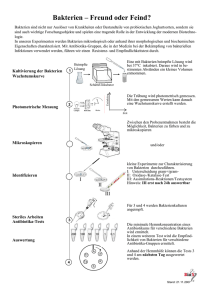
Herstellung und Untersuchung von Hydrogelen als Biofilm
Werbung

Gerhard-Mercator-Universität - Gesamthochschule Duisburg Fachbereich 6 ( Chemie - Geographie ) Herstellung und Untersuchung von Hydrogelen als Biofilm-Modelle Diplomarbeit II zur Erlangung des akademischen Grades eines Diplom-Chemikers vorgelegt von Martin Strathmann aus Gelsenkirchen Bearbeitungszeit: vom 08.12.1998 bis 07.06.1999 Betreuer: Prof. Dr. Hans-Curt Flemming Fachgebiet: Aquatische Mikrobiologie ERKLÄRUNG: Hiermit erkläre ich, daß ich die Arbeit selbständig verfaßt habe. Die verwendeten Quellen sowie die verwendeten Hilfsmittel sind vollständig angegeben. Duisburg, den 07.06.1999 __________________________ Unterschrift ii Vorwort Ich danke Herrn Prof. Dr. Hans-Curt Flemming für die Ausgabe des interessanten Themas für meine Diplomarbeit, für die freundliche Aufnahme in seiner Arbeitsgruppe und für die Ermöglichung der Durchführung der Arbeiten im Fachgebiet Aquatische Mikrobiologie an der Gerhard-Mercator-Universität Duisburg. Mein besonderer Dank gilt Dipl.-Biol. Thomas Griebe für die Betreuung der Arbeit. Er stand mir immer mit zahlreichen Ratschlägen und tatkräftiger Unterstützung zur Seite. Ebenfalls möchte ich mich bei Dr. Jost Wingender bedanken, der mir mit seinem fachlichen Wissen und seiner Diskussionsbereitschaft weiterhelfen konnte. Christiane Seibert, der guten Seele des Sekretariats, möchte ich dafür danken, daß sie stets alle Verwaltungsaufgaben bereitwillig erledigt hat und immer mit einem netten Wort für Aufmunterung sorgte. Bedanken möchte ich mich auch bei Astrid Dannehl und Alexander Rode, mit denen mir das Arbeiten im Labor viel Freude bereitete. Den Mitarbeitern des Fachgebietes „Angewandte Physikalische Chemie“, insbesondere Herrn Prof. Dr. W. Borchard und Frau Eva Selic, danke ich für die Unterstützung bei den Quellungsversuchen. Der Firma E. Merck, Darmstadt sei für die großzügige Bereitstellung der benötigten Agarose gedankt. iii Inhaltsverzeichnis Inhaltsverzeichnis ZUSAMMENFASSUNG ...................................................................................................................................1 1 EINLEITUNG....................................................................................................................................................2 1.1 BIOFILME .........................................................................................................................................................2 1.1.1 Biofilme als mikrobielle Lebensform .....................................................................................................2 1.1.2 Die Biofilmentwicklung .........................................................................................................................3 1.1.3 Zusammensetzung von Biofilmen...........................................................................................................5 1.1.4 Struktur von Biofilmen...........................................................................................................................6 1.2 POLYMERNETZWERKE, GELE UND HYDROGELE ..............................................................................................6 1.2.1 Was ist ein Netzwerk?............................................................................................................................7 1.2.2 Was ist ein Gel?.....................................................................................................................................8 1.3 THEORETISCHE VORÜBERLEGUNGEN...............................................................................................................9 1.3.1 Auswahl des Polymers zur Gelbildung ..................................................................................................9 1.3.2 Agarose..................................................................................................................................................9 1.3.3 Einflußfaktoren bei der Herstellung von Agarose-Beads ....................................................................11 1.4 ZIEL DER ARBEIT ...........................................................................................................................................13 1.5 HERANGEHENSWEISE .....................................................................................................................................14 2 MATERIAL UND METHODEN ...................................................................................................................16 2.1 AUSGANGSVERBINDUNGEN ...........................................................................................................................16 2.2 BAKTERIENSTÄMME ......................................................................................................................................17 2.3 KENNDATEN DER ANALYSENSYSTEME ..........................................................................................................18 2.3.1 Konfokales Laser-Raster-Mikroskop ...................................................................................................18 2.3.2 Fluoreszenz-Mikroskop .......................................................................................................................18 2.3.3 Stereomikroskop ..................................................................................................................................18 2.3.4 sonstige Geräte ....................................................................................................................................18 2.4 HERSTELLUNG VON AGAROSE-BEADS ...........................................................................................................19 2.4.1 Herstellung superporöser Agarose-Beads...........................................................................................19 2.4.2 Herstellung homogener Agarose-Beads ..............................................................................................20 2.4.3 Herstellung kompakter, superporöser Agarose-Körper ......................................................................20 2.5 PRÄPARATIONEN FÜR MIKROSKOPISCHE UNTERSUCHUNGEN ........................................................................20 2.5.1 Stereomikroskopische Untersuchungen...............................................................................................20 2.5.2 CLSM-Untersuchungen .......................................................................................................................21 2.6 CHARAKTERISIERUNG DER AGAROSE-BEADS ................................................................................................22 2.6.1 Bead-Durchmesser ..............................................................................................................................22 2.6.2 Superporen-Durchmesser ....................................................................................................................22 2.6.3 Superporen-Anteil................................................................................................................................23 2.6.4 Bestimmung des Löslichkeitsverhaltens...............................................................................................23 2.6.5 Bestimmung des Quellungsverhaltens .................................................................................................24 2.7 AGAROSE-BEADS MIT EINGEBETTETEN MIKROORGANISMEN ........................................................................25 2.7.1 Vorbereitung der Bakteriensuspension................................................................................................25 2.7.2 Einbettung von Mikroorganismen in Agarose-Beads ..........................................................................26 2.7.3 Bestimmung von Position und physiologischem Zustand der Bakterien .............................................26 2.7.4 Kultivierung von Bakterien in Agarose-Beads ....................................................................................28 2.8 HERSTELLUNG VON BEAD-SCHICHTEN ..........................................................................................................28 2.8.1 Fixierung der Agarose-Beads..............................................................................................................28 2.8.2 Bestimmung der dreidimensionalen Struktur der Bead-Schichten ......................................................29 2.9 BIOZIDEINWIRKUNG.......................................................................................................................................29 2.9.1 Natriumhypochlorit .............................................................................................................................29 2.9.2 Quantifizierung der inaktivierten und überlebenden Zellen ................................................................30 2.10 STATISTISCHE AUSWERTUNG.........................................................................................................................30 iv Inhaltsverzeichnis 3 ERGEBNISSE..................................................................................................................................................31 3.1 HOMOGENE AGAROSE-BEADS .......................................................................................................................31 3.1.1 Variation der Rührgeschwindigkeit .....................................................................................................32 3.1.2 Variation der Agarose-Konzentration .................................................................................................33 3.1.3 Löslichkeitsverhalten der homogenen Beads.......................................................................................35 3.2 SUPERPORÖSE AGAROSE-BEADS ...................................................................................................................36 3.2.1 Porenstruktur der Beads......................................................................................................................36 3.2.2 Variation des Bead-Durchmessers ......................................................................................................38 3.2.3 Variation der Porosität........................................................................................................................39 3.2.4 Löslichkeitsverhalten der porösen Beads ............................................................................................40 3.3 QUELLUNGSVERHALTEN DER AGAROSE-BEADS ............................................................................................41 3.4 HERSTELLUNG VON SCHICHTEN AUS AGAROSE-BEADS .................................................................................43 3.5 HERSTELLUNG SUPERPORÖSER AGAROSE-KÖRPER .......................................................................................44 3.6 EINBETTUNG VON BAKTERIEN IN BEADS .......................................................................................................46 3.6.1 Überprüfung der Vitalität der Bakterien im Verlauf der Bead-Herstellung ........................................46 3.6.2 Agarose-Beads mit immobilisierten Bakterien ....................................................................................47 3.6.3 Kultivierung von Bakterien in Agarose-Beads ....................................................................................48 3.7 SCHUTZWIRKUNG DER EINBETTUNG ..............................................................................................................50 4 DISKUSSION...................................................................................................................................................53 4.1 HERSTELLUNG UND CHARAKTERISIERUNG VON AGAROSE-BEADS ................................................................53 4.1.1 Bead-Herstellung.................................................................................................................................53 4.1.2 Löslichkeits- und Quellungsverhalten .................................................................................................54 4.2 EINBETTUNG, KULTIVIERUNG UND UNTERSUCHUNG VON BAKTERIEN IN AGAROSE-BEADS .........................55 4.2.1 Immobilisierung von Bakterien in Agarose-Beads ..............................................................................55 4.2.2 Vermehrung von immobilisierten Bakterien in Agarose-Beads...........................................................56 4.3 SCHUTZWIRKUNG DER EINBETTUNG ..............................................................................................................58 4.4 BEWERTUNG DER ENTWICKELTEN BIOFILM-MODELLE ..................................................................................60 4.4.1 Agarose-Beads.....................................................................................................................................60 4.4.2 Bead-Schichten und Agarose-Körper ..................................................................................................60 4.4.3 Einbettung von Mikroorganismen .......................................................................................................61 4.4.4 Gesamtbeurteilung...............................................................................................................................61 4.5 AUSBLICK ......................................................................................................................................................62 5 LITERATURVERZEICHNIS........................................................................................................................63 v Abkürzungen Abkürzungen AO CLSM CTC CTF DPD EPS NB PBS PI PIA Acridinorange Confocal Laser Scanning Microscopy (konfokale Laser-Raster-Mikroskopie) 5-Cyano-2,3-ditolyl-tetrazoliumchlorid CTC Formazan N,N-Diethyl-1,4-phenylendiaminsulfat extrazelluläre polymere Substanz Nutrient Broth Phosphatgepufferte Kochsalzlösung (phosphate buffered saline) Propidiumiodid Pseudomonas-Isolierungs-Agar Symbole d mx n P s Durchmesser arithmetischer Mittelwert der Größe x Anzahl der Meßwerte statistische Sicherheit (in %) Standardabweichung vi Zusammenfassung Zusammenfassung Die vorliegende Arbeit befaßt sich mit einem neuen Konzept für ein Biofilm-Modell auf der Basis eines Hydrogels sowie dessen Herstellung und Untersuchung. Dieses soll dazu dienen, ausgewählte Aspekte der Biofilm-Forschung gezielt zu simulieren und zu bearbeiten. Dabei ist es das Ziel, die natürlichen Eigenschaften eines Biofilmes auf einfache Weise möglichst genau und gut reproduzierbar zu simulieren. Für physiko-chemische Versuche ist hierbei die Modellierung der typischen Poren- und Kanalstruktur von Biofilmen wichtig. Für weitergehende Untersuchungen ist zusätzlich die Einbettung von lebenden Mikroorganismen in steuerbarer Zelldichte und deren Kultivierung notwendig. Es wird eine Methode zur Herstellung von homogenen und porösen Agarose-Beads (bead (engl.) = Perle) sowie von porösen Agarose-Körpern vorgestellt. Mit dieser Methode konnten homogene Beads im Durchmesserbereich von ca. 100 bis 500 µm hergestellt werden. Poröse Beads konnten mit einem mittleren Bead-Durchmesser von etwa 260 µm und mit Poren von 10 bis 80 µm Durchmesser (im Mittel 28 µm) erhalten werden. Anhand dieser Technik werden die Variationsmöglichkeiten der Reaktionsparameter und die damit verbundenen Auswirkungen auf das Produkt aufgezeigt. Die Charakterisierung der Agarose-Beads erfolgte mit dem Lichtmikroskop. Die Porenstrukturen wurden mit Hilfe der konfokalen Laser-Raster-Mikroskopie aufgeklärt. Zur Simulation verschiedener Bakterienpopulationen wurde ein Verfahren zur Einbettung von Bakterienzellen in die Agarose-Matrix entwickelt. Eine Immobilisierung war in homogenen und porösen Beads sowie in porösen Agarose-Körpern möglich. Im Vergleich zu allgemein aus der Biotechnologie bekannten Immobilisierungsmethoden ist es im Rahmen der vorliegenden Arbeit erstmals gelungen, Mikroorganismen in eine poröse Gelmatrix einzubetten. Die Aufklärung der dabei erhaltenen Zellverteilung wurde mit Hilfe der Laser-RasterMikroskopie durchgeführt. Außerdem wurde der physiologische Zustand der immobilisierten Bakterien untersucht und daraus Rückschlüsse auf eventuelle Schädigungen im Verlauf der Herstellung gezogen. Es ergab sich, das ca. 35 % der immobilisierten P. aeruginosa-Zellen, vermutlich durch die nötige Reaktionstemperatur von 47 °C, geschädigt waren. Das Wachstumsverhalten der eingebetteten Bakterien wurde untersucht. Es war möglich, die Bakterien in den Beads bis hin zur Ausbildung von Mikrokolonien zu vermehren. Es zeigte sich dabei, daß es bei den homogenen Beads zu einer Substratlimitierung im Inneren der Beads kommt und eine Zellvermehrung nur nahe der Bead-Oberfläche stattfand. Die porösen Beads zeigten ein homogenes Auftreten der Zellvermehrung über den Bead-Querschnitt, welche vermutlich auf eine gleichmäßigere Nährstoffversorgung durch den konvektiven Transport in den Poren zurückzuführen ist. Zur Untersuchung der Schutzwirkung der künstlichen Gelmatrix auf die eingebetteten Bakterien in Anlehnung an die Schutzwirkung der natürlichen Schleimmatrix wurde die desinfizierende Wirkung eines Biozids auf die eingeschlossenen Organismen im Vergleich zur Zellsuspension untersucht. Beim Einwirken von Natriumhypochlorit (0.5 mg/l, 30 min) konnte eine Zunahme der membrangeschädigten Zellen um etwa eine log-Stufe beobachtet werden. Im Falle der immobilisierten Zellen fiel die Schädigung innerhalb der ersten 20 Minuten dabei signifikant geringer aus. Dies entspricht den Beobachtungen an Mikroorganismen in natürlichen Biofilmen, wo eine Schutzwirkung der Gelmatrix gegenüber Bioziden bekannt und bereits eingehend untersucht ist. Ein reproduzierbares Biofilm-Modell mit variabler Steuerung der Porenstruktur und einer Möglichkeit zur gezielten Einbettung von Bakterien wurde entwickelt. 1 1 Einleitung 1 Einleitung 1.1 Biofilme 1.1.1 Biofilme als mikrobielle Lebensform Biofilme bestehen aus Mikroorganismen, welche sich an einer Grenzfläche anheften. Alle Oberflächen, biologische (z.B. Pflanzen und Tiere) sowie abiotische (z.B. Metall, Mineralien, Kunststoffe und Glas), können durch Bakterien besiedelt werden, sobald ausreichend Feuchtigkeit vorhanden ist. Die Mikroorganismen in Biofilmen sind in einer selbst produzierten organischen Matrix eingebunden. Diese Matrix, aus stark hydratisierten extrazellulären polymeren Substanzen (EPS), kann darüber hinaus auch anorganische Partikel einschließen oder organische Substanzen sorbieren. Mikrobielle Biofilme sind ubiquitär (Costerton et al., 1987; Flemming, 1991). Es ist inzwischen allgemein akzeptiert, daß die meisten Mikroorganismen in aquatischen Systemen, Sedimenten und Böden immobilisiert an Oberflächen existieren und dort mikrobielle Biofilme bilden (Flemming, 1994). Costerton et al. (1995) definieren einen Biofilm als eine in eine Matrix eingehüllte bakterielle Population, die entweder an eine Grenzschicht oder aneinander angeheftet ist. Hierin sind auch mikrobielle Flocken o.ä. eingeschlossen. Im Biofilm zu leben, scheint für Mikroorganismen in vielen Bereichen ökologische Vorteile gegenüber der planktonischen Lebensweise zu bieten, bei der sie sich als Einzelzellen in Suspension befinden. Einige dieser Vorteile sind in der folgenden Übersicht zusammengestellt: • Akkumulation von Nährstoffen (Fletcher, 1991), besonders in oligotropher Umgebung von Bedeutung (Marshall, 1985) • Akkumulation von Stoffwechselprodukten, welche von anderen Spezies genutzt werden können • Schutz vor - Bioziden - Salzbelastungen - pH-Extremen - hydraulischer Belastung • Ausbildung von Mikrokonsortien: - Symbiose verschiedener Arten (z.B. bei Nitrifikanten) - Nutzung schwer abbaubarer Substrate durch Zusammenarbeit verschiedener Spezies (Costerton und Boivin, 1991) - Schaffung ökologischer Nischen (z.B. anaerobe Bereiche unter aerobem Oberflächenbiofilm) • Gentransfer Es sind jedoch auch einige Nachteile mit der sessilen Lebensweise im Biofilm verbunden. So können einzelne Organismen etwa in Bereiche eines Biofilms gelangen, in dem ungünstige Überlebensbedingungen herrschen. Es ist z.B. möglich, daß aerobe Mikroorganismen durch weitere Zunahme der Besiedelung an der Biofilmoberfläche in einen anaeroben Bereich an der Basis des Biofilms geraten. 2 1 Einleitung 1.1.2 Die Biofilmentwicklung Für die Entstehung von Biofilmen sind folgende Voraussetzungen notwendig: • Existenz einer Grenzfläche • Vorhandensein von Wasser (Feuchtigkeit) • Anwesenheit von Mikroorganismen • Angebot von Nährstoffen Sind alle diese Faktoren gegeben, so ist die Ausbildung eines Biofilms möglich. Trotz der Komplexität der verschiedenen Wechselwirkungen bei seiner Entstehung, lassen sich generell drei verschiedene Phasen bei diesem Prozeß unterscheiden. Eine graphische Darstellung dieser Stadien ist in Abbildung 1 (nach Characklis, 1990) wiedergegeben. Abbildung 1: Phasen der Biofilmentwicklung (nach Characklis, 1990) 1.1.2.1 Induktionsphase Der erste Schritt bei der Ausbildung eines mikrobiellen Bewuchses ist der sogenannte „conditioning film“. Dieser ist Ergebnis der Adsorption von Makromolekülen (z.B. Huminstoffe, Polysaccharide oder Proteine) und hydrophoben Molekülen aus der wäßrigen Phase an die Grenzfläche (Marshall, 1984, 1992). Dieser Film beeinflußt die Ladung der Oberfläche und die freie Energie der Grenzschicht. Die Zelloberfläche von Mikroorganismen tritt mit diesem „conditioning film“ in Wechselwirkung. Die adsorbierten Stoffe können als Nährstoffe verwendet werden und fördern die Adhäsion von Bakterien, in einigen Fällen können sie auch aufwuchshemmend wirken (Pringle und Fletcher, 1986). Es folgt die Primäradhäsion von Mikroorganismen. Der Transport von Mikroorganismen an die Oberfläche erfolgt in turbulent durchströmten wäßrigen Systemen bis an die laminare Grenzschicht durch Konvektion. Bis an die Grenzfläche gelangen die Bakterien dann durch verschiedene Mechanismen, wie etwa Diffusion, Brown’sche Molekularbewegung, elektrostatische Anziehung, Chemotaxis oder Mikrowirbel (Characklis et al., 1990). 3 1 Einleitung ZoBell (1943) vermutete bereits, daß zwei Schritte an der Primäradhäsion beteiligt sind. Zunächst eine reversible Anheftung an die Oberfläche, die dann bei längerer Aufenthaltszeit von einer irreversiblen Immobilisierung gefolgt wird. Im Zustand der reversiblen Anheftung wird noch Brown’sche Molekularbewegung der Mikroorganismen beobachtet und sie können leicht wieder von der Oberfläche abgelöst werden. Bei der irreversiblen Adhäsion sind die Organismen fest auf der Oberfläche immobilisiert, bilden extrazelluläre polymere Substanzen und beginnen zu wachsen. Die Dauer der Induktionsphase wird in entscheidender Weise durch die Oberflächenrauhigkeit, die „biologische Affinität“ der Oberfläche, die auftretenden Scherkräfte und dem Zustand der Mikroorganismen beeinflußt. Sie kann zwischen einigen Stunden in stark belasteten Systemen und bis zu Jahren in stark oligotrophen Systemen betragen. 1.1.2.2 Wachstumsphase Nach der irreversiblen Adhäsion der Zellen an die Grenzfläche beruht die Akkumulation des Biofilms auf zwei Faktoren: zum einem dem Wachstum der Primärbesiedler und zum anderen der Anheftung weiterer Mikroorganismen (Marshall, 1984). Für die Adsorption von Zellen oder abiotischen Partikeln aus der flüssigen Phase sind jetzt nicht mehr die Oberflächeneigenschaften der Grenzfläche ausschlaggebend, da die Oberfläche der bereits bestehenden Besiedlung die Eigenschaften der ursprünglichen Grenzfläche maskiert. Die Akkumulation des Biofilms wird während der Wachstumsphase hauptsächlich durch das Wachstum und die Ausbildung von Mikrokolonien, insbesondere Mikrokonsortien, bestimmt. Daher ist besonders die Nährstoffversorgung in dieser Phase ein limitierender Faktor (Characklis, 1990). Sie wird in entscheidender Weise durch Transportprozesse in der Schleimmatrix beeinflußt. 1.1.2.3 Plateauphase Die Plateauphase bezeichnet das Erreichen eines Gleichgewichtszustandes in der Biofilmentwicklung. In dieser Phase halten sich Zuwachs- und Ablösevorgänge die Waage. Das bedeutet, die durch Wachstum oder Adhäsion hinzukommende Menge Biomasse wird durch stoßweise Ablösung von Biofilm-Fetzen („sloughing off”) (Rittmann, 1989) oder durch kontinuierlichen Abtrag von Zellen (Erosion) wieder entfernt. Für das Einstellen der Gleichgewichtsdicke des Biofilms sind hierbei in erster Linie das Zellwachstum und die Scherkräfte im System verantwortlich. Bei einem hohen Nährstoffangebot und entsprechend hoher Wachstumsrate kann sich in einem System mit geringen Scherkräften ein Dicker und rauher Biofilm bilden. In Systemen mit hohen Scherkräften und somit hohen Abrasionsraten bilden sich dünnere aber dafür dichtere und relativ glatte Biofilme aus (Characklis et al., 1990). Für die Dicke eines Biofilms kann darüber hinaus die Anwesenheit von höheren Organismen (z.B. Protozoen) von Bedeutung sein. Sie können durch Abweiden („grazing“) des Biofilms sein Wachstum begrenzen und seine Oberflächenbeschaffenheit beeinflussen. 4 1 Einleitung 1.1.3 Zusammensetzung von Biofilmen Im allgemeinen können die Komponenten eines Biofilms wie folgt unterteilt werden: • Wasser • extrazelluläre polymere Substanzen (EPS) („Schleim“) • Mikroorganismen • sorbierte Substanzen • eingelagerte Partikel Der größte Anteil eines Biofilms besteht aus Wasser. Der Wassergehalt kann zwischen 50 und 98 % variieren. Der organische Anteil der Biofilmmasse besteht wiederum zum größten Teil aus den extrazellulären polymeren Substanzen (zwischen 60 und 98% der Trockenmasse) (Christensen und Characklis, 1990; Wingender et al., 1999). In diese EPS-Gelmatrix sind die Mikroorganismen und abiotische Partikel eingebettet und immobilisiert. Die EPS sind organische Polymere und bestehen hauptsächlich aus Polysacchariden (Sutherland, 1984), enthalten aber auch deutliche Anteile an Proteinen, Glycoproteinen, Lipiden und Nucleinsäuren (Schaule et al., 1993a; Costerton und Irvin, 1981). Im Unterschied zu den Verhältnissen bei mikrobiellen Kapseln handelt es sich bei der EPS-Matrix in Biofilmen um Zellausscheidungen, die nicht fest an die Zelloberfläche gebunden sind. In der Regel produzieren Bakterienstämme spezifische EPS (Christensen und Characklis, 1990). Diese enthalten Polysaccharide aus Wiederholungseinheiten von einfachen Zuckern wie Glucose, Galactose, Mannose, Rhamnose, sowie Glucuron- und Galacturonsäure (Sutherland, 1984). Es ist aber auch die Produktion von unspezifischer EPS möglich, die häufig z.B. aus einfachen Homopolysacchariden (Dextran, Curdlan) oder Heteropolysacchariden (Alginat o. ä.) besteht. Die EPS-Matrix prägt in entscheidender Weise die Eigenschaften des Biofilms. Sie verleiht im z.B. die Eigenschaften eines Molekularsiebs, einer Diffusionsbarriere, eines Ionenaustauschers und eines Sorbens (Geesey, 1982; Flemming, 1995b; Flemming et al., 1996). Die hohe Bedeutung der EPS für einen Biofilm läßt sich aus der folgenden Zusammenstellung erkennen. Es ist eine Übersicht über die Prozesse und Eigenschaften gegeben, die durch EPS beeinflußt werden: • Bildung einer Gelmatrix - immobilisiert und fixiert Mikroorganismen zueinander - heftet den Biofilm auf die Grenzfläche - bildet eine Diffusionsbarriere • Bindung von Ionen durch geladene Gruppen • Anheftung und Einbettung von Partikeln aus der flüssigen Phase • Bindung von Wasser (bis zu 98%) • Zehrung von oxidierenden Substanzen (z.B. Biozide) • geben die mechanische Stabilität • verursachen die viskoelastischen Eigenschaften • erzeugen hydrophile Oberflächen 5 1 Einleitung 1.1.4 Struktur von Biofilmen Frühere Überlegungen über den strukturellen Aufbau von Biofilmen basierten auf der Vorstellung, daß Bakterien in eine flächige, kompakte Matrix aus EPS eingebettet sind (Center for Biofilm Engineering, 1994). Dieses Modell konnte aber viele experimentell beobachtete Phänomene nicht erklären. Erst durch neuere, zerstörungsfreie Meßmethoden, wie z.B. die Konfokale Laser Scanning Mikroskopie (CLSM), konnten genauere Erkenntnisse über die Struktur von natürlichen Biofilmen gewonnen werden. Bei früheren Untersuchungen, z.B. bei elektronenmikroskopischen Techniken, mußten Biofilme dehydratisiert werden, wodurch es nicht möglich war, Rückschlüsse auf die dreidimensionale Struktur im hydratisierten Zustand zu ziehen. Mit der CLSM-Technik erzielte Ergebnisse führten zu dem in Abbildung 2 gezeigten schematischen Strukturbild, welches als besondere Eigenschaften von natürlichen Biofilmen eine ausgedehnte Kanalstruktur und große Inhomogenitäten der Matrix erkennen läßt (Costerton et al., 1994). Abbildung 2: Aus CLSM Daten gewonnene Strukturvorstellung eines Biofilms (nach Costerton et al., 1994) Natürliche Biofilme mit diesen komplexen Strukturen sind jedoch nicht einfach zu handhaben und nicht auf einfache Weise reproduzierbar anzuzüchten. Zur Aufklärung von einigen Einzelaspekten wäre es daher wünschenswert, ein Modellsystem zu haben, mit dem Biofilme simuliert werden können. 1.2 Polymernetzwerke, Gele und Hydrogele Es wurde bereits im Vorhergehenden darauf eingegangen, daß die EPS von Biofilmen zu einem großen Teil aus Polysacchariden und anderen Polymeren besteht und bis zu 98 % Wasser enthalten kann. Für ein Biofilm-Modell erscheint es daher sinnvoll, ein Polymer-Hydrogel als Basiskomponente einzusetzen. Im Folgenden sind einige Grundlagen über die Netzwerk- und Gelbildung zusammengestellt. 6 1 Einleitung 1.2.1 Was ist ein Netzwerk? Im Bereich der Polymerchemie spricht man von Netzwerken bei Systemen von zwei- oder dreidimensionalen makromolekularen Ketten, welche untereinander über Knotenpunkte verknüpft sind. Die Begriffe „polymeres Netzwerk“ und „vernetzte Polymere“ werden oft synonym verwendet. „Polymeres Netzwerk“ wird jedoch für Produkte aus der StufenwachstumsPolymerisation von multifunktionellen Monomeren bevorzugt und „vernetzte Polymere“ für solche Systeme, die durch nachträgliche Vernetzung vorgeformter Polymerketten entstehen. Prinzipiell können alle chemischen oder physikalischen Bindungen vernetzend wirken, wobei nach der Art der Bindung zwischen chemisch bzw. physikalisch vernetzten Polymeren unterschieden wird. Die mechanischen Eigenschaften eines Netzwerks werden nur wenig von der Natur der Bindungen bestimmt. Sie werden hauptsächlich durch die Zahl und die Verteilung der Knotenpunkte pro Volumeneinheit des Netzwerks, den sogenannten Vernetzungsgrad, bestimmt. Die rheologischen und thermischen Eigenschaften eines Netzwerks werden hingegen auch von der Natur der Bindungen beeinflußt. • Chemisch vernetzte Polymere: Netzwerke dieser Art entstehen bei chemischen Reaktionen, wie z. B. radikalische Polymerisation oder Kondensationspolymerisation, durch die Ausbildung von kovalenten Bindungen. Kennzeichen chemisch verknüpfter Netzwerke ist ihre Stabilität gegenüber Lösemitteln und Temperatur, weshalb sie auch als "irreversible" Netzwerke bezeichnet werden. Bei erhöhten Temperaturen zeigen solche Netzwerke häufiger das Verhalten weicher aber elastischer Feststoffe als das viskoser Flüssigkeiten (Ferry, 1970). Hierfür ist vulkanisierter Kautschuk das bekannteste Beispiel. Bei der EPS von Biofilmen sind hingegen keine kovalenten Bindungen am Zusammenhalt der EPS-Matrix beteiligt (Flemming, 1996). • Physikalisch vernetzte Polymere: Bei den über physikalische Bindungen verknüpften Netzwerken handelt es sich im allgemeinen um sogenannte thermoreversible Gele. In Abhängigkeit von der Temperatur oder auch der Menge zugefügtem Lösemittel erfolgt bei diesen Polymer/Lösemittel-Systemen ein reversibler Gel-Sol-Übergang (Stockmayer, 1944). Beispiele für physikalische Bindungen, die zu einer Vernetzung führen, sind in Abbildung 3 dargestellt. Abbildung 3: Darstellung der Bildung eines Netzwerkes durch a) Verschlaufungen, b) kristalline Bereiche und c) Ausbildung von glasigen Bereichen. (nach Rehage, 1977) 7 1 Einleitung Prinzipiell können durch die folgenden Möglichkeiten physikalische Netzwerke entstehen: 1. Mechanische Verschlaufung oder Verhakung der Polymerketten untereinander (Rehage, 1977). Hierbei können die Ketten mit der Zeit voneinander abrutschen und es handelt sich daher um ein nicht permanentes Netzwerk. 2. Partielle Bildung von Doppelhelices aus zwei Einzelsträngen. Bekanntes Beispiel hierfür ist das System Gelatine/Wasser, bei dem eine thermoreversible Vernetzung durch Ausbildung von Doppelhelices erfolgt (Cox, 1976). 3. Ausbildung von kristallinen Bereichen (Rehage und Halboth, 1968). 4. Amorphe Domänen bzw. glasartige Erstarrung von Teilbereichen in Blockcopolymeren (Rehage, 1977; Rehage und Borchard, 1973). 5. Wechselwirkungen zwischen den makromolekularen Ketten, wie z. B. Wasserstoffbrückenbindungen, van der Waals Kräfte und Coulomb-Wechselwirkungen. Diese sind auch für die Netzwerkbildung in der EPS verantwortlich (Flemming, 1996) Diese Möglichkeiten für physikalische Netzwerkverknüpfungen bringen im Gegensatz zu kovalenten Bindungen (≈200 kJ·mol-1) nur eine verhältnismäßig kleine Bindungsenergie (≈20 kJ·mol-1) mit sich. Durch die hohe Vernetzungsdichte, z.B. bei Wasserstoffbrückenbindungen in Doppelhelices, werden jedoch ähnlich hohe Brutto-Bindungsstärken wie bei kovalenten Bindungen erreicht. 1.2.2 Was ist ein Gel? Als Gele bezeichnet man allgemein disperse Systeme aus mindestens zwei Komponenten, die zumeist aus einem festen, kolloid zerteilen Stoff mit langen oder stark verzweigten Teilchen (z.B. ein chemisch oder physikalisch verknüpftes Polymernetzwerk) und einer Flüssigkeit als Dispersionsmittel bestehen. Meistens handelt es sich bei der Flüssigkeit um Wasser. In diesem Fall wird dann von einem Hydrogel gesprochen. In dem Dispersions- bzw. Quellungsmittel quellen die Polymernetzwerke bis zu einem Gleichgewichtsvolumen auf. Durch die Natur der Verknüpfungspunkte, die in einem Netzwerk vorliegen, wird unterschiedliches Quellverhalten der Gele beobachtet: Bei physikalisch verknüpften Netzwerken wird im allgemeinen ein Gel-Sol-Übergang gefunden. In Abhängigkeit von der Temperatur und/oder der Quellmittelmenge bildet das Polymer ein Gel, kann aber auch ohne Zerstörung der einzelnen Polymerketten in den Solzustand gebracht werden. Das Polymer/Quellmittel-System bildet dann eine Lösung. Dieser Vorgang ist reversibel, weshalb diese Gele auch als thermoreversible Gele bezeichnet werden (Stockmayer, 1944). Bei chemisch verknüpften Netzwerken hingegen, ist ein Gel-Sol-Übergang lediglich unter irreversiblem Abbau der Netzwerkstruktur zu erreichen. Das Netzwerk ist nur bis zu einem bestimmten Grad in der Lage Quellmittel aufzunehmen, dem sogenannten Sättigungs- oder Gleichgewichtsquellungsgrad. Kennzeichen für den Gelzustand eines chemisch verknüpften Netzwerks ist die Beibehaltung der äußeren Form des Netzwerks sowie die Eigenschaft der Gummielastizität. Physikalisch betrachtet besitzt ein Gel sowohl elastische als auch viskose Eigenschaften, das heißt es ist viskoelastisch (Rehage, 1977). 8 1 Einleitung 1.3 Theoretische Vorüberlegungen Aus der Biotechnologie sind mehrere Verfahren zur Herstellung von Polymer-Beads bekannt (Gustavsson und Larsson, 1996; Mosbach und Nilsson, 1987; Ekman und Lindahl, 1989; Cook et al., 1991; Weaver, 1981). Im Folgenden soll eine Übersicht der Auswahlkriterien und Einflußfaktoren bei der Bead-Herstellung gegeben werden. 1.3.1 Auswahl des Polymers zur Gelbildung Die Auswahl der verwendeten Gelsubstanz zur Modellierung der EPS-Matrix von Biofilmen erfolgt nach verschiedenen Gesichtspunkten. Zum einen sollen die Eigenschaften der natürlichen EPS-Substanzen möglichst gut wiedergegeben werden. Außerdem muß die Herstellung der Beads mit den genannten Eigenschaften einfach und kostengünstig möglich sein. Die Anforderungen, die im einzelnen an das Polymermaterial gestellt werden, sind von Leenen (1996) zusammengefaßt worden und in Tabelle 1 angegeben. Tabelle 1: Auswahlkriterien für das Material zur Immobilisierung von Bakterien (nach Leenen, 1996) Auswahlkriterium Löslichkeit in Wasser Abbaubarkeit mechanische Stabilität Netzwerkbildung Diffusionmöglichkeit für niedermolekulare Wachstum von Bakterien Immobilisationsverfahren Toxizität Kosten gewünschte Eigenschaft gering gering hoch Ionenbindung und Wasserstoffbrückenbindung hoch möglich einfach, im Labor durchführbar nicht toxisch gering Natürliche Gele entsprechen den gestellten Anforderungen eher als synthetische Gele, da diese die in Tabelle 1 zusammengestellten Anforderungen besser erfüllen. Zu den natürlichen Gelbildnern gehören z. B. Xanthan, Agarose und Alginat. Die von Leenen (1996) genannten Auswahlkriterien und gewünschten Eigenschaften werden mit Agarose weitgehend erfüllt. 1.3.2 Agarose Agarose ist ein gelierfähiges Polysaccharid, welches aus Meeresalgen der Klasse Rhodophyceae gewonnen wird. Die chemische Struktur besteht idealisiert aus sich abwechselnd wiederholenden Einheiten von β-1,3-verknüpfter D-Galaktopyranose und α-1,4-verknüpfter 3,6-Anhydro-LGalaktopyranose (Römpp, 1995). Diese Agarobiose-Basiseinheit bildet Polymerketten mit einer durchschnittlichen Molekularmasse von 120.000 Dalton, entsprechend etwa 400 AgarobioseEinheiten. In Abbildung 4 ist die Struktur der Agarobiose-Einheit dargestellt. 9 1 Einleitung O HO CH2OH HO O O O O OH n Abbildung 4: Agarobiose-Einheit Einige Vorzüge von Agarose haben Cook et al. (1991) in ihrer Arbeit über die Einbettung von biologischem Material in Agarose-Beads zusammengefaßt: 1. Agarose benötigt keine Polymerisationsreaktion wie z.B. Polyacrylamid. Es entfällt somit die Zugabe von Polymerisationsreagenzien, die unter Umständen auf Bakterien toxisch wirken. 2. Agarose benötigt im Gegensatz zu anderen Gelmedien wie z.B. Alginat oder Carrageenan, nicht die Anwesenheit von Kationen für die Gelbildung. Die Abwesenheit dieser Kationen vereinfacht Untersuchungen z.B. mit spektroskopischen Methoden. 3. Die durch diese Kationen, insbesondere Calcium hervorgerufenen Beeinflussungen der Zellfunktionen werden bei Agarose vermieden. Erste Überlegungen für den Mechanismus der Gelbildung von Agarose wurde von Rees (1969, 1972) angestellt und später von Arnott et al. (1974) experimentell überprüft. Beteiligt ist ein Wechsel von einer zufälligen Knäulstruktur zu Bereichen mit einer Doppelhelix-Struktur zu Beginn der Gelbildung. Im weiteren Fortschritt bilden sich dann Bündel von parallel angeordneten Doppelhelices. Die dabei im Agarose-Gel entstehenden Diffusionsporen, haben abhängig von der Konzentration und dem Vernetzungsgrad der Agarose, eine Größe von typischer Weise 100 bis 300 nm (Griess, 1989). Einer der wichtigsten Faktoren für die erfolgreiche Verwendung von Agarose als Gelbildner, ist das Erreichen von hohen Gelstärken schon bei niedrigen Agarose-Konzentrationen (1-6%). Die Gelstärke ist definiert als die Kraft, ausgedrückt in g/cm², die nötig ist, um ein Gel mit einer definierten Konzentration durch Druck zu zerstören. Die Energie, die nötig ist um ein Agarose-Gel aufzuschmelzen nimmt mit steigender AgaroseKonzentration zu (steigende Schmelztemperatur). Die Temperatur bei dem sich ein aufgeschmolzenes Agarose-Gels wieder verfestigt (Geltemperatur), nimmt ebenfalls mit steigender Gelkonzentration zu. Das Agarose-Polysaccharid enthält in Abhängigkeit von seiner Herkunft einen unterschiedlichen Anteil an Methylgruppen. Das Ausmaß dieser natürlichen Methylierung ist direkt proportional zur Geltemperatur (FMC, 1988). Unerwarteterweise haben synthetisch methylierte Agarosen niedrigere statt höhere Geltemperaturen und die Geltemperatur ist umgekehrt proportional zum Grad der synthetischen Methylierung. Außerdem sind die Agarobiose-Einheiten teilweise mit neutralen oder geladenen Gruppen, wie z.B. Sulfat, Methyl oder Pyruvat, an C-6 substituiert. Die Charakteristik der gesamten Agarose-Struktur, wie etwa die Flexibilität der Glykosidbindungen, die Abfolge der Grundbausteine und das Auftreten von Substituenten, bestimmt weitestgehend die physikalischen Eigenschaften des Agarose-Gels, insbesondere die Gel- und Schmelztemperatur, sowie die Gelstärke (FMC, 1988). Durch gezielte Auswahl des Grundmaterials können diese Eigenschaften bestimmt und auf die Anforderungen an das spätere Gel abgestimmt werden. 10 1 Einleitung 1.3.3 Einflußfaktoren bei der Herstellung von Agarose-Beads 1.3.3.1 Welches Ausgangsmaterial ist für die Herstellung von Beads und die Einbettung von Partikeln (Bakterien) geeignet? Bei der verwendeten Agarose ist darauf zu achten, daß diese eine möglichst niedrige GelTemperatur besitzt. Dies ermöglicht, daß die Agarose-Lösung noch bei relativ niedrigen Temperaturen flüssig bleibt und somit auch wärmeempfindliche Bakterien zugegeben und immobilisiert werden können. Hierbei muß allerdings berücksichtigt werden, daß mit einer niedrigen Gel- und Schmelztemperatur ebenfalls eine niedrige Gelstabilität einhergeht. 1.3.3.2 Welchen Einfluß hat die Konzentration von Agarose auf die Porosität und Stabilität des Gels? In der Literatur werden zur Einbettung von Bakterien in Agarose Konzentrationen im Bereich von 0.5 bis 6 % w/v Agarose in Wasser verwendet (Gustavsson und Larsson, 1996; Mosbach und Nilsson, 1987; Ekman und Lindahl, 1989; Cook et al., 1991; Weaver, 1981). Bei Verfahren, die nach der Emulsionstechnik arbeiten, finden Agarose-Lösungen von 5 bis 6 % w/v Verwendung. Methoden mit Sprüh- und Tropftechniken werden mit niedrigeren Konzentrationen (0.5 bis 3 % w/v) durchgeführt. Bei der in dieser Arbeit angewendeten Emulsionstechnik ist es sinnvoll, die AgaroseKonzentration ebenfalls in einen Bereich von 1 bis 2 % zu senken, da dies eher den typischen EPS-Konzentrationen in natürlichen Biofilmen entspricht (Flemming, 1995a). 1.3.3.3 Welches Lösemittel ist als äußere Phase für die Emulsion bei der Herstellung von Beads geeignet? Zur Ausbildung der Beads wird die wäßrige Agarose-Lösung in eine äußere Phase eingetragen und durch Rühren dispergiert. Bei der Auswahl der zu verwendenden äußeren Phase ist zu beachten, daß diese Substanz leicht zu handhaben, leicht von den Beads abzutrennen, für die immobilisierten Bakterien unschädlich und biologisch nicht abbaubar ist. Häufig werden hierfür Öle wie z.B. Paraffinöl, Siliconöl, Sojabohnen-Öl oder andere Speiseöle verwendet. Diese haben jedoch den Nachteil, daß sie zum Teil biologisch angreifbar sind. Das Abtrennen des Öls von den Beads ist eine weitere Schwierigkeit. Hierbei ist häufig nach dem Dekantieren oder Sieben eine weitere Reinigung der Beads nötig. Gustavsson und Larsson (1996) verwenden bei ihren Versuchen Cyclohexan als äußere Phase. Durch den hohen Dampfdruck des Cyclohexans können nach dem Abtrennen der Beads enthaltene Cyclohexan-Reste durch einfaches Verdunsten entfernt werden. Allerdings ist durch den hohen Dampfdruck und das höhere Gefahrenpotential der apparative Aufwand bei der Herstellung der Emulsion höher. Zudem ist vorher die Verträglichkeit der immobilisierten Bakterien mit Cyclohexan zu überprüfen. Eine weitere Möglichkeit ist die Verwendung einer wäßrigen Polyethylenglycol-Lösung als äußere Phase. Dies hätte den Vorteil, daß auf ein organisches Lösemittel verzichtet werden könnte. Beispiele dieser Art werden von Ekman und Lindahl (1989) für Alginat, Stärke, Carrageenan, Dextran, Pectin und Fibrin beschrieben. 11 1 Einleitung Bei der Abwägung der Vor- und Nachteile der genannten Alternativen erscheint die Verwendung von Cyclohexan als äußerer Phase am geeignetsten. Dieses Verfahren stellt einen guten Kompromiß zwischen einer einfachen Durchführung und guten Abtrennbarkeit der Beads einerseits und einem erhöhten apparativen Aufwand andererseits dar. Die Verwendung von Öl würde einen erheblichen Reinigungsaufwand erfordern. Die Methode unter Verwendung von Polyethylenglycol-Lösung ist hingegen erheblich Aufwendiger. 1.3.3.4 Welchen Einfluß hat die Immobilisationstechnik auf das Überleben der Bakterien in den Beads? Da lebende Mikroorganismen immobilisiert werden sollen ist zu untersuchen, in welchem Ausmaß die verwendeten Chemikalien bei der Bead-Herstellung und die nötigen Reaktionsbedingungen die Vitalität der eingeschlossenen Mikroorganismen beeinflussen. Zur Überprüfung ist es z.B. möglich, die Bakterien in den Beads in Abhängigkeit von ihrem physiologischem Zustand durch bestimmte Färbetechniken kenntlich zu machen, zu differenzieren und diese mikroskopisch zu erfassen (z.B. mit dem CLSM). Im Vorfeld kann die toxische Wirkung der Chemikalien auf Bakterien z.B. mit Hilfe des Leuchtbakterientests überprüft werden. Hierbei ist auch zu berücksichtigen, daß die eingebetteten Bakterien durch die umgebende Gelmatrix gegen die Einwirkung von toxischen Substanzen geschützt werden. 1.3.3.5 Müssen Emulgatoren und/oder Stabilisatoren eingesetzt werden? Fast alle Emulsions-Techniken unter Verwendung von Ölen kommen ohne Emulgatoren oder andere Zusätze aus. Bei der von Gustavsson und Larsson (1996) angewandten Methode in Cyclohexan werden jedoch Tween 80 (Polyoxyethylen-sorbitanmonooleat) und Span 85 (Ethylenglycol-dimethacrylat) eingesetzt. Die am Emulgierprozeß beteiligten Vorgänge sind in der Abbildung 5 in Form eines Schemas chronologisch dargestellt. Abbildung 5: Vorgänge beim Emulgierprozeß (nach Schubert und Armbruster, 1989) 12 1 Einleitung Häufig ist es bei Emulsionen notwendig, Stabilisatoren in Form von festen Partikeln (z.B. Kaolin) zuzusetzen, um eine Zusammenlagerung der dispergierten Tröpfchen zu verhindern und so die gewünschte Tröpfchengröße zu erzielen. Feine Feststoffpartikel, die sich an der Grenzfläche zwischen der öligen und wäßrigen Phase anlagern, können eine Tropfenkoaleszenz verhindern (Schubert und Armbruster, 1989). Voraussetzung hierfür ist eine nicht vollständige Benetzbarkeit der Partikel durch die innere Phase. Es könnte aber sinnvoll sein, diese Feststoffpartikel auch zuzugeben, wenn sie für die Stabilisierung der Emulsion nicht unbedingt nötig sind. Sie lagern sich auf der Oberfläche der Beads an und könnten so z. B. die Modellfunktion für absorbierte Korrosionsprodukte bilden. 1.4 Ziel der Arbeit In den letzten Jahren hat die Erforschung von Biofilmen in vielen Bereichen der Wissenschaft und Technik stark an Bedeutung gewonnen (Flemming, 1994; Meyer-Reil, 1996). Besonders von Interesse sind hierbei die Aufklärung der Struktur von Biofilmen, die Untersuchung von Transportprozessen im Biofilm und die Charakterisierung der immobilisierten Mikroorganismen. Die Verwendung von natürlichen Biofilmen ist aus praktischen Gründen nur bedingt geeignet, da diese häufig nicht in den benötigten Mengen und nicht einheitlich genug zur Verfügung stehen. Das Anzüchten von Biofilmen auf Trägermaterialien in speziellen Reaktoren im Labor ist sehr schwierig und zeitaufwendig. Es ist daher der Wunsch entstanden, künstliche Modell-Biofilme mit bestimmbaren Eigenschaften und guter Reproduzierbarkeit herzustellen, an denen grundlegende Untersuchungen durchgeführt werden können. Einfache Modelle bedienten sich bislang flächig aufgebrachter Polysaccharid-Filme, welche die EPS-Matrix darstellten, in die die gewünschten Mikroorganismen eingebettet wurden. Dieses Modell ist aber für viele Untersuchungen unzulänglich, da die natürliche Poren- und Kanalstruktur (Costerton et al., 1994) mit ihren typischen Auswirkungen auf das Diffusionsverhalten und den Stofftransport im Biofilm nicht wiedergegeben wird. Wesentliche Aspekte von natürlichen Biofilmen, wie z.B. die Aufnahme von Nährstoffen und Partikeln über diese Kanäle und Poren sowie die physikalischen Eigenschaften dieser unregelmäßigen Oberflächen, können mit solchen Modellen nicht simuliert werden. Ziel der Arbeit ist daher die Herstellung von Modell-Biofilmen, welche den charakteristischen Eigenschaften von natürlichen Biofilmen möglichst genau entsprechen, aber im Labor mit geringem Aufwand, möglichst variablen Eigenschaften und einer hohen Reproduzierbarkeit herstellbar sind. Im Rahmen dieser Diplomarbeit wurden Hydrogele mit eingebetteten Bakterien hergestellt und es wurde daran untersucht, inwieweit hiermit die gewünschten Kriterien erfüllt werden können. Folgende Anforderungen wurden dabei an das Modellsystem gestellt: • • • • • optisch transparent (für die Mikroskopie) Poren und Kanäle (>10 µm) Einbettung von Bakterien und anderen Partikeln möglich als Film mit definierter Dicke auftragbar reproduzierbar 13 1 Einleitung 1.5 Herangehensweise Die nachträgliche Erzeugung von Poren und Kanälen in der gewünschten Größe (> 5 µm) in einem flächigen Hydrogel ist nicht mit vertretbarem Aufwand möglich. Das Erzeugen von Poren in dieser Größe ist auf chemischem Wege nicht realisierbar. Mechanisch eingebrachte Kanäle verheilen aufgrund der Fließeigenschaften des Hydrogels wieder in kurzer Zeit. Eine Lösungsmöglichkeit liegt in der Verwendung von Schichten aus Hydrogel-Kugeln (Beads) mit immobilisierten Partikeln und Bakterien. Der schematische Aufbau eines solchen Gel-Beads ist in Abbildung 6 dargestellt. Hierbei werden Hydrogel-Beads im Größenbereich von 50 bis 500 µm, wie in Abbildung 7 skizziert, als Mono- oder Multilayer auf eine Oberfläche aufgebracht. Die bei der zu erwartenden dichtesten Kugelpackung der Beads entstehenden Zwischenräume (Zwickel) simulieren die Kanalstruktur des Biofilms näherungsweise. Durch Variation des Beaddurchmessers kann auf die Größe der Kanäle Einfluß genommen werden. Darüber hinaus ist eine Diffusion in das Polymernetzwerk der Gel-Beads möglich. Bead (Polysaccharid-Gel) immobilisierte Bakterien (lebend) 50-500 µm Abbildung 6: Gel-Bead mit eingebetteten Bakterien Oberfläche (transparent) Oberfläche (transparent) Abbildung 7: Monolayer (links) und Multilayer mit definierter Dicke (rechts) aus Gel-Beads mit Bakterien 14 1 Einleitung Eine weitere Verbesserung dieses Biofilm-Modells ist durch die Herstellung und Verwendung der in Abbildung 8 dargestellten „superporösen“ Hydrogel-Beads denkbar. Agarose mit Diffusionsporen Kanal Immobilisierte Bakterien Abbildung 8: Schematische Darstellung von immobilisierten Bakterien in Agarose-Beads mit "superporösen" Kanalstrukturen Hierbei lassen sich neben den Zwickel-Kanälen durch eine Emulsionstechnik mit Phasenumkehr weitere Kanalstrukturen im Größenbereich von etwa 10 bis 50 µm direkt in die Beads (100 - 500 µm) einbringen. Im weiteren Verlauf dieser Arbeit werden diese künstlich erzeugten, polymerfreien Kanalstrukturen im Größenbereich von 10 bis 50 µm auch als „Superporen“ bzw. die damit hergestellten Agarose-Beads als „superporös“ bezeichnet. Die im Polymernetzwerk des AgaroseGels intrinsisch vorhandenen Poren im Größenbereich von 100 bis 300 nm werden zur Abgrenzung davon „Diffusionsporen“ genannt. 15 2 Material und Methoden 2 Material und Methoden 2.1 Ausgangsverbindungen Tabelle 2: verwendete Chemikalien Substanzname Hersteller Formel Best.-Nr. Agarose (mittlere Elektroendoosmose) Merck Für die Elektrophorese 116801 Cyclohexan, zur Synthese Merck 822268 C6H12 Merck Ethanol 96% reinst DAB C2H6O 100971 Glycerin wasserfrei, z. A. Fluka 1,2,3-Propantriol 49770 C3H8O3 Merck Kaliumdihydrogen104873 phosphat, z.A. KH2PO4 Katalase aus Rinderleber Sigma C-40 Mowiol 40-88 Aldrich (Polyvinylalkohol) 32,459-0 Natriumchlorid, z.A. Baker NaCl 0278 Merck di-Natriumhydrogen106580 phosphat-Dihydrat, z.A. Na2HPO4·2H2O Natriumhypochlorit-Lsg. Riedel-de Haën NaClO 13440 Natriumthiosulfat wasserfrei Merck Na2S2O3 106512 Span 85, zur Synthese Merck C60H108O8 840124 TRIZMA Sigma Tris[hydroxymethyl]aminometha T-3253 FW Dichte Smp. [g/mol] [g/cm³] [°C] - 60-90 Gefahren- R-Sätze symbol Sdp. [°C] S-Sätze - - - - 84.16 0.78 6.5 80.7 F 11 9-16-33 46.07 0.79 -117 78 F 11 7-16 92.10 1.262 - - - - - 136.09 2.34 253 - - - - - - - - - - - ca. 127000 58.44 - - - - - - 2.16 800 1461 - 36/37/38 26-36 177.99 2.1 95 - - - - 74.44 1.25 - - C 31-34 28-45-50.1 158.11 - - - - - - 957.68 0.95 - > 100 - - - 157.6 - - - - 36/37/38 26-36 Merck 822187 - 1.08 - - - - - Wasserstoffperoxid (30%) Merck 108597 H 2O 2 34.01 1.11 -26 107 C 34 3-2636/3739-45 nhydrochlorid C4H11NO3·HCl Tween 80, zur Synthese Polyoxyethylensorbitanmonoolea t Tabelle 3: verwendete Lösungen Bezeichnung isotonische Kochsalzlösung Phosphatgepufferte Kochsalzlösung (PBS), pH 7.54 Tris-HCl-Puffer, pH 8.5 Zusammensetzung: NaCl NaCl KH2PO4 Na2HPO4 · 2 H2O C4H11NO3·HCl 8.2 g 9.0 g 0.27 g 1.43 g 31.5 g auf 1000 ml deionisiertes Wasser auf 1000 ml deionisiertes Wasser auf 1000 ml deionisiertes Wasser 16 2 Material und Methoden Tabelle 4: verwendete Farbstoffe Substanzname Formel Acridinorange CTC (5-Cyano-2,3-ditolyltetrazoliumchlorid) C16H15N5Cl Kongorot C32H22N6Na2O6S2 LIVE/DEAD® BacLight™ Bacterial Viability Kit • Propidiumiodid • SYTO 9 Malachitgrün-Oxalat C48H50N4O4 · 2 C2H2O4 Methylblau C37H27N3Na2O9S3 Orange G Certistain C16H10N2Na2O7S2 Propidiumiodid 95-98% TLC C27H34N4I2 Hersteller Best.-Nr. Merck 114281 Polysciences Europe 19292 Merck 101340 Molecular Probes L-7012 Merck 15942 Merck 116316 Merck 115925 Sigma P4170 FW Dichte Smp. Sdp. Gefahren- R-Sätze symbol [g/mol] [g/cm³] [°C] [°C] 438.09 - S-Sätze - 311.8 - - - - - - 696.68 - - - - - 25 - - - - - - - 927.02 - 159 - Xn 21/22 24/25 799.8 - - - - - - 452.37 - - - - - - 668.4 - - - Xn 46-36/37/38 45-2636/37/39-22 Tabelle 5: verwendete Nährmedien Bezeichnung Pseudomonas-Isolierungs-Agar (PIA) Yeast Extract Nutrient Broth-Medium (NB) Hersteller Best.-Nr. Difco 0927-17-1 Difco 0127-17-9 Difco 0003-17-8 enthaltene Komponenten Bacto Peptone MgCl2 K2SO4 Irgasan Bacto Agar Hefeextrakt Bacto Beef Extract Bacto Peptone Menge auf 1000 ml deionisiertes Wasser 20 g 1,4 g 10 g 25 mg 13,6 g 16 g 3g 5g 2.2 Bakterienstämme Tabelle 6: verwendete Bakterienstämme Stamm Pseudomonas aeruginosa SG81R1 Phänotyp spontane, nichtmukoide Revertante, abgeleitet vom Stamm SG81 Herkunft Referenz Grobe et al., 1994 Stammsammlung Uni-Duisburg, Aquatische Mikrobiologie 17 2 Material und Methoden 2.3 Kenndaten der Analysensysteme 2.3.1 Konfokales Laser-Raster-Mikroskop - Konfokales Laser Scanning Modul LSM 510 der Firma Carl Zeiss Jena - gekoppelt an ein inverses Mikroskop „Axiovert 100 M BP“, Zeiss - Argon-Laser: 458, 488, 514 nm (25mW) - Helium-Neon-Laser: 543 nm (1mW) und Helium-Neon-Laser: 633 nm (5mW) - Objektive: Plan-Neofluar 10x / 0.30, Plan-Neofluar 20x / 0.50, Plan-Neofluar 40x / 1.30 Öl, C-Apochromat 40x / 1.20 W. Korr., Plan-Apochromat 63x /1.40 Öl DIC - Zeiss LSM 510 Software Release 2.02 - Steuerrechner: SIEMENS SCENIC PRO M7 2.3.2 Fluoreszenz-Mikroskop Laborlux S, Fa. Leitz - Objektive: - Okular - Photo-Okular PL Fluotar 10x / 0.30; 25x / 0.60; 40x / 0.70; 100x / 1.32 Öl 2x Periplan 10x/18, 1x Strichplatte 10x10 Felder (100 µm bei 10x) Periplan 10x/18 2.3.3 Stereomikroskop Leica MZ 6, Fa. Leica - Objektiv: - Okular - Photo-Okular - Photoeinrichtung verstellbar von 0.63x bis 4.0x 10x/21B 10x Leica MPS 60 mit Einstellfernrohr 1% 2.3.4 sonstige Geräte - Magnetrühr-/Heizplatte - Rührwerk: - UV/VIS-Photometer: - Chlor-Photometer-Kit: - Thermostateinheit: - Wasserbad: - Wasserbad-Rundschüttler: - Zentrifuge: IKA RCT basic mit Temperatursensor IKA ETS-D4 IKA RW 20 DZM.n Milton Roy Company, Spectronic 601 und Shimadzu UV-1202 SWAN Chematest 20 HAAKE Thermostat DC-1 mit Kühlbad K20 GFL 1013, Gesellschaft für Labor-Technik mbH GFL 1092, Gesellschaft für Labor-Technik mbH Heraeus Biofuge fresco (Weitere laborübliche Kleingeräte sind hier nicht aufgeführt.) 18 2 Material und Methoden 2.4 Herstellung von Agarose-Beads 2.4.1 Herstellung superporöser Agarose-Beads In einem auf 98 °C temperierten Wasserbad wird Agarose-Pulver (0.5 bis 3.0 g) in 50 ml deionisiertem Wasser vollständig gelöst, in ein auf 60 °C thermostatisiertes Reaktionsgefäß überführt und eine ebenfalls auf 60 °C vorgewärmte Lösung von 1.5 ml Tween 80 in 25 ml Cyclohexan unter Rühren (1000 U·min-1) zugegeben. Es bildet sich eine viskose, weiße Öl-inWasser Emulsion. Anschließend wird eine auf 60 °C temperierte Lösung von 8.0 ml Span 85 in 200 ml Cyclohexan unter Rühren (500 U·min-1) unter Ausbildung einer Wasser-in-Öl Emulsion zugegeben. Nach 10 Minuten wird die Thermostat-Einheit des Reaktionsgefäßes auf 20 °C eingestellt und so der Reaktionsansatz unter die Geltemperatur der Agarose abgekühlt. Das Reaktionsgemisch wird entnommen und der Cyclohexanüberstand dekantiert. Die zurückbleibenden Agarose-Beads werden viermal mit je 500 ml deionisiertem Wasser gewaschen und durch Dekantieren abgetrennt. Die Verfahrensschritte zur Darstellung von superporösen Agarose-Beads sind in Abbildung 9 dargestellt. Die ca. 98 °C heiße Agarose-Lösung wird in den Reaktor gegeben und auf 60 °C temperiert. Das Cyclohexan mit Tween 80 wird zugegeben. Es bildet sich eine Emulsion von Cyclohexan-Tröpfchen in der AgaroseLösung. Das Cyclohexan mit Span 85 wird zugegeben. Es bildet sich eine Emulsion von Agarose-Tropfen in Cyclohexan. Die Tropfen der Agarose-Lösung enthalten weiterhin die Cyclohexan-Tröpfchen aus dem vorherigen Schritt welche langsam herauswandern und so die Kanäle ausformen. Abbildung 9: Phasen bei der Herstellung von superporösen Agarose-Beads 19 2 Material und Methoden 2.4.2 Herstellung homogener Agarose-Beads In einem auf 98 °C temperierten Wasserbad wird Agarose-Pulver (0.5 bis 6.0 g) in 100 ml deionisiertem Wasser vollständig gelöst, in ein auf 60 °C thermostatisiertes Reaktionsgefäß überführt und eine ebenfalls auf 60 °C vorgewärmte Lösung von 8.0 ml Span 85 in 200 ml Cyclohexan unter Rühren (500 U·min-1) unter Ausbildung einer Wasser-in-Öl Emulsion zugegeben. Nach etwa 10 Minuten wird die Thermostat-Einheit des Reaktionsgefäßes auf 20 °C eingestellt und so der Reaktionsansatz unter die Geltemperatur der Agarose abgekühlt. Das Reaktionsgemisch wird entnommen und der Cyclohexanüberstand dekantiert. Die zurückbleibenden Agarose-Beads werden viermal mit je 500 ml deionisiertem Wasser gewaschen und durch Dekantieren abgetrennt. 2.4.3 Herstellung kompakter, superporöser Agarose-Körper In einem auf 98 °C temperierten Wasserbad wird Agarose-Pulver (0.5 bis 3.0 g) in 100 ml deionisiertem Wasser gelöst. Nach vollständiger Auflösung der Agarose wird diese Lösung in ein auf 60 °C thermostatisiertes Reaktionsgefäß überführt und eine ebenfalls auf 60 °C vorgewärmte Lösung von 1.5 ml Tween 80 in 25 ml Cyclohexan unter Rühren (1000 U·min-1) zugegeben. Es bildet sich eine viskose, weiße Öl-in-Wasser Emulsion. Diese Emulsion wird in einen auf 60 °C vorgewärmten Glasbehälter (z.B. eine Glas-Petrischale) gegeben und durch Abkühlen unter die Geltemperatur verfestigt. Auf diese Weise wird ein kompakter AgaroseKörper mit einer Superporen-Struktur erhalten. Die in diesen Superporen befindliche organische Phase kann durch ausgiebiges Waschen in reichlich deionisiertem Wasser entfernt werden. 2.5 Präparationen für mikroskopische Untersuchungen Für die mikroskopischen Untersuchungen der Agarose-Beads und -Schichten, insbesondere mit dem CLSM, erfolgten Färbungen mit fluoreszierenden und nichtfluoreszierenden Farbstoffen. 2.5.1 Stereomikroskopische Untersuchungen Zur besseren Visualisierung und Kontrastierung der Agarose-Matrix im Lichtmikroskop wurden Färbungen mit Orange G, Methylblau, und Acridinorange durchgeführt. Hierzu wurden die zu untersuchenden Proben in einer 1 %-igen Lösung (w/v) des jeweiligen Farbstoffes in deionisiertem Wasser 15 Minuten gefärbt und anschließend dreimal mit reichlich deionisiertem Wasser gewaschen. Die Proben wurden ohne weitere Vorbehandlungen auf Objektträger aufgebracht und direkt mit einem Stereomikroskop betrachtet. Die Bilddokumentation erfolgte mit Hilfe einer Leica Photoeinrichtung. 20 2 Material und Methoden 2.5.2 CLSM-Untersuchungen Für die Aufnahme der Agarose-Strukturen mit dem CLSM erfolgten Färbungen mit Acridinorange (AO), Propidiumiodid (PI) und Kongorot. 2.5.2.1 Acridinorange-Färbung Zur Färbung der Agarose-Beads mit AO werden ca. 0.5 g Bead-Material in einem EppendorfReaktionsgefäß mit 1.0 ml einer Lösung von 10 mg/l AO in deionisiertem Wasser versetzt, eine Minute geschüttelt, 5 Minuten bei 10000 g abzentrifugiert und der Überstand abdekantiert. Es werden 2.0 ml deionisiertes Wasser zugegeben, eine Minute geschüttelt und ebenfalls abzentrifugiert und dekantiert. Die so präparierten Beads wurden wie unter 2.5.2.4 beschrieben eingebettet und anschließend untersucht. 2.5.2.2 Propidiumiodid-Färbung Es wird eine Lösung von 10 mg/l PI in deionisiertem Wasser verwendet. Die Färbeprozedur gleicht der von AO (2.5.2.1). Es wird jedoch zweimal mit deionisiertem Wasser gewaschen. 2.5.2.3 Kongorot-Färbung Die Färbung mit Kongorot erfolgt mit einer 1 %-igen Lösung (w/v) von Kongorot in deionisiertem Wasser. Die Färbeprozedur gleicht der von AO (2.5.2.1). Es wird jedoch zweimal mit deionisiertem Wasser gewaschen. 2.5.2.4 Einbettung der Proben Die Proben für die Konfokalmikroskopie werden zum Schutz gegen Austrocknung und zur Konservierung der Präparate in ein Einbettungsmedium eingeschlossen. Hierfür kommt das Fluoreszenz-Einschlußmittel Mowiol 40-88 (ein Polyvinylalkohol) zum Einsatz, welches wie im Folgenden beschrieben, vorbereitet wird. Es wird eine Mischung von 2.4 g Mowiol, 6.0 g Glycerin und 6.0 g deionisiertem Wasser hergestellt und für 2 h bei Raumtemperatur stehengelassen. Anschließend werden 12.0 ml einer 0.2 molaren Lösung von Tris[hydroxymethyl]aminomethanhydrochlorid (pH 8.5) zugegeben, 10 min bei 50 °C inkubiert und 15 min bei 5000 g zentrifugiert. Der klare, honigartige Überstand wird aliquotiert und bei –20 °C aufbewahrt. Zur Verwendung wird ein Aliquot aufgetaut und zum baldmöglichsten Verbrauch bei 4 °C gelagert. Eingeschlossene Präparate sind bei 4 °C im Dunkeln haltbar. 21 2 Material und Methoden 2.6 Charakterisierung der Agarose-Beads 2.6.1 Bead-Durchmesser Mit Hilfe der Lichtmikroskopie lassen sich der Durchmesser und die Form der einzelnen Beads bestimmen. Hiermit läßt sich auf einfache Weise feststellen, ob man mit dem angewendeten Verfahren in der Lage ist, reproduzierbar kugelförmige Beads in einem steuerbaren, möglichst engen Größenbereich herzustellen. Hierzu wird eine repräsentative Teilmenge der bei einem Versuch hergestellten Agarose-Beads mit einem Mikroskop unter Verwendung einer Strichplatte vermessen sowie die Form der Beads qualitativ bestimmt. Für den Durchmesser wird ein Durchschnittswert, Maximal-, Minimalwert und Standardabweichung berechnet. Es erfolgt darüber hinaus eine grafische Analyse der Häufigkeitsverteilungen sowie eine Überprüfung der Normalverteilung mit dem Statistikprogramm SPSS. 2.6.2 Superporen-Durchmesser Mit dem konfokalen Laser-Scanning-Mikroskop werden konfokale Bildschnitte durch angefärbte Agarose-Beads aufgenommen und die Größe der erkennbaren Kanalstrukturen mit der Bildverarbeitungssoftware vermessen. Hierzu wird eine Probe der Agarose-Beads wie unter 2.5.2 gefärbt und eingebettet. Das Präparat wird bei 200-facher Vergrößerung mit dem CLSM betrachtet. Die für die jeweiligen Fluoreszenzfarbstoffe verwendeten Anregungs- und Detektionswellenlängen sowie die Filtereinstellungen sind in Tabelle 7 zusammengestellt. Darüber hinaus ist es möglich einen Stapel von Bildschnitten durch ein Bead aufzunehmen (z-stack) und mit Hilfe der LSM-Software eine dreidimensionale Rekonstruktion des Beads zu berechnen. Hierbei lassen sich die Oberfläche der Beads, sowie die Kanaleingänge betrachten. Tabelle 7: Verwendete CLSM-Einstellungen zur Bestimmung der Porengrößen Farbstoff Ex.,max [nm] Acridinorange Kongorot Propidiumiodid 490 500 480 Em.,ma x [nm] 525 500 Anregung [nm] Filter 543 543 488 LP 530 LP 530 BP 505-530 22 2 Material und Methoden 2.6.3 Superporen-Anteil Der Superporen-Anteil der porösen Agarose-Beads wird durch die im ersten Schritt zu der Agarose-Lösung zugegebenen Menge an organischer Phase bestimmt. Zur experimentellen Bestimmung des Porenanteils in den Beads werden bei den in Abschnitt 2.6.2 mit dem CLSM aufgenommenen Bildschnitten die Flächenverhältnisse von AgaroseMaterial und Porenraum bestimmt. Dies erfolgt durch Ausmessen der Flächen der AgaroseMatrix und der Kanäle mit der Bildverarbeitungssoftware. In Abbildung 10 bis Abbildung 12 ist die Vorgehensweise zur Bestimmung der Flächenanteile schematisch dargestellt. Die zugehörigen Flächen werden von der LSM-Software automatisch angegeben. Hieraus ergibt sich der Porenanteil in den hergestellten superporösen Agarose-Beads, welcher idealer Weise nicht von dem anfangs eingesetzten Verhältnis der Cyclohexan-Phase zur Agarose-Lösung abweichen sollte. Abbildung 10: Bildschnitt durch ein Abbildung 11: Fläche des Agarose- Abbildung 12: Fläche des Porenporöses AgaroseMaterials zum Ausraumes zum AusBead, (CLSM-Aufmessen markiert. messen markiert. nahme, gefärbt mit Kongorot) 2.6.4 Bestimmung des Löslichkeitsverhaltens Zur Überprüfung der Beständigkeit der Gel-Beads soll das Löslichkeitsverhalten der Beads gegenüber der wäßrigen Phase getestet werden. Ein Aliquot der Gel-Beads (ca. 2.0 g) wird in 100 ml deionisiertem Wasser in einem Erlenmeyerkolben bei 130 Umdrehungen auf einem Schütteltisch geschüttelt. In regelmäßigen Abständen wird eine Probe der Beads entnommen, der Durchmesser und die Größenverteilung ermittelt (wie unter 2.6.1). Durch das Aufnehmen einer Zeitreihe lassen sich Veränderungen des Bead-Durchmessers und der Größenverteilung erkennen und so Rückschlüsse auf das Auflösungsverhalten der AgaroseBeads ziehen. 23 2 Material und Methoden 2.6.5 Bestimmung des Quellungsverhaltens Für die Bestimmung der Volumenquellungsgrade wurde eine von Selic (1999) entwickelte Quellungsapparatur verwendet, welche die Online-Messung von Volumenquellungsgraden in Abhängigkeit von der Zeit ermöglicht. Der schematische Aufbau der Apparatur ist in Abbildung 13 dargestellt. Abbildung 13: Schematische Darstellung der Quellungsapparatur (nach Selic, 1999) Gemessen wird der Hub eines Stempels in einem Zylinder mit genau definierten Abmessungen, der durch die Volumenänderung der eingeschlossenen, quellenden Substanz verursacht wird. Die Probe befindet sich im Zylinder auf einem Metallnetz, das fest auf einer Sintermetallplatte aufliegt. Die Sintermetallplatte ist in einer Edelstahlschale angeschraubt, über die das Quellungsmittel seitlich hinzugegeben wird. Quillt die Probe bei Kontakt mit dem Quellmittel, wird der Stempel aufgrund der Volumenänderung uniaxial angehoben. Das Auftreten von Haftreibungseffekten, welches zu ruckartigen Bewegungen des Stempels führen würde, wird durch eine leichte, oszillierende Bewegung des Stempels verhindert. Der Hub des Stempels wird mit einem induktiven Wegaufnehmer gemessen, der über eine Analog-/Digital-Wandlerkarte mit einem Computer verbunden ist. Die Aufnahme der Meßdaten wird online durchgeführt. 24 2 Material und Methoden Die Waagenkonstruktion, die das Gewicht des Stempels annähernd aufhebt, garantiert eine freie Quellung der Probe. Daher kann man davon ausgehen, daß die Beads während der Quellung ihre annähernde Kugelform beibehalten und folglich keine Veränderung des Zwickelvolumenanteils auftritt. Dies ist eine wesentliche Voraussetzung für die Berechnung des Volumenquellungsgrades. Für die Ermittlung von Quellungsgraden unter Verwendung der beschriebenen Volumenquellungsapparatur werden ca. 0.1 bis 0.5 g Substanz eingewogen. Die Probe wird gleichmäßig auf dem Metallnetz verteilt und der Stempel in den Zylinder eingesetzt. Das Löse- bzw. Quellmittel gelangt aus einem externen Behälter, unter Ausnutzung des „Prinzips der kommunizierenden Röhren“, in die Edelstahlschale und über die Sintermetallplatte in Kontakt mit der Probe. Die Meßdatenakquisition wird von Hand gestartet, wenn das Quellmittelniveau die Probe erreicht hat. Der induktive Wegaufnehmer mißt mit einer zu definierenden Frequenz die relative Position des Stempels zum Zeitpunkt t. Die relativen Höhen des Stempels werden vom Wegaufnehmer in Form von Spannungswerten angegeben und mit einem Hilfsprogramm in Wegstrecken umgerechnet. In gleicher Weise läßt sich auch die Entquellung der hergestellten Gel-Beads unter einer äußeren Belastung untersuchen. Hierbei wird das Gewicht des Stempels nicht durch die Waagenkonstruktion ausgeglichen, sondern so eingestellt, daß ein definierter Druck auf die eingeschlossene Probe ausgeübt wird. Mit dem Wegaufnehmer wird dann die Volumenabnahme in Abhängigkeit von der Zeit registriert. 2.7 Agarose-Beads mit eingebetteten Mikroorganismen 2.7.1 Vorbereitung der Bakteriensuspension Es wird ein Einzelkolonie-Ausstrich von Pseudomonas aeruginosa SG81R1 auf PseudomonasIsolierungs-Agar (PIA) durchgeführt und 24 h bei 30 °C bebrütet. Der Bewuchs wird mit einem Metallspatel abgenommen und in 2 ml einer 0.14 M NaCl-Lösung suspendiert. Die Zellzahl der Suspension wird mit Hilfe einer Thoma-Zählkammer bestimmt. Hierfür wurden Verdünnungen von 1:100 und 1:1000 gewählt. Die Kleinstquadrate der verwendeten ThomaZählkammer haben eine Größe von 0.05 x 0.05 mm. Die Kammertiefe beträgt 0.1 mm. Als Kammerfaktor ergibt sich somit 4·106. Es werden jeweils fünf zufällig ausgewählte Kleinstquadrate in vier Gesichtsfeldern ausgezählt. Die Gesamtzellzahl der Suspension (in Zellen pro ml) ergibt sich aus dem Mittelwert der in den Feldern ausgezählten Bakterien, multipliziert mit dem Kammerfaktor und dem Verdünnungsfaktor. Es wird mikroskopisch überprüft, daß die Bakterien in der Suspension vereinzelt vorliegen und keine Aggregate bilden. 25 2 Material und Methoden 2.7.2 Einbettung von Mikroorganismen in Agarose-Beads Eine Einbettung von Bakterien wurde sowohl in homogenen als auch in porösen Agarose-Beads, sowie in kompakten Agarose-Körpern durchgeführt. Hierzu wird ein Teil der hergestellten Bakteriensuspension unter Rühren zu der im Reaktionsgefäß temperierten Agarose-Lösung gegeben. Die im Einzelnen gewählten Parameter sind in Tabelle 8 aufgeführt. Das Zugabevolumen sollte jedoch möglichst gering gehalten werden, um eine Temperaturabsenkung durch die Zugabe auszuschließen. Die Temperatur während des gesamten Reaktionsverlaufes wird auf 47 °C gesenkt, um eine Schädigung der Bakterien zu minimieren. Tabelle 8: Versuchsparameter bei der Einbettung von Bakterien Versuch homogene Beads poröse Beads poröse Agarose-Stücke Agarose-Lösung Bakteriensuspension Konzentration Menge Zellzahl [%] 4 4 4 [ml] 100 50 50 [Zellen/ml] 4,7·109 4,7·109 4,7·109 Zugabemenge [µl] 500 250 250 resultierende Zelldichte (rechnerisch ) [Zellen/ml] 2,3·107 2,3·107 2,3·107 Der übrige Versuchsablauf entspricht den unter 2.4.1 bis 2.4.3 gemachten Angaben. Bei allen Reaktionsschritten wurde jedoch Anstelle des deionisierten Wassers eine isotonische Kochsalzlösung (0.14 mol/l NaCl) verwendet um einen hydrodynamischen Schock der Bakterien zu verhindern. 2.7.3 Bestimmung von Position und physiologischem Zustand der Bakterien Durch das Anfärben mit verschiedenen, spezifischen Fluoreszenzfarbstoffen ist eine Unterscheidung zwischen Bakterien, Polysaccharid, Partikeln und Kanälen/Hohlräumen möglich. Durch spezielle Färbetechniken ist es auch möglich, die Vitalität der Mikroorganismen bzw. den Zustand der Membran sichtbar zu machen. Dies ist z.B. mit dem kommerziell erhältlichen LIVE/DEAD® BacLight™ Bacterial Viability Kit von Molecular Probes (Kombination der Farbstoffe SYTO 9 und PI) durchführbar. 26 2 Material und Methoden 2.7.3.1 CTC-Färbung Zur Fluoreszenzmarkierung der stoffwechselaktiven Zellen mit CTC werden ca. 0.5 g BeadMaterial in einem Eppendorf-Reaktionsgefäß mit 1.0 ml einer 1 mM CTC-Lösung (Schaule et al., 1993b) in 0.14 M NaCl-Lösung versetzt, eine Minute geschüttelt und 3 h im Dunkeln bei Raumtemperatur inkubiert. Stoffwechselaktive Zellen reduzieren das CTC gemäß der nachstehenden Gleichung zu Formazankristallen (CTF), welche eine starke Fluoreszenz ( Ex.,max = 450 nm, Em.,max = 630 nm) aufweisen. Das unreduzierte CTC absorbiert über 400 nm nicht und zeigt keine Fluoreszenz. CN N N H3C N CN N H3C H N N C N N CH3 CH3 Der Überstand wird bei 10000 g abzentrifugiert und dekantiert. Die so präparierten Beads können wie unter 2.5.2.4 beschrieben eingebettet und anschließend untersucht werden. Das Präparat wird hierzu bei 200-facher Vergrößerung mit dem CLSM betrachtet und ein konfokaler Bildschnitt durch die Beads aufgenommen. Die Anregung erfolgt bei 488 nm mit einem ArgonLaser. Die Detektion erfolgt unter Verwendung des LP 560 Filters und des Nebenfarbteilers NFT 570. 2.7.3.2 SYTO 9/Propidiumiodid-Färbung Das von Molecular Probes erhältliche „LIVE/DEAD® BacLight™ Bacterial Viability Kit“ ermöglicht durch eine Doppelfärbung eine Differenzierung von Bakterienzellen in Bezug auf eine vorhandene Membranschädigung. Das Kit enthält den grün fluoreszierenden DNAbindenden Farbstoff SYTO 9 ( Ex.,max = 480 nm, und den rot Em.,max = 500 nm) fluoreszierenden DNA-bindenden Farbstoff Propidiumiodid ( Ex.,max = 490 nm, Em.,max = 635 nm). Beide Farbstoffe unterscheiden sich in ihrer Membrangängigkeit. Mit SYTO 9 lassen sich sowohl intakte Bakterien wie auch geschädigte Zellen markieren. Propidiumiodid kann jedoch nur beim Vorliegen einer Membranschädigung in die Zelle eindringen und konkurriert dort mit SYTO 9 um die Bindungsplätze an der Nucleinsäure (Molecular Probes, 1996; Lawrence et al., 1997). So ergibt sich beim Anfärben mit einer Mischung aus SYTO 9 und Propidiumiodid, daß Bakterien mit einer intakten Zellmembran grün fluoreszieren, Bakterien mit einer geschädigten Membran hingegen rot fluoreszieren. Zur Doppelfärbung mit dem LIVE/DEAD® BacLight™ Bacterial Viability Kit werden gleiche Mengen der beiden gelieferten Komponenten (A: 3.34 mM SYTO 9 in DMSO; B: 20 mM Propidiumiodid in DMSO) gemischt und 3 µl dieser Lösung zu 1 ml Bakteriensuspension (Molecular Probes, 1996) bzw. zu ca. 0.5 g Beads in 0.5 ml deionisiertem Wasser gegeben. Es wird gründlich gemischt und 15 Minuten bei Raumtemperatur im Dunkeln inkubiert. Die Beads werden abgetrennt und wie unter 2.5.2.4 beschrieben eingebettet. Mit dem CLSM können konfokale Bildschnitte der so präparierten Beads aufgenommen werden. Die Anregung erfolgt bei 488 nm (Argon-Laser). Die Detektion für die PropidiumiodidFluoreszenz erfolgt mit dem LP 560 Filter (NFT 570) und für die SYTO 9-Fluoreszenz mit dem BP 505-530 Filter. Die Bildschnitte wurden unter 200-facher Vergrößerung aufgenommen. 27 2 Material und Methoden 2.7.4 Kultivierung von Bakterien in Agarose-Beads Zur Überprüfung der Lebens- und Wachstumsfähigkeit von immobilisierten Bakterien in den Beads sowie zur Beobachtung der Ausbildung von Mikrokolonien werden Agarose-Beads mit eingebetteten Bakterien in eine Nährlösung gegeben und bebrütet. Hierzu werden etwa 0.5 g Bead-Material mit immobilisierten Bakterien (P. aeruginosa) aus 2.7.2 in 3 ml Nährlösung gegeben. Als Nährmedium wurde eine Lösung von 0.2 g/l Nutrient Broth-Medium in deionisiertem Wasser verwendet. Für eine Versuchsreihe werden z.B. fünf dieser Proben bei 30 °C bebrütet und in regelmäßigen Zeitabständen (z.B. alle 24 h) die Beads einer Probe entnommen und untersucht. Es erfolgt eine Überprüfung der Beads auf sichtbare Kolonien mit dem Stereomikroskop. Darüber hinaus werden Färbungen mit SYTO 9 und PI in Kombination (LIVE/DEAD® BacLight™ Bacterial Viability Kit, Molecular Probes) sowie mit CTC durchgeführt und eine anschließende Betrachtung mit dem CLSM vorgenommen (siehe Abschnitt 2.7.3). 2.8 Herstellung von Bead-Schichten Zur Modellierung eines künstlichen Biofilms ist es notwendig, die Agarose-Beads auf einem Träger flächig zu fixieren. Im Folgenden sind hierfür mögliche Verfahren angegeben, sowie die angewandten Untersuchungsmethoden aufgeführt. 2.8.1 Fixierung der Agarose-Beads Für die Fixierung der Beads auf einem Träger, z.B. einem Objektträger, wurden verschiedene Techniken angewandt: • Auf einen Glas-Objektträger aufgebrachte Beads wurden durch kurzes Erwärmen über einer Bunsenbrennerflamme fixiert. Hierbei können jedoch nur einlagige Bead-Schichten hergestellt werden. Darüber hinaus ist das Verfahren für Beads mit eingebetteten Bakterien nur bedingt einsetzbar, da die Mikroorganismen durch die kurzzeitige Temperaturbelastung geschädigt werden können. • Es wurden die auf einem Glas-Objektträger aufgebrachten Beads mit einigen Tropfen einer verdünnten Agarose Lösung fixiert. Hierzu werden 0.5 % (w/v) Agarose in deionisiertem Wasser bei 98 °C im Wasserbad gelöst und anschließend auf 45 °C temperiert. Mit dieser Lösung können die Agarose-Beads auf einem Träger angeheftet werden. Mit dieser Methode wurden sowohl einlagige, wie auch mehrlagige Bead-Schichten erzeugt. Mit diesen Methoden war es ebenfalls möglich, Filme, Schnitte oder Stücke der in Abschnitt 2.4.3 hergestellten kompakten, superporösen Agarose auf einem Träger zu fixieren. 28 2 Material und Methoden 2.8.2 Bestimmung der dreidimensionalen Struktur der Bead-Schichten Die Struktur und räumliche Anordnung der Beads in den Schichten kann, wie bereits bei den Einzelbeads beschrieben, mit dem Stereomikroskop betrachtet oder nach einer Färbung mit dem CLSM untersucht werden. Hierzu werden die fixierten Beads durch Eintauchen der Träger in die unter 2.5.1 bzw. 2.5.2 verwendeten Farbstoff-Lösungen angefärbt und durch Spülen mit deionisiertem Wasser gewaschen. Die Aufnahme der konfokalen Bildschnitte erfolgt bei 100-facher und 200-facher Vergrößerung unter Anwendung der in Abschnitt 2.6.2 in Tabelle 7 angegebenen Parameter. 2.9 Biozideinwirkung Zur Überprüfung, ob sich die Einbettung der Bakterien auf die Wirkung eines Biozids auswirkt, wird eine vergleichende Untersuchung zwischen einer Zellsuspension und homogenen sowie porösen Beads durchgeführt. Dabei soll insbesondere Untersucht werden, ob die Einbettung der Zellen in die Gelmatrix eine Schutzwirkung bezüglich des Einwirkens von Bioziden hat. 2.9.1 Natriumhypochlorit Es werden jeweils 0.5 g Bead-Material mit PBS pH 7.54 auf 1.0 ml aufgefüllt und anschließend mit 1.0 ml Natriumhypochlorit-Lösung (1 mg/l freies Chlor in PBS pH 7.54) versetzt. Es ergibt sich somit eine Endkonzentration von 0.5 mg/l freiem Chlor. Die verwendeten Beads besitzen eine Zelldichte von etwa 109 Zellen/cm3. Nach verschiedenen Reaktionszeiten (1 bis 60 Minuten) bei 25 °C werden zur Inaktivierung des Chlors 100 µl Natriumthiosulfat-Lösung (100 mg/ml) zugegeben (Endkonzentration 5 mg/ml). Alle verwendeten Lösungen wurden auf 25 °C vortemperiert Zur Kontrolle der Chlorzehrung durch das Agarose-Material wird zusätzlich die Chlorkonzentration in einer Probe mit Agarose-Beads ohne Bakterien über den Reaktionszeitraum quantifiziert. Die Bestimmung der Chlorkonzentrationen erfolgte durch Messung der Absorption bei 290 nm. Als Vergleich wird eine Bakteriensuspension von P. aeruginosa in PBS pH 7.54 ((3.5 0.2)·108 Zellen/ml) in gleicher Weise behandelt. Zur Herstellung der Zellsuspension wird eine Kolonie von P. aeruginosa SG81R1 von der Stammplatte in 1 ml isotonischer NaCl-Lösung suspendiert, auf Pseudomonas-Isolierungs-Agar (PIA) ausplattiert und 72 h bei 30 °C bebrütet. Der Bewuchs wird abgenommen und in PBS pH 7.54 suspendiert. Der Zelltiter wird mit Hilfe einer Thoma-Zählkammer bestimmt und die Suspension entsprechend auf die Endzellzahl verdünnt. Zur Kontrolle der Chlorzehrung durch die verwendeten Lösungen und Glasgeräte wird eine Blindprobe von 1 ml PBS pH 7.54 verwendet und auch mit 1 ml der Hypochlorit-Lösung behandelt. Die Chlorkonzentration in dieser Probe wird zu Beginn und am Ende des Untersuchungszeitraumes überprüft. In einem zweiten Versuchsansatz wird die Zehrung des freien Chlors über den Reaktionszeitraum bestimmt. Dies erfolgt an einer Zellsuspension, an porösen Agarose-Beads 29 2 Material und Methoden mit eingebetteten Bakterien und an Agarose-Beads ohne Bakterien. Der Gehalt an freiem Chlor wird photometrisch mittels DPD-Methode (N,N-Diethyl-1,4-phenylendiaminsulfat) bestimmt. Dadurch wird der störende Effekt der Bakterien in der Suspension vermieden, welcher eine Absorptionsmessung bei 290 nm behindern würde. Die Durchführung wird entsprechend dem Vorgehen zur Untersuchung der Membranschädigung vorgenommen. Die Probenvolumina werden jedoch auf das 5-Fache erhöht, da für die photometrisch Bestimmung 10 ml Probenvolumen (statt 2 ml) benötigt werden. 2.9.2 Quantifizierung der inaktivierten und überlebenden Zellen Die Bestimmung des physiologischen Zustandes der mit Bioziden behandelten Bakterien erfolgte anhand der Erfassung der Membranschädigung der Zellen. Hierzu wurden die Zellen in den Beads mit SYTO 9 und PI in Kombination (LIVE/DEAD® BacLight™ Bacterial Viability Kit, Molecular Probes) angefärbt und mit dem Laser-Raster-Mikroskop untersucht. Zur Auswertung der Inaktivierungsleistung wird der mit Propidiumiodid gefärbte Anteil an der Gesamtzahl aller Bakterien (SYTO 9) bestimmt. Die Auswertung des Suspensionsversuches erfolgte durch Färbung mit SYTO 9 und PI in Kombination (LIVE/DEAD® BacLight™ Bacterial Viability Kit, Molecular Probes) in Suspension und Auszählung am Fluoreszenz-Mikroskop. 2.10 Statistische Auswertung Zur Beurteilung der erhaltenen Ergebnisse wurde eine statistische Auswertung vorgenommen. Zum Vergleich der berechneten Mittelwerte wurde Student’s t-Test angewendet. Dabei wird objektiv beurteilt, ob sich zwei Mittelwerte mx1 und mx2 unter Berücksichtigung ihrer Fehlerbreite signifikant unterscheiden. Hierzu ist die Prüfgröße TAU gemäß der folgenden Formel zu berechnen: TAU = m x1 − m x 2 (n1 − 1) * s1 + (n2 − 1) * s 2 n1 + n 2 − 2 2 2 * n1 * n 2 n1 + n2 Durch den Vergleich des berechneten Wertes für TAU mit den entsprechenden Tabellenwerten (Kaiser und Mühlbauer, 1983) kann beurteilt werden, ob zwischen den betrachteten Mittelwerten ein hochsignifikanter, ein signifikanter, ein wahrscheinlicher oder kein feststellbarer Unterschied besteht. Die Resultate dieses Tests sind Basis der Ergebnisbeurteilung und darauf folgender Entscheidungen, deren Schärfe von der gewählten statistischen Sicherheit (P) abhängt. Im Rahmen dieser Arbeit wurde eine statistische Sicherheit von P = 95 % gefordert. 30 3 Ergebnisse 3 Ergebnisse 3.1 Homogene Agarose-Beads In einem Vorversuch wurde die Herstellung von homogenen Agarose-Beads nach Abschnitt 2.4.2 mit zwei unterschiedlichen Rührwerkzeugen getestet. Die Verwendung eines Überkopfrührwerks mit Plattenrührer erwies sich als ungeeignet, da sich hiermit keine ausreichenden Scherkräfte aufbringen ließen um eine gleichmäßige Emulgierung der AgaroseTropfen zu gewährleisten. Mit einem 4-flügeligen Flügelrührer wurden bessere Ergebnisse erzielt. Es ließ sich in einem weiten Geschwindigkeitsbereich eine gleichmäßige Tropfenzerteilung und eine gute Emulsionsbildung erreichen. Alle nachfolgenden Versuche wurden daher mit dem Flügelrührer durchgeführt. Die Ergebnisse des Vorversuches sind in Tabelle 9 dargestellt. Tabelle 9: Vorversuch zur Auswahl des Rührwerkzeuges Rührer Rührgeschwindigkeit [U·min-1] Flügelrührer 500 Plattenrühre 500 r md 326,1 288,6 Bead-Durchmesser (d) [µm] dmin dmax s 120 780 149,9 50 1400 228,8 n 100 100 Vertrauensbereich (P = 95%) ± 29,7 ± 45,3 In Abbildung 14 und Abbildung 15 sind die Größenverteilungen der Beads aus beiden Experimenten graphisch dargestellt. Die eingezeichnete Kurve gibt jeweils die Normalverteilungsfunktion wieder. Es läßt sich erkennen, daß der Durchmesser der mit dem Plattenrührer hergestellten Beads wesentlich ungleichmäßiger ist und über einen erheblich größeren Bereich streut. Abbildung 14: Beadgrößenverteilung bei Herstellung mit dem Flügelrührer (500 U·min-1) Abbildung 15: Beadgrößenverteilung bei Herstellung mit dem Plattenrührer (500 U·min-1) 31 3 Ergebnisse 3.1.1 Variation der Rührgeschwindigkeit Zur Regelung des mittleren Bead-Durchmessers hat sich die bei der Herstellung verwendete Rührgeschwindigkeit als geeignet erwiesen. Um die Abhängigkeit des mittleren Durchmessers von der Rührgeschwindigkeit zu ermitteln, wurde in einer Versuchsreihe die Herstellung von homogenen Beads bei unterschiedlichen Geschwindigkeiten durchgeführt. Die Durchführung wurde entsprechend Abschnitt 2.4.2 vorgenommen. In Tabelle 10 sind die verwendeten Rührgeschwindigkeiten sowie die Daten der damit erzeugten Beads zusammengestellt. Tabelle 10: Bead-Durchmesser und Verteilung in Abhängigkeit von der Rührgeschwindigkeit Rührgeschwindigkeit [U·min-1] 400 500 700 900 1100 Bead-Durchmesser (d) [µm] md s n 162,9 100 409,9 149,9 100 326,1 64,9 100 206,5 69,1 100 159,3 59,9 100 133,8 Vertrauens -bereich (P = 95%) ± 32,3 ± 29,7 ± 12,9 ± 13,7 ± 11,9 t-Test 3,79 7,32 4,98 2,79 Die statistische Betrachtung (Abschnitt 2.10) dieser Ergebnisse zeigte, daß sich die Mittelwerte des Bead-Durchmessers für die Rührgeschwindigkeiten 400 bis 1100 U·min-1 hochsignifikant unterscheiden und die beobachteten Unterschiede nicht als zufällig angesehen werden können. Abbildung 16 und Abbildung 17 zeigen zwei typische Aufnahmen von den dabei hergestellten Beads. Abbildung 16: Stereomikroskopische Aufnahme von homogenen Agarose-Beads (500 U·min-1) Abbildung 17: Stereomikroskopische Aufnahme von homogenen Agarose-Beads (1100 U·min-1) 32 3 Ergebnisse Trägt man die ermittelten mittleren Bead-Durchmesser gegen die verwendete Rührgeschwindigkeit auf, so ergibt sich der in Abbildung 18 graphisch dargestellte Zusammenhang. 500 450 Mittelwert des Bead-Durchmessers [µm] 400 350 300 250 200 150 100 50 0 0 200 400 600 800 1000 1200 1400 -1 Rührgeschwindigkeit [U•min ] Abbildung 18: Abhängigkeit des Bead-Durchmessers von der Rührgeschwindigkeit Berücksichtigt man die maximal mit dem vorhandenen Rührwerk mögliche Rührgeschwindigkeit von 2000 U·min-1, so läßt sich durch Extrapolation des Kurvenverlaufes der bei Beibehaltung der anderen Versuchsparameter kleinstmögliche, herstellbare BeadDurchmesser auf ca. 65 µm abschätzen. Der größtmögliche mittlere Bead-Durchmesser ist bei 400 U·min-1 nahezu erreicht. Bei geringeren Rührgeschwindigkeiten erfolgt keine genügende Durchmischung der Phasen und es tritt eine Phasentrennung aufgrund der Tropfenkoaleszenz (Zusammenfließen von Tropfen) ein. 3.1.2 Variation der Agarose-Konzentration Zur Simulation von verschiedenen EPS-Konzentrationen des Modell-Biofilms läßt sich die Konzentration der zur Bead-Herstellung eingesetzten Agarose-Lösung variieren. Um den Einfluß der Agarose-Konzentration auf die Beschaffenheit der daraus hergestellten Beads zu überprüfen, wurde eine Versuchsreihe mit unterschiedlichen Konzentrationen durchgeführt. Die Herstellung der Beads wurde entsprechend der in Abschnitt 2.4.2 angegebenen Vorschrift durchgeführt. Die Emulgierung erfolgte mit dem Flügelrührer bei 600 U·min-1. In Tabelle 11 sind die gewählten Konzentrationen und die Daten der damit produzierten Beads zusammengestellt. 33 3 Ergebnisse Tabelle 11: Bead-Durchmesser und Verteilung in Abhängigkeit von der Agarose-Konzentration AgaroseKonzentration [% (w/v)] 0,5 1,0 2,0 3,0 4,0 Bead-Durchmesser (d) [µm] md s n 40,1 100 102,7 48,9 100 125,6 71,5 100 191,2 86,4 100 241,0 138,3 100 291,1 Vertrauens -bereich (P = 95%) ± 8,0 ± 9,7 ± 14,2 ± 17,1 ± 27,4 t-Test 3,62 7,57 4,44 3,07 Für die Mittelwerte der Bead-Durchmesser bei Agarose-Konzentrationen von 0.5 bis 3% konnte ein hochsignifikanter, bzw. zwischen dem Mittelwert für 3% und 4% ein signifikanter Unterschied nachgewiesen werden (Abschnitt 2.10). In Abbildung 19 sind die ermittelten Wertepaare sowie die sich daraus ergebende Abhängigkeit des mittleren Bead-Durchmessers von der Agarose-Konzentration aufgetragen. 350,0 mittlerer Bead-Durchmesser [µm] 300,0 250,0 200,0 150,0 100,0 50,0 0,0 0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5 Agarose-Konzentration [%] (w/v) Abbildung 19: Zusammenhang zwischen Agarose-Konzentration und mittlerem Bead-Durchmesser Es läßt sich erkennen, daß der mittlere Bead-Durchmesser erwartungsgemäß mit steigender Agarose-Konzentration zunimmt, da mit der höheren Konzentration des Polysaccharids eine höhere Viskosität der Lösung einhergeht und daher die Emulsionsbildung größere Scherkräfte erfordert. Bei gleicher Rührgeschwindigkeit und somit gleichen Scherkräften führt dies zwangsläufig zu größeren Beads. 34 3 Ergebnisse Bei den praktischen Versuchen zur Bead-Herstellung bei unterschiedlichen Konzentrationen wurde deutlich, daß sich Beads aus Agarose-Lösungen von ca. 0.5 bis 6% Agarose (w/v) mit der angewendeten Methode reproduzierbar herstellen lassen. Niedrigere Konzentrationen sind, aufgrund der damit verbundenen geringen mechanischen Stabilität des Agarose-Gels, nicht möglich. Hinzu kommt, daß Agarose in diesen niedrigen Konzentrationen nicht mehr zur Ausbildung einer Gelphase neigt. Agarose-Lösungen mit mehr als 6% (w/v) Agarose sind hingegen bereits so hochviskos, daß sie sich mit den zur Verfügung stehenden Rührwerkzeugen nicht zufriedenstellend verarbeiten lassen. Die Produktion von Agarose-Beads mit höheren Konzentrationen erscheint in Hinblick auf das Biofilm-Modell-Konzept auch wenig sinnvoll, da sich die EPS-Konzentrationen in natürlichen Biofilmen ebenfalls fast ausschließlich in diesem niedrigen Konzentrationsbereich (<5%) bewegen. 3.1.3 Löslichkeitsverhalten der homogenen Beads Zur Überprüfung der Beständigkeit des Hydrogel-Netzwerkes im wäßrigen Medium wurde das Auflöseverhalten der Agarose-Beads mit Hilfe des in Abschnitt 2.6.4 angegebenen Versuches untersucht. Hiermit soll sichergestellt werden, daß sich die hergestellten Beads im wäßrigen Milieu nicht auflösen. Die ermittelten Werte sind in Tabelle 12 zusammengetragen. Tabelle 12: Löslichkeitsverhalten der homogenen Beads Zeit [h] 0 0,5 24 48 120 288 Bead-Durchmesser (d) [µm] md 201,1 208,6 209,3 207,4 208,9 208,2 s 66,0 69,9 106,3 97,1 90,9 122,2 n 100 100 100 100 100 100 Vertrauens -bereich t-Test (P = 95%) 13,1 13,9 21,1 19,3 18,0 24,2 0,78 0,06 0,13 0,11 0,05 Zwischen sämtlichen bestimmten Mittelwerten konnte bei einer statistischen Auswertung (Abschnitt 2.10) kein signifikanter Unterschied feststellt werden. Die Unterschiede können aufgrund des vorliegenden Datenmaterials somit als rein zufällig angesehen werden. Dennoch zeigt sich ein leichter Anstieg des Mittelwertes innerhalb der ersten 0.5 h, welcher wahrscheinlich auf ein Quellungs-Phänomen zurückzuführen ist. Die Meßpunkte im weiteren chronologischen Verlauf zeigen, daß sich keine Auflösungserscheinungen nachweisen lassen. 35 3 Ergebnisse 3.2 Superporöse Agarose-Beads Eine Verbesserung des Kugelmodells aus homogenen Agarose-Beads wurde durch die Herstellung von porösen Agarose-Beads möglich. Diese weisen im Gegensatz zu den homogenen Beads eine ausgedehnte Kanalstruktur auf, welche bei der Modellierung eines künstlichen Biofilms, neben den entstehenden Zwickelräumen, zu einer weiteren Simulation eines verzweigten Poren- und Kanalsystems in einem Biofilm beiträgt. Superporöse AgaroseBeads wurden erstmals von Gustavsson und Larsson (1996) als stationäre Phase für chromatographische Anwendungen hergestellt. Die Nutzung solcher superporösen Strukturen wird im Rahmen der vorliegenden Arbeit erstmals auf die Modellierung von Biofilmen angewendet. 3.2.1 Porenstruktur der Beads Zur Untersuchung der Superporen-Struktur der Beads wurden verschiedene Proben, teilweise nach Anfärbung (Abschnitt 2.5.1), stereomikroskopisch betrachtet, sowie konfokale Bildschnitte gemäß Abschnitt 2.6.2 mit dem CLSM (Abschnitt 2.5.2) erstellt. Zwei typische Bilder, die mit dem Stereomikroskop aufgenommen wurden, sind in den folgenden Abbildungen gezeigt: Porenöffnungen Abbildung 20: Stereomikroskopische Aufnahme von porösen Agarose-Beads (1100 / 500 U·min-1, Hellfeld) Abbildung 21: Stereomikroskopische Aufnahme von porösen Agarose-Beads (1000 / 500 U·min-1, Dunkelfeld) Auf den Bildern läßt sich die poröse Struktur der Beads erkennen. Insbesondere die Porenöffnungen an der Bead-Oberfläche sind deutlich sichtbar. In Abbildung 20 sind zur Verdeutlichung zwei Porenöffnungen markiert worden (rote Pfeile). Man erkennt auch, daß es aufgrund der Kanalbildung zu Verformungen der Beads kommt, so daß diese nicht mehr ideal rund geformt sind, sondern teilweise ovale bis längliche Formen aufweisen. 36 3 Ergebnisse Die Bestimmung der Superporen-Größe und des Superporen-Anteils erfolgte anhand der erstellten konfokalen Bildschnitte. Ein Beispiele für einen solchen Bildschnitt ist in Abbildung 22 dargestellt. Abbildung 22: Bildschnitt durch ein poröses Agarose-Bead (1000 / 500 U·min-1), (CLSM-Aufnahme, gefärbt mit AO) Mit Hilfe der Meßfunktion der Zeiss LSM-Software ließen sich die Porengröße sowie die Fläche des Bead-Materials und des Porenraumes ausmessen (vergl. hierzu Abschnitt 2.6.3) und der flächenbezogene Porenanteil berechnen. Für die untersuchten Beads ergaben sich die in Tabelle 13 zusammengestellten Werte. Tabelle 13: Porengröße und Porenanteil in superporösen Agarose-Beads Rührgeschw. (Schritt 1 / 2) [U·min-1] 1000 / 600 1000 / 500 1100 / 600 1100 / 500 Porengröße [µm] mx 28,4 27,9 27,6 27,0 s 15,9 14,1 14,2 13,2 Vertrauens Poren-bereich anteil n 100 100 100 100 (P = 95%) ± 3,16 ± 2,80 ± 2,82 ± 2,63 [%] 34,2 29,3 36,4 31,3 t-Test 0,24 0,31 0,38 0,46 Da die statistische Überprüfung (Abschnitt 2.10) zwischen sämtlichen Mittelwerten keine signifikanten Unterschiede erkennen ließ, ist es erlaubt, die Daten der einzelnen Meßreihen zusammenzufassen und einen gemeinsamen Mittelwert anzugeben. Für die Porengröße und den Porenanteil erhält man demnach die in Tabelle 14 angegebenen Mittelwerte. 37 3 Ergebnisse Tabelle 14: gemittelte Porengröße und Porenanteil in superporösen Agarose-Beads Porengröße [µm] mx 27,7 min 8,3 max 81,4 s 14,3 Vertrauens -bereich n 400 (P = 95%) ± 1,42 Porenanteil [%] mx s 32,8 3,1 Vertrauens -bereich n 4 (P = 95%) ± 4,3 Wie bereits in Abschnitt 2.6.3 erwähnt, wird der Porenanteil experimentell durch die der Agarose-Lösung zugegebenen Menge an Cyclohexan-Phase bestimmt. Das Volumenverhältnis von Agarose-Lösung zu Cyclohexan-Phase im ersten Schritt der Emulsionsbildung betrug in den durchgeführten Versuchen einheitlich 2:1. Daher sollte theoretisch ein Porenanteil von 33.3 %, gemessen am gesamten Bead (Agarose-Material plus Superporen), gefunden werden. Der in Tabelle 14 angegebene, experimentell ermittelte Wert des Porenanteils von 32.8 %, stimmt damit sehr gut überein. Der minimal kleinere Porenanteil läßt sich dadurch erklären, daß nicht der gesamte Anteil der suspendierten Cyclohexan-Tröpfchen zur Porenbildung beiträgt. Bei der Ausbildung der Beads im zweiten Schritt liegt ein Teil der Cyclohexan-Einschlüsse bereits an der Oberfläche der suspendierten Beads und vereinigt sich mit der umgebenden CyclohexanPhase. 3.2.2 Variation des Bead-Durchmessers Eine Steuerung des Durchmessers der porösen Agarose-Beads durch die entsprechende Variation der Rührgeschwindigkeit ließ sich nicht erreichen. Entgegen der bei homogenen Beads über einen weiten Bereich wählbaren Rührgeschwindigkeit ist diese bei den superporösen Beads durch das angewandte Verfahren der doppelten Emulsionsbildung weitestgehend vorgegeben. Im zweiten Schritt der Bead-Herstellung (vergl. Abbildung 9) muß die Emulsion aus dem ersten Schritt so zu einer Emulsion zerteilt werden, daß eine gleichmäßige Ausbildung von porösen Beads gewährleistet ist. Eine zu hohe Rührgeschwindigkeit führt aufgrund der damit verbundenen hohen Scherkräfte zu einer Zerstörung der in Schritt 1 gebildeten Emulsion. Die damit erhaltene Mischung von Agarose-Lösung, Cyclohexan, Span 85 und Tween 80 wird zu einer einzigen, unbrauchbaren Emulsion „zerrührt“. In Abbildung 23 ist eine Aufnahme dieser Emulsion wiedergegeben, welche bei Anwendung einer Rührgeschwindigkeit von 700 U·min-1 erhalten wurde. Man erkennt deutlich, daß es weder zur Ausbildung von Beads noch zu einer verwendbaren Superporen-Struktur kommt. Abbildung 24 zeigt poröse Beads eines gelungenen Versuches bei einer Rührgeschwindigkeit von 500 U·min-1. Wie bei den homogenen Beads ist auch eine Mindestgeschwindigkeit einzuhalten, um eine ausreichend homogene Zerteilung der ersten Emulsion in Form von Beads zu erreichen. In der Praxis hat sich für das in Abschnitt 2.4.1 angegebene Verfahren ein nur sehr schmaler Bereich der möglichen Rührgeschwindigkeiten von 500 bis 600 U·min-1 ergeben. In Tabelle 15 sind einige Daten der unter diesen Voraussetzungen hergestellten superporösen Beads zusammengestellt. Die erhaltenen Mittelwerte des Bead-Durchmessers wiesen keinen statistisch signifikanten Unterschied auf (Abschnitt 2.10). 38 3 Ergebnisse Abbildung 23: Stereomikroskopische Aufnahme eines mißglückten Versuches zur Herstellung poröser Beads (1100 / 700 U·min-1) Abbildung 24: Stereomikroskopische Aufnahme von porösen Agarose-Beads (1000 / 500 U·min-1) Tabelle 15: Bead-Durchmesser und Verteilung von superporösen Agarose-Beads Rührgeschw. (Schritt 1 / 2) [U·min-1] 1000 / 600 1000 / 500 1100 / 600 1100 / 500 Bead-Durchmesser (d) [µm] md 265,4 250,0 253,7 269,0 s 119,4 141,5 123,0 132,4 n 100 100 100 100 Vertrauens -bereich (P = 95%) ± 23,7 ± 28,1 ± 24,4 ± 26,3 t-Test 0,83 0,85 0,68 0,98 3.2.3 Variation der Porosität Es hat sich gezeigt, daß auch die Größe der Superporen nicht über die jeweils angewendete Rührgeschwindigkeit gesteuert werden kann, da auch hier die Methode der doppelten Emulsionsbildung enge Grenzen für die Variation der Rührgeschwindigkeit setzt. Im ersten Schritt, bei der Herstellung der „Cyclohexan in Agarose“-Emulsion, muß die Rührgeschwindigkeit mindestens so hoch gewählt werden, daß eine Verteilung von ausreichend kleinen Cyclohexan-Tröpfchen in der Agarose-Lösung entsteht. Sind die Tropfen zu groß, tritt im weiteren Verlauf der Herstellung eine zu frühe Tropfenkoaleszenz auf, so daß das eingeschlossene Cyclohexan bereits vor der endgültigen Formierung der Beads im nächsten Verfahrensschritt aus der Agarose heraustritt. Als Folge hiervon würden nahezu homogene Beads entstehen. Wählt man die Rührgeschwindigkeit hingegen zu hoch, erhält man eine feine, zu stabile Verteilung des Cyclohexans in der Agarose-Lösung, welche bei der Bead-Bildung bestehen bleibt und nicht zur Ausbildung der Superporen führt. In den durchgeführten Versuchen haben sich Rührgeschwindigkeiten von 1000 bis 1200 U·min-1 bewährt. Es läßt sich lediglich der Porenanteil in den Beads durch die zugegebene Menge an Cyclohexan im ersten Emulsions-Schritt beeinflussen. (siehe hierzu auch 3.2.1) 39 3 Ergebnisse 3.2.4 Löslichkeitsverhalten der porösen Beads Zur Absicherung, daß das Hydrogel-Netzwerk der superporösen Beads im wäßrigen Medium beständig ist und die Beads sich nicht auflösen, wurde das Löslichkeitsverhalten der AgaroseBeads gemäß Abschnitt 2.6.4 überprüft. Dabei wurden die in Tabelle 16 zusammengestellten Ergebnisse erhalten. Tabelle 16: Löslichkeitsverhalten der porösen Beads Zeit [h] Bead-Durchmesser (d) [µm] Vertrauens -bereich t-Test 0 md 197,3 s 89,1 n 100 (P = 95%) ± 17,7 0,5 242,2 101,6 100 ± 20,2 3,32 24 243,9 113,3 100 ± 22,5 0,11 48 243,0 115,3 100 ± 22,9 0,06 120 244,9 101,0 100 ± 20,0 0,12 288 245,0 97,4 100 ± 19,3 0,01 Wie schon bei den homogenen Beads (Abschnitt 3.1.3) zeigte der Bead-Durchmesser auch bei den superporösen Agarose-Beads im Verlauf des Versuches keine signifikanten Unterschiede. Eine Auflösung der superporösen Beads in wäßriger Phase ist somit nicht feststellbar. Der signifikante Anstieg (Abschnitt 2.10) der mittleren Beadgröße innerhalb der ersten 30 Minuten ist, wie bereits bei den homogenen Beads, sehr wahrscheinlich auf einen Quellungsprozeß der verwendeten und zuvor bereits einige Tage gelagerten Beads zurückzuführen. 40 3 Ergebnisse 3.3 Quellungsverhalten der Agarose-Beads Für die Untersuchung des Quellungsverhaltens der Agarose-Beads wurden, mit der in Abschnitt 2.6.5 vorgestellten Quellungsapparatur, Messungen des Volumenquellungsgrades in Abhängigkeit von der Zeit vorgenommen. Mit Hilfe dieses Parameters kann beurteilt werden, in welchem Ausmaß die Agarose-Beads in der Lage sind reversibel Wasser aus ihrem GelNetzwerk abzugeben und wieder aufzunehmen. Der Versuch zeigt auch, wie hoch das Wasserbindungsvermögen der Beads ist und wieviel Wasser selbst bei mehrtägigem Trocknen im Gel zurückgehalten werden kann. Die Versuche wurden für homogene und poröse Agarose-Beads an jeweils zwei ausgewählten Proben durchgeführt. Hierzu wurden Proben der Agarose-Beads bei 30 °C acht Tage getrocknet. Für die Messungen wurden jeweils 0.5 g getrocknetes Bead-Material eingesetzt. Die Datenakquisition erfolgte mit einer Meßrate von 1 Hz. Die Daten der verwendeten Agarose-Beads sowie die erhaltenen Ergebnisse sind in Tabelle 17 zusammengestellt. Tabelle 17: Volumenquellungsgrade von Agarose-Beads Probe homogene Beads (500 U·min-1) homogene Beads (900 U·min-1) poröse Beads (1100/600 U·min-1) poröse Beads (1100/500 U·min-1) mittlerer Durchmesser [µm] 326,1 ± 149,9 159,3 ± 69,1 253,7 ± 123,0 269,0 ± 132,4 GleichgewichtsVolumenquellungsgrad QV,e 5,92 5,87 7,07 7,18 Das Quellungsverhalten der Agarose-Beads zeigt für alle vier untersuchten Proben qualitativ einen vergleichbaren Verlauf über den Versuchzeitraum. Bereits nach ca. 10 Minuten ist der Zustand der Gleichgewichtsquellung erreicht. Es wird deutlich, daß die porösen Agarose-Beads im Vergleich zu den homogenen Agarose-Beads im Gleichgewicht einen höheren Volumenquellungsgrad aufweisen. Um die im Vorhergehenden gemachten Aussagen zu verifizieren, wurde für die gefundenen Werte ein „t-Test für den Vergleich von Paardifferenzen“ durchgeführt. Die dabei berechneten Ergebnisse und die erhaltenen Aussagen sind in Tabelle 18 zusammengestellt. Es wird dabei bestätigt, daß zwischen den Werten des Volumenquellungsgrades für homogene und poröse Beads ein hochsignifikanter Unterschied besteht (Abschnitt 2.10). Für die paarweise verglichenen Werte der homogenen bzw. porösen Beads mit den Beads der gleichen Art kann kein signifikanter Unterschied festgestellt werden. Die gefundenen Unterschiede können hier als zufällig angesehen werden. 41 3 Ergebnisse Tabelle 18: t-Test für die Unterschiede des Volumenquellungsgrades Vergleich mittlere Paardifferenz t-Test gefolgerter Unterschied mD sD n homogene Beads untereinander 0,140 0,106 720 1,893 nicht signifikant poröse Beads untereinander 0,005 0,113 720 1,304 nicht signifikant 1,220 0,235 720 139,8 hochsignifikant 1,074 0,230 720 129,2 hochsignifikant -1 poröse Beads (1100/600 U·min ) mit homogenen Beads (500 U·min1 ) poröse Beads (1100/500 U·min-1) mit homogenen Beads (900 U·min1 ) In Abbildung 25 sind zum Vergleich jeweils die Volumenquellungsgrade von homogenen und porösen Agarose-Beads gegen die Zeit aufgetragen. 8,0 7,0 Volumenquellungsgrad QV(t) 6,0 5,0 4,0 3,0 poröse Beads (1100/500 U/min) homogene Beads (900 U/min) 2,0 1,0 0 1 2 3 4 5 6 7 8 9 10 11 12 Zeit [min] Abbildung 25: Volumenquellungsgrade ausgewählter Agarose-Beads, (Vergleich homogene / poröse Beads) 42 3 Ergebnisse 3.4 Herstellung von Schichten aus Agarose-Beads Um eine realitätsnahe Modellierung und eine praxisgerechte Anwendbarkeit des ModellBiofilms auf Basis des Bead-Modells zu ermöglichen, ist eine Fixierung der Gel-Beads auf einem Trägermaterial notwendig. Dadurch wird die flächige Ausbreitung eines Biofilms nachempfunden. Die Dicke der Schichten läßt sich dabei prinzipiell einstellen (s. u.). Darüber hinaus wird durch die entstehenden Zwickelräume das gewünschte Porensystem modelliert. Im praktischen Versuch zeigten sich beide in Abschnitt 2.8.1 genannten Techniken als anwendbar. Die Fixierung einer einlagig auf einem Glas-Objektträger ausgestrichenen Bead-Schicht durch kurzes hindurchziehen durch eine Bunsenbrennerflamme erwies sich als eine einfache und schnell durchführbare Methode. Wie bereits erwähnt ist hierbei jedoch mit einer Schädigung etwaig eingebetteter Mikroorganismen zu rechnen, so daß diese Vorgehensweise nur eingeschränkt, z.B. für physikalische Untersuchungen (Rheologie, Diffusionsmessungen etc.), nutzbar ist. Mit dieser Technik können Schichtdicken im Bereich von ca. 70 bis 450 µm erhalten werden, was dem Bead-Durchmesser der verwendeten Beads entspricht. Das Anheften von flächig auf einem Glas-Objektträger ausgestrichenen Agarose-Beads durch Auftropfen einer verdünnte Agarose-Lösung ist im Gegensatz zu der vorherigen Methode für ein- und mehrlagige Bead-Schichten einsetzbar. Es können Schichten von ca. 70 µm bis hin zu mehreren Millimetern angefertigt werden. Außerdem ist kein beeinträchtigender Einfluß auf die immobilisierten Bakterien zu erwarten, da die maximal auftretende Temperaturbelastung bei der Fixierung nicht über der Temperatur bei der Bead-Herstellung liegt und darüber hinaus nur sehr kurzzeitig auftritt. Im Abbildung 26 ist ein konfokaler Bildschnitt durch eine Bead-Schicht dargestellt, welcher durch Fixierung mit einer 0.5 %-igen Agarose-Lösung hergestellt wurde. Es läßt sich zwischen den eigentlichen Beads das zur Fixierung aufgetropfte Agarose-Material erkennen, welches die einzelnen Beads an ihren Kontaktstellen aneinander heftet (siehe Pfeile). Abbildung 26: konfokaler Schnitt durch eine fixierte Bead-Schicht, (CLSM, gefärbt mit Kongorot) (Pfeile: zur Fixierung aufgetropftes Agarose-Material an den Kontaktstellen) 43 3 Ergebnisse 3.5 Herstellung superporöser Agarose-Körper Das zuvor beschriebene Biofilm-Modell aus Schichten von fixierten Agarose-Beads zeigt in der Modellierung von flächigen Biofilmen noch einige Schwächen. So müssen die einzelnen Beads nachträglich zu einer Schicht angeordnet und fixiert werden. Diese Fixierung stellt dabei den wesentlichen Nachteil dieser Methode dar, da eine Hitzefixierung etwaig eingebettete Organismen schädigen kann (vergl. hierzu Abschnitt 3.4), bzw. eine Fixierung mit AgaroseLösung im Fall von porösen Agarose-Beads einen Teil der Poren wieder verschließen kann. Als Alternative zur Herstellung des porösen Agarose-Materials in Bead-Form wurde daher die Modellierung von zusammenhängenden, superporösen Agarose-Körpern und -Schichten durchgeführt. Diese superporösen Agarose-Materialien weisen bereits ein weitreichendes Porensystem auf und können auch in größeren Dimensionen hergestellt werden. In der vorliegenden Arbeit wurden sie in Form von beliebig dimensionierten Stücken bis in den Zentimeterbereich oder als Film von ca. 1 mm Dicke und mehr hergestellt. Die Durchführung ist in Abschnitt 2.4.3 beschrieben. Im Folgenden sind Aufnahmen der hergestellten Proben zusammengestellt. Hierfür wurden Schnitte des Materials mit einer Stärke von etwa 0.5 mm mit einem Skalpell angefertigt und stereomikroskopisch betrachtet. In Abbildung 27 ist ein Schnitt eines superporösen Agarose-Stücks wiedergegeben. Man erkennt deutlich die poröse Struktur des Hydrogels. Bei den teilweise sichtbaren, rundlichen Gebilden handelt es sich um eingeschlossene Blasen. Diese sind jedoch nicht fest in die Gelmatrix eingebunden, sondern können durch äußeres Einwirken innerhalb des Gels bewegt werden. Dies ist ein weiterer Hinweis auf die offenporige, flexible Struktur des Agarose-Gels. Der in Abbildung 28 gezeigte, bei der Präparation „zerrissene“ Schnitt, bestätigt ebenfalls die zusammenhängende aber dennoch poröse und von Kanälen durchzogene Struktur des hergestellten Materials. Abbildung 27: Schnitt eines porösen Agarose-Stücks (Stereomikroskop, gefärbt mit AO, Hellfeld) Abbildung 28: „Zerrissener“ Schnitt eines porösen Agarose-Stücks (Stereomikroskop, gefärbt mit Methylblau, Hellfeld) 44 3 Ergebnisse Ein Beispiel für einen gegossenen, porösen Agarose-Film ist in Abbildung 29 zu sehen. Der Film weist ebenfalls die typische Superporen-Struktur innerhalb des Hydrogel-Netzwerkes auf. Diese erstreckt sich von der Basis des Filmes bis an die Oberfläche. Die Aufnahme- und Beleuchtungstechnik führt dazu, daß das hervorstehende Agarose-Material an der Schnittfläche hell erscheint und die Poren im „Schatten“ liegen. Die Poren selbst sind mit Wasser gefüllt und zeigen nur einen geringen Kontrast. In Abbildung 29 sind einige Porenöffnungen zur Verdeutlichung mit roten Pfeilen markiert. Lediglich an der Oberfläche selbst ist eine dichtere, feiner strukturierte Porenmatrix zu erkennen. Dies läßt sich auf die Vorgänge beim Herstellungsprozeß zurückführen. Während der Formierung der Kanäle durch das Austreten der eingeschlossenen Cyclohexan-Tröpfchen bildet sich auf dem Agarose-Film ein Überstand an Cyclohexan. Anders als bei den porösen Beads, wird die Wanderrichtung der Tropfen hier hauptsächlich durch den Auftrieb des Cyclohexans bestimmt. Die an der entstehenden Phasengrenzfläche, zwischen Agarose und Cyclohexanüberstand, herrschenden Bedingungen sind vermutlich für die Veränderung der GelStruktur verantwortlich. Abbildung 29: Querschnitt eines porösen Agarose-Films (Stereomikroskop, gefärbt mit AO, Hellfeld) Die Pfeile markieren beispielhaft einige Porenöffnungen 45 3 Ergebnisse 3.6 Einbettung von Bakterien in Beads Die vorhergehend beschriebenen Agarose-Beads und -Schichten simulieren näherungsweise die EPS-Matrix eines Biofilms. Sie sind jedoch nur für bestimmte Fragestellungen, z.B. rheologische Untersuchungen, anwendbar. Für Untersuchungen unter Einbeziehung biologischer Effekte ist es nötig, auch die Bakterien eines Biofilms zu berücksichtigen. Das bedeutet, daß mit einer geeigneten Methode Bakterienzellen in die Agarose-Matrix eingebettet werden müssen. Als Ausgangsmaterial für die Immobilisierung von Bakterien in Agarose-Beads fand eine Zellsuspension des nichtmukoiden Stammes SG81R1 von P. aeruginosa Anwendung, welche entsprechend Abschnitt 2.7.1 vorbereitet wurde. Es wurde eine Zelldichte der Stammsuspension von (4,7 0,5)·109 Zellen/ml ermittelt. 3.6.1 Überprüfung der Vitalität der Bakterien im Verlauf der Bead-Herstellung Um die Auswirkung der einzelnen Reaktionseinflüsse auf die Vitalität der Bakterien zu untersuchen, wird eine modellhafte Überprüfung anhand einer Bakteriensuspension vergleichbarer Zelldichte durchgeführt. Dazu werden Aliquote der Bakteriensuspension (je 1 ml) gezielt den verschiedenen Einflußfaktoren ausgesetzt und die Schädigung der Bakterien durch Anfärbung mit SYTO 9 und PI in Kombination (LIVE/DEAD® BacLight™ Bacterial Viability Kit, Molecular Probes) und Auswertung am CLSM quantifiziert. Für diesen Versuch wird eine Verdünnung der Bakterien-Stammsuspension von 1:100 in 0.14 M NaCl verwendet. Dies entspricht einer Zelldichte von 4,7·107 Zellen/ml wie sie auch in etwa bei der Einbettung von Bakterien in Agarose-Beads erhalten wird. Die bestimmten Werte sind in Tabelle 19 zusammengestellt. Die angegebenen Zellzahlen sind die ausgezählten Zellen, welche sich in einem mit dem CLSM aufgenommenen Bildausschnitt (Gesichtsfeld) befanden. Tabelle 19: Schädigung der Bakterien in Abhängigkeit von den Bedingungen bei der Bead-Herstellung Einwirkender Reaktionsfaktor Zahl der SYTO 9 gefärbten Zellen*) 421 12 429 19 441 13 Zahl der PI gefärbten Zellen*) 81 4 85 6 93 4 Anteil der PI t-Test gefärbten Zellen unbehandelt 19,2 0,3 % Suspendieren mit 2 ml 19,8 0,9 % 1,05 Suspendieren mit 2 ml 20,9 1,3 % 2,02 Cyclohexan und 0,1 ml Span 85 20 Minuten bei 47 °C 424 11 146 9 34,4 1,8 % 14,52 *) Zellzahlen in einem CLSM-Gesichtsfeld bei 200-facher Vergrößerung; jeweils Mittelwerte aus drei Messungen Lediglich zwischen der mit Cyclohexan und Span 85 behandelten Probe und der wärmebehandelten Probe konnte ein signifikanter Unterschied festgestellt werden (Abschnitt 2.10). Es zeigt sich daher, daß fast ausschließlich die Temperaturbelastung eine Schädigung der Zellmembran hervorruft. Die Einwirkung der zur Herstellung der Beads notwendigen Chemikalien führt hingegen nicht zu einer signifikanten Veränderung der Membranpermeabilität. 46 3 Ergebnisse 3.6.2 Agarose-Beads mit immobilisierten Bakterien Zur Einbettung von P. aeruginosa in das Agarose-Material wurde entsprechend den in Abschnitt 2.7.2 genannten Vorschriften vorgegangen. Die Herstellung der Agarose-Beads in homogener und poröser Form, sowie von porösen Agarose-Stücken ließ sich bei Zugabe der Bakteriensuspension in Bezug auf die Emulsionsbildung ohne sichtbare Veränderungen durchführen. Es zeigten sich ebenfalls keine Auswirkungen auf die Beadgrößenverteilung oder die Porenausbildung. Die Beads mit immobilisierten Bakterien wurden wie in Abschnitt 2.7.3 beschrieben, mit SYTO 9 und PI in Kombination (LIVE/DEAD® BacLight™ Bacterial Viability Kit, Molecular Probes), bzw. mit CTC angefärbt. Mit dem CLSM erfolgte eine Aufnahme von konfokalen Bildschnitten durch die Beads, um die Verteilung und den physiologischen Zustand der Zellen zu bestimmen. Einige typische Aufnahmen sind in Abbildung 30 und Abbildung 31 gezeigt. Die Auszählungsergebnisse der Fluoreszenzfärbungen sind in Tabelle 20 angegeben. Abbildung 30: P. aeruginosa SG81R1 in einem homogenen Agarose-Bead, (CLSM, grün: SYTO 9 / rot: PI) Abbildung 31: P. aeruginosa SG81R1 in einem porösen Agarose-Bead, (CLSM, grün: SYTO 9 / rot: PI) Tabelle 20: Zustand der Bakterien nach dem Einbettungsprozeß Agarose- SYTO 9 gefärbte Beads Zellen*) [Zellen/mm²] PI gefärbte Zellen*) [Zellen/mm²] homogen 3714 447 1247 82 Anteil der PI gefärbten Zellen*) [%] 33,7 2,1 poröse 2633 398 1004 167 38,1 3,3 CTC gefärbte Zellen*) [Zellen/mm²] 2578 233 1780 86 Anteil der CTC gefärbten Zellen*) [%] 69,4 4,6 67,6 3,1 *) jeweils Mittelwerte aus drei Messungen 47 3 Ergebnisse Rein qualitativ konnte festgestellt werden, daß die immobilisierten Bakterien in allen Fällen gleichmäßig über die Hydrogel-Matrix verteilt waren. Die Bakterien lagen hierbei in der Regel als vereinzelte Zellen vor. Eine signifikant unterschiedliche Verteilung der Zellen zwischen homogenen und porösen Beads konnte nicht festgestellt werden. Der Anteil an Propidiumiodid gefärbten Zellen in den untersuchten Beads liegt mit 33.7 % bei den homogenen Beads, bzw. 38.1 % bei den porösen Beads in einem ähnlichen Bereich. Es zeigt sich hierbei eine gute Übereinstimmung mit dem durchgeführten Vorversuch, in dem die Schädigung der Bakterien durch die einzelnen Reaktionseinflüsse untersucht wurde (Abschnitt 3.6.1). Dort wurde ein Anteil von 34.4 % an Propidiumiodid gefärbten Zellen in einer Zellsuspension gefunden, nachdem diese für 20 Minuten auf 47 °C erwärmt wurde. Für den Anteil stoffwechselaktiver Zellen (CTC-positiv) ergibt sich ebenfalls eine gute Übereinstimmung (69.4 bzw. 67.6 %), die auch mit den Ergebnissen der PI-Färbung gut korreliert. Es bestätigt sich demnach, daß die Schädigung der Bakterien bei der Einbettung durch die hierbei nötige Reaktionstemperatur verursacht wird. Eine Modifikation des Herstellungsverfahrens ist daher wünschenswert (vergl. hierzu 4.2.1). 3.6.3 Kultivierung von Bakterien in Agarose-Beads Um die Kultivierbarkeit der immobilisierten Bakterien und die Art der dabei entstehenden Zellanordnung in der Hydrogel-Matrix zu untersuchen, wurden Proben des Bead-Materials mit eingebetteten Bakterien in ein Nährmedium gegeben und bebrütet. Für die Kultivierung der immobilisierten Bakterien wurde ein Medium von 200 mg/l Nutrient Broth ausgewählt. Es wurde eine Färbung mit SYTO 9 und PI in Kombination (LIVE/DEAD® BacLight™ Bacterial Viability Kit, Molecular Probes) durchgeführt und anschließend konfokale Bildschnitte der Agarose-Beads am CLSM aufgenommen (Abschnitt 2.7.3). Die erhaltenen Ergebnisse sind in Tabelle 21 wiedergegeben und in Abbildung 32 graphisch aufgetragen. Tabelle 21: Kultivierung in Nutrient Broth-Medium (200 mg/l) Zeit [h] 0 24 48 72 96 SYTO 9 gefärbter Flächenanteil*) [%] 0,37 0,76 1,68 3,69 6,25 0,083 0,076 0,251 0,935 0,890 PI gefärbter Flächenanteil*) [%] 0,091 0,075 0,112 0,120 0,135 0,0243 0,0056 0,0314 0,0172 0,0023 *) jeweils Mittelwerte aus drei Messungen Bei der Auswertung der Bildschnitte wurde jeweils die Fläche des Querschnittes durch ein Bead ausgemessen und die Fläche der darin enthaltenen angefärbten Zellen mit Hilfe der LSMSoftware bestimmt. Die Angabe der fluoreszierenden Fläche erfolgt als prozentualer Wert in Bezug auf die Beadfläche um eine Vergleichbarkeit zu gewährleisten. Ein Auszählen der Zellen war in diesem Versuch nicht möglich, da es hier zur Ausbildung von Zellaggregaten gekommen ist, welche sich unter den möglichen Aufnahmebedingungen nicht in Einzelzellen differenzieren ließen. 48 3 Ergebnisse 8,0% PI 7,0% SYTO 9 Flächenanteil gefärbter Zellen 6,0% 5,0% 4,0% 3,0% 2,0% 1,0% 0,0% 0 12 24 36 48 60 72 84 96 108 Zeit [h] Abbildung 32: Zellzahlen nach Kultivierung in Nutrient Broth-Medium (200 mg/l) In Abbildung 33 ist eine Aufnahme der SYTO 9 und Propidiumiodid gefärbten Zellen in einem homogenen Bead nach 96 h Bebrütung bei 30 °C in Nutrient Broth-Medium (4 g/l) gezeigt. Abbildung 34 zeigt ein in gleicher Weise aufgenommenes Bild eines porösen Beads welches unter den selben Bedingungen bebrütet wurde. Es wird deutlich, daß im Fall der homogenen Agarose-Beads ein Zellwachstum überwiegend im äußeren Bereich der Beads stattfindet. Bei den porösen Beads ist hingegen ein relativ gleichmäßiges Wachstum über die gesamte Schnittfläche zu beobachten. Bei einer in gleicher Weise durchgeführten Bebrütung in Hefeextrakt (160 mg/l, 30 °C) über 6 Tage konnte nur eine geringe Zunahme der Gesamtzellzahl (SYTO 9) um den Faktor 2.4 festgestellt werden. Die stoffwechselaktiven Zellen (CTC-positiv) haben in dieser Zeit in vergleichbarem Maße zugenommen (Faktor 2.2). Die Zahl der PI gefärbten Zellen blieb konstant. Dies läßt darauf schließen, daß es in diesem Fall aufgrund einer unzureichenden Nährstoffzufuhr oder fehlender Stoffwechseledukte nur zu einer geringen Vermehrung der Bakterien gekommen ist. Da die Zellzahlen der SYTO 9 und CTC gefärbten Bakterien während der Bebrütung um etwa die gleiche Größenordnung zugenommen haben, ist nicht von einer Revitalisierung bereits von Beginn an eingebetteter aber atmungsinaktiver Zellen auszugehen. 49 3 Ergebnisse Abbildung 33: homogenes Bead mit P. aeruginosa nach Kultivierung in NB-Medium (200 mg/l, 96 h, 30 °C), (CLSM, grün: SYTO 9 / rot: PI) Abbildung 34: poröses Bead mit P. aeruginosa nach Kultivierung in NB-Medium, (200 mg/l, 96 h, 30 °C) (CLSM, grün: SYTO 9 / rot: PI) 3.7 Schutzwirkung der Einbettung Es ist bekannt, daß Mikroorganismen durch ihre Einbettung in der EPS-Matrix im Biofilm einen Schutz vor toxischen Substanzen erfahren (LeChevallier et al., 1988; Grobe et al., 1994; Morton et al., 1998). Es soll daher überprüft werden, ob das entwickelte Biofilm-Modell diese Schutzwirkung, z.B. gegen ein Biozid, ebenfalls aufweist. Für diesen Versuch wurde Natriumhypochlorit als Biozid ausgewählt, da es ein typisches Desinfektionsmittel in der Praxis ist und bereits gute Kenntnisse über die Wirkung auf Bakterien vorliegen (Wingender et al., 1998; Grobe, 1996). Zur Untersuchung der Membranschädigung von P. aeruginosa SG81R1 durch ein Biozid wurden wie in Abschnitt 2.9.1 beschrieben, eine Zellsuspension sowie in Agarose-Beads immobilisierte Bakterien mit Natriumhypochlorit-Lösung behandelt. Für die Bestimmung des Membranzustandes nach der Einwirkung wurde eine Färbung mit SYTO 9 und PI in Kombination (LIVE/DEAD® BacLight™ Bacterial Viability Kit, Molecular Probes) durchgeführt. Die Auswertung wurde für die Zellsuspension mit einem FluoreszenzMikroskop und für die Agarose-Beads mit dem CLSM vorgenommen. In beiden Fällen wurde der Anteil Propidiumiodid gefärbter Zellen ermittelt. Die daraus erhaltenen Daten sind in Tabelle 22 zusammengetragen. Abbildung 35 zeigt die Zunahme membrangeschädigter Zellen im Falle der Zellsuspension und der porösen Beads zum Vergleich in graphischer Form. 50 3 Ergebnisse Tabelle 22: Membranschädigung durch Natriumhypochlorit (0,5 mg/l freies Chlor) Zeit [min] 0 0.5 2 4 6 10 20 30 1) Zellsuspension 11,8 0,9 49,7 2,9 60,3 3,2 64,8 2,6 69,0 2,1 71,0 1,5 81,3 1,4 Anteil PI gefärbter Zellen [%] homogene Agarose-Beads2) poröse Agarose-Beads2) 12,0 1,2 13,4 1,0 48,1 3,3 40,2 4,1 60,2 4,2 49,3 1,6 63,0 3,2 57,0 2,2 70,2 4,4 59,3 4.7 75,1 5,5 71,9 2,6 78,7 2,3 79,1 4,5 1) jeweils Mittelwerte aus 20 Messungen 2) jeweils Mittelwerte aus fünf Messungen 100 membrangeschädigte Zellen [%] 90 80 70 60 50 40 30 poröse Beads 20 Zellsuspension 10 0 0 5 10 15 Zeit [min] 20 25 30 Abbildung 35: Zunahme des Anteils membrangeschädigter Zellen nach Behandlung mit NatriumhypochloritLösung, (0.5 mg/l freies Chlor) Aus dem Diagramm wird deutlich, daß für die untersuchten Fälle (Zellsuspension und poröse Beads) vergleichbare Inaktivierungskinetiken gefunden werden. Ausgehend von den anfänglich im Bereich von 11.8 bis 13.4 % liegenden Anteilen membrangeschädigter Zellen wird nach 30 Minuten in allen Fällen eine ähnliche Zunahme auf einen Anteil von 79.1 bis 81.3 % PI gefärbter Bakterien beobachtet. Lediglich im Bereich von 0.5 bis 20 Minuten ließ sich ein signifikant unterschiedliches Verhalten bei der Einwirkung von Hypochlorit auf die beiden Proben erkennen (Abschnitt 2.10). Dabei ließ sich anfänglich eine geringere Wirkung auf die eingebetteten Zellen feststellen, welche vermutlich auf eine Schutzwirkung der Gelmatrix zurückzuführen ist (vergl. hierzu Abschnitt 4.3). 51 3 Ergebnisse Für die Zehrung des freien Chlors über den Reaktionszeitraum haben sich bei der photometrischen Messung mittels DPD-Methode die in Tabelle 23 zusammengestellten Werte ergeben. Diese sind in Abbildung 36 graphisch aufgetragen. Tabelle 23: Zehrung des freien Chlors Zeit [min] 0 1 2 5 10 20 30 freies Chlor [% der Anfangskonzentration (0.5 mg/l)] Zellsuspension poröse Agarose-Beads poröse Agarose-Beads mit Bakterien ohne Bakterien 100,0 100,0 100,0 52,9 56,1 94,4 37,9 33,8 25,8 90,3 26,5 18,2 83,3 13,2 12,1 80,6 10,3 4,6 77,8 100% porösen Beads mit Bakterien Zellsuspension Agarose-Beads ohne Bakterien Chlorkonzentration (% der Startkonzentration) 90% 80% 70% 60% 50% 40% 30% 20% 10% 0% 0 5 10 15 20 25 30 Zeit [min] Abbildung 36: Zeitabhängigkeit der Zehrung des freien Chlors (100% entsprechen einer Anfangskonzentration von 0.5 mg/l freiem Chlor) Auch hier zeigt sich zwischen Bakterien in Suspension und immobilisiert in Agarose-Beads eine gute Übereinstimmung des Chlorzehrungs-Verhaltens. Die als Vergleich vermessenen AgaroseBeads ohne Bakterien verursachen ebenfalls eine Zehrung des freien Chlors, welche aber erheblich geringer als die durch die Bakterien hervorgerufene Zehrung ist. 52 4 Diskussion 4 Diskussion 4.1 Herstellung und Charakterisierung von Agarose-Beads 4.1.1 Bead-Herstellung Im Rahmen der vorliegenden Arbeit konnten, wie in Abschnitt 3.1 und 3.2 gezeigt wurde, homogene sowie superporöse Agarose-Beads reproduzierbar und in ihren Eigenschaften steuerbar hergestellt werden. Für die homogenen Agarose-Beads hat sich ergeben, daß der Bead-Durchmesser über die angewendete Rührgeschwindigkeit, daher über die aufgebrachten Scherkräfte, gezielt beeinflußt werden konnte. Mit steigender Rührgeschwindigkeit ging eine Abnahme des mittleren BeadDurchmessers einher. Dies entspricht den von Gustavsson (1998) gewonnenen Erkenntnissen, wobei dort für den Zusammenhang zwischen Bead-Durchmesser und Rührgeschwindigkeit nur eine entsprechende Tendenz angegeben wurde. Im Unterschied zu Gustavsson (1998) konnte eine Steuerung des Durchmessers der superporösen Beads und der Porengröße über die Rührgeschwindigkeit nicht erreicht werden (vergl. hierzu Abschnitt 3.2.2). Hierzu sind evtl. Modifikationen am Reaktoraufbau, insbesondere am Rührer nötig, um über das gesamte Reaktionsvolumen definiert gleichmäßigere Scherkräfte aufbringen zu können. Darüber hinaus konnten homogene Agarose-Beads mit unterschiedlichen Konzentrationen (0.5 bis 6 % w/v) erzeugt werden (Abschnitt 3.1.2). Agarose- Abbildung 37 zeigt eine Aufnahme der von Gustavsson und Larsson (1996) für chromatographische Anwendungen hergestellten Beads, welche in ihrem mikroskopischen Erscheinungsbild deutliche Ähnlichkeiten mit den Beads aus der eigenen Arbeiten (vergl. Abschnitt 3.2.1) aufweisen. Die quantitativen Merkmale der hergestellten porösen AgaroseBeads stimmten ebenfalls weitestgehend mit den von Gustavsson und Larsson (1996) dargestellten Beads überein. In Tabelle 24 sind einige Werte gegenübergestellt. Abbildung 37: Mikroskopische Aufnahme von homogenen und porösen Beads (Gustavsson und Larsson, 1996) 53 4 Diskussion Tabelle 24: Vergleich von Eigenschaften von porösen Agarose-Beads Eigenschaft Bead-Durchmesser [µm] Porengröße [µm] Porenanteil [%] vorliegende Arbeit 150 - 400 27,7 ± 14,3 32,8 ± 3,1 Gustavsson und Larsson (1996) 200 - 500 ~30 ~40 Es wird deutlich, daß sowohl der Durchmesser der Beads als auch Durchmesser und Anteil der Poren in der selben Größenordnung liegen. Hierbei muß jedoch beachtet werden, daß der von Gustavsson und Larsson (1996) benutzte Reaktoraufbau nicht beschrieben wurde und durchaus von dem in dieser Arbeit verwenden abweichen kann. Bei der Bewertung der in Tabelle 24 zusammengestellten Daten muß außerdem berücksichtigt werden, daß die im Rahmen dieser Arbeit ermittelten Werte für Porengröße und –Anteil unter Zuhilfenahme eines CLSM bestimmt wurden. Dadurch war eine eingehende Betrachtung und Vermessung der Porenstruktur auch im Inneren der Beads möglich. Gustavsson und Larsson (1996) haben lediglich eine Betrachtung und Ausmessung am Lichtmikroskop vorgenommen. 4.1.2 Löslichkeits- und Quellungsverhalten In Abschnitt 3.1.3 bzw. 3.2.4 wurde gezeigt, daß weder homogene noch poröse Agarose-Beads erkennbare Auflösungserscheinungen in deionisiertem Wasser zeigen. Dies stimmt mit den Ergebnissen der Quellungsversuche überein, in denen die Volumenquellungsgrade gegen einen Gleichgewichtswert liefen und ebenfalls keine weiteren Auflösungserscheinungen festgestellt wurden. Für die Messungen des Volumenquellungsgrades (Abschnitt 3.3) der Agarose-Beads hat sich ergeben, daß die homogenen Beads unabhängig von ihrem mittleren Bead-Durchmesser auf den gleichen Gleichgewichts-Volumenquellungsgrad aufquellen. Daraus läßt sich folgern, daß sowohl bei kleinem als auch bei großem Bead-Durchmesser, auch unter Berücksichtigung der Streubreite des Bead-Durchmessers, der Anteil des Zwickelvolumens identisch ist. Dies entspricht auch der Erwartung, da bei angenommener „dichtester Kugelpackung“ das entstehende Zwickelvolumen unabhängig vom Kugeldurchmesser einen konstanten Anteil hat. Für die porösen Beads wurde ein um etwa 20 % höherer Gleichgewichts-Volumenquellungsgrad ermittelt. Da, wie bereits zuvor angegeben, der Bead-Durchmesser keinen Einfluß auf das Quellungsverhalten hat, muß es eine Ursache geben, die in der Anwesenheit der Poren begründet ist. Eine sinnvolle Erklärung für den erhöhten Volumenquellungsgrad im Vergleich zu den homogenen Beads ist, daß die Porenstruktur im getrockneten Zustand „zusammenfällt“ und erst während des Quellungsvorganges wieder ihre ursprünglichen Ausmaße annimmt. Bei gleichem Ausgangsvolumen an getrockneten Beads entsteht so im Fall der porösen Beads ein höheres Endvolumen, auch wenn das Quellungsausmaß des eigentlichen Agarose-Gels vergleichbar ist, da das Volumen der dann mit Wasser gefüllten Poren hinzukommt. Die für die homogenen Beads ermittelten Gleichgewichts-Volumenquellungsgrade liegen im Bereich von in der Literatur angegebenen Werten. So berichten Rochas et al. (1996), daß entquollene (getrocknete) Agarose-Gele auf das 5-fache Volumen aufgequollen werden können und dabei ihr ursprüngliches Volumen wiedererlangen. 54 4 Diskussion 4.2 Einbettung, Kultivierung und Untersuchung von Bakterien in Agarose-Beads 4.2.1 Immobilisierung von Bakterien in Agarose-Beads Durch Zugabe von entsprechenden Zellsuspensionen bei der Bead-Herstellung ließen sich homogene und poröse Agarose-Beads mit eingebetteten Bakterien auf einfache Weise herstellen. Die Zellen lagen hierbei in der Regel vereinzelt und homogen verteilt vor. Die Untersuchung des physiologischen Zustandes der Zellen nach der Einbettungsprozedur wurde anhand des Anteils membrangeschädigter Zellen (quantifiziert über Färbung mit PI) und des Anteils atmungsinaktiver Zellen (quantifiziert über Färbung mit CTC; CTC-negative Zellen) bestimmt. Es ergab sich hierbei, daß etwa 35 % der zugegebenen Zellen, bezogen auf die Gesamtzellzahl (quantifiziert über Färbung mit SYTO 9), geschädigt waren. Die Werte für eine über Färbung mit SYTO 9 und PI in Kombination nachgewiesene Membranschädigung korrelierten gut mit dem als stoffwechselinaktiv (CTC-negativ) bestimmten Bakterienanteil (Abschnitt 3.6.2). Im Vergleich dazu, wurde bei den eingesetzten, unbehandelten Bakteriensuspensionen ein Anteil membrangeschädigter Zellen von ca. 10 % festgestellt. Es wird vermutet, daß die deutliche Zunahme membrangeschädigter Zellen, bzw. die Abnahme atmungsaktiver Zellen auf die Reaktionseinflüsse bei der Herstellungsprozedur zurückzuführen ist. Als Anhaltspunkt hierfür sind die Arbeiten von Heinemeyer und Geuenich (1997) zu nennen, in der die Temperaturabhängigkeit der Wiederfindung bei der Keimzahlbestimmung mittels Gußplattenverfahren untersucht wurde. Einige Ergebnisse, die Heinemeyer und Geuenich (1997) bei der Bestimmung der Koloniezahlen einer E. coli-Suspension unter Verwendung von Gußagar verschiedener Temperaturen ermittelt haben sind in Abbildung 38 dargestellt. Es wird deutlich, daß bereits bei Temperaturen im Bereich nahe 50 °C etwa um 20 % niedrigere Keimnachweisraten erhalten werden. Man könnte also bezüglich der Zellschädigung bei der Einbettung in Agarose-Beads von einer vergleichbaren Temperaturabhängigkeit ausgehen. Abbildung 38: Nachweis von E. coli bei verschiedenen Temperaturen des Gußagars und daraus resultierender Trendberechnung, (Heinemeyer und Geuenich, 1997) In Übereinstimmung damit konnte mit den durchgeführten Vorversuchen (Abschnitt 3.6.1) nachvollzogen werden, daß die Zellschädigung bei der Einbettung von P. aeruginosa-Zellen in Beads durch die nötige Temperatur von 47 °C während der Bead-Herstellung verursacht wird. Die Temperaturempfindlichkeit ist je nach Bakterienart unterschiedlich, so daß aus dem hier mit P. aeruginosa durchgeführten Versuch keine allgemeinen Schlußfolgerungen gezogen werden 55 4 Diskussion können. Es läßt sich jedoch sagen, daß es durch die Verwendung einer anderen Agarose mit niedrigerer Geltemperatur möglich ist, die Reaktionstemperatur entsprechend zu senken und so vermutlich die Schädigung der Bakterien zu minimieren. Über die Vor- und Nachteile der Verwendung verschiedener Agarosesorten mit Geltemperaturen von 15 bis 40 °C zur BeadHerstellung wurde von Cook et al. (1991) berichtet. Die in der vorliegenden Arbeit verwendete Agarose (Merck 116801) wies eine Geltemperatur von 40 °C auf und ist somit für die Einbettung temperaturempfindlicher Mikroorganismen eher als weniger geeignet anzusehen. 4.2.2 Vermehrung von immobilisierten Bakterien in Agarose-Beads Es wurde eine Kultivierung der in Agarose eingebetteten P. aeruginosa-Bakterien für homogene und poröse Beads durchgeführt (Abschnitt 3.6.3). Zur Unterstützung des Wachstums wurde das universelle Nährmedium „Nutrient Broth“ eingesetzt. Bei der Kultivierung in NB-Medium (200 mg/l, 30 °C) war bereits nach 96 Stunden eine 17fache Zunahme der Gesamtzellzahl feststellbar. Aus der Wuchskurve wurde ungefähr eine Verdoppelung der Zellzahl innerhalb von 24 Stunden erkennbar. Die Auswertung der Anordnung der Mikrokolonien nach der Bebrütung ergab dabei deutlich einen charakteristischen Unterschied zwischen den homogenen und porösen Beads. Bei den homogenen Agarose-Beads war eine Zellvermehrung nur in den peripheren Bereichen nahe der Bead-Oberfläche erkennbar. Die Bakterien im Bead-Inneren zeigten auch nach mehrtägiger Bebrütung (96 h) keine merklichen Veränderungen in Bezug auf Zellwachstum oder Zellvermehrung. Im Fall der porösen Beads war im Gegensatz dazu eine Formierung von Mikrokolonien gleichmäßig über den gesamten Bead-Querschnitt feststellbar (vergl. hierzu Abbildung 33 und Abbildung 34). Dies ist ein Hinweis auf eine lokale Zellteilung und Vermehrung bis hin zur Ausbildung von Mikrokolonien. Als Ursache hierfür ist zu vermuten, daß die Nährstoffzufuhr ins Zentrum der Beads in den homogenen Beads durch die Diffusionslimitierung des Polymers eingeschränkt wird. Hinzu kommt, daß die peripher angeordneten Bakterien im Diffusionsweg liegen und durch ihren Nährstoffverbrauch um die hineindiffundierenden Nährstoffe konkurrieren. Im Fall der porösen Beads ist dieses Verhalten nicht zu beobachten, da durch die Kanäle ein ausreichender Nährstofftransport aufgrund des möglichen konvektiven Transportes gewährleistet zu seien scheint. (Bezüglich des konvektiven Transportes siehe auch Abschnitt 4.3) Untersuchungen und Berechnungen zur Diffusionslimitierung von Nährstoffen in homogenen Beads und den daraus resultierenden Folgen auf das Wachstumsverhalten der eingebetteten Bakterien wurden von Wijffels et al. (1995) durchgeführt. Die Ergebnisse von Wijffels et al. (1995) bestätigen, daß es in homogenen Beads mit immobilisierten Bakterien zur Ausbildung eines Substratgradienten kommt, welcher eine stark heterogene Verteilung der entstehenden Mikrokolonien zur Folge hat. In Abbildung 39 sind einige der Ergebnisse von Wijffels et al. (1995) dargestellt. Dort wurde das Wachstumsverhalten von Nitrosomonas europaea in homogenen N-Carrageenan-Beads untersucht. Es zeigte sich ebenfalls, daß ein Wachstum der Zellen bereits von Beginn an nur in der äußeren Schicht der Beads zu beobachten war. Mit fortschreitender Kultivierung kam es zur Ausbildung einer stark besiedelten, relativ scharf abgegrenzten oberflächennahen Schicht und im Inneren der Beads wurde keine Zunahme oder gar ein Rückgang der Biomasse festgestellt. 56 4 Diskussion Abbildung 39: Beobachtete (Punkte) und berechnete (Linien) Biomassenkonzentrations-Profile in verschiedenen Stadien des Wachstums von immobilisierten Nitrosomonas europaea in Beads. Die Berechnungen wurden nach dem “colony expansion model” unter Berücksichtigung von Zellaustrag erstellt. (Wijffels et al., 1995) Eine weitere Arbeit zur Ausbildung von Stoffgradienten in Biofilmen wurde von Wimpenny und Kinniment (1995) durchgeführt. Sie beschreiben, in welcher Weise das Entstehen solcher Gradienten von den Transportprozessen wie Diffusion oder konvektivem Transport in Poren beeinflußt wird. Unter anderem wurden von Wimpenny und Kinniment (1995) die in Abbildung 40 dargestellten Werte für die Verteilung von Zellen in P. aeruginosa Biofilmen ermittelt. Auch hier zeigt sich, daß die höchste Zelldichte nur wenig unter der Biofilmoberfläche zu finden ist und bereits wenig darunter drastisch abfällt. Wimpenny und Kinniment (1995) führen dieses Verhalten ebenfalls auf einen durch Diffusions-Reaktions-Wechselwirkungen (Summe der Effekte von Diffusionslimitierung, Nährstoffverbrauch und Reaktion mit der Matrix) entstandenen Substratgradienten zurück. Abbildung 40: Zellverteilung über den Querschnitt eines “steady state” P. aeruginosa Biofilms, (Wimpenny und Kinniment, 1995) 57 4 Diskussion 4.3 Schutzwirkung der Einbettung Bei der Behandlung von P. aeruginosa SG81R1 mit Natriumhypochlorit (0.5 mg/l freies Chlor) konnten für die Zellsuspension sowie für in homogenen und porösen Agarose-Beads immobilisierten Zellen vergleichbare Inaktivierungskinetiken beobachtet werden. Innerhalb der Einwirkzeit (30 Minuten) wurde dabei eine Abnahme der membranintakten Zellen um etwa eine Zehnerpotenz festgestellt. Aus den in Abbildung 35 dargestellten Kurvenverläufen läßt sich entnehmen, daß die in Suspension vorliegenden Bakterien bereits nach kürzerer Einwirkdauer eine Membranschädigung aufweisen als in Agarose-Beads immobilisierte Zellen. Die in Agarose-Beads eingebetteten Bakterien erfahren wie die Bakterien in natürlichen Biofilmen einen Schutz durch die umgebende Gelmatrix (Wingender et al., 1998; Morton et al., 1998 Grobe et al., 1994) und werden, wie aufgrund der vermuteten Diffusionslimitierung im Agarose-Gel zu erwarten, in den anfänglichen 20 Minuten erkennbar langsamer inaktiviert. Es erscheint jedoch auf den ersten Blick widersprüchlich, daß hierbei die Bakterien in den homogenen Beads im Vergleich zu den porösen Beads schneller geschädigt werden, obwohl hier ein stärkerer Effekt der Diffusionshemmung zu erwarten wäre. Eine Erklärung für diesen Widerspruch findet man, wenn man die bereits in Abschnitt 3.6.3 gewonnenen Erkenntnisse zu Hilfe nimmt. Dort wurde gezeigt, daß sich in den homogenen Agarose-Beads die Bakterienkolonien nach der Kultivierung fast ausschließlich an der Peripherie der Beads befinden (vergl. Abbildung 33). Es ist zu vermuten, daß Bakterien in diesen äußeren Bereichen bei der Behandlung mit Natriumhypochlorit schneller eine Schädigung erfahren, als es trotz des möglichen konvektiven Transportes in den porösen Beads möglich ist. Untersuchungen zu den Möglichkeiten von diffusivem und konvektivem Transport in superporösen Agarose-Beads wurden bereits von Gustavsson et al. (1998) durchgeführt. Es wurde beispielsweise der konvektive Transport von gefärbten Hefezellen durch die Superporen in Agarose-Beads verfolgt. Eine der dabei entstandenen Aufnahmen, aus der sich eine Wanderungsgeschwindigkeit von etwa 6 µm·s-1 entnehmen läßt, ist in Abbildung 41 wiedergegeben. Hiermit ließ sich die vorhandene Möglichkeit des konvektiven Stofftransportes eindeutig nachweisen. Abbildung 41: Position einer gefärbten Hefezelle während der Durchwanderung eines porösen Agarose-Beads in Zeitabständen von einer Sekunde (Bead-Durchmesser 150 µm, Porendurchmesser 30 µm, Hefezellen-Duchmesser 4 µm, Hefezelle gefärbt mit Anilin-Blau), (Gustavsson et al., 1998) 58 4 Diskussion Zur Diffusion von Chlor in Modell-Biofilmen und der Reaktion mit den Komponenten wurden Untersuchungen von Chen und Stewart (1996) durchgeführt. Es wurde untersucht, inwieweit hineindiffundierendes Chlor durch Reaktion mit der Zellmasse (P. aeruginosa ERC1) oder der EPS-Modellsubstanz (Agarose) reagiert und ob es dadurch zu einer Ausbildung eines Gradienten der Chlorkonzentration kommt. Chen und Stewart (1996) haben gefunden, daß das Chlor sowohl mit der Zellmasse als auch mit der Agarose reagiert und es so zu einer Zehrung kommt. Die Reaktion mit der Agarose ist jedoch um zwei Größenordnungen geringer. Da die Zehrung des Chlors durch Reaktion mit der Zellmasse ein relativ schneller Prozeß ist, kommt es so zu einer Diffusionslimitierung. In Abbildung 42 ist ein von Chen und Stewart erstelltes Diagramm wiedergegeben, aus dem die zeitabhängige Chlorzehrung einer Zellsuspension von P. aeruginosa ERC1 in Abhängigkeit von der Anfangskonzentration an Biomasse zu entnehmen ist. Abbildung 42: Kinetische Daten für die Reaktion von Chlor mit der Biomasse in P. aeruginosa ERC1-Suspensionen Es sind verschiedenen Anfangskonzentrationen an Biomasse angegeben (Chen und Stewart, 1996) Für die in Abschnitt 3.7 in Abbildung 36 angegebenen Werte für die Chlorzehrung durch Bakterien in Suspension, Agarose-Beads mit eingebetteten Bakterien und Agarose-Beads ohne Zellen läßt sich eine gute Übereinstimmung mit den von Chen und Stewart (1996) gemachten Angaben erkennen. Daß der Effekt der Diffusionslimitierung des Chlors in den Untersuchungen der vorliegenden Arbeit nicht beobachtet werden konnte, liegt wie bereits erwähnt vermutlich daran, daß genau diese Limitierung bei den porösen Beads durch den möglichen konvektiven Transport vermieden wird. Bei den homogenen Beads konnte die Beobachtung der Diffusionslimitierung nicht gemacht werden, da die Bakterienkolonien fast ausschließlich nahe der Oberfläche der Beads lokalisiert waren. Darüber hinaus ist auffallend, daß selbst nach einer Einwirkzeit von 30 Minuten bei allen im Rahmen der vorliegenden Arbeit durchgeführten Versuchen zur Desinfektionswirkung von Natriumhypochlorit (0.5 mg/l freies Chlor) auf P. aeruginosa SG81R1 sowohl in Suspension wie auch immobilisiert in Beads ein Anteil überlebender Bakterien von einheitlich etwa 20 % zu beobachten war. Hierfür können unter anderem Resistenzmechanismen von P. aeruginosa verantwortlich sein (Morton et al., 1998; Grobe, 1996; Vess et al., 1993; Seyfried und Fraser, 1980). Es ist jedoch auch möglich, daß eine weitere Desinfektion der restlichen Zellen nicht möglich war, da die Restkonzentration an freiem Chlor aufgrund der fast vollständigen Chlorzehrung über den Versuchszeitraum zu diesem Zeitpunkt bereits zu niedrig lag. 59 4 Diskussion 4.4 Bewertung der entwickelten Biofilm-Modelle Es ist im Rahmen der vorliegenden Arbeit gelungen, homogene und poröse Agarose-Beads sowie poröse Agarose-Stücke und -Filme herzustellen, P. aeruginosa-Zellen in der Gelmatrix zu immobilisieren sowie diese Bakterien darin zu kultivieren. Bei einer Bewertung der damit erstellten Biofilm-Modelle muß jedoch immer beachtet werden, daß die Struktur, die Zusammensetzung und die damit verbundenen physiko-chemischen Eigenschaften eines Biofilms sehr unterschiedlich sein können und z. B. vom Biofilmalter, den Umweltfaktoren (z.B. Scherkräfte) und der Nährstoffsituation abhängen. Es kann folglich nicht ein einziges Biofilm-Modell geben, mit dem alle natürlichen Biofilme beschrieben werden können. (Eine Übersicht der verschiedenen Biofilm-Strukturmodelle ist von Wingender und Flemming (1999) zusammengestellt worden.) Es ist daher stets zu berücksichtigen, welches System modelliert werden soll, bzw. in wie weit das entwickelte Biofilm-Modellkonzept an die grundlegend unterschiedlichen Strukturen angepaßt werden kann. 4.4.1 Agarose-Beads In der vorangegangenen Diskussion wurde bereits gezeigt, daß es mit den hergestellten homogenen Agarose-Beads möglich ist, die Verhältnisse in einem dichten konfluenten Biofilm zu simulieren, der dem Modell von Marshall und Blainey (1991) entspricht, und die für dieses Modell typische Ausbildung von Substratgradienten und die heterogene, „schichtenartige“ Biomasseverteilung gut wiedergibt (vergl. Abschnitt 4.2.2). Unter Verwendung der porösen Agarose-Beads mit eingebetteten Bakterien läßt sich das von Costerton (1994) aufgestellte „Mushroom“-Modell, welches sich u.a. durch die mit Wasser gefüllten Kanalstrukturen auszeichnet, näherungsweise simulieren. Durch den in diesen Kanälen, bzw. in den Superporen der Agarose-Beads, möglichen konvektiven Transport wird eine Limitierung von Substraten etc. vermieden oder zumindest gemindert. Die in die porösen Agarose-Beads eingebrachten Kanäle im Größenbereich von ca. 10 bis 80 µm (im Mittel 28 µm) sind vergleichbar mit Kanalstrukturen wie sie auch in natürlichen oder in angezüchteten Biofilmen beobachtet wurden (Costerton, 1994). 4.4.2 Bead-Schichten und Agarose-Körper Die einzelnen Agarose-Beads geben als Modellsystem die Verhältnisse eines natürlichen Biofilms prinzipiell näherungsweise wieder, wobei aber die typischen Eigenschaften von flächigen Biofilmen nicht realistisch simuliert werden können. Hierbei werden einige Phänomene von Biofilmen, wie z.B. Schichtstrukturen oder Substratgradienten, nur schematisch durch die Verhältnisse in den Kugelschalen repräsentiert. Um auch den makroskopischen Vorgaben eines natürlichen Biofilms, z.B. der flächigen Besiedelung einer Oberfläche, gerecht zu werden, wurde die Möglichkeit der Fixierung der Beads auf einem Trägermaterial verwirklicht (Abschnitt 3.4). Durch diese Vorgehensweise kann eine Oberflächenbesiedelung durch einen Biofilm simuliert werden. Die bei der Fixierung als Bead-Schicht entstehenden Zwickelräume geben hierbei die Kanalstruktur annähernd wieder. 60 4 Diskussion Es wurde jedoch bereits in Abschnitt 3.4 darauf eingegangen, daß die Fixierungstechniken einige Nachteile haben. So können bei der Hitzefixierung der Beads eingebettete Zellen geschädigt werden. Mit Hilfe der Fixierung der Beads durch das Auftropfen von verdünnter, warmer Agarose-Lösung kann diese Schädigung zwar vermieden werden, jedoch wird bei dieser Methode ein Teil der erzeugten Poren und der Zwickelräume durch die Agarose-Lösung verschlossen. Darüber hinaus ist es mit dem Modell der Bead-Schichten nur möglich, BiofilmDicken zu modellieren, die mindestens dem Bead-Durchmesser entsprechen. Bei einem Monolayer läge die Untergrenze daher bei ca. 70 µm. Als Alternative hierzu wurde die Herstellung von porösem Agarose-Material, erstmals auch mit eingebetteten Zellen, als Film oder in Stücken entwickelt. Dieses Agarose-Material ist mit einem ausgedehnten Porensystem durchzogen, welches dem der superporösen Beads gleicht. Es kann aber im Gegensatz zu den Beads direkt in Form von Schichten und Filmen erhalten werden, die zwischen etwa 0.5 mm und 2 cm dick sind und in ihrer flächigen Ausbreitung nur von den apparativen Möglichkeiten begrenzt sind. Die Nachteile, die bei der Fixierung von BeadSchichten auftreten, werden hierbei prinzipiell vermieden. Um Biofilme zu modellieren, deren Dicke unter den direkt erzeugbaren 0.5 mm liegen, ist es möglich die hergestellten superporösen Agarose-Stücke nachträglich in dünne Schichten zu zerschneiden. Hierbei wäre z.B. die Anwendung eines Mikrotoms denkbar, mit dem Schnittdicken bis hinab in den µm-Bereich möglich sind. 4.4.3 Einbettung von Mikroorganismen Bakterienzellen konnten auf einfache Weise in die Agarose-Matrix eingebettet werden. Die Zelldichte im Agarose-Material läßt sich dabei nach Wunsch variieren und so verschieden dicht populierte Biofilme simulieren. Durch die Möglichkeit der Kultivierung der Bakterien in den Beads läßt sich die Ausbildung von Mikrokolonien steuern und verschiedene Wachstumsphasen nachstellen. Über die Auswahl verschiedener Nährmedien läßt sich hierbei auch der Einfluß des Substratangebotes auf die Entwicklung der Mikroorganismen im Modell-Biofilm studieren. Es ist aus einigen Untersuchungen bekannt, daß Bakterien in natürlichen Biofilmen z.B. in ihren physiologischen Eigenschaften grundlegend von der planktonischen Form des gleichen Stammes abweichen können (Goodman und Marshall, 1995; Davies et al., 1993; Wingender und Flemming, 1999). Es ist beispielsweise von Davies et al. (1993) gezeigt worden, daß die Produktion von Exopolysacchariden, z. B. Alginat bei P. aeruginosa, in Abhängigkeit von der Anheftung in Biofilmen reguliert wird. Es bleibt daher fraglich, inwieweit diese mikrobiologischen und physiologischen Besonderheiten von in Biofilmen sessil lebenden Mikroorganismen auch für die im Modell-Biofilm eingebetteten Bakterien zutreffend sind. 4.4.4 Gesamtbeurteilung Es stellt sich abschließend die Frage, in welchem Ausmaß das entwickelte BiofilmModellkonzept von in Agarose eingebetteten Mikroorganismen ein brauchbares Biofilm-Modell repräsentiert. Anhand der in dieser Arbeit zusammengetragenen Ergebnisse läßt sich folgern, daß zumindest einige physiko-chemische Eigenschaften von natürlichen Biofilmen gut modelliert werden können. Hierbei sind insbesondere die Struktur (von homogen bis porös) und die daraus hervorgehenden Möglichkeiten von Diffusion und Konvektion sowie das damit einhergehende etwaige Auftreten von Diffusionshemmungen oder Substratlimitierungen zu nennen. 61 4 Diskussion Darüber hinaus konnte anhand der durchgeführten Löslichkeitsversuche die Stabilität des physikalischen Gel-Netzwerkes und mit den Quellungsversuchen das hohe Wasserbindungsvermögen der Beads nachgewiesen werden, wie es auch aus Untersuchungen an realen Biofilmen bekannt ist (Ophir und Gutnick, 1994). Die Einwirkung von Natriumhypochlorit hat darüber hinaus gezeigt, daß auch die Schutzwirkung der EPS auf Bakterien in natürlichen Biofilmen gegenüber der Einwirkung von toxischen Substanzen durch das Modell näherungsweise simuliert wird. 4.5 Ausblick Wie bereits in der vorausgegangenen Diskussion an einigen Stellen erwähnt wurde, läßt sich an vielen Stellen des im Rahmen dieser Arbeit neu vorgestellten Biofilm-Modells mit weiteren Forschungsarbeiten ansetzen. Einige für die Fortführung der Entwicklung dieses Biofilm-Modells in Hinblick auf eine Optimierung und bessere Charakterisierung als sinnvoll erachtete Versuche sind im folgenden zusammengestellt: • Die Verwendung einer anderen Agarose mit niedrigerer Geltemperatur für die Einbettung der Bakterien wäre zur Minimierung der Zellschädigung zu überprüfen. • Es wäre zu überprüfen, ob sich durch eine Veränderung der Herstellungsprozedur der porösen Beads eine steuerbare Variation der Porengröße erreichen ließe. Hierbei ist insbesondere der Einfluß der Rührapparatur und der Emulgatoren zu untersuchen. • Die Ergebnisse der Untersuchungen zum physiologischen Zustandes der Bakterien mittels LIVE/DEAD® BacLight™ Bacterial Viability Kit (Molecular Probes) könnten mit unabhängigen Methoden überprüft werden. • Es könnte die Immobilisierung anderer Organismen, die Einbettung anderer Substanzen (z.B. Korrosionsprodukte) oder die Zugabe weiterer EPS-typischer Komponenten (z.B. Proteine) durchgeführt und deren Einflüsse auf die Bakterien und auf die physiko-chemischen Eigenschaften des Gel-Netzwerkes untersucht werden. • Eine geeignete Möglichkeit zur exakten Steuerung der Schichtdicken bei der Herstellung von Bead-Schichten und porösen Agarose-Filmen wäre zu entwickeln. • Weitergehende Untersuchungen zur Rheologie der Agarose-Materialien wären denkbar. • Darüber hinaus ist die Durchführung praxisnaher Versuche (z.B. Desinfektion) im Vergleich zu realen Biofilmen zur Validierung des Biofilm-Modells möglich. Abschießend kann gesagt werden, daß mit dem aufgezeigten Konzept eine gute Grundlage für die Modellierung von Biofilmen geschaffen wurde. Es steht ein Methode zur Verfügung, mit der reproduzierbar und auf einfache Weise Modell-Biofilme mit steuerbaren Eigenschaften hergestellt werden können. Es besteht somit die Möglichkeit, mit relativ geringen Zeit- und Arbeitsaufwand eine definierte Probensubstanz für eine Vielzahl von Untersuchungsmethoden bereitzustellen, was für bestimmte Fragestellungen eine gute Alternative zur Anzüchtung von Biofilmen in speziellen Reaktoren darstellt. Darüber hinaus stellt insbesondere die Einbettung von Bakterien in poröse Agarose-Beads eine neue Entwicklung dar, die neben der Verwendung als Biofilm-Modell auch für biotechnologische Anwendungen von Bedeutung seien kann. 62 5 Literaturverzeichnis 5 Literaturverzeichnis Arnott, S., Fulmer, A., Scott, W.E., Dea, I.C.M., Moorhouse, R., Rees, D.A., J. Mol. Biol. 1974, 90, 269 – 284. Center for Biofilm Engineering News 1994, Vol. 2 No. 1, S. 1 – 2. Characklis, W.G., „Biofilm processes“, in: Biofilms, (Hrsg.: Characklis, W.G., Marshall, K.C.), John Wiley & Sons, New York, 1990, S. 195 – 232. Characklis, W.G., Turakhai, M.H., Zelver, N., „Transport and interfacial transfer phenomena“, in: Biofilms, (Hrsg.: Characklis, W.G., Marshall, K.C.), John Wiley & Sons, New York, 1990, S. 265 – 340. Chen, X., Stewart, P.S., „Chlorine penetration into artificial biofilm is limited by a reactiondiffusion interaction“, Environ. Sci. Technol. 1996, 30, 2078 – 2083. Christensen, B.E., Characklis, W.G., „Physical and chemical properties of biofilms“, in: Biofilms, (Hrsg.: Characklis, W.G., Marshall, K.C.), John Wiley & Sons, New York, 1990, S. 93 – 130. Cook, R.B., Provonchee, R.B., Nochumson, S., „Agarose beads, preferably incorporating biological materials”, U. S. Patent 5.053.332 1991. Costerton, J.W., Irvin, R.T., „The bacterial glycocalyx in nature and disease“, Ann. Rev. Microbiol. 1981, 35, 299 – 324. Costerton, J.W., Cheng, K.-J., Geesey, G.G., Ladd, T.I., Nickel, J.C., Dasgupta, M., „Bacterial biofilms in nature and disease“, Ann. Rev. Microbiol. 1987, 41, 435 – 464. Costerton, J.W., Boivin, J., „Biofilms and corrosion“, in: Biofouling and biocorrosion in industrial water systems, (Hrsg.: Flemming, H.-C. und Geesey, G.G.) Springer Verlag, Heidelberg, 1991, S. 195 – 204. Costerton, J.W., Lewandowski, Z., DeBeer, D., Caldwell, D., Korber, D., James, G., „Biofilms, the customized microniche“, J. Bacteriol. 1994, 176, 2137 – 2142. Costerton, J.W., Lewandowski, Z., Caldwell, D.E., Korber, D.R., Lappin-Scott, H.M., „Microbial biofilms“, Ann. Rev. Microbiol. 1995, 49, 711 – 745. Cox, R.C., Photographic gelatine II, Academic Press, London, 1995. Davies, D.G., Chakrabarty, A.M., Geesey, G.G., „Exopolysaccharide production in biofilms: substratum activation of alginate gene expression by Pseudomonas aeruginosa“, Appl. Environ. Microbiol. 1993, 59, 1181 – 1186. Ekman, B., Lindahl, A., „Method for producing small, spherical polymer particles“, U. S. Patent 4.822.535 1989. Ferry, J.D., Viscoelastic properties of polymers, John Wiley & Sons, New York, 1970, S. 431 – 465. Flemming, H.-C., „Biofilms as a particular form of microbial life“, in: Biofouling and biocorrosion in industrial water systems, (Hrsg.: Flemming, H.-C. und Geesey, G.G.) Springer Verlag, Heidelberg, 1991, S. 3 – 9. 63 5 Literaturverzeichnis Flemming, H.-C., Biofilme, Biofouling und mikrobielle Schädigung von Werkstoffen, Habilitationsschrift, in: Stuttgarter Berichte zur Siedlungswasserwirtschaft, Band 129, Oldenbourg Verlag, München, 1994. Flemming, H.-C., „Biofilme und mikrobielle Materialzerstörung“, in: Mikrobielle Materialzerstörung und Materialschutz, (Hrsg.: Brill, H.), Gustav Fischer Verlag, Stuttgart, 1995a, S. 28. Flemming, H.-C., „Sorption sites in biofilms“, Wat. Sci. Technol. 1995b, 32, 27 – 33. Flemming, H.-C., „The forces that keeps biofilms together“, DECHEMA Monographs 1996, 133, 311 – 316. Flemming, H.-C., Schmitt, J., Marshall, K.C., „Sorption properties of biofilms“, in: Environmental behaviour of sediments, (Hrsg.: Calmano, W., Förstner, U.), Lewis Publishers, Chelsea, 1996, S. 115 – 157. Fletcher, M.M., „The physiological activity of bacteria attached to solid surfaces“, Adv. Microb. Physiol. 1991, 32, 53 – 85. FMC Corp., The agarose monograph, 4th ed., FMC Corp. Publ., 1988. Geesey, G.G., „Microbial exopolymers: Ecological and economic considerations“, ASM News 1982, 48, 9 – 14. Goodman, A.E., Marshall, K.C., „Genetic responses of bacteria at surfaces“, in: Microbial Biofilms, (Hrsg.: Lappin-Scott, H.M., Costerton, J.W.), Cambridge University Press, Cambridge, 1995, S. 80 – 98. Griess, G.A., Biopolymers 1989, 28, 1475 – 1484. Grobe, S., Fiedler, S., Wingender, J., Overath, H., „Wirkung von Chlor und Wasserstoffperoxid auf schleimbildende Bakterien aus wasserführenden Systemen”, in: Vom Wasser, 1994, 83, S. 407 – 424 Grobe, S., „Bedeutung des extrazellulären Polysaccharids Alginat für die Resistenz aquatischer Stämme von Pseudomonas aeruginosa gegenüber Chlor“, Dissertation aus dem IWW, Mülheim an der Ruhr, 1996. Gustavsson, P.-E., Larsson, P.-O., „Superporous agarose, a new material for chromatography“, J. Chromatogr. A 1996, 734, 231 – 240. Gustavsson, P.-E., „Superporous agarose – a new material for chromatography“, Dissertation, Lund University Sweden, 1998. Hartmeier, W., Immobilisierte Biokatalysatoren – Eine Einführung, Springer Verlag, Berlin/Heidelberg, 1986. Heinemeyer, E.-A., Geuenich, H.H., „Mikrobiologischer Ringtest“, in: DVGW-Schriftenreihe Wasser, 1997, Nr. 91, S. 373 – 387. Kaiser, R.E., Mühlbauer, J.A., Elementare Tests zur Bibliographisches Institut, Mannheim/Wien/Zürich, 1983. Beurteilung von Meßdaten, Lawrence, J.R., Korber, D.R., Wolfaardt, G.M., Caldwell, D.E., „Analytical imaging and microscopy techniques“, in: Manual of environmental microbiology, (Hrsg.: Hurst, C.J.) ASM Press, Washington D.C., 1997, S. 29 – 51. LeChevallier, M.W., Cawthon, C.D., Lee, R.G., „Inactivation of biofilm bacteria“, Appl. Environ. Microbiol. 1988, 54, 2492 – 2499. 64 5 Literaturverzeichnis Leenen, E.J.T.M., dos Santos, V.A.P., Grolle, K.C.F., Tramper, J., Wijffels, R.H., „Characteristics of and selection criteria for support materials for cell immobilization in wastewater treatment“, Wat. Res. 1996, 30 (12), 2985 – 2996. Marshall, K.C., Blainey, B., „Role of bacterial adhesion in biofilm formation and biocorrosion“, in: Biofouling and biocorrosion in industrial water systems, (Hrsg.: Flemming, H.-C. und Geesey, G.G.) Springer Verlag, Heidelberg, 1991, S. 8 – 45. Marshall, K.C., Microbial adhesion and aggregation, Springer Verlag, Berlin, 1984. Marshall, K.C., „Bacterial adhesion in oligotrophic habitats“, Microb. Sci. 1985, 2, 321 – 326. Marshall, K.C., „Biofilms: an overview of bacterial adhesion, activity and control at surfaces“, ASM News 1992, 58, 202 – 207. Meyer-Reil, L.A., „Ökologie mikrobieller Biofilme“, in: Ökologie der Abwasserorganismen, (Hrsg.: Lemmer, H., Griebe, T., Flemming, H.-C.) Springer Verlag, Heidelberg, 1996, S. 25 – 42. Molecular Probes, „LIVE/DEAD® BacLight™ Bacterial Viability Kits – Product information sheet“, Molecular Probes Inc., 1996. Morton, L.H.G., Greenway, D.L.A., Gaylarde, C.C., Surman, S.B., „Consideration of some implications of the resistance of biofilms to biocides“, International Biodeterioration & Biodegradation 1998, 41, 247 – 259. Mosbach, K., Nilsson, K., „Method of encapsulating biomaterial in bead polymers“, U. S. Patent 4.647.536 1987. Ophir, T., Gutnick, D.L., „A role for exopolysaccharides in the protection of microorganisms from desiccation”, Appl. Environ. Microbiol. 1994, 60, 740 - 745. Pringle, J.H., Fletcher, M.M., „Influence of substratum hydration and adsorbed macromolecules on bacterial attachment to surfaces“, Appl. Environ. Microbiol. 1986, 51, 1321 – 1325. Rees, D.A., „Structure, conformation, and mechanism in the formation of polysaccharide gels and networks”, Advan. Carbohyd. Chem. 1969, 24, 267 – 332. Rees, D.A., „Shapely polysaccharides”, Biochem J. 1972, 126, 257 – 273. Rehage, G., Borchard, W., The thermodynamics of the glasy state, Applied Science Publishers Ltd., England, 1973. Rehage, G., Halboth, H., Makromol. Chem. 1968, 119, 235. Rehage, G., „Thermodynamische Eigenschaften von Gelen“, Ber. Bunsenges. Phys. Chem. 1977, 81 (10), 969. Rittmann, B.E., „Detachment from biofilms“, in: Structure and function of biofilms, (Hrsg.: Characklis, W.G., Wilderer, P.), John Wiley & Sons, New York, 1989, S. 49 – 58. Rochas, C., Hecht, A.M., Geissler, E., „Swelling properties of agarose gels“, J. Chem. Phys. 1996, 93, 850 – 857. Römpp Chemie Lexikon, 9. Aufl., Georg Thieme Verlag, Stuttgart/New York, 1995. Schaule, G., Flemming, H.C., Poralla, K., „Forces involved in primary adhesion of Pseudomonas diminuta to filtration membranes“, 6th Int. Conf. on Microb. Ecol. 1993a, Sept. 8-12, Barcelona. 65 5 Literaturverzeichnis Schaule, G., Flemming, H.C., Ridgway, H.F., „The use of CTC in the quantification of respiratory active bacteria in biofilms“, Appl. Environ. Microb. 1993b, 59, 3850 – 3857. Schubert, H., Armbruster, H., „Prinzipien der Herstellung und Stabilität von Emulsionen“, Chem.-Ing.-Tech. 1989, 61, 701 – 711. Selic, E., Dissertation Gerhard-Mercator-Universität Duisburg, 1999. Seyfried, P.L., Fraser, D.J., „Persistence of Pseudomonas aeruginosa in chlorinated swimming pools“, Can. J. Microbiol. 1980, 26, 350 – 355. Stockmayer, W., J. Chem. Phys. 1944, 12, 125. Sutherland, I.W., „Microbial polysaccharides – their role in microbial adhesion in aqueous systems“, CRC Crit. Rev. Microbiol. 1984, 10, 173 – 201. Vess, R.W., Anderson, R.L., Carr, J.H., Bond, W.W., Favero, M.S., „The colonization of solid PVC surfaces and the acquisition of resistance to germicides by water micro-organisms“, J. Appl. Bacteriol. 1993, 74, 215 – 221. Vorlop, K.-D., „Entwicklung von Verfahren zur Polymerfixierung von Mikroorganismen und Anwendung der Biokatalysatoren zur Spaltung von Penicillin G und Synthese von L-Tryptophan“, Dissertation TU Braunschweig, 1984. Weaver, J.C., „Process for isolating microbiologically active material“, U. S. Patent 4.399.219 1981. Wijffels, R.H., de Gooijer, C.D., Schepers, A.W., Beuling, E.E., Mallée, L.F., Tramper, J., „Dynamic modeling of immobilized Nitrosomonas europaea: Implemetation of diffusion limitation over expanding microcolonies“, Enzyme Microb. Technol. 1995, 17, 462 – 471. Wimpenny, J.W.T., Kinniment, S.L., „Biochemical reactions and the establishment of gradients within biofilms“, in: Microbial Biofilms, (Hrsg.: Lappin-Scott, H.M., Costerton, J.W.), Cambridge University Press, Cambridge, 1995, S. 99 – 117. Wingender, J., Grobe, S., Fiedler, S., Flemming, H.-C., „The effect of extracellular polysaccharides on the resistance of Pseudomonas aeruginosa to chlorine and hydrogen peroxide“, in: Biofilms in aquatic systems, (Hrsg.: Keevil, C., Holt, D., Dow, C., Godfree, A.), Royal Society of Chemistry, Cambridge, 1998, im Druck. Wingender, J., Neu, T., Flemming, H.-C., Bacterial extracellular polymer substances, Springer Verlag, Heidelberg, 1999. Wingender, J., Flemming, H.-C., „Autoaggregation of Microorganisms: flocs and biofilms“, in: Biotechnology, (Hrsg.: Winter, J.), Wiley-VCH, Weinheim, 1999, Vol. 11a, S. 65 – 83. ZoBell, C.E., „The effect of solid surfaces upon bacterial activity“, J. Bacteriol. 1943, 46, 39 – 56. 66