Instrumentelle Analytik Massenspektrometrie MS Seite 4.2.2
Werbung

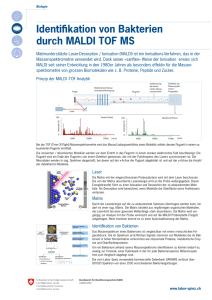
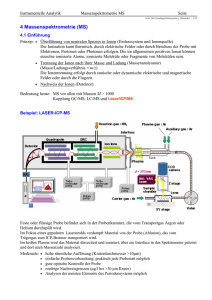
Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 1/18 4.2.2 Ionenquellen Das Massenspektrum eines Moleküls hängt sehr stark von der Art der Fragmentierung und damit von der Art der Ionisierung ab. harte Ionisation, starke Fragmentierung Elektronenstoß-Ionisation (EI) Molekülpeak oft nicht zu registrieren Chemische Ionisation (CI) Elektrospray-Ionisation (ESI) Laser Desorption (MALDI) Fast Atom Bombardment (FAB) weiche Ionisation Desorption: Direkte Bildung gasförmiger Ionen aus festen oder flüssigen Proben Die beiden neueren Verfahren ESI und MALDI sind die wichtigsten Verfahren in der Untersuchung von großen Biomolekülen in der Proteinanalytik und Sequenzierung von Peptiden. 4.2.2.1 Harte Ionisation (Elektronenstoß-Ionisation EI ) Wichtigste und am häufigsten verwendete Methode für flüchtige Analyten mit m < 1000 amu Prinzip: Die Ionisation erfolgt durch den Beschuss der gasförmigen Moleküle mit thermischen Elektronen hoher Energie bei stark vermindertem Druck (ca.10−5 mbar). Als Elektronenquelle dient eine Glühkathode aus Wolfram oder Rhenium. Beschleunigungs Spannung 1-10 kV Abb.: Schema einer Ionenquelle mit Elektronenstoßionisation Abb.: Ionenausbeute als Funktion der Primärenergie der Elektronen Standard-Elektronenenergie: 70 eV -5 Ionenausbeute gering (ca. 10 ) reproduzierbare Fragmentierung Ionisierungsenergie für organ. Verbind. ca. 10 eV Anregungsenergie ca. 5 eV Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 2/18 Mechanismus der Ionisierung und Fragmentierung bei der EI 1) M + e- M+ + 2e- Primärionisation der Analytmoleküle zu Radikalkationen mit hoher Überschussenergie (Elektron wird entfernt, bevorzugt eines der freien Elektronenpaare bei vorhandenen Heteroatomen oder einer Doppelbindung). M + e- M- Elektroneneinfang mit Bildung negativer Ionen (sehr selten: M-/M+ = 1/1000) Überschussenergie von ca. 5-10 eV in M+ (SchwingungsRotationsanregung) führt zur gewünschten Fragmentierung. (Spaltung einer C-C Bindung benötigt nur ca. 2 eV.) * 2) Innerhalb einiger µs zerfallen die hochangeregten Molekülionen in Primärfragmente. Zwei Möglichkeiten: - Molekül-Radikalkation zerfällt in Molekülkation + Neutralradikal - Molekül-Radikalkation zerfällt in Radikalkation + Neutralmolekül M+ A+ + B M+ A + B+ M+ C+ + D Fragmentierung in Kation und Neutralradikal Fragmentierung in Radikalkation plus Neutralteilchen (Verlust) 3) Die Primärfragmente haben meist noch genügend innere Energie, um weiter zu zerfallen. In einer Kette von mitunter sehr komplexen Fragmentierungs- und Umordnungsreaktionen entstehen so weitere Fragmente. Abb.: Schematischer Fragmentierungsmechanismus eines org. Moleküls bei der EI Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 3/18 Wichtige Fragmentierungsreaktionen Terminologie / Symbolik Molekülkation/Radikalkation: M+, M+, [M]+, M ┐+ Fragmentbildung: Homolyse: Bindungsbruch durch Wanderung des Elektrons Zeichen: ; Heterolyse: Bindungsbruch durch Wanderung eines Elektronenpaares Zeichen: keine Aussage über Mechanismus Beispiel: Allylspaltung 1) Primärionisation 2) Homolyse Spaltung geht von der Radikalstelle aus 2’)Heterolyse Spaltung geht von der Ladung aus ; Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 4/18 1) Allylspaltung Leichte Spaltung einer Bindung in Nachbarschaft zu einer Doppelbindung (Allylstellung). 2) Benzylspaltung Benzylfragment bildet sich besonders leicht, wenn die Probe eine Benzylgruppe enthält. Fragment stabilisiert sich durch Umlagerung in Tropylium-Kation. 3) Alkylspaltung Die Stabilität von Carbokationen nimmt in der Reihe CH3+ < RCH2+ < R2CH+ < R3C+ zu. Die Spaltung an Verzweigungspunkten der Kohlenstoffkette ist daher am wahrscheinlichsten. Die positive Ladung verbleibt bevorzugt am höchstsubstituierten C-Atom. 4) -Spaltung Die C-Bindung in -Stellung (eine Bindung Abstand) zu einer funktionellen Gruppe (z.B. Carbonylgruppe, Heteroatome) wird gespalten. Grund: freie Elektronenpaare der funkt. Gruppe stabilisieren die positive Ladung des Radikalions. -Spaltungen treten mit großer Wahrscheinlichkeit in Carbonylverbindungen, Ethern, und in Nachbarschaft zu OH-, SH- und Aminogruppen auf. 5) Decarbonylierung Kationen, die durch -Spaltung aus einer Carbonylgruppe entstanden sind, spalten sehr leicht ein (polares) CO-Neutral-Molekül ab. Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 5/18 Wichtige Umlagerungsreaktionen 6) Onium-Reaktion Folgereaktion der bei der -Spaltung gebildeten Ammonium-, Oxonium-, Sulfonium-, etc. Ionen. Dabei wird ein H-Atom aus dem an das Heteroatom gebundenen Rest, unter Abspaltung dieses Restes auf das Heteroatom übertragen. 7) Retro-Diehls-Adler-Reaktion Ein Sechsring (Cyclohexen) mit einer Doppelbindung zerfällt in ein Dien und in ein Dienophil. (Umkehrung der Diels-Alder-Reaktion) 8) McLafferty-Umlagerung Vorrausetzung ist eine Doppelbindung, von der eine mindestens dreiatomige Kette abzweigt. Ein H-Atom wird über einen “Sechsring-Übergangszustand“ auf die Doppelbindung übertragen. Gleichzeitig wird ein (substituiertes) Ethenmolekül abgespalten. Zusammenfassung: Eine Fragmentierung ist immer dann günstig, wenn das entstehende Radikal bzw. Kation stabil ist. Die Abspaltung eines neutralen Moleküls (z.B. CO, C2H4) ist günstig. Besonders bevorzugt sind Spaltungen neben mesomeren Systemen im Molekül (Benzyl-, Allyl-). Ebenso bevorzugt sind Spaltungen neben Heteroatomen (O, N, S ), insbesondere in -Stellung. Dabei wird meist der schwerere Substituent eliminiert. Gesättigte Kohlenwasserstoffe spalten besonders an Verzweigungen des Gerüsts (Alkylspaltung). Bei Cyclohexan-verwandten Verbindungen (Sechsring mit einer Doppelbindung) ist eine Retro-Diels-Alder-Reaktion möglich. Eine McLafferty Umlagerung ist bei allen Verbindungen möglich, bei denen ein H-Atom über einen Sechsring-Übergangszustand an eine Doppelbindung übertragen werden kann. Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 6/18 Beispielspektren zur Fragmentierung (Elektronenstoßionisation) Das Tropylium spaltet weiter ein Ethin (C2H2) ab und wird zum Cyclopentadienyl (m/z = 65). Ebenso spaltet der Benzolring (m/z = 77) weiter ein neutrales Ethinmolekül ab. Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 7/18 4.2.2.2 Weiche (sanfte) Ionisation Mit weichen Ionisierungsmethoden wird weniger Energie auf die Moleküle übertragen. Moleküle fragmentieren nicht so schnell und der Molpeak kann registriert werden. 1) Chemische Ionisation (CI) Methode der Wahl bei flüchtigen Substanzen, die keinen Molpeak zeigen (m bis 1000 amu). Prinzip: In die Ionisierungskammer wird zusätzlich ein “Reaktandgas“ (meist Methan, Isobutan oder Ammoniak) im Überschuss eingeleitet. Die Elektronen stoßen dann hauptsächlich mit dem Reaktandgas zusammen. Diese aktivierten Reaktandgasionen können dann mit den eigentlichen Probenmolekülen reagieren (Protonierung des Analyten bei Stoßwechselwirkung). Die dampfförmige Probe strömt senkrecht zur Bildebene in die Ionenquelle Reaktionen in der Ionenquelle Primär: CH4 + e- CH4+ + 2eCH4 + e- CH3+ + H + 2eCH4 + e- CH2+ + H2 + 2e- Ionisation des Reaktandgases (z.B. Methan) CH4+ + CH4 CH5+ + CH3 CH3+ + CH4 C2H5+ + H2 CH2+ + 2CH4 C3H5+ + 2H2 + H Sekundärionen durch H-Transfer Unter einem Druck von 1 hPa des Reaktandgases werden also hauptsächlich folgende Ionen gebildet: CH5+ , C2H5+ , C3H5+ (protoniertes Methan, Äthan, Propan mit Massenzahlen 17, 29 und 41), die dann mit den Analytmolekülen reagieren und auch im Spektrum als Peaks erscheinen. Sekundär: Protonentransfer: CH5+ + M CH4 + [M+H]+ C2H5+ + M C2H4 + [M+H]+ stabiles Quasimolekül: M M + 1 Masse: M M + 1 Alkyladdition : C2H5+ + M (M)C2H5+ C3H5+ + M (M)C3H5+ Masse: M M + 29 Masse: M M + 41 Hydridabspaltung : CH5+ + M CH4 + H2 + [M-H]+ C2H5+ + M C2H5 + [M-H]+ Masse: M M - 1 Masse: M M - 1 Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 8/18 Abb.: Kombinierte Chemische Ionisations- und Elektronenstoß-Ionisations-Quelle Der Elektronenstrahl verläuft senkrecht zur Bildebene. 2) Atmosphärendruck Chemische Ionisation (APCI) (atmospheric pressure chemical ionisation) Thermospray-Ionenquelle zur LC/MS-Kopplung Heizung (200-600 °C) “Stickstoffvorhang“ Skimmer zum Massenanalysator Einlass 0.2-2 ml/min Durchmesser 100µm Zerstäubergas (N2) Make-up-Gas (H2O ..) Korona-Entlading (1 - 2 kV) Vakuumpumpe 1. Mechanismus (chemische Ionisation) Die Probenlösung wird zerstäubt und in einer beheizten Röhre verdampft. Eine Korona-Entladung durch ein extrem inhomogenes Feld an der Spitze einer Nadel erzeugt (unmittelbar vor dem Probeneinlass des MS) hochenergetische Primärelektronen. Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 9/18 Die freigesetzten Elektronen setzen eine Kette von Reaktionen in Gang, bei der letztlich positive Analytionen erzeugt werden. Fragmentierung spielt keine Rolle. N2 + eN2+ + 2N2 N4+ + H2O H2O+ + H2O N2+ + 2eN4+ + N2 H2O+ +2N2 H3O+ + OH(H2O)n H3O+ + M [M+H]+ + H2O (Bildung von Quasimolekülion durch Protontransfer) 2. Mechanismus (Ladungsrückstands-Ionisation) Die Elektronen aus der Korona-Entladung wechselwirken mit den Molekülen des Lösungsmittels, des Make-up-Gases (H2O, NH3, ..) und den Probenmolekülen. Reaktion: Tropfen Mikrotropfen Cluster Analytion (siehe Skizze) Da die Prozesse bei Atmosphärendruck ablaufen, finden genügend viele Wechselwirkungen mit den Probenmolekülen statt, um ausreichend viele Ionen zu erzeugen. Fragmentierungen spielen nur eine untergeordnete Rolle. APCI-Techniken sind sanfte Ionisationsprozesse und sehr gut geeignet für die Analyse polarer und nichtpolarer sowie labiler Komponenten mittlerer Molmasse. Es können fast alle pharmazeutischen Verbindungen analysiert werden. Die Methode ist auch gut geeignet zur Spurenanalytik von Luftschadstoffen. Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 10/18 3) Elektrospray Ionisation (ESI) Beim ESI-Vefahren wird eine Lösung des Analyten (10-3 bis 10-5 mol/l) bei Atmosphärendruck aus einer Kapillare in ein starkes elektrisches Feld versprüht. Dabei erfolgt eine pos. Ladungsübertragung. Je nach Spraykapillare und Flussrate unterscheidet man: mikro-ESI: Versprühung aus einer Stahlkapillare (Ø ca. 0,1 mm) mit Flussraten von 1-5 µg/min. Oft direkte Kopplung zu HPLC. Pumpe erforderlich. nano-ESI: Versprühung aus ausgezogenen Glaskapillaren (Ø ca. 1µm) mit Flussraten von ca. 20 nl/min. Keine Pumpe erforderlich. Aufbau einer ESI Quelle Da die Versprühung unter Normaldruckbedingungen erfolgt, ist der Transfer des Analyten zum Hochvakuum des Massenspektrometers auch hier sehr aufwendig. Rasche und feine Zerstäubung der an der Kapillarspitze austretenden Lösung in hoch geladene Initial-Tröpfchen aufgrund der hohen Feldstärke an der Spitze. Transfer der geladenen Tröpfen über eine geheizte Transferkapillare (Ø 100-500 µm) zur Vorvakuumstufe. Aufheizung der Tröpfchen und Desolvatisierung in der Vorvakuum- und der nachfolgenden Hochvakuumstufe. Beim Erreichen der Öffnung zum Massenanalysator haben sich durch vollständige Desolvatisierung freie Ionen gebildet. Die Effizienz der Ionenbildung (0,01 bis 0,1 vergl. EI 0,00001) wird zusätzlich erhöht, wenn gegen den Spraystrom ein Stickstoffstrom fließt. Mechanismus der Ionenfreisetzung An der Kapillarspitze wird die Flüssigkeitsoberfläche mit positiven Ladungsträgern angereichert. Die Ladungen werden zur negativen Gegenelektrode gezogen und bilden dabei den sog. Taylor Konus, der aus der Balance zwischen elektr. Feld und Oberflächenspannung resultiert. Ab einer gewissen Distanz erfolgt eine Destabilisierung und es werden Tropfen mit Durchmessern von ca. 2-10 µm und positiver Überschussladung in einem stabilen Spray emittiert. Das Lösungsmittel in den Tropfen verdampft und die Tröpfchen schrumpfen. Die Oberflächenladungsdichte nimmt zu. Bei Erreichen einer kritischen Größe bilden sich Ausstülpungen und es werden viele kleine Tröpfen freigesetzt, die nur ca. 2% der Masse, aber ca. 15% der Ladung des Muttertropfens tragen. Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 11/18 Der Vorgang wiederholt sich bis schließlich nur noch einzelne Tropfen von ca. 1 nm Ø vorliegen, die das Analytmolekül in einer Solvathülle enthalten. Ist das Lösungsmittel schließlich ganz verdampft, bleiben Ionen mit einer großen Anzahl von Ladungen übrig. Für Protein-Ionen gilt grob: eine Ladung/1000 amu. Leistungsmerkmale der ESI Besonderheit der ESI: Bildung mehrfach geladener Ionen bei M > 1000 Da. Es können alle herkömmlichen Analysatoren verwendet werden (außer TOF). Massenspektrometer mit geringem m/z-Messbereich (einige 1000) können benutzt werden, um Moleküle mit Molgewichten über 100000 zu bestimmen. Warum? Viele Signale zu einem Molekülion (mit verschiedenen Ladungen) tragen zur Verbesserung der Genauigkeit und Verlässlichkeit der Methode bei. Beispiel: ESI-Spektrum einer unbekannten Substanz mit Molekülgewicht M Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 12/18 Das Signal bei m1 (m/z = 1274.4) trägt z Ladungen (oder genauer gesagt ist z-fach protoniert). Daraus folgt also: m1 M z z M m1 z z Das Signal bei m2 (m/z = 991.4) trägt 4 Ladungen mehr (4 Protonen mehr). Daraus folgt: m2 M ( z 4) z4 m1 z z m2 ( z 4) z 4 M m2 ( z 4) z 4 z 13,9986 14 M m1 z z 1274,4 14 14 17828 4.2.2.3 Ionisation nicht verdampfbarer Proben (Bombardierungs-Ionisierungen; Desorptionsmethoden) Mit der Entwicklung der Bombardisierungs-Ionisation (ca. 1980) wurde die Anwendbarkeit der Massenspektrometrie grundsätzlich erweitert. Mit diesen “Desorptionsmethoden“ war es nun möglich, gasförmige Ionen von nichtflüchtigen, hochmolekularen Makromolekülen zu analysieren. Verfahren: FAB (Fast Atom Bombardment) MALDI (Matrix-Assisted Laser Desorption Ionisation) FIB/SIMS (Fast Ion Bombardment = Secondary Ion Mass Spectrometry) 1) Fast Atom Bombardment (FAB) Prinzip (Barber 1980): Die Probe wird in einer schwer flüchtigen, flüssigen Matrix (meist Glyzerin) molekular gelöst und auf dem FAB-Target (Metallträger) in die Ionenquelle gebracht. Beschuss der Probe mit neutralen Atomen (Ar, Xe) führt zur Desorption und Ionisation der Probe (z.B. Peptide, Nucleotide). Die keV-Primärteilchen werden durch Ladungsaustausch in der FAB-Kanone erzeugt. Xe + e- Xe+ + 2eXe+ Beschleunigung Xe+schnell Xe+schnell +Xe Xeschnell + Xe+ Es entsteht ein kontinuierlicher, relativ intensiver Ionenstrom, weil die vom Primärstrahl zerstäubte Oberfläche des Matrixtropfens durch Nachfließen ständig erneuert wird. Abb.: Schema einer FAB-Ionenquelle ("Fast Atom Bombardment") Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 13/18 Mechanismus der Desorption und Ionisation: Beim Eindringen in die flüssige Matrix geben die keV-Primärpartikel in einer Stoßkaskade ihre kinetische Energie ab, wobei sich bis zu 150 Å tiefe Krater in der Tropfenoberfläche bilden. Pro Primärpartikel werden innerhalb von ca. 10-10 s etwa 1000 Matrixmoleküle mit den darin gelösten Analytmolekülen desorbiert (impact cavities). Die kinetische Energie des auftreffenden Primärteilchens führt dabei zu einer kurzzeitigen thermischen Überhitzung (thermal spike). In der Folge bildet sich ein Übergangsbereich zwischen flüssiger Matrix und dem Vakuum (selvedge region), in dem ein hoher Druck (Aufheizen) und eine hohe Teilchendichte vorliegt1. Nachfolgend sinkt die innere Energie der thermisch angeregten Moleküle stark, da die Ausdehnungsarbeit bei der Expansion ins Vakuum aus dem Reservoir der Inneren Energie der Analyt- und Matrixmoleküle aufgebracht wird (intermolekulare Wechselwirkungsenergie). Die desorbierten Analytmoleküle kühlen bei der (weiteren) Expansion stark ab. Es verbleibt damit wenig Überschussenergie bei den generierten Ionen, so dass nur Quasimolekül-Kationen ([M+H]+, [M+Na]+ etc.) auftreten. Fragmentierungen spielen wegen der geringen Überschussenergie keine Rolle. heiße Stoßkaskade Abb.: Vorgänge bei verschiedenen Desorptionsmethoden (FAB, MALDI, FIB/SIMS) Kennzeichen von FAB-Spektren FAB-MS-Spektren zeigen einen für die Matrix charakteristischen Hintergrund, z.B. Ionenserien des Gycerins: [(Gly)n+H]+ , n = 1-15. mehrfach geladene Ionen werden bei FAB-MS selten beobachtet. 1 Die Ausdehnung von der flüssigen Phase in das Vakuum erfolgt ähnlich wie die Ausbreitung der Stosswelle bei einem Überschallstrahl (supersonic jet). Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 14/18 2) Matrix-Assisted Laser Desorption/Ionisation (MALDI) Sanfte Ionisierung sehr schwerer Moleküle (m bis 500000 amu) Prinzip (Hillenkamp und Kara, Münster 1987) Die zu untersuchende Probe wird mit einer konzentrierten Lösung der Matrix gemischt und auf einem metallischen Probenhalter getrocknet. Beim Trocknen werden die gelösten Analytmoleküle in die kristallisierende Matrix eingebaut. Nach der Überführung ins Vakuum wird die feste Matrix/Analyt-Mischung mit kurzen Laserimpulsen (0,5 - 20 ns UV, 5 - 200 ns IR) beschossen, wobei gasförmige Ionen freigesetzt werden. Mechanismus der Desorption/Ionisation: Mechanismus ähnlich wie bei der FAB aber keine Erneuerung der Matrixoberfläche. Bei der Verwendung von UV-Lasern (meist N2-Laser 337 nm oder Nd:YAG-Laser 266/355 nm) erfolgt Resonanzanregung der Elektronen des aromatischen Systems der Matrixmoleküle. Die Energie relaxiert innerhalb von ps in Schwingungszustände (IC: S1 S0*). Dabei erfolgt auch ein Energieübertrag auf die Analytmoleküle (rapid heating). Das ultraschnelle Aufheizen führt dazu, dass die mit der Fragmentierung (Spaltung) der Analytmoleküle konkurrierende Verdampfung überwiegt. Neben einem unerwünschten Matrixuntergrund entstehen überwiegend Molekülionen und Quasimolekül-Ionen (meist protoniert oder deprotoniert) der Form: [nM mY]k z.B. [M]+ , [M+H]+, [M-H]+ , [2M+H]+ , [M+2H]+ , [M+H]2+ , etc. Funktion der Matrix beim MALDI-Prozess Die Matrix isoliert die Analytmoleküle voneinander und schützt sie vor zu hoher Laserenergie. Die Analytmoleküle sollen bei der Laserwellenlänge nicht absorbieren! Die Matrixsubstanz stellt die zur Desorption der Analytmoleküle notwendige Energie zur Verfügung und liefert Protonen oder abstrahiert diese zur Ionisation des Analyten. Protonenübergang Abb.: Prinzip des MALDI-Prozesses (Matrix-Assisted-Laser-Desorption/Ionisation) Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 15/18 Wichtige Matrixsubstanzen Matrixmoleküle für die UV-Laseranregung müssen eine chromophore Gruppe im UV-Bereich besitzen, z.B. aromatische Systeme. Nikotinsäure DHB: 2,5 Dihydroxybenzoesäure Super-DHB: (2-Hydroxy-5-methoxybenzoesäure) MALDI-TOF MALDI wird üblicherweise in Kombination mit TOF durchgeführt. Verbesserte Auflösung und Massenbereich durch “verzögerte Extraktion” (Delayed Extraction) der Ionen (DE-MALDI-TOF). Abb.: Prinzip der verzögerten Ionenextraktion beim linearen TOF (DE-MALDI-TOF) Zur Zeit t = 0 werden die Analytmoleküle durch Laserbeschuss vom Probenträger freigesetzt, besitzen aber unterschiedliche Geschwindigkeiten. Nach der Zeit t = t1 wird das Beschleunigungspotential zwischen Probenteller und Gitter angelegt. (Hinweis: funktioniert nur, wenn das Potential zum Gitter hin flacher wird - warum?) ursprünglich schnelle Ionen befinden sich näher am Gitter und durchlaufen daher ein geringeres Potentialgefälle bei geringerer Feldstärke. Fokussierung Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 16/18 4.2.3 Detektoren Umwandlung des Ionenstromes nach dem Massenanalysator in einen elektrisch messbaren Strom. Ionenstrom: 10-9 bis 10-18 A : großer Dynamikbereich erforderlich Verstärkungsfaktor 106 - 108 notwendig Gebräuchliche Verfahren: Faraday Becher (Faraday Cup FC): Ionen fallen auf einen “Auffänger” und geben ihre Ladung an diesen ab. Der Entladungsstrom wird elektrisch verstärkt. Sekundär-Elektronen-Vervielfacher (SEV): Elektronen lösen Sekundärelektronen aus, die kaskadenartig verstärkt werden. Micro-Channel-Plate (MCP): Zweidimensionale Anordnung von vielen mikroskopischen SEV-Einheiten. 4.2.3.1 Faraday Becher Der Faraday-Becher wird zum direkten Auffangen der Ionen verwendet. Ein angeschlossenes Elektrometer misst damit direkt die Ladung der Ionen. Ionen aus dem Massenanalysator werden direkt in elektrischen Strom umgewandelt. Neutralisierung der Ladungen unabhängig von Masse und Energie Nachteile: -14 mittlere Empfindlichkeit (10 A) geringe Zeitauflösung (ms) 4.2.3.2 Sekundär-Elektronen-Vervielfacher (SEV) SEVs sind ähnlich konstruiert wie Photomultiplier. Sie bestehen aus einer Kaskade von Dynoden, die durch einen Lawineneffekt der Sekundärelektronen die eintreffende Ladung der Ionen verstärken. Auf die Konversionsdynode (1) auftreffende Ionen erzeugen Elektronen. Elektronenvervielfachung in einer Reihe weiterer Dynoden (2...11) An der Anode (A) wird der Elektronenstrom (Verstärkung bis 108) aufgefangen. -8 Zeitauflösung: ca. 10 s Nachweisgrenze: 1 Ion/ 10 s Nachteile: Vervielfachung ist abhängig von - Ionenmasse, - Ionenart, - Ionenenergie, Kontaminierung der Dynodenoberfläche Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 17/18 "Dynolyte" Photomultiplier (Gekapselter SEV) Ein sog. Dynolyte Photomultiplier arbeitet mit einem versiegelten SEV und kann deshalb nicht mit Ionen kontaminiert werden. Ionen aus dem Massenspektrometer werden an der Konversionsdynode in Elektronen konvertiert. Diese Elektronen werden beschleunigt und treffen auf einen Phosphor unmittelbar vor einem Photomultilpier. Die Photonen aus dem Phosphor treffen auf die Photokathode und lösen Elektronen aus. Verstärkung des Elektronenstromes in der nachfolgenden Dynodenkaskade. KonversionsDynode ( 2 kV) Photoelektronen Licht Elektronen Ionen Phosphor (+ 6..10 kV) Photomultiplier Elektronenabweisgitter Abb.: Schema eines hochempfindlichen Ionendetektors (Dynolyte) Channeltron (Channel Electron Multilier CEM “Posthorn“-Detektor) Ein Channeltron ist eine hornförmige Dynoden-Struktur die im Inneren mit einer Elektronenemittierenden Schicht (z.B. Antimon) überzogen ist. Ionen aus dem Massenspektrometer werden so abgelenkt, dass sie beim Auftreffen auf die Widerstandsschicht Elektronen freisetzen. Die hornförmige Bauweise ergibt eine kontinuierlich arbeitende Dynode. Der Potentialabfall entlang der Widerstandsschicht beschleunigt die Sekundärelektronen, welche durch einen Lawineneffekt vervielfacht werden. Nachweis von einzelnen Ionen möglich. Abb.: Schema einer kontinuierlich arbeitenden Dynode (Channeltron) Instrumentelle Analytik Massenspektrometrie MS Seite N151_MS_IonenquellenDetektoren_b_BAneu.doc - 18/18 4.2.3.3 Microchannel-Plate (MCP) Ein Channelplate ist eine zweidimensionale Elektronenverstärkungseinheit, die aus mehreren Millionen einzelnen Channeltrons (Innendurchmesser ca. 10 µm, Länge ca. 0,5 mm) besteht, welche innen mit einer Elektronen-emittierenden Schicht überzogen sind. An die Channeltron-Kapillaren wird eine hohe Spannung (einige kV) angelegt, so dass die Sekundärelektronen beschleunigt und vervielfacht werden. Die Anode (oder ein nachgeschalteter Phosphorschirm) kann ortsauflösend ausgelesen werden. Durch den kurzen Weg der Elektronen von der Kathode zur Anode werden Zeitauflösungen im 100 ps-Bereich erzielt. hohe Empfindlichkeit bzw. Verstärkung: (50 mV/ Ion) ortsabhängige Detektion möglich (z.B. bei Sektorfeldgeräten Erhöhung der Auflösung) Nachweis von einzelnen Ionen möglich. Abb.: Einzelansicht einer Kapillare einer Microchannel-Plate Abb.: Querschnitt und Schema einer Microchannel-Plate (MCP)