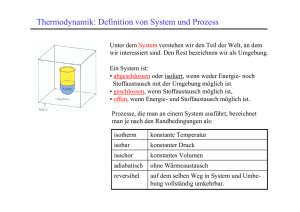
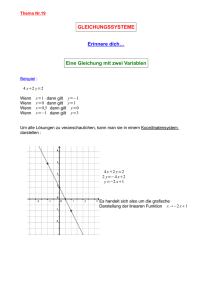
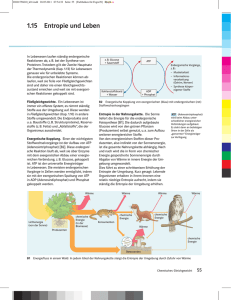
7 Druck und Temperatur
Werbung

- 40 - 7 Druck und Temperatur Bei der Einführung der Gibbs-Euler-Funktion (7.1) war angenommen worden, dass alle unabhängigen Größen außer der Entropie bereits in anderen Bereichen der Physik oder Chemie definiert worden waren. Bei allen intensiven Größen unter Einschluß von Druck und Temperatur muss die Übereinstimmung mit den entsprechenden Größen aus anderen Bereichen gezeigt werden. Insbesondere ist daher zu zeigen, dass der thermodynamische Druck und die thermodynamische Temperatur (7.2), (7.3) mit dem in der Mechanik definierten Druck bzw. der gasthermometrischen Temperatur entsprechend Gl. (1.2.8) übereinstimmen. Um dieses zeigen zu können, wird angenommen, dass die thermodynamische Energie E (oder die Innere Energie U) der gleichbenannten mechanischen Größe entspricht. 7.1 Druck Zuerst soll der Zusammenhang zwischen dem thermodynamischen Druck und der thermodynamischen Kraft untersucht werden. Wir benutzen das in Abb. 18 gezeigte abgebildete, zusammengesetzte isolierte System in einem Raum ohne Gravitationsfeld. Das Gas mit dem Druck p bildet ein System. Der masselose, volumenlose und reibungsfreie Kolben bildet die Wand zwischen den Systemen. Auf ihn wirkt die Feder F mit der Kraft K, die das 2. System bilden. Die Feder sei eine Spezialanfertigung. Ihre Kraft sei immer gerade so groß, dass sie der vom Gas über den Druck auf den Kolben ausgeübten Kraft das Gleichgewicht hält. Sie sei weiterhin ideal. Darunter versteht Abb. 19 Feder mit Abb. 18 Gaskompresman eine Feder, bei der eine einmal angeregte Masse sion Schwingung unendlich lange fortdauert (siehe Abb. 19). Auch hier möge kein Gravitationsfeld vorhanden sein. Der Schwingungsprozess im isolierten System ist offensichtlich reversibel und daher )S = 0. Da die Masse zumindest an den beiden Totpunkten die gleiche Entropie aufweisen muss, ist die Entropie dieser idealen Feder unabhängig von ihrer Dehnung. Mit ähnlichen Verfahren kann man die Isentropie vieler idealer, d. h. reibungsfreier, mechanischer Prozesse nachweisen. Bei dem Prozess mit dem Gas handelt es sich wegen der kraftfreien Verschiebbarkeit des Kolbens um einen reversiblen und daher isentropen Prozess. Da die Feder und der Kolben ihre Entropie nicht ändern, muss die des Gases auch konstant bleiben. Von der GFFF verbleibt daher (7.1.1) wobei sich p und V auf das Gas, K und r auf die Feder beziehen. Wegen der Parallelität von K und r genügen skalare Größen in der Gleichung. Einführung der Kolbenfläche A ergibt (7.1.2) Hier muss man mit den Vorzeichen aufpassen. V wächst bei der Ausdehnung des Gases, r wird jedoch - 41 kleiner, d. h. es gilt (7.1.3) Aus Gl. (7.1.2) folgt wie beim mechanischen Druck (7.1.4) Jetzt muss noch nachgewiesen werden, dass die thermodynamische Kraft der mechanischen entspricht. In der Mechanik gilt (7.1.5) oder (7.1.6) Was wird bei der partiellen Differenziation der Federenergie konstant gehalten? Sicher die Stoffmenge, der Impuls, der Drehimpuls und & wie wir festgestellt haben & die Entropie. Gl. (7.1.6) stimmt daher mit der thermodynamischen Definition der Kraft überein. Wegen Gl. (7.1.4) gilt dies auch für den Druck. Wir stellen daher fest: Der thermodynamische Druck eines Gleichgewichtssystems kann mit jedem geeigneten mechanischen Druckmessgerät bestimmt werden. 7.2 Temperatur Jetzt soll der Nachweis geführt werden, dass die thermodynamische und die gasthermometrische Temperatur übereinstimmen. Dazu wird wie folgt vorgegangen. Die Gibbssche Fundamentalform wird für konstante Stoffmengen umgeschrieben. (7.2.1) dU wird durch das totale Differenzial von U(V,T) ersetzt. (7.2.2) Weiterhin wird der Schwarzsche Satz auf das Differenzial (6.13) der Freien Energie angewendet. (7.2.3) Vergleich mit Gl. (7.2.2) ergibt schließlich (7.2.4) Für Stoffe mit (MU/MV)T = 0 gilt daher (7.2.5) Integration dieser Differenzialgleichung liefert (7.2.6) wobei C eine frei wählbare Konstante ist. Für Stoffe mit (MU/MV)T = 0 ist daher bei konstantem Volumen der Druck proportional zur thermodynamischen Temperatur. Das Experiment zeigt nun, dass für bestimmte Gase, die Idealen Gase, (MU/MV)T = 0 gilt (siehe Kap. 10), d. h. bei diesen Gasen ist bei - 42 konstantem Volumen der Druck proportional zur Temperatur. Gasthermometer arbeiten nun nach der Messvorschrift p % T, so dass die gasthermometrische Temperatur mit der thermodynamischen Temperatur übereinstimmt, wenn für beide Temperaturskalen die Proportionalitätskonstante C geeignet& z. B. durch die Festsetzung T = 273,16 K für die Temperatur des Tripelpunkts von Wasser & festgelegt wird. Die thermodynamische Temperatur eines Systems im Gleichgewicht kann daher mit jedem geeigneten Gasthermometer gemessen werden. Bei der Messung wird davon Gebrauch gemacht, dass die Temperaturen von zwei Körpern im Gleichgewicht übereinstimmen. 8 Arbeit und Wärme Die Prozessgrößen Arbeit und Wärme erlauben für viele Zwecke eine sehr praktische Beschreibung von Systemwechselwirkungen. Wir verwenden dazu 2 Systeme entsprechend Abb. 20 und machen die Annahme: Das System N ist geschlossen und im Gleichgewicht. Alle Prozesse in ihm verlaufen reversibel. Die Einschränkung auf ein reversibles zweites System erlaubt eine einfache Beschreibung der Wechselwirkungen. Das zweite System stellt sozusagen das Messinstrument für die Wechselwirkungen mit dem ersten System Abb. 20 Zwei wechselwirkende Systeme dar, das selbst keinen Einschränkungen bezüglich chemischer Reaktionen oder irreversibler Prozesse unterliegt. Bei Isolation des zusammengesetzten Systems gilt (8.1) (8.2) Der Summenterm mit den chemischen Potenzialen verschwindet, da sich das zweite System nach Voraussetzung im Gleichgewicht befindet, d. h. auch jede chemische Reaktion in ihm. Wegen Gl. (5.4.8) muss daher der Summenterm für die Reaktanden verschwinden. Für nicht reagierende Komponenten verschwindet er, da kein Materiefluss erlaubt ist. Für einen Prozess zwischen den Zuständen 1 und 2 gilt: (8.3) Jetzt werden folgende Definitionen getroffen. (8.4) (8.5) Die Größen A und Q werden als Arbeit und Wärme bezeichnet. Gl. (8.3) kann daher in der folgender Form geschrieben werden: Für ein geschlossenes System gilt - 43 (8.6) Vom Standpunkt des Systems aus gesehen sagt man auch, am System wird Arbeit (die Innere Energie wird vergrößert und AN ist positiv) oder vom System wird Arbeit (die Innere Energie wird kleiner und AN ist negativ) geleistet. Entsprechend gilt für die Wärme: In das System fließt Wärme (die Innere Energie wird vergrößert und QN ist positiv) oder aus dem System fließt Wärme (die Innere Energie wird kleiner und QN ist negativ). Unter der Bedingung, dass beide Systeme geschlossen und im Gleichgewicht sind, dürfen anstelle der gestrichenen Größen in den Gl. (8.4) und (8.5) auch die Systemgrößen verwendet werden. Die Differenziale der Inneren Energie in Gl. (8.1) werden entsprechend der Gibbsschen Fundamentalform ausgeschrieben. (8.7) Die Summenterme mit den chemischen Potenzialen verschwinden aus den oben genannten Gründen. Es verbleibt daher (8.8) Integration liefert (8.9) Analog zu den Gl. (8.4) und (8.5) setzt man daher (8.10) (8.11) Gl. (8.6) bleibt gültig. (8.12) Ein Beispiel für ein System, bei dem Gl. (8.4) gültig ist, Gl. (8.10) jedoch nicht, ist die schnelle Kompression eines Gases. Im Fall der schnellen Kompression entstehen Druckunterschiede im Gas und Gl. (8.10) verliert ihren Sinn. Gl. (8.4) bleibt gültig, wobei pN = K/A den von außen auf den Kolben wirkenden Druck darstellt. Ein System bzw. die das System umgebende Wand wird als adiabatisch bezeichnet, wenn zwischen System und Umgebung kein Wärmefluss erfolgt. Ein Prozess, der bei konstanter Entropie anläuft, wird als isentrop bezeichnet. Ein adiabatischer und reversibler Prozess in einem geschlossenem System ist isentrop. Der Nachweis wird mit Gl. (8.11) erbracht, die unter den vorausgesetzten Bedingungen gültig ist. Ein Verschwinden von Q für jeden Teil des Prozessweges lässt sich wegen T > 0 nur dadurch erreichen, dass die Integralgrenzen zusammenfallen, d. h. der Prozess isentrop verläuft. Der Begriff Arbeit wird auch für andere Wechselwirkungen, als die in Gl. (8.10) definierte Volumenarbeit verwendet. So gibt es z. B. die "normale" Arbeit - 44 - (8.13) und die elektrische Arbeit (8.14) Wie bestimmt man in einem real durchgeführten Experiment, die in ein System geflossene Arbeit und Wärme? Für die Arbeit verwendet man die Gl. (8.4), (8.10) oder (8.13), die messtechnisch leicht zugängliche Größen enthalten. Für die Wärme ist die Anwendung der Definitionsgleichungen (8.5) oder (8.11) schlecht praktikabel. Am Ende von Kap. 4 war ein Verfahren besprochen worden, mit dem man mit einer Heizwicklung definiert Wärme in ein System fließen lassen konnte. Es gilt (8.15) und daher (8.16) Gl. (8.6) bzw. (8.12) stellen den Ausgangspunkt der traditionellen Darstellung der Thermodynamik dar. Man betrachtet Energie und Arbeit als durch die Mechanik vorgegebene Größen. Gl. (8.12) stellt dann die Definitionsgleichung für die Wärme dar. Gl. (8.12) ist dem Anfänger sicher leichter zugänglich als die GFF. Der große Nachteil dieser Darstellung ist, dass eine direkte Herleitung der GFF aus Gl. (8.12) nicht möglich ist. Gl. (8.11) als Differenzial geschrieben lautet (8.17) Das sieht nach einem sehr einfachen Berechnungsverfahren für die Entropie aus! Leider weist diese Gleichung eine Reihe Schwierigkeiten auf. Die erste Schwierigkeit besteht darin, dass dQ kein Differenzial ist und auch die Division durch T kein Differenzial daraus machen kann. Auf der linken Seite der Gleichung steht jedoch ein Differenzial. Da dQ kein Differenzial ist, schreibt man auch anstelle von dQ häufig *Q oder pQ. Dies macht die Sache aber nicht besser. Eine zweite Schwierigkeit besteht darin, dass Gl. (8.17) bei irreversiblen Prozessen versagt. Denken Sie z. B. an den Temperaturausgleich zwischen zwei Körpern. Von außen wird keine Wärme zugeführt; trotzdem erhöht sich die Entropie des aus den beiden Körpern zusammengesetzten Systems. Gl. (8.17) muss daher entsprechend den für Gl. (8.11) getroffenen Voraussetzungen auf reversible Prozesse beschränkt werden. Um Gl. (8.17) bei irreversiblen Prozessen verwenden zu können, wird wie folgt verfahren. Die Entropie ist eine Zustandsfunktion. Es ist daher für die Entropieänderung bei einem Prozess völlig egal, auf welchem Weg dieser Prozess durchgeführt wird. Es müssen nur Anfangs- und Endzustand für beide Prozesse übereinstimmen. Wählt man nun einen reversiblen Ersatzprozess mit gleichem Anfangs- und Endzustand wie der irreversible Prozess, so darf die Berechnung mit (8.17) durchgeführt werden. Als Beispiel wollen wir die bereits diskutierte in Kap. 4.1 diskutierte Entropieänderung für den Temperaturausgleich zwischen zwei identischen Körpern mit den Temperaturen TN und TO zu ansehen. Der reversible Ersatzprozess besteht darin, dass wir unendlich viele Wärmereservoire zwischen den Temperaturen TN und TO zur Verfügung stellen, so dass wir den wärmeren Körper bei p = const. - 45 reversibel auf die Endtemperatur abkühlen und den kälteren reversibel aufwärmen können. Die entsprechende Rechnung führt zum gleichen Ergebnis wie in Kap. 12, wo die Berechnung auf einem anderen Weg durchgeführt wird. Läuft ein Prozess reversibel und isotherm ab, so darf die Temperatur vor das Integral gezogen werden, und es gilt (8.18) Arbeit und Wärme sind keine Zustandsgrößen. Wir vergleichen hierzu die Expansion eines Idealen Gases in ein evakuiertes und isoliertes Gefäß mit einem konstanten Volumen (I) mit einer Expansion unter isothermen, reversiblen Bedingungen (II). Beim Prozess I gilt: (8.19) Beim Prozess II gilt wegen (MU/MV)T = 0 für ein Ideales Gas (siehe auch Kap. 10): (8.20) und daher mit Q und A 0 (8.21) Stimmen die Anfangszustände des Gases bei beiden Prozessen und die Endvolumina überein, so stimmen auch die Endzustände überein, da sich die Temperatur bei beiden Prozesse nicht ändert. Arbeit und Wärme unterscheiden sich also, obwohl die Zustände des Systems identisch sind. Diese Prozesse sind auch gute Beispiele für Entropieberechnungen mit Gl. (8.17). Wendet man Gl. (8.17) unzulässigweise auf den irreversiblen Prozess der Gasexpansion in das Vakuum mit Q = 0 an, so ergibt sich das offensichtlich falsche Ergebnis )S = 0. Der Vorgang ist ja irreversibel und die Entropie muss zunehmen. Richtig ist dagegen die Anwendung beim reversiblen Ersatzprozess der isothermen Expansion (siehe Kap. 12). - 46 - 9 Innere Energie und Enthalpie als Funktionen der Temperatur Im Kapitel 3 war die Gibbs-Euler-Funktion (9.1) eingeführt worden. Als unabhängige Größen enthält sie die Entropie, das Volumen und die Stoffmengen. Auf der anderen Seite gibt es die praktische Messtechnik, für die es sehr schwer ist, die Entropie konstant zu halten oder definiert zu verändern. Zur Ausführung von Messungen ist es erheblich sinnvoller, folgende Funktionen zu untersuchen (9.2) (9.3) Die Differenziale dieser Funktionen lauten für einen einheitlichen Bereich (9.4) (9.5) Hier haben die Differenzialquotienten (MU/MV) und (MU/Mni) wegen der anderen Konstanthaltungen eine andere Bedeutung als in der GFF; insbesondere sind die (MU/Mni) nicht die chemischen Potenziale. Die Abhängigkeit der Inneren Energie und der Enthalpie von der Temperatur wird in diesem Kapitel, die Abhängigkeit vom Volumen und den Stoffmengen bei chemischen Reaktionen in den Kapiteln 10 bzw. 11 behandelt. 9.1 Molwärmen Unter den Molwärmen versteht man die Differenzialquotienten (9.1.1) Die klein geschriebenen Symbole u und h zeigen, dass 1 mol der reinen Substanz oder Mischung betrachtet werden soll. Der Name Molwärme bei konstantem Volumen bzw. konstantem Druck ist in den frühen Zeiten der Thermodynamik entstanden: Man führte 1 mol einer Substanz bei konstantem Volumen oder Druck Wärme zu. Die für eine Temperaturerhöhung von 1 K notwendige Wärme wurde als Molwärme bezeichnet. Gegen diese Bezeichnung spricht auch, dass die Energiezufuhr mit allen Energieformen und nicht nur mit Wärme erfolgen darf; so wird für Messzwecke die Energie üblicherweise in Form elektrischer Energie zugeführt. Geeignetere Namen wären: molare Energie- bzw. Enthalpiekapazität. Was ist unter der Konstanthaltung der ni in Gl. (9.1.1) in chemisch komplexen Gleichgewichtssystemen, wie z. B. Wasser, zu verstehen? Während der Temperaturänderung ist es sicherlich nicht möglich, die Stoffmengen der einzelnen Assoziate konstant zu halten. In solchen Fällen betrachtet man 1 mol der fiktiven Substanz H2O & in welcher Form sie auch immer vorliegen möge & und untersucht ein geschlossenes System. - 47 9.1.1 Molwärmen von Gasen Abb. 21 zeigt die Temperaturabhängigkeit der Molwärme cL für einige Gase bei niedrigen Drücken. Aufgetragen ist cL/R gegen die Temperatur T. Die Größe cL/R ist dimensionslos und es ergeben sich so offensichtlich in vielen Fällen glatte Werte. Die Edelgase weisen unabhängig von der Temperatur cL/R = 3/2 auf. Wasserstoff und Stickstoff ergeben bei höheren Temperaturen 5/2. Wasser ergibt einen Wert oberhalb von 3. Ein tieferes Verständnis für dieses Verhalten wird erst mit den Mitteln der Statistischen Thermodynamik möglich sein. Hier soll nur folgendes erwähnt werden. Für jeden molekularen Bewegungsfreiheitsgrad beträgt Abb. 21 Molwärmen einiger Gase als f(T) cL = R/2. Einatomige Gase, wie die Edelgase: 3 Translationsfreiheitsgrade, cL = 3/2 R. Zweiatomige Gase, wie H2 und N2: 3 Translations- und 2 Rotationsfreiheitsgrade, cL = 5/2 R. Die Rotation um die Moleküllängsachse darf hierbei nicht mitgezählt werden. Bei tiefen Temperaturen sind beim Wasserstoff die Rotationen noch nicht "voll angeregt". Das ergibt die Temperaturabhängigkeit. Wegen des höheren Trägheitsmoments und des höheren Siedepunkts wird dieser Effekt bei Stickstoff nicht beobachtet. Bei 3- und mehratomigen Gase ist die Temperaturabhängigkeit kompliziert. 9.1.2 Molwärmen von Festkörpern Die Molwärmen von Festkörpern sind stark temperaturabhängig. Bei hohen Temperaturen streben die Werte bei Elementen gegen cL/R = 3. In einem Festkörper können die Atome nur Schwingungen ausführen. Ist die Temperatur hoch genug, so sind diese Schwingungen voll angeregt. Für jedes Atom sind 3 Schwingungsfreiheitsgrade zu zählen, die wegen der beiden Energieformen in der Schwingung (potentielle und kinetische Energie) doppelt zu zählen sind. Der Grenzwert für hohe Temperaturen ist daher (9.1.2.1) Abb. 22 Molwärme von Elementen Diese Gleichung wird als Dulong-Petitsche Regel bezeichnet. Bei Verbindungen sind die Molwärmen der einzelnen Elemente zu addieren (Kopp-Neumannsche Regel). (9.1.2.2) wobei n die Zahl der Atome im Molekül darstellt. Bei sehr tiefen Temperaturen ist cL proportional zu T 3. Dieses nach Debye benannte Gesetz ist auch mit Hilfe der Statistischen Thermodynamik erklärbar. Abb. 23 Molwärmen bei tiefen Temperaturen - 48 9.1.3 cp & cL Bisher wurde nur die Molwärme cL bei konstantem Volumen diskutiert. Diese Größe ist auch von der Theorie her leichter verständlich, da in der Molwärme cp bei konstantem Druck noch der Energieaufwand steckt, den die Substanz an die Umgebung bei ihrer Ausdehnung abgibt. Die Differenz cp & cL gibt gerade diesen Energieaufwand an. Im Experiment ist jedoch die Bestimmung von cp wesentlich einfacher. Wir wollen diese Differenz berechnen, die auch für andere Zwecke wichtig ist. Es gilt (9.1.3.1) (9.1.3.2) (9.1.3.3) (9.1.3.4) Zur weiteren Rechnung benutzen wir Gl. (7.2.4) (9.1.3.5) Damit folgt: (9.1.3.6) (9.1.3.7) (9.1.3.8) Für ein Ideales Gas gilt nach Gl. (1.2.11) (9.1.3.9) und daher (9.1.3.10) (9.1.3.11) Die Differenz der beiden Molwärmen entspricht gerade der allgemeinen Gaskonstante. Die Gleichung wird bei der Diskussion der adiabatischen Kompression Verwendung finden. 9.1.4 cL als Funktion von L und cp als Funktion von p Die Berechnung ist eine typische Anwendung des Schwarzschen Satzes in der Thermodynamik. Unter Verwendung von Gl. (7.2.4) findet man - 49 - (9.1.4.1) (9.1.4.2) In analoger Weise findet man (9.1.4.3) Für Ideale Gase verschwinden beide Abhängigkeiten. 9.2 U und H als Funktionen der Temperatur Aus den Definitionen der Molwärmen findet man durch Integration (9.2.1) Mit der Annahme cL bzw. cp = const. erhält man daraus (9.2.2) Für den Fall des Debyeschen Gesetzes findet man (9.2.3) Vielfach werden Molwärmen in Tabellen in Form von Reihenentwicklungen angegeben: (9.2.4) Zur Berechnung der Temperaturabhängigkeit von u bzw. h ist dann eine Integration entsprechend Gl. (9.2.1) durchzuführen. 9.3 Umwandlungsenergien, Umwandlungsenthalpien Die Gleichungen (9.2.1ff) gestatten die Berechnung der Energie bzw. Enthalpie nur für den Fall, dass die Temperaturänderung nicht über eine Phasenumwandlung hinweg erfolgt. Die dargestellte Enthalpiekurve, die bei einer Substanz mit einer Phasenumwandlung beobachtet wird, kann mit dem in Kap. 11.6.1 beschriebenen Gerät gemessen werden. Sie weist bei der Temperatur TU der Phasenumwandlung einen Sprung auf, da der Substanz für die Phasenumwandlung (z. B. Verdampfung) Energie bzw. Enthalpie zugeführt werden muss. Eine Beschreibung des Sprunges durch eine Molwärme ist nicht sinnvoll. Für den Fall unterschiedlicher Bereiche ist das Differenzial (9.5) auch anders zu schreiben. Bei kon- Abb. 24 Verlauf der Enthalpie über eine stantem p und T gilt: Phasenumwandlung hinweg - 50 - (9.3.1) wobei N die Niedertemperaturphase und O die Hochtemperaturphase darstellen sollen. Die Phasenumwandlung wird nun wie eine Reaktion durch den Umsatz > beschrieben (9.3.2) (9.3.3) Die Größe (9.3.4) wird als Umwandlungsenthalpie bezeichnet. Sie stellt die Enthalpieänderung für die Umwandlung von 1 mol Substanz dar. Sie entspricht der Differenz der molaren Enthalpien der beteiligten Phasen. Man unterscheidet nach der Art der Phasenumwandlung: Tab. 9.3.1 Phasenumwandlungsenthalpien Prozess Name der Umwandlungsenthalpie Symbol Name der Enthalpie für den Umkehrprozess fest 6 flüssig Schmelzenthalpie )HS Kristallisationsenthalpie flüssig 6 gasförmig Verdampfungsenthalpie )HV Kondensationsenthalpie fest 6 gasförmig Sublimationsenthalpie )HSu & fest 1 6 fest 2 Umwandlungsenthalpie )HU Umwandlungsenthalpie In der Nähe des Tripelpunkts kann man durch eine geringfügige Änderung des Drucks statt über die Flüssigkeit direkt vom Festkörper in die Gasphase kommen. Der 1. Hauptsatz verlangt dann (9.3.5) Den Verdampfungsprozess (bzw. den Sublimationsprozess) bei konstantem Druck kann man in 2 Schritte zerlegen. Die Verdampfungsenergie bei konstantem Volumen dient zur Überwindung der Wechselwirkung in der Flüssigkeit; bei konstantem Druck ist dann noch eine zusätzliche Kompressionsenergie p)VV gegen den äußeren Druck p zu leisten. )VV ist die Volumenveränderung bei der Verdampfung von 1 mol. Insgesamt muss also gelten (9.3.6) Auch aus der formalen Thermodynamik lässt sich diese Gleichung gewinnen. Das Verfahren wird bei der Ableitung der Gl. (11.1.10) bzw. (11.1.11) beschrieben. Verläuft die Verdampfung bei sehr viel niedrigerer Temperatur als der kritischen, so gilt (9.3.7) (9.3.8) d. h. die Verdampfungsenthalpie und -energie unterscheiden sich gerade um RT. - 51 - 10 Innere Energie als Funktion des Volumens, Enthalpie als Funktion des Drucks, Gaskompression bzw. -expansion Zuerst soll der bereits angesprochene Unterschied zwischen den beiden Differenzialquotienten (MU/MV)S und (MU/MV)T in Gl. (3.3) bzw. (9.4) diskutiert werden, wobei hier und in den folgenden Gleichungen die Konstanthaltung der ni nicht mitgeschrieben wird. In Gl. (3.3) gilt (10.1) In Gl. (9.4) gilt dagegen nach Gl. (7.2.4) (10.2) Für ein Ideales Gas verschwindet die rechte Seite von Gl. (10.2), d. h. für ein Ideales Gas gilt (10.3) während (MU/MV)S nicht verschwindet. Die Expansion bzw. Kompression von Gasen, mit denen wir uns hier ausschließlich beschäftigen wollen, kann unter unterschiedlichen Bedingungen erfolgen. 1) Expansion in ein evakuiertes Gefäß mit konstantem Volumen unter Systemisolation. 2) Expansion gegen einen verschiebbaren Kolben unter isothermen und reversiblen Bedingungen. 3) Expansion gegen einen verschiebbaren Kolben unter adiabatischen und reversiblen Bedingungen. 4) Expansion & meistens wird hierbei der Begriff Entspannung benutzt & durch ein Drosselventil unter adiabatischen Bedingungen (Joule-Thomson-Effekt). Die Expansion in ein evakuiertes Gefäß mit konstantem Volumen unter isothermen Bedingungen ist von geringerem Interesse. Die vier beschriebenen Expansionsarten, wobei es sich bei den Fällen 2 bis 4 auch um Kompressionen handeln kann, werden in den folgenden Kapiteln abgehandelt. 10.1 Adiabatische Expansion in ein evakuiertes Gefäß mit konstantem Volumen Die ersten Experimente hierzu wurden im 19. Jahrhundert von Gay-Lussac und von Joule durchgeführt. Die Expansion wird meistens durch Öffnen eines Ventils zwischen den Volumina, die das Gas enthalten bzw. evakuiert sind, bewerkstelligt. Während des Druckausgleichs entsteht durch Expansion des ausströmenden Gases und Kompression des bereits ausgeströmten Gases eine komplizierte TemperaturAbb. 25 Gasexpansion in ein Vakuum verteilung im System. Eine Messung der Gastemperatur vor und nach dem Experiment ergab & mit der damals möglichen Genauigkeit & keinen Unterschied. Mit den heute zur Verfügung stehenden Methoden findet man nur bei Idealen Gasen keine Temperaturunterschiede. Für Ideale Gase lässt sich daher der folgende Schluß ziehen. Da das Gesamtsystem isoliert ist, gilt (10.1.1) oder dU = 0. Da keine Temperaturänderung beobachtet wird, gilt T = const. Insgesamt ergibt daher das - 52 Experiment für ein Ideales Gas, dessen Volumen sich im Gegensatz zum Gesamtvolumen ändert: (10.1.2) wie oben bereits durch eine Berechnung mit der Idealen Gasgleichung gezeigt wurde. Bei einem Idealen Gas ist bei konstanter Temperatur die Innere Energie unabhängig vom Volumen. Für ein vander-Waals-Gas findet man (10.1.3) (10.1.4) d. h. (Mu/ML)T entspricht dem Binnendruck des van-der-Waals-Gases. Entsprechend findet man für die Enthalpie bei einem Idealen Gas (10.1.5) wobei das Analogon zur Gl. (10.2) Kapitel 12.1 entnommen wird. (Mh/Mp)T für ein van-der-Waals-Gas ergibt einen relativ komplizierten Ausdruck. Wie groß ist die Temperaturänderung, die bei der Expansion eines realen Gases in ein evakuiertes Gefäß entsteht? Am einfachsten geht man von der GFF aus und setzt dU = 0. (10.1.6) Dies oder die entsprechende Gl. des mathematischen Teils führen zu (10.1.7) Die Herleitung wurde mit wenig Kommentar durchgeführt, da es im Kap. 10.4 eine ausführlich besprochene Herleitung des wichtigeren Analogons mit der Enthalpie gibt. In Analogie zu dem dort besprochenen Effekt kann man den hier diskutierten als isoenergetische Expansion bezeichnen. Wegen der für den technischen Einsatz günstigeren Eigenschaften der isenthalpischen Drosselung bei der Abkühlung von Gasen wird die isoenergetische Expansion technisch nicht eingesetzt. Für Ideale Gase verschwindet (MT/ML)u. 10.2 Isotherme, reversible Expansion gegen einen verschiebbaren Kolben Das Experiment soll mit einem Idealen Gas durchgeführt werden. Die Arbeit beträgt dann (10.2.1) Die Arbeit ist positiv für den Kompressionsfall (V2 < V1) und negativ für den Expansionsfall (V2 > V1). Sie hängt beim Idealen Gas nicht von den absoluten Volumen- und Druckwerten, sondern nur von den Verhältnissen ab. Sie ist bei gegebenem Volumenverhältnis und gegebener Temperatur unabhängig von der Art des Idealen Gases. Die Einschränkung auf eine reversible Expansion ist aus folgenden - 53 Gründen notwendig. Eine Irreversibilität wird z. B. bei einer Gaskompression mit einer sehr hohen Geschwindigkeit vorliegen. Unter diesen Bedingungen wird zu Beginn nur das Gas in der Kolbennähe komprimiert, da sich das weiter hinten befindliche Gas wegen der endlichen Ausbreitungsgeschwindigkeit der Kompression erst später in Bewegung setzen wird. Es kommt daher zu einer ungleichen Druckverteilung im Gas, so dass Gl. (10.2.1) nicht mehr gültig sein kann. Im isothermen Fall ändert sich die Innere Energie des Gases nicht und die GFF lautet: (10.2.2) Das was an Kompressionsenergie &p dV vom Gas aufgenommen wird, muss als entropische Energie T dS oder Wärme an ein Wärmereservoir wieder abfließen. Durch Integration von Gl. (10.2.2) oder direkt aus Gl. (8.12) findet man (10.2.3) 10.3 Adiabatische, reversible Expansion gegen einen verschiebbaren Kolben Die adiabatische Expansion eines Gases lässt sich mit unterschiedlichen Methoden bewerkstelligen. a) Das System erhält eine Umhüllung, die den Wärmetransport verhindert (Wärmeisolierung, DewarGefäß) b) Die Expansion wird so schnell durchgeführt, dass der Wärmefluss vernachlässigbar wird. Zu schnell darf der Kolben jedoch auch nicht bewegt werden, da man sonst zum Fall des Kap. 10.1 gelangt. Die GFF lautet (10.3.1) Unter den genannten Bedingungen (adiabatisch und reversibel) bleibt die Entropie konstant. Es ergibt sich daher für 1 mol (10.3.2) Der Term mit der Molwärme cL bei einem Prozess mit variablem L und p (!) resultiert aus dem totalen Differenzial von U (Gl. (9.4)) für ein Ideales Gas bei konstanter Stoffmenge. Für ein Ideales Gas folgt (10.3.3) Die Integration der Differenzialgleichung ergibt (10.3.4) oder (10.3.5) Durch Einführung des Verhältnisses 6 = cp/cL erhält man (10.3.6) und (10.3.7) Mit Hilfe der Idealen Gasgleichung wird daraus die Poissonsche Gleichung (10.3.8) die auch noch in einer p,T-Darstellung geschrieben werden kann - 54 (10.3.9) Wann gilt pL = const., wann gilt pL6 = const.? Die Ideale Gasgleichung pL = RT gilt als Zustandsgleichung immer. Die anderen Gleichungen gelten nur für bestimmte Prozesse im isothermen Fall (10.3.10) im adiabatischen Fall (10.3.11) Ein Vergleich der isothermen und adiabatischen Kompression zeigt die Abbildung. Die Steilheit der Adiabaten nimmt mit 6 zu (einatomiges Gas: 6 = cp/cL = 5/3; zweiatomiges Gas: 6 = 7/5). Die stärkere Abnahme des Drucks bei der adiabatischen Expansion wird durch die Abnahme der Temperatur bewirkt. Die Poissonsche Gleichung wird viel in der Technik benutzt (Kompressoren, Verbrennungsmotoren). Die vom Gas unter adiabatischen Bedingungen geleistete Arbeit und die Änderung der Inneren Energie erhält man durch Integration von (10.3.2) (10.3.12) Abb. 26 Isotherme und Adiabate von Gasen für n mol Gas. )T ist die Differenz zwischen End- und Anfangstemperatur. 10.4 Isenthalpische Drosselung Im Unterschied zum vorherigen, auch adiabatischen Fall erfolgt hier die Entspannung in einem Drosselventil oder einer porösen Membran. Weiterhin handelt es sich um einen kontinuierlichen Prozess mit strömenden Gasen. Das Drosselventil oder die Membran sollen nur die Herabsetzung des Drucks bewirken. Das Gas soll keine kinetische Energie hinter der Membran aufweisen, wie beispielsweise nach einer Durchströmung von Löchern in einem Blech. Der Versuch, den Entspannungsvorgang in der Drossel mit der GFF zu untersuchen, würde die Berechnung unnötig komplizieren. Es ist einfaAbb. 27 Isenthalpische Drosselung cher, eine Energiebilanz für das Gas vor und hinter der Drossel aufzustellen. Das Gesamtsystem besteht aus den beiden Gasmengen N und O. In dieses System fließt Kompressionsenergie aus der Umgebung hinein (Motor hinter dem Kolben N) bzw. heraus (Generator hinter dem KolbenO). Wir wollen eine gewisse Gasmenge ins Auge fassen und die von der Umgebung am System geleisteten Arbeiten berechnen. Für diese Arbeiten gilt wegen des konstanten Drucks pNVN bzw. pOVO. Der für das System resultierende Energiegewinn führt zur Erhöhung der Inneren Energie, wobei vorausgesetzt wird, dass kein Energietransport durch Wärmeleitung über die Drossel hinweg auftritt. (10.4.1) oder (10.4.2) Die Enthalpie ändert sich also während des Prozesses nicht. Daher rührt auch der Name isenthalpischer Drosseleffekt; eine andere Bezeichnung ist Joule-Thomson-Effekt. Der Joule-Thomson-Effekt wird in der Technik zur Abkühlung und Verflüssigung von Gasen benutzt. - 55 Zur Berechnung der auftretenden Temperaturerniedrigung schreiben wir das totale Differenzial von h(T,p) auf und setzen für den isenthalpischen Fall dh = 0 (10.4.3) Die daraus folgende Gleichung ist auch direkt aus der entsprechenden Gleichung im mathematischen Teil herleitbar. (10.4.4) (MT/Mp)h ist die interessierende Größe; sie wird als Joule-Thomson-Koeffizient * bezeichnet. Mit Gl. (12.1.16), die das Analogon zu Gl. (7.2.4) darstellt (10.4.5) erhält man (10.4.6) Für ein Ideales Gas findet man * = 0. Für ein van-der-Waals-Gas ist die vollständige Berechnung kompliziert. Einfacher geht es mit der van-der-Waals-Gleichung in der Virialform (10.4.7) Damit ergibt sich (10.4.8) (10.4.9) Offensichtlich ist sowohl * < 0 als auch * > 0 möglich. Die Temperatur bei der * = 0 wird, wird als Inversionstemperatur TI bezeichnet. (10.4.10) Die Inversionstemperatur entspricht also der doppelten Boyle-Temperatur (s. Gl. (1.3.3.12)). Der JouleThomson-Effekt wird großtechnisch zur Kühlung von Gasen und zur Gasverflüssigung eingesetzt. Das technische Verfahren besteht aus folgenden Schritten: - 56 1) Gaskompression 2) Kühlung des durch die Kompression erhitzten Gases mit Luft oder Kühlwasser. 3) Abkühlung durch Entspannung in einem Drosselventil. 4) Zur Tiefkühlung wird ein Gegenstromverfahren benutzt, bei dem die abgekühlten Gase neu zuströmendes Gas vor der Drossel vorkühlen (siehe Abb. 28). Voraussetzung für die Anwendung des Joule-Thomson-Verfahren ist * = (MT/Mp)h > 0, da bei einer Druckerniedrigung eine Abkühlung auftreten soll. Viele Gase wie O2 und N2 erfüllen diese Bedingung bereits bei Raumtemperatur. H2 muss dagegen auf 224 K und He sogar auf 35 K vorgekühlt werden. Abb. 28 Gasverflüssigung - 57 - 11 Reaktionsenthalpien und Reaktionsenergien 11.1 Definitionen, Zusammenhang zwischen )H und )U Chemische Reaktionen werden üblicherweise bei konstantem Druck durchgeführt. In den meisten Fällen entspricht der Druck einfach dem Luftdruck, der mit ausreichender Genauigkeit als konstant und mit 1 bar angesetzt werden kann. Wir werden daher fast vollständig auf eine Diskussion der Inneren Energie verzichten. Bei einer chemischen Reaktion ändert sich nun der letzte Term der Gl. (9.5) (11.1.1) Wir wollen bei unserem Gedankenexperiment p und T konstant halten, d. h. wir führen die abzugebende Enthalpie unter Konstanthaltung von T in ein anderes System über, z. B. in einen Thermostaten. Ähnlich wie bei der Diskussion der Gleichgewichte sind die dni nicht frei variabel, sondern hängen über die stöchiometrischen Koeffizienten <i der Reaktion (11.1.2) mit dem Umsatz > wie folgt zusammen (11.1.3) Mit Gl. (11.1.1) ergibt sich damit bei konstantem p und T: (11.1.4) Unter der Reaktionsenthalpie wird nun (11.1.5) verstanden. Bei einer Festkörperreaktion, bei der die einzelnen Komponenten als reine Substanzen vorliegen, ist Gl. (11.1.5) einfach zu verstehen. Die Reaktionsenthalpie, d. h. die Enthalpieänderung bei einem Umsatz von <i mol aller Ri, entspricht der Summe aller molaren Enthalpien multipliziert mit den stöchiometrischen Koeffizienten. Die Vorzeichen der stöchiometrischen Koeffizienten bewirken, dass die molaren Enthalpien der Edukte und Produkte mit dem richtigen Vorzeichen eingehen. Entsprechend definieren wir die Reaktionsenergie (11.1.6) Zu den Einheiten von )H und )U ist folgendes zu bemerken. Da die stöchiometrischen Koeffizienten einheitenfrei sind, weist der Umsatz wegen Gl. (11.1.3) die Einheit "mol" auf. )H und )U weisen daher die Einheit J/mol bzw. kJ/mol auf. Die Vorzeichen der beiden Reaktionsgrößen sind durch die GFF festgelegt. Fließt bei konstant gehaltener Temperatur Enthalpie bzw. Innere Energie aus dem System ab, so sind )H bzw. )U negativ (exotherme Reaktion); andernfalls sind sie positiv (endotherme Reaktion). Der Zusammenhang zwischen )H und )U lässt sich aus einer Überlegung, wie sie bereits bei der Diskussion der Differenz der Molwärmen cp und cL durchgeführt wurde, gewinnen. Analog finden wir für den isothermen Fall - 58 (11.1.7) und führen das Differenzial von U(V,>) bei T = const. ein (11.1.8) Bildung der Ableitung M/M> bei konstantem p und T ergibt (11.1.9) wobei (MV/M> ) die bei einem Formelumsatz auftretende Volumenänderung ist, die im folgenden mit )V bezeichnet wird. (11.1.10) a) Zuerst soll eine Reaktion zwischen Idealen Gasen untersucht werden. Es gilt (11.1.11) Formal lässt sich der Term p)V wie folgt berechnen (mit etwas Nachdenken geht es auch ohne Rechnung) (11.1.12) Die Endformel für den Fall Idealer Gase lautet daher (11.1.13) )< ist die (vorzeichenbehaftete) Summe der stöchiometrischen Koeffizienten der Reaktionsgleichung. Beispiel: N2 + 3 H2 6 2 NH3 )< = &2 b) Bei Reaktionen in kondensierter Phase ist )V so klein, dass der letzte Term in Gl. (11.1.10) i. a. vernachlässigt werden kann, d. h. )H = )U. c) Entsteht ein Ideales Gas bei einer Reaktion in kondensierter Phase, so wird man nach der vorangehenden Diskussion nur den Term p )V zu berücksichtigen haben, d. h. (11.1.14) wobei <g der stöchiometrische Koeffizient des Gases ist. Ein Beispiel zu c): Ca CO3 6 Ca O + C O2 Bei 25 C ist die Reaktionsenthalpie bekannt. Sie beträgt )H = 178,1 kJ/mol. Die Reaktion wird jedoch unter diesen Bedingungen kaum ablaufen. Für die Reaktionsenergie findet man )U = (178,1 & 1 @ 8,314 @ 298,15 @ 10&3) kJ/mol = (178,1 & 2,5) kJ/mol = 175,6 kJ/mol. 11.2 Temperaturabhängigkeit von )H und )U Die Temperaturabhängigkeiten von )H und )U lassen sich in einfacher Weise durch eine Vertauschung der Differenziationsreihenfolge berechnen. - 59 - (11.2.1) Analog gilt (11.2.2) Cp und CV sind die Wärmekapazitäten (besser Enthalpie- bzw. Energiekapazität) und )Cp und )CV sind die Änderungen bei einem Formelumsatz, d. h. es wird nicht auf 1 mol bezogen, sondern auf einen Formelumsatz. Bestehen zwischen den Reaktionspartnern des Systems keine Wechselwirkungen, d. h. bei Reaktion Idealer Gase, Festkörperreaktionen und Reaktionen in idealen Lösungen, so lassen sich die )C aus den Molwärmen berechnen. (11.2.3) Es sind also die Molwärmen mit den vorzeichenbehafteten stöchiometrischen Koeffizienten zu multiplizieren. Die Integration von Gl. (11.2.1) ergibt unter Verwendung von (11.2.3) (11.2.4) und mit der Annahme cp,i f(T) findet man (11.2.5) Diese Gleichung ist auch unter dem Namen "Kirchhoffscher Satz" bekannt. Bei einer Temperaturabhängigkeit der Molwärmen muss die Integration mit Gl. (11.2.4) ausgeführt werden. Die vorstehende Ableitung ist ein typisches Beispiel für eine formale thermodynamische Argumentation. Das ist der moderne, heute übliche Weg. Früher hat man den Beweis anders geführt. Dazu wird die Reaktion bei zwei verschiedenen Temperaturen mit entsprechenden Abkühlungen und Erwärmungen im Kreis geführt, so dass nach diesem Prozess wieder der Ausgangszustand entsteht und daher )H = 0 ist. Die Enthalpiebilanz für diesen Kreisprozess ist: (11.2.6) oder (11.2.7) Wir erhalten das gleiche Ergebnis. Die Voraussetzungen, unter denen das Ergebnis gültig ist, sind jedoch bei vielen Kreisprozessen schlechter erkennbar als bei der formalen Rechnung. Gl. (11.2.4) bzw. (11.2.5) sind für viele technische Anwendungen wichtig, da die Reaktionsenthalpien meistens nur für 25 C tabelliert sind und Reaktionen oft bei höheren Temperaturen durchgeführt werden. - 60 Ein Beispiel für eine Berechnung: 2 H2 + O2 6 2 H2Og Die Reaktionsenthalpie bei 25 C findet man in Tabellenwerken (siehe Anhang). )H25/ = &483,6 kJ/mol Es soll )H bei 350 K berechnet werden. Für die Molwärmen findet man in Tabellen: = 28,8; = 29,1; = 33,6 J K&1 mol&1 )Cp = 2@33,6 & 2@28,8 & 29,1 = &19,5 J K&1 mol&1 )H350 = &483,6 & 19,5 (350 & 298,15)@10&3 = &484,6 kJ/mol 11.3 Heßscher Satz Der Heßsche Satz besagt: Die Reaktionsenthalpien und -energien sind unabhängig vom Weg, auf dem die Reaktion durchgeführt wird. Ein Nachweis dieses Satzes ist eigentlich nicht notwendig, da die Enthalpie eine Zustandsfunktion ist und dementsprechend die Enthalpiedifferenz zwischen den Zuständen Produkte und Edukte einen festen Wert aufweisen muss. Die Reaktionsenthalpie entspricht dieser Enthalpiedifferenz und muss daher unabhängig vom Weg sein. Die Argumentation gilt in gleicher Weise für die Reaktionsenergie. Etwas anschaulicher ist vielleicht die folgende Argumentation. Zuerst wird die Behauptung bewiesen: Die Reaktionsenthalpien für eine Reaktion und ihre Rückreaktion müssen bis auf das Vorzeichen übereinstimmen. Annahme: )HHin &)HRück. Man führt die Reaktion im Kreis und findet für den Gesamtprozess )H = )HHin + )HRück 0. Das verletzt den Erhaltungssatz für die Enthalpie bei p = const. Es muss also gelten )HHin = &)HRück. Jetzt wird der Heßsche Satz bewiesen. Annahme: )HI )HII. Man lässt die Reaktion I rückwärts laufen und führt den Prozess im Kreis. (11.3.1) Das verletzt jedoch den Erhaltungssatz für die Enthalpie. Die Annahme der Ungleichheit von )HI und )HII ist unzulässig. Die beiden Reaktionsenthalpien müssen gleich sein. 11.4 Reaktionsenthalpien aus Verbrennungsenthalpien Der Heßsche Satz erlaubt die Bestimmung von Reaktionsenthalpien für Reaktionen, die direkt nicht durchgeführt werden können. Als Beispiel wollen wir Reaktionsenthalpien aus Verbrennungsenthalpien bestimmen. Dabei wählen wir die nebenstehend dargestellten Wege. Da die Verbrennungsprodukte der Edukte und Produkte übereinstimmen, gilt (11.4.1) Der Index c kommt von "combustion". Die Größen der rechten Seite werden noch in einer Summe zusammengefasst (11.4.2) Sind also die Verbrennungsenthalpien )Hic aller Reaktionspartner bekannt, so kann die Reaktions- - 61 enthalpie nach Gl. (11.4.2) bestimmt werden. Dieses ist insbesondere für organische Verbindungen ein wichtiges Verfahren, da die meisten Reaktionen organischer Verbindungen nicht vollständig ablaufen, aber fast alle organischen Verbindungen gut verbrennbar sind. Eine Schwierigkeit besteht darin, dass die Reaktionsenthalpie aus einer Differenz großer Zahlen berechnet wird. Die Verbrennungsenthalpien müssen daher sehr genau bekannt sein. 11.5 Reaktionsenthalpien aus Bildungsenthalpien Wären die absoluten Enthalpiewerte der Reaktionspartner bekannt und dürften & wie schon früher diskutiert & jegliche Wechselwirkungen zwischen den Reaktionspartnern vernachlässigt werden, so findet man mit Gl. (11.1.5) (11.5.1) wobei die hi die molaren Enthalpien der Reaktionspartner sind. Die absoluten Enthalpiewerte, d. h. die für die Erzeugung der Reaktionspartner aus dem Vakuum heraus aufzuwendenden Enthalpien, stehen nun nicht zur Verfügung. Zur Verfügung stehen in Tabellenwerken die Bildungsenthalpien )HiB. Das sind die Reaktionsenthalpien für die Bildung der Verbindungen aus den Elementen unter Standardbedingungen. Die Standardbedingungen sind 25 C und (leider) 1 atm, d. h. 1,01325 bar. Wenn nicht anders vermerkt, beziehen sich die Daten immer auf die unter diesen Bedingungen stabilen Modifikationen. Wie erhält man die Reaktionsenthalpien aus den Bildungsenthalpien? Analog zur Bestimmung aus den Verbrennungsenthalpien findet man (11.5.2) (11.5.3) Vorausgesetzt wird dabei, dass die zwischen den Reaktionspartnern möglichen Wechselwirkungen vernachlässigt werden können. Genauer gesagt: sowohl zwischen den Edukten als auch zwischen den Produkten soll es keine Mischungsenthalpien geben. Der Vergleich von Gl. (11.5.1) und (11.5.3) zeigt, dass man für diesen Zweck mit den Bildungsenthalpien wie mit den absoluten Enthalpien rechnen kann, da die Absolutwerte der Elemententhalpien bei der Rechnung herausfallen. Dieses Verfahren funktioniert natürlich nicht mehr, wenn Kernumwandlungen zugelassen werden! Ein Blick in die Tabelle im Anhang zeigt, dass für alle Elemente (natürlich!) )HB = 0 gilt. Die bei Kernumwandlungen auftretenden Enthalpieunterschiede sind definitionsgemäß nicht erfaßt. Wie erhält man die Bildungsenthalpien? Für viele anorganische und fast alle organischen Verbindungen gelingt das über die Verbrennungsenthalpien der Elemente und der entsprechenden Verbindung nach Gl. (11.4.2). Zusätzlich gibt es im Bereich der Anorganischen Chemie noch viele Reaktionen, die vermessen werden können, da diese oft schnell und quantitativ ablaufen. Auch die Kombination mehrerer Reaktionen über den Heßschen Satz kann die interessierende Bildungsreaktion ergeben. Ein Beispiel für die Bestimmung einer Reaktionsenthalpie: die Hydrierungsenthalpie von Acetylen zu Ethan C2H2 + 2 H2 6 C2H6 - 62 (11.5.4) 11.6 Kalorimeter Kalorimeter (von lat. calor: Wärme) sind Apparaturen, mit denen Molwärmen, Reaktionsenthalpien, Umwandlungsenthalpien usw. bestimmt werden können. 11.6.1 Bestimmung von Molwärmen Im Prinzip ist ein dafür geeignetes Kalorimeter bereits bei der Diskussion der GFFF in Kap. 4 beschrieben worden. Der Aufbau ist im Grund einfach, im Detail jedoch wegen unterschiedlicher und teilweise extremer Anforderungen (Gase, Festkörper; Messungen bei 1 K, 2000 K, 0,1 bar, 10 kbar) sehr kompliziert. Ein Gerät für Flüssigkeiten und Festkörper bei Normaldruck und nicht zu extremen Anforderungen bei der Temperatur zeigt Abb. 33. Wichtig sind die adiabatischen Wände (Vakuum, Verspiegelung), die den Energieaustauch zwischen Probengefäß und Umgebung weitgehend verhindern. Die Molwärme bei konstantem Druck wird mit diesem Gerät wie folgt bestimmt. Man Abb. 33 Kalorimeter lässt über die Heizwicklung elektrische Energie in die Probe fließen und bestimmt die Temperaturänderung der Probe. Bei konstantem Druck gilt (11.6.1.1) Bleiben die Temperaturänderungen bei einem Messvorgang so klein, dass cp als konstant angesehen werden kann, folgt (11.6.1.2) und (11.6.1.3) wobei EEl die während eines Experiments in das Kalorimeter fließende elektrische Energie darstellt. Der Anteil der Wärmekapazität des Probengefäßes ist mit einem zweiten Versuch zu bestimmen. Für Gase kann ein derartiges Kalorimeter nicht verwendet werden, da die Wärmekapazität des Gases im Vergleich zum Probengefäß zu klein wäre. Für Gase werden daher Kalorimeter mit einem kontinuierlichen Gasstrom eingesetzt. Durch das mehrfache Abb. 34 Kalorimeter für Gase Gegenstromverfahren verhindert man einen - 63 Energieabfluss nach außen. Die Auswertung ergibt sich aus Gl. (11.6.1.2) durch Differenziation nach der Zeit zu (11.6.1.4) und (11.6.1.5) wobei N die von der Heizwicklung abgegebene elektrische Leistung bedeutet. Die Temperaturdifferenz kann mit einem Thermoelement bestimmt werden. 11.6.2 Bestimmung von Reaktionsenthalpien in kondensierter Phase Vor der Reaktion müssen die Edukte getrennt in das Kalorimeter eingebracht werden. Das Reaktionsgefäß muss diese Trennung, die anschließende Vereinigung der Edukte und den sicheren Einschluss der Produkte erlauben. Einige Reaktionsgefäße zeigen die Abbildungen 35 und 36. In den drei ersten Reaktionsgefäßen kann eine der Flüssigkeiten durch einen Festkörper ersetzt werden. Abb. 35 Reaktionskalorimeter Abb. 36 Reaktionskalorimeter Bei der Ampulle wird die die Reaktanden trennende Glasfläche mit der u. U. ummantelten Stahlkugel durchschlagen. Bei dem Rohrsystem wird die Glasspitze des inneren Rohrs durch Andrücken gegen das äußere Rohr zerbrochen. Das letzte Gefäß ist eine Verbrennungsbombe, in welcher der Pressling mit der Zündwicklung lokal kurz erhitzt wird, so dass die Verbrennung in dem unter einem Druck von etwa 20 bar stehenden Sauerstoff eingeleitet wird. Die Reaktionsgefäße werden in das eigentliche Kalorimeter eingesetzt. Eine mögliche Form ist das in Abb. 37 dargestellte Wasserkalorimeter. Zur Bestimmung der Reaktionsenthalpie soll eigentlich der Druck konstant gehalten werden. Dies kann in einigen Reaktionsgefäßen, z. B. dem Rohrreaktor, eingehalten werden. In vielen Fällen sind die Gefäßvolumina über den Reaktanden evakuiert und weisen dann den Dampfdruck der Reaktanden auf. In fast allen Fällen können die Unterschiede zwischen )H und )U bei Reaktionen in kondensierter Phase vernachlässigt werden kann. Die Auswertung ergibt sich nach Gl. (9.5) bei p = const. zu (11.6.2.1) - 64 (11.6.2.2) Abb. 37 Wasserkalorimeter Abb. 38 Wärmeflusskalorimeter Das negative Vorzeichen ist richtig, da bei MT/M> > 0 )H negativ sein muss. Die Wärmekapazität des Kalorimeters samt Reaktionsgefäß wird in einem zweiten Versuch durch die Zufuhr elektrischer Energie bestimmt. Schließlich soll noch ein Wärmeflusskalorimeter erwähnt werden, das insbesondere für den Bereich hoher Temperaturen eingesetzt werden kann. Bei diesem Kalorimeter wird gerade ein definierter Wärmefluss zwischen Reaktionsgefäß und einem auf der Ausgangstemperatur befindlichen, dickwandigen Metallrohr zugelassen. Die Temperaturdifferenz zwischen dem Reaktionsgefäß bzw. einem dieses direkt umschließenden dünnwandigen Metallrohr und dem äußeren Rohr wird durch eine große Zahl hintereinander geschalteter Thermoelemente gemessen. Abb. 39 zeigt den Verlauf dieser Temperaturdifferenz )T nach der Reaktion in Abhängigkeit von der Zeit. Die unter dieser Kurve liegende Fläche ist proportional zur freigesetzten Enthalpie. Die Eichung des Kalorimeters kann entweder mit einer elektrischen Heizung oder durch Vermessung einer Abb. 39 Temperaturverlauf im WärReaktion mit bekannter Reaktionsenthalpie vorgenommen meflusskalorimeter werden. - 65 - 12 Entropie Nach der kurzen Diskussion in Kap. 3 wollen wir uns mit Freude an die ausführliche Behandlung der unanschaulichsten Größe der Thermodynamik begeben. 12.1 Entropie als Funktion von p, V, T für reine Phasen Die GFF ergibt (12.1.1) wenn alle anderen extensiven Größen, insbesondere V und n, konstant gehalten werden. Unter diesen Bedingungen folgt für 1 mol (12.1.2) und durch Division (12.1.3) Wem die Division nicht gefällt, der kann auch das totale Differenzial von s(T,V) hinschreiben und mit Gl. (12.1.2) mit dem gleichen Ergebnis vergleichen. Analog zu Gl. (12.1.3) gilt bei konstantem Druck (12.1.4) Die Ableitungen (MS/MV)T und (MS/Mp)T erhalten wir aus den Differenzialen der Freien Energie und der Freien Enthalpie (12.1.5) (12.1.6) durch Anwendung des Schwarzschen Satzes (12.1.7) (12.1.8) Diese Gleichungen werden als Maxwellsche Beziehungen bezeichnet. Die Gl. (12.1.3) und (12.1.7) sowie (12.1.4) und (12.1.8) kann man in zwei totalen Differenzialen zusammenfassen (12.1.9) (12.1.10) Diese beiden Gleichungen erlauben es uns, die beiden bereits häufig verwendeten Gleichungen für (MU/MV)T bzw. (MH/Mp)T abzuleiten. Für (MU/MV)T ist das zwar bereits in Kap. 7.2 erfolgt; aus Symmetriegründen erfolgt es hier noch einmal. Die GFF wird umgeschrieben und entsprechend wird mit dem Differenzial der Enthalpie verfahren - 66 (12.1.11) (12.1.12) du und dh werden durch die Differenziale von u(T,L) und h(T,p) ersetzt. (12.1.13) (12.1.14) Der Vergleich mit (12.1.9) und (12.1.10) ergibt (12.1.15) (12.1.16) Zuerst wollen wir die Gleichungen (12.1.9) und (12.1.10) auf ein Ideales Gas anwenden. Wir erhalten mit (Mp/MT) = R/L und (ML/MT) = R/p (12.1.17) (12.1.18) Bei Annahme temperaturunabhängiger Molwärmen ergibt die Integration (12.1.19) (12.1.20) Im Prinzip genügt eine dieser Gleichungen zur Berechnung der Entropieänderung bei beliebigen Prozessen in Idealen Gasen, da L2/L1 und p2/p1 zusammenhängen. Ersetzt man die Volumina in der oberen Gleichung durch die aus dem Idealen Gasgesetz folgenden Ausdrücke, ergibt sich die untere Gleichung. Mit )s = 0 ergeben sich aus Gl. (12.1.19) und (12.1.20) direkt die Poissonschen Gleichungen (10.3.7) und (10.3.9). Für den Fall realer Gase und kondensierter Phasen wollen wir uns nur die Temperaturabhängigkeit bei konstantem Druck ansehen. Aus (12.1.4) folgt (12.1.21) Für den Fall temperaturunabhängiger Molwärmen folgt - 67 - (12.1.22) Ähnlich wie bei der Enthalpie gilt diese Formel nur innerhalb einer Phase und darf nicht über eine Phasenumwandlung hinweg verwendet werden. Die Änderung der Entropie über eine Phasenumwandlung hinweg wird als Umwandlungsentropie (Schmelzentropie, Verdampfungsentropie) bezeichnet. Sie lässt sich aus der Umwandlungsenthalpie bzw. -energie berechnen. Es gilt bei konstantem Druck bei der Umwandlungstemperatur TU (12.1.23) Für die Umwandlung gilt dnN = &dnO und im Gleichgewicht ist :N = :O, so dass die beiden letzten Terme entfallen. (12.1.24) oder (12.1.25) oder (12.1.26) Diese Gleichung gilt nur für die Umwandlung im Gleichgewicht! Im Prinzip ist es mit den Gleichungen (12.1.21) und (12.1.26) möglich, falls die entsprechenden Daten zur Verfügung stehen, die Entropie jeder Verbindung bezogen auf den Entropiewert bei 0 K zu berechnen (12.1.27) Die Untersuchung der GFF zeigt nun, dass alle intensiven und extensiven Größen & außer der Entropie & absolut festgelegte Größen sind. Es ist daher zu erwarten, dass dieses auch für die Entropie gilt. Planck hat aufgrund experimenteller Erkenntnisse und theoretischer Überlegungen den Vorschlag gemacht, die Nullpunktsentropie S0 für reine, ideal aufgebaute Kristalle Null zu setzen. Diese Annahme wird auch als 3. Hauptsatz der Thermodynamik bezeichnet. Wegen des engen Zusammenhangs dieser Annahme mit Überlegungen aus der Statistischen Thermodynamik wollen wir uns kurz die Begründung aus der Statistischen Thermodynamik ansehen. Die Quantenmechanik zeigt, dass die Materie Energie nicht kontinuierlich aufnehmen kann, sondern dass die Energiezustände der Atome und Moleküle i. a. gequantelt, d. h. & wenn auch u. U. sehr eng & diskontinuierlich sind. Unter der thermodynamischen Wahrscheinlichkeit W versteht man die Zahl der Möglichkeiten, ein thermodynamisch definiertes System über diese Energiezustände zu verteilen. Dabei sollen alle Fälle gezählt werden, die mit dem gegebenen thermodynamischen Zustand verträglich sind. S ist nun eine unbekannte Funktion der thermodynamischen Wahrscheinlichkeit (12.1.28) Die funktionelle Abhängigkeit lässt sich nun relativ einfach mit folgender Methode bestimmen. Man stelle zwei Systeme (mit S1, W1 und S2, W2) nebeneinander. Dann gilt für das Gesamtsystem - 68 (12.1.29) (12.1.30) Das Produkt ergibt sich aus der Unabhängigkeit der Wahrscheinlichkeiten. Mit Gl. (12.1.28) folgt daraus (12.1.31) Die gemischte partielle Ableitung nach W1 und nach W2 ergibt (12.1.32) oder (12.1.33) Die Lösung dieser Differenzialgleichung ist (12.1.34) Die Durchrechnung eines Beispiels, z. B. ein Gas in einem vorgegebenen Volumen, ergibt für a die Boltzmann-Konstante k = R/NA. Um eine Übereinstimmung mit dem dritten Hauptsatz zu erhalten, muss die Konstante C verschwinden. (12.1.35) Diese berühmte Gleichung wurde zuerst von Boltzmann diskutiert. Hat man nun einen reinen, ideal aufgebauten Kristall, so gibt es bei 0 K nur eine Anordnungsmöglichkeit W, d. h. S = 0. Schwieriger wird der Fall beim Vorliegen von Isotopengemischen. Für einen aus einer Isotopenmischung aufgebauten Kristall muss es die bereits kurz diskutierte Mischungsentropie geben, d. h. S0 0. In der Chemie bleibt nun diese Isotopenmischung i. a. erhalten, d. h. dieser Mischungsterm bleibt bei allen Prozessen erhalten und muss daher nicht berücksichtigt werden. Man darf daher auch für diesen Fall S0 = 0 setzen. Ein berühmtes Beispiel für einen experimentellen Hinweis auf die Entropien bei 0 K ist die Untersuchung am Schwefel. Vom Schwefel gibt es eine rhombische Tieftemperaturmodifikation und eine monokline Hochtemperaturmodifikation. Der Umwandlungspunkt liegt bei 368,5 K. Die Hochtemperaturmodifikation ist bei tiefen Temperaturen so stabil, dass die Molwärmen mit den beschriebenen Verfahren bis zum Umwandlungspunkt bestimmt werden können. Man findet am Umwandlungspunkt nach einer Auswertung des Experiments mit Gl. (12.1.21) (12.1.36) Am Umwandlungspunkt gilt daher (12.1.37) Andererseits lässt sich die Umwandlungsentropie direkt aus der Umwandlungsenthalpie mit Gl. (12.1.26) bestimmen (12.1.38) d. h. )So = 0,13 J K&1mol&1 bei einem geschätzten Fehler von ±0,65 J K&1mol&1. Innerhalb der experimentellen Genauigkeit gilt daher )So = 0. Dies hat man auch für eine Reihe anderer Umwandlungen und Reaktionen gefunden, so dass die Annahme So = 0 zulässig ist. Es gibt jedoch eine Reihe von Verbindungen, für die dieses nicht zutrifft. Ein Beispiel dafür ist das kristalline Kohlenmonoxid. In einem ideal aufgebauten Kristall sollten die CO-Moleküle mit einer definierten Orientierung der CO-Achse im Kristall vorliegen. In einem Realkristall sind jedoch die CO- - 69 Moleküle nicht einheitlich ausgerichtet, sondern statistisch orientiert eingebaut. Dies hängt damit zusammen, dass der Energieunterschied für die unterschiedlichen Einbaurichtungen im Kristall sehr viel kleiner als kT bei der Kristallisation ist. Bei tiefen Temperaturen ist dann eine Drehung der Moleküle im Kristall nicht mehr möglich. Für diesen Fall lässt sich die Nullpunktsentropie relativ einfach aus Gl. (12.1.35) bestimmen. Jedes CO-Molekül kann 2 Lagen einnehmen. Für NA Moleküle ist daher die thermodynamische Wahrscheinlichkeit (12.1.39) (12.1.40) Experimentell findet man auch einen Wert in dieser Größenordnung. Die Entropiewerte werden für viele Zwecke benötigt. Sie sind daher für viele Verbindungen mit großem experimentellen Aufwand (siehe oben) bestimmt oder mit Hilfe der Statistischen Thermodynamik berechnet worden und werden in Tabellenwerken für Standardbedingungen angegeben (siehe Anhang). 12.2 Mischungsentropie Während sich U, H und V bei der Herstellung einer idealen Mischung (z. B. einer Mischung aus nicht wechselwirkenden Idealen Gasen) additiv verhalten, kann das bei der Entropie nicht der Fall sein, da die Entropie nach (12.1.35) unterschiedlich sein muss, wenn zwei Gase getrennt oder gemischt ein gegebenes Volumen erfüllen. Für die Berechnung der Mischungsentropie verwenden wir zwei Ideale Gase 1 und 2 mit gleicher Temperatur und gleichem Druck. p, T n1, V1 p, T n2, V2 Es muss gelten n1/n2 = V1/V2. Entfernt man die Trennwand, so läuft der irreversible und damit entropieerhöhende Mischungsvorgang ab. Zur Berechnung wird ein reversibler Prozess mit identischem Anfangs- und Endzustand durchgeführt. Da die Entropie eine Zustandsfunktion ist, ist dies erlaubt. Schritt I Isotherme Expansion beider Gase in reinem Zustand auf V = V1 + V2 (12.2.1) Schritt II Die beiden Gase werden mit dem in Abb. 40 dargestellten Gerät ineinander geschoben: Der Platte P1 ist eine semipermeable Membran, die nur das Gas 1 durchlässt; P2 lässt nur das Gas 2 durch. Die Gase lassen sich mit diesem Gerät ohne Kraftaufwand ineinander schieben. Der Vorgang ist adiabatisch und reversibel; daher gilt )S = 0. Die Mischungsentropie besteht also nur darin, dass Abb. 40 Gerät mit semipermeabler Membran jedes Gas das Gesamtvolumen V einnehmen darf. Gl. (12.2.1) wird noch durch Einführung der Molenbrüche in der Mischung x1 = n1/n = V1/V und x2 umgeschrieben (12.2.2) Für 1 mol der Mischung finden wir schließlich - 70 (12.2.3) Die Gleichung kann leicht auf Vielkomponentensysteme erweitert werden (12.2.4) Da xi < 1 gilt, folgt grundsätzlich )s > 0. Diese Aussage ist offensichtlich richtig, da die Entropie bei diesem irreversiblen Prozess zunehmen muss. Die Gleichung kann auch für flüssige Mischungen verwendet werden, wenn sich die Mischung ideal verhält. Dies ist der Fall, wenn ähnliche Verbindungen miteinander gemischt werden. 12.3 Entropieänderung beim Temperaturausgleich zwischen zwei Körpern An dieser Stelle wollen wir noch dieses im Kap. 4.1 begonnene Problem endgültig behandeln. Die Entropieänderungen für jeden Einzelkörper lässt sich mit (12.1.22) berechnen. Beim Vorliegen identischer Körper N und O wird die Endtemperatur T0 = (TN+TO)/2 betragen. Die Gesamtentropieänderung ist daher (12.3.1) (12.3.2) wobei Cp die Wärmekapazität eines der Körper ist. Im Logarithmus steht nun das Verhältnis des arithmetischen zum geometrischen Mittel der Ausgangstemperaturen. Dieses Verhältnis ist für ungleiche Temperaturen immer größer als 1, d. h. )S > 0, wie es sich für einen irreversiblen Prozess gehört. Abb. 41 Temperaturausgleich zwischen zwei Körpern - 71 - 13 Prozesse in isolierten zusammengesetzten Systemen 13.1 Allgemeines Prozesse in isolierten zusammengesetzten Systemen können nur so ablaufen, dass sich ein Term in der GFFF auf Kosten eines anderen ändert. Es gilt in einem aus zwei SystemenN und O zusammengesetzten isolierten System (13.1.1) Bei dem Prozess ist auf die Einhaltung des zweiten Hauptsatzes zu achten. Der erste Hauptsatz wird durch Gl. (13.1.1) grundsätzlich erfüllt. Alle Prozesse gemäß Gl. (13.1.1.), bei denen sich die Entropie nicht ändert, sind daher grundsätzlich möglich. Beispiele dafür sind: 1) Kinetische Energie in Verschiebungsenergie (senkrecht nach oben geworfener Ball) 2) Deformationsenergie in Rotationsenergie (Unruh einer Uhr) 3) Deformationsenergie in Verschiebungsenergie und kinetische Energie (Masse an einer Feder im Gravitationsfeld) 4) Elektrische Energie in Verschiebungsenergie (Entladung eines Kondensators über einen Elektromotor, der mit einer Seilwinde ein Gewicht im Gravitationsfeld der Erde hebt) Falls die Umwandlung nicht vollständig erfolgt, ist das eine Folge mehr oder weniger großer Anteile anderer, meist irreversibler Prozesse, wie Luftreibung, bleibende Verformung von Federn, ohmsche Widerstände usw. Gl. (13.1.1) bleibt gültig; in solchen Fällen wird Entropie produziert und der Term TdS muss berücksichtigt werden. Falls bei der Umwandlung Entropie erzeugt wird, ist die Umwandlung mit dem 2. Hauptsatz verträglich und daher auch vollständig möglich. Beispiele dafür sind: 1) Kinetische Energie in entropische Energie (Autobremse) 2) Elektrische Energie in entropische Energie (Heizlüfter) Die entropische Energie fließt dann zumeist als Wärme in die Umgebung ab. Falls bei einer Umwandlung Entropie vernichtet werden würde, ist die Umwandlung grundsätzlich unmöglich. Beispiele dafür sind: Wärme in alle "entropiefreien" Energieformen wie elektrische Energie, Verschiebungsenergie usw. Es ist noch nie beobachtet worden, dass ein Stein unter Abkühlung nach oben springt, obwohl das mit Gl. (13.1.1) verträglich wäre. Der technisch wichtige Prozess der Umwandlung von Wärme in Arbeit ist daher nur mit Kunstgriffen möglich und erlaubt i. a. nur geringe Wirkungsgrade (siehe nächstes Kapitel). Kompliziert ist die Umwandlung chemischer Energie in andere Energieformen. Die Ablauffähigkeit chemischer Reaktionen wird in Kap. 16 diskutiert werden. Eine Möglichkeit zur Nutzung chemischer Energie besteht darin, durch einen Verbrennungsprozess Wärme zu erzeugen und diese mit dem leider niedrigen Wirkungsgrad weiter umzusetzen. Eine weitere Möglichkeit besteht in der direkten Umsetzung in elektrische Energie in einer Brennstoffzelle (Kap. 26.3). Dies kann theoretisch zu Wirkungsgraden über 100 % (!) führen, da u. U. sogar Wärme aus der Umgebung mit verwendet werden kann. Bei einer Brennstoffzelle für die Reaktion C + O2 6 CO2 beträgt der theoretische Wirkungsgrad bei 1000 C etwa 200 %! Leider ist dieses Verfahren wegen technischer Schwierigkeiten heute noch nicht einsetzbar. Im Kapitel 10.2 war gezeigt worden, dass es bei einer isothermen Gasexpansion eine vollständige Umwandlung von Wärme in Arbeit gibt. Der vermeintliche Widerspruch zum 2. Hauptsatz ist keiner, da durch die Gasexpansion ein Entropiegewinn von )S = Q/T erfolgt und damit die Entropieerniedrigung des Wärmereservoirs um )S = Q/T gerade aufgehoben wird. Für eine Wärmekraftmaschine lässt - 72 sich dieses Umwandlungsverfahren nicht einsetzen, da das Gas unter Einsatz der gerade gewonnenen Arbeit wieder komprimiert werden müsste. Auch eine Umgehung dieser Rück-Kompression durch Verwendung neu zuströmender Luft von 1 bar und Expansion auf einen kleineren Druck führt & wie man leicht einsehen kann & nicht zum Ziel. 13.2 Wirkungsgrad von Wärmekraftmaschinen Die bisherigen Erkenntnisse gestatten in relativ einfacher Weise, den Wirkungsgrad von Wärmekraftmaschinen (WKM) zu berechnen. Die von der WKM abgegebene elektrische Energie oder Verschiebungsenergie soll als Arbeit A bezeichnet werden. Die WKM benutzt Wärmereservoire. Das sind Systeme, in die bei einer bestimmten Temperatur Wärme hineinoder herausfließen kann, ohne dass sich die Temperatur des Systems ändert. Eine sehr große Menge Wasser stellt ein Wärmereservoir dar. Eine WKM, die nur durch einen Wärmefluss aus einem einzigen Wärmereservoir Arbeit leistet, kann es nicht geben (s. Kap. 4.1). Eine WKM muss daher mindestens mit zwei Wärmereservoiren, einem Hochtemperatur- (HTWR) und einem Niedertemperaturwärmereservoir (NTWR), arbeiten. Als Wirkungsgrad definieren wir die Größe (13.2.1) wobei in dieser und den folgenden Gleichungen die Vorzeichen von der WKM aus gesehen werden sollen. Das negative Vorzeichen vor A verhindert, dass der Wirkungsgrad eine negative Größe wird. Wir untersuchen jetzt folgende Behauptung: Alle reversibel und zyklisch arbeitenden WKM zwischen zwei Wärmereservoiren mit gegebenen Temperaturen haben den gleichen Wirkungsgrad. Nachweis: Falls es eine reversibel und zyklisch arbeitende WKM mit einem größeren oder Abb. 42 Wärmekraftmakleineren (!) Wirkungsgrad gäbe, führt das zu einem Widerspruch. Dazu schine lässt man zwei WKM mit unterschiedlichen Wirkungsgraden "gegeneinander" arbeiten. Die WKM mit dem größeren Wirkungsgrad erzeugt viel Arbeit und gibt wenig Wärme an das NTWR ab. Mit der zweiten, umgedreht laufenden Maschine wird die wenige Abwärme aus dem NTWR mit einem hohen (!) Wirkungsgrad wieder in das HTWR gepumpt. Im Endeffekt ist so Arbeit durch Wärmeentzug aus einem Wärmereservoir entstanden. 1. WKM (0 = 80%): Q2 = 100 J, A = &80 J, Q1 = &20 J 2. WKM (0 = 50%): Q2 = &40 J, A = 20 J, Q1 = 20 J Gesamtprozess: Q2 = 60 J, A = &60 J Das ist nicht erlaubt (siehe oben). Zur Berechnung des Wirkungsgrads faßt man die WKM und die beiden Reservoire zu einem System zusammen, in dem ein adiabatischer und reversibler, d. h. isentroper, Prozess abläuft. Vergleicht man nun den Systemzustand mit dem nach einem Zyklus, so haben sich der Zustand und damit die Entropie der WKM nicht geändert (d. h. ja gerade zyklisch). Bezeichnet man die Entropieänderungen der WR bei den Temperaturen T1 und T2 mit )S1 und )S2, so gilt z. B.: (13.2.2) und daher mit Gl. (8.18) (13.2.3) Weiterhin gilt aufgrund des 1. Hauptsatzes - 73 (13.2.4) Aus diesen beiden Gleichungen folgt (13.2.5) und schließlich (13.2.6) für jede reversibel und zyklisch arbeitende WKM. Eine WKM, mit der dieses prinzipiell durchführbar ist, werden wir weiter unten kennenlernen. In der Praxis werden durch Irreversibilitäten (Reibungsverluste, Verluste durch endliche Wärmeleitfähigkeiten) geringere Wirkungsgrade erreicht. Die Übertragung der Diskussion am Anfang dieses Kapitels auf eine WKM mit teilweise irreversiblen Prozessen zeigt, dass der Wirkungsgrad für eine derartige WKM zwischen 0 und dem in Gl. (13.2.6) angegebenen Wirkungsgrad liegen muss. Bei einer Wärmepumpe interessiert das Verhältnis der bei der höheren Temperatur abgegebenen Wärme zur eingesetzten Arbeit. Als Wirkungsgrad wird daher (13.2.7) definiert. Ist die Differenz T2 & T1 klein, so lassen sich hohe Wirkungsgrade 0P >> 1 erzielen. Bei einem Kühlaggregat interessiert das Verhältnis der bei der niedrigen Temperatur entzogenen Wärme zur eingesetzten Arbeit. Als Wirkungsgrad wird daher (13.2.8) definiert. Mit Gl. (13.2.4) folgt aus Gl. (13.2.6) (13.2.9) 13.3 Carnot-Prozess Eine Realisierung einer reversiblen, zyklischen Wärmekraftmaschine, die zwischen zwei Wärmereservoiren arbeitet, ist der CarnotProzess (Sadi Carnot, franz. Ingenieur, Anfang 18. Jahrhundert). Er besteht aus einer Abfolge von vier Schritten, die in Abb. 43 und der folgenden Tabelle beschrieben sind. Der Prozess wird hier mit 1 mol eines Idealen Gases durchgeführt. Abb. 43 Carnotprozess - 74 Tab. 13.3.1 Arbeiten und Wärmen beim Carnot-Prozess A Q2 Schritt Prozess 1 Isotherme Expansion bei T2 von V21 auf V22 2 Adiabatische Expansion von T2, V22 auf T1, V12 0 3 Isotherme Kompression bei T1 von V12 auf V11 0 4 Adiabatische Kompression von T1, V11 auf T2, V21 0 E Gesamtprozess Q1 0 0 0 Alle Vorzeichen werden von der WKM aus gesehen. Bei der Berechnung der sich insgesamt ergebenden Arbeit wird die aus folgender Überlegung resultierende Gleichheit der Volumenverhältnisse verwendet: (13.3.1) Durch Division dieser zwei Gleichungen folgt: (13.3.2) Der Wirkungsgrad einer Carnot-Maschine (13.3.3) stimmt natürlich mit dem oben berechneten Wirkungsgrad (Gl. (13.2.5)) überein. 13.4 Thermodynamische Temperaturskala Gl. (13.2.3) kann auf die Form (13.4.1) gebracht werden. Diese Gleichung bzw. Gl. (13.2.6) geben prinzipiell die Möglichkeit, Verhältnisse von Temperaturen über Energiemessungen zu bestimmen. Zur Festlegung des absoluten Wertes muss ein Wert festgesetzt werden. Dies ist der Wert T = 273,16 K für den Tripelpunkt des Wassers (dazu siehe Kap. 5.5 und 17.5). In der traditionellen Darstellung der Thermodynamik und in gesetzlichen Vorschriften werden diese Gleichungen als Definition der thermodynamischen Temperatur bezeichnet. Da diese Gleichungen aus der erheblich fundamentaleren Gl. (3.7) folgen, führt dies zum gleichen Ergebnis wie die Definition mit Gl. (3.7). Natürlich wird keiner ernsthaft versuchen, aus dem Wirkungsgrad einer niemals idealen WKM oder - 75 gemäß Gl. (3.7) Temperaturen zu bestimmen. Bei der Diskussion der Idealen Gasgleichung hatten wir bereits festgestellt, dass sich präzise Gasthermometer über die Proportionalität p % T bei V = const. aufbauen lassen und gezeigt, dass die damit bestimmten Temperaturen mit der thermodynamischen Temperatur übereinstimmen. Da auch der Umgang mit Gasthermometern aufwändig ist, hat man in der ITS-90 (Internationale Temperaturskala 1990) eine Reihe von Fixpunkten durch Vermessung mit Gasthermometern festgelegt (neben dem Tripelpunkt von Wasser bei 273,16 K werden darin z. B. der Tripelpunkt von Neon (24,5561 K) und der Erstarrungspunkt von Zink (692,677 K) festgelegt), zwischen denen im Bereich mittlerer Temperaturen mit Widerstandsthermometern interpoliert wird. Bei den Widerstandsthermometern wird die Temperatur über die Temperaturabhängigkeit eines Platinwiderstands bestimmt. Die Hierarchie der Temperaturskalen sieht daher wie folgt aus: 1) Thermodynamische Temperatur gemäß Gl. (3.7). 2) Temperatur entsprechend Gl. (13.2.6) bzw. (13.4.1). Die so definierte Temperatur stimmt mit der Temperatur entsprechend 1) überein. 3) Gasthermometrische Temperatur entsprechend Gl. (1.2.8). Die so definierte Temperatur stimmt mit der Temperatur entsprechend 1) überein. 4) Internationale Temperaturskala 1990. Diese stimmt weitgehend (im Bereich mittlerer Temperaturen besser als 1 mK), aber nicht exakt mit der Temperatur entsprechend 1) überein. 13.5 Sprache und naturwissenschaftliche Erkenntnisse Die Sprache hat sich lange vor den Naturwissenschaften entwickelt. Jede Sprache enthält Konstruktionen, die den naturwissenschaftlichen Erkenntnisstand der Zeit ihrer Entstehung widerspiegeln. Man hat lange Zeit die Begriffe "Wärme" und "Temperatur" nicht auseinander halten können. So bezeichnet man einen Körper als "warm" und man "erwärmt" ihn, wobei letzteres nicht ausdrücken soll, dass man ihm Wärme zuführt, sondern seine Temperatur erhöht. "Erwärmen" und "Abkühlen" sollten durch "Zutempern" und "Abtempern" ersetzt werden und "warm" und "kalt" durch "temp" und "untemp". Dies lässt sich heute wohl nicht mehr erreichen. Wärme ist eine Prozessgröße. Deswegen sind Aussagen, wie z. B. "Dem System wird Wärme entzogen" nicht korrekt, da sie implizieren, dass das System Wärme enthält. Korrekt & aber nicht üblich & ist dagegen die Aussage: Die Änderung der Energie des Systems erfolgt durch Wärme. Überträgt man den Begriff "Wärmereservoir" oder noch schlimmer "Wärmespeicher" auf den Fall des Drucks, so gäbe es kein Druckreservoir, sondern ein "Volumenarbeitsreservoir" oder das Wärmereservoir müsste Temperaturreservoir heißen. - 76 - 14 Freie Enthalpie Die Freie Enthalpie wurde in Kap. 6 eingeführt. Für das Differenzial wurde der Ausdruck (14.1) gefunden. Durch Vergleich mit dem totalen Differenzial von G(p,T,ni) findet man (14.2) Der Index nj ni heißt: es sollen alle Stoffmengen außer ni konstant gehalten werden. Diese Gleichungen gestatten es uns, die Abhängigkeit der Freien Enthalpie von p und T zu ermitteln. Die Druckabhängigkeit ergibt sich bei konstanter Temperatur mit (14.3) zu (14.4) Für ein Ideales Gas ergibt sich (14.5) Die Temperaturabhängigkeit bei konstantem Druck ist komplizierter (14.6) (14.7) Da S selbst schon eine komplizierte Funktion der Temperatur ist, soll das hier nicht weiter verfolgt werden. Bei der Diskussion der Temperaturabhängigkeit der Gleichgewichtskonstante des MWG werden wir uns noch einmal damit befassen müssen. Wie ändert sich G bei einer Phasenumwandlung? (14.8) Erfolgt die Umwandlung bei einem vorgegebenen Druck bei der Gleichgewichtstemperatur, so ist dT = dp = 0, :N = :O und dnN = &dnO, d. h. (14.9) Eine andere Sicht ergibt sich aus der Definition (14.10) (14.11) wegen Gl. (12.1.26). Die Freie Enthalpie ist die ideale Größe, um Gleichgewichte bei p,T = const. zu untersuchen. Im Gleichgewicht muss dG = 0 sein. Wie ändert sich G beim Mischen von Gasen? Wir wollen uns hier wie bei der Entropie auf die Untersuchung von idealen Gasmischungen beschränken. Für den irreversiblen Prozess wird )G sicher - 77 0 sein. Wie gerade zuvor gilt bei p,T = const: (14.12) Mit )S nach Gl. (12.2.4) ergibt sich (14.13) Für den Mischungsvorgang Idealer Gase gilt )g < 0 im Gegensatz zur Entropie, die ansteigt. G wird im Gleichgewicht minimal; daher muss dGp,T bei irreversiblen Prozessen < 0 werden. Für viele Zwecke ist es nützlich, die Arbeit berechnen zu können, die von einem System für eine bestimmte Zustandsänderung unter isothermen Bedingungen maximal geliefert werden kann. Diese maximale Arbeit erhält man bei einer reversiblen Prozessführung. Würde für eine bestimmte Zustandsänderung ein anderer Prozessweg mehr als diese reversible Arbeit ergeben, so könnte man eine Maschine konstruieren, die Wärme vollständig in Arbeit verwandelt. Dazu wird die Zustandsänderung auf dem alternativen Prozessweg durchgeführt und so viel Arbeit (und wenig Wärme, um dem 1. Hauptsatz Genüge zu tun) gewonnen. In einem zweiten Schritt wird mit dem reversiblen Prozessweg diese Wärme mit wenig Arbeit in das System zurückgeschoben. Es wäre daher möglich, aus einem Wärmereservoir Wärme zu entnehmen und zu 100 % in Arbeit umzuwandeln. Dies widerspricht dem zweiten Hauptsatz. Für einen reversiblen, isothermen Prozess gilt (14.14) oder (14.15) oder (14.16) Die Änderung der Freien Energie entspricht unter isothermen Bedingungen der reversiblen und daher auch der maximalen Arbeit. Aus diesem Grund wurde auch die Bezeichnung "Freie" Energie gewählt: Sie enthält den unter isothermen Bedingungen in Arbeit umsetzbaren Anteil der gesamten Energie. Für die Freie Enthalpie gibt es eine entsprechende Beziehung für isotherme und isobare Bedingungen. Unter der Arbeit AN sollen alle Arbeiten außer der Volumenarbeit verstanden werden, wie beispielsweise die elektrische Arbeit, die Verschiebungsarbeit usw. Aus Gl. (14.15) folgt durch Addition von p)V (14.17) wobei hier unter AN alle interessierenden Arbeiten subsummiert werden sollen. Unter isothermen und isobaren Bedingungen steht auf der linken Seite gerade )G, d. h. (14.18) Die Änderung der Freien Enthalpie ergibt unter isothermen und isobaren Bedingungen gerade die reversible Arbeit unter Ausschluß der Volumenarbeit. Das Prinzip von Le Chatelier und Braun Das Prinzip von Le Chatelier und Braun macht eine Aussage über die Richtung der in einem Gleichgewichtssystem auftretenden Prozesse, wenn dieses Gleichgewichtssystem von außen gestört wird. Das Prinzip lautet: Ein Gleichgewichtssystem reagiert auf einen äußeren Zwang so, dass es diesen Zwang zu vermindern versucht. Die vorsichtige Formulierung mit "versucht" drückt aus, dass die Verminderung des Zwangs nicht immer gelingt. Führt man beispielsweise einem System mit einem chemischen Gleichgewicht Wärme zu und erhöht so die Temperatur, so läuft im System die endotherme - 78 Reaktion (siehe Beispiel 2) ab. Ob sich der Zwang "Temperaturerhöhung" vermindert oder nicht, hängt davon ab, wie die Ankopplung des Systems an die Umgebung ist. Erfolgt diese Ankopplung durch einen Thermostaten, so erniedrigt sich die Temperatur trotz Ablaufs der endothermen Reaktion natürlich nicht. Eine etwas allgemeinere, aber vielleicht nicht so griffige Formulierung des Prinzips ist die folgende. Jedes System im Gleichgewicht reagiert auf eine Änderung einer der das Gleichgewicht bestimmenden Größen so, dass wenn die Reaktion des Systems allein erfolgen würde, sie eine Änderung der gestörten Größe in umgekehrter Richtung bewirken würde. Hier soll der Nachweis des Prinzips für zwei Beispiele geführt werden. 1) Zwei Phasen & beispielsweise zwei feste Modifikationen einer Verbindung & sollen sich bei der unter dem angelegten Druck vorliegenden Umwandlungstemperatur im Gleichgewicht befinden. Das System wird jetzt durch eine geringe Erhöhung des angelegten Drucks gestört. Der Phasenumwandlungsprozess wird wie eine Reaktion durch den Umsatz > beschrieben. (14.19) Jetzt wird das totale Differenzial der Freien Umwandlungsenthalpie (14.20) bei konstanter Temperatur gebildet (14.21) )V ist das Reaktionsvolumen, d. h. die Volumenänderung bei einem Umsatz von 1 mol. Der Druck wird jetzt langsam geändert, so dass das System dieser Änderung folgen kann und sich immer im Gleichgewicht befindet. Für jeden Druck gilt unter isothermen Bedingungen entsprechend Gl. (6.14) (14.22) Ändert man daher den Druck und lässt das Gleichgewicht sich dauernd einstellen, so gilt für diesen Prozess (14.23) und daher mit Gl. (14.21) (14.24) oder (14.25) Nun ist G als Funktion von > bei p, T = const. im Gleichgewicht minimal, d. h. M2G/M> 2 ist positiv. Daher ändert sich > mit steigendem p so, wie es das Vorzeichen von -)V vorgibt. Die Änderung von - 79 > ist daher positiv, wenn )V negativ ist, d. h. das Volumen bei der Umwandlung abnimmt. Unter der Annahme, dass die Dichten der beiden festen Phasen nicht vom Druck abhängen, gilt für das Systemvolumen (14.26) und daher (14.27) d. h. eine Druckerhöhung bewirkt unabhängig vom Vorzeichen von )V eine Volumenverminderung durch die Phasenumwandlung. 2) Eine Reaktion befinde sich im Gleichgewicht. Dem System wird Wärme zugeführt und damit die Temperatur erhöht. Wie verschiebt sich das Gleichgewicht? Bei konstantem Druck gilt analog zum Vorgehen beim vorhergehenden Beispiel (14.28) und daher im Gleichgewicht (14.29) wobei die letzte Manipulation nur für ein System im Gleichgewicht wegen (14.30) möglich ist. > ändert sich daher mit steigender Temperatur so, wie das Vorzeichen von )H ist, d. h. bei einer endothermen Reaktion läuft die "Hinreaktion" ab - dafür wird nämlich Wärme aus der Umgebung benötigt - und umgekehrt. Wie im vorigen Beispiel kann die Rechnung auch noch einen Schritt weitergeführt werden. Bei Vernachlässigung der Abhängigkeit der Systementhalpie von der Temperatur über die Molwärmen gilt (14.31) und daher (14.32) d. h. die Systementhalpie steigt bei einer Temperaturerhöhung durch Ablauf der entsprechenden Reaktion immer an. Die Gültigkeit des Prinzips von Le Chatelier und Braun kann durch eine Herleitung mit den allgemeinen intensiven und extensiven Größen > und X und einem beliebigen thermodynamischen Potenzial unter Verlust des letzten Rests an Anschaulichkeit sehr allgemein nachgewiesen werden und ist dann von außerordentlich großer Tragweite. - 80 Der Name "Braun" wird häufig bei der Bezeichnung des Prinzips weggelassen, obwohl die Arbeit von Braun in Z. Phys. Chem. 1887 erschien und die von Le Chatelier erst ein Jahr später. - 81 - 15 Chemisches Potenzial Wir hatten in Kap. 5.3 und 5.4 das chemische Potenzial als wichtige Größe zur Beschreibung von Gleichgewichten kennengelernt, bei denen sich die Stoffmengen ändern. Für diesen Zweck benötigen wir die Abhängigkeit des chemischen Potenzials von p, T und der Zusammensetzung in Mischungen. Weiterhin wird für die Berechnung von Gleichgewichten die Kenntnis der Differenziale d:i und d(:i/T) von Nutzen sein. 15.1 Chemisches Potenzial reiner Phasen Aus Gl. (14.2) folgt für eine reine Phase (n1 = n) (15.1.1) Für reine Phasen entspricht (MG/Mn)p,T der molaren Freien Enthalpie g. Aus Gl. (14.2) ergeben sich damit die Beziehungen (15.1.2) und (15.1.3) Für ein reines Ideales Gas resultiert daraus bei T = const. (15.1.4) (15.1.5) Setzt man insbesondere p1 dem Standarddruck ps (i. a. ps = 1,013 bar) gleich, folgt (15.1.6) :s entspricht dem chemischen Potenzial bei p = ps, da dann das logarithmische Glied verschwindet. Für das Differenzial von :/T finden wir (15.1.7) Der zweite Term ist einfach zu berechnen. Für den ersten gilt (15.1.8) so dass sich insgesamt ergibt (15.1.9) 15.2 Chemisches Potenzial in idealen Mischphasen Analog zu Gl. (15.1.1) erhält man für eine Mischphase - 82 - (15.2.1) Derartige Größen werden im Gegensatz zu molaren Größen in reinen Phasen als partielle molare Größen bezeichnet. Das chemische Potenzial entspricht daher der partiellen molaren Freien Enthalpie. Da nun (MG/Mp) und (MG/MT) bekannt sind, kann man durch Vertauschung der Differenziationsreihenfolge die entsprechenden Differenzialquotienten für :i berechnen (15.2.2) Bei der Differenziation sind alle ni konstant zu halten. si und Li sind die partielle molare Entropie und das partielle molare Volumen. (15.2.3) Am Beispiel des partiellen molaren Volumens soll die Bedeutung der partiellen molaren Größen veranschaulicht werden. Man nehme eine so große Menge der Mischung, dass der Zusatz eines Mols der Komponente i die Konzentrationen nur unwesentlich verändert. Dann entspricht die nach diesem Zusatz auftretendende Volumenveränderung dem partiellen molaren Volumen Li. Mit Hilfe von Gl. (15.2.2) können wir das totale Differenzial von :i(p,T) bei fester Zusammensetzung formulieren: (15.2.4) Analog findet man (15.2.5) Als nächstes wollen wir die Abhängigkeit von der Zusammensetzung in einer idealen Gasmischung untersuchen. In einem Gedankenexperiment trennen wir N O durch eine nur für die Komponente i durchlässige Membran T T die Mischung mit dem Partialdruck pi und das reine Gas p1, p2, ..., pi pi unter dem gleichen Druck pi. Da die Membran für die x1, x2, ..., xi xi = 1 Komponente i durchlässig ist, steht das Gas i in den zwei Volumina im Gleichgewicht und es gilt (15.2.6) wegen der Gleichgewichtsbedingung und Gl. (15.1.6). Die Striche können jetzt weggelassen werden, da nur noch im System N definierte Größen auftreten. (15.2.7) :is ist das chemische Potenzial des reinen Gases i beim Standarddruck. Eine Umstellung mit p = p1 + p2 + ... ergibt - 83 - (15.2.8) und mit pi /p = xi folgt (15.2.9) Oft werden noch die beiden ersten Glieder der rechten Seite zu einem neuen Glied, dem Standardpotenzial :o für die reine Phase beim Gesamtdruck p, zusammengefasst (15.2.10) Damit folgt (15.2.11) Die letzte Gleichung enthält dann nur die Abhängigkeit von der Zusammensetzung des Gases. Die Gesamtdruckabhängigkeit steckt bereits im :io. Gl. (15.2.11) gestattet die Berechnung des Differenzials von :i(T, p, xi). Um die Berechnung zu vereinfachen, wird aus dem zweiten Term von (15.2.11) die Temperatur entfernt (15.2.12) und das Differenzial mit Hilfe von Gl. (15.1.9) gebildet (15.2.13) hio und Lio stellen die molare Enthalpie und das molare Volumen für die reine Komponente i dar. Wegen der zentralen Bedeutung der Gl. (15.2.7) bis (15.2.13) soll noch ein zweiter Weg zu ihnen gezeigt werden. Da sowohl der Zusammenhang zwischen : und G als auch die Freie Mischungsenthalpie bekannt sind, muss eine Ableitung dieser Gleichungen daraus möglich sein. Für die Mischung von zwei Idealen Gasen gilt nach Gl. (14.13) (15.2.14) Jetzt wird M/Mn1 gebildet (15.2.15) (15.2.16) (15.2.17) und wir erhalten das Gl. (15.2.11) entsprechende Ergebnis für den Fall einer idealen Gasmischung aus zwei Komponenten. Wie kommt man nun zu einer Gleichung für das chemische Potenzial in flüssigen Mischungen? Der - 84 zu (15.2.11) führende Weg ist nur für die Gasphase beschreitbar. Auch der Weg zu Gl. (15.2.17) ist nur über die Gasphase möglich, da die Freie Mischungsenthalpie nur für die Gasphase zugänglich ist. Von der Thermodynamik her unterscheiden sich nun die Gasphase und die flüssige Phase nicht grundsätzlich. Der gravierende Unterschied ist die in der flüssigen Phase etwa um den Faktor 1000 größere Teilchendichte. Dies bewirkt in der flüssigen Phase erheblich stärkere Wechselwirkungen als in der Gasphase. Grundsätzlich gibt es jedoch kein Argument gegen die Benutzung von Gl. (15.2.11) in der flüssigen Phase. Die in Gl. (15.2.11) auftretenden Größen sind in der flüssigen Phase definiert. Die flüssige Mischung muss sich nur ideal verhalten. Diese Bedingung ist in Flüssigkeiten wegen der grundsätzlich höheren Wechselwirkung der Teilchen deutlich schwerer als in der Gasphase zu erfüllen. An dieser Stelle ist es von Nutzen, den Begriff "ideal" genauer zu diskutieren. Die für ein Ideales Gas geforderten Voraussetzungen können in der flüssigen Phase sicherlich nicht vorliegen. Es kann keine "ideale" Flüssigkeit geben, da das Eigenvolumen und das eingenommene Volumen vergleichbar sind und auch merkliche Wechselwirkungen vorhanden sein müssen, um den Zusammenhalt der Flüssigkeit zu gewährleisten. Es kann nur noch ideale Mischungen geben. Darunter wollen wir Mischungen verstehen, bei denen die Größen V, U und H beim Mischungsvorgang bei entsprechenden Konstanthaltungen additiv sind (Xm = X1 + X2) und für welche die Gl. (12.2.4), (14.13) und (15.2.11) für die Mischungsentropie, die Freie Mischungsenthalpie bzw. das chemische Potenzial gelten. Diese Forderungen sind nicht unabhängig voneinander; so folgt z. B. die Forderung für die Enthalpie aus der Forderung für die Innere Energie und das Volumen. Bei H und U sagt man auch, dass die Mischungsenthalpie und die Mischungsenergie im idealen Fall verschwinden. Wir sollten jetzt einen Blick ins mikroskopische Geschehen bei der Herstellung einer Mischung aus den Komponenten 1 und 2 werfen. Offensichtlich ist es wichtig, dass die Wechselwirkungen W11, W22 und W12 übereinstimmen oder zumindest W12 = (W11+W22)/2 gilt. Andernfalls treten Temperaturänderungen bei der Mischung auf. Dies führt sicher zu Abweichungen von der Freien Mischungsenthalpie idealer Systeme, da )H in Gl. (14.12) nicht mehr verschwindet. Eine Auswahl von Systemen, die in der flüssigen Phase weitgehend ideale Mischungen bilden, ist die folgende: Mischungen optischer Antipoden, Mischungen von Komponenten mit unterschiedlichen Isotopen, Mischungen von benachbarten Gliedern in homologen Reihen, stark verdünnte Lösungen bezüglich der gelösten Komponente. Die zuletzt aufgeführten Lösungen befolgen zwar Gl. (15.2.11), jedoch nicht die oben angegebene Additivität einiger Größen. Für Mischungen von Komponenten, die sich in der Polarität, der Molekülgröße oder dem Vorliegen spezifischer Wechselwirkungen (z. B. Wasserstoffbrückenbindungen) auch nur mäßig unterscheiden, muss von nichtidealem Verhalten ausgegangen werden. Das Verhalten derartiger realer Systeme wird in einer späteren Vorlesung (PC III) behandelt werden. Im Prinzip lässt sich Gl. (15.2.11) auch für ideale feste Mischungen verwenden. Hierzu ist jedoch festzustellen, dass bereits die Bildung von festen Mischungen (Mischkristalle) die Ausnahme darstellt (siehe Kap. 20.3 und 21.2), und die für die Idealität zu fordernde Ähnlichkeit der Komponenten noch strenger als bei den Flüssigkeiten ist. Mischkristalle von Komponenten mit unterschiedlichen Isotopen sind ein Beispiel für ideale Systeme. Ist das chemische Potenzial einer Komponente in einer idealen oder realen Zweikomponentenmischung bekannt, so lässt sich das der anderen Komponente berechnen. Für das Differenzial der Freien Enthalpie gilt für vielkomponentige Systeme bei p,T = const. (15.2.18) Diese Gleichung wird jetzt über die Stoffmengen bei fester Zusammensetzung integriert (15.2.19) - 85 Bildung des Differenzials und Vergleich mit (15.2.18) liefert (15.2.20) (15.2.21) Dieser Zusammenhang wird als Gibbs-Duhemsche Gleichung bezeichnet. Entsprechende Gleichungen lassen sich aus den Differenzialen der thermodynamischen Potenziale für viele andere Größen finden. Aus der Mathematik ist dieser Zusammenhang als Eulersches Theorem bekannt. Für eine zweikomponentige Mischung gilt (15.2.22) (15.2.23) (15.2.24) Trägt man :1 und :2 gegen x1 auf, so müssen die Steigungen an der Stelle x1 = 0,5 bis auf das Vorzeichen übereinstimmen. Die Auflösung von Gl. (15.2.24) nach d:1 und Integration liefert schließlich (15.2.25) (15.2.26) Ist :2 = f(x1) bekannt, so lässt sich :1 mit Hilfe dieser Gleichung bestimmen. Für ideale Mischungen entsteht daraus die bereits bekannte Abhängigkeit. Wichtig ist die Gleichung für die Untersuchung realer Mischungen. - 86 - 16 Chemisches Gleichgewicht 16.1 Massenwirkungsgesetz (MWG) In Kap. 5 und 6 war gezeigt worden, dass für die Reaktion (16.1.1) im chemischen Gleichgewicht bei beliebigen Konstanthaltungen (16.1.2) gilt. Die Gleichgewichtsbedingung gilt unabhängig davon, ob die Ri in einer Phase (homogenes Gleichgewicht) oder in unterschiedlichen Phasen (heterogenes Gleichgewicht) vorliegen. Zuerst wollen wir uns aber auf die Gasphase beschränken und ideales Verhalten annehmen. Mit Gl. (15.2.7) finden wir (16.1.3) (16.1.4) wobei A das zu G analoge Zeichen für ein Produkt ist. (16.1.5) Den ersten Teil dieser Gleichung bezeichnet man als Massenwirkungsgesetz (MWG). Kp & die Konstante des MWG & ist für eine gegebene Reaktion und eine gegebene Temperatur eine Konstante, die nicht von den Partialdrücken und vom Gesamtdruck (siehe rechten Teil der Gl. (16.1.5)) abhängt. Die pi sind die Partialdrücke im Gleichgewicht. Ist (16.1.6) so befindet sich die Reaktion nicht im Gleichgewicht und bewegt sich mit mehr oder weniger großer Reaktionsgeschwindigkeit auf das Gleichgewicht zu. Über die Reaktionsgeschwindigkeit kann die Thermodynamik keine Aussage machen. Kp ist in der Schreibweise von Gl. (16.1.5) einheitenfrei, da jeder Partialdruck durch den Standarddruck dividiert wird. Dieses an und für sich korrekte Verfahren führt leider zu einer Reihe von Schwierigkeiten. In der einheitenfreien Schreibweise enthält die Angabe von Kp keinen Hinweis mehr, ob als Standarddruck 1 atm, 1 bar oder 1 Pa verwendet wurde. Mit unterschiedlichen Standarddrücken ergeben sich jedoch unterschiedliche Kp-Werte. Z. B. kürzen sich bei der Gasreaktion (16.1.7) bei der Umrechnung von bar in Pa die Umrechnungsfaktoren 105 im Produkt nur teilweise heraus und es bleibt ein Faktor 105 im Zähler stehen, d. h. Kp ändert sich um den Faktor 105. Unabhängig von den verwandten Einheiten sind die Gleichgewichtskonstanten nur für den Fall )< = 0. Bei den Übungsaufgaben wird daher entweder pÈ angegeben oder die einheitenbehaftete Form des Massenwirkungsgesetzes - 87 (16.1.8) verwendet. Statt der Drücke werden in der Gasphase oft Molenbrüche verwendet. (16.1.9) Das MWG lässt sich daher auch mit Molenbrüchen formulieren (16.1.10) Kx ist grundsätzlich einheitenfrei. Kx hängt vom Gesamtdruck entsprechend Gl. (16.1.10) ab. Bei Reaktionen mit gleicher Molekülzahl rechts und links ()< = 0) entfällt die Druckabhängigkeit. Das mit Molenbrüchen formulierte MWG lässt sich auch für die flüssige Phase verwenden. Eine von der vorhergehenden Ableitung in der Gasphase unabhängige Ableitung erhält man aus der Gleichgewichtsbedingung G<i:i = 0 und Gl. (15.2.11), die beide auch in der flüssigen Phase gültig sind. Natürlich muss auch hier die bei Gl. (15.2.11) vorausgesetzte Idealität vorliegen. Schließlich wollen wir noch das Konzentrationsmaß ci = ni/V in der Gasphase einführen. (16.1.11) (16.1.12) Das MWG lässt sich daher auch mit Konzentrationen formulieren. (16.1.13) Im Prinzip könnte man die Klammer in der zweiten Gleichung mit Gl. (16.1.11) weiter vereinfachen. ps und cs sind jedoch festgelegte Größen mit inkompatiblen Einheiten und folgen daher dem Zusammenhang (16.1.11) nicht. Bei der Umrechnung von Kp in Kc muss daher auf die Umrechnung der Einheiten geachtet werden! Das bereits bei der Diskussion von Kp Gesagte gilt auch für Kc: Wegen der Unsicherheit bzgl. der Einheit von cÈ (1 mol/dm3 oder 1 mol/m3) ist cÈ anzugeben oder das MWG ist einheitenbehaftet (16.1.14) zu formulieren. Die Gl. (16.1.13) und (16.1.14) können auch für chemische Gleichgewichte in der flüssigen und festen Phase verwendet werden. Als nächstes wollen wir uns mit dem Zusammenhang zwischen Kp und den chemischen Potenzialen unter Standardbedingungen befassen (s. Gl. (16.1.5)) (16.1.15) oder (16.1.16) - 88 Damit dies eine ordentliche Gleichung ist, muss Kp dimensionslos sein. Diese wichtige Gleichung der Thermodynamik gestattet es, Gleichgewichtskonstanten aus Reaktionsgrößen zu berechnen. )Gs ist die Freie Reaktionsenthalpie unter Standardbedingungen. )Gs < (bzw. >) 0 heißt, die entsprechende Verbindung ist stabil (instabil oder metastabil) gegen einen Zerfall in die Elemente, wenn alle Reaktanden unter Standardbedingungen vorliegen. Beispiele für Verbindungen mit )Gs > 0 sind Ozon, Ethen, Benzol und Ethin. Benzol kann jedoch sogar ohne Gegenwart von 1 atm Wasserstoff beliebig lange ohne Zerfall gelagert werden, da die Zerfallskinetik gehemmt ist. Derartige Verbindungen werden als metastabil bezeichnet. Natürlich ist für Verbindungen mit )Gs > 0 eine direkte Synthese aus den Elementen ausgeschlossen. Viele Tabellenwerke (siehe Anhang) enthalten analog zu den Bildungsenthalpien die Freien Bildungsenthalpien )GB unter Standardbedingungen. Ganz analog zu den Reaktionsenthalpien lassen sich die Freien Reaktionsenthalpien unter Standardbedingungen nach (16.1.17) berechnen. Woher stammen die )GB-Werte? Dazu leiten wir die Definitionsgleichung für G und > ab und finden für die Bildungsreaktion unter Standardbedingungen (16.1.18) Sind also die entsprechenden Bildungsenthalpien und -entropien bekannt, so kann )GB berechnet werden. Beispielsweise gilt für Wasser unter Standardbedingungen (16.1.19) was mit dem im Tabellenanhang angegebenen Wert übereinstimmt. Die Bildungsentropie lässt sich aus den nach Kap. 12.1 ermittelten Entropien der Reaktionspartner berechnen: (16.1.20) Schwierigkeiten mit der Mischungsentropie gibt es nicht, da die Entropie eines Gases in einer idealen Mischung nur eine Funktion des Partialdrucks ist, und dieser ist unter Standardbedingungen festgelegt. Zusätzlich verlangt auch der Standardzustand reine Komponenten. Im Prinzip ist die Angabe von )GB neben )HB und den Entropien in Tabellenwerken unnötig, aber sehr praktisch. Als Beispiel soll die Gleichgewichtskonstante für die Dimerisierungsreaktion (16.1.21) in der Gasphase bei der Standardtemperatur berechnet werden. Es gilt (16.1.22) und daher (16.1.23) wobei der Standarddruck entsprechend der Tabelle 1 atm entspricht. Wegen der für den Chemiker zentralen Bedeutung des MWG wollen wir uns noch eine zweite Herleitung dieses Gesetzes ansehen. Dazu schreiben wir die Freie Reaktionsenthalpie für eine Reaktion auf, die sich nicht im Gleichgewicht befindet. - 89 - (16.1.24) (16.1.25) wobei wir den Zusammenhang gi = :i und die Abhängigkeit des chemischen Potenzials in idealen Gasmischungen vom Partialdruck verwendet haben. Diese Gleichung zeigt folgendes: a) Gilt für alle i pi = pÈ, so wird natürlich )G = )GÈ b) Im Gleichgewicht muss unter isobaren und isothermen Bedingungen dG und damit auch )G verschwinden. Die rechte Seite ergibt dann das MWG und den Zusammenhang von )GÈ mit Kp. )G stellt also ein Maß für die Abweichung vom Gleichgewicht dar. In einigen Lehrbüchern wird daher für &)G die besondere Bezeichnung "Affinität" eingeführt. 16.2 Berechnung von Gleichgewichtsdrücken mit Hilfe des MWGs a) Es sei Kp für eine Gasphasenreaktion bekannt. Weiterhin sollen alle Partialdrücke im Gleichgewicht bis auf den einer Komponente bekannt sein. Das MWG wird nach diesem Partialdruck aufgelöst. Einsetzen der bekannten pi und Kp führt zum Ergebnis. b) Es sei Kp für eine Gasphasenreaktion bekannt. Das Reaktionsgefäß wird mit festgelegten Partialdrücke opi der Edukte gefüllt. Danach erfolgt die Reaktion zum Gleichgewicht im konstant gehaltenen Volumen. Die Berechnung ist vom Rechenaufwand her bei Reaktionen, an denen viele Komponenten beteiligt sind, aufwändig. An der Reaktion mögen n Komponenten beteiligt sein. Wir führen den Umsatz > ein. Insgesamt gibt es dann n+1 Unbekannte. Andererseits gibt es als Gleichungen das MWG und n Bilanzgleichungen pi = fi(>). Die Bilanzgleichungen erhält man wie folgt: (16.2.1) Mit Hilfe des Idealen Gasgesetzes berechnet man daraus die Drücke (16.2.2) Einsetzen in das MWG ergibt (16.2.3) Diese Gleichung enthält nur noch eine Unbekannte und ist daher lösbar. Die Auflösung des Polynoms vom Grade Max(&E<i, Edukte , E<i, Produkte) nach >N kann aufwändig sein. Die Partialdrücke pi werden dann mit Gl. (16.2.2) berechnet. In vielen Fällen lässt sich die Einführung des Umsatzes vermeiden. Liegt eines der Produkte vor der Reaktion mit dem Partialdruck 0 vor, so kann man diesen Partialdruck als Umsatzvariable benutzen. Das Produkt, für das dies zutreffen möge, sei Rk. Der Zusammenhang mit dem Umsatz ergibt sich aus Gl. (16.2.2) (16.2.4) Für die anderen Komponenten ist dann im Gleichgewicht (16.2.5) - 90 Einsetzen in das MWG ergibt (16.2.6) Auch das Glied mit i = k ist in dieser Gleichung korrekt enthalten, da opk nach Voraussetzung verschwindet und das Verhältnis der stöchiometrischen Koeffizienten 1 wird. Gl. (16.2.6) ist das kürzeste Verfahren, um das Problem zu lösen. Vermeintlich einfachere Eigenkonstruktionen sind mit Sicherheit falsch. Mit solchen Fehlrechnungen gewinnt man jedoch viel Erfahrung. Im folgenden soll ein Beispiel durchgerechnet werden. Die Gleichgewichtsreaktion sei A + 3B W C + 2D; die Gleichgewichtskonstante betrage 4 bar &1; vor Einstellung des Gleichgewichts sollen die Drücke pA = 1 und pB = 2 bar betragen. Die Reaktion soll bei konstantem Volumen ablaufen. Gl. (16.2.3) ergibt (16.2.7) oder (16.2.8) Diese Bestimmungsgleichung für >N wird nun mit Hilfe irgendeines Verfahrens (Intervallschachtelung, Regula falsi, Newtonsches Näherungsverfahren, Solve-Funktion eines Taschenrechners oder entsprechendes Programm auf einem PC) gelöst. Für das Beispiel gibt es zwei reelle Lösungen >N = 0,472 005 und 1,311 558 bar, wovon die zweite physikalisch sinnlos ist, da damit die Partialdrücke von A und B negativ werden würden. Die erste Lösung ergibt mit Hilfe von Gl. (16.2.2) folgende Partialdrücke: pA = 0,527 995 pB = 0,583 985 pC = 0,472 005 pD = 0,944 010 bar Zur Kontrolle sollten die gefundenen Werte immer in das MWG eingesetzt werden. Hier ergibt sich für das Produkt der Wert 4,000 045 bar &1. Die entsprechende Rechnung mit Hilfe von Gl. (16.2.6) und k = D ergibt (16.2.9) Der Vergleich mit Gl. (16.2.7) zeigt, dass pD = 2>N gilt und die beiden Gleichungen sich entsprechen. c) Bei den sogenannten offenen Systemen durchströmt ein Gemisch den Reaktor, reagiert dort und verlässt den Reaktor. Ein Beispiel ist die Umsetzung von SO2 und O2 zu SO3 an einem Katalysator in einem von dem Gemisch durchströmten Rohrreaktor. Im Gegensatz zum Fall b) ist jetzt nicht mehr das Volumen konstant, sondern es soll p = const. angenommen werden. Für diesen Fall ist die Berechnung der Partialdrücke nun nicht mehr so einfach wie im Fall b). Am günstigsten ist es, das mit Molenbrüchen formulierte MWG zu verwenden. (16.2.10) (16.2.11) - 91 (16.2.12) (16.2.13) Weiterhin wollen wir noch folgendes Maß für den Umsatz definieren (16.2.14) d. h. den auf die anfängliche Stoffmenge bezogenen Umsatz. Damit wird aus (16.2.13), wobei Zähler und Nenner durch on geteilt werden (16.2.15) Dies ist eine Bestimmungsgleichung für .. Die Molenbrüche in der Gleichgewichtsmischung erhält man direkt aus dem Ausdruck in der Klammer in (16.2.15) und die Partialdrücke pi = xi p berechnet man nach (16.2.16) Gl. (16.2.15) darf auch für homogene Gleichgewichte in der flüssigen und festen Phase verwendet werden. 16.3 Heterogenes Gleichgewicht Da die Physikochemiker nicht viel von der restlichen Chemie verstehen, wollen wir wieder das bereits diskutierte Beispiel Ca CO3, s W Ca Os + CO2, g verwenden. Für die Formulierung des MWG ist folgendes Verfahren zwar nicht sehr elegant, aber sicher richtig. In der Gasphase gilt (16.3.1) pCaO und pCaCO3 sind die extrem kleinen Partialdrücke in der Gasphase! Im Gleichgewicht stehen diese Gase auch im Gleichgewicht mit den festen Phasen, d. h. die Partialdrücke entsprechen den Dampfdrücken der festen Phasen. Es gilt daher (16.3.2) d. h. für eine gegebene Temperatur ist pCaO festgelegt. Wir können daher pCaO und pCaCO3 in die Konstante KpN einbeziehen und erhalten (16.3.3) Ein etwas entartetes, aber sicher korrektes MWG! In manchen Lehrbüchern der Anorganischen Chemie findet man als Argument für das Streichen der Größen pCaO und pCaCO3 in Gl. (16.3.1), die Größen sind klein und können daher gestrichen werden. Das ist Unsinn! Das Streichen einer kleinen, multiplikativ eingehenden Größe macht eine Gleichung total - 92 falsch. Nur kleine additive Größen dürfen gestrichen werden. Unsere Argumentation zeigt, dass der Wert der Größe völlig unerheblich ist. Auf die Konstanz kommt es an! Eine andere und bessere Sicht des heterogenen Gleichgewichts ist die folgende: Wir gehen auf die chemischen Potenziale zurück (16.3.4) Die Komponente Rk soll jetzt in der festen Phase vorliegen. Bei der Diskussion der Herleitung dieser Gleichung hatten wir festgestellt, dass dann das :k für die entsprechende Phase zu verwenden ist. Ist diese feste Phase nun rein, d. h. kein Mischkristall, so ist das chemische Potenzial nur eine Funktion der Temperatur und sicher unabhängig von der Zusammensetzung der Gasphase. Das entsprechende chemische Potenzial tritt daher im Exponenten von Gl. (16.1.15) auf; es entsteht jedoch kein Druckterm im MWG, d. h. man erhält wieder Gl. (16.3.3). Etwas anders ist die Lage, wenn ein heterogenes Gleichgewicht zwischen einer flüssigen Mischung und einer gasförmigen Phase vorliegt, wobei eine Komponente fast nur in der flüssigen Phase auftreten soll. Hier wird man mit Vorteil die Formulierung mit den Molenbrüchen verwenden. Gl. (16.3.4) ergibt auch für den heterogenen Fall (16.3.5) wobei die xi in den entsprechenden Phasen zu verwenden sind. 16.4 Temperaturabhängigkeit der Gleichgewichtskonstante Kp Gl. (16.1.16) enthält den Zusammenhang zwischen Kp und der Freien Reaktionsenthalpie )GÈ unter Standardbedingungen. Die Freie Reaktionsenthalpie hängt selbst von der Temperatur ab. Zur Feststellung dieses Zusammenhangs wird die Definitionsgleichung für G wie bereits früher nach > abgeleitet. (16.4.1) (16.4.2) )HÈ und )SÈ sind die Reaktionsenthalpie bzw. -entropie der reinen Komponenten unter dem Standarddruck, wobei die Temperatur aber frei variabel sein soll. Mit diesen Gleichungen ist es nun möglich, die interessierende Temperaturabhängigkeit von Kp zu bestimmen. (16.4.3) (16.4.4) Diese Gleichung wird als "van't Hoffsche Reaktionsisobare" bezeichnet. Der Begriff "isobare" bezieht sich auf die Konstanthaltung des Drucks bei der Differenziation nach der Temperatur. Für den hier diskutierten Fall der idealen Systeme ist dies ohne Belang, da Kp sowieso nicht vom Druck abhängt. Anstelle von )HÈ darf auch )H, die Reaktionsenthalpie unter den vorliegenden Bedingungen, in Gl. (16.4.4) verwendet werden, da in idealen Systemen beide Größen übereinstimmen, da sowohl die Mischungsenthalpien als auch (MH/Mp)T verschwinden. Die Temperaturabhängigkeiten von Kx und Kc lassen sich aus Gl. (16.4.4) und (16.1.10) bzw. (16.1.13) berechnen. Für Kx ergibt sich bei p = const. eine zu Gl. (16.4.4) analoge Gleichung. Für Kc ist der Fall etwas komplizierter, da der Zusammenhang mit Kp selbst temperaturabhängig ist. I. a. kann man jedoch diese Abhängigkeit vernachlässigen, so dass auch für diesen Fall die van't Hoffsche Reaktionsisobare - 93 mit ausreichender Genauigkeit angesetzt werden kann. Auch für flüssige und feste Phasen lässt sich die mit Kx bzw. Kc formulierte van't Hoffsche Reaktionsisobare anwenden. Für nicht zu große Temperaturbereiche kann nun )H als unabhängig von der Temperatur angenommen werden. Die Integration von Gl. (16.4.4) ergibt dann (16.4.5) Trägt man daher ln Kp gegen 1/T auf, so sollte sich eine lineare Abhängigkeit mit der Steigung &)H/R ergeben. Abb. 44 zeigt die Änderung von Kp mit der Temperatur für endo- und exotherme Reaktionen. Die Richtung der Änderung ist bereits mit dem Prinzip von Le Chatelier und Braun vorraussagbar; der quantitative Zusammenhang ergibt sich jedoch erst aus der van't Hoffschen Reaktionsisobare. Gl. (16.4.5) wird in der Praxis für zwei Dinge genutzt. 1) Ist Kp = f (T) bekannt, so lässt sich daraus die Reaktionsenthalpie bestimmen. 2) Ist Kp bei einer Temperatur & z. B. bei der Standardtemperatur & bekannt und soll für eine Abb. 44 van’t Hoffsche Reaktionsisobare andere Temperatur berechnet werden, so gelingt das bei bekannter Reaktionsenthalpie. Dazu wird (16.4.5) für zwei Temperaturen angesetzt und die Differenz gebildet (16.4.6) Ist die Annahme der Konstanz von )H unzureichend, so werden i. a. nicht an Gl. (16.4.5) Verbesserungen angebracht, sondern man geht auf die Ausgangsgleichung (16.1.16) und (16.4.2) zurück und erhält die Ulichschen Näherungen. Es gilt immer exakt (16.4.7) d. h., wenn )HÈ und )SÈ bei der interessierenden Temperatur bekannt sind, lässt sich )GÈ und damit Kp exakt berechnen. 1. Näherung: Alle cp sollen verschwinden. (16.4.8) In dieser Gleichung steckt wenigstens in T noch die korrekte Abhängigkeit. 2. Näherung: Alle cp sind 0, aber sollen unabhängig von T sein. Mit den bekannten Gleichungen für die Temperaturabhängigkeit von )H und )S finden wir (16.4.9) In der dritten, hier nicht aufgeführten Ulichschen Näherung wird schließlich ein linearer Ansatz cp = a + b T für die Molwärmen verwendet. - 94 16.5 Druckabhängigkeit der Gleichgewichtskonstante Kx Wie wir gesehen haben, ist Kp bei idealen Systemen nicht abhängig vom Druck. Kx ist nach Gl. (16.1.10) abhängig vom Druck. Dies wird bei technischen Anwendungen, z. B. bei der AmmoniakSynthese, ausgenutzt. Für Lösungen darf die Druckabhängigkeit nach Gl. (16.1.10) natürlich nicht verwendet werden. Für Lösungen wird wie folgt verfahren. Aus den Gleichgewichtsbedingungen (16.5.1) und der Abhängigkeit der chemischen Potenziale idealer Lösungen vom Molenbruch (16.5.2) wobei sich o auf die reine Phase mit xi = 1 bezieht, erhält man nach dem für die Gasphase durchgeführten Verfahren (16.5.3) wobei )Go die Freie Reaktionsenthalpie für die reinen Phasen ist. Diese Gleichung erlaubt, die Druckabhängigkeit zu berechnen (16.5.4) wobei )Vo die Änderung des Volumens bei einem Formelumsatz unter den obigen Standardbedingungen ist. )Vo wird höchstens einige 10 cm3 betragen. Dies ergibt (16.5.5) Die Änderungen von Kx für Gleichgewichte in Lösungen bei Drücken im Bereich einiger bar kann daher vernachlässigt werden. Größere Effekte erreicht man erst bei Drücken von einigen kbar. 16.6 Experimentelle Bestimmung der Gleichgewichtskonstante K a) Direkte Messungen In vielen Fällen können die Partialdrücke der Reaktionspartner in der Gasphase oder die Konzentrationen in Lösungen direkt gemessen werden. Dafür kommen in erster Linie spektroskopische Verfahren (UV-, IR-, NMR-Spektroskopie) in Frage. Das Messverfahren darf dabei die Gleichgewichtszusammensetzung nicht ändern. b) Bei Gleichgewichtsreaktionen, bei denen sich die Molekülzahl ändert (z. B. Dissoziationsreaktionen vom Typ A W 2 B), reicht schon die Bestimmung der Gasdrucks bei einer Reaktion in einem konstanten Volumen zur Berechnung der Partialdrücke aus. c) Einfrieren von Gleichgewichten Bei Reaktionen, die nur bei hohen Temperaturen an Katalysatoren ablaufen, ist es oft möglich, die Gleichgewichtsmischung nach Kontakt mit dem Katalysator so schnell abzukühlen, dass die Molenbrüche der Reaktionspartner erhalten bleiben (Einfrieren des Gleichgewichts). Die bei a) genannte Einschränkung entfällt dann und die Gasmischung kann bei Raumtemperatur einer Gasanalyse, z. B. mit der Gaschromatographie, unterworfen werden. d) Für Ionenreaktionen in wässrigen Lösungen lassen sich häufig elektrochemische Zellen aufbauen, an denen Spannungen gemessen werden können. Dies erlaubt in vielen Fällen die direkte Bestimmung von )GÈ mit hoher Genauigkeit (siehe Kap. 24.5). e) Für Reaktionen, bei denen Ionen entstehen oder verschwinden, kann oft die sehr einfache Messung der elektrischen Leitfähigkeit zur Konzentrationsbestimmung herangezogen werden. - 95 16.7 Schwache Säuren und Basen und deren Salze Als Beispiel für eine Anwendung des MWGs sollen die Dissoziationsgleichgewichte dieser Verbindungen in wässrigen Lösungen untersucht werden. 16.7.1 Dissoziationskonstanten Unter schwachen Säuren/Basen versteht man Säuren/Basen, die in wässriger Lösung nur teilweise dissoziieren. (16.7.1.1) (16.7.1.2) wobei HS und BOH allgemeine Symbole für Säuren und Basen darstellen. Im Gegensatz zu den starken Säuren/Basen, wie HCl und NaOH, findet man bei diesen Verbindungen auch die undissoziierte Form in der wässrigen Lösung. Obwohl bei der Dissoziation Ionen entstehen und schon bei mäßig konzentrierten Ionen-Lösungen nichtideales Verhalten hervorgerufen wird, ist hier das Problem nicht so schwerwiegend, da die bei höheren Konzentrationen schwache Dissoziation die Ionenkonzentrationen relativ gering hält. In der folgenden Tabelle sind die Dissoziationskonstanten (16.7.1.3) einiger organischer Säuren bei 25 oC als pKS-Werte (16.7.1.4) dargestellt. KS ist dimensionslos; KS' dagegen nicht! Säure pKS Säure pKS Säure pKS Ameisensäure 3,75 Benzoesäure 4,19 Phenol 9,99 Essigsäure 4,75 2-Hydroxybenzoes. 3 2-Chlorphenol 8,49 Propionsäure 4,87 3-Hydroxybenzoes. 4,08 4-Chlorphenol 9,18 Monochloressigs. 2,85 4-Hydroxybenzoes. 4,58 2-Methylphenol 10,2 Dichloressigsäure 1,48 2-Nitrobenzoesäure 2,17 2-Nitrophenol 7,23 Trichloressigsäure 0,7 3-Nitrobenzoesäure 3,49 2,4-Dinitrophenol 4,09 "-Hydroxypropions. 3,86 4-Nitrobenzoesäure 3,44 2,4,6-Trinitrophenol 0,29 $-Hydroxypropions. 4,51 2-Aminobenzoes. 6,97 Dimethylessigsäure 4,86 4-Aminobenzoes. 4,92 Trimethylessigsäure 5,05 CH3COOD 5,23 Ein Vergleich der aliphatischen Carbonsäuren zeigt, dass die Säurestärke der Ameisensäure durch die - 96 Substitution am "-Kohlenstoff mit Methylgruppen abnimmt. Die Methylgruppe wirkt als Elektrondonator und hemmt die Abgabe des H+. In der Sprache der Organiker ist das der +I-Effekt (I von induktiver Effekt). Substitution mit Chlor oder einer Hydroxylgruppe, die als Elektronakzeptor wirken (&I-Effekt), ziehen Elektronendichte ab und erleichtern die H+-Abgabe. Bei der Trichloressigsäure ergibt sich so eine drastische Erhöhung des pKS-Wertes. Am Beispiel der beiden Hydroxypropionsäuren erkennt man die Wirkung unterschiedlicher Abstände des Substituenten zum Wasserstoffatom. Die elektronischen Strukturen von CH3COOH und CH3COOD stimmen vollständig überein. Die schwächere Säurestärke der deuterierten Verbindung kommt dadurch zustande, dass das Deuteron wegen seiner größeren Masse langsamer in seinem Potenzialtopf schwingt und wegen seiner kleineren Nullpunktsenergie ½h< etwas tiefer in diesem Potenzialtopf sitzt und daher schwerer ablösbar ist. Die substituierten Benzoesäuren zeigen eine Abhängigkeit der Säurestärke von der Art und Stellung des Substituenten. Neben dem I-Effekt spielt hier der M-Effekt (Mesomerie-Effekt) eine Rolle. Weist ein Substituent ein freies Elektronenpaar an dem Atom auf, das an den Benzolring gebunden ist, so wird dieses Elektronenpaar partiell in den Ring geschoben. Dieser +M-Effekt erhöht die Elektronendichte im Ring und verringert die Säurestärke. Weist dagegen der Substituent eine zum Ring konjugierte Doppelbindung und ein endständiges, elektronegatives Atom an dieser Doppelbindung auf, so wird Elektronendichte aus dem Ring herausgezogen (&M-Effekt). M- und I-Effekt überlagern sich und wirken teilweise mit unterschiedlichen Vorzeichen. Die Säurestärken der Hydroxy-, Nitro- und Amino-benzoesäuren lassen sich durch den &I-Effekt, &MEffekt bzw. +M-Effekt erklären. Die unterschiedlich starke Wirkung bei der 2- und 4-Substitution wird durch die Überlagerung des M- und I-Effekts verursacht, wobei letzterer in der 4-Position erheblich schwächer wirkt. Bei den 2-Hydroxybenzoesäuren tritt darüber hinaus eine Stabilisierung der Anionen durch die Bildung von Wasserstoffbrücken in den Anionen auf. Die Wirkung der Substituenten bei den Phenolen ist ähnlich wie bei der Benzoesäure. Organische Basen (Amine, Anilinderivate) dissoziieren in wässriger Lösung entsprechend (16.7.1.5) Das zugehörige MWG ist (16.7.1.6) Die weitgehend konstante Wasserkonzentration wird in KBO einbezogen und ergibt die zur Dissoziationskonstante der Säure analoge Basizitätskonstante (16.7.1.7) - 97 Bei den Basen ist die Wirkung der Substituenten umgekehrt wie bei den Säuren: Der +M-Effekt und der +IEffekt erhöhen die Basenstärke wie in der Tabelle mit einer kleinen Auswahl von Anilinderivaten bei 25 oC gezeigt wird. Base pKB Anilin 9,37 2-Chloranilin 11,35 3-Chloranilin 10,48 4-Nitroanilin 13,00 4-Methylanilin 8,92 16.7.2 Dissoziationsgrad, pH-Wert Unter dem Dissoziationsgrad " versteht man den Anteil der dissoziierten Moleküle an der Gesamtzahl. Für die Säuredissoziation gilt (16.7.2.1) wobei co die Gesamtkonzentration ("Einwaagekonzentration" oder analytische Konzentration) der Säure darstellt. Als Maß für die Konzentration der Wasserstoffionen wird der pH-Wert (16.7.2.2) eingeführt. Diese logarithmische Definition komprimiert den viele Zehnerpotenzen umfassenden Konzentrationsbereich auf handliche Zahlen. Die Definition (16.7.2.2) gilt nur für verdünnte Lösungen, die hier auch nur von Interesse sind. Die im folgenden durchgeführte Berechnung der Wasserstoffionenkonzentration und des Dissoziationsgrades schwacher Säuren bei gegebener Säurekonzentration ist für viele Zwecke wichtig. Die exakte Berechnung ist leider etwas aufwändig; in den meisten Fällen darf man jedoch mit sehr einfachen Näherungen arbeiten. Folgende Gleichungen stehen für die Berechnung der Säuredissoziation zur Verfügung. 1) Das MWG (16.7.2.3) 2) Eine Bilanzgleichung für den Zerfall der Säure. (16.7.2.4) 3) H+-Ionen werden auch durch die Dissoziation des Wassers geliefert. (16.7.2.5) 4) Die drei Bestimmungsgleichungen reichen für die Bestimmung der vier Unbekannten nicht aus. Die fehlende Gleichung erhält man aus der "Elektroneutralitätsbedingung" der Ionenlösung: Die Ladungen aller Kationen und Anionen müssen sich aufheben. (16.7.2.6) Die Auflösung dieses Gleichungssystems gelingt wie folgt. Gl. (3) wird nach cOH& aufgelöst und in Gl. (4) eingesetzt. Die neue Gl. (4) nach cS& auflösen und in die Gl. (1) und (2) einsetzen. Es entstehen dann zwei Gleichungen mit zwei Unbekannten, aus denen sich cHS eliminieren lässt. Die daraus - 98 resultierende Gleichung (16.7.2.7) ist dritten Grades in cH+ und daher sehr unübersichtlich und für die meisten Anwendungen nutzlos. Immerhin kann man mit ihr den pH-Wert einer 10&8-molaren Essigsäurelösung berechnen (pH 6,98), was mit der jetzt herzuleitenden Näherung dann nicht mehr gelingt. Zu den Näherungsgleichungen kann man von Gl. (16.7.2.7) aus gelangen. Einfacher ist es jedoch, die Näherungen gleich bei den Ausgangsgleichungen einzuführen. Für die Näherungsrechnung wird angenommen, dass die durch die Wasserdissoziation erzeugte H+-Konzentration gegenüber der aus der Säuredissoziation stammenden vernachlässigt werden kann. Dadurch braucht Gl. (3) nicht mehr berücksichtigt zu werden und Gl. (4) wird zu (16.7.2.8) was auch ohne die Elektroneutralitätsbedingung einsichtig ist. Gl. (2) und (4) werden in Gl. (1) eingesetzt. (16.7.2.9) Nimmt man jetzt noch an, dass der Dissoziationsgrad klein ist, d. h. cH+ n co, so folgt (16.7.2.10) und der Dissoziationsgrad ergibt sich zu (16.7.2.11) (16.7.2.12) Diese Gleichungen ergeben natürlich nur sinnvolle Resultate, wenn die oben getroffenen Annahmen zutreffen (Dissoziationsgrad einer Säure bei co < KSN ?). Eine etwas genauere Gleichung für den Dissoziationsgrad erhält man aus Gl. (16.7.2.9) mit Gl. (16.7.2.1). (16.7.2.13) oder (16.7.2.14) Es entsteht so eine quadratische Gleichung für ". Für die Basen findet man analog (16.7.2.15) und daher - 99 - (16.7.2.16) und (16.7.2.17) Eine sehr elegante Darstellung zur Bestimmung des pH-Wertes in Abhängigkeit vom Dissoziationsgrad findet man wie folgt. Für die Säuredissoziation gilt nach Gl. (16.7.2.1) und (16.7.2.4) (16.7.2.18) Einige Umstellungen ergeben (16.7.2.19) Weiterhin folgt durch Logarithmieren des Säure-MWGs (16.7.2.20) oder (16.7.2.21) d. h. es ergibt sich eine für alle Säuren universelle Funktion. Der Dissoziationsgrad kann z. B. durch Verdünnung der Lösung oder durch Zusatz einer Base (dazu siehe Kap. 16.7.3) verändert werden. Abb. 47 Dissoziationsgrad und pHBei der Dissoziation mehrbasiger Säuren treten eine Reihe Wert Besonderheiten auf. Bei zweibasigen Säuren H2S sind die MWGs für die Dissoziation in den beiden Dissoziationsstufen (16.7.2.22) (16.7.2.23) Die zweistufige Dissoziation in einem Schritt ergibt sich durch Multiplikation der Gleichungen (16.7.2.24) - 100 Entsprechend ist das )Gs für die zweistufige Dissoziation die Summe aus den )Gs-Werten für die einzelnen Dissoziationsstufen. In der Tabelle sind die Dissoziationskonstanten für einige organische, zweibasige Säuren bei Säure pKS,1 pKS,2 25 oC zusammengestellt. Oxalsäure 1,23 4,19 Durch die negative Ladung des Anions der ersten Dissoziationsstufe wird die DissoziationsMalonsäure 2,83 5,69 konstante für die zweite Stufe stark herabgeBernsteinsäure 4,16 5,61 setzt. Dieser Effekt ist um so deutlicher, je geringer der Abstand der Carboxylgruppen ist. Bei Glutarsäure 4,31 5,41 der Oxalsäure unterscheiden sich die Dissoziationskonstanten um nahezu drei Zehnerpotenzen, bei der Glutarsäure um etwas mehr als eine Zehnerpotenz. Für den Zusammenhang zwischen pH-Wert und Dissoziationsgrad darf man Gl. (16.7.2.21) für die einzelnen Stufen nur dann verwenden, wenn die pKS-Werte genügend weit auseinander liegen, was z. B. bei H2S (hier ist jetzt wirklich Schwefelwasserstoff mit pKS = 7,04/11,96 gemeint) und Phosphorsäure (pKS = 2,00/6,69 für die beiden ersten Stufen) der Fall ist. Liegen die Dissoziationskonstanten dagegen sehr viel näher beieinander, so wird die Berechnung des pH-Wertes kompliziert, da die Dissoziation in erster Stufe noch nicht vollständig ist, wenn die in zweiter Stufe bereits beginnt. Für den allgemeinen Fall hat man die Gl. (16.7.2.22) und (16.7.2.23) mit vier Unbekannten & bei Vernachlässigung der Wasserdissoziation & zur Verfügung. Des weiteren gibt es die Bilanzgleichung für die Säure (16.7.2.25) und die Elektroneutralitätsgleichung (16.7.2.26) Dies erlaubt die hier nicht durchgeführte Berechnung aller interessierenden Konzentrationen. 16.7.3 Salze schwacher Säuren mit starken Basen, Mischungen von schwachen Säuren mit starken Basen Die Berechnung der pH-Werte von Mischungen schwacher Säuren mit starken Basen ist für die Untersuchung von Puffern wichtig und erlaubt die Berechnung der pH-Werte beim Titrieren einer schwachen Säure mit einer starken Base. Die Eigenschaften des Salzes dieser Säure und Base fallen als Spezialfall der allgemeinen Rechnung mit an. Der umgekehrte Fall (schwache Base/starke Säure, z. B. NH4Cl) lässt sich analog lösen und wird hier nicht gesondert behandelt. Eine Mischung der schwachen Säure HS mit der starken Base KOH, wobei K+ ein allgemeines Kation darstellt, enthält die Ionen und undissoziierten Moleküle H+, S&, HS, OH&, K+ Es gibt daher fünf unbekannte Konzentrationen, denen eine entsprechende Anzahl Gleichungen gegenüberstehen muss. Von den unbekannten Konzentrationen wird cK+ durch die Einwaagekonzentration cB der Base festgelegt. Die restlichen Unbekannten entsprechen den bereits beim Fall der Dissoziation der schwachen Säure auftretenden Konzentrationen. Auch die zur Verfügung stehenden Gleichungen sind identisch bis auf Gl. (4), die hier (16.7.3.1) lautet. Im Prinzip ist die Lösung dieses Gleichungssystems wie bei den schwachen Säuren möglich, wobei der entstehende Ausdruck - 101 - (16.7.3.2) noch etwas komplizierter ist. Abb. 48 zeigt die mit einem Programm berechnete Abhängigkeit des pHWerts bei der Titration einer 10&2-molaren Säurelösung, wobei als Dissoziationskonstante der Säure 10&5 mol/dm3 angenommen wurde. Man erkennt den Pufferbereich bei cB . 0,005 mol/dm3, d. h. den Bereich, in dem Salz und Säure nebeneinander vorliegen, und den Bereich des Salzes bei cB = 0,01 mol/dm3, der für die Titration von Interesse ist. Für eine Berechnung mit der Hand muss ein Verfahren gefunden werden, dass die Komplexität des allgemeinen Ausdrucks deutlich verringert. Es soll zuerst der pH-Wert-Bereich untersucht werden, in dem die Base unterschüssig vorliegt, d. h. man kann dieses System als Lösung eines Salzes mit überschüssiger Säure betrachten. Da dieser Bereich deutlich im Sauren liegt, darf die Wasserdissoziation vernachlässigt werden. Das Salz ist vollständig dissoziiert und durch die hohe S& -Konzentration wird die Säuredissoziation fast vollstän- Abb. 48 pH-Wert bei Zusatz einer Base dig unterbunden. Dies führt zu einer Möglichkeit, in der Dissoziationsgleichung der Säure die darin auftretenden Konzentrationen mit den bekannten Größen zu verknüpfen. Es gilt (16.7.3.3) Aus cH+ wird der pH-Wert berechnet, cS& entspricht der Salzkonzentration und cHS der Säurekonzentration bei einer aus Salz und Säure zusammengesetzten Mischung, d. h. (16.7.3.4) oder (16.7.3.5) Die Pufferwirkung dieses Salz/Säure-Gemischs beruht darauf, dass der Zusatz einer starken Base bzw. Säure das Verhältnis cSalz/cSäure verändert, das nur logarithmisch zum pH-Wert beiträgt. Die Pufferwirkung ist auf Zusätze von Stoffmengen beschränkt, wie sie in der Lösung vorliegen. Setzt man so viel einer starken Säure zu, dass das Salz vollständig zum Salz der starken Säure umgesetzt wird und die Konzentration der schwachen Säure entsprechend erhöht wird, so ist die Pufferwirkung aufgehoben. Die Pufferkapazität sowohl gegen den Zusatz starker Säuren als auch den starker Basen ist optimal für cSalz = cSäure. Unter diesen Bedingungen ist auch die Pufferwirkung optimal, d. h. die Steigung in der obigen Abb. minimal. Es gilt für die Ableitung von Gl. (16.7.3.5) - 102 - (16.7.3.6) Das Minimum, d. h. der Punkt geringster Steigung, liegt daher bei cS = 2cB oder cSalz = cSäure. Es fällt auf, dass die funktionelle Abhängigkeit im Pufferbereich und die Funktion in der vorhergehenden Abb. sehr ähnlich aussehen. Dies hat folgenden Grund. In der vorhergehenden Abb. war der pH-Wert als Funktion des Dissoziationsgrades aufgetragen. Dabei war nicht diskutiert worden, wie man diesen Dissoziationsgrad einstellt. Der pH-Wert bzw. der Dissoziationsgrad wird durch Zugabe einer starken Base zur Lösung der schwachen Säure eingestellt. Genau dieses Verfahren beschreibt Gl. (16.7.3.5). Der Dissoziationsgrad ist per definitionem cS&/cS und das entspricht weitgehend dem Verhältnis cB/cS. Gl. (16.7.3.5) kann man daher auch in der Form (16.7.3.7) geschrieben werden. Gl. (16.7.2.21) ist abgesehen von der vernachlässigten Wasserdissoziation exakt; Gl. (16.7.3.5) ist es nicht mehr. Der pH-Wert des reinen Salzes, d. h. am Äquivalenzpunkt der Titrationskurve, ist mit Gl. (16.7.3.5) nicht mehr zugänglich (cSäure = 0!). Zur Berechnung dieses pH-Wertes wird wie folgt verfahren. Die alkalische Reaktion des Salzes wird durch die Hydrolyse nach (16.7.3.8) bedingt. Das MWG lautet (16.7.3.9) Bezeichnet man mit ( den Hydrolysegrad, so folgt aus dieser Gleichung (16.7.3.10) und daher für den Fall ( n 1 (16.7.3.11) Die Wasserstoffionenkonzentration beträgt (16.7.3.12) und daher (16.7.3.13) Der Term ½pKW ergibt den pH-Wert des reinen Wassers. Die Hydrolyse ergibt eine um so alkalischere Lösung, je schwächer die Säure ist und je größer die Salzkonzentration ist. Für den diskutierten Fall gilt (16.7.3.14) Weitere einfach zugängliche Werte der Kurve des obigen Diagramms sind der pH-Wert der reinen schwachen Säure und der Bereich der stark alkalischen Lösungen, für die (16.7.3.15) - 103 gilt. Mit etwas modifizierten Methoden kann man den Fall eines Salzes einer schwachen Säure mit einer schwachen Base behandeln. Je nach der relativen Schwäche von Säure und Base entstehen hier leicht saure oder basische Lösungen. Dieser Fall soll hier nicht im einzelnen behandelt werden. 16.7.4 Ampholyte, Zwitterionen Substanzen, die sich sowohl wie Säuren als auch wie Basen verhalten können, werden als amphotere Elektrolyte oder Ampholyte (von griechisch amphoteros: beiderseitig und griechisch lysis: Trennung) bezeichnet. Beispiele sind die Aminosäuren, wie Glycin NH2CH2COOH, und viele Metallhydroxide, wie Al(OH)3. Alle Ampholyte können je nach pH-Wert als Kationen oder Anionen vorliegen. Im Bereich mittlerer pH-Werte liegen sie entweder als neutrale Substanzen vor oder bilden Innere Salze oder Zwitterionen mit zwei Ladungen verschiedenen Vorzeichens, wie beispielsweise NH3+CH2COO&. Die Bildung eines Zwitterions ist nur dann möglich, wenn die basische und saure Gruppe räumlich getrennt sind und sich nicht wie beim Al(OH)3 an einem Atom befinden. Weiterhin dürfen die Säureund Basenstärke nicht zu gering sein, da sonst & wie z. B. bei den Aminophenolen & im Bereich mittlerer pH-Werte die Dissoziation zu gering ist. Für den allgemein mit HXOH bezeichneten Ampholyten gibt es & abgesehen von der jetzt nicht angenommenen Bildung eines Zwitterions & folgende Dissoziationsmöglichkeiten (16.7.4.1) (16.7.4.2) mit den MWGs 1) (16.7.4.3) 2) (16.7.4.4) Daneben gibt es noch die Bilanzgleichung 3) (16.7.4.5) wobei cA die Einwaagekonzentration des Ampholyten darstellt, und die Wasserdissoziation 4) (16.7.4.6) und die Elektroneutralitätsbedingung 5) (16.7.4.7) Die vollständige Rechnung führt auf eine komplizierte Gleichung 3. Grades. Belässt man jedoch die an und für sich unbekannte Konzentration cHXOH in der Endgleichung, d. h. Gl. (3) wird nicht berücksichtigt, so erhält man aus (1) und (2) (16.7.4.8) Addition von KW ergibt (16.7.4.9) (16.7.4.10) Division ergibt - 104 - (16.7.4.11) wobei das Kürzen der Klammer wegen der Elektroneutralitätsbedingung möglich ist. Schließlich wird noch die OH&-Konzentration in die H+-Konzentration umgerechnet und es entsteht (16.7.4.12) Ist KSN = KBN, so folgt cH+ = (KW)½, d. h. pH 7. Ist die Dissoziationskonstante der Säure höher als die der Base, so entsteht eine saure Lösung. Die explizite Berechnung eines pH-Wertes mit Gl. (16.7.4.12) ist nur dann möglich, wenn sowohl die Säure- als auch die Basedissoziation so gering sind, dass Gl. (16.7.4.5) mit cHXOH = cA gilt. Legt man an eine "normale" Elektrolytlösung ein elektrisches Feld, so wandern Säureanion bzw. Basekation in diesem Feld auch bei teilweiser Dissoziation. Bei den Ampholyten kann man den pHWert durch Säure- bzw. Basezusatz so einstellen, dass der Ampholyt im Mittel kein Ladung trägt, d. h. die Konzentrationen von XOH& und HX+ übereinstimmen. Dieser pH-Wert wird als isoelektrischer Punkt (was heißt hier Punkt!?) bezeichnet. Am isoelektrischen Punkt ist die Wanderungsgeschwindigkeit des Ampholyten vernachlässigbar klein, da sich die Wanderung des Kations und Anions wegen der im Mittel gleichen Konzentration und der weitgehend übereinstimmenden Geschwindigkeiten aufheben. Den pH-Wert des isoelektrischen Punkts findet man aus den Gl. (1) und (2), die nach diesen Konzentrationen aufgelöst und gleichgesetzt werden. (16.7.4.13) oder (16.7.4.14) oder (16.7.4.15) oder (16.7.4.16) Sind die Säure- und Basestärke gleich, so liegt der isoelektrische Punkt bei pH 7, sonst geht die Differenz der pK-Werte ein, wobei bei einer starken Säure die saure Dissoziation durch Zusatz von Säure zurückgedrängt werden muss. Am isoelektrischen Punkt ist die Ampholytdissoziation minimal. Dies ist aus dem folgenden Diagramm ersichtlich. Für dieses Diagramm wurde der gesamte Dissoziationsgrad (16.7.4.17) berechnet. Diesen erhält man ohne Lösung des gesamten Gleichungssystems wie folgt. In die Bilanzgleichung (3) werden die nach den Konzentrationen cXOH& bzw. cHX+ aufgelösten Dissoziationsgleichgewichte (1) und (2) eingesetzt. Dies führt zu - 105 - (16.7.4.18) (16.7.4.19) und (16.7.4.20) Diese Gleichung zeigt die Symmetrie bezüglich der Säure- und Basedissoziation. Für die Berechnung der Funktionen im Diagramm wurde dann noch die OH&-Konzentration über die Wasserdissoziation eliminiert. Für die im Diagramm dargestellten Funktionen wurde KS = KB gewählt. Der an den Kurven angegebene Parameter ist der pKS-Wert. Demnach gibt es Ampholyte, bei denen am isoelektrischen Punkt durch geringe Dissoziationskonstanten fast nichts dissoziiert ist. In diesem Fall wandert der Ampholyt auch in einem gewissen pH-Wert-Bereich um den isoelektrischen Punkt nicht. Sind dagegen die Dissoziationskonstanten größer, so bleibt der Ampholyt nur am isoelektrischen pH-Wert stehen. Die für Abb. 49 Dissoziationsgrad von Ampholyten den Ampholyten mit pKS = 8 bei pH 7 auftretende Dissoziation wird durch die Dissoziation als Säure oder als Base bewirkt. Eine Zwitterionenbildung kann nicht auftreten, da sie in den Gl. (1) bis (5) nicht berücksichtigt wurde. Bei einem realen Ampholyten wäre dies jedoch sicherlich der Fall. Gibt es in einem Molekül räumlich getrennt eine saure und eine basische Gruppe, so können sich Zwitterionen bilden. Dieses wird in Lösung dann auftreten, wenn die beiden Gruppen nicht zu weit voneinander entfernt sind und die beiden Dissoziationskonstanten nicht zu gering sind. Die Dissoziation einer Gruppe hilft durch die entstehende Ladung der anderen Gruppe bei der Dissoziation. Bei den Aminosäuren liegen die Moleküle bereits im Kristall als Zwitterionen vor. Ein Hinweis dafür sind die hohen Schmelzpunkte von 230 oC und höher. Am isoelektrischen Punkt liegt bei den Verbindungen mit Zwitterionenbildung kein undissoziierter Ampholyt, sondern das Zwitterion vor. Es gibt jetzt die beiden Gleichgewichte (16.7.4.21) (16.7.4.22) d. h. hier wird der andere Extremfall im Vergleich zu den Gl. (16.7.4.1) und (16.7.4.2), wo keinerlei Zwitterionenbildung vorliegen sollte, angenommen. Für den Grundkörper des Zwitterions wurde die Bezeichnung XY gewählt, um die räumliche Trennung anzudeuten. Für die beiden Gleichgewichte gilt unter Einbezug der Wasserkonzentration in die Gleichgewichtskonstante (16.7.4.23) - 106 - (16.7.4.24) Da auch die restlichen Gleichungen (3), (4) und (5), wobei die letzte wegen der Gesamtladung 0 des Zwitterions auch erhalten bleibt, identisch sind, gelten die bereits hergeleiteten Ergebnisse. Auch die Überlegungen zum isoelektrischen Punkt bleiben gültig, da das Zwitterion nach außen neutral ist.