Molekulargenetik I
Werbung

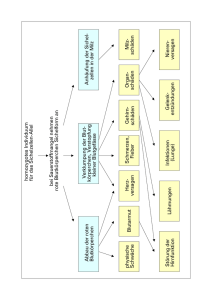
Humangenetik Molekulargenetik Teil 1 Vorlesungsmitschrift SS 05 1 Humangenetik: Molekulargenetik - Teil I „Ein Gen ist ein DNA-Abschnitt, der für ein funktionelles Produkt codiert.“ Das humane Genom: - Größe des Genoms: Anzahl der Gene: Gendichte: Gengröße (im Durchschnitt): Exons pro Gen (im Durchschnitt): Exongröße (im Durchschnitt): 2,8 Mrd. Basenpaare (bp) 20.000 – 25.000 1 Gen / 130.000 bp 27.000 bp 9 (1 bis 363) 120 bp (10 bis 7600 bp) Monogene Vererbung: Sind relativ selten (5-10% der Fälle): Mutationen des Gens weisen meist eine geringe Phänotyp-Variabilität auf, unterliegen einer geringen Modifikation durch die Umwelt Rezessive vs. Dominante Mutationen / Erkrankungen Rezessiv: - Heterozygot nicht betroffen - ‚loss of function’ - z.B. SMA (Verlust des SMN1 Gens) Dominant: - Heterozygot betroffen - ‚gain of function’ - Creutzfeld-Jacob (Mutationen im PrP Gen) Vererbungmodi: [Allel ‚A’ bzw. ‚X’ = gesund Allel ‚a’ bzw. ‚x’ = krank] Autosomal dominant: - betroffene Person hat i.d.R ein betroffenes Elternteil - beide Geschlechter betroffen - Nachkommen von Betroffenen haben ein a priori Erkrankungsrisiko von 50% A A - A A/A A/A A/A = 50% A/a = 50% a A/a A/a gesund krank z.B. : o Neurofibromatose Typ I o Chorea Huntington 2 Autosomal rezessiv: - betroffene Person hat i.d.R nicht betroffene Eltern - beide Geschlechter betroffen - beide Eltern sind i.d.R. Anlageträger (Carrier) - A priori Erkrankungsrisiko der Nachkommen: 25% A a A A/A A/a a A/a a/a A/A = 25% gesund, kein Anlageträger A/a = 50% gesund, Anlageträger a/a = 25% krank - z.B. : o Cystische Fibrose (CF) o Spinale Muskelatrophie (SMA) X-chromosomal dominant: - Betroffener Vater: o alle Töchter sind betroffen; Söhne sind NIE betroffen Keine Vater-Sohn Transmission X X - x Y X/x X/Y (krank) X/x X/Y (krank) Betroffene Mutter: o geschlechtsunabhängiges a priori Erkrankungsrisiko 50% X x X X/X x/X Y X/Y x/Y o selten! z.B. Rett-Syndrom o Ornithin-Transkarbamylase-Defekt X-chromosomal rezessiv: - keine Vater-Sohn Transmission - Mütter sind meist assymptomatische Anlageträger - Vorwiegend männliche Familienangehörige betroffen X x - X X/X X/x Y X/Y x/Y x/Y = kranke Männer, Erkrankungsrisiko der Männer 50% z.B.: o Rot-Grün Blindheit / Schwäche o Muskeldystrophie Duchenne / Becker 3 Y-chromosomal - sehr selten! Azoospermie Männliche Infertilität - nur Männer sind betroffen - alle Söhne eines Vaters erkranken Begriffe: - - - - Penetranz: o Anteil in %, mit dem sich ein Gendefekt phänotypisch manifestiert; bei unvollständiger Penetranz werden häufig Generationen übersprungen. Heterogenität: o Gleiche oder ähnliche Krankheitsbilder können durch Mutationen in verschiedenen Genen verursacht werden o Standartbeispiel Taubheit: Über 50 Gene/Genorte bekannt Pleiotropie: o Eine Mutation kann scheinbar unabhängige phänotypische Merkmale verursachen o Beispiel: Marfan-Syndrom (AD, Fibrillin-1 Gen): generelle Bindegewebserkrankung, verschiedene Organsysteme sind betroffen (Auge, Skelett, kardiovaskuläres System...) Variable Expressivität: o Variable phänotypische Manifestation trotz identischer Mutation o häufig bei autosomal dominanten Erkrankungen o Beispiel: Syndactylie Typ I Inzidenz: o Anzahl der Neuerkrankungsfälle einer bestimmten Erkrankung innerhalb eines bestimmten Zeitraums Z.B.: SMA 70 Neuerkrankungen / Jahr Prävalenz: o Anzahl der Fälle einer bestimmten Erkrankung zu einem bestimmten Zeitpunkt Z.B:. SMA etwa 5.000 SMA-Patienten (zur Zeit) - A priori Wiederholungsrisiko bei monogenen Erkrankungen - Beispiel 1: o Cystische Fibrose o wird autosomal rezessiv vererbt o beide Eltern sind heterozygote Anlageträger o die Wahrscheinlichkeit, dass das Kind ein krankes Allel von einem Elternteil bekommt ist 50% (Vater (A/a) 50% ‚A’ und 50% ‚a’ und Mutter (A/ a) 50% A und 50% a) o Rechnung: ½ x ½ = ¼ 25% 4 - Beispiel 2: o Spinale Muskelatrophie Typ III o wird autosomal rezessiv vererbt o Heterozygotenfrequenz 1:50 o Rechnung: 1 x 1/50 x ½ = 1/100 1% 1 weil der kranke Mann homozygot ist und das kranke Allel zu 100% weitergeben wird. (100% = 1). 1/50 weil die Ehefrau des Erkrankten mit einer Wahrscheinlichkeit von 1/50 heterozygot ist (Heterozygotenfrequenz). ½ weil, wenn die Ehefrau des Erkrankten nun wirklich heterozygote Trägerin ist (also A/a), sie das kranke Allel zu 50% (½) an das Kind weitergeben wird. - Beispiel 3: o Ausrechnung der Heterozygotenwahrscheinlichkeit Die Frage lautet also: Wie hoch ist die Wahrscheinlichkeit, dass der Sohn heterozygoter Anlageträger ist? o Vater und Mutter heterozygot o Rechunug: A a A A/A A/a a A/a a/a o a/a können wir ausschließen, weil der Sohn phänotypisch keine Anzeichen für eine Erkrankung zeigt, könnte also heterozygot betroffen sein. o Es bleiben also nur noch A/a, A/a, A/A übrig. Diese Allele könnte der ‚gesunde’ Sohn also haben. davon sind aber nur 2 heterozygot (A/a) also 2 von 3 o d.h. er ist mir einer Wahrscheinlichkeit von 2/3 heterozygot Heterozygotenwahrscheinlichkeit: 2/3 5 - Beispiel 4: o Cystische Fibrose (autosomal rezessiv) o Heterozygotenfrequenz 1:25 o Rechnung: 2/3 x ½ x 1/25 x ½ = 1/150 2/3 weil: Heterozygotenwahrscheinlichkeit des „gesunden“ Sohnes (s.Beispiel 3) ½ weil: wenn er („gesunde“ Sohn) heterozygot ist, gibt er das kranke Allel mit einer Wahrscheinlichkeit von ½ (50%) ab. 1/25 weil: Heterozygotenfrequenz, d.h. die Ehefrau ist mit einer Wahrscheinlichkeit von 1/25 heterozygote Anlageträgerin. ½ weil: wenn sie (Ehefrau) heterozygot ist, gibt sie das kranke Allel mit einer Wahrscheinlichkeit von ½ (50%) ab. - Beispiel 5: o Cystische Fibrose (autosomal rezessiv) o Heterozygotenfrequenz 1:25 o Rechnung: ½ x 1/25 x ½ = 1/100 in diesem Beispiel ist es schon bekannt, dass der phänotypisch gesunde Sohn heterozygoter Anlageträger ist. ½ weil: Sohn ist heterozygot und gibt Allel mit Wahrscheinlichkeit ½ weiter 1/25 weil: Heterozygotenfrequenz der Ehefrau ½ weil: wenn Ehefrau krank, dann wird das kranke Allel mit Wahrscheinlichkeit ½ weitergegeben 6 Zustandsformen eines Gens - Genlocus = Position eines Gens auf dem Chromosom Allel = eine oder mehrere alternative Form eines Genlocus Wildtyp Allel Mutiertes Alles a. Heterozygot = verschiedene Allele an einem Genlocus b. Homozygot = identische Allele an einem Genlocus c. Hemizygot = nur 1 Allel vorhanden (z.B. X-chromosomal bei Männern) - Polymorphismus = Mindestens 2 verschiedene Allele an einem Genlocus Allele müssen mind. Eine Frequenz von 1% in der Population aufweisen. Varianten = keine Auswirkung auf den Phänotyp Mutation = Auswirkung auf den Phänotyp - Genotyp-Phänotyp Korrelation - Allelische Heterogenität: o Unterschiedliche Mutationen in ein und demselben Gen verursachen identischen Phänotyp Genetische Variabilität: o Mutationen in ein und demselben Gen verursachen unterschiedliche Phänotypen Genetische (Locus-) Heterogenität: o Mutationen in verschiedenen Genen verursachen den gleichen Phänotyp Pleiotropie: o Mutation verursacht eine scheinbar unzusammenhängende phänotypische Expression Beispiele von autosomal-dominanten Erkrankungen Familiäre Hypercholesterinämie - häufigste, autosomal-dominante Erkrankung Prävalenz: 1:200 bis 1:1000 LDL-Cholesterin ist auf das Zwei- bis Dreifache erhöht. LDL transportiert Chlesterin, das für die Biosynthese von Steroidhormonen benötigt wird. - bei Homozygotie treten in den ersten Lebensjahren auf: o Xanthome (Hautveränderungen) o Artheriosklerose o Herzinfarkt o Porzellanweiße Arcus lipoides am Außenrand der Iris o Lebenserwartung 20J bei Heterozygotie: o Lebenserwartung 40-60J - 7 - basiert auf Mutation im LDL-Rezeptorgen dadurch werden keine oder nicht funktionsfähige Rezeptoren gebildet Homozygote können keine normalen Rezeptoren bilden Heterozygote haben verminderte Rezeptoren - LDL-Rezeptorgen ist am kurzen Arm des Chromosoms 19 - verschiedene Mutationen (wie z.B. Punktmutationen, Deletionen, Insertionen...) können entlang des gesamten Gens zur Hypercholesterinämie führen. allelische Heterogenität Chorea Huntington - neurodegenerative Erkrankung mit autosomal-dominantem Erbgang schwere fortschreitende Erkrankung des Nervensystems Ursache: Degenration der Neurone im Striatum und Cortex Häufigkeit 1:10.000 Verlust motorischer und intellektueller Kontrolle Beginn: ca. 35-40 Lebensjahr (hohe Variabilität: 7-70J) Molekulare Ursache: o Gen für Chorea Huntington liegt auf dem kurzen Arm des Chromosoms 4 o Dieses Chorea-Huntington-Gen (HD-Gen) ist ein expandierendes CAG-Repeat (CAG-CAG-CAG-CAG-CAG-.....) Bei gesunden <36 Repeats 36-41 Repeats Prämutation je mehr Repeats umso schwerer der Grad der Erkrankung Normal Chorea Huntington Akkumulation von kernhaltigen und cytoplasmatischen Aggregaten in den Neuronen toxischer Effekt Degeneration der Neurone o Gain-of function Mutataion o Verlust eines HD-Gens verursacht keinen Phänotyp CAG-Tripletts werden bei der Proteinsynthese in die Aminosäure Glutamin übersetzt. alle Krankheiten mit CAG-Repeats neurodegenerative Störungen 8 Myotone Dystrophie (MD) - autosomal-dominant Häufigkeit: 1:8000 Aktive und passive Myotonie Fortschreitende Muskelschwäche Schluckstörungen Herzrhythmusstörungen Katarakt Hypersomnie Endikrinologische Störungen (wie Diabetes mellitus und Hypogonadismus) - CTG-Expansion in der 3’nicht -translatierenden Region des DMPK-Gens (Chromosom 19q 13,3) Variable Expression durch Repeatverlängerung o Normal 5-35 Repeats o Prämutation 35-50 Repeats o Erkrankt 50-2000 Repeats Antizipation und variabler Schweregrad bei MD Immer maternaler Herkunft - - Achondroplasie - Autosomal-dominant Häufigkeit: 1:15.000 disproportionierter Kleinwuchs stark verkürzte Extremitäten (v.a. proximal) relativ kurze Finger Dreizackhand (vermehrter Abstand zw. 3. und 4. Finger) großer Kopf, vorgewölbte Stirn tiefe Nasenwurzel deutliche Lordose fast normale Sitzhöhe Ursache: o Mutationen im Fibroblasten-Growth-factor-Rezeptor III (FGFR III) Dieses FGFR III-Protein hat3 Domänen 1. Extrazelluläre Immunglobulin Domäne thanatophore Dysplasie 2. Transmembran Domäne G380R Achondroplasie 3. Tyrosin.Kinase Domäne N540K Hypochondroplasie Kraniosynostose genetische Variabilität: Mutationen in ein und demselben Gen verursachen unterschiedliche Phänotypen 4 verschiedene Phänotypen 9 Marfan-Syndrom - autosomal-dominant generalisierte Bindegewebsschwäche (Störung der Kollagensynthese) Defekt im Fibrillin-Gen Disproportionierter Hochwuchs Trichterbrust Lange schmale Extremitäten Arachnodaktylie überstreckbare Gelenke und Sehnen Kardiovaskuläre Veränderungen o Mitralklappenprolaps o Aortenaneurysma Linsenluxation und andere Augenfehler Pleiotropie: Eine Mutation kann scheinbar unabhängige phänotypische Merkmale verursachen Neurofibromatose TYP I (von Recklinghausen-Krankheit) - autosomal-dominant Häufigkeit 1:3000 Cafe-au-lait-Flecken Neurofibrome Axillary- oder Inguinal-Freckling o sommersprossenartige Hautveränderungen in den Achselhöhlen und Leisten - Irishämatome (Lisch-Knötchen) Weitere Symptome: - Skoliose - Pseudoarthrose - Makrozephalie Neurofibrome können in verschiedenen Organen auftreten Funktionsstörungen z.B.: o psychomotorische Retardierung o Kleinwuchs o kraniofaziale Dysmorphiezeichen auch hier Pleiotropie - verantwortliche Gen auf Chromosom 17 o NF1-Gen 10 führt zu sekundären