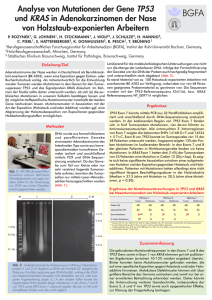
Untersuchungen über den Einfluss von p53
Werbung

Aus der Klinik für Chirurgie der Universität zu Lübeck Direktor: Prof. Dr. med. H.-P. Bruch Untersuchungen über den Einfluss von p53-Mutationen auf die Expression des Vascular Endothelial Growth Factors (VEGF) in humanen kolorektalen Karzinomen Inauguraldissertation zur Erlangung der Doktorwürde der Universität zu Lübeck - Aus der Medizinischen Fakultät - vorgelegt von Steffen W. Pfarr aus Wesel Lübeck 2005 1. Berichterstatter: Prof. Dr. med. Rainer Broll 2. Berichterstatter: Prof. Dr. med. Alexander Herold Tag der mündlichen Prüfung: 13.01.2006 Zum Druck genehmigt, Lübeck den 13.01.2006 2 Inhaltsverzeichnis 1 Einleitung 6 1.1 Der Angiogenesefaktor Vascular Endothelial Growth Factor (VEGF) 7 1.1.1 Molekulare und biologische Eigenschaften von VEGF 8 1.1.2 Rezeptor und Signaltransduktion des VEGF 10 1.1.3 VEGF-Expression in Neoplasien 12 1.2 Das Tumorsuppressorgen p53 13 1.2.1 Das p53-Gen 13 1.2.2 Eigenschaften des Genproduktes Wildtyp-p53-Protein (Wt-p53) 14 1.2.3 Mutiertes p53 (Mut-p53) 16 1.2.4 p53-Mutationen und kolorektale Tumore 17 1.3 Zusammenhang zwischen VEGF und p53 18 1.4 Zielsetzung 19 2 Material und Methoden 21 2.1 Patienten 21 2.2 Histologie 22 2.3 Gewebevorbereitung 23 2.4 Ermittlung der VEGF-Konzentration in den Gewebeproben 23 2.4.1 Proteinextraktion 23 2.4.2 Ermittlung der Gesamtproteinkonzentration 24 2.4.3 VEGF-Konzentrationsmessung 25 2.5 Mutationsanalyse im p53-Gen der Gewebeproben 27 2.5.1 DNA-Extraktion 27 2.5.2 Polymerase Kettenreaktion (Polymerase Chain Reaction, PCR) 30 2.5.2.1 Gelelektrophorese mit zeitlichem Temperaturgradienten, (Temporal Temperature Gradient Electrophoresis, TTGE) und Gelelektrophorese mit einem Denaturans Gradienten Gel (Denaturing Gradient Gel Electrophoresis, DGGE) 34 2.5.3 Sequenzierung 41 2.5.3.1 Klonierung der amplifizierten Homoduplex-DNA 42 2.5.3.2 Durchführung der Sequenzierung 45 2.6 Statistik 48 3 3 Ergebnisse 49 3.1 Alterationen des p53-Gens im Normal- und Tumorgeweben 49 3.2 Lokalisation und Art der p53-Mutationen 50 3.3 Art der nachgewiesenen Mutationen 53 3.4 VEGF-Konzentration im Normal- und im Tumorgewebe 54 3.5 VEGF-Konzentration im Normal- und Tumorgewebe von Patienten 55 mit und ohne Mutation im p53-Gen 3.6 VEGF-Konzentration im Tumorgewebe bei Patienten mit Lokalisation der Mutation im Exon 5,6,7 oder 8 3.7 57 VEGF-Konzentration im Tumorgewebe mit und ohne p53-Mutation in Abhängigkeit vom Geschlecht 3.8 58 VEGF-Konzentration im Tumorgewebe mit und ohne Mutation im p53-Gen getrenntnach Lokalisation des Tumors 3.9 VEGF-Konzentration im Tumorgewebe mit und ohne Mutation im p53-Gen unterteilt in Dukes-Stadien 3.10 59 60 VEGF-Konzentration im Tumorgewebe zwischen Patienten mit und ohne Mutation beiunterschiedlicher Invasionstiefe des Tumors nach dem TNM-System 3.11 62 VEGF-Konzentrationen bei Patienten mit Mutationen in Codons, die für die DNA-bindende Struktur und die Tertiärstruktur des p53-Proteins kodieren 63 4 Diskussion 65 4.1 Häufigkeit von p53-Mutationen und Mutationstypen 65 4.2 VEGF Konzentration im Tumor- und Normalgewebe 70 4.3 VEGF Konzentration bei Tumoren mit und ohne Mutation im p53-Gen 71 4.4 Schlussfolgerung 74 5 Zusammenfassung 76 6 Literaturverzeichnis 79 7 Anhang 96 7.1 Abkürzungen in alphabetischer Reihenfolge 96 7.2 Materialien 96 4 7.3 Geräte 104 7.4 Analysedaten 107 7.5 Abbildungsverzeichnis 112 7.6 Tabellenverzeichnis 112 8 Danksagung 114 9 Lebenslauf 115 5 1 Einleitung Die Entstehung von kolorektalen Karzinomen ist ein kontinuierlicher Prozess. Die zugrunde liegenden genetischen Alterationen treten in einer bestimmten zeitlichen Reihenfolge auf und beeinflussen die biologischen Eigenschaften der Tumore [37,178]. Die Tumorgenese aus gesunder Schleimhaut über ein Adenom bis hin zum Karzinom in Abhängigkeit der genetischen Alterationen beschrieben Vogelstein et al. [172] mit der Adenom-Karzinom-Sequenz. Als molekulargenetische Grundlage gilt in diesem Modell die Akkumulation von Mutationen in dem Genom einer einzigen Zelle oder eines kleinen Zellverbandes. Aus diesen Genomveränderungen resultieren entweder eine Aktivierung von Onkogenen oder die Inaktivierung von Tumorsuppressorgenen [37]. Zu den Tumorsuppressorgenen werden das APC-(Adenopolyposis Coli)Gen, das DCC-(Deleted in Colorectal Cancer)Gen sowie das p53Tumorsuppressorgen gezählt [91]. Onkogene vermitteln im aktiven Zustand einen Proliferationsreiz, die physiologische Funktion der Tumorsuppressorgene besteht darin, das unkontrollierte Zellwachstum zu unterdrücken. Die veränderte Aktivität der Suppressorgene stellt einen entscheidenden Schritt in der Tumorprogression dar. Eine Störung in dem Verhältnis dieser Regulatoren kann zu pathologischem Zellwachstum im Sinne eines malignen Prozesses führen. Zur Induktion einer Transformation der Zelle vom physiologischen zum malignen Zustand wird die Aktivierung mehrerer Onkogene benötigt [95,99]. Qualitative und quantitative Veränderungen auf der Ebene der Transkription und Translation, die zur veränderten Expression oder zum Verlust der transkriptionellen Regulation führen, beeinflussen die Funktion oder die Menge der Genprodukte. Das Tumorsuppressorgen p53 wird hauptsächlich durch missense-Mutation oder Deletion in einem oder beiden Allelen inaktiviert. Dadurch unterscheidet sich dieses von anderen Tumorsuppressorgenen, die regelmässig durch Deletionen oder nonsense-Mutationen inaktiviert werden, oder von Onkogenen der ras-Familie, die durch Mutationen in wenigen gut definierten Kodons aktiviert werden [9]. Mutationen im p53-Gen treten regelmässig in kolorektalen Karzinomen auf [9]. Entgegen dem 1990 postulierten Modell von Fearon und Vogelstein [37], welches sukzessive genetische Veränderungen bei kolorektalen Malignomen (CRC, colo rectal cancer), in den Genen APC, k-ras, DCC und p53 beschreibt, scheint aus heutiger Sicht die Summe der Mutationen und deren zeitliche Reihenfolge für die Tumorgenese relevant zu sein [7,24,172]. Folgende Schemata der Mutationsabfolge werden bei kolorektalen Tumoren als Grundlage vorgeschlagen: Mutation in k-ras und folgend in APC, SMAD 2/4, p53 [7], oder zuerst eine Mutation im APC, folgend COX-2-(Cyclooxygenase 2) Überexpression, anschließend eine Mutation in k-ras, p53 und 18q Allel-Verlust (Genlokus für u.a. DCC/SMAD 2/4) [24]. 6 Neben dem Mutationsmuster der Tumore ist auch der, zum Teil durch die Mutationen bedingte, Einfluss von Wachstumsfaktoren entscheidend für das weitere Proliferationsverhalten. Es zeigte sich ein supportiver Einfluss des „Vascular Endothelial Growth Factors“ (VEGF) auf die Progression von Adenomen zu kolorektalen Karzinomen. Die VEGF-Expression ist während der Progression vom Adenom zum Karzinom in kolorektalen Tumoren schrittweise verstärkt [84,93]. 1.1 Der Angiogenesefaktor Vascular Endothelial Growth Factor (VEGF) Der Vascular Endothelial Growth Factor ist ein potenter mitogener und angiogener Faktor, welcher selektiv auf vaskuläre Endothelzellen wirkt [39,58,122] und möglicherweise auch die Chemotaxis von Monozyten stimuliert [38,43,57,88]. Die Entstehung von neuen Blutgefäßen aus schon vorhandenem Gefäß-Endothelium ist eine wichtige Komponente der Angiogenese. Der 1989 entdeckte VEGF [39,58], auch als „Vascular Permeability Factor“ (VPF) [26] und Vasculotropin [122] bekannt, ist ein fundamentaler Regulator der physiologischen und pathologischen Angiogenese [43]. Im Rahmen der Tumorprogression wird das zu versorgende Tumorvolumen größer. Für viele Neoplasieentitäten gilt, daß zu Beginn des Wachstums die Tumorzellen nach dem „cooptedvessel-model“ vorhandene Kapillaren als nutritive Gefäße rekrutieren. Das „besetzte“ Gefäßsystem unterliegt im Verlauf nicht der tumorinduzierten Angiogense. Diese Gefäße ziehen sich aus dem Versorgungsgebiet ab einem bestimmten Zeitpunkt im Sinne einer „host-defenceReaktion“ zurück [72,73]. Wird nun ein kritischer Punkt erreicht, an dem die Perfusion durch das noch vorhandene, den Tumor umgebende Gefäßnetz nicht mehr ausreicht, kann das Wachstum der neoplastischen Zellen im Tumorverbund nicht über ein Volumen von 1-2mm3 hinaus expandieren. Erlangt der Tumor über angiogene Mechanismen wie z.B. ein verändertes Expressionsmuster des antiangiogenen Angiopoietin-2 oder dem Agonisten VEGF die Potenz zur Gefäßneubildung, nimmt sein Wachstum jedoch exponentiell zu [46,47,56,72]. Es zeigt sich eine gesteigerte VEGF-mRNA-Expression und vermehrte VEGF-Rezeptor-mRNA in den zu den Tumoren benachbarten Endothelzellen [162]. Die VEGF-Expression ist in avaskulären Arealen von Tumoren besonders stark. Hypoxie induziert die VEGF-Expression und stimuliert bzw. reguliert somit die Tumorangiogenese, unabhängig von den einzelnen genetischen Mutationen, die zur Transformation geführt haben. Der Tumorwachstumsvorteil durch die VEGF-Expression scheint eine Konsequenz aus der parakrinen Stimulation der Angiogenese zu sein [162]. Zusätzlich zu seiner mitogenen Aktivität beeinflusst VEGF die Glukoseaufnahme und die Produktion von Gewebefaktoren, Kollagenasen und PlasminogenAktivatoren in Endothelzellen [165]. 7 1.1.1 Molekulare und biologische Eigenschaften von VEGF Das humane VEGF-Gen ist auf dem Chromosom 6p21.1 lokalisiert. Die kodierende Sequenz spannt sich über einen Bereich von etwa 16kb. Das Gen besteht aus 8 Exons, getrennt durch 7 Introns [43,76,82,124,165,167]. Durch alternatives Verspleissen der mRNA können mindestens fünf verschiedene Isoformen des VEGF-Proteins entstehen. Diese werden je nach Anzahl der Aminosäuren bezeichnet (VEGF121, VEGF145, VEGF 165, VEGF189 und VEGF206) und setzen sich aus den Produkten der Exons des VEGF-Gens wie in Abbildung 1 zu sehen zusammen [43, 76,82,124,165,167]. Abbildung 1: Die fünf verschiedenen Isoformen des humanen VEGF entstanden durch Translation alternativ verspleisster VEGF-mRNA. Exon I kodiert für ein Signalpeptid, das wie die Produkte der Exons II-V und des Exon VIII in allen Isoformen präsent ist. Die durch die Exons III und IV kodierten Abschnitte werden für die Bindung an VEGFR-1 und VEGFR-2 benötigt. Die Isoformen unterscheiden sich durch die An- oder Abwesenheit der durch die Exon VI- und Exon VII- kodierten Aminosäuren, welche hauptsächlich die Heparinbindung vermitteln bzw. für die Interaktion mit Neuropilin-1 verantwortlich sind [n. 112,134]. Das als Dimer vorliegende VEGF-Protein besitzt in Abhängigkeit von der Anzahl der Aminosäuren ein Molekulargewicht von 45-46kDa [41], das sich auf zwei Untereinheiten mit jeweils etwa 23kDa verteilt [26]. Alle Isoformen verfügen über eine für sekretierte Proteine typische N-terminale Sequenz. Sie unterscheiden sich in ihrer Fähigkeit, Heparin zu binden. Während VEGF121 kaum Heparin bindet, ist für VEGF189 und VEGF206 eine starke und für VEGF165 sowie VEGF145 eine mittelgradige Affinität für Heparin nachgewiesen [42,43,114]. Nur VEGF121 liegt ausschliesslich in löslicher Form vor, VEGF189 und VEGF206 hingegen sind in bioaktiver Form subepithelial an Heparin enthaltende Proteoglykane in der extrazellulären Matrix (ECM) gebunden. Aus der ECM können diese mit Hilfe von Heparin und Plasmin durch Spaltung am COOH-Terminus freigesetzt werden [43,77,117]. Die beiden Isoformen VEGF145 8 und VEGF165 liegen sowohl in löslicher Form als auch gebunden an die ECM vor [42,124]. VEGF165 ist die dominierende Isoform, welche in einer Reihe von normalen und transformierten Zellen produziert wird. Transkripte, die für VEGF121 und VEGF189 kodieren, werden in der Mehrzahl der Zellen und Gewebe gefunden, die das VEGF-Gen exprimieren [40]. Das VEGFProtein kann durch zwei Mechanismen für die endothelialen Zellen verfügbar werden, als frei lösliches Protein (VEGF121/165) oder als Folge einer Protease-Aktivierung und Spaltung der längeren Isoformen [43]. Die VEGF-Expression ist unter hypoxischen [121,152,153] aber auch unter hypoglykämischen [20,153] Bedingungen erhöht. Dies geschieht über zwei Mechanismen, zum einen über vermehrte Transkription z.B. über die Aktivierung durch HIF-1 [20,158], zum anderen über eine Stabilisierung der mRNA [158]. Das VEGF-Gen kann durch einige Wachstumsfaktoren und Zytokine induziert [38] oder in bestimmten Tumorentitäten durch die gesteigerte Expression des ras-, raf-, src- oder mutierten p53-Gens reguliert werden [57,61,111,128,176]. Auch die Akkumulation von Adenosin, welche unter hypoxischen Bedingungen auftritt, ist an der Induktion der Transkription des VEGF-Gens beteiligt [102]. Über den aktivierten Adenosin A2Rezeptor kommt es zu einer erhöhten intrazellulären cAMP-Konzentration. Diese steigert die VEGF-mRNA-Konzentration über einen Proteinkinase A vermittelten Signalweg [160]. Eine Aktivierung des Onkogens c-Src zeigt ebenfalls einen positiven Einfluss auf die hypoxieinduzierte Hochregulation der VEGF-Gen Expression [111]. Die biologischen Wirkungen von VEGF treten spezifisch nur an vaskulären Endothelzellen auf, unabhängig von der Lokalisation des Gefässbettes oder der Spezies. Dabei wirkt VEGF über einen parakrinen Mechanismus auf die Endothelzellen [9,26,41,114]. An Gefässendothelzellen bewirkt VEGF Migration und Mitose in vitro [41,58] sowie Angiogenese und erhöhte Permeabilität in vivo [26,98,121,143]. Der mitogene Effekt in vitro ist halbmaximal bei VEGFKonzentrationen von 2-3pM und maximal bei 22pM [41]. In Übereinstimmung mit der Hypothese, dass im Zuge der Angiogenese eine begrenzte extrazelluläre Proteolyse vonnöten ist [158], induziert VEGF die Expression von Plasminogen-Aktivator (PA) und PlasminogenAktivator-Inhibitor in Endothelzellen [20]. Die Plasminogen-Aktivierung und Generierung des Plasmins spielen eine wichtige Rolle in der Angiogenesekaskade. Die Koinduktion dieser Faktoren produziert ein Umfeld, welches Migration und Aussprossen von Endothelzellen ermöglicht [176]. Generierung des bioaktiven VEGF, das durch proteolytische Spaltung entsteht, scheint besonders wichtig in der Mikroumgebung eines Tumors zu sein. In Tumornähe wird eine erhöhte Konzentration von Proteasen, z.B. des Plasminogen-Aktivators nachgewiesen [133,156]. Dvorak [35] vermutete, dass eine Steigerung der mikrovaskulären Permeabilität für Proteine ein bedeutender Schritt in der Angiogenese ist. Die VEGF-abhängige Phospholipase-D (PLD) Aktivität ist direkt an der Kontrolle der DNATranskription beteiligt; diese beeinflusst die Synthese des Zytoskelettes, welches einen 9 weiteren wichtigen Schritt in der VEGF vermittelten Neoangiogenese darstellt [4]. Die verschiedenen Isoformen von VEGF mit ihren unterschiedlichen Eigenschaften bezüglich Heparinbindung und Bindung an die extrazelluläre Matrix scheinen in unterschiedlicher Weise die Angiogenese zu induzieren. Zelllinien aus der Maus, die nur einzelne Isoformen exprimierten, waren nicht fähig, ein voll funktionsfähiges Gefäßnetz zu bilden. Einzig VEGF164, in der Maus analog dem humanen VEGF165, konnte eine angiogene Antwort induzieren. VEGF120, analog dem humanen VEGF121, konnte nur Arme umgebender Gefäße rekrutieren, ohne eine Vaskularisierung des Tumors zu gewährleisten. VEGF188, analog dem humanen VEGF189, bewirkte eine ausgedehnte Mikrovaskularisierung des Tumors ohne ausreichende Versorgung durch größere Gefäße [62]. VEGF beeinflusst neben der Angiogenese auch das Überleben der Endothelzellen. So führte es bei in-vitro Experimenten an Endothelzellen zur Inhibition der Apoptose bei gleichzeitig erhöhter Expression der antiapoptotischen Proteine Bcl-2 und A1 [53]. Auch auf das Überleben von Karzinomzellen scheint VEGF einen Einfluss zu haben. So konnte in vitro bei Erhöhung der VEGF-Konzentration eine Verminderung der Apoptoserate von Adenokarzinomzellen aus einem metastasierenden Mammakarzinom festgestellt werden. Es fand sich ebenfalls eine erhöhte bcl2-Expression. Im Gegenzug hatte eine Inkubation mit Anti-VEGF-Antikörpern eine Erhöhung der Apoptoserate zur Folge [119]. 1.1.2 Rezeptoren und Signaltransduktion des VEGF Die Expression der VEGF-Rezeptoren Flt-1 (VEGFR-1) und Flk-1/KDR (VEGFR-2) ist begrenzt auf das vaskuläre Endothel [39,114]. Hypoxie und Hypoglykämie sind wie bei VEGF wirksame Mediatoren, die Genexpression der VEGF-Rezeptoren zu erhöhen [168]. VEGF selbst kann die Expression von VEGFR-2 und VEGFR-1 in humanen Endothelzellkulturen hochregulieren. Auch andere Mediatoren wie TNF-α, oder TGF-β beeinflussen die Expression beider VEGFRezeptoren [43]. VEGF bindet mit hoher Affinität an seine Rezeptoren. Die Dissoziationskonstanten betragen für Flt-1 Kd = 16pM und für Flk-1/KDR Kd = 760pM [170,174]. Die Bindung von VEGF165 an VEGFR-2 kann dabei durch Neuropilin-1, einem Rezeptor der Collapsin/Semaphorin-Familie, welcher als Co-Rezeptor wirkt, verstärkt werden [154]. Neuropilin-1 präsentiert VEGF165 dem Flk-1/KDR Rezeptor mit dem Ergebnis, dass die Effektivität der Flk-1/KDR vermittelten Signaltransduktion verstärkt wird [43]. Beide Rezeptoren sind transmembranöse Tyrosinkinasen mit sieben immunglobulinartigen extrazellulären Domänen (ECD), einer transmembranösen Domäne und einer intrazellulären Tyrosinkinase-Domäne mit einer „Kinase Insert Domain“ [55,127,164]. Ein zusätzliches Mitglied der RTK (Rezeptor-Tyrosin-Kinasen)-Familie mit sieben Ig-ähnlichen Domänen in der ECD ist Flt-4 (VEGFR-3). Flt-4 ist jedoch kein wirklicher Rezeptor 10 für VEGF, bindet aber einen Liganden mit dem Namen VEGF-C oder VEGF-related peptide (VRP). VEGF-C zeigt eine Wirkung als Regulator der Lymphgefäßentwicklung [43,81]. Ähnlich wie andere Wachstumsfaktoren mit transmembranösen Tyrosinkinaserezeptoren unterzieht sich auch der VEGF-Rezeptor einer Liganden-induzierten Dimerisierung [165]. Die LigandenRezeptor-Interaktion führt bei VEGFR-2 zu einer Reihe von Effekten, die bei VEGFR-1 nicht oder nur in geringem Ausmaße auftreten [55,174]. So führt die Stimulation von VEGFR-2 durch VEGF in vitro zur Autophosphorylierung. Tyrosinreste in der zytoplasmatischen KDR-Domäne sind das Ziel der Auto- oder Transphosphorylierung. In vivo führt eine Aktivierung von Phospholipase C- (PLC- ) und PI(3)K sowie erhöhter intrazellulärer Calciumspiegel und vermehrt freigesetzter von Willebrand Faktor zu gesteigerter Angiogenese und zur Erhöhung der Gefäßpermeabilität [18,55,127,165]. Dabei spielt möglicherweise das „VEGF receptor associated protein“ (VRAP) eine Rolle, das an der intrazellulären Domäne an die „Kinase Insert Domain“ des VEGFR-2 bindet und selbst mit der PLC- assoziiert ist [182]. In vitro ist bei Stimulation von VEGFR-2 eine ausgeprägte Änderung der Zellmorphologie, eine Reorganisation des Aktins, Mitogenese und Chemotaxis zu beobachten [174]. All dieses tritt bei Stimulation von VEGFR-1 nicht auf, wenn auch die Stimulation von VEGFR-1 in vitro ebenfalls zur Autophosphorylierung des Rezeptors führt, allerdings in einem weitaus geringerem Ausmaße als bei VEGFR-2 [174]. In vivo kommt es nicht zu den oben bei VEGFR-2 beschriebenen Effekten [55]. Das durch VEGF induzierte Überleben von Endothelzellen durch Hemmung der Apoptose wird über VEGFR-2, wahrscheinlich unter Einbeziehung der PI3Kinase, vermittelt [54]. VEGFR-2 scheint somit der Rezeptor zu sein, der für die Angiogenese entscheidend ist. Sowohl VEGFR-1 als auch VEGFR-2 werden in Endothelzellen von Gefäßen, die maligne Tumore versorgen, verstärkt exprimiert [20,120,176,177]. Der zur Zeit einzig bekannte endogen exprimierte VEGF-Inhibitor ist eine alternative Spleissversion des FLT-1 VEGF-Rezepors. Die Spleissversion führt zu einer verkürzten löslichen Form (sFLT), welche aus der Retention eines Introns innerhalb der mRNA resultiert. Die entsprechende an diesem Intron nicht prozessierte mRNA wird bis zum ersten im Leseraster liegenden Stopcodon translatiert. Das sFLT-1 Protein beinhaltet die N-terminalen sechs der sieben extrazellulären Ig-ähnlichen Domänen welche mit den 31-Intron kodierten Aminosäuren der C-terminalen Sequenz fusionieren. sFLT-1 ist ein Heparin-bindendes Protein, das VEGF mit der gleichen Affinität und annähernd äquivalenter Spezifität wie der FLT-1Rezeptor bindet. Es wird durch die vaskulären endothelialen Zellen exprimiert und kann deren mitogene Antwort auf VEGF hemmen. Durch Heterodimerisierung mit den extrazellulären Ligandenbindungsregionen der VEGF-Rezeptoren werden die Rezeptoren vor der TyrosinTransphosphorylierung geschützt und damit auch die Signaltransduktion und die funktionelle Aktivierung gehemmt. Die erhöhte Expression von sFLT-1 durch Tumorzellen inhibiert in vivo 11 das Wachstum der soliden Tumore, bzw. die metastatische Aussaat in die Lymphknoten und verlängert das Überleben der Versuchsgruppe [57,85]. 1.1.3 VEGF-Expression in Neoplasien Die Annahme, dass das Tumorwachstum angiogeneseabhängig ist und dass VEGF das Aussprossen von Gefäßen fördert, wird durch Untersuchungen unterstützt, welche eine positive Korrelation zwischen der Vaskularisierung humaner Tumore und der VEGF-mRNA-Expression zeigten [38]. Es wurde eine erhöhte VEGF-mRNA-Expression in den Tumorzellen, nicht jedoch in den Endothelzellen gefunden. In den Endothelzellen konnten jedoch erhöhte VEGF-ProteinKonzentrationen nachgewiesen werden [20,96,163,177]. Ebenso trat in den Tumorzellen sowie in den tumorassoziierten Endothelzellen eine erhöhte Konzentration von mRNA, welche für VEGFR-1 und VEGFR-2 kodiert, auf [20]. Innerhalb von Karzinomen bestehen lokale Unterschiede in der VEGF-Konzentration. In Glioblastoma multiforme und anderen Tumorentitäten mit einem hohen Anteil an nekrotischen Komponenten sind die VEGF-mRNAKonzentrationen in ischämischen Tumorzellen nahe den nekrotischen Arealen hoch [20,38,152]. Bei Magenkarzinomen findet sich eine positive Korrelation zwischen VEGF-Expression und Metastasierung, wobei Patienten mit erhöhter VEGF-Konzentration im Karzinom eine schlechtere Prognose bezüglich der Überlebenszeit haben [103]. Verschiedenen Studien wiesen auch bei kolorektalen Karzinomen eine erhöhte VEGFExpression nach [19,20,96]. In Adenomen konnte hingegen keine erhöhte VEGF-Expression gezeigt werden [96]. Die Expression von VEGF und VEGFR-2 scheint bei metastasierenden kolorektalen Karzinomen stärker zu sein als bei nicht-metastasierenden kolorektalen Karzinomen. Gleichzeitig korreliert die Expression von VEGF und VEGFR-2 mit dem Ausmaß der Neovaskularisation [162]. Die VEGF-Konzentration im Serum kann bei kolorektalen Karzinomen gemeinsam mit dem carcinoembryonalen Antigen (CEA) für die Marker-Diagnostik eingesetzt werden. Die Serumkonzentration von VEGF korreliert nicht nur mit dem Volumen bzw. der Größe des Tumors, sondern zeigt auch einen positiven Zusammenhang mit der Überlebenszeit der Patienten mit kolorektalen Karzinomen [19]. Hieraus lässt sich eine Korrelation zwischen der Prognose der Patienten mit kolorektalen Karzinomen und dem Ausmaß der Angiogenese im Tumorgewebe erkennen [84]. In acht humanen Kolonkarzinomzelllinien und 30 humanen Lebermetastasen von kolorektalen Karzinomen wurden erhöhte Konzentrationen von VEGFR2/KDR und Flt-1 mRNA in tumorendothelialen Zellen im Vergleich zur Kontrolle (aus gesundem Lebergewebe) bzw. dem umgebenden Gewebe nachgewiesen [177]. Aus diesen Ergebnissen bzw. Beobachtungen können Therapieansätze abgeleitet werden, die an den verschiedenen 12 Punkten der VEGF-vermittelten Angiogenese inhibierend ansetzen. VEGF-Antikörper blockieren die VEGF-induzierte Endothelzellproliferation in vitro [90,125]. In vivo gelang im Mausmodell eine Suppression des Tumorwachstums mittels VEGF-Antikörpern bei etablierten Tumoren, zudem kam es zur Reduktion der Tumormasse, sowie zur Verringerung der Gefäßdichte [177]. Mittlerweile sind diese Therapieoptionen auch auf den Menschen übertragen worden, indem Anti-VEGF-Antikörper [90], Rezeptorantagonisten oder dominant negative Rezeptormutanten in der Antitumortherapie eingesetzt werden können [108]. Derzeit ergeben sich Therapieoptionen durch eine passive Immuntherapie mit Avastin, einem rekombinanten humanen monoklonalem IgG1 Antikörper gegen VEGF. Auch zeigen aktive Immuntherapien mit Impfungen unter Verwendung von Endothelzellen, dentritischen Zellen oder DNA sowie verschiedene Ansätze zur Inhibition der Ligand-Rezeptor-Interaktion und der Signaltransduktion, welche zur Therapie von kolorektalen Karzinome eingesetzt werden, antiproliferative Effekte. Nach Prüfung in verschiedenen klinischen Studien ist Avastin als Therapieoption in den USA zur Therapie zugelassen [57,65,75]. Es konnte eine Verlängerung der mittleren Überlebenszeit und Reduktion der Tumorgröße, Vaskularisierung und Tumorzellproliferation im Tiermodell dargestellt werden [81,90,145,164], des weiteren hemmen Inhibitoren der Tyrosinkinaseaktivität von VEGFR-2 die Mitogenese in vitro sowie die Neovaskularisation in vivo [33,96,141,164]. 1.2 Das Tumorsuppressorgen p53 1.2.1 Das p53-Gen Das p53-Gen ist auf dem Chromosom 17p13 lokalisiert. Es erstreckt sich über 20kb der genomischen DNA und enthält 11 Exons, die zu einer 2,6kb mRNA transkribiert und verspleisst werden [99]. Die Expression des p53-Gens wird über Cis-Elemente reguliert. Das Tumorsuppressorgen besitzt keine TATAA oder CAAT Box. Die Transkription startet von der Basenposition –114 am 3‘-Ende des Exon 1. Der p53-Promotor besitzt ein Helix-Schleife-HelixErkennungsmotiv (CACGTG) in Position +70 in Exon 1. An dieser Erkennungsstelle binden cMyc/Max Heterodimere und aktivieren die Transkription des p53-Promotors [33]. Die transkriptionale Aktivierung wird durch das humane MDM2-(murine double minus-2 protein) Protein negativ reguliert [33,99]. Das p53-Gen ist ein universeller Sensor des genotoxischen Stresses der Zelle. Dieses Tumorsuppressorgen kodiert für ein im Zellkern lokalisiertes Phosphoprotein mit antikarzinogener Eigenschaft. Der Verlust der physiologischen p53-Aktivität auf Grund einer 13 Mutation führt zur zellulären Immortalität und prädisponiert diese Zellen für weitere onkogene Mutationen [59,67,92,99]. 1.2.2 Eigenschaften des Wildtyp-p53-Proteins (Wt-p53) Das humane 53kD grosse p53-Protein umfasst 393 Aminosäuren und ist unterteilt in 3 strukturelle und funktionelle Domänen (s. Abbildung 2). Die ersten 42 Aminosäuren am NTerminus stellen eine transkriptionelle Aktivator-Domäne dar, welche als positiver basaler transkriptioneller Regulator der Genexpression wirkt [99]. Die Tetramerisierungsdomäne am Cterminalen Ende ist mit der DNA-Bindungsdomäne durch einen flexiblen 37 Aminosäurereste umfassenden Strang (AS 287-323) verbunden. Die Aminosäuren 324-355 werden für die Oligomerisation des Proteins benötigt [99]. P53 interagiert über eine hydrophobe Bindungsstelle an seiner N-terminalen Domäne als negativer Regulator der transkriptionellen Aktivität der Zellen [99]. Es beeinflusst u.a. die Transkription des humanen hsp70-Gens, durch Interaktion mit dem CCAAT-Bindungsfaktor (CBS). Durch diese Protein-Protein-Interaktion wird die Transkription des hsp70-Gens negativ beeinflusst. Das p53-Protein bindet an einen minimalen Promotor mit einer TATA-Sequenz, wodurch TATA-Box-Gene negativ reguliert werden können [33]. Die DNA-bindende Domäne im Bereich der Aminosäuren 102-292 von p53 unterscheidet sich von der anderer bekannter DNAbindender Proteine durch eine grosse ß-Sandwich-Struktur, welche die Orientierung und die Positionierung zur Interaktion mit der DNA erlaubt [136]. Die ß-Sandwich-Struktur bildet das Gerüst für eine Faltblattstruktur und für zwei anti-parallele ß-Schleifen. Die korrekte Ausrichtung zur DNA-Bindung hängt von der Seitenketteninteraktion zwischen den parallelen ß-Schleifen und der tetrahedralen Koordination eines Zinkatoms zwischen Cys176 und His179 in Schleife 2 und Cys238 und Cys242 in Schleife 3 ab. Das native p53-Protein ist in Lösung ein Tetramer. Die Struktur beinhaltet Dimere mit zwei ß-Bögen und zwei α-Helices. Zwei Dimere werden durch eine grosse hydrophobe Oberfläche der beiden Helix-Paare zusammengehalten, welche ein 4Helix-Bündel bilden. Die Möglichkeit des Wt-p53, einen antiproliferativen Effekt auf die Zelle auszuüben, ist an den Wt-Konformationsstatus mit einem hohen Grad an Phosphorylierung gebunden. Das Wt-p53-Protein kann seine Konformation und Phosphorylierung abhängig vom Zellzyklus der Zelle verändern [33]. 14 Abbildung 2: p53-Protein. Oben: Aminosäurenpositionen der in humanen Tumoren nachgewiesenen Mutationen, welche die Funktion des p53-Proteins beeinflussen. Graue Boxen I-V repräsentieren die evolutionär hoch konservierten Sequenzen. Die vertikalen Linien kennzeichnen die am häufigsten gefundenen Stellen für einen Aminosäureaustausch in humanen Tumoren. Die zugrundeliegenden Mutationen konzentrieren sich in den konservierten Regionen II–V. Unten: Strukturelle Organisation des p53-Proteins. Phosphorylierungsstellen verschiedener Kinasen, gekennzeichnet durch P. MDM2 Protein bindet am N-Terminus und unterdrückt die transkriptionelle Aktivierung von p53 als Teil der normalen Kontrolle der p53-Funktion. Die Aktivität von p53 wird ebenfalls inhibiert z.B. durch die Bindung von viralen Proteinen wie E6 des humanen Papillomavirus und E1b von Adenoviren [n. 69,92]. Das Tumorsuppressorprotein ist ein multifunktionaler Transkriptionsfaktor, welcher bei der Kontrolle der Zellzyklusprogression, der DNA-Integrität und des Zellüberlebens bei agentienbedingten DNA-Schäden über die Regulation der Apoptose, der Suppression der Zellproliferation und des G1/S- oder G2-Zellzyklusarrests eine Rolle spielt [2,9,92,99]. Die Induktion der Apoptose durch Überexpression des Wt-p53-Proteins ist in humanen Kolontumorzelllinien und Zelllinien anderer Tumore gezeigt worden [25,33,101,148]. Die p53-induzierte Apoptose wird über die positive transkriptionale Regulation des Bax- und IGF-BP3-Gens vermittelt. BAX beschleunigt die Apoptose, indem es mit Bcl2 Heterodimere bildet und dadurch die Bcl2-Funktion als Antagonist der Apoptose inhibiert. Bcl2Überexpression hemmt die p53-vermittelte Apoptose [25,99,101,130,148]. Das humane p53 zeigt sich als Substrat von CDC2. Die primäre Phosphorylierungsstelle ist Serin-315. An dieser Position wird das Protein sowohl von p60-cdc2- als auch von Cyclin B- 15 cdc2-Enzymen phosphoryliert. Die Phosphorylierung durch CDC2 reguliert die antiproliferative Aktivität des p53 [78]. Gene bzw. deren Produkte, die über p53-bindende Elemente hochreguliert werden (GADD45, p21WAF-1/CIP1 und MDM2 oder Thrombospondin), sind negative Regulatoren der metastatischen Aussaat oder der Angiogenese [29,136]. GADD45 ist an der Stimulation der DNAReparaturvorgänge beteiligt und wird als Antwort auf einen DNA-Schaden induziert. Es bindet an PCNA (proliferating cell nuclear antigen), welches ein DNA-Polymerase prozessiver Faktor ist, und kann somit den Zellzyklusarrest auslösen bzw. die DNA-Replikation inhibieren [99,136,173]. Der p53-vermittelte G1/S-Zellzyklusarrest ist assoziiert mit der Transaktivierung des p21WAF1/CIP1 -Gens. Der Zellzyklus wird durch aktiviertes p53 und Modulation der p21WAF-1/CIP1 Transkription reguliert. In Abhängigkeit zum DNA-Schaden steigt die Konzentration des p21Transkripts proportional zum p53-Anstieg an [2,31,66,109,181,185]. p21 bindet an verschiedene Cycline und Cdk-Komplexe sowie an die C-terminale Domäne des PCNA. Diese p21-Bindungen führen zum Stop der DNA-Replikation. Neben diesem Mechanismus existiert ein p21-unabhängiger, p53-vermittelter G1-Arrest [99,173]. MDM-2, ein Onkogenprodukt, welches selbst unter regulatorischem Einfluss des p53 steht (s.o.), führt über den Abbau von p53-Protein zu einer Inaktivierung der p53-vermittelten Transkription und wirkt als autoregulatorische Feedback-Schleife für die p53-Aktivität (s. Bindungsstellen in Abbildung 2). MDM-2 bildet einen engen Komplex mit p53 über eine hydrophobe Tasche, in der sich eine durch p53 geformte amphiphatische Helix mit hydrophober Oberfläche einfügt. Es agiert sowohl mit mutiertem als auch mit Wt-p53 und inhibiert die negativ regulatorischen Effekte von p53 [11,33,83,146,183,186]. Die p53-Protein-Konzentration in einer Zelle wird durch ständige Proteolyse auf einem konstant niedrigen Niveau gehalten mit einer Halbwertszeit (HWZ) des Proteins von 20min [68,99]. Das p53-Protein bindet an DNA-Reste und Reparaturstellen oder an interne Deletionsschleifen. Die intrazelluläre Erhöhung der Wt-p53-Konzentrationen ist proportional zum Umfang des aquirierten DNA-Schadens der Zelle. Zusätzlich zum Genomschaden kann Hypoxie die Wt-p53Protein-Konzentration steigern und das p53-Protein aktivieren. Durch die Aktivierung des Wtp53 kann so in Abhängigkeit des zytotoxischen Einflusses über die Apoptose eine negative Wachstumsregulation transformierter bzw. potentieller maligner Zellen vermittelt werden [99]. 1.2.3 Mutiertes p53 (Mut-p53) Die Alteration des p53-Gens sind v.a. Punktmutationen in den stark evolutionär erhaltenen Regionen (s. Abbildung 2, graue Boxen), mit komplett oder partiell veränderter p53Proteinstruktur [33]. Die Mehrzahl der Mutationen im p53-Gen gehen mit einem veränderten 16 p53-Protein einher und verhindern die sequenzspezifische Bindung des Proteins an die DNA. Mutationen in der DNA-bindende Domäne sind v.a. missense-Mutationen, welche mit der Entwicklung von humanen Tumoren einhergehen. Im Bereich des N-Terminus (AS 1-50) in der sogenannten Aktivierungsdomäne finden sich prolinreichen Folgeabschnitt 63-92) 40,7%, (AS 50,0% in missense-Mutationen, der in sequenzspezifischen dem DNA- Bindungsdomäne (AS 100-300) 76,6%, in der Tetramerisierungsdomäne (AS 323-358) 34,2% missense- und 40,2% nonsense-Mutationen und im C-Terminus (AS 363-393) 50% missenseMutationen [136,155]. Insgesamt wurden 73% der hauptsächlich missense-Mutationen in den ‚hot spot’-Exons 5-8 in den evolutionär hochkonservierten Regionen gefunden (s. Kapitel 1.2.4) [44]. Cho et al. [23] klassifizierten drei Mutationstypen, welche in humanen Karzinomen die DNA-Bindungsstelle von p53 beeinflussen. Klasse I der Mutationen betreffen die Aminosäuren der DNA-bindenden Oberfläche von p53. Durch Veränderungen in Arg248 und Arg273 kommt es zum Verlust der Protein-DNAKontaktpunkte. Diese stellen ‚hot spots’ für p53-Mutationen dar. Klasse II-Mutationen beeinträchtigen die korrekte Orientierung der DNA-bindenden Oberfläche. Diese in Arg175 und Arg249 lokalisierten Mutationen beeinflussen die Verbindung zwischen dem Proteingerüst und der Bindungsoberfläche. Klasse III-Mutationen stören die Ausrichtung des p53-Proteingerüsts. Dieses führt zur Destabilisierung der Tertiärstruktur, mit einer Veränderung der gesamten DNA- Bindungsdomäne [50,60,68,136]. Das mutierte p53-Protein scheint stabiler zu sein als der Wildtyp. Nach einer Alteration des p53Gens mit konsekutiv verändertem Protein, folgt eine erhöhte intrazelluläre Konzentration des Proteins, da die HWZ in transformierten Zellen (in vitro) auf über 24h u.a. durch die Phosphorylierung der MDM-2 Bindungsstellen verlängert und die Translation der p53-mRNA in den Zellen gesteigert wird [68,99]. Daraus resultiert eine Akkumulation des Proteins im Zellkern [115,131]. Das Mut-p53-Protein wirkt über eine Komplexbildung und Oligomerisierung direkt auf Wt-53-Protein inaktivierend [87]. Zusätzlich könnten neben der veränderten Struktur des Mutp53-Proteins auch eine Veränderungen der intrazellulären Rezeptoren des p53-Proteins mögliche wachstumsstimulierende Funktionen erklären [33]. 1.2.4 p53-Mutationen und kolorektale Tumore Für humane Kolonkarzinome resultiert eine genetische Instabilität aus einem Allelverlust auf Chromosom 17. Die Mehrzahl dieser Karzinome enthalten zudem im korrespondierenden p53Allel eine Punktmutation [8,37]. Die Anwesenheit von Mut-p53-Protein korreliert mit dem Verlust des korrespondierenden Wt-p53-Allels: In Kolonkarzinomzellen liegt die Koexistenz von Wt- und Mut-Allel in einer Zelle bei unter 30%. Karzinomzellen mit nur einem Allel weisen in 91% ein 17 Mut-p53-Protein auf. Der Verlust der Heterozygozität zeigt sich entweder (in 75% der Kolonkarzinome) in einem Chromosomenverlust des 17p Chromosoms oder (bei 86% der Tumore) in einer Punktmutation (missense-Mutation) in einem p53-Allel [8,9,44]. Die p53-Mutationen finden sich selten in frühen Tumorstadien (Adenome). Mit dem Übergang vom benignen zum malignen Wachstum (Karzinome) treten Mutationen entsprechend häufiger auf [9,33,37,44]. Sowohl in in-vivo Untersuchungen, als auch die Beobachtung, die in vitro an kolorektalen Zellkulturen gemacht wurden legen die Vermutung nahe, dass das Wt-p53-Gen einen Tumorsuppressor darstellt, welcher kolorektales Tumorwachstum inhibiert [9]. Der Wildtypverlust ist ein genetisches Schlüsselereignis, das unweigerlich zu Malignität führt [33]. Die häufigste Mutationsart in CRC sind missense-Mutationen (73%) in den ‚hot spot’-Regionen (HSR) der Exons 5-8 (s. Abbildung 2) [44]. 52% der Punkmutationen wurden in den Codons 175, 248 und 283 gefunden. Kolorektale Tumore wiesen in 87% Transitionen und nur in 13% Transversionen auf. Die häufigste Transition an CpG-Dinukleotiden war GC AT mit 65%. missense-Mutationen, die auf solche Transitionen zurückgehen, fanden sich zu 25% in kolorektalen Karzinomen [10,33,44,74,113]. In 39-69% der CRC wurde eine p53-Protein-Überexpression nachgewiesen [28,32]. Bei der Aufteilung der Tumore entsprechend ihrer Dukes-Stadien zeigte sich eine deutliche Zunahme der Mutationen in den beiden letzten Stadien (Dukes A-B: 55%, Dukes C-D: 67%) [74]. 1.3 Zusammenhang zwischen VEGF und p53 In kolorektalen [161] und gastralen [138] Malignomen wurde ein Zusammenhang zwischen p53Proteinakkumulation und einer erhöhten VEGF-Konzentration mittels immunhistologischen Nachweismethoden dargestellt. In Tumoren der Prostata [142], Lunge [5] und Niere [97] hingegen konnte diese Korrelation nicht bestätigt werden. Andere Studien zeigten, dass onkogene Transformationen mit aktiviertem ras, src oder raf-1 als Verbindung zwischen der Tumorgenese und der Angiogenese zur gesteigerten Expression von VEGF führen [61,111,128]. Mutiertes p53 scheint in kolorektalen Karzinomen die VEGF-Expression zu induzieren. So zeigten 61,5% der untersuchten Karzinome eine Überexpression von p53Protein. In allen Karzinomen mit positiver p53-Immunhistologie wurde eine erhöhte VEGFExpression nachgewiesen. Mit dem Dysplasiegrad erhöhte sich auch die VEGF-Konzentration in den untersuchten Tumorproben. Diese Untersuchungsergebnisse legen die Vermutung nahe, dass es in Abhängigkeit des Tumorstadiums zu einer verstärkten Induktion der VEGFExpression in kolorektalen Tumoren kommt [93]. 18 In einigen Tumorentitäten besteht eine Korrelation zwischen der erhöhten VEGF-Expression und der p53-Proteinexpression. Diese geht signifikant mit dem Vaskularisationsgrad des Tumors und der Metastasierungspotenz einher [49,161]. In in vitro Versuchen mit Hep3B (Hepatom)- und RKO (humanes kolorektales Karzinom)-Zellen ging parallel mit dem Verlust der Wt-p53-Funktion eine gesteigerten Expression von VEGF einher. Jedoch konnte bei Hep3B-Zellen keine signifikante Korrelation zwischen der VEGFmRNA-Expression und den Wt- oder Mut-p53-Proteinkonzentrationen gefunden werden. Auch die hypoxieinduzierte physiologische VEGF-Expression wird nicht durch Wt-p53 beeinflusst [3]. Jedoch zeigten Zellen, die die Funktionen des Wt-p53-Proteins verloren hatten, eine erhöhte Hypoxieresistenz. Hieraus kann sich eine Selektion von nicht-apoptosefähigen, hypoxieresistenten Zellen ergeben, welche zum dominanten genotypischen Zelltyp im Tumor werden können [107]. Der Wechsel zu einem angiogenetischen Phänotyp ist eine fundamentale Veränderung im neoplastischen Wachstum und der Tumorprogression. Wt-p53 trägt bei zur MDM2-vermittelten proteosomalen Degradierung eines heterodimeren Transkriptionsfaktors, der Untereinheit des „hypoxia-inducible factor“ (HIF-1), der den zellulären Energiemetabolismus und die Angiogenese in Abhängigkeit zur Sauerstoffdeprivation regelt. Ein Verlust des Wt-p53 in Tumorzellen steigert die Konzentration von HIF-1α und die HIF-1 abhängige transkriptionale Aktivierung des VEGF-Gens in Abhängigkeit zur Hypoxie und erhöht die Neovaskularisation und das Wachstum im Tumorxenograft. Dies zeigt, dass die von HIF-1 abhängige Antwort auf Hypoxie über den Verlust der p53-Funktion als Kofaktor zum angiogenetischen Wechsel während der Tumorgenese beiträgt [129]. Die Überproduktion von Wt-p53-Protein reduziert in vitro dosisabhängig die endogene VEGF-mRNA über die VEGF-Promotor-Aktivität. So wurde in der humanen Kolonkarzinomzelllinie SW620 nach Transfektion mit dem Wt-p53-Gen eine reduzierte Konzentration des VEGF-Proteins und seiner mRNA nachgewiesen [16]. Da Wt-p53-Protein, aber nicht Mut-p53 die Expression von Inhibitoren der Angiogenese stimuliert, stellen Zellen mit Wt-p53 im Gegensatz zu jenen mit Mut-p53 einen antiangiogenetischen Phänotyp dar [31]. Für Mut-p53 Ala135-Val besteht ein direkter Zusammenhang zwischen der Mutation und der Regulation der VEGF-Expression. So wurde in NIH-3T3-Zellen mit diesem Mutationstyp die Expression des VEGF-Gens über eine aktivierte Proteinkinase C (PKC) induziert [89]. 19 1.4 Zielsetzung Mutationen im p53-Gen und erhöhte VEGF-Konzentration im Tumorgewebe sind wichtige Faktoren beim Tumorwachstum und der Progression von kolorektalen Karzinomen. Die bis heute veröffentlichten Ergebnisse lassen vermuten, dass diese Faktoren nicht unabhängig voneinander die Tumorprogression beeinflussen. In humanen kolorektalen Tumorzelllinien zeigte sich in vitro ein Zusammenhang zwischen der Wt-p53 Funktion als Suppressor der VEGF-Expression. In vivo wurde unter anderem durch immunhistologische Verfahren eine Korrelation zwischen erhöhter VEGF-Konzentration und p53-Protein-Überexpression in kolorektalen Karzinomen nachgewiesen. Dieses führte zu der Hypothese, dass ein direkter regulatorischer Einfluss von mutiertem p53 auf die VEGF-Expression besteht. In der vorliegenden Arbeit sollte untersucht werden, ob bei Patienten mit kolorektalen Karzinomen eine bestimmte Mutation im p53-Gen mit einer erhöhter VEGF-Konzentration im Tumorgewebe einhergeht. Es ergeben sich die folgenden Fragestellungen: 1) Besteht ein Unterschied in der VEGF-Konzentration zwischen Tumorgewebe mit bzw. ohne Mutation im p53-Gen? 2) Beeinflussen unterschiedliche Mutationstypen bzw. Mutationen in bestimmten ‚hot spot’Exons die VEGF-Konzentration? 3) Beeinflussen Mutationstypen bzw. Mutationen in der kodierenden Sequenz a) für die DNA-bindende Domäne oder b) in der für die Tertiärstruktur des p53-Proteins die VEGF-Konzentration? 4) Besteht ein Unterschied in der VEGF-Expression zwischen Tumoren mit bzw. ohne Mutation im p53-Gen bei zusätzlicher Unterscheidung nach klinischen sowie pathologischen Daten wie Geschlecht, Tumorlokalisation (Kolon/Rektum), Tumorstadium (Dukes A-D) und T-Kategorie der TNM-Klassifikation als Kriterium der Invasionstiefe des Tumors? Die Ergebnisse sollen zum weiteren Verständnis der Progression von kolorektalen Karzinomen und deren Beeinflussung durch die Angiogenese beitragen. 20 2 Material und Methoden Die Zahlen in den eckigen Klammern verweisen u.a. auf die Liste der Materialien und Geräte im Anhang unter Punkt 7.2. und 7.3. 2.1 Patienten Die in der Untersuchung verwendeten Gewebeproben wurden Patienten der Klinik für Chirurgie (Direktor: Prof. Dr. H.-P. Bruch) des Universitätsklinikum Schleswig-Holstein Campus Lübeck entnommen, welche im Zeitraum von Februar 1996 bis September 1997 an einem kolorektalen Adenokarzinom operiert worden waren. Das Resektat wurde nach Entnahme direkt in das Institut für Pathologie (Direktor: Prof. Dr. Feller) gebracht und dort Gewebeblöcke sowohl aus der Region des Karzinoms (Tumorgewebe, Tumorprobe) als auch aus dem gesunden angrenzenden Darmabschnitt (Normalgewebe, Normalprobe) entnommen. Diese Gewebeblöcke konnten ohne weitere Behandlung sofort nach Entnahme in flüssigem Stickstoff bei -196°C eingefr oren werden. Ebenfalls wurde durch das Institut für Pathologie eine Aufarbeitung des OP-Resektates nach der damals gültigen TNMKlassifikation [71] durchgeführt. Die Studie wurde von der Ethikkommission der Medizinischen Fakultät der Universität zu Lübeck unter dem Aktenzeichen 33-96 vom 25. Mai 1996 genehmigt. Die Gewebeproben von 52 Patienten, 21 (40,4%) weibliche und 31 (59,6%) männliche wurden untersucht. 31 (59,6%) der Tumore waren im Kolon lokalisiert und 21 (40,4%) im Rektum. Im TNM-System ist folgende Verteilung festzustellen: von 52 Tumoren waren 4 Tumore (7,7%) klassifiziert als T1, 6 Tumore (11,5%) als T2, 33 Tumore (63,5%) als T3 und 9 Tumore (17,3%) als T4. Die 52 Tumore unterteilten sich in die folgenden Dukes-Stadien: 9 (17,3%) im Stadium Dukes A, 17 (32,7%) im Stadium Dukes B, 14 (26,9%) im Stadium Dukes C und 12 (23,1%) im Stadium Dukes D. Das Alter der Patienten zur Zeit der Operation lag zwischen 36 und 92 Jahren, der Altersmedian betrug 67 Jahre (s. Tabelle I). 21 Tabelle I: Charakterisierung des Patientenkollektivs Patienten n 52 in % 100,00 Geschlecht: Frauen 21 Männer 31 40,4 59,6 Lokalisation des Tumors Kolon 31 Rektum 21 59,6 40,4 Staging nach pTNM T1 4 T2 6 T3 33 T4 9 7,7 11,5 63,5 17,3 N0 N1 N2 N3 28 12 11 1 53,8 23,1 21,2 1,9 M0 M1 40 12 76,9 23,1 Klassifikation nach Dukes Dukes A 9 Dukes B 17 Dukes C 14 Dukes D 12 17,3 32,7 26,9 23,1 Altersverteilung Median Range Jahre 67 36-92 2.2 Histologie Von jeder Gewebeprobe wurde durch einen histologischen Schnitt geprüft, ob es sich tatsächlich um Tumor- oder Normalgewebe handelte. Weiterverwendet werden konnten nur Gewebeproben, die im Falle der Tumorproben mindestens 80% Tumorgewebe respektive im Falle der Normalgewebsproben keine Tumoranteile enthielten. Vorbereitung der Proben: Zunächst wurden die Gewebeproben nach Standardmethoden in Formalin fixiert und in Paraffin eingebettet, um anschließend 4µm Schnitte auf dem Mikrotom [7.3.5] anzufertigen. Aufgetragen auf Objektträger [7.2.42] trockneten die Schnitte im Anschluss. 22 Färbung der histologischen Schnitte: Die bei Raumtemperatur durchgeführte Hämatoxylin-Eosin-Färbung der Gewebeschnitte folgte dem allgemein bekannten Protokoll. 2.3 Gewebevorbereitung Von den in flüssigem Stickstoff gelagerten Gewebeproben der Patienten konnten unter der sterilen Werkbank [7.3.17] mittels in flüssigem Stickstoff gekühltem Skalpell und Pinzette jeweils zwei 50mg schwere Proben des Normal- bzw. des Tumorgewebes separiert werden. Um eine Materialverschleppung der Proben untereinander zu verhindern, wurde für jede Gewebeprobe eine neue Arbeitsfläche aus Aluminiumfolie verwendet, sowie eine Reinigung der benutzten Instrumente zwischen der Bearbeitung zweier Proben durch Ausglühen und Behandlung mit 100% Ethanol [7.2.18] durchgeführt. Nach der Portionierung lagerten die Proben bis zur weiteren Verwendung in 2-ml-Eppendorf-Cups in flüssigem Stickstoff. Die asservierten Proben wurden nachfolgend einer Proteinextraktion zur VEGF-Konzentrationsbestimmung und einer DNA-Gewinnung zur p53-Mutationsanalyse zugeführt. 2.4 Ermittlung der VEGF-Konzentration in den Gewebeproben 2.4.1 Proteinextraktion Für die Bestimmung der VEGF-Konzentration in den Gewebeproben mussten von diesen zunächst Proteinextrakte hergestellt werden. Die folgenden Arbeitsgänge fanden unter ständiger Kühlung mit Eis statt. Die Gewebeproben (50mg) wurden jeweils in einem 2-ml-Eppendorf-Reaktionsgefäß mit 400µl Proteinextraktionspuffer (PE-Puffer) [7.2.47] nach Takano et al. [163] überschichtet und mit dem Ultraturrax® [7.3.3] für 1min bei maximaler Geschwindigkeit zerkleinert und homogenisiert, bis makroskopisch keine Gewebestücke mehr zu erkennen waren. Anschließend wurde das Homogenisat durch Zugabe von 600µl PE-Puffer auf ein Endvolumen von 1ml gebracht. Hieran schloss sich eine Zentrifugation der Proben bei 22.900g und 4°C über 20min in der Zentrifuge [7.3.19] an. Je 250µl des Überstandes, welcher den Proteinextrakt darstellte, wurden in 1,5-mlEppendorf-Cups aliquotiert und bei -80°C gelagert. 23 Zwischen den Arbeitsgängen mit unterschiedlichen Gewebeproben erfolgte eine Reinigung des Ultraturrax® nacheinander mit 10% SDS [7.2.52], 1M NaOH [7.2.39], 70% Ethanol [7.2.20] und aqua bidest., um ein Verschleppen von Probenmaterial zu vermeiden. 2.4.2 Ermittlung der Gesamtproteinkonzentration Als Bezugsgröße für die VEGF-Konzentration diente die nach Bradford [17] ermittelte GesamtProtein-Konzentration in den Extrakten. Die im ELISA gemessene VEGF-Konzentration konnte so auf die Gesamtproteinkonzentration bezogen werden, dies ermöglichte den Vergleich der einzelnen Proben untereinander. Bei der Proteinkonzentrationsbestimmung nach Bradford wird die durch unspezifische Bindung an v.a. basische Aminosäuren hervorgerufene Verschiebung des Absorptionsmaximums des Farbstoffes Coomassie Brilliant Blue G250 von 465 zu 595nm ausgenutzt. Erstellen und Messen der Standardreihe: Je 5, 10, 15, 20 und 40µl der BSA-Lösung [7.2.36] wurden in jeweils zwei 1,5-ml-EppendorfReaktionsgefäße pipettiert und mit je 80, 75, 70, 65 und 45µl 0,15M NaCl [7.2.37] sowie mit je 15µl PE-Puffer [7.2.47] auf ein Endvolumen von 100µl aufgefüllt. Dies entsprach einer Menge von 2,5, 5,0, 7,5, 10,0 und 20,0µg BSA/Ansatz. Zwei weitere Ansätze mit je 85µl 0,15M NaCl und 15µl PE-Puffer dienten als Leerwert. Alle Ansätze wurden mit 1ml Coomassie Brilliant Blue Lösung [7.2.6], 1:5 verdünnt mit aqua bidest. (ultrafiltriert), versetzt und gründlich gemischt. Nach einer Inkubationszeit von 10min bei Raumtemperatur konnten die Ansätze der Standardreihe in Plastik-Halbmikroküvetten [7.2.27] pipettiert und die Extinktion im Spektrophotometer [7.3.11] bei 595nm gemessen werden. Die Auftragung der erhaltenen Werte gegen die BSA-Menge pro Ansatz (in µg) ergab die Standardkurve. Messung der Proteinextrakte: Die Proteinextrakte lagerten auf Eis, nachdem sie für 30s anzentrifugiert worden waren. Aus 50µl Proteinextrakt und 150µl PE-Puffer [7.2.47] wurde eine 1:4 Verdünnung des Proteinextraktes erstellt. Zur dreifachen Bestimmung wurden je 5µl (I), 10µl (II) und 15µl (III) der verdünnten Proteinextrakt-Lösung in Eppendorf-Cups pipettiert und mit jeweils 10 (I), 5 (II) und 0 (III) µl PE-Puffer sowie je 85µl 0,15M NaCl auf ein Endvolumen von jeweils 100µl vermischt. Nachdem die Proben mit 1ml Coomassie Brilliant Blue Lösung [7.2.6] (1:5 verdünnt mit aqua bidest., ultrafiltriert) gründlich vermischt wurden und 10min ruhten, konnte die Extinktion analog zur Standardreihe gemessen werden. 24 Das verwendete Spektrophotometer [7.3.11] berechnete automatisch die Proteinmenge anhand der zuvor erstellten Standardkurve. Gemäss des eingesetzten Volumens in µl wurde das Ergebnis durch 5 (I), 10 (II) bzw. 15µl (III) geteilt und entsprechend der Vorverdünnung mit 4 multipliziert, woraus sich die Proteinkonzentration des unverdünnten Proteinextraktes (in µg Protein/µl) ergab. Zur weiteren Berechnung wurde aus den Proben (I), (II) und (III) der Median verwendet. 2.4.3 VEGF-Konzentrationsbestimmung Die Bestimmung der VEGF-Konzentration erfolgte mittels des quantitativen Sandwich-ELISA (Enzyme Linked Immuno Sorbent Assay) Quantikine® Human VEGF Immunoassay [7.2.49], welcher nach den Herstellerangaben verwendet wurde. Bei dem Quantikine® Human VEGF Immunoassay werden monoklonale „capture“-Antikörper aus der Maus gegen VEGF165, welche an die Titerplatte gebunden sind, verwendet. Der polyklonale „Detektions“-Antikörper gegen VEGF165 stammt aus dem Kaninchen und ist mit Meerrettich-Peroxidase konjugiert. Im Färbungsschritt wird eine Substratlösung, hier Wasserstoff-Peroxid und Tetramethylbenzidin zugegeben, die durch die Meerrettich-Peroxidase in ein gefärbtes Produkt umgesetzt wird. Diese Reaktion wird zu einem definierten Zeitpunkt mittels Schwefelsäure gestoppt und die Intensität des Farbstoffes photometrisch ermittelt. Vorbereitung der Reagenzien: Der Waschpuffer [7.2.49] (20ml, 25x konzentriert) wurde mit aqua bidest. auf 500ml aufgefüllt und so auf einfache Konzentration verdünnt. Der lyophilisierte VEGF-Standard [7.2.49] musste in 1ml Kalibrier-Verdünnungslösung RD6U [7.2.49] gelöst und 15min leicht geschüttelt werden. Die so erreichte Konzentration entsprach 2000pg VEGF/ml. Zudem wurden die Farbreagenzien A und B [7.2.49] kurz vor der Färbung im Verhältnis 1:1 gemischt. Erstellung der Standardreihe: Die Standardreihe umfasste einen Bereich von 0 bis 2000pg VEGF/ml, wobei der unverdünnte VEGF-Standard (2000pg VEGF/ml) [7.2.49] als höchster Wert und die Kalibrierverdünnungslösung RD6U [7.2.49] als Leerwert dienten. Die dazwischen liegenden Ansätze mit den Konzentrationen 31,3, 62,5, 125,0, 250,0, 500,0 und 1000,0pg VEGF/ml wurden durch Verdünnen des Standards mit der Kalibrierverdünnungslösung RD6U gemäss dem in der Tabelle II angeführtem Schema erhalten. Jeder Standard wurde zweifach eingesetzt. 25 Vorbereitung der Proben: Nach dem Anzentrifugieren lagerten die Proteinextrakte auf Eis. Eine 1:10 Verdünnung der Proteinextrakte aus den Tumorgewebeproben aus 20µl Proteinextrakt und 180µl Proteinextraktionspuffer [7.2.47] sowie die unverdünnten Proteinextrakte der Normalgewebeproben wurden in die Messung eingesetzt. Von jedem Proteinextrakt erfolgte eine Doppelbestimmung. Tabelle II: Pipettierschema zur Erstellung der Standardreihe des VEGF-ELISAs Ansatz VEGF-Standard (2000 pg/ml), in µl RD6U Verdünnungslösung in µl 1 2 3 4 5 6 7 8 0 500 500 0 0 0 0 0 500 0 500 500 500 500 500 50 0 0 0 500 500 500 500 500 (3) (4) (5) (6) (7) 0 2000,0 1000,0 500,0 250,0 125,0 62,5 31,3 Volumen aus vorhergehendem Ansatz (Nr.), in µl VEGF-Konzentration, in pg/ml Die Durchführung des ELISA-Protokolls: Nachdem 50µl Assay-Verdünnungslösung RD1W in jede Vertiefung der Microwell-Platte [7.2.49] pipettiert worden waren, konnten 100µl Standard- bzw. Proben-Ansatz hinzugegeben werden. Es folgte das Abkleben der Wells und eine Inkubation bei Raumtemperatur für 2h. Nach dieser Zeit wurde die Flüssigkeit aus den Wells abgesaugt und diese dreimal mit jeweils 400µl Wasch-Puffer [7.2.49] gewaschen. Anschließend wurden 200µl VEGF-Konjugat [7.2.49] in jedes Probenwell pipettiert. Wiederum wurden die Wells abgeklebt und 2h bei Raumtemperatur inkubiert. An diesen Schritt schloss sich erneutes Absaugen der Lösungen und dreimaliges Waschen mit jeweils 400µl Wasch-Puffer an. Nun konnten 200µl Substratlösung, bestehend aus den Farbreagentien A und B [7.2.49], in jede Vertiefung pipettiert werden. Es schloss sich erneutes Abkleben der Mikrotiterplatte und Inkubation bei Raumtemperatur für 25min an. Dieser Färbevorgang endete durch die Zugabe von 50µl StopLösung [7.2.49] in jede Vertiefung. Innerhalb der nächsten 30min sollte die Extinktion bei 450nm mit dem Photometer [7.3.7] gemessen werden. Das verwendete Photometer [7.3.7] erstellte eine Standardreihe und bestimmte gleichzeitig die Konzentration in den Proben durch Vergleich der gemessenen Extinktion der Probe mit der Standardreihe (Easy Fit, Version 3.45, SLT Lab Instruments, Crailsheim). Aus den Extinktionen der doppelt bestimmten Proben wurde der Mittelwert gebildet. Die VEGF-Konzentrationen der Standardreihe sowie der Proben wurde in pg/ml angegeben. Die VEGF-Konzentration der 1:10 26 verdünnten Proben (nur T-Proben) wurde mit dem Faktor 10 multipliziert und ergab so die Konzentration von VEGF/ml der entsprechenden Proteinextrakte. Berechnung der VEGF-Konzentration einer Probe: Die VEGF-Konzentration einer Probe (in pg/ml) wurde auf die Gesamt-Proteinkonzentration (in g/ l) dieser Probe bezogen. Formel: [VEGF]Probe X (in pg/ml) / [Protein]Probe X (in µg/µl) = [VEGF]Probe X (in pg VEGF/mg Protein) 2.5 Mutationsanalyse im p53-Gen der Gewebeproben 2.5.1 DNA-Extraktion Aus allen 52 Normal- und den korrespondierenden Tumorgewebeproben wurde die DNA mit Hilfe des QIAGEN Blood & Cell Culture DNA Kits [7.2.25] isoliert. Grundlage der Methode des Kits ist die Möglichkeit der Isolierung von chromosomaler DNA mit einer Größe von 20-150kb direkt aus dem Gewebe, basierend auf einem Puffersystem zur vorsichtigen Lyse von Zellen und/oder Zellkernen. Der Vorgang der DNA-Extraktion mittels Anionen-Austausch-Chromatographie bedient sich dem Prinzip, dass negativ geladene Phosphatgruppen der DNA an positiv geladenen Gruppen der QUIAGEN-Anionen-AustauscherHarzsäule binden. Das Verfahren arbeitet mit Hilfe des Gravitationsflusses. Zur Aufreinigung des Produktes werden RNA, Proteine, Zelldedritus und niedrigmolekulare Verunreinigungen durch Puffer mit hoher Salzkonzentration ausgewaschen. Die gebundene genomische DNA wird mit einem Elutionspuffer gelöst. Die Nachbearbeitung des Eluats umfasst das Fällen, Konzentrieren und Waschen sowie das Resuspendieren der DNA, um eine optimale Konzentration für die Weiterverwendung zu erhalten. Vorbereitung der Gewebeproben: In sterile 15ml Cellstar PP-Röhrchen mit Schraubdeckel [7.2.9] wurden 19µl RNase A [7.2.25] in 9,5ml Puffer G2 [7.2.25] verdünnt. Hiernach konnte die in flüssigem Stickstoff gelagerte Gewebeprobe (50mg) hinzugeben und mit dem Homogenisator Ultraturrax® [7.3.3] bei maximaler Stufe homogenisiert werden, bis makroskopisch keine Gewebereste mehr zu erkennen waren. Diese Arbeitsschritte fanden unter ständiger Kühlung mit Eis statt. Nach Zugabe von 0,5ml Protease-Lösung (20mg/ml) [7.2.25] wurde der Ansatz gründlich gemischt und 2h im Wasserbad [7.3.16] bei 50°C unter ständig em Rütteln inkubiert. 27 Anschließend folgte eine Zentrifugation des Homogenisates bei 4°C und 5000g über 20min in der Zentrifuge [7.3.19]. Die daraufhin entstandene obere trübe Schicht von wenigen Millilitern, bzw. der Bodensatz als makroskopische Verunreinigung wurden verworfen und das makroskopisch klare Homogenisat, welches die mittlere Schicht bildete, verwendet. Protokoll der DNA-Isolation: Um eine Kontamination und den Einfluss von ubiquitären DNAsen auf die isolierte DNA zu vermeiden, fanden die folgenden Arbeitsschritte unter der sterilen Werkbank [7.3.17] statt. Nach der Equilibrierung der Säule (QIAGEN Genomic 100/G Tip [7.2.25]) mit 4ml Puffer QBT [7.2.25] wurde die wie oben beschrieben homogenisierte und proteolysierte Probe auf die Säule pipettiert. Nachdem die Probenflüssigkeit vollständig durch die Säule gelaufen war, wurden Verunreinigungen durch dreimaliges Auftragen von je 7,5ml Puffer QC [7.2.25] ausgewaschen. Danach konnte das Auffanggefäß ausgewechselt und die DNA mit 5ml Puffer QF [7.2.25] (auf 50°C erwärmt) eluiert werden. Zur Präzipitation der DNA wurde dem Eluat 3,5ml -20°C kaltes Isopropanol [7.2.29] hinzugefügt. Nach der Inkubation des Gemisches für 10min bei Raumtemperatur konnte der Ansatz bei 4°C mit 5000g über 25min in der Zentrifuge [7.3.19] zentrifugiert und der Überstand dekantiert werden. Das bei der Zentrifugation entstandene Pellet entsprach der gefällten DNA, welche noch zweimal mit Ethanol gewaschen werden musste. Zur Aufreinigung folgte jeweils die Überschichtung des Pellets mit 2ml 70% Ethanol [7.2.20] (auf -20°C gekühlt), im Anschluss vorsichtiges Mischen, Zentrifugation bei 4°C mit 5000g über 10min in der Zentrifuge [7.3.19] und Dekantieren des Überstandes sowie Trocknung des Pellets bei Raumtemperatur über 10min. Zur Lösung der DNA im Puffer wurde das Pellet in 250µl TE-Puffer [7.2.61] resuspendiert und über 2h bei 55°C im Wasserbad [7.3.16] inkubiert. DNA-Konzentrationsbestimmung: Um die DNA in definierter Konzentration in die PCR einsetzen zu können, musste die DNAKonzentration in der DNA-Lösung gemessen und die Probe anschließend mit TE-Puffer verdünnt werden. Mit 10µl DNA-Lösung und 90µl TE-Puffer [7.2.61] wurde zunächst eine 1:10 Verdünnung hergestellt. Messung der DNA-Proben: Zur Bestimmung der DNA-Konzentration in den Proben wurde ihre Extinktion bei 260 und 280nm sowie zur Hintergrundkorrektur bei 320nm mit dem Spektrophotometer [7.3.11] 28 gemessen. Das verwendete Photometer berechnete automatisch die DNA-Konzentration unter Verwendung folgender Formel nach Warburg und Christian [175]: [DNA] (in µg/ml) = (-36.0 x A280) + (62.9 x A260) Des Weiteren wurde zur Bestimmung des Reinheitsgrades der DNA-Lösung der Quotient aus Extinktion bei 260nm und 280nm durch das Spektrophotometer berechnet. Für eine hoch reine DNA-Probe sollte dieser etwa 1,8 betragen. Um die gewünschte DNA-Konzentration von 100ng/µl zur Weiterverwendung in der PCR zu erhalten, wurde die unverdünnte DNA-Lösung entsprechend dem jeweils berechneten Verdünnungsfaktor mit TE-Puffer [7.2.61] verdünnt. Verdünnungsfaktor = ([DNA] ng/µl gemessen x 10) / [DNA] ng/µl verdünnt Kontrollgel der DNA: Das Vorhandensein von DNA in der Lösung wurde zusätzlich in einem Agarosegel (8,5cm x 7,8cm x 1,0cm; Länge x Breite x Dicke) überprüft. Diesem war Ethidiumbromid [7.2.21] beigefügt, welches ein in die DNA-interkalierender Fluoreszenzfarbstoff ist, der bei einer Wellenlänge von 260–360nm zur Fluoreszenz angeregt wird. Als positive Kontrolle und Standard wurde der Molekulargewichtsmarker VI [7.2.13] verwendet. Zur Herstellung des 1%igen (w/v) Agarosegels [7.2.2] wurden 0,5g Agarose in 50ml 1x TAEPuffer [7.2.56] suspendiert und durch dreimaliges Aufkochen in dem Mikrowellengerät [7.3.6] gelöst. Nach Abkühlen unter ständigem Rühren auf etwa 55°C konnten das noch flüssige Gel in ein 50ml Cellstar PP-Röhrchen [7.2.9] umgefüllt und 5µl Ethidiumbromidlösung [7.2.21] hinzupipettiert werden. Es folgte der blasenfreie Giessvorgang in den Gelschlitten der Minigelkammer [7.3.1]. Das Gel kühlte innerhalb von ca. 20min auf Raumtemperatur ab, härtete aus und wurde mit 1x TAE-Puffer [7.2.56] überschichtet. Zuletzt konnte das Gel mit den Proben in die durch den Spacer freigehaltenen Probentaschen beladen werden. Je 10µl jeder DNA-Probe (entsprechend 1µg DNA) wurden in einer Mikrotiterplatte mit je 1µl Laufmarker [7.2.33] vermischt und von diesem Ansatz 10µl in die Beladungstaschen des Gels eingesetzt. Laufbedingungen: - Laufzeit: 1h - Spannung: 80V (konstant) 29 Auswertung des Gels: Das Gel wurde auf dem UV-Transilluminator [7.3.15] unter UV-Licht mit einer Wellenlänge von 312nm untersucht. Eine fluoreszierende Bande zeigte das Vorhandensein von DNA in der Lösung an. Keine Bande führte zur erneuten Gelkontrolle. Bei einem zweiten negativen Ergebnis wurde das DNA-Isolat verworfen und eine erneute DNA-Extraktion der Probe durchgeführt. 2.5.2 Polymerase Kettenreaktion (Polymerase Chain Reaction, PCR) Das Prinzip der PCR basiert auf der Aktivität thermostabiler DNA-Polymerasen, welche in vitro unter Verwendung sequenzspezifischer Primer (16-24 Basen grosse Oligonukleotide), aus einem Gemisch unterschiedlicher DNA-Moleküle ein bestimmtes Fragment beliebig amplifizieren können. Die verwendete SAWADY Pwo-DNA-Polymerase, ein 90-kD-Protein, ist eine hochprozessive 5’-3’-DNA-Polymerase mit einer zusätzlichen 3’-5’-Exonuklease-Aktivität, dem sog. „Korrekturlesen“. Durch diese „proofreading“-Aktivität weist die Pwo-Polymerase die niedrigste Fehlerrate der thermostabilen Polymerasen auf. Ein einzelner DNA-Synthesezyklus ist in folgende drei Phasen bestimmter Reaktionstemperatur und Dauer entsprechend dem zu amplifizierendem DNA-Abschnitt unterteilt (s.u.). Mehrere solcher DNA-Synthesezyklen können direkt aneinandergereiht werden, wobei die DNA-Fragmente in jedem Zyklus jeweils verdoppelt werden. Die Amplifizierung der DNA durch die PCR erfolgt daher exponentiell. Phase 1: Thermische Denaturierung der Doppelstrang-DNA (94°C, 30-120s) Phase 2: Bindung der Primer an die Einzelstrang-DNA (37°-70°C, 30-120s) Phase 3: Elongation der Primer in 5´-3´Richtung (72°C bei Verwendung von Pwo-Polymerase, 90-180s) Untersucht wurden die Exons 5-8 des p53-Gens. Diese entsprechen den ‚hot spot‘-Regionen für Mutationen in kolorektalen Karzinomen (s. Kapitel 1.2.3) [74]. Das Exon 5 wurde in zwei Abschnitte unterteilt, welche sich im Bereich der Codons 156-160 überlappten: Exon 5-1 und Exon 5-2. Dies war nötig, da die DNA dieses Exons ein komplexeres Schmelzverhalten mit zwei Temperaturplateaus aufweist, und so für die Folgemethoden der Gelelektrophorese mit zeitlichem Temperaturgradienten (Temporal Temperature Gradient Electrophoresis, TTGE) bzw. der Gelelektrophorese mit einem Denaturans Gradienten (Denaturing Gradient Gel Electrophoresis, DGGE) unterteilt werden musste. Die in dieser Arbeit zur Amplifikation verwendeten Primer sind von Anderson und Börrisson beschrieben worden (s. Tabelle III) [6]. Zur weiteren Verwendung in der Mutationsanalyse mittels TTGE bzw. DGGE, wurden die Amplifikate der Exons 5-1, 6, 7 und 8 über den Antisense-Primer bzw. im Falle des Abschnitts 30 5-2 über den Sense-Primer mit GC-Klammern („gc-clamp“, Guanin-Cytosinreiche Sequenz) versehen. Für jeden der zu untersuchenden fünf DNA-Abschnitte wurden unterschiedliche PCR-Bedingungen optimiert und jede Probe zur Doppelbestimmung zweimal amplifiziert. Die Primer-Sequenzen [7.2.45] sind in Tabelle III aufgeführt: a Tabelle III: Angaben zu den verschiedenen Primern und den PCR-Produkten Primer PrimerSequenzlänge Primer-Sequenz Exon 5-1 sense 18bp 5’ - CTC TGT CTC CTT CCT CTT - 3’ 5’ - CGC CCC GCC GCG CCC CGC GCC Exon 5-1 antisense 59bp Länge des amplifizierten Fragmentes amplifizierte Codons 191bp Codon: 126-160 176bp Codon: 156-184 96bp Codon: 189-215 192bp Codon: 225-259 191bp Codon: 265-300 CGT CCC GCC GCC CCC GCC CGT GTG ACT GCT TGT AGA TG - 3’ 5’ - CGC CCG CCG CGC CCC GCG CCC Exon 5-2 sense Exon 5-2 antisense Exon 6 sense 60bp GTC CCG CCG CCC CCG CCC GTT CCA CAC CCC CGC CCG GCA - 3’ 20bp 5’ - CTC CAG CCC CAG CTG CTC AC - 3’ 19bp 5’ - CAC TGA TTG CTC TTA GGT C – 3’ 5’ - CGC CCG CCG CGC CCC GCG CCC Exon 6 antisense 57bp Exon 7 sense 20bp GTC CCG CCG CCC CCG CCC AGT TGC AAA CCA GAC CTC - 3’ 5’ - TGG GCC TGT GTT ATC TCC TA - 3’ 5’ - CGC CCG CCG CGC CCC GCG CCC Exon 7 antisense 60bp Exon 8 sense 20bp GTC CCG CCG CCC CCC CCG CCC GGC AAG TGG CTC CTG ACC TGG – 3’ 5’ - ATC CTG AGT AGT GGT AAT CT – 3’ 5’ – CGC CCG CCG CGC CCC GCG CCC Exon 8 antisense a 60bp GTC CCG CCG CCC CCG CCC GTA CCT CGC TTA GTG CTC CCT – 3’ basierend auf mRNA-Sequenz No NM_000546 (Genbank) PCR mit Pwo-Peqlab PCR System: Vorbereitung: Für die PCR wurde das Pwo-Polymerase PCR System der Firma Peqlab [7.2.48] verwendet und alle Pipettierschritte mit Filtertips [7.2.7] durchgeführt. Die 1mM Stammlösungen der Primer [7.2.45] wurden 1:40 verdünnt: 1 Teil Sense-Primer : 1 Teil Antisense-Primer : 38 Teile aqua bidest., steril. Die Konzentration nach Verdünnung betrug somit 25µM für jeden Primer. Aufgrund der Exonuklease-Aktivität der Pwo-Polymerase, waren die einzelnen Komponenten auf zwei Mastermixe (I und II, s. Tabelle IVa/b und Va/b) auf Eis angesetzt, so dass Primer und Polymerase zunächst getrennt vorlagen. 31 PCR-Mastermix I: Tabelle IVa: Mastermix I für die Exons 5-1, 6, 7, 8 Reagenz dNTP, 20mM [7.2.14] Konzentration Volumen pro im PCR Ansatz PCR Ansatz 200µM 0,5µl 600nM 1,2µl ad 24µl 22,3µl senseantisense Primergemisch je 25µM aqua bidest., steril Tabelle IVb: Mastermix I für das Exon 5-2 Reagenz Glycerin 10%, steril [7.2.26] dNTP, 20mM [7.2.14] Konzentration Volumen pro im PCR Ansatz PCR Ansatz 0,1% 0,5µl 200µM 0,5µl 600nM 1,2µl ad 24µl 19,6µl senseantisense Primergemisch je 25µM aqua bidest., steril PCR-Mastermix II: Tabelle Va: Mastermix II für die Exons 5-1, 6, 7, 8 Reagenz Pwo-DNA-Polymerase (1 U/µl) [7.2.48] 10x Peqlab Reaktionspuffer mit MgSO4 [7.2.48] aqua bidest., steril Konzentration Volumen pro im PCR Ansatz PCR Ansatz 2,5U/Ansatz 2,5µl 1x Puffer mit 2mM MgSO4 ad 25µl 5µl 17,5µl 32 Tabelle Vb: Mastermix II für das Exon 5-2 Konzentration Volumen pro im PCR Ansatz PCR Ansatz 2,5U/Ansatz 2,5µl 2fach 5µl MgSO4(25mM) 1,5mM 1,5µl Aqua bidest., steril ad 25µl 16,0µl Reagenz Pwo-DNA-Polymerase (1 U/µl) [7.2.48] 10x Peqlab Reaktionspuffer ohne MgSO4 [7.2.48] Die PCR-Cups wurden auf Eis nach folgendem Schema beladen: 1. 24µl Mastermix I 2. 1µl DNA Probe (=100ng DNA) 3. 25µl Mastermix II Die PCR-Ansätze für die Exons 5-1, 6, 7, und 8 unterschieden sich nur durch die verwendeten Primer. Im Falle des Exons 5-2 wurde dem Mastermix I steriles Glycerin [7.2.26] zugesetzt und für den Mastermix II die MgSO4-Konzentration optimiert. Beide Optimierungen dienten zur Erhöhung der Spezifität der Primerbindung. Bei jedem PCR-Lauf wurde eine Negativkontrolle verwendet, indem einem Ansatz statt der DNA-Probe 1µl steriles aqua bidest. hinzupipettiert wurde. Es folgte der Einsatz in den Thermocycler [7.3.12] und Start des jeweiligen PCRProgramms. Reaktionsbedingen für die PCR: 1x 95°C, 2min (Denaturierung, „hot start“) Dieser Schritt war für alle untersuchten Exons gleich. Bei den folgenden Schritten wurden die Bedingungen des Primerannealing und der Zykluszahl für die einzelnen Exons durch Testreihen optimiert (s. Tabelle VI). Tabelle VI: Reaktionsbedingungen der PCR für die untersuchten Exons des p53-Gens Exon Zykluszahl Denaturierung Annealing Elongation 5-1 30x 95°C, 75s 60°C, 75s 72°C, 60s 5-2 35x 95°C, 75s 58°C, 75s 72°C, 60s 6 30x 95°C, 75s 56°C, 75s 72°C, 60s 7 30x 95°C, 75s 65°C, 75s 72°C, 60s 8 35x 95°C, 75s 55°C, 75s 72°C, 60s 33 An die Amplifikation schloss sich jeweils eine Elongation von 1x 72°C für 7min an. Nach Beendigung der PCR wurden die PCR-Produkte auf 4°C abgekühlt und im Agarosegel kontrolliert (s. u.), anschließend in den PCR Cups bei -20°C bis zur weiteren Verwendung gelagert. Bei allen amplifizierten Exons waren das Bearbeitungsprotokoll, die PCR-Negativkontrolle sowie die Kontrolle der PCR-Produkte standardisiert. Kontrolle der PCR Produkte: Zur Kontrolle der PCR-Produkte wurde ein 1,5% (w/v) Agarose Gel verwendet. Das Prozedere der Gelherstellung sowie der Laufbedingungen war identisch mit dem im Abschnitt DNAProduktkontrolle (s. Kapitel 2.5) beschriebenen. Vorbereitung der Proben: 10µl des PCR-Produktes wurden auf einer Mikrotiterplatte mit je 1µl Laufmarker [7.2.33] vermischt und 10µl des Gemisches in die Taschen des Gels pipettiert. Als Längenstandard dienten 5µl des DNA-Molekulargewichtsmarker VI [7.2.13], welcher ebenfalls mit 1µl Laufmarker vermischt in das Gel eingesetzt wurde. Auswertung des Gels: Das Gel wurde auf dem UV-Transilluminator [7.3.15] unter UV-Licht mit einer Wellenlänge von 312nm betrachtet. Durch fluoreszierendes Ethidiumbromid visualisierten sich die PCRProdukte. Es wurden jeweils die Negativprobe (als Kontaminationsausschluss) auf ein negatives Signal sowie die PCR-Produkte auf ein positives Signal kontrolliert. Die Größe der PCR-Produkte wurden zudem mit den Fragmentlängen des DNA-Molekulargewichtsmarker VI [7.2.13] verglichen. Bei fehlendem Signal in einem der PCR-Produkte oder falscher Position im Vergleich zum Längenstandard folgte eine zweite Gelkontrolle. Bei gleichem Ergebnis wurde die PCR für diese Probe wiederholt. Bei Auftreten einer DNA-Bande in der Negativprobe wurden alle Ansätze einer PCR-Reihe verworfen und wiederholt. 34 2.5.3 Gelelektrophorese mit zeitlichem Temperaturgradienten (Temporal Temperature Gradient Electrophoresis, TTGE) sowie mit einem Denaturans Gradienten Gel (Denaturing Gradient Gel Electrophoresis, DGGE) Die TTGE basiert auf dem Prinzip, dass doppelsträngige DNA in denaturierendem Milieu in Abhängigkeit von der Basensequenz bei einer bestimmten Temperatur schmilzt und somit aus doppelsträngiger DNA (double stranded DNA, dsDNA) zunächst ein aufgespaltenes Molekül und dann, bei weiterem Ansteigen der Temperatur einzelsträngige DNA (single stranded DNA, ssDNA) entsteht. Sowohl das aufgespaltene Molekül als auch die ssDNA weisen ein anderes Laufverhalten auf als die dsDNA. Da die jeweiligen Schmelztemperaturen von der Basensequenz abhängig sind, bewirkt eine Änderung der Basenabfolge, wie dies z.B. bei Punktmutationen der Fall ist, eine Änderung in der Schmelztemperatur [45]. Bei der TTGE wird die DNA in einem denaturierenden Polyacrylamid-Gel aufgetrennt, welchem Harnstoff als Denaturans zugesetzt ist, um die Schmelztemperatur der zu untersuchenden DNA zu senken. Die Elektrophorese läuft über eine definierte Zeit, wobei die Temperatur kontinuierlich erhöht wird. In Abhängigkeit von der Basenabfolge beginnt die DNA aufzuschmelzen und ändert somit ihr Laufverhalten über die Zeit. Da die mutierte DNA (Punktmutationen, Insertionen, Deletionen, Frameshiftmutationen) ein anderes Laufverhalten aufweist als die Wildtyp-DNA, ist das sichtbare Ergebnis ein differierendes Bandenmuster, anhand dessen sich eine Mutation identifizieren lässt. Ähnlich der molekularen Grundlage der TTGE arbeitet die DGGE. Bei dieser Methode wird eine Änderung des Konformationszustandes der DNA in Abhängigkeit der Basenabfolge durch einen Gradienten der denaturierenden Agentien Formamid und Harnstoff erreicht. Die Temperatur ist während des gesamten Gellaufes konstant. Bei Proben mit dem untersuchten Allel in homozygotem Zustand wird bei der TTGE und der DGGE eine Homoduplex-Bande erhalten. Liegen Wildtyp und mutierte DNA nebeneinander vor, wie es bei heterozygotem Zustand des untersuchten Allels oder bei einem Gemisch von mutierter und nicht-mutierter DNA der Fall ist, zeigen sich im Idealfall vier Banden in der TTGE bzw. DGGE. Zwei Banden stellen jeweils die Homoduplizes des Wildtyps bzw. der mutierten DNA dar, die beiden weiteren Banden die Heteroduplizes. Diese sind während der vorangegangenen PCR durch wiederholtes Aufschmelzen und Renaturieren von einem Wildtyp und einem mutiertem DNA-Strang entstanden und weisen untereinander ein unterschiedliches Laufverhalten in der TTGE bzw. DGGE auf (s. Abbildung 3). Um die Unterschiede im Laufverhalten zu erhöhen und damit die Auftrennung der Banden zu verbessern, wird ein möglichst frühes Aufschmelzen des mutierten Abschnittes sowie ein möglichst lang erhaltener partiell aufgeschmolzenem Zustand der dsDNA angestrebt. Durch die hohe Bindungsenergie der GC-Klammer des PCR-Produktes ist zu ihrem Aufschmelzen eine hohe Temperatur in der 35 TTGE oder entsprechend eine hohe Konzentration an Denaturantien in der DGGE erforderlich. Daher schmilzt die GC-Klammer als letztes auf und sichert so möglichst lange einen partiell aufgetrennten Zustand der DNA [13,14,180]. Abbildung 3: Laufverhalten der DNA in der Denaturans Gradient Gelelektrophorese, DGGE, [modifiziert n. 13] Zur Festlegung der Laufbedingungen muss die zu untersuchende Basensequenz bekannt sein. Um einen approximativen Temperaturbereich für die Auftrennung der DNA in der TTGE/DGGE zu bestimmen, wurde für die zu untersuchenden Exons eine Berechnung der Schmelztemperatur sowie des Schmelzverhalten online mit Hilfe des Poland Servers des Instituts für Physikalische Biologie der Heinrich-Heine Universität Düsseldorf [123,157] durchgeführt. Der berechnete Temperaturbereich diente als Grundlage, den Schmelztemperaturbereich eines Exons einzugrenzen. Der Bereich wurde mit einer für das entsprechende Exon heterozygoten Kontrolle (Gemisch aus Wildtyp- und mutierter DNA) in einer Gelelektrophorese mit räumlichen Temperaturgradient, der sogenannten Temperature 36 Gradient Gel Electrophoresis (TGGE) verifiziert [137]. Bei dieser Methode wurde ein Gel mit konstantem denaturierendem Milieu verwendet. Über dem Gel liegt ein Temperaturgradient, der über die Zeit konstant ist. Die Laufrichtung der Probe verläuft senkrecht zum Temperaturgradienten. Man erhält eine sigmoide Kurve, aus der man auf das Laufverhalten der Probe bei unterschiedlichen Temperaturen rückschliessen kann. Die Kurve teilt sich im Optimalfall in drei verschiedene Abschnitte und bei einem Gemisch aus mutierter und nichtmutierter DNA in vier verschiedene Einzelkurven. Diese repräsentieren die beiden Homo- sowie die beiden Heteroduplizes mit ihrem unterschiedlichen Laufverhalten. Für den Temperaturgradienten in der TTGE interessant ist der Temperaturbereich des Gels, bei dem der Abstand zwischen den vier Kurvenverläufen in Laufrichtung der Probe am grössten ist. Hier zeigt das Laufverhalten zwischen den beiden Allelen den grössten Unterschied. Dieser Anteil der Kurve deckt sich mit dem Bereich der grössten Steigung der sigmoiden Kurve, welcher den Schmelzbereich der DNA markiert (s. Abbildung 4). Abbildung 4: Schema der Temperaturgradient-Gelelektrophorese, TGGE, Tmin bzw. Tmax entspricht der Start- bzw. der Endtemperatur des Schmelzvorganges, zwischen den Symbolen +/- liegt eine definierte Spannung an [modifiziert n. 184]. Im Vorversuch betrug dieser Bereich für die untersuchten Exons: Tabelle VII: Temperaturbereiche der einzelnen Exons in der TGGE Exon Tmin Tmax 5-1 51°C 61°C 5-2 58°C 64°C 6 48°C 56°C 7 56°C 66°C 8 56°C 68°C 37 Mit diesen Ergebnissen als Ausgangspunkt konnten mehrere Versuche durchgeführt werden, in denen die Temperaturbereiche, die Temperatursteigerungsrate (ramp rate) bzw. die denaturierenden Gradienten und die Laufzeit variiert wurden, bis ein Ergebnis mit optimaler Trennung der Banden in der TTGE erzielt werden konnte. Positivkontrollen [7.2.32]: Als Positivkontrolle wurden DNA-Isolate aus den in Tabelle VIII beschriebenen Zelllinien mit bekannten Mutationen im p53-Gen verwendet. DNA aus dem Blut eines gesunden Spenders diente als Negativkontrolle. Die DNA sowohl der Positiv- als auch der Negativkontrollen wurde mittels PCR analog den untersuchten Tumor- und Normalproben verarbeitet. Tabelle VIII: Positivkontrollen für die Mutationsanalyse der untersuchten Exons des p53-Gens Exon Zellreihe Charakterisierung Mutation in Codon Basenaustausch Literatur 5 Capan-1 Adenokarzinom Pankreas 159 GCC CTC 70 6 BXPC-3 Adenokarzinom Pankreas 220 TAT TGT 51 7 DLD-1 Adenokarzinom Kolon 241 TCC TTC 135 8 SW480 Adenokarzinom Kolon 273/309 CGT CAT/CCC CCT 113,135,179 TTGE/DGGE-System: Das DCode Universal Mutation Detection System [7.3.2] ist eine Gelkammer, in der das Gel senkrecht in einem mit Puffer gefüllten Becken hängt, während eine Temperatureinheit über einen Heizstab und einen Rührer die Temperatur des Puffers und damit des Gels kontinuierlich steuert. Vor Beginn des Gellaufes wird die Start- und Zieltemperatur sowie die Temperatursteigerungsrate über die Zeit (in °C/h) eingegeben. Die Laufzeit wird über den Spannungsgeber [7.3.10] kontrolliert. Vorbereitung des TTGE-Gels: Entsprechend Tabelle IX ist aus Acrylamid/Bisacrylamid [7.2.1], TAE-Puffer [7.2.58], Harnstoff [7.2.28] und aqua bidest. eine Stammlösung angesetzt worden, von der 30ml für ein Gel von 16,0cm x 16,0cm x 0,1cm (Länge x Breite x Dicke) benötigt wurden. Tabelle IX: Stammlösung für das TTGE-Gel Reagenz Volumen 40% Acrylamid/Bis TAE Puffer, 50x Harnstoff aqua bidest. 10ml 1ml 16,8g ad 40ml Konzentration in der Stammlösung 10% 1,25x 7M 38 Zu 30ml der Stammlösung wurden in einem 50ml Becherglas 270µl 10% APS [7.2.3] und 27µl TEMED [7.2.60] pipettiert. Nach ausreichendem Vermischen wurde das noch flüssige Gel in eine 50ml Spritze aufgezogen und zwischen die Glasplatten, welche im herstellereigenen Gelgiessständer eingespannt waren, gegeben. Nach 1h Polymerisationsdauer konnte das Gel in den Tank des DCode Universal Mutation Detection Systems gehängt werden, der zuvor mit 7 Litern TAE Puffer, 1,25x konzentriert [7.2.57], gefüllt und auf die Starttemperatur vorgeheizt war. Vorbereitung des DGGE-Gels: Für die Analyse des Exons 5-2 und 6 wurde wie folgt ein 10%iges DGGE-Gel hergestellt. Als erster Schritt mussten 25ml einer 10%igen Acrylamidlösung mit 75% Denaturans angesetzt werden, indem 18,75ml einer Lösung mit 100% Denaturans (s. Tabelle Xa) mit 6,25ml einer Lösung ohne Denaturans (0%ig, s. Tabelle Xb) gründlich gemischt wurden. Zu Beginn wurden 15ml der 75%igen Denaturanslösung, 135µl 10% APS [7.2.3] und 13,5ml TEMED [7.2.60] gemischt. Dieses Gemisch konnte dann zwischen die vorbereiteten Glasplatten gegeben werden. Im Anschluss daran konnte mit dem Gradientengiesssystem des DCode Systems der Gel Gradient des Gels gegossen werden. Hierzu wurden 9ml der 75% Denaturanslösung und 9ml einer 30% Denaturanslösung, die zuvor aus 2,7ml 100% Denaturanslösung und 6,3ml 0% Denaturanslösung hergestellt worden war, kontinuierlich vermischt und zwischen die Glasplatten auf das polymerisierte Gel mit 75%igen Denaturanslösung gegeben. Beiden Ansätzen war vorher 100µl 10% APS und 10µl TEMED zugegeben worden. Nach einer Polymerisationszeit von einer Stunde wurde das Gel analog dem Gel der TTGE in den Tank des DCode Universal Mutation Detection System [7.3.2] gehängt. Tabelle Xa: 10% Acrylamid-Lösung mit 100% Denaturans Acrylamid-Lösung ml/Ansatz Endkonzentrationen 40% Acrylamid/Bis [7.2.1] 25ml 10% 50x TAE-Puffer [7.2.58] 2ml 1x Formamid (deionisiert) [7.2.24] 40ml 40% Harnstoff [7.2.28] 42g 7M aqua bidest. ad Endvolumen 100ml 39 Tabelle Xb: 10%ige Acrylamid-Lösung mit 0% Denaturans Acrylamid-Lösung ml/Ansatz Endkonzentrationen 40% Acrylamid/Bis [7.2.1] 25ml 10% 50x TAE-Puffer [7.2.58] 2ml 1x aqua bidest. ad Endvolumen 100ml Vorbereitung der Proben: Je 3µl des PCR Produktes [2.5.2] wurden mit 5µl des TTGE-Laufmarkers [7.2.34] und 2µl des TAE-Puffers, 1,25x konzentriert, [7.2.57] in einer Mikrotiterplatte gemischt und in die Taschen des auf die Starttemperatur vorgeheizten Geles pipettiert. Die Laufbedingungen der Gele zur Auftrennung der PCR-Produkte für die untersuchten Bereiche sind in Tabelle XI aufgeführt. Tabelle XI: Laufbedingungen der TTGE für die Exons 5-1,7,8 und der DGGE für die Exons 5-2 und 6 Laufzeit Spannung/V Temperatur in Heizrate (ramprate) in h (konstant) Tmin°C-T max°C in °C/h TTGE 4 160 53- 63 2,5 5-2 DGGE 5 160 58 konstant 6 DGGE 5 160 58 konstant 7 TTGE 6 160 56- 66 1,7 8 TTGE 4 160 50- 59 2,25 Exon Gelart 5-1 Gellauf: Nachdem die vorbereiteten Proben in die Taschen des auf Tmin vorgeheizten (TTGE) bzw. auf Arbeitstemperatur befindlichen (DGGE) Gels pipettiert worden waren, konnte der Gellauf unter den für das zu untersuchende Exon bestimmten Bedingungen gestartet werden. Auswertung des Gels: Die DNA-Banden in den TTGE- bzw. DGGE-Gele wurden mittels Silberfärbung, die zu einer unspezifischen Färbung von DNA führt, visualisiert [modifiziert n. 137]. Färbung des Gels: Die Durchführung der Färbung erfolgte bei Raumtemperatur gemäss folgendem Protokoll. Zunächst wurde das Gel dreimal jeweils für 5min in frischer Fixierlösung [7.2.23] fixiert. Hieran schloss sich der Färbevorgang für 15min in der Silbernitratlösung [7.2.53] an. Nachdem das Gel zweimalig mit aqua bidest. und darauffolgend zweimalig mit Entwicklerlösung [7.2.16] gespült worden war, wurde es für 3, 5 und 12min in jeweils erneuerter Entwicklerlösung inkubiert. Der Färbevorgang endete nach 5min Inkubieren in einer Natriumbikarbonatlösung [7.2.40]. Im 40 Anschluss daran folgte ein kurzes Waschen mit aqua bidest. zur Entfernung restlicher Reagenzien und die Verpackung des Gels durch luftdichtes Verschweissen in Kunststofffolie. Alle Inkubationen mit den Lösungen fanden auf dem Schwenktisch [7.3.8] statt. Auswertung des Gels: Das Bandenmuster der Tumor- und Normalgewebsproben wurde jeweils mit denen der Positivund der Negativkontrolle eines Gellaufes verglichen. Da Positiv- und Negativkontrolle homozygot für das untersuchte Exon waren, ergab sich in der TTGE bzw. DGGE jeweils eine einzelne Bande (entsprechend der Homoduplex), die sich untereinander in der Position unterschieden. Erwartet wurde bei den Proben der Patienten im Falle einer Mutation bei heterozygotem Gemisch der Probe für die untersuchten Exons insgesamt vier Banden (entsprechend den zwei Homo- und den zwei Heteroduplizes; s. auch Ergebnisse Abbildung 6). 2.5.4 Sequenzierung Zur Charakterisierung der Mutation wurden die im Laufverhalten von der Negativprobe abweichenden Homoduplizes aus dem TTGE- bzw. DGGE-Gel einer Klonierung und anschließend einer Sequenzierung zugeführt. Dazu wurden die DNA-Banden aus dem Gel eluiert, durch eine PCR amplifiziert und mittels TOPO-Klonierung [7.2.31] in den Vektor pCR®Blunt II-TOPO® inseriert. Die Plasmidinserts durchliefen ein Gel im Sequenzanalysegerät [7.3.9], das mittels Laserabtastung die Basenabfolge des untersuchten DNA-Abschnittes ermittelt. Isolierung der DNA-Banden aus dem TTGE-Gel bzw. DGGE-Gel: Die DNA-Banden wurden mit einem Skalpell aus dem Gel ausgeschnitten, das Acrylamid-DNAGemisch manuell zerkleinert und in einem Eppendorf-Cup mit 30µl Elutionspuffer [7.2.15] überschichtet. Die Eppendorf-Cups wurden bei 37°C über 2h im Thermomixer [7.3.13] unter ständigem Schütteln inkubiert und im Anschluss für 15min mit 22.900g bei 18°C in der Zentrifuge [7.3.18] zentrifugiert. Der flüssige Überstand, welcher die eluierten Homoduplizes enthielt, konnte in Eppendorf-Cups abpipettiert und der Bodensatz verworfen werden [180]. Zur Amplifizierung der aus dem Acrylamidgel eluierten DNA wurde eine PCR mit den exonspezifischen Primern und Zyklusbedingungen durchgeführt (s. PCR p53 Exon 5-8, 2.5.2). Die PCR-Produkte wurden mittels Agarosegel kontrolliert (s. PCR-Produktkontrolle, 2.5.2). 41 2.5.4.1 Klonierung der amplifizierten Homoduplex-DNA Bei dem Zero Blunt® TOPO® PCR Cloning [7.2.31] werden in einem Ein-Schritt-Verfahren bluntend-PCR-Produkte, die durch eine „proofreading“ Polymerase (z.B. Pwo) erzeugt werden, in einen Plasmid-Vektor inseriert. Der im verwendeten Klonierungs-Kit enthaltene linearisierte Plasmidvektor (pCR®-Blunt II-TOPO®) hat an den 3’-Phosphatgruppen jedes DNA-Stranges Topoisomerase I aus dem Vaccinia Virus kovalent gebunden. Diese Topoisomerase bindet dsDNA an einer spezifischen Stelle und spaltet das Phosphodiester Rückgrat nach einer 5’CCCTT-Sequenz in einem Strang [151]. Die Energie der Phosphodieesterbindung wird in der Bildung einer kovalenten Bindung zwischen der 3’-Phosphatgruppe des gespaltenen Stranges und einem Tyrosylrest (Tyr-274) der Topoisomerase I konserviert. Die Phospho-Tyrosyl Bindung zwischen DNA und Enzym kann später von der 5’-Hydroxylgruppe des gespaltenen Stranges angegriffen werden, somit die Reaktion rückgängig gemacht und die Topoisomerase abgelöst werden [150]. Diese Reaktion wird bei der TOPO® -Klonierung ausgenutzt, um sehr effizient PCR-Produkte zu klonieren. In dem Vektor pCR®-Blunt II TOPO ist das Gen ccdB [12], welches für einen Gyrase-Hemmer kodiert und so das Wachstum der meisten Escherichia coliStämme verhindert, an den C-Terminus des LacZα-Fragmentes fusioniert. Die Insertion eines PCR-Produktes unterbricht die Expression der LacZα-ccdB-Fusion. Diese erlaubt eine direkte Selektion von Rekombinanten nach einer Transformation, da die Zellen, welche Vektor ohne Insertion enthalten, abgetötet werden. Auf die Anwesenheit des Vektors in den Zellen kann durch die Anwesenheit von Genen gegen Zeocin oder Kanamycin selektiert werden. Für die Transformation der TOPO® Klonierungs-Produkte wurden chemisch kompetente Zellen des Bakterienstammes Escherichia coli TOP10 One Shot®verwendet [80]. Durchführung der TOPO®- Klonierung: Vorbereitung: Das Wasserbad [7.3.16] wurde auf 42°C vorgewärmt, u nd das SOC-Medium [7.2.54] auf Raumtemperatur gebracht. LB-Platten [7.2.35] mit 50µg/ml Kanamycinsulfat [7.2.30] waren 30min auf 37°C vorgewärmt. Pro Klonierungsansatz wu rde eine Einheit der One Shot® Zellen auf Eis aufgetaut. Klonierungsreaktion: In einem Eppendorf-Reaktionsgefäß wurden 4µl des blunt end PCR-Produktes sowie1µl der im Klonierungs-Kit enthaltenen Salzlösung [7.2.31] und 1µl TOPO Vektor zusammenpipettiert. Nach einer Inkubationszeit von 5min bei Raumtemperatur konnte der Reaktionsansatz auf Eis gegeben werden. 42 Transformation: 2µl des Klonierungs-Ansatzes wurden zu einer Einheit chemisch kompetenter Escherichia coli One Shot® TOP10 hinzugegeben und vermischt. Es folgte eine Inkubation des Ansatzes auf Eis für 5min. Daran schloss sich ein Hitze-Schock der Zellen für 30s bei 42°C an. Im Anschluss folgte ein direkter Transfer auf Eis. Die nächsten Schritte waren die Zugabe von 250µl des SOC-Medium [7.2.54] bei Raumtemperatur und nachfolgendes horizontales Schütteln bei 200rpm und 37°C für 1h. Von jedem Transformationsan satz wurden 100µl auf eine vorgewärmte LB+Kanamycin-Platte ausplattiert und über Nacht bei 37°C inkubiert. Analyse der Klone: Sechs der nach der Transformation auf dem Selektivmedium gewachsenen Kolonien waren zur Aufreinigung auf LB+Kanamycin-Platten ausgestrichen und von jeweils einer der auf diesen Platten gewachsenen Einzelkolonien Sektorenausstriche angefertigt worden. Diese dienten als Ausgangsmaterial zum Beimpfen der für die Plasmidisolierung (Mini- oder Maxiprep) benötigten Bakterienkulturen. Plasmidisolierung: Miniprep: Zur Isolierung analytisch verwendbarer Mengen Plasmid-DNA wurde der NucleoSpin® Kit der Firma Macherey-Nagel [7.2.41] verwendet. Diese Methode basiert auf der Bindung von DNA an eine Silica-Membran. Jeweils 5ml LB-Medium mit 50µg/ml Kanamycin in 15ml GreinerRöhrchen wurden mit einer auf den Sektorenausstrichen gewachsenen Einzelkolonie beimpft und über Nacht bei 37°C unter Schütteln inkubiert. Durch Zentrifugation bei 3500g und 4°C wurden die Zellen pelletiert. Nachdem der Überstand dekantiert war, konnten die Zellen in 250µl Puffer A1 [7.2.41] resuspendiert und in 1,5ml-Eppendorf-Reaktionsgefäße überführt werden. Danach wurden 250µl Puffer A2 [7.2.41] hinzugefügt und die Reaktionsgefäße sechs- bis achtmal umgedreht. Nach 5min Inkubation bei Raumtemperatur wurden 300µl Puffer A3 [7.2.41] hinzugegeben, erneut sechs- bis achtmal gewendet und dann 10min bei 4°C und 22.000g zentrifugiert. Im Anschluss wurde der die Plasmid-DNA enthaltende Überstand auf die vorbereiteten NucleoSpin® Plasmid-Säulen, welche auf 2ml Eppendorfgefäße aufgesetzt worden waren, aufgetragen. Diese wurden mit 22.000g für 1min in einer Tischzentrifuge zentrifugiert. Nach Verwerfen des Filtrats wurde die Säule mit 600µl Puffer A4 [7.2.41] gewaschen und erneut mit 22.000g 1min zentrifugiert. Nachdem die Säulen auf 1,5mlEppendorf-Reaktionsgefäße überführt worden waren, wurden zur Elution der DNA 50µl Puffer AE [7.2.41] aufgetragen, 1min bei Raumtemperatur inkubiert und anschließend 1min bei 22.000g zentrifugiert. Eine erneute Elution mit 50µl Puffer AE erhöhte die Ausbeute um 15-20%, verdünnte jedoch die DNA-Lösung. Die Proben wurden mit einem Endvolumen von 10µl (9µl Aliquot und 1µl Puffer AE 10x konzentriert [7.2.41]) aliquotiert. 43 Zur Kontrolle der isolierten Plasmide wurde mit diesen ein Restriktionsverdau mit dem Enzym EcoRI durchgeführt. Da der Klonierungsvektor beidseitig der Insertionsstelle für das zu klonierende Fragment jeweils eine Erkennungssequenz für EcoRI besitzt, wurde ein inseriertes Fragment durch diesen Verdau herausgeschnitten. Hierzu wurden jeweils 9µl Plasmid-DNA mit 1µl 10x konzentriertem Puffer H [7.2.15] und 1µl EcoRI-Lösung (2,5U) gemischt und 2h bei 37°C inkubiert. Die Kontrolle erfolgte durch Agaros e-Gel-Elektrophorese (s. PCR- Produktkontrolle 2.5.2). Hierbei gab sich eine erfolgreich verlaufende Klonierung an dem Auftreten zweier Banden, von denen eine dem Vektor und die andere dem inserierten PCRFragment entsprach, zu erkennen. Zwei der nach EcoRI-Verdau als Insert-tragend identifizierte Klone dienten als Ausgangsmaterial für eine Maxiprep-Plasmidisolierung unter Verwendung des QUIAGEN Plasmid Maxi Kit 100 [7.2.44]. Maxiprep: Die zur Aufreinigung eingesetzten Säulen sind Anionenaustauscher, von denen die PlasmidDNA durch einen Hochsalzpuffer eluiert wird. Zunächst wurde eine Vorkultur vorbereitet. Dazu wurden 10ml LB-Medium+50µg/ml Kanamycin [7.2.30] mit einer transformierten E. coli-Kolonie beimpft und 5 bis 6h lang bei 37°C stark geschüttelt. Mit je 5ml Vorkultur konnten dann 100ml LB-Medium+50µg/ml Kanamycin inokuliert und ca. 16h lang unter starkem Schütteln bei 37°C i nkubiert werden. 1,7ml dieser Kulturen wurden entnommen und mit 300µl sterilem Glycerin [7.2.26] gemischt und dieses als Stammkultur bei -80°C eingefroren. Der Rest der Hau ptkultur wurde gleichmässig in vier 50ml Greiner-Reaktionsröhrchen überführt und 15min lang bei 4°C mit 3.400g zentrifugiert. Nachdem der Überstand dekantiert war, konnte das Zell-Pellet in je 2,5ml P1-Puffer [7.2.44] pro Gefäß resuspendiert werden. Nach dem Vereinigen der Suspensionen wurden diese mit 10ml P2Puffer [7.2.44] versetzt. Nach sechsmaligem Umdrehen des Gefäßes schloss sich 5min Inkubation bei Raumtemperatur an. Nach Zugabe von 10ml 4°C kaltem P3-Puffer [7.2.44] wurde wieder durch sechsmaliges Umdrehen gemischt und dann 20min auf Eis inkubiert. Im Anschluss daran folgte eine Zentrifugation für 45min bei 3.400g und 4°C. Während dieser Zeit waren die DNA-Säulen (Qiagen-tip 500) des Kits mit 10ml QBT-Puffer [7.2.44] äquilibriert worden, und im Anschluss daran konnte die DNA-Lösung auf die Säulen aufgetragen werden. Nach zweimaligem Waschen mit 30ml QC-Puffer [7.2.44] wurde die Säule auf ein 50ml Reaktionsgefäß überführt und die Plasmid-DNA mit 15ml QF-Puffer [7.2.44] eluiert. Die DNA wurde bei Raumtemperatur durch Zugabe von 0,7 Volumenanteilen (=10,5ml) 2-Propanol [7.2.46] gefällt. Im Anschluss wurde 45min bei 3.400g und 4°C zentrifugiert. Der Überstand wurde dekantiert und das DNA-Pellet mit 5ml 70%igem Ethanol [7.2.20] gewaschen. Anschließend wurde für weitere 30min bei 3.400g und 4°C zentrifugiert, danach der Überstand vorsichtig dekantiert, und die DNA als Pellet bei Raumtemperatur getrocknet. Die DNA wurde in 44 100µl TE-Puffer [7.2.61] resuspendiert und nach Inkubation über Nacht bei 4°C in 1,5mlEppendorf-Reaktionsgefäße überführt. Nach einer 1:100 Verdünnung der DNA-Lösung konnte die DNA-Konzentration photometrisch wie unter Kapitel 2.5.1 beschrieben gemessen werden. Der Zielwert der DNA-Konzentration lag über 3mg/ml. Nach abschließender Kontrolle der Plasmide durch Agarose-Gelelektrophorese (s. PCR-Produktkontrolle 2.5.2) wurden diese bei 20°C bis zur weiteren Verarbeitung gelagert. 2.5.4.2 Sequenzierung In der vorliegenden Arbeit kam die „Cycle-Sequenzierungs“-Methode, eine Abwandlung der ursprünglich von Sanger et al. (1977) [139] beschriebenen Didesoxynukleotid-KettenabbruchSequenzierungsmethode, unter Verwendung von am 5‘-Ende mit einem IR-Farbstoff markierten Primern zur Anwendung. Für die Sequenzreaktion wurde ein DNA-Sequenzierungskit mit Thermo-Sequenase und 7-Desaza-dGTP der Firma Amersham Pharmacia Biotech [7.2.62] verwendet. Als Primer wurden folgende IRD800-markierte Oligonukleotide, deren Sequenz im Bereich des Polylinkers des Plasmides pCR®-Blunt II TOPO [7.2.31] zu finden ist, von der Firma MWG Biotech [7.2.45] bezogen. Primerangabe für die Cycle-Sequenzierung: Primer 1: M13Universal(-21), 18mer: 5’-TGT AAA ACG ACG GCC AGT-3’ Primer 2: M13Reverse (-29), 18mer: 5’-CAG GAA ACA GCT ATG ACC-3’ Zunächst wurde für jede Sequenzierungsreaktion der in Tabelle XII aufgeführte Ansatz in PCRCups zusammenpipettiert. Tabelle XII: Mastermix für Sequenzierungs-PCR Reagenzien für Sequenzierungs-PCR µl/Ansatz DNA (aus Maxiprep, 1 µg/µl) 1µl Primer [2mM] 1µl DMSO [7.2.12] 0,5µl aqua bidest., steril 10,5µl Von diesem Ansatz waren jeweils in 4 Ansätzen 3µl mit jeweils 1µl der vier dNTP/ddNTPGemische [7.2.14] in 0,2ml PCR-Reaktionsgefäßen [7.2.43] vermischt und anschließend mit 20µl “Chill-out Liquid Wax” [7.2.10] überschichtet worden. 45 Alle Pipettierschritte fanden auf Eis statt. Die Sequenzierungs-Ansätze wurden in den Thermocycler [7.3.12] eingesetzt und den in Tabelle XIII aufgeführten Reaktionsbedingungen ausgesetzt: Tabelle XIII: Reaktionsbedingungen für die Cycle-Sequenzierung Zykluszahl Zeit Temperatur 1x 1min 45s 95°C 12x jeweils 15s 95°C, 55°C, 70°C 13x jeweils 20s 95°C, 55°C, 70°C 1x 40s 95°C Anschließend wurden die Reaktionsansätze mit jeweils 3µl Stop-Lösung [7.2.55] versetzt und bis zur weiteren Verwendung bei -20°C aufbewahrt. Sequenzierungsgel: Entsprechend Tabelle XIV wurden die Reagenzien gemischt, das Gel mit einer 50ml-Spritze aufgezogen und unter Zuhilfenahme eines Membran-Filters (0.2µM) blasenfrei zwischen die nach Anleitung des Herstellers montierten Glasplatten des Sequenzierers gegeben. Das Gel hatte eine Größe von 41,0cm x 25,5cm x 0,25cm (Höhe x Breite x Dicke). Als Platzhalter für den für die Auftragung der Proben verwendeten „Haifischzahnkamm“ wurde ein rechteckiger Vorkamm eingesetzt. Tabelle XIV: Zusammensetzung des Gels für die Sequenzierung Reagenz Volumen/Menge RapidGel XL Sol 40% [7.2.51] 7,5ml Harnstoff [7.2.29] 21g 10x TBE Long Run Puffer [7.2.60] 5ml aqua bidest., steril 28ml DMSO [7.2.12] 500µl TEMED [7.2.61] 50µl APS, 10% [7.2.3] 350µl Nach 1,5h Polymerisation konnte das Gel in den automatischen Infrarot-Fluoreszens DNASequenzierer [7.3.9] eingesetzt und mit oberer und unterer Pufferkammer verbunden werden. Als Laufpuffer diente einfach konzentrierter TBE-Puffer [7.2.59]. Nach Kalibrierung des Lasers wurde zunächst ein Vorlauf von ca. 20min unter Sequenzierungsbedingungen gestartet, um Verunreinigungen aus dem Gel, die die Sequenzierung stören könnten, zu entfernen. Dann 46 wurde der „Haifischzahn-Kamm“, der es ermöglichte 64 Ansätze, d.h. 16 Proben gleichzeitig aufzutrennen, eingesetzt und je 1µl der Sequenzierungsansätze in der Basenreihenfolge A-CG-T aufgetragen. Nach erneuter Laserjustierung wurde der Gellauf unter folgenden Bedingungen gestartet: Spannung: 1500V (konstant) Stromstärke: 35mA (zu Beginn) Temperatur: 45°C Laufzeit: 12h Auswertung der Sequenzierung: Die Sequenzdaten wurden mit Hilfe des Programms Base Image IR Data Collection, Version 2.31 (Li-Cor, Nebraska, USA, 1992-95) aufgezeichnet und gespeichert sowie anschließend mit dem Analyseprogramm Base Image IR Image Analysis, Version 4.0 (Li-Cor, Lincoln, Nebraska, USA, 1997) ausgewertet. Die erhaltenen Sequenzdaten wurden durch Vergleich mit der Wildtyp-DNA (EMBL:U94788) mit Hilfe des multiplen Alignment Programms Clustal W online beim European Bioinformatics Institute (http://www.ebi.uc.uk) auf Mutationen hin untersucht. Von jedem nach TTGE/DGGE als mutiert identifizierten Fragment wurden routinemässig jeweils 2 Klone untersucht, wobei jeweils beide DNA-Stränge durch Verwendung von M13-Universalund M13-Reverse-Primer [7.2.45] sequenziert wurden. Bei unterschiedlichen Ergebnissen wurden weitere Klone des jeweiligen Fragmentes zur Sequenzierung herangezogen. 2.6 Statistik Die Stichprobenschätzung erfolgte zu Beginn der Doktorarbeit nach der Datenlage der Literatur zu diesem Thema. Die Stichprobengröße betrug 52. Zur Berechnung der Signifikanz des Unterschiedes in der VEGF-Konzentration zwischen Tumor- und Normalgewebe wurde der Wilcoxon-Test angewendet. Die Signifikanz der Unterschiede in der VEGF-Konzentration im Tumorgewebe zwischen Tumoren mit und ohne Mutation im p53-Gen, unterteilt in verschiedene Gruppen (s. Kapitel 3), wurde mit Hilfe des Mann-Whitney-U Test berechnet. Als signifikant wurde ein p-Wert von p≤0.05 gewertet. Der Wilcoxon-Test sowie der Mann-Whitney-U Test wurden mit „SPSS for Windows“, Version 9.0.1, Fa. SPSS Inc., Chicago, USA, durchgeführt. 47 Abbildung 5: Boxploterklärung Definitionen des Boxplot [169]: Die Innere Eingrenzung ist definiert durch: - Quartile, oberhalb bzw. unterhalb + 1,5 x F-Spanne Werte ausserhalb der Inneren Eingrenzung sind als Ausreisser definiert. Die Unterscheidung ausserhalb und weit ausserhalb wird getroffen durch zusätzliche Grenzen, die wie folgt definiert sind: - Quartile, oberhalb + 3 x F-Spanne - Quartile, unterhalb - 3 x F-Spanne 48 3 Ergebnisse 3.1 Alterationen des p53-Gens im Normal- und Tumorgewebe Die ‚hot spot‘-Regionen für Mutationen des p53-Gens in den Exon 5-8 wurden mit der TTGE für die Exons 5-1, 7 und 8, und mit der DGGE für die Exons 5-2 und 6 auf Mutationen hin untersucht (s. Kapitel 2.5.3). Die Abbildung 6 zeigt beispielhaft das Bandenmuster einer TTGE mit der Positiv- (Zelllinie SW480) und der Negativkontrolle in den Spuren 1 und 2. War keine Mutation vorhanden, so trat analog der Negativprobe (Spur 2) nur eine Bande auf, entsprechend der Homoduplex aus homozygoter Wildtyp-DNA. Bei der Zelllinie SW480 (Positivkontrolle, Spur 1) mit einer Mutation im Exon 8 (Codon 273: CGT->CAT) ist ebenfalls nur eine Bande zu sehen. Sie entspricht der Homoduplex aus homozygot mutierter DNA und hat eine kürzere Laufstrecke als die Bande der Negativprobe. In den Spuren 4 (T83) und 10 (T125) sind jeweils vier Banden zu erkennen. Die beiden Banden mit der längsten Laufstrecke entsprechen den Homoduplizes aus Wildtyp- respektive mutierter DNA. Darüber, mit kürzerer Laufstrecke, sind die Heteroduplizes aus Wildtyp- und mutierter DNA zu erkennen, mit unterschiedlichen Laufverhalten. 1 2 3 4 5 6 7 9 11 Abbildung 6: TTGE-Gel zum Nachweis von Mutationen im Exon 8 des p53-Gens. Bedingungen: 10% Acrylamid, 7M Harnstoff, 160V, Temperaturbereich 53-63°C, Heizrate 2°C/h, 5h Aufgetragen wurden Amplifikationsprodukte aus einer PCR mit Exon-8-spezifischen Primern und genomischer DNA aus Spur 1: kolorektale Karzinom-Zelllinie SW480 [Positiv-Kontrolle: Mutation in Codon 273 (CGT CAT)]; Spur 2: Blut eines gesunden Spenders (Negativ-Kontrolle); Spuren 3-11: kolorektale Tumorgewebe. Proben, die von der Negativ-Kontrolle abweichende Banden aufwiesen, wurden als mutiert identifiziert, siehe Spur 4 und 10. Mit Hilfe der TTGE bzw. der DGGE wurden in 26 der 52 untersuchten Tumorproben insgesamt 29 Abweichungen von der Wildtyp-Sequenz in den untersuchten Exons des p53-Gens detektiert (s. Tabelle XV). 25 Proben trugen jeweils eine Mutation und eine Probe (T169) zwei 49 Mutationen. Bei zwei Proben (T188 und T217) wurde zusätzlich zu einer Mutation im Tumor ein Polymorphismus in Codon 213 sowohl im Tumor- als auch im Normalgewebe nachgewiesen. In die Auswertung der Mutationshäufigkeit, Transversionen, Transitionen und Mutationen in den einzelnen Stadien wurden alle 26 Tumorproben mit Mutation einbezogen. Der Polymorphismus wurde nicht als Mutation gewertet. Im korrespondierendem Normalgewebe der 26 Tumorproben mit Mutation fand sich keine Mutation in den untersuchten Exons. In den Proben N188 und N217 konnte ebenfalls der Polymorphismus nachgewiesen werden. Tabelle XV: Häufigkeiten der p53-Mutation in den Exon 5-8 in % aller Mutation N nein 26 50,00 ja 26 50,00 3.2 Tumore (n=52) Lokalisation und Art der p53-Mutationen Die Mutationsanalyse durch die TTGE bzw. DGGE ergab die in Tabelle XVI dargestellte Verteilung der Mutationen auf die fünf untersuchten Exons. Tabelle XVI: Verteilung der detektierten Mutationen auf die Exons 5-8 Lokalisation der Mutation in % der p53 n positiven Tumore in % aller Tumore (n=52) (n=26) p53ex5 8 30,76 15,38 p53ex6 4 15,38 7,69 p53ex7 7 26,92 13,46 p53ex8 10 38,46 19,23 In der Sequenzierung konnten die 29 gefundenen Mutationen 14 verschiedenen Codons zugeordnet werden (s. Tabelle XVII). 15 dieser Mutationen (51,72%) lagen in den ‚hot spot‘Codons 175, 245, 248, 273 und 282 des p53-Gens (s. Abbildung 2). Es zeigte sich, dass die Mutationen in der Mehrzahl der Fälle (n=26) einen Aminosäurenaustausch zur Folge hatte (vgl. Tabellen XVIII bis XXI). In zwei Proben wurde ein Polymorphismus in Codon 213 (s. Tabelle XIX) nachgewiesen. Eine Probe trug eine stille Mutation (s. Tabelle XVIII). 50 Tabelle XVII: Verteilung der Mutationen im p53-Gen Exon 5-8 in den 26 Tumorproben auf die einzelnen Codons a Lokalisation in Codon n 135 171 173 175 a 193- 201 Deletion a 213 Polymorphismus 220 234 245 248 266 273 280 282 285 4 1 1 2 1 2 1 1 2 4 1 3 1 4 1 in % aller Mutationen (n=26) 15,38 3,85 3,85 7,70 3,85 7,70 3,85 3,85 7,70 15,38 3,85 11,54 3,85 15,38 3,85 in % aller Tumoren (n=52) 7,69 1,92 1,92 3,85 1,92 3,85 1,92 1,92 3,85 7,68 1,92 5,77 1,92 7,69 1,92 s. Tabelle XIX In acht Tumorproben traten Mutationen im Exon 5 auf (s. Tabelle XVIII), von denen vier im Codon 135 lagen und zu einer Sequenzänderung von TGC im Wildtyp nach TTC führten. In zwei Fällen wurde im Codon 175 eine Mutation von CGC zu CAC detektiert. Eine Mutation in Exon 5 betraf das Codon 171 (GAG GGG). In einer weiteren Probe war das Codon 173 an der dritten Position von GTG zu GTT mutiert. Bei dieser Mutation handelte es sich somit um eine "stille" Mutation, da beide Sequenzen für die gleiche Aminosäure (Val) kodieren (s. Tabelle XVIII). Tabelle XVIII: Mutationen in Exon 5 (n=8) Codon Anzahl der Mutationen Wt-Sequenz Mut-Sequenz Wt-Aminosäure Mut-Aminosäure 135 4 TGC TTC Cys Phe 171 1 GAG GGG Glu Gly 173 1 GTG GTT Val Val 175 2 CGC CAC Arg His In vier Tumorproben wurde eine Sequenzänderung im Exon 6 nachgewiesen (s. Tabelle XIX). Dabei handelte es sich in zwei Fällen um einen bekannten seltenen Polymorphismus im Codon 213 [21], welcher abweichend von der Wildtyp-Sequenz CGG zur Sequenz CGA führte. Dieser Polymorphismus wurde auch in den korrespondierenden Normalgewebeproben gefunden. In einer Tumorprobe wurde eine Deletion der Codons 193 bis 201 mit dem Verlust von 8 51 Basentripletts nachgewiesen, welche für die Aminosäuren His, Leu, Ile, Arg, Val, Glu, Gly, Asn, Leu kodieren. Das durch die Deletion entstandene Basentriplett kodiert für die Aminosäure Leu. Eine weitere Tumorprobe besaß eine Punktmutation im Codon 220 von Wt TAT nach Mut TGT. Tabelle XIX: Mutationen in Exon 6 (n=4) Codon Anzahl der Mutationen Wt-Sequenz Mut-Sequenz 193-CAT CTT ATC 193-201 1 CGA GTG GAA Wt-Aminosäure Mut-Aminosäure His, Leu, Ile, Arg, 193 -CTG- 201 GGA AAT TTG-201 Val, Glu, Gly, Asn, Leu Leu 213 2 CGA CGG Arg Arg 220 1 TAT TGT Tyr Cys Insgesamt sieben Tumorproben trugen eine Mutation in Exon 7. Davon war in vier Proben das Codon 248 mutiert, in drei Fällen von CGG zu CAG und einmal von CGG zu TGG. In zwei Tumorproben waren die Mutationen im Codon 245 lokalisiert. Die Sequenz veränderte sich in einem Fall vom Wildtyp GGC zu AGC, im anderen von GGC zu GAC. Eine weitere Mutation zeigte sich im Codon 234 mit einer Triplettänderung von TAC zu CAC (s. Tabelle XX). Tabelle XX: Mutationen in Exon 7 (n=7) Codon Anzahl der Mutationen Wt-Sequenz Mut-Sequenz Wt-Aminosäure Mut-Aminosäure 234 1 TAC CAC Tyr His 245 1 GGC AGC Gly Ser 245 1 GGC GAC Gly Asp 248 3 CGG CAG Arg Gln 248 1 CGG TGG Arg Trp Von den zehn Tumorproben mit einer Mutation in Exon 8 trugen vier eine Mutation im Codon 282 mit einer Sequenzänderung vom Wildtyp CGG zu TGG auf. In drei Proben war das Codon 273 mutiert, wovon zweimal eine Mutation von Wt CGT zu CAT und einmal eine zu TGT stattgefunden hatte. Jeweils eine Probe trug eine Mutation in Codon 266 Wt GGA zu Mut GAA, Codon 280 mit Wt AGA zu Mut ATA und Codon 285 von Wt GAG zu Mut AAG (s. Tabelle XXI). 52 Tabelle XXI: Mutationen in Exon 8 (n=10) Codon Anzahl der Mutationen Wt-Sequenz Mut-Sequenz Wt-Aminosäure Mut-Aminosäure 266 1 GGA GAA Gly Glu 273 2 CGT CAT Arg His 273 1 CGT TGT Arg Cys 280 1 AGA ATA Arg Ile 282 4 CGG TGG Arg Trp 285 1 GAG AAG Glu Lys 3.3 Art der nachgewiesenen Mutationen In Tabelle XXII sind die Mutationsergebnisse nach ihrer Art aufgetrennt, d.h. ob es sich um den Austausch einer Purinbase durch eine Purinbase bzw. einer Pyrimidinbase durch eine Pyrimidinbase (Transition) handelte oder aber um den Austausch einer Purinbase durch eine Pyrimidinbase bzw. umgekehrt (Transversion). Bei der Mehrzahl der Fälle (75,86%) war eine Transition aufgetreten. Es zeigte sich, dass eine Transition, die zu einer Änderung der Nukleotidpaarung von GC zu AT mit 11 Fällen (37,93%) die häufigste Mutation in dem untersuchten Tumorkollektiv darstellte. Mutationen, welche eine Veränderung der Nukleotidabfolge von CG zu TA zeigten traten mit 6 (20,69%), AT zu GC mit 4 Fällen (13,79%) und TA zu CG in einem Fall (3,45%) auf. Acht der 11 Transitionen (72,73%) GC zu AT und alle 6 Transitionen von CG nach TA lagen im Bereich von CpG Dinukleotiden (s. Tabelle XXIII). Transversionen, die zu einer Änderung des Nukleotidpaares GC zu TA führten wurden in 6 Fällen nachgewiesen. In einem Fall bestand die Mutation aus einer Deletion in Exon 6 (vgl. Tabelle XIX). 53 Tabelle XXII: Verteilung der Mutationsart in den untersuchten Exons des p53-Gen Art der Mutation Gesamt- Exon 5 Exon 6 Exon 7 Exon 8 Ereignisse in % GC/AT 2 0 5 4 11 37,93 CG/TA 0 0 1 5 6 20,69 AT/GC 1 a 3 0 0 4 13,79 TA/CG 0 0 1 0 1 3,45 5 0 0 1 6 20,69 0 1 0 0 1 3,45 8 4 7 10 29 100 Ereignisse n Transition Transversion GC/TA Deletion Gesamtmutationen a davon 2mal Polymorphismus Tabelle XXIII: Transitionen in CpG Dinukleotiden Art der Mutation Exon 5 Exon 6 Exon 7 Exon 8 GesamtEreignisse n GC/AT CG/TA 2 0 0 0 4 1 2 5 8 6 3.4 Ereignisse in % auf Gesamtmutation 27,59 20,69 VEGF-Konzentration im Normal- und Tumorgewebe Normalgewebe: Die mediane VEGF-Konzentration in den 52 ausgewerteten Proben betrug 82,87pg VEGF/mg Protein (Range: 26,66-743,11pg VEGF/mg Protein). Der Mittelwert lag bei 72,41pg VEGF/mg Protein mit einer Standardabweichung (SD) von 128,58pg VEGF/mg Protein (s. Tabelle XXIV und Abbildung 7). Tumorgewebe: Die Probe T145 wurde in sämtliche VEGF-Berechnungen nicht mit einbezogen, da sie einen Ausreisser darstellte (22552,71pg VEGF/mg Protein). Bei den 51 ausgewerteten Tumorgewebeproben betrug der Median 887,43pg VEGF/mg Protein (Range 113,91-5642,11pg VEGF/mg Protein) und der Mittelwert 1138,58pg VEGF/mg Protein mit einer Standardabweichung 1176,07pg VEGF/mg Protein. Der Vergleich der VEGF-Konzentration von Normal- und Tumorgewebe ergab einen hoch signifikanten Unterschied (s. Tabelle XXIV und Abbildung 7). Es zeigte sich im Tumorgewebe in 92,3% der Proben eine höhere VEGF-Konzentration (VEGF T/VEGF N >1). 54 als im Normalgewebe a Tabelle XXIV: VEGF-Konzentration im Normal- und Tumorgewebe a Proben n Median Mittelwert SD Range N 52 82,87 72,41 128,58 26,66 bis 743,11 T 51 887,43 1138,58 1176,07 113,91 bis 5642,11 in pg VEGF/mg Protein Abbildung 7: VEGF-Konzentration im Normal- und Tumorgewebe 55 p p<0,001 3.5 VEGF-Konzentration im Normal- und im Tumorgewebe von Patienten mit und ohne Mutation im p53-Gen Die VEGF-Konzentration im Tumorgewebe bei Patienten ohne Mutation im p53-Gen unterschied sich nicht signifikant von der bei Patienten mit Mutation (s. Tabelle XXV und Abbildung 8). a Tabelle XXV: VEGF-Konzentration im Tumorgewebe von Patienten mit und ohne Mutation im p53-Gen im Tumorgewebe n a Mutation p53-Gen Median Mittelwert SD Range 25 Nein 897,01 1239,79 1373,83 117,28 bis 5642,11 26 Ja 816,31 1049,29 967,41 113,91 bis 4370,12 p 0,925 in pg VEGF/mg Protein Abbildung 8: VEGF-Konzentration im Tumorgewebe mit und ohne Mutation im p53-Gen Ebenso unterschied sich die VEGF-Konzentration im Normalgewebe bei Patienten ohne Mutation im Tumor nicht signifikant von der bei Patienten mit p53-Mutation im Tumorgewebe (siehe Tabelle XXVI und Abbildung 9). 56 a Tabelle XXVI: VEGF-Konzentration im Normalgewebe von Patienten mit und ohne Mutation im p53-Gen im Tumorgewebe n Mutation p53-Gen Median Mittelwert SD Range 26 nein 70,89 128,56 164,49 26,66 bis 743,11 26 ja 86,96 110,91 80,66 36,71 bis 428,63 p 0,257 a in pg VEGF/mg Protein Abbildung 9: VEGF-Konzentration im Normalgewebe mit und ohne Mutation in Exon 5-8 im korrespondierendem Tumorgewebe 3.6 VEGF-Konzentration im Tumorgewebe bei Patienten mit Lokalisation der Mutation im Exon 5, 6, 7 oder 8 Die Unterteilung der Fälle nach Lokalisation der Mutation in den verschiedenen untersuchten Exons ergab keinen signifikanten Unterschied in den entsprechenden VEGF-Konzentrationen im Vergleich zu den aller anderen Tumorproben (s. Tabelle XXVII). Die Gruppe der Tumore welche eine Mutation in Exon 7 trugen, besaß zwar mit 344,08pg VEGF/mg Protein (Median) eine um 61% niedrigere VEGF-Konzentration als die mediane VEGF-Konzentration aller Tumorproben (887,42pg VEGF/mg Protein), dieser Unterschied war jedoch nicht signifikant (p=0,071). 57 a Tabelle XXVII: VEGF-Konzentration im Tumorgewebe von Patienten mit Mutation im Exon 5, 6, 7 und 8 a b b Exon n Median Mittelwert SD Range p 5 8 758,36 881,65 572,45 201,86 bis 1715,55 0,836 6 4 2837,5 1711,42 2167,46 113,91 bis 4370,12 0,700 7 7 344,08 470,74 299,39 113,91 bis 954,06 0,071 8 10 1161,56 1530,45 1321,48 234,29 bis 4370,12 0,209 in pg VEGF/mg Protein; Unterschied jeweils im Vergleich zu allen übrigen Tumorproben 3.7 VEGF-Konzentration im Tumorgewebe mit und ohne p53-Mutation in Abhängigkeit vom Geschlecht Unter den 52 untersuchten Patienten waren 21 Frauen (38,9%) und 31 Männer (59,6%). Es wurden 51 Proben in Bezug auf die VEGF-Konzentration ausgewertet (s. Kapitel 3.1). Bei den Frauen fand sich bei 10 Tumorproben (50% der Frauen) und bei den Männern bei 16 Tumorproben (51,61% der Männer) eine Mutation. In beiden Gruppen war kein signifikanter Unterschied in der VEGF-Konzentration im Tumorgewebe mit und ohne Mutation zu finden (s. Tabelle XXVIII und Abbildung 10). a Tabelle XXVIII: Unterschied in der VEGF-Konzentration im Tumorgewebe von Patienten mit und ohne Mutation im p53-Gen, getrennt nach Geschlecht a Geschlecht n weiblich 10 weiblich Mutation Median Mittelwert SD Range nein 1045,32 1277,3 1321,7 221,01 bis 4644,2 10 ja 533,02 1278,26 1413,9 234,29 bis 4370,1 männlich 15 nein 893,11 1214,79 1452,9 117,28 bis 5642,1 männlich 16 ja 868,1 906,18 550,97 113,91 bis 1866,5 p53-Gen in pg VEGF/mg Protein 58 p 0,705 0,968 Abbildung 10: VEGF-Konzentration im Tumorgewebe mit und ohne Mutation im p53-Gen bei Frauen und Männern 3.8 VEGF-Konzentration im Tumorgewebe mit und ohne Mutation im p53-Gen getrennt nach Lokalisation des Tumors Von den untersuchten Tumoren waren 30 (57,7%) im Kolon und 21 (40,4%) im Rektum lokalisiert. Bei 13 der im Kolon lokalisierten Tumore (43,3%; 25,0% aller Tumore) wurde eine Mutation im p53-Gen detektiert. Ebenso fanden sich bei 13 Tumoren mit der Lokalisation im Rektum (61,9%; 25,0% aller Tumore) Mutationen. Sowohl bei den im Kolon als auch im Rektum lokalisierten Tumoren bestand kein signifikanter Unterschied (p>0,05) in der VEGFKonzentration zwischen Tumoren mit und ohne Mutation (s. Tabelle XXIX und Abbildung 11). 59 a Tabelle XXIX: VEGF-Konzentration im Tumorgewebe von Patienten mit und ohne Mutation im p53-Gen, getrennt nach Lokalisation des Tumors Mittelwert SD Range nein 893,11 1132,00 1359,42 158,28 bis 5642,11 13 ja 887,43 1304,59 1255,44 113,91 bis 4370,12 Rektum 8 nein 1169,55 1468,86 1469,29 117,28 bis 4644,21 Rektum 13 ja 732,88 793,98 482,07 201,86 bis 1522,72 n Kolon 17 Kolon a Mutation Median Lokalisation p53-Gen p 0,601 0,346 in pg VEGF/mg Protein Abbildung 11: VEGF-Konzentration im Tumorgewebe mit und ohne Mutation im p53-Gen bei Lokalisation des Tumors im Kolon bzw. Rektum 3.9 VEGF-Konzentration im Tumorgewebe mit und ohne Mutation unterteilt in Dukes-Stadien Bei 2 von 9 Tumoren in Stadium A (22,2 %), 7 von 17 Tumoren in Stadium B (41,18%), 9 von 14 Tumoren in Stadium C (64,3%) und 8 von 12 Tumoren in Stadium D (66,67%) liess sich eine Mutation im p53-Gen nachweisen. In allen vier Stadien fand sich kein signifikanter Unterschied in der VEGF-Konzentration im Tumorgewebe zwischen Tumoren mit und solchen ohne Mutation (s. Tabelle XXX, Tabelle XXXI und Abbildung 12). 60 Tabelle XXX: Mutationen im p53-Gen getrennt nach Dukes-Stadien Dukes Stadium Mutation n p53-Gen %-Mutationen Exon 5 Exon 6 Exon 7 Exon 8 in dem Stadium A 9 2 2 0 0 0 22,2 B 17 7 2 1 0 4 41,2 C 14 9 4 1 3 2 64,3 D 12 8 0 0 4 4 66,7 a Tabelle XXXI: VEGF-Konzentration im Tumorgewebe von Patienten mit und ohne Mutation getrennt nach Dukes-Stadien Dukes Stadium a n Mutation p53-Gen Median Mittelwert SD Range A 7 nein 407,36 954,19 918,95 221,01 bis 2340,72 A 2 ja 334,97 334,97 188,25 201,86 bis 468,09 B 10 nein 1228,83 1059,90 741,77 117,28 bis 2042,86 B 7 ja 1435,69 1603,66 1315,26 237,36 bis 4370,12 C 5 nein 897,01 1509,65 1782,84 158,28 bis 4644,21 C 9 ja 552,33 624,25 451,51 113,91 bis 1320,88 D 4 nein 357,39 2056,10 3107,00 168,81 bis 5642,11 D 7 ja 954,06 1310,00 989,45 284,29 bis 3029,37 in pg VEGF/mg Protein 61 p 0,380 0,558 0,257 0,732 Abbildung 12: VEGF-Konzentration in Tumorgewebe mit und ohne Mutation im p53-Gen in bei Einteilung der Tumore in die Dukes-Stadien 3.10 VEGF-Konzentration im Tumorgewebe bei Patienten mit und ohne Mutation im p53-Gen bei unterschiedlicher Invasionstiefe des Tumors nach dem TNM-System Von den 52 untersuchten Tumoren waren nach dem TNM-System [71] 4 (7,7%) Tumore in T1, 7 (13,5%) in T2, 32 (61,5%) in T3 und 9 (17,3%) in T4 klassifiziert. Keiner der 4 T1 Tumore, aber 3 der 7 (42,9%) T2 Tumore, 17 der 32 (53,1%) T3 Tumore sowie 6 der 9 (66,7%) T4 Tumore wiesen eine Mutation im p53-Gen auf (s. Tabelle XXXII). Tabelle XXXII: Mutationen im p53-Gen, getrennt nach T-Kategorie der TNM-Klassifikation TKategorie n Mutation p53-Gen % der Mutationen bezogen auf Tumoren in der T-Kategorie 1 4 0 0 2 7 3 42,9 3 32 17 53,1 62 4 9 6 66,7 Der Unterschied in der VEGF-Konzentration im Tumorgewebe zwischen Tumoren mit und ohne Mutation war weder bei T1 noch bei T2, T3 oder T4 Tumoren signifikant (s. Tabelle XXXIII und Abbildung 13). a Tabelle XXXIII: VEGF-Konzentration im Tumorgewebe von Patienten mit und ohne Mutation im p53-Gen bei unterschiedlicher Invasionstiefe des Tumors nach dem TNM-System TNMStadium a n Mutation p53-Gen Median Mittelwert SD Range T1 4 nein 334,87 743,09 892,94 225,30 bis 2077,30 T1 0 ja - - - - T2 4 nein 1742,97 2087,79 1912,46 221,01 bis 4644,20 T2 3 ja 468,09 658,27 575,58 201,86 bis 1304,90 T3 15 nein 926,32 953,05 675,4 117,28 bis 2042,90 T3 17 ja 848,78 1103,97 987,14 234,29 bis 4370,10 T4 3 nein 357,39 2109,52 3059,34 329,06 bis 5642,10 T4 6 ja 630,74 1089,86 1150,98 113,91 bis 3029,40 in pg VEGF/mg Protein 63 p - 0,289 0,874 0,439 Abbildung 13: VEGF-Konzentration im Tumorgewebe mit und ohne Mutationen im p53-Gen bei Einteilung der Tumore nach den T-Stadien der TNM-Klassifikation 3.11 VEGF-Konzentrationen bei Patienten mit Mutationen in Codons, die für die DNA-bindende Struktur und die Tertiärstruktur des p53-Proteins kodieren Mutationen der Codons 248 und 273, welche beide im Wildtyp für Arginin kodieren, führen zur Veränderung der DNA-Bindungsregion des p53-Proteins. Dagegen haben Mutationen mit resultierendem Aminosäureaustausch in den Codons 175, 245, 249 oder 282 Auswirkung auf die Tertiärstruktur des p53-Proteins. Beide Veränderungen des Genproduktes führen dazu, dass das p53-Protein seine Wirkung zum Teil oder gänzlich verliert (s. Einleitung 1.2.3) [95]. Von den 26 Tumoren mit einer Mutation im p53-Gen, zeigten 13 einen Basenaustausch in den oben genannten Codons (s. Tabelle XVII). Eine Mutation in den Codons 248 und 273 besaßen insgesamt 7 Tumorproben (vgl. Tabellen XX und XXI). Die VEGF-Konzentrationen in diesen 7 Tumoren wiesen im Vergleich zu den 19 Tumoren mit Mutationen in anderen Codons des p53-Gen keinen signifikanten Unterschied (p>0,05) auf. 64 a Tabelle XXXIV: VEGF-Konzentration im Tumorgewebe von Patienten mit einer Mutation im Codon 248 oder 273 des p53-Gens Mutation n Codon 248 Median Mittelwert SD Range P oder 273 a 7 ja 552,33 669,58 433,88 267,83 bis 1435,69 19 nein 887,43 1189,18 1077,20 113,91 bis 4370,12 0,37 in pg VEGF/mg Protein In insgesamt 8 Tumoren wurde eine Mutation in den Codons 175, 245 oder 282 gefunden (s. Tabelle XVII),. Die VEGF-Konzentration in den entsprechenden Tumoren war etwa um das 1,6 fache erhöht im Vergleich zu der der 18 Tumoren, welche Mutationen in anderen Codons des p53-Gen aufwiesen (s. Tabelle XXXV). Allerdings war dieser Unterschied mit einem p-Wert von 0,085 nicht signifikant. a Tabelle XXXV: VEGF-Konzentration bei Patienten mit einer Mutation in Codon 175, 245 oder 282des p53-Gens Mutation n Codon 175, Median Mittelwert SD Range p 245 oder 282 a 8 ja 1104,46 1639,1 1403,46 307,42 bis 4370,12 18 nein 668,08 787,16 573,41 113,91 bis 1842,58 in pg VEGF/mg Protein 65 0,085 4 Diskussion Mutationen im p53-Gen werden als spätes Ereignis in der Tumorprogression kolorektaler Karzinome angesehen. Entsprechend der Adenom-Karzinom-Sequenz von Vogelstein et al. [172] treten Mutationen im p53-Gen erst im Übergang von Adenomen zu Karzinomen auf. Dabei scheint die Mutation im p53-Gen über den Verlust der Apoptose und dem Zusammenspiel weiterer Mutationen (k-ras, DCC, APC) einen Auslösermechanismus darzustellen, welcher zu einem malignen Zellwachstum führt. Ein angiogenetischer Phänotyp ist die Grundlage für die nutritive Versorgung und das Wachstumspotenzial von proliferierenden Tumoren. Hierbei kommt dem VEGF als angiogenen Wachstumsfaktor eine bedeutende Rolle bei der Ausbildung eines Gefäßnetzes in vielen Tumoren, inklusive CRC, zu [19,161,162]. Für Onkogene wie die der ras-Familie wurden in invitro Versuchen zum einen ein direkter positiver Einfluss auf die VEGF-Expression zum anderen ein synergistischer Effekt, der die Expression von VEGF über die Hypoxie zusätzlich verstärkt, beobachtet [63]. In in-vitro Studien konnte ein negativer Einfluss des Wild-Typ p53-Proteins auf die Angiogenese nachgewiesen werden. Dieser beruhte auf einer Stimulation der TSP-1-Expression (Thrombospondin-1) und einer dosisabhängigen Herunterregelung der Promotoraktivität des VEGF-Gens [16,30,110]. In Gentransferstudien an den humanen Kolonkrebszellinen SW620 und KM12L4 konnte deutlich gezeigt werden, dass in Karzinomzellen transferiertes Wt-p53 die VEGF-Expression herunterregelt und so zu einer Inhibierung der tumorinduzierten Angiogenese führt [16]. Immunhistologische Untersuchungen an verschiedenen Tumorentitäten zeigten eine Korrelation von erhöhten intrazellulären p53-Proteinkonzentrationen, bedingt durch eine Verlängerung der Halbwertszeit bei mutiertem p53-Genstatus, und erhöhten VEGF-Konzentrationen [15,84,100,161]. Weiterhin wurden molekulare Mechanismen beschrieben, durch die mutiertes p53-Protein über 12-O-tetra-Decanylphorbol-13-Acetat (Phorbolester) und Proteinkinase C die VEGF-Expression direkt steigert [52,89]. In der vorliegenden Arbeit sollte die Frage beantwortet werden, ob bei kolorektalen Tumoren in vivo ein Zusammenhang zwischen dem Mutationsstatus des p53-Gens und der VEGFExpression besteht. 66 4.1 Häufigkeit von p53-Mutationen und Mutationstypen In dieser Arbeit wurde mit Hilfe der TTGE bzw. DGGE bei 26 (50%) der 52 untersuchten kolorektalen Karzinomen eine Mutation im p53-Gen festgestellt. Dieses Ergebnis liegt in der Spannweite der in der Literatur beschriebenen Mutationshäufigkeiten, welche in Abhängigkeit vom Stichprobenumfang und Detektionsverfahren zwischen 29% und 61% liegen [1,27,32,51,79,86,149]. Die Spannbreite der Ergebnisse ist zum einen durch nicht-einheitliche Verfahren zur Detektion der Mutation (RFLP; SSCP; TTGE; CDGE; DGGE) mit unterschiedlicher Sensitivität und zum anderen durch eine unterschiedliche Selektion der Stichproben (untersuchte Exons, Stadien der Karzinome und Tumorentitäten) erklärbar. Die verwendeten Normalgewebeproben wurden aus makroskopisch gesunden Darmarealen entnommen und wiesen in der mikroskopischen Untersuchung (s. Kapitel 2) keine Tumoranteile auf. Bei den hier verwendeten Proben wurden im Normalgewebe in keinem Fall eine Mutation im p53-Gen nachgewiesen. Bei zwei der 52 Patienten konnte in den Normalgewebsproben und in dem korrespondierenden Tumorgewebe ein bekannter Polymorphismus im Codon 213 Exon 6 des p53-Gens nachgewiesen werden [21]. Anzumerken ist jedoch, daß grundsätzlich jedoch trotz negativem Mutationsstatus für p53 die Möglichkeit einer anderen zeitlich früheren Genmutationen, in Anlehnung an die AdenomKarzinom-Sequenz von Vogelstein et al. [172] besteht. Diese können auf Grund der in dieser Arbeit durchgeführten Analyse nicht ausgeschlossen werden. Die Mutationshäufigkeiten verteilten sich auf die einzelnen Dukes-Stadien wie folgt: Stadium A 22,2%, Stadium B 41,2%, Stadium C 64,3% und Stadium D 66,7%. Innerhalb der 26 Tumoren mit mutiertem p53-Gen waren 30,7% der Mutationen im Exon 5, 7,7% im Exon 6, 26,9% im Exon 7 und 38,5% im Exon 8 lokalisiert. Wie oben bereits erwähnt, lag in zwei Proben der Polymorphismus in Codon 213 vor [21]. Eine Probe wies eine Deletion im Bereich der Codons 193-201 (Exon 6) auf. Im Gegensatz zu diesen Ergebnissen zeigte eine andere Untersuchung an 43 kolorektalen Karzinomen, welche die DGGE zur Detektion einsetzte, eine Mutationsfrequenz im p53-Gen von 60,5%. Die Mutationsereignisse waren Punktmutationen, Mikroinsertionen oder Deletionen. Bei den 24 Punktmutationen wurde eine Verteilung von 25% in Exon 5, 16,7% in Exon 6, 25% in Exon 7 und 29,2% in Exon 8 nachgewiesen [64]. In einer weiteren Studie mit einer Fallzahl von 109 Proben und einer Vorselektion der Stichprobe von 59,6% der Tumoren in Dukes B und 40,4% Dukes C Tumoren, konnte bei den 40 im p53-Gen mutierten kolorektalen Karzinomen (37%) eine Verteilung von 41% in Exon 5, 12% in Exon 6, 15% in Exon 7 und 32% in Exon 8 mittels SSCP detektiert werden [32]. Die unterschiedlichen Ergebnisse könnten darin begründet liegen, dass die DukesStadienverteilung im Krankengut auch durch den Zeitpunkt der Diagnosestellung bzw. der 67 Selektion der Operationsgruppe mitbeeinflusst wird. Frühe bzw. kleine Tumore (Dukes A) werden nicht oder selten klinisch detektiert und dadurch einer operativen Sanierung zugeführt. Hingegen sind Tumoren des Stadiums Dukes D z.T. schwer oder nicht mehr nach kurativen Ansätzen zu operieren oder präsentieren ausgedehnte lokoregionale Metastasierungen oder Fernmetastasen. Dadurch wird das Kollektiv der Proben in den Studien beeinflusst: Tumoren der Dukes-Stadien A und D sind daher seltener als z.B. Tumoren der Kategorie Dukes B oder C. Abgesehen von diesen Einflussfaktoren vor der Untersuchung der Proben, zeigen sich bei dem Vergleich der drei Studien in Abhängigkeit der Dukes-Stadienverteilung und der Detektionsmethode Differenzen in der Mutationsrate und Verteilung der Tumoren in den DukesStadien. Diese unterschiedlichen Ergebnisse können als Effekt bekannter Grundlagen gedeutet werden, bei denen sich bei fortschreitender Tumorprogression eine Zunahme der Mutationenereignisse präsentiert. Im speziellen treten p53-Mutationen als zeitliches Ereignis vermehrt im Übergang von Adenomen zu Karzinomen oder in einem noch späteren Stadium der Progression auf. Die hier gefunde Verteilung der Mutationsereignisse auf die einzelnen Exons weicht von der in anderen Studien an anderen Tumorentitäten beobachteten z.T. stark ab. So verteilten sich z.B. die in 17 (16,5%) von 109 untersuchten Mammakarzinomen mittels PCR und CDGE detektierten Mutationen im p53 Gen zu 52,9% auf das Exon 5, zu 41,2% auf das Exon 7 und nur zu 17,7% auf das Exon 8. Das Exon 6 wurde nicht analysiert [166]. In 17 nicht-kleinzelligen-Bronchialkarzinomen bei denen eine Mutation mittels PCR-SSCP nachgewiesen worden war, ergab sich eine Verteilung der Mutationsereignisse von 38,8% im Exon 5, 29,4% in Exon 7 und 11,8% in Exon 8. Auch in dieser Arbeit wurde das Exon 6 nicht untersucht [171]. In der p53-Mutations-Datenbank der "International Agency for Research on Cancer" (IARC) [155] sind Mutationen mit Schwerpunkt im Bereich der Codons der sequenzspezifischen DNABindungsdomäne zwischen Codon 100 und 300 aufgelistet (s. www-p53.iarc.fr). Unter den somatische Mutationen weisen die Codons 175, 213, 220, 245, 248, 249, 273 und 282 eine deutliche Akkumulation der Mutationsereignisse auf. Diese Codons liegen in den evolutionär hoch konservierten Regionen (s. Abbildung 2). Diese Aussage trifft für die untersuchte Tumorentität kolorektales Karzinom zu. Inwiefern Mutationen im p53-Gen auch bei anderen Tumorentitäten auf wenige Exons, bzw. auch auf die evolutionär hoch konservierten Regionen beschränkt sind ist fraglich. Nigro et al. [113] beschrieben 1989 lediglich, dass in verschiedenen Tumorentitäten überhaupt p53-Mutationen auftreten. Hingegen postulierten Hollstein et al. 1991, dass mehr als 80% der p53 Mutationen in den Exon 5, 7 und 8 lokalisiert sind, aber auch in anderen Bereichen des Gens Mutationen auftreten würden, dann bevorzugt im Exon 6 [74]. 68 Unter Berücksichtigung potenzieller Einflussgrößen, wie die Sensitivität der Analysemethode oder der Stichprobenauswahl besteht möglicherweise bei unterschiedlichen Tumorentitäten ein spezielles, vielleicht sogar charakteristisches Verteilungsmuster der Mutationen in dem DNAAbschnitt der Exon 5 bis 8 (AS 100-300) im p53-Gen. So konnten bei Lungentumoren (NSCLC) [171] einen Fokussierung auf Exon 5, bei oralen Plattenepithelkarzinomen [105] auf Exon 5 und 8, bei Plattenepithelkarzinomen des Kopf/Hals-Bereiches [132] auf Exon 5 und bei Mammakarzinomen [166] auf Exon 7 und 8 festgestellt werden. In der in dieser Arbeit untersuchten Stichprobe wurden Punktmutationen in den Exons 5-8 in 13 verschiedenen Codons detektiert. In einem Fall kam es im Bereich der Codons 193-201 (Exon 6) zu einer Deletion. Veränderungen innerhalb der ‚hot spot’-Codons (s. Kapitel 1.2.3) zeigten sich mit 15,4% in Codon 135 (Cys Phe), 248 (3 Proben mit Arg Glu, 1 Probe mit Arg Trp) und 282 (Arg Trp). Weiterhin traten 11,5% der Mutationen im Codon 273 (2 Proben mit Arg His und 1 Probe mit Arg Cys), und den Codons 175 (Arg His) und 245 (Gly Ser und Gly Asp) mit 7,7% auf. Bei 75,86% der Punktmutationen handelte es sich um Transitionen. Der Schwerpunkt der Nukleotidveränderung lag bei der Transition von GC AT mit 37,93%, hingegen kamen die Nukleotidveränderungen CG TA mit 20,69% und AT GC mit 13,79% weniger häufig vor (s. auch Tabelle XXII/XXIII). Acht der 11 Transitionen (72,73%) GC zu AT und alle 6 Transitionen von CG nach TA lagen im Bereich von CpG Dinukleotiden (s. Tabelle XXIII). Transversionen, die zu einer Änderung des Nukleotidpaares GC zu TA führten wurden in 6 Fällen nachgewiesen. Es wurde keine G C Substitution gefunden. Costa et al. [27] untersuchten die Exon 5-9 des p53-Gen in kolorektalen Karzinomen mittels SSCP. Dabei zeigte sich eine Gesamtmutationsrate von 60,5% (22 von 38 Tumoren). Unter den Transitionen (87% der Ereignisse) trat mit 65% am häufigsten eine GC AT Transition (an CpG Dinukleotiden) auf. Weitere Ereignisse (an nicht CpG-Dinukleotiden) waren Veränderungen von GC AT (18%), GC TA (13%) und AT GC (4%). Insgesamt wurden 53% der Mutationen in den drei ‚hot spot’-Codons 175, 248, 282 gefunden. Im Vergleich mit der Studie von Costa et al. [27] war in der vorliegenden Untersuchung GC TA mit 20,69% das zweit häufigste Ereignis, im Gegensatz zeigte sich jedoch die Transition von AT GC mit 13,79% (4% bei Costa et al. [27]). Weiterhin fanden sich in dieser Arbeit nur 38,5% der Mutationen in den angegeben ‚hot spot’-Codons. Interessanterweise lagen bei Costa et al. die Mutationsereignisse in den ‚hot spot’-Codons um 14,5% höher. Hollstein et al. [74] äußerten schon 1991 unter Einbezug mehrerer Studienergebnisse [lt. 8,10,113,135], dass das Mutationsspektrum zwischen einzelnen Malignomentitäten differiert. In Kolonkarzinomen, neben Tumoren des ZNS (Zentralnervensystem) und Lymphomen ist die Transition an CpG 69 Dinukleotiden von GC AT die häufigste Substitution. Diese Transition trat in Kolontumoren mit 79% am häufigsten auf. Als zweithäufigste Transition kam AT GC mit 15% vor [74]. Ein Erklärungsansatz für kolorektale Karzinome könnte sein, dass die Transitionen von GC AT durch Nahrungsmittel- bzw. Konservierungmittelanteile wie z.B. der Nitrosaminsäure ausgelöst werden. Sehr lange schon wird postuliert, dass dieses Agens, welches in Nahrungsmitteln vorkommt, eine karzinogene Substanz darstellt. Weiterhin sind CpG Dinukleotide häufig Mutationen im Sinne einer Transition unterworfen, denn Cytosin ist häufig methyliert. Dieses führt zu hoher Mutagenität über eine Desaminierung des Cytosin zum Thymidin [22]. In dieser Arbeit wurde keine G C (GC/CG) Substitution gefunden, so dass sich die Frage stellt, ob die Mutationsanalyse mittels TTGE zur Detektion solcher Substitutionen sensitiv genug ist. In der Literatur wird für kolorektale Tumoren das Auftreten von G C Mutationen mit 3% angegeben. Für solide Tumore allgemein liegt die Häufigkeit solcher Veränderungen bei 9% [74]. Tatsächlich können G C (GC/CG) Substitutionen in der TTGE übersehen werden, wenn keine Heteroduplexbildung stattgefunden hat, da der Laufunterschied der beiden Homoduplizes sehr gering ist [36]. Dies wäre eine mögliche Fehlerquelle, es ist aber davon auszugehen, dass bei jeder untersuchten Probe mit vorliegender Mutation durch die vorhergehende PCR eine Heteroduplexbildung stattgefunden hat. Die Verteilung der Mutationsereignisse auf die verschiedenen Codons deckten sich zum Teil mit Ergebnissen anderer Studien, welche ebenfalls kolorektale Karzinome untersuchten [27,74]. Da jedoch die Fallzahl, die Analysemethode der Mutationen in den Exons und die weitere Aufbereitung differieren, ist ein direkter Vergleich wohl kaum zulässig. Bei einer Studie an 33 Plattenepithelkarzinomen konnten 12 Tumore mit einer Mutation im p53Gen detektiert werden (36,4%). Bei der Analyse der einzelnen Codons konnte, entgegen dieser Arbeit, keine Akkumulation von Mutationsereignissen in den Codons festgestellt werden, weiterhin gab es keine deutlich hervorgehobenen Mutationsereignisse wie Deletion, Transition, oder Insertion [132]. In einer Untersuchung an NSCLC-Karzinomen von Ambs et al. [5], wurde bei 14 von 27 Tumoren (51,9%) eine Mutation im p53-Gen nachgewiesen. Das häufigste Mutationsereignis war mit 28,6% eine Transversion von G T (GC/TA) und mit der selben Häufigkeit die Transition von C T (CG/TA). Am zweithäufigsten war der Austausch von G A (GC/AT) mit 21,4%. Ähnliche Ergebnisse ergaben sich auch durch Auswertung verschiedener Einzelstudien [22]. Dieses sind weitere Ergebnisse welche die Hypothese verstärken, dass in Abhängigkeit der zugrunde liegenden Noxe bzw. dem Mutationsmechanismus, dem originären Gewebe der Neoplasie und der Tumorentität, jeweils ein anderes Mutationsmuster sowohl im Bereich der Exons als auch des Basenaustausches zu Grunde liegt [22,74,116]. Analysen der Datenbank der IARC beschreiben diese „Mutationsidentität“ der einzelnen Karzinomentitäten. Die Betreiber der Datenbank demonstrieren in Analysen 70 einen Zusammenhang zwischen dem Ursprungsorgan des Tumors, den histologischen Kriterien und dem Tumorstadium gegenüber charakteristischen Mutationstypen in Bezug auf die Basenabfolge und der Lokalisation der Mutation im Genom [155]. 4.2 VEGF-Konzentration im Tumor- und Normalgewebe In dieser Arbeit lag die VEGF-Konzentration im Normalgewebe im Median bei 82,9 und im Mittelwert (MW) bei 72,4pg VEGF/mg Protein. Im Tumorgewebe wurden weitaus höhere Werte von 887,4 (Median) und 1138,6pg VEGF/mg Protein (MW) festgestellt. Die mediane VEGFKonzentration war somit im Tumorgewebe um den Faktor 15,7 gegenüber dem Normalgewebe erhöht. In einer Studie von Broll et al., in der 38 kolorektale Karzinome untersucht worden waren, lag die VEGF-Konzentration im Tumor im Mittelwert bei 984pg VEGF/mg Protein und im Normalgewebe bei 89pg VEGF/mg Protein [19]. Diese Daten decken sich mit denen der vorliegenden Arbeit. Zudem zeigte sich in beiden Untersuchungen eine große Spannbreite der VEGF-Konzentration sowohl im Normalgewebe als auch im Tumorgewebe. Das gesamte Bearbeitungsprotokoll der Proben war in beiden Studien identisch, und zur Bestimmung der VEGF165-Konzentration wurde der gleiche ELISA-Kit verwendet. Auch Linderholm et al. demonstrierten in einer Studie aus dem Jahr 2001, in der die VEGF165Konzentration in 224 primären Mammakarzinomen mit Hilfe des Quantikine-ELISA-Kits ermittelt worden war, eine große Spannbreite der VEGF-Konzentration in den Tumoren von 7,5 – 9084pg/mg, wobei der Median bei 256,4pg VEGF/mg lag [100]. Damit lag die mediane VEGFKonzentration in den Mammakarzinomen nur bei etwa 29% des in unserer Studie für kolorektale Karzinome ermittelten Wertes. Diese Diskrepanz kann möglicherweise in der Wahl zweier verschiedener Tumorentitäten begründet sein. Untersuchungen an ZNS-Tumoren zeigten einen deutlichen Unterschied zwischen dem hoch malignen Glioblastom mit im MW 6281,6pg VEGF/mg Protein zu den niedrig malignen Tumoren wie dem anaplastischem Astrozytom mit MW 493,6pg VEGF/mg Protein, dem low grade Astrozytom mit 600,4pg VEGF/mg Protein (MW) bzw. zum ZNS-Normalgewebe mit 25,8pg VEGF/mg Protein (MW) [163]. In der genannten Studie wurde die Konzentration des VEGF121 unter Verwendung eines VEGF-ELISAs gemessen. Ein Expressionsunterschied von VEGF mit deutlich erhöhten Werten im Tumor- gegenüber dem Normalgewebe wurde auch als Ergebnis anderer Studien, welche jedoch die VEGF-Expression immunhistochemisch oder auf mRNA-Ebene durch in situ Hybridisierung untersuchten, beschrieben [20,85,97,163]. Dieser Befund unterstützt die These, dass VEGF in proliferierenden Tumoren verstärkt exprimiert wird und so die Neovaskularisation eines proliferierenden Malignoms fördert. Gewebehypoxie und 71 Hypoglykämie, wie sie bei proliferierendem Gewebe aber insuffizienter Vaskularisation vorkommen, stellen einen starken Reiz zur vermehrten VEGF-Expression dar [20,121,140,152,153]. Diese Einflussgrößen, welche die VEGF-Konzentration im Gewebe erhöhen, haben eine direkte Auswirkung auf jede Studie die sich mit expansiv wachsenden Tumoren beschäftigt. Die angeführten Ergebnisse lassen vermuten, dass die VEGF-Expression in Tumoren, welche in oder aus einem Gewebe mit hoher Vaskularisationsdichte und hoher Grundexpression von VEGF entstehen, à priori eine deutlich höhere Konzentration von VEGF im Tumor aufweisen. Denkbar ist auch der Zusammenhang zwischen hoher Malignität und daraus resultierend schneller Wachstumstendenz unter gleichzeitig schlechtem Gewebe-Perfusionsverhältnis. Die unterschiedliche Größe der untersuchten Tumore, mit unterschiedlichen Anteilen an nekrotischem und hypoxischem Zentrum sowie unterschiedlich gut vaskularisierter Peripherie werden die Ergebnisse der VEGF-Konzentration stark beeinflussen. Diese verschiedenen Gewebebereiche können VEGF in unterschiedlichem Ausmaß exprimieren, je nach Ausmaß der Hypoxie (hohe Expression in hypoxischen, niedrige Expression in gut vaskularisierten Bereichen) und der Vitalität der Zellen (niedrige Expression in nekrotischem Zentrum). Je nachdem, aus welchem Bereich des Tumors die jeweiligen Gewebeproben entnommen sind, können sich für ein und denselben Tumor somit unterschiedliche VEGF-Konzentrationen ergeben. Gleichfalls kann die mittlere VEGF-Konzentration einer Stichprobe durch Überwiegen des einen oder anderen Tumorbereichs beeinflusst sein. 4.3 VEGF Konzentration bei Tumoren mit und ohne Mutation im p53-Gen Verschiedene Studien geben Anlass zu der Hypothese, dass p53 einen direkten Einfluss auf die VEGF-Expression hat. So beschreibt Fontanini et al. [48], dass die Expression von Bcl-2 und p53-Proteinen die Regulation der VEGF Expression in NSCLC gegensätzlich kontrollieren und somit einen Einfluss auf die Entwicklung der Tumorangiogenese nehmen. Bouvet et al. [16] beschrieben in vitro an Kolonkarzinomzellinien einen negativen Effekt der Expression von VEGF durch ein durch Adenoviren verändertes Wt-p53-Protein. Dieser Effekt wirkt sich im Verlauf inhibitorisch auf die Angiogenese bzw. Neoangiogenese aus. In der Vergangenheit beschäftigte sich keine Untersuchung mit dem Einfluss von Mutationen in den ‚hot spot’-Codons des p53-Gens auf die VEGF-Proteinkonzentration (in pg/mg), bzw. mit der Frage nach Veränderung der VEGF-Expression bei Mutationen in den Codons welche für die Tertiärstruktur bzw. die DNA-Bindungsregion des p53-Protein kodieren. Ergebnisse verschiedener Studien an Plattenepithelkarzinomen des Kopf/Hals-Bereiches [132], an Magenkarzinomen [104,138] und an nicht-kleinzelligen Bronchialkarzinomen [49] stellten eine signifikante Korrelation zwischen der p53-Proteinkonzentration und der VEGF-Expression in Tumoren fest. Die zitierten Studien 72 untersuchten die VEGF-Expression mit immunhistologischen Verfahren. Eine Studie [132] verwendete zur Analyse des p53-Status eine PCR-SSCP. Hingegen wurden in den anderen Studien auch für den p53-Status eines Tumors lediglich immunhistologische Verfahren verwendet, welche eine erhöhte p53- Proteinakkumulation nachweisen können. Diese Methode der Konzentrationsbestimmung von p53 kann jedoch nicht eine physiologische Überexpression von einer Konzentrationserhöhung auf Grund einer Mutation im p53-Gen diskriminieren, da einer erhöhten p53-Proteinkonztration in einer Zelle zwei Mechanismen zugrunde liegen können, zum einen eine verlängerte HWZ, zum anderen eine vermehrte Expression. Erhöhte Konzentrationen des p53-Proteins ohne Mutation im Gen treten z.B. auch bei Strangbrüchen der dsDNA durch - oder UV-Strahlung auf. Ein weiterer Stimulator sowohl für die p53-Expression und Aktivierung als auch der VEGFExpression ist eine zelluläre Hypoxie [99,152]. Diese Grundlagen lassen daran zweifeln ob der Versuchsaufbau der o.g. Studien überhaupt eine Aussage bezüglich der Interaktion von p53 mit VEGF zulässt. Zudem unterliegen beide Proteine getrennt einer Regulation durch den hypoxischen zellulären Stress, welcher ein häufiger Zustand in expansiv wachsenden Tumoren ist [99,153]. Ähnliche Studienschwachpunkte gelten auch für Untersuchungen an kolorektalen Karzinomen durch Kang et al. (163 Patientenproben) und Takahashi et al. (93 Patientenproben). Beide Untersuchungen demonstrierten eine signifikante Korrelation zwischen immunhistochemisch detektierter erhöhter VEGF-Konzentration und einer p53- Proteinüberexpression [84,161]. Ergebnisse an weiteren Tumorentitäten aus anderen Untersuchungen deuten auf eine erhöhte VEGF-Expression in Verbindung mit einer Mutation im p53-Gen hin, wobei in den Studien z.T. eine erhöhte p53-Proteinkonzentration mit einer Mutation gleichgesetzt wird [49,104,132,138,161]. In dieser Arbeit konnte das Postulat der vorgängig zitierten Studien nicht für die untersuchte Stichprobe der 52 kolorektalen Karzinome in vivo bestätigt werden. So bestand keine signifikante Korrelation zwischen der VEGF-Konzentration und einer p53-Gen-Mutation im Tumorgewebe. Auch die Unterteilung der Tumore in Subgruppen nach Geschlecht der Patienten, Lokalisation des Tumors oder Stadien nach Dukes zeigte hinsichtlich der VEGFExpression keinen signifikanten Unterschied zwischen Tumoren mit und ohne Mutation im p53Gen. Ebenfalls wurde kein signifikanter Unterschied in der VEGF-Konzentration festgestellt bei der Einteilung der Tumoren nach Mutationen in den einzelnen Exons und Tumoren, welche ausserhalb dieses untersuchten Exons p53-Mutationen aufwiesen. Die Tumore mit einer Mutation in Exon 7 wiesen von allen untersuchten Exons die niedrigste VEGF-Konzentration auf. Es konnte in dieser Arbeit ein Trend in der VEGF-Konzentration im Tumorgewebe zwischen Patienten mit Mutation im Exon 7 im Vergleich zu allen Tumorproben mit einem p-Wert von 0,071 durch den Mann-Whitney-U-Test ermittelt werden (s. Tabelle XXVII). In Exon 7 sind die Codons 245 und 248, welche für die Tertiärstruktur und für die DNA- 73 Bindungsregion des p53-Proteins kodieren, enthalten. Da die Stichprobe bzw. die Untergruppen zu den einzelnen Exons zu klein ist, kann kein eindeutiger Zusammenhang zwischen der Mutation und möglichen Regulationsmechanismen postuliert werden. In der Literatur wurde für Patienten mit kolorektalen Karzinomen mit Mutation im Exon 7 eine kürzere postoperative Überlebenszeit beschrieben [79]. In der zitierten Studie kumulieren die Mutationen im Exon 7 in den Dukes Stadien C und D, bzw. Patienten mit einer Mutation im Exon 7 zeigten eine verkürzte Überlebenszeit. Es bleibt jedoch unklar ob die Mutation im Exon 7 eine rasche Tumorprogredienz verursacht und so die Anzahl an Dukes C und D Tumoren erhöht ist oder ob unabhängig von der Mutation die postoperative Patientenüberlebenszeit allein durch ungünstige Ausgangssituation eines Dukes C bzw. D Tumors bestimmt wird. In einer Untersuchung an NSCLC zeigte sich, dass auch Mutationen in Exon 5 einen prognostisch ungünstigen Faktor in Bezug auf die Überlebenszeit darstellten [171]. Vergleicht man diese Studien mit den Ergebnissen der vorliegenden Arbeit, konnte ebenfalls eine Fokussierung der Mutationen in Exon 7 auf die Stadien Dukes C und D festgestellt werden. Die Beobachtung tumorspezifischer Mutationen mit weiterführenden Effekten könnte mit dem in Kapitel 4.1 schon genannten entitätsspezifischem Mutationsmuster erklärt werden und als weites tumorspezifisches Charakteristikum gedeutet werden. Kang et al. [84] und Kern et al. [86] beschreiben eine prognostisch ungünstige Konstellation bei hohen p53-Proteinkonzentrationen bzw. Mutation im p53-Gen mit gleichzeitig erhöhten VEGFKonzentrationen im Tumor. Möglicherweise können diese Ergebnisse für kolorektale Karzinome als prognostisch ungünstige Konstellation zwischen der Mutation im p53-Gen mit konsekutiv erhöhten p53-Proteinkonzentrationen und gesteigerter VEGF-Expression oder aber spezielle Mutationen so z.B. im Exon 7 gedeutet werden. Beide Konstellationen zeigen eine schlechtere Überlebensrate der Patienten insbesondere, da für die erste eine verstärkte metastatische Aussaat beschrieben wird [84,86]. Über welchen Mechanismus eine Mutation im Exon 7 zu einer schlechteren Prognose der Patienten führt, sollte in weiteren Studien untersucht werden. Ebenfalls warum gerade bei Mutationen in Exon 7 die VEGF-Konzentration am geringsten ist. Erklärungsansätze bei denen man von einem einfachen Zusammenhang zwischen vermehrter Metastasierung über die Neoangiogenese in Kombination mit Mutation im Exon 7 ausgeht, lassen sich nicht durch die Ergebnisse dieser Arbeit unterstützen. Da in dieser Arbeit durch die Gruppenbildung die Stichprobengröße der einzelnen Kriterien noch weiter verringert wurde, ist die Fallzahl zu gering, um eine eindeutige Aussage zu erhalten. Bei der Analyse der Mutationen in den für die DNA-Bindungsregion bzw. die Tertiärstruktur des Proteins kodierenden Codons, zeigte sich kein signifikanter Unterschied der VEGFKonzentration im Vergleich zu den Tumoren mit Mutation an anderer Position im p53-Gen bzw. im Vergleich zu allen anderen Tumoren mit und ohne Mutation in den untersuchten Exons des 74 p53-Gens. Allerdings besteht auch hier in den Ergebnissen ein Trend zwischen der VEGFKonzentration von Tumoren mit Mutation in den Codons welche für die Tertiärstruktur kodieren gegenüber den anderen Tumoren mit p53-Mutation. Der p-Wert lag bei dieser Stichprobe bei 0,085. Auch hier ist möglicherweise die Fallzahl mit einer Gesamtstichprobe von 26 Tumoren mit p53-Mutation und nur 8 Tumoren mit Mutation in dem betreffenden Codon zu gering, um ein eindeutig signifikantes Ergebnis zu erhalten. In Anbetracht dieser Ergebnisse wäre eine Wirkung des veränderten Proteins auf die Regulationsmechanismen der VEGF-Expression denkbar. Da die Mutation im p53-Gen in kolorektalen Karzinomen ein spätes Ereignis in der Tumorprogression darstellt, könnten andere, zeitlich frühere Mutationen in Onkogenen oder anderen Tumorsuppressorgenen an der Regulation der VEGF-Expression beteiligt sein. Möglich wären eigenständige, direkte oder synergistische Wirkungen mit mutiertem p53-Protein auf die VEGF-Regulation. Für kolorektale Karzinomen wurde z.B. in vitro [7] und für Lungentumoren in vivo [94] eine signifikante Korrelation zwischen einer Mutation im k-ras Gen und der VEGF-Expression beschrieben. Weiter wurde an verschiedenen Zellreihen eine Korrelation zwischen der Überexpression von v-Src und VEGF gefunden [110]. Die Einflussgröße anderer Mutationen konnten in dieser Arbeit nicht berücksichtigt werden. Neben dem Mutationsstatus in Onkogenen und Tumorsuppressorgenen nehmen auch der Oxygenierungs- und CO2-Retentionszustand einen signifikanten Einfluss auf die VEGF Konzentration. Weiterhin können aus dem Serum stammende oder parakrine Wachstumsfaktoren und Zytokine wie z.B. „keratinocyte growth factor“ (fibroblast growth factor7), „epidermal growth factor, „tumor necrosis factor“, „transforming growth factor-1“, und Interleukin-1, auf die VEGF-Expression stimulierend wirken (s. Einleitung 1.2). Alle diese Faktoren zeigten auch einen Einfluss auf die VEGF-Konzentration in Zellkulturen und Tumorgewebe [118,165]. Diese einzelnen Faktoren konnten bei der Untersuchung einer Korrelation zwischen dem p53-Mutationsstatus und der VEGF-Konzentration in kolorektalen Tumoren nicht berücksichtigt werden. 4.4 Schlussfolgerungen Die in dieser Untersuchung gefundenen Daten lassen keinen signifikanten Effekt der p53Mutation in kolorektalen Karzinomen auf die Expression von VEGF erkennen. Möglicherweise liegt der beschriebenen Assoziation zwischen Überexpression von p53-Protein und VEGFKonzentration in kolorektalen Karzinomen ein p53-unabhängiger Mechanismus zugrunde. Vorstellbar ist, dass das reziproke Expressionsverhalten zwischen Wt-p53-Protein und VEGFProtein in Tumoren durch mehrere koexistente zelluläre Regelkreise beeinflusst wird. Da Hypoxie die Expression von Wt-p53 verstärkt [99], werden Zellen sensibilisiert für die 75 hypoxieinduzierte Apoptose. Durch die Mutationen, welche die Funktion des p53-Protein alterieren, fällt der Mechanismus der hypoxieinduzierten Apoptose aus. Dies kann die Grundlage für einen hypoxieresistenten Zelltyp im proliferierenden Zellverband, der im größeren Masse VEGF exprimiert darstellen [99]. Die p53-induzierte Apoptose wird über die positive transkriptionale Regulation des Bax- und IGF-BP3-Gens vermittelt. BAX beschleunigt die Apoptose, in dem es mit Bcl2 Heterodimere bildet und dadurch die Bcl2-Funktion inhibiert. Bcl2Überexpression hemmt die p53-vermittelte Apoptose. Bax bindet an Bcl2 und ist Antagonist dieses negativen Einflusses auf die Apoptose [24,95,98,126]. Zudem wurde für VEGF ebenfalls ein Mechanismus beschrieben, in dem über Bcl2 ein antiapoptotischer Effekt ausgelöst wird [119]. Diese Ergebnisse lassen auf eine Effektschleife schliessen, die Fontanini et al. 1998 beschrieb, bei der bcl2 und p53 möglicherweise die Tumorangiogenese in NSCLC über VEGF regulieren [48]. Dies würde zur weiteren Induktion der Neoangiogenese führen. Mutationen im p53-Gen beeinflussen negativ die antiangiogenetische Wirkung von Thrombospondin-1 [30,31]. Es gibt Hinweise, das eine Verbidung bzw. Wirkung zwischen Mutp53 und der Ras/MAPK Endstrecke über das hHSP70 (humanes Hitzeschockprotein 70) besteht, was zu einer Induktion der VEGF Expression in humanen kolorektalen Karzinomen führt [34,106]. Für zeitlich frühere Mutationen wie z.B. der Mutation in der ras-Familie wurden direkte positive Regelmechanismen der VEGF-Expression beschrieben. Fasst man die Ergebnisse aus der Literatur zusammen unter Berücksichtigung der negativen Korrelation zwischen dem p53-Mutationsstatus und der VEGF-Konzentration aus dieser Arbeit, so könnte die Assoziation zwischen der erhöhten VEGF-Konzentrationen und der p53- Proteinkonzentration als Ergebnis einer vermehrten Hypoxie in Tumorzellen gesehen werden. Möglich sind auch komplexere Regelmechanismen des p53 auf Wachstumsfaktoren inklusive dem VEGF über einen noch nicht identifizierten zellulären Regelmechanismus. Dies scheint möglich, da p53 oder seine Liganden an einer grossen Zahl von molekularen Regelkreisen teilnehmen [99,126,147]. In der Zukunft könnten Studien zu diesem Thema mit Analyse der VEGF- sowie der Thrombospondin-Expression in Korrelation zum p53-Mutationsstatus in den ‚hot spot’-Regionen des p53-Gens an einer ausreichend grossen Stichprobe zum Verständnis der Neoangiogenese in kolorektalen Karzinomen führen. Dieses würde zum weiteren Verständnis des komplexen zellulären Zusammenspiels von positiven und negativen Faktoren der Tumorprogression beitragen. Mit dem Verständnis der molekularen Mechanismen der Angiogenese bei kolorektalen Karzinomen bietet sich die Möglichkeit das gewonnene Wissen in den klinischen Alltag bei der Behandlung von Karzinompatienten weiter einfliessen zu lassen. Aus dem Grundlagenwissen ergeben sich neue und spezifischere Strategien zur Therapie von u.a. malignen Neoplasien. VEGF war in der Vergangenheit Ziel verschiedener präklinischer Therapieansätze, um über die 76 Angiogenese auf die Proliferation von Tumoren einzuwirken. Zum Einsatz kamen Antikörper gegen VEGF [88,125] und VEGFR-2 [144,159] sowie Hemmung der Tyrosinkinaseaktivität von VEGFR-2 [141,144]. Die Effekte dieser Therapieansätze wirkten sich auf die Tumorexpansion sowie auf die Reduktion der Tumormasse, vermittelt über eine Reduktion der Vaskularisation des Tumors, mit verlängertem Überleben der Versuchstiere aus. Derzeit steht mit Avastin eine für den Menschen zugelassene Substanz zur Verfügung (s. auch Kapitel 1.1.3). Auf dem Gebiet des expansiven Tumorwachstums, vermittelt und unterstützt durch die Angiogenese durch eine erhöhte VEGF-Expression bedarf es weiterer Forschungen, um das komplexe Zusammenspiel von Wachstumsfaktoren und genetischen Defekten zu klären. Besonders die vielfältigen Interaktionen und Wirkungen des p53-Gens und dessen Genproduktes, sowie die Steuerung von Wachstumsfaktoren speziell die des VEGF und dessen Expression wird in der Zukunft weiter zu analysieren sein. 5 Zusammenfassung Mutationen im p53-Gen spielen eine wichtige Rolle bei der Progression von humanen kolorektalen Karzinomen. Das Wachstum eines soliden Karzinoms ist allerdings nicht nur durch den Verlust der Suppressorgenfunktion und anderen onkogenen Mutationen bestimmt, sondern auch von der Fähigkeit, eine ausreichende Vaskularisation des Malignoms zu induzieren und ein nutritives Gefäßnetz auszubilden. VEGF kommt als parakrinem Wachstumsfaktor, der selektiv auf Endothelzellen wirkt, eine entscheidende Rolle in diesem Prozess zu. Der Neoangiogenesefaktor wird in kolorektalen Karzinomen wie auch in anderen Karzinomen verstärkt exprimiert. Es zeigte sich in vitro ein Zusammenhang zwischen der Wildtyp p53 Funktion als Suppressor der VEGF-Expression in humanen kolorektalen Tumorzellinien. In vivo wurde durch immunhistologische Verfahren ein Zusammenhang zwischen erhöhter VEGF-Konzentration und p53-Protein Überexpression in kolorektalen Karzinomen postuliert, und die Vermutung über einen direkten Einfluss von mutiertem p53 auf die VEGF-Expression geäussert. Mit dieser Studie an kolorektalen Karzinomen sollten folgende Fragen beantwortet werden: 77 1) Besteht ein Unterschied in der VEGF-Konzentration zwischen Tumorgewebe mit bzw. ohne Mutation im p53-Gen? 2) Beeinflussen unterschiedliche Mutationstypen bzw. Mutationen in bestimmten ‚hot spot’Exons die VEGF-Konzentration? 3) Beeinflussen Mutationstypen bzw. Mutationen in der kodierenden Sequenz a) für die DNA-bindende Domäne oder b) in der für die Tertiärstruktur des p53-Proteins die VEGF-Konzentration? 4) Besteht ein Unterschied in der VEGF-Expression zwischen Tumoren mit bzw. ohne Mutation im p53-Gen bei zusätzlicher Unterscheidung nach klinischen sowie pathologischen Daten wie Geschlecht, Tumorlokalisation (Kolon/Rektum), Tumorstadium (Dukes A-D) und T-Kategorie der TNM-Klassifikation als Kriterium der Invasionstiefe des Tumors? Um diese Frage zu beantworten, wurden Gewebeproben von 52 Patienten (w 21/ m 31) mit kolorektalen Adenokarzinomen (Stadium A: n=9, B: n=17, C: n= 14, D: n=12) analysiert. Bei jedem Patienten wurden dabei sowohl im Tumor als auch im gesunden Gewebe mittels ELISA die VEGF-Konzentration in pg VEGF/mg Protein bestimmt sowie mit Hilfe von PCR und Temporal Temperature Gradient Electrophoresis (TTGE), bzw. Denaturing Gradient Gel Electrophoresis (DGGE) nach einer Mutation im p53-Gen gesucht und, soweit vorhanden, durch Sequenzierung charakterisiert. In 26 von 52 (50%) Tumorproben wurde eine Mutation im p53-Gen identifiziert. Die in dieser Arbeit vorliegenden Ergebnisse zeigen, dass die VEGF-Konzentration bei kolorektalen Karzinomen im Tumorgewebe gegenüber dem gesunden kolorektalen Gewebe signifikant erhöht ist (p<0.001, VEGF-Median: Tumor 887,43, Normalgewebe 82,87 in pg VEGF/mg Protein). Es bestand keine signifikante Korrelation zwischen der VEGF-Konzentration im Tumorgewebe und dem Mutationsstatus im p53-Gen Exon 5-8 (p>0,05). Die VEGF-Konzentration im Tumorgewebe bei Tumoren mit Mutation im p53-Gen unterschied sich nicht signifikant von der bei Tumoren ohne eine solche Mutation. Ebenfalls fand sich bei Untersuchung der Stichprobe nach Untergruppen (z.B. Geschlecht, Lokalisation des Tumors, Lokalisation der Mutation in Exon 5-8 des p53-Gen, in Codon 248 und 273 kodierend für die DNA-Bindungsregion bzw. Codon 175, 245 und 282, mit Einfluss auf die Tertiärstruktur des Genproduktes, Stadium des Tumors nach Dukes und T-Kategorie der TNMKlassifikation) kein signifikanter Unterschied in der VEGF-Konzentration zwischen Tumoren mit und solchen ohne Mutation im p53-Gen (p>0,05). 78 Die Neoangiogenese als massgeblicher Faktor des Größenwachstums und der Tumorprogression in kolorektalen Karzinomen wird weiter zu analysieren sein, um nicht nur die zugrundeliegenden molekularbiologischen Mechanismen zu verstehen, sondern auch um das therapeutisches Potenzial einer spezifischen Tumortherapie zu vergrößern. Weiter scheint es sinnvoll das Mutationsmuster in kolorektalen Tumoren und damit den Einfluss auf die Tumorprogression, die Wachstumstendenz und wachstumsfördernden Faktoren zu untersuchen. Dieses kann Hinweise für die Prognose der Patienten geben. Im klinischen Alltag könnten Antikörper gegen Produkte der Onkogene und Tumorsuppressorgene, die das Mutationsmuster charakterisieren als Tumormarker oder Screeningmethode eingesetzt werden. Auch ist eine Gentherapie mittels Vektoren zur Regulation wichtiger Schlüsselgene der Tumorprogression eine hoffnungsvolle Strategie Karzinomleiden. 79 zur Bekämpfung von humanen 6 Literaturverzeichnis 1 Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR Clues to the pathogenesis of familial colorectal cancer Science 1993; 260: 812-16 2 Abrahamson JL, Lee JM, Bernstein A Regulation of p53-mediated apoptosis and cell cycle arrest by steel factor Mol and Cell Biol 1995; 15:6953-60 3 Agani F, Kirsch DG, Friedman SL, Kastan MB, Semenze G p53 does not repress hypoxia-induced transcription of the vascular endothelial growth factor gene Cancer Res 1997; 57: 4474-77 4 Ahmed A, Elvin P, Kerr DJ, Wakelam MJO Vascular endothelial growth factor stimulates protein kinase C-dependent phospholipase d activity in endothelial cells Lab Invest 1996; 75: 427-37 5 Ambs S, Bennet WP, Merrriam WG, Ogunfusika MO, Oser SM, Khan MA, Jones RT, Harris CC Vascular endothelial growth factor and nitric oxide synthase expression in humanen lung cancer and the relation to p53 Br J Cancer 1998; 78: 233-9 6 Anderson TI, Börrisson AL Alterations of the TP53 gene as a potential prognostic marker in breast cancer Diagn Mol Pathol 1995; 4: 203-11 7 Arends JW Molecular interactions in the Vogelstein model of colorectal carcinoma J Pathol 2000; 190: 412-16 8 Baker SJ Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas Science 1989; 244: 217 9 Baker SJ, Markowitz S Suppression of human colorectal carcinoma cell growth by wild-type p53 Science 1990; 249: 912-15 10 Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, Hamilton S, Vogelstein B p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis Cancer Res 1990; 50: 7717-22 11 Barak Y, Juven T, Haffner R, Oren M mdm2 expression is induced by wild type p53 activity EMBO J 1993; 12: 461-68 12 Bernard P, Gabant P, Bahassi EM, Coutrier M Positive selection vectors using the F plasmid ccdB killer gene Gene 1994; 148: 71-74 80 13 Bio-Rad Handbuch The Dcode Universal Mutation Detection System 1994 14 Borresen AL, Hovig E Constant denaturant gel elctrophoresis as a rapid screening technique for p53 mutations Proc Nat Acad Sci 1991; 88: 405-09 15 Bosari S, Viale G, Roncalli M, Graziani D, Borsani G, Lee AK, Coggi G p53 gene mutations, p53 protein accumulation and compartmentalization in colorectal adenocarcinoma Am J Pathol 1995; 147: 790-8 16 Bouvet M, Ellis LM, Nishizaki M, Fujiwara T, Liu W, Bucana CD, Fang B, Lee JJ, Roth JA Adenovirus-mediated wild-type p53 gene transfer down-regulates vascular endothelial growth factor expression and inhibits angiogenesis in human colon cancer Cancer Res 1998; 58: 2288-92 17 Bradford MM A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding Anal Biochem 1976, 72: 248-54 18 Brock TA, Dvorak HF, Senger DR Tumor-secreted vascular permeability factor increases cytosolic Ca2+ and von Willebrand factor release in human endothelial cells Am J Pathol 1991; 138: 213-21 19 Broll R, Erdmann H, Duchrow M, Oevermann E, Schwandner O, Markert U, Bruch HP, Windhovel U Vascular endothelial growth factor (VEGF) -- a valuable serum tumor marker in patients with colorectal cancer? Eur J Surg Oncol 2001; 27: 37-42 20 Brown LF, Berse B, Jackman RW, Tognazzi K, Manseau EJ, Senger DR, Dvorak HF Expression of vascular permeability factor (vascular endothelial growth factor) and its receptors in adenocarcinomas of the gastrointestinal tract Cancer Res 1993; 53: 4727-35 21 Carbone D, Chiba I, Mitsudomi T Polymorphism at codon 213 within the p53 gene Oncogen 1991; 6: 1691-92 22 Caron de Fromentel C, Soussi T TP53 tumor suppressor gene: a model for investigating human mutagenesis Genes Chromosomes Cancer 1992; 4: 1-15 23 Cho Y, Gorina S, Jeffrey PD, Pavletich NP Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations Science 1994; 265: 346-55 81 24 Chung DC The genetic basis of colorectal cancer: insights into critical pathways of tumorigenesis Gastroenterology 2000; 119: 854-65 25 Clark LJ, MacKenzie K, Parkinson EK Elevated levels of the p53 tumour suppressor protein in the basal layer of recurrent laryngeal papillomas Clin Otolaryngol 1993; 18: 63-5 26 Connolly DT, Heuvelman DM, Nelson R, Olander JV, Eppley BL, Delfino JJ, Siegel NR, Leimgruber RM, Feder J Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis J Clin Invest 1989; 84: 1470-78 27 Costa A, Marasca R, Valentinis B, Savarino M, Franda A, Silvestrini R, Torelli G p53 Gen point mutations in relation to p53 nuclear protein accumulation in colorectal carcinoma J Pathol 1995; 176: 45-53 28 Cripps KJ, Purdie CA, Carder PJ A study of stabilization of p53 protein versus point mutation in colorectal carcinomas Oncogene 1994, 9: 23739-43 29 Dameron KM, Volpert OV The p53 tumor suppressor gene inhibits angiogenesis by stimulating the production thrombospondin Cold Spring Harb Symp Quant Biol 1994; 59: 483-9 of 30 Dameron KM, Volpert OV, Tainsky MA, Bouck N Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1 Science 1994; 256: 1582-4 31 Datto MB, Li Y, Panus JF Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism Proc Natl Acad Sci 1995; 92: 5545-49 32 Dix B, Robbins P, Carello S Comparison of p53 gene and protein overexpression in colorectal carcinomas Br J Cancer 1994; 60: 585-90 33 Donehower LA, Bradley A The tumor suppressor p53 Biochimica et Biophysica Acta 1993; 115: 181-205 34 Dundas SR, Lawrie LC, Rooney PH, Murray GI Mortalin is over-expressed by colorectal adenocarcinomas and correlates with poor survival J Pathol 2005; 205: 74-81 35 Dvorak HF Tumors Wounds that do not heal. Similarities between tumor stroma generation and wound healing N Engl J Med 1986; 315: 1650-59 82 36 Farnleitner AH, Kreuzinger N, Kavka GG, Grillenberger S, Rath J, Mach RL Comparative analysis of denaturing gradient gel electrophoresis and temporal temperature gradient gel electrophoresis in separating Escherichia coli uidA amplicons differing in single base substitutions Lett Appl Microbiol 2000; 30: 427-31 37 Fearon ER, Vogelstein B A genetic model for colorectal tumorigenesis Cell 1990; 61: 759-67 38 Ferrara N, Davis-Smyth T The biology of vascular endothelial growth factor Endocrine Reviews 1997; 18: 4-25 39 Ferrara N, Henzel WJ Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells Biochem Biophys Res Commun 1989; 161: 851-58 40 Ferrara N, Houck KA, Jakeman LB, Winer J, Leung DW The vascular endothelial growth factor family of polypeptides J Cell Biochem 1991; 47: 211-18 41 Ferrara N, Leung DW, Cachianes G, Winer J, Henzel WJ Purification and cloning of vascular endothelial growth factor secreted by folliculostellate cells Methods Enzymol 1991; 198: 391-405 42 Ferrara N Role of vascular endothelial growth factor in regulation of physiological angiogenesis Am J Physiol Cell Physiol 2001; 280: 1358-66 43 Ferrara N Role of vascular endothelial growth factor in regulation of physiological angiogenesis Kidney International 1999; 56: 794-814 44 Finlay CA Normal and malignant growth control by p53 Cancer Treat Res 1993; 63: 327-44 45 Fisher SG, Lerman LS DNA fragments differing by single base-pair substitutions are seperated in denaturing gradient gels: correspondence with melting theory Proc Nat Acad Sci 1983; 80: 1579-83 46 Folkman J What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 1990; 80: 4-6 47 Folkman J, Watson K, Ingber D, Hanahan D Induction of angiogenesis during the transition from hyperplasia to neoplasia Nature 1989; 339:58-61 83 48 Fontanini G, Boldrini L, Vignati S, Chiné S, Basolo F, Silvestri V, Lucchi M, Mussi A, Angeletti CA, Bevilacqua G Bcl2 and p53 Regulate Vascular Endothelial Growth Factor (VEGF)-mediated Angiogenesis in Non-small Cell Lung Carcinoma Eur J Cancer 1998; 34: 718-23 49 Fontanini G, Vignati S, Mussi A, Calcinai A, Boldrini L, Chiné S, Silvestri V, Angeletti CA, Basolo F, Bevilacqua G Neoagiogenesis and p53 protein in lung cancer: their prognostic role and their relation with vascular endothelial growth factor expression Br J Cancer 1997; 75: 1295-1301 50 Forrester K, Lupold SE, Ott VL, Chay CH, Band V, Wang XW, Harris CC Effects of p53 mutants on wild-type p53-mediated transactivation are cell type dependent Oncogene 1995; 10: 2103-11 51 Furuwatari C, Yagi A, Yamagami O, Ishikawa M, Hidaka E, Ueno I, Furihata K, Ogiso Y, Katsuyama T A comprehensive system to explore p53 mutations Am J Clin Pathol 1998; 110: 368-73 52 Gasparini G p53 and angiogenesis J Clin Oncol 1995; 13: 1830 53 Gerber HP, Dixit V, Ferrara N Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells J Biol Chem 1998; 273: 13313-16 54 Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N VEGF regulates endothelial cell survival by the phosphatidylinositol-3'-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation J Biol Chem 1998; 273: 30336-43 55 Gille H, Kowalski J, Li B, LeCouter J, Moffat B, Zioncheck TF, Pelletier N, Ferrara N Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants J Biol Chem 2001; 276: 3222-30 56 Gimbrone MA Jr, Leapman SB, Cotran RS, Folkman J Tumor dormancy in vivo by prevention of neovascularization J Exp Med 1972; 136: 261-76 57 Goldman CK, Kendall R, Cabrera G, Soroceanu L, Heike Y, Gillespie GY, Siegal GP, Mao X, Bett AJ, Huckle WR, Thomas KA, Curiel DT Paracrine expression of a native soluble vascular endothelial growth factor receptor inhibits tumor growth, metastasis, and mortality rate Proc. Natl. Acad. Sci 1998; 95: 8795-8800 58 Gospodarowicz D, Abraham JA, Schilling J Isolation and characterization of a vascular endothelial cell mitogen produced by pituitary-derived folliculo stellate cells 84 Proc Natl Acad Sci 1989; 86: 7311-15 59 Greenblatt MS, Bennett WP, Hollstein M, Harris CC Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis Cancer Res 1994; 54: 4855-78 60 Greenblatt MS, Grollman AP, Harris CC Deletions and insertions in the p53 tumor suppressor gene in human cancers: confirmation of the DNA polymerase slippage/misalignment model Cancer Res 1996; 56: 2130-6 61 Grugel S, Finkenzeller G, Weindel K, Barleon B, Marme D, Both v-H-ras and v-raf stimulate expression of the vascular endothelial growth factor in NIH 3T3 cells J Biol Chem 1995; 270: 25915-19 62 Grunstein J, Masbad JJ, Hickey R, Giordano F, Johnson RS Isoforms of vascular endothelial growth factor act in a coordinated fashion to recruit and expand tumor vasculature Mol Cell Biol 2000; 20: 7282-91 63 Grunstein J, Roberts JW, Mathieu-Costello O, Hanahan D, Johnson RS Tumor-derived Expression of Vascular Endothelial Growth Factor Is a Critical Factor in Tumor Expansion and Vascular Function Cancer Research 1999; 59: 1592-98 64 Guldberg P, Nedergaard T, Nielsen HJ, Olsen AC, Ahrenkiel V, Zeuthen J Single-step DGGE-based mutation scanning of the p53 gene: Application to genetic diagnosis of colorectal cancer Human Mutation 1997; 9: 348-55 65 Hadj Tahar A. Bevacizumab for advanced colorectal cancer Issues Emerg Health Technol 2004; 63: 1-4 66 Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases Cell 1993; 75: 805-16 67 Harris CC p53 tumor suppressor gene: from the basic research laboratory to the clinic--an abridged historical perspective Carcinogenesis 1996; 17: 1187-98 68 Harris CC Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies Natl Cancer Inst 1996; 88: 1442-55 69 Harris CC p53: at the crossroads of molecular carcinogenesis and risk assessment Science 1993; 262: 1980 85 70 Hejna M, Hamilton G, Brodowicz T, Haberl I, Fiebiger WC, Scheithauer W, Virgolini I, Kostler WJ, Oberhuber G, Raderer M Serum levels of vasoactive intestinal peptide (VIP) in patients adenocarcinomas of the gastrointestinal tract Anticancer Res 2001; 21: 1183-7 with 71 Hermanek P, Sobin LH TNM classification of malignant tumors fourth edition, 2nd revision, Springer Verlag; Berlin 1992 72 Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF Science 1999; 284:1994-8 73 Holash J, Wiegand SJ, Yancopoulos GD New model of tumor angiogenesis: dynamic balance between vessel regression and growth mediated by angiopoietins and VEGF Oncogene 1999; 18: 5356-62 74 Hollstein M, Sidransky D, Vogelstein B, Harris CC p53 mutations in human cancers Science 1991; 253: 49-53 75 Hou J, Tian L, Wie Y Immunotherapy of tumor by targeting angiogenesis Sci China C Life Sci 2004; 47: 545-52 76 Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA Mol Endocrinol 1991; 5: 1806-14 77 Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms J Biol Chem 1992; 267: 26031-37 78 Hunter T Braking the cycle Cell 1993; 75: 839-41 79 Iniesta P, Vega FJ, Caldes T, Massa M, de Juan C, Cerdan FJ, Sanchez A, Lopez JA, Torres AJ, Balibrea JL, Benito M p53 exon 7 mutations as a predictor of poor prognosis in patients with colorectal cancer Cancer Lett 1998; 130: 153-60 80 Invitrogen Invitrogen Zero Blunt TOPO PCR Cloning Kit Manual Version F 02/2001, Carlsbad, Carlifornia, USA 81 Jeltsch M, Kaipainen A, Joukov V, Meng X, Lakso M, Rauvala H, Awartz M, Fukumura D, Jain RK, Alitalo K Hyperplasia of lymphatic vessels in VEGF-C transgenic mice 86 Science 1997; 276: 1423-25 82 Jingjing L, Xue Y, Agarwal N, Roque RS Human Muller cells express VEGF183, a novel spliced variant of vascular endothelial growth factor Invest Ophthalmol Vis Sci 1999; 40: 752-59 83 Juven T, Barak Y, Zauberman A, George DL, Oren M Wild type p53 can mediate sequence-specific transactivation of an internal promoter within the mdm2 gene Oncogene 1993; 8: 3411-16 84 Kang SM, Maeda K, Onoda N, Chung YS, Nakata B, Nishiguchi Y, Sowa M Combined analysis of p53 and vascular endothelial growth factor expression in colorectal carcinoma for determination of tumor vascularity amd liver metastasis Int J Cancer 1997; 74: 502-7 85 Kendal RL, Thomas KA Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor Proc. Natl. Acad. Sci. 1993; 90: 10705-09 86 Kern A, Taubert H, Scheele J, Rudroff C, Mothes H, Kappler M, Bartel F, Richter KK Association of p53 mutations, microvessel density and neoangiogenesis in pairs of colorectal cancers and corresponding liver metastases Int J Oncol 2002; 21: 243-9 87 Kern SE, Kinzler KW, Baker SJ, Nigro JM, Rotter V, Levine AJ, Friedman P, Prives C, Vogelstein B Mutant p53 proteins bind DNA abnormally in vitro Oncogene 1991; 6: 131-6 88 Keyt BA, Nguyen HV, Berleau LT, Duarte CM, Park J, Chen H, Ferrara N Identification of Vascular Endothelial Growth Factor Determinants for Binding KDR and FLT-1 Receptors J Biol Chem 1996; 271: 5638-46 89 Kieser A, Weich HA, Brandner G, Marme D, Kolch W Mutant p53 potentiates protein kinase-C induction of vascular endothelial growth factor expression Oncogene 1994; 9: 963-69 90 Kim KJ, Li B, Winer J, Armanini M, Gilett N, Phillips HS, Ferrara N Inhibition of vascular endothelial growth factor-induced angiogenesis supresses tumor growth in vivo Nature 1993; 362: 841-44 91 Klein G Multistep emancipation of tumors from growth control: can it be cured step? Bioessays 1990; 12: 347-350 92 Ko L, Prives C p53: puzzle and paradigm Genes Dev 1996; 10: 1054-72 87 in a single 93 Kondo Y, Arii S, Furutani M, Isigami S, Mori A, Onodera H, Chiba T, Imamura M Implication of vascular endothelial growth factor and p53 status for angiogenesis in noninvasive colorectal carcinoma Cancer 2000; 88: 1820-7 94 Konishu T, Huang CL, Adachi M, Taki T, Inufusa H, Kodama K, Kohno N, Miyake M The K-ras gene regulates vascular endothelial growth factor gene expression in nonscmall cell lung cancers Int J Oncol 2000; 16: 501-11 95 Land H, Parada LF Cellular Oncogenes and multistep carcinogenesis Science 1983; 222: 771-778 96 Lee JC, Chow NH, Wang ST, Huang SM Prognostic value of vascular endothelial growth factor expression in colorectal cancer patients Eur J Cancer 2000; 36: 748-53 97 Lee JS, Kim HS, Jung JJ, Park CS, Lee MC Expression of vascular endothelial growth factor in renal cell carcinoma and the relation to angiogenesis and p53 protein expression J Surg Oncol 2001; 77: 55-60 98 Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N Vascular endothelial growth factor is a secreted angiogenic mitogen Science 1989; 246: 1306-09 99 Levine AJ p53, the cellular gatekeeper for growth and division Cell 1997; 88: 323-331 100 Linderholm BK, Lindahl T, Holmberg L, Klaar S, Lennerstrand J, Henriksson R, Bergh J The expression of vascular endothelial growth factorcorrelates with mutant p53 and poor prognosis in human breast cancer Cancer Res 2001; 61: 2256-2260 101 Lowe SW, Ruley HE Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis Genes Dev 1993; 7: 535-45 102 Madan A, Curtin PT A 24-base pair sequence 3' to the human erythropoietin contains a hypoxiaresponsive transcriptional enhancer Proc Natl Acad Sci USA 1993; 90: 3928-32 103 Maeda K, Chung YS, Ogawa Y, Takatsuka S, Kang SM, Ogawa M, Sawada T, Sowa M Prognostic value of vascular endothelial growth factor expression in gastric carcinoma Cancer 1996; 77: 858-63 88 104 Maeda K, Kang SM, Onoda N, Ogawa M, Sawada T, Nakata B, Kato Y, Chung YS, Sowa M Expression of p53 and vascular endothelial growth factor associated with tumor angiogenesis and prognosis in gastric cancer Oncology 1998; 55: 594-99 105 Maeda T, Matsumura S, Hiranuma H, Jikko A, Furukawa S, Ishida T, Fuchiata H Expression of vascular endithelial growth factor in human oral squamous cell carcinoma: its association with tumor progression and p53 gene status J Clin Pathol 1998; 51: 771-5 106 Marchetti A, Cecchinelli B, D'Angelo M, D'Orazi G, Crescenzi M, Sacchi A, Soddu S p53 can inhibit cell proliferation through caspase-mediated cleavage of ERK2/MAPK Cell Death Differ 2004; 11:596-607 107 Mazure NM, Chen EY, Yeh P, Laderoute KR, Giaccia AJ Oncogenic transformation an hypoxia synergistically act to modulate vascular endothelial growth factor expression Cancer Res 1996; 56: 3436-40 108 Millauer B, Shawyer LK, Plate KH, Risau W, Ullrich A Glioblastoma growth inhibited in vivo by a dominant negative Flk-1 mutant Nature 1994; 367: 576 109 Momand J, Zambetti GP, Olson DC, George D, Levine AJ The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation Cell 1992; 69: 1237-45 110 Mukhopadhyay D, Tsiokas L, Sukhatme VP Wild-type p53 and v-Src exert opposing influences on human vascular endothelial growth factor gene expression Cancer Res 1995; 55: 6161-65 111 Mukhopadhyay D, Tsiokas L, Zhou X-M, Foster D, Brugge JS, Sukhatme VP Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation Nature 1995; 375: 577-81 112 Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z Vascular endothelial growth factor (VEGF) and its receptors FASEB J 1999; 13: 9-22 113 Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, Bigner SH, Davidson N, Baylin S, Devilee P Mutations in the p53 gene occur in diverse human tumour types Nature 1989; 7: 705-8 114 Olander JV, Connolly DT, DeLarco JE Specific binding of vascular permeability factor to endothelial cells Biochem Biophys Res Commun 1991; 175: 68-76 115 Oren M, Maltzman W, Levine AJ Post-translational regulation of the 54k cellular tumor antigen in normal and transfected cells 89 Mol Cell Biol 1981; 1: 101-10 116 Ory K, Legros Y, Auguin C, Soussi T Analysis of the most representative tumour-derived p53 mutants reveals that changes in protein conformation are not correlated with loss of transactivation or inhibition of cell proliferation EMBO J 1994; 13: 3496-504 117 Park JE, Keller GA, Ferrara N The vascular endothelial growth factor isoforms (VEGF): differential deposition into the subepithelial extracellular matrix and bioactivity of extra cellular matrix- bound VEGF Mol Cell Biol 1993; 4: 1317-26 118 Pertovaara L, Kaipainen A, Mustonen T, Orpana A, Ferrara N, Saksela O, Alitalo K Vascular endothelial growth factor is induced in response to transforming growth factor-beta in fibroblastic and epithelial cells J Biol Chem 1994; 269: 6271-74 119 Pidgeon GP, Barr MP, Harmey JH, Foley DA, Bouchier-Hayes DJ Vascular endothelial growth factor (VEGF) upregulates BCL-2 apoptosis in human and murine mammary adenocarcinoma cells Br J Cancer 2001; 85: 273-78 and inhibits 120 Plate KH, Breier G, Weich H, Mennel HD, Risau W Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms Int J Cancer 1994; 59: 520-29 121 Plate KH, Breier G, Weich HA, Risau W Vascular endothelial growth factor is a potential tumor angiogenesis factor in human gliomas in vivo Nature 1992; 359: 845-48 122 Plouet J, Schilling J, Gospodarowicz D Isolation and characterization of a newly identified endothelial cell mitogen produced by AtT-20 cells EMBO J 1989; 8: 3801-6 123 Poland D Recursion relation generation of probability macromolecules with long range correlations Biopolymers 1974; 13: 1859-71 profiles for specific-sequence 124 Poltorak Z, Cohen T, Sivan R, Kandelis Y, Spira G, Vlodavsky I, Keshet E, Neufeld G VEGF145, a secreted vascular endothelial growth factor isoform that binds to extracellular matrix J Biol Chem 1997; 272: 7151-58 125 Presta LG, Chen H, O'Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N Humanization of a an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders Cancer Res 1997; 57: 4593-99 90 126 Prives C, Hall PA The p53 pathway J Pathol 1999; 187: 112-26 127 Quinn TP, Peters KG, De Vries C, Ferrara N, Williams LT Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium Proc Natl Acad Sci 1993; 90:7533-37 128 Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel R Mutant ras oncogenes up-regulate VEGF/VPF expression: implicates induction and inhibition of tumor angiogenesis Cancer Res 1995; 55: 4574-80 for 129 Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A Regulation of tumor angiogenesis by p53-induced degradation od hypoxiainducible factor 1alpha Genes & development 2000; 14: 34-44 130 Reed J Bcl-2 and the regulation of programmed cell death J Cell Biol 1994; 124: 1-6 131 Reich NC, Oren M, Levine AJ Two distinct mechanisms regulate the levels of a cellular tumor antigen p53 Mol Cell Biol 1983; 3: 2143-50 132 Riedel F, Gotte K, Schwalb J, Schafer C, Hormann K Vascular endothelial growth factor expression correlates with p53 mutations and angiogenesis in squamous cell carcinoma of the head and neck Acta Otolaryngol 2000; 120: 105-11 133 Rifkin DB, Moscatelli D, Bizik J, Quarto N, Blei F, Dennis P, Flaumenhaft R, Mignatti P Growth factor control of extracellular proteolysis Cell Differ Dev 1990; 32: 313-18 134 Robinson CJ, Stringer SE The splice variants of vascular endothelial growth factor (VEGF) and their receptors J Cell Sci 2001; 114: 853-65 135 Rodrigues NR, Rowan A, Smith MEF, Kerr IB, Bodmer WF, Gannon JV, Lane DP p53 mutations in colorectal cancer Proc Natl Acad Sci 1990; 87: 7555-59 136 Rolley N, Butcher S Specific DNA binding by different classes of human p53 mutants Oncogene 1995; 11: 763-70 137 Rosenbaum V, Riesner D Temperature-gradient gel elctrophoresis. Thermodynamic analysis of nucleic acids and proteins in purified form and in cellular extracts Biophys Chem 1987; 26: 235-46 138 Saito H, Tujitani S, Ikeguchi M, Maeta M, Kaibara N 91 Neoangiogenesis and relationship to nuclear p53 accumulation and vascular endothelial growth factor expression in advanced gastric carcinoma Oncology 1999; 57: 164-72 139 Sanger F, Nickeln S, Coulson AR DNA sequenzing with chain-terminating inhibitors Proc Natl Acad Sci 1977; 74: 5463-7 140 Satake S, Kuzuya M, Miura H, Asai T, Ramos MA, Muraguchi M, Ohmoto Y, Iguchi A Up-regulation of vascular endothelial growth factor in response to glucose deprivation Biol Cell 1998; 90: 161-68 141 Schlaeppi JM, Wood JM Targeting vascular endothelial growth factor (VEGF) for anti-tumor therapy, by antiVEGF neutralizing monoclonal antibodies or by VEGF receptor tyrosine- kinase inhibitors Cancer Metastasis Rev 1999; 18: 473-81 142 Schlechte H, Lenk SV, Löning T, Schnorr D, Rudolh BD, Ditscherlein G, Loening SA p53 tumour suppressor gene mutations in benign prostatic hyperplasia and prostatic cancer Eur Urol 1998; 34: 433-40 143 Senger DR, Van de Water L, Brown LF, Nagy JA, Yeo KT, Yeo TK, Berse B, Jackman RW, Dvorak AM, Dvorak HF Vascular permeability factor (VPF, VEGF) in tumor biology Cancer Metastasis Rev 1993; 12: 3003-24 144 Shaheen RM, Ahmad SA, Liu W, Reinmuth N, Jung YD, Tseng WW, Drazan KE, Bucana CD, Hicklin DJ, Ellis LM Inhibited growth of colon cancer carcinomatosis by antibodies to vascular endothelial and epidermal growth factor receptors Br J Cancer 2001; 85: 584-89 145 Shaheen RM, Tseng WW, Vellagas R, Liu W, Ahmad SA, Jung YD, Reinmuth N, Drazan KE, Bucana CD, Hicklin DJ, Ellis LM Effects of an antibody to vascular endothelial growth factor receptor-2 on survival, tumor vascularity, and apoptosis in a murine model of colon carcinomatosis Int J Oncol 2001; 18: 221-26 146 Shaulsky G, Goldfinger N Nuclear localization is essential for the activity of p53 protein Oncogene 1991; 6: 2055-65 147 Shaulsky G, Goldfinger N, Ben-Ze’ev A, Rotter V Nuclear accumulation of p53 Protein is mediated by several nuclear localizations signals and plays a role in tumorigenesis Mol and Cell Biol. 1990; 10: 6565-77 148 Shaw P, Bovey R, Tardy S, Sahli R, Sordat B, Costa J Induction of apoptosis by wild-type p53 in a human colon tumor-derived cell line Proc Natl Acad Sci 1992; 89: 4495-99 149 Shi CY, Phang TW, Wee A, Ngoi SS, Lin Y, Li B, Ong CN, Lee HP 92 Mutations of the tumour suppressor gene p53 in colorectal and hepatocellular carcinomas Ann Acad Med 1995; 24: 204-10 150 Shuman S Novel approch to molecular cloning and polynucleotide synthesis using Vaccinia DNA topoisomerase J Biol Chem 1994; 269: 32678-84 151 Shuman S Recombination mediated by vaccinia virus DNA topoisomerase I in Escherichia coli is sequence specific Proc Natl Acad Sci 1991; 88: 10104-08 152 Shweiki D, Itin A, Soffer D, Keshet E Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis Nature 1992; 359: 843-45 153 Shweiki D, Neeman M, Itin A, Keshet E Induction of vascular endothelial growth factor expression by hypoxia and by glucose deficiency in multicell spheroids: implications for tumor angiogenesis Proc Natl Acad Sci 1995; 92: 768-72 154 Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M Neuropilin-1 is expressed by endothelial and tumor cells as isoform-specific receptor for vascular endothelial growth factor Cell 1998; 92: 735-45 155 Hainaut P, Hernandez T, Robinson A, Rodriguez-Tome P, Flores T, Hollstein M, Harris CC, Montesano R IARC Database of p53 gene mutations in human tumors and cell lines: updated compilation, revised formats and new visualisation tools. Nucleic Acids Res 1998; 26: 205-13 156 Sreenath T, Matrisian LM, Stetler-Stevenson W, Gattoni-Celli S, Pozzatti RO Expression of matrix metalloproteinases in transformed rat cell lines of high and low metastatic potential Cancer Res 1992; 52: 4942-47 157 Steger G Thermal denaturation of double-stranded nucleic acids: prediction of temperatures critical for gradient gel electrophoresis and polymerase chain reaction Nucleic Acids Res 1994; 22: 2760-68 158 Stein I, Neeman M, Shweiki D, Itin A, Keshet E Stabilization of vascular endothelial growth factor mRNA hypoglycemia and coregulation with other ischemia-induced genes Mol Cell Biol 1995; 15: 5363-68 159 by hypoxia and Strawn LM, McMahon G, App H, Schreck R, Kuchler WR, Longhi MP, Hui TH, Tang C, Levitzki A, Gazit A, Chen I, Keri G, Orfi L, Risau W, Flamme I, Ullrich A, Hirth KP, Shawyer LK Flk-1 as a target for tumor growth inhibition Cancer Res 1996; 56: 3540-45 93 160 Takagi H, King GL, Robinson GS, Ferrara N, Aiello LP Adenosine mediates hypoxic induction of vascular endothelial growth factor in retinal pericytes and endothelial cells Invest Opthalmol Vis Sci 1996; 37: 2165-76 161 Takahashi Y, Bucana CD, Cleary KR, Ellis LM P53, vessel count, and vascular endothelial growth factor expression in human colon cancer Int J Cancer 1998; 79: 34-38 162 Takahashi Y, Kitadai Y, Bucana CD, Cleary KR, Ellis LM Expression of vascular endothelial growth factor and ist receptor (KDR) correlates with vascularity, metastasis, and proliferation in human colon cancer Cancer Res 1995; 55: 3964-68 163 Takano S, Yoshii Y, Kondo S, Suzuki H, Maruno T, Shirai S, Nose T Concentration of vascular endothelial growth factor in the serum and tumor tissue of brain tumor patients Cancer Res 1996; 56: 2185-90 164 Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, Bohlen P Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor Biochem Biophys Res Commun 1992; 187: 1579-86 165 Thomas KA Vascular endothelial growth factor, a potent and selective angiogenic agent J Biol Chem 1996; 271: 603-06 166 Thorlacius S, Boerresen AL, Eyfjoerd JE Somatic p53 mutations in human breast carcinomas in an icelandig population: a prognostic factor Cancer Res 1993; 53: 1637-41 167 Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, Abraham JA The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through altrnative exon splicing J Biol Chem 1991; 266: 11947-54 168 Tuder RM, Flook BE, Voelkel NF Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia J Clin Invest 1995; 95: 1798-1807 169 Turkey Statistik, Boxplot 1970 170 Vaisman N, Gospodarowicz D, Neufeld G Characterization of the receptors for vascular endothelial growth factor J Biol Chem 1990; 265: 19461-66 171 Vega FJ, Iniesta P, Caldés T, Sánches, López JA, de Juan C, Diaz-Rubio E, Torres A, Balibrea JL, Benito M 94 p53 exon 5 mutations as a prognostic indicator of shorted survival in non-small-cell lung cancer Br J Cancer 1997; 76: 44-51 172 Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL Genetic alterations during colorectal-tumor development N Engl J Med 1988; 319: 525-32 173 Waga S, Hannon GJ The p21 inhibitor of interaction with PCNA Nature 1994; 369: 574-8 cyclin-dependent kinases controls DNA replication by 174 Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor J Biol Chem 1994; 269: 26988-95 175 Warburg O, Christian W Isolierung und Kristallisation des Gärungsferment Enolase Biochem Z 1942; 310: 384-421 176 Warren RS, Yuan H, Matli MR, Ferrara N, Donner DB Induction of Vascular Endothelial Growth Factor by Insulin-like Growth Factor 1 in Colorectal Carcinoma J Biol Chem 1996; 271: 29483-8 177 Warren RS, Yuan H, Matli MR, Gillett NA, Ferrara N Regulation by vascular endothelial growth factor of human colon cancer tumorigenesis in a mouse model of experimental liver metastasis J Clin Invest 1995; 95: 1789-97 178 Weinberg RA Oncogenes and the molecular biology of cancer J Cell Biol 1983; 97: 1661-62 179 Weiss J Mutation and expression of the p53 gene in malignant melanoma cell lines Int J Cancer 1993; 54: 693-9 180 Wiese U, Wulfert M Scanning for mutations in the human prion protein open reading frame by temporal temperature gradient gel electrophoresis Electrophoresis 1995; 16: 1851-60 181 Wu GS, El-Diery WS p53 and chemosensitivity Nat Med 1996; 2: 255-6 182 Wu LW, Mayo LD, Dunbar JD, Kessler KM, Ozes ON, Warren RS, Donner DB VRAP is an adaptor protein that binds KDR, a receptor for vascular endothelial growth factor J Biol Chem 2000; 275: 6059-62 183 Wu X, Bayle JH, Olson D, Levine AJ 95 The p53-mdm-2 autoregulatory feedback loop Genes Dev 1993; 7: 1126-32 184 Wulfert M, Manual Whatman Biometra TGGE The TGGE System: A fast method for differentiation between wild type and mutant DNA of the human prion protein gene 1996 185 Xiong Y, Khang H p21 is an universal inhibitor of cyclin kinases Nature 1993; 366: 701-4 186 Zietz C, Rössle M, Haas C, Sendelhofert A, Hirschmann A, Stürzl M, Löhrs U MDM-2 oncoprotein overexpression, p53 gene mutation, and VEGF up-regulation in angiosarcomas Am J Path 1998; 153: 1425-33 96 7 Anhang 7.1 Abkürzungen in alphabetischer Reihenfolge Abkürzung Erläuterung 5-JÜR 5-Jahres Überlebensrate APC AS Bcl-2 BSA c-AMP COX-2 CRC DCC DGGE Adenopolyposis Coli–Gen Aminosäure Proto-Onkogen im B-Zell-Leukämie-Genlokus Chromosom 18 bovines Serum Albumin zyklisches Adenosinmonophosphat Cyclooxygenase 2 colorectal carcinoma, kolorektale Karzinome deleted in colorectal cancer–Gen denaturing gradient gel electrophoresis, Gelelektrophorese mit chemischem Gradienten deoxy Nukleotid Triphosphat double strandend DNA, doppelsträngige DNA extrazelluläre Domäne extrazelluläre Matrix enzyme linked immuno sorbent assay fetal liver kinase fms-like tyrosine kinase GTPase activating Protein, GTPase aktivierendes Protein Guanidinnukleotid Diphosphat Guanidinnucleotide dissociation stimulator, Guanidinnukleotid Dissoziations-Stimulator Guanidinnukleotid Triphosphat Stunde Hypoxie-induzierter-Faktor 1 Kilo Basen Kilo Dalton kinase-insert domain containing receptor Onkogen Molar mitogen activated protein kinase MAPK oder ERK Kinase maximal minimal Minute milli Molar messenger RNA (Ribonukleinsäure) Medizinische Universität zu Lübeck mutierter Typus Mittelwert Normal-... Nanometer nicht signifikant nach optische Dichte open reading frame, offener Leserahmen vom Start- bis Stop-Codon Plasminogen-Aktivator dNTP dsDNA ECD ECM ELISA flk flt GAP GDP GDS GTP h HIF-1 kb kDa KDR k-ras M MAPK MEK max min (Zahl)min mM mRNA MUL Mut MW N nm n.s. n. OD ORF PA 97 PCR PE-Puffer PGE2 PI(3)K PIP PLC-γ PLD pM ras raf RTK src s s. SD ssDNA SMAD 2/4 T TGF TGGE TNF TSP TTGE UKL VEGF VEGFR vgl. VPF VRAP v-Src Wt µM ZNS 7.2 Polymerase Chain Reaction, Polymerase Kettenreaktion Proteinextraktionspuffer Prostaglandin E2 Phosphatidyl Inositol 3 Kinase Phosphoinositol-Phosphat Phospholipase C-γ Phospholipase D piko Mol Onkogen Onkogen Rezeptor-Tyrosin-Kinasen Onkogen Sekunde siehe Standardabweichung single stranded DNA, einzelsträngige DNA mothers against decapentaplegic homolog 2/4; Transkriptionsfaktor von u.a. TGF-beta Tumor-... tumor growth factor, Tumorwachstumsfaktor temperature gradient gel electrophoresis, Gelelektrophorese mit räumlichem Temperaturgradienten Tumornekrosefaktor Thrombospondin temporal temperature gradient electrophoresis, Gelelektrophorese mit zeitlichem Temperaturgradienten Universitätsklinik Lübeck vascular endothelial growth factor VEGF receptor vergleiche vascular permeability factor VEGF receptor associated protein Tumorsuppressorgen Wildtyp 10-6 Meter Zentrales Nervensystem Materialien 7.2.1 Acrylamid/Bis, 40% 40% (w/v) Acrylamid und N’N’ Methylen-bis-Acrylamid, Verhältnis vom Monomer zum Quervernetzer: 37,5:1 Fa. Bio-Rad, Hercules, California; USA 7.2.2 Agarosegel, 1% eigene Herstellung 1% (w/v) Agarose, Fa. Invitrogen Corp., Carlsbad, California; USA TAE-Puffer, 1x [7.2.56] 7.2.3 Ammoniumpersulfat, 10% eigene Herstellung 10% (w/v) APS, in aqua bidest., steril Fa. Sigma Chemical Co, St. Louis; USA 98 7.2.4 Bacto® Tryptone Becton Dickinson and Company Sparks, MD 21152 USA 38800 Le Pont de Claix ; France 7.2.5 Bacto® Yeast Extract DIFCO Laboratories Detroit MI 48232-7058; USA 7.2.6 Bio-Rad Protein Assay Coomassie Brilliant Blue G250 Lösung Fa. Bio-Rad, Hercules, California; USA 7.2.7 Biosphere Filtertips Filtertips, steril, 10 µl Filtertips, steril, 100µl Filtertips, steril, 1000µl Fa. Sarstedt, Nuembrecht; Deutschland 7.2.8 Bitek Agar Becton Dickinson Microbiology System Becton Dickinson and Company, France S.A. 8800 Le Pont de Claix; France 7.2.9 Cellstar PP-Röhrchen 15ml, mit Schraubdeckel, steril 50ml, mit Schraubdeckel, steril Fa. Greiner Labortechnik GmbH, Solingen; Deutschland 7. 2.10 Chill out wax Fa. Biozym, Hessisch Oldendorf; Deutschland 7.2.11 Deckgläser 24 x 24 x 0,1 mm Fa. Menzel Gläser, Braunschweig; Deutschland 7. 2.12 DMSO, Dimethylsulfoxid Fa. Sigma Chemical Co, St. Louis; USA 7.2.13 DNA-Molekulargewichtsmarker VI 154-2176 bp, 25µg/µl; 50mg Fa. Roche Diagnostics GmbH, Mannheim; Deutschland 7. 2.14 DNA-Polymerisation Mix dNTP-Gemisch, 20mM Fa. Amersham Pharmacia Biotech, Piscatanay, New Jersey; USA 7.2.15 EcoR I, Restriktionsendonuklease Puffer H (Pufferangaben vertraulich), 20U/µl Fa. Amersham Biosciences, Freiburg; Deutschland 7. 2.16 Elutions-Puffer eigene Herstellung (nach: Separation of small DNA fragments by conventional gel electrophoresis, in: Current Protocols in Molecular Biology [178], Kapitel 2.7.4, Suppl. 45): 99 10mM Tris (Fa. Bio-Rad, Hercules, California; USA) 50mM NaCl (Fa. Merck KGaA, Darmstadt; Deutschland) 1mM EDTA (Fa. Merck KGaA, Darmstadt; Deutschland) mit HCL auf pH 7,5 eingestellt 7. 2.17 Entwicklerlösung eigene Herstellung 1,5% (w/v) NaOH (Fa. Merck KGaA, Darmstadt; Deutschland) 0,4% (v/v) Formaldehyd 35-40% (Fa. Merck KGaA, Darmstadt; Deutschland) aqua bidest. 7. 2.18 Ethanol, 100% vergällt Apotheke der Universitätsklinik Lübeck 7. 2.19 Ethanol, 96% eigene Herstellung 96% (v/v) Ethanol 100% p.a. [7.2.18] aqua bidest 7. 2.20 Ethanol, 70% eigene Herstellung 70% (v/v) Ethanol 100% p.a. [7.2.18] aqua bidest 7. 2.21 Ethidiumbromidlösung, 10mg/ml Fa. Bio-Rad, Hercules, California; USA 7. 2.22 Eukitt Einschlussmittel für histologische Schnitte Fa. O. Kindler GmbH & Co, Freiburg; Deutschland 7. 2.23 Fixierlösung eigene Herstellung 10% (v/v) Methanol 100%, p.a. (Fa. JT Baker, Deventer; Niederlande) 0,5% (v/v) Essigsäure 100% (Fa. Merck KGaA, Darmstadt; Deutschland) aqua bidest. 7.2.24 Formamid 99,9%, pH 7,0 Micro Select for Molecular Biology Fa. Fluka Chemie GmbH, Buchs; Schweiz 7.2.25 Genomic Kit QIAGEN Blood & Cell Culture DNA Kits RNase A (100mg/ml; 7000U/ml) 1 Unit katalysiert die Hydrolyse von RNA mit einer Geschwindigkeitskonstante ersten Grades von 1,0 bei 25°C, pH 5,0 QIAGEN Protease: 28mg lyophilisierte Protease, 45 mU / mg Protein, gelöst in 1,4ml sterilem H2Obidest = 20mg/ml 1 mU ist die Aktivität, die pro Minute folin-positive Aminosäure und Peptide freisetzt, die 1µmol Tyrosin entsprechen Puffer G2: 800mM Guanidino Hydrochlorid 30mM Tris Cl, pH 8,0 30mM EDTA, pH 8,0 5% Tween-20 0,5% Triton X-100 (Benzyltrimethylammoniumhydroxid) 100 - - - - Puffer QBT: 750mM NaCl 50mM MOPS (3-(N-morpholino)Propan Sulfon Säure), pH 7,0 15% (v/v) Isopropanol 0,15% (v/v) Triton X-100 (Benzyltrimethylammoniumhydroxid) Puffer QC: 1,0M NaCl 50mM MOPS (3-(N-morpholino)Propan Sulfon Säure), pH 7,0 15% Isopropanol Puffer QF: 1,25M NaCl 50mM Tris Cl, pH 8,5 15% Isopropanol QIAGEN Genomic 100/G Tips: Anionen-Austauscherharz-Säulen Fa. QIAGEN GmbH, Hilden; Deutschland 7.2.26 Glycerin 10%, steril eigene Herstellung Verdünnung Glycerin (99,2%, steril) 1:10 mit aqua bidest. (steril) auf Endkonzentration 10% Apotheke der Universitätsklinik Lübeck 7.2.27 Halbmikroküvetten, 1,5ml PMMA Plastibrand® , Fa. Brand; Deutschland 7.2.28 Harnstoff Fa. Bio-Rad, Hercules, California; USA 7.2.29 Isopropanol, 100%, p.a. Fa. Merck KGaA, Darmstadt; Deutschland 7.2.30 Kanamycin-Monosulfat Fa. Sigma Chemical Co, St. Louis; USA 7.2.31 Kit des pCR®- Blunt II-TOPO® Zero Blunt® TOPO® PCR Cloning Kit, Firma Invitrogen, Carlsbad, Californien; USA pCR®-Blunt II-TOPO® 10ng/µl Plasmid DNA in: 50% Glycerin 50mM Tris-HCL, pH 7,4 1mM EDTA 2mM DTT 0,1% Triton X-100 100µg/ml BSA Bromphenolblau dNTP Mix 12,5mM dATP 12,5mM dCTP 12,5mM dGTP 12,5mM dTTP pH 8,0 Salzlösung 1,2M NaCl 0,06M MgCl2 SOC Medium 2%Trypton 7.2.32 Kontroll-DNA Positivkontrolle Zelllinien: 101 Capan-1 Zelllinie aus humanem Pankreaskarzinom mit Mutation in p53 Exon 5 Codon 159 GCC CTC Deutsche Sammlung von Mikroorganismen und Deutschland Zellkulturen, Braunschweig; BXPC-3 Zelllinie aus humanem Pankreaskarzinom mit Mutation in p53 Exon 6 Codon 220 TAT TGT ATCC, Manassas, UA; USA DLD-1 Zelllinie aus humanem Kolon Adenokarzinom mit Mutation in p53 Exon 7 Codon 241 TCC TTC Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig; Deutschland SW 480 Zellinie aus humanem Kolonkarzinom mit Mutation in p53 Exon 8 Codon 273: CGT CAT Codon 309: CCC TCC Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig; Deutschland Negativ Kontrolle DNA aus Eigenblutspende des Verfassers der Dissertationsarbeit 7. 2.33 Laufmarker (Agarosegel) 10x konzentriert eigene Herstellung 40% (w/v) Saccharose (Fa. Merck KGaA, Darmstadt; Deutschland) 0,25% (w/v) Xylencyanol (Fa. Serva, Heidelberg; Deutschland) 0,1M EDTA, pH 8,0 7.2.34 Laufmarker (TTGE-Gel), 2x konzentriert eigene Herstellung 0,05% (w/v) Bromphenolblau (Fa. Merck KGaA, Darmstadt; Deutschland) 0,05% (w/v) Xylencyanol (Fa. Serva, Heidelberg) 70% (v/v) Glycerol (Fa. Caesar und Loretz, Hilden; Deutschland) aqua bidest. 7.2.35 LB-Platten eigene Herstellung, 10g NaCl [7.2.38], 10g Bacto Trypton [7.2.4], 5g Yeast Extract [7.2.5], ad 1 l aqua bidest., pH 7,5 mit 1M NaOH [7.2.39], 15g Agar [7.2.8] 7.2.36 Micro-Standard Bovines Serum Albumin, 1mg/ml in 0,15M NaCl und 0,05% Natriumazid (Konservierungsmittel) eingesetzt in 1:2 Verdünnung mit NaCl Lösung, 0,15M [7.2.37], entsprechend einer Konzentration von 0,5mg/ml 102 Fa. Sigma Chemical Co, St. Louis; USA 7.2.37 NaCl-Lösung, 0,15M eigene Herstellung 0,15M NaCl (Fa. Merck KGaA, Darmstadt; Deutschland) aqua bidest. (ultrafiltriert) 7.2.38 NaCl p.a. Fa. Merck KGaA, Darmstadt; Deutschland 7.2.39 NaOH-Lösung, 1M eigene Herstellung 1M NaOH (Fa. Merck KGaA, Darmstadt; Deutschland) aqua bidest. 7.2.40 Natriumbikarbonatlösung eigene Herstellung 0,75% (w/v) NaHCO3 (Fa. Merck KGaA, Darmstadt; Deutschland) aqua bidest. 7.2.41 NucleoSpin® Kit Pufferangaben vom Hersteller vertraulich. Firma Macherey-Nagel, Düren; Deutschland 7.2.42 Objektträger Super Frost Plus, 25x75x1 mm Fa. Menzel Gläser, Braunschweig; Deutschland 7.2.43 PCR-Reaktionsgefässe ThermoTubeTM PCR Tubes 0,2ml Fa. PeQlab, Erlangen; Deutschland 7.2.44 Plasmid Purifikation Maxi Kit Quiagen ® Puffer: P1: Resuspensionspuffer 50mM Tris-HCl, pH 8,0; 10mM EDTA; 100µg/ml RNase A P2: Lysepuffer 200mM NaOH, 1% SDS (w/v) P3: Neutralisationspuffer 3,0 mM K-Acetat, pH 5,5 QBT: Equilibrierungspuffer 750mM NaCl ; 50mM MOPS, pH 7,0 ; 15% Isopropanol (v/v); 0,15% Triton ® X-100 (v/v) QC: Waschpuffer 1M NaCl; 50mM MOPS, pH 7,0 15% Isopropanol (v/v) QF: Eluierungspuffer 103 1,25M NaCl; 50mM Tris-Cl, pH 8,5; 15% Isopropanol (v/v) TE: 10mM Tris-HCl, pH 8,0; 1mM EDTA Fa. QIAGEN GmbH, Hilden; Deutschland 7.2.45 Primer (basierend auf der Sequenz der p53mRNA mit der Genebank Acc. No NM_000546) lyophilisiert, lösen zu einer Endkonzentration von 1mM in sterilem aqua bidest. Primersequenzen s. Tabelle III PCR für TTGE, DGGE: p53ex 5-1 sense: Sequenzlänge 18 bp p53ex 5-1 antisense: Sequenzlänge 59 bp Länge des amplifizierten Genproduktes: 191 bp p53ex 5-2 sense: Sequenzlänge 60 bp p53ex 5-2 antisense: Sequenzlänge 20 bp Länge des amplifizierten Genproduktes: 176 bp p53ex 6 sense: Sequenzlänge 19 bp p53ex 6 antisense: Sequenzlänge 57 bp Länge des amplifizierten Genproduktes: 96 bp p53ex7 sense: Sequenzlänge 20 bp p53ex7 antisense: Sequenzlänge 60 bp Länge des amplifizierten Genproduktes: 192 bp p53ex8 sense: Sequenzlänge 20 bp p53ex8 antisense: Sequenzlänge 60 bp Länge des amplifizierten Genproduktes: 191 Sequenzierung: Primer 1: M13universal(-21), 18mer: 5’-TGT AAA ACG ACG GCC AGT-3’ Primer 2: M13Reverse (-29), 18mer: 5’-CAG GAA ACA GCT ATG ACC-3’ mit IRD 800 Farbstoff am 5’-Ende alle Primer: Fa. MWG Biotech, Ebersberg; Deutschland 7.2.46 Propanol (2-Propanol) Apotheke der Universität Lübeck 7.2.47 Proteinextraktionspuffer eigene Herstellung (nach: [163]) 25 mM Tris, pH 7,4 (Fa. Bio-Rad, Hercules, California, USA) 100 mM NaCl (Fa. Merck KGaA, Darmstadt; Deutschland) 20 mM NH4HCO3 (Fa. Merck KGaA, Darmstadt; Deutschland) 104 7.2.48 Pwo-Peqlab PCR System Enzym Mix: 3,5x103 U/ml 1 Unit entspricht der Enzymmenge, die benötigt wird, um 20nmol dNTP in 60min bei 65°C in säureunlösliche Substanz zu überführen Expand HF Puffer, 10x konzentriert, 15mM MgCl2, genaue Zusammensetzung nach Angabe des Herstellers vertraulich Fa. Roche Diagnostics GmbH, Mannheim; Deutschland 7.2.49 Quantikine Kit VEGF Microwell-Platte, 96 (12x8) wells aus Polystyrol, beschichtet mit monoklonalen Antikörpern aus der Ratte gegen VEGF VEGF-Konjugat, polyklonaler Antikörper gegen VEGF mit Meerrettich-Peroxidase konjugiert VEGF-Standard, humanes rekombinantes VEGF165 in gepufferter Proteinlösung, lyophilisiert (2000 pg) Assay-Verdünnung RD1W, gepufferte Proteinlösung Kalibrier-Verdünnung RD6U, Tierserum Waschpuffer, 25x konzentriert, gepuffertes Surfactant Farbreagenz A, Wasserstoffperoxid Farbreagenz B, Tetramethylbenzidin (Farbreagenz A und B 1:1 gemischt ergeben die Substratlösung) Stop-Lösung, 2N Schwefelsäure Fa. R&D Systems, Abingdon, Oxon; UK 7. 2.50 Rapid Gel XL, 40% Fa. Amersham Pharmacia Biotech, Piscatanay, New Jersey; USA 7. 2.51 Sawady Pwo DNA Polymerase Pwo-Polymerase, 1U/µl 1 Unit entspricht der Enzymmenge, die benötigt wird, um 10nmol dNTP in 30min bei 70°C in säureunlösliche Substanz zu überführen Reaktionspuffer, 10x konzentriert: 100mM Tris-HCL (pH 8,8) 50mM (NH4)2SO4 250mM KCl 20mM MgSO4 Fa. Peqlab, Erlangen; Deutschland 7.2.52 SDS-Lösung eigene Herstellung 10% (w/v) SDS (Fa. Sigma Chemical Co, St. Louis; USA) aqua bidest. 7. 2.53 Silbernitratlösung eigene Herstellung 0,1% (w/v) AgNO3 (Fa. Fluka Chemie GmbH, Buchs; Schweiz) aqua bidest. 7.2.54 SOC-Medium Fa. Invitrogen Corp., Carlsbad, California; USA 10g Bacto-Trypton 2,5 g Yeast-Extrakt 1ml % M NaCl-Lsg. 0,41ml 3M KCl-Lsg. pH 7,0 ad 500ml aqua bidest., autoklavieren 105 kalt hinzugeben nach steriler Filtration: 2,5ml 2M MgCL2 5ml 1M MgSO4 4,5ml 40% Glukose 7.2.55 Stop Solution 0,5M EDTA, 0,2ml Formamid, deionisiert, 9,5ml Bromphenol-Blau, 10mg aqua bidest. ad 10ml 7. 2.56 TAE-Puffer, 1x konzentriert eigene Herstellung 2% (v/v) TAE-Puffer, 50x [7.2.58] aqua bidest. 7.2.57 TAE-Puffer, 1,25x konzentriert eigene Herstellung 2,5% (v/v) TAE-Puffer, 50x [7.2.58] aqua bidest. 7.2.58 TAE-Puffer, 50x konzentriert eigene Herstellung 2M Trisbase (Fa. Sigma Chemical Co, St. Louis, USA) 5,7% (v/v) Eisessig (Fa. Merck KGaA, Darmstadt; Deutschland) 10mM EDTA (Fa. Merck KGaA, Darmstadt; Deutschland) aqua bidest., pH 8,0 7. 2.59 TBE Long Run Buffer, 10x eigene Herstellung 1340mM Tris 450mM Borsäure 25mM EDTA aqua bidest. eingesetzt als TBE Long Run Buffer, 1x, nach 1 :10 Verdünnung mit aqua bidest. bei 50°C ca. pH 8,5 7. 2.60 TEMED Fa. Gibco BRL 7.2.61 TE-Puffer eigene Herstellung 10mM Tris-HCl 7,5pH(Fa. Bio-Rad, Hercules, California, USA) 1mM EDTA (Fa. Merck KGaA, Darmstadt; Deutschland) aqua bidest. 7. 2.62 Thermosequenase Kit dNTP/ddNTP-Gemische: - A Reagenz: Tris-HCl, pH 9,5, MgCl2, Tween 20, Nonidet P-40, 2-Mercaptophenol, dATP, dCTP, 7-deaza-dGTP, dTTP, ddATP, thermostabile Pyrophosphatase, Thermo Sequenase DNA Polymerase - C Reagenz: 106 Tris-HCl, pH 9,5, MgCl2, Tween 20, Nonidet P-40, 2-Mercaptophenol, dATP, dCTP, 7-deaza-dGTP, dTTP, ddCTP, thermostabile Pyrophosphatase, Thermo Sequenase DNA Polymerase - G Reagenz: Tris HCl, pH 9,5, MgCl2, Tween 20, Nonidet P-40, 2-Mercaptophenol, dATP, dCTP, 7-deaza-dGTP, dTTP, ddGTP, thermostabile Pyrophosphatase, Thermo Sequenase DNA Polymerase - T Reagenz: Tris HCl, pH 9,5, MgCl, Tween 20, Nonidet P-40, 2-mercaptophenol, dATP, dCTP, 7-deaza-dGTP, dTT, ddTTP, thermostabile Pyrophosphatase, Thermo Sequenase DNA-Polymerase Fa. Amersham Pharmacia Biotech, Piscatanay, New Jersey, USA 7.3 Geräte in alphabetischer Reihenfolge 7.3.1 Gelkammer (Agarosegel) Minigelkammer Fa. cti, Idstein; Deutschland 7.3.2 Gelkammer (TTGE/DGGE) DCode Universal Mutation Detection System Fa. Bio-Rad, Hercules, California; USA 7.3.3 Homogenisator Ultra Turrax T25 basic Fa. IKA Labortechnik, Staufen; Deutschland 7.3.4 Mikroskop Axioskop Fa. Carl Zeiss, Jena; Deutschland 7.3.5 Mikrotom Mikrotom Jung SM 2000 R Fa. Leica, Wetzlar; Deutschland 7.3.6 Mikrowellengerät Fa. Siemens, Berlin; Deutschland 7.3.7 Photometer (ELISA) 340 ATTC Fa. SLT Lab Instruments, Crailsheim, jetzt: Tecan Austria, Salzburg; Österreich 7.3.8 Schwenktisch KS 250 basic Fa. IKA Labortechnik, Staufen; Deutschland 7.3.9 Sequencer DNA Sequencer Model 4000L Fa. LiCOR, Lincoln, Nebraska; USA 7.3.10 Spannungsgeber Bio-Rad Power Pac 300 107 Fa. Bio-Rad, Hercules, California; USA 7.3.11 Spectrophotometer Beckmann DU 640 Fa. Beckmann, Fullerton, California; USA 7.3.12 Thermocycler Primus 96 plus Fa. MWG Biotech, Ebersberg; Deutschland 7.3.13 Thermomixer Thermomixer compact Fa. Eppendorf, Köln; Deutschland 7.3.14 TGGE Gelkammer Fa. Whatman Biometra® , Göttingen; Deutschland 7.3.15 UV-Transilluminator Fa. MWG Biotech, Ebersberg; Deutschland 7.3.16 Wasserbad Type 850208, Nr. 1550 Fa. Braun, Melsungen; Deutschland 7.3.17 Werkbank, steril Class II Type A/B3, Nuaire Lagbard Sicherheitswerkbank (NU 425-400E) Fa. Nuaire, Plymouth, MN, USA 7.3.18 Zentrifuge Biofuge 22R Fa. Heraeus (Sepatech), Osterode; Deutschland 7.3.19 Zentrifuge Rotanta/TR Fa. Hettich, Tuttlingen; Deutschland 108 7.4 Analysedaten Klinisch pathologische Eigenschaften der 52 untersuchten Kolonkarzinompatienten 109 Nummer Alter bei OP 77 80 83 85 90 91 92 100 105 116 121 122 123 124 125 127 129 131 132 135 136 137 138 141 142 143 144 145 156 167 168 169 171 172 173 175 182 183 188 189 192 194 196 204 206 211 215 217 223 231 232 236 65 74 61 60 81 67 58 70 59 63 67 73 70 75 73 57 81 56 64 68 64 65 86 74 68 56 73 82 55 86 63 84 52 59 55 74 70 66 67 55 77 74 76 85 75 52 62 36 52 45 67 92 0=weiblich, 1=männlich 0 1 1 0 1 1 0 1 0 1 1 0 1 1 0 1 0 1 1 0 1 1 0 1 1 0 1 0 0 0 1 1 1 1 0 1 1 0 1 1 1 1 0 0 1 0 1 0 0 1 1 0 1=Kolon, 2=Rektum 2 2 1 2 1 2 2 2 1 1 2 1 1 2 1 1 2 1 1 2 2 1 1 1 2 1 1 1 2 1 1 2 2 2 2 2 1 1 1 2 1 1 2 2 1 2 1 1 1 1 1 1 VEGF-Konzentration 110 T N M 3 2 3 4 3 3 3 1 3 4 3 3 4 3 3 2 3 3 4 3 3 1 2 3 1 4 3 3 2 3 3 3 3 3 2 3 2 4 4 3 3 3 3 4 3 3 3 3 4 1 3 3 1 0 1 0 0 0 2 0 0 1 0 0 2 0 1 0 0 1 2 2 0 0 0 2 0 2 2 1 1 0 1 3 0 0 0 1 0 0 1 0 0 0 1 2 1 2 2 0 2 0 1 0 0 0 0 0 0 0 1 0 0 1 0 0 1 0 1 0 0 0 1 1 0 0 0 0 0 1 1 1 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 1 0 1 0 0 0 0 0 Dukes Grading c a c b b b d a b d b b d b d a b c d d b a a c a d d d c b c c b b a c a d c b b b c c d c d b c a c b 2 2 2 2 2 2 2 2 2 2 3 2 2 2 2 2 2 2 3 2 2 2 2 2 2 2 2 2 2 2 2 3 2 2 2 2 2 2 3 2 2 2 2 3 2 3 2 2-3 3 1 2 2 Nummer 77 80 83 85 90 91 92 100 105 116 121 122 123 124 125 127 129 131 132 135 136 137 138 141 142 143 144 145 156 167 168 169 171 172 173 175 182 183 188 189 192 194 196 204 206 211 215 217 223 231 232 236 N-Proben: VEGF T-Proben: VEGF N-Proben: VEGF T-Proben: VEGF Konzentration Konzentration Konzentration Konzentration (pg/ml (pg/ml (in pg VEGF / mg (in pg VEGF / mg Proteinextrakt) Proteinextrakt) Protein) Protein) 175,85 809,65 66,04 234,29 175,32 424,67 88,35 201,86 119,50 528,50 76,01 237,36 173,40 805,40 50,19 329,06 259,38 1418,74 110,99 1836,99 180,84 294,21 234,15 213,38 167,16 1559,43 73,08 597,96 142,51 280,58 51,90 407,36 191,28 1827,20 62,99 1322,64 430,05 8075,90 197,30 1866,52 236,25 3065,56 428,63 1304,87 247,16 1955,23 102,81 1135,02 153,82 15432,98 53,36 5642,11 94,85 3842,67 52,34 1435,69 123,22 3430,30 55,87 1842,58 54,42 5983,90 35,17 2340,72 116,49 3295,52 39,97 1383,49 150,99 561,20 44,13 158,28 525,68 1559,45 132,00 954,06 478,24 664,49 109,77 344,08 142,51 451,39 54,32 117,28 139,68 983,32 54,36 262,39 123,50 885,95 26,66 221,01 159,95 1623,51 60,58 552,33 223,61 6486,24 52,94 2077,31 455,94 4914,61 160,06 3029,37 211,59 2001,82 175,81 848,78 309,63 9396,96 743,11 22552,71 232,38 1935,09 557,72 4644,21 601,62 1695,48 189,66 381,67 259,58 3969,06 174,81 897,01 66,45 561,20 66,72 732,88 114,47 2740,76 81,54 1836,58 112,09 2835,19 43,83 1056,79 181,62 1152,64 59,13 468,09 109,70 2634,56 36,71 783,84 117,89 1737,08 98,88 1145,23 196,58 547,45 153,70 357,39 269,68 219,30 206,01 113,91 110,47 2026,80 85,57 1522,72 246,48 1630,52 174,82 1715,55 112,18 545,51 102,25 887,43 157,01 2521,04 94,87 1320,88 124,86 274,30 53,36 267,83 56,91 440,70 51,32 168,81 156,04 1541,07 84,20 955,62 111,32 569,10 85,16 284,29 150,23 5007,81 94,86 4370,12 176,28 353,92 104,56 307,42 70,57 515,35 30,38 225,30 284,70 1278,53 124,36 893,11 190,16 2436,40 78,78 2042,86 Mutationsnachweis im p53-Gen Exon 5-8 111 N-Proben: Mutation in der T-Proben: Mutation in der TTGE/DGGE (0=nein, TTGE/DGGE (0=nein, Nummer 1=ja) 1=ja) 77 0 1 80 0 1 83 0 1 85 nicht untersucht 0 90 nicht untersucht 0 91 nicht untersucht 0 92 0 1 100 nicht untersucht 0 105 nicht untersucht 0 116 0 1 121 0 1 122 nicht untersucht 0 123 nicht untersucht 0 124 0 1 125 0 1 127 nicht untersucht 0 129 nicht untersucht 0 131 nicht untersucht 0 132 0 1 135 0 1 136 nicht untersucht 0 137 nicht untersucht 0 138 nicht untersucht 0 141 0 1 142 nicht untersucht 0 143 0 1 144 0 1 145 nicht untersucht 0 156 nicht untersucht 0 167 nicht untersucht 0 168 nicht untersucht 0 169 0 1 171 nicht untersucht 0 172 0 1 173 0 1 175 0 1 182 nicht untersucht 0 183 nicht untersucht 0 188 0 1 189 0 1 192 0 1 194 0 1 196 0 1 204 0 1 206 nicht untersucht 0 211 nicht untersucht 0 215 0 1 217 0 1 223 0 1 231 nicht untersucht 0 232 nicht untersucht 0 236 nicht untersucht 0 Charakterisierung der Mutation durch Sequenzierung: 112 CodonSequenz Wt TGC GAG TGC GTG TGC TGC CGC CGC CodonSequenz Mut TTC GGG TTC GTT TTC TTC CAC CAC Mutation in Exon: ProbenNr.: Codon: 5 5 5 5 5 5 5 5 80 169 173 175 189 192 196 223 135 171 135 173 135 135 175 175 6 121 220 6 172 Deletion 193- 201 6 6 188 217 213 213 7 7 7 7 7 7 7 92 132 135 169 188 204 215 245 248 248 245 234 248 248 GGC CGG CGG GGC TAC CGG CGG AGC CAG CAG GAC CAC CAG TGG Gly Arg Arg Gly Tyr Arg Arg Ser Gln Gln Asp His Gln Trp 8 8 8 8 8 8 8 8 8 8 77 83 116 124 125 141 143 144 194 217 285 280 282 273 266 273 282 273 282 282 GAG AGA CGG CGT GGA CGT CGG CGT CGG CGG AAG ATA TGG CAT GAA TGT TGG CAT TGG TGG Glu Arg Arg Arg Gly Arg Arg Arg Arg Arg Lys Ile Trp His Glu Cys Trp His Trp Gly 7.5 Wt-AS Mut-AS Cys Glu Cys Val Cys Cys Arg Arg Phe Gly Phe Val Phe Phe His His TAT TGT Tyr 193 -CAT CTT ATC His, Leu, Ile, CGA GTG 193- CTG – Arg, Val, 201 GAA GGA Glu, Gly, AAT TTGAsn, Leu 201 CGA CGG Arg CGA CGG Arg Cys Leu Arg Arg Abbildungsverzeichnis Abbildung 1: Die fünf verschiedenen Isoformen des VEGF Abbildung 2: Mutationen und Eigenschaften des humanen p53-Protein Abbildung 3: Laufverhalten der DNA in der Denaturans Gradient Gelelektrophorese, DGGE Abbildung 4: Schema der Zeit Gradient Gelelektrophorese, TGGE Abbildung 5: Boxploterklärung Abbildung 6: TTGE-Gel zum Nachweis von Mutationen im Exon 8 des p53-Gen Abbildung 7: VEGF-Konzentration in Normal- und Tumorgewebe Abbildung 8: VEGF-Konzentration in Tumorgewebe mit und ohne Mutation im p53-Gen Abbildung 9: VEGF-Konzentration im Normalgewebe mit und ohne Mutation in Exon 5-8 im korrespondierendem Tumorgewebe Abbildung 10: VEGF-Konzentration im Tumorgewebe mit und ohne Mutation im p53-Gen bei Frauen und Männern Abbildung 11: VEGF-Konzentration im Tumorgewebe mit und ohne Mutation im p53-Gen in bei Lokalisation des Tumors im Kolon bzw. Rektum Abbildung 12: VEGF-Konzentration in Tumorgewebe mit und ohne Mutation im p53-Gen in 113 bei Einteilung der Tumore in die Dukes-Stadien Abbildung 13: VEGF-Konzentration im Tumorgewebe mit und ohne Mutationen im p53-Gen bei Einteilung der Tumore nach den T-Stadien der TNM-Klassifikation 7.6 Tabellenverzeichnis Tabelle I: Charakterisierung des Patientenkollektivs Tabelle II: Pipettierschema zur Erstellung der Standardreihe des VEGF-ELISAs Tabelle III: Angaben zu den verschiedenen Primern und den PCR-Produkten Tabelle IVa: Mastermix I für die Exons 5-1,6,7,8 Tabelle IVb: Mastermix I für das Exon 5-2 Tabelle Va: Mastermix II für die Exons 5-1,6,7,8 Tabelle Vb: Mastermix II für das Exon 5-2 Tabelle VI: Reaktionsbedingungen der PCR für die untersuchten Exons des p53-Gens Tabelle VII: Temperaturbereiche der einzelnen Exons in der TGGE Tabelle VIII: Positivkontrollen für die Mutationsanalyse der untersuchten Exons des p53-Gens Tabelle IX: Stammlösung für das TTGE-Gel Tabelle Xa: 10% Acrylamid-Lösung mit 100% Denaturans Tabelle Xb: 10% Acrylamid-Lösung mit 0% Denaturans Tabelle XI: Laufbedingungen der TTGE für die Exons 5-1,7,8 und der DGGE für die Exons 5-2 und 6 Tabelle XII: Mastermix für Sequenzierungs-PCR Tabelle XIII: Reaktionsbedingungen für die Cycle-Sequenzierung Tabelle XIV: Zusammensetzung des Gels für die Sequenzierung Tabelle XV: Häufigkeiten der p53-Mutation in den Exon 5-8 Tabelle XVI.: Verteilung der detektierten Mutationen auf die Exons 5-8 Tabelle XVII: Verteilung der Mutationen im p53-Gen Exon 5-8 in den 26 Tumorproben auf die einzelnen Codons Tabelle XVIII: Mutationen in p53 Exon 5 Tabelle XIX: Mutationen in p53 Exon 6 Tabelle XX: Mutationen in p53 Exon 7 Tabelle XXI: Mutationen in p53 Exon 8 Tabelle XXII: Verteilung der Mutationsart in den untersuchten Exons des p53-Gen Tabelle XXIII: Transitionen in CpG Dinukleotiden Tabelle XXIV: VEGF-Konzentration im Normal- und Tumorgewebe Tabelle XXV: VEGF-Konzentration im Tumorgewebe bei Patienten mit und ohne Mutation im p53-Gen im Tumorgewebe Tabelle XXVI: VEGF-Konzentrationa im Normalgewebe von Patienten mit und ohne Mutation im p53-Gen im Tumorgewebe Tabelle XXVII: VEGF-Konzentration im Tumorgewebe bei Patienten mit Mutation im Exon 5, 6, 7 und 8 Tabelle XXVIII: Unterschied in der VEGF-Konzentration im Tumorgewebe bei Patienten mit und ohne Mutation im p53-Gen, getrennt nach Geschlecht Tabelle XXIX: VEGF-Konzentration im Tumorgewebe bei Patienten mit und ohne Mutation im p53-Gen, getrennt nach Lokalisation des Tumors Tabelle XXX: Mutationen im p53-Gen, getrennt nach Dukes-Stadien Tabelle XXXI: VEGF-Konzentration im Tumorgewebe bei Patienten mit und ohne Mutation getrennt, nach Dukes-Stadien Tabelle XXXII: Mutationen im p53-Gen, getrennt nach T-Kategorie der TNM-Klassifikation Tabelle XXXIII: VEGF-Konzentration im Tumorgewebe bei Patienten mit und ohne Mutation im p53-Gen bei unterschiedlicher Invasionstiefe des Tumors nach dem TNM-System Tabelle XXXIV: VEGF-Konzentrationa im Tumorgewebe von Patienten mit einer Mutation im Codon 248 oder 273 des p53-Gens Tabelle XXXV: VEGF-Konzentrationa bei Patienten mit einer Mutation in Codon 175, 245 oder 282 des p53-Gens 114 8 Danksagung Hiermit möchte ich folgenden Personen meinen besonderen Dank aussprechen: Hr. Prof. Dr. R. Broll, Forschungslabor der Klinik für Chirurgie, Universitätsklinikum SchleswigHolstein, Campus Lübeck, für die freundliche Überlassung des Themas dieser Arbeit, die Bereitstellung des Arbeitsplatzes und die stets hilfreiche Unterstützung in allen Stadien meiner Arbeit Fr. Dr. U. Windhövel, für die stets freundliche und hilfreiche Unterstützung in allen Stadien meiner Arbeit sowie die Betreuung und Hilfe während der praktischen Arbeit Hr. PD Dr. H.-J. Friedrich, Institut für Biometrie und Statistik des Universitätsklinikums Schleswig-Holstein, Campus Lübeck, für die hilfreiche Beratung in den Bereichen der Statistik und der kritischen Ergebnisbetrachtung Fr. Elke Gheribi, Fr. Regina Kaatz, Fr. Vera Grobleben, Fr. Gisela Grosser-Pape und allen anderen Mitarbeitern des Forschungslabors der Klinik für Chirurgie, Universitätsklinikum Schleswig-Holstein, Campus Lübeck, für die tatkräftige Unterstützung und das gute Arbeitsklima Hr. Dr. Menke, ehemals Mitarbeiter des Instituts für Pathologie der Christian Albrecht Universitätsklinikum Schleswig-Holstein, Campus Kiel, für die Unterstützung im Bereich der TGGE und der Entwicklung einer geeigneten Färbetechnik für Polyacrylamidgele Hr. Prof. Dr. Stürzbecher, Institut für Humangenetik, Universitätsklinikum Schleswig-Holstein, Campus Lübeck, für Anregungen im Bereich der TTGE und bei der Auswahl der DNAKontrollen Hr. Dr. Till Milde, für die Zeit als Kommilitone und Freund über all die Jahre und den gemeinsamen Weg Meinen Eltern für die stetige und unerlässliche Unterstützung Fr. B.-L. Matthiessen und Fr. Dr. med. C. Küchlin für die Unterstützung, Anregung und Hilfe Hr. PD Dr. med. F. W. Tischendorf für die Beratung bei formellen Aspekten 115 9 Lebenslauf Curriculum Vitae Name: Geburtsdatum: Geburtsort: Familienstand: Adresse: Steffen W. Pfarr 24.06.1973 Wesel ledig Moosmattstr. 34 CH-6005 Luzern Tel.: +41 79 331 8010 e-mail: [email protected] Schulbildung: Besuch der Martin-Luther-Grundschule, Brilon Besuch des Gymnasium Petrinum, Brilon 1979- 1983 1983- 1992 Ausbildung: Ausbildung zum examinierten Krankenpfleger, Marsberg Oktober 1992September 1995 Studium: Humanmedizinstudium, Medizinische Universität Lübeck Ärztliche Vorprüfung Erstes Staatsexamen Gasthörer an der University of the Witwatersrand Johannesburg Oktober 1999 Zweites Staatsexamen Drittes Staatsexamen seit Oktober 1995 September 1997 August 1998 SeptemberMärz 2001 Juni 2002 Assistenzarzt: Assistenzarzt Chirurgie, PD Dr. med. R. Babst Kantonsspital Luzern Chirurgie A, Luzern Schweiz seit Juli 2004 Ausbildungsstelle in der allgemeinen Chirurgie, Dr. med. M. Lüdi Am Kantonsspital Obwalden, Sarnen Schweiz Januar 2003Juni 2004 Praktisches Jahr: I. Tertial: Innere Medizin, kardiologische Abteilung/ Intensivstation, Prof. Dr. med. H. Katus Medizinische Universität Lübeck II. Tertial: Chirurgie, Prof. Dr. med. M. von Flüe, Handchirurgie/Plastische Chirurgie, Dr. med. U. v. Wartburg/ Dr. med. E. Fritsche Kantonsspital Luzern/ Schweiz. III. Tertial: Zahn- Mund- Kiefer- Gesicht- Chirurgie, Prof. Dr. A. Pogrel, UCSF San Francisco/ USA und Prof. Dr. dent., Dr. med. v. Domarus, Medizinische Universität Lübeck 116 Aus-/ Weiterbildung: Jahr 2002: 25.-28. November: Weiterbildungsblock: Strahlenschutz in der Röntgendiagnostik, Gesundheitswesen und Wirtschaftlichkeit, Ethik und Berufsrecht, Notfallmedizin. Neustadt Schleswig-H./D 18. Dezember: „Brauchen wir noch die kontrollierte maschinelle Beatmung?“ Ärztekammer Hamburg/D Jahr 2003: 27. März: Workshop zur Untersuchungstechnik, Diagnostik, Therapie und Nachbehandlung von Schulterverletzungen Nottwil/CH 21.-24. Mai Jahreskongress der Schweizer Gesellschaft für Chirurgie und der Schweizerischen Gesellschaft für Thorax-,Herz- und Gefässchirurgie St. Gallen/CH 15. Juni: Erwerb der Sachkundezeiten Anwendungsgebiet Notfalldiagnostik 03.-06. September: 39th National Congress of Swiss Society of Plastic, Reconstruktiv and Aesthetic Surgery as well as Swiss ISAPS Course, Lugano/CH 23. Oktober: Workshop zur Untersuchungstechnik, Diagnostik, Therapie und Nachbehandlung von Knieverletzungen Nottwil/CH 05.-08. November: Kombinierter Grund- und Spezialkurs im Strahlenschutz (Fachkunde nach Röntgenverordnung) Landesärztekammer Schleswig- H./D 14.-19. Dezember: AO Kurs: Prinzipien der operativen Frakturbehandlung, Davos/CH Jahr 2004: Frühjahr: 4 Tage Hospitation in der Abteilung für Plastische Chirurgie, Handchirurgie, Intensiveinheit für Schwerbrandverletzte Universitätsklinikum Schleswig-Holstein Campus Lübeck 05. Februar: Handgelenksverletzungen- Gips oder OP Nottwil/CH 28. Februar- 5. März: 21st internationaler Workshop for Gastrointestinal Surgery, Davos/CH 23.-24. April: 28. April: 4. Ostseesymposium, Hand-Brust-Plastische Chirurgie Ästhetische-Rekonstruktive-Mammaplastik inkl. Übungen, Neustadt Schleswig-H./D Verletzungen des Handgelenkes- Primärdiagnostik und weitere therapeutische Schritte 17. Juni: Beurteilung und Versorgung proximaler Femurfrakturen Nottwil/CH 10. Juli: Notfallsonokurs KLS/Luzern CH 09. September: Gipskurs: Gipstechnik und praktische Übungen KSL/Luzern CH November: Schweizer Chirurgie Basisexamen absolviert 117 Veröffentlichungen: Präsentation der Arbeit auf dem Deutschen Krebskongress 2002 in Berlin, Abstract: S. Pfarr, T. Milde, U. Windhoevel, M. Duchrow, H.- P. Bruch, R. Broll: Correlation between VEGF tissue level and Ki-ras or p53 mutations in human colorectal carcinoma, J Cancer Res Clin Oncol, Suppl. to Vol. 128 (200), S.145. Präsentation der Arbeit auf den 6. Chirurgischen Forschungstagen in Köln, Abstract: U. Windhövel, S. W. Pfarr, T. Milde, H.-P. Bruch, R. Broll Correlation between VEGF tissue level an mutations of the p53 gene in human colorectal carcinomas Präsentation der Ergebnisse European Surgery Acta Chirurgica Austria, Abstract: 26th Seminar of the Austrian Society for Surgical Research, 2002 in Gosau R. Broll, T. Milde, S. Pfarr, O. Schwandner, U. Windhövel: Beeinflussen Mutationen im k-ras Onkogen die Expression des Vascular- Endothelial- GrowthFactors (VEGF) in kolorektalen Karzinomen? Eur. Surg. Vol. 34; Suppl. No. 189, S. 34; 2002 Praktische Tätigkeiten: Famulatur in der kardiologischen Abteilung der Medizinischen Klinik II, MUL, Prof. Dr. med. H. Katus Frühjahr 1998 Famulatur in der neurochirurgischen Klinik der Friedrich-Alexander Universität, Erlangen- Nürnberg Prof. Dr. med. R. Fahlbusch Herbst 1998 Praxisfamulatur radiologischen/ nuklearmedizinischen Praxis, Maria-Hilf-Krankenhaus in Brilon, Dr. med. R. Krämer Frühjahr 1999 Praxisfamulatur allgemeinmedizinischen Praxis Dr. med. B. Walters in Brilon. Frühjahr 1999 Famulatur in der chirurgischen Klinik Helen Joseph Hospital, Herbst 1999 Univerity of the Witwatersrand, Johannesburg, Prof. Dr. med. GJ Oettle Famulatur in der plastischen Chirurgie der Fachklinik Hornheide- Münster, Dr. med. H. Tilkorn Frühjahr 2000 Sonstiges: regelmässige Wochenend- und Nachtdienste im Krankenhaus Süd der Hansestadt Lübeck 1999- 2001 Präparationsarbeit für Fortgeschrittene Sommer 1997 Grundkurs Medizinische Ethik Kurs der Medizinischen Informatik und Statistik Kurs der Sportmedizin (2 Semester) Notfallmedizinisches Kolloquium (2 Semester) OP- Kurs der Klinik für Kiefer- u. Gesichtschirurgie Kurs der Plastisch- chirurgische Indikationen und Anatomie, mit Training des mikrochirurgischen Arbeiten der Plastischen Chirurgie 1995/96 1997 1998/99 1998/99 1999 2000 Steffen W. Pfarr Luzern den 01.01.2005 118