4 Übergangsmetall-katalysierte Reaktionen
Werbung

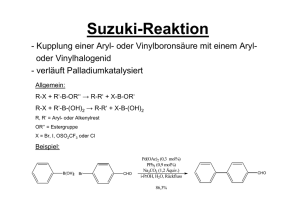
4 Übergangsmetall-katalysierte Reaktionen 48 4.1 Allgemeines 49 4.1.1 Oxidationsstufen 49 4.1.2 Koordinative Sättigung, 18e- -Regel 49 4.1.3 Einteilung der Liganden 50 4.1.4 Bindungsmodelle 51 4.2 Mechanismen metallorganischer Reaktionen 52 4.2.1 Ligandenaustauschprozesse 52 4.2.2 Oxidative Addition / Reduktive Eliminierung 53 4.2.3 Insertion / β-Hydrideliminierung a) CO-Insertion b) Alken-Insertion c) Alkin-Insertion 54 54 54 54 4.2.4 Nucleophiler Angriff an koordinierten Liganden a) Carbonylkomplexe b) Acylkomplexe c) π-Komplexe 55 55 55 55 4.2.5 Transmetallierung 57 4.3 Synthetische Anwendungen 57 4.3.1 Synthetische Anwendungen von Übergangsmetallhydriden 1) Homogene katalytische Hydrierung 2) Hydrometallierung 57 57 61 4.3.2 Synthetische Anwendungen von σ-Komplexen a) C-M-σ-Komplexe durch Transmetallierung von Carbanionen b) C-M-σ-Komplexe durch Hydrometallierung c) σ-Komplexe durch oxidative Addition / Transmetallierung d) σ-Komplexe durch oxidative Addition / Insertion 62 62 63 65 68 4.3.3 Synthetische Anwendungen von Carbenkomplexen 70 4.3.4 Synthetische Anwendungen von Alken- und Dienkomplexen 72 4.3.5 Synthetische Anwendungen von π-Allyl-Komplexen 74 Literatur zur Übergangskatalyse 77 V Periodensystem der Elemente 4. Übergangsmetall-katalysierte Reaktionen 48 48 frühe Übergangsmetalle hauptsächlich stöchiometrisch → Carbonylreaktionen katalytische Prozesse → Sharpless-Oxidierung Lewis-Säure-Katalyse späte Übergangsmetalle hauptsächlich katalytische Reaktionen Ausnahme: Cu → Cuprate 4.1 Allgemeines Reaktionen spielen sich in der Koordinationssphäre der Metalle ab → Metallzentren und elektronische Umgebung bestimmen Verlauf. Für Verständnis wichtig: • Oxidationsstufe des Metalls • Anzahl der d-Elektronen • Koordinationszahl des Metalls • freie Koordinationsstellen am Metall 4.1.1 Oxidationsstufen Ladung die am Metall verbleibt, wenn alle Liganden mit abgeschlossener Elektronenschale, d.h. mit e−-Paar entfernt würden. Beispiele: Halogenide M−Cl ⇒ M+ + Cl− Hydride M−H ⇒ M+ + H− Carbonylkomplexe M−C≡O ⇒ M + CO Phosphinkomplexe Olefinkomplexe M− Allylkomplexe M−PPh3 ⇒ M + PPh3 − + ⇒ M + M ⇒ Μ + 4.1.2 Koordinative Sättigung, 18 e−-Regel Bestimmung der e−-Zahl anhand Periodensystem und Oxidationszahl 18 e−-Regel: Bei einkernigen diamagnetischen Komplexen ist die Summe der d- e− und der e− die die Liganden beisteuern maximal 18. Verbindungen mit 18 e− (maximale Ligandenzahl) sind koordinativ gesättigt! Verbindungen mit < 18 e− sind koordinativ ungesättigt → wichtig für katalytische Prozesse! Metall benötigt freie Koordinationsstellen um Substrat zu binden!!!! 49 4.1.3 Einteilung der Liganden formal 3 Familien: Anionen, Neutralliganden, (Kationen) → Oxidationsstufen-Formalismus Anionische Liganden: R− > Ar− > H− > R-CO− > X− (X− = Hal−, CN−) Donorfähigkeit: Liganden mit mehreren funktionellen Gruppen → Mehrelektronenliganden Beispiel: Allylgruppe C3H5− M M 1 M η η 3 η 3 (2e−) (4e−) (4e−) 1 2 2 Koordinationsstellen (KS) Beispiel: Cyclopentadienyl-Ligand C5H5− M M 1 M η η 3 η 5 (2e−) (4e−) (6e−) 1 2 3 Koordinationsstellen (KS) Neutral-Liganden: Beispiel: Phosphane (PR3) Amine (NR3) gute σ-Donatoren → erhöhen Elektronendichte am Metall Beispiel: CO, Olefine, Isonitrile: gute π-Akzeptoren → erniedrigen Elekt.-dichte am Metall Kationische Liganden: sehr selten, Bsp: NO+ → Nitrosylkomplexe 50 Abzählen der Elektronen: Beispiele: + Ph3P PPh3 Rh Fe OC OC FeII: Cp−: CO: : Cl Pd Cl Ph3P Pd Cl 6 e− (d6) 6 e− 2 e− x 2 2 e− RhI: 8 e− (d8) Cl− : 2 e− Ph3P: 2 e− x 3 PdII : 8 e− (d8) C3H3−: 4 e− Cl− : 2 e− Cl : 2 e− − − 18 e (gesättigt) 16 e− (ungesättigt) 16 e (ungesättigt) 18 e− -Komplexe: koordinativ gesättigt → unreaktiv 16 e− -Komplexe: koordinativ ungesättigt → reaktiv 4.1.4 Bindungsmodelle Übergangsmetalle: (teilweise) besetzte d-Orbitale, leere s,p-Orbitale Liganden: besetzte s, p, sp-Hybridorbitale, (teilweise) unbesetzte π*-Orbitale Orbitale gleicher Symmetrie können überlappen → Bindung σ-Donor-Bindung 2 Arten von Bindung π-Akzeptor-Bindung σ-Donorbindungen: - bei Liganden mit freien e−-Paaren (R3P, R3N, H−, R−) spn-HybridLigand + „dsp“-HybridMetall → erhöht e−-Dichte am Metall - π-Systeme (Alkene, Alkine, Arene, CO, Isonitrile) sowohl σ-Donor als auch π-Akzeptor σ-Bindung M leeres "dsp" besetztes π-Orbital 51 π-Akzeptorbindungen: 2 Arten a) seitengebundene (side-on) Akzeptoren Beispiel: Alkene, Alkin π-Rückbindung σ-Donor-π-Akzeptor-Komplex π M σ M besetztes δ-Orbital π leeres π*-Prbital b) endgebunden (edge-on) Akzeptoren Beispiel: CO, RNC π-Rückbindung σ-Donor-π-Akzeptor-Komplex π M M besetztes δ-Orbital σ π leeres π*-Prbital 4.2 Mechanismen metallorganischer Reaktionen 4.2.1 Ligandenaustauschprozesse extrem wichtig: M-L + L’ M-L’ + L Beispiel: inaktiver 18e− -Komplex Abspaltung Ligand aktiver 16e− -Komplex Substratbindung Eliminierung Produktkomplex Umsetzung inaktiver 18e− -Komplex 52 4.2.2 Oxidative Addition / Reduktive Eliminierung Grundlage: Leichter Wechsel der Oxidationsstufe am Metall Prinzip: B Oxidative Addition Mn + A−B n+2 M Reduktive Eliminierung A Vorteil: Es müssen nicht unbedingt dieselben Reste reduktiv entfernt werden die zuvor oxidativ addiert wurden! → wichtig für metallorganische Kupplungsreaktionen → Kreuzkupplungen wichtige Systeme: Ni0 → NiII Pd0 → PdII (d10 → d8 ) RhI → RhIII IrI → IrIII (d8 → d6 ) Oxidative Addition: Beispiel: Vaska-Komplex Ph3P Ph3P CO Ir Cl d8 16 e− -Komplex koordinativ ungesättigt CO Ir + A B PPh3 Cl A B PPh3 d6 18 e− -Komplex koordinativ gesättigt Palette an Substraten sehr breit → viele Anwendungen Polare Elektrophile: H−X, R−X, RCO−X Unpolare Elektrophile: H−H, R−H, R3Si−H Mehrfachbindungssysteme: Alkene, Alkine (A und B bleiben verbunden) „Oxidative Addition“ beschreibt Phänomen, keine Aussage über Mechanismus Reduktive Eliminierung: meist letzter Schritt eines Katalyse-Zyklus → extrem wichtig → Grundlage der meisten C−C-Knüpfungen Reduktive Eliminierung erfolgt immer aus der syn-Position → stereospezifisch! 53 4.2.3 Insertion / β-Hydrideliminierung formal wird ungesättigter Ligand Y (CO, RNC, Alken, Alkin) in benachbarte cis-Metallσ-Bindung insertiert. Prinzip: L Z Y M Insertion koordin. gesättigt M Y Z L koordin. ungesättigt M Y Z koordin. gesättigt Für Z = H, Alkyl wird neue C−H bzw. C−C-Bindung gebildet → reversibel Besitzt Z chirales Zentrum → Konfigurationserhalt a) CO-Insertion: wichtiger Prozess → Synthese von Aldehyden, Ketonen, Carbonsäurederivaten Carbonylierung, mit Z = H: Hydroformylierung Rückreaktion: Decarbonylierung R CO M Insertion R M C L L R M C O O Wanderungstendenz: η1-Allyl ≥ Et > Me > PhCH2 > Vinyl ≥ Aryl ~ ROCH2 > HOCH2 b) Alken-Insertion: geht gut bei M−C und M−H Bindungen (katalytische Hydrierung) Rückreaktion: β-Hydrideliminierung (für R = H) → Zersetzung von Alkylmetall-Komplexen R R M Insertion β-Hydridel. (R = H) stereospezifisch syn M Wanderungstendenz: Η >> R, Vinyl, Aryl > RCO >> RO, R2N c) Alkin-Insertion: analog: R R M Insertion M stereospezifisch syn 54 4.2.4 Nucleophiler Angriff an koordinierten Liganden Angriff an ungesättigten Verbindungen sehr wichtiger Prozess a) Carbonylkomplexe (Angriff am CO von Carbonylkomplexen) - + LnM C O - R R LnM C O - LnM C O + R−Li LnM C O E+ R O E R R reduktive Eliminierung LnM C O LnM C O E + E Carbenkomplexe Aldehyde Ketone Carbonsäureder. (stöchiometrische Umsetzungen) b) Acylkompexe Schlüsselschritt vieler Metall-katalysierter Acylierungsreaktionen Beispiel: X L O Pd C Ar O Ar + ROH C OR + L2Pd + HX L c) π-Komplexe extrem wichtig Reaktivität koordinierter Liganden: > > > > >> > > 55 • Alken-Komplexe (η2-Komplexe) - anti-Addition: R R Nu − M + Nu M Bildung einer C−M und C−Nu-Bindung -syn-Addition: bei Cl−, AcO−, R− möglich R R + Nu− M M R M Nu Nu • Dien-Komplexe (η4-Komplexe) Pd: Angriff des Nukleophils an der sterisch weniger gehinderten Position → Allylkomplex Prinzip: Nu - Nu Pd Cl Pd Cl - − 2 Cl +2L Nu Pd + Cl Cl L L • π-Allyl-Komplexe (η3-Komplexe) sehr wichtig → Pd-Chemie Prinzip: Pd R 0 Pd + R X −X − Pd + R X π-Allyl-Pd-Komplex π-Komplex 1 Nu - Pd 0 + R Pd R Nu Nu π-Komplex 2 optisch aktive Substrate: Retention (2x Inversion) Beispiel: L Ph OAc L4Pd − AcO − 2L - Pd + L Ph Nu - Ph + L 4Pd Nu 56 4.2.5 Transmetallierung Übertragung eines Restes R von Hauptgruppenmetall M auf Übergangsmetall (ÜM) M‘ R M + M'X R M' + MX Hauptgruppenmetall sollte elektropositiver sein als Übergangsmetall wichtig: Kombination Transmetallierung mit anderen Reaktionen (oxid. Add. etc.) Übertragung zweier Reste R und R‘ auf ein ÜM → Kreuzkupplung durch red. Eliminierung Transmetallierung in Regel geschwindigkeitsbestimmender Schritt 4.3 Synthetische Anwendungen 4.3.1 Synthetische Anwendungen von Übergangsmetallhydriden ÜM-Hydride spielen zentrale Rolle bei Hydrierung, Hydrometallierung und Hydroformylierung Herstellungsverfahren: LnM + H 2 oxid. Add M M H β-H-Elim O (H) LnM-X + M'H H M Transmet. H a) Homogene katalytische Hydrierung Oxidative Addition von H2 homogene Hydrierung empfindlicher und selektiver als heterogene Hydrierung Katalysatoren (2 Klassen) Monohydrid-Komplexe Dihydrid-Komplexe → unterschiedliche Reaktivität und Selektivität • Monohydridkomplexe Beispiel: Rh(H)(PPh3)3CO selektiver Hydrierkatalysator für endständige Alkene • Dihydridkomplexe genereller anwendbar, besser untersucht Beispiel: RhCl(PPh3)3 Wilkinson-Katalysator 57 Katalysecyclus: Monohydridkomplexe H L Rh L L CO −L H H L redukt. Elim. R H R Rh CO L R H H H L L Rh L H H Rh β-HElimin. L R CO CO R β-H-Elimin. oxidative Addition Insertion R L H2 Rh CO H L Katalysecyclus: Dihydridkomplexe (vereinfacht) L L Rh Cl L −L H redukt. Elim. schnell H R L (S) H2 Rh Cl L oxidative Addition R H H L L Rh Cl H Rh Cl H H (S) L (S) Insertion langsam R H L H Rh Cl L R 58 Reaktivität der Alkene: R R > > R > R > R R R asymmetrische Hydrierungen: Verwendung chiraler Liganden O PPh2 O PPh2 Ph2P P PPh2 OMe Diop N P OMe O O PPh2 PPh2 Dipamp BPPM Chiraphos 59 Beispiel: Synthese von Aminosäuren P *P E Rh + E E = COOR + S Ph NHAc + + NH P *P S Rh P diastereomere Komplexe Ph O O *P NH Rh E Ph geschwindigkeitsbestimmender Schritt E P H * * Ph O Rh E P O H P H Rh P + + NH NH H Ph E H * + Rh P H + NH P Ph P H * NH Rh E H P O O Ph E NHAc Ph H 98% E NHAc Ph H 2% Vorraussetzung für hohe ee’s: 1) Diastereomere Komplexe müssen sehr unterschiedlich reagieren 2) Zwischen beiden Komplexen muss schnelles Gleichgewicht sein 60 b) Hydrometallierung Hydrozirkonierung Schwartz Reagenz H Cp2Zr Cl Beispiele: H ZrClCp2 Cp2ZrHCl Cp2ZrHCl R R ZrClCp2 H Cp2ZrHCl ZrClCp2 Angew. Chem. 1976, 88, 402. Hydrostannylierung Alkine: stereospezifische syn-Addition, aber Gemisch von Regioisomeren Beispiele: SnBu3 Bu3SnH BnO BnO Pd(PPh3)4 BnO + SnBu3 J. Org. Chem. 1990, 55, 1867. Vinylstannane: wichtige Substrate für Kreuzkupplungsreaktionen (Stille-Kupplungen) geht auch mit Alkene und α,β-ungesättigten Carbonylverbindungen OH OH O O O O O O Bu3SnH Pd(PPh3)4 OH O OH O Tetrahedron Lett. 1982, 477. 61 Mechanismus: SnBu3 H R L H Pd R R Bu3Sn O L oxidative Addition L L H Bu3SnH Pd O H3O+ L L redukt. Elim. H L Pd O H Bu3Sn L Insertion R H L Pd O R Bu3Sn O 4.3.2 Synthetische Anwendungen von σ-Komplexen enthalten Metall-C-σ-Bindung Herstellung: R' M' + MX R M + Transmetallierung Insertion R' nucl. Angriff + Nu M Hydrometallierung M H + M oxidat. Addition R' X + M0 teilweise auch Kombinationen a) C-M-σ σ-Komplexe durch Transmetallierung von Carbanionen (Li, Mg) wichtig: Chemie der Kupferverbindungen (Cuprate) Gilman-Cuprate: 2 RLi + CuI R2CuLi wichtige Umsetzungen: 1,4-Additionen, Epoxidöffnungen, mit Säurechloriden → Ketonen aber alle stöchiometrisch! 62 Kreuzkupplungen: Beispiel: Stephens-Castro-Kupplung (ebenfalls stöchiometrisch) Ar I + Cu R Ar R + CuI Sonagashira-Kupplung: Katalytisch in Cu und Pd CuI, NEt3 + AcHN PdCl2(PPh3)2 I 95% DMF, ∆ COOMe AcHN COOMe Tetrahedron 1992, 48, 3239. Mechanismus: Pd(PPh3)4 2 PPh3 Pd(PPh3)2 R R' R oxidative Addition reduktive Eliminierung I R = Ar, Vinyl R R (PPh3)2Pd (PPh3)2Pd I R' CuI, NEt3 Transmet. CuI Cu R' R' NEt3H+ I− 63 b) C-M-σ σ-Komplexe durch Hydrometallierung H H M H + M M M M H H Beispiel: Pd-katalysierte Cycloisomerisierung von Eninen O Pd2(dba)3 CHCl3 HOAc + R R dba = Ph Ph R J. Am. Chem. Soc. 1991, 113, 636. Mechanismus: H H Pd "HPdX" Insertion Produkte Pd Pd H R H R H R Addition und Eliminierung von Pd−H erfolgt stereospezifisch syn (cis) Beispiele: Mehrfachcyclisierung E E E X E X Pd2(dba)3 CHCl3 57% PPh3, HOAc - "HPdX" "HPdX" E E E E E X X E X PdX PdX XPd H J. Am. Chem. Soc. 1991, 113, 701; 1993, 115, 9421. OR OR Pd2(dba)3 CHCl3 AsPh3, HOAc 86% R R R R J. Am. Chem. Soc. 1991, 113, 701; 1993, 115, 9421. 64 c) σ-Komplexe durch oxidative Addition / Transmetallierung Transmetallierung: meist Transfer eines Restes R von Hauptgruppen- auf Übergangsmetall → oft geschwindigkeitsbestimmender Schritt wichtiger Prozess: + R X Pd(0) R Pd Ni(0) R Ni X X Nur Aryl- und Vinylhalogenide → Retention Alkylhalogenide → σ-Alkylkomplex → β-Hydrideleminierung Reaktivität: I > OTf > Br >> Cl Mechanismus und Reaktionsmöglichkeiten: Kreuzkupplung mit CO-Insertion R CO c R R' R' Pd(0) R X c R Pd a R' R CO Pd II CO d II R Pd R' b R Pd II II X R' R CO Pd II X M R CO Pd II R' b R' DiorganoMetall-Komplex M X M: Mg, Zr, Zn, Sn, B, Al, Si, Cu Teilschritte: a) Oxidative Addition b) Transmetallierung c) Reduktive Eliminierung d) Insertion extrem gut entwickelt: Pd(0)-katalysierte Kupplungen Katalysator: Pd(PPh3)4 oder in situ z.B. aus Pd2(dba)3 und PPh3 bzw. Reduktion: PdCl2(PPh3)2 Dibal-H 2 PPh3 Pd(PPh3)4 PPh3 Pd(OAc)2 65 • Zink: Zinkorganyle transmetallieren sehr gut → auch funktionalisierte Substrate Beispiel: Ph IZn I BocHN COOBn O PhCOCl Zn/Cu 60° BocHN COOBn (Ph3P)4Pd BocHN 70% COOBn J. Org. Chem. 1992, 57, 3398. • Zirkonium: Negishi-Kupplung Vinylzirkonium-Verbindungen leicht durch Hydrozirkonierung (Schwartz-Reagenz) erhältlich Beispiel: Br Cp2Zr(H)Cl RO RO RO E [Zr] Pd(0) E 70% Tetrahedron Lett. 1978, 1027. endständige Zr-Verbindungen kuppeln gut, interne kuppeln nicht. • Bor: Suzuki-Kupplung Bororganyle durch Hydroborierung Transmetallierung B → Pd zuerst problematisch Abhilfe: Umsetzung im Basischen Beispiel: I PdCl2, dppf I 9-BBN NaOH B OR H . OR OR PPh2 B 9-BBN : dppf : Fe PPh2 Tetrahedron Lett. 1992, 33, 2571. ebenfalls gut: aromatische Boronsäuren → Herstellung funktionalisierter Aromaten 66 B(OH)2 CONEt2 + CONEt2 CONEt2 Pd(PPh3)4 aq. Na2CO3 DME, ∆ Br CONEt2 • Zinn: Stille Kupplung eine der besten Methoden → viele Anwendungen Transmetallierung: > R C C > Ar > R CH CH Vinylzinn: Konfigurationserhalt CH2 CH CH2 , Bn >> Alkyl Allylzinn: Allylumlagerung Prinzipiell lassen sich alle Substrate kuppeln, die oxidativ an Pd addieren. Beispiel: Säurechloride O R R Cl O n O Bu3Sn Pd(0), CO O O n 32-70% 110° O Tetrahedron 1992, 48, 2957. Beispiel: Vinyl-, Aryliodide und –triflate O F O O F COOR + TfO LiCl, THF, ∆ N O N SnBu3 F COOR 2% PdCl2(PPh3)2 88% F Tetrahedron Lett. 1990, 31, 1837. Geschwindigkeitsbestimmender Schritt: Transmetallierung → Koppeln mit Carbonylierung Beispiel: O O + I Pd(PPh3)4 Bu3Sn 63% 4 bar CO O J. Am. Chem. Soc. 1984, 106, 6417. 67 d) σ-Komplexe durch oxidative Addition / Insertion • CO-Insertion: geht sehr gut (auch bei Normaldruck) auch intramolekular → eine der besten Methoden um Carbonylgruppe einzuführen. Beispiel: OR OR RO PdCl2(PPh3)2 4 bar CO OH RO O K2CO3, DMF I 78% O J. Org. Chem. 1982, 47, 3630. • Alken-Insertion: Heck-Reaktion im Prinzip analog, Alkene etwas weniger reaktionsfähig Mechanismus: HX H Pd II R X Pd(0) e a X R' R R Pd II X d II R Pd H H R' H b X c R Pd II R' a: Oxidative Addition b: Koordination c: Insertion d: β-Hydrideliminierung e: reduktive Eliminierung X R' Insertion: schwierigster Schritt verläuft cis selektiv ebenso wie β-H-Eliminierung → Regioselektivität bei cyclischen Sys. Katalyator: entweder Pd(0) oder Pd(II) mit Reduktionsmittel (PPh3) Reaktion toleriert viele funktionellen Gruppen → breite Anwendung Beispiel: O O OTf N + O N R PdCl2, PPh3 NEt3, DMF, 60° R N 96% O N Heterocycles 1987, 26, 355. 68 Beispiel: intramolekulare Variante OSiR3 OTf OSiR3 Pd(PPh3)4 91% LiCl, Li2CO3 Acta. Chem. Scand. 1992, 46, 597. Beispiel: asymmetrische Variante + ArOTf O Pd(OAc)2 (R)-Binap O Insertion β-H-Elim. PdH H Pd Pd Ar H Ins. β-H-Elim. O 40 - 60% > 96% ee Ar O Ar H O Ar Tetrahedron Lett. 1992, 33, 1485 Beispiel: Dominoreaktionen Pd I Pd I oxidative I O CO-Insertion Addition n n n 5% PdCl2(PPh3)2 40 bar CO, NEt3 MeOH, MeCN, 100° AlkenInsertion O COOMe O Pd PdI O MeOH n O hoher n n O geringer CO-Druck − HPdI CO-Druck − HPdI I n J. Org. Chem. 1988, 53, 913. R R 3% Pd(PPh3)4 I MeCN, NEt 3 , ∆ 76% J. Am. Chem. Soc. 1990, 112, 8590. 69 4.3.3 Synthetische Anwendungen von Carbenkomplexen 2 Typen: Fischer-Carbene Schrock-Carbene OMe Beispiele: Cp2Ti (CO)5M R elektrophile Carbene Heteroatom subst. nucleophile Carbene Olefinierung (→) • ÜM-katalysierte Zersetzung von Diazoverbindungen Katalysatoren: Cu(I)- und Pd(II)-Salze, Rh(OAc)4 Beispiel: Cyclopropanierung O O COOMe Rh2(OAc)4 N2 RO COOMe RO 83%, 69% ds J. Org. Chem. 1992, 57, 441. Beispiel: asym. Cyclopropanierung O O R R' R Rh2L*4 O O R' N2 30 - 88% 72 - 94% ee L* = E N − O J. Am. Chem. Soc. 1991, 113, 1423 Keine geeignete Doppelbindung vorhanden → Insertion in C−H, O−H, N−H Beispiel: O E OH N2 Rh2(OAc)4 O O 71% E J. Chem. Soc. Perkin 1 1988, 1417. 70 • Metathese meist Metall-katalysierte Umsetzungen von Alkenen (Austausch von Alkenen) Prinzip: R' + R R' Katalysator + R R ≠ R’: Kreuzmetathese Ringöffnungsmetathese Ringschlußmetathese n (ROMP) (RCM) gängige Katalysatoren: Cl iPr iPr Cl N F3C F3C PCy3 Ph O F3C F3C Ru PCy3 Ru Cl Mo O O Ph Cl PCy3 Schrock Grubbs Hoveyda Mechanismus: Ringschlussmetathese LnM MLn MLn MLn viele präparative Anwendungen, starke Entwicklung Beispiel: OBn N O OBn OBn Grubbs-Kat. N ∆ OBn 80% O Tetrahedron Lett. 1996, 52, 7251. 71 4.3.4 Synthetische Anwendungen von Alken- und Dienkomplexen Durch Koordination: Umkehrung der Reaktivität des Alkens Nucl. Angriff an koord. Alken fundamenteller Prozess wichtige Metalle: Fe → stöchiometrisch, Pd → katalytisch • Alken-Komplexe Katalysatoren: PdCl2-Komplexe (MeCN, PhCN) Prinzip: Nu R Cl R Nu − Pd L2PdCl2 + L Cl R Cl Pd − Cl− L Nu β-H-Elim. R L H Pd Folgereaktionen Cl Pd(0) + HCl Beachte: Pd(II) nötig für Koordination. Nach nucl. Angriff entsteht Pd(0) → Reoxidation Beispiel: Wacker-Oxidation (Nucl. Addition von H2O) OHC OHC O PdCl2 / CuCl 59% O2, H2O O O Mechanismus: HX Pd(0) e H Pd II O2 2 CuCl2 2 CuCl a Cl II HO L2PdCl2 d R' b L II Cl Pd O II c HO R' L Cl R' Cl Pd Cl R' R' a: Oxidative Addition b: Koordination c: Insertion / Nucl. Angriff d: β-Hydrideliminierung e: reduktive Eliminierung Nur monosubstituierte Alkene → Methylketone Auch andere O-Nucleophile möglich 72 Beispiel: Cyclisierung mit CO-Insertion Pr OMe OMe Pr 5% PdCl2 2 eq. CuCl2 OH O 87% COOMe 1 atm CO MeOH O O Pd(II) OMe MeOH OMe Pr O Pr O CO Pd Pd O O O J. Am. Chem. Soc. 1983, 105, 2034. • Dien-Komplexe hoch interessant da Bildung von π-Allylkomplexen (→) Prinzip: Nu1 − X− Pd X − X− Pd X Nu1 Nu1 Nu2− Nu1− Nu2 Nu2 Pd X Pd(0) Beispiel: Bäckvall-Oxidation cis- oder trans-Diacetat je nach Reaktionsbedingungen Pd(OAc)2 OAc L OAc − Pd(0) AcO Pd AcO Pd OAc AcO L Cl − OAc Reoxidation: − Pd(0) − AcO + AcO OAc Benzochinon, MnO2 Pd Cl L J. Org. Chem. 1984, 49, 4619; J. Am. Chem. Soc 1990, 112, 5160. 73 4.3.5 Synthetische Anwendungen von π-Allyl-Komplexen Im Prinzip von fast allen Metallen Komplexe bekannt wichtig: Pd(II)-Komplexe Herstellung: Pd X Pd PdH H 0 II PdX PdX2 Pd MgX X PdX2 Pd L Nu− Nu Pd Umsetzung von Allylsubstraten extrem viele Anwendungen allgemeiner Mechanismus: R Nu Pd(0) X a Nu PdX R Pd d Pd b Nu− L RM c + Pd f Pd L e L X Pd R − MX L Verschiedene Katalysatoren und Vorstufen, Bsp: Pd(PPh3)4 oder a: Oxidative Addition b: π−σ−π-Isomerisierung c: Ligandenaustausch d: Nucleophiler Angriff e: Τransmetallierung f: reduktive Eliminierung Cl Pd Pd Cl • Allylische Alkylierung vor allem mit „weichen Nucleophilen“ Stereochemischer Verlauf: Retention (2 x Inversion) Beispiel: Ph LnPd Ph OAc Nu− Nu Pd L Ph L Abgangsgruppen, Reaktivität: Cl > OCOOR > OAc >> OH 74 → selektive Umsetzung von Bisallylsubstraten Beispiel: OH OH Pd(OAc)2, PPh3 O2N OAc SO2Ph Base O2N SO2Ph ClCOOMe O2N SO2Ph OCOOMe Pd(OAc)2, PPh3 O2N OAc SO2Ph Base OAc J. Org. Chem. 1992, 57, 1588. effizient: Cyclisierungen Beispiel: OAc AcO Pd(PPh3)4 O 79% NaH OAc O E E H Tetrahedron Lett. 1992, 33, 3527, 3531. O O Pd(OAc)2 SO2Ph SO2Ph P(OiPr)3 O OH 92% SO2Ph SO2Ph OR OR Tetrahedron Lett. 1986, 27, 5695. modern: Asymmetrische Katalyse → Verwendung chiraler Liganden Beispiel: − Ph Ph LnPd E E Ph Ph O OAc E N E 74 - 98% 98.5% ee Ph2P Angew. Chem. 1993, 105, 614; Tetrahedron Lett. 1993, 34, 1769. geht auch mit Heteronucleophilen 75 Beispiel: Allylschutzgruppen: OH OH Pd(PPh3)4 SR N PPh3 O O N + N 92% O N H O SR O OH Tetrahedron Lett. 1987, 28, 4371. • Allylierung via Transmetallierung Beispiel: O OAc + Bu3Sn OBn Pd(PPh3)4 OBn DMF, LiCl 60°, 48h 81% O J. Org. Chem. 1990, 55, 3019. • π-Allylkomplexe als Intermediate Leichte Insertion von Alkenen, Alkinen und CO in π-Allylkomplexe Beispiel: Cyclisierungen Ts N AcO Ts N Pd2(dba)3 PPh3, CO MeOH, AcOH 77% COOMe Pd(0) Ts N MeOH Ts Ts N N Pd Pd Pd O J. Am. Chem. Soc. 1986, 108, 284. 76 Literatur Übergangsmetallkatalyse Allgemeine Lehrbücher: L. Hegedus: Organische Synthese mit Übergangsmetallen, Wiley-VCH C. Elschenbroich, A. Salzer: Organometallchemie, Teubner Aktuelle Reviews : Reaktionen über Carbenkomplexe: Catalytic enantioselective cyclopropanation of olefins using carbenoid chemistry, Synthesis 1997, 137. New aspects of catalytic asymmetric cyclopropanation, Tetrahedron 1998, 54, 7919. Recent advances in asym. catalytic metal carbene transformations, Chem. Ber. 1998, 911. Olefine carbonylation catalysis with cationic Palladium complexes, Chem. Ber./Recl. 1997, 130, 1557. Catalytic applications of transition metals in organic synthesis, J. Chem. Soc. Perkin I, 1998, 3637. Ring closing metathesis of nitrogen containing compounds, Aldrichimica Acta, 1999, 32, 75. Olefine metathesis and beyond Angew. Chem. Kreuzkupplungen: Palladium-catalyzed reactions of organotin compounds, Synthesis 1992, 803. The Stille reaction, Organic Reactions, 1997, 50, 1. The intramolecular Stille Rreaction, J. Chem. Soc. Perkin I, 1999, 1235. Recent advances in the cross coupling reactions of organoboron derivatives, J. Organomet. Chem. 1999, 576, 147. Heck-Reaktionen: Enantioselective Heck Reactions, Nachr. Chem. Tech. Lab. 1994, 42, 270. Heck-Reaktionen, Angew. Chem. 1994, 106, 2473. Allylische Alkylierungen / π-Allylchemie: Asym. transition metal catalyzed allylic alkylations, Chem. Rev. 1996, 96, 395. Transition metal catalyzed carbocyclization in organic synthesis, Chem. Rev. 1996, 96, 635. Allylic Amination, Chem. Rev. 1998, 98, 1689. Transition metal catalyzed cycloisomerizations, Synlett, 1998, 1. 77