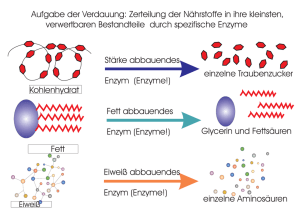
1_01.QXD NEU - Universität des Saarlandes
Werbung

Enzyme an der Schnittstelle zwischen Natur und Technik – Screening nach neuen Biokatalysatoren von Andreas Tholey und Elmar Heinzle Enzyme sind leistungsfähige Biokatalysatoren Nahezu alle Eigenschaften, die einen lebenden Organismus charakterisieren, werden von Proteinen beeinflusst. Diese – unabhängig von ihrer Funktion – einheitlich aus einem Satz von 20 Aminosäuren aufgebauten biologischen Makromoleküle transportieren und speichern Stoffe und Energie, dienen als Gerüstsubstanzen beim Aufbau biologischer Strukturen und sind essentielle Bestandteile bei der Steuerung der Replikation und der Interaktion von Zellen. Eine der wichtigsten Aufgaben von Proteinen stellt die Katalyse biochemischer Prozesse dar. Diese speziellen Proteine werden Enzyme genannt. Sämtliche Prozesse in lebenden Systemen beruhen auf der Abfolge chemischer Reaktionen. Viele dieser Reaktionen könnten aber ohne die Katalyse durch Enzyme gar nicht ablaufen. Dabei senken diese Biokatalysatoren – genauso wie klassische chemische Katalysatoren – nur die Aktivierungsenergie der Reaktion ab, beeinflussen das Gleichgewicht der Reaktion jedoch nicht. Die Wirkung der Enzyme beruht dabei auf verschiedenen Prinzipien. So bewirken sie beispielsweise, dass zwei Reaktionspartner in eine günstige räumliche 46 Enzyme werden bereits heute in vielen Bereichen zur Synthese von verschiedenen Chemikalien und pharmazeutisch aktiven Substanzen verwendet. Die Entwicklung und Modifikation geeigneter Biokatalysatoren ist daher ein wichtiger Zweig der Biotechnologie. In der Arbeitsgruppe Technische Biochemie an der Universität des Saarlandes werden Methoden ausgearbeitet, welche die Suche nach den effektivsten Enzymen erleichtern sollen. Diese Methoden stellen dabei gleichzeitig eine wichtige Erweiterung des Repertoirs an Werkzeugen für die biomedizinische Forschung dar. In diesem Artikel geben wir einen kurzen Überblick über aktuelle Entwicklungen und stellen eigene Änsatze auf diesem Gebiet kurz vor. Nähe zueinander gebracht werden, oder dass chemische Übergangszustände bei der Reaktion stabilisiert werden. Enzyme besitzen, wie alle Proteine, eine definierte dreidimensionale Struktur, die auch ihre Aktivität beeinflusst. In dieser dreidimensionalen Struktur können in den sogenannten aktiven Zentren auch Zonen geschaffen werden, in denen ein anderes Milieu vorherrscht als in der meist wässrigen Umgebung der Zelle. Dieses Schaffen einer speziellen Mikroumgebung – zum Beispiel einer hydrophoben Tasche – führt dann dazu, dass auch solche chemische Reaktionen ablaufen können, die in der wässrigen Umgebung nicht möglich wären. Im Gegensatz zu klassischen anorganischen Katalysatoren vereinen Enzyme eine Reihe ein- zigartiger Eigenschaften in sich. Dies macht sie unter anderem interessant für die Anwendung im technischen Bereich, in der pharmazeutischen Industrie, der Medizin und im Umweltschutz. Die Bedingungen enzymatisch katalysierter Umsetzungen sind in der Regel schonender als die klassischer chemischer Reaktionen. Damit können solche biotechnisch geführten Prozesse oft unter umweltfreundlicheren und zugleich ökonomisch günstigeren Bedingungen durchgeführt werden [1]. Im Laufe der Evolution entwickelten sich Enzyme für eine große Anzahl unterschiedlichster chemischer Reaktionen. Diese Biokatalysatoren zeichnen sich dabei durch eine hohe Selektivität und Spezifität aus. Für die Anwendungen in der Chemie sind hierbei insbesondere UniversitŠt des Saarlandes die Regio- und die Enantioselektivität wichtig. Durch die Regiospezifität – das heisst, die Fähigkeit, Moleküle nur an bestimmten Stellen zu bearbeiten – entfallen im Gegensatz zu chemischen Reaktionen oft zeitund materialaufwendige Schritte zum Schutz anderer Funktionalitäten in eben diesem Molekül. Eine der wichtigsten Eigenschaften von Enzymen ist ihre Fähigkeit, Reaktionen enantioselektiv durchführen zu können, dass heißt, sie können gezielt eine stereochemisch reine, chirale Verbindung darstellen. Solche Verbindungen sind sehr wichtig zum Beispiel bei der Synthese von Pharmazeutika. Dagegen stellt die chemische Synthese solcher enantiomerenreiner Verbindungen auch heute noch hohe Anforderungen an die Chemie. Weitere günstige Eigenschaften enzymatischer Umsetzungen sind die oft hohen Ausbeuten und vor allem die Vermeidung von Nebenprodukten. nicht-natürlichen Oberflächen zu optimieren [2]. Weiterhin werden bei solchen Prozessen oft nicht-natürliche Substrate umgesetzt, was die Ausbeuten solcher Reaktionen meist negativ beeinflusst. Die Neuentwicklung oder Modifikation schon vorhandener Enzyme verläuft in mehreren Schritten (vgl. Abbildung 1). Zunächst einmal muss die Fragestellung genau definiert werden, das heisst, es muss klar sein, mit welchen Bedingungen eine neuer Katalysator im Produktionsprozess zurechtkommen muss und welche Substanzklassen umgesetzt werden sollen. Danach erfolgt die eigentliche Modifikation, zumeist mit molekularbiologischen Methoden. Dabei wird in den meisten Fällen nicht ein einzelnes neues Enzym geschaffen, sondern es werden gleich ganze Bibliotheken neuer Biokatalysatoren erzeugt. In einem letzten Schritt erfolgt dann das Screening dieser Bibliotheken oder Enzymbänke nach dem für die jeweilige Fragestellung effektivsten Kandidaten [3]. Das Screening kann aber auch in der Weise erfolgen, dass ein bestimmtes Enzym auf seine Fähigkeit getestet werden soll, welche Substrate es umsetzen kann oder – und dies ist insbesondere in der Wirkstoffforschung wichtig – ob man aus einer Substanzbibliothek geeignete Inhibitoren für dieses Enzym finden kann. Diese Substanzbibliotheken werden dabei zumeist mit Methoden der kombinatorischen Chemie hergestellt. Im Folgenden sollen die drei Hauptschritte der Entwicklung und Modifikation von Enzymen angesprochen werden, wobei der Schwerpunkt auf der Besprechung neuer Screening-Me- Abb. 1: Schritte bei der Entwicklung eines neuen Biokatalysators Probleme beim technischen Einsatz von Enzymen In der Natur wurden Enzyme so selektioniert, dass sie in einer definierten, zumeist wässrigen Umgebung in einem Netzwerk mit vielen anderen Enzymen und biologischen Strukturen und mit definierten Substraten optimal arbeiten. Sie sind in der Regel dabei nicht in Bezug auf Stabilität, speziell unter den anspruchsvollen Reaktionsbedingungen, wie sie in chemischtechnologischen Prozessen auftreten, selektioniert worden. Um diese Biokatalysatoren also technisch nutzen zu können, ist es notwendig, sie in Bezug auf Temperaturstabilität, den Einsatz in organischen Lösungsmitteln, Reaktionen direkt in flüssigen Substraten oder den Kontakt mit magazin forschung 1/2001 47 thoden liegt, die derzeit unter anderem in der Arbeitsgruppe Technische Biochemie an der Universität des Saarlandes entwickelt werden. Wie können neue oder modifizierte Biokatalysatoren hergestellt werden? Eine enzymkatalysierte Umsetzung stellt oft nur eine Stufe in einer Sequenz von Syntheseschritten dar. Dies muss bereits in der frühestmöglichen Entwicklungsphase für einen neuen Biokatalysator beachtet werden. Das Design neuer Enzyme kann dabei auf rationalem oder evolutionärem Wege erfolgen. In jedem Fall bedeutet jedoch die Kenntnis der dreidimensionalen Struktur des jeweiligen Vorläuferenzmys eine deutliche Erleichterung auf dem Wege zum effektiven, maßgeschneiderten Biokatalysator. Dabei spielen in zunehmende Maße auch Erkenntnisse aus der Untersuchung von Struktur-AktivitätsBeziehungen (structure-activity relationships, SAR) in Kombination mit Methoden der Bioinformatik eine wichtige Rolle. Quellen für neue Biokatalysatoren können bislang unentdeckte oder wenig erforschte Enzyme aus natürlichen Quellen, wie zum Beispiel aus Mikroorganismen, sein [4]. Moderne Methoden zur Veränderung von Enzymstrukturen durch Mutationen beginnen mit der Identifizierung und Sequenzierung des entsprechenden codierenden Gens. Für eine wachsende Zahl von Mikroorganismen sind komplette Gensequenzen in Datenbanken gespeichert, während die Aufklärung der genetischen Informationen aus höheren Organismen wie zum Beispiel Pilzen 48 meist viel aufwendiger ist. Die Enzyme werden in Gastzellen (host) wie B. subtilis, E. coli oder Hefen kloniert und exprimiert. Um die Enzymaktivität leichter bestimmen zu können ist es dabei oft wünschenswert, die entsprechenden Proteine an der Zelloberfläche dieser Organismen zu präsentieren. Nachdem ein geeigneter Gastorganismus gefunden ist, können verschiedene Methoden eingesetzt werden, um die eigentlichen Veränderungen am Enzym vorzunehmen. Dabei stehen Methoden der Mutagenese [4], des gene-shufflings [5], der gerichteten Evolution [6, 7] oder kombinatorische Methoden [8] zur Verfügung. Neben der direkten Veränderung der Primärstruktur – also der Abfolge der einzelnen Aminosäuren – im Enzym gibt es weitere Möglichkeiten, um Biokatalysatoren für den Einsatz unter den oben beschriebenen Bedingungen zu optimieren. So führt zum Beispiel die Quervernetzung von Enzymen (crosslinked-enzyme crystals, (CLEC) [9]) oder die Immobilisierung an geeigneten Trägersubstanzen [10] oft zu einer deutlichen Steigerung der Stabilität der Enzyme. Bei derartigen Experimenten zur Veränderung oder Neusynthese von Enzymen wird meist eine grosse Anzahl von Mutanten erzeugt – eine sogenannte Enzymbank – aus deren Vielfalt nun in einem Screening-Prozess die effektivsten Katalysatoren ausgewählt werden müssen. Die Ergebnisse dieser Screening-Prozedur fließen dann wiederum in das Design einer nächsten Generation weiter verbesserter Biokatalysatoren ein, bis nach mehrmaligen Durchlaufen dieses iterativen Prozesses die ge- wünschten Eigenschaften realisiert sind. Das Screening Der Screening-Prozess kann in drei unterschiedliche Stufen unterteilt werden. In der ersten Stufe wird eine schnelle und qualitative Identifizierung katalytisch aktiver Enzyme für eine bestimmte Umsetzung angestrebt. Diese Verbindungen werden dann in einer zweiten Stufe in einem halb-quantitativen Test weiter selektioniert, wobei die effektivsten Kandidaten die dritte Stufe des quantitativen Screenings erreichen. Hier werden dann im günstigsten Fall gleich die kinetischen Konstanten mit bestimmt. Im Prinzip gibt es zwei verschiedene Arten des Screenings. Die erste betrifft das Screenen einer Enzymbank nach dem effektivsten Biokatalysator. Die Zweite ist das Screenen nach geeigneten Substraten oder Inhibitoren für ein bestimmtes Enzym. Die Kenntnis geeigneter Substrate oder inhibierender Substanzen kann ihrerseits wichtige Erkenntnisse über katalytische Mechanismen von Enzymen liefern, zum Beispiel durch die Kombination der bereits oben erwähnten Struktur-AktivitätsBeziehungen und bioinformatischen Methoden und Studien im Bereich des molecular modeling. Die Optimierung von Enzymen richtet sich nach den Erfordernissen der späteren Anwendung. Für industrielle Prozesse sind beispielsweise schnelle Reaktionsabläufe, hohe Enzymstabilitäten und vor allem die Regio- und Enantiospezifität die wichtigsten Kriterien, wohingegen die Affinität zu bestimmten Substraten oder die Substratspezifität mehr im Hintergrund stehen. Bei der Wahl der BedinUniversitŠt des Saarlandes Prof. Dr.-Ing. ELMAR HEINZLE, geb. 1949 in Götzis, Österreich, studierte an der Technischen Universität in Graz Technische Chemie, Studienzweig Biochemie. Nach dem Diplom promovierte er dort am Institut für Biotechnologie. Ab 1978 war er Assistent am Technisch Chemischen Labor der ETH Zürich. Ab 1985 war er für zwei Jahre Leiter des Labors für Biotechnologie und Biochemie der Forschungsgesellschaft Joanneum in Graz, kam dann wieder an die ETH und habilitierte 1994 an der Abteilung Chemie im Lehrgebiet Biochemische Reaktionstechnik. Seit Juni 1997 ist er Professor für Technische Biochemie an der Universität des Saarlandes. An der Universität des Saarlandes forscht er im Bereich der frühen Entwicklung biotechnologischer Prozesse an der Entwicklung neuer Biokatalysatoren. Neue Möglichkeiten der Detektion (Optik, Massenspektrometrie) kombiniert mit Modellierung und Simulation der ablaufenden Prozesse werden für Screeningverfahren eingesetzt. Dies geschieht einerseits mit einstufigen enzymkatalysierten Reaktionen und andererseits mit ganzen Zellen, in denen komplexe metabolische Netzwerke studiert werden. gungen in einem Screening ist auch zu berücksichtigen, in welchem Reaktionsumfeld die Enzyme später eingesetzt werden, ob also beispielsweise mit isolierten, gereinigten Enzymen gearbeitet werden soll, oder ob die Reaktionen in ungereinigten Zelllysaten oder gar in ganzen Zellen durchgeführt werden. Aus Gründen des Effektivität, des hohen Probendurchsatzes (high throughput screening) und des Umweltschutzes – hier sei der möglichst gering zu haltende Einsatz organischer Lösungsmittel als Beispiel genannt – ist es erstrebenswert, dass Screeningprozeduren schnell ablaufen, sehr sensitiv und spezifisch, miniaturisierbar und automatisierbar sind. Oft sind jedoch nicht alle dieser Anforderungen in einem Prozess realisierbar. Analytische Methoden Analytische Methoden für das Screening können in drei Klassen eingeteilt werden [3]: nichtinvasive Methoden, direkt-invasive Methoden und Methoden, die mit Trennmethoden gekoppelt sind. Nicht-invasive Methomagazin forschung 1/2001 den beruhen auf der Wechselwirkung des Analyten mit elektromagnetischer Strahlung. In diese Klasse lassen sich alle optischen Methoden wie die Messung der Absorption von Licht (UV/Vis-Spektroskopie) sowie die kernmagnetische Resonanz (nuclear magnetic resonance, NMR) einordnen. Invasive Methoden wie die später zu besprechende Massenspektrometrie, erfordern eine Probenahme. Chromatographische und elektrophoretische Trennmethoden sind ihrerseits zur Detektion mit einer der Methoden der nicht-invasiven beziehungsweise invasiven Detektion gekoppelt. Optische Methoden Die klassische Methode zur Analyse von Enzymaktivitäten ist die UV/Vis-Spektroskopie. Sie beruht auf der Änderung des Absorptionsverhaltens eines Substrates während des Reaktionsprozesses, was sowohl durch die Veränderung oder die Freisetzung eines Chromophors – einer Gruppierung, welche die Fähigkeit besitzt, Licht in einem bestimmten Wellenlängenbe- reich zu absorbieren – verursacht werden kann. Eine Vielzahl von Enzymreaktionen kann so durch die Messung des Absorptionsverhaltens eines Substrates oder eines essentiellen Cofaktors spektrophotometrisch analysiert werden. Klassische Beispiele hierfür sind die Oxidoreduktasen, die NAD/NADH als Cofaktor verwenden oder Acetyltransferasen, bei denen der Umsatz des Acetyl-Coenzym A direkt photometrisch bestimmt werden kann. Spektrophotometrische Tests bieten eine Reihe von Vorteilen. So können sie in Mikrotiterplatten oder als Filterpapier-Assays durchgeführt werden. Beides erlaubt einen hohen Probendurchsatz bei gleichzeitig geringen Proben- und Lösungsmittelmengen. Des Weiteren ist ihre Durchführung einfach, der apparative Aufwand ist gering und die Prozeduren lassen sich einfach automatisieren. Dennoch sind diese Methoden nicht universell einsetzbar. So kann beispielsweise die biologische oder chemische Matrix – also die Umgebung, in der die Reaktion abläuft – die Messergebnisse durch Streuung oder zu starke Eigenabsorption stören oder eine Messung gar unmöglich machen. Oft sind diese Test daher nur mit aufgereinigten Enzymen durchführbar, wohingegen bereits in Zelllysaten Probleme auftreten. Ein weiteres Problem stellen Reaktionen dar, bei denen weder Cofaktoren, noch Substrate oder Produkte eine chromophore Gruppe besitzen. In solchen Fällen wurden oft künstliche Substratanaloga eingesetzt, um die Enzymreaktion verfolgen zu können. Dabei besteht jedoch sehr leicht die Gefahr, dass ein solches Substratanalogon zu vollständig irreführenden Ergebnissen führt, oder dass die Ergebnisse sich zumin49 dest nicht vollständig auf die realen Substrate übertragen lassen. So führte beispielsweise der Einsatz von o-Nitrophenyl-β-DGalactopyranosid, einem oft benutzten Substratanalogon zur Messung der Aktivität der β-Galactosidase, zur Bestimmung vollständig anderer kinetischer Konstanten als denjenigen, die beim Umsatz des realen Substrates Lactose gemessen werden konnten [11]. Diese Probleme können unter anderem durch unterschiedliche räumliche Ausdehnung von Substrat und Substratanalogon oder unterschiedliche hydrophile/hydrophobe oder ladungsbasierte Interaktionen zwischen Substratanalogon und Enzym beziehungsweise Substrat und Enzym verursacht werden. Daher besteht in vielen Fällen ein Bedarf an der Entwicklung neuer Screening-Techniken. Trotz dieser beschriebenen Nachteile lassen sich mit spektrophotometrischen Methoden des Screenings beeindruckende Fortschritte erzielen. So war es möglich, die Enantioselektivität einer Lipase durch Mutation mittels der sogenannten ep-PCR (error prone-polymerase chain reaction) in nur vier Generationen von einem Enantiomerenüberschuss (enantiomeric excess, ee) von 2% auf über 88% zu steigern [12]. Das Screening erfolgte dabei durch die parallele Messung des Umsatzes der jeweils enantiomerenreinen Substrate in Mikrotiterplatten, wobei die Absorptionsänderung – und damit der Reaktionsfortschritt – für die beiden Enantiomere direkt photometrisch miteinander verglichen werden konnte. Neben der UV/Vis-Spektroskopie stellen auf Messung der Fluoreszenz basierende Tests eine weitere Schlüsseltechno50 Abb.2: pH-stat- und pH-Dyn-Assay zum Screenen von Enzymreaktionen, bei denen es zur Bildung oder dem Verbrauch von Säure kommt logie zum Screenen von Enzymen dar. Die Messung der Fluoreszenz ist etwa 1000 mal empfindlicher als vergleichbare Absorptionsmessungen, was diese Methode insbesondere zur Lösung von Problemen interessant macht, bei denen nur minimale Probenmengen vorhanden sind, so zum Beispiel im medizinisch-pharmazeutischen Bereich. Wegen des deutlich geringeren Substanzbedarfs kommt es auch seltener zu Problemen, die durch die geringe Löslichkeit verschiedener Substanzen verursacht werden. Dennoch gibt es auch bei fluoreszenz-basierten Messsystemen einige Schwierigkeiten, welche die Einsatzmöglichkeiten einschränken. So treten die bereits oben besprochenen Probleme beim Einsatz nicht-natürlicher, mit fluorophoren Gruppen markierter Substrate auf. Weitere Probleme ergeben sich aus der Anwesenheit von Gruppen, welche die Fluoreszenzsignale abschwächen oder gar löschen können (quenching) beziehungsweise aus der Autofluoreszenz der Matrix, welche die eigentlichen Signale überdecken und maskieren kann. Aus diesen Gründen ist es besser anstelle der Intensität der Fluoreszenz entweder die Lebensdauer eines Fluoreszenzsignals, die Energieübertragung auf ein geeigneteres Fluorophor oder die Anisotropie zu messen [13]. Unter Beachtung dieser Prämissen konnte eine Reihe verschiedenster Tech- niken entwickelt werden, um mittels fluorimetrischer Methoden die Aktivität von Enzymen sowie die Interaktionen von Biomolekülen untereinander zu studieren [14]. Bei einer Reihe enzymatisch katalysierter Reaktionen wird Säure – in Form von Protonen – verbraucht oder gebildet. Die daraus resultierende Veränderung des pH-Wertes kann daher als sensitiver Monitor für den Fortschritt einer solchen Reaktion verwendet werden. Bei der sogenannten pH-stat-Methode wird Säure oder Base zum Reaktionsansatz zupipettiert, um einen konstanten pH-Wert, gemessen mit konventionellen Glaselektroden oder pH-Indikatoren, zu gewährleisten (vgl. Abbildung 2 a) [15]. In der Literatur wird beschrieben, dass beim Einsatz von pH-Indikatoren mit ähnlichen pKaWerten wie die der Substrate beziehungsweise der Produkte lineare Abhängigkeiten zwischen Reaktionsfortschritt und Absorptionsänderung der Indikatoren bestehen [16]. Diese Methode besitzt jedoch den Nachteil, das Indikatorsysteme mit ähnlichen pKa-Werten verfügbar sein müssen. Daher arbeiten Gernot John und Svenja Weiß in unserer Gruppe zur Zeit an Alternativen zu dieser Methode [17]. Der genannte Nachteil kann dadurch umgangen werden, dass die Dynamik des gesamten analytischen Systems UniversitŠt des Saarlandes unter Verwendung aller Stoffbilanzgleichungen und eines kinetischen Modells vollständig modelliert/simuliert wird (Abb. 2 b). Mit diesem sogenannten pH-dyn-Assay ist es möglich, durch Methoden der numerischen Simulation und der Parameterschätzung die kinetischen Parameter einer Reaktion ohne vorherige Linearisierung zu bestimmen. In Zusammenarbeit mit der Gruppe von Prof. I. Klimant an der Universität Regensburg sowie zwei kleineren mittelständischen Betrieben werden in einem von der Bundesstiftung Umwelt geförderten Projekt neuartige Mikrotiterplatten entwickelt, bei denen der Indikator am Boden der Platte immobilisiert ist. Dazu wird ein fluoreszierender pH-Indikator in einer dünnen Polymerschicht am Boden der Mikrotiterplatte immobilisiert (Abbildung 3). Änderungen der Fluoreszenzintensität oder der Lebensdauer können dann einfach in einem kommerziell erhältlichen Reader (Abbildung 4) gemessen und somit zur Bestimmung der pH-Änderung und damit zur Bestimmung des Reaktionsverlaufs verwendet werden. Durch den Einsatz von Abb. 3: Mikrotiterplatte mit immobilisiertem Indikator. (Mit freundlicher Genehmigung der Presens GmbH, Regensburg) Ruthenium-Diimin-Komplexen als Indikatoren ist es bislang möglich, dieses Prinzip auch zur Bestimmung der Konzentration von gelöstem Sauerstoff und Kohlendioxid und zur Quantifizierung verschiedener Kationen und von Zuckern einzusetzen [18]. Ein Ziel dieser Entwicklungen ist unter anderem die Anzucht von Säugerzellen auf solchen Platten, die dann als Ersatz für Tiermodelle dienen können. Dabei können zum Beispiel nach Gabe einer pharmazeutisch wirksamen Substanz aus den mit Sensoren gemessenen Änderungen des pH oder des gelösten Sauerstoffs Veränderungen der Aktivität der Zellen bestimmt werden. Massenspektrometrie als neues Tool im Screening Eine der aktuellsten Entwicklungen im Bereich des Screening betrifft den Einsatz der Massenspektrometrie (MS). Bei dieser Methode ist es notwendig, eine Probe der zu screenenden Substanz zu entnehmen, weshalb Abb. 4: a) Reader zum Auslesen von Mikrotiterplatten. b) Mikrotiterplatte im Reader. Abgebildet ist eine zusätzliche Piezopumpe zum Zudosieren kleinster Mengen von Flüssigkeiten magazin forschung 1/2001 51 Abb. 5: Prinzip der Elektrospray-Ionisation sie zur Klasse der direkt-invasiven Screening-Methoden zu zählen ist. Die Entwicklung schonender Ionisationstechniken wie der Elektrospray-Ionisation (ESI) [19] und der matrixunterstützten Laserdesorptions-Ionisation (matrix assisted laser desorption/ ionization, MALDI) [20] hat die Massenspektrometrie in den Blickpunkt des Interesses in fast allen Bereichen der Biochemie und der Biotechnologie gerückt. Die Massenspektrometrie bietet eine Reihe von Vorteilen, die sie zu einer idealen Methode für ein Screening von Biokatalysatoren machen. Die Messungen sind sehr schnell, benötigen nur kleinste Probenmengen im Nano- oder Picomolbereich und können weitestgehend automatisiert werden. Desweiteren bieten sie die Möglichkeit der Kopplung mit vorgeschalteten Trennprozeduren wie der Flüssigkeitschromatographie oder elektrophoretischen Methoden. Sowohl die ESI-MS als auch die MALDI-MS bieten die Möglichkeit, nicht nur die Substrate und Produkte einer Enzymreaktion zu messen, sondern man kann auch den Biokatalysator selbst untersuchen. So können beispielsweise während der Reaktion auftretende kovalente Veränderungen an den Enzymen, die zu einem Aktivititätsverlust 52 oder gar der Zerstörung des Katalysators führen können, relativ schnell detektiert werden. So wurden in unserer Gruppe von Gerhard Treitz mittels MALDI-Massenspektrometrie oxidative Veränderungen von Oxidasen während ihres Einsatzes als Biokatalysatoren untersucht. Das Prinzip der ElektrosprayIonisation besteht darin, dass ein Analyt in einem Lösungsmittel aufgenommen und über eine feine Kapillare in ein elektrisches Feld unter Normaldruck gesprüht wird (vgl. Abbildung 5). Dabei laden sich die Lösungsmitteltropfen elektrisch auf. Das Lösungsmittel verdampft, die Ladungsdichte auf den kleiner werdenden Tropfen wird immer größer, bis es schließlich durch die elektrostatische Abstoßung zur sogenannten Coulomb-Ex- plosion kommt. Der Tropfen „explodiert“ und bildet nun viele kleine Tropfen, die den beschrieben Vorgang mehrmals wieder durchlaufen, bis schließlich die nackten, desolvatisierten Analytionen vorliegen. Diese werden ins Hochvakuum überführt, beschleunigt und mit einem geeigneten Massenanalysator nach ihrem Masse-zu-Ladungsverhältnis aufgetrennt. Nach der Detektion dieser Ionen kann an einem PC das Massenspektrum aufgezeichnet werden. Die Methode der ESI-MS konnte mehrfach erfolgreich zur Quantifizierung von Substraten und Produkten von enzymatisch katalysierten Reaktionen angewendet werden. Weiterhin ist es auch gelungen, durch den Einsatz von isotopenmarkierten Substanzen in der Form von pseudo-Enantiomeren oder pseudo-prochiralen Verbindungen die Enantioselektivität solcher Biokonversionen zu bestimmen [21]. Daneben wird die ESI-MS auch in hohem Maße beim Screening von kombinatorisch erzeugten Substanzbibliotheken in der Wirkstoffforschung eingesetzt. Ein Forschungsschwerpunkt in unserer Gruppe stellt die Entwicklung von Methoden des Screenings mittels MALDI-MS Dr. ANDREAS THOLEY, geb. 1968, studierte in Saarbrücken Chemie und arbeitete während seiner Diplomarbeit an der Entwicklung von Modellpeptiden zum Studium posttranslationaler Proteinmodifikationen. Während der Promotion am Deutschen Krebsforschungszentrum in Heidelberg beschäftigte er sich mit den strukturellen Konsequenzen der Proteinphosphorylierung und der Entwicklung massenspektrometrischer Methoden zur Identifizierung von Phosphorylierungsstellen in Proteinen. Zur Zeit arbeitet er in der Arbeitsgruppe von Prof. Heinzle (Technische Biochemie) an der Entwicklung von massenspektrometrischen Methoden zum Screenen von Biokatalysatoren und zur Proteomanalyse. UniversitŠt des Saarlandes vakuum mit einem Laser beschossen, wobei jeder Schuss nur einige Nanosekunden dauert. Dadurch verdampfen Matrix und Analyt und werden dabei gleichzeitig schonend ionisiert. Abb. 6: Prinzip der MALDI-MS. a) Matrixkristalle (2,5-Dihydroxybenzoesäure) in einer Mikroskopaufnahme (100-fach). b) Prinzip der MALDI-Ionisation. c) Target zur Aufnahme von 384 Proben. d) MALDI-Massenspektrometer in der Abteilung Technische Biochemie dar. Bei dieser Art der MS wird der Analyt mit einem kleinen organischen Molekül – der Matrix – auf einem Metalltarget co- kristallisiert (vgl. Abbildung 6). Das Matrix/Analyt-Gemisch wird dann in der Probenkammer des Massenspektrometers unter Hoch- Diese Ionisation ist so schonend (die meiste Energie wird von der in großem Überschuss vorliegenden Matrix absorbiert und dann auf den Analyten übertragen), dass selbst große Proteine oder gar Antikörper ohne Fragmentierung, das heißt ohne Aufbrechen von kovalenten Bindungen, mit dieser Methode analysiert werden können. Es können aber auch niedermolekulare Verbindungen ohne Fragmentierung ionisiert werden, was die MALDI-MS zusammen mit der relativ großen Toleranz gegenüber in der Probe enthaltenen Salzen (zum Beispiel Puffersustanzen) zu einem idealen Werkzeug für das Screening macht. Jedoch gilt es dabei noch eine Reihe von Schwie- Abb. 7: Quantifizierung mittels MALDI-MS. a) Modellreaktion: Lipasekatalysierte Umsetzung von racemischem 1-Phenylethylamin (rac-PEA) zum enantiomerenreinen 2-Methoxy-N-[(1R)-1-phenylethyl]-acetamid (MET). b) MALDIKalibrationsgerade zur Quantifizierung von PEA unter Verwendung eines deuterierten internen Standards. c) Vergleich einer Enzymkinetik gemessen mittels Gaschromatographie und MALDI-MS. magazin forschung 1/2001 53 rigkeiten zu überwinden, die insbesondere die Quantifizierung betreffen. Das Hauptproblem stellt die inhomogene Verteilung von Matrix und Analyt in einem Probenspot dar. Dies führt dazu, dass die Intensitäten der Signale keine ausreichend genaue Schuss-zu-Schuss-Reproduzierbarkeit aufweisen. Der einfachste Weg, um dieses Problem zu umgehen, ist das Analysieren eines Probenspots an mehreren Stellen mit anschließender Addition der Einzelsignale sowie der Einsatz geeigneter interner Standards. Dabei werden bevorzugt Substanzen verwendet, die möglichst hohe molekulare Homologie zum Analyten aufweisen, wie zum Beispiel mit Deuterium markierte Verbindungen [22]. Auf diese Art und Weise war es uns möglich, eine lineare Korrelation zwischen den relativen Signalintensitäten und der relativen Konzentration des Analyten zu messen (vgl. Abbildung 7). Als Modellsystem untersuchte MinJung Kang in unserer Gruppe in einem von der BASF finanziell unterstützten Projekt eine Lipase-katalysierte Umsetzung von racemischem 1-Phenylethylamin (PEA) zum 2-Methoxy-N-[(1R)1-phenylethyl]-acetamid (MET). Durch diese Messungen war es weiterhin möglich, die kinetischen Konstanten der enzymkatalysierten Umsetzung zu bestimmen, wobei die Werte sehr gut mit denen durch klassische Messungen mittels Gaschromatographie übereinstimmten. In einigen Fällen ist eine direkte Messung niedermolekularer Verbindungen nur nach vorhergehender Derivatisierung der Analyten möglich, da die Verbindungen entweder zu flüchtig sind und im Hochvakuum verdampfen, bevor die eigentliche Messung beginnt oder dass sie sich wegen ihres molekularen 54 Das Screenen von Biokatalysatoren stellt auch ein Teilprojekt des neuen von der Landesregierung geförderten Programms „Enzyme – Tools, Targets, Therapheutics“ an der Universität des Saarlandes dar. In diesem interdisziplinären Forschungsverbund stehen die Entwicklung von leistungsfähigen Biokatalysatoren und von potenten Inhibitoren für therapeutische Zwecke im Zentrum des Interesses. Weitere Informationen sind im Internet unter der Adresse:“http://www.uni-saarland.de/fak8/heinzle/research/ ETTT_Projekte.htm” verfügbar. Aufbaus nicht direkt ionisieren lassen. In den meisten Fällen können für diese Derivatisierungen jedoch einfachste organische Reaktionen durchgeführt werden, so zum Beispiel die Bildung von Oximen aus flüchtigen Aldehyden. Umgekehrt gelang es Klaus Hollemeyer aus unserer Gruppe in einer Kooperation mit Andreas Speicher aus der Organischen Chemie, mittels MALDI-MS ohne den Einsatz einer zusätzlichen Matrix in Zelllysaten aus Moosen halogenierte Naturstoffe nachzuweisen [23]. Damit konnte indirekt die Enzymaktivität von bis dato in diesen Organismen nicht bekannten Haloperoxidasen nachgewiesen werden. Screening mit Trennmethoden Neben den oben erwähnten Methoden des Screenings gibt es noch eine Reihe weiterer Verfahren, so zum Beispiel chromatographische und elektrophoretische Methoden. Diese sind jedoch oft sehr zeitaufwendig und verwenden in den meisten Fällen die oben genannten Methoden zur Detektion. Dennoch sind sie, speziell zur Analyse der Enantiomerenreinheit, von Interesse. Diese auf Trennoperationen beruhenden Methoden sind insbesondere in der letzten, quantitativen Stufe eines Screening-Prozesses anwendbar, wenn nur noch die effektivsten Kandidaten der ersten Screening-Stufen miteinander verglichen werden sollen. Ausblick In Zukunft werden Enzyme eine zunehmend wichtige Rolle bei industriellen Prozessen spielen. Daher ist die Entwicklung von Methoden zum Screening dieser Biokatalysatoren ein sehr wichtiges Gebiet in der Biotechnologie. Die wichtigsten Aufgaben werden in den nächsten Jahren die Automatisierung und die Miniaturisierung der Methoden sein. Ein Ziel dieser Bemühungen kann dabei das sogenannte „Labor auf einem Chip“ sein. Weiterhin wichtig ist die Verknüpfung des Screenings mit der Bioinformatik. Dies betrifft zum einen die intelligente Auswertung der enormen Datenmassen und zum anderen die Umsetzung und Verwendung der bei einem Screeningprozess gewonnenen Erkenntnisse in der Entwicklung neuer Generationen von Biokatalysatoren. Literatur [1] Faber, K. (2000). Biotransformations in organic chemistry, 4th edn. Springer, Berlin, Heidelberg, New York. [2] Buchholz, K. & Kasche, V. (1997). Biokatalysatoren und UniversitŠt des Saarlandes Enzymtechnologie, Wiley-VCH, Weinheim. 326-334. [3] Tholey, A. & Heinzle, E. (2001). Adv. Biochem. Eng. Biotechnol., im Druck. [11] Wallenfels, K. & Weil, R. (1972). In: Boyer, P.D. (ed) The Enzymes, 3rd edn., Vol VII, Academic Press, New York, S. 617. [4] Marrs, B., Delagrave, S. & Murphy, D. (1999). Curr. Opin. Microbiol., 2, 241-245. [12] Reetz, M.T. & Jaeger, K.E. (2000). Chem. Eur. J., 6, 407412. [5] Stemmer, W.P.C. (1994). Nature, 370, 389-391. [13] Pope, A.J., Haupts, U.M. & Moore, K.J. (1999). Drug Discovery Today, 4, 350-362. [6] Petrounia, I.P. & Arnold, F.H. (2000). Curr. Opin. Biotechnol., 11, 325-330. [7] Arnold, F.H. & Volkov, A.A. (1999). Curr. Opin. Chem. Biol., 3, 54-59. [8] Altreuther, D.H. & Clark, D.S. (1999). Curr. Opin. Biotechnol., 10, 130-136. [9] DeSantis, G. & Jones, J.B. (1999). Curr. Opin. Biotechnol., 10, 324-330. [10] Tischer, W. & Kasche, V. (1999). Trends Biotechnol., 17, magazin forschung 1/2001 [14] Mere, L., Bennett, T., Coassin, L., England, P., Hamman, B., Rink, T., Zimmerman, S. & Negelescu, P. (1999). Drug Discovery Today, 4, 363-369. [15] Pantel, S. (1987). Anal. Chim. Acta, 199, 1-14. [16] Janes, L.E., Löwendahl, C. & Kazlauskas, R.J. (1998). Chem. Eur. J., 4, 2324-2331. [17] John G.T. & Heinzle, E. (2001). Biotechnol. Bioeng., 72, 620-627. [18] Wolfbeis, O.S., Klimant, I., Werner, T., Huber, C., Kosch, U., Krause, C., Neurauter, G. & Dürktop, A. (1998). Sensors and Actuators B51, 17-24. [19] Fenn, J.B., Mann, M., Meng, C.K., Wong, S.F. & Whitehouse, C.M. (1989). Science 246, 64-71. [20] Karas, M. & Hillenkamp, F. (1988). Anal. Chem., 60, 22992301. [21] Reetz, M.T., Becker, M.H., Klein, H.W. & Stöckigt, D. (1999). Angew. Chem., 111, 1872-1875. [22] Kang, M.J., Tholey, A. & Heinzle, E. (2000). Rapid Commun. Mass Spectrom., 14, 1972-1978. [23] Speicher, A., Hollemeyer, K. & Heinzle, E. (2001). Rapid Commun. Mass Spectrom., 15, 124-127. 55