchen, wurden Antikörper gegen das rekombinante GST-X-tag-Protein in Kaninchen generiert. Die beiden Proteine wurden nativ mittels Glutathion Sepharose im Batch-Verfahren aufgereinigt und mittels reduziertem Glutathion eluiert. Dieses wurde durch eine Dialyse gegen 1x PBS entfernt. Das Protein konnte im Western Blot mit den beiden Antikörpern anti-GST und anti-tag detektiert werden, woraufhin eine Immunisierung von 2 Kaninchen von der Firma Biogenes in Berlin durchgeführt wurde. Mit 1mg Protein wurden 2 verschiedene Kaninchen immunisiert. Sowohl die Präimmun- als auch die Antiseren wurden durch die Firma Biogenes im ELISA auf natürliche Reaktivitäten gegen die Proteine oder das Vektorprotein und generierte Antikörper gestestet. In Tabelle 2-12 ist das Immunisierungsschema der Kaninchen dargestellt. Tabelle 2-12: Immunisierungsschema der Firma Biogenes Nach diesem Schema wurden zwei Kaninchen durch die Firma Biogenes immunisiert. GST: GlutathionS-Transferase. Antigen Präimmunserum Antikörpername Protein Nativ 0.Tag Immunisierung 7.Tag Boost 14.Tag Boost 28.Tag Boost/Blutung 35.Tag Blutung 70.Tag Boost 77.Tag Blutung GAGE-7b P5101 P5102 K5101 K5102 GST-GAGE-7b-tag x x x x x x x / 25mL x / 25mL x / 25mL x / 25mL x x 10mL 10mL MATERIAL UND METHODEN 68 Das gelieferte Antiserum wurde mit 0,02% Thimerosal stabilisiert und bei –20°C gelagert. Die Antiseren vom 77. Tag wurden zusätzlich viermal über eine Säule, an die GST-tag-Protein gebunden war, aufgereinigt, um GST- und tag-Antikörper, die bei der Immunisierung aufgetreten waren zu eliminieren. Zentrales Tierlabor des DKFZ Da die Immunisierung von Kaninchen mit rekombinantem GST-X-tag-Protein durch die Firma Biogenes keine spezifischen Antikörper ergab und festgestellt wurde, dass der GST-Teil des Fusionsproteins sehr immunogen ist, wurde zur Immunisierung von einem Kaninchen im zentralen Tierlabor des DKFZ abgespaltenes GAGE-7b-tag-Protein verwendet. Das Protein lag in 1x PBS und steril (Sterilisierung durch Gamma-Strahlung) vor. Das Immunisierungsschema kann Tabelle 2-13 entnommen werden. Immunisierungen erfolgten sowohl subkutan in den Rückenbereich des Kaninchens als auch intravenös in die Ohrvene. Tabelle 2-13: Im Zentralen Tierlabor des DKZF verwendetes Immunisierungsschema Nach diesem Schema wurde ein Kaninchen im zentralen Tierlabor des DKFZ immunisiert. Antigen Präimmunserum Antikörpername Protein 0.Tag 28.Tag Nativ, abgespalten Immunisierung 1. Boost 38.Tag bzw. 39.Tag 45.Tag bzw. 40.Tag Blutentnahme Ausblutung GAGE-7b P80 K80 = anti-GAGE-7b GAGE-7b-tag 750µg subkutan 400µg subkutan 300µg intravenös 15mL 105mL = 57mL Serum Vor jeder Immunisierung wurden aus einer Ohrvene ca. 15mL Blut entnommen, das nach seiner Gerinnung für 10min bei 2500UPM, Rotor 2150, zentrifugiert wurde, um Serum zu erhalten. Zur Verstärkung der Immunreaktion auf das Fremdprotein wurden bei der Immunisierung 750µg GAGE-7b-tag-Protein 1:2 mit komplettem Freund’schen Adjuvans vermischt und subkutan injiziert. Um die Immunreaktion des Kaninchens zusätzlich zu boosten, wurden nach 28 Tagen 400µg GAGE-7b-tag-Protein in einer 1:2 Verdünnung mit inkomplettem Freund’schen Adjuvans subkutan und 300µg reines Protein in die Ohrvene appliziert. Ein ausreichend hoher Titer an Antikörpern wurde für das Kaninchen nach dem 1. Boost nachgewiesen, so dass das Kaninchen ohne weiteren Boost ausgeblutet werden konnte. 2.7.15 ELISA Zur Evaluierung der Kaninchenseren zu verschiedenen Zeitpunkten während der Immunisierung wurde ein ELISA durchgeführt (siehe 2.6.4). Soweit nicht anders erwähnt wurden 100µL MATERIAL UND METHODEN 69 Lösung pro Loch verwendet, und alle Inkubationsschritte erfolgten für 1h bei 37°C. Eine ELISA96 Lochplatte wurde ÜN bei 4°C oder 1h bei 37°C mit 0,5µg aufgereinigtem, rekombinantem Protein in 1x PBS beschichtet. Nach dem dreimaligen Eintauchen der Platte in eine mit PBS-T gefüllte Plastikwanne und dem Ausklopfen auf einem Papierhandtuchstapel (Waschschritt) wurden unspezifische Bindungsstellen mit 0,2% Casein in 1x PBS (180µL pro Loch) blockiert. Das Serum wurde in einer Verdünnungsreihe in 1x PBS mit der Platte inkubiert. Als Negativkontrolle wurde 1x PBS verwendet. Auf jeweils einen Waschschritt folgte die Inkubation der Platte mit Ziege anti-Kaninchen Sekundärantikörper (1:10.000 in 1x PBS) und die Farbreaktion mit TMB und H2O2 in Substratpuffer. Nach 5- bis 8-minütiger Inkubation der Platte im Dunkeln wurde die Reaktion bei entsprechender Färbung mit Schwefelsäure gestoppt. Die Extinktion bei einer Wellenlänge von 450nm wurde in einem ELISA-Lesegerät gemessen. 2.7.16 Aufreinigung von Antikörpern aus Kaninchenantiserum Um polyklonale, monospezifische Antikörper zu erhalten, wurde eine Aufreinigung der Antikörper aus dem Antiserum mittels NHS-aktivierter Sepharose durchgeführt. Zunächst wurden NHS-aktivierte Sepharose-Säulen entweder mit GST-tag-Protein oder mit Protein, das zur Immunisierung verwendet wurde, beladen. Anschließend wurde das Antiserum zunächst über die GST-tag-Säule aufgereinigt, um unspezifische GST- oder tag-Antikörper zu entfernen. Der Durchfluss dieser ersten Aufreinigung wurde im Anschluss für die Aufreinigung der spezifischen Antikörper mittels einer Säule, an die das zur Immunisierung verwendete Protein gebunden war, benutzt. Die Säule wurde während der gesamten Prozedur manuell bei Rt betrieben. Nach der Elution der spezifischen Antikörper von der 2. Säule wurde die Konzentration der Antikörperlösung bestimmt und die Lösung ÜN bei 4°C gegen 0,3M NaCl dialysiert. Aliquots der dialysierten Antikörperlösung wurden bei –80°C gelagert. 2.7.16.1 Herstellung einer NHS-aktivierten Sepharose-Säule Zunächst wurde eine Plastiksäule bis zu einem Bettvolumen von 0,5mL mit NHS-aktivierten Sepharose-Beads gefüllt. Um das Säulenmaterial zu aktivieren wurden die Beads 3x 2min mit 2SV eiskalter 1mM HCl gespült und mit 10SV Kopplungspuffer äquilibriert. Das zu bindende Protein wurde 1:2 mit Kopplungspuffer verdünnt oder ÜN gegen den Kopplungspuffer dialysiert und in 2-3SV mit den Beads für 1h inkubiert, wobei die Säule alle 15min umgerührt wurde. Ungebundenes Protein wurde in 6SV Kopplungspuffer am Ende der Inkubationszeit aufgefangen und für die Bestimmung der Kopplungseffizienz zurückgehalten. Im Anschluss wurde die Säule abwechselnd mit 3x 2SV NHS-Puffer A und NHS-Puffer B je dreimal gewaschen. Eine Inkubation mit Puffer A war für 30min als mittlerer Waschschritt erforderlich. Nachdem das MATERIAL UND METHODEN 70 Protein durch die unterschiedlichen Waschschritte fest an die Beads gebunden war, wurde die Säule mit 40SV Bindungspuffer für die Bindung der Antikörper aus dem Antiserum äquilibriert. Zur Lagerung wurde die Säule bei 4°C unter 20% EtOH aufbewahrt. Die Kopplungseffizienz der Säule wurde wie folgt berechnet: Effizienz = wobei A− B 100% A A = OD280 vor x VolumenProteinlösung vor B = OD280 nach x VolumenProteinlösung nach x Verdünnung 2.7.16.2 Antikörper-Aufreinigung aus Kaninchenantiserum Die NHS-aktivierte Sepharose-Säule mit dem jeweils gebundenen Protein wurde mit 40SV Bindungspuffer äquilibriert. Das 1:2 mit Bindungspuffer verdünnte Kaninchenantiserum wurde dreimal mit einer Flussrate von 1mL/min über die Säule gegeben. Um die Bindungen zwischen den Proteinen und Antikörpern zu festigen, wurde die Säule 1h lang mit dem Serum inkubiert. Anschließend wurde die Säule mit bis zu 100SV NHS-Waschpuffer gewaschen, bis die OD280nm einen konstanten Wert knapp über 0 erreicht hatte. Die Säule wurde mit NHS-Elutionspuffer für 5min inkubiert und die Antikörper in 1mL Aliquots eluiert. Nach der Bestimmung der Proteinkonzentration wurden die Peakfraktionen gepoolt und ÜN bei 4°C gegen den NHSWaschpuffer dialysiert. Nach der Antikörper-Elution wurde die Säule mit Bindungspuffer äquilibriert und unter 20% EtOH bei 4°C gelagert. 2.7.17 Bestimmung von Antikörpern in Serum mittels Western Blot Um Antikörper in Serum von Patienten und Kontrollpersonen mittels Western Blot zu detektieren, wurde zunächst eine SDS-PAGE mit dem rekombinanten, aufgereinigten Protein durchgeführt. Pro Spur wurden 1-2µg Protein aufgetragen (siehe 2.6.3). Das Protein wurde mittels Western Blot auf eine Nitrocellulosemembran transferiert und 1,5h mit 5% Milchpulver (w/v) in 1x PBS-T blockiert. Die Inkubation der 1:50 in 1xPBS verdünnten Seren mit schmalen Membranstreifen erfolgte ÜN bei Rt in Costar Cluster Tubes, speziellen Serumröhrchen. Nach der Inkubation der Membranstreifen mit sekundärem, Esel anti-humanem Antikörper (1:10.000 in 0,5% Milchpulver (w/v) in 1x PBS-T) für 1h bei Rt wurden die gebundenen Serumantikörper mittels DAB (3mg/mL 1x PBS) als braune Banden visualisiert. MATERIAL UND METHODEN 71 2.7.18 Western Blot Die Analyse der Proteinexpression erfolgte unter Verwendung der generierten Antikörper mittels Western Blot. Die Methode wurde wie unter 2.6.3 beschrieben durchgeführt. Pro Spur wurden 40µg Protein aus Normalgewebe aufgetragen und gleiche Volumina (20µL) an Proteinlösung, die aus Zellpellets (1x 106 Zellen) von Zelllinien isoliert wurde. 2.7.18.1 Proteinisolierung Zellpellets aus 1x 107 Zellen wurden in 200µL 0,1% Trition-X-100 lysiert und 20min auf Eis inkubiert. Durch Zugabe von 200µL 50mM Tris-HCl pH 7,5 wurden bei einer anschließenden Zentrifugation (13.000UPM, 10min, 4°C) unlösliche Zellbestandteile gefällt. Das Protein befand sich im Anschluss im Überstand und konnte für weitere Experimente verwendet werden. 2.7.18.2 Proteinproben Mit den generierten Antikörpern wurde Protein von Normalgeweben der Firmen Clontech, Palo Alto, USA (Brustdrüse, Haut, Leber, Magen, Milz, Plazenta, Testis, Thymus und Uterus) und BioCat, Heidelberg, D (Gehirn, Knochenmark, Kolon, Luftröhre, Skelettmuskulatur und Prostata) untersucht. Zudem wurde Protein von Zelllinien, Keratinozyten, Melanozyten und MM-Patienten (siehe 2.7.5) verwendet. 2.7.19 Immunhistologie Die immunhistochemische Färbung von Zellen mit Antikörpern dient der Proteinlokalisation in der Zelle. Färbungen wurden sowohl an Zytospins von Tumorzelllinien, Keratinozyten und Melanozyten (siehe 2.7.5) als auch an Paraffinschnitten von Testis, normaler Haut und 40 MMPatienten durchgeführt. 2.7.19.1 Zytospins Zytospins wurden unter Verwendung einer Zytospin-Zentrifuge mit passenden Einsätzen hergestellt. 100µL Zellsuspension wurde mit einer Zellzahl von ungefähr 1x 106 Zellen/mL in Halterungen gegeben, die auf Objektträger aufgebracht und für 4min bei 700UPM zentrifugiert wurden. Überschüssige Zelllösung wurde mit Hilfe von in die Halterung eingepasstem Filterpapier entfernt. Anschließend wurden die Objektträger mit den aufzentrifugierten Zellen für 15min bei Rt in 3,7% Formaldehyd fixiert. Diese Fixierung gewährleistete eine Lagerung über längere Zeit bei -20°C. MATERIAL UND METHODEN 72 Die Zytospins wurden nach dem Auftauen 10min in eiskaltem Aceton fixiert und anschließend 5min in 10mM Citratpuffer pH 6,0 in der Mikrowelle bei 350Watt aufgekocht. Nach dem Abkühlen wurden die Zellhaufen mit einem DakoCytomation Stift umrandet und 3x 2min in 1x PBS gewaschen. Danach liefen alle Schritte in einer Feuchtkammer bei Rt ab, wenn nicht anders erwähnt. Unspezifische Avidin- oder Biotin-Bindungen wurden durch eine je 15-minütige Inkubation mit Avidin D- und Biotin-Lösung des Avidin/Biotin blocking Kits blockiert. Ein weiterer Blockierungsschritt folgte nach einem Waschschritt von 3x 5min in 1xPBS. Die Präparate wurden mit 5% Ziegenserum in 1x PBS für 30min blockiert, um die Bindung von unspezifischen Antikörpern zu unterbinden. Der Primärantikörper (GAGE-7b 2µg/mL) wurde in 1% Ziegenserum ÜN mit den Zytospins inkubiert. Als interne Kontrolle wurde anstatt des Primärantikörpers 5% Ziegenserum und als Negativkontrolle wurde Präimmunserum verwendet. Nach einem erneuten Waschschritt (3x 5min, 1x PBS) wurde der Sekundärantikörper des ABCSystems in 10% Ziegenserum für 30min bei 37°C mit den Zellen inkubiert. Dieser Sekundärantikörper erkennt Kaninchenantikörper und ist Biotin-gekoppelt. Das Avidin und die Biotin-gekoppelte, AP des „AP Rabbit“ IgG ABC Kits wurden nach Angaben des Herstellers angesetzt. Nach einem Waschschritt erfolgte eine 30-minütige Inkubation mit dem 30min zuvor angesetzten Reagenz. Nach erneutem Waschen wurden die Proben kurz mit Tris-HCl pH 8,5 abgespült, um den richtigen pH für die anschließende Färbung herzustellen. Diese erfolgte mittels des AP-Substrat-Kits für 10-30min nach Herstellerangaben. Die gefärbten Zytospins wurden nach 3x 5min Waschen in 1x PBS und H2O mit Hämatoxylin für 5-10min gegengefärbt und durch fließendes Wasser gebläut. Zur Konservierung wurden die Zytospins mit Histogel eingedeckt. 2.7.19.2 Paraffinschnitte Gewebeschnitte von MM-Patienten, Testis und normaler Haut wurden in 4% Formaldehyd fixiert, dehydriert und in Paraffin eingebettet. Zu Beginn der immunhistologischen Untersuchung wurden die Paraffinschnitte für 3x 5 min in 100% Xylol inkubiert, um das Paraffin zu entfernen. Anschließend wurden die Schnitte in einer absteigenden Ethanol-Reihe (100%, 96%, 80%) rehydriert. Die nachfolgenden Inkubationsschritte wurden entsprechend des Protokolls der Zytospins durchgeführt. Die Konzentration des Primärantikörpers GAGE-7b betrug 0,4µg/mL. 2.7.20 Zellkultur Um Kontaminationen mit Bakterien, Hefen oder Pilzen zu vermeiden, wurden alle Arbeiten mit Zelllinien unter einer sterilen Werkbank und mit sterilen Medien und Arbeitsmaterialien durchgeführt. Die Zellen wurden in RPMI 1640-Medium, das 1% Penicillin/Streptomycin, 2% MATERIAL UND METHODEN 73 L-Glutamin und 10% Fetales Kälberserum (Vollmedium) enthielt, bei 37°C und 5% CO2 kultiviert. Da die meisten Melanomzelllinien adherente Zellen sind, wurden sie zur Subkultivierung mit Trypsin/EDTA behandelt. Alle CTCL-Zelllinien sind Suspensionszellen, so dass sie durch Zentrifugation (900UPM, 10min, 15°C) und anschließender Aufteilung subkultiviert werden konnten. Die Zelllinie HaCat wurde ebenfalls in Vollmedium kultiviert, dessen Basismedium jedoch aus DMEM bestand. Melanozyten wurden in Melanocyte Growth Medium, das mit SupplementMix supplementiert wurde, kultiviert. Um die Zellzahl zu bestimmen wurde der automatische Zellzähler Casy verwendet. ERGEBNISSE 74 3 ERGEBNISSE Ziel dieser Arbeit war es zum Einen, die Häufigkeit von Serumantikörpern bei Tumorpatienten und Kontrollpersonen gegen eine Vielzahl von Antigenen zu bestimmen und zu vergleichen. Die auf dieser Basis gewonnenen Daten sollten zur Bewertung der Verwendung von spezifischen Serumantikörperantworten in Diagnostik, Monitoring und Prognostik dienen. Zum Anderen wurde ein Cancer Testis Antigen (CTA), GAGE-7b, näher charakterisiert. In der vorliegenden Arbeit wurde die Serumantikörperantwort gegen zahlreiche Tumorantigene, die zuvor in der Arbeitsgruppe identifiziert worden waren, untersucht. Zunächst wurde ein Serumantikörper-Detektionsarray (SADA) etabliert, der auf der „Serological analysis of recombinantly expressed copy deoxyribonucleic acid (cDNA) clones“ (SEREX)-Methode basiert. Hiermit wurden zahlreiche Seren von kutanen T-Zell Lymphom (CTCL), malignes Melanom (MM) Patienten und von Kontrollen (Ctrl) auf ihre Reaktivität gegen 42 Tumorantigene gestestet. Ein zweites System zur Bestimmung der Seroreaktivität gegen ausgewählte Tumorantigene stellt die Multiplex-Serologie dar, die von Dr. Tim Waterboer in der Arbeitsgruppe (AG) von Dr. Michael Pawlita am Deutschen Krebsforschungszentrum (DKFZ) für virale Antigene entwickelt wurde. Sieben CTA, drei Tumor-assoziierte Antigene (TAA) und zwei Tumorsuppressorgene wurden in diesem System auf ihre Serumantikörperantwort in CTCL-, MM-Patienten und Ctrl untersucht. Vier virale Antigene wurden als interne Kontrolle zur Zuordnung von Seren zu Patienten mitgeführt, da sich für diese Antigene kaum Schwankungen in der Serumantikörperantwort über die Zeit ergeben. Schließlich wurde ein polyklonaler Antikörper gegen das CTA GAGE-7b hergestellt, um dieses auf Proteinebene im CTCL und MM zu charakterisieren. Im folgenden sollen zunächst die Ergebnisse, die unter Verwendung der SADA-Methode erhalten wurden, dargestellt werden, gefolgt von den mit der Multiplex-Serologie generierten Daten, und abschließend sollen die Ergebnisse zur Charakterisierung von GAGE-7b beschrieben werden. ERGEBNISSE 75 3.1 SADA Da es die SEREX-Methode nur erlaubt, ein Serum mit einem Klon zu testen, wurde auf ihrer Grundlage und in Anlehnung an Arbeiten von M. Scanlan (Scanlan et al. 2001; Scanlan et al. 2002) die SADA-Methode etabliert. Sie ermöglicht die parallele Bestimmung von Serumantikörperantworten eines Serums gegen mehrere Antigene. Es wurden 100 Seren von CTCL-Patienten, 18 Seren von MM-Patienten und 100 Seren von Ctrl auf ihre Reaktivität gegen 42 Antigene getestet. 3.1.1 Aufbau des SADA Zwei Membranen mit jeweils unterschiedlichen Antigenen wurden hergestellt, die stets eine Positivkontrolle (Membran1: PINCH, Membran 2: UVEAL Autoantigen (UACA)) und eine Negativkontrolle (blauer Phage ohne Insert im Vektor) aufwiesen. Auf Membran 1 befanden sich 20 verschiedene Antigene und auf Membran 2 waren 22 unterschiedliche Antigene angeordnet, die in früheren SEREX-Screens an einer Lymphom-, Testis-, Zelllinien- und MelanomPhagenbank identifiziert wurden (siehe Tabelle 2-4). Eine positive Antikörperreaktion wurde einem Antigen zugeordnet, wenn die Reaktion dieses Antigens auf der Membran eindeutig stärker violett war als die der Negativkontrolle. Jedes Antigen war als Duplikat auf jeder Membran gespottet, und jedes Serum wurde zweimal getestet. Eine positive Seroreaktivität wurde nur gewertet, wenn bei beiden Versuchen eine Positivität aller 4 zusammengehörender Plaques vorhanden war. Kam es zu unstimmigen Ergebnissen wurde das Serum ein drittes Mal getestet und bei positiven Ergebnissen auf zwei der drei Membranen als positiv sonst als negativ gewertet. 3.1.2 Eigenschaften der ausgewählten Antigene Die im SADA verwendeten Antigene wurden anhand ihrer serologischen Reaktivität in sekundären SEREX-Experimenten, der Ergebnisse im virtuellen Northern und ihres Zusammenhangs mit Krebserkrankungen laut Literatur ausgewählt (Eichmuller et al. 2001; Ehlken et al. 2004; Fellenberg et al. 2004; Hartmann 2004; Hartmann et al. 2004). In Tabelle 3-1 sind die Expression der Antigene auf „messenger“ Ribonukleinsäure (mRNA)-Ebene sowie wichtige Eigenschaften aus aktueller Literatur und Datenbanken (NCBI, Entrez Gene, SwissProt) dargestellt. ERGEBNISSE Tabelle 3-1: 76 mRNA Expression und Eigenschaften der SADA-Antigene Diese Tabelle beschreibt die Expression auf mRNA-Ebene (D: Differentiell, Ub: Ubiquitär, CTA: Cancer Testis Antigen) und Eigenschaften des jeweiligen Antigens. mRNA: „Messenger“ Ribonukleinsäure, GBP: Guanylat-bindendes Protein, GTP: Guanosintriphophosphat, GDP: Guanosindiphosphat, GMP: Guanosinmonophosphat, PBMC: Periphere Blut mononukleäre Zellen, CTCL: Kutanes T-Zell Lymphom, MM: Malignes Melanom, PARD3: „Partitioning defective 3 homolog“, KRAB: Krüppel-assoziierte Box, MAGE: Melanom Antigen, DPK: “Dendritic cell derived protein kinase”, ERK: Extrazelluläre regulatorische Proteinkinase, JNK: c-Jun N-terminale Kinase, SAPK: Stress-aktivierte Proteinkinase, DNEL-2: “Dynein heavy chain”, PAX9: “Paired Box 9”, BF-1: „Brain factor 1“, EST: „Expressed sequence tag“, RAP: Retinoblastom-assoziiertes Protein, DNA: Desoxyribonukleinsäure, NFκB: Nukleärer Faktor kappa B, P-TEPb: Positiver transkriptioneller Elongationsfaktor b, BRCA1: „Breast Cancer 1“, BAX: B-Zell Leukämie/Lymphom 2 (BCL2)-assoziiertes X Protein, LAR: „Leukocyte antigen-related“, EEF1A1: Eukaryotischer Translations-Elongationsfaktor 1 alpha 1, PTI-1: Prostata Tumor-induzierendes Gen 1, NFATC3: Nukleärer Faktor von aktivierten T-Zellen 3, AP-1: Aktivierungsprotein-1, HIV-1: Humanes Immundefizienz Virus Typ 1, FHL2: Vier-und-ein-halb LIM Protein 2, NP220: Nukleäres Protein 220, HPV: Humanes Papillomavirus, MAP2: Mikrotubuliassoziiertes Protein 2. Antigen, homologes Gen mRNA Expression Eigenschaften D GBP-5a, GBP-5ta (HD-CL-05) - GBPs sind große GTPasen, die GTP, GDP und GMP binden und die Hydrolyse von GTP zu GDP und GMP katalysieren. - In Normalgeweben werden beide Varianten auf Proteinebene nur in PBMC exprimiert. - GBP-5ta wird in CTCL-Tumorgewebe exprimiert. - Mehrere CTCL- und MM-Zelllinien exprimieren beide GBP-Varianten. - In CTCL-Zelllinien nicht durch Interferon induzierbar. - Isoprenylierungsregion bei GBP-5ta nicht vorhanden, da die letzten 97 Aminosäuren im Vergleich zu GBP-5a/b fehlen. pGAGEt, GAGE-3 (Van den Eynde et al. 1995; Cheung et al. 1999; Zambon et al. CTA 2001) - Multigenfamilie, die in Normalgeweben nur in Testis und in zahlreichen Neoplasien exprimiert wird. - Abgesehen von GAGE-1 unterscheiden sich die Familienmitglieder nur durch einzelne Nukleotidsubstitutionen. - Alle Familienmitglieder besitzen das Epitop zur Erkennung durch zytotoxische T-Zellen. - GAGE Expression ist mit schlechter Prognose in MM und Ösophaguskarzinom korreliert. - Am N-terminalen Ende um 25 Aminosäuren verkürzt (pGAGEt = pGAGE „truncated“). se1-1, GCC2 (Luke et al. 2003) Ub - GCC2 ist ein peripheres Membranprotein mit Verbindung zum Trans-Golgi-Netzwerk. - GCC2 ist Brefeldin A-sensitiv. - se1-1 fehlt eine GRIP-Domäne gegenüber dem homologen Gen. - se1-1 ist ein überexprimiertes Tumorantigen in Zelllinien und mögliches Onkogen. se2-1, SCP1 (de Vries et al. 2005; Neumann et al. 2005) CTA - Teil des synaptonemalen Komplexes, der für die korrekte Paarung der homologen Chromosomen während der Meiose wichtig ist. - Expression in vielen verschiedenen Neoplasien. - Sycp1-Null Mäuse sind unfruchtbar. - Antikörperantworten und ein Epitop, das von dendritischen Zellen präsentiert wird, sind bekannt. ERGEBNISSE 77 Antigen, homologes Gen mRNA Expression Eigenschaften se2-5, PARD3ta (Joberty et al. 2000) Ub - Im Vergleich zu PARD3 fehlt PARD3ta die erste der 3 PDZ-Domänen. - PARD3 ist an der Signaltransduktion während der asymmetrischen Zellteilung und polarisiertem Wachstum beteiligt. - Par6 verbindet Par3 und atypische Proteinkinase C mit Cdc42, wobei dieser Komplex für die Bildung von normalen epithelialen „Tight junctions“ an epithelialen Zell-Zell-Kontakten wichtig ist. - Differentielles Spleißen führt zu maligner Überexpression. - PARD3ta ist ein mögliches Onkogen. HD-CL-12, ZNF195 (Hussey et al. 1997) Ub - ZNF195 ist ein DNA-bindendes Protein, das möglicherweise die Regulation der Transkription beeinflusst. Die ca. 75 Aminosäuren zählende Krüppel-assoziierte Box (KRAB)-Domäne ist eine potente Repressionsdomäne, die abhängig von der DNA-Bindung ist. - HD-CL-12 fehlt die N-terminale Krüppel-assoziierte Box (KRAB)-Domäne. - ZNF195 enthält zahlreiche Cystein-Histidin-ähnliche Zinkfinger, die ein Zinkion binden können. pMAGE-A9ag, MAGE-A9 (De Plaen et al. 1994; Serrano et al. 1999) CTA - Den 12 Mitgliedern der MAGE-A-Multigenfamilie ist die MAGE-Domäne gemeinsam. - Expression in zahlreichen Tumorentitäten und in Normalgewebe nur in Testis und Plazenta, jedoch geringer im Vergleich mit anderen MAGE-A Genen. CTA cTAGE-1 (Usener et al. 2003b) - Nur in der gesunden Testis und in zahlreichen Neoplasien (CTCL, Kopf-Hals-Karzinome) exprimiert. - Serumantikörperantwort in CTCL-Patienten beschrieben. CTA cTAGE-5a (Usener et al. 2003b) - Wird auf Proteinebene nur in der gesunden Testis und in zahlreichen Neoplasien (CTCL, MM, Brusttumore) exprimiert. - Serumantikörperantwort in CTCL-Patienten beschrieben. se20-7, GOLGA4 (Kooy et al. 1992; Funaki et al. 1996) Ub - GOLGA4 ist ein peripheres Membranprotein, das mit dem Trans-Golgi-Netzwerk verbunden ist. - GOLGA4 besitzt möglicherweise eine Funktion im vesikulären Transport vom trans-Golgi-Apparat. - Autoantigen, Autoantikörper im Serum von Patienten mit Sjoegren Syndrom und Hepatitis B. - GOLGA4 besitzt eine GRIP-Domäne. du20-10L4, CKAP2 (Maouche-Chretien et al. 1998; Bae et al. 2003) Ub - CKAP2 ist ein mögliches Lymphozyten-spezifisches Differenzierungsantigen. - Erhöhte Expression von CKAP2/se20-10 in primären Magen-Adenokarzinomen und CTCL. D HD-CL-04, KIAA0555 - KIAA-Gene kodieren für große Proteine aus Gehirngewebe. - Funktion ist noch unbekannt. HD-CL-03, retSDR2 (Brereton et al. 2001; Chai et al. 2003) Ub - In vitro wandelt retSDR2 Androstan-3-alpha und 17-beta-diol in Androsteron um. Dies lässt eine Rolle im Androgenmetabolismus während der Steroidgenese vermuten. - HD-CL-03 ist ein Tumor-assoziiertes Antigen im CTCL. - Hohe Expression von retSDR2 in Zellen mit Steroidmetabolismus. - Hohe Expression von RetSDR2 in Retina, Pankreas, Niere, Leber, Lunge, Dünndarm, Gebärmutter etc. Ub HD-CL-09, DPK - HD-CL-09 weist einen Verlust der ersten 300 Aminosäuren im Vergleich zu DPK auf. - DPK verhindert die basale Aktivität der Jun Kinase. - Epidermaler Wachstumsfaktor wirkt als negativer Regulator auf DPK. - Bei Überexpression von DPK kann eine Aktivierung von ERK1/ERK2 und JNK/SAPK erfolgen. - Regulierung von Transkriptionsfaktoren für Proliferation, Transformation, Differenzierung durch DPK. - Hohe Expression in peripheren Blutleukozyten, Thymus, Milz, Niere, Skelettmuskel, Herz und Leber. ERGEBNISSE 78 Antigen, homologes Gen mRNA Expression Eigenschaften HD-MM-01, DNEL-2 (Milisav und Affara 1998) D - DNEL-2 kodiert für die schwere Kette des Dyneins, welches einen Mikrotubulus-assoziierten MotorProtein-Komplex darstellt, der vor allem für die Bewegung von Flagellen und Spermien zuständig ist. - Zusammenhang mit Infertilität bei Männern wird vermutet, da DNEL-2 in Testis exprimiert ist. - DNEL-2 könnte im Kartagener Syndrom eine Rolle spielen. HD-MM-02, PLU-1 (Tan et al. 2003) CTA - Expression in Tumorgewebe von Brustkrebspatienten, Brustkrebs- und Melanomzelllinien. - PLU-Domäne ermöglicht Bindung zweier Transkriptionsfaktoren („Paired Box 9“ (PAX9) und „Brain factor 1“(BF-1)) und somit die Unterdrückung der Transkription. - Im Mausmodell spielen Plu-1-Bf-1/Pax9 eine wichtige Rolle in der Embryogenese. - PLU-1-PAX9 Proteininteraktionen könnten eine Rolle in der Brutskrebsentwicklung spielen. - Zelluläre und humorale Immunantworten gegen PLU-1 sind beschrieben. Ub HD-MM-03, Rbbp2h1a - Rbb2h1a ist identisch zu PLU-1, besitzt jedoch ein längeres 5’-Ende (1080bp). - Interaktionspartner des Retinoblastom-Proteins. - Serumantikörperantworten gegen HD-MM-03 und HD-MM-02 lassen sich vermutlich kaum unterscheiden, da die Proteine eine sehr hohe Homologie aufweisen. D se57-1 - Homologes EST, welches durch den respiratorischen Syncytialvirus induziert wird. - Möglicher Entzündungsmarker. D (?) se89-1, RAP140 - se89-1 weist einen Verlust von 178 Aminosäuren N-terminal im Vergleich zu RAP140 sowie ein Fehlen von 20 Aminosäuren in der kodierenenden Sequenz auf. - Funktion ist noch unbekannt. HD-MM-48, RIF1 Variante 2 (Silverman et al. 2004) Ub - In der S-Phase ist RIF1 als Reaktion auf DNA-Schädigung wichtig für die Hemmung der Zellzyklusprogression. - In der Telophase ist RIF1 an kondensierten Chromosomen und in der Anaphase an Mikrotubuli der mitotischen Spindel lokalisiert. - Expression von RIF1 in Sertoli-Zellen, frühen primären Spermatozyten und Oozyten sowie embryonalen Stammzellen. Verlust der Expression während der Zelldifferenzierung. HD-MM-08, HEXIM-1 (Ouchida et al. 2003; Stone et al. 2003; Wittmann et al. Ub 2003; Michels et al. 2004) - HEXIM-1 ist ein Inhibitor des Transkriptionsfaktors NFκB und primärer Bindungspartner des Transkriptionsfaktors positiver transkriptioneller Elongationsfaktor b (P-TEPb), der eine wichtige Rolle in der globalen Kontrolle des Zellwachstums und der Zelldifferenzierung spielt. - Hemmung der Brustkrebszellproliferation durch HEXIM-1. - HEXIM-1 Expression ist in Brustkrebsgewebe erniedrigt, und Östrogene erniedrigen die HEXIM-1 Expression. - HEXIM-1 ist ein intronloses Gen, das im Nukleus lokalisiert ist. - Hexamethylen-bis-Acetamid-induzierte Expression in vaskulären glatten Muskelzellen. - Lokalisation von HEXIM-1 auf Chromosom 17q21 in einer Region mit Her2/neu und BRCA1. HD-MM-16, Humanin (Hashimoto et al. 2001a; Hashimoto et al. 2001b; Tajima Ub et al. 2002; Guo et al. 2003) - Humanin verhindert als neuroprotektiver Faktor den durch Gene der familiären Alzheimer-Erkrankung und beta-Amyloid bedingten Zelltod von Neuronen. - Humanin verhindert die Translokation von BAX vom Zytosol zu den Mitochondrien. - Humanin ist hoch homolog zur 16S mitochondrialen ribosomalen RNA. - Hohe Expression von Humanin (24 Aminosäuren) in Herz, Skelettmuskel, Niere und Leber sowie geringere Expression in Gehirn und Gastrointestinaltrakt. ERGEBNISSE 79 Antigen, homologes Gen mRNA Expression Eigenschaften HD-MM-19, ZNF133 (Margolin et al. 1994; Heiskanen et al. 2000) Ub - ZNF133 ist ein Transkriptionsfaktor der Zinkfingerprotein-Familie. - ZNF133 enthält eine KRAB-Domäne mit beiden Subdomänen (A und B) am N-Terminus. - Es konnte eine erhöhte ZNF133 Expression in einer Neuroblastomzelllinie nachgewiesen werden. HD-MM-23, LAR-interagierendes Protein 1a/b (Serra-Pages et al. 1995; Serra- Ub Pages et al. 1998) - Zugehörigkeit zur Familie der LAR Protein Tyrosin Phosphatase-interagierender Proteine (alphaLiprine). - LAR transmembrane Protein Tyrosin Phosphatasen sind wichtig in der Führung von Axonen und der Entwicklung von Brustdrüsen. - LAR-interagierendes Protein 1a/b spielt eine Rolle in Zell-Matrix-Kontakten und der Adhesionsvermittelten Signaltransduktion. HD-CL-02, Inositol-Polyphosphat-5-Phosphatase (Laxminarayan et al. 1994) Ub - Inositol-Polyphosphat-5-Phosphatase ist das hauptsächliche Isoenzym, das die Dephosphorylierung des Calcium-ausschüttenden Botenstoffes Inositol-1,4,5-Triphosphat katalysiert, somit inaktiviert und das Signal eines Neurotransmitters beendet. - Die hohe Inositol-Polyphosphat-5-Phosphatase Expression in den Purkinje Zellen des Gehirns ist begründet auf der zentralen Rolle in der Signaltransduktions-Kaskade im zentralen Nervensystem. HD-CL-08, EEF1A1 (Ditzel et al. 2000; Sang Lee et al. 2002; Talapatra et al. Ub 2002; Mansilla et al. 2005) - EEF1A1 ist die alpha Untereinheit des Elongationsfaktor 1 Komplexes. - Im Vergleich zu EEF1A1 fehlen HD-CL-08 die ersten 77 Aminosäuren. - EEF1A1 ist in zahlreichen Neoplasien überexprimiert (Prostata, Pankreas, Kolon, Lunge und Magen). - EEF1A1 reguliert die GTP-abhängige Bindung von Aminoacyl-tRNA an die 80S Ribosomen während der Proteinsynthese. - EEF1A1 ist ein Autoantigen in 66% aller Patienten mit Felty Syndrom. - Das Prostata Tumor-induzierende Gen 1 (PTI-1) stellt eine verkürzte Variante von EEF1A1 dar und ist ein Onkogen. - Überexpression von EEF1A1 führt zur selektiven Resistenz gegenüber Apoptose, die durch Stress des endoplasmatischen Retikulums verursacht wird. HD-CL-14, NFATC3 (Hoey et al. 1995; Masuda et al. 1995; Graef et al. 2001) Ub - HD-CL-14 kodiert für ein Intron (chromosomale Sequenz) von NFATC3. - NFATC3 spielt eine Rolle in T-Zellen für die Expression von Zytokingenen (v.a. IL-2) nach Stimulation der T-Zellen durch ein Antigen. - NFATC3 enthält eine Rel homologe Domäne, die wichtig für die Bindung von AP-1 ist. - NFATC3 ist im Mausembryo wichtig für die Angiogenese. HD-CL-17, Hefe ribosomales Protein S28 Homolog (Kenmochi et al. 1998) Ub - Hohe Ähnlichkeit zu ribosomalem Protein S23, das zur 40S Untereinheit der Ribosomen zählt. - Mehrere Pseudogene zum ribosomalen Protein S23 innerhalb des Genoms sind beschrieben. HD-CL-24, SART3 (Yang et al. 1999; Liu et al. 2002; Sasatomi et al. 2002) Ub - SART3 ist ein RNA-bindendes nukleäres Protein. - SART3 ist ein verbreitetes Tumorantigen, das Tumor-spezifische zytotoxische T-Zellen in Krebspatienten (Ösophagus, Kolon) bildet und für eine spezifische Immuntherapie verwendet werden könnte. - SART3 spielt eine Rolle im mRNA-Spleißen. - SART3 könnte ein zellulärer Faktor für die Genexpression von HIV-1 und die virale Vermehrung sein. - SART3 wird im Zytoplasma von allen proliferierenden normalen und malignen Zellen exprimiert sowie im Nukleus maligner Tumorzelllinien und vieler Tumorgewebe (MM, Leukämie). Ub HD-CL-30, Klon RP11-14I17 - Funktion ist noch unbekannt. se20-4, CDA-1 (Chai et al. 2001) Ub - CDA-1 ist ein Autoantigen, welches Antikörper im Serum von Patienten hervorruft. - CDA-1 ist ein negativer Regulator des Zellwachstums, wobei die Aktivität durch die Höhe der Expression und die Phosphorylierung überwacht wird. ERGEBNISSE 80 Antigen, homologes Gen mRNA Expression Eigenschaften se33-1, NP220 (Ng et al. 2002) D - NP220 ist ein DNA-bindendes Protein, das an Cytidin Cluster in doppelsträngiger DNA bindet. - Se33-1 ist ein überexprimiertes Tumor-assoziiertes Antigen. - Vier-und-ein-halb LIM Protein 2 (FHL2) ist ein Proteinbindungspartner von NP220 und scheint für die Differenzierung von Herzmuskelzellen wichtig zu sein. Ub se37-2, UBE3A - UBE3A ist ein Autoantigen und Teil des Ubiquitin Protein Degradationssystems. - Eine vererbte UBE3A Gendeletion führt zum Angelman Syndrom mit schweren motorischen und intellektuellen Störungen. - Die Reaktion von UBE3A mit dem E6 Protein von HPV16/18 bewirkt die Ubiquitin-abhängige Proteolyse von p53. pMAGE-A3ag, MAGE-A3 (Gaugler et al. 1994; Kruit et al. 2005; Ma et al. 2005; CTA Zhang et al. 2005a; Zhang et al. 2005b) - Verbreitetes Tumorantigen, das eine Expression in Normalgewebe auf Testis beschränkt aufweist und von zytotoxischen T-Zellen erkannt wird. - Wird in zahlreichen Neoplasien (MM, Lunge, Leber, Brust) exprimiert. - Verwendung als Antigen in der Krebstherapie mittels Peptid-Impfungen, Impfungen mit MAGE-A3beladenen dendritischen Zellen. Ub HD-CL-41, STK17b (Sanjo et al. 1998) - C-terminale Domäne ist wichtig für die Funktion als positiver Regulator der Apoptose. HD-CL-49, NUDC (Angus et al. 2002; Zhang et al. 2002; Aumais et al. 2003; Wei Ub et al. 2006) - NUDC besitzt eine Funktion in der Neurogenese, neuronaler Wanderung und während der Mitose bei der korrekten Bildung der mitotischen Spindeln sowie der Zell-Proliferation. - NUDC besitzt eine erhöhte Expression in sich teilenden hämatopoietischen Vorläuferzellen und Zellen von Leukämie-Patienten, erniedrigte Expression während der Reifung von Erythrozyten Vorläuferzellen. - Vermutlich ist NUDC an der Entstehung von Leukämien beteiligt. - NUDC spielt eine Rolle in der Megakaryogenese, Zytopoiese und Thrombopoiese. HD-CL-50, MAPK6 (Zhu et al. 1994; Meloche et al. 1996; Coulombe et al. 2003) Ub - MAPK6 zählt zu der Serin/Threonin Protein Kinase-Familie. - MAPK6-Aktivierung durch eine Protein Phosphorylierungs-Kaskade als Antwort auf extrazelluläre Signale führt zur Phosphorylierung von Mikrotubuli-assoziiertem Protein 2 (MAP2). - MAPK6 wird in Fibroblasten nach der Stimulation mit Phorbolestern/Serum aktiviert. - Relativ unstabiles MAPK6 Protein, welches eine höhere Halbwertszeit, Stabilität und somit eine Anhäufung während der Differenzierung von Muskelzellen aufweist. HD-CL-51, PS1D (Gueydan et al. 2002) Ub - PS1D zählt zu der Hülle der 60S ribosomalen Untereinheit in Eukaryonten. - PS1D enthält ein Zinkfingermotiv und assoziiert mit der 60S ribosomalen Untereinheit. - Funktion ist noch unbekannt. Ub HD-CL-52, BM-009 - Funktion ist noch unbekannt. HD-CL-53, PTP4A2 (Rommens et al. 1995; Zhao et al. 1996; Wang et al. 2002) Ub - PTP4A2 zählt zur Familie der Protein Tyrosin Phosphatasen. - PTP4A2 besitzt eine erhöhte Expression in Prostatatumorgewebe. - PTP4A2 ist ein Signalmolekül in vielen zellulären Prozessen. - PTP4A2 Überexpression ist mit transformiertem Phänotyp assoziiert, was eine Rolle in der Tumorgenese vermuten lässt. Ub HD-CL-54, Klon RP11-621B3 - Chromosomale Sequenz ohne bekannt Funktion. ERGEBNISSE 81 3.1.3 Validierung des SADA: Phagenmobilität und Monoklonalität Der erste Schritt für die Herstellung der SADA-Membranen bestand in der Übertragung der Phagenlösungen mittels eines Stempels aus einer 96 Lochplatte auf Agarplatten. Hierfür wurde der Stempel mehrfach in die 96 Lochplatte eingetaucht. Eine 96 Lochplatte inklusive Stempel wurde höchstens bis zu drei Tage verwendet, um Kontaminationen mit beispielsweise Pilzen oder Bakterien zu vermeiden. Um sicherzustellen, dass bei dem mehrfachen Eintauchen des Stempels in die Phagenlösungen keine Phagen in benachbarte Löcher verschleppt wurden, wurde ein Experiment mit einem Immunglobulin G (IgG)-Phagen durchgeführt. Das Proteinprodukt dieses Phagenklons reagiert direkt mit dem sekundären Antikörper ohne Bindung eines Serumantikörpers, wobei ein sehr starkes Signal entsteht. Ein IgG-Phagenklon wurde in zwei unabhängigen Ansätzen mittels eines Stempels mehrfach auf eine Membran übertragen. Um das Loch des IgG-Phagen befanden sich Löcher mit anderen Phagenklonen oder SM-Puffer. Nach der Entwicklung der Membranen konnte der IgG-Klon auf der Membran nur an der gestempelten Stelle detektiert werden. Somit kam es zu keiner Kreuzkontamination zwischen verschiedenen Phagen während des mehrfachen Stempelns und das „Springen“ der Phagen zwischen verschiedenen Löchern der 96 Lochplatte konnte ausgeschlossen werden. Im SADA wurden die Phagenlösungen zur Herstellung der SADA-Membranen auf einen Titer von ungefähr 2000 Phagen pro µL eingestellt. Als Basis dienten Phagenstocklösungen mit einem Titer von über 2000 Pfu/µL. Diese Lösungen wurden dadurch hergestellt, dass mehrere Phagenplaques in SM-Puffer aufgenommen und die Phagen durch Schütteln aus dem Agar gelöst wurden. Da mehrere Phagenplaques gepickt wurden, bestand die Gefahr, dass die Monoklonalität der Phagenlösungen nicht mehr gegeben war. Um dies zu überprüfen, wurden jeweils Phagengebrauchslösungen zweier Phagenklone von jeder Membran (se1-1, cTAGE-1, HD-MM-48 und HD-CL-53) auf ihre Monoklonalität getestet. Drei Phagenplaques einer Agarplatte des jeweiligen Phagenklons wurden gepickt, einer in vivo Exzision unterzogen und die somit erhaltenen Plasmide mit Restriktionsenzymen gespalten. Die Spaltprodukte wurden in einer gelelektrophoretischen Kammer aufgetrennt. Wie aus Abbildung 3-1 ersichtlich ist konnte für alle drei Probespaltungen des Plasmids des jeweiligen Phagenklons dasselbe Spaltmuster nachgewiesen. Somit ist die Monoklonalität nach der Vereinigung mehrerer Plaques eines Klons weiterhin vorhanden. ERGEBNISSE M 1 2 3 4 5 6 7 8 82 9 10 11 12 M M 13 14 15 16 17 18 19 20 21 M 3000bp 2000bp 3000bp 2000bp 1000bp 1000bp 500bp 500bp se1-1 Abbildung 3-1: HD-CL-53 HD-MM-48 Blauer Ph. cTAGE-1 Gelelektrophoretische Auftrennung der mit Restriktionsenzymen gespaltenen Plasmide von vier SADA-Klonen nach einer in vivo Exzision Die gelelektrophoretische Auftrennung der gespaltenen Plasmide nach der in vivo Exzision weist identische Spaltmuster in den Spuren 1-6 (se1-1), 7-12 (HD-CL-53), 13-15 (HD-MM-48), 16-18 (blauer Phage) und 19-21 (cTAGE-1) nach. M: Marker, bp: Basenpaare. 3.1.4 Validierung des SADA: Sensitivität Die Serumverdünnung von 1:100 im SADA wurde entsprechend der üblichen Serumverdünnung im SEREX (Sahin et al. 1995; Eichmuller et al. 2001) gewählt. Zur Bestätigung der Verwendbarkeit dieser Verdünnung im SADA wurden identische Serumproben eines CTCLPatienten jeweils in einer Verdünnung von 1:50, 1:100, 1:200, 1:400, 1:800 und 1:1000 unabhängig von einander präabsorbiert und im SADA getestet. Für die Serumverdünnungen 1:50, 1:100 und 1:200 waren die Antigene se2-5, se20-7 und UACA (Positivkontrolle) positiv. Bei einer Verdünnung von 1:1000 wurde keine Reaktion eines Antigens mit dem Serum des Patienten detektiert (Abbildung 3-2). Dementsprechend war eine Serumverdünnung von 1:100 eine Verdünnung, bei der noch alle Antigene, gegen welche Serumantikörper gebildet wurden, mit der SADA-Methode detektiert werden konnten. ERGEBNISSE A B C Abbildung 3-2: 83 D Entwickelte SADA-Membranen verschiedener Serumverdünnungen Unterschiedliche Serumverdünnungen wurden mit den SADA-Membranen inkubiert und anschließend entwickelt. Positive Reaktionen sind mit Pfeilen gekennzeichnet. A: Membran 1, Serumverdünnung 1:50, positiv sind se2-5 und se20-7; B: Membran 1, Serumverdünnung 1:1000, keine positive Reaktion; C: Membran 2, Serumverdünnung 1:200, positiv ist UACA; D: Membran 2, Serumverdünnung 1:1000, keine positive Reaktion. 3.1.5 Validierung des SADA: Reproduzierbarkeit Um die Reproduzierbarkeit der Präabsorption der Seren sowie der SADA-Methode selbst zu testen, wurden je zwei identische Serumproben von 5 Seren von MM-Patienten unabhängig von einander präabsorbiert. Zunächst wurden die Seren an einer mechanischen und anschließend an einer lytischen Säule präabsorbiert. Zudem erfolgte eine weitere Präabsorption der Seren mittels sechs kleiner lytischer Membranen. Zwei der zehn Seren mussten zusätzlich noch mit 3-4 großen lytischen Membranen präabsorbiert werden. Im Anschluss wurden die Seren an einer LymphomPhagenbank auf die erfolgreiche Präabsorption getestet. Keine Reaktion des Serums mit den Phagenplaques bestätigte eine erfolgreiche Präabsorption. Jedes Serum wurde in zwei unabhängigen Experimenten an je einer Membran 1 und 2 getestet. Für alle Patienten ergab sich eine Übereinstimmung hinsichtlich der Serumantikörperantwort der beiden präabsorbierten Serumproben für beide Membranen. ERGEBNISSE 84 3.1.6 Seroreaktivitäten aller SADA-Antigene gegen CTCL-, MM-Patienten und Kontrollpersonen Alle 42 Antigene wurden auf ihre Seroreaktivität mit den drei Kollektiven CTCL-, MMPatienten und Kontrollpersonen getestet. Ein statistisch signifikanter Unterschied von entweder CTCL-Patienten und Kontrollgruppe oder MM-Patienten und Kontrollgruppe wurde mit dem „Fisher’s Exact Test“ berechnet. Ein p-Wert von unter 0,05 wies einen statistisch signifikanten Unterschied der Gruppen aus. Mehrere repräsentative, entwickelte SADA-Membranen sind beispielhaft in Abbildung 3-3 dargestellt. Auffällig hinsichtlich der Seroreaktivitäten war zunächst, dass die Häufigkeit an Serumantikörperantworten deutlich niedriger lag als in bisher durchgeführten sekundären SEREX-Untersuchungen (Tabelle 3-2). Insgesamt lagen die Serumantikörperantwort- Häufigkeiten zwischen 0% bis 72%. Gegen sechs Antigene (cTAGE-1, cTAGE-5a, se89-1, HDCL-50, HD-CL-51, HD-CL-54) konnten in diesem Testsystem in keinem Kollektiv Serumantikörper detektiert werden. Das Antigen se20-7 kann als Autoantigen bezeichnet werden, da es in 67% der CTCL-Patienten, 72% der MM-Patienten und 65% der Ctrl und somit in allen drei Kollektiven häufig erkannt wurde. Fünf von 42 Antigenen (Guanylat-bindendes Protein (GBP)-5a, GBP-5ta, HD-MM-48, HDMM-19 und HD-CL-02) wiesen einen statistisch signifikanten Unterschied in der Häufigkeit ihrer Serumantikörperantworten auf. GBP-5a reagierte mit 0% der Kontrollseren, 5% der CTCLSeren und 17% der MM-Seren. Statistisch signifikant unterschieden sich die Kontroll- von der MM-Gruppe (p=0,0031). Für GBP-5ta wurde eine Seroreaktivität von 0% in Ctrl, 5% in CTCLPatienten und 28% in MM-Patienten ermittelt. Ein statistisch signifikanter Unterschied von p=0,00005 der beiden Gruppen Ctrl und MM-Patienten wurde berechnet. HD-MM-48 konnte eine Serumantikörperantwort in 3% der Ctrl, 20% der CTCL-Patienten und 17% der MMPatienten auslösen. Für dieses Antigen besteht ein statistisch signifikanter Unterschied sowohl zwischen den Kontroll- und CTCL- (p=0,0002) als auch den Kontroll- und MM-Patienten (p=0,0449). Im Gegensatz zu den bisher beschriebenen Antigenen induzierte HD-MM-19 auch im Kontroll-Kollektiv eine hohe Häufigkeit an Serumantikörperantworten mit annähernd 40%. In CTCL-Patienten wurden in fast der Hälfte aller Patienten Antikörper gegen dieses Antigen nachgewiesen, und in MM-Patienten in 83% der Fälle. Somit ergab sich ein statistisch signifikanter Unterschied (p=0,0006) zwischen der Kontroll- und der MM-Gruppe. HD-CL-02 schließlich war in seiner Serumantikörperantwort statistisch signifikant unterschiedlich im Vergleich der Kollektive Kontroll- und CTCL-Patienten (0%, 6%, p=0,0289). Auch im Kollektiv der MM-Patienten wurden Serumantikörperantworten bestimmt (6%). ERGEBNISSE 85 Membran 1 Membran 2 A B CTCL C D Ctrl E F MM Abbildung 3-3: Repräsentative SADA-Membranen verschiedener Seren Membranen A und B wurden mit CTCL-Patientenseren, C und D mit Kontrollseren und E und F mit MM-Patientenseren inkubiert. Positive Serumantikörperantworten sind für die Antigene A: se1-1, se2-5, se20-7; B: UVEAL Autoantigen (UACA), HD-MM-23; C: se2-1; D: HD-MM-48; E: GBP-5a, GBP-5ta, se1-1, se2-1, se2-5, se20-7, du20-10L4, se57-1; F: UACA, HD-MM-23 ersichtlich (siehe Pfeile). CTCL: Kutanes T-Zell Lymphom, Ctrl: Kontrollen, MM: Malignes Melanom. Im MM-Kollektiv konnten keine Serumantikörper gegen MAGE-A3 und MAGE-A9 detektiert werden. Serumantikörper gegen diese beiden CTA wurden in 1% (MAGE-A3) und 0% (MAGEA9) der Ctrl und 4% und 8% der CTCL-Patienten nachgewiesen. Serumantikörper gegen pGAGEt wurden in allen 3 Kollektiven gemessen (Ctrl. 1%, CTCL 3%, MM 6%). Neben se20-7 und HD-MM-19 wurden hohe Serumantikörper-Häufigkeiten für das Antigen HDMM-23 detektiert (Ctrl 30%, CTCL 21%, MM 50%). Für dieses Antigen liegen die Seroreaktivitäten in Ctrl höher als in der CTCL-Gruppe. 37 von 42 Antigenen hatten eine Serumantikörper-Häufigkeit von weniger gleich 5% in Ctrl, wohingegen 31 von 42 Antigenen im CTCL-Kollektiv und 25 von 42 Antigenen im MMKollektiv diese Häufigkeit aufwiesen. Insgesamt wurde festgestellt, dass beim Vergleich der Serumantikörper-Häufigkeiten größer gleich 5% mehr Antigene im MM-Kollektiv (15) im Vergleich zum CTCL-Kollektiv (6) reaktiv waren. Dies deutet auf eine höhere Immunogenität des MM im Vergleich zum CTCL hin. ERGEBNISSE Tabelle 3-2: 86 Seroreaktivitäten aller SADA-Antigene, statistische Auswertung und Ergebnisse im sekundären SEREX Die Tabelle gibt die Seroreaktivitäten aller SADA-Antigene in Prozent für die Kollektive CTCL-, MMPatienten und Kontrollpersonen an. Die statistische Auswertung erfolgte anhand des „Fisher’s Exact Test“. Ein statistisch signifikanter Unterschied zweier Kollektive basiert auf einem p-Wert < 0,05. Antigen GBP-5a GBP-5ta pGAGEt se1-1 se2-1 se2-5 HD-CL-12 pMAGE-A9ag cTAGE-1 cTAGE-5a se20-7 du20-10L4 HD-CL-04 HD-CL-03 HD-CL-09 HD-MM-01 HD-MM-02 HD-MM-03 se57-1 se89-1 HD-MM-48 HD-MM-08 HD-MM-16 HD-MM-19 HD-MM-23 HD-CL-02 HD-CL-08 HD-CL-14 HD-CL-17 HD-CL-24 HD-CL-30 se20-4 se33-1 se37-2 pMAGE-A3ag HD-CL-41 HD-CL-49 HD-CL-50 HD-CL-51 HD-CL-52 HD-CL-53 HD-CL-54 Ctrl CTCL (n=100) (n=100) 0% 0% 1% 19% 2% 12% 0% 0% 0% 0% 65% 0% 0% 0% 1% 0% 0% 0% 2% 0% 3% 3% 3% 39% 30% 0% 1% 2% 0% 0% 2% 3% 0% 0% 1% 0% 1% 0% 0% 2% 2% 0% 5% 5% 3% 17% 8% 14% 1% 4% 0% 0% 67% 4% 0% 2% 7% 0% 1% 0% 5% 0% 20% 6% 9% 48% 21% 6% 2% 2% 2% 5% 5% 3% 1% 1% 8% 2% 0% 0% 0% 3% 1% 0% MM (n=18) 17% 28% 6% 28% 17% 17% 0% 0% 0% 0% 72% 6% 6% 0% 0% 6% 6% 6% 11% 0% 17% 0% 0% 83% 50% 6% 0% 0% 0% 0% 0% 0% 0% 0% 0% 0% 0% 0% 0% 0% 0% 0% p-Wert CTCL Ctrl 0,0002 0,0289 - p-Wert MM Ctrl 0,0031 0,00005 0,0449 0,0006 - SEREX Ctrl SEREX CTCL 0% (0/14) 18% (2/11) 0% (0/14) 56% (9/16) n.g. 16% (3/19) 0% (0/5) 50% (5/10) 0% (0/10) 22% (4/18) 0% (0/5) 30% (3/10) 0% (0/8) 33% (4/12) n.g. 22% (4/18) 0% (0/17) 33% (10/30) 0% (0/14) 15% (3/20) 0% (0/5) 30% (3/10) 0% (0/10) 11% (2/18) 0%(0/7) 33% (7/21) 0% (0/6) 48% (10/21) 0% (0/8) 41% (7/17) n.g. n.g. n.g. n.g. n.g. n.g. 0% (0/5) 33% (3/9) 0% (0/9) 55% (12/22) n.g. n.g. n.g. n.g. n.g. n.g. n.g. n.g. n.g. n.g. 0% (0/5) 19% (4/21) 0% (0/8) 6% (1/16) 0% (0/8) 25% (3/12) 50% (4/8) 33% (4/12) 0% (0/6) 14% (1/7) 0% (0/6) 14% (1/7) 0% (0/5) 30% (3/10) 0% (0/10) 29% (5/17) 0% (0/10) 29% (5/17) n.g. 6% (1/18) 0% (0/6) 43% (3/7) 0% (0/6) 43% (3/7) 0% (0/6) 14% (1/7) 0% (0/6) 29% (2/7) 0% (0/6) 43% (3/7) 0% (0/6) 14% (1/7) 0% (0/6) 14% (1/7) SEREX MM n.g. n.g. n.g. n.g. n.g. n.g. n.g. 20% (3/15) 7% (1/15) 0% (0/15) n.g. n.g. n.g. n.g. n.g. 8% (1/12) 17% (2/12) 17% (2/12) n.g. n.g. n.g. n.g. 8% (1/12) 25% (3/12) 17% (2/12) n.g. n.g. n.g. n.g. n.g. n.g. n.g. n.g. n.g. 27% (4/15) n.g. n.g. n.g. n.g. n.g. n.g. n.g. Seroreaktivitäten aller SADA-Antigene früherer sekundärer SEREX-Untersuchungen sind aufgeführt (Eichmuller et al. 2001; Ehlken et al. 2004; Fellenberg et al. 2004; Hartmann 2004; Hartmann et al. 2004). Ctrl: Kontrollen, n.g.: nicht getestet, -: statistisch nicht signifikant unterschiedlich, n: Anzahl, ERGEBNISSE 87 CTCL: Kutanes T-Zell Lymphom, MM: Malignes Melanom, SADA: Serumantikörper-Detektionsarray, GBP: Guanylat-bindendes Protein, MAGE: Melanom Antigen. Desweiteren induzierten 14 Antigene (GBP-5a, GBP-5ta, HD-CL-12, pMAGE-A9ag, du2010L4, HD-CL-03, HD-MM-02, HD-MM-03, HD-CL-02, HD-CL-17, HD-CL-24, se33-1, se371, HD-CL-41) keine Serumantikörperantwort in den Ctrl, wohingegen entweder im CTCL- und oder im MM-Kollektiv Serumantikörperantworten detektiert werden konnten Obwohl diese Serumantikörperantworten in den Patientenkollektiven zumeist sehr gering waren, können diese Antigene als serologisch tumorspezifisch bezeichnet werden. Im Vergleich zu früheren Ergebnissen im sekundären SEREX ist festzustellen, dass die Frequenzen der Seroreaktivitäten in Ctrl bis auf die vier Ausnahmen se1-1, se2-5, se20-7, HDCL-17 sehr ähnlich sind. Bei der Betrachtung des CTCL-Kollektivs liegen die Frequenzen der Seroreaktivitäten im SADA deutlich unter denjenigen im sekundären SEREX. Ausnahmen sind die Antigene se20-7, HD-CL-08 und pMAGE-A3ag. Für diejenigen Antigene, die für ein MMKollektiv sowohl im SADA als auch im sekundären SEREX evaluiert wurden, ergeben sich deutliche Unterschiede für die Antigene HD-MM-19 und HD-MM-23. Allerdings muss beachtet werden, dass die Kollektivgrößen sehr unterschiedlich waren. ERGEBNISSE 88 3.2 ELISA und Multiplex-Serologie Die Bestimmung der Serumantikörperantwort unter Verwendung der SEREX- und SADAMethode ist sehr zeitaufwendig. Deshalb wurde in dieser Arbeit ein weiteres System getestet, das Serumantikörperantworten in einem Hochdurchsatzverfahren misst: die Luminex-Technologie – die ein Multiplex-System ist. Sieben CTA, drei TAA und zwei Tumorsuppressorgene wurden in diesem System auf ihre Serumantikörperantwort in 144 CTCL-, 191 MM-Patienten, 100 Kontrollpersonen und 25 immunbehandelten Tumorpatienten untersucht. Zudem dienten vier virale Antigene als interne Ctrl zur Überprüfung der Zugehörigkeit von Seren zu Patienten. Die Multiplex-Serologie stellt eine Weiterentwicklung des GST-X-tag-„Enzyme-linked immunosorbent assay“ (ELISA) dar. Beide Systeme wurden in der Gruppe von Dr. M. Pawlita am DKFZ für virale Antigene entwickelt. Der ELISA ist der zur Zeit meistgebrauchte immunologische Test, um den Immunstatus eines Patienten zu untersuchen und wird in diesem Zusammenhang zumeist als „Antibody Capture Assay“ verwendet. Um allerdings noch mehr Seren gleichzeitig mit einer großen Anzahl von Antigenen testen zu können, entwickelte Dr. T. Waterboer die Multiplex-Serologie als Hochdurchsatzverfahren. Der GST-X-tag-ELISA und die Multiplex-Serologie besitzen einen entscheidenden Vorteil zu anderen klassischen „Antibody Capture Assays“. Rekombinant hergestellte Antigene werden direkt auf der Platte bzw. an Beads gebunden und dabei aufgereinigt, weshalb eine aufwendige Aufreinigung der Proteine im Vorfeld entfällt. Beide Methoden beruhen auf der Bindung der Glutathion-S-Transferase (GST) an Glutathion, welches kovalent an Casein gekoppelt, auf eine solide Plastikoberfläche oder an Beads gebunden vorliegt (Abbildung 3-4). In einer Ein-Schritt-Aufreinigung (In-situ Affinitätsaufreinigung) werden die Antigene direkt aus dem bakteriellen Lysat entweder auf der ELISA-Platte oder in der Beadsuspension von den restlichen Lysatbestandteilen abgetrennt (Sehr et al. 2001; Waterboer 2005; Waterboer et al. 2005). In der vorliegenden Arbeit wurde die Luminex-Technologie erstmals zur Bestimmung von Serumantikörpern gegen humane Selbstantigene etabliert und eingesetzt. 3.2.1 Die Grundlage der Multiplex-Serologie Die Basis der Multiplex-Serologie ist die Luminex-Technologie (xMAPTM), deren Grundeinheit Polystyrol-Kügelchen (Beads) sind, die einen Durchmesser von 5,6µm besitzen. Da die Beads mit zwei verschiedenen Fluoreszenz-Farbstoffen gefüllt sind, lassen sich 100 distinkte Beadsorten unterscheiden. Diese Beads werden in einem Luminex-Analysegerät untersucht, das weitgehend einem Durchflusszytometer mit zwei Lasern entspricht. Die beiden in die Beads gefüllten Fluoreszenz-Farbstoffe werden mit Licht einer Wellenlänge von 635nm ERGEBNISSE 89 (Klassifizierungs-Laser) angeregt. Die Identifizierung der Beadsorte erfolgt durch die Detektion der Emissionsmaxima der beiden Farbstoffe. Der grüne (Reporter-)Laser erlaubt die Quantifizierung der Reporter-Fluoreszenz durch eine Anregung der Beads bei 532nm (Abbildung 3-4). A Streptavidin-RPhycoerythrin B HRP-/Biotin-gekoppelter Sekundärer Antikörper Serum Antikörper t ag GST GST GST GST casein casein ELISA Platte Abbildung 3-4: t ag Tumorantigen als GST-tag-Fusionsprotein Glutathion-Casein Bead Schematische Darstellung von „GST-capture-ELISA“ (A) und MultiplexSerologie (B) Glutathion-Reste sind als grüne Pfeilspitzen dargestellt. An ELISA-Platten bindet das GC mittels Physicosorption, wohingegen es kovalent an Luminex-Beads gekoppelt wird. Das Glutathion-Casein bindet zunächst an die ELISA-Platte oder das Bead. Anschließend wird das Tumorantigen in-situ affinitätsaufgereinigt. Mittels eines HRP-gebundenen oder biotinylierten sekundären Antikörpers werden die gebundenen Serumantikörper erkannt und über eine Farbreaktion oder Streptavidin-Phycoerythrin detektiert. HRP: „Horseradish peroxidase“, GC: Glutathion-Casein. Die Beadsuspension eines Lochs der 96 Lochplatte wird so lange vermessen, bis von jeder vorhandenen Beadsorte mindestens 100 einzelne Beads analysiert werden konnten. Die Bindungsreaktionen auf jeder Beadsorte werden als Median der Reporter-FluoreszenzIntensitäten („mean fluorescence intensity“ – MFI) der 100 analysierten Einzelbeads errechnet. Im Vergleich von GST-X-tag-ELISA und Multiplex-Serologie wurde im Wesentlichen die feste Phase ausgetauscht. Für jedes Antigen wurde ein Loch einer ELISA-Platte durch eine LuminexBeadsorte ersetzt (Abbildung 3-5). Zu Beginn fand eine in-situ Affinitätsaufreinigung der GSTFusionsproteine aus dem Bakterienlysat statt. Kovalentes Koppeln von Glutathion-Casein (GC) an die Beads ermöglichte die direkte Bindung von GST-Fusionsprotein, und ungebundenes Bakterienprotein konnte durch Waschen der Beads entfernt werden. Unterschiedliche GCBeadsorten wurden mit verschiedenen Antigenen beladen (eine Sorte pro Antigen), separat gewaschen und schließlich zu einer Beadmischung vereint. Diese Beadmischung wurde in einer ERGEBNISSE 90 96 Lochplatte mit Filterboden (Waschplatte) mit gegen GST-tag- und E. coli-Protein präabsorbiertem Serum inkubiert. Ungebundene Serumproteine wurden durch das Anlegen eines Vakuums an die Waschplatte entfernt, wobei die Beads vom Filter zurückgehalten wurden. Nach erneutem Waschen der Beads wurden gebundene Serumantikörper durch biotinylierten Sekundärantikörper und nach einem weiteren Waschschritt durch Streptavidin-R-Phycoerythrin detektiert. Nach dem Einsetzen der Waschplatte in den xy-Rastertisch des LuminexAnalysegeräts saugte die Hohlnadel sukzessive aus jedem der 96 Löcher das entsprechende Volumen an Lösung auf. Anschließend wurde jede Beadsuspension nach und nach im Analysegerät vermessen. Abbildung 3-5: Schematische Darstellung des Luminex-Analysegeräts Zu Beginn wird eine Mischung verschiedener Beadsorten (verschieden farbige Beads) mittels einer Hohlnadel aus einem Loch einer 96 Lochplatte aufgesogen (nicht dargestellt). Die Beadmischung wird in einer Mikrokapillare vereinzelt und in einer Durchflussküvette unter Verwendung von zwei Lasern analysiert (Quelle: www.luminexcorp.com). Im Vergleich zum GST-X-tag ELISA hat die Luminex-Technologie die folgenden Vorteile: o Erhöhter Durchsatz o Serumsparend, da mit einer Probe theoretisch die Antikörperantwort gegen bis zu 100 Antigene parallel gemessen werden kann. o Mehrfachbestimmungen bzw. Wiederholungen entfallen, da pro Antigen mindestens 100 Messwerte erzeugt werden, deren Median als MFI-Wert berechnet wird. 3.2.2 Verwendete Antigene in der Multiplex-Serologie In Tabelle 3-3 sind alle humanen, in der Multiplex-Serologie verwendeten Antigene beschrieben. Die Antigene MAGE-A1, -A3, -A9, LAGE-1a, GAGE-7b, CAMEL und cTAGE-5a N-terminal und C-terminal wurden in den pGEX-4T-3-tag-Vektor kloniert. Dr. F. Fellenberg führte diese ERGEBNISSE 91 Klonierungsarbeiten für die Antigene se70-2, se57-1 und GBP-5ta durch sowie Dr. P. Sehr und Dr. C. Michael für die Tumorsuppressorgene p53 und p16 und die viralen Antigene BK, JC, LPV und Humanes Papillomavirus 1 (HPV1). Alle Tumorantigene (X) wurden als Fusionsproteine mit N-terminaler GST und C-terminalem Markerepitop (tag) als GST-X-tagProteine bakteriell exprimiert. Der Sinn des einheitlichen Markerepitops (tag) am C-Terminus besteht darin, dass eine vergleichende Quantifizierung aller eingesetzter Fusionsproteine im GST-X-tag-ELISA möglich ist. Tabelle 3-3: Übersicht über die verwendeten humanen Tumorantigene In der Tabelle sind Übersichtsdaten in Bezug auf mRNA, kodierendes Protein („open reading frame“ – ORF), Homologien, mRNA Expression und chromosomale Lokalisation der in der Multiplex-Serologie verwendeten humanen Tumorantigene und Tumorsuppressorgene aufgeführt. bp: Basenpaare, AS: Aminosäuren, RSV: „Respiratory syncytial virus“, EST: „Expressed sequence tag“, CTA: Cancer Testis Antigen, D: Differentiell, U: Ubiquitär. Tumorantigen/ mRNA Tumor[bp] suppressorgen ORF [AS] Homologes Gen Homologes EST mRNA Chromosom Expression MAGE-A1 1722 310 MAGE-A1 - CTA Xq28 MAGE-A3 1753 314 MAGE-A3 - CTA Xq28 MAGE-A9 1824 315 MAGE-A9 - CTA Xq28 LAGE-1a 766 180 LAGE-1a - CTA Xq28 GAGE-7b 526 117 GAGE-7b - CTA Xp11.2-4 CAMEL 993 117 CAMEL - CTA Xq28 se70-2 1592 306 Neu „Large cell D 13q33.3-34 carcinoma, adipose“ se57-1 3997 336 Neu RSV-induziertes EST D 18q21 GBP-5ta 1981 489 Guanylat-bindendes Protein - D 1p22.2-3 cTAGE-5a Nterminal 2778 408 Neu - CTA 14q13.3 cTAGE-5a Cterminal 2778 369 Neu - CTA 14q13.3 p53 2629 393 p53 - U 17p13.1 p16 1163 156 p16 - U 9p21 3.2.3 Qualitätskontrolle der Antigene und der Multiplex-Analyse Da, wie bereits beschrieben, die Multiplex-Serologie eine Weiterentwicklung des GST-X-tagELISA ist, wurde die Verwendbarkeit der Antigene für das Luminex-Verfahren im Vorfeld durch einen GST-X-tag-ELISA ermittelt. Wurde im ELISA eine Sättigungskurve für die ERGEBNISSE 92 Antigene ermittelt und die Sättigung unterhalb von 25µg pro Loch erreicht, waren die Antigene für den Einsatz in der Multiplex-Serologie geeignet (Abbildung 3-6). A: GST-Antikörper 2,5 OD 450nm 2,0 1,5 1,0 GST MAGE-A3 CAMEL GAGE-7b cTAGE-5a C-terminal se57-1 0,5 0,0 0 20 40 MAGE-A1 MAGE-A9 LAGE-1a cTAGE-5a N-terminal GBP-5ta se70-2 60 80 100 Gesamtprotein pro Loch [µg] B: tag-Antikörper 2,5 OD 450nm 2,0 1,5 1,0 GST MAGE-A3 CAMEL GAGE-7b cTAGE-5a C-terminal se57-1 0,5 0,0 0 20 40 MAGE-A1 MAGE-A9 LAGE-1a cTAGE-5a N-terminal GBP-5ta se70-2 60 80 100 Gesamtprotein pro Loch [µg] Abbildung 3-6: Titration der Antigene für die Multiplex-Serologie im GST-X-tag-ELISA Mit Glutathion-Casein beschichtete ELISA-Platten wurden mit Lysaten transformierter Bakterien inkubiert, die GST-tag- oder GST-X-tag-Fusionsproteine produziert hatten. Die Menge des Lysatproteins wurde von 100µg bis 0,05µg pro Loch titriert. Gebundenes Antigen wurde mittels polyklonalem GSTAntikörper (A) oder monoklonalem tag-Antikörper (B) quantifiziert. Dargestellt ist eine repräsentative Antigentitration für alle selbst hergestellten Antigene. Laut den Sättigungskurven sind alle Antigene zur Verwendung in der Multiplex-Serologie geeignet. OD: Optische Dichte. Hierfür wurden die Proteinlysate titriert und in Duplikaten aufgetragen. Mit Hilfe von Antikörpern gegen GST wurde gebundenes Fusionsprotein gemessen. Der tag-Antikörper erkennt den C-Terminus und eignet sich daher zur Bestimmung von Volle-Länge-Protein. Für ERGEBNISSE 93 alle selbst hergestellten Antigene wurden Sättigungen in ähnlicher Höhe mit beiden Antikörpern nachgewiesen. Alle Antigene waren für den Einsatz in der Multiplex-Serologie geeignet. Während der Durchführung der Multiplex-Analyse wurden interne Kontrollen mitgeführt. Bei der Begutachtung der MFI-Werte wurden zunächst für jedes Serum die MFI-Werte gegen alle Antigene inklusive GST-tag gemittelt. Dieser für jedes Serum berechnete MFI-Mittelwert musste größer als 20 MFI sein, sonst bestand der Verdacht, dass kein Serum in das entsprechende Loch pipettiert worden war. Für alle untersuchten Antigene traf diese Bedingung zu. Des Weiteren wurde ein Loch ohne Serum und ein Loch ohne sekundären Antikörper jedoch ansonsten mit allen verwendeten Reagenzien angesetzt. Die Mittelwerte der MFI-Werte aller Antigene für diese beiden Löcher sollten ebenfalls unter 20 MFI liegen. Dies traf mit einem Mittelwert von 3 MFI für das Loch ohne sekundären Antikörper zu. Für das Loch ohne Serum ergab sich ein MFIMittelwert von 37, der etwas erhöht ist und den Hintergrundwert ergibt. Da ansonsten alle anderen internen Kontrollen geeignet erschienen, wurde dieses Ergebnis als Ausreißer betrachtet. Schließlich wurde mittels Antikörpern gegen in der Multiplex-Serologie verwendete Antigene überprüft, ob die Bindung von Antigen an die jeweils ausgewählte Beadsorte erfolgreich und richtig war. Dies erfolgte mit drei Antikörpern (gegen die Antigene MAGE-A9, GAGE-7b und cTAGE-5a), da nur gegen diese Antigene Antikörper zur Verwendung in der MultiplexSerologie vorlagen. Der MAGE-A9-Antikörper reagierte stark gegen das Antigen MAGE-A9 bei einer Verdünnung von 1:1000 mit 3294 MFI. Zusätzlich wurden schwächere Reaktionen gegen MAGE-A1 (67 MFI) und MAGE-A3 (225 MFI) detektiert. Der GAGE-7b-Antikörper reagierte stark gegen GAGE-7b mit 1604 MFI bei einer Verdünnung von 1:100. cTAGE-5a wurde vom cTAGE-5a-Antikörper bei einer Verdünnung von 1:50 mit 902 MFI identifiziert, wobei der Antikörper nur das C-terminale Proteinende erkannte. Der tag-Antikörper stellte die letzte interne Kontrolle dar. Mit diesem Antikörper wurde die Verteilung von Voll-Länge-Protein auf den Beads überprüft. Der MFI-Mittelwert für diesen Antikörper lag bei 2570 MFI. Dies bedeutet, dass die durchgeführte Multiplex-Serologie die Voraussetzungen für einen erfolgreichen Assay erfüllte und die erhaltenen MFI-Werte valide waren. 3.2.4 Festlegung der Cut-off-Werte und statistische Auswertung Zur Festlegung der Cut-off-Werte für alle verwendeten Antigene wurde eine unabhängige Gruppe von 100 Kontrollseren herangezogen. Der Mittelwert plus zweifacher Standardabweichung der Antikörperantworten dieser Ctrl wurde als Cut-off für alle humanen Antigene definiert. Für die viralen Antigene wurde der Cut-off-Wert auf 250 MFI bzw. 100 MFI ERGEBNISSE 94 festgelegt (Waterboer et al. 2005). Daraus ergaben sich die in Tabelle 3-4 dargestellten Cut-offWerte. Tabelle 3-4: Cut-off-Werte für alle Antigene In dieser Tabelle sind die Cut-off-Werte für alle in der Multiplex-Serologie verwendeten Antigene dargestellt. Für alle humanen Antigene wurde der Cut-off-Wert anhand eines unabhängigen KontrollKollektivs von 100 Kontrollpersonen als Mittelwert plus zweifacher Standardabweichung bestimmt. Die Cut-off-Werte für die viralen Antigene wurden festgelegt. MFI: „Mean fluorescence intensity“. Antigen Cut-off-Wert [MFI] Antigen Cut-off-Wert [MFI] MAGE-A1 102 cTAGE-5a N-terminal 1781 MAGE-A3 366 cTAGE-5a C-terminal 2564 MAGE-A9 256 p53 1403 LAGE-1a 85 p16 636 GAGE-7b 59 BK 250 CAMEL 169 JC 250 se70-2 752 LPV 250 se57-1 649 HPV1 100 GBP-5ta 1488 Der prozentuale Anteil an positiven Seren pro Kollektiv wurde anhand der Cut-off- und MFIWerte aller Seren berechnet. Zur statistischen Auswertung der Multiplex-Serologie Ergebnisse wurden die prozentualen Anteile an positiven Seren der unterschiedlichen Patientenkollektive und Stadiengruppen gegen das Kontroll-Kollektiv unter Verwendung des „Fisher’s Exact Tests“ verglichen. Ein statistisch signifikanter Unterschied lag vor, wenn der p-Wert zwischen dem Kontroll-Kollektiv und einem Patienten- oder Stadien-Kollektiv unterhalb von 0,05 berechnet wurde. Um einen statistisch signifikanten Unterschied der verschiedenen Stadiengruppen gegenüber dem Kontroll-Kollektiv zu sichern, musste neben dem oben erwähnten p-Wert ebenfalls der p-Wert, der über alle Stadien und das Kontroll-Kollektiv berechnet wurde unterhalb von 0,05 liegen. Sonst konnte nur eine Tendenz festgestellt werden. Nachfolgend sind die prozentual positiven Seren pro Antigen zum einen in Säulendiagrammen dargestellt. Zum anderen werden zur graphischen Darstellung der prozentualen Verteilung von Seren im Verhältnis zu den entsprechenden MFI-Werten kumulative Distributionen verwendet. In dieser besonderen Darstellung werden die erhaltenen Messwerte zunächst nach ihrer Größe sortiert. Im Anschluss wird jedem Messwert ein Platz auf einer Skala von 1 bis 100% zugewiesen. Die x-Achse wird als prozentuale Achse gewählt, wobei sich die Seren mit den höchsten MFI-Werten in der linken Hälfte der Achse befinden. Die y-Achse repräsentiert die ERGEBNISSE 95 Skala der MFI-Werte der Seren. Nach dem Auftragen aller Punkte für die Seren einer bestimmten Kohorte werden diese verbunden. Demnach kann nach Eintragung des Cut-offWertes des entsprechenden Antigens abgelesen werden, wieviel Prozent der Seren oberhalb des Cut-off-Wertes liegen und somit positiv sind. Zusätzlich kann anhand dieser Graphik bestimmt werden, wie viele Seren einer Kohorte prozentual oberhalb eines bestimment MFI-Wertes liegen. Im Folgenden sollen zunächst die Ergebnisse für das MM- und anschließend für das CTCLKollektiv dargestellt werden. Abschließend werden die Ergebnisse für zwei unterschiedliche Immunisierungsstrategien in CTCL-Patienten aufgeführt. 3.2.5 Untersuchung der Serumantikörperantwort gegen die verwendeten Antigene in einem MM-Kollektiv Antikörper gegen die unter 3.2.2 beschriebenen humanen und viralen Antigene wurden in 191 MM-Patienten sowie 100 Ctrl untersucht. Von den 191 MM-Patienten befanden sich 48 Patienten in Stadium I, 49 Patienten in Stadium II und jeweils 47 Patienten in Stadium III und IV. Für acht der 191 MM-Patienten konnten zudem Daten von bis zu drei aufeinander folgenden Serumabnahmen ausgewertet werden. Somit konnte ein Vergleich der Antikörperantworten in verschiedenen Kollektiven und eine Beurteilung der Antikörperantwort in einigen MM-Patienten über die Zeit durchgeführt werden. Bei Betrachtung der kumulativen Distributionen in Abbildung 3-7, Abbildung 3-8, Abbildung 3-9, Abbildung 3-10 und Abbildung 3-11 fällt für die Antigene MAGE-A1 (A, B), A3 (C, D), A9 (E, F), GAGE-7b (C, D), CAMEL (E, F), se57-1 (C, D) und p16 (A, B) auf, dass nur für einige wenige MM-Seren MFI-Werte oberhalb der Werte von Kontroll-Personen und oberhalb des jeweiligen Cut-off-Wertes gemessen wurden. Dies gilt sowohl für das gesamte MMKollektiv als auch für die verschiedenen MM-Stadien-Kollektive insbesondere für höhere Stadien. Hingegen bestätigen Abbildung 3-9 und Abbildung 3-10, dass die MFI-Werte der Patientenkollektive für die Antigene se70-2 (A, B), GBP-5ta (E), cTAGE-5a N- (A) und Cterminal (C, D) sowie p53 (E) unterhalb denjenigen des Kontroll-Kollektivs lagen sobald nur die MFI-Werte oberhalb des Cut-off-Wertes betrachtet werden. Für die Antigene GBP-5ta (Abbildung 3-9 F), cTAGE-5a N-terminal (Abbildung 3-10 B) und p53 (Abbildung 3-10 F) wurden für MM-Patienten einzelner Stadien-Kollektive MFI-Werte oberhalb des KontrollKollektivs nachgewiesen. Lediglich LAGE-1a unterschied sich deutlich von den anderen Antigenen im Vergleich der Antikörperantworten zwischen den verschiedenen Kollektiven. In Abbildung 3-8 A ist der signifikante Unterschied zwischen dem gesamten MM-Kollektiv und dem Kontroll-Kollektiv ersichtlich. Ca. 10% aller MM-Seren besitzen einen höheren MFI-Wert ERGEBNISSE 96 im Vergleich zu den Kontrollseren, der zudem oberhalb des Cut-off-Wertes von 85 MFI liegt. In Abbildung 3-8 B ist der Vergleich des Kontroll-Kollektivs mit allen MM-Stadien dargestellt. Auch für die verschiedenen Stadien sind deutliche Abgrenzungen zwischen Ctrl und MMPatienten zu erkennen. Ca. 8% der MM-Patienten in den Stadien II und III sowie ca. 20% der MM-Patienten in Stadium IV besitzen einen deutlich höheren MFI-Wert oberhalb des Cut-OffWertes im Vergleich zu allen Kontrollpersonen. Es wurde eine eindeutig stadienabhängige Serumantikörperantwort in den MM-Patienten gegen LAGE-1a nachgewiesen, die statistisch signifikant ist. Desweiteren befinden sich die Patienten mit den höchsten gemessenen MFIWerten im Stadium IV, welches eine Unterscheidung der verschiedenen Stadien möglich machen könnte. Anhand der kumulativen Distributionen ist auf den ersten Blick erkennbar, dass für die Antigene MAGE-A1, A3, A9, LAGE-1a, GAGE-7b und CAMEL die höchsten detektierbaren MFI-Werte im MM-Kollektiv und bei näherer Betrachtung im Stadium IV gemessen wurden. Hingegen wurden die höchsten MFI-Werte für die Antigene se70-2, GBP-5ta, cTAGE-5a N- und Cterminal sowie p53 im Kontroll-Kollektiv gemessen. Die Antigene se57-1 und p16 wiesen die höchsten MFI-Werte in den MM-Patienten der Stadien I und II auf. Demnach wurden hohe MFIWerte in den Patientenkollektiven vor allem für CTA mit Ausnahme von cTAGE-5a gemessen. Laut „Fisher`s Exact Test“ konnten statistisch signifikante Unterschiede zwischen MM-Patienten und Kontrollpersonen für die Antigene MAGE-A3, LAGE-1a, se70-2 und se57-1 berechnet werden (Anhang 1). Ein Trend zu einem signifikanten Unterschied konnte für GAGE-7b festgestellt werden. Die prozentual positiven Seren aller MM-Kollektive im Vergleich mit dem Kontroll-Kollektiv sind in grafischer Form in Abbildung 3-12 aufbereitet. Bemerkenswert für das Antigen LAGE-1a ist, dass sechs der 47 MM-Patienten in Stadium IV Antikörpertiter von mehr als 10000 MFI gebildet hatten. Diese Titer liegen bereits im Bereich der Reaktivitäten gegen virale Antigene. Zudem traten bei zwei der 47 Patienten Antikörper gegen mehrere Antigene auf. Eine Patientin besaß gleichzeitig hohe Antikörperantworten gegen MAGE-A1 (18275 MFI), MAGE-A3 (18000 MFI), MAGE-A9 (14310 MFI), LAGE-1a (17818 MFI) und GAGE-7b (5653 MFI) und eine andere Patientin gegen MAGE-A1 (6788 MFI), MAGE-A3 (10823 MFI), MAGE-A9 (299 MFI) und LAGE-1a (4733 MFI). Für Patienten in Stadium IV traten gleichzeitig Antikörper gegen mehrere Antigene der MAGE-Gruppe auf. Hierzu zählten vier von 47 Stadium IV Patienten im Vergleich zu keinem oder einem Patienten der anderen Stadien. ERGEBNISSE Ctrl (n=100) MM (n=191) Cut-off (102 MFI) 18000 16000 14000 12000 10000 8000 6000 4000 2000 0 B 20000 MAGE-A1 MFI MFI A 20000 0,1 1 10 97 Ctrl (n=100) 18000 16000 14000 12000 10000 8000 6000 4000 2000 0 100 MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (102 MFI) MAGE-A1 0,1 1 Ctrl (n=100) MM (n=191) Cut-off (366 MFI) 18000 16000 14000 12000 10000 8000 6000 4000 2000 0 D 20000 MAGE-A3 MFI MFI C 20000 0,1 1 10 100 MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (366 MFI) MAGE-A3 0,1 1 12000 F 16000 100 MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (366 MFI) 12000 10000 MFI 8000 Ctrl (n=100) 14000 MAGE-A9 10000 MFI 10 Seren [%] Ctrl (n=100) MM (n=191) Cut-off (256 MFI) 14000 100 Ctrl (n=100) 18000 16000 14000 12000 10000 8000 6000 4000 2000 0 Seren [%] E 16000 10 Seren [%] Seren [%] 8000 6000 6000 4000 4000 2000 2000 MAGE-A9 0 0 0,1 1 10 100 0,1 Seren [%] Abbildung 3-7: 1 10 100 Seren [%] Darstellung der kumulativen Distribution des Kontroll- und MMKollektivs für MAGE-A1, A3 und A9 In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für jedes Antigen ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnten statistisch signifikant erhöhte Seroreaktivitäten der MM-Patienten des Stadiums IV gegenüber der Kontroll-Gruppe für MAGE-A3 berechnet werden (p = 0,0128). Ctrl: Kontrollen, MM: Malignes Melanom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. ERGEBNISSE A 25000 Ctrl (n=100) MM (n=191) Cut-off (85 MFI) 20000 98 B 25000 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (85 MFI) 20000 MFI MFI LAGE-1a 15000 15000 10000 10000 5000 5000 LAGE-1a 0 0 0,1 1 10 100 0,1 1 Seren [%] C 6000 Ctrl (n=100) MM (n=191) Cut-off (85 MFI) 5000 3000 4000 GAGE-7b 3000 2000 2000 1000 1000 0 0 1 10 100 0,1 1 Seren [%] E 4000 3000 2000 4000 3000 2500 1500 1000 1000 500 500 0 0 10 100 CAMEL 2000 1500 1 0,1 1 Seren [%] Abbildung 3-8: 100 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (169 MFI) 3500 MFI 2500 MFI F CAMEL 0,1 10 Seren [%] Ctrl (n=100) MM (n=191) Cut-off (169 MFI) 3500 100 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (59 MFI) 5000 MFI MFI D 6000 GAGE-7b 4000 0,1 10 Seren [%] 10 100 Seren [%] Darstellung der kumulativen Distribution des Kontroll- und MMKollektivs für LAGE-1a, GAGE-7b und CAMEL In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für jedes Antigen ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnte ein statistisch signifikanter Unterschied zwischen Kontroll-Gruppe und MM-Patienten (p = 0,0034), Kontroll -Gruppe und MM-Patienten des Stadiums III (p = 0,0302) und IV (p = 0,0007) für LAGE-1a berechnet werden, wobei die Seroreaktivitäten der Patienten jeweils erhöht waren. Für GAGE7b waren die Seroreaktivitäten der Patienten in Stadium II tendentiell gegenüber den Ctrl erhöht. Ctrl: Kontrollen, MM: Malignes Melanom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. ERGEBNISSE A 5000 Ctrl (n=100) MM (n=191) Cut-off (752 MFI) 3500 3000 2500 2000 1500 B 5000 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (752 MFI) 4500 se70-2 MFI MFI 4500 4000 99 1000 500 0 4000 3500 3000 se70-2 2500 2000 1500 1000 500 0 0,1 1 10 0,1 100 1 5000 Ctrl (n=100) MM (n=191) Cut-off (649 MFI) 4000 se57-1 D 6000 4000 3000 se57-1 3000 2000 2000 1000 1000 0 0 0,1 1 10 100 0,1 1 Seren [%] E 10000 8000 7000 F MFI 6000 5000 4000 10000 7000 6000 3000 2000 1000 0 1000 0 10 100 GBP-5ta 5000 4000 3000 2000 1 0,1 Seren [%] Abbildung 3-9: 100 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (1488 MFI) 9000 8000 GBP-5ta 0,1 10 Seren [%] Ctrl (n=100) MM (n=191) Cut-off (1488 MFI) 9000 MFI 100 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (649 MFI) 5000 MFI MFI C 6000 10 Seren [%] Seren [%] 1 10 100 Seren [%] Darstellung der kumulativen Distribution des Kontroll- und MMKollektivs für se70-2, se57-2 und GBP-5ta In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für jedes Antigen ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnten für se70-2 statistisch signifikant erniedrigte Seroreaktivitäten der MM-Patienten (p = 0,0398) gegenüber der Kontroll-Gruppe berechnet werden. Für se57-1 waren die Seroreaktivtäten der MM-Patienten in Stadium II statistisch signifikant erhöht gegenüber der Kontroll-Gruppe (p = 0,0149). Ctrl: Kontrollen, MM: Malignes Melanom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. ERGEBNISSE A 8000 Ctrl (n=100) MM (n=191) Cut-off (1781 MFI) 7000 6000 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (1781 MFI) 6000 5000 MFI 4000 8000 7000 cTAGE-5a N-terminal 5000 MFI B 100 4000 3000 3000 2000 2000 1000 1000 0 cTAGE-5a N-terminal 0 0,1 1 10 100 0,1 1 D 10000 cTAGE-5a C-terminal MFI MFI Ctrl (n=100) MM (n=191) Cut-off (2564 MFI) 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 0,1 1 10 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (2564 MFI) 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 100 cTAGE-5a C-terminal 0,1 1 Seren [%] E 8000 6000 4000 8000 6000 5000 3000 2000 2000 1000 1000 0 0 10 Seren [%] 100 p53 4000 3000 1 100 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (1403 MFI) 7000 MFI 5000 MFI F p53 0,1 10 Seren [%] Ctrl (n=100) MM (n=191) Cut-off (1403 MFI) 7000 100 Seren [%] Seren [%] C 10000 10 0,1 1 10 100 Seren [%] Abbildung 3-10: Darstellung der kumulativen Distribution des Kontroll- und MMKollektivs für cTAGE-5a N- und C-terminal sowie p53 In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für jedes Antigen ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnte für kein abgebildetes Antigen ein statistisch signifikanter Unterschied zwischen den MM-Patienten und der Kontroll-Gruppe berechnet werden. Ctrl: Kontrollen, MM: Malignes Melanom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. ERGEBNISSE A 3500 Ctrl (n=100) MM (n=191) Cut-off (636 MFI) 3000 B 3500 p16 2000 1500 2500 Ctrl (n=100) MM Stadium I (n=48) MM Stadium II (n=49) MM Stadium III (n=47) MM Stadium IV (n=47) Cut-off (636 MFI) 2000 p16 3000 MFI MFI 2500 1500 1000 1000 500 500 0 101 0 0,1 1 10 100 Seren [%] 0,1 1 10 100 Seren [%] Abbildung 3-11: Darstellung der kumulativen Distribution des Kontroll- und MMKollektivs für p16 In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für p16 ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnte für p16 kein statistisch signifikanter Unterschied zwischen den MM-Patienten und der Kontroll-Gruppe berechnet werden. Ctrl: Kontrollen, MM: Malignes Melanom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. Antikörper gegen MAGE-A1 konnten in 7,3% der MM-Patienten und in 6,0% der Ctrl detektiert werden. In Stadium I fanden sich in 10,4% der MM-Patienten, in Stadium II in 4,1%, in Stadium III in 4,3% und in Stadium IV 10,6% Serumantikörperantworten gegen MAGE-A1 (Abbildung 3-12 und Anhang 2). Ein statistisch signifikanter Unterschied zwischen Ctrl und Patienten konnte nicht berechnet werden (siehe Anhang 1). Gegen MAGE-A3 besaßen 8,4% der MM-Patienten und 5,0% der Ctrl Antikörper. 4,2% der MM-Patienten in Stadium I, 2,0% in Stadium II, 8,5% in Stadium III und 19,1% in Stadium IV waren seropositiv für MAGE-A3. Ein statistisch signifikanter Unterschied (p = 0,0128) für eine höhere Frequenz an seropositiven MAGE-A3-Seren im Kollektiv der MM-Patienten in Stadium IV gegenüber den Ctrl konnte bestimmt werden. 2,0% der Kontrollpersonen und 4,2% der MM-Patienten wiesen Antikörper gegen MAGE-A9 auf. Ähnliche Antikörperfrequenzen unter 10% wurden für Stadium I MM-Patienten mit 2,1%, Stadium II mit 2,0%, Stadium III mit 4,3% und Stadium IV mit 8,5% gemessen. Für dieses Antigen konnte kein statistisch signifikanter Unterschied zwischen den verschiedenen Kollektiven bestimmt werden. LAGE-1a-Antikörper wurden in 3,0% der Ctrl und in 13,6% der MM-Patienten nachgewiesen. Laut „Fisher’s Exact Test“ wurden statistisch signifikant erhöhte Antikörperfrequenzen in MMPatienten gegenüber Ctrl gemessen (p = 0,0034). In Stadium I MM-Patienten konnten in 10,4% ERGEBNISSE 102 der Fälle Antikörper detektiert werden sowie in 10,2% in Stadium II, 12,8% in Stadium III und 21,3% in Stadium IV (Anhang 2). Neben den statistisch signifikant erhöhten Antikörperantworten im Gesamtkollektiv bestätigte sich die signifikante Erhöhung der Antikörperfrequenzen in Stadium III (p = 0,0302) und Stadium IV (p = 0,0007). 1,0% der Kontrollpersonen und 5,8% aller MM-Patienten waren seropositiv für GAGE-7b. Antikörperantworten unter 10% ergaben sich ebenfalls für die Patientenkollektive der verschiedenen Stadien (I: 4,2%, II: 8,2%, III: 4,3%, IV: 6,4%). Ein Trend hin zu signifikant erhöhten Antikörperantworten gegen GAGE-7b in MM-Patienten des Stadiums II konnte analysiert werden (p = 0,0404). Der p-Wert war nicht statistisch signifikant, da keine Signifikanz im Vergleich aller Stadien mit dem Kontroll-Kollektiv berechnet wurde (Anhang 1). Ein MM-Patient im Gegensatz zu keiner Kontrollperson bildete Antikörper gegen CAMEL. Ebenso wurden keine Serumantikörper gegen CAMEL in MM-Patienten des Stadiums I und III detektiert. Nur in 2,0% der Patienten des Stadiums II und 2,1% in Stadium IV wurden Serumantikörper identifiziert. Aufgrund der niedrigen Serumantikörperantworten konnte kein statistisch signifikanter Unterschied für CAMEL zwischen den Kollektiven festgestellt werden. Für das Antigen se70-2 fanden sich in 3,0% der Ctrl Antikörper, jedoch nicht in MM-Patienten. Daraus ergaben sich statistisch signifikant erhöhte se70-2-Antikörper in Kontrollpersonen im Vergleich zum MM-Gesamtkollektiv (p = 0,0398). se57-1 Antikörper wurden in 1,0% der Kontrollpersonen gegenüber 3,7% der MM-Patienten gemessen. Hingegen besaßen 10,2% der MM-Patienten des Stadiums II Antikörper gegen se57-1 (p = 0,0149), woraus sich ein statistisch signifikanter Unterschied berechnen ließ. In Stadium I (2,1%), Stadium III (0%) und Stadium IV MM-Patienten (2,1%) wurden weniger häufig Antikörper gegen dieses Antigen nachgewiesen. Das Antigen GBP-5ta erzeugte Antikörper in gleicher Häufigkeit in MM-Patienten (3,1%) und in Ctrl (3,0%). Weder MM-Patienten in Stadium I noch in Stadium IV wiesen Antikörper gegen GBP-5ta auf. MM-Patienten in Stadium II (6,1%) und IV (6,4%) besaßen leicht höhere Antikörperfrequenzen, die jedoch allesamt nicht statistisch signifikant erhöht gegenüber den Ctrl waren (Anhang 1 und Anhang 2). ERGEBNISSE A: Prozentuale Seroreaktivitäten KontrollGruppe versus gesamtes MM-Kollektiv 103 Ctrl (n=100) MM (n=191) p16 p53 cTAGE-5a C-terminal cTAGE-5a N-terminal Antigen GBP-5ta se57-1 se70-2 * CAMEL GAGE-7b LAGE-1a ** MAGE-A9 MAGE-A3 MAGE-A1 0% 2% 4% 6% 8% 10% 12% 14% 16% Seren über cut-off [%] B: Prozentuale Seroreaktivitäten KontrollGruppe versus verschiedene MMStadien p16 Ctrl (n=100) MM-St I (n=48) p53 MM-St II (n=49) MM-St III (n=47) MM-St IV (n=47) cTAGE-5a C-terminal cTAGE-5a N-terminal Antigen GBP-5ta se57-1 * se70-2 CAMEL GAGE-7b * LAGE-1a * *** MAGE-A9 MAGE-A3 * MAGE-A1 0% 5% 10% 15% 20% 25% Seren über cut-off [%] Abbildung 3-12: Vergleich der prozentualen Seroreaktivität der Kontrollen mit allen MMPatienten (A) und den verschiedenen MM-Stadiengruppen (B) Die Säulendiagramme stellen die prozentuale Seroreaktivität der einzelnen Personengruppen gegen die humanen Antigene dar. Die mit Stern (*) gekennzeichneten Antigene sind laut „Fisher’s Exact Test“ ERGEBNISSE 104 statistisch signifikant unterschiedlich zwischen der Kontroll-Gruppe und dem jeweiligen MM-Kollektiv. Dies trifft auf die Antigene MAGE-A3, LAGE-1a, se70-2 und se57-1 zu. Für GAGE-7b konnte laut Anhang 1 nur ein Trend nachgewiesen werden. *: 0,05 > p > 0,01; **: 0,01 > p > 0,001; ***: 0,001 > p. Ctrl: Kontrollen, MM-St I: Malignes Melanom Stadium I, n: Anzahl getesteter Seren. Bei der Betrachtung der Antikörperantworten gegen das Antigen cTAGE-5a wurden der Nterminale und C-terminale Teil unterschieden. Höhere Serumantikörper-Frequenzen wurden für den N-terminalen Teil nachgewiesen. Jedoch konnte für beide Teile kein statistisch signifikanter Unterschied zwischen MM-Patienten und Ctrl bestimmt werden. Gegen beide Antigene produzierten 2,0% der Ctrl Antikörper. MM-Patienten wiesen in 2,6% (N-terminal) und 0,5% (C-terminal) der Fälle Antikörper auf. Kein Stadium I Patient hatte Antikörper gegen einen der beiden Teile. Der N-terminale Teil wurde von keinem Stadium II, 4,3% Stadium III und 6,4% Stadium IV MM-Patienten erkannt. Der C-terminale Teil wurde von 2,0% der Stadium II MMPatienten und keinem Patienten in Stadium III und IV erkannt. Für die Antigene p53 und p16 konnte kein statistisch signifikanter Unterschied zwischen MMPatienten und Kontrollpersonen berechnet werden, da ähnliche Serumantikörperantworten für alle Kollektive gemessen wurden. Für p53 wurden folgende Werte bestimmt: Ctrl: 3,0%, MM: 2,1%, MM I: 0%, MM II: 2,0%, MM III: 4,3%, MM IV: 2,1%. Für p16 wurden die angegebenen Werte erhalten: Ctrl: 2,0%, MM: 2,1%, MM I: 4,2%, MM II: 2,0%, MM III: 2,1%, MM IV: 0% (Anhang 2). Zusammenfassend bedeutet dies für die Beurteilung der Ergebnisse anhand der kumulativen Distributionen und der statistischen Auswertung, dass Serumantikörper gegen die untersuchten Antigene mit Ausnahme von LAGE-1a in einem MM-Kollektiv eher selten per se nachgewiesen werden können. Die Antikörper-Häufigkeiten liegen zumeist unterhalb von 10%. Kontrollpersonen haben teilweise bereits Serumantikörper gegen die verwendeten Antigene gebildet. Hingegen sind Serumantikörper gegen LAGE-1a gerade in Stadium IV Patienten deutlich gegenüber Kontrollpersonen erhöht. Ein eindeutig stadienabhängiges Auftreten an LAGE-1a Serumantikörpern wurde nachgewiesen. Verläufe von acht MM-Patienten Ein Antikörper-Monitoring war bei acht MM-Patienten möglich. Es wurden zwei bis drei Seren untersucht, deren Abnahmedatum bis zu 171 Wochen auseinander lag. Die zeitlich erste Serumabnahme wurde als Woche 0 definiert. Von einem Patienten konnten drei Serumproben, von den sieben weiteren Patienten jeweils zwei Serumproben untersucht werden. Fünf Patienten schritten im Zeitraum der Serumabnahmen im Stadium fort („progressive disease“, PD, Patienten #M1, #M3 - #M5 und #M8), für zwei Patienten blieb das Stadium gleich („stable disease“, SD, ERGEBNISSE 105 Patienten #M6 und #M7) und für einen Patienten verbesserte sich das Stadium („partial response“, PR, Patient #M2). In Abbildung 3-13 sind die Serumantikörperantworten exemplarisch für einige humane Antigene (LAGE-1a, se57-1 und cTAGE-5a C-terminal) und alle viralen Antigene angegeben. Eine Serokonversion, die durch mindestens eine Verdopplung oder eine Halbierung der Serumantikörperantwort gekennzeichnet ist, wobei mindestens ein Wert über dem Cut-off-Wert liegen muss, wurde bei Patient #M3 nachgewiesen. Dieser schritt über einen Zeitraum von 98 Wochen von Stadium I bis IV fort. Diese Tatsache spiegelte sich in der Serumantikörperantwort gegen LAGE-1a wider, welche von 0 auf 13820 MFI signifikant anstieg. Obwohl für MAGE-A9 kein MFI-Wert über dem Cut-off-Wert lag, ist auch hier ein Trend der Zunahme der Antikörper zu erkennen (5 auf 139 MFI). Zudem lagen die gemessenen MFI-Werte gegen cTAGE-5a Cterminal für diesen Patienten mit 2563 auf 2531 MFI nur knapp unter dem Cut-off-Wert und blieben über den untersuchten Zeitraum hinweg gleich. Für Patient #M2 konnten Antikörper gegen se57-1 nachgewiesen werden, die sich kaum über den Zeitraum von 144 Wochen veränderten (2262 auf 1781 MFI). Patient #M6 in Stadium III hatte gleichbleibende Antikörper gegen p16 (935 auf 654 MFI) über den Verlauf von 171 Wochen. Für Patient #M7 in Stadium IV wurde ein Anstieg unterhalb Cut-off von 589 auf 1316 MFI für Antikörper gegen cTAGE-5a Nterminal über den Verlauf von 33 Wochen gemessen. Schließlich verringerte sich die Seroreaktivität von Patient #M8 gegen cTAGE-5a C-terminal über einen Zeitraum von 36 Wochen von 1615 auf 289 MFI, wobei auch diese MFI-Werte wiederum unter dem Cut-off Wert lagen. Dieser Patient schritt in der Stadieneinteilung von III nach IV fort. Keine signifikanten Antikörper-Veränderungen über die Zeit wurden für alle anderen Antigene analysiert. Die gemessenen MFI-Werte lagen unter den Cut-off-Werten der jeweiligen Antigene. Die viralen Antigene erzeugten deutlich höhere Serumantikörperantworten (BK bis zu 20000 MFI, vgl. Skala Abbildung 3-13) als die humanen Antigene. Für die viralen Antigene konnte keine Serokonversion nachgewiesen werden. Dies entspricht den Erfahrungen der Arbeitsgruppe von Dr. M. Pawlita am DKFZ (persönliche Mitteilung). Daher bestätigte sich die Verwendbarkeit der Antikörperantworten gegen die viralen Antigene als Fingerprint. ERGEBNISSE 106 A 16000 LAGE-1a se57-1 cTAGE-5a C-terminal 14000 12000 LAGE-1a, se57-1, cTAGE-5a C-terminal MFI 10000 8000 6000 4000 2000 #M1 (PD) #M2 (PR) #M3 (PD) #M4 (PD) #M5 (PD) #M6 (SD) #M7 (SD) 0 wo (III) 36 wo (IV) 0 wo (IV) 33 wo (IV) 0 wo (III) 171 wo (III) 0 wo (III) 21 wo (IV) 0 wo (II) 87 wo (III) 0 wo (I) 98 wo (IV) 0 wo (II) 144 wo (I) 0 wo (I) 38 wo (III) 65 wo (IV) 0 #M8 (PD) Patient [#], Zeit nach erster Serumabnahme [wo] und Stadium B 25000 BK, JV, LPV, HPV1 MFI 20000 BK JC LPV HPV1 15000 10000 5000 #M1 (PD) #M2 (PR) #M3 (PD) #M4 (PD) #M5 (PD) #M6 (SD) 0 wo (III) 36 wo (IV) 0 wo (IV) 33 wo (IV) 0 wo (III) 171 wo (III) 0 wo (III) 21 wo (IV) 0 wo (II) 87 wo (III) 0 wo (I) 98 wo (IV) 0 wo (II) 144 wo (I) 0 wo (I) 38 wo (III) 65 wo (IV) 0 #M7 (SD) #M8 (PD) Patient [#], Zeit nach erster Serumabnahme [wo] und Stadium Abbildung 3-13: Monitoring der Serumantikörperantwort von acht MM-Patienten Die Serumantikörperantwort von acht MM-Patienten in unterschiedlichen Stadien der Erkrankung wurde während unterschiedlicher Zeiträume (33 bis 171 Wochen) betrachtet. Die erste untersuchte Serumabnahme wurde mit Woche 0 betitelt. In Abbildung A wurden drei der humanen Antigene ausgewählt und exemplarisch die Serumantikörperantworten über die Zeit dargestellt. Die Cut-off-Werte sind mit unterbrochenen Linien und dem Farbcode des jeweiligen Antigens eingezeichnet. Der Patient ERGEBNISSE 107 #M3 durchlief eine deutliche Serokonversion für LAGE-1a, die mit der Verschlechterung des Stadiums einhergeht. Patient #M2 besaß ungefähr gleich bleibende Antikörper gegen se57-1. In Abbildung B sind die Veränderungen der Serumantikörperantworten über die Zeit gegen alle viralen Antigene dargestellt. Für die viralen Antigene konnte keine Serokonversion nachgewiesen werden, weshalb diese als Fingerprint dienen. #M1: Patient 1, wo: Wochen, (I): Stadium I, MFI: „Mean fluorescence intensity“, PD: “Progressive disease”, PR: “Partial response”, SD: “Stable disease”. 3.2.6 Untersuchung der Serumantikörperantwort gegen die verwendeten Antigene in einem CTCL-Kollektiv Antikörper gegen die unter 3.2.2 beschriebenen humanen und viralen Antigene wurden in 144 CTCL-Patienten sowie 100 Kontrollpersonen untersucht. Von den 144 CTCL-Patienten befanden sich 55 Patienten in Stadium I, jeweils 14 Patienten in Stadium II und III und 16 Patienten in Stadium IV. Informationen über die Zuordnung zu einem Stadium waren nur für 99 CTCL-Patienten vorhanden, die entsprechend zur Evaluierung der Ergebnisse für die StadienKollektive herangezogen wurden. Die übrigen CTCL-Patienten wurden nur im Gesamtkollektiv berücksichtigt. Für zwölf der 144 CTCL-Patienten konnten zudem Daten von bis zu 4 aufeinander folgenden Serumabnahmen ausgewertet werden. Somit konnte ein Vergleich der Antikörperantworten in verschiedenen Kollektiven und ein Monitoring der Antikörperantwort in CTCL-Patienten über die Zeit durchgeführt werden Die kumulativen Distributionen in Abbildung 3-14 und Abbildung 3-15 stellen grafisch alle Antigene dar, für die die MFI-Werte der verschiedenen CTCL-Kollektive zumeist über denjenigen des Kontroll-Kollektivs und des Cut-off-Wertes lagen. Dies galt für die Antigene MAGE-A1 (A, B), A3 (C, D), A9 (E, F), LAGE-1a (A, B), GAGE-7b (C, D) und CAMEL (E, F). Für die Antigene se57-1 (Abbildung 3-16 C und D), cTAGE-5a N-terminal (Abbildung 3-17 A und B) und p53 (Abbildung 3-17 E und F) wurden ähnliche Profile der Serumantikörperantworten in den CTCL-Kollektiven im Vergleich zum Kontroll-Kollektiv gemessen. Für diese drei Antigene lagen die Kurven der MFI-Werte der Patienten und Ctrl fast aufeinander und waren kaum unterscheidbar. Die Antigene se70-2 (Abbildung 3-16 A und B), GBP-5ta (Abbildung 3-16 E und F), cTAGE-5a C-terminal (Abbildung 3-17 C und D) und p16 (Abbildung 3-18 A und B) schließlich bildeten höhere Serumantikörperantworten im KontrollKollektiv im Vergleich zu den CTCL-Kollektiven. Für diese Antigene verliefen die Kurven der CTCL-Patienten unterhalb der Kurve des Kontroll-Kollektivs. Neben dem Verlauf der Kurven für CTCL-Patienten und Kontrollpersonen kann anhand der kumulativen Distributionen abgelesen werden, welches Kollektiv die höchsten MFI-Werte aufwies. Damit können Stadiengruppen identifiziert werden, in denen sehr hohe Antikörperantworten induziert wurden. Die Antigene MAGE-A1, A3, LAGE-1a, CAMEL und ERGEBNISSE 108 cTAGE-5a N-terminal erzeugten die höchsten MFI-Werte in der Gruppe der CTCL-Patienten, die sich in Stadium I befanden. MAGE-A9, GAGE-7b und p53 induzierten die höchsten MFIWerte in CTCL-Patienten der Stadiengruppe IV. Allein die MFI-Werte des Antigens se57-1 waren in CTCL-Patienten in Stadium II am höchsten. Für die restlichen Antigene (se70-2, GBP5ta, cTAGE-5a C-terminal und p16) wurden die höchsten MFI-Werte in der Kontroll-Gruppe gemessen. In den CTCL-Patienten-Kollektiven konnte für kein Antigen eine eindeutig stadienabhängige Induktion von Serumantikörpern gemessen werden (vgl. LAGE-1a im MMKollektiv). Allerdings erzeugten wiederum vor allem die CTA hohe MFI-Werte in den CTCLPatienten. Dies weist auf die Immunogenität dieser Antigene hin. Um die CTCL-Kollektive mit dem Kontroll-Kollektiv vergleichen zu können, wurde der „Fisher`s Exact Test“ angewandt. Statistisch signifikant erhöhte Antikörperantworten in CTCLPatienten in Stadium IV gegenüber den Kontrollpersonen wurden nur für das CTA MAGE-A9 berechnet (Anhang 1). Ein Trend zu erhöhten Serumantikörper-Häufigkeiten konnte für Patienten in Stadium III im Vergleich zu den Kontrollpersonen für MAGE-A1 bestimmt werden. In grafischer Form sind diese Ergebnisse in Abbildung 3-19 dargestellt. Allerdings ist hinsichtlich der Berechnung der statistischen Signifikanz zu bedenken, dass die Anzahl an Seren in den vier Stadiengruppen sehr unterschiedlich und teilweise gering mit unter 20 Seren pro Stadium für die Stadien II bis IV war. Gegen MAGE-A1 konnten Antikörper in 10,4% aller CTCL-Patienten und 6,0% der Ctrl nachgewiesen werden (Anhang 2). 10,9% der CTCL-Patienten in Stadium I, 7,1% in Stadium II und 6,3% in Stadium IV besaßen Antikörper gegen dieses CTA. Signifikant erhöhte Serumantikörper konnten in 28,6% der CTCL-Patienten in Stadium III gegenüber den Ctrl identifiziert werden (p = 0,0199). Jedoch ist hier nur ein Trend erkennbar, da die statistische Signifikanz über alle CTCL-Kollektive und das Kontroll-Kollektiv nicht erfüllt war. Antikörper gegen MAGE-A3 konnten in 4,9% aller CTCL-Patienten und in 5,0% der Ctrl detektiert werden. In Stadium I wurden in 3,6% der CTCL-Patienten und in Stadium IV in 12,5% Serumantikörperantworten gegen MAGE-A3 gemessen (Abbildung 3-19). Keine Serumantikörper wurden in Patienten in Stadium II und III identifiziert. Ein statistisch signifikanter Unterschied zwischen Ctrl und Patienten konnte nicht berechnet werden (siehe Anhang 1). Gegen MAGE-A9 besaßen 4,9% der CTCL-Patienten und 2,0% der Ctrl Antikörper. CTCLPatienten in Stadium I, II und III waren seronegativ für MAGE-A9. Hingegen wurden MAGEA9-Antikörper in einem Viertel aller CTCL-Patienten in Stadium IV beobachtet, so dass ein ERGEBNISSE 109 statistisch signifikanter Unterschied (p = 0,0032) zwischen dem CTCL- und dem KontrollKollektiv bestimmt wurde. Für alle folgenden Antigene konnte laut „Fisher’s Exact Test“ kein statistisch signifikanter Unterschied zwischen den einzelnen Kollektiven berechnet werden. Ähnliche Frequenzen an Serumantikörperantworten wurden im Gesamt-CTCL-Kollektiv (4,9%) im Vergleich zum Kontroll-Kollektiv (3,0%) für LAGE-1a gemessen. Nur in Stadium I CTCLPatienten (10,9%) wurden Serumantikörper gegen LAGE-1a nachgewiesen, wohingegen in keinem anderen Stadium Serumantikörper vorhanden waren (Anhang 2). 1,0% der Kontrollpersonen und 2,8% aller CTCL-Patienten wiesen Antikörper gegen GAGE-7b auf. Antikörper-Frequenzen unter 10% wurden ebenfalls für Stadium I CTCL-Patienten mit 1,8% und Stadium IV mit 6,3% gemessen. Keine Antikörper lagen für dieses CTA in Stadium II und III Patienten vor. Für CAMEL konnten Antikörper nur im Gesamt-CTCL-Kollektiv (2,8%) und in Stadium I (3,6%) detektiert werden. Alle anderen Kollektive waren seronegativ für CAMEL. Aufgrund der geringen Antikörper-Frequenzen von CAMEL in allen Kollektiven wurde kein statistisch signifikanter Unterschied berechnet. 0,7% aller CTCL-Patienten und eine Kontrollperson bildeten Antikörper gegen se70-2. CTCLPatienten in Stadium I besaßen zu 1,8% Serumantikörper gegen se70-2, wohingegen keine Antikörper in Patienten in Stadium II bis IV gefunden wurden. Für das Antigen se57-1 wurden in 1,0% der Kontrollpersonen und 0,7% aller CTCL-Patienten Antikörper gemessen. Neben der Seronegativität der CTCL-Patienten in Stadium I, III und IV, bildeten Patienten in Stadium II in 7,1% der Fälle Serumantikörperantworten gegen se57-1. GBP-5ta-Antikörper wurden in 3,0% der Ctrl und 3,5% aller CTCL-Patienten gemessen. Eine ähnliche Antikörper-Häufigkeit wurde mit 3,6% für CTCL-Patienten in Stadium I bestätigt, wohingegen Patienten in Stadium II bis IV seronegativ waren (Anhang 2). Die Antigene cTAGE-5a N- und C-terminal hatten ein ähnliches Antikörperprofil in CTCLPatienten und Ctrl. Beide erzeugten Antikörper von derselben Frequenz in Ctrl (2,0%) und in CTCL-Patienten in Stadium III (7,1%). Weder CTCL-Patienten in Stadium II noch in Stadium IV wiesen Antikörper gegen beide Antigene auf. Alle CTCL-Patienten (2,8%) und Patienten in Stadium I (3,6%) wiesen Antikörper gegen den N-terminalen Anteil des Antigens auf, jedoch nicht gegen den C-terminalen Teil. ERGEBNISSE A 4000 Ctrl (n=100) CTCL (n=144) Cut-off (102 MFI) 3500 3000 2500 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (102 MFI) 2000 MAGE-A1 2500 MAGE-A1 1500 MFI MFI B 110 2000 1000 1500 1000 500 500 0 0 0,1 1 10 100 0,1 1 10 Seren [%] C 4000 Ctrl (n=100) CTCL (n=144) Cut-off (366 MFI) 3500 3000 2000 Ctrl (n=100) 3500 CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (366 MFI) 3000 2500 MFI MFI D 4000 MAGE-A3 2500 2000 1500 1500 1000 1000 500 500 MAGE-A3 0 0 0,1 1 10 0,1 100 1 E 4000 Ctrl (n=100) CTCL (n=144) Cut-off (256 MFI) 3500 3000 10 100 10 100 Seren [%] Seren [%] F 2500 MAGE-A9 2000 1500 1500 1000 1000 500 500 0 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (256 MFI) 3000 MFI 2000 4000 3500 MAGE-A9 2500 MFI 100 Seren [%] 0 0,1 1 10 100 0,1 Seren [%] 1 Seren [%] Abbildung 3-14: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für MAGE-A1, A3 und A9 In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für jedes Antigen ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnten für MAGE-A1 tendentiell erhöhte Seroreaktivitäten der CTCL-Patienten im Stadium III gegnüber der Kontroll-Gruppe berechnet werden (p = 0,0199). Für MAGE-A9 wurden statistisch signifikant erhöhte Seroreaktivitäten der CTCL-Patienten in Stadium IV gegenüber der Kontroll-Gruppe berechnet (p = 0,0032). Ctrl: Kontrollen, CTCL: Kutanes T-Zell Lymphom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. ERGEBNISSE A 14000 Ctrl (n=100) CTCL (n=144) Cut-off (85 MFI) 12000 B 14000 10000 LAGE-1a 8000 6000 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (85 MFI) 12000 MFI MFI 10000 111 LAGE-1a 8000 6000 4000 4000 2000 2000 0 0 0,1 1 10 100 0,1 1 Seren [%] Ctrl (n=100) CTCL (n=144) Cut-off (59 MFI) 900 800 700 600 500 400 300 200 100 0 D 1000 GAGE-7b 0,1 1 10 100 0,1 1 F CAMEL 2000 MFI MFI 2500 100 Seren [%] Ctrl (n=100) CTCL (n=144) Cut-off (169 MFI) 3000 10 Ctrl (n=100) CTCL Stadium I (n=55) 900 CTCL Stadium II (n=14) CTCL Stadium III (n=14) 800 CTCL Stadium IV (n=16) 700 Cut-off (59 MFI) 600 GAGE-7b 500 400 300 200 100 0 Seren [%] E 3500 100 Seren [%] MFI MFI C 1000 10 1500 1000 500 450 400 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (169 MFI) 350 300 250 200 150 CAMEL 100 50 0 500 0 0,1 1 10 100 0,1 1 Seren [%] 10 100 Seren [%] Abbildung 3-15: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für LAGE-1a, GAGE-7b und CAMEL In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für jedes Antigen ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnte für kein abgebildetes Antigen ein statistisch signifikanter Unterschied zwischen den CTCL-Patienten und der Kontroll-Gruppe berechnet werden. Ctrl: Kontrollen, CTCL: Kutanes T-Zell Lymphom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. ERGEBNISSE Ctrl (n=100) CTCL (n=144) Cut-off (752 MFI) 4500 4000 3500 3000 2500 2000 1500 1000 500 0 B 5000 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (752 MFI) 4500 se70-2 MFI MFI A 5000 112 4000 3500 3000 se70-2 2500 2000 1500 1000 500 0 0,1 1 10 100 0,1 1 Seren [%] C 3500 Ctrl (n=100) CTCL (n=144) Cut-off (649 MFI) 3000 2500 100 3500 3000 se57-1 2000 1500 10 100 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (649 MFI) D 4000 2500 se57-1 MFI MFI 10 Seren [%] 2000 1500 1000 1000 500 500 0 0 0,1 1 10 100 0,1 1 Seren [%] Ctrl (n=100) CTCL (n=144) Cut-off (1488 MFI) 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 F 10000 GBP-5ta MFI MFI E 10000 Seren [%] 0,1 1 10 100 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (1488 MFI) 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 GBP-5ta 0,1 Seren [%] 1 10 100 Seren [%] Abbildung 3-16: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für se70-2, se57-1 und GBP-5ta In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für jedes Antigen ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnte für kein abgebildetes Antigen ein statistisch signifikanter Unterschied zwischen den CTCL-Patienten und der Kontroll-Gruppe berechnet werden. Ctrl: Kontrollen, CTCL: Kutanes T-Zell Lymphom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. ERGEBNISSE A 9000 7000 Ctrl (n=100) CTCL (n=144) Cut-off (1781 MFI) 6000 cTAGE-5a N-terminal 5000 B 9000 4000 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (1781 MFI) 8000 7000 6000 MFI MFI 8000 113 4000 3000 3000 2000 2000 1000 1000 0 cTAGE-5a N-terminal 5000 0 0,1 1 10 100 0,1 1 Seren [%] C 10000 D 10000 cTAGE-5a C-terminal MFI MFI 7000 6000 5000 4000 3000 2000 1000 0 0,1 1 10 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (2564 MFI) 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 100 cTAGE-5a C-terminal 0,1 1 Seren [%] E 14000 10000 F 14000 100 10000 p53 8000 6000 10 100 8000 p53 6000 4000 4000 2000 2000 0 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (1403 MFI) 12000 MFI MFI 10 Seren [%] Ctrl (n=100) CTCL (n=144) Cut-off (1403 MFI) 12000 100 Seren [%] Ctrl (n=100) CTCL (n=144) Cut-off (2564 MFI) 9000 8000 10 0 0,1 1 10 Seren [%] 100 0,1 1 Seren [%] Abbildung 3-17: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für cTAGE-5a N- und C-terminal sowie p53 In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für jedes Antigen ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnte für kein abgebildetes Antigen ein statistisch signifikanter Unterschied zwischen den CTCL-Patienten und der Kontroll-Gruppe berechnet werden. Ctrl: Kontrollen, CTCL: Kutanes T-Zell Lymphom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. ERGEBNISSE Ctrl (n=100) CTCL (n=144) Cut-off (636 MFI) 900 MFI 800 700 600 500 400 p16 B 1000 MFI A 1000 300 200 100 0 0,1 1 10 114 Ctrl (n=100) CTCL Stadium I (n=55) CTCL Stadium II (n=14) CTCL Stadium III (n=14) CTCL Stadium IV (n=16) Cut-off (636 MFI) 900 800 700 600 500 400 300 200 100 0 100 p16 0,1 1 Seren [%] 10 100 Seren [%] Abbildung 3-18: Darstellung der kumulativen Distribution des Kontroll- und des CTCLKollektivs für p16 In den Abbildungen sind die Seren in prozentualer Verteilung gegen die entsprechenden MFI-Werte dargestellt. Der Cut-off-Wert für p16 ist als unterbrochene Linie eingezeichnet. Laut „Fisher’s Exact Test“ konnte für p16 kein statistisch signifikanter Unterschied zwischen den CTCL-Patienten und der Kontroll-Gruppe berechnet werden. Ctrl: Kontrollen, CTCL: Kutanes T-Zell Lymphom, n: Anzahl getesteter Seren, MFI: „Mean fluorescence intensity“. Das Tumorsuppressorgen p53 induzierte in 3,0% der Kontrollpersonen und 2,1% aller CTCLPatienten Antikörper. Seropositive Seren wurden zudem in 3,6% der Stadium I und 6,3% der Stadium IV Patienten gemessen. Entsprechend wurden keine Antikörper in Stadium II und III Patienten nachgewiesen. Schließlich konnten für p16 nur Antikörper in 2.0% der Ctrl identifiziert werden, wohingegen kein CTCL-Patient seropositiv für p16 war (Anhang 2). Zusammenfassend kann anhand der kumulativen Distributionen und der statistischen Auswertung festgestellt werden, dass ähnlich wie bereits für das MM-Kollektiv Serumantikörper gegen die untersuchten Antigene in einem CTCL-Kollektiv eher selten per se nachgewiesen werden können. Die Antikörper-Häufigkeiten liegen zumeist unterhalb von 10%. Kontrollpersonen haben teilweise bereits Serumantikörper gegen die verwendeten Antigene gebildet. Ein Antigen, das eindeutig stadienabhängig Serumantikörper in CTCL-Patienten induziert, konnte bei dieser Untersuchung nicht identifiziert werden. ERGEBNISSE A: Prozentuale Seroreaktivitäten KontrollGruppe versus gesamtes CTCL-Kollektiv 115 Ctrl (n=100) p16 CTCL (n=144) p53 cTAGE-5a C-terminal cTAGE-5a N-terminal Antigen GBP-5ta se57-1 se70-2 CAMEL GAGE-7b LAGE-1a MAGE-A9 MAGE-A3 MAGE-A1 0% 2% 4% 6% 8% 10% 12% Seren über cut-off [%] B: Prozentuale Seroreaktivitäten KontrollGruppe versus verschiedene CTCLStadien Ctrl (n=100) p16 CTCL-St I (n=55) p53 CTCL-St II (n=14) CTCL-St III (n=14) cTAGE-5a C-terminal CTCL-St IV (n=16) cTAGE-5a N-terminal Antigen GBP-5ta se57-1 se70-2 CAMEL GAGE-7b LAGE-1a MAGE-A9 ** MAGE-A3 MAGE-A1 * 0% 5% 10% 15% 20% 25% 30% Seren über cut-off [%] Abbildung 3-19: Vergleich der prozentualen Seroreaktivität der Kontrollen mit allen CTCL-Patienten (A) und den verschiedenen CTCL-Stadiengruppen (B) Die Säulendiagramme stellen die prozentuale Seroreaktivität der einzelnen Personengruppen gegen die humanen Antigene dar. Die mit Stern (*) gekennzeichneten Antigene sind laut „Fisher’s Exact Test“ statistisch signifikant unterschiedlich zwischen der Kontroll-Gruppe und dem jeweiligen CTCL- ERGEBNISSE 116 Kollektiv. Dies trifft auf das Antigen MAGE-A9 zu. Für MAGE-A1 konnte laut Anhang 1 nur ein Trend zu erhöhten Serumantikörper-Häufigkeiten in MM-Patienten gegenüber Ctrl gemessen werden *: 0,05 > p > 0,01, **: 0,01 > p > 0,001. Ctrl: Kontrollen, CTCL-St I: Kutanes T-Zell Lymphom Stadium I, n: Anzahl getesteter Seren. Verläufe von zwölf CTCL-Patienten Ein Antikörper-Monitoring war bei zwölf nicht immununbehandelten CTCL-Patienten möglich. Es wurden zwei bis vier Seren pro Patient untersucht, die Zeiträume von bis zu 398 Wochen überspannen. Die zeitlich erste Serumabnahme wurde als Woche 0 definiert. Von vier Patienten war das Stadium zu Beginn und zum Ende des Abnahmezeitraumes bekannt. Bei zwei dieser Patienten erfolgte eine Stadienverschlechterung (#C5, #C10) und zwei Patienten verblieben in demselben Stadium (#C11, #C12). Acht der zwölf Patienten waren seronegativ für alle humanen Tumorantigene und Tumorsuppressorgene (#C1, #C4 - #C7, #C9, #C11 #C12). Zu dieser Gruppe zählten ebenfalls die Patienten mit bekannten Stadien. Allerdings wurde für Patient #C5 ein Anstieg der Antikörper gegen BK und für Patient #C12 ein Abfall der Antikörper gegen HPV1 jeweils im Sinne einer Serokonversion nachgewiesen. Diese Veränderungen in der Serumantikörperantwort der viralen Antigene standen jedoch in keinem Zusammenhang mit einer Veränderung der Serumantikörperantwort gegen humane Antigene. Patient #C2 wies einen signifikanten Abfall der Antikörper gegen MAGE-A1 über 287 Wochen (von 241 auf 120 MFI, Serokonversion) und einen signifikanten Anstieg der Antikörper gegen MAGE-A3 von 272 auf 859 MFI (Serokonversion) auf. Dieser Patient befand sich zum Zeitpunkt 0 in Stadium Ib, das Stadium nach 287 Wochen ist jedoch unbekannt. Für Patient #C3 konnte ein signifikanter Anstieg der Antikörper gegen p53 von 137 auf 12361 MFI in einem Zeitraum von 52 Wochen analysiert werden (Abbildung 3-20). Der Patient befand sich bei der letzten Serumabnahme im Stadium IVc. Eine Korrelation der Zunahme an p53-Antikörpern mit einem Übergang in Stadium IV ist bei diesem Patienten leider nicht zu treffen, da die Angabe des Stadiums zum Zeitpunkt 0 fehlt. Patient #C8, der sich zu Beginn im Stadium Ic befand, besaß Antikörper gegen MAGE-A1 über 147 Wochen. Seine Antikörper gegen LAGE-1a nahmen signifikant von 5700 auf 2325 MFI ab. Auch für diesen Patienten ist das Stadium nach 147 Wochen nicht bekannt. Patient #C10 schließlich schritt über 200 Wochen von Stadium Ib nach IIIc fort. Dies spiegelte sich in einer signifikanten Antikörperzunahme gegen cTAGE-5a C-terminal (1159 auf 2773 MFI) wider. Bei vier von zwölf CTCL-Patienten konnte demnach eine Serokonversion hinsichtlich der Serumantikörperantwort gegen humane Antigene im Verlauf der Erkrankung festgestellt werden, die zumindest für einen Patienten mit einer Verschlechterung des Stadiums korreliert. ERGEBNISSE A 117 14000 MAGE-A3 LAGE-1a p53 cTAGE-5a C-terminal 12000 MFI 10000 MAGE-A3, LAGE-1a, cTAGE-5a C-terminal, p53 8000 6000 4000 2000 #C1 #C2 #C3 #C4 #C5 (PD) #C6 #C7 (SD) #C8 #C9 #C10 (PD) #C11 (SD) 0 wo (Ib) 261 wo (Ib) 0 wo (IVa) 39 wo (IVa) 0 wo (Ib) 200 wo (IIIc) 0 wo (Ib) 17 wo 0 wo (Ic) 147 wo 0 wo (Ib) 315 wo (Ib) 0 wo 33 wo (Ic) 85 wo 0 wo (Ib) 192 wo (IIIc) 0 wo (Ib) 62 wo (Ic) 285 wo 398 wo 0 wo 52 wo (IVc) 0 wo (Ib) 287 wo 0 wo (Ib) 103 wo 0 #C12 (SD) Patient [#], Zeit nach erster Abnahme [wo], Stadium und klinisches Bild B 16000 14000 12000 MFI 10000 BK JC LPV HPV1 BK, JC, LPV, HPV1 8000 6000 4000 2000 #C1 #C2 #C3 #C4 #C5 (PD) #C6 #C7 (SD) #C8 #C9 #C10 (PD) #C11 (SD) 0 wo (Ib) 261 wo (Ib) 0 wo (IVa) 39 wo (IVa) 0 wo (Ib) 200 wo (IIIc) 0 wo (Ib) 17 wo 0 wo (Ic) 147 wo 0 wo (Ib) 315 wo (Ib) 0 wo 33 wo (Ic) 85 wo 0 wo (Ib) 192 wo (IIIc) 0 wo (Ib) 62 wo (Ic) 285 wo 398 wo 0 wo 52 wo (IVc) 0 wo (Ib) 287 wo 0 wo (Ib) 103 wo 0 #C12 (SD) Patient [#], Zeit nach erster Abnahme [wo], Stadium und klinisches Bild Abbildung 3-20: Monitoring der Serumantikörperantwort von zwölf CTCL-Patienten Die Serumantikörperantwort von zwölf CTCL-Patienten in unterschiedlichen Stadien der Erkrankung wurde während unterschiedlicher Zeiträume (17 bis 398 Wochen) betrachtet. Die erste untersuchte Serumabnahme wurde mit Woche 0 betitelt. In Abbildung A wurden vier der humanen Antigene ausgewählt und exemplarisch die Serumantikörperantworten über die Zeit dargestellt. Die Cut-off-Werte sind mit unterbrochenen Linien und dem Farbcode des jeweiligen Antigens eingezeichnet. Patient #C2 wies einen signifikanten Anstieg der Antikörper gegen MAGE-A3 auf, ebenso wie Patient #C3 gegen p53 und Patient #C10 gegen cTAGE-5a C-terminal. Antikörper gegen LAGE-1a nahmen signifikant in Patient #C8 ab. In Abbildung B sind die Veränderungen der Serumantikörperantworten über die Zeit gegen alle viralen Antigene dargestellt. Eine Serokonversion konnte für BK in Patient #C5 und für HPV1 in Patient #C12 gemessen werden. #C1: Patient 1, wo: Wochen, (I): Stadium I, MFI: „Mean fluorescence intensity“, ERGEBNISSE 118 CTCL: Kutanes T-Zell Lymphom, PD: “Progressive disease”, PR: “Partial response”, SD: “Stable disease”. Zusammenfassend ist in den vorliegenden Studien festzustellen, dass sowohl bei CTCL- als auch bei MM-Patienten, die keine Immuntherapie erfahren haben, eine Veränderung des Serumantikörperprofils oder eine Serokonversion relativ selten stattfinden. Das Serumantikörperprofil ist somit relativ stabil über die Zeit gesehen. Falls es jedoch zu einer dramatischen Serokonversion bzw. Veränderung der Serumantikörper kommt, ist dies teilweise mit einer Veränderung des Stadiums korreliert (vgl. Patient #M3 Verläufe der acht MMPatienten). Allein für LAGE-1a wurde in MM-Patienten ein stadienabhängiges Serumantikörperprofil nachgewiesen, welches für Therapieverlaufskontrollen verwendet werden könnte. ERGEBNISSE 119 3.2.7 Monitoring der Serumantikörperantwort in immunbehandelten CTCLPatienten Für an CTCL erkrankte Patienten werden immunmodulierende Behandlungen als vielversprechende Therapien diskutiert (Muche and Sterry 2002). Um mögliche Effekte verschiedener Immuntherapien auf CTCL-Patienten zu untersuchen, wurden Seren auf ihre Seroreaktivität gegen alle in Tabelle 3-4 aufgeführten humanen und viralen Antigene mittels der Multiplex-Serologie getestet. Für diese Untersuchung konnten 66 Seren von 19 Patienten, die mit einem rekombinanten Adenovirus als Träger von Interferon-γ (IFN-γ) behandelt wurden, herangezogen werden. Zudem wurden 45 Seren von fünf Patienten, die mit einem handelsüblichen Masernimpfstoff immunisiert wurden, auf ihre humorale Immunantwort getestet. Von den Patienten standen jeweils Seren vor, während und nach den jeweiligen Immunisierungen zur Verfügung. Um die Veränderung der Serumantikörperantwort richtig werten zu können, wurden folgende Kriterien festgelegt: Eine Serokonversion liegt erst vor, wenn sich die Antikörperantwort gegen ein Antigen über die Zeit mindestens verdoppelt oder mindestens halbiert. Zu beachten ist, dass hierbei mindestens ein MFI-Wert über dem Cut-offWert liegen muss. Adenovirale Immuntherapie Neunzehn CTCL-Patienten wurden mit einem nicht-replizierenden Adenovirus Typ 5 Vektor immunisiert, der ein humanes IFN-γ cDNA Insert trägt. Die Patienten erhielten in den ersten drei Wochen der Behandlung einmal wöchentlich eine Injektion in den Tumor. In der vierten Woche erfolgte keine Injektion. Diese Art der Immunisierung im Verlauf von vier Wochen wurde in bis zu vier Zyklen durchgeführt. Elf der 19 immunbehandelten Patienten wiesen keine Antikörper gegen eines der humanen Antigene auf. Jedoch konnte für Patient #008 eine eindeutig positive Serokonversion detektiert werden. Für diesen Patienten stiegen die Antikörper gegen MAGE-A3 von 30 über 1539 bis zu finalen 798 MFI an. Sieben Patienten hatten gleich bleibende Serumantikörper gegen unterschiedliche Antigene (Tabelle 3-5, Abbildung 3-21). Patient #003 besaß Antikörper gegen GBP-5ta und cTAGE-5a N-terminal (zwei Seren von vier seropositiv). Patient #004 erkannte mit gleich bleibenden Serumantikörpern die Antigene MAGE-A3 und A9. LAGE-1a hatte Antikörper in Patient #006 und GAGE-7b in Patient #009 bereits vor der Immuntherapie induziert. Gegen CAMEL und GBP-5ta (ein Serum von drei Seren seropositiv) konnten Antikörper im Serum von Patient #014 identifiziert werden. Patient #018 schließlich bildete gleich bleibende Antikörper gegen die vier Antigene MAGE-A1, A3, A9 und LAGE-1a. Jedoch konnte kein einheitlicher Effekt der adenoviralen Immuntherapie auf die humorale ERGEBNISSE 120 Immunantwort der Patienten erkannt werden. Die Auswertung der klinischen Parameter und des klinischen Verlaufs liegen zur Zeit nur für die Patienten #001 bis #009 vor. Eine vollständige Regression konnte für drei Patienten (#002, #006, #009) detektiert werden. Wie oben beschrieben besaßen #006 und #009 gleich bleibende Antikörpertiter ohne Serokonversion. Eine partielle Regression wurde für die Patienten #001 und #008 registriert. Einzig Patient #008 wies eine Serokonversion auf. Die Patienten #003 bis #005 und #007 befanden sich in einem stabilen Zustand ihrer Erkrankung. Der stabile Krankheitszustand der Patienten #003 und #004 spiegelte sich in konstanten Antikörpertitern wider (Dummer et al. 2004). Die Serumantikörperantworten gegen die viralen Antigene waren für alle Patienten bis auf die Patienten #011, #015 und #020 gleich bleibend (Abbildung 3-21). Für Patient #011 wurde ein Anstieg der Serumantikörper gegen BK und HPV1 detektiert und für Patient #015 gegen JC. Die Antikörperantworten von Patient #020 stiegen gegen die Antigene BK, JC und LPV an, wobei in einem Wiederholungstest diese Anstiege nicht mehr detektiert werden konnten. Somit ist von einem Pipettierfehler auszugehen. Für eine verlässliche statistische Auswertung war die Anzahl der getesteten Seren zu gering. Tabelle 3-5: Veränderung der Serumantikörperantwort gegen alle Antigene in 19 immunbehandelten CTCL-Patienten im Verlauf der Therapie Neunzehn CTCL-Patienten wurden mit einem adenoviralen Vektor, der das humane Interferon (IFN)-γ Gen trägt, immunisiert und die Serumantikörperantwort analysiert. Während des Monitorings der Serumantikörperantwort wurden vor allem gleich bleibende Antikörpertiter (→,→1 ein Serum von drei Seren seropositiv,→2 vier Seren von sechs seropositiv,→3 zwei Seren von vier seropositiv) und für ein humanes Antigen (MAGE-A3) eine positive Serokonversion (↑) nachgewiesen. PR: „Partial response“, CR: „Complete response”, SD: “Stable disease”. Patient MAGE- MAGE- MAGE- LAGE- GAGE- CAMEL se70- se57- GPB- cTAGE-5a cTAGE-5a p53 p16 Klinisches A1 A3 A9 1a 7b 2 1 5ta N-terminal C-terminal Bild # 001 PR # 002 CR # 003 # 004 # 005 # 006 # 007 # 008 # 009 # 010 # 011 # 013 - → ↑ - → - - → → - - - - → - → - # 014 # 015 # 016 # 017 # 018 → → → → - → - - - →1 - # 019 # 020 - - - - - - - - → - 2 3 - - - SD SD SD CR SD PR CR - - - - - - - - - - - ERGEBNISSE 121 A 6000 MAGE-A3 CAMEL GBP-5ta 5000 MAGE-A3, CAMEL, GBP-5ta MFI 4000 3000 2000 #003 #005 #006 #007 #008 #009 #010 #011 #016 #017 #018 #019 0 41 0 40 75 110 152 215 0 38 #015 0 42 77 106 133 169 197 224 252 294 #014 #013 0 34 64 133 0 326 0 43 78 104 140 0 42 0 62 0 69 102 144 0 32 0 36 0 29 66 93 0 33 #004 0 33 62 90 #002 0 36 64 #001 0 35 64 105 X 41 91 140 0 0 36 1000 #020 Patienten und Tage nach Vakzinierung B 25000 BK JC LPV HPV1 20000 BK, JC, LPV, HPV1 MFI 15000 10000 #004 #005 #006 #007 #008 #009 #010 #011 #013 #016 #017 #018 #019 0 41 0 40 75 110 152 215 0 38 #015 0 42 77 106 133 169 197 224 252 294 #014 0 34 64 133 0 326 0 43 78 104 140 0 42 0 62 0 69 102 144 0 32 0 36 0 29 66 93 0 33 0 35 64 105 #003 0 33 62 90 #002 0 36 64 #001 X 41 91 140 0 0 36 5000 #020 Patienten [#] und Tage nach Vakzinierung Abbildung 3-21: Darstellung der Serumantikörperantwort der 19 immunbehandelten CTCL-Patienten im Verlauf der Therapie In Abbildung A und B sind die Serumantikörperantworten aller 19 CTCL-Patienten, die mit einem adenoviralen Vektor, der das humane Interferon (IFN)-γ-Gen trägt, immunisiert wurden, im Verlauf der Therapie dargestellt. Mit Ausnahme von Patient #002 (X) sind alle Seren am Tag 0 vor Beginn der Immuntherapie entnommen worden. Der Zeitraum des Monitorings der Patienten beträgt bis zu 234 Tage nach der ersten Immunisierung. Cut-off-Werte der humanen Antigene sind mit unterbrochenen Linien entsprechend des Farbcodes des jeweiligen Antigens eingezeichnet. In A sind die Serumantikörperantworten exemplarisch für die humanen Antigene MAGE-A3, CAMEL und GBP-5ta zusammengefasst, in B für alle viralen Antigene. Eine positive Serokonversion wurde in Patient #008 gegen MAGE-A3 nachgewiesen. In verschiedenen Patienten konnten gleich bleibende ERGEBNISSE 122 Antikörperantworten gegen MAGE-A1, A3, A9, LAGE-1a, GAGE-7b, CAMEL, GBP-5ta und cTAGE5a N-terminal gemessen werden. Über die Zeit wurden positive Serokonversionen einiger viraler Antigene in den Patienten #011, #015 und #020 gemessen. MFI: „Mean fluorescence intensity“, CTCL: Kutanes T-Zell Lymphom. Masernimpfstoff Fünf Patienten wurden mit dem Lebendimpfstoff des Edmonston-Zagreb Masernvirus behandelt. Der Lebendimpfstoff wurde direkt in den Tumor appliziert, nachdem eine neun Millionen Einheiten Dosis an IFN-α zweimalig subkutan verabreicht worden war. Diese Vorbehandlung fand jeweils 72 und 24 Stunden vor der eigentlichen Lebendimpfstoff-Injektion statt. Jeder Behandlungszyklus bestand aus zwei Injektionen in den Tumor an Tag vier und 17 der Therapie, wobei zwei unterschiedliche Dosierungen an Lebendimpfstoff gewählt wurden (Heinzerling et al. 2005). Für die Patienten, die mit dem Masernimpfstoff immunisiert wurden, wurden ausschließlich gleich bleibende Antikörperantworten gemessen, sowohl für die humanen als auch für die viralen Antigene (Tabelle 3-6). Für keinen Patienten fand eine Serokonversion in Form eines Anstiegs oder eines Abfalls der Serumantikörperantwort statt. Tabelle 3-6: Veränderung der Serumantikörperantwort gegen alle Antigene in fünf immunbehandelten CTCL-Patienten im Verlauf der Therapie Fünf CTCL-Patienten wurden mit einem handelsüblichen Masernimpfstoff immunisiert und die Serumantikörperantwort analysiert. Während des Monitorings der Serumantikörperantwort wurden ausschließlich gleich bleibende Antikörpertiter (→,→1 sechs Seren von zwölf Seren seropositiv,→2 ein Serum von elf Seren seropositiv) nachgewiesen. CTCL: Kutanes T-Zell Lymphom, MAGE: Melanom Antigen, HPV: Humanes Papillomavirus, PR: „Partial response“, CR: „Complete response”, MR: “Minor response”. Patient MAGE- MAGE- MAGE- LAGE- GAGE- CAMEL se70- se57- GBP- cTAGE-5a cTAGE-5a p53 p16 Klinisches Bild A1 A3 A9 1a 7b 2 1 5ta N-terminal C-terminal →2 # 0001 PR 1 → # 0002 → CR # 0003 PR # 0004 MR # 0005 → → PR Zwei der fünf Patienten wiesen keine Antikörper gegen humane Antigene auf. Patient #0005 bildete gleich bleibende Serumantikörper gegen die humanen Antigene MAGE-A3 und A9. Nur ein Serum von elf von Patient #0001 war positiv für p53. Antikörper gegen CAMEL waren vor und nach der Therapie bei Patient #0002 vorhanden sowie Antikörper gegen GBP-5ta bei der Hälfte von 12 Seren (Abbildung 3-22). Allerdings ist bei Patient #0002 eine eindeutige Tendenz zum Anstieg verschiedener Serumantikörper zu erkennen, wenn auch teilweise unterhalb des Cut-off-Wertes oder nicht gemäß der Definition einer Serokonversion. ERGEBNISSE 123 A 3000 MAGE-A3 CAMEL GBP-5ta cTAGE-5a N-terminal 2500 MAGE-A3, CAMEL, GBP-5ta, cTAGE-5a N-terminal MFI 2000 1500 1000 500 # 0001 # 0002 (PR) (CR) # 0003 # 0004 (MR) (PR) 0 10 11 16 24 31 0 13 14 21 27 34 38 41 42 48 56 0 1 6 14 22 0 6 12 15 16 56 58 61 65 68 69 90 0 14 21 28 32 91 92 98 105 109 113 0 # 0005 (PR) Patienten und Tage nach Vakzinierung 25000 B 20000 BK, JC, LPV, HPV1 MFI 15000 BK JC LPV HPV1 10000 5000 0 14 21 28 32 91 92 98 105 109 113 0 6 12 15 16 56 58 61 65 68 69 90 0 1 6 14 22 0 13 14 21 27 34 38 41 42 48 56 0 10 11 16 24 31 0 # 0001 # 0002 (PR) (CR) # 0003 (PR) # 0004 (MR) # 0005 (PR) Patienten und Tage nach Vakzinierung Abbildung 3-22: Darstellung der Serumantikörperantwort der fünf mit einem Masernimpfstoff behandelten CTCL-Patienten im Verlauf der Therapie In Abbildung A und B sind die Serumantikörperantworten der fünf CTCL-Patienten, die mit einem handelsüblichen Masernimpfstoff immunisiert wurden, im Verlauf der Therapie dargestellt. Allen Patienten wurde vor der Immuntherapie (Tag 0) Blut entnommen. Der Zeitraum des Monitorings der Patienten beträgt bis zu 113 Tage nach der ersten Immunisierung. Cut-off-Werte der humanen Antigene sind mit unterbrochenen Linien entsprechend des Farbcodes des jeweiligen Antigens eingezeichnet. In A sind die Serumantikörperantworten exemplarisch für die humanen Antigene MAGE-A3, CAMEL, GBP- ERGEBNISSE 124 5ta und cTAGE-5a N-terminal zusammengefasst, in B für alle viralen Antigene. Nur für Patient # 0002 lässt sich eine tendenziell ansteigende Antikörperantwort der Antigene CAMEL, GBP-5ta und cTAGE-5a N-terminal erkennen. MFI: „Mean fluorescence intensity“, CTCL: Kutanes T-Zell Lymphom, PR: „Partial response“, CR: „Complete response”, MR: “Minor response”. Für cTAGE-5a N-terminal wurde in Patient #0002 ein deutlicher Anstieg der Antikörper von 84 auf 408 MFI gemessen. Auch für CAMEL ist dieser Anstieg von 1889 auf 2400 MFI zu erkennen. Für GBP-5ta steigen die Antikörper leicht von 1408 auf 1759 MFI an. Dieser Trend des Anstiegs von Serumantikörpern korreliert mit dem Krankheitsverlauf des Patienten. Heinzerling et al. (2005) beschrieben im Verlauf der Therapie bei diesem Patienten eine komplette Remission des mit dem Masernimpfstoff behandelten Plaque sowie ein Ansprechen einer weiteren Läsion, die im Anschluss an die Remission des ersten Plaques behandelt wurde. Einzig bei Patient #0003 konnte eine vollständige Remission des behandelten Plaques erzielt werden. Drei der anderen Patienten wiesen eine partielle Remission auf. Für Patient #0004 konnte nur ein geringfügiges Ansprechen auf die Therapie nachgewiesen werden. ERGEBNISSE 125 3.3 Expressionsanalyse des Cancer Testis Antigens GAGE-7b Das CTA GAGE-7b sollte in dieser Arbeit hinsichtlich seiner Expression an verschiedenen Zelllinien und Gewebeproben von MM-Patienten untersucht werden. Dies erfolgte auf mRNAEbene mittels Reverse Transkriptase-Polymerasekettenreaktion (RT-PCR) und auf Proteinebene mittels polyklonaler Antikörper, die hierfür in Kaninchen generiert wurden. Für die Herstellung der Antikörper musste zunächst aufgereinigtes Protein gewonnen werden. 3.3.1 Identifizierung von GAGE-7b und Homologie der GAGE-Familie Zunächst wurde GAGE-1 unter der Verwendung von Tumor-reaktiven, autologen zytotoxischen T-Lymphozyten (CTL) an der MM-Zelllinie MZ2-Mel identifiziert (Van den Eynde et al. 1995). Das antigene Peptid YRPRPRRY wurde von reaktiven CTL auf dem HLA Klasse I Molekül Cw6 erkannt und führte zur Lyse der Tumorzellen. GAGE-1, 2 und 8 kodieren für dieses Peptid. Die durchgeführte Expressionsanalyse ergab eine Cancer Testis-spezifische Expression. Ein anderer CTL-Klon reagierte gegen das HLA Klasse I A29-präsentierte Epitop YYWPRPRRY, welches aus dem gleichen Bereich des GAGE-Proteins stammt (De Backer et al. 1999). Dieses Epitop wird von den Antigenen GAGE-3, -4, -5, -6, -7 und –7b kodiert. Somit konnten an cDNA-Banken insgesamt 9 verschiedene GAGE-mRNAs isoliert werden (Van den Eynde et al. 1995; Chen et al. 1998; De Backer et al. 1999). In Abbildung 3-23 ist eine Analyse mit dem HUSAR-Programm „Clustal“ zur Verdeutlichung der hohen Homologie der GAGE-Familie auf Proteinebene dargestellt. 3.3.2 Expressionsanalyse von GAGE-7b mRNA Zwei verschiedene Primerpaare wurden für die Expressionsanalyse verwendet (De Backer et al. 1999). Es wurde darauf verzichtet, Normalgewebe auf mRNA-Ebene zu testen, da bekannt ist, dass alle GAGE-Gene CTA sind. Für den Nachweis von GAGE-1, 2 und 8 wurden der Forward Primer vde44 und der Reverse Primer vde24 verwendet, die ein Produkt der Länge 244bp ergeben. Ein Produkt gleicher Größe wurde für den Nachweis von GAGE-3 bis 7 und 7b unter Verwendung des Forward Primers vv-1 und des Reverse Primers vde24 gewählt. In Abbildung 3-24 ist ein repräsentatives Agarosegel der RT-PCR mit verschiedenen Zelllinien unter Verwendung beider Primerpaare dargestellt. ERGEBNISSE 126 GAGE-1.PEP GAGE-2.PEP GAGE-3.PEP GAGE-4.PEP GAGE-5.PEP GAGE-6.PEP GAGE-7.PEP GAGE-7b.pep GAGE-8.PEP 1 ~MSWRGR.ST ~MSWRGR.ST MNLSRGKSTY ~MSWRGRSTY ~MSWRGRSTY ~MSWRGRSTY ~MSWRGRSTY ~MSWRGRSTY ~MSWRGR.ST YRPRPRRYVE YRPRPRRYVE YWPRPRRYVQ YWPRPRRYVQ YWPRPRRYVQ YWPRPRRYVQ YWPRPRRYVQ YWPRPRRYVQ YRPRPRRYVE PPEMIGPMRP PPEMIGPMRP PPEVIGPMRP PPEMIGPMRP PPEVIGPMRP PPEVIGPMRP PPEMIGPMRP PPEMIGPMRP PPEMIGPMRP EQFSDEVEPA EQFSDEVEPA EQFSDEVEPA EQFSDEVEPA EQFSDEVEPA EQFSDEVEPA EQFSDEVEPA EQFSDEVEPA EQFSDEVEPA 50 TPEEGEPATQ TPEEGEPATQ TPEEGEPATQ TPEEGEPATQ TPEEGEPATQ TPEEGEPATQ TPEEGEPATQ TPEEGEPATQ TPEEGEPATQ GAGE-1.PEP GAGE-2.PEP GAGE-3.PEP GAGE-4.PEP GAGE-5.PEP GAGE-6.PEP GAGE-7.PEP GAGE-7b.pep GAGE-8.PEP 51 RQDPAAAQEG RQDPAAAQEG RQDPAAAQEG RQDPAAAQEG RQDPAAAQEG RQDPAAAQEG RQDPAAAQEG RQDPAAAQEG RQDPAAAQEG EDEGASAGQG EDEGASAGQG EDEGASAGQG EDEGASAGQG EDEGASAGQG EDEGASAGQG EDEGASAGQG EDEGASAGQG EDEGASAGQG PKPEADSQEQ PKPEAHSQEQ PKPEADSQEQ PKPEADSQEQ PKPEADSQEQ PKPEADSQEQ PKPEAHSQEQ PKPEAHSQEQ PKPEADSQEQ GHPQTGCECE GHPQTGCECE GHPQTGCECE GHPQTGCECE GHPQTGCECE GHPQTGCECE GHPQTGCECE GHPQTGCECE GHPQTGCECE 100 DGPDGQEMDP DGPDGQEMDP DGPDGQEMDP DGPDGQEMDP DGPDGQEMDP DGPDGQEVDP DGPDGQEMDP DGPDGQEMDP DGPDGQEMDP GAGE-1.PEP GAGE-2.PEP GAGE-3.PEP GAGE-4.PEP GAGE-5.PEP GAGE-6.PEP GAGE-7.PEP GAGE-7b.pep GAGE-8.PEP 101 PNPEEVKTPE PNPEEVKTPE PNPEEVKTPE PNPEEVKTPE PNPEEVKTPE PNPEEVKTPE PNPEEVKTPE PNPEEVKTPE PNPEEVKTPE EEMRSHYVAQ EGEKQSQC~~ EGEKQSQC~~ EGEKQSQC~~ EGEKQSQC~~ EGEKQSQC~~ EGEKQSQC~~ EGEKQSQC~~ EGEKQSQC~~ TGILWLLMNN ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ 140 CFLNLSPRKP ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ ~~~~~~~~~~ Abbildung 3-23: Multiples Alignment der GAGE-Proteine Unter Verwendung der Software „Clustal“ des HUSAR Bioinformatik-Programms des DKFZ wurde dieses multiple Alignment der Proteine der 9 Familienmitglieder der GAGE-Familie angefertigt. Blau: 100% Identität der Aminosäuren innerhalb der Spalte, rot: mindestens 75% Identität der Aminosäuren innerhalb der Spalte, grün: mehr als 50% Identität der Aminosäuren in der Spalte. 3000bp 3000bp 250bp 2 3 4 5 6 1000bp 1000bp 500bp 500bp 1 3000bp 1000bp 1000bp A 3000bp M M 7 8 500bp 250bp 500bp 250bp 9 10 B 1 2 3 4 5 6 7 8 9 10 11 12 13 14 M Abbildung 3-24: Repräsentative RT-PCR mit beiden GAGE-Primerpaaren cDNA von CTCL- und MM-Zelllinien sowie MM-Geweben wurde verwendet. Als Positivkontrolle diente Testis- und UKVR-Mel-02-cDNA, als Negativkontrolle Wasser. In A wurde das Primerpaar für GAGE-1, 2, 8 und in B für GAGE-3 bis 7b verwendet. Es ist jeweils eine Bande in Höhe von 244bp zu sehen. A Gewebe von MM-Patienten, Spur 1:MM10, 2: MM9, 3: MM6, 4: MM2, 5: MM7, 6: MM4, 7: MM3, 8: MM5, 9: Wasser, 10: UKVR-Mel-02, M: Marker. B CTCL- und MM-Zelllinien, Spur 1: CouL3, 2: MeWo: 3: Ma-Mel-47, 4: Ma-Mel-12, 5: Ma-Mel-52, 6: Ma-Mel-04, 7: SeAx, 8: Ma-Mel-11, 9: Ma-Mel-05, 10: Ma-Mel-37b, 11: Ma-Mel-16, 12: Wasser, 13: UKRV-Mel-02, 14: Testis, M: Marker, bp: Basenpaare. ERGEBNISSE 127 Die Expression der beiden GAGE-Gruppen wurde an cDNA von 5 CTCL-, 17 MM- sowie zwei Kontrollzelllinien (Melanozyten, HaCat) und Gewebeproben von 8 MM-Patienten untersucht. Die Ergebnisse der RT-PCR sind in Tabelle 3-7 zusammengefasst. Tabelle 3-7: Expressionsanalyse der beiden GAGE-Gruppen in Zelllinien und Geweben In der Tabelle sind die positiven Zelllinien und Gewebe aufgeführt. Die Gesamtzahl der getesteten Proben ist in Klammern (n) angegeben. Die Einzelergebnisse der RT-PCR sind in Anhang 3 dargestellt. CTCL: Kutanes T-Zell Lymphom, MM: Malignes Melanom, n: Anzahl, RT-PCR: Reverse TranskriptasePolymerasekettenreaktion. GAGE-Gruppe GAGE-1, 2, 8 GAGE-3 bis 7b Positive RT-PCR Kontrollzelllinien CTCL-Zelllinien MM-Zelllinien (n=2) (n=5) (n=17) 0 (0%) 0 (0%) 12 (71%) 0 (0%) 0 (0%) 14 (82%) MM-Gewebe (n=8) 3 (38%) 4 (50%) Weder GAGE-1, 2, 8 noch GAGE-3 bis 7b wurden in den untersuchten Kontroll- und CTCLZelllinien exprimiert. GAGE-3 bis 7b waren mit 82% in den MM-Zelllinien und 50% in den MM-Geweben häufiger exprimiert als GAGE-1, 2, 8 (71% und 38%). 3.3.3 Antikörper-Herstellung mittels GST-GAGE-7b-tag-Fusionsprotein GAGE-7b wurde in den GST-tag-Vektor kloniert und rekombinant exprimiert. Aus 6L Expressionskultur wurden 2mg Protein, welches nativ im Batch-Verfahren über Glutathion Sepharose aufgereinigt wurde, gewonnen. Dieses Protein wurde im Western Blot mit den GSTund tag-Antikörpern und im Coomassie-Gel überprüft. Da eine ausreichende Reinheit erhalten wurde, konnte das Protein abschließend gegen 1x phosphatgepufferte Salzlösung (PBS) dialysiert werden, um das Glutathion aus dem Elutionspuffer zu entfernen. Mit dem dialysierten Protein wurden zwei Kaninchen bei der Firma Biogenes immunisiert und die Antikörper K5101 und K5102 hergestellt. Beide Antikörper wurden anschließend im Western Blot (15% Polyacrylamidgel) in einer Verdünnung von 1:5000 evaluiert. Wie aus Abbildung 3-25 ersichtlich wurden keine spezifischen Antikörper gegen GAGE-7b in den immunisierten Kaninchen gebildet. Es ist keine spezifische Bande, die das GAGE-7b-Protein bei 25 kilo Dalton (kDa) (laut HUSAR 13kDa) darstellt, in Testis zu sehen. Zudem reagierte das Antiserum gegen andere GST-X-tag-Proteine, die rekombinant in Bakterien exprimiert wurden, was auf die Bildung von GST- und tag-Antikörper hindeutet. Aufgrund dieser Ergebnisse wurde die Aufreinigung des rekombinanten Proteins abgeändert. Um die Bildung von GST-Antikörpern zu verhindern, wurde die Abspaltung des GST-Proteinteils und ein sehr reines Protein angestrebt. ERGEBNISSE A M 1 2 3 4 5 6 7 8 9 B 202kDa 128kDa 80kDa 202kDa 128kDa 80kDa 37,9kDa 37,9kDa 30,5kDa 30,5kDa 19kDa 128 M 1 2 3 4 5 6 7 8 9 19kDa 6,8kDa 6,8kDa Abbildung 3-25: Analyse der Antikörper K5101 und K5102 im Western Blot Sowohl das Antiserum K5101 (A) als auch das Antiserum K5102 (B) reagierten stark gegen andere GSTX-tag-Fusionsproteine. Keine spezifische Reaktion wurde in Testis bei einer Verdünnung von 1:5000 gemessen. GAGE-7b sollte durch eine Bande bei 25kDa detektiert werden. Die folgenden Proteine stammen aus rekombinanter Expression in Bakterien, Zelllinien oder gesunden Spendern, M: Marker, Spur 1: GST-tag, 2: GST-tag, 3: GST-LAGE-1a-tag, 4: GST-LAGE-1a-tag, 5: SeAx, 6: MyLa, 7: Testis, 8: GST-GAGE-7b-tag, 9: GST-GAGE-7b-tag, kDa: kilo Dalton, GST: Glutathion-S-Transferase. 3.3.4 Evaluierung der optimalen Expressions- und Aufreinigungsbedingungen für das rekombinante GST-GAGE-7b-tag-Protein Um die optimalen Expressionsbedingungen für das rekombinante GST-GAGE-7b-tag-Protein zu erhalten, wurde jeweils 1mL Bakteriensuspension einer 250mL Expressionskultur vor und nach der Induktion der Proteinbildung mit Isopropyl-β-D-thiogalactosid (IPTG) entnommen. Die Induktion wurde jeweils über Nacht durchgeführt. Die pelletierten Bakterien wurden direkt in Probenpuffer für den Western Blot aufgenommen und lysiert. Zum Vergleich der Bedingungen wurden gleiche Volumina aller Proben in einem Coomassie-Gel aufgetragen. Das GST-GAGE7b-tag-Protein wurde bei 45kDa als dicke Bande detektiert und im Western Blot mit den Antikörpern anti-GST und anti-tag identifiziert (Ergebnisse nicht dargestellt). Laut Abbildung 3-26 ist die deutliche Induktion von GST-GAGE-7b-tag-Protein in allen Fraktionen nach der Proteininduktion durch IPTG-Zugabe als dicke Bande bei 45kDa erkennbar. Bei einer IPTGKonzentration von 0,5mM und einer Induktion bei Raumtemperatur (Rt) wurde mehr Protein als bei 17°C gewonnen und weniger Abbruchbanden als bei 37°C erzeugt. Unterschiedliche IPTGKonzentrationen erbrachten bei 17°C vergleichbare Proteinmengen. Deshalb wurde eine mittlere IPTG-Konzentration von 0,25mM zur Induktion gewählt. Zudem wurde später festgestellt, dass sowohl eine Induktion über Nacht als auch eine Induktion für 6h ein äquivalentes Ergebnis ergibt, so dass fortan die rekombinante Proteinproduktion für 6h bei Rt mittels 0,25mM IPTG induziert wurde. ERGEBNISSE M 1 2 3 4 5 6 129 7 8 9 10 M 250kDa 150kDa 100kDa 75kDa 250kDa 150kDa 100kDa 75kDa 50kDa 50kDa 37kDa 37kDa 25kDa 20kDa 25kDa 20kDa Induktion vor nach vor nach vor IPTG [mM] 0.1 0.1 0.25 0.25 0.5 Temperatur [°C] 17 17 17 17 17 nach vor nach vor nach 0.5 0.5 0.5 0.5 0.5 17 Rt Rt 37 37 Abbildung 3-26: Coomassie-Gel zur Bestimmung der optimalen Expressionsbedingungen für GST-GAGE-7b-tag In dieser Abbildung ist ein Coomassie-Gel (15% Polyacrylamidgel) zur Bestimmung der optimalen Expressionsbedingungen für GST-GAGE-7b-tag-Protein dargestellt. In allen Spuren wurden gleiche Probevolumina des Bakterienlysats aufgetragen. Der Zeitpunkt der Probenentnahme, die Isopropyl-β-Dthiogalactosid (IPTG)-Konzentration und die Temperatur sind der dargestellten Tabelle zu entnehmen. Das GST-GAGE-7b-tag-Protein ist durch Pfeile gekennzeichnet. Rt: Raumtemperatur, kDa: kilo Dalton, M: Marker, GST: Glutathion-S-Transferase. Die Art der Bakterienlyse, French Press im Vergleich zu Sonifizierung inklusive Homogenisieren, wurde unter Verwendung des GST-tag-Proteins evaluiert. 1L GST-tagProteinlysat wurde in zwei gleiche Teile geteilt. Ein Teil wurde dreimal in der French Press lysiert, wohingegen der andere Teil sonifiziert, mit Lysozym, Triton-X-100 und 1,4-Dithiotreitol (DTT) behandelt und homogenisiert wurde. Abbildung 3-27 verdeutlicht, dass in den Eluaten nach der French Press deutlich mehr eluiertes GST-tag-Protein enthalten ist und dass in allen vier Eluaten nach der French Press hohe GST-tag-Proteinmengen messbar sind. Das GST-tagProtein wurde bei 26kDa detektiert. Deshalb wurden alle Bakterienlysate fortan mittels French Press aufgearbeitet. Aus zahlreichen Vorversuchen war ersichtlich, dass GST-X-tag-Fusionsprotein nicht an die Glutathion Sepharose gebunden wurde und mit dem Durchfluss, auch nach mehrmaligem Inkubieren der Sepharose mit demselben Proteinlysat, entfernt wurde. Um den Einfluss des pHs während der Bindung der GST-X-tag-Proteine an die Glutathion Sepharose zu analysieren wurden neben dem üblichen pH von 7,4 zwei andere pH-Werte von 6,5 und 8,0 in den Bakterienlysaten gewählt. Da im Lysat ausreichend GST-GAGE-7b-tag-Protein enthalten war, konnte dieses Protein an die Glutathion Sepharose binden (Abbildung 3-27). Bei einem Bindungs-pH von 6,5 wurde mehr Protein gebunden als bei einem pH von 8,0. Die Fraktionen ERGEBNISSE 130 der Eluate mit einem Bindungs-pH von 6,5 enthalten deutlich mehr Protein als pH8,0 Eluate. In Folge dessen wurde ein pH von 6,5 in den Bakterienlysaten für die Bindung der Proteine an die Glutathion Sepharose gewählt. A M 1 2 3 4 B 5 6 7 M 50kDa 50kDa 37kDa 37kDa 50kDa 37kDa 25kDa 25kDa 20kDa 25kDa 20kDa M 1 2 3 4 5 6 7 20kDa Abbildung 3-27: Coomassie-Gel zum Vergleich der Lyse von GST-tag-Protein in der French Press gegenüber Sonifizierung und Homogenisieren (A) und zur Bestimmung des optimalen pH-Werts während der Bindung von GSTGAGE-7b-tag-Protein an Glutathion Sepharose (B) A In diesem Coomassie-Gel (15% Polyacrylamidgel) wird die Lyse von Bakterien mittels French Press verglichen mit Sonifizierung und Homogenisieren. Das aufgereinigte, von der Glutathion SepharoseSäule eluierte GST-tag-Protein wurde bei einer Größe von 26kDa detektiert und ist vermehrt in den Eluaten des mit der French Press erhaltenen Proteins vorhanden. M: Marker, Spuren 1-4: French Press, 1: Eluat 1, 2: Eluat 2, 3: Eluat 3. 4:Eluat 4, Spuren 5-7: Sonifizierung und Homogenisieren, 5: Eluat 2, 6:Eluat 3, 7: Eluat 4, kDa: kilo Dalton. B Im Coomassie-Gel (15% Polyacrylamidgel) ist der Unterschied zwischen der Bindung von GST-GAGE-7b-tag-Protein an Glutathion Sepharose bei einem pH-Wert des Lysats von 6,5 und 8,0 dargestellt. GST-GAGE-7b-tag-Protein bindet bei einem pH von 6,5 vermehrt an die Glutathion Sepharose. Entsprechend wird mehr Protein eluiert, wobei gleiche Probevolumina aufgetragen wurden. GST-GAGE-7b-tag-Protein ist bei 45kDa detektierbar. M: Marker, Spur 1: Geklärtes Lysat, 2: Durchfluss bei pH6,5, 3: Durchfluss bei pH8,0, 4: gebundenes Protein pH6,5, 5: gebundenes Protein pH8,0, 6: Eluat pH6,5, 7: Eluat pH8,0. GST: Glutathion-S-Transferase. 3.3.5 Aufreinigung des GST-GAGE-7b-tag-Proteins an Glutathion Sepharose und Abspaltung des GST-Fusionspartners mittels Thrombin Eine 3L Expressionskultur des rekombinanten GST-GAGE-7b-tag-Proteins wurde 6h bei Rt mit 0,25mM IPTG induziert, die Bakterienpellets in insgesamt 40mL 1x PBS pH6,5 aufgenommen und bis zur Verarbeitung in der French Press eingefroren. Die 40mL Bakterienlysat wurden drei Mal in der French Press aufgeschlossen und anschließend direkt über eine Glutathion SepharoseSäule aufgereinigt. Hierbei wurde das eisgekühlte Bakterienlysat bei Rt drei Mal über die Säule gegeben, um Fusionsprotein zu binden. Zur Entfernung von ungebundenem Protein wurde die Säule intensiv mit PBS gewaschen. Der GAGE-7b-tag-Fusionsteil wurde unter Verwendung von 100 Units (U) Thrombin während einer zweistündigen Inkubation bei Rt abgespalten. Das lösliche GAGE-7b-tag-Protein konnte anschließend in zwei bis drei Elutionsschritten mit PBS ERGEBNISSE 131 erhalten werden. Schließlich lagen zumeist zwei Mal je 3mL abgespaltenes Protein von ca. 1mg/mL Konzentration vor. Wie in Abbildung 3-28 dargestellt, war eine deutliche Bande mit GST-GAGE-7b-tag-Fusionsprotein (ca. 45kDa) im aufgeschlossenen Bakterienlysat vor und nach Zentrifugation vorhanden. Allerdings befanden sich ebenfalls ein Anteil an unlöslichem Fusionsprotein und einigen Abbauprodukten im Pellet. Deutlich kann man in Spur vier das an die Säule gebundene Fusionsprotein erkennen. Nach der Abspaltung lag nur noch GST-Protein an der Säule gebunden vor (Spur 5), das abgespaltene Protein befand sich in Lösung und war als 26kDa große Bande sichtbar (Spur 6). Jedoch wurde GST-Protein in der Proteinlösung nach GST-Abspaltung nachgewiesen (Spur 6, 25kDa), so dass eine weitere Aufreinigung notwendig wurde. GST-Protein sollte möglichst gänzlich entfernt werden, damit es bei einer Immunisierung zu keiner Bildung von unspezifischen GST-Antikörpern kommen würde. M 1 2 3 4 5 6 75kDa 50kDa 37kDa 25kDa 20kDa 15kDa Abbildung 3-28: Coomassie-Gel zur Aufreinigung von GST-GAGE-7b-tag und Abspaltung des GST-Fusionspartners Dieses repräsentative Coomassie-Gel (15% Polyacrylamidgel) stellt die Aufreinigung von GST-GAGE7b-tag und die Abspaltung des GST-Fusionspartners von GAGE-7b-tag mittels Thrombin dar. In den Spuren 1-3 wurden ca. 10µg Protein und in den Spuren 4-6 ca. 1µg Protein aufgetragen. Das GSTGAGE-7b-tag-Protein wird in den Spuren 1-4 bei ca. 45kDa, das GST-Protein in den Spuren 4-6 bei ca. 25kDa und das GAGE-7b-tag-Protein in Spur 6 bei 26kDa detektiert. M : Marker, Spur 1: E. coli Lysat, 2: geklärtes E. coli Lysat, 3: Pellet, 4: GST-GAGE-7b-tag-Protein gebunden an Glutathion Sepharose, 5: GST-Protein gebunden an Glutathion Sepharose nach Thrombinabspaltung von GAGE-7b-tag, 6: Abgespaltenes lösliches Protein. kDa: kilo Dalton, GST: Glutathion-S-Transferase. 3.3.6 Zusätzliche Aufreinigung des GAGE-7b-tag-Proteins mittels Anionenaustauschchromatographie (MonoQ) und Identifizierung von GAGE-7b mittels MALDI-TOF Mittels Anionenaustauschchromatographie sollten verunreinigende Proteine aus der Lösung des abgespaltenen GAGE-7b-tag-Proteins entfernt werden. Hierzu wurden die ersten beiden ERGEBNISSE 132 Elutionen nach der Abspaltung des GAGE-7b-tag-Proteins vom GST-Fusionspartner verwendet. Diese beiden Elutionen wurden vereint und mit Puffer A 1:10 verdünnt, um eine Salzkonzentration von ungefähr 15mM Natriumchlorid (NaCl) zu erhalten. Nach der Bindung der Proteine an die MonoQ Säule wurde ein Salzgradient von 15mM bis 1M NaCl verwendet, um die Proteine aufzutrennen. Das Eluat wurde in Fraktionen von 1mL gesammelt. Anhand des Chromatogramms der Proteinauftrennung (Abbildung 3-29) wurde entschieden, welche Fraktionen in einem Coomassie-Gel untersucht wurden. Im Chromatogramm ist deutlich zu erkennen, dass kein Reinheitspeak und somit keine optimale Auftrennung der Proteinlösung erzielt werden konnte. Sechs Peaks laufen zu Beginn der Auftrennung bei einer Salzkonzentration von 200 bis 400mM NaCl ineinander. Jedoch konnten einige Proteine (Peaks bei Fraktion 26, 30 und 35) abgetrennt werden. Da aus dem Chromatogramm keine eindeutige Auftrennung erkennbar war, musste ein Coomassie-Gel die Güte der Auftrennung während der Ionenaustauschchromatographie aufklären. NWGAGE101:1_UV1_280nm NWGAGE101:1_UV2_260nm NWGAGE101:1_Conc NWGAGE101:1_Fractions mAU %B 80 600 60 400 40 200 20 0 Waste 3 105.0 4 5 6 7 8 110.0 9 101112 13 14 15 115.0 16 17 18 19 20 21 120.0 22 23 24 25 26 125.0 27 28 29 30 31 130.0 32 33 34 35 36 135.0 37 38 39 0 ml Abbildung 3-29: Chromatogramm der Ionenaustauschchromatographie für GAGE-7b-tag In diesem repräsentativen Chromatogramm ist die Auftrennung der GAGE-7b-tag-Proteinlösung an einer MonoQ Säule dargestellt. Auf der x-Achse sind die Anzahl der bereits durch die Säule geflossenen mL an Lösung und Puffer und die gesammelten Fraktionen aufgeführt. Die y-Achse gibt die milli „absorption units“ (mAU) an. Die rote Kurve stellt das UV-Spektrum bei 260nm und die blaue Kurve das Spektrum bei 280nm dar. Der NaCl-Gradient wird durch die grüne Gerade und an der rechten Achse in %B angegeben. NaCl: Natriumchlorid. Zur Untersuchung des Ergebnisses der Ionenaustauschchromatographie in einem Coomassie-Gel (15% Polyacrylamidgel) wurden die Proben vor der Chromatographie sowie Fraktionen, die prominente Peaks im Chromatogramm darstellen ausgewählt. Die so erzeugten Coomassie-Gele ERGEBNISSE 133 sind in Abbildung 3-30 dargestellt. In Spur 1 und 2 sind die Banden von GAGE-7b-tag bei 26kDa und darunter bei 25kDa für GST sichtbar. In der stark verdünnten Proteinlösung des Durchlaufs durch die MonoQ Säule sind auch bei maximalem Volumenauftrag keine nicht an die Säule bindenden Proteine erkennbar. Ab Spur 7 sind unterschiedliche Fraktionen, die während der Elution der gebundenen Proteine mittels Salzgradienten aufgefangen wurden, aufgetragen. In den Fraktionen 15, 16 und 18 (Spuren 10-12 und 15-17) ist kein GST-Protein detektierbar und die Bande für GAGE-7b-tag-Protein ist prominent. Die Bande unterhalb von 25kDa wurde in der „Matrix assisted laser desorption/ionisation-time of flight“ (MALDI-TOF) Analyse untersucht. Die Bande bei 75kDa in Fraktion 30 (Spur 22) stellt ein abgetrenntes Protein dar. Somit wurden die Fraktionen 15, 16 und 18 (Spuren 10-12 und 15-17) eindeutig als Fraktionen mit ausreichend aufgereinigtem Protein identifiziert und nach ihrer Vereinigung mittels Vakuumzentrifugation eingeengt. Schließlich konnten aus 3L Bakterienlysat ca. 1,5mg aufgereinigtes und abgespaltenes GAGE-7b-tag-Protein erhalten werden. M 1 2 3 4 5 M 6 7 8 9 10 11 12 13 14 M 15 16 17 18 19 20 21 22 23 75kDa 50kDa 37kDa 25kDa 20kDa 15kDa Abbildung 3-30: Coomassie-Gele der Proben und Fraktionen der Ionenaustauschchromatographie In diesen Coomassie-Gelen (15% Polyacrylamidgele) sind die Proben vor und Fraktionen nach der Ionenaustauschchromatographie dargestellt. Für die Spuren 1 und 2 wurden ca. 5µg Protein aufgetragen. In den restlichen Spuren wurden gleiche Probevolumina von 10µL (Spuren 10-13, 15-19) und 20µL (Spuren 3-9, 14, 20-23) aufgegeben. Spur 1+2: GAGE-7b-tag 1.+2. Elution, 3+4: 1.+2. Elution 1:10 verdünnt mit Puffer A, 5+6: Durchlauf 1+2, 7-14: Fraktion 9, 12, 13, 15, 16, 18, 19, 22, 15-17: Fraktion 15, 16, 18 1:10 verdünnt mit 1xPBS, 18-23: Fraktion 20, 21, 24, 26, 30, 35, kDa: kilo Dalton, M: Marker. In der Arbeitsgruppe von Dr. M. Schnölzer in der zentralen Proteinanalytik des DKFZ wurde die Analyse des abgespaltenen Proteins mittels MALDI-TOF durchgeführt. Hierzu wurden die Banden bei 26 und 22,5kDa aus dem denaturierten Coomassie-Gel ausgeschnitten, mit Trypsin verdaut und massenspektrometrisch untersucht. Die Bande bei 26kDa konnte als GAGE-7b-tag identifiziert werden. Mittels Peptidmassen-Fingerprint Analyse unter Verwendung von MALDITOF-MS und massenspektrometrischer Sequenzierung mit MALDI „Post source decay ERGEBNISSE 134 fragmentation“ wurden drei eindeutig GAGE-7b-tag zuzuordnende Peptide nachgewiesen: STYYWPR (m/z 972,47), STYYWPRPRR (m/z 1225,60) sowie QSQCVDKPPTPPPEPET (m/z 1906,85; C: carbamidomethyliert). Die Analyse der Bande bei 22,5kDa konnte nur das GAGE7b-tag zugehörige Peptid STYYWPRPRR in sehr geringer Konzentration bestätigen. Somit ist die untere Bande höchstwahrscheinlich GAGE-7b-tag. Für eine eindeutige, zuverlässige Zuordnung sollten zwei Peptide des jeweiligen Proteins identifiziert werden. 3.3.7 Immunisierung eines Kaninchens mit aufgereinigtem GAGE-7b-tag-Protein Im zentralen Tierlabor des DKFZ wurde ein Kaninchen mit dem wie unter 3.3.5 und 3.3.6 beschrieben aufgereinigten, abgespaltenen GAGE-7b-tag-Protein immunisiert. Zunächst wurde das Kaninchen K80 mit 750µg Protein subkutan immunisiert und über vier Wochen beobachtet. Anschließend wurde K80 mit insgesamt 700µg Protein sowohl subkutan als auch intravenös geboostet. Nach 10 Tagen wurde dem Tier Blut entnommen, und ein ELISA mit Präimmunserum, Serum vier Wochen nach der Immunisierung bzw. vor dem 1. Boost sowie dem Serum 10 Tage nach dem 1. Boost durchgeführt. Die Ergebnisse dieser Untersuchung sind in Abbildung 3-31 dargestellt. Aus den Titrationskurven ist deutlich ersichtlich, dass K80 vor der Immunisierung keine Antikörper gegen GAGE-7b-tag-Protein besaß, da das Präimmunserum selbst bei einer Verdünnung von 1:200 eine optische Dichte (OD) bei 450nm von unter 0,1 aufwies. Desweiteren wurden innerhalb der vier Wochen nach Immunisierung Antikörper gebildet (Kurve K80 Serum vor dem 1. Boost), die nach dem Boost deutlich zunahmen. Ein Antikörpertiter von 1:6400 konnte ermittelt werden, woraufhin K80 am 17. Tag nach dem 1. Boost ausgeblutet wurde. ERGEBNISSE 135 3,0 K80 Präimmunserum K80 Serum vor dem 1. Boost K80 Serum 10 Tage nach dem 1. Boost 2,5 1,5 OD 450nm 2,0 1,0 0,5 0,0 1:25600 1:12800 1:6400 1:3200 1:1600 1:800 1:400 1:200 Verdünnung Abbildung 3-31: Titration der Kaninchenseren vor und nach Immunisierung im ELISA Diese Abbildung stellt die Titration des Präimmunserums des Kaninchen 80 (K80), des Serums vier Wochen nach der Immunisierung mit GAGE-7b-tag-Protein und vor dem 1. Boost und des Serums 10 Tage nach dem 1. Boost mittels ELISA dar. Verdünnungen von 1:200 bis 1:25600 wurden als Titrationsstufen gewählt. Nach dem 1. Boost ist ein deutlicher Anstieg der spezifischen GAGE-7bAntikörper im Antiserum gegenüber dem Präimmunserum, das auch bei einer 1:200 Verdünnung eine vernachlässigbar niedrige Reaktion besitzt, zu erkennen. OD 450nm: Optische Dichte bei 450nm, K80: Kaninchen 80. 3.3.8 Isolierung der spezifischen GAGE-7b-Antikörper aus dem Antiserum Zur Isolierung der monospezifischen GAGE-7b-Antikörper aus dem Kaninchenantiserum wurden jeweils eine N-hydroxysuccinimid (NHS)-aktivierte Sepharose-Säule mit gebundenem GST-tag-Protein und eine Säule mit gebundenem GAGE-7b-tag-Protein hergestellt. Die Effizienz der Bindung von GST-tag-Protein an die NHS-aktivierte Sepharose-Säule lag bei 97%, so dass insgesamt 3,88mg Protein gebunden werden konnten. Für GAGE-7b-tag lag die Effizienz bei 66%, und 0,7mg GAGE-7b-tag-Protein wurde an die Säule gebunden. Um die spezifischen Antikörper gegen GAGE-7b isolieren zu können, wurden 10mL Serum, das nach der Ausblutung von K80 gewonnen wurde, über die NHS-aktivierte Sepharose-Säule mit gebundenem GST-tag aufgereinigt. Gebundene GST- bzw. tag-Antikörper wurden anschließend eluiert. Das über die 1. Säule (GST-tag) geflossene Serum wurde anschließend über eine NHS- ERGEBNISSE 136 aktivierte Sepharose-Säule mit gebundenem GAGE-7b-tag-Protein gegeben. Das Serum nach dem Durchlauf durch beide Säulen sollte weniger Antikörper enthalten. Die spezifischen Antikörper gegen GAGE-7b konnten von der 2. Säule (GAGE-7b-tag) eluiert werden. Eine durchschnittliche Proteinkonzentration von 0,1mg/mL an monospezifischem GAGE-7bAntikörper sowie ein Titer von 1:2560 konnten erzielt werden. In einem ELISA wurde das Serum vor, während und nach der Säulenaufreinigung, sowie die eluierten Antikörper untersucht (Abbildung 3-32). Für den ELISA wurde identisch aufgereinigtes Protein wie bereits für die Immunisierung verwendet. Wie in Abbildung 3-32 dargestellt, nahm die Antikörperkonzentration im Serum K80 vor der Säulenaufreinigung, nach der 1. Säule (GST-tag) und nach der 2. Säule (GAGE-7b-tag) deutlich ab. Insbesondere die Bindung von Antikörpern an die 2. Säule entfernte Antikörper aus dem Serum K80. Dies wird anhand der Erniedrigung der OD bei 450nm um 0,5 deutlich. 2,0 1,8 1,6 OD 450nm 1,4 1,2 1,0 0,8 Serum K80 (1:2 vorverdünnt) 0,6 0,4 Serum K80 nach NHS-GST-tag-Säule Serum K80 nach NHS-GAGE-7b-tag-Säule 0,2 tag-AK GAGE-7b-AK 0,0 unverdünnt 1:20 1:40 1:80 1:160 1:320 1:640 1:1280 Verdünnung Abbildung 3-32: Titration des Antiserums und der isolierten Antikörper nach den unterschiedlichen Aufreinigungsschritten Mittels ELISA und unter Verwendung des in der Immunisierung eingesetzten aufgereinigten GAGE-7btag-Proteins wurde eine Titration des K80 Antiserums und der isolierten Antikörper nach unterschiedlichen Aufreinigungsschritten durchgeführt. Titrationsstufen von unverdünntem Serum bis zu einer Verdünnung von 1:1280 wurden gewählt. Das Antiserum von K80 vor Aufreinigung 1:2 verdünnt, das Antiserum nach Aufreinigung über eine NHS-Säule (GST-tag-Protein), das Antiserum nach Aufreinigung über eine NHS-Säule (GAGE-7b-tag-Protein), und die eluierten tag-Antikörper sowie die eluierten GAGE-7b-Antikörper wurden untersucht. tag-Antikörper wurden entfernt. GAGE-7bAntikörper wurden aufgereinigt. AK: Antikörper, OD 450nm: Optische Dichte bei 450nm, K80: Kaninchen 80, NHS: N-hydroxysuccinimid, GST: Glutathion-S-Transferase. ERGEBNISSE 137 Die Analyse der eluierten tag- und GAGE-7b-Antikörper wies nach, dass tag-Antikörper aus dem Antiserum entfernt werden konnten. Somit bildete K80 Antikörper gegen das C-terminale tag-Ende des Proteins. Diese sind jedoch gegen ein nicht-humanes Protein gerichtet. Deshalb sollten diese Antikörper in Untersuchungen an humanem Material nicht störend sein. Theoretisch konnten mittels der verwendeten GST-tag-NHS-aktivierte Sepharose-Säule gebildete GST-Antikörper entfernt werden, falls bei der Ionenaustauschchromatographie eine minimale Konzentration an GST-Protein im aufgereinigten Protein enthalten gewesen sein sollte. Die eluierten monospezifischen GAGE-7b-Antikörper reagierten eindeutig mit dem auf der ELISA-Platte gebundenen GAGE-7b-tag-Protein. Ein ähnlicher Kurvenverlauf wurde für die GAGE-7b-Antikörper und das Serum vor der Aufreinigung ermittelt. Das Serum nach dem Durchfluss durch beide Säulen enthielt noch Antikörper gegen das aufgereinigte GAGE-7b-tagProtein. Dies weist auf eine Sättigung der Säulen mit Antikörpern hin. 3.3.9 Spezifität des GAGE-7b-Antikörpers Um die Spezifität des aufgereinigten monospezifischen GAGE-7b-Antikörpers zu überprüfen, wurde der Antikörper sowohl im Western Blot, in der Immunzytologie als auch in der MultiplexSerologie getestet. Western Blot Der aufgereinigte monospezifische GAGE-7b-Antikörper wurde im denaturierenden Western Blot (15% Polyacrylamidgel) gegen verschiedene GST-Fusionsproteine sowie verschiedene Verdünnungen an aufgereinigtem GAGE-7b-tag-Protein getestet (Abbildung 3-33). Der monospezifische Antikörper erkannte bei einer Verdünnung von 1:200 bzw. einer Konzentration von 1µg/mL die GAGE-7b-tag Bande bei ca. 26kDa sowie eine GAGE-7b-Bande bei 22,5kDa (Spur 1). Ansonsten reagierte der Antikörper schwach gegen drei Proteine bei ca. 35, 55 und 75kDa, die zu den E. coli-Proteinen zählen könnten. Diese Banden wurden ebenfalls im GSTGAGE-7b-tag-Lysat (Spur 2) erkannt. In Spur 2 wurde zudem eine eindeutige Reaktion des Antikörpers gegen GST-GAGE-7b-tag-Protein bei ca. 45kDa nachgewiesen. Der Antikörper reagierte sehr schwach gegen MAGE-A9-tag-Protein bei einer Größe von ca. 43kDa (Spur 3). In allen Spuren detektierte der Antikörper eine Bande bei ca. 75kDa, die vermutlich dem E. coliProtein Hsp70 zuzuordnen ist. Dieses Protein wird während der Proteinexpression gebildet, ist jedoch prokaryontenspezifisch. Die ausgewählten GST-Fusionsproteine besitzen ein Molekulargewicht von 70 bis 80kDa. Die Reaktion des Antikörpers ist vermutlich gegen das Cterminale tag-Ende gerichtet. Eine Ausnahme bildet GST-LAGE-1a-tag-Protein, das ein Molekulargewicht von 47kDa besitzt. Der Antikörper erkannte in Spur 9 eine schwache Bande ERGEBNISSE 138 dieses Proteins sowie vermutlich das prokaryontenspezifische E. coli Protein HSP70. Dies deutet ebenfalls auf einen Restgehalt an tag-Antikörpern hin. Die Untersuchung unterschiedlicher Verdünnungen an aufgereinigtem Protein mit dem GAGE-7b-Antikörper bestimmte eine Nachweisgrenze von 15ng GAGE-7b-tag-Protein (Verdünnung 1:100). Ab einer Proteinmenge von 1,5ng (Verdünnung 1:1000) erkannte der Antikörper das aufgereinigte Protein nicht mehr. A M 1 2 3 4 5 6 7 8 9 B 250kDa 250kDa 100kDa 75kDa 100kDa 75kDa 50kDa 50kDa 37kDa 37kDa GST-GAGE-7b-tag 25kDa M 1 2 3 4 25kDa 20kDa 20kDa GAGE-7b-tag GAGE-7b-tag 15kDa 15kDa Abbildung 3-33: Western Blot an verschiedenen GST-Fusionsproteinen unter Verwendung des monospezifischen GAGE-7b-Antikörpers A In diesem Western Blot (15% Polyacrylamidgel) wurden ca. 0,1µg aufgereinigtes, rekombinantes GAGE-7b-tag-Protein sowie 0,1µg andere rekombinante GST-Fusionsproteine aufgetragen. Zur Detektion wurde der GAGE-7b-Antikörper in einer 1:200 Verdünnung (1µg/mL) eingesetzt. Der aufgereinigte monospezifische Antikörper reagierte deutlich mit GAGE-7b-tag und GST-GAGE-7b-tagProtein sowie einigen unspezifischen E. coli Banden. Es bestehen schwache Reaktionen mit den GSTFusionsproteinen vermutlich aufgrund des tag-Anteils. Spur 1: GAGE-7b-tag, 2: GST-GAGE-7b-tag, 3: MAGE-A9-tag, 4: GST-MAGE-A9-tag, 5: GST-MAGE-A1-tag, 6: GST-MAGE-A3-tag, 7: GST-GBP5ta-tag, 8: GST-tag, 9: GST-LAGE-1a-tag. B Der GAGE-7b-Antikörper (1:200, 1µg/mL) erkennt das aufgereinigte GAGE-7b-tag-Protein bis zu einer Verdünnung von 1:100. Dies entspricht einer Proteinmenge von 15ng. Ab einer Verdünnung von 1:1000 wird das aufgereinigte Protein nicht mehr erkannt. Spur 1, 2, 3, 4: Aufgereinigtes GAGE-7b-tag-Protein (1,5mg/mL) 1:10, 1:100, 1:1000 und 1:10000 verdünnt. M: Marker, kDa: kilo Dalton, GST: Glutathion-S-Transferase. Immunzytologie In der Immunzytologie erkannte der GAGE-7b-Antikörper die Zelllinie UKRV-Mel-27, die in Abbildung 3-34 dargestellt ist. Diese positive Reaktion konnte nach der Präabsorption des Antikörpers mit aufgereinigtem Antigen vollständig aufgehoben werden. Das Antigen besaß dieselbe Qualität wie das zur Immunisierung verwendete Antigen. Ein mol Antikörper wurde mit 2 mol Antigen für 30min präabsorbiert und anschließend ÜN bei RT mit den Zellen inkubiert. Somit konnte bestätigt werden, dass der Antikörper GAGE-7b spezifisch erkennt, das GAGE-7btag-Protein bindet und keine anderen unspezifischen Antikörper vorhanden sind, die mit ERGEBNISSE 139 humanen Proteinen reagieren. Entsprechend der Färbung in Abbildung 3-34 A ist GAGE-7b im Cytosol der Zellen lokalisiert. Innerhalb einer Zelllinie sind GAGE-7b-positive und GAGE-7bnegative Zellklone nachweisbar. Eine Erklärung hierfür ist, dass diese MM-Zelllinie aus der Kultivierung von Biopsiematerial einer kutanen Metastase eines MM-Patienten stammt, und hier die Vermehrung mehrerer Zellklone möglich ist. Zur internen Kontrolle wurde die Zelllinie UKRV-Mel-27 mit dem 1:100 verdünnten K80 Präimmunserum gestestet. Diese Verdünnung entspricht der Verdünnung des aufgereinigten Antikörpers. Nach der Gegenfärbung der Zellen wurde keine positive Reaktion des Präimmunserums mit der GAGE-7b-positiven Zelllinie nachgewiesen. Diese Untersuchung bestätigt die Spezifität des GAGE-7b-Antikörpers. A: UKRVMel-27, GAGE-7bAntikörper 2µg/mL, 40x A Abbildung 3-34: Überprüfung der B: UKRVMel-27, GAGE-7bAntikörper 2µg/mL, Präabsorption mit Antigen 1:2, 40x B Spezifität des GAGE-7b-Antikörpers mittels Immunzytologie Mittels Immunzytologie wurden zwei Zytospins der GAGE-7b-positiven Zelllinie UKRV-Mel-27 untersucht. In A wurde eine Färbung mit 2µg/mL GAGE-7b-Antikörper durchgeführt. In B wurde die Färbung unter vorheriger Präabsorption des GAGE-7b-Antikörpers mit GAGE-7b-tag-Antigen in einer 1:2 Verdünnung parallel vorgenommen. Beide Zytospins wurden mit einer 40-fachen Vergrößerung betrachtet und fotografiert. Multiplex-Serologie Der GAGE-7b-Antikörper wurde ebenfalls in der Multiplex-Serologie verwendet, da er auf diese Weise gleichzeitig gegen 17 verschiedene Antigene getestet und die Bindung von GAGE-7b-tagProtein an die entsprechende Beadsorte bestätigt werden konnte. In diesem System wurde der Antikörper eine Stunde mit GST-tag-Protein präabsorbiert und in einer Verdünnung von 1:100 (2µg/mL) eingesetzt. Schließlich wurde der MFI-Wert, der mit dem GAGE-7b-Antikörper gegen das GST-tag-Protein erzielt wurde (7 MFI), von den MFI-Werten von allen anderen Antigenen subtrahiert, um die Netto MFI-Werte zu erhalten. Die somit berechneten MFI-Werte sind in Tabelle 3-8 festgehalten. Der GAGE-7b-Antikörper reagierte ausschließlich mit dem GSTGAGE-7b-tag-Protein. Die MFI-Werte von 22 und 21, die jeweils für das GST-MAGE-A9-tagund GST-LAGE-1a-tag-Protein berechnet wurden, liegen im Rahmen der Hintergrundreaktion. ERGEBNISSE 140 Sie weisen nicht auf eine Reaktion des Antikörpers mit diesen beiden Proteinen hin. Der niedrigste Cut-off-Wert, der in dieser Arbeit verwendet wurde und negative von positiven Seren unterschied, lag bei 59 MFI (siehe Tabelle 3-4). Dementsprechend sind Reaktionen erst ab ca. 50 MFI als positiv zu werten. Nach dem Nachweis der Spezifität des GAGE-7b-Antikörpers in Western Blot, Immunzytologie und Multiplex-Serologie wird deutlich, dass der Antikörper sowohl denaturiertes als auch natives sowie rekombinant hergestelltes Protein erkennt und entsprechend in verschiedenen Testsystemen verwendet werden kann. Im Western Blot wurde der Antikörper in einer Verdünnung von 1:200 (1µg/mL), in der Immunzytologie von 1:50 bis 1:100 (2µg/mL) und in der Multiplex-Serologie in einer Verdünnung von 1:100 (2µg/mL) verwendet. Tabelle 3-8: Verwendung des GAGE-7b-Antikörpers in der Multiplex-Serologie Diese Tabelle führt die MFI-Werte, die die Reaktion des GAGE-7b-Antikörpers mit jedem einzelnen Antigen in der Multiplex-Serologie darstellen, auf. Die MFI-Werte bestätigen, dass der Antikörper ausschließlich das GAGE-7b-Antigen bindet. Reaktionen von ungefähr 20 MFI liegen innerhalb der Hintergrundreaktion und sind somit vernachlässigbar. MFI: „Mean fluorescence intensity“. Antigen MFI-Wert Antigen MFI-Wert Antigen MFI-Wert MAGE-A1 0 se70-2 0 JC 0 MAGE-A3 0 se57-1 0 LPV 0 MAGE-A9 21 GBP-5ta 0 HPV1 0 LAGE-1a 22 cTAGE-5a Nterminal 0 p53 0 GAGE-7b 1597 cTAGE-5a Cterminal 0 p16 0 CAMEL 0 BK 0 3.3.10 Expressionsanalyse des Proteins GAGE-7b Um die Expression des Proteins GAGE-7b zu untersuchen, wurde der wie oben beschrieben selbst generierte GAGE-7b-Antikörper im Western Blot sowie in der Immunzytologie und -histologie eingesetzt. Expression in Kontrollgewebe Um das CTA-Profil von GAGE-7b auf Proteinebene zu bestätigen, wurden 15 verschiedene Kontrollgewebe mit dem generierten GAGE-7b-Antikörper im Western Blot (15% Polyacrylamidgel) untersucht. Der Antikörper wurde in einer Verdünnung von 1:200 (1µg/mL) eingesetzt und detektierte eine spezifische Bande bei 26kDa. In Testis- und in Gehirngewebe wurde weiterhin eine sehr schwache Bande bei ca. 90kDa detektiert. In Testisgewebe könnte ERGEBNISSE 141 diese Bande ein GAGE-Protein-Aggregat darstellen, das nicht denaturiert wurde. Im Gehirngewebe ist die Bande nur sehr schwach erkennbar und somit vernachlässigbar. In Abbildung 3-35 ist ein repräsentativer Western Blot dargestellt. Der Antikörper erkannte GAGE7b ausschließlich in Testisgewebe und keinem anderen Normalgewebe. Dies bestätigt, dass GAGE-7b auch auf Proteinebene ein CTA ist. Der Western Blot unter Verwendung eines βAktin-Antikörpers verdeutlicht, dass von allen Normalgeweben gleiche Proteinmengen aufgetragen wurden. A M 1 2 3 4 5 6 7 8 9 B 250kD 100kD 75kD 250kD 100kD 75kD 50kD 50kD 37kD 37kD 25kD 20kD 15kD 10kD M 1 2 3 4 5 6 7 8 9 25kD 20kD 15kD Abbildung 3-35: Western Blot an Kontrollgewebe mit GAGE-7b-Antikörper Ca. 40µg Kontrollprotein wurden zur Untersuchung im Western Blot (15% Polyacrylamidgel) mit den Antikörpern GAGE-7b (1:200) (A) und β-Aktin (1:5000) (B) aufgetragen. GAGE-7b konnte als Bande bei 26kDa und β-Aktin als Bande bei 45kDa nachgewiesen werden. Die GAGE-7b Expression in Normalgeweben ist auf Testis beschränkt. A und B Spur 1: Plazenta, 2: Brustdrüse, 3: Knochenmark, 4: Testis, 5: Haut, 6: Leber, 7: Thymus, 8: Magen, 9: Gehirn. kDa: kilo Dalton, M: Marker. Expression in Zelllinien und Tumorgewebe Insgesamt wurden 24 Zelllinien auf GAGE-7b-Expression mittels Western Blot (15% Polyacrylamidgel) untersucht. Als internes Kontrollprotein wurde β-Aktin verwendet. Alle Kontroll- (Melanozyten und HaCat) sowie alle CTCL-Zelllinien waren GAGE-7b-negativ (Abbildung 3-36). Hingegen konnte eine GAGE-7b-Expression in zehn von 17 MM-Zelllinien als Bande von 26kDa nachgewiesen werden. Dies bedeutet, dass mehr als 50% aller MMZelllinien GAGE-7b auch auf Proteinebene exprimieren. Zusätzlich wurden neun Tumorgewebe mit dem GAGE-7b-Antikörper im Western Blot (15% Polyacrylamidgel) getestet. Da die Proteinkonzentration der aus den Tumorgeweben erhaltenen Proteinlösungen gering war, konnten nur sehr schwache Banden im Western Blot detektiert werden (Abbildung 3-37, Spur 9 und 11). In zwei MM-Tumorgeweben (22%) konnte GAGE-7b nachgewiesen werden. Obwohl die malignen Gewebe zur Proteinisolierung nicht denjenigen entsprachen, die zur Zelllinienetablierung verwendet wurden, wurde GAGE-7b sowohl in ERGEBNISSE 142 Tumorgewebe- als auch Zelllinienprotein der Zelllinien Ma-Mel-37b und -47 exprimiert. Von weiteren vier Tumorgeweben konnten die entsprechenden Zelllinien innerhalb der durchgeführten Untersuchungen auf GAGE-7b-Expression getestet werden. A M 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 26kDa GAGE 1µg/mL 45kDa β-Actin 1:5000 B M 1 2 3 4 5 6 7 26kDa GAGE 1µg/mL 45kDa β-Actin 1:5000 Abbildung 3-36: Western Blot an verschiedenen Zelllinien mit GAGE-7b-Antikörper Ca. 40µg Zelllinienprotein wurden zur Untersuchung im Western Blot (15% Polyacrylamidgel) mit den Antikörpern GAGE-7b (1:200) und β-Aktin (1:5000) aufgetragen. GAGE-7b konnte als Bande bei 26kDa und β-Aktin als Bande bei 45kDa nachgewiesen werden. 58% der MM-Zelllinien waren GAGE-7bpositiv, wohingegen alle CTCL- sowie Kontroll-Zelllinien GAGE-7b-negativ waren. A Spur 1: UKRVMel-02, 2: UKRV-Mel-06a, 3: UKRV-Mel-14a, 4: UKRV-Mel-21a, 5: UKRV-Mel-27, 6: UKRV-Mel31, 7: SK-Mel-023, 8: Ma-Mel-04, 9: Ma-Mel-05, 10: Ma-Mel-11, 11: Ma-Mal-12, 12: Ma-Mel-16, 13: Ma-Mel-21, 14: Ma-Mel-37b, 15: Ma-Mel-47, 16: Ma-Mel-52, 17:MeWo. B Spur 1: Melanozyten, 2: HaCat, 3: Cou-L3, 4: HH, 5: HuT78, 6: MyLa, 7: SeAx. kDa: kilo Dalton, M: Marker, MM: Malignes Melanom, CTCL: Kutanes T-Zell Lymphom. Ein MM-Patient reagierte weder im Tumorgewebe noch in der entsprechenden Zelllinie (UKRVMel-06a) positiv gegen GAGE-7b. Für zwei weitere MM-Patienten wurde keine GAGE-7bExpression in Western Blot Untersuchungen des Tumorgewebes und der Zelllinie nachgewiesen (Ma-Mel-05 und Ma-Mel-12), jedoch GAGE-7b-Positivität auf mRNA-Ebene in Gewebe und Zelllinie. Ein weiterer auf diese Weise evaluierter MM-Patient war GAGE-7b-positiv auf mRNA- und Proteinebene für die entsprechende Zelllinie (UKRV-Mel-27), jedoch das Tumorgewebe war GAGE-7b-negativ (vgl. Anhang 3). ERGEBNISSE M 1 2 3 4 5 6 7 8 143 9 10 11 26kDa GAGE 1µg/mL 45kDa β-Actin 1:5000 Abbildung 3-37: Western Blot an MM-Tumorgewebe mit GAGE-7b-Antikörper Da die Konzentration an Tumorgewebeprotein gering war, wurden ca. 10µg Protein im Western Blot (15% Polyacrylamidgel) aufgetragen. Die Blots wurden mit den Antikörpern GAGE-7b (1:200) und βAktin (1:5000) untersucht. GAGE-7b konnte als Bande bei 26kDa und β-Aktin als Bande bei 45kDa nachgewiesen werden. 22% der MM-Tumorgewebe waren sehr schwach GAGE-7b-positiv (Spur 9 und 11). Als Vergleich wurde Protein der Zelllinien aufgetragen, die von denselben Patienten stammen wie das schwach positive Tumorgewebe (Spur 8 und 10). Spur 1-7: Tumorgewebe von MM-Patienten 1-7, 8: Protein der Zelllinie Ma-Mel-37b, die von MM-Patient 8 stammt, 9: Tumorgewebe von MM-Patient 8, 10: Protein der Zelllinie Ma-Mel-47, die von MM-Patient 9 stammt, 10: Tumorgewebe von MM-Patient 9. kDa: kilo Dalton, M: Marker, MM: Malignes Melanom. Zelluläre Lokalisation von GAGE-7b Immunzytologie Neben den Untersuchungen der Expression von GAGE-7b im denaturierenden Western Blot wurde die Expression in der Immunzytologie und somit einer nativen Form des Proteins getestet. Insgesamt konnten Zytospins von 22 verschiedenen Zelllinien hergestellt werden, wobei 16 MM-, fünf CTCL-Zelllinien und Melanozyten untersucht wurden. Der GAGE-7b-Antikörper wurde in einer Verdünnung von 1:100 (2µg/mL) eingesetzt. Als Kontroll-Zelllinie dienten Melanozyten, die nicht mit dem Antikörper reagierten. In den untersuchten CTCL-Zelllinien konnte keine GAGE-7b Expression nachgewiesen werden. 43,8% der MM-Zelllinien (7/16) konnten in der Immunzytologie als GAGE-7b-positiv identifiziert werden (Tabelle 3-9, Anhang 3). Bis auf eine Ausnahme (Ma-Mel-12) waren alle Zelllinien, die in der Immunzytologie eine positive Reaktion mit dem GAGE-7b-Antikörper aufwiesen, ebenfalls im Western Blot GAGE7b-positiv. Wie bereits in 3.3.9 beschrieben ist GAGE-7b zytoplasmatisch lokalisiert. In der Mehrheit der untersuchten Zelllinien wurden sowohl GAGE-7b-positive als auch GAGE-7bnegative Zellklone nachgewiesen, so dass zumeist von heterogenen Zellkulturen ausgegangen werden kann. Abschließend wird angenommen, dass der generierte GAGE-7b-Antikörper sowohl GAGE-3, 4, 5, 6, 7 und 7b erkennt, da diese Proteine sich wie unter 1.5.1 und 3.3.1 aufgeführt nur durch einzelne Nukleotidsubstitutionen unterscheiden. ERGEBNISSE 144 Immunhistologie Um eine Aussage über die GAGE-7b Expression in Gewebeschnitten treffen zu können, wurde der GAGE-7b-Antikörper mit einer Konzentration von 0,4µg/mL in der Immunhistologie verwendet. Bei der Untersuchung von Paraffinschnitten von 40 MM-Patienten konnten in 21 von 40 Patienten (53%) GAGE-7b-positive Zellen detektiert werden (Tabelle 3-9). GAGE-7bpositive Zellen wurden jedoch hauptsächlich als einzelne Zellen oder Zellhaufen nachgewiesen. Normale Haut war GAGE-7b-negativ, wohingegen Spermatogonien in gefärbten Schnitten von normaler Testis vom GAGE-7b-Antikörper gefärbt wurden. Dies bestätigt zusätzlich den CTA Charakter von GAGE-7b auf Proteinebene. Tabelle 3-9: Zusammenfassung der Ergebnisse der Expressionsanalyse von GAGE-7b In dieser Tabelle sind zusammenfassend alle Ergebnisse, die mit dem GAGE-7b-Antikörper erhalten werden konnten dargestellt. Zudem ist die Verwendung des Antikörpers sowohl im Western Blot als auch in der Immunzytologie und -histologie aufgeführt. MM: Malignes Melanom, CTCL: kutanes T-Zell Lymphom, n: Anzahl, Ctrl: Kontrolle, neg.: negativ, pos.: positiv. Western Blot Zelllinie GAGE -7b Ctrl (n=2) MM (n=17) Anwen -dung Positiv Immunzytologie Immunhistologie Zelllinie Gewebe Gewebe CTCL (n=5) Ctrl (n=15) inkl. Testis MM (n=9) Ctrl (n=1) 1:200 (1µg/mL) MM (n=16) CTCL (n=5) 1:100 (2µg/mL) Ctrl Pos. Neg. Testis Haut MM (n=40) 1:500 (0,4µg/mL) 0 10 0 1 2 0 7 0 (0%) (58,8%) (0%) (4,4%, Testis positiv) (22,2%) (0%) (43,8%) (0%) Pos. Neg. 21 (53%) In Abbildung 3-38 sind sowohl immunzytologische als auch -histologische Untersuchungen unter Verwendung des GAGE-7b-Antikörpers zusammengefasst. ERGEBNISSE A 145 B C E F H I K L Haut Testis B C D Präimmunserum G Präabsorbierter GAGE-7b-Antikörper H J Abbildung 3-38: Immunzytologische und –histologische Untersuchungen mit GAGE-7bAntikörper Immunzytologische und -histologische Färbungen mit dem GAGE-7b-Antikörper an Zytospins sind in AF und an Paraffinschnitten in G-L dargestellt. MM-Zellen der Zelllinie UKRV-Mel-27 wurden durch den GAGE-7b-Antikörper in A, B, C und E gefärbt. Hingegen färbte das Präimmunserum von K80 keine MM-Zellen (D). Die Spezifität des GAGE-7b-Antikörpers konnte durch die Präabsorption des Antikörpers mit rekombinantem GAGE-7b-tag-Protein (2 mol GAGE-7-tag + 1 mol GAGE-7bAntikörper) in F nachgewiesen werden. Färbungen in MM-Gewebe sind in einzelnen Zellen (G, H) und in Zellhaufen (I, J) erkennbar. Normale Haut ist GAGE-7b-negativ (K), wohingegen normale Testis GAGE7b-positiv ist. Maßstab: 50µm. K80: Kaninchen 80. ERGEBNISSE 146 3.3.11 Serumantikörperantworten gegen GAGE-7b Neben der Bestimmung der Serumantikörperantwort gegen GAGE-7b unter Verwendung der SADA-Methode und Multiplex-Serologie wurde eine dritte Methodik herangezogen. Im Anschluss an eine “sodium dodecyl sulphate polyacrylamide gel electrophoresis” (SDS-PAGE, 15%) mit aufgereinigtem GAGE-7b-tag-Protein und dem Blotten der Proteine auf eine Nitrozellulose-Membran, wurden anstelle eines polyklonalen Antikörpers Serumantikörper mit der Membran inkubiert. Um einen hohen Durchsatz an Seren zu erzielen, wurden die Membranen in kleine Streifen zerteilt und mit dem entsprechenden Serum (1:50) inkubiert. Das Binden von Serumantikörpern an das aufgereinigte Protein wurde mittels Farbreaktion (braune Bande) mit Diaminobenzidin (DAB) nachgewiesen. Die somit erhaltenen positiv reagierenden Seren wurden zur endgültigen Bestätigung der Antigen-Antikörper-Reaktion nochmals in einem Western Blot und Chemilumineszenz-Detektion gestestet (Abbildung 3-39). Auf diese Weise konnten für GAGE-7b 36 Kontroll-, 72 MM- und 34 CTCL-Seren getestet werden. Im KontrollKollektiv konnte keine positive Reaktion gegen GAGE-7b nachgewiesen werden. Vier MM(5,6%, positive Reaktion in Abbildung 3-39 Spur 10) und zwei CTCL-Patienten (5,9%) besaßen Antikörper gegen dieses Antigen. Zahlreiche Seren detektierten eine Bande bei 12,5kDa (Abbildung 3-39 Spur 3). Vermutlich handelt es sich um ein E. coli-Protein, das nicht abgetrennt werden konnte. Antikörper gegen E. coli-Proteine sind häufig im Serum von Menschen vorhanden infolge der ständigen Abwehr von Bakterien durch das körpereigene Immunsystem. A M 1 2 3 4 5 B M 1 2 3 4 5 6 100kDa 75kDa 50kDa 37kDa 25kDa 20kDa 15kDa 25kDa 20kDa 15kDa 10kDa 10kDa 7 8 9 10 11 12 M 100kDa 75kDa 50kDa 37kDa 25kDa 20kDa 15kDa 10kDa Abbildung 3-39: Serumantikörperantworten gegen GAGE-7b ermittelt mit Western Blot und DAB (A) sowie Chemilumineszenz (B) Der Nachweis von GAGE-7b-Antikörpern im Serum erfolgte mittels Western Blot und den Detektionssystemen Diaminobenzidin (A) und Chemilumineszenz (B). Es wurde 1µg aufgereinigtes GAGE-7b-tag-Protein pro Spur aufgetragen. In A fallen positive Reaktionen von Antikörpern mit geblotteten Proteinen durch eine gelb bis braune Bande bei 26kDa (Spur 3, siehe Pfeil, Antikörperreaktion gegen unspezifisches geblottetes Protein) und in B durch eine schwarze Bande (Spur 10, siehe Pfeil, Antikörperreaktion gegen GAGE-7b-Protein) auf. In diesen repräsentativen Blots wurden Kontrollseren (A) und MM-Seren (B) untersucht. A Spur 1-5: Kontrollen 1-5. B Spur 1-12: Malignes Melanom 1-12. M: Marker, kDa: kilo Dalton. DISKUSSION 147 4 DISKUSSION In den letzten Jahren steigt die Zahl an Krebserkrankungen weltweit an. Dennoch gibt es für viele Krebsarten keine umfassende und adäquate Therapie, so dass neben der Inzidenz auch die Mortalität einen ansteigenden Trend aufweist. Neben operativen Maßnahmen und Chemotherapie stehen den Medizinern heute verschiedene Immuntherapieansätze zur Verfügung, um betroffene Patienten zu behandeln. Eine Vielzahl dieser Immuntherapien benötigt Zielstrukturen, die tumorspezifisch oder zumindest Tumor-assoziiert sind sowie eine spezifische Immunantwort in Tumorpatienten auslösen können. Zudem können tumorspezifische Strukturen in der Diagnose, Prognose, dem Monitoring von Krebserkrankungen und einer Therapieentscheidung Verwendung finden. Deshalb wurde in den letzten beiden Jahrzehnten intensiv an der Identifizierung und möglichen Anwendung von Tumorantigenen geforscht. Ziel dieser Arbeit war es, bereits identifizierte Cancer Testis Antigene (CTA) und Tumorassoziierte Antigene (TAA) hinsichtlich ihrer Seroreaktivität in Kontrollen (Ctrl) und Patienten zweier Hauttumorentitäten zu untersuchen. Dies erfolgte mittels zweier Untersuchungsansätze, dem Serumantikörper-Detektionsarray (SADA) und der Multiplex-Serologie. Des Weiteren wurde das CTA GAGE-7b auf Desoxyribonukleinsäure (DNA)- und Proteinebene hinsichtlich seiner Expression in beiden dermalen Tumorentitäten charakterisiert. Im Folgenden soll zunächst die Verwendung von Tumorantigenen in Prognose, Diagnose und Immuntherapien im Allgemeinen dargestellt werden. Anschließend werden die in dieser Arbeit erzielten Ergebnisse der humoralen Immunantwort gegen und Expression von bestimmten Tumorantigenen diskutiert. 4.1 Verwendung von Tumorantigenen Tumorantigene können sowohl für Prognose-, Monitoring- und Diagnoseverfahren als auch in Immuntherapien eingesetzt werden. Um Antigene in der Krebsdiagnostik zu verwenden, müssen sie nicht tumorspezifisch exprimiert, hingegen jedoch spezifisch immunogen sein. Immuntherapien mit solchen Zielstrukturen (Vakzinierungen) bei Krebspatienten setzen immunologische Spezifität voraus, um Autoimmuneffekten vorzubeugen. Sind membranständige Tumorantigene das Ziel einer Krebstherapie, können monoklonale Antikörper gegen diese Tumorantigene entwickelt und eingesetzt werden. DISKUSSION 148 4.1.1 Prognose und Diagnose Antigene, die serologisch erkannt werden, können als Tumormarker dienen, sofern Antikörper nur in Patienten, nicht aber in gesunden Kontrollpersonen nachgewiesen werden. Zumeist steht die Höhe der serologischen Reaktivität in direktem Zusammenhang mit der Eignung des Antigens als Marker. Im Gegensatz zur Immuntherapie müssen die Antigene für die diagnostische Tumorserologie, zur Beurteilung der Erkrankung oder zur Früherkennung von Rezidiven nicht tumorspezifisch exprimiert sein (Old und Chen 1998; Sahin et al. 1999). Die Verwendung von Tumorantigenen als spezifische Marker soll eine frühere Diagnose und eine bessere Unterscheidung von Patienten mit aggressiven Formen von anderen Erscheinungsbildern ermöglichen. Insbesondere die Antigene Her2/neu und p53 wurden hinsichtlich ihrer Antikörperantworten in verschiedenen Krebserkrankungen untersucht. Hohe Her2/neu Antikörper-Titer wurden häufiger in neu diagnostizierten Brustkrebspatienten identifiziert im Vergleich zu einer Kontrollgruppe (Disis et al. 1997). Ein signifikant höheres Auftreten von Antikörpern gegen p53 wurde in Krebspatienten im Vergleich zu Kontrollen berichtet (Shimada et al. 2003). 4.1.2 Immuntherapie Herkömmliche Krebstherapien basieren zumeist weder auf einer gezielten noch auf einer gesicherten Vernichtung aller Krebszellen in einem Patienten und sind daher oft nur palliativer Natur. Deshalb zielen neuere Therapieansätze auf eine Stärkung der Tumorimmunantwort ab, da es Hinweise darauf gibt, dass viele und sogar womöglich alle Krebserkrankungen immunogen sind (Sahin et al. 1995). Das körpereigene Immunsystem besitzt das Potential, neoplastische Zellen erkennen und anschließend lysieren zu können, ebenso wie auch fremde oder virusinfizierte Zellen entdeckt und eliminiert werden können. Dies ist bereits bei verschiedenen Tumoren nachgewiesen worden (Hammad et al. 2003; Kageshita et al. 2003; Mangel et al. 2003). Da neben der T-Zellantwort auch humorale Immunantworten gegen Tumorzellen beschrieben sind (Sahin et al. 1995), kann sowohl die zelluläre als auch die humorale Immunantwort durch gezielte Aktivierung des Immunsystems induziert werden. Vorteile einer Immuntherapie liegen eindeutig in einem eingeschränkteren Nebenwirkungsspektrum und einer geringeren systemischen Toxizität im Vergleich zu Chemo- oder Radiotherapie (Nencioni et al. 2004). Immuntherapien in Form von Vakzinierungen müssen die Unterdrückung einer Immunantwort durch den Tumor überwinden und effektiv sowie langfristig vorbeugen, ohne eine Autoimmunität im Patienten hervorzurufen (Finn 2003). DISKUSSION 149 4.1.2.1 Nicht-spezifische Immuntherapie Tumore durch eine unspezifische Immunstimulation des Patienten-eigenen Immunsystems zu bekämpfen, ist ein in den letzten Jahrzehnten intensiv untersuchter Ansatz. Hier werden spezifische Zytokine als Adjuvantien und als Therapieform in der Behandlung von Krebserkrankungen eingesetzt. Zytokine sind Proteine, die von einer Vielzahl von Zellen gebildet und sezerniert werden sowie durch Rezeptorbindung als interzelluläre Mediatoren zur Aktivierung von anderen Zellen beitragen (Pschyrembel 1998). Das Zytokinprofil in der Umgebung eines Tumors entscheidet über die Art der Immunabwehr, mit der der Organismus gegen die neoplastischen Zellen vorgeht (Chouaib et al. 1997). In verschiedenen in vitro und in vivo Untersuchungen konnten bislang sowohl antiproliferative als auch immunsupprimierende Effekte von Zytokinen gemessen werden. Insbesondere die Wirkungen von Interferon (IFN)-α und -γ, Interleukin (IL)-2 und Tumor Nekrosis Faktor (TNF)-α finden in der Behandlung von verschiedenen Krebserkrankungen Anwendung. Für IFN-α und IL-2 konnten klinische Ansprechraten von 13% bis 50% in malignen Melanom (MM)- und kutanen T-Zell Lymphom (CTCL)-Patienten detektiert werden (Asadullah et al. 2002). Verschiedene Therapiestrategien (z.B. Hoch-Dosis-Therapie, Chemotherapie plus IL-2) wurden bereits verwendet. Hierbei führten die Hoch-Dosis-IL-2-Therapien jedoch zu einer sehr hohen Toxizität (Kragel et al. 1990). Schließlich erfolgt die Anwendung von Zytokinen ebenfalls indirekt über die Gabe von genmodifizierten Tumorzellen, die in der Lage sind, diese Zytokine zu sezernieren. Ein klinischer Erfolg kann häufig durch die Kombination einer Therapie mit diesen Immunmodulatoren verstärkt werden (Salgaller und Lodge 1998). 4.1.2.2 Spezifische Immuntherapie Die Expression von spezifischen Antigenen auf Krebszellen stellt die Grundlage von spezifischen Immuntherapien dar. In diesem Zusammenhang ist die Identifizierung von neuen TAA von großer Bedeutung, die ebenfalls eine stetig zunehmende Anwendung in Diagnostik und Prognostik finden. Die geeignetsten Tumorantigene sind Patienten-spezifisch (z.B. Mutationsantigene), Tumor-spezifisch (z.B. bcr/abl) oder aber Gewebe-spezifisch (z.B. CTA) und wurden zuvor nicht vom Immunsystem erkannt. Bei der Vakzinierung von Mäusen mit Tumorantigenen und gleichzeitig nicht-mutierten Selbstantigenen konnte eine höhere anti-Tumor Aktivität erzielt werden als mit Tumorantigenen allein (Nishikawa et al. 2001). Spezifische Immuntherapien für Krebspatienten dienen dazu, die Immunabwehr des Patienten auf spezifische Zielstrukturen z.B. in Form von Tumorantigenen zu richten. Diese Vakzinierungen müssen die immunologische Toleranz überwinden und T-Zellen aktivieren, die eine niedrige Affinität zu DISKUSSION 150 Antigenen besitzen, die auf Tumorzellen exprimiert werden (Pardoll 1998). Grundsätzlich können zwei Arten von Immuntherapien unterschieden werden: Antikörper- und Vakzintherapie. Antikörpertherapie Die Lyse von Tumorzellen, die von monoklonalen Antikörpern spezifisch erkannt werden, kann sowohl durch direkte als auch indirekte Mechanismen erfolgen. Zu den direkten Effekten zählen die Aktivierung der Zell-eigenen Apoptose (Trauth et al. 1989), die Blockierung von Wachstumsfaktor-Rezeptoren (Baselga und Mendelsohn 1994) und eine Anti-Idiotyp-Antwort (Bhattacharya-Chatterjee et al. 2000). Die indirekten Mechanismen umfassen die Antikörperabhängige zelluläre Zytotoxizität (ADCC) (Steplewski et al. 1983) und die lokale Aktivierung der Komplementkaskade (Ballare et al. 1995). Für Krebserkrankungen sind bisher drei monoklonale Antikörper zugelassen. Rituximab (Handelsname: MabThera) ist gegen das B-Zell Oberflächenantigen „Cluster of Differentiation“ (CD) 20 gerichtet und wird bei Non-Hodgkin Lymphomen (NHL) angewendet. In ersten klinischen Studien wurden mit Rituximab vielversprechende Ergebnisse hinsichtlich der Ansprechrate erzielt (McLaughlin et al. 1998). Trastuzumab (Handelsname: Herceptin) findet Anwendung bei Mammakarzinomen und richtet sich gegen das überexprimierte Her2/neu Membranprotein. Die Kombination von Herceptin und Chemotherapie verlängerte in klinischen Studien signifikant das progressionsfreie Intervall und erzielte eine höhere Überlebensrate im Vergleich zu herkömmlichen Chemotherapieansätzen (Slamon et al. 2001). Gemtuzumab (Handelsname: Mylotarg) wird bei rezidivierender akuter myeloischer Leukämie angewendet und erkennt CD33 (Glennie und van de Winkel 2003). Zusätzlich wurde in einigen Therapieansätzen die Antikörperantwort verstärkt, in dem die monoklonalen Antikörper an Toxine (z.B. radioaktive Isotope oder bakterielle Toxine) gekoppelt wurden oder spezifische Sekundärantikörper mit gekoppelten Toxinen produziert wurden (Clynes et al. 2000; Flieger et al. 2000). Schließlich kommen ebenfalls bispezifische Antikörper in klinischen Studien zum Einsatz, die sowohl gegen ein Tumorantigen auf der Krebszelle als auch einen Rezeptor auf einer Effektorzelle (z.B. CD20 auf B-Zellen oder CD3 auf T-Zellen) gerichtet sind (De Gast et al. 1995). Vakzinierungsstrategien Im Gegensatz zu den Antikörpertherapien hat eine Vakzinierungsstrategie generell die aktive Beteiligung und Unterstützung des Immunsystems zum Ziel, damit eine gegen die Tumorzellen DISKUSSION gerichtete Immunantwort hervorgerufen werden 151 kann. Verschiedene Ansätze dieser Impfstrategien können unterschieden werden. In klinischen Studien wurden Peptid-Vakzinierungen vor allem an MM-Patienten unter Verwendung z.B. des MAGE-3.A1 Peptids getestet. Bei sieben Patienten wurden signifikante Tumorregressionen und bei zwei dieser Patienten komplette Regressionen nachgewiesen (Marchand et al. 1999). Gentherapien in Form von Vakzinierungen mit „copy“ DNA (cDNA), die für ein TAA kodiert, unter Verwendung von Plasmid- oder viraler Vektoren, stellen eine weitere Möglichkeit der spezifischen Tumorbekämpfung dar (Naitoh und Belldegrun 1998). Neben der Peptid- und Gen-Vakzinierung werden Immunzellen selbst zur Immunbehandlung herangezogen. Antigen-präsentierende Zellen (APC), zumeist in Form von dendritischen Zellen (DC), werden mit spezifischen Antigenen oder Tumorlysaten von außen beladen. Bei fünf von 16 MM-Patienten konnte eine Regression von Metastasen in mehreren Organen nach einer Vakzinierung mit DC, die mit Peptiden oder Tumorlysat beladen wurden, gemessen werden (Nestle et al. 1998). Da die Beladung der DC mit Peptiden humanem Leukozyten Antigen (HLA) abhängig erfolgt, können nur spezifische und zuvor identifizierte Epitope verwendet werden. Andere Ansätze versuchen APC mittels rekombinanter Viren oder Bakterien zu infizieren zur Aktivierung von Lymphozyten (Xiang et al. 1996; Paschen et al. 2000). Die Fusion von autologen Tumorzellen und APCs soll zur Präsentation von autologen Antigenen an der Oberfläche der APC eingesetzt werden (Stuhler und Walden 1994). Erste Studien mit diesem Therapieansatz ergaben eine gute Resonanz bei MM-Patienten (Trefzer et al. 2000). Vakzinierungen mit autologen Gedächtniszellen stehen ebenfalls in der Diskussion (Feuerer et al. 2001). Um prämaligne Erkrankungen oder Krebs im Frühstadium identifizieren und Antworten auf Immuntherapien monitorieren zu können, werden quantitative Verfahren zur Bestimmung von Tumor-spezifischen Antikörper-Immunreaktionen immer häufiger als klinische Instrumente verwendet. Zwei mögliche methodische Ansätze, die in dieser Arbeit verwendet wurden, werden im Folgenden diskutiert. 4.2 SADA Der SADA wurde in dieser Arbeit als eine von zwei methodischen Ansätzen zur Bestimmung von Immunreaktionen im Serum verwendet. Antikörperantworten wurden in Kontrollen und einem CTCL- und MM-Kollektiv bestimmt. Alle untersuchten Antigene hatten entweder laut DISKUSSION 152 Literatur oder im „serological analysis of recombinantly expressed cDNA clones“ (SEREX) positive Ergebnisse und eine humorale Immunantwort zumeist ausschließlich in Tumorpatienten erbracht. Die etablierte Methode basiert auf der SEREX-Methode und Arbeiten von M. Scanlan (Scanlan et al. 2001; Scanlan et al. 2002). Um eine Eignung der untersuchten Tumorantigene anhand der gemessenen Seroreaktivität in Diagnostik und Prognostik an sich oder aber als Kombination von mehreren Tumorantigenen bewerten zu können, wurde die Häufigkeit der serologischen Reaktivität herangezogen. Insgesamt wurden 42 Antigene an 218 Seren getestet. 4.2.1 Validierung des SADA Bei der Etablierung einer neuen Methode ist eine Validierung der Methode in gewissem Umfang angebracht. Für den SADA wurden die Parameter Phagenmobilität, Monoklonalität, Sensitivität und Reproduzierbarkeit evaluiert. Da Phagen eine hohe Mobilität besitzen und durch Lösungen leicht verschleppt werden können, wurde getestet, ob beim Stempeln der Phagenlösungen eine Kontamination eines Phagenstammes mit einem anderen möglich sei. Hierzu wurde ein Immunglobulin G (IgG)-Klon verwendet, der eine starke Reaktion mit dem sekundären Antikörper im SADA-System besitzt. Es konnte eindeutig belegt werden, dass ein „Springen“ oder Übertreten der Phagen von einem zum anderen Loch der 96 Lochplatte und somit eine Vermischung der Phagenstämme beim mehrfachen Stempeln nicht auftritt. Dies ist entscheidend für die Monoklonalität der Phagenklone. Die Monoklonalität der Phagenlösungen konnte auch nach dem Picken von mehreren Phagenplaques unter Verwendung von in vivo Exzision und reverse Transkriptase Polymerasekettenreaktion (RT-PCR) exemplarisch für vier verwendete Phagenklone bewiesen werden. Somit wurden ausschließlich monoklonale Phagenlösungen mit eindeutig definierten Phagenklonen verwendet. Die Sensitivität der SADA-Methode wurde bestimmt, um die in Anlehnung an die SEREXMethode gewählte Verdünnung von 1:100 zu bestätigen. Die Untersuchung eines Serums eines CTCL-Patienten in fünf Verdünnungen von 1:50 bis 1:1000 belegte deutlich, dass eine 1:100 Verdünnung in einem adäquaten Verdünnungsbereich liegt. Bei den Verdünnungen 1:50, 1:100 und 1:200 wurden dieselben Antigene von Antikörpern aus dem Serum erkannt. Ab einer Verdünnung von 1:400 wurden weniger Antigene erkannt und bei einer Verdünnung von 1:1000 wurde kein Antigen mehr erkannt. Somit stellt eine 1:1000 Verdünnung eine eindeutig zu hohe Verdünnung dar, um Serumantikörper mittels SADA detektieren zu können. Da die 1:100 Verdünnung in einem adäquaten Verdünnungsbereich liegt, kann im Gegensatz zu einer 1:50 Verdünnung Serum gespart werden. Ein direkter Vergleich mit der SEREX-Methode kann DISKUSSION 153 aufgrund der identischen Serumverdünnung erfolgen. Die Sensitivität liegt laut diesem Test bei einer Verdünnung von 1:200. Die Reproduzierbarkeit der SADA-Methode wurde durch die unabhängige Präabsorption von jeweils zwei identischen Serumproben von fünf MM-Patienten belegt. Die Seren wurden an verschiedenen Tagen nach demselben Schema präabsorbiert. Beim Test der Seren mittels SADA wurden identische Antikörperreaktionen nach erfolgreicher Präabsorption nachgewiesen. Dies bedeutet, dass die Präabsorption der Seren keinen Einfluss auf das Ergebnis hat und die Methode reproduzierbar ist. 4.2.2 Untersuchung von humoralen Immunantworten mittels SADA In dieser Arbeit wurden erstmalig Seren von CTCL- und MM-Patienten im Vergleich zu gesunden Ctrl mittels SADA auf ihre humorale Immunantwort gegen 42 SEREX-definierte Antigene getestet. Hierzu wurden die titrierten Phagenlösungen von 42 Antigenen sowie einem Negativ- und einem Positivphagenklon auf einen E. coli Bakterienrasen aufgespottet und inkubiert. Die gebildeten Phagenplaques, die das rekombinant exprimierte Protein des jeweiligen Phagenklons darstellten, wurden auf Nitrozellulosemembranen geblottet, die Membranen blockiert, mit Serum und einem Zweitantikörper inkubiert und schließlich mit Detektionsreagenz gefärbt. Zwei Membranen mit in Duplikaten geblotteten Proteinspots von jeweils 20 bzw. 22 Antigenklonen und einem Negativ- sowie einem Positivklon wurden hergestellt. Die Serumantikörper-Häufigkeiten in den drei untersuchten Kollektiven lagen zwischen 0% und 72%. Gegen die Antigene cTAGE-1, cTAGE-5a, se89-1, HD-CL-50, 51 und 54 wurden in keinem der drei Kollektive Antikörperantworten nachgewiesen. In sekundären SEREXUntersuchungen hingegen reagierten alle sechs Antigene mit Antikörperantworten über 10%. Dieser deutliche Unterschied in der Antikörperreaktion könnte auf die Anwesenheit von Negativ- und Positivkontrolle zusammen mit dem Antigen auf einer Membran zurückzuführen sein. Zudem wurden andere und eine bedeutend höhere Anzahl an zufällig ausgewählten Seren verwendet. Ein größeres Kollektiv spiegelt möglicherweise die Verteilung von reaktiven und nicht-reaktiven Ctrl und Patienten besser wider als kleine Kollektive wie sie im SEREX verwendet wurden. Der Klon se20-7 kann als Autoantigen bezeichnet werden, da er im CTCLKollektiv zu 67%, im MM-Kollektiv zu 72% und in den Ctrl zu 65% erkannt wurde. Dieser Klon könnte in zukünftigen SADA-Projekten als Positivkontrolle dienen, da Serumantikörperantworten in über 50% aller Seren zu erwarten sind. DISKUSSION 154 Auffallend ist ebenfalls, dass die Serumantikörper-Häufigkeiten gegen die CTA MAGE-A3, A9 und GAGE-t sehr gering waren. Im MM-Kollektiv wurden keine Antikörper gegen die MAGEAntigene detektiert, obwohl MAGE-Antigen Expression in MM-Patienten beschrieben wurde (Brasseur et al. 1995; Dalerba et al. 1998; Roeder et al. 2005). Ein Grund hierfür könnte sein, dass diese Antigene eine eher schwache humorale Immunantwort auslösen und die Antikörpertiter, falls Antikörper vorhanden sind, zu niedrig für eine Detektion im SADA sind. Bereits in der Literatur beschriebene Antikörperuntersuchungen kamen zu ähnlich niedrigen Ergebnissen unter Verwendung der „Enzyme linked immunosorbent assay“ (ELISA)-Methode (Stockert et al. 1998). Vierzehn Antigene (Guanylat-bindendes Protein (GBP)-5a, GBP-5ta, HD-CL-12, pMAGEA9ag, du20-10L4, HD-CL-03, HD-MM-02, HD-MM-03, HD-CL-02, HD-CL-17, HD-CL-24, se33-1, se37-1, HD-CL-41) wiesen ausschließlich Antikörperantworten in den Patientenkollektiven auf, so dass sie als serologisch tumorspezifisch bezeichnet werden können. Jedoch waren die bestimmten Serumantikörperantworten gegen diese Antigene in den Patientenkollektiven zumeist eher gering mit 1% bis 28%. Zwischen den SADA- und zuvor durchgeführten sekundären SEREX-Ergebnissen ist ein gewisser Unterschied nachweisbar. Die mittels SEREX untersuchten Ctrl besaßen keine Serumantikörper gegen 29 im SADA verwendete Antigene mit Ausnahme von HD-CL-17. Im SADA hingegen wurden keine Antikörper gegen 21 Antigene in Kontrollseren detektiert. Elf der im SADA an Kontrollseren getesteten Antigene wurden bisher nicht im sekundären SEREX untersucht. Im SADA wurden für alle anderen Antigene Antikörper-Häufigkeiten in den Kontrollen von 1% bis 39% gemessen. Viele der SADA-Antigene, die im SADA und im sekundären SEREX mit CTCL-Seren getestet wurden, wiesen höhere Antikörper-Häufigkeiten im sekundären SEREX im Vergleich zum SADA auf. In sekundären SEREX-Untersuchungen unter Verwendung von MM-Seren wurden nur HD-MM-Antigene sowie die cTAGE-Antigene getestet. Auch für diese Antigene sind leicht höhere Antikörper-Häufigkeiten bei Untersuchungen im sekundären SEREX vorhanden. Diese Unterschiede könnten wiederum auf die höhere Anzahl an Seren und die Anwesenheit von Positiv- und Negativkontrolle auf der SADA-Membran zurückzuführen sein. Schließlich wurde nachgewiesen, dass beim Vergleich der Serumantikörper-Häufigkeiten größer gleich 5% mehr Antigene im MM-Kollektiv (15) im Vergleich zum CTCL-Kollektiv (6) reaktiv waren. Dies deutet auf eine höhere Immunogenität des MM im Vergleich zum CTCL hin. Die hohe Immunogenität des MM wurde bereits in der Literatur beschrieben (Kawakami et al. 2000; Anichini et al. 2004). DISKUSSION 155 Serumantikörperantworten in CTCL-Patienten 30 von 42 Antigenen besaßen Serumantikörper-Häufigkeiten von weniger gleich 5% im Kontroll- und CTCL-Kollektiv. 22 dieser 30 Antigene hatten keine Immunreaktion in der Kontroll-Gruppe hervorgerufen. Die zwölf anderen Antigene erzeugten Antikörperantworten von 6% bis 67% in den CTCL-Patienten und 12% bis 39% in den Ctrl. Für zwei von 42 Antigenen wurde ein statistisch signifikanter Unterschied zwischen den Serumantikörper-Häufigkeiten von CTCL-Patienten und der Kontroll-Gruppe gemessen. HDMM-48 ist mit 3% Antikörper-Häufigkeit zwar nicht serologisch tumorspezifisch. Jedoch ist die Reaktivität in den CTCL-Patienten mit 20% deutlich unterschiedlich von den Ctrl. Das heißt jeder fünfte CTCL-Patient bildete Antikörper gegen dieses Antigen, so dass es in ein Panel von Antigenen zur Diagnose und Monitoring aufgenommen werden könnte. Die Anzahl an positiven Kontrollseren ist mit 3% eher vernachlässigbar. Ein weiteres Antigen, das einen statistisch signifikanten Unterschied in seiner Reaktivität zwischen CTCL-Patienten und Ctrl besitzt ist HC-CL-02. Dieses Antigen ist serologisch tumorspezifisch und erzeugte Antikörper in 6% der CTCL-Patienten. HD-CL-02 ist sicherlich aufgrund seiner serologischen Tumorspezifität interessant und sollte in weiteren Screening-Studien zur Zusammenstellung eines Panels an Antigenen enthalten sein. Aufgrund der statistischen Berechnungen werden im Folgenden nur die soeben genannten Antigene genauer beschrieben. HD-MM-48 Dieser in CTCL- und MM-Patienten stark reaktive Phagenklon ist homolog zum Rezeptorinteragierenden Faktor 1 Variante 2 (RIF1). Dieser Phagenklon wurde zuvor nicht in sekundären SEREX-Untersuchungen analysiert. Das homologe nukleäre Protein RIF1 spielt eine wichtige Rolle bei der Hemmung der Zellzyklusprogression in der S-Phase nach DNA Schädigung (Silverman et al. 2004). In der Telophase lokalisiert RIF1 an kondensierte Chromosomen, wohingegen es in der Anaphase an Mikrotubuli der mitotischen Spindel nachgewiesen wird. Expression von RIF1 wurde in Sertoli-Zellen, frühen primären Spermatozyten und Oozyten sowie embryonalen Stammzellen detektiert. Jedoch nimmt die RIF1 Expression bis hin zu ihrem Verlust während der Zelldifferenzierung ab. Eine Reduzierung der RIF1 Expression unter Verwendung von „small interfering“ Ribonukleinsäure (siRNA) führte zu einer höheren Radiosensitivität. DISKUSSION 156 HD-CL-02 HD-CL-02 ist homolog zur Homo sapiens mRNA für Inositol-1,4,5-Trisphosphat 5-Phosphatase (Hartmann et al. 2004). Dieses Membran-assoziierte Enzym katalysiert die Dephosphorylierung des Botenstoffes Inositol-1,4,5-Triphosphat, welches zur Inaktivierung des Botenstoffes führt (Laxminarayan et al. 1994). Der Botenstoff Inositol-1,4,5-Triphosphat reguliert die Mobilisierung von intrazellulärem Calcium. HD-CL-02 stellt einen verkürzten Klon mit einer veränderten C-terminalen Proteinregion dar, der auf mRNA-Ebene in 80% der CTCL-Gewebe exprimiert ist. In sekundären SEREX-Analysen erzeugte HD-CL-02 mit 19% fast in einem Fünftel aller CTCL-Patienten Antikörper, wohingegen keine Antikörperreaktion in Ctrl detektiert werden konnten. Somit war dieser Klon sowohl im SEREX wie auch im SADA tumorspezifisch. Dies ist eine gute Voraussetzung für weitere serologische Untersuchungen an verschiedenen Patientenkollektiven. Immunogenität dieses ubiquitären Proteins kann von seinem gegenüber dem homologen Protein veränderten C-terminalen Ende stammen. Die Inositol-1,4,5Trisphosphat 5-Phosphatase ist in den Purkinje Zellen des Gehirns stark exprimiert (De Smedt et al. 1994). Das Enzym könnte eine Rolle bei der Zellproliferation spielen, da Patienten mit akuter und chronischer Leukämie nur eine geringe oder keine Enzymaktivität aufwiesen (Mengubas et al. 1994). Serumantikörperantworten in MM-Patienten 25 von 42 Antigenen induzierten keine Serumantikörper in MM-Patienten. Die restlichen 17 Antigene hingegen wiesen Antikörper-Häufigkeiten von mehr als 5% auf. Bei der Berechnung des statistisch signifikanten Unterschieds zwischen Ctrl und MM-Patienten konnten vier dieser Antigene identifiziert werden. GBP-5a reagierte mit keiner Ctrl, jedoch mit 17% aller MM-Seren. Ähnlich verhielt sich auch GBP-5ta, das ebenfalls serologisch tumorspezifisch getestet wurde und 28% aller MM-Patienten zur Bildung von Antikörpern angeregt hatte. Wie bereits für den Vergleich von Ctrl und CTCLKollektiv war der Vergleich von Ctrl und MM-Kollektiv hinsichtlich der Serumantikörperantworten gegen HD-MM-48 statistisch signifikant mit 17% der MM-Patienten gegenüber 3% der Ctrl. Schließlich wurde für HD-MM-19 ein statistisch signifikanter Unterschied zwischen Kollektiven identifiziert. Allerdings erzeugte dieses Antigen eine sehr hohe Reaktivität mit annähernd 40% in Ctrl. Dieses Antigen ist, obwohl 83% der MM-Patienten Antikörper gebildet hatten, nicht für die Diagnosestellung geeignet, da es vermutlich zu viele falsch positive Ergebnisse erzeugen würde. Hingegen ist die hohe Reaktivität in MM-Patienten DISKUSSION 157 wirklich bemerkenswert. Im Folgenden sollen die bisher noch nicht genauer beschriebenen Antigene näher erläutert werden. GBP-5a und GBP-5ta GBP-5 stellt ein neues Gen der GBP-Familie dar, dessen drei Spleißvarianten in zwei Proteine, GBP-5a/b und GBP-5ta, translatiert werden. Am C-terminalen Ende ist GBP-5ta um 97 Aminosäuren gegenüber GBP-5a/b verkürzt. Eine hohe Immunogenität von GBP-5ta von 50% in CTCL-Patienten wurde mittels SEREX bestimmt. Von Interesse ist, dass GBP-5ta im SEREX immunogener war als das Volle-Länge-Protein. Auf „messenger“ RNA (mRNA)-Ebene sind alle drei Varianten in Normalgeweben differentiell sowie GBP-5ta/-5b zu 26% - 58% in CTCLTumorgewebe und zu 45% in MM exprimiert. Auf Proteinebene wird eine Expression aller drei Varianten nur in peripheren Blut mononukleären Zellen (PBMC) nachgewiesen, wobei die Expression von GBP-5ta sehr schwach ist. In diesem Zusammenhang wurde GBP-5ta als PBMC-spezifisches Tumorantigen identifiziert. In CTCL-Tumorgewebe wurde nur GBP-5ta nachgewiesen, wohingegen im MM hauptsächlich GBP-5a/b detektiert wurde. In Untersuchungen von Zelllinien des CTCL und MM wurden die GBP-5 Proteine perinukleär und intrazellulär lokalisiert. In den untersuchten CTCL-Zelllinien wurde weder auf mRNA- noch auf Proteinebene eine induzierte Expression der GBP-5 Proteine nach INF-γ Behandlung gemessen. Dies deutet auf eine IFN-γ-Resistenz der Zelllinien hin (Fellenberg et al. 2004). Alle drei GBP-5 Proteine sind auf dem Chromosom 1p22 lokalisiert, welches vielfach in der Literatur als Region mit chromosomalen Veränderungen im Zusammenhang mit malignen Transformationen beschrieben wurde (Parada et al. 1998; Cigudosa et al. 1999; Dave et al. 2002). Schließlich könnte GBP-5ta ein potentielles Onkogen darstellen, da GBP-5ta gegenüber GBP-1 einen Verlust der C-terminalen Region aufweist. Bei GBP-1 bewirkt diese Region den antiproliferativen Effekt auf Endothelzellen (Guenzi et al. 2001). HD-MM-19 HD-MM-19 ist zu 99% homolog zum Zinkfingerprotein 133 (ZNF133), welches zur größten Gruppe von Transkriptionsfaktoren zählt (Ehlken et al. 2004). Die „Krüppel-associated Box (KRAB)“ ist eine stark konservierte Region von 75 Aminosäuren am N-Terminus von etwa einem Drittel der Krüppel-Klasse Cys2His2-Zinkfingerproteine. 25% von 12 MM-Patienten erzeugten in sekundären SEREX-Untersuchungen eine Antikörperreaktion gegen dieses ubiquitär exprimierte Antigen nach einer Immunisierung mit entweder IL-7 oder IL-2. Einer der drei Patienten besaß bereits Antikörper vor der Immunisierung. CTCL-Patienten und Ctrl wurden im sekundären SEREX nicht analysiert. DISKUSSION 158 Die KRAB-Domäne wird durch zwei Untereinheiten, der A- und B-Unterdomäne, gebildet. Laut Margolin et al. können KRABs potente transkriptionelle Repressionsdomänen darstellen (Margolin et al. 1994). Schließlich konnte in Mikroarray-Untersuchungen eine erhöhte Expression von ZNF133 in einer Neuroblastomzelllinie nachgewiesen werden (Heiskanen et al. 2000). 4.2.3 SADA als Methode zur Bestimmung der Antikörperantwort In der Literatur sind vier Untersuchungen unter Verwendung der SADA-Methode beschrieben, die neben SADA auch als „Spot assay“ oder „Serological mini-arrays of recombinant tumor antigens“ (SMARTA) bezeichnet wird. Die Untersuchungen von Devitt et al. wurden mit Membranen, die in dieser Arbeit angefertigt wurden, durchgeführt. In der SMARTA-Studie wurde ausschließlich die Etablierung und Optimierung der Methode selbst anhand von neun Tumorantigenen beschrieben (Lagarkova et al. 2003). Scanlan et al. identifizierten 94 Brustkrebs-assoziierte Antigene, von welchen 40 Antigene mittels des „Spot assays“ an Seren von 25 Brust-, 19 Dickdarm-, 15 Lungen-, 15 Gebärmutterund 15 Speiseröhrenkrebspatienten aufgrund ihrer Reaktion mit ausschließlich Krebspatientenseren getestet wurden (Scanlan et al. 2001). Die berichteten AntikörperHäufigkeiten lagen ähnlich wie die der SADA-Ergebnisse dieser Arbeit unter 20%. Die Antigene mit den höchsten Häufigkeiten waren NY-BR-53 (homolog zu LAGE-1) mit annähernd 20% Reaktivität in Brustkrebspatienten, einem reaktiven Serum jeweils im Dickdarm- und Gebärmutterkrebskollektiv und zwei reaktiven Seren jeweils im Lungen- und Speiseröhrenkrebskollektiv. Zwei weitere häufiger reaktive Antigene stellten NY-BR-94 (homolog zu p53, 3 von 19 Seren von Dickdarmkrebspatienten positiv) und NY-BR-98 (homolog zu MAGE D, 2 von 25 Seren von Brustkrebspatienten positiv) dar (Scanlan et al. 2001). In einer weiteren Studie untersuchten Scanlan et al. ein Panel von 77 SEREX-definierten Tumorantigenen mit Seren von 74 Dickdarmkrebspatienten und 75 Ctrl (Scanlan et al. 2002). Dreizehn der getesteten Antigene wiesen Seroreaktionen ausschließlich in Krebspatienten auf, so dass nur diese Antigene weiter untersucht wurden. Auch hier lagen die analysierten Antikörperreaktivitäten für alle 77 Antigene unter 20%. Das Antigen NY-CO-13 (homolog zu p53) hatte die stärkste immunologische Reaktion mit Antikörpern in 15% der Patienten. 8% der Patienten reagierten mit Antikörpern auf die Antigene NY-CO-42 (homolog zu Thyroidhormon Rezeptor-interagierendes Protein 4 (TRIP4)) und MAGE-A3. Desweiteren wurden Antikörper gegen diese dreizehn Antigene mit dem Stadium der Erkrankung korreliert. Hier war eine DISKUSSION 159 Abnahme an Antikörper-Häufigkeit gegen mindestens eines der dreizehn Antigene von Phase I (71%) über Phase II (36%) zu Phase III (14%) auffällig. In Phase IV wurden wieder höhere Antikörper-Häufigkeiten (52%) identifiziert (Scanlan et al. 2002). Devitt et al. analysierten Serumantikörperantworten unter Verwendung desselben Panels von Antigenen, welches in dieser Arbeit mittels SADA untersucht wurde. Seren von 35 Nierenzellkarzinom (RCC)-Patienten und 15 Ctrl wurden für diese Studie herangezogen (Devitt 2004; Devitt et al. 2006). Für zwei Antigene konnte ein statistisch signifikanter Unterschied in der Antikörperantwort von RCC-Patienten und Ctrl nachgewiesen werden. 23% der RCCPatienten besaßen Antikörper gegen se2-5, wohingegen keine Antikörper in Ctrl identifiziert wurden. 40% aller RCC-Patienten wiesen Antikörper gegen HD-MM-19 auf, jedoch nur 7% aller Ctrl. Da in dieser Studie andere Kontrollseren verwendet wurden als in der vorliegenden Arbeit, sind die Ergebnisse hinsichtlich der Antwort der Kontrollseren gegen se2-5 und HDMM-19 in dieser Studie im Gegensatz zur vorliegenden Arbeit unterschiedlich. Dies könnte auf die Anzahl und Auswahl der Seren zurückzuführen sein. Gegen die vier Antigene se1-1, se2-1, se20-4 und HD-MM-19 wurden höhere Antikörperantworten in RCC-Patienten als in Ctrl gemessen. Die acht Antigene HD-MM-03, cTAGE-5a, se2-5, se89-1, se57-1, du20-10L4, GAGE-t und HD-MM-08 wurden immunologisch tumorspezifisch erkannt. Gegen 28 Antigene konnten keine Antikörper im Serum detektiert werden. Se20-7 konnte ebenso wie in dieser Arbeit als Autoantigen identifiziert werden, da 100% der RCC-Patienten und Ctrl Antikörper gegen dieses Antigen gebildet hatten. HD-MM-23 wurde mit Antikörper-Häufigkeiten von 100% in Ctrl und 97% in RCC-Patienten ebenfalls häufig von Seren erkannt (Devitt 2004; Devitt et al. 2006). 4.2.4 Vergleich von SADA und SEREX Beim Vergleich der SADA- und SEREX-Methode fällt zunächst auf, dass beim SADA vergleichsweise viele Antigene, das heißt bis zu 22 verschiedene Antigene plus Negativ- und Positivkontrolle, gleichzeitig mit einem Serum getestet werden können. Dies stellt eindeutig den Vorteil des SADA gegenüber der SEREX-Methode dar, bei der pro Platte nur ein Phagenklon mit einem Serum getestet werden kann. Zusätzlich kann ein Vergleich zwischen Färbung des Antigenspots mit der Negativ- und Positivkontrolle durchgeführt werden. Somit ist eine vergleichende und bessere Beurteilung der Positivität eines Antigens möglich als beim SEREX. Beim SADA ist die Stelle eines Antigens auf der Membran definiert, wohingegen auf der SEREX-Membran nur zwischen reaktiven und nichtreaktiven Plaques unterschieden werden kann. Erst nach der Isolierung des entsprechenden SEREX-Plaques und einer in vivo Exzision DISKUSSION 160 kann eindeutig die Identität eines Plaques festgestellt werden. In sekundären SEREXUntersuchungen wird zumeist eine heterogene Phagenlösung mit dem Phagenklon und einem Phagen ohne Insert (Negativkontrolle) eingesetzt. Auf der Platte jedoch kann nicht festgestellt werden, welche Plaques welchem Phage zuzuordnen sind. Ein deutlicher Unterschied zwischen den beiden Methoden besteht in der Bildung der Phagenplaques. Beim SEREX lässt sich ein Phagenplaque auf der Platte auf ein einziges Bakterium, das von einem einzigen Phagenklon infiziert wurde, zurückführen. Beim SADA hingegen werden ca. 2µL Phagenlösung eines Phagenklons mit einem Titer von ca. 2000 Phagen pro µL mittels Stempel auf den Bakterienrasen übertragen. Dies bedeutet, dass viele Bakterien an der Auftragstelle mit einem Phagenklon infiziert werden und somit viele Plaques nebeneinander entstehen. Teilweise wurde beim SEREX eine helle Hofbildung um die Phagenplaques bei der Färbung der entwickelten Membranen beschrieben. Dieser Effekt wurde bei den SADA-Membranen nicht festgestellt. Somit wurde durch die Entstehung von vielen Phagenplaques an einer Stelle keine Auslöschung der Färbung durch Randeffekte ermittelt. 4.2.5 SADA - Fazit und Ausblick Zusammenfassend ist festzustellen, dass die gemessenen humoralen Immunantworten sowohl in CTCL- als auch in MM-Patienten hinsichtlich ihrer Frequenz in den verschiedenen Kollektiven niedrig sind und selten über 20% der entsprechenden Kohorte ausmachen. Die Frequenz der Antikörperantworten in MM-Patienten ist zumeist höher als in den CTCL-Patienten. Dies ist vermutlich auf die höhere Immunogenität des MM selbst zurückzuführen. Die Antikörperantworten gegen CTA liegen in allen Kollektiven unter 10%. Dies entspricht Ergebnissen von E. Stockert, die unter Verwendung eines ELISAs ähnlich geringe Frequenzen an Antikörperantworten gegen CTA in MM-Patienten nachwies. Schließlich lagen alle gemessenen Antikörperfrequenzen in CTCL-Patienten unterhalb der Frequenz, die in sekundären SEREX-Untersuchungen bestimmt wurden. Ursachen hierfür können in einem größeren, zufällig ausgewählten Serenkollektiv im SADA und im direkten Vergleich von Antikörperreaktionen gegen den Phagenklon und den Negativ- sowie Positivklon liegen Generell ist festzustellen, dass mittels SADA im Vergleich zu SEREX mehr Antigene gleichzeitig und parallel mit einem Serum getestet werden können. Jedoch ist SADA eine aufwendige Methode, da die Präabsorption der Seren zeitaufwendig und teuer ist. Zudem dauert ein vollständiger Test inklusive Wiederholung vier Arbeitstage, so dass eine Anwendung in der Klinikroutine wohl eher unwahrscheinlich ist. Des Weiteren wurden in dieser Untersuchung mittels SADA nur zwei Phagenklone für das CTCL und vier Phagenklone für das MM DISKUSSION 161 identifiziert, die statistisch signifikant unterschiedlich zwischen Ctrl und dem jeweiligen Patientenkollektiv waren, so dass sie in Prognostik und Diagnostik eingesetzt werden könnten. Für eine relevante Vorhersage wäre jedoch ein Panel von mindestens zehn Antigenen wünschenswert, die in weiteren SEREX- und SADA-Screens identifiziert sowie überprüft werden müssten. Schließlich wäre ein direkter Vergleich von denselben Seren im SEREX und SADA unter Verwendung derselben Antigene sinnvoll, um präzisere Aussagen über die Vergleichbarkeit beider Methoden zu ermöglichen. Antigene, die auf diese Art und Weise als relevant für Diagnostik und Prognostik bestätigt würden, wären ebenfalls ideale KandidatenAntigene zum Einsatz in der nachfolgend diskutierten Multiplex-Serologie. 4.3 Multiplex-Serologie Für die diagnostische Tumorserologie, zur Beurteilung von Krebserkrankungen oder zur Früherkennung von Rezidiven wird der ELISA unter Verwendung von tumor-assoziierten, tumorspezifischen und nicht-tumorspezifischen Antigenen seit Jahrzehnten eingesetzt (Old und Chen 1998; Sahin et al. 1999). Die Entwicklung des ELISA erfolgte, um viele Seren in kurzer Zeit zu testen. Durch die einfache Handhabung eignet sich diese Methode ausgezeichnet für einen Einsatz sowohl in der Forschung als auch in der Klinikroutine. Dr. Peter Sehr (DKFZ, Heidelberg) entwickelte den Glutathion-S-Transferase (GST) „capture“ ELISA, der den Einsatz von Proteinen in einem ELISA-System in ihrer nativen Konformation ohne vorherige Aufreinigung erlaubt (Sehr et al. 2001). Dieser methodische Ansatz ist sehr sensitiv und spezifisch und kann mit jedem löslichen, rekombinant hergestellten GST-Fusionsprotein durchgeführt werden. Der einzige Nachteil ist jedoch die Beschränkung auf ein Antigen pro Loch sowie ein hoher Serumverbrauch und Zeitaufwand. Seit Ende der 80er Jahre wird der Begriff „Multiplex“ im Zusammenhang mit der PCR benutzt. Heute bedeutet „Multiplex“, dass die Reaktion von einer Probe mit mehreren Bindungspartnern untersucht wird. Seit Mitte der 90er Jahre sind Suspensions-Arrays erhältlich, die meistens auf Polystyrol-Kügelchen (Beads) basieren, die sich durch ihre Größe oder Farbe voneinander unterscheiden. Die Beads dienen hierbei als feste Phase für Bindungsassays, wobei die Identifikation mittels Durchflusszytometrie erfolgt. Obwohl die Bead-Population auf maximal 100 verschiedene Beads beschränkt ist, erlaubt dieses System einen hohen Durchsatz (Templin et al. 2004). Das xMAP-System, das Ende der 90er Jahre von der Firma Luminex auf den Markt gebracht wurde, wurde in der vorliegenden Arbeit verwendet. Es beruht auf bis zu 100 unterschiedlich farbkodierten Beads. Die Auswertung erfolgt mit einem speziellen Durchflusszytometer, dem Luminex Analyzer. Das xMAP-System ist derzeit das kommerziell DISKUSSION 162 als auch wissenschaftlich erfolgreichste System. Dr. Tim Waterboer (DKFZ, Heidelberg) entwickelte die Multiplex-Serologie auf der Grundlage des xMAP-Systems für die Analyse von komplexen Antikörperantworten inklusive einer in situ Aufreinigung der rekombinant exprimierten Antigene direkt auf den Beads (Waterboer 2005; Waterboer et al. 2005). In bisherigen xMAP-basierten Publikationen wurden beispielsweise Zytokine (Prabhakar et al. 2002), monoklonale Antikörper (Seideman und Peritt 2002), Antikörper gegen virale Antigene (Opalka et al. 2003) und Autoantigene (Gilburd et al. 2004) im Serum untersucht. In dieser Arbeit wurde das xMAP-System als Multiplex-Hochdurchsatzverfahren angewendet, um Antikörperantworten gegen sieben CTA, drei TAA und zwei Tumorsuppressorgene in 144 CTCL-, 191 MM-Patienten, 100 Kontrollpersonen und 25 immunbehandelten Tumorpatienten zu untersuchen. Vier virale Antigene dienten als interne Ctrl zur Überprüfung der Zugehörigkeit von Seren zu Patienten. Insgesamt wurden zwölf humane und vier virale Antigene an 460 Seren getestet. Ähnlich wie die Untersuchungen mittels SADA diente die Untersuchung von Antikörperantworten mittels Multiplex-Serologie der Analyse der Eignung der untersuchten Tumorantigene in Diagnostik und Prognostik an sich oder aber als Kombination von mehreren Tumorantigenen. 4.3.1 Qualitätskontrolle und statistische Auswertung In dieser Arbeit wurden erstmalig Seren von CTCL- und MM-Patienten im Vergleich zu gesunden Ctrl mittels Multiplex-Serologie auf ihre humorale Immunantwort gegen zwölf humane und vier virale Antigene untersucht. Die kovalent an Glutathion-Casein (GC) gekoppelten Beads wurden von Dr. T. Waterboer zur Verfügung gestellt, da diese sehr stabil sind und über Monate im Kühlschrank gelagert werden können (Waterboer 2005). An jede GC-Beadsorte wurde ein rekombinant hergestelltes GSTFusionsprotein direkt gebunden. Dies ging mit einer in situ Aufreinigung der GSTFusionsproteine aus dem Bakterienlysat einher. Die gewaschenen 17 verschiedenen Beadsorten wurden zu einer Mischung vereinigt und in einer 96 Lochplatte mit Filterboden mit gegen GSTtag- und E. coli-Protein präabsorbiertem Serum inkubiert. Ungebundene Serumproteine wurden durch Waschen der Beads entfernt. Gebundene Serumantikörper wurden durch Inkubation mit biotinyliertem Sekundärantikörper und anschließender Streptavidin-R-Phycoerythrin-Reaktion markiert. Die Detektion der Antigen-Antikörper-Reaktion erfolgte schließlich im LuminexAnalysegerät. Die Beadsorte wird unter Verwendung eines Klassifizierungs-Lasers identifiziert und die Quantifizierung der Bindungsreaktion erfolgt unter Verwendung eines Reporter-Lasers. DISKUSSION 163 Pro Beadsorte wurden 100 Beads vermessen, und die Reporter-Fluoreszenz-Intensitäten als Median der 100 Einzelbeads in „mean fluorescence intensity“ (MFI) berichtet. Zur Qualitätskontrolle der Multiplex-Serologie wurden zwei verschiedene Ansätze gewählt. Zunächst wurden die rekombinant exprimierten GST-Fusionsproteine, die im Bakterienlysat vorlagen, im GST „capture“ ELISA mittels der GST- und tag-Antikörper untersucht. Hierzu wurde der GST „capture“ ELISA durchgeführt und eine zwölfstufige Verdünnungsreihe jedes GST-Fusionsproteins hergestellt. Die Untersuchung jedes Antigens mit beiden Antikörpern bewies eindeutig, dass alle Proteine in voller Länge exprimiert waren. Zusätzlich wurden alle GST-Fusionsproteine mittels GST- und tag-Antikörper im Western Blot analysiert. Hier konnte für alle Antigene die volle Länge jedes GST-Fusionsproteins neben einigen Abbauprodukten nachgewiesen werden. In einem zweiten Ansatz wurden interne Ctrl während der Durchführung der MultiplexSerologie mitgeführt. Hierzu diente der tag-Antikörper, mittels welchem die Bindung von VolleLänge-Protein an die Beads überprüft wurde. Diese Bindung konnte für alle Antigene nachgewiesen werden. Zudem wurde der Mittelwert aller MFI-Werte gegen alle Antigene inklusive GST-tag für jedes Serum berechnet. Für alle untersuchten Seren lag dieser Mittelwert über 20 MFI. Weiterhin wurde je ein Loch ohne Serum und ohne sekundären Antikörper als interne Ctrl herangezogen. Der MFI-Mittelwert über alle Antigene lag für das Loch ohne sekundären Antikörper wie erwartet unter 20 MFI. Für das Loch ohne Serum ergab sich mit 37 MFI ein leicht erhöhter Wert, der eine geringe Hintergrundreaktion verdeutlichte. Schließlich wurden drei spezifische Antikörper zur Bestätigung der Bindung des richtigen Antigens an die entsprechende Beadsorte in der Multiplex-Serologie eingesetzt. Für die Antigene cTAGE-5a Cterminal, GAGE-7b und MAGE-A9 wurde nachgewiesen, dass sie an die entsprechend ausgewählte Beadsorte gebunden waren. Für alle anderen verwendeten Antigene lagen keine in der Multiplex-Serologie verwendbaren Antikörper bei der Durchführung der Analyse vor. Zur Beurteilung der Seroreaktivitäten gegen jedes Antigen mussten seropositive von seronegativen Reaktionen unterschieden werden. Im allgemeinen wird ein diskriminatorischer Schwellenwert (Cut-off) anhand des Mittelwerts plus x-facher Standardabweichung der Seroreaktivitäten einer seronegative Kontroll-Gruppe berechnet (Stockert et al. 1998; Stone et al. 2003; Zhang et al. 2003; Goodell und Disis 2005; Strid et al. 2007). In dieser Arbeit dienten 100 unabhängige Kontrollseren von einer Blutbank als Kontroll-Gruppe. Für die vier in dieser Arbeit verwendeten viralen Antigene ist diese Methode der Cut-off-Wert-Bestimmung jedoch problematisch, weil sehr viele Erwachsene Antikörper gegen die verwendeten Antigene (BK, JC, LPV, humanes Papillomavirus (HPV) 1) besitzen. Deshalb wurde der Cut-off-Wert für jedes DISKUSSION 164 virale Antigen anhand von Voruntersuchungen festgesetzt (Waterboer 2005). Für alle humanen Antigene wurde der Cut-off-Wert als Mittelwert plus zweifacher Standardabweichung der Seroreaktivitäten berechnet. Diese Vorgehensweise ist bereits in der Literatur für HER-2/neu (Goodell und Disis 2005) und Salmonellen Lipopolysaccharid (Strid et al. 2007) beschrieben. Für die humanen Antigene wurde eine andere Bestimmung des Cut-off-Wertes im Vergleich zu den viralen Antigenen gewählt. Denn nur wirklich reaktive Seren sollten identifiziert werden, um eine fundierte Aussage über die Verwendung der Antigene in Diagnose, Prognose oder Monitoring treffen zu können. Allerdins handelt es sich letztlich jedoch um Autoantigene. Die Darstellung der Reaktivitäten aller Seren einer Kohorte erfolgte durch kumulative Distributionen. Bei dieser grafischen Darstellung kann nach Einzeichnung des Cut-off-Wertes direkt die Prozentzahl an reaktiven Seren, das heißt Seren über Cut-off, abgelesen werden. Statistisch ausgewertet wurden die erhaltenen Ergebnisse unter Verwendung des „Fisher’s Exact Tests“. Zwei unterschiedliche Serumkategorien wurden verwendet: reaktive und nicht-reaktive Seren beziehungsweise seropositive oder seronegative Patienten und Ctrl. Nach der Bestimmung der prozentualen Anteile an reaktiven und nicht-reaktiven Seren in jedem Kollektiv, wurden die Reaktivitäten der unterschiedlichen Kollektive anhand des „Fisher’s Exact Tests“ verglichen. Sobald ein Signifikanzniveau von unter 0,05 bei der Bewertung eines Tests ermittelt wurde, wurde dieses Ergebnis als statisch signifikant unterschiedlich gewertet. Serumverläufe wurden als signifikant ansteigend gewertet, wenn eine der nachfolgenden Seroreaktivitäten eine doppelte Reaktivität aufwies und zudem über dem Cut-off-Wert lag. Ein signifikanter Abfall bestand in mindestens einer Halbierung der Seroreaktivität bei einer nachfolgenden Seroreaktivität, solange die erste Seroreaktivität über dem Cut-off-Wert des jeweiligen Antigens lag. 4.3.2 Humorale Immunantworten von MM-Patienten Häufigkeiten von Antikörperantworten gegen sieben CTA, drei TAA, zwei Tumorsuppressorgene und vier virale Antigene wurden mittels Multiplex-Serologie in insgesamt 191 MM-Patienten untersucht. Die grafische Auswertung erfolgte anhand von kumulativen Distributionen. Die statistische Beurteilung wurde nach der Bestimmung eines Cut-off-Wertes anhand eines unabhängigen Kontroll-Kollektivs für jedes Antigen unter Verwendung des „Fisher’s Exact Tests“ vorgenommen. Die Analyse der MM-Patienten im Vergleich zu den Kontrollpersonen ergab eindeutig, dass Serumantikörperreaktionen gegen die verwendeten CTA, TAA und Tumorsuppressorgene nur sporadisch auftreten und zumeist unter 10% liegen. Antikörperantworten wurden auch in DISKUSSION 165 Kontrollpersonen detektiert. Somit sind alle CTA und TAA außer CAMEL immunologisch nicht tumorspezifisch. Allerdings traten die höchsten, gemessenen MFI-Werte in MM-Patienten vor allem in Stadium IV auf. Dies traf für die Antigene MAGE-A1, A3, A9, LAGE-1a, GAGE-7b und CAMEL zu. Entsprechend könnte gerade bei Patienten in Stadium IV eine Abgrenzung gegenüber Patienten anderer Stadien anhand MFI-Werten von teilweise mehr als 10000 MFI erfolgen. Einschränkend muss jedoch erwähnt werden, dass Stadium IV Patienten bereits anhand des klinischen Bildes von Patienten anderer Stadien unterscheidbar sind. Des Weiteren wurden gerade in Stadium IV Patienten häufiger Antikörper gegen mehrere CTA in einem Patienten nachgewiesen. Diese Häufung von Antikörpern gegen verschiedene CTA war außerordentlich deutlich bei zwei der 47 Patienten in Stadium IV ausgeprägt. Statistisch signifikante Unterschiede zwischen Patienten- und Kontroll-Kollektiv wurden für die Antigene MAGE-A3, LAGE-1a, se70-2 und se57-1 berechnet. Patienten in Stadium IV besaßen statistisch signifikant häufiger Antikörper gegen MAGE-A3 (19,1%) gegenüber der Ctrl (6%). Statistisch signifikant häufiger bildeten Patienten des Stadiums II (10,2%) Antikörper gegen se57-1 gegenüber der Ctrl (1%). Für GAGE-7b konnte ein Trend zu einem statistisch signifikanten Unterschied zwischen Antikörper-Häufigkeiten in MM-Patienten des Stadiums II (8,2%) und Ctrl (1%) erkannt werden. Für se70-2 hingegen wurden statistisch signifikant mehr Antikörperantworten im Kontroll-Kollektiv (3%) gegenüber dem Patientenkollektiv (0%) gemessen. Für alle anderen Antigene ließen sich keine statistischen Unterschiede zwischen Kontrollpersonen und Patienten ermitteln. LAGE-1a stellte sich in dieser Arbeit als sehr wertvolles CTA hinsichtlich Serumantikörperantworten in MM-Patienten dar. Als einziges CTA aller untersuchter Antigene demonstrierte LAGE-1a eine stadienabhängige Serumantikörper-Häufigkeit. Bereits der Unterschied an Serumantikörpern zwischen Ctrl (3%) und allen 191 MM-Patienten (13,6%) ist statistisch signifikant. Des Weiteren wurden statistisch signifikant erhöhte AntikörperHäufigkeiten in Stadium III (12,8%) und Stadium IV Patienten (21,3%) nachgewiesen. Ein Einsatz dieses Antigens in der Prognostik und eventuell sogar in der Diagnostik (Stadium I 10,4%) von MM-Erkrankungen wäre anhand dieser Untersuchungen sinnvoll. Nachfolgend soll das CTA LAGE-1a besonders hinsichtlich humoraler Immunantworten näher beschrieben werden. Das Antigen LAGE-1 zählt aufgrund seines Expressionsprofils zu den CTA und wurde 1998 zum ersten Mal mittels einer differentiellen RT-PCR-Analyse an einer MM-Zelllinie beschrieben (Lethe et al. 1998). Ein Jahr zuvor wurde das CTA NY-ESO-1 mittels SEREX an einer Ösophaguskarzinombank identifiziert, zu welchem LAGE-1 zu 84% homolog ist (Chen et al. DISKUSSION 166 1997). Dadurch sind Kreuzreaktionen zwischen den beiden Proteinen, die hintereinander auf Chromosom Xq28 lokalisiert sind, nicht auszuschließen. Zudem hat LAGE-1 mit LAGE-1a und –1b zwei Spleißvarianten, und CAMEL, ein kürzeres Produkt von einem alternativen Leserahmen, zählt ebenfalls zu dieser Genfamilie (Lethe et al. 1998; Aarnoudse et al. 1999). Vor einigen Jahren wurde das ubiquitäre ESO-3, das zu weniger als 50% homolog zu NY-ESO-1 ist, identifiziert (Alpen et al. 2002). Die Funktion von LAGE-1 und NY-ESO-1 ist bisher noch ungeklärt. LAGE-1 Expression wird häufig zusammen mit jener von NY-ESO-1 bei RT-PCRUntersuchungen detektiert. Für ungefähr ein Drittel aller Tumore wurden individuelle Expressionsmuster berichtet (Vaughan et al. 2004; Bolli et al. 2005). Auch andere CTA wie beispielsweise MAGE-Antigene werden mit NY-ESO-1 koexprimiert (Bolli et al. 2005). NYESO-1 und LAGE-1 sind in vielen Neoplasien z.B. Mamma-, Kolon- und Prostatakarzinom exprimiert (Lethe et al. 1998). Im MM wurden Expressionen von 24% bis 43% berichtet (Lethe et al. 1998; Sahin et al. 2000; Prasad et al. 2004; Vaughan et al. 2004). Dies würde die hohe Reaktivität der MM-Seren, die in dieser Arbeit gemessen wurde, erklären. Die NHL-Gewebe waren NY-ESO-1- und LAGE-1-negativ (Lethe et al. 1998; Claus et al. 2005). Wie unter 3.2.6 und 4.3.3 beschrieben wurden kaum seropositive CTCL-Patienten identifiziert. Antikörperantworten wurden bisher oft gegen NY-ESO-1 untersucht, da es ein sehr immunogenes Antigen ist und das Auftreten von Antikörpern in vielen Neoplasien mit seiner Expression einhergeht. Höchstwahrscheinlich sind diese Antikörper auch gegen LAGE-1 reaktiv aufgrund der hohen Proteinhomologie. Im MM und Ovarialkarzinom wurden Antikörper in 5% 15% aller Patienten gemessen (Stockert et al. 1998; van Rhee et al. 2005). In fortgeschrittenem MM wurden bei ca. 50% der Patienten mit NY-ESO-1-positiven Tumoren Antikörper gegen NY-ESO-1 detektiert (Jager et al. 2000b). Fünfzehn von 42 Seren (35,7%) von Patienten mit sporadischem Schilddrüsenkarzinom waren NY-ESO-1-positiv (Maio et al. 2003). Die hohe Immunogenität von NY-ESO-1 wird durch eine häufig simultane Antwort des humoralen und zellulären Immunsystems bestätigt. In einem MM-Patienten wurden Antikörper gegen NY-ESO1 gemeinsam mit HLA-A2/NY-ESO-1-spezifischen zytotoxischen T-Lymphozyten (CTL) im Blut nachgewiesen (Jager et al. 1998). Dieses parallele Auftreten von Antikörpern und CTL traf ebenfalls auf zehn von 36 Patienten mit fortgeschrittenen Krebserkrankungen zu. Kein Patient mit NY-ESO-1-negativem Tumor wies Antikörper auf (Jager et al. 2000b). In einer weiteren Studie wurde nachgewiesen, dass NY-ESO-1 Antikörper auf der Anwesenheit von NY-ESO-1exprimierenden Tumoren und dem Verlauf der Erkrankung in Abhängigkeit von NY-ESO-1 basieren (Jager et al. 1999). Für NY-ESO-1/LAGE-1 sind mittlerweile eine Vielzahl von Haupthistokompatibilitätskomplex (MHC) I präsentierten CD8+ Epitopen, HLA-A2 (Jager et al. 1998) oder HLA-A31 (Wang et al. 1998) präsentiert, bekannt. CD4+ T-Zellepitope sind DISKUSSION 167 ebenfalls identifiziert worden (Zeng et al. 2000; Mandic et al. 2003). Aufgrund der CTA Expression und Immunogenität von NY-ESO-1/LAGE-1 wurden bereits einige Immuntherapien unter Verwendung von NY-ESO-1 durchgeführt. In den ersten Studien wurden HLA-A2spezifische NY-ESO-1-Epitope in Verbindung mit Adjuvantien intradermal injiziert, und CD8+ T-Zellantworten konnten nachgewiesen werden (Jager et al. 2000a; Chen et al. 2005). Volle Länge NY-ESO-1 Protein wurde in Verbindung mit dem Adjuvans ISCOMatrixTM in klinischen Studien eingesetzt (Davis et al. 2004). Da komplexe Immunantworten mit Antikörperbildung und CD8+ und CD4+ T-Zellantworten nachgewiesen wurden, befinden sich die Studien zwischenzeitlich in Phase II. Somit stellt NY-ESO-1/LAGE-1 ein aussichtsreiches Antigen für Immuntherapien in Krebspatienten dar. Im Zuge der Untersuchung von MM-Patienten konnten Serumverläufe von acht MM-Patienten mittels Multiplex-Serologie getestet werden. Von den Patienten lagen jeweils zwei bis drei aufeinander folgende Serumproben vor, so dass Zeiträume von bis zu 171 Wochen untersucht werden konnten. Wie bereits im Kollektiv analysiert, traten Antikörperantworten über Cut-off auch in den Serumverläufen weniger häufig auf. Einzig die Antigene LAGE-1a, se57-1, cTAGE5a N-terminal und p16 hatten Antikörperantworten in den acht untersuchten Patienten hervorgerufen. Relativ hohe MFI-Werte, jedoch unter Cut-off, wurden ebenfalls für cTAGE-5a C-terminal identifiziert. Nur für Patient #M3 wurde eine Serokonversion gemessen. Die Antikörper gegen LAGE-1a stiegen von 0 auf über 13000 MFI signifikant an. Dieser enorme Antikörperanstieg korrelierte mit dem Krankheitsbild des Patienten, der im Zeitraum von 98 Wochen von Stadium I nach Stadium IV fortschritt. Dieser Patient stellt ein ausgezeichnetes Beispiel einer erhöhten Tumorlast im Zuge der Krankheitsverschlechterung dar. In diesem Zusammenhang wäre es sicherlich interessant gewesen, den Tumor hinsichtlich LAGE-1a zu charakterisieren. Leider lag dieses Material nicht vor. Mit Ausnahme von MAGE-A9, für welches der Antikörpertiter von 5 auf 139 MFI zunahm, wiesen andere Antigene keine Antikörperanstiege auf. Für zwei weitere Patienten konnten Antikörperantworten gemessen werden. Patient #M2, Stadium IV, besaß gleichbleibende Antikörper gegen se57-1 genauso wie Patient #M6, Stadium III, gegen p16. Somit wird auch bei der Untersuchung von Serumverläufen deutlich, dass Patienten individuelle Antikörperantworten besitzen und diese nicht generell mit dem Stadium korrelieren. Einzig LAGE-1a bildete in diesem Punkt eine Ausnahme für einen Patienten. Jedoch ist ersichtlich, dass vorhandene Antikörperantworten über lange Zeiträume hinweg stabil bleiben können. Deutlich wurde ebenfalls, dass virale Antigene eine bedeutend höhere Antikörperantwort, gemessen in MFI-Werten, von bis zu annähernd 20000 MFI erzeugen können. Dies ist eindeutig auf die hohe Immunogenität dieser Antigene, die DISKUSSION dem Körper fremd sind, zurückzuführen. Für 168 einen optimalen Vergleich mit Serumantikörperverläufen von gesunden Personen wären Multiplex-Untersuchungen an Seren von gesunden Personen über ähnliche Zeiträume wie die der MM-Patienten angeraten. 4.3.3 Humorale Immunantworten von CTCL-Patienten Entsprechend der Untersuchung der humoralen Immunantwort von MM-Patienten wurde die Häufigkeit von Antikörperantworten in 144 CTCL-Patienten mittels Multiplex-Serologie an dem in dieser Arbeit verwendeten Antigenpanel untersucht. Ähnliche Aussagen wie für das MM-Kollektiv trafen auch für das CTCL-Kollektiv zu. Serumantikörperreaktionen gegen die verwendeten CTA, TAA und Tumorsuppressorgene waren im CTCL-Kollektiv nur sporadisch vorhanden und lagen zumeist unter 10%. Die höchsten, gemessenen MFI-Werte in CTCL-Patienten wurden vor allem in Stadium I für die Antigene MAGE-A1, A3, LAGE-1a, CAMEL und cTAGE-5a N-terminal sowie in Stadium IV für die Antigene MAGE-A9, GAGE-7b und p53 gemessen. Somit erzeugten wiederum die CTA hohe MFI-Werte. Einschränkend muss jedoch erwähnt werden, dass die Stadiengruppengröße unterschiedlich und gerade für Stadium II bis IV sehr klein war (n=14-16). Gleichzeitiges Auftreten von Antikörpern gegen mehrere Antigene in einem Patienten wurde in den CTCLPatienten für höchstens vier CTA nachgewiesen. Die MFI-Werte lagen deutlich unterhalb von 10000 MFI. Nur für jeweils einen Patienten in Stadium I (12172 MFI, LAGE-1a) und Stadium IV (12361 MFI, p53) wurden hohe MFI-Werte gemessen. Diese Ergebnisse weisen auf die höhere Immunogenität des MM hin. Schließlich konnte im CTCL-Kollektiv kein Antigen identifiziert werden, welches stadienabhängig Serumantikörper in Patienten induziert. Ein statistisch signifikanter Unterschied zwischen Patienten- und Kontroll-Kollektiv wurde einzig für das Antigen MAGE-A9 berechnet. Patienten in Stadium IV besaßen statistisch signifikant häufiger Antikörper gegen MAGE-A9 (25%) gegenüber den Ctrl (2%). Allerdings waren Patienten der Stadien I bis III seronegativ für MAGE-A9. Deshalb wäre MAGE-A9 wohl primär für prognostische jedoch weniger für diagnostische Untersuchungen geeignet. Für MAGE-A1 konnte ein Trend zu statistisch signifikant erhöhten Antikörper-Häufigkeiten in CTCL-Patienten des Stadiums III (28,6%) gegenüber Ctrl (6%) nachgewiesen werden. Für alle anderen Antigene ließen sich keine statistisch signifikanten Unterschiede zwischen Ctrl und Patienten ermitteln. Interessant ist, dass LAGE-1a ausschließlich in MM-Patienten starke und stadienabhängige Antikörperreaktionen hervorrief. Nur knapp 5% der CTCL-Patienten waren LAGE-1a-seropositiv. LAGE-1a-seropositive Patienten beschränkten sich auf Stadium I. Da nur DISKUSSION 169 die MAGE-Antigene statistisch signifikante Unterschiede im Vergleich von CTCL-Patienten und Ctrl aufwiesen, soll diese Antigengruppe im Folgenden näher beschrieben werden. Die MAGE-Antigenfamilie stellt eine der größten CTA-Gruppen des X Chromosoms dar, zu welcher die MAGE-A, B und C Antigene zählen. Die biologische Funktion der MAGE-Antigene ist noch weitgehend unbekannt. Einige Studien weisen jedoch auf eine regulatorische Rolle hin (Nagao et al. 2003; Laduron et al. 2004). Mittlerweile sind 12 Mitglieder der MAGE-A-Familie bekannt, die hinsichtlich ihrer Expression in den verschiedensten Tumorentitäten untersucht wurden (De Plaen et al. 1994). Diese Daten beschränken sich jedoch vor allem auf MAGE-A1, A3, A4, A6, A10 und A12. In einer Studie von Akcakanat et al. in Patienten mit Speiseröhrenkrebs wurde die Proteinexpression von MAGE-A-Antigenen (A1, A2, A3, A4, A6, A10 und A12) in 52% (111/213) aller Patienten gemessen (Akcakanat et al. 2006). In kutanen MM war MAGE-A3 bis zu 65% exprimiert (De Plaen et al. 1994). Eine hohe Expression von MAGE-A1 von 80% wurde in Leberkarzinomen beschrieben (Yamashita et al. 1996), wohingegen myelomonozytische Leukämien MAGE-A-negativ waren (Chambost et al. 1993). In nicht-kleinzelligem Lungenkrebs wurde in ungefähr 45% der Patienten eine Genexpression von MAGE-A1 oder A3 gemessen (Gure et al. 2005). Des Weiteren wurden NY-ESO-1 und MAGEA3 als Marker für eine schlechte Prognose in nicht-kleinzelligem Lungenkrebs identifiziert (Gure et al. 2005). Untersuchungen von Antikörperantworten gegen die MAGE-Antigene sind bisher eher selten durchgeführt worden. Antikörperantworten gegen NY-ESO-1, MAGE-A1, A3 und einige andere CTA wurden mittels ELISA an Seren von Sarkompatienten getestet. Zwei von 54 Patienten (4%) wiesen Antikörper gegen NY-ESO-1 auf. Antikörper gegen andere CTA wurden nicht nachgewiesen (Lee et al. 2003). Diese Untersuchungen bestätigen wiederum, die in dieser Arbeit ermittelten Ergebnisse sowie Studien von Stockert et al. (Stockert et al. 1998). Schließlich wurde insbesondere MAGE-A3 häufig in klinischen Vakzinierungsstudien eingesetzt. Eine Verbesserung des klinischen Krankheitsbildes wurde häufig nur in einem kleinen Prozentsatz der Patienten nachgwiesen (10%) (Marchand et al. 1999; Thurner et al. 1999). In einer Phase I/II Studie, die ein rekombinantes MAGE-A3 Fusionsprotein mit Lipoprotein D von H. influenzae verwendete, wurden anti-MAGE-A3 CD4+ T-Lymphozyten in einem von fünf MM-Patienten, die eine klinische Antwort auf die Therapie aufwiesen, detektiert (Kruit et al. 2005). Antikörper gegen MAGE-A3 wurden nur in einem von 24 MM-Patienten nach der Vakzinierung und in keinem von 26 MM-Patienten vor der Vakzinierung gemessen (Kruit et al. 2005). Hoon et al. immunisierten MM-Patienten mit einer MAGE-A1 exprimierenden Melanomzell-Vakzine. Antikörperantworten gegen MAGE-A1 wurden mittels eines ELISA überwacht. IgG Antikörperantworten von 57% der 53 immunisierten MM-Patienten DISKUSSION 170 nahmen signifikant zu (Hoon et al. 1995). Spontane Immunantworten gegen MAGE-A3 vermittelt durch CTL sind mit ca. 5% sehr selten in MM-Patienten (Hanagiri et al. 2006). Serumverläufe konnten ebenfalls für CTCL-Patienten mittels Multiplex-Serologie untersucht werden. Hierbei wurden Serumantikörperantworten von zwölf Patienten über bis zu 398 Wochen analysiert. Auch in diesen Patienten waren Antikörperantworten individuell und eher selten. Serokonversionen traten wie bereits im Kollektiv der MM-Patienten eher selten auf. Die Antigene MAGE-A1, A3, LAGE-1a, cTAGE-5a C-terminal und p53 hatten Antikörper in vier (#C2, #C3, #C8 und #C10) der zwölf Patienten induziert. Serokonversionen wurden für diese vier Patienten gemessen, wobei leider für drei der vier Patienten entweder das Stadium zu Beginn oder am Ende des Abnahmezeitraumes nicht vorlag. Patient #C2 war über fast 290 Wochen seropositiv für MAGE-A1 und wies eine positive Serokonversion für MAGE-A3 auf (272 auf 859 MFI). Patient #C8 war seropositiv für MAGE-A1 und über 147 Wochen wurde eine negative Serokonversion für LAGE-1a (5700 auf 2325 MFI) gemessen. Eine eindeutige Korrelation von Antikörperantwort und Krankheitsstadium konnte für Patient #C10 berichtet werden. Über einen Zeitraum von 200 Wochen nahmen die Antikörper gegen cTAGE-5a Cterminal signifikant zu (1159 auf 2773 MFI), und das klinische Bild des Patienten verschlechterte sich von Stadium Ib nach IIIc. Eine Untersuchung des Tumors hinsichtlich cTAGE-5a Expression wäre nützlich gewesen, um die Tumorlast und Antigenexpression mit dem Krankheitsverlauf optimal korrelieren zu können. Auch hier lag das entsprechende Material leider nicht vor. In zukünftigen Untersuchungen ist es angeraten, Antigenexpression und Antikörperantwort parallel zu testen sowie Serumverläufe mit mehr Serumabnahmen in Patienten inklusive aller Patienteninformationen und Serumverläufe von Ctrl zu testen. Die signifikanteste positive Serokonversion wurde für Patient #C3 gegen p53 (137 auf 12361 MFI) in einem Zeitraum von einem Jahr gemessen. Der Patient befand sich am Ende des Abnahmezeitraumes in Stadium IVc. Im Folgenden soll das Antigen p53 noch etwas näher mit Fokus auf humorale Immunantworten beschrieben werden. Das Protein p53 wird durch das Gen TP53 kodiert. Dieses Tumorsuppressorgen, das seit Jahren im Fokus der Krebsforschung steht, ist ein nukleäres Phosphoprotein. Es spielt eine entscheidende Rolle in der Zellzykluskontrolle zur Erhaltung der Integrität des Genoms und Erkennung sowie Beseitigung von DNA-Schäden. Unter normalen Bedingungen ist der Transkriptionsfaktor p53 nicht aktiv. Beim Auftreten eines DNA-Schadens oder anderer onkogener Stresssignale wird p53 mittels Checkpoint-Kinasen und DNA-abhängiger Proteinkinase phosphoryliert und damit aktiviert. Die Zelle wird entsprechend in G1 oder G2 zur DNA-Reparatur arretiert. Nach erfolgreicher Reparatur wird der Zellzyklus fortgesetzt; nach DISKUSSION 171 erfolgloser Reparatur wird in der Zelle die Apoptose eingeleitet. Da bei etwa der Hälfte aller Tumoren somatische Mutationen des TP53-Gens auftreten, kann mutiertes p53 seine zentrale Kontrollfunktion nicht erfüllen, so dass es trotz DNA-Schäden zur Teilung und Vermehrung mutierter Zellen kommt (Vousden und Lu 2002). Aufgrund der Heterogenität der p53 Mutationen sind Antikörperantworten leichter zu evaluieren als T-Zellantworten. Entsprechend gibt es zahlreiche Studien über immunologische Antworten gegen p53 in den verschiedensten Tumorentitäten. Antikörperantworten wurden in präoperativen Seren von 255 Kolonkarzinompatienten untersucht. Eine Häufigkeit von 25% wurde hier ermittelt, wobei das Auftreten von Antikörpern mit schlechter Prognose gerade bei Patienten in frühen Stadien, korrelierte (Houbiers et al. 1995). Antikörperantworten von ungefähr 30% in Kolonkarzinompatienten wurden ebenfalls von anderen Gruppen nachgewiesen (Angelopoulou et al. 1997; Hammel et al. 1997; Hammel et al. 1999). In einer Studie von Shimada et al. wurden 1085 Krebspatienten unterschiedlicher Entitäten auf Antikörper gegen p53 untersucht (Shimada et al. 2003). 20% aller Patienten hatten Antikörper gebildet. Häufigkeiten von 0% bis 32% abhängig von der Krebserkrankung wurden gemessen (Hals und Kopf 32%, Speiseröhre 30%, Kolon 24%, Gebärmutter 23%, Gebärmutterhals 19%, Brust 18%, Prostata 17%, Galle 17%, Lunge 14%, Blase 12%, Magen 11%, Pankreas 11%, Eierstöcke 7%, Gliom 6%, Leber 0%). In 5% der Kontrollpersonen wurden Antikörper gegen p53 identifiziert. Der Einsatz von Antikörpern gegen p53 als diagnostisches Werkzeug in Lungen- und Brustkrebs wird durch die folgenden Studien unterstrichen. Lubin et al. berichteten von zwei starken Rauchern, die eine Antikörperantwort gegen p53 ohne Anzeichen eines Lungentumors besaßen. Im Folgenden entwickelten beide Raucher p53-positive Lungenkarzinome, so dass Antikörper gegen p53 in diesem Zusammenhang als diagnostische Biomarker identifiziert wurden (Lubin et al. 1995). Allerdings konnte die Verwendung von p53 als diagnostischer und prognostischer Marker bei Lungenkrebs von Mitsudomi et al. nicht bestätigt werden (Mitsudomi et al. 1998). In einer Studie von Green et al. wurden Antikörper gegen p53 häufiger in Frauen (11%), die noch keinen Brustkrebs entwickelt jedoch eine familiäre Brustkrebsvorbelastung hatten, identifiziert als in Kontrollpersonen (1%) (Green et al. 1994). In Ovarialkarzinomen wurde p53 als prognostischer Biomarker identifiziert (Goodell et al. 2006). Goodell et al. ermittelten, dass das Gesamtüberleben signifikant höher für Patienten, die Antikörper gegen p53 gebildet hatten, war gegenüber Patienten ohne Antikörper gegen p53. Antikörper gegen p53 wurden in 6% der Patienten in Stadium I und II und in 30% der Patienten in Stadium III und IV gemessen (Goodell et al. 2006). Zusammenfassend kann festgestellt werden, dass Antikörperantworten gegen p53 mit einer Häufigkeit von zumeist niedriger als 35% in Krebspatienten auftreten. Die in dieser Arbeit gemessenen Antikörper-Häufigkeiten von 2-3% bei MM- und CTCL-Patienten liegen DISKUSSION 172 deutlich unter der Antikörper-Häufigkeit von Kolonkarzinom- oder Brustkrebspatienten. Dies könnte auf das weniger häufige Auftreten von Mutationen im TP53-Gen im MM und CTCL zurückzuführen sein. Die gemessene Antikörper-Häufigkeit von 3% in Ctrl lässt sich hingegen gut mit Werten aus der Literatur vergleichen. 4.3.4 Humorale Immunantworten von immunbehandelten CTCL-Patienten Neben der Untersuchung von Immunantworten in Patientenkollektiven ohne immunmodulatorische Vorbehandlung wurden Seren von immunbehandelten Patienten mittels Multiplex-Serologie unter Verwendung aller Antigene getestet. Pro Patient wurden ein Serum vor der Behandlung und mehrere Seren nach wiederholten Immunisierungen untersucht. In einer Impfstudie wurden 19 CTCL-Patienten mit einem rekombinanten Adenovirus immunisiert (Dummer et al. 2004). Die Adenoviridae besitzen ein doppelsträngiges, lineares Genom und infizieren bevorzugt Epithelzellen des Pharynx, des Dünndarmes sowie die Konjunktivalzellen. Der in der klinischen Studie unter der Leitung von Prof. Dummer verwendete rekombinante Adenovirus wurde mittels Klonierungstechnologie verändert. Das humane IFN-γ-Gen ist in die E1 Region des Adenovirus 5 integriert, so dass E1 und E3 deletiert sind und der Adenovirusvektor sich nicht unabhängig vermehren kann. Das vom Zytomegalo Virus (CMV)-Promoter gesteuerte IFN-γ-Gen wird mittels des Adenovirus in die humanen Zellen transfiziert. An der Injektionsstelle kommt es anschließend zu einer lokalen IFN-γ Expression. In dieser Impfstudie wurde der IFN-γ-exprimierende Adenovirus direkt in die Tumorläsion injiziert, um das Zytokinmilieu des Tumors positiv in Richtung einer T-Helferzelle 1 (TH1)-Typ Immunantwort zu beeinflussen. In einer zweiten Impfstudie wurden fünf CTCL-Patienten mit dem handelsüblichen Masernimpfstoff behandelt (Heinzerling et al. 2005). Das Masernvirus zählt zu den Paramyxoviridae und besitzt einzelsträngige RNA in Negativorientierung. Es infiziert Makrophagen, Endothelzellen, Mono- und Lymphozyten. Im Verlauf der akuten Erkrankung eliminieren vor allem CTL virusinfizierte Zellen. Zudem werden virusspezifische CD4+ TZellen vom TH2 Typ aktiviert. Diese sezernieren Zytokine, die Makrophagen und zusätzliche TZellen aktivieren und die Proliferation von B-Zellen induzieren. Der Masernimpfstoff stellt einen attenuierten Lebendimpfstoff dar, der vor allem CD4+ T-Zellen vom TH2-Typ aktiviert (Bedford 2003). Da das CTCL vorwiegend auf der Proliferation von TH2-Zellen beruht, besteht das Ziel dieser Immunisierungsstrategie in einer Virus-induzierten anti-Tumor Immunantwort. DISKUSSION 173 Bei der Betrachtung der Antikörperantworten in beiden Immuntherapiestudien ist festzustellen, dass unter Verwendung der bestimmten Cut-off-Werte sowohl bei den Patienten in der Adenovirusstudie als auch der Masernimpfstoffstudie ungefähr die Hälfte aller untersuchter Patienten keine Antikörper gegen die humanen Antigene aufwies. In der Adenovirusstudie traf dies für elf von 19 und in der Masernimpfstoffstudie für zwei von fünf Patienten zu. Insgesamt wurden hauptsächlich stabile Antikörperantworten über die Zeit gemessen. Sieben Patienten der Adenovirusstudie und drei Patienten der Masernimpfstoffstudie besaßen Antikörper gegen verschiedene Antigene vor und während der Studie. Antikörperantworten traten gegen alle humanen Antigene mit der Ausnahme von se70-2, se57-1 und p16 auf (siehe Tabelle 3-5 und Tabelle 3-6). Einzig für Patient #008 der Adenovirusstudie wurde eine Serokonversion anchgewiesen. Die Antikörper gegen p53 stiegen von 30 über 1539 bis auf 798 MFI an. Das klinische Bild dieses Patienten war durch eine partielle Regression gekennzeichnet. Für die drei Patienten #002, #006 und #009 wurde keine Serokonversion detektiert. Eine vollständige Regression wurde für diese drei Patienten nachgewiesen. Hingegen wurden gleichbleibende Antikörpertiter gegen GAGE7b für Patient #006 und #009 gemessen. Während der Erstellung dieser Arbeit waren nur für neun der 19 Patienten klinische Daten erhältlich. Mit dieser begrenzten Information konnte kein einheitlicher Effekt der adenoviralen Immuntherapie auf die humorale Immunantwort der Patienten der Adenovirusstudie erkannt werden. Dies bedeutet, dass die erhaltenen Ergebnisse wenig Aufschluss über systemische Effekte der Immunisierung geben. Ähnlich verhielt es sich ebenfalls für die Patienten der Masernimpfstoffstudie, für welche keine Serokonversion festgestellt wurde. Einzig Patient #0002 wies eine eindeutige Tendenz zum Anstieg verschiedener Serumantikörper auf. Die Antikörper gegen cTAGE-5a N-terminal (84 auf 408 MFI), CAMEL (1889 auf 2400 MFI) und GBP-5ta (1408 auf 1759 MFI) stiegen teilweise stark an. Jedoch lagen die Zunahmen entweder unterhalb des Cut-off-Wertes oder waren geringer als eine Verdopplung des MFI-Wertes. Trotz der nicht-Signifikanz dieser gemessenen Antikörperanstiege korreliert der beschriebene Trend mit dem Krankheitsverlauf des Patienten. Einzig dieser Patient hatte eine komplette Regression des mit dem Masernimpfstoff behandelten Plaques sowie ein Ansprechen einer weiteren Läsion, die im Anschluss an die Remission des ersten Plaques behandelt wurde. Für die vier anderen Patienten der Masernimpfstoffstudie wurde eine partielle Regression (#0001, #0003 und #0005) des behandelten Plaques oder nur ein geringes Ansprechen (#0004) beschrieben. Für eine zuverlässige und statistisch auswertbare Beurteilung der Antikörperantworten der untersuchten immunbehandelten CTCL-Patienten müsste eine größere Anzahl an Seren und DISKUSSION 174 Antigenen getestet werden. Jedoch ist es möglich, dass die Antikörperantwort keine Aussage über den immunmodulierenden Effekt zulässt, sondern die Immunantwort nur auf Ebene der CD8+ und/oder der CD4+ T-Zellen nachweisbar ist. 4.3.5 Vergleich von Multiplex-Serologie und SADA/SEREX Beim Vergleich der Ergebnisse für die humanen Antigene in der Multiplex-Serologie, im SADA und im SEREX (Tabelle 4-1) unterscheidet sich insbesondere die SEREX-Analyse gegenüber den Analysen mittels Multiplex-Serologie und SADA. Jedoch ist der Vergleich über alle Methoden und alle drei Kollektive nur für das Antigen cTAGE-5a möglich. Für alle anderen Antigene wurden nicht alle Methoden an allen Kollektiven durchgeführt. Die Antigene MAGEA1, LAGE-1a, CAMEL, se70-2, p53 und p16 wurden nicht im SADA eingesetzt. Im SEREX wurden die Antigene CAMEL, p53 und p16 nicht und die Antigene MAGE-A1, A3, A9, LAGE1a, GAGE-7b, se70-2, se57-1 und GBP-5ta nur an ein bis zwei Patientenkollektiven untersucht. Dies ist vor allem darauf zurückzuführen, dass SEREX eine aufwendige Methode ist und gerade für die CTA das Kontroll-Kollektiv häufig nicht untersucht wurde aufgrund der ausschließlichen Expression der CTA in der immunprivilegierten Testis. Es ist kein Vergleich der drei Methoden für die Antigene CAMEL, p53 und p16 möglich. Einschränkend muss zudem erwähnt werden, dass der im SADA und SEREX verwendete GAGE-t Phagenklon eine verkürzte Variante des Gens GAGE-3 ist. Da jedoch GAGE-3 bis –7b für dasselbe Protein kodieren, sollten Antikörperantworten gegen diese Proteine ähnlich sein. Während in der Multiplex-Serologie 0 - 6% der Kontrollseren (n = 100) und im SADA 0 – 2% (MAGE-A3, A9, GAGE-7b, se57-1, GBP-5ta, cTAGE-5a, n = 100) positiv waren, reagierte im SEREX kein Kontrollserum (n = 5 – 14) gegen die im SEREX untersuchten Antigene se70-2, se57-1, GBP-5ta und cTAGE-5a. Das heißt, dass alle im SEREX an Kontrollseren untersuchten Antigene serologisch tumorspezifisch waren, dies jedoch nicht in den Analysen mittels Multiplex-Serologie und SADA bestätigt werden konnte. Im SADA wurden in keinem Kollektiv Antikörper gegen cTAGE-5a gemessen. Hingegen wurden im SEREX Antikörper in 15% der CTCL-Patienten (n = 20) identifiziert. Keine Antikörper gegen cTAGE-5a wurden im SEREX gegen MM-Patienten (n = 15) und Ctrl (n = 14) detektiert. Mittels Multiplex-Serologie wurden Antikörper-Häufigkeiten zwischen 0,5 und 2,8% gegen cTAGE-5a in Ctrl, MM- und CTCL-Patienten gemessen, die jedoch bedeutend geringer sind als im SEREX. Ein entscheidender Unterschied könnte die Größe der Kollektive sein und die Unterschiedlichkeit der Blutspender in den Kollektiven, die jeweils für SADA, SEREX und Multiplex-Serologie verwendet wurden. DISKUSSION 175 Im Vergleich der Antikörperantworten gegen die CTA ergaben sich insbesondere im Vergleich von SEREX und Multiplex-Serologie deutliche Unterschiede. 27% der MM-Patienten besaßen laut SEREX Antikörper gegen MAGE-A1, hingegen nur 7,3% laut Multiplex-Serologie. Deutliche Unterschiede im Vergleich der Antikörperantworten im MM-Kollektiv ergaben sich ebenfalls für die CTA MAGE-A3 (SEREX: 27%, Multiplex: 8,4%, SADA: 0%), MAGE-A9 (SEREX: 20%, Multiplex: 4,2%, SADA: 0%) und se57-1 (SADA: 11%, Multiplex: 3,7%). Vergleichbare Antikörperantworten wurden im MM-Kollektiv für die Antigene LAGE-1a (SEREX: 13%, Multiplex: 13,6%) und GAGE-7b (SADA: 6%, Multiplex: 5,8%) nachgewiesen. Ähnliche Antikörperantworten gegen MAGE-A1 wurden für CTCL-Patienten im SEREX mit 6% und in der Multiplex-Serologie mit 10,4% bestimmt. Ebenfalls vergleichbare Ergebnisse wurden für MAGE-A3 (SEREX: 6%, SADA: 8%, Multiplex: 4,9%) und LAGE-1a (SEREX: 0%, Multiplex: 4,9%) im CTCL-Kollektiv gemessen. Unterschiede bei der Betrachtung des CTCL-Kollektivs waren für die Antigene MAGE-A9 (SEREX: 22%, SADA: 4%, Multiplex: 4,9%), GAGE-7b (SEREX: 16%, SADA: 3%, Multiplex: 2,8%), se70-2 (SEREX: 16%, Multiplex: 0,7%) und se57-1 (SEREX: 33%, SADA: 5%, Multiplex: 0,7%) ersichtlich. Große Unterschiede in der Antikörperantwort je Methode wurden für das Antigen GBP-5ta getestet. Während im SADA 28% der MM-Patienten Antikörper aufwiesen, waren in der Multiplex-Serologie nur 3,1% der MM-Patienten Antikörper-positiv. CTCL-Patienten hatten laut SEREX sogar zu 56% Antikörper gegen GBP-5ta gebildet, wohingegen im SADA nur 5% und in der Multiplex-Serologie nur 3,5% der CTCL-Patienten Antikörper besaßen. DISKUSSION Tabelle 4-1 176 Vergleich der Seroreaktivitäten von Ctrl, MM- und CTCL-Patienten in der Multiplex-Serologie, im SADA und SEREX Die Tabelle stellt die Ergebnisse der serologischen Untersuchungen mittels Multiplex-Serologie (Multi), SADA (Serumantikörper-Detektionsarray) und sekundärem „serological analysis of recombinantly expressed cDNA clones“ (SEREX) mit allen humanen Antigenen dar, die in dieser Arbeit verwendet wurden (Eichmuller et al. 2001; Fellenberg et al. 2004). Untersucht wurden Seren von kutanes T-Zell Lymphom (CTCL)-, malignes Melanom (MM)-Patienten und Kontrollen (Ctrl). *Im SADA und SEREX wurde nur ein Klon für cTAGE-5a verwendet. Aufgrund der Proteingröße musste dieser Klon in zwei Teilen kloniert werden, so dass in der Multiplex-Serologie zwei Glutathion-S-Transferase (GST)Fusionsproteine eingesetzt wurden. n: Anzahl, n.g.: nicht getestet. % Positive (n) Methode Multi Tumorantigen Kontrollseren MM-Seren CTCL-Seren MM-Seren CTCL-Seren 6,0 (100) 7,3 (191) 10,4 (144) MM-Seren CTCL-Seren Tumorantigen Kontrollseren MM-Seren CTCL-Seren MM-Seren CTCL-Seren n.g. n.g. n.g. n.g. n.g. 3,0 (100) 0,0 (191) 0,7 (144) n.g. n.g. n.g. SEREX n.g. 27 (15) 6 (18) n.g. 13 (15) 0 (19) 1,0 (100) 5,8 (191) 2,8 (144) GAGE-7b 1 (100) 6 (18) 3 (100) 1,0 (100) 3,7 (191) 0,7 (144) se57-1 2 (100) 11 (18) 5 (100) 0 (14) n.g. 16 (19) cTAGE-5a N-terminal* 2,0 0 0 (100) (100) (14) 2,6 0 0 (191) (18) (15) 2,8 0 15 (144) (100) (20) p16 2,0 (100) 2,1 (191) 0,0 (144) SADA MAGE-A3 5,0 1 (100) (100) n.g. 8,4 0 27 (191) (18) (15) 4,9 8 6 (144) (100) (18) se70-2 Tumorantigen Kontrollseren n.g. Multi LAGE-1a 3,0 (100) 13,6 (191) 4,9 (144) Tumorantigen Kontrollseren SEREX MAGE-A1 Tumorantigen Kontrollseren SADA n.g. n.g. n.g. n.g. n.g. n.g. Multi SADA SEREX MAGE-A9 2,0 0 (100) (100) n.g. 4,2 0 20 (191) (18) (15) 4,9 4 22 (144) (100) (18) CAMEL n.g. n.g. 16 (19) 0 (5) 0,0 (100) 1,0 (191) 2,8 (144) n.g. 33 (9) 3,0 (100) 3,1 (191) 3,5 (144) cTAGE-5a C-terminal* 2,0 0 0 (100) (100) (14) 0,5 0 0 (191) (18) (15) 0,7 0 15 (144) (100) (20) 3,0 (100) 2,1 (191) 2,1 (144) n.g. n.g. n.g. n.g. n.g. n.g. GBP-5ta 0 (100) 28 (18) 5 (100) 0 (14) n.g. 56 (16) p53 n.g. n.g. n.g. n.g. n.g. n.g. DISKUSSION 177 Um die dargestellten Unterschiede zwischen den Multiplex-Serologie-, SADA- und SEREXErgebnissen erklären zu können, werden nachfolgend die Komponenten „Protein“, „Serum“ und „Auswertung“ der Methoden miteinander verglichen. Mögliche Unterschiede zwischen SADA und SEREX wurden bereits unter 4.2.4 diskutiert. Im SEREX und SADA liegt die kodierende Region zusammen mit der „untranslated region“ (UTR) des Antigens im pBluescript-Vektor vor. Die genaue Translation der Proteine ist unbekannt. Es ist somit möglich, dass neben dem „open reading frame“ (ORF) auch kleine Proteinfragmente der UTR translatiert werden können, die immunogen sind. Für die MultiplexSerologie wurde die kodierende Sequenz im Leseraster in den Expressionsvektor pGEX-4T-3tag kloniert. Laut Western Blot Analyse wurde ein definiertes Protein exprimiert. Dieses Protein ist N-terminal durch das 26 kilo Dalton (kDa) große GST-Protein verlängert. Eventuell könnten immunogene Epitope durch das GST-Fusionsprotein sterisch behindert werden. Dies könnte gerade für kleine Proteine (< 20 kDa) zutreffen. Ein weiterer möglicher Grund für die höheren Reaktivitäten im SEREX im Vergleich zur Multiplex-Serologie, könnte in der Nativität des Proteins liegen. Während das Antigen im SEREX nativ vorliegt, ist das Antigen in der Multiplex-Serologie durch den Bakterienaufschluß in der French Press hohen Scherkräften ausgesetzt, die Einfluss auf die Konformation des Proteins haben könnten. Zudem unterscheidet sich die Art der Blockierung von unspezifischen Bindungen. Im SEREX/SADA wird die geblottete Membran mit einer Milchpulverlösung inkubiert, damit unspezifische Bindungsstellen abgesättigt werden. In der Multiplex-Serologie hingegen, wird dem Seruminkubationspuffer eine Mischung aus Polyvinylalkohol, Polyvinylpyrrolidon und Superchemiclock zugesetzt. Dadurch könnte ein Unterschied im Zustand des Antigens im SEREX/SADA gegenüber der MultiplexSerologie bedingt sein. Im SEREX/SADA sind die Seren 1:100 verdünnt und aufwendig gegen E. coli- und Phagenproteine präabsorbiert. In der Multiplex-Serologie werden die Seren in der Verdünnung 1:50 eingesetzt und 1h mit 2mg/mL E. coli-Lysat bei Raumtemperatur (Rt) deutlich kürzer präabsorbiert. Beim Vergleich der verwendeten Seren lässt sich feststellen, dass im SEREX meist Seren aus späteren Krankheitsstadien untersucht wurden, die meist höhere Antikörpertiter besitzen (Talpur et al. 2002). Sowohl in der Multiplex-Serologie als auch im SADA wurde hingegen eine wesentlich größere Anzahl an zufällig zusammengestellten Seren aus allen Krankheitsstadien und Altersgruppen getestet, welches die geringere Anzahl an positiven Seren erklären könnte (Abendstein et al. 2000). Unterschiede in der Auswertung bestehen zwischen SEREX/SADA und Multiplex-Serologie. Die SEREX/SADA-Analyse erfolgt visuell durch den Vergleich positiver und negativer Plaques DISKUSSION 178 auf der jeweiligen Membran. Die Ergebnisermittlung in der Multiplex-Serologie findet im Luminex-Gerät objektiv unter Verwendung des Reporter-Lasers statt. Der Antigen-spezifische Cut-off-Wert entscheidet über die Positivität eines Serums (siehe 2.6.5.6). Die visuelle Analyse bei SEREX/SADA birgt mehr Fehlerquellen in sich durch die subjektive Sichtweise des Bearbeiters (Verhältnis der positiven zu den negativen Plaques, ungleiche Entwicklung etc.) als die Cut-off-Bewertung. Andererseits ist die Festlegung eines Cut-off-Wertes für die untersuchten Autoantigene ebenfalls diskussionsfähig. Da die Reaktivitäten gegen Autoantigene im Vergleich zu Reaktivitäten gegen z.B. virale Antigene sehr niedrig sind, ist es fraglich, ob die berechneten Cut-off-Werte nicht zu hoch gewählt wurden und damit reaktive Seren übersehen werden. Dies könnte für cTAGE-5a der Fall sein, da hier sehr hohe Cut-off-Werte berechnet und ausgewählt wurden (1781 und 2564 MFI). Schließlich stellten Lagarkova et al. die These auf, dass SEREX sensitiver als ein wenn auch einfacherer und robusterer ELISA (somit letztlich auch Multiplex-Serologie) sei (Lagarkova et al. 2003). Demnach wäre die SEREX-Methode besser für schwach immunogene Antigene geeignet, im Gegensatz zum ELISA, der eine ideale Methode für immunogene Antigene darstellt. 4.3.6 Multiplex-Serologie im Vergleich zu ELISA in der Krebsdiagnostik Die Multiplex-Serologie kombiniert ein gut etabliertes Proteinantigensystem inklusive in situ Affinitätsaufreinigung mit einem Hochdurchsatz-Suspensionsarray. Die eindeutigen Vorteile dieser Methode gegenüber dem konventionellen ELISA sind bedeutend geringerer Zeitaufwand, weniger Normalisierungsaufwand, Durchführung von großen komplexen seroepidemiologischen Studien in kurzer Zeit und Serumersparnis. In Studien von Waterboer et al. wurde zudem nachgewiesen, dass die Multiplex-Serologie für HPV 16 E7 sensitiver war als der GST „capture“ ELISA (Waterboer 2005; Waterboer et al. 2005). In einer Studie von Komatsu et al. wurde das Luminex-System auf seine Verwendung zur Detektion von Antikörpern gegen Peptide der Proteine „Squamous cell carcinoma antigen recognized by T-cells“ (SART), Lymphozytenprotein Tyrosinkinase (Lck) und Adenosindiphosphat-Ribosyltransferase (ART) untersucht (Komatsu et al. 2004). Dieses System soll laut den Autoren bei Vakzinierungsstudien einsetzbar sein. Eine ähnliche Sensitivität wie bereits vorhandene ELISA-Systeme konnte ermitelt werden. Auch hier überwiegen die Vorteile der Luminex-Technologie gegenüber dem ELISA-System. Andere zwischenzeitlich verwendete Ansätze, die jedoch hauptsächlich der Identifizierung von Antigenen dienen, beinhalten 2D Western Blot Analysen (Le Naour et al. 2001) und Protein Microarrays (Qiu et al. 2004). Hier handelt es sich jedoch um planare DISKUSSION 179 Systeme, die teilweise sehr aufwändig sind. Anders als Infektionen, bei denen Patienten vor allem auf ein immundominantes Antigen reagieren, besteht Krebs aus einer Mischung von verschiedenen Antigenen. Deshalb bilden Krebspatienten zumeist Antikörper gegen verschiedene Antigene, wobei ein Antigen jedoch nicht in allen Tumoren exprimiert wird. Somit liegen die meisten Antikörperantworten gegen Selbstantigene um die 15-20%. Entsprechend ist es äußerst wichtig und interessant in zukünftigen Analysen den Multiplex-Ansatz zu verwenden, um die Antikörperantworten gegen mehrere Antigene gleichzeitig bestimmen zu können. Dies erhöht die Sensitivität und Spezifität der Analysen deutlich (Anderson und LaBaer 2005). Die bislang entwickelten ELISA, die Antikörperantworten gegen Tumorantigene in Krebspatienten analysierten, wiesen keine hohen Antikörperhäufigkeiten nach. Stockert et al. untersuchten Antikörper gegen die Tumorantigene NY-ESO-1, MAGE-1, 3, MelanA, Tyrosinase und SSX in 234 Seren von Krebspatienten und 70 Ctrl (Stockert et al. 1998). NY-ESO-1 bildete die häufigsten Antikörperantworten mit 9,4% (12/127) der MM-, 12,5% (4/32) der Ovarialkarzinom-, 4,2% (1/24) der Lungenkrebsseren und 7,7% (2/26) der Seren von Brustkrebspatienten (Stockert et al. 1998). Kein Patient war seropositiv für MelanA und Tyrosinase. Drei Patienten besaßen Antikörper gegen MAGE-1, zwei gegen MAGE-3 und einer gegen SSX2. Damit wurden Reaktivitäten gegen NY-ESO-1 in MM-Patienten signifikant häufiger nachgewiesen als gegen die MAGE-Antigene, obwohl die Expression der MAGEAntigene in MM-Gewebe höher ist. Vergleichbar mit den Multiplex-Serologie Ergebnissen dieser Arbeit liegen gegen die untersuchten Tumorantigene nur schwache und niedrig frequente humorale Immunantworten vor. Ähnlich niedrige Antikörperantworten wurden gegen das CTA OY-TES-1 in epithelialen Ovarialkarzinompatienten (10%) und gegen TA90 (12%) in einer MM-Patientengruppe gemessen (Litvak et al. 2004; Tammela et al. 2006). Litvak et al. identifizierten IgM Antikörper gegen TA90 in 12% der MM-Patienten, die nach einer operativen Tumorentfernung in Stadium Ib und IIa innerhalb von sieben Jahren an Stadium IV Symptomen starben, und in 62% der MM-Patienten, die innerhalb von 10 Jahren keine Krankheitssymptome entwickelten (Litvak et al. 2004). IgG Antikörper wurden in 74% beider Patientengruppen ermittelt. Zhang et al. untersuchten Antikörperantworten in 527 Krebspatienten gegen ein Panel von sieben Tumorantigenen: c-myc, p53, Cyclin B1, p62, Koc, Insulin-ähnlicher Wachstumsfaktor 2 mRNA Bindungsprotein 1 (IMP1) und Survivin (Zhang et al. 2003). Die Antikörper-Häufigkeit gegen einzelne dieser Antigene in den sechs untersuchten Tumorentitäten (Brust-, Lungen-, Kolon-, Magen-, Prostata- und Leberkrebs) lag zumeist unter 20%. Bei der Betrachtung des kompletten Antigenpanels erhöhte sich die Antikörperhäufigkeit auf 44-68%. Des Weiteren besaßen Brust-, Lungen- und Prostatakrebspatienten ein charakteristisches DISKUSSION 180 Reaktivitätsprofil (Zhang et al. 2003). Höhere Häufigkeiten von Antikörperantworten konnten gegen die extrazelluläre Proteinkinase A in verschiedenen Tumorentitäten (90%) und in Ctrl (12%) gemessen werden (Nesterova et al. 2006). 4.3.7 Multiplex-Serologie – Fazit und Ausblick Aufgrund der erzielten Ergebnisse ist festzuhalten, dass die verwendeten Antigene für die Multiplex-Serologie zumindest bedingt geeignet sind und eindeutig Antikörperantworten gegen Autoantigene detektiert werden können. Da die Multiplex-Serologie die Messung von bis zu 100 verschiedenen Antigenen mit einem Serum zulässt, ist eine Eignung für die klinische Routine generell möglich und empfehlenswert. Die Methode benötigt sehr wenig Serum, lässt sich innerhalb von ein bis zwei Tagen durchführen und benötigt aufgrund der Messung von 100 Beads pro Antigen und der Mitführung von internen Ctrl keine Wiederholung der Untersuchung. Dies bedeutet, dass pro Tag 100 Antigene an 1000 Seren gemessen werden können. Die Festlegung von sinnvollen Cut-off-Werten für Autoantigene ist notwendig. In dieser Arbeit wurden die Antikörperantworten von MM- und CTCL-Patientenkollektiven gegenüber Ctrl untersucht. In den MM-Patienten erzeugte einzig LAGE-1a eine eindeutig stadienabhängige Induktion von Antikörpern in den Patienten, die besonders in Stadium IV ausgeprägt war. Somit könnte LAGE-1a das erste CTA darstellen, das in der MultiplexSerologie als diagnostisches und prognostisches humanes Tumorantigen für MM-Patienten nützlich sein und in der Klinik Anwendung finden könnte. Für das CTCL-Kollektiv wurde kein Antigen identifiziert, das stadienabhängig Antikörperantworten hervorruft. Nur CTCL-Patienten des Stadiums IV besaßen statistisch signifikant häufiger Antikörper gegen MAGE-A9 gegenüber Ctrl. Die Untersuchung von Serumverläufen in MM- und CTCL-Patienten ergab bei einem MMPatienten eine Korrelation von Progression und Zunahme an LAGE-1a Antikörpern sowie bei einem CTCL-Patienten von Progression und Zunahme an cTAGE5a C-terminal Antikörpern. Weitere Untersuchungen von Serumverläufen wären in diesem Zusammenhang angebracht. Bei der Betrachtung der Antikörperantworten in immunstimulierten CTCL-Patienten war festzustellen, dass kaum Antikörperantworten gegen die untersuchten Antigene gebildet wurden. Nur in einem Patienten wurde eine Serokonversion für das Antigen p53 identifiziert. Diese Serokonversion korrelierte mit einer partiellen Regression des Patienten. Im Allgemeinen waren die Antikörperhäufigkeiten mit zumeist unter 10% eher selten. Nur das CTA CAMEL wies eine immunologische Tumorspezifität auf. Schließlich erzeugten die verwendeten Tumorantigene zumeist eindeutig niedrigere Serumantikörper-Häufigkeiten als virale Antigene. Eine Erklärung DISKUSSION 181 für diese Ergebnisse liegt in der Natur der Antigene selbst, da virale Antigene vom Körper als fremd, Tumorantigene jedoch als körpereigen angesehen werden. Die Untersuchungen von Antikörperantworten gegen humane Tumorantigene mittels MultiplexSerologie befinden sich noch in den Anfängen. In Zukunft sollten noch mehr Tumorantigene in diesem System getestet werden, um ein relevantes und optimales Antigenpanel zusammenstellen zu können. Nützliche und sinnvolle Folgeanalysen könnten folgende MM- und CTCL-Kollektive beinhalten: IFN-γ behandelte Patienten, Patienten aus Studien mit verschiedenen Tumorantigenen z.B. MAGE-Antigene, LAGE-1a und/oder NY-ESO-1, weitere Verläufe von Patienten sowie Verläufe von Ctrl als Vergleich und Patienten nach einer operativen Entfernung des Tumors mit und ohne Rezidiv in Verbindung mit der Untersuchung der Antigenexpression im Tumor. 4.4 Das Cancer Testis Antigen GAGE-7b Eine Voraussetzung für eine erfolgreiche Entwicklung von Antigen-spezifischen Tumortherapien ist eine eingehende Charakterisierung der verwendeten Antigene und ihrer Funktion. Spezifische Funktionen konnten bisher nur wenigen Tumorantigenen zugeordnet werden wie z.B. SCP-1 (Meuwissen et al. 1997) und OY-TES-1 (Baba et al. 1994). Die CTA stellen eine wichtige Gruppe an Antigenen dar, die vom Immunsystem auf Krebszellen erkannt werden können. Zu den CTA zählt unter anderem die Familie der GAGE-Antigene, die wie viele der CTA auf dem X-Chromosom lokalisiert sind. Die GAGE-Gene wurden in den 90er Jahren identifiziert (Van den Eynde et al. 1995; De Backer et al. 1999). Die GAGE-Familie bestand ursprünglich aus neun Mitgliedern, die drei Gruppen zugeordnet werden konnten. Mittlerweile sind 16 Familienmitglieder bekannt (Gjerstorff und Ditzel, 2008). GAGE-2 bis 7, 7b und 8 weisen eine Proteinhomologie von 95% auf, wohingegen GAGE-1 gegenüber den anderen GAGE-Proteinen eine Homologie von 80% besitzt. HLA-A- und -C-präsentierte Epitope wurden bereits für GAGE beschrieben (Van den Eynde et al. 1995; De Backer et al. 1999). Nah verwandte Gene zu GAGE sind Prostatakarzinom-assoziiertes Antigen (PAGE) und XAGE. Beide Antigenfamilien wurden mittels differentieller Expressionsanalysen an einem Prostatakarzinom Modell, mittels Computeranalysen von Prostata „expressed sequence tags“ (ESTs) und Krebs-assoziierter ESTs identifiziert (Brinkmann et al. 1998; Chen et al. 1998; Vasmatzis et al. 1998; Brinkmann et al. 1999). Die GAGE-, PAGE- und XAGE-Antigene sind durch eine Krebs-spezifische Expression gekennzeichnet. Alle Antigene sind nicht auf eine Tumorentität beschränkt. DISKUSSION 182 In dieser Arbeit wurde die Expression des Antigens GAGE-7b im MM und CTCL auf Proteinebene unter Verwendung eines eigens zu diesem Zweck generierten Antikörpers in Zelllinien und Tumorgewebe näher charakterisiert. Mittels dieser Untersuchungen sollte schließlich der Wert dieses CTAs für Immuntherapien beim MM und CTCL abgeschätzt werden. 4.4.1 Spezifität des Antikörpers Drei unterschiedliche Ansätze wurden verwendet, um die Spezifität des GAGE-7b-Antikörpers zu testen. Durch diese ausführliche und aussagekräftige Analyse des Antikörpers konnte seine Spezifität zweifelsfrei belegt werden. Anhand der Analyse des Antikörpers mit verschiedenen Methoden wurde festgestellt, dass der Antikörper sowohl denaturiertes als auch natives und rekombinantes sowie Wildtyp-Protein erkennt. Antikörperkonzentrationen von 1 – 2µg/mL wurden eingesetzt. Zunächst wurde der aufgereinigte Antikörper in einer Konzentration von 1µg/mL im denaturierenden Western Blot gegen verschiedene GST-Fusionsproteine getestet. Dabei konnte eine eindeutige Reaktion des Antikörpers gegen das GST-GAGE-7b-tag-, das GAGE-7b-tagsowie das verkürzte GAGE-7b-tag-Protein analysiert werden. Allerdings wurde ebenfalls ein Restgehalt an tag-Antikörpern und Antikörpern gegen E. coli-Proteine wie Hsp70 festgestellt. Die Anwesenheit dieser Antikörper neben den GAGE-7b-spezifischen Antikörpern ist jedoch unproblematisch, da der GAGE-7b-Antikörper in Untersuchungen an humanem Material Anwendung findet. Hier spielen E. coli- und SV40-Virus-Proteine keine Rolle. Neben der Untersuchung an GST-Fusionsproteinen wurde der Antikörper ebenfalls an einer Verdünnungsreihe des aufgereinigten GAGE-7b-tag-Proteins im Western Blot getestet. Im Western Blot betrug die Nachweisgrenze von GAGE-7b-Protein 15ng. In immunzytologischen Untersuchungen wurde GAGE-7b im Cytosol der Zellen detektiert. Innerhalb Zellen einer Zelllinie wurde eine heterogene GAGE-7b-Expression nachgewiesen. Da diese Zelllinien aus Biopsiematerial eines Tumorpatienten stammen, ist das Auftreten von verschiedenen Zellklonen mit unterschiedlichen Antigenexpressionsmustern möglich. Die Inkubation von aufgereinigtem GAGE-7b-Antikörper und aufgereinigtem GAGE-7b-tag-Protein führte zur Bindung von Antigen und Antikörper, wodurch die Färbung einer GAGE-7b-positiven Zelllinie vollständig verhindert wurde. Dies ist ein eindeutiger Nachweis der Spezifität des Antikörpers. Zudem reagierte das Präimmunserum des immunisierten Kaninchens bei einer Verdünnung von 1:100 nicht mit einer GAGE-7b-positiven Zelllinie. Schließlich konnte die Spezifität des GAGE-7b-Antikörpers ebenfalls in der Multiplex-Serologie nachgewiesen werden. Der Antikörper (2µg/mL) wurde parallel gegen 17 GST-Fusionsproteine DISKUSSION 183 getestet. Da in diesen Analysen zusätzlich E. coli- und GST-tag-Antikörper aus dem zu untersuchenden Material entfernt werden, ist diese Methode vermutlich am aussagekräftigsten hinsichtlich der Spezifität des GAGE-7b-Antikörpers. Die Reaktion des GAGE-7b-Antikörpers mit den GST-GAGE-7b-tag-Beads betrug 1597 MFI und ist somit spezifisch für das GAGE-7bAntigen. Andere CTA und TAA sowie Tumorsuppressorgene wurden nicht erkannt. Beim Vergleich der Untersuchungen der GAGE-7b-Expression auf mRNA- und Proteinebene wurde festgestellt, dass der generierte Antikörper vermutlich zumindest die Antigene GAGE-3 bis 7b erkennt. Dies ist auf die hohe Homologie von über 95% dieser Proteine zurückzuführen. Deshalb impliziert eine Expression von GAGE-7b auf Proteinebene im folgenden stets die Expression aller GAGE-Antigene (Bazhin et al. 2007). 4.4.2 Expressionsanalyse von GAGE-7b Zunächst wurde die Expression des Antigens GAGE-7b auf mRNA-Ebene mittels RT-PCR in Kontroll-, CTCL- und MM-Zelllinien sowie MM-Gewebe untersucht. Hierbei ließ die Auswahl an möglichen Primersequenzen nur eine Unterscheidung zwischen GAGE-1, 2, 8 und GAGE-3 bis 7b zu (De Backer et al. 1999). Wie bereits durch in der Literatur beschriebene Untersuchungen erwartet, wurde keine Expression von GAGE-Antigenen in den beiden Kontrollzelllinien (Melanozyten und HaCat) nachgewiesen. Die CTCL-Zelllinien waren ebenfalls GAGE-negativ. Die GAGE-Expression der MM-Zelllinien und –Gewebe betrug bis zu 82%. GAGE-1, 2 und 8-Expression wurde zu 71% in MM-Zelllinien und zu 38% in MMGewebe gemessen. GAGE-3 bis 7b wurde in 82% der MM-Zelllinien und 50% der MM-Gewebe nachgewiesen. Ähnlich hohe Expressionen sind bereits in der Literatur beschrieben. Van den Eynde et al. wiesen GAGE-1 bis 6 in keiner Lymphomzelllinie (n=6), jedoch in 61% der MMZelllinien (n=74), in 13% der primären (n=39) und in 36% der metastasierten MM-Gewebe nach. GAGE-1 und 2 wurden in 13% der primären und 28% der metastasierten MM-Gewebe sowie in 54% der MM-Zelllinien (n=74) gemessen (Van den Eynde et al. 1995). De Backer et al. konnten eine GAGE-1, 2, 8- und/oder GAGE-3 bis 7b-Expression in 42% primärer kutaner MM (n=79) und 54% metastasierter kutaner MM (n=211) detektieren (De Backer et al. 1999). Häffner et al. untersuchten die GAGE-Expression in 41 CTCL-Geweben, wobei GAGE-mRNA lediglich für einen Sézary Syndrom Patienten nachgewiesen werden konnte (Haffner et al. 2002). Somit ist festzustellen, dass GAGE-Gene häufig in MM-Zelllinien und –Geweben, jedoch eher selten in CTCL-Zelllinien und –Geweben exprimiert sind. Damit beschränkt sich die Relevanz von GAGE auf mRNA-Ebene auf das MM. DISKUSSION 184 Zur Untersuchung der Expression von GAGE-7b auf Proteinebene wurden Western Blot Analysen unter Verwendung des innerhalb dieser Arbeit generierten monospezifischen Antikörpers durchgeführt. Zunächst konnte die CTA-spezifische Expression von GAGE-7b in Testis anhand der Untersuchung von 15 verschiedenen Kontrollgeweben auch auf Proteinebene bestätigt werden. Die spezifische Bande für GAGE-7b bei 26kDa wurde ausschließlich im Testisgewebe detektiert. Die untersuchten 24 Zelllinien hatten unterschiedliche GAGE-7b-Profile. Die MM-Zelllinien waren zu mehr als 50% (10/17) GAGE-7b-positiv, wohingegen keine Kontrollzelllinie (HaCat und Melanozyten) und keine CTCL-Zelllinie (n=5) GAGE-7b-positiv war. Dies unterstreicht die Ergebnisse auf mRNA-Ebene dahingehend, dass GAGE-7b auch auf Proteinebene für das MM interessant zu sein scheint, hingegen jedoch keine Rolle im CTCL spielt. In 22% der untersuchten MM-Tumorgewebe (2/9) wurde GAGE-7b nachgewiesen. Die GAGE7b-positiven Tumorgewebe stammen von zwei Patienten, aus deren Tumorgewebe ebenfalls GAGE-7b-positive Zelllinien etabliert werden konnten. Ein Vergleich der GAGE-7b-Expression zwischen den Tumorgeweben und zugehörigen Zelllinien war für vier weitere MM-Patienten möglich. Das Tumorgewebe wie auch die etablierte Zelllinie eines der vier MM-Patienten waren GAGE-7b-negativ. Weitere zwei MM-Patienten exprimierten GAGE-7b in Tumorgewebe und Zelllinie auf mRNA-Ebene jedoch nicht auf Proteinebene. Ein letzter Patient war GAGE-7bpositiv hinsichtlich Expression auf mRNA-Ebene und auf Proteinebene in der Zelllinie und GAGE-7b-negativ auf Proteinebene im Tumorgewebe. Unterschiede der Expression auf mRNAund Proteinebene könnten auf eine fehlende Translation der mRNA zurückzuführen sein. Unterschiede der Expression zwischen aus Tumorgewebe etablierten Zelllinien und Tumorgewebe selbst könnten auf die Heterogenität des Tumors zurückzuführen sein. Es ist durchaus möglich, dass ein Teil des Tumorgewebes CTA-positiv auf Proteinebene ist, ein anderer Teil des Tumors jedoch nicht. Immunzytologische Untersuchungen wurden an 22 verschiedenen Zellllinien durchgeführt. Melanozyten, die als Kontrollzelllinie dienten, waren eindeutig GAGE-7b-negativ. Gerade für einen Einsatz von GAGE als mögliches Immuntherapeutikum ist diese Tatsache entscheidend, da keine Normalgewebe in der Haut durch einen Therapieansatz mit der Zielstruktur GAGE geschädigt würden. Wie bereits in den Untersuchungen mittels Western Blot wurde auch in der Immunzytologie keine GAGE-7b-positive CTCL-Zelllinie (n=5) identifiziert. Entsprechend scheint der Einsatz von GAGE als Immuntherapeutikum im CTCL fraglich. Während dieser DISKUSSION 185 Arbeit standen keine Gewebe von CTCL-Patienten zur Verfügung, um diese Frage eingehender an Tumormaterial zu evaluieren. Knapp 44% der MM-Zelllinien (7/16) waren GAGE-7b-positiv. Die GAGE-7b-Expression war zytoplasmatisch lokalisiert und erwies sich als heterogen. Dies bedeutet, dass sowohl GAGE-7b-positive als auch GAGE-7b-negative Zellklone in einer immunzytologischen Analyse vorhanden waren. Beispiele hierfür liegen auch in der Literatur vor (Akcakanat et al. 2006). Alle MM-Zelllinien, die in der Immunzytologie positiv mit dem Antikörper reagierten, waren ebenfalls im Western Blot positiv. Eine Ausnahme hierzu bildete die Zelllinie Ma-Mel-12. Diese Zelllinie war GAGE-3 bis 7b-positiv in RT-PCRUntersuchungen, negativ in Western Blot Untersuchungen an der Zelllinie und an Tumorgewebe sowie positiv in immunzytologischen Untersuchungen. Diese unterschiedlichen Ergebnisse könnten darauf zurückzuführen sein, dass im denaturierenden Western Blot einige Epitope durch den Antikörper nicht erkannt, jedoch in der Immunzytologie detektieren werden können. Eine andere Möglichkeit wäre, dass die GAGE-7b-Expression sehr heterogen in diesem Tumorpatienten war. Entsprechend ließ sich zwar eine GAGE-7b-positive Zelllinie etablieren. Zur Aufarbeitung des Tumorgewebes selbst jedoch wurde Gewebe aus einer anderen Region des Tumors verwendet, welches eine bedeutend geringere GAGE-7b-Expression aufwies, die im Western Blot nicht mehr nachweisbar ist. Neben der Analyse der GAGE-7b-Expression auf Proteinebene an Zelllinien wurden Tumorgewebe von MM-Patienten untersucht. Die Hälfte der Paraffingewebeschnitte war GAGE-7b-positiv (21/40). Allerdings wurden GAGE-7b-positive Zellen nur vereinzelt oder in Zellhaufen nachgewiesen. Dieses Expressionsverhalten ähnelt der heterogenen Expression von GAGE-7b in den MM-Zelllinien. Die GAGE-7b-Negativität der Melanozyten als Zellen der Haut wurde durch Untersuchungen an normaler Haut bestätigt. In Schnitten von normaler Testis wurden die Spermatogonien als GAGE-7b-positive Zellen identifiziert und GAGE-7b ebenfalls anhand dieser Untersuchungen als CTA bestätigt. Untersuchungen der GAGE-Expression auf Proteinebene sind bisher nur selten in der Literatur beschrieben. Akcakanat et al. berichteten von Analysen der CTA GAGE, NY-ESO-1, MAGE-A und SSX auf Proteinebene bei 213 Speiseröhrenkrebspatienten (Akcakanat et al. 2006). SSX-Expression wurde nicht detektiert, wohingegen GAGE-Expression in 20% der Patienten, MAGE-AExpression in 52% und NY-ESO-1-Expression in 21% der Patienten gemessen wurde. Zur Detektion von GAGE in immunhistologischen Untersuchungen wurde ein monoklonaler GAGEAntikörper der Firma BD Biosciences verwendet. Die Färbung der GAGE-positiven Gewebe war ebenfalls heterogen, zytoplasmatisch und nukleär. Die GAGE-Expression war statistisch DISKUSSION 186 signifikant erhöht in Patienten, die älter als 64 Jahre waren und Tumoren größer als 60mm sowie in der abdominalen Speiseröhre aufwiesen. Jedoch wurde keine Korrelation gefunden zwischen CTA-Expression und Krankheitsfortschritt oder Überlebensrate. Des Weiteren wurde eine Korrelation zwischen der MAGE-A- und GAGE-Expression sowie der GAGE- und NY-ESO-1Expression beschrieben. Somit kann man von einer hohen Koexpression der MAGE-A-Proteine mit anderen CTA wie GAGE-Proteinen sprechen (Akcakanat et al. 2006). Gjerstorff et al. generierten einen Maus-Hybridom monoklonalen Antikörper, führten eine Charakterisierung des Antikörpers durch und untersuchten sowohl Kontroll- als auch Tumorgewebe (Gjerstorff et al. 2006). Bei der Untersuchung der GAGE-Expression auf Proteinebene in Kontrollgeweben, wurde die Expression von GAGE-1 bis 8 in Testis bestätigt. Eine GAGE-Expression wurde zum ersten Mal in postfötalen Oozyten nachgewiesen, die wie Spermatogonien zu immunprivilegierten Zellen zählen. Insgesamt wurden 256 Tumorgewebe aus 22 Tumorentitäten getestet. Zu den Tumorentitäten mit den meisten zur Verfügung stehenden Geweben zählten Brust- und Lungenkrebs sowie MM. GAGE-Expression wurde in 12% (5/43) der Brustkrebsgewebe, 16% (10/64) der Lungenkrebsgewebe und 17% (4/24) der MM-Gewebe nachgewiesen. Diese Expression ist deutlich geringer als die in dieser Arbeit berichteten Ergebnisse. Die Anzahl an untersuchten MM-Geweben war nur halb so groß. Diese Ergebnisse könnten auf eine unterschiedliche Sensitivität der Antikörper zurückzuführen sein. Die Expression in den Gewebeschnitten war stark heterogen, zytoplasmatisch und teilweise sehr stark im Nukleus repräsentiert. Die GAGE-Expression in zwei Lymphomgeweben war negativ, welches sich mit den negativen Ergebnissen dieser Arbeit in den CTCL-Zelllinien deckt. Zudem berichteten die Autoren über eine Assoziation der GAGE-Expression mit dem Phenotyp, jedoch nicht mit dem Genotyp einer distinkten Zellpopulation. Die Hypothese, dass GAGE-Proteine Marker für Stammzellen und Kontrollproteine der Keimzell-Genexpression sein könnten wird diskutiert. Schließlich wurde der Antikörper mit dem kommerziell erhältlichen Antikörper von BD Biosciences verglichen. Es wurde ein vergleichbares Färbeverhalten berichtet (Gjerstorff et al. 2006). In einer dritten Studie wurden MM-Gewebe auf GAGE-Expression getestet unter Verwendung des GAGE-Antikörpers von BD Biosciences. Alle 19 analysierten Naevi waren GAGE- sowie MAGE-A1-, A4- und NY-ESO-1-negativ (Luftl et al. 2004). Fünf der 26 (19%) untersuchten primären kutanen MM waren GAGE-positiv. Zudem wurden 19% der kutanen MM positiv getestet für MAGE-A1, 35% für MAGE-A4, 15% für MAGE-A3, 31% für NY-ESO-1 und 38% für MAGE-C1. Zwanzig der 26 Gewebe waren mindestens für ein CTA positiv, so dass die Sensitivität der CTA-Immunreaktivität in MM-Gewebeschnitten mit der Anzahl an CTA zunahm DISKUSSION 187 (Luftl et al. 2004). Die untersuchten CTA wiesen auf eine maligne Transformation der kutanen MM hin. Die Expressionshäufigkeit jedes einzelnen CTA war eher gering. Laut den Autoren steht die optimale Kombination von CTA für immunhistologische Analysen zur Diskussion. 4.4.3 Serologische Untersuchung von GAGE-7b Neben SADA und Multiplex-Serologie wurde eine dritte Methode zur Bestimmung von spezifischen Antikörpern gegen Tumorantigene im Serum von Ctrl und Patienten verwendet. Im Anschluss an eine Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese des aufgereinigten GAGE-7b-tag-Proteins wurde das geblottete Protein mit Serum inkubiert, um auf diese Weise vorhandene Serumantikörper nachzuweisen. Diese Methode stellt sicherlich keine vergleichbare Hochdurchsatzmethode ähnlich der Multiplex-Serologie dar, ist jedoch mit zwei Tagen Dauer schneller als die SADA-Methode. Positiv reagierende Seren wurden zusätzlich mittels Chemilumineszenz bestätigt. In diesem methodischen Ansatz konnte kein GAGE-7b-positives Kontrollserum ermittelt werden. 5,9% der 34 untersuchten CTCL-Seren und 5,6% der 72 MMSeren waren GAGE-positiv. Die erhaltenen Ergebnisse bestätigen sowohl die SADA (Ctrl: 1%, MM: 6%, CTCL: 3%) als auch die Multiplex-Serologie Ergebnisse (Ctrl: 1%, MM: 5,8%, CTCL: 2,8%). Für alle drei Methoden wurden Kontrollseren aus demselben Serenpool verwendet, wobei die verwendeten Seren im SADA und in der Multiplex-Serologie identisch waren. Jedoch war das jeweils positiv reagierende Serum nicht identisch (SADA: BB36-39, Multiplex: BB15-59). Das MM-Kollektiv war für jede Methode unterschiedlich, so dass hier einzig die Häufigkeit der Seropositivität verglichen werden kann. Diese ist wie bereits erläutert mit ungefähr 6% in allen Methoden sehr ähnlich. Der CTCL-Serenpool war ebenfalls für alle Methoden gleich, so dass ein Vergleich angestellt werden kann. Jede Methode identifizierte andere Seren als seropositiv, so dass hier keine Übereinstimmung herrscht. Bezüglich der SADA-Methode könnte dies damit erklärt werden, dass ein in der cDNA-Sequenz verkürzter Klon verwendet wurde und somit eventuell vorhandene antigene Strukturen des volle-LängeProteins verloren gegangen sind. Allerdings sollten die in dieser Methode identifizierten positiven Seren auch mit den anderen Methoden als positiv detektiert werden. Zudem gibt es bei der SADA-Methode keine Kontrolle des induzierten Proteins. Es wäre theoretisch möglich, dass auch Proteine der UTR abgelesen werden und hier Antikörperreaktionen auftreten. Im Western Blot könnten durch die Denaturierung des Proteins antigene Strukturen sowohl entstehen als auch verschwinden. In der Multiplex-Serologie könnte eventuell der GST-Teil einen Teil des GAGE-7b-Proteins verdecken und somit schwerer zugänglich für Serumantikörper machen. Zusammenfassend ist festzustellen, dass die Häufigkeit an Immunreaktivitäten gegen GAGE in Kontrollen sehr gering oder nicht vorhanden ist, jedoch einige MM- und CTCL-Patienten DISKUSSION 188 Antikörper ausbilden. Die Häufigkeit an humoralen Immunreaktionen gegen GAGE in Patienten sind jedoch mit unter 10% so gering, dass eine aussagekräftige Diagnose allein anhand dieser Immunreaktion nicht möglich erscheint. Da CTCL-Patienten Antikörper gegen GAGE besitzen, wären weitere Analysen hinsichtlich GAGE-Expression in CTCL-Gewebe interessant. Untersuchungen der humoralen Immunantwort gegen GAGE sind bisher noch sehr selten in der Literatur beschrieben. Wadle et al. untersuchten die Immunantwort gegen verschiedene CTA in Pankreaskrebspatienten (Wadle et al. 2006). Hierfür wurde die „rekombinant exprimierte Antigene auf Hefeoberfläche“-(RAYS)-Technologie angewendet, die der SEREX-Methode ähnelt. 14% der Seren waren „synaptonemal complex protein“ (SCP-1)-positiv, wohingegen alle Seren GAGE-negativ waren. 4.4.4 GAGE-7b als Ziel einer Immuntherapie Aufgrund der in dieser Arbeit dokumentierten Ergebnisse sowie Berichten in der Literatur stellt die GAGE-Antigenfamilie eine interessante Proteinfamilie für Tumorimmuntherapien dar. Da die Expression der GAGE-Gene sowohl auf mRNA- als auch auf Proteinebene CTA-spezifisch ist, würden Immuntherapien nur Krebszellen angreifen ohne normales Gewebe zu schädigen. Die gerringe Immunogenität des Antigens selbst könnte mittels Immuntherapien verstärkt werden. Immuntherapien sind am erfolgreichsten, wenn die Antigenexpression homogen und hoch im Tumorgewebe ist. Dies trifft laut der Ergebnisse dieser Arbeit jedoch nicht zu. Dementsprechend gibt es zwei Ansätze, diese Hürde zu überwinden. Zum einen kann eine Immuntherapie verschiedene Antigene als Zielstruktur definieren oder zum anderen die Antigenexpression erhöhen und anschließend immuntherapeutisch wirken. Der zweite Ansatz konnte in einer Studie von Bazhin et al. für GAGE auf Zelllinien-Ebene unter Verwendung eines Histondeacetylase Inhibitors und DNA Methyltransferase Inhibitors nachgewiesen werden. Eine MM-Zelllinie wurde mit dem DNA Methyltransferase Inhibitor 5-Aza-2’-Deoxycytidin und dem Histondeacetylase Inhibitor Trichostatin A behandelt. 5-Aza-2’-Deoxycytidin führt zu einer Promotordemethylierung vieler CTA-Gene und verursacht eine erhöhte mRNA Expression. Trichostatin A inhibiert die Histondeacetylase spezifisch und stimuliert die Genexpression ausgewählter Gene wie CTA. Unter Verwendung der Western Blot und ELISA-Methode konnte die GAGE-Expression nach 5-Aza-2’-Deoxycytidin-Behandlung um 200% erhöht werden. Eine Erhöhung um das vierfache wurde nach Kombination von 5-Aza-2’-Deoxycytidin und Trichostatin A berichtet (Bazhin et al. 2007). Drei weitere Zelllinien wurden mit diesen Inhibitoren behandelt. In immunhistologischen Untersuchungen wurde eine erhöhte Zahl an GAGE-positiven Zellen detektiert. Durch die Behandlung von MM-Zelllinien mit DISKUSSION 189 Histondeacetylase und Methyltransferase Inhibitoren könnte eine homogene GAGE-Expression erzielt werden. Die Übertragung dieser Ergebnisse auf den Menschen muss in klinischen Studien nachgewiesen werden und steht momentan noch aus. Grundlage dieser Ergebnisse waren Studien von Sigalotti et al. Demnach ist die Promotermethylierung von CTA für die starke Heterogenität der Expression von CTA in Tumoren verantwortlich. Die Expression aller untersuchter CTA wurde durch 5-Aza-2’Deoxycytidin in CTA-negativen Zellklonen induziert (Sigalotti et al. 2004). Entsprechend könnte 5-Aza-2’-Deoxycytidin ein Wirkstoff sein, der die therapeutische Effizienz einer multiCTA-Immuntherapie in MM-Patienten verbessert. In RCC-Zelllinien konnte die Expression von GAGE-1 bis 6 ebenfalls hochreguliert werden (Coral et al. 2002). Die Expression von GAGE-1 bis 6 konnte in MM-Xenografts von 5-Aza-2’-Deoxycytidin-behandelten Mäusen nachgewiesen werden. Kontrollmäuse wiesen hingegen keine Expression auf (Coral et al. 2006). Zudem wurde berichtet, dass die Transkription der CTA mit der Hypomethylierung der CTA Promoter korreliert (De Smet et al. 2004). Erste klinische Phase I Studien zur Verwendbarkeit und Sicherheit von 5-Aza-2’-Deoxycytidin in Krebspatienten wurden bereits durchgeführt (Samlowski et al. 2005; Gollob et al. 2006). Schließlich wurde für ein Antigen der GAGE-Familie, GAGE-7c, eine mögliche Funktion nachgewiesen. Cilensek et al. berichteten, dass GAGE-7c eine Funktion in der Tumorgenese hat. Durch eine GAGE-7c-Expression wurde die Epithelialkarzinom Zelllinie HeLa resistent gegen Apoptose und Krebstherapien wie Radio- und Chemotherapie (Cilensek et al. 2002). Dies betraf apoptotische Ereignisse, die durch Fas oder IFN-γ initiiert wurden. Im Fas- Signaltransduktionsweg wurde der Einfluss von GAGE-7c auf die Apoptose nach der Caspase-8Aktivierung und vor der poly-ADP-Ribose-Polymerase (PARP)-Spaltung lokalisiert. GAGE-7bpositive Zelllinien waren ebenfalls resistent gegen Fas-induzierte Apoptose (Cilensek et al. 2002). GAGE-7b und 7c unterscheiden sich in zwei Aminosäuren an den Positionen 50 und 87. Somit könnten sich GAGE-7b- und 7c-positive Tumorzellen der T-Zell-induzierten Apoptose entziehen und zur Tumorprogression durch Proliferation beitragen. Entsprechend wäre eine zielgerichtete Immuntherapie gegen GAGE eine Möglichkeit, GAGE-positive Tumore sensitiv gegen Chemotherapeutika wie Taxol und γ-Bestrahlung zu machen. 4.4.5 GAGE-7b – Fazit und Ausblick Die in dieser Arbeit durchgeführte Charakterisierung von GAGE-7b im MM und CTCL ergänzt das bereits in der Literatur beschriebene Wissen über die Bedeutung der CTA. Die Ergebnisse dieser Arbeit stellen deutlich dar, dass GAGE-7b eine hohe Expression in MM-Zelllinien und DISKUSSION 190 eine mittlere Expression in MM-Geweben besitzt. In immunhistologischen Analysen wurde eine heterogene Expression von GAGE-7b in MM-Tumorgewebe ermittelt. In weitergehenden Untersuchungen von Bazhin et al. konnte eine starke Induktion der GAGE-7b-Expression in mit 5-Aza-2’-Deoxycytidin behandelten Zelllinien nachgewiesen werden (Bazhin et al. 2007). Für das CTCL ist GAGE-7b nicht relevant, da in CTCL-Zelllinien keine GAGE-7b-Expression nachgewiesen werden konnte. Aufgrund der beschränkten Expression von GAGE auf immunprivilegierte Gewebe und der weitverbreiteten Expression in verschiedenen Tumorentitäten hat GAGE das Potential für ein optimal geeignetes Therapieziel in der Onkologie. Um dieses Potential noch deutlicher hervorzuheben, sollten Expressionsanalysen auf Proteinebene für verschiedenste Tumorentitäten durchgeführt werden. Interessant wäre hier zum einen der Vergleich von Ergebnissen, die mittels PCR und mittels Immunhistologie erhoben werden. Ein Vergleich der Ergebnisse von immunhistologischen Untersuchungen mittels polyklonalem und monoklonalem Antikörper wäre hier ebenfalls von Interesse. Des Weiteren müsste die Funktion von GAGE untersucht werden. Eine Annäherung an die Aufklärung der Funktion von GAGE kann über die genaue Lokalisation von GAGE in der Zelle z.B. mittels FACS-Untersuchungen erfolgen in Verbindung mit der Suche nach Bindungspartnern. Bindungspartner könnten durch die Immobilisierung von GSTGAGE-Protein an eine Glutathion-Säule, Inkubation mit Zelllysat und anschließende Charakterisierung von gebundenen Proteinen erfolgen. Zudem könnte die Zellproliferation nach Zugabe des GAGE-Antikörpers zu bestimmten Zellsystemen untersucht werden. Auf der Basis der Analysen von Bazhin et al. wäre es sicherlich sinnvoll, klinische Studien mit einer Kombination von GAGE-Antikörpern und 5-Aza-2’-Deoxycytidin durchzuführen, um den Erfolg einer Therapie beurteilen zu können, während welcher die Expression des Zielantigens hochreguliert wird. 4.5 CTA und TAA in der Krebstherapie Das Ziel von Immuntherapien ist die Stärkung der Immunantwort gegen den Tumor unter Verwendung von spezifischen Strukturen z.B. in Form von Antigenen. CTA sind von einem immunologischen Standpunkt aus ausgezeichnete Ziele für Immuntherapien, da sie in vielen Tumorentitäten exprimiert sind und in Normalgeweben ausschließlich in Testis vorkommen. Die Testis selbst ist für Immunzellen nicht zugänglich, da es keinen direkten Kontakt zwischen Keim- und Immunzellen gibt, und die MHC I Oberflächenexpression auf den Keimzellen fehlt (Barker und Billingham 1977; Tomita et al. 1993). Bislang stimulieren viele Immuntherapien den zellulären CD8+ T-Zellarm mit weniger befriedigenden Resultaten (Wang 2001), die auf DISKUSSION 191 eine fehlende Induktion der humoralen Immunantwort zurückgeführt werden (Nakatsura et al. 2002). Eine effektive CTL-Immunantwort gegen Tumorantigene benötigt jedoch die Unterstützung von CD4+ T-Helferzellen (Bennett et al. 1997). In aktuellen Studien wird eine erfolgreiche Immunantwort immer im Zusammenhang mit einer CD4+ T-Helferzellantwort diskutiert (Hung et al. 1998; Toes et al. 1999). Neben der MHC Klasse II-Präsentation exogener und endogener Proteine durch APC konnte eine MHC Klasse I-Präsentation derselben Proteine über eine „Cross“-Präsentation nachgewiesen werden. Dieses APC-„Cross-Priming“ induziert über CD4+ T-Lymphozyten eine humorale und über CD8+ T-Lymphozyten eine zelluläre Immunantwort. Es wurde berichtet, dass intrazelluläre Proteine mittels Autophagie prozessiert werden können und somit in den MHC Klasse II Präsentationsweg gelangen (Dengjel et al. 2005; Paludan et al. 2005; Zhou et al. 2005). Hierbei sind Endosomen und Lysosomen beteiligt. „Cross“-Präsentation wurde für virale Proteine (Gooding und Edwards 1980) und Tumorantigene sowie Selbstantigene (Huang et al. 1994; Kurts et al. 1996) berichtet. Beim CTCL, einer Neoplasie der CD4+ Immunzellen, könnte die Aktivierung einer CD8+ TZellantwort beeinträchtigt sein. In Diskussion steht bis heute, ob nur die TH1-Immunzellen, die eine zelluläre Immunantwort induzieren, oder auch die TH2-Immunzellen, die eine humorale Immunantwort induzieren, eine effektive Anti-Tumor-Immunantwort hervorrufen. Zudem werden mittlerweile multi-Epitop Immuntherapien, die zwei oder mehr Antigene als Zielstrukturen haben, diskutiert (Wadle et al. 2006). Die Therapie des MM ist vor allem durch die operative Entfernung des Tumorgewebes mit teilweise sich anschließender adjuvanter Therapie in Form von Chemotherapeutika und Immunmodulatoren gekennzeichnet. Für fortgeschrittene Stadien werden Chemotherapien und Immuntherapien empfohlen. Vor allem bei Immuntherapien kommen CTA und TAA zum Einsatz. Zahlreiche Ansätze von Immuntherapien z.B. Peptide, beladene DC unter Verwendung der CTA MAGE-A1 und A3 sowie NY-ESO-1 sind bisher beschrieben worden (Hoon et al. 1995; Marchand et al. 1999; Jager et al. 2000a). Auch das in dieser Arbeit näher charakterisierte CTA GAGE-7b stellt durch seine Expression in MM-Tumorgeweben ein vielversprechendes Ziel für Immuntherapien dar. Durch die Kombination mit 5-Aza-2’-Deoxycytidin wird die Expression dieses Antigens deutlich erhöht, welches den Angriff auf den Tumor in einer möglichen Immuntherapie intensivieren würde. Deshalb wären Studien zur Verwendung von Therapien mit GAGE-7b-Peptiden oder beladenen DC in Kombination mit 5-Aza-2’Deoxycytidin angebracht. Möglich wäre auch, dass die Expression anderer CTA parallel erhöht würde, so dass eine Immuntherapie gegen eine Vielzahl an CTA gleichzeitig möglich wäre. DISKUSSION 192 In der Therapie des CTCL werden vor allem Psoralen plus ultraviolettes Licht, Radiotherapie und topische Medikamente eingesetzt. In fortgeschrittenen Stadien finden Chemotherapie, Retinoide und Interferone sowie Interleukine Anwendung. Mittlerweile werden therapeutische Antikörper, Immuntherapien in Form von Vakzinierungen und Histondeacetylase Inhibitoren in der Therapie des CTCL verwendet. Allerdings sind bisher kaum Ansätze unter Verwendung von CTA und TAA beschrieben. In dieser Arbeit konnte kein eindeutiges antigenes Ziel für das CTCL identifiziert werden. Jedoch sind zahlreiche Antigene, die im CTCL relevant sein könnten beschrieben, so dass weitere Untersuchungen in diese Richtung angebracht sind (Usener et al. 2003b; Hartmann et al. 2004). 4.6 CTA und TAA in der Krebsdiagnostik und -prognostik Neben Serumbiomarkern standen Antikörper gegen bestimmte immunogene Ziele jeher im Fokus der Diagnostik. Antikörper sind stabil im Serum, hochspezifisch, haben eine Halbwertszeit von mehr als sieben Stunden und können schnell mit charakterisierten Sekundärreagenzien und validierbaren Methoden gemessen werden (Anderson und LaBaer 2005). Zudem sind Antikörper bereits im Serum amplifiziert, wenn auch nur geringe Mengen an immunogenem Protein vorhanden sind. Die Höhe der Immunantwort gegen Krebs ist zumeist geringer als gegenüber Infektionserregern. Die mögliche Zahl an Tumorantigenen ist ähnlich groß wie das gesamte Tumorproteom inklusive aller Varianten (Anderson und LaBaer 2005). Wenn Antikörper gegen ein Tumorantigen durch SEREX oder SADA detektiert werden, ist eine immunologische Relevanz der Antigene per se vorhanden, da beide Methoden hohe Antikörpertiter voraussetzen. Ein hoher Titer an spezifischen Antikörpern setzt die Hilfe von CD4+ T-Zellen voraus. SEREX und SADA spiegeln demnach das CD4+ T-Zell-Repertoire der Tumorerkrankung wider (Jager et al. 1998; Old und Chen 1998). Humorale und zelluläre Immunantwort in SEREX und Immunantworten durch CTL korrelieren für einige Antigene (Sahin et al. 1995; Chen et al. 1997). In Diskussion steht mittlerweile, dass parallel ablaufende Immunantworten für eine effektivere Immunantwort verantwortlich sind (Nishikawa et al. 2001). Einige Studien haben bereits für andere Antigenklassen nachgewiesen, dass die Detektion eines Panels an Serumantikörpern ein wertvolles diagnostisches Werkzeug darstellen könnte. Wang et al. verwendeten 22 Phagenpeptide als Screeningtest für Prostatakrebs (Wang et al. 2005). Diese Peptide stammen aus kodierenden und nicht-kodierenden Regionen von Prostatakrebsgewebe und fanden in einem Phagenproteinmicroarray Anwendung. Mit 88,2% Spezifität und 81,6% Sensitivität konnten Seren von Prostatakrebspatienten von Ctrl unterschieden werden (Wang et al. 2005). In einem Proteinmicroarray wurden 65 Antigene zur Unterscheidung zwischen DISKUSSION 193 Gebärmutterhalskrebspatienten und Ctrl verwendet (Chatterjee et al. 2006). Die Sensitivität lag hier bei 55% und die Spezifität bei 98%. Bisher wurden Immunantworten gegen CTA und TAA per se nur sehr selten gemessen, so dass von einer niedrigen Immunogenität der CTA und TAA unter Normalbedingungen auszugehen ist. Dies könnte auf die Verarbeitung dieser Antigene im Proteasom zurückzuführen sein. Hoch organisierte Proteine, die einen großen Anteil an α-helikalen Strukturen haben, scheinen der Entfaltung der Proteine im Proteasom zu widerstehen. Somit können diese Proteine schlechter verarbeitet und damit auch präsentiert werden. Zu dieser Proteingruppe zählen unter anderem die CTA-Antigengruppen MAGE, „Synaptonemal complex protein“ (SCP) und SART. NY-ESO-1, welches in dieser Arbeit hohe Antikörperhäufigkeiten in MM-Patienten in der MultiplexSerologie hatte, gehört nicht zu dieser Gruppe, da es deutlich weniger α-helikale Strukturen besitzt (Kirkin et al. 2002). Generell zählen die CTA und TAA zu den Autoantigenen und somit zu den körpereigenen Proteinen. In Gesunden werden autoreaktive T-Zellen, die ein durch APC präsentiertes Selbstpeptid erkennen, entweder im Thymus eliminiert oder in der Peripherie inaktiviert. Im Tumor kann eine Fehlfunktion dieses Mechanismus vorliegen, so dass es zu Antikörper- und T-Zell-vermittelten Immunreaktionen gegen körpereigene Strukturen bzw. Antigene kommen kann (Janeway et al. 2002). Diese Reaktionen gegen Selbstpeptide stellen jedoch eher schwächere Immunantworten im Vergleich zu Fremdpeptiden von Bakterien oder Viren dar (Houghton et al. 2001). Es ist nicht bekannt, ob Autoantikörper die Immunüberwachung widerspiegeln oder einen Einfluss auf die klinische Entwicklung des Patienten haben könnten (Anderson und LaBaer 2005). Jedoch verschwanden IgG Antikörper gegen die Antigene KU-MEL1 und CDX2 nach einer Behandlung und nachfolgender guter Prognose von Patienten (Kiniwa et al. 2001; Ishikawa et al. 2003). Im Fokus der Diagnostik und Prognostik des MM steht die immunhistologische Untersuchung von Tumorgewebe unter Verwendung von Antikörpern gegen Differenzierungsantigene wie S100, Melan A und Tyrosinase (Busam und Jungbluth 1999; Orchard 2000). Verschiedene serologische Tumormarker wie S100β, Laktatdehydrogenase und „melanoma inhibitory activity“ sind ebenfalls bekannt (Djukanovic et al. 2001; Bottoni et al. 2003). Allerdings ist bisher kein spezifischer und sensitiver diagnostischer und prognostischer Marker für das MM identifiziert worden. Bislang spielen CTA und TAA eine eher untergeordnete Rolle in der Diagnostik und Prognostik des MM. In dieser Arbeit konnte kein eindeutiger, diagnostischer Marker bestimmt werden. Jedoch stellen die Antikörper gegen LAGE-1a einen ausgezeichneten prognostischen Marker dar. Antikörper gegen NY-ESO-1, zu welchem LAGE-1a hoch homolog ist, wurden bereits für MM-Patienten beschrieben, jedoch noch nicht in einem ähnlich großen Kollektiv. DISKUSSION 194 Neben LAGE-1a sollten ebenfalls MAGE-A3, se57-1 und GAGE-7b in weiteren serologischen Studien eingeschlossen werden. Die Messung von Antikörpern gegen GBP-5a, 5ta und HD-MM48 sollte auch in Betracht gezogen werden. Zusätzlich sollte die Expression von HD-MM-48 im MM untersucht werden. Zudem sollten weitere Antigene identifiziert werden, um ein mindestens 10 Antigene umfassendes Panel zusammenstellen zu können. Die Diagnostik und Prognostik des CTCL stützen sich bisher hauptsächlich auf klinische und (immun-) histologische Untersuchungen, wobei CTA und TAA bisher keine große Rolle spielen. Oberflächenmarker, β-Ketten-Rearrangement des T-Zellrezeptors in Lymphknoten, maligne Zellen und verschiedene Proteine wie Neopterin und β2-Mikroglobulin im Blut werden ebenfalls zur Analyse der Erkrankung herangezogen (Ralfkiaer 1994; Guitart und Kaul 1999; Hassel et al. 2004). Antikörper im Blut von CTCL-Patienten wurden bisher nur selten untersucht (Dummer et al. 2004). Zur Diagnose werden zur Zeit hauptsächlich monoklonale Antikörper, die gegen die Differenzierungs- und Proliferations-assoziierten Antigene Ki-1/CD30, Ki67 und IL2R/CD25 gerichtet sind, in immunhistologischen Untersuchungen oder lösliche Formen dieser Antigene im Serum verwendet (Nadali et al. 1995; Vonderheid et al. 1998; Kanavaros et al. 2001). Allerdings ermöglicht nur die kombinierte Auswertung der Antikörperanwesenheit in Verbindung mit der Expression der Antigene im Patientengewebe eine korrekte Klassifizierung. Anhand der Untersuchungen in dieser Arbeit wären HD-MM-48, HD-CL-02 und MAGE-A9 interessante Antigene für ein Panel, gegen welches Antikörperantworten in CTCL-Patienten untersucht werden sollten. Diese Antigene sollten weiterhin näher im CTCL charakterisiert werden. Zudem sollten weitere Antigene hinsichtlich ihrer Antikörperantworten in CTCL-Patienten untersucht werden, um ein Panel von mindestens 10 relevanten Antigenen zusammenstellen zu können. ZUSAMMENFASSUNG 195 5 ZUSAMMENFASSUNG Essentielle Voraussetzungen für die Entwicklung von spezifischen Immuntherapien und Verfahren zur Diagnose und Prognose von Tumorerkrankungen sind die Kenntnis und Charakterisierung von Tumorantigenen. Potentielle Antigene für immuntherapeutische Strategien und Diagnoseverfahren sollten möglichst tumorspezifisch sein. Zudem sollten Hinweise auf bereits im Patienten induzierte Immunantworten z.B. in Form von Antikörper- oder T-Zellantworten bestehen. In dieser Arbeit wurden Antikörperantworten in Patienten des kutanen T-Zell Lymphoms (CTCL), des malignen Melanoms (MM) und von Kontrollen (Ctrl) mittels zweier methodischer Ansätze untersucht sowie das Tumorantigen GAGE-7b unter Verwendung eines in einem Kaninchen generierten polyklonalen Antikörpers charakterisiert. Um Antikörper-Häufigkeiten für zahlreiche Tumorantigene parallel zu bestimmen, wurde ein Serumantikörper-Detektionsarray (SADA) etabliert. Bei der Untersuchung von 42 Tumorantigenen wurden für 14 Antigene Antikörper nur in Patienten jedoch nicht in Ctrl detektiert. Die Serumantikörper-Häufigkeiten der Antigene GBP-5ta, GBP-5a, HD-MM-48 und HD-MM-19 im MM-Kollektiv war signifikant höher als im Kontroll-Kollektiv. Dies traf auch für die Antigene HD-MM-48 und HD-CL-02 im Vergleich von CTCL- und Kontroll-Kollektiv zu. Insgesamt waren die Antikörperantwort-Häufigkeiten mit zumeist unter 10% relativ gering, welches die Verwendung der entsprechenden Antigene als diagnostische Ziele einschränkt. Ein weiteres Verfahren zur Bestimmung der Immunreaktion von 13 Cancer-Testis- und Tumorassoziierten Antigenen in verschiedenen Kollektiven wurde in Form der Multiplex-Serologie verwendet. In einem MM-Kollektiv wurden Antikörper gegen LAGE-1a signifikant häufiger identifiziert im Vergleich zu einem Kontroll-Kollektiv. Zusätzlich war diese höhere AntikörperHäufigkeit in MM-Patienten stadienabhängig. Für die Antigene MAGE-A3, GAGE-7b und se571 wurden ebenfalls höhere Antikörper-Häufigkeiten in MM-Patienten gemessen. AntikörperTiter von MM- und CTCL-Patienten veränderten sich über die Zeit selten, so dass Serokonversionen nur vereinzelt nachgewiesen werden konnten. Für MAGE-A1 und A9 konnten für verschiedene Stadien signifikant höhere Reaktivitäten im CTCL- im Vergleich zum KontrollKollektiv ermittelt werden. Interessante Ansatzpunkte für zukünftige Analysen wären Kollektive, die mit Interferon-γ, Tumor- oder dendritischen Zellen behandelt oder in Immuntherapien gegen spezifische Tumorantigene aufgenommen wurden. Erstmalig konnte ein umfassend beschriebener polyklonaler Kaninchen-Antikörper gegen GAGE-7b hergestellt werden, der im Western Blot und in der Immunhistologie verwendet wurde. Im Western Blot konnte nachgewiesen werden, dass 59% der untersuchten MMZelllinien GAGE-7b exprimierten, wohingegen kein Protein in CTCL-Zelllinien und in Kontrollgeweben mit Ausnahme von Testis detektiert werden konnte. Dieses Expressionsverhalten konnte in immunzytologischen Untersuchungen bestätigt werden. Immunhistologisch exprimierten 53% der MM-Gewebe GAGE-7b. Des Weiteren konnten in 6% der MM- und CTCL-Patienten Antikörper gegen GAGE-7b nachgewiesen werden, während keine Antikörper in Ctrl gefunden wurden. Aufgrund der erhaltenen Ergebnisse erkennt der GAGE-7b Antikörper wohl alle GAGE-Familienmitglieder. Zukünftige Untersuchungen unter Verwendung dieses Antikörpers können die Aufklärung funktioneller Aspekte der GAGEExpression in Tumorerkrankungen ermöglichen. SUMMARY 196 6 SUMMARY Essential prerequisites for the development of specific immunotherapies and procedures for diagnosis and prognosis of tumor diseases are the knowledge and characterization of tumor antigens. Potential antigens for immunotherapeutic strategies and diagnosis tools should be tumor-specific. Moreover, indications for immune responses in patients e.g. antibody or T cell responses should be available. In this thesis, antibody responses of patients of cutaneous T cell lymphoma (CTCL), melanoma (MM) and controls were investigated using two different methods. In addition, the tumor antigen GAGE-7b was characterized using a rabbit polyclonal antibody. In order to test antibody frequencies of various tumor antigens in parallel, a serum antibody detection array (SADA) was established. Testing 42 tumor antigens, antibodies against 14 antigens were only detectable in patients but not in controls. Serum antibody frequencies of antigens GBP-5ta, GBP-5a, HD-MM48 and HD-MM-19 were significantly higher in MM patients compared to controls. The same was true for antigens HD-MM-48 and HD-CL-02 comparing CTCL patients and controls. In summary, antibody frequencies were relatively low ranging most frequently from 0% to 10% which limits the use of the respective antigens as diagnostic targets. An additional method to measure immunoreactivities of 13 cancer testis and tumor-associated antigens in different groups named multiplex serology was used. Antibodies against LAGE-1a were detected significantly more frequently in MM patients in comparison to controls. In addition, this higher antibody frequency in MM patients was stage dependent. For antigens MAGE-A3, GAGE-7b and se57-1 a higher antibody frequency in MM patients was measured likewise. Antibody titers of MM and CTCL patients rarely changed over time, thus seroconversions were seen only sporadically. Significantly higher reactivities could be identified for MAGE-A1 and A9 in CTCL patients of different stages compared to controls. In future, analyzing patients immunized with interferon-γ, tumor or dendritic cells or immunotherapy targeting specific tumor antigens would be of high interest. For the first time, a comprehensively characterized rabbit polyclonal antibody against GAGE-7b was generated and used in Western Blot and immunohistology. In Western Blot analysis, 59% of all MM cell lines tested expressed GAGE-7b, whereas no protein was detected in both CTCL cell lines and control tissues except testis. This expression pattern was confirmed by immunocytology. Immunohistological tests confirmed that 53% of MM tissues are positive for GAGE-7b protein expression. Moreover, antibodies against GAGE-7b could be identified in 6% of MM and CTCL patients whereas controls had no detectable antibody responses. On the basis of the achieved results, the GAGE-7b antibody recognizes probably all GAGE family members. Future analysis using this antibody may permit the elucidation of functional aspects of GAGE expression in tumor diseases. LITERATURVERZEICHNIS 197 7 LITERATURVERZEICHNIS Aarnoudse, C.A., van den Doel, P.B., Heemskerk, B. und Schrier, P.I. 1999. Interleukin-2induced, melanoma-specific T cells recognize CAMEL, an unexpected translation product of LAGE-1. Int J Cancer, 82(3): 442-8. Abendstein, B., Marth, C., Muller-Holzner, E., Widschwendter, M., Daxenbichler, G. und Zeimet, A.G. 2000. Clinical significance of serum and ascitic p53 autoantibodies in epithelial ovarian carcinoma. Cancer, 88(6): 1432-7. Abrams, J.T., Vonderheid, E.C., Kolbe, S., Appelt, D.M., Arking, E.J. und Balin, B.J. 1999. Sezary T-cell activating factor is a Chlamydia pneumoniae-associated protein. Clin Diagn Lab Immunol, 6(6): 895-905. Aguirre, A.J., Bardeesy, N., Sinha, M., Lopez, L., Tuveson, D.A., Horner, J., Redston, M.S. und DePinho, R.A. 2003. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev, 17(24): 3112-26. Akcakanat, A., Kanda, T., Tanabe, T., Komukai, S., Yajima, K., Nakagawa, S., Ohashi, M. und Hatakeyama, K. 2006. Heterogeneous expression of GAGE, NY-ESO-1, MAGE-A and SSX proteins in esophageal cancer: Implications for immunotherapy. Int J Cancer, 118(1): 123-8. Alpen, B., Gure, A.O., Scanlan, M.J., Old, L.J. und Chen, Y.T. 2002. A new member of the NYESO-1 gene family is ubiquitously expressed in somatic tissues and evolutionarily conserved. Gene, 297(1-2): 141-9. Anderson, K.S. und LaBaer, J. 2005. The sentinel within: exploiting the immune system for cancer biomarkers. J Proteome Res, 4(4): 1123-33. Angelopoulou, K., Stratis, M. und Diamandis, E.P. 1997. Humoral immune response against p53 protein in patients with colorectal carcinoma. Int J Cancer, 70(1): 46-51. Angus, S.P., Fribourg, A.F., Markey, M.P., Williams, S.L., Horn, H.F., DeGregori, J., Kowalik, T.F., Fukasawa, K. und Knudsen, E.S. 2002. Active RB elicits late G1/S inhibition. Exp Cell Res, 276(2): 201-13. Anichini, A., Vegetti, C. und Mortarini, R. 2004. The paradox of T-cell-mediated antitumor immunity in spite of poor clinical outcome in human melanoma. Cancer Immunol Immunother, 53(10): 855-64. Armstrong, B.K. und Kricker, A. 2001. The epidemiology of UV induced skin cancer. J Photochem Photobiol B, 63(1-3): 8-18. Asadullah, K., Docke, W.D., Haeussler, A., Sterry, W. und Volk, H.D. 1996. Progression of mycosis fungoides is associated with increasing cutaneous expression of interleukin-10 mRNA. J Invest Dermatol, 107(6): 833-7. Asadullah, K., Sterry, W. und Trefzer, U. 2002. Cytokines: interleukin and interferon therapy in dermatology. Clin Exp Dermatol, 27(7): 578-84. Atillasoy, E.S., Seykora, J.T., Soballe, P.W., Elenitsas, R., Nesbit, M., Elder, D.E., Montone, K.T., Sauter, E. und Herlyn, M. 1998. UVB induces atypical melanocytic lesions and melanoma in human skin. Am J Pathol, 152(5): 1179-86. Aumais, J.P., Williams, S.N., Luo, W., Nishino, M., Caldwell, K.A., Caldwell, G.A., Lin, S.H. und Yu-Lee, L.Y. 2003. Role for NudC, a dynein-associated nuclear movement protein, in mitosis and cytokinesis. J Cell Sci, 116(Pt 10): 1991-2003. LITERATURVERZEICHNIS 198 Autier, P. und Dore, J.F. 1998. Influence of sun exposures during childhood and during adulthood on melanoma risk. EPIMEL and EORTC Melanoma Cooperative Group. European Organisation for Research and Treatment of Cancer. Int J Cancer, 77(4): 533-7. AWMF. 2005. Interdisziplinäre kurzgefasste Leitlinien der Deutschen Krebsgesellschaft und der Deutschen Dermatologischen Gesellschaft: Malignes Melanom der Haut. http://www.uniduesseldorf.de/WWW/AWMF/ll/index.html. Baba, T., Niida, Y., Michikawa, Y., Kashiwabara, S., Kodaira, K., Takenaka, M., Kohno, N., Gerton, G.L. und Arai, Y. 1994. An acrosomal protein, sp32, in mammalian sperm is a binding protein specific for two proacrosins and an acrosin intermediate. J Biol Chem, 269(13): 10133-40. Bae, C.D., Sung, Y.S., Jeon, S.M., Suh, Y., Yang, H.K., Kim, Y.I., Park, K.H., Choi, J., Ahn, G. und Park, J. 2003. Up-regulation of cytoskeletal-associated protein 2 in primary human gastric adenocarcinomas. J Cancer Res Clin Oncol, 129(11): 621-30. Bakker, A.B., Schreurs, M.W., de Boer, A.J., Kawakami, Y., Rosenberg, S.A., Adema, G.J. und Figdor, C.G. 1994. Melanocyte lineage-specific antigen gp100 is recognized by melanoma-derived tumor-infiltrating lymphocytes. J Exp Med, 179(3): 1005-9. Balch, C.M., Buzaid, A.C., Soong, S.J., Atkins, M.B., Cascinelli, N., Coit, D.G., Fleming, I.D., Gershenwald, J.E., Houghton, A., Jr., Kirkwood, J.M., McMasters, K.M., Mihm, M.F., Morton, D.L., Reintgen, D.S., Ross, M.I., Sober, A., Thompson, J.A. und Thompson, J.F. 2001. Final version of the American Joint Committee on Cancer staging system for cutaneous melanoma. J Clin Oncol, 19(16): 3635-48. Balch, C.M., Urist, M.M., Karakousis, C.P., Smith, T.J., Temple, W.J., Drzewiecki, K., Jewell, W.R., Bartolucci, A.A., Mihm, M.C., Jr., Barnhill, R. und et al. 1993. Efficacy of 2-cm surgical margins for intermediate-thickness melanomas (1 to 4 mm). Results of a multiinstitutional randomized surgical trial. Ann Surg, 218(3): 262-7; discussion 267-9. Ballare, C., Barrio, M., Portela, P. und Mordoh, J. 1995. Functional properties of FC-2.15, a monoclonal antibody that mediates human complement cytotoxicity against breast cancer cells. Cancer Immunol Immunother, 41(1): 15-22. Banchereau, J., Briere, F., Caux, C., Davoust, J., Lebecque, S., Liu, Y.J., Pulendran, B. und Palucka, K. 2000. Immunobiology of dendritic cells. Annu Rev Immunol, 18: 767-811. Barker, C.F. und Billingham, R.E. 1977. Immunologically privileged sites. Adv Immunol, 25: 154. Barnhill, R.L., Roush, G.C., Titus-Ernstoff, L., Ernstoff, M.S., Duray, P.H. und Kirkwood, J.M. 1992. Comparison of nonfamilial and familial melanoma. Dermatology, 184(1): 2-7. Baselga, J. und Mendelsohn, J. 1994. Receptor blockade with monoclonal antibodies as anticancer therapy. Pharmacol Ther, 64(1): 127-54. Bastian, B.C. 2003. Hypothesis: a role for telomere crisis in spontaneous regression of melanoma. Arch Dermatol, 139(5): 667-8. Bauer, J. und Garbe, C. 2003. Acquired melanocytic nevi as risk factor for melanoma development. A comprehensive review of epidemiological data. Pigment Cell Res, 16(3): 297-306. Bazhin, A.V., Wiedemann, N., Schnolzer, M., Schadendorf, D. und Eichmuller, S.B. 2007. Expression of GAGE family proteins in malignant melanoma. Cancer Lett, 251(2): 25867. Bedford, H. 2003. Measles: the disease and its prevention. Nurs Stand, 17(24): 46-52; quiz 54-5. LITERATURVERZEICHNIS 199 Bennett, S.R., Carbone, F.R., Karamalis, F., Miller, J.F. und Heath, W.R. 1997. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med, 186(1): 65-70. Berke, Z., Andersen, M.H., Pedersen, M., Fugger, L., Zeuthen, J. und Haurum, J.S. 2000. Peptides spanning the junctional region of both the abl/bcr and the bcr/abl fusion proteins bind common HLA class I molecules. Leukemia, 14(3): 419-26. Bernengo, M.G., Quaglino, P., Novelli, M., Cappello, N., Doveil, G.C., Lisa, F., De Matteis, A., Fierro, M.T. und Appino, A. 1998. Prognostic factors in Sezary syndrome: a multivariate analysis of clinical, haematological and immunological features. Ann Oncol, 9(8): 85763. Bhattacharya-Chatterjee, M., Chatterjee, S.K. und Foon, K.A. 2000. Anti-idiotype vaccine against cancer. Immunol Lett, 74(1): 51-8. Bilbe, G., Delabie, J., Bruggen, J., Richener, H., Asselbergs, F.A., Cerletti, N., Sorg, C., Odink, K., Tarcsay, L., Wiesendanger, W. und et al. 1992. Restin: a novel intermediate filamentassociated protein highly expressed in the Reed-Sternberg cells of Hodgkin's disease. Embo J, 11(6): 2103-13. Bliss, J.M., Ford, D., Swerdlow, A.J., Armstrong, B.K., Cristofolini, M., Elwood, J.M., Green, A., Holly, E.A., Mack, T., MacKie, R.M. und et al. 1995. Risk of cutaneous melanoma associated with pigmentation characteristics and freckling: systematic overview of 10 case-control studies. The International Melanoma Analysis Group (IMAGE). Int J Cancer, 62(4): 367-76. Bocchia, M., Korontsvit, T., Xu, Q., Mackinnon, S., Yang, S.Y., Sette, A. nd Scheinberg, D.A. 1996. Specific human cellular immunity to bcr-abl oncogene-derived peptides. Blood, 87(9): 3587-92. Bolli, M., Schultz-Thater, E., Zajac, P., Guller, U., Feder, C., Sanguedolce, F., Carafa, V., Terracciano, L., Hudolin, T., Spagnoli, G.C. und Tornillo, L. 2005. NY-ESO-1/LAGE-1 coexpression with MAGE-A cancer/testis antigens: a tissue microarray study. Int J Cancer, 115(6): 960-6. Bosserhoff, A.K. und Buettner, R. 2002. Expression, function and clinical relevance of MIA (melanoma inhibitory activity). Histol Histopathol, 17(1): 289-300. Bottoni, U., Izzo, P., Richetta, A., Mannooranparampil, T.J., Devirgiliis, V., Del Giudice, M., Reale, M.G., Frati, L. und Calvieri, S. 2003. S100 serum level: a tumour marker for metastatic melanoma. Melanoma Res, 13(4): 427-9. Brasseur, F., Rimoldi, D., Lienard, D., Lethe, B., Carrel, S., Arienti, F., Suter, L., Vanwijck, R., Bourlond, A., Humblet, Y. et al. 1995. Expression of MAGE genes in primary and metastatic cutaneous melanoma. Int J Cancer, 63(3): 375-80. Brereton, P., Suzuki, T., Sasano, H., Li, K., Duarte, C., Obeyesekere, V., Haeseleer, F., Palczewski, K., Smith, I., Komesaroff, P. und Krozowski, Z. 2001. Pan1b (17betaHSD11)-enzymatic activity and distribution in the lung. Mol Cell Endocrinol, 171(1-2): 111-7. Brichard, V., Van Pel, A., Wolfel, T., Wolfel, C., De Plaen, E., Lethe, B., Coulie, P. und Boon, T. 1993. The tyrosinase gene codes for an antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med, 178(2): 489-95. Brinkmann, U., Vasmatzis, G., Lee, B. und Pastan, I. 1999. Novel genes in the PAGE and GAGE family of tumor antigens found by homology walking in the dbEST database. Cancer Res, 59(7): 1445-8. LITERATURVERZEICHNIS 200 Brinkmann, U., Vasmatzis, G., Lee, B., Yerushalmi, N., Essand, M. und Pastan, I. 1998. PAGE1, an X chromosome-linked GAGE-like gene that is expressed in normal and neoplastic prostate, testis, and uterus. Proc Natl Acad Sci U S A, 95(18): 10757-62. Bunn, P.A., Jr., Ihde, D.C. und Foon, K.A. 1986. The role of recombinant interferon alfa-2a in the therapy of cutaneous T-cell lymphomas. Cancer, 57(8 Suppl): 1689-95. Burg, G., Kempf, W., Häffner, A., Nestle, F., Hess-Schmid, M., Doebbeling, U., Mueller, B. und Dummer, R. 1997. Cutaneous lymphomas. Curr Probl Dermatol, 9: 137-204. Burg, G., Kerl, H., Przybilla, B. und Braun-Falco, O. 1984. Some statistical data, diagnosis, and staging of cutaneous B-cell lymphomas. J Dermatol Surg Oncol, 10(4): 256-62. Busam, K.J., Iversen, K., Berwick, M., Spagnoli, G.C., Old, L.J. und Jungbluth, A.A. 2000. Immunoreactivity with the anti-MAGE antibody 57B in malignant melanoma: frequency of expression and correlation with prognostic parameters. Mod Pathol, 13(4): 459-65. Busam, K.J. und Jungbluth, A.A. 1999. Melan-A, a new melanocytic differentiation marker. Adv Anat Pathol, 6(1): 12-8. Chai, Z., Brereton, P., Suzuki, T., Sasano, H., Obeyesekere, V., Escher, G., Saffery, R., Fuller, P., Enriquez, C. und Krozowski, Z. 2003. 17 beta-hydroxysteroid dehydrogenase type XI localizes to human steroidogenic cells. Endocrinology, 144(5): 2084-91. Chai, Z., Sarcevic, B., Mawson, A. und Toh, B.H. 2001. SET-related cell division autoantigen-1 (CDA1) arrests cell growth. J Biol Chem, 276(36): 33665-74. Chambost, H., Brasseur, F., Coulie, P., de Plaen, E., Stoppa, A.M., Baume, D., Mannoni, P., Boon, T., Maraninchi, D. und Olive, D. 1993. A tumour-associated antigen expression in human haematological malignancies. Br J Haematol, 84(3): 524-6. Chatterjee, M., Mohapatra, S., Ionan, A., Bawa, G., Ali-Fehmi, R., Wang, X., Nowak, J., Ye, B., Nahhas, F.A., Lu, K., Witkin, S.S., Fishman, D., Munkarah, A., Morris, R., Levin, N.K., Shirley, N.N., Tromp, G., Abrams, J., Draghici, S. und Tainsky, M.A. 2006. Diagnostic markers of ovarian cancer by high-throughput antigen cloning and detection on arrays. Cancer Res, 66(2): 1181-90. Chen, M.E., Lin, S.H., Chung, L.W. und Sikes, R.A. 1998. Isolation and characterization of PAGE-1 and GAGE-7. New genes expressed in the LNCaP prostate cancer progression model that share homology with melanoma-associated antigens. J Biol Chem, 273(28): 17618-25. Chen, Q., Jackson, H., Shackleton, M., Parente, P., Hopkins, W., Sturrock, S., MacGregor, D., Maraskovsky, E., Tai, T.Y., Dimopoulos, N., Masterman, K.A., Luke, T., Davis, I.D., Chen, W. und Cebon, J. 2005. Characterization of antigen-specific CD8+ T lymphocyte responses in skin and peripheral blood following intradermal peptide vaccination. Cancer Immun, 5: 5. Chen, Y.T., Scanlan, M.J., Sahin, U., Tureci, O., Gure, A.O., Tsang, S., Williamson, B., Stockert, E., Pfreundschuh, M. und Old, L.J. 1997. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc Natl Acad Sci U S A, 94(5): 1914-8. Cheung, I.Y., Cheung, N.K., Ghossein, R.A., Satagopan, J.M., Bhattacharya, S. und Coit, D.G. 1999. Association between molecular detection of GAGE and survival in patients with malignant melanoma: a retrospective cohort study. Clin Cancer Res, 5(8): 2042-7. Cheung, I.Y., Chi, S.N. und Cheung, N.K. 2000. Prognostic significance of GAGE detection in bone marrows on survival of patients with metastatic neuroblastoma. Med Pediatr Oncol, 35(6): 632-4. LITERATURVERZEICHNIS 201 Chiari, R., Hames, G., Stroobant, V., Texier, C., Maillere, B., Boon, T. und Coulie, P.G. 2000. Identification of a tumor-specific shared antigen derived from an Eph receptor and presented to CD4 T cells on HLA class II molecules. Cancer Res, 60(17): 4855-63. Choi, Y.C. und Chae, C.B. 1993. Demethylation of somatic and testis-specific histone H2A and H2B genes in F9 embryonal carcinoma cells. Mol Cell Biol, 13(9): 5538-48. Chomez, P., De Backer, O., Bertrand, M., De Plaen, E., Boon, T. und Lucas, S. 2001. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res, 61(14): 5544-51. Chouaib, S., Asselin-Paturel, C., Mami-Chouaib, F., Caignard, A. und Blay, J.Y. 1997. The hosttumor immune conflict: from immunosuppression to resistance and destruction. Immunol Today, 18(10): 493-7. Cigudosa, J.C., Parsa, N.Z., Louie, D.C., Filippa, D.A., Jhanwar, S.C., Johansson, B., Mitelman, F. und Chaganti, R.S. 1999. Cytogenetic analysis of 363 consecutively ascertained diffuse large B-cell lymphomas. Genes Chromosomes Cancer, 25(2): 123-33. Cilensek, Z.M., Yehiely, F., Kular, R.K. und Deiss, L.P. 2002. A member of the GAGE family of tumor antigens is an anti-apoptotic gene that confers resistance to Fas/CD95/APO-1, Interferon-gamma, taxol and gamma-irradiation. Cancer Biol Ther, 1(4): 380-7. Clark, W.H., Jr., From, L., Bernardino, E.A. und Mihm, M.C. 1969. The histogenesis and biologic behavior of primary human malignant melanomas of the skin. Cancer Res, 29(3): 705-27. Claus, R., Almstedt, M. und Lubbert, M. 2005. Epigenetic treatment of hematopoietic malignancies: in vivo targets of demethylating agents. Semin Oncol, 32(5): 511-20. Clemente, C.G., Mihm, M.C., Jr., Bufalino, R., Zurrida, S., Collini, P. und Cascinelli, N. 1996. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer, 77(7): 1303-10. Clynes, R.A., Towers, T.L., Presta, L.G. und Ravetch, J.V. 2000. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med, 6(4): 443-6. Cochran, A.J., Huang, R.R., Guo, J. und Wen, D.R. 2001. Current practice and future directions in pathology and laboratory evaluation of the sentinel node. Ann Surg Oncol, 8(9 Suppl): 13S-17S. Cooper, S., Erickson, A.L., Adams, E.J., Kansopon, J., Weiner, A.J., Chien, D.Y., Houghton, M., Parham, P. und Walker, C.M. 1999. Analysis of a successful immune response against hepatitis C virus. Immunity, 10(4): 439-49. Coral, S., Sigalotti, L., Altomonte, M., Engelsberg, A., Colizzi, F., Cattarossi, I., Maraskovsky, E., Jager, E., Seliger, B. und Maio, M. 2002. 5-aza-2'-deoxycytidine-induced expression of functional cancer testis antigens in human renal cell carcinoma: immunotherapeutic implications. Clin Cancer Res, 8(8): 2690-5. Coral, S., Sigalotti, L., Colizzi, F., Spessotto, A., Nardi, G., Cortini, E., Pezzani, L., Fratta, E., Fonsatti, E., Di Giacomo, A.M., Nicotra, M.R., Natali, P.G., Altomonte, M. und Maio, M. 2006. Phenotypic and functional changes of human melanoma xenografts induced by DNA hypomethylation: Immunotherapeutic implications. J Cell Physiol, 207(1): 58-66. Cory, S. und Adams, J.M. 2002. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer, 2(9): 647-56. Coulie, P.G., Brichard, V., Van Pel, A., Wolfel, T., Schneider, J., Traversari, C., Mattei, S., De Plaen, E., Lurquin, C., Szikora, J.P., Renauld, J.C. und Boon, T. 1994. A new gene LITERATURVERZEICHNIS 202 coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. J Exp Med, 180(1): 35-42. Coulombe, P., Rodier, G., Pelletier, S., Pellerin, J. und Meloche, S. 2003. Rapid turnover of extracellular signal-regulated kinase 3 by the ubiquitin-proteasome pathway defines a novel paradigm of mitogen-activated protein kinase regulation during cellular differentiation. Mol Cell Biol, 23(13): 4542-58. Cox, A.L., Skipper, J., Chen, Y., Henderson, R.A., Darrow, T.L., Shabanowitz, J., Engelhard, V.H., Hunt, D.F. und Slingluff, C.L., Jr. 1994. Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science, 264(5159): 716-9. Dalerba, P., Ricci, A., Russo, V., Rigatti, D., Nicotra, M.R., Mottolese, M., Bordignon, C., Natali, P.G. und Traversari, C. 1998. High homogeneity of MAGE, BAGE, GAGE, tyrosinase and Melan-A/MART-1 gene expression in clusters of multiple simultaneous metastases of human melanoma: implications for protocol design of therapeutic antigenspecific vaccination strategies. Int J Cancer, 77(2): 200-4. Daniotti, M., Oggionni, M., Ranzani, T., Vallacchi, V., Campi, V., Di Stasi, D., Torre, G.D., Perrone, F., Luoni, C., Suardi, S., Frattini, M., Pilotti, S., Anichini, A., Tragni, G., Parmiani, G., Pierotti, M.A. und Rodolfo, M. 2004. BRAF alterations are associated with complex mutational profiles in malignant melanoma. Oncogene, 23(35): 5968-77. Darlington, S., Siskind, V., Green, L. und Green, A. 2002. Longitudinal study of melanocytic nevi in adolescents. J Am Acad Dermatol, 46(5): 715-22. Dave, B.J., Nelson, M., Pickering, D.L., Chan, W.C., Greiner, T.C., Weisenburger, D.D., Armitage, J.O. und Sanger, W.G. 2002. Cytogenetic characterization of diffuse large cell lymphoma using multi-color fluorescence in situ hybridization. Cancer Genet Cytogenet, 132(2): 125-32. Davies, H., Bignell, G.R., Cox, C., Stephens, P., Edkins, S., Clegg, S., Teague, J., Woffendin, H., Garnett, M.J., Bottomley, W., Davis, N., Dicks, E., Ewing, R., Floyd, Y., Gray, K., Hall, S., Hawes, R., Hughes, J., Kosmidou, V., Menzies, A., Mould, C., Parker, A., Stevens, C., Watt, S., Hooper, S., Wilson, R., Jayatilake, H., Gusterson, B.A., Cooper, C., Shipley, J., Hargrave, D., Pritchard-Jones, K., Maitland, N., Chenevix-Trench, G., Riggins, G.J., Bigner, D.D., Palmieri, G., Cossu, A., Flanagan, A., Nicholson, A., Ho, J.W., Leung, S.Y., Yuen, S.T., Weber, B.L., Seigler, H.F., Darrow, T.L., Paterson, H., Marais, R., Marshall, C.J., Wooster, R., Stratton, M.R. und Futreal, P.A. 2002. Mutations of the BRAF gene in human cancer. Nature, 417(6892): 949-54. Davis, I.D., Chen, W., Jackson, H., Parente, P., Shackleton, M., Hopkins, W., Chen, Q., Dimopoulos, N., Luke, T., Murphy, R., Scott, A.M., Maraskovsky, E., McArthur, G., MacGregor, D., Sturrock, S., Tai, T.Y., Green, S., Cuthbertson, A., Maher, D., Miloradovic, L., Mitchell, S.V., Ritter, G., Jungbluth, A.A., Chen, Y.T., Gnjatic, S., Hoffman, E.W., Old, L.J. und Cebon, J.S. 2004. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc Natl Acad Sci U S A, 101(29): 10697-702. De Backer, O., Arden, K.C., Boretti, M., Vantomme, V., De Smet, C., Czekay, S., Viars, C.S., De Plaen, E., Brasseur, F., Chomez, P., Van den Eynde, B., Boon, T. und van der Bruggen, P. 1999. Characterization of the GAGE genes that are expressed in various human cancers and in normal testis. Cancer Res, 59(13): 3157-65. De Gast, G.C., Van Houten, A.A., Haagen, I.A., Klein, S., De Weger, R.A., Van Dijk, A., Phillips, J., Clark, M. und Bast, B.J. 1995. Clinical experience with CD3 x CD19 bispecific antibodies in patients with B cell malignancies. J Hematother, 4(5): 433-7. LITERATURVERZEICHNIS 203 De Plaen, E., Arden, K., Traversari, C., Gaforio, J.J., Szikora, J.P., De Smet, C., Brasseur, F., van der Bruggen, P., Lethe, B., Lurquin, C. et al. 1994. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics, 40(5): 360-9. De Plaen, E., Lurquin, C., Lethe, B., van der Bruggen, P., Brichard, V., Renauld, J.C., Coulie, P., Van Pel, A. und Boon, T. 1997. Identification of genes coding for tumor antigens recognized by cytolytic T lymphocytes. Methods, 12(2): 125-42. De Smedt, F., Verjans, B., Mailleux, P. und Erneux, C. 1994. Cloning and expression of human brain type I inositol 1,4,5-trisphosphate 5-phosphatase. High levels of mRNA in cerebellar Purkinje cells. FEBS Lett, 347(1): 69-72. De Smet, C., De Backer, O., Faraoni, I., Lurquin, C., Brasseur, F. und Boon, T. 1996. The activation of human gene MAGE-1 in tumor cells is correlated with genome-wide demethylation. Proc Natl Acad Sci U S A, 93(14): 7149-53. De Smet, C., Loriot, A. und Boon, T. 2004. Promoter-dependent mechanism leading to selective hypomethylation within the 5' region of gene MAGE-A1 in tumor cells. Mol Cell Biol, 24(11): 4781-90. de Vries, F.A., de Boer, E., van den Bosch, M., Baarends, W.M., Ooms, M., Yuan, L., Liu, J.G., van Zeeland, A.A., Heyting, C. und Pastink, A. 2005. Mouse Sycp1 functions in synaptonemal complex assembly, meiotic recombination, and XY body formation. Genes Dev, 19(11): 1376-89. de Wit, N.J., van Muijen, G.N. und Ruiter, D.J. 2004. Immunohistochemistry in melanocytic proliferative lesions. Histopathology, 44(6): 517-41. Deichmann, M., Benner, A., Bock, M., Jackel, A., Uhl, K., Waldmann, V. und Naher, H. 1999. S100-Beta, melanoma-inhibiting activity, and lactate dehydrogenase discriminate progressive from nonprogressive American Joint Committee on Cancer stage IV melanoma. J Clin Oncol, 17(6): 1891-6. Deichmann, M., Kahle, B., Moser, K., Wacker, J. und Wust, K. 2004. Diagnosing melanoma patients entering American Joint Committee on Cancer stage IV, C-reactive protein in serum is superior to lactate dehydrogenase. Br J Cancer, 91(4): 699-702. Demunter, A., Stas, M., Degreef, H., De Wolf-Peeters, C. und van den Oord, J.J. 2001. Analysis of N- and K-ras mutations in the distinctive tumor progression phases of melanoma. J Invest Dermatol, 117(6): 1483-9. Dengjel, J., Schoor, O., Fischer, R., Reich, M., Kraus, M., Muller, M., Kreymborg, K., Altenberend, F., Brandenburg, J., Kalbacher, H., Brock, R., Driessen, C., Rammensee, H.G. und Stevanovic, S. 2005. Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci U S A, 102(22): 7922-7. Depil, S., Roche, C., Dussart, P. und Prin, L. 2002. Expression of a human endogenous retrovirus, HERV-K, in the blood cells of leukemia patients. Leukemia, 16(2): 254-9. Devitt, G. 2004. Immunomic analysis of human renal cell carcinoma, Universität Karlsruhe, Karlsruhe, 139 pp. Devitt, G., Meyer, C., Wiedemann, N., Eichmuller, S., Kopp-Schneider, A., Haferkamp, A., Hautmann, R. und Zoller, M. 2006. Serological analysis of human renal cell carcinoma. Int J Cancer, 118(9): 2210-9. Disis, M.L., Calenoff, E., McLaughlin, G., Murphy, A.E., Chen, W., Groner, B., Jeschke, M., Lydon, N., McGlynn, E., Livingston, R.B. und et al. 1994. Existent T-cell and antibody immunity to HER-2/neu protein in patients with breast cancer. Cancer Res, 54(1): 16-20. LITERATURVERZEICHNIS 204 Disis, M.L., Pupa, S.M., Gralow, J.R., Dittadi, R., Menard, S. und Cheever, M.A. 1997. Hightiter HER-2/neu protein-specific antibody can be detected in patients with early-stage breast cancer. J Clin Oncol, 15(11): 3363-7. Ditzel, H.J., Masaki, Y., Nielsen, H., Farnaes, L. und Burton, D.R. 2000. Cloning and expression of a novel human antibody-antigen pair associated with Felty's syndrome. Proc Natl Acad Sci U S A, 97(16): 9234-9. Djukanovic, D., Hofmann, U., Sucker, A. und Schadendorf, D. 2001. Melanoma tumour markers S100B and MIA: evaluation of stability in serum and blood upon storage and processing. Br J Dermatol, 145(6): 1030-1. Dobbeling, U., Dummer, R., Laine, E., Potoczna, N., Qin, J.Z. und Burg, G. 1998. Interleukin-15 is an autocrine/paracrine viability factor for cutaneous T-cell lymphoma cells. Blood, 92(1): 252-8. Dreno, B., Claudy, A., Meynadier, J., Verret, J.L., Souteyrand, P., Ortonne, J.P., Kalis, B., Godefroy, W.Y., Beerblock, K. und Thill, L. 1991. The treatment of 45 patients with cutaneous T-cell lymphoma with low doses of interferon-alpha 2a and etretinate. Br J Dermatol, 125(5): 456-9. Dummer, R., Häffner, A.C., Hess, M. und Burg, G. 1996a. A rational approach to the therapy of cutaneous T-cell lymphomas. Onkologie, 19: 226-230. Dummer, R., Hassel, J.C., Fellenberg, F., Eichmuller, S., Maier, T., Slos, P., Acres, B., Bleuzen, P., Bataille, V., Squiban, P., Burg, G. und Urosevic, M. 2004. Adenovirus-mediated intralesional interferon-gamma gene transfer induces tumor regressions in cutaneous lymphomas. Blood, 104(6): 1631-8. Dummer, R., Heald, P.W., Nestle, F.O., Ludwig, E., Laine, E., Hemmi, S. und Burg, G. 1996b. Sezary syndrome T-cell clones display T-helper 2 cytokines and express the accessory factor-1 (interferon-gamma receptor beta-chain). Blood, 88(4): 1383-9. Dummer, R., Michie, S.A., Kell, D., Gould, J.W., Haeffner, A.C., Smoller, B.R., Warnke, R.A. und Wood, G.S. 1995. Expression of bcl-2 protein and Ki-67 nuclear proliferation antigen in benign and malignant cutaneous T-cell infiltrates. J Cutan Pathol, 22(1): 11-7. Duncan, L.M., Deeds, J., Cronin, F.E., Donovan, M., Sober, A.J., Kauffman, M. und McCarthy, J.J. 2001. Melastatin expression and prognosis in cutaneous malignant melanoma. J Clin Oncol, 19(2): 568-76. Duncan, L.M., Deeds, J., Hunter, J., Shao, J., Holmgren, L.M., Woolf, E.A., Tepper, R.I. und Shyjan, A.W. 1998. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer Res, 58(7): 1515-20. Duvic, M., Hymes, K., Heald, P., Breneman, D., Martin, A.G., Myskowski, P., Crowley, C. und Yocum, R.C. 2001. Bexarotene is effective and safe for treatment of refractory advancedstage cutaneous T-cell lymphoma: multinational phase II-III trial results. J Clin Oncol, 19(9): 2456-71. Duvic, M., Talpur, R. und Chiao, J. 2003. Phase II trial of oral suberoylanilide hydroxamic acid (SAHA) for cutaneous T-cell lymphoma (CTCL) and peripheral T-cell lymphoma (PTCL). Blood, 102: 179a. Edelson, R.P., Heald, P., Perez, M. und Rook, A. 1991. Photopheresis update. Prog Dermatol, 25(3): 1-6. Edwards, R.H., Ward, M.R., Wu, H., Medina, C.A., Brose, M.S., Volpe, P., Nussen-Lee, S., Haupt, H.M., Martin, A.M., Herlyn, M., Lessin, S.R. und Weber, B.L. 2004. Absence of BRAF mutations in UV-protected mucosal melanomas. J Med Genet, 41(4): 270-2. LITERATURVERZEICHNIS 205 Ehlken, H., Schadendorf, D. und Eichmuller, S. 2004. Humoral immune response against melanoma antigens induced by vaccination with cytokine gene-modified autologous tumor cells. Int J Cancer, 108(2): 307-13. Eichmuller, S., Usener, D., Dummer, R., Stein, A., Thiel, D. und Schadendorf, D. 2001. Serological detection of cutaneous T-cell lymphoma-associated antigens. Proc Natl Acad Sci U S A, 98(2): 629-34. Eichmuller, S., Usener, D., Thiel, D. und Schadendorf, D. 2003. Tumor-specific antigens in cutaneous T-cell lymphoma: expression and sero-reactivity. Int J Cancer, 104(4): 482-7. Elwood, J.M. und Jopson, J. 1997. Melanoma and sun exposure: an overview of published studies. Int J Cancer, 73(2): 198-203. Engvall, E. und Perlman, P. 1971. Enzyme-linked immunosorbent assay (ELISA). Quantitative assay of immunoglobulin G. Immunochemistry, 8(9): 871-4. Falk, K., Rotzschke, O., Stevanovic, S., Jung, G. und Rammensee, H.G. 1991. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature, 351(6324): 290-6. Fellenberg, F., Hartmann, T.B., Dummer, R., Usener, D., Schadendorf, D. und Eichmuller, S. 2004. GBP-5 splicing variants: New guanylate-binding proteins with tumor-associated expression and antigenicity. J Invest Dermatol, 122(6): 1510-7. Feuerer, M., Beckhove, P., Bai, L., Solomayer, E.F., Bastert, G., Diel, I.J., Pedain, C., Oberniedermayr, M., Schirrmacher, V. und Umansky, V. 2001. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat Med, 7(4): 452-8. Finn, O.J. 2003. Cancer vaccines: between the idea and the reality. Nat Rev Immunol, 3(8): 63041. Fiszer, D. und Kurpisz, M. 1998. Major histocompatibility complex expression on human, male germ cells: a review. Am J Reprod Immunol, 40(3): 172-6. Flieger, D., Renoth, S., Beier, I., Sauerbruch, T. und Schmidt-Wolf, I. 2000. Mechanism of cytotoxicity induced by chimeric mouse human monoclonal antibody IDEC-C2B8 in CD20-expressing lymphoma cell lines. Cell Immunol, 204(1): 55-63. Forus, A., Berner, J.M., Meza-Zepeda, L.A., Saeter, G., Mischke, D., Fodstad, O. und Myklebost, O. 1998. Molecular characterization of a novel amplicon at 1q21-q22 frequently observed in human sarcomas. Br J Cancer, 78(4): 495-503. Foss, F.M., Koc, Y., Stetler-Stevenson, M.A., Nguyen, D.T., O'Brien, M.C., Turner, R. und Sausville, E.A. 1994. Costimulation of cutaneous T-cell lymphoma cells by interleukin-7 and interleukin-2: potential autocrine or paracrine effectors in the Sezary syndrome. J Clin Oncol, 12(2): 326-35. Funaki, T., Fujiwara, T., Hong, H.S., Misumi, Y., Nishioka, M. und Ikehara, Y. 1996. Identification and characterization of a 230-kDa Golgi-associated protein recognized by autoantibodies from a patient with HBV hepatitis. Cell Struct Funct, 21(1): 63-72. Garbe, C. 1997a. Epidemiologie des Hautkrebses. In: C. Garbe, R. Dummer, R. Kaufmann und W. Tilgen (Editors), Dermatologische Onkologie. Springer Verlag, Berlin, Heidelberg, New York, pp. 40-56. Garbe, C. 1997b. Melanozytäre Nävi und Melanomrisiko. Leitlinien für die Betreuung. In: C. Garbe, R. Dummer, R. Kaufmann und W. Tilgen (Editors), Dermatologische Onkologie. Springer Verlag, Berlin, Heidelberg, New York, pp. 215-130. LITERATURVERZEICHNIS 206 Garbe, C. und Blum, A. 2001. Epidemiology of cutaneous melanoma in Germany and worldwide. Skin Pharmacol Appl Skin Physiol, 14(5): 280-90. Gaugler, B., Brouwenstijn, N., Vantomme, V., Szikora, J.P., Van der Spek, C.W., Patard, J.J., Boon, T., Schrier, P. und Van den Eynde, B.J. 1996. A new gene coding for an antigen recognized by autologous cytolytic T lymphocytes on a human renal carcinoma. Immunogenetics, 44(5): 323-30. Gaugler, B., Van den Eynde, B., van der Bruggen, P., Romero, P., Gaforio, J.J., De Plaen, E., Lethe, B., Brasseur, F. und Boon, T. 1994. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J Exp Med, 179(3): 921-30. Gentilini, P., Laffi, G., La Villa, G., Romanelli, R.G., Buzzelli, G., Casini-Raggi, V., Melani, L., Mazzanti, R., Riccardi, D., Pinzani, M. und Zignego, A.L. 1997. Long course and prognostic factors of virus-induced cirrhosis of the liver. Am J Gastroenterol, 92(1): 6672. Georgieva, J., Milling, A., Orfanos, C.E. und Geilen, C.C. 2002. Magnetic bead RT-PCR: establishment of a new method for detecting circulating melanoma cells. Melanoma Res, 12(4): 309-17. Gerhardt, A., Usener, D., Keese, M., Sturm, J., Schadendorf, D. und Eichmuller, S. 2004. Tissue expression and sero-reactivity of tumor-specific antigens in colorectal cancer. Cancer Lett, 208(2): 197-206. Gilboa, E. 1999. The makings of a tumor rejection antigen. Immunity, 11(3): 263-70. Gilburd, B., Abu-Shakra, M., Shoenfeld, Y., Giordano, A., Bocci, E.B., delle Monache, F. und Gerli, R. 2004. Autoantibodies profile in the sera of patients with Sjogren's syndrome: the ANA evaluation--a homogeneous, multiplexed system. Clin Dev Immunol, 11(1): 53-6. Gjerstorff, M.F., Johansen, L.E., Nielsen, O., Kock, K. und Ditzel, H.J. 2006. Restriction of GAGE protein expression to subpopulations of cancer cells is independent of genotype and may limit the use of GAGE proteins as targets for cancer immunotherapy. Br J Cancer, 94(12): 1864-73. Gjerstorff, M.F. und Ditzel, H.J. 2008. An overview of the GAGE cancer/testis antigen family with the inclusion of newly identified members. Tissue Antigens, 71(3): 187-92. Glennie, M.J. und van de Winkel, J.G. 2003. Renaissance of cancer therapeutic antibodies. Drug Discov Today, 8(11): 503-10. Gollob, J.A., Sciambi, C.J., Peterson, B.L., Richmond, T., Thoreson, M., Moran, K., Dressman, H.K., Jelinek, J. und Issa, J.P. 2006. Phase I trial of sequential low-dose 5-aza-2'deoxycytidine plus high-dose intravenous bolus interleukin-2 in patients with melanoma or renal cell carcinoma. Clin Cancer Res, 12(15): 4619-27. Goodell, V. und Disis, M.L. 2005. Human tumor cell lysates as a protein source for the detection of cancer antigen-specific humoral immunity. J Immunol Methods, 299(1-2): 129-38. Goodell, V., Salazar, L.G., Urban, N., Drescher, C.W., Gray, H., Swensen, R.E., McIntosh, M.W. und Disis, M.L. 2006. Antibody immunity to the p53 oncogenic protein is a prognostic indicator in ovarian cancer. J Clin Oncol, 24(5): 762-8. Gooding, L.R. und Edwards, C.B. 1980. H-2 antigen requirements in the in vitro induction of SV40-specific cytotoxic T lymphocytes. J Immunol, 124(3): 1258-62. Graef, I.A., Chen, F., Chen, L., Kuo, A. und Crabtree, G.R. 2001. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell, 105(7): 86375. LITERATURVERZEICHNIS 207 Gratzl, M. 2002. Haut. In: L.C. Junqueira, J. Carneiro und R.O. Kelley (Editors), Histologie. Springer Verlag, Berlin, Heidelberg, New York, pp. 311-324. Green, J.A., Mudenda, B., Jenkins, J., Leinster, S.J., Tarunina, M., Green, B. und Robertson, L. 1994. Serum p53 auto-antibodies: incidence in familial breast cancer. Eur J Cancer, 30A(5): 580-4. Greene, M.H., Clark, W.H., Jr., Tucker, M.A., Kraemer, K.H., Elder, D.E. und Fraser, M.C. 1985. High risk of malignant melanoma in melanoma-prone families with dysplastic nevi. Ann Intern Med, 102(4): 458-65. Groves, F.D., Linet, M.S., Travis, L.B. und Devesa, S.S. 2000. Cancer surveillance series: nonHodgkin's lymphoma incidence by histologic subtype in the United States from 1978 through 1995. J Natl Cancer Inst, 92(15): 1240-51. Guenzi, E., Topolt, K., Cornali, E., Lubeseder-Martellato, C., Jorg, A., Matzen, K., Zietz, C., Kremmer, E., Nappi, F., Schwemmle, M., Hohenadl, C., Barillari, G., Tschachler, E., Monini, P., Ensoli, B. und Sturzl, M. 2001. The helical domain of GBP-1 mediates the inhibition of endothelial cell proliferation by inflammatory cytokines. Embo J, 20(20): 5568-77. Gueydan, C., Wauquier, C., De Mees, C., Huez, G. und Kruys, V. 2002. Identification of ribosomal proteins specific to higher eukaryotic organisms. J Biol Chem, 277(47): 45034-40. Guitart, J. und Kaul, K. 1999. A new polymerase chain reaction-based method for the detection of T-cell clonality in patients with possible cutaneous T-cell lymphoma. Arch Dermatol, 135(2): 158-62. Guo, B., Zhai, D., Cabezas, E., Welsh, K., Nouraini, S., Satterthwait, A.C. und Reed, J.C. 2003. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature, 423(6938): 456-61. Gure, A.O., Chua, R., Williamson, B., Gonen, M., Ferrera, C.A., Gnjatic, S., Ritter, G., Simpson, A.J., Chen, Y.T., Old, L.J. und Altorki, N.K. 2005. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res, 11(22): 8055-62. Gure, A.O., Stockert, E., Arden, K.C., Boyer, A.D., Viars, C.S., Scanlan, M.J., Old, L.J. und Chen, Y.T. 2000. CT10: a new cancer-testis (CT) antigen homologous to CT7 and the MAGE family, identified by representational-difference analysis. Int J Cancer, 85(5): 726-32. Haffner, A.C., Tassis, A., Zepter, K., Storz, M., Tureci, O., Burg, G. und Nestle, F.O. 2002. Expression of cancer/testis antigens in cutaneous T cell lymphomas. Int J Cancer, 97(5): 668-70. Halaban, R. 1991. Growth factors and tyrosine protein kinases in normal and malignant melanocytes. Cancer Metastasis Rev, 10(2): 129-40. Hall, W.W., Liu, C.R., Schneewind, O., Takahashi, H., Kaplan, M.H., Roupe, G. und Vahlne, A. 1991. Deleted HTLV-I provirus in blood and cutaneous lesions of patients with mycosis fungoides. Science, 253(5017): 317-20. Hamberg, A.P., Korse, C.M., Bonfrer, J.M. und de Gast, G.C. 2003. Serum S100B is suitable for prediction and monitoring of response to chemoimmunotherapy in metastatic malignant melanoma. Melanoma Res, 13(1): 45-9. LITERATURVERZEICHNIS 208 Hammad, A.M., Paris, G.R., van Heuven, W.A., Thompson, I.M. und Fitzsimmons, T.D. 2003. Spontaneous regression of choroidal metastasis from renal cell carcinoma. Am J Ophthalmol, 135(6): 911-3. Hammel, P., Boissier, B., Chaumette, M.T., Piedbois, P., Rotman, N., Kouyoumdjian, J.C., Lubin, R., Delchier, J.C. und Soussi, T. 1997. Detection and monitoring of serum p53 antibodies in patients with colorectal cancer. Gut, 40(3): 356-61. Hammel, P., Leroy-Viard, K., Chaumette, M.T., Villaudy, J., Falzone, M.C., Rouillard, D., Hamelin, R., Boissier, B. und Remvikos, Y. 1999. Correlations between p53-protein accumulation, serum antibodies and gene mutation in colorectal cancer. Int J Cancer, 81(5): 712-8. Hanagiri, T., van Baren, N., Neyns, B., Boon, T. und Coulie, P.G. 2006. Analysis of a rare melanoma patient with a spontaneous CTL response to a MAGE-A3 peptide presented by HLA-A1. Cancer Immunol Immunother, 55(2): 178-84. Hartmann, T.B. 2004. Identifizierung und Charakterisierung von neuen Tumor-assoziierten Genen des malignen Melanoms und des kutanen T-Zell Lymphoms, Ruprecht-KarlsUniversität, Heidelberg, 123 pp. Hartmann, T.B., Thiel, D., Dummer, R., Schadendorf, D. und Eichmuller, S. 2004. SEREX identification of new tumour-associated antigens in cutaneous T-cell lymphoma. Br J Dermatol, 150(2): 252-8. Hashimoto, Y., Niikura, T., Ito, Y., Sudo, H., Hata, M., Arakawa, E., Abe, Y., Kita, Y. und Nishimoto, I. 2001a. Detailed characterization of neuroprotection by a rescue factor humanin against various Alzheimer's disease-relevant insults. J Neurosci, 21(23): 923545. Hashimoto, Y., Niikura, T., Tajima, H., Yasukawa, T., Sudo, H., Ito, Y., Kita, Y., Kawasumi, M., Kouyama, K., Doyu, M., Sobue, G., Koide, T., Tsuji, S., Lang, J., Kurokawa, K. und Nishimoto, I. 2001b. A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Abeta. Proc Natl Acad Sci U S A, 98(11): 6336-41. Hassel, J.C., Meier, R., Joller-Jemelka, H., Burg, G. und Dummer, R. 2004. Serological immunomarkers in cutaneous T cell lymphoma. Dermatology, 209(4): 296-300. Hauschild, A., Michaelsen, J., Brenner, W., Rudolph, P., Glaser, R., Henze, E. und Christophers, E. 1999. Prognostic significance of serum S100B detection compared with routine blood parameters in advanced metastatic melanoma patients. Melanoma Res, 9(2): 155-61. Healy, E., Belgaid, C., Takata, M., Harrison, D., Zhu, N.W., Burd, D.A., Rigby, H.S., Matthews, J.N. und Rees, J.L. 1998. Prognostic significance of allelic losses in primary melanoma. Oncogene, 16(17): 2213-8. Hehl, E.M. 2001. Opinion on the use of the antitumor drug trastuzumab (Herceptin) in patients with metastatic breast cancer in the county Mecklenburg-Vorpommern. Int J Clin Pharmacol Ther, 39(11): 503-6. Heinzerling, L., Kunzi, V., Oberholzer, P.A., Kundig, T., Naim, H. und Dummer, R. 2005. Oncolytic measles virus in cutaneous T-cell lymphomas mounts antitumor immune responses in vivo and targets interferon-resistant tumor cells. Blood, 106(7): 2287-94. Heiskanen, M.A., Bittner, M.L., Chen, Y., Khan, J., Adler, K.E., Trent, J.M. und Meltzer, P.S. 2000. Detection of gene amplification by genomic hybridization to cDNA microarrays. Cancer Res, 60(4): 799-802. LITERATURVERZEICHNIS 209 Herndon, J.M., Stuart, P.M. und Ferguson, T.A. 2005. Peripheral deletion of antigen-specific T cells leads to long-term tolerance mediated by CD8+ cytotoxic cells. J Immunol, 174(7): 4098-104. Herne, K.L., Talpur, R., Breuer-McHam, J., Champlin, R. und Duvic, M. 2003. Cytomegalovirus seropositivity is significantly associated with mycosis fungoides and Sezary syndrome. Blood, 101(6): 2132-6. Herrmann, J.J., Roenigk, H.H., Jr., Hurria, A., Kuzel, T.M., Samuelson, E., Rademaker, A.W. und Rosen, S.T. 1995. Treatment of mycosis fungoides with photochemotherapy (PUVA): long-term follow-up. J Am Acad Dermatol, 33(2 Pt 1): 234-42. Hoey, T., Sun, Y.L., Williamson, K. und Xu, X. 1995. Isolation of two new members of the NFAT gene family and functional characterization of the NF-AT proteins. Immunity, 2(5): 461-72. Hofmann, I., Schnolzer, M., Kaufmann, I. und Franke, W.W. 2002. Symplekin, a constitutive protein of karyo- and cytoplasmic particles involved in mRNA biogenesis in Xenopus laevis oocytes. Mol Biol Cell, 13(5): 1665-76. Hogdall, E.V., Hogdall, C.K., Christensen, L., Glud, E., Blaakaer, J., Bock, J.E., Vuust, J., Norgaard-Pedersen, B. und Kjaer, S.K. 2002. Distribution of p53 codon 72 polymorphisms in ovarian tumour patients and their prognostic significance in ovarian cancer patients. Anticancer Res, 22(3): 1859-64. Hoon, D.S., Yuzuki, D., Hayashida, M. und Morton, D.L. 1995. Melanoma patients immunized with melanoma cell vaccine induce antibody responses to recombinant MAGE-1 antigen. J Immunol, 154(2): 730-7. Hoppe, R.T. 1991. The management of mycosis fungoides at Stanford--standard and innovative treatment programmes. Leukemia, 5 Suppl 1: 46-8. Hoppe, R.T., Medeiros, L.J., Warnke, R.A. und Wood, G.S. 1995. CD8-positive tumorinfiltrating lymphocytes influence the long-term survival of patients with mycosis fungoides. J Am Acad Dermatol, 32(3): 448-53. Houbiers, J.G., van der Burg, S.H., van de Watering, L.M., Tollenaar, R.A., Brand, A., van de Velde, C.J. und Melief, C.J. 1995. Antibodies against p53 are associated with poor prognosis of colorectal cancer. Br J Cancer, 72(3): 637-41. Houghton, A.N., Gold, J.S. und Blachere, N.E. 2001. Immunity against cancer: lessons learned from melanoma. Curr Opin Immunol, 13(2): 134-40. Huang, A.Y., Golumbek, P., Ahmadzadeh, M., Jaffee, E., Pardoll, D. und Levitsky, H. 1994. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science, 264(5161): 961-5. Hufton, S.E., Moerkerk, P.T., Brandwijk, R., de Bruine, A.P., Arends, J.W. und Hoogenboom, H.R. 1999. A profile of differentially expressed genes in primary colorectal cancer using suppression subtractive hybridization. FEBS Lett, 463(1-2): 77-82. Hung, K., Hayashi, R., Lafond-Walker, A., Lowenstein, C., Pardoll, D. und Levitsky, H. 1998. The central role of CD4(+) T cells in the antitumor immune response. J Exp Med, 188(12): 2357-68. Hunt, D.F., Henderson, R.A., Shabanowitz, J., Sakaguchi, K., Michel, H., Sevilir, N., Cox, A.L., Appella, E. und Engelhard, V.H. 1992. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science, 255(5049): 1261-3. LITERATURVERZEICHNIS 210 Hussey, D.J., Parker, N.J., Hussey, N.D., Little, P.F. und Dobrovic, A. 1997. Characterization of a KRAB family zinc finger gene, ZNF195, mapping to chromosome band 11p15.5. Genomics, 45(2): 451-5. Hussussian, C.J., Struewing, J.P., Goldstein, A.M., Higgins, P.A., Ally, D.S., Sheahan, M.D., Clark, W.H., Jr., Tucker, M.A. und Dracopoli, N.C. 1994. Germline p16 mutations in familial melanoma. Nat Genet, 8(1): 15-21. Ishikawa, T., Fujita, T., Suzuki, Y., Okabe, S., Yuasa, Y., Iwai, T. und Kawakami, Y. 2003. Tumor-specific immunological recognition of frameshift-mutated peptides in colon cancer with microsatellite instability. Cancer Res, 63(17): 5564-72. Jager, E., Chen, Y.T., Drijfhout, J.W., Karbach, J., Ringhoffer, M., Jager, D., Arand, M., Wada, H., Noguchi, Y., Stockert, E., Old, L.J. und Knuth, A. 1998. Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med, 187(2): 265-70. Jager, E., Gnjatic, S., Nagata, Y., Stockert, E., Jager, D., Karbach, J., Neumann, A., Rieckenberg, J., Chen, Y.T., Ritter, G., Hoffman, E., Arand, M., Old, L.J. und Knuth, A. 2000a. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci U S A, 97(22): 12198-203. Jager, E., Nagata, Y., Gnjatic, S., Wada, H., Stockert, E., Karbach, J., Dunbar, P.R., Lee, S.Y., Jungbluth, A., Jager, D., Arand, M., Ritter, G., Cerundolo, V., Dupont, B., Chen, Y.T., Old, L.J. und Knuth, A. 2000b. Monitoring CD8 T cell responses to NY-ESO-1: correlation of humoral and cellular immune responses. Proc Natl Acad Sci U S A, 97(9): 4760-5. Jager, E., Stockert, E., Zidianakis, Z., Chen, Y.T., Karbach, J., Jager, D., Arand, M., Ritter, G., Old, L.J. und Knuth, A. 1999. Humoral immune responses of cancer patients against "Cancer-Testis" antigen NY-ESO-1: correlation with clinical events. Int J Cancer, 84(5): 506-10. Janeway, C.A., Travers, P., Walport, M. und Shlomchik, M.J. 2002. Immunologie, 5. Spektrum Akadedmischer Verlag, Heidelberg Berlin, 777 pp. Joberty, G., Petersen, C., Gao, L. und Macara, I.G. 2000. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol, 2(8): 531-9. Kageshita, T., Inoue, Y. und Ono, T. 2003. Spontaneous regression of congenital melanocytic nevi without evidence of the halo phenomenon. Dermatology, 207(2): 193-5. Kamb, A., Shattuck-Eidens, D., Eeles, R., Liu, Q., Gruis, N.A., Ding, W., Hussey, C., Tran, T., Miki, Y., Weaver-Feldhaus, J. et al. 1994. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet, 8(1): 23-6. Kanavaros, P., Bai, M., Stefanaki, K., Poussias, G., Rontogianni, D., Zioga, E., Gorgoulis, V. und Agnantis, N.J. 2001. Immunohistochemical expression of the p53, mdm2, p21/Waf1, Rb, p16, Ki67, cyclin D1, cyclin A and cyclin B1 proteins and apoptotic index in Tcell lymphomas. Histol Histopathol, 16(2): 377-86. Kanavaros, P., Ioannidou, D., Tzardi, M., Datseris, G., Katsantonis, J., Delidis, G. und Tosca, A. 1994. Mycosis fungoides: expression of C-myc p62 p53, bcl-2 and PCNA proteins and absence of association with Epstein-Barr virus. Pathol Res Pract, 190(8): 767-74. Kawakami, Y., Dang, N., Wang, X., Tupesis, J., Robbins, P.F., Wang, R.F., Wunderlich, J.R., Yannelli, J.R. und Rosenberg, S.A. 2000. Recognition of shared melanoma antigens in LITERATURVERZEICHNIS 211 association with major HLA-A alleles by tumor infiltrating T lymphocytes from 123 patients with melanoma. J Immunother, 23(1): 17-27. Kenmochi, N., Kawaguchi, T., Rozen, S., Davis, E., Goodman, N., Hudson, T.J., Tanaka, T. und Page, D.C. 1998. A map of 75 human ribosomal protein genes. Genome Res, 8(5): 50923. Kerl H, S.W. 1987. Classification and staging. In: S.W. Burg G (Editor), EORTC/BMFT Cutaneous Lymphoma Project Group: Recommendations for staging and therapy of cutaneous lymphomas, pp. 1-10. Kessler, J.F., Jones, S.E., Levine, N., Lynch, P.J., Booth, A.R. und Meyskens, F.L., Jr. 1987. Isotretinoin and cutaneous helper T-cell lymphoma (mycosis fungoides). Arch Dermatol, 123(2): 201-4. Kikuchi, A., Nishikawa, T., Ikeda, Y. und Yamaguchi, K. 1997. Absence of human Tlymphotropic virus type I in Japanese patients with cutaneous T-cell lymphoma. Blood, 89(5): 1529-32. Kim, E.J., Hess, S., Richardson, S.K., Newton, S., Showe, L.C., Benoit, B.M., Ubriani, R., Vittorio, C.C., Junkins-Hopkins, J.M., Wysocka, M. und Rook, A.H. 2005. Immunopathogenesis and therapy of cutaneous T cell lymphoma. J Clin Invest, 115(4): 798-812. Kim, Y.H., Liu, H.L., Mraz-Gernhard, S., Varghese, A. und Hoppe, R.T. 2003. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol, 139(7): 857-66. Kiniwa, Y., Fujita, T., Akada, M., Ito, K., Shofuda, T., Suzuki, Y., Yamamoto, A., Saida, T. und Kawakami, Y. 2001. Tumor antigens isolated from a patient with vitiligo and T-cellinfiltrated melanoma. Cancer Res, 61(21): 7900-7. Kirkin, A.F., Dzhandzhugazyan, K.N. und Zeuthen, J. 2002. Cancer/testis antigens: structural and immunobiological properties. Cancer Invest, 20(2): 222-36. Kittler, H., Pehamberger, H., Wolff, K. und Binder, M. 2002. Diagnostic accuracy of dermoscopy. Lancet Oncol, 3(3): 159-65. Knox, S., Hoppe, R.T., Maloney, D., Gibbs, I., Fowler, S., Marquez, C., Cornbleet, P.J. und Levy, R. 1996. Treatment of cutaneous T-cell lymphoma with chimeric anti-CD4 monoclonal antibody. Blood, 87(3): 893-9. Komatsu, N., Shichijo, S., Nakagawa, M. und Itoh, K. 2004. New multiplexed flow cytometric assay to measure anti-peptide antibody: a novel tool for monitoring immune responses to peptides used for immunization. Scand J Clin Lab Invest, 64(6): 535-45. Kondoh, N., Nishina, Y., Tsuchida, J., Koga, M., Tanaka, H., Uchida, K., Inazawa, J., Taketo, M., Nozaki, M., Nojima, H., Matsumiya, K., Namiki, M., Okuyama, A. und Nishimune, Y. 1997. Assignment of synaptonemal complex protein 1 (SCP1) to human chromosome 1p13 by fluorescence in situ hybridization and its expression in the testis. Cytogenet Cell Genet, 78(2): 103-4. Kooy, J., Toh, B.H., Pettitt, J.M., Erlich, R. und Gleeson, P.A. 1992. Human autoantibodies as reagents to conserved Golgi components. Characterization of a peripheral, 230-kDa compartment-specific Golgi protein. J Biol Chem, 267(28): 20255-63. Kragel, A.H., Travis, W.D., Steis, R.G., Rosenberg, S.A. und Roberts, W.C. 1990. Myocarditis or acute myocardial infarction associated with interleukin-2 therapy for cancer. Cancer, 66(7): 1513-6. LITERATURVERZEICHNIS 212 Kruit, W.H., van Ojik, H.H., Brichard, V.G., Escudier, B., Dorval, T., Dreno, B., Patel, P., van Baren, N., Avril, M.F., Piperno, S., Khammari, A., Stas, M., Ritter, G., Lethe, B., Godelaine, D., Brasseur, F., Zhang, Y., van der Bruggen, P., Boon, T., Eggermont, A.M. und Marchand, M. 2005. Phase 1/2 study of subcutaneous and intradermal immunization with a recombinant MAGE-3 protein in patients with detectable metastatic melanoma. Int J Cancer, 117(4): 596-604. Kupper, T.S. und Fuhlbrigge, R.C. 2004. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol, 4(3): 211-22. Kurts, C., Heath, W.R., Carbone, F.R., Allison, J., Miller, J.F. und Kosaka, H. 1996. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med, 184(3): 923-30. Laduron, S., Deplus, R., Zhou, S., Kholmanskikh, O., Godelaine, D., De Smet, C., Hayward, S.D., Fuks, F., Boon, T. und De Plaen, E. 2004. MAGE-A1 interacts with adaptor SKIP and the deacetylase HDAC1 to repress transcription. Nucleic Acids Res, 32(14): 434050. Lagarkova, M.A., Koroleva, E.P., Kuprash, D.V., Boitchenko, V.E., Kashkarova, U.A., Nedospasov, S.A. und Shebzukhov, Y.V. 2003. Evaluation of humoral response to tumor antigens using recombinant expression-based serological mini-arrays (SMARTA). Immunol Lett, 85(1): 71-4. Lamerz, R. 1997. AFP isoforms and their clinical significance (overview). Anticancer Res, 17(4B): 2927-30. Lauritzen, A.F., Vejlsgaard, G.L., Hou-Jensen, K. und Ralfkiaer, E. 1995. p53 protein expression in cutaneous T-cell lymphomas. Br J Dermatol, 133(1): 32-6. Laxminarayan, K.M., Chan, B.K., Tetaz, T., Bird, P.I. und Mitchell, C.A. 1994. Characterization of a cDNA encoding the 43-kDa membrane-associated inositol-polyphosphate 5phosphatase. J Biol Chem, 269(25): 17305-10. Le Naour, F., Misek, D.E., Krause, M.C., Deneux, L., Giordano, T.J., Scholl, S. und Hanash, S.M. 2001. Proteomics-based identification of RS/DJ-1 as a novel circulating tumor antigen in breast cancer. Clin Cancer Res, 7(11): 3328-35. Lee, P.P., Yee, C., Savage, P.A., Fong, L., Brockstedt, D., Weber, J.S., Johnson, D., Swetter, S., Thompson, J., Greenberg, P.D., Roederer, M. und Davis, M.M. 1999. Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med, 5(6): 677-85. Lee, S.Y., Obata, Y., Yoshida, M., Stockert, E., Williamson, B., Jungbluth, A.A., Chen, Y.T., Old, L.J. und Scanlan, M.J. 2003. Immunomic analysis of human sarcoma. Proc Natl Acad Sci U S A, 100(5): 2651-6. Leiter, U., Schmid, R.M., Kaskel, P., Peter, R.U. und Krahn, G. 2000. Antiapoptotic bcl-2 and bcl-xL in advanced malignant melanoma. Arch Dermatol Res, 292(5): 225-32. Lennette, E.T., Winberg, G., Yadav, M., Enblad, G. und Klein, G. 1995. Antibodies to LMP2A/2B in EBV-carrying malignancies. Eur J Cancer, 31A(11): 1875-8. Lethe, B., Lucas, S., Michaux, L., De Smet, C., Godelaine, D., Serrano, A., De Plaen, E. und Boon, T. 1998. LAGE-1, a new gene with tumor specificity. Int J Cancer, 76(6): 903-8. Ley, R.D. 1997. Ultraviolet radiation A-induced precursors of cutaneous melanoma in Monodelphis domestica. Cancer Res, 57(17): 3682-4. LITERATURVERZEICHNIS 213 Litvak, D.A., Gupta, R.K., Yee, R., Wanek, L.A., Ye, W. und Morton, D.L. 2004. Endogenous immune response to early- and intermediate-stage melanoma is correlated with outcomes and is independent of locoregional relapse and standard prognostic factors. J Am Coll Surg, 198(1): 27-35. Liu, Y., Li, J., Kim, B.O., Pace, B.S. und He, J.J. 2002. HIV-1 Tat protein-mediated transactivation of the HIV-1 long terminal repeat promoter is potentiated by a novel nuclear Tat-interacting protein of 110 kDa, Tip110. J Biol Chem, 277(26): 23854-63. Lubin, R., Zalcman, G., Bouchet, L., Tredanel, J., Legros, Y., Cazals, D., Hirsch, A. und Soussi, T. 1995. Serum p53 antibodies as early markers of lung cancer. Nat Med, 1(7): 701-2. Luftl, M., Schuler, G. und Jungbluth, A.A. 2004. Melanoma or not? Cancer testis antigens may help. Br J Dermatol, 151(6): 1213-8. Luke, M.R., Kjer-Nielsen, L., Brown, D.L., Stow, J.L. und Gleeson, P.A. 2003. GRIP domainmediated targeting of two new coiled-coil proteins, GCC88 and GCC185, to subcompartments of the trans-Golgi network. J Biol Chem, 278(6): 4216-26. Lundin, J., Osterborg, A., Brittinger, G., Crowther, D., Dombret, H., Engert, A., Epenetos, A., Gisselbrecht, C., Huhn, D., Jaeger, U., Thomas, J., Marcus, R., Nissen, N., Poynton, C., Rankin, E., Stahel, R., Uppenkamp, M., Willemze, R. und Mellstedt, H. 1998. CAMPATH-1H monoclonal antibody in therapy for previously treated low-grade nonHodgkin's lymphomas: a phase II multicenter study. European Study Group of CAMPATH-1H Treatment in Low-Grade Non-Hodgkin's Lymphoma. J Clin Oncol, 16(10): 3257-63. Lurquin, C., Van Pel, A., Mariame, B., De Plaen, E., Szikora, J.P., Janssens, C., Reddehase, M.J., Lejeune, J. und Boon, T. 1989. Structure of the gene of tum- transplantation antigen P91A: the mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell, 58(2): 293-303. Ma, J.H., Sui, Y.F., Ye, J., Huang, Y.Y., Li, Z.S., Chen, G.S., Qu, P., Song, H.P. und Zhang, X.M. 2005. Heat shock protein 70/MAGE-3 fusion protein vaccine can enhance cellular and humoral immune responses to MAGE-3 in vivo. Cancer Immunol Immunother, 54(9): 907-14. Macaluso, M., Russo, G., Cinti, C., Bazan, V., Gebbia, N. und Russo, A. 2002. Ras family genes: an interesting link between cell cycle and cancer. J Cell Physiol, 192(2): 125-30. Maier, T., Tun-Kyi, A., Tassis, A., Jungius, K.P., Burg, G., Dummer, R. und Nestle, F.O. 2003. Vaccination of patients with cutaneous T-cell lymphoma using intranodal injection of autologous tumor-lysate-pulsed dendritic cells. Blood, 102(7): 2338-44. Maio, M., Coral, S., Sigalotti, L., Elisei, R., Romei, C., Rossi, G., Cortini, E., Colizzi, F., Fenzi, G., Altomonte, M., Pinchera, A. und Vitale, M. 2003. Analysis of cancer/testis antigens in sporadic medullary thyroid carcinoma: expression and humoral response to NY-ESO1. J Clin Endocrinol Metab, 88(2): 748-54. Mandic, M., Almunia, C., Vicel, S., Gillet, D., Janjic, B., Coval, K., Maillere, B., Kirkwood, J.M. und Zarour, H.M. 2003. The alternative open reading frame of LAGE-1 gives rise to multiple promiscuous HLA-DR-restricted epitopes recognized by T-helper 1-type tumorreactive CD4+ T cells. Cancer Res, 63(19): 6506-15. Mangel, J., Barth, D., Berinstein, N.L. und Imrie, K.R. 2003. Spontaneous regression of Hodgkin's disease: two case reports and a review of the literature. Hematology, 8(3): 1916. LITERATURVERZEICHNIS 214 Mansilla, F., Hansen, L.L., Jakobsen, H., Kjeldgaard, N.O., Clark, B.F. und Knudsen, C.R. 2005. Deconstructing PTI-1: PTI-1 is a truncated, but not mutated, form of translation elongatin factor 1A1, eEF1A1. Biochim Biophys Acta, 1727(2): 116-24. Mao, X., Lillington, D., Scarisbrick, J.J., Mitchell, T., Czepulkowski, B., Russell-Jones, R., Young, B. und Whittaker, S.J. 2002. Molecular cytogenetic analysis of cutaneous T-cell lymphomas: identification of common genetic alterations in Sezary syndrome and mycosis fungoides. Br J Dermatol, 147(3): 464-75. Maouche-Chretien, L., Deleu, N., Badoual, C., Fraissignes, P., Berger, R., Gaulard, P., Romeo, P.H. und Leroy-Viard, K. 1998. Identification of a novel cDNA, encoding a cytoskeletal associated protein, differentially expressed in diffuse large B cell lymphomas. Oncogene, 17(10): 1245-51. Marchand, M., van Baren, N., Weynants, P., Brichard, V., Dreno, B., Tessier, M.H., Rankin, E., Parmiani, G., Arienti, F., Humblet, Y., Bourlond, A., Vanwijck, R., Lienard, D., Beauduin, M., Dietrich, P.Y., Russo, V., Kerger, J., Masucci, G., Jager, E., De Greve, J., Atzpodien, J., Brasseur, F., Coulie, P.G., van der Bruggen, P. und Boon, T. 1999. Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int J Cancer, 80(2): 21930. Margolin, J.F., Friedman, J.R., Meyer, W.K., Vissing, H., Thiesen, H.J. und Rauscher, F.J., 3rd 1994. Kruppel-associated boxes are potent transcriptional repression domains. Proc Natl Acad Sci U S A, 91(10): 4509-13. Marolleau, J.P., Baccard, M., Flageul, B., Rybojad, M., Laroche, L., Verola, O., Brandely, M., Morel, P. und Gisselbrecht, C. 1995. High-dose recombinant interleukin-2 in advanced cutaneous T-cell lymphoma. Arch Dermatol, 131(5): 574-9. Marti, R.M., Estrach, T., Reverter, J.C. und Mascaro, J.M. 1991. Prognostic clinicopathologic factors in cutaneous T-cell lymphoma. Arch Dermatol, 127(10): 1511-6. Masuda, E.S., Naito, Y., Tokumitsu, H., Campbell, D., Saito, F., Hannum, C., Arai, K. und Arai, N. 1995. NFATx, a novel member of the nuclear factor of activated T cells family that is expressed predominantly in the thymus. Mol Cell Biol, 15(5): 2697-706. Matsuzaki, Y., Hashimoto, S., Fujita, T., Suzuki, T., Sakurai, T., Matsushima, K. und Kawakami, Y. 2005. Systematic identification of human melanoma antigens using serial analysis of gene expression (SAGE). J Immunother, 28(1): 10-9. McGregor, J.M., Crook, T., Fraser-Andrews, E.A., Rozycka, M., Crossland, S., Brooks, L. und Whittaker, S.J. 1999. Spectrum of p53 gene mutations suggests a possible role for ultraviolet radiation in the pathogenesis of advanced cutaneous lymphomas. J Invest Dermatol, 112(3): 317-21. McLaughlin, P., Grillo-Lopez, A.J., Link, B.K., Levy, R., Czuczman, M.S., Williams, M.E., Heyman, M.R., Bence-Bruckler, I., White, C.A., Cabanillas, F., Jain, V., Ho, A.D., Lister, J., Wey, K., Shen, D. und Dallaire, B.K. 1998. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol, 16(8): 2825-33. Meloche, S., Beatty, B.G. und Pellerin, J. 1996. Primary structure, expression and chromosomal locus of a human homolog of rat ERK3. Oncogene, 13(7): 1575-9. Mengubas, K., Jabbar, S.A., Nye, K.E., Wilkes, S., Hoffbrand, A.V. und Wickremasinghe, R.G. 1994. Inactivation of calcium ion-regulating inositol polyphosphate second messengers is impaired in subpopulations of human leukemia cells. Leukemia, 8(10): 1718-25. LITERATURVERZEICHNIS 215 Meschede, W., Zumbach, K., Braspenning, J., Scheffner, M., Benitez-Bribiesca, L., Luande, J., Gissmann, L. und Pawlita, M. 1998. Antibodies against early proteins of human papillomaviruses as diagnostic markers for invasive cervical cancer. J Clin Microbiol, 36(2): 475-80. Meuwissen, R.L., Meerts, I., Hoovers, J.M., Leschot, N.J. und Heyting, C. 1997. Human synaptonemal complex protein 1 (SCP1): isolation and characterization of the cDNA and chromosomal localization of the gene. Genomics, 39(3): 377-84. Michels, A.A., Fraldi, A., Li, Q., Adamson, T.E., Bonnet, F., Nguyen, V.T., Sedore, S.C., Price, J.P., Price, D.H., Lania, L. und Bensaude, O. 2004. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. Embo J, 23(13): 2608-19. Middleton, M.R., Grob, J.J., Aaronson, N., Fierlbeck, G., Tilgen, W., Seiter, S., Gore, M., Aamdal, S., Cebon, J., Coates, A., Dreno, B., Henz, M., Schadendorf, D., Kapp, A., Weiss, J., Fraass, U., Statkevich, P., Muller, M. und Thatcher, N. 2000. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol, 18(1): 158-66. Milisav, I. und Affara, N.A. 1998. A potential human axonemal dynein heavy-chain gene maps to 17q25. Mamm Genome, 9(5): 404-7. Milne, E., Johnston, R., Cross, D., Giles-Corti, B. und English, D.R. 2002. Effect of a schoolbased sun-protection intervention on the development of melanocytic nevi in children. Am J Epidemiol, 155(8): 739-45. Mitsudomi, T., Suzuki, S., Yatabe, Y., Nishio, M., Kuwabara, M., Gotoh, K., Hatooka, S., Shinoda, M., Suyama, M., Ogawa, M., Takahashi, T. und Ariyoshi, Y. 1998. Clinical implications of p53 autoantibodies in the sera of patients with non-small-cell lung cancer. J Natl Cancer Inst, 90(20): 1563-8. Miyashiro, I., Kuo, C., Huynh, K., Iida, A., Morton, D., Bilchik, A., Giuliano, A. und Hoon, D.S. 2001. Molecular strategy for detecting metastatic cancers with use of multiple tumorspecific MAGE-A genes. Clin Chem, 47(3): 505-12. Molin, L., Thomsen, K., Volden, G. und Groth, O. 1981. Photochemotherapy (PUVA) in the pretumour stage of mycosis fungoides: a report from the Scandinavian Mycosis Fungoides Study Group. Acta Derm Venereol, 61(1): 47-51. Molin, L., Thomsen, K., Volden, G., Jensen, P., Knudsen, E., Nyfors, A. und Schmidt, H. 1987. Retinoids and systemic chemotherapy in cases of advanced mycosis fungoides. A report from the Scandinavian Mycosis Fungoides Group. Acta Derm Venereol, 67(2): 179-82. Moreau-Aubry, A., Le Guiner, S., Labarriere, N., Gesnel, M.C., Jotereau, F. und Breathnach, R. 2000. A processed pseudogene codes for a new antigen recognized by a CD8(+) T cell clone on melanoma. J Exp Med, 191(9): 1617-24. Morton, D.L., Wen, D.R., Foshag, L.J., Essner, R. und Cochran, A. 1993. Intraoperative lymphatic mapping and selective cervical lymphadenectomy for early-stage melanomas of the head and neck. J Clin Oncol, 11(9): 1751-6. Muche, J.M., Lukowsky, A., Asadullah, K., Gellrich, S. und Sterry, W. 1997. Demonstration of frequent occurrence of clonal T cells in the peripheral blood of patients with primary cutaneous T-cell lymphoma. Blood, 90(4): 1636-42. Muche, J.M. und Sterry, W. 2002. Vaccination therapy for cutaneous T-cell lymphoma. Clin Exp Dermatol, 27(7): 602-7. Mulcahy, M.F. und Benson, A.B., 3rd 1999. The role of carcinoembryonic antigen monitoring in management of colorectal cancer. Curr Oncol Rep, 1(2): 168-72. LITERATURVERZEICHNIS 216 Munger, K., Scheffner, M., Huibregtse, J.M. und Howley, P.M. 1992. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv, 12: 197-217. Murray, R.J., Kurilla, M.G., Brooks, J.M., Thomas, W.A., Rowe, M., Kieff, E. und Rickinson, A.B. 1992. Identification of target antigens for the human cytotoxic T cell response to Epstein-Barr virus (EBV): implications for the immune control of EBV-positive malignancies. J Exp Med, 176(1): 157-68. Musholt, T.J., Goodfellow, P.J., Scheumann, G.F., Pichlmayr, R., Wells, S.A., Jr. und Moley, J.F. 1997. Differential display in primary and metastatic medullary thyroid carcinoma. J Surg Res, 69(1): 94-100. Nadali, G., Vinante, F., Stein, H., Todeschini, G., Tecchio, C., Morosato, L., Chilosi, M., Menestrina, F., Kinney, M.C., Greer, J.P. und et al. 1995. Serum levels of the soluble form of CD30 molecule as a tumor marker in CD30+ anaplastic large-cell lymphoma. J Clin Oncol, 13(6): 1355-60. Nagao, T., Higashitsuji, H., Nonoguchi, K., Sakurai, T., Dawson, S., Mayer, R.J., Itoh, K. und Fujita, J. 2003. MAGE-A4 interacts with the liver oncoprotein gankyrin and suppresses its tumorigenic activity. J Biol Chem, 278(12): 10668-74. Naitoh, J. und Belldegrun, A. 1998. Gene therapy for prostate cancer. Prostate Cancer Prostatic Dis, 1(4): 189-196. Nakatsura, T., Senju, S., Ito, M., Nishimura, Y. und Itoh, K. 2002. Cellular and humoral immune responses to a human pancreatic cancer antigen, coactosin-like protein, originally defined by the SEREX method. Eur J Immunol, 32(3): 826-36. Navas, I.C., Algara, P., Mateo, M., Martinez, P., Garcia, C., Rodriguez, J.L., Vanaclocha, F., Barrientos, N., Iglesias, L., Sanchez, L., Piris, M.A. und Ortiz-Romero, P. 2002. p16(INK4a) is selectively silenced in the tumoral progression of mycosis fungoides. Lab Invest, 82(2): 123-32. Nencioni, A., Gruenbach, F., Patrone, F. und Brossart, P. 2004. Anticancer vaccination strategies. Ann Oncol, 15 Suppl 4: iv153-60. Neri, A., Fracchiolla, N.S., Roscetti, E., Garatti, S., Trecca, D., Boletini, A., Perletti, L., Baldini, L., Maiolo, A.T. und Berti, E. 1995. Molecular analysis of cutaneous B- and T-cell lymphomas. Blood, 86(8): 3160-72. Nesterova, M.V., Johnson, N., Cheadle, C., Bates, S.E., Mani, S., Stratakis, C.A., Khan, I.U., Gupta, R.K. und Cho-Chung, Y.S. 2006. Autoantibody cancer biomarker: extracellular protein kinase A. Cancer Res, 66(18): 8971-4. Nestle, F.O., Alijagic, S., Gilliet, M., Sun, Y., Grabbe, S., Dummer, R., Burg, G. und Schadendorf, D. 1998. Vaccination of melanoma patients with peptide- or tumor lysatepulsed dendritic cells. Nat Med, 4(3): 328-32. Neumann, F., Wagner, C., Preuss, K.D., Kubuschok, B., Schormann, C., Stevanovic, S. und Pfreundschuh, M. 2005. Identification of an epitope derived from the cancer testis antigen HOM-TES-14/SCP1 and presented by dendritic cells to circulating CD4+ T cells. Blood, 106(9): 3105-13. Ng, E.K., Chan, K.K., Wong, C.H., Tsui, S.K., Ngai, S.M., Lee, S.M., Kotaka, M., Lee, C.Y., Waye, M.M. und Fung, K.P. 2002. Interaction of the heart-specific LIM domain protein, FHL2, with DNA-binding nuclear protein, hNP220. J Cell Biochem, 84(3): 556-66. Nickoloff, B.J. 1988. Light-microscopic assessment of 100 patients with patch/plaque-stage mycosis fungoides. Am J Dermatopathol, 10(6): 469-77. LITERATURVERZEICHNIS 217 Nickoloff, B.J., Basham, T.Y., Merigan, T.C. und Morhenn, V.B. 1984. Antiproliferative effects of recombinant alpha- and gamma-interferons on cultured human keratinocytes. Lab Invest, 51(6): 697-701. Nishikawa, H., Tanida, K., Ikeda, H., Sakakura, M., Miyahara, Y., Aota, T., Mukai, K., Watanabe, M., Kuribayashi, K., Old, L.J. und Shiku, H. 2001. Role of SEREX-defined immunogenic wild-type cellular molecules in the development of tumor-specific immunity. Proc Natl Acad Sci U S A, 98(25): 14571-6. Old, L.J. 2001. Cancer/testis (CT) antigens - a new link between gametogenesis and cancer. Cancer Immun, 1: 1. Old, L.J. und Chen, Y.T. 1998. New paths in human cancer serology. J Exp Med, 187(8): 11637. Olsen, E.A., Rosen, S.T., Vollmer, R.T., Variakojis, D., Roenigk, H.H., Jr., Diab, N. und Zeffren, J. 1989. Interferon alfa-2a in the treatment of cutaneous T cell lymphoma. J Am Acad Dermatol, 20(3): 395-407. Oltra, S., Martinez, F., Orellana, C., Grau, E., Fernandez, J.M., Canete, A. und Castel, V. 2004. Minimal residual disease in neuroblastoma: to GAGE or not to GAGE. Oncol Res, 14(6): 291-5. Opalka, D., Lachman, C.E., MacMullen, S.A., Jansen, K.U., Smith, J.F., Chirmule, N. und Esser, M.T. 2003. Simultaneous quantitation of antibodies to neutralizing epitopes on virus-like particles for human papillomavirus types 6, 11, 16, and 18 by a multiplexed luminex assay. Clin Diagn Lab Immunol, 10(1): 108-15. Orchard, G.E. 2000. Comparison of immunohistochemical labelling of melanocyte differentiation antibodies melan-A, tyrosinase and HMB 45 with NKIC3 and S100 protein in the evaluation of benign naevi and malignant melanoma. Histochem J, 32(8): 475-81. Ouchida, R., Kusuhara, M., Shimizu, N., Hisada, T., Makino, Y., Morimoto, C., Handa, H., Ohsuzu, F. und Tanaka, H. 2003. Suppression of NF-kappaB-dependent gene expression by a hexamethylene bisacetamide-inducible protein HEXIM1 in human vascular smooth muscle cells. Genes Cells, 8(2): 95-107. Overwijk, W.W., Lee, D.S., Surman, D.R., Irvine, K.R., Touloukian, C.E., Chan, C.C., Carroll, M.W., Moss, B., Rosenberg, S.A. und Restifo, N.P. 1999. Vaccination with a recombinant vaccinia virus encoding a "self" antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4(+) T lymphocytes. Proc Natl Acad Sci U S A, 96(6): 2982-7. Overwijk, W.W. und Restifo, N.P. 2000. Autoimmunity and the immunotherapy of cancer: targeting the "self" to destroy the "other". Crit Rev Immunol, 20(6): 433-50. Palmer, J.S., Duffy, D.L., Box, N.F., Aitken, J.F., O'Gorman, L.E., Green, A.C., Hayward, N.K., Martin, N.G. und Sturm, R.A. 2000. Melanocortin-1 receptor polymorphisms and risk of melanoma: is the association explained solely by pigmentation phenotype? Am J Hum Genet, 66(1): 176-86. Paludan, C., Schmid, D., Landthaler, M., Vockerodt, M., Kube, D., Tuschl, T. und Munz, C. 2005. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science, 307(5709): 593-6. Pancake, B.A., Zucker-Franklin, D. und Coutavas, E.E. 1995. The cutaneous T cell lymphoma, mycosis fungoides, is a human T cell lymphotropic virus-associated disease. A study of 50 patients. J Clin Invest, 95(2): 547-54. LITERATURVERZEICHNIS 218 Pape, K.A., Merica, R., Mondino, A., Khoruts, A. und Jenkins, M.K. 1998. Direct evidence that functionally impaired CD4+ T cells persist in vivo following induction of peripheral tolerance. J Immunol, 160(10): 4719-29. Parada, L.A., Hallen, M., Tranberg, K.G., Hagerstrand, I., Bondeson, L., Mitelman, F. und Johansson, B. 1998. Frequent rearrangements of chromosomes 1, 7, and 8 in primary liver cancer. Genes Chromosomes Cancer, 23(1): 26-35. Pardoll, D.M. 1998. Cancer vaccines. Nat Med, 4(5 Suppl): 525-31. Paschen, A., Dittmar, K.E., Grenningloh, R., Rohde, M., Schadendorf, D., Domann, E., Chakraborty, T. und Weiss, S. 2000. Human dendritic cells infected by Listeria monocytogenes: induction of maturation, requirements for phagolysosomal escape and antigen presentation capacity. Eur J Immunol, 30(12): 3447-56. Pawson, R., Catovsky, D. und Schulz, T.F. 1996. Lack of evidence of HHV-8 in mature T-cell lymphoproliferative disorders. Lancet, 348(9039): 1450-1. Pfreundschuh, M., Shiku, H., Takahashi, T., Ueda, R., Ransohoff, J., Oettgen, H.F. und Old, L.J. 1978. Serological analysis of cell surface antigens of malignant human brain tumors. Proc Natl Acad Sci U S A, 75(10): 5122-6. Piekarz, R.L., Robey, R.W., Zhan, Z., Kayastha, G., Sayah, A., Abdeldaim, A.H., Torrico, S. und Bates, S.E. 2004. T-cell lymphoma as a model for the use of histone deacetylase inhibitors in cancer therapy: impact of depsipeptide on molecular markers, therapeutic targets, and mechanisms of resistance. Blood, 103(12): 4636-43. Pollock, P.M., Harper, U.L., Hansen, K.S., Yudt, L.M., Stark, M., Robbins, C.M., Moses, T.Y., Hostetter, G., Wagner, U., Kakareka, J., Salem, G., Pohida, T., Heenan, P., Duray, P., Kallioniemi, O., Hayward, N.K., Trent, J.M. und Meltzer, P.S. 2003. High frequency of BRAF mutations in nevi. Nat Genet, 33(1): 19-20. Polsky, D., Bastian, B.C., Hazan, C., Melzer, K., Pack, J., Houghton, A., Busam, K., CordonCardo, C. und Osman, I. 2001. HDM2 protein overexpression, but not gene amplification, is related to tumorigenesis of cutaneous melanoma. Cancer Res, 61(20): 7642-6. Polsky, D., Melzer, K., Hazan, C., Panageas, K.S., Busam, K., Drobnjak, M., Kamino, H., Spira, J.G., Kopf, A.W., Houghton, A., Cordon-Cardo, C. und Osman, I. 2002. HDM2 protein overexpression and prognosis in primary malignant melanoma. J Natl Cancer Inst, 94(23): 1803-6. Prabhakar, U., Eirikis, E. und Davis, H.M. 2002. Simultaneous quantification of proinflammatory cytokines in human plasma using the LabMAP assay. J Immunol Methods, 260(1-2): 207-18. Prasad, M.L., Jungbluth, A.A., Patel, S.G., Iversen, K., Hoshaw-Woodard, S. und Busam, K.J. 2004. Expression and significance of cancer testis antigens in primary mucosal melanoma of the head and neck. Head Neck, 26(12): 1053-7. Pschyrembel 1998. Klinisches Wörterbuch, 258. Walter de Gruyter. Qin, J.Z., Kamarashev, J., Zhang, C.L., Dummer, R., Burg, G. und Dobbeling, U. 2001. Constitutive and interleukin-7- and interleukin-15-stimulated DNA binding of STAT and novel factors in cutaneous T cell lymphoma cells. J Invest Dermatol, 117(3): 583-9. Qiu, J., Madoz-Gurpide, J., Misek, D.E., Kuick, R., Brenner, D.E., Michailidis, G., Haab, B.B., Omenn, G.S. und Hanash, S. 2004. Development of natural protein microarrays for diagnosing cancer based on an antibody response to tumor antigens. J Proteome Res, 3(2): 261-7. LITERATURVERZEICHNIS 219 Querfeld, C., Rosen, S.T., Guitart, J. und Kuzel, T.M. 2005. The spectrum of cutaneous T-cell lymphomas: new insights into biology and therapy. Curr Opin Hematol, 12(4): 273-8. Quiros, P.A., Jones, G.W., Kacinski, B.M., Braverman, I.M., Heald, P.W., Edelson, R.L. und Wilson, L.D. 1997. Total skin electron beam therapy followed by adjuvant psoralen/ultraviolet-A light in the management of patients with T1 and T2 cutaneous Tcell lymphoma (mycosis fungoides). Int J Radiat Oncol Biol Phys, 38(5): 1027-35. Ralfkiaer, E. 1994. Controversies and discussion on early diagnosis of cutaneous T-cell lymphoma. Phenotyping. Dermatol Clin, 12(2): 329-34. Ramsay, D.L., Halperin, P.S. und Zeleniuch-Jacquotte, A. 1988. Topical mechlorethamine therapy for early stage mycosis fungoides. J Am Acad Dermatol, 19(4): 684-91. Ramsay, D.L., Lish, K.M., Yalowitz, C.B. und Soter, N.A. 1992. Ultraviolet-B phototherapy for early-stage cutaneous T-cell lymphoma. Arch Dermatol, 128(7): 931-3. Reynolds, S.R., Albrecht, J., Shapiro, R.L., Roses, D.F., Harris, M.N., Conrad, A., ZeleniuchJacquotte, A. und Bystryn, J.C. 2003. Changes in the presence of multiple markers of circulating melanoma cells correlate with clinical outcome in patients with melanoma. Clin Cancer Res, 9(4): 1497-502. Roeder, C., Schuler-Thurner, B., Berchtold, S., Vieth, G., Driesch, P., Schuler, G. und Luftl, M. 2005. MAGE-A3 is a frequent tumor antigen of metastasized melanoma. Arch Dermatol Res, 296(7): 314-9. Rogers, G.S. und Braun, S.M. 2002. Prognostic factors. Dermatol Clin, 20(4): 647-58, viii-ix. Rommens, J.M., Durocher, F., McArthur, J., Tonin, P., LeBlanc, J.F., Allen, T., Samson, C., Ferri, L., Narod, S., Morgan, K. et al. 1995. Generation of a transcription map at the HSD17B locus centromeric to BRCA1 at 17q21. Genomics, 28(3): 530-42. Rook, A.H. und Heald, P. 1995. The immunopathogenesis of cutaneous T-cell lymphoma. Hematol Oncol Clin North Am, 9(5): 997-1010. Rook, A.H., Wood, G.S., Yoo, E.K., Elenitsas, R., Kao, D.M., Sherman, M.L., Witmer, W.K., Rockwell, K.A., Shane, R.B., Lessin, S.R. und Vonderheid, E.C. 1999. Interleukin-12 therapy of cutaneous T-cell lymphoma induces lesion regression and cytotoxic T-cell responses. Blood, 94(3): 902-8. Rook, A.H., Zaki, M.H., Wysocka, M., Wood, G.S., Duvic, M., Showe, L.C., Foss, F., Shapiro, M., Kuzel, T.M., Olsen, E.A., Vonderheid, E.C., Laliberte, R. und Sherman, M.L. 2001. The role for interleukin-12 therapy of cutaneous T cell lymphoma. Ann N Y Acad Sci, 941: 177-84. Rosen, S.T. nd Foss, F.M. 1995. Chemotherapy for mycosis fungoides and the Sezary syndrome. Hematol Oncol Clin North Am, 9(5): 1109-16. Rosenberg, S.A. 1996. Development of cancer immunotherapies based on identification of the genes encoding cancer regression antigens. J Natl Cancer Inst, 88(22): 1635-44. Rosenberg, S.A., Packard, B.S., Aebersold, P.M., Solomon, D., Topalian, S.L., Toy, S.T., Simon, P., Lotze, M.T., Yang, J.C., Seipp, C.A. und et al. 1988. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med, 319(25): 1676-80. Rubinfeld, B., Albert, I., Porfiri, E., Munemitsu, S. und Polakis, P. 1997. Loss of beta-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res, 57(20): 4624-30. LITERATURVERZEICHNIS 220 Russell-Jones, R. und Whittaker, S. 1999. T-cell receptor gene analysis in the diagnosis of Sezary syndrome. J Am Acad Dermatol, 41(2 Pt 1): 254-9. Sahin, U., Koslowski, M., Tureci, O., Eberle, T., Zwick, C., Romeike, B., Moringlane, J.R., Schwechheimer, K., Feiden, W. und Pfreundschuh, M. 2000. Expression of cancer testis genes in human brain tumors. Clin Cancer Res, 6(10): 3916-22. Sahin, U., Tureci, O., Chen, Y.T., Seitz, G., Villena-Heinsen, C., Old, L.J. und Pfreundschuh, M. 1998. Expression of multiple cancer/testis (CT) antigens in breast cancer and melanoma: basis for polyvalent CT vaccine strategies. Int J Cancer, 78(3): 387-9. Sahin, U., Türeci, Ö. und Pfreundschuh, M. 1999. Vom Immunsystem erkennbare Antigene auf menschlichen Malignomen. Onkologe, 5: 659-667. Sahin, U., Tureci, O., Schmitt, H., Cochlovius, B., Johannes, T., Schmits, R., Stenner, F., Luo, G., Schobert, I. und Pfreundschuh, M. 1995. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci U S A, 92(25): 11810-3. Saiki, I., Dunegan, M.A., Fann, A.V. und Koff, W.C. 1986. Regulatory effects on macrophages of human recombinant interferons-alpha. J Interferon Res, 6(5): 603-11. Saleh, M.N., LeMaistre, C.F., Kuzel, T.M., Foss, F., Platanias, L.C., Schwartz, G., Ratain, M., Rook, A., Freytes, C.O., Craig, F., Reuben, J., Sams, M.W. und Nichols, J.C. 1998. Antitumor activity of DAB389IL-2 fusion toxin in mycosis fungoides. J Am Acad Dermatol, 39(1): 63-73. Salgaller, M.L. und Lodge, P.A. 1998. Use of cellular and cytokine adjuvants in the immunotherapy of cancer. J Surg Oncol, 68(2): 122-38. Samlowski, W.E., Leachman, S.A., Wade, M., Cassidy, P., Porter-Gill, P., Busby, L., Wheeler, R., Boucher, K., Fitzpatrick, F., Jones, D.A. und Karpf, A.R. 2005. Evaluation of a 7-day continuous intravenous infusion of decitabine: inhibition of promoter-specific and global genomic DNA methylation. J Clin Oncol, 23(17): 3897-905. Sander, C.A., Simon, M., Puchta, U., Raffeld, M. und Kind, P. 1996. HHV-8 in lymphoproliferative lesions in skin. Lancet, 348(9025): 475-6. Sang Lee, J., Gyu Park, S., Park, H., Seol, W., Lee, S. und Kim, S. 2002. Interaction network of human aminoacyl-tRNA synthetases and subunits of elongation factor 1 complex. Biochem Biophys Res Commun, 291(1): 158-64. Sanjo, H., Kawai, T. und Akira, S. 1998. DRAKs, novel serine/threonine kinases related to death-associated protein kinase that trigger apoptosis. J Biol Chem, 273(44): 29066-71. Sarris, A.H., Esgleyes-Ribot, T., Crow, M., Broxmeyer, H.E., Karasavvas, N., Pugh, W., Grossman, D., Deisseroth, A. und Duvic, M. 1995. Cytokine loops involving interferongamma and IP-10, a cytokine chemotactic for CD4+ lymphocytes: an explanation for the epidermotropism of cutaneous T-cell lymphoma? Blood, 86(2): 651-8. Sasatomi, T., Suefuji, Y., Matsunaga, K., Yamana, H., Miyagi, Y., Araki, Y., Ogata, Y., Itoh, K. und Shirouzu, K. 2002. Expression of tumor rejection antigens in colorectal carcinomas. Cancer, 94(6): 1636-41. Sauter, E.R., Yeo, U.C., von Stemm, A., Zhu, W., Litwin, S., Tichansky, D.S., Pistritto, G., Nesbit, M., Pinkel, D., Herlyn, M. und Bastian, B.C. 2002. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res, 62(11): 3200-6. Scanlan, M.J., Chen, Y.T., Williamson, B., Gure, A.O., Stockert, E., Gordan, J.D., Tureci, O., Sahin, U., Pfreundschuh, M. und Old, L.J. 1998. Characterization of human colon cancer antigens recognized by autologous antibodies. Int J Cancer, 76(5): 652-8. LITERATURVERZEICHNIS 221 Scanlan, M.J., Gout, I., Gordon, C.M., Williamson, B., Stockert, E., Gure, A.O., Jager, D., Chen, Y.T., Mackay, A., O'Hare, M.J. und Old, L.J. 2001. Humoral immunity to human breast cancer: antigen definition and quantitative analysis of mRNA expression. Cancer Immun, 1: 4. Scanlan, M.J., Simpson, A.J. und Old, L.J. 2004. The cancer/testis genes: review, standardization, and commentary. Cancer Immun, 4: 1. Scanlan, M.J., Welt, S., Gordon, C.M., Chen, Y.T., Gure, A.O., Stockert, E., Jungbluth, A.A., Ritter, G., Jager, D., Jager, E., Knuth, A. und Old, L.J. 2002. Cancer-related serological recognition of human colon cancer: identification of potential diagnostic and immunotherapeutic targets. Cancer Res, 62(14): 4041-7. Scarisbrick, J.J., Whittaker, S., Evans, A.V., Fraser-Andrews, E.A., Child, F.J., Dean, A. und Russell-Jones, R. 2001. Prognostic significance of tumor burden in the blood of patients with erythrodermic primary cutaneous T-cell lymphoma. Blood, 97(3): 624-30. Schadendorf, D., Ugurel, S., Schuler-Thurner, B., Nestle, F.O., Enk, A., Brocker, E.B., Grabbe, S., Rittgen, W., Edler, L., Sucker, A., Zimpfer-Rechner, C., Berger, T., Kamarashev, J., Burg, G., Jonuleit, H., Tuttenberg, A., Becker, J.C., Keikavoussi, P., Kampgen, E. und Schuler, G. 2006. Dacarbazine (DTIC) versus vaccination with autologous peptidepulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol, 17(4): 56370. Schmekel, K., Meuwissen, R.L., Dietrich, A.J., Vink, A.C., van Marle, J., van Veen, H. und Heyting, C. 1996. Organization of SCP1 protein molecules within synaptonemal complexes of the rat. Exp Cell Res, 226(1): 20-30. Schuchter, L., Schultz, D.J., Synnestvedt, M., Trock, B.J., Guerry, D., Elder, D.E., Elenitsas, R., Clark, W.H. und Halpern, A.C. 1996. A prognostic model for predicting 10-year survival in patients with primary melanoma. The Pigmented Lesion Group. Ann Intern Med, 125(5): 369-75. Sehr, P., Zumbach, K. und Pawlita, M. 2001. A generic capture ELISA for recombinant proteins fused to glutathione S-transferase: validation for HPV serology. J Immunol Methods, 253(1-2): 153-62. Seideman, J. und Peritt, D. 2002. A novel monoclonal antibody screening method using the Luminex-100 microsphere system. J Immunol Methods, 267(2): 165-71. Serrano, A., Lethe, B., Delroisse, J.M., Lurquin, C., De Plaen, E., Brasseur, F., Rimoldi, D. und Boon, T. 1999. Quantitative evaluation of the expression of MAGE genes in tumors by limiting dilution of cDNA libraries. Int J Cancer, 83(5): 664-9. Serrano, M., Hannon, G.J. und Beach, D. 1993. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature, 366(6456): 704-7. Serra-Pages, C., Kedersha, N.L., Fazikas, L., Medley, Q., Debant, A. und Streuli, M. 1995. The LAR transmembrane protein tyrosine phosphatase and a coiled-coil LAR-interacting protein co-localize at focal adhesions. Embo J, 14(12): 2827-38. Serra-Pages, C., Medley, Q.G., Tang, M., Hart, A. und Streuli, M. 1998. Liprins, a family of LAR transmembrane protein-tyrosine phosphatase-interacting proteins. J Biol Chem, 273(25): 15611-20. Shankaran, V., Ikeda, H., Bruce, A.T., White, J.M., Swanson, P.E., Old, L.J. und Schreiber, R.D. 2001. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature, 410(6832): 1107-11. LITERATURVERZEICHNIS 222 Shimada, H., Ochiai, T. und Nomura, F. 2003. Titration of serum p53 antibodies in 1,085 patients with various types of malignant tumors: a multiinstitutional analysis by the Japan p53 Antibody Research Group. Cancer, 97(3): 682-9. Shimakage, M., Sasagawa, T., Kawahara, K., Yutsudo, M., Kusuoka, H. und Kozuka, T. 2001. Expression of Epstein-Barr virus in cutaneous T-cell lymphoma including mycosis fungoides. Int J Cancer, 92(2): 226-31. Sigalotti, L., Fratta, E., Coral, S., Tanzarella, S., Danielli, R., Colizzi, F., Fonsatti, E., Traversari, C., Altomonte, M. und Maio, M. 2004. Intratumor heterogeneity of cancer/testis antigens expression in human cutaneous melanoma is methylation-regulated and functionally reverted by 5-aza-2'-deoxycytidine. Cancer Res, 64(24): 9167-71. Silverman, J., Takai, H., Buonomo, S.B., Eisenhaber, F. und de Lange, T. 2004. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev, 18(17): 2108-19. Slamon, D.J., Leyland-Jones, B., Shak, S., Fuchs, H., Paton, V., Bajamonde, A., Fleming, T., Eiermann, W., Wolter, J., Pegram, M., Baselga, J. und Norton, L. 2001. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med, 344(11): 783-92. Slater, D.N. 1991. Cutaneous lymphoproliferative disorders: an assessment of recent investigative techniques. Br J Dermatol, 124(4): 309-23. Sparrow, L.E., Soong, R., Dawkins, H.J., Iacopetta, B.J. und Heenan, P.J. 1995. p53 gene mutation and expression in naevi and melanomas. Melanoma Res, 5(2): 93-100. Stahlecker, J., Gauger, A., Bosserhoff, A., Buttner, R., Ring, J. und Hein, R. 2000. MIA as a reliable tumor marker in the serum of patients with malignant melanoma. Anticancer Res, 20(6D): 5041-4. Stang, A. und Jockel, K.H. 2003. Changing patterns of skin melanoma mortality in West Germany from 1968 through 1999. Ann Epidemiol, 13(6): 436-42. Statistisches Bundesamt Deutschland 2003. www.destatis.de. Steinwaerder, D.S., Carlson, C.A. und Lieber, A. 2001. Human papilloma virus E6 and E7 proteins support DNA replication of adenoviruses deleted for the E1A and E1B genes. Mol Ther, 4(3): 211-6. Steplewski, Z., Lubeck, M.D. und Koprowski, H. 1983. Human macrophages armed with murine immunoglobulin G2a antibodies to tumors destroy human cancer cells. Science, 221(4613): 865-7. Sterry, W., Gellrich, S., Audring, H., Schulze, P. und Jahn, S. 1997. Diagnostik und Klassifikation kutaner Lymphome. In: C. Garbe, R. Dummer, R. Kaufmann and W. Tilgen (Editors), Dermatologische Onkologie. Springer Verlag, Berlin, Heidelberg, New York, pp. 427-435. Stockert, E., Jager, E., Chen, Y.T., Scanlan, M.J., Gout, I., Karbach, J., Arand, M., Knuth, A. und Old, L.J. 1998. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens. J Exp Med, 187(8): 1349-54. Stone, B., Schummer, M., Paley, P.J., Thompson, L., Stewart, J., Ford, M., Crawford, M., Urban, N., O'Briant, K. und Nelson, B.H. 2003. Serologic analysis of ovarian tumor antigens reveals a bias toward antigens encoded on 17q. Int J Cancer, 104(1): 73-84. Stott, F.J., Bates, S., James, M.C., McConnell, B.B., Starborg, M., Brookes, S., Palmero, I., Ryan, K., Hara, E., Vousden, K.H. und Peters, G. 1998. The alternative product from the LITERATURVERZEICHNIS 223 human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. Embo J, 17(17): 5001-14. Strid, M.A., Dalby, T., Molbak, K. und Krogfelt, K.A. 2007. Kinetics of the human antibody response against non-typhoid Salmonella determined by LPS ELISA. Clin Vaccine Immunol. Stuhler, G. und Walden, P. 1994. Recruitment of helper T cells for induction of tumour rejection by cytolytic T lymphocytes. Cancer Immunol Immunother, 39(5): 342-5. Sturm, R.A., Box, N.F. und Ramsay, M. 1998. Human pigmentation genetics: the difference is only skin deep. Bioessays, 20(9): 712-21. Sundram, U., Harvell, J.D., Rouse, R.V. und Natkunam, Y. 2003. Expression of the B-cell proliferation marker MUM1 by melanocytic lesions and comparison with S100, gp100 (HMB45), and MelanA. Mod Pathol, 16(8): 802-10. Tajima, H., Niikura, T., Hashimoto, Y., Ito, Y., Kita, Y., Terashita, K., Yamazaki, K., Koto, A., Aiso, S. und Nishimoto, I. 2002. Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer's disease-related insults. Neurosci Lett, 324(3): 227-31. Talapatra, S., Wagner, J.D. und Thompson, C.B. 2002. Elongation factor-1 alpha is a selective regulator of growth factor withdrawal and ER stress-induced apoptosis. Cell Death Differ, 9(8): 856-61. Talpur, R., Lifshitz, O., Breuer-Mcham, J. und Duvic, M. 2002. Increased serum immunoglobulin levels are common in mycosis fungoides and Sezary syndrome. J Am Acad Dermatol, 47(5): 685-91. Tammela, J., Uenaka, A., Ono, T., Noguchi, Y., Jungbluth, A.A., Mhawech-Fauceglia, P., Qian, F., Schneider, S., Sharma, S., Driscoll, D., Lele, S., Old, L.J., Nakayama, E. und Odunsi, K. 2006. OY-TES-1 expression and serum immunoreactivity in epithelial ovarian cancer. Int J Oncol, 29(4): 903-10. Tan, K., Shaw, A.L., Madsen, B., Jensen, K., Taylor-Papadimitriou, J. und Freemont, P.S. 2003. Human PLU-1 Has transcriptional repression properties and interacts with the developmental transcription factors BF-1 and PAX9. J Biol Chem, 278(23): 20507-13. Taniura, H., Matsumoto, K. und Yoshikawa, K. 1999. Physical and functional interactions of neuronal growth suppressor necdin with p53. J Biol Chem, 274(23): 16242-8. Templin, M.F., Stoll, D., Bachmann, J. und Joos, T.O. 2004. Protein microarrays and multiplexed sandwich immunoassays: what beats the beads? Comb Chem High Throughput Screen, 7(3): 223-9. Tensen, C.P., Vermeer, M.H., van der Stoop, P.M., van Beek, P., Scheper, R.J., Boorsma, D.M. und Willemze, R. 1998. Epidermal interferon-gamma inducible protein-10 (IP-10) and monokine induced by gamma-interferon (Mig) but not IL-8 mRNA expression is associated with epidermotropism in cutaneous T cell lymphomas. J Invest Dermatol, 111(2): 222-6. Thangavelu, M., Finn, W.G., Yelavarthi, K.K., Roenigk, H.H., Jr., Samuelson, E., Peterson, L., Kuzel, T.M. und Rosen, S.T. 1997. Recurring structural chromosome abnormalities in peripheral blood lymphocytes of patients with mycosis fungoides/Sezary syndrome. Blood, 89(9): 3371-7. Thews, G., Mutschler, E. und Vaupel, P. 1999. Anatomie, Physiologie, Pathophysiologie des Menschen, 5. Wissenschaftliche Verlagsgesellschaft mbH Stuttgart, 751-761 pp. LITERATURVERZEICHNIS 224 Thurner, B., Haendle, I., Roder, C., Dieckmann, D., Keikavoussi, P., Jonuleit, H., Bender, A., Maczek, C., Schreiner, D., von den Driesch, P., Brocker, E.B., Steinman, R.M., Enk, A., Kampgen, E. und Schuler, G. 1999. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med, 190(11): 1669-78. Toes, R.E., Ossendorp, F., Offringa, R. und Melief, C.J. 1999. CD4 T cells and their role in antitumor immune responses. J Exp Med, 189(5): 753-6. Tomita, Y., Kimura, M., Tanikawa, T., Nishiyama, T., Morishita, H., Takeda, M., Fujiwara, M. und Sato, S. 1993. Immunohistochemical detection of intercellular adhesion molecule-1 (ICAM-1) and major histocompatibility complex class I antigens in seminoma. J Urol, 149(3): 659-63. Trauth, B.C., Klas, C., Peters, A.M., Matzku, S., Moller, P., Falk, W., Debatin, K.M. und Krammer, P.H. 1989. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science, 245(4915): 301-5. Trefzer, U., Weingart, G., Chen, Y., Herberth, G., Adrian, K., Winter, H., Audring, H., Guo, Y., Sterry, W. und Walden, P. 2000. Hybrid cell vaccination for cancer immune therapy: first clinical trial with metastatic melanoma. Int J Cancer, 85(5): 618-26. Tucker, M.A. und Goldstein, A.M. 2003. Melanoma etiology: where are we? Oncogene, 22(20): 3042-52. Tureci, O., Sahin, U., Vollmar, E., Siemer, S., Gottert, E., Seitz, G., Parkkila, A.K., Shah, G.N., Grubb, J.H., Pfreundschuh, M. und Sly, W.S. 1998. Human carbonic anhydrase XII: cDNA cloning, expression, and chromosomal localization of a carbonic anhydrase gene that is overexpressed in some renal cell cancers. Proc Natl Acad Sci U S A, 95(13): 760813. UICC 1993. TNM Supplement 1993. In: P. Hermanek, D.E. Henson, R.V.P. Hutter, B. O'Sullivan and L.H. Sobin (Editors), A commentary on uniform use. Springer Verlag, Berlin, Heidelberg, New York, pp. 85-87. Usener, D., Gerhardt, A., Schadendorf, D. und Eichmuller, S. 2003a. Seroreactivity against MAGE-A and LAGE-1 proteins in melanoma patients. Br J Dermatol, 149(2): 282-8. Usener, D., Schadendorf, D., Koch, J., Dubel, S. und Eichmuller, S. 2003b. cTAGE: a cutaneous T cell lymphoma associated antigen family with tumor-specific splicing. J Invest Dermatol, 121(1): 198-206. Valverde, P., Healy, E., Jackson, I., Rees, J.L. und Thody, A.J. 1995. Variants of the melanocyte-stimulating hormone receptor gene are associated with red hair and fair skin in humans. Nat Genet, 11(3): 328-30. Valverde, P., Healy, E., Sikkink, S., Haldane, F., Thody, A.J., Carothers, A., Jackson, I.J. und Rees, J.L. 1996. The Asp84Glu variant of the melanocortin 1 receptor (MC1R) is associated with melanoma. Hum Mol Genet, 5(10): 1663-6. Van den Eynde, B., Peeters, O., De Backer, O., Gaugler, B., Lucas, S. und Boon, T. 1995. A new family of genes coding for an antigen recognized by autologous cytolytic T lymphocytes on a human melanoma. J Exp Med, 182(3): 689-98. Van Den Eynde, B.J., Gaugler, B., Probst-Kepper, M., Michaux, L., Devuyst, O., Lorge, F., Weynants, P. und Boon, T. 1999. A new antigen recognized by cytolytic T lymphocytes on a human kidney tumor results from reverse strand transcription. J Exp Med, 190(12): 1793-800. LITERATURVERZEICHNIS 225 van den Oord, J.J., Maes, A., Stas, M., Nuyts, J., Battocchio, S., Kasran, A., Garmyn, M., De Wever, I. und De Wolf-Peeters, C. 1996. CD40 is a prognostic marker in primary cutaneous malignant melanoma. Am J Pathol, 149(6): 1953-61. van der Bruggen, P., Traversari, C., Chomez, P., Lurquin, C., De Plaen, E., Van den Eynde, B., Knuth, A. und Boon, T. 1991. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science, 254(5038): 1643-7. van Rhee, F., Szmania, S.M., Zhan, F., Gupta, S.K., Pomtree, M., Lin, P., Batchu, R.B., Moreno, A., Spagnoli, G., Shaughnessy, J. und Tricot, G. 2005. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood, 105(10): 3939-44. Vasmatzis, G., Essand, M., Brinkmann, U., Lee, B. und Pastan, I. 1998. Discovery of three genes specifically expressed in human prostate by expressed sequence tag database analysis. Proc Natl Acad Sci U S A, 95(1): 300-4. Vaughan, H.A., St Clair, F., Scanlan, M.J., Chen, Y.T., Maraskovsky, E., Sizeland, A., Old, L.J. und Cebon, J. 2004. The humoral immune response to head and neck cancer antigens as defined by the serological analysis of tumor antigens by recombinant cDNA expression cloning. Cancer Immun, 4: 5. Volkenandt, M., Schmidt, M., Konz, B., Gummer, M., Hein, R., Plewig, G. und Holzel, D. 1999. Klinisch-epidemiologische Daten von Patienten mit malignen Melanomen aus dem Bereich des Tumorzentrums München von 1977-1997. Hautarzt, 50(7): 470-8. Vonderheid, E.C., Tan, E.T., Kantor, A.F., Shrager, L., Micaily, B. und Van Scott, E.J. 1989. Long-term efficacy, curative potential, and carcinogenicity of topical mechlorethamine chemotherapy in cutaneous T cell lymphoma. J Am Acad Dermatol, 20(3): 416-28. Vonderheid, E.C., Zhang, Q., Lessin, S.R., Polansky, M., Abrams, J.T., Bigler, R.D. und Wasik, M.A. 1998. Use of serum soluble interleukin-2 receptor levels to monitor the progression of cutaneous T-cell lymphoma. J Am Acad Dermatol, 38(2 Pt 1): 207-20. Vousden, K.H. und Lu, X., 2002. Live or let die: the cell's response to p53. Nat Rev Cancer, 2(8): 594-604. Wadle, A., Kubuschok, B., Imig, J., Wuellner, B., Wittig, C., Zwick, C., Mischo, A., Waetzig, K., Romeike, B.F., Lindemann, W., Schilling, M., Pfreundschuh, M. und Renner, C. 2006. Serological immune response to cancer testis antigens in patients with pancreatic cancer. Int J Cancer. Wang, F., Bade, E., Kuniyoshi, C., Spears, L., Jeffery, G., Marty, V., Groshen, S. und Weber, J. 1999a. Phase I trial of a MART-1 peptide vaccine with incomplete Freund's adjuvant for resected high-risk melanoma. Clin Cancer Res, 5(10): 2756-65. Wang, Q., Holmes, D.I., Powell, S.M., Lu, Q.L. und Waxman, J. 2002. Analysis of stromalepithelial interactions in prostate cancer identifies PTPCAAX2 as a potential oncogene. Cancer Lett, 175(1): 63-9. Wang, R.F. 2001. The role of MHC class II-restricted tumor antigens and CD4+ T cells in antitumor immunity. Trends Immunol, 22(5): 269-76. Wang, R.F., Appella, E., Kawakami, Y., Kang, X. und Rosenberg, S.A. 1996. Identification of TRP-2 as a human tumor antigen recognized by cytotoxic T lymphocytes. J Exp Med, 184(6): 2207-16. Wang, R.F., Johnston, S.L., Zeng, G., Topalian, S.L., Schwartzentruber, D.J. und Rosenberg, S.A. 1998. A breast and melanoma-shared tumor antigen: T cell responses to antigenic peptides translated from different open reading frames. J Immunol, 161(7): 3598-606. LITERATURVERZEICHNIS 226 Wang, R.F., Robbins, P.F., Kawakami, Y., Kang, X.Q. und Rosenberg, S.A. 1995. Identification of a gene encoding a melanoma tumor antigen recognized by HLA-A31-restricted tumorinfiltrating lymphocytes. J Exp Med, 181(2): 799-804. Wang, R.F., Wang, X., Atwood, A.C., Topalian, S.L. und Rosenberg, S.A. 1999b. Cloning genes encoding MHC class II-restricted antigens: mutated CDC27 as a tumor antigen. Science, 284(5418): 1351-4. Wang, R.F., Wang, X. und Rosenberg, S.A. 1999c. Identification of a novel major histocompatibility complex class II-restricted tumor antigen resulting from a chromosomal rearrangement recognized by CD4(+) T cells. J Exp Med, 189(10): 165968. Wang, X., Yu, J., Sreekumar, A., Varambally, S., Shen, R., Giacherio, D., Mehra, R., Montie, J.E., Pienta, K.J., Sanda, M.G., Kantoff, P.W., Rubin, M.A., Wei, J.T., Ghosh, D. und Chinnaiyan, A.M. 2005. Autoantibody signatures in prostate cancer. N Engl J Med, 353(12): 1224-35. Wasik, M.A., Vonderheid, E.C., Bigler, R.D., Marti, R., Lessin, S.R., Polansky, M. und Kadin, M.E. 1996. Increased serum concentration of the soluble interleukin-2 receptor in cutaneous T-cell lymphoma. Clinical and prognostic implications. Arch Dermatol, 132(1): 42-7. Waterboer, T. 2005. Multiplex-Serologie - Simultane Analyse von Antikörper-Antworten gegen humane Papillomviren (HPV) mit Protein-Arrays, Ruprecht-Karls-Universität, Heidelberg, 187 pp. Waterboer, T., Sehr, P., Michael, K.M., Franceschi, S., Nieland, J.D., Joos, T.O., Templin, M.F. und Pawlita, M. 2005. Multiplex human papillomavirus serology based on in situpurified glutathione s-transferase fusion proteins. Clin Chem, 51(10): 1845-53. Wei, M.X., Yang, Y., Ge, Y.C. und Xu, P. 2006. Functional characterization of hNUDC as a novel accumulator that specifically acts on in vitro megakaryocytopoiesis and in vivo platelet production. J Cell Biochem, 98(2): 429-39. Weinlich, G., Bitterlich, W., Mayr, V., Fritsch, P.O. und Zelger, B. 2003. Metallothioneinoverexpression as a prognostic factor for progression and survival in melanoma. A prospective study on 520 patients. Br J Dermatol, 149(3): 535-41. Weinstock, M.A. und Horm, J.W. 1988. Mycosis fungoides in the United States. Increasing incidence and descriptive epidemiology. Jama, 260(1): 42-6. Wheatley, K., Ives, N., Hancock, B. und Gore, M. 2002. Interferon as adjuvant treatment for melanoma. Lancet, 360(9336): 878. Whiteman, D.C., Whiteman, C.A. und Green, A.C. 2001. Childhood sun exposure as a risk factor for melanoma: a systematic review of epidemiologic studies. Cancer Causes Control, 12(1): 69-82. Whittaker, S.J., Marsden, J.R., Spittle, M. und Russell Jones, R. 2003. Joint British Association of Dermatologists and U.K. Cutaneous Lymphoma Group guidelines for the management of primary cutaneous T-cell lymphomas. Br J Dermatol, 149(6): 1095-1107. Willemze, R., Jaffe, E.S., Burg, G., Cerroni, L., Berti, E., Swerdlow, S.H., Ralfkiaer, E., Chimenti, S., Diaz-Perez, J.L., Duncan, L.M., Grange, F., Harris, N.L., Kempf, W., Kerl, H., Kurrer, M., Knobler, R., Pimpinelli, N., Sander, C., Santucci, M., Sterry, W., Vermeer, M.H., Wechsler, J., Whittaker, S. und Meijer, C.J. 2005. WHO-EORTC classification for cutaneous lymphomas. Blood, 105(10): 3768-85. LITERATURVERZEICHNIS 227 Willemze, R., Kerl, H., Sterry, W., Berti, E., Cerroni, L., Chimenti, S., Diaz-Perez, J.L., Geerts, M.L., Goos, M., Knobler, R., Ralfkiaer, E., Santucci, M., Smith, N., Wechsler, J., van Vloten, W.A. und Meijer, C.J. 1997. EORTC classification for primary cutaneous lymphomas: a proposal from the Cutaneous Lymphoma Study Group of the European Organization for Research and Treatment of Cancer. Blood, 90(1): 354-71. Wittmann, B.M., Wang, N. und Montano, M.M. 2003. Identification of a novel inhibitor of breast cell growth that is down-regulated by estrogens and decreased in breast tumors. Cancer Res, 63(16): 5151-8. Wolfel, T., Hauer, M., Schneider, J., Serrano, M., Wolfel, C., Klehmann-Hieb, E., De Plaen, E., Hankeln, T., Meyer zum Buschenfelde, K.H. und Beach, D. 1995. A p16INK4ainsensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science, 269(5228): 1281-4. Xiang, Z.Q., Yang, Y., Wilson, J.M. und Ertl, H.C. 1996. A replication-defective human adenovirus recombinant serves as a highly efficacious vaccine carrier. Virology, 219(1): 220-7. Yamashita, N., Ishibashi, H., Hayashida, K., Kudo, J., Takenaka, K., Itoh, K. und Niho, Y. 1996. High frequency of the MAGE-1 gene expression in hepatocellular carcinoma. Hepatology, 24(6): 1437-40. Yang, D., Chertov, O. und Oppenheim, J.J. 2001. The role of mammalian antimicrobial peptides and proteins in awakening of innate host defenses and adaptive immunity. Cell Mol Life Sci, 58(7): 978-89. Yang, D., Nakao, M., Shichijo, S., Sasatomi, T., Takasu, H., Matsumoto, H., Mori, K., Hayashi, A., Yamana, H., Shirouzu, K. und Itoh, K. 1999. Identification of a gene coding for a protein possessing shared tumor epitopes capable of inducing HLA-A24-restricted cytotoxic T lymphocytes in cancer patients. Cancer Res, 59(16): 4056-63. Yazdi, A.S., Medeiros, L.J., Puchta, U., Thaller, E., Flaig, M.J. und Sander, C.A. 2003. Improved detection of clonality in cutaneous T-cell lymphomas using laser capture microdissection. J Cutan Pathol, 30(8): 486-91. Young, A.M., Marsden, J., Goodman, A., Burton, A. und Dunn, J.A. 2001. Prospective randomized comparison of dacarbazine (DTIC) versus DTIC plus interferon-alpha (IFNalpha) in metastatic melanoma. Clin Oncol (R Coll Radiol), 13(6): 458-65. Yuen, M.F. und Lai, C.L. 2005. Serological markers of liver cancer. Best Pract Res Clin Gastroenterol, 19(1): 91-9. Zackheim, H.S. 1994. Topical carmustine (BCNU) for patch/plaque mycosis fungoides. Semin Dermatol, 13(3): 202-6. Zackheim, H.S., Kashani-Sabet, M. und Amin, S. 1998. Topical corticosteroids for mycosis fungoides. Experience in 79 patients. Arch Dermatol, 134(8): 949-54. Zambon, A., Mandruzzato, S., Parenti, A., Macino, B., Dalerba, P., Ruol, A., Merigliano, S., Zaninotto, G. und Zanovello, P. 2001. MAGE, BAGE, and GAGE gene expression in patients with esophageal squamous cell carcinoma and adenocarcinoma of the gastric cardia. Cancer, 91(10): 1882-8. Zeng, G., Touloukian, C.E., Wang, X., Restifo, N.P., Rosenberg, S.A. und Wang, R.F. 2000. Identification of CD4+ T cell epitopes from NY-ESO-1 presented by HLA-DR molecules. J Immunol, 165(2): 1153-9. LITERATURVERZEICHNIS 228 Zerp, S.F., van Elsas, A., Peltenburg, L.T. und Schrier, P.I. 1999. p53 mutations in human cutaneous melanoma correlate with sun exposure but are not always involved in melanomagenesis. Br J Cancer, 79(5-6): 921-6. Zhang, J.Y., Casiano, C.A., Peng, X.X., Koziol, J.A., Chan, E.K. und Tan, E.M. 2003. Enhancement of antibody detection in cancer using panel of recombinant tumorassociated antigens. Cancer Epidemiol Biomarkers Prev, 12(2): 136-43. Zhang, M.Y., Huang, N.N., Clawson, G.A., Osmani, S.A., Pan, W., Xin, P., Razzaque, M.S. und Miller, B.A. 2002. Involvement of the fungal nuclear migration gene nudC human homolog in cell proliferation and mitotic spindle formation. Exp Cell Res, 273(1): 73-84. Zhang, Y., Renkvist, N., Sun, Z., Schuler-Thurner, B., Glaichenhaus, N., Schuler, G., Boon, T., van der Bruggen, P. und Colau, D. 2005a. A polyclonal anti-vaccine CD4 T cell response detected with HLA-DP4 multimers in a melanoma patient vaccinated with MAGE3.DP4-peptide-pulsed dendritic cells. Eur J Immunol, 35(4): 1066-75. Zhang, Y., Sun, Z., Nicolay, H., Meyer, R.G., Renkvist, N., Stroobant, V., Corthals, J., Carrasco, J., Eggermont, A.M., Marchand, M., Thielemans, K., Wolfel, T., Boon, T. und van der Bruggen, P. 2005b. Monitoring of anti-vaccine CD4 T cell frequencies in melanoma patients vaccinated with a MAGE-3 protein. J Immunol, 174(4): 2404-11. Zhao, Z., Lee, C.C., Monckton, D.G., Yazdani, A., Coolbaugh, M.I., Li, X., Bailey, J., Shen, Y. und Caskey, C.T. 1996. Characterization and genomic mapping of genes and pseudogenes of a new human protein tyrosine phosphatase. Genomics, 35(1): 172-81. Zhou, D., Li, P., Lin, Y., Lott, J.M., Hislop, A.D., Canaday, D.H., Brutkiewicz, R.R. und Blum, J.S. 2005. Lamp-2a facilitates MHC class II presentation of cytoplasmic antigens. Immunity, 22(5): 571-81. Zhu, A.X., Zhao, Y., Moller, D.E. und Flier, J.S. 1994. Cloning and characterization of p97MAPK, a novel human homolog of rat ERK-3. Mol Cell Biol, 14(12): 8202-11. Zuo, L., Weger, J., Yang, Q., Goldstein, A.M., Tucker, M.A., Walker, G.J., Hayward, N. und Dracopoli, N.C. 1996. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet, 12(1): 97-9. ANHANG 229 8 ANHANG Anhang 1: Darstellung der p-Werte für alle humanen Antigene in der MultiplexSerologie laut „Fisher’s Exact Test“ Ctrl: Kontrollen, MM: Malignes Melanom, MM I: MM Stadium I, CTCL: Kutanes T-Zell Lymphom, CTCL I: CTCL Stadium I, n.e.: nicht evaluierbar. Antigen Kollektiv Ctrl – MM Ctrl – MM I – MM II – MM III – MM IV Ctrl – MM I Ctrl – MM II Ctrl – MM III Ctrl – MM IV Ctrl - CTCL Ctrl – CTCL I – CTCL II – CTCL III – CTCL IV Ctrl – CTCL I Ctrl – CTCL II Ctrl – CTCL III Ctrl – CTCL IV MAGE -A1 0,8092 0,5464 MAGE -A3 0,3475 0,0198 MAGEA9 0,5026 0,3300 p-Wert LAGE1a 0,0034 0,0088 GAGE -7b 0,0638 0,1657 CAMEL se70-2 0,5473 0,3743 0,0398 0,4309 0,3363 1,0000 1,0000 0,3295 0,2549 0,1052 1,0000 0,6640 0,4676 0,0128 1,0000 0,5386 1,0000 1,0000 0,5930 0,0828 0,3154 0,0024 0,1128 0,1151 0,0302 0,0007 0,5327 0,1941 0,2457 0,0404 0,2397 0,0964 0,6512 0,5498 n.e. 0,3289 n.e. 0,3197 0,1464 0,3884 0,5512 0,5511 0,5514 0,5514 0,3080 1,0000 0,3483 1,0000 0,0199 1,0000 1,0000 1,0000 1,0000 0,2476 0,5392 1,0000 1,0000 0,0032 0,0688 0,6724 1,0000 1,0000 1,0000 1,0000 1,0000 0,2579 0,1244 n.e. n.e. n.e. 1,0000 1,0000 1,0000 1,0000 Antigen Kollektiv se57-1 GBP5ta Ctrl – MM Ctrl – MM I – MM II – MM III – MM IV Ctrl – MM I Ctrl – MM II Ctrl – MM III Ctrl – MM IV Ctrl - CTCL Ctrl – CTCL I – CTCL II – CTCL III – CTCL IV Ctrl – CTCL I Ctrl – CTCL II Ctrl – CTCL III Ctrl – CTCL IV 0,2708 0,0265 1,0000 0,1390 p-Wert cTAGE- cTAGE5a N5a Cterminal terminal 1,0000 0,2724 0,1484 1,0000 0,5450 0,0149 1,0000 0,5387 1,0000 0,3130 0,5512 0,3952 0,3846 0,5514 1,0000 1,0000 1,0000 1,0000 0,5930 0,3276 1,0000 0,5573 1,0000 1,0000 1,0000 1,0000 0,5695 0,3015 0,5512 1,0000 0,6549 1,0000 0,6913 0,8223 0,5956 1,0000 1,0000 1,0000 0,1670 0,7208 1,0000 0,2315 1,0000 1,0000 1,0000 1,0000 1,0000 1,0000 0,6155 1,0000 0,3276 1,0000 0,5392 1,0000 0,3276 1,0000 1,0000 1,0000 1,0000 0,4524 0,5392 1,0000 1,0000 1,0000 p53 p16 0,6949 0,7456 1,0000 0,8472 ANHANG Anhang 2: 230 Darstellung der positiven Seroreaktivitäten für alle humanen Antigene in der Multiplex-Serologie Ctrl: Kontrollen, MM: Malignes Melanom, MM I: MM Stadium I, CTCL: Kutanes T-Zell Lymphom, CTCL I: CTCL Stadium I. Antigen Kollektiv Ctrl MM MM I MM II MM III MM IV CTCL CTCL I CTCL II CTCL III CTCL IV (n) (100) (191) (48) (49) (47) (47) (144) (55) (14) (14) (16) Antigen Kollektiv (n) Ctrl MM MM I MM II MM III MM IV CTCL CTCL I CTCL II CTCL III CTCL IV (100) (191) (48) (49) (47) (47) (144) (55) (14) (14) (16) MAGE -A1 6,0% 7,3% 10,4% 4,1% 4,3% 10,6% 10,4% 10,9% 7,1% 28,6% 6,3% MAGE -A3 5,0% 8,4% 4,2% 2,0% 8,5% 19,1% 4,9% 3,6% 0,0% 0,0% 12,5% se57-1 GBP5ta 1,0% 3,7% 2,1% 10,2% 0,0% 2,1% 0,7% 0,0% 7,1% 0,0% 0,0% 3,0% 3,1% 0,0% 6,1% 6,4% 0,0% 3,5% 3,6% 0,0% 0,0% 0,0% MAGEA9 2,0% 4,2% 2,1% 2,0% 4,3% 8,5% 4,9% 0,0% 0,0% 0,0% 25,0% Positive LAGE1a 3,0% 13,6% 10,4% 10,2% 12,8% 21,3% 4,9% 10,9% 0,0% 0,0% 0,0% Positive cTAGE- cTAGE5a N5a Cterminal terminal 2,0% 2,0% 2,6% 0,5% 0,0% 0,0% 0,0% 2,0% 4,3% 0,0% 6,4% 0,0% 2,8% 0,7% 3,6% 0,0% 0,0% 0,0% 7,1% 7,1% 0,0% 0,0% GAGE -7b 1,0% 5,8% 4,2% 8,2% 4,3% 6,4% 2,8% 1,8% 0,0% 0,0% 6,3% CAMEL se70-2 0,0% 1,0% 0,0% 2,0% 0,0% 2,1% 2,8% 3,6% 0,0% 0,0% 0,0% 3,0% 0,0% 0,0% 0,0% 0,0% 0,0% 0,7% 1,8% 0,0% 0,0% 0,0% p53 p16 3,0% 2,1% 0,0% 2,0% 4,3% 2,1% 2,1% 3,6% 0,0% 0,0% 6,3% 2,0% 2,1% 4,2% 2,0% 2,1% 0,0% 0,0% 0,0% 0,0% 0,0% 0,0% ANHANG Anhang 3: 231 GAGE-7b Expressionsanalyse n.g.: nicht getestet. Zelllinie MeWo Ma-Mel-04 Ma-Mel-05 Ma-Mel-11 Ma-Mel-12 Ma-Mel-16 Ma-Mel-21 Ma-Mel-37b Ma-Mel-47 Ma-Mel-52 SK-Mel-023 UKRV-Mel-02 UKRV-Mel-06a UKRV-Mel-14a UKRV-Mel-21a UKRV-Mel-27 UKRV-Mel-31 Melanozyten HaCat Cou-L3 HH HuT78 MyLa SeAx MM-Gewebe MM1 MM2 MM3 MM4 MM5 MM6 MM7 MM8 MM9 MM10 RT-PCR GAGE-1, -2, -8 GAGE-3 bis –7b + + + + + + + + + + + + + + + + + + + + + + + + + + RT-PCR GAGE-1, -2, -8 GAGE-3 bis –7b n.g. n.g. + + + + + + n.g. n.g. + + Western Blot Zelllinie Gewebe n.g. n.g. + n.g. + n.g. + n.g. + + + + n.g. + n.g. + n.g. n.g. + n.g. + + n.g. n.g. n.g. n.g. n.g. n.g. n.g. n.g. Immunzytochemie + n.g. + + + + + + n.g. - Western Blot Korrespondierende Zelllinie + n.g. + - Ma-Mel-37b n.g. n.g. Ma-Mel-05 n.g. UKRV-Mel-06a Ma-Mel-12 Ma-Mel-47 n.g. UKRV-Mel-27 LEBENSLAUF 232 LEBENSLAUF Name: Wiedemann geb. Gottstein Vorname: Nicole Staatsangehörigkeit: deutsch Geburtsdatum: 11.01.1976 Geburtsort: Pforzheim Familienstand: verheiratet Schulbildung und Studium 1982 – 1986 Grundschule Neulingen-Bauschlott 1986 – 06/1995 Hildagymnasium Pforzheim Allgemeine Hochschulreife 06/1995 10/1995 – 09/1996 Studium der Rechtswissenschaften, Universität Heidelberg 08/2001 – 04/2002 Diplomarbeit, Hugh Sinclair Human Nutrition Unit, School of Food Biosciences, University of Reading, UK Thema: “Molecular mechanisms by which isoflavones protect against coronary artery disease – studies in cultured cells 10/1996 – 06/2002 Studium der Ernährungswissenschaft, Universität Hohenheim Diplom 06/2002 06/2002 – 03/2006 Promotion unter Betreuung von Frau Prof. Dr. Christiane Bode, Institut für Biologische Chemie und Ernährungswissenschaft, Fachgebiet Ernährungsphysiologie, Universität Hohenheim durchgeführt bei Herrn Prof. Dr. Stefan Eichmüller, Klinische Kooperationseinheit für DermatoOnkologie, Arbeitsgruppe Tumorantigene, Deutsches Krebsforschungszentrum, Heidelberg Seit 03/2006 Wissenschaftliche Referentin, Analytical Development Biotech Products, Merck KGaA, Darmstadt EIDESTATTLICHE ERKLÄRUNG 233 EIDESTATTLICHE ERKLÄRUNG Hiermit erkläre ich an Eides statt, dass ich die vorliegende Arbeit selbständig und nur mit den angegebenen Quellen und Hilfsmitteln und der zitierten Literatur angefertigt habe. Wörtlich oder inhaltlich übernommene Stellen wurden als solche gekennzeichnet. Diese Dissertation wurde weder in gleicher noch in ähnlicher Form in einem anderen Promotionsverfahren vorgelegt. Nicole Wiedemann Rheinhausen, den 22.06.2008






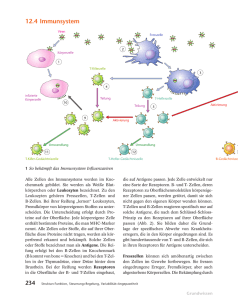




![(Microsoft PowerPoint - Blut3 [Kompatibilit\341si m\363d])](http://s1.studylibde.com/store/data/001460679_1-7cc1bea38c64f816011e1d199e4f776d-300x300.png)