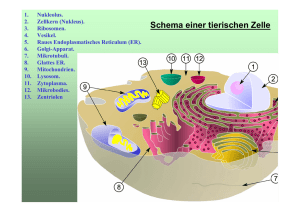
1. Einleitung - repOSitorium
Werbung

Struktur, Funktion und Regulation der Plasmamembran V-ATPase von Manduca sexta Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften des Fachbereiches Biologie/Chemie an der Universität Osnabrück Vorgelegt von Markus Huß aus Augsburg Osnabrück, 2001 Inhaltsverzeichnis Inhaltsverzeichnis 1. Einleitung 1 1.1. Ionentransport-ATPasen und ihre Funktion in der Membran 1 1.2. Die V-ATPase im Mitteldarm von M. sexta 5 1.3. Regulation von V-ATPasen 7 1.4. Die Charakterisierung von ATPasen mit Hilfe von Inhibitoren 10 1.5. Aufgabenstellung 12 2. Material 13 2.1. Chemikalien, Enzyme und sekundäre Antikörper 13 2.2. Puffer und Medien 14 2.3. Versuchstiere 15 3. Methoden 16 3.1. 16 3.1.1. 3.1.2. 3.1.3. 3.1.4. 3.1.5. 3.1.6. 3.1.7. 3.1.8. 3.1.9. 3.1.10. 3.1.11. 3.1.12. 3.1.13. 3.1.14. 3.1.15. 3.1.16. 3.1.17. 3.1.18. 3.1.19. 3.1.20. 3.1.21. 3.1.22. Biochemische Methoden Präparation von angereicherten und hochreinen GobletzellApikalmembranen Reinigung des V1Vo Holoenzyms aus angereicherter GCAM oder aus Mitteldarmrohhomogenat Reinigung des Vo Komplexes aus angereicherter GCAM oder aus Mitteldarmrohhomogenat Reinigung des V1 Komplexes aus Mitteldarm SDS-PAGE Proteinbestimmung Western Blot Reinigung von Antikörpern Immunfärbung Coomassie-Färbung Silber-Färbung Markierung mit 14C-DCCD Markierung mit 125J-Concanamycin A Isoelektrische Fokussierung mit anschließender 2-D Elektrophorese Röntgenkleinwinkelstreuung Matrix unterstützte Laserdesorptions-Ionisations-Massenspektroskopie (MALDI-MS) N-terminale Sequenzierung ATPase-Aktivitätstest und Phosphatbestimmung Bestimmung und Entfernung endogener Nukleotide Behandlung des V1 Komplexes mit chaotropen Ionen (KI-Stripping) Messung des ATP-getriebenen Protonentransports Rekonstitution des V1Vo Holoenzyms in Liposomen 16 17 18 18 19 20 21 21 22 23 23 23 24 24 25 25 26 26 27 28 28 29 Inhaltsverzeichnis 3.1.23. 3.1.24. 3.2. 3.2.1. 3.2.2. 3.2.3. 3.2.4. 3.2.5. 3.2.6. 3.2.7. 3.2.8. 3.2.9. 3.2.10. Messung kapazitiver Ströme am blacklipid Bilayer Auftrennung von Subkomplexen des V1Vo Holoenzyms Molekularbiologische Methoden Polymerase-Kettenreaktion (PCR) Phenolextraktion und Ethanolfällung Plasmid DNA Präparation Spaltung von DNA durch Restriktionsenzyme Agarosegelelektrophorese von DNA Ligation und Transformation Herstellung kompetenter Zellen Sequenzierung von DNA Klonierung, Expression und Reinigung von rekombinanten Teilen der B Untereinheit im pMal-c2-System Herstellung der rekombinanten Untereinheit C im pET System 29 30 30 30 31 31 31 32 32 32 33 33 35 4. Ergebnisse 36 4.1. 36 4.1.1. 4.1.2. 4.1.3. 4.1.4. 4.1.5. 4.2. 4.2.1. 4.2.2. 4.2.3. 4.2.4. 4.3. 4.3.1. 4.3.2. 4.3.3. 4.3.4. 4.4. 4.4.1. 4.4.2. 4.4.3. 4.4.4. Die Struktur der V-ATPase aus M. sexta Optimierung der V1Vo Holoenzym Präparation Entdeckung der Untereinheit a bei M. sexta Die Untereinheit B und die Identifizierung ihrer putativen Isoform B´ als Hsp60 Identifizierung der Untereinheit D Analyse aller Untereinheiten der V-ATPase durch MALDI-MS Der V1 Komplex Modifikation der Präparation des V1 Komplexes Die Untereinheitenzusammensetztung des V1 Komplexes in der 2-D Gelelektrophorese Untersuchung der Untereinheit C im V1 Komplex Behandlung des V1 Komplexes mit chaotropen Ionen Der Vo Komplex Reinigung des Vo Komplexes Identifizierung der 26 kDa Bande des Vo Komplexes Markierung der plecomakrolidbindenden Untereinheit der V-ATPase Die Wirkung neuer Makrolide Funktion und Regulation des V1Vo Holoenzyms und des V1 Komplexes Einfluß von Alkoholen und Detergenzien auf die Mg-ATPase Aktivität des V1 Komplexes Einfluß von Methanol auf das V1Vo Holoenzym Abhängigkeit der Ca-abhängigen Hydrolyse-Aktivität des V1 Komplexes vom verwendeten Nukleosidtriphosphat Einfluß von Ca-ADP, Mg-ADP und Pi auf die Hydrolyse von ATP durch das V1Vo Holoenzym und den V1 Komplex 36 40 42 45 45 49 49 52 53 59 61 61 65 66 73 76 76 78 79 80 Inhaltsverzeichnis 4.4.5. 4.4.6. 4.4.7. 4.4.8. 4.4.9. Bestimmung der endogen gebundenen Nukleotide des V1Vo Holoenzyms und des V1 Komplexes Dissoziation des V1Vo Holoenzyms in vitro Einfluß von DCCD und Concanamycin auf die Mg2+- und Ca2+ATPase-Aktivität des V1Vo Holoenzyms Messung des Protonentransports an GCAM-Vesikeln mit Mg2+und Ca2+-ATP als Substrat Messungen kapazitiver Ströme des rekonstituierten V1Vo Holoenzyms 85 87 91 93 94 5. Diskussion 95 5.1. 95 5.1.1. 5.1.2. 5.1.3. 5.1.4. 5.1.5. 5.1.6. 5.1.7. 5.2. 5.2.1. 5.2.2. 5.2.3. 5.2.4. 5.3. 5.3.1. 5.3.2. 5.3.3. Die Struktur der V-ATPase aus M. sexta Das V1Vo Holoenzym Der Vo Komplex Der V1 Komplex Die putative Isoform der Untereinheit B gehört zu den Hsp60-Proteinen Gehört die Untereinheit C zum V1 Komplex? Ist die Untereinheit E bei V-ATPasen das funktionelle Homolog zur Untereinheit der F-ATPasen? Die räumliche Struktur des V1 Komplexes Die Aktivität der V-ATPase Ca2+ ist kein vollwertiger Ersatz für Mg2+ Warum ist der isolierte V1 Komplex inaktiv? Die V-ATPase-Aktivität wird durch ADP gehemmt Mg-ATP spielt eine wichtige Rolle bei der Dissoziation der V-ATPase in den V1 und den Vo Komplex Makrolid Antibiotika als Inhibitoren der V-ATPase Die Identifizierung der plecomakrolidbindenden Untereinheit der V-ATPase Gibt es eine gemeinsame Bindestelle für Makrolide in V-ATPasen? Die V-ATPase von M. sexta: Ein Modellsystem für die Osteoporoseforschung 95 96 99 100 101 102 104 106 106 107 109 110 112 112 118 120 6. Zusammenfassung 122 7. Literaturverzeichnis 124 8. Publikationen 133 9. Abkürzungsverzeichnis 134 10. Lebenslauf 135 11. Danksagung 136 Einleitung 1. Einleitung 1.1. Ionentransport-ATPasen und ihre Funktion in der Membran Für die Funktion einer Minimalzelle ist, neben einem grundlegenden Stoffwechselzyklus und einer replikationsfähigen Matrize, eine Membran erforderlich, welche die ganze Zelle umschließt und sie gegen ihre Umgebung abgrenzt (T. Ganti, 1971). Diese Membran muß die Fähigkeit zu einem kontrollierten Austausch von Ionen und Molekülen zwischen dem Cytosol und dem extrazellulär Medium besitzen. Da der Austausch häufig entgegen einem elektrochemischen Gradienten erfolgen muß, sind spezielle Proteine in die Membran integriert, die diese Transportprozesse energetisieren. Ist der Transport direkt an die Hydrolyse von ATP gekoppelt, nennt man ihn primären Transport, wird er dagegen durch eine primär erzeugte ionenmotorische Kraft über der Membran angetrieben spricht man von sekundärem Transport (Harold, 1986). Bei den für den primären Transport zuständigen Membranproteinen handelt es sich um Ionentransport-ATPasen, die nach Pedersen & Carafoli (1987) in die drei Klassen der P-, F- und V-ATPasen eingeteilt werden können, wobei die letzteren beiden sich von einem gemeinsamen Vorfahren ableiten lassen (Nelson & Taiz, 1989). Bei den P-ATPasen handelt es sich um die am einfachsten gebauten Vertreter der Ionentransport-ATPasen (Pedersen & Carafoli, 1987). In der Regel bestehen sie aus einer, manchmal auch aus zwei oder mehr Untereinheiten (Geering, 2000). Sie heißen P-ATPasen, da sie während der Hydrolyse von ATP ein Aspartyl-Phosphat-Intermediat bilden, also kurzzeitig phosphoryliert werden; aus diesem Grunde ist eine Hemmung ihrer Enzymaktivität durch das Phosphatanalogon ortho-Vanadat möglich. Das wohl bekannteste Mitglied dieser Klasse ist die Na+/K+-ATPase, die in der basolateralen Membran fast jeder tierischen Zelle vorkommt. Sie energetisiert über den von ihr erzeugten Na+-Gradienten die sekundär-aktive Aufnahme von Glukose und Aminosäuren, ist an der Regulation des Zellvolumens und an der elektrischen Erregbarkeit von Nervenzellen beteiligt. Einige weitere Vertreter der P-ATPasen sind die Ca2+-ATPasen in der Membran des sarcoplasmatischen Retikulums, die H+/K+ATPase in der Magenschleimhaut von Wirbeltieren, die H+-ATPasen in der Plasmamembran von Pflanzen und Pilzen und die Kdp-ATPasen in der Cytoplasmamembran von Eubakterien (Pedersen & Carafoli, 1987). 1 Einleitung Die zweite Klasse der Ionentransport-ATPasen sind die F-ATPasen, welche in der inneren Mitochondrienmembran, in der Thylakoidmembran von Chloroplasten und in der Plasmamembran von Bakterien vorkommen. Dort fungieren sie als ATP-Synthasen und nutzen eine durch Elektronentransportketten aufgebaute protonenmotorische Kraft (proton motive force, pmf) zur Synthese von ATP aus ADP und Phosphat (Mitchell, 1966). Die bakterielle F-ATPase kann unter anaeroben Bedingungen auch in die entgegengesetzte Richtung arbeiten und durch ATP-Hydrolyse einen Protonengradienten über der Membran aufbauen, der zu sekundären Transportprozessen genutzt werden kann. Der Aufbau der FATPasen ist wesentlich komplexer als bei den P-ATPasen. Mit einer Molekularmasse von bis zu 800 kDa setzen sie sich aus einem peripheren F1 Komplex und einem membranständigen Fo Komplex zusammen. Vom F1 Komplex leitet sich auch der Name der ganzen Klasse ab, da er unter der Bezeichnung Faktor 1 erstmals als Bestandteil der ATP-Synthase erkannt und isoliert wurde (siehe Pedersen & Carafoli, 1987). Der Fo Komplex wiederum erhielt seinen Namen auf Grund der Sensitivität der mitochondrialen F-ATPase gegenüber dem Antibiotikum Oligomycin (siehe Pedersen & Carafoli, 1987). Weitere wirksame Inhibitoren sind unter anderem NaN3 und N´,N´-Dicyclohexylcarbodiimid (DCCD). Die F-ATPase besteht, bei einem ihrer am einfachsten gebauten Vertreter aus dem Bakterium E. coli, aus den F1 Untereinheiten und und den Fo Untereinheiten a, b und c in der Stöchiometrie a, b2 und c12 (Deckers-Hebestreit & Altendorf, 1996, Fillingame et al., 1998). Der Fo Komplex der F-ATPasen von Mitochondrien und Chloroplasten ist ungleich komplexer aufgebaut und besteht aus bis zu 8 verschiedenen Untereinheiten (McCarty, 1992). Die Stöchiometrie der Untereinheit c scheint bei den F-ATPasen zwischen verschiedenen Spezies zu variieren. So wurde für den Fo Komplex aus Chloroplasten 14 Kopien der Untereinheit c identifiziert (Seelert et al., 2000), für den Fo Komplex aus Hefe jedoch nur 10 (Stock et al., 1999). Weiterhin gibt es Beobachtungen, dass die Stöchiometrie der Untereinheit c bei E. coli in Abhängigkeit von der angebotenen Kohlenstoffquelle variieren kann (Schemidt et al., 1998). Die Struktur des F1 Komplexes und von Teilen des Fo Komplexes ist inzwischen durch Röntgenkristallographie bestimmt worden (Abrahams et al., 1994, Stock et al., 1999, Gibbons et al., 2000). Die drei - und die drei -Untereinheiten sind alternierend um die zentrale -Untereinheit angeordnet, die mit dem Ring der c Untereinheiten des Fo Komplexes in Verbindung steht. Eine durch die pmf angetriebene Rotation dieses Rings wird über die asymmetrisch geformte -Untereinheit in den F1 Komplex übertragen und führt dort zu einer zyklischen Konformationsänderung der katalytischen -Untereinheiten, 2 Einleitung wie sie zur ATP-Synthese nach dem binding-change Mechanismus erforderlich ist (Boyer, 1989; Junge et al., 1997). Mittlerweile konnte diese Rotation der -Untereinheit und des Rings aus c Untereinheiten experimentell bestätigt werden (Duncan et al., 1995; Sabbert et al., 1996; Noji et al., 1997, 1999; Pänke et al., 2000; Tsunoda et al., 2000). Die dritte Klasse der Ionentransport-ATPasen bilden die V-ATPasen. Sie erhielten ihren Namen aufgrund ihrer erstmaligen Beschreibung in der Vakuolenmembran der Hefe (siehe Pedersen & Carafoli, 1987) und zeichnen sich durch ihre hohe Empfindlichkeit gegenüber den Plecomakroliden Bafilomycin und Concanamycin aus, durch die sie bereits im nanomolaren Bereich gehemmt werden (Überblick in Dröse & Altendorf, 1997). V-ATPasen sind elektrogene Protonenpumpen, die in bestimmten Endomembranen aller Eukaryonten und in den Plasmamembranen einer ganzen Reihe tierischer Zellen vorkommen (Gogarten et al., 1992; Gluck, 1992; Anraku et al., 1992). Auch in der Zellmembran von Prokaryonten wurden ATPasen vom V-Typ nachgewiesen, allerdings translozieren sie dort Na+-Ionen statt H+Ionen (Takase et al., 1993). Im Gegensatz zu den mit ihnen verwandten F-ATPasen arbeiten V-ATPasen ausschließlich als ATP-abhängige Ionenpumpen. Bei Eukaryonten erfolgt der Protonentransport immer vom Cytosol in das entsprechende Zellorganell oder den Extrazellulärraum. Sie generieren dabei eine pmf, welche für die unterschiedlichsten sekundär aktiven Transportprozesse genutzt wird. In Abhängigkeit von den entsprechenden Antiportern oder Symportern und dem entsprechenden Gegenion kann sich der pH-Wert des transKompartiments ins Saure oder Alkalische verschieben oder er bleibt neutral (Harvey, 1992). Wird kein Gegenion transportiert, entsteht eine Spannung über der Membran. In Endosomen, Vakuolen und Lysosomen ist eine Ansäuerung jeweils entscheidend für deren Funktion. Durch die Senkung des pH-Wertes werden in Lysosomen Proteasen aktiviert oder in Endosomen die Liganden von ihren Rezeptoren dissoziiert (Mellman, 1992). In Plasmamembranen übernehmen die V-ATPasen ebenfalls die Aufgabe der Protonensekretion, wie zum Beispiel bei der Regulation des Säure/Base-Haushalts im proximalen und distalen Tubulus der Säugerniere (Gluck et al., 1992) oder bei der Auflösung der Knochenmatrix durch Osteoklasten (Blair, 1998). Darüber hinaus nehmen sie in vielen tierischen Epithelien die bisher weniger bekannte Aufgabe wahr, sekundär aktive Transportprozesse zu energetisieren. In tierischen Zellen erfolgt dies normalerweise durch das von der Na+/K+ATPase generierte Na+-Potential. Sind die Zellen aber einer Na+-armen Umgebung ausgesetzt, wie z. B. im Mitteldarm von pflanzenfressenden Insekten oder im Epithel der Froschhaut und den Kiemen von Süßwasserfischen, wird das benötigte Ionen-Potential nicht 3 Einleitung mit einem Na+-Gradienten erzeugt (Wieczorek et al., 1991; Harvey B. J., 1992; Wieczorek et al., 1999). In diesen Fällen erzeugt eine V-ATPase ein Protonenpotential. Wie bereits erwähnt, sind die F- und V-ATPasen miteinander verwandt und haben mehrere strukturelle Merkmale gemeinsam. So lassen sich auch die V-ATPasen in einen peripheren, katalytischen und einen membranständigen, protonentranslozierenden Komplex unterteilen, welche in Analogie zu den F-ATPasen als V1 und Vo Komplex bezeichnet werden. Der V1 Komplex setzt sich aus den Untereinheiten A3, B3, C, D, E1-3, F, G1-3 und H zusammen, wobei die Stöchiometrie der Untereinheiten E und G (wie durch die Indices angedeutet) noch nicht geklärt ist (Arai et al., 1988; Supekova et al., 1995; Xie, 1996; siehe auch Abb. 3). Die Untereinheiten A und B des V1 Komplexes weisen eine mehr als 25%ige Sequenzhomologie zu den Untereinheiten und der F-ATPasen auf (Nelson & Taiz, 1989). Bei der Untereinheit A handelt es sich um die katalytisch aktive Untereinheit, die ebenso wie die Untereinheit der F-ATPasen die Glycin-reiche Nukleotid-bindende Sequenz GXXXXGKT/S, das sogenannte Walker-Motiv enthält (Walker et al., 1982). Außerdem enthält sie zwei hochkonservierte Cysteine, wodurch die V-ATPasen sensitiv für SH-Gruppen modifizierende Substanzen wie zum Beispiel N-Ethylmaleimid (NEM) sind (Moriyama & Nelson, 1987). Die Untereinheit B wird in Analogie zu ihrem Pendant der -Untereinheit der F-ATPasen als regulatorische Untereinheit bezeichnet. Sie besitzt zwar kein Walker-Motiv, kann aber das ATP-Analogon 3-O-(4-Benzoyl)Benzoyladenosin-5´-triphosphat (BzATP) binden (Manolson et al., 1985; Vasilyeva & Forgac, 1996). Die restlichen V1-Untereinheiten C bis H, welche den Stiel der V-ATPase bilden, haben keinerlei Sequenzhomologien zu FATPase Untereinheiten. Auf jeden Fall ist jede der Untereinheiten wichtig für die korrekte Funktion der V-ATPase, da in Hefe gezeigt wurde, dass sie entweder essentiell für die Assemblierung und/oder die Aktivität sind (Stevens & Forgac, 1997). Da für die Kopplung von ATP-Hydrolyse und Protonentranslokation in Analogie zu F-ATPasen auch für VATPasen ein Rotationsmechanismus angenommen wird, interessiert besonders, welche der Stieluntereinheiten den Part der -Untereinheit übernimmt; sowohl für die Untereinheit E als auch für die Untereinheit D gibt es entsprechende Hinweise (Bowman et al., 1995; Nelson et al., 1995). Der Vo Komplex besteht in seiner minimalen Ausstattung aus den Untereinheiten a, d und c, wobei von der Untereinheit c, welche auch als Proteolipid bezeichnet wird, die Isoformen c´ und c´´ vorkommen können (Arai et al., 1988; Hirata et al., 1997). Die Untereinheit c ist homolog zur Untereinheit c der F-ATPasen und offensichtlich durch eine Genduplikation aus einem gemeinsamen Vorfahren entstanden (Mandel et al., 1988). Im Vergleich zu ihrem 4 Einleitung Pendant bei den F-ATPasen hat sie deshalb eine doppelt so große Molekularmasse, vier statt zwei Membrandurchgänge und liegt auch nur in sechs Kopien pro Vo Komplex vor (Noumi et al., 1991; Nelson, 1992). Das Proteolipid ist vermutlich die entscheidende Untereinheit für die Protonentranslokation über die Membran, da die Bindung von DCCD an ein Glutamat in der vierten Transmembranhelix diesen komplett unterbindet, wobei nur eine der Untereinheiten c ein DCCD-Molekül gebunden haben muß (Arai et al., 1988; Forgac, 1998). Außerdem nimmt man an, dass es sich beim Ring aus den c Untereinheiten, wiederum in Analogie zu den FATPasen, um den membranständigen Teil des Rotors handelt (Forgac, 1998). Die Untereinheit a wird als mögliche Bindestelle des V-ATPase-Inhibitors Bafilomycin und als Hauptbestandteil des Stators diskutiert (Zhang et al., 1994; Forgac, 1998). Für die Untereinheit d gibt es noch keine konkreten Hinweise auf eine spezielle Funktion, es konnte nur gezeigt werden, dass sie ebenfalls wichtig für die Assemblierung der V-ATPase ist (Bauerle et al., 1993). 1.2. Die V-ATPase im Mitteldarm von M. sexta Eine der am besten untersuchten V-ATPasen befindet sich im Mitteldarmepithel der Raupe des Tabakschwärmers M. sexta, wo das von ihr erzeugte Transmembranpotential für die Energetisierung aller Transportprozesse verantwortlich ist. Das einschichtige Epithel des Mitteldarms besteht aus nur zwei differenzierten Zelltypen, den Columnar- oder Säulenzellen und den Goblet- oder Becherzellen (Abb. 1). Es enthält keine Na+/K+-ATPase (Jungreis & Vaughan, 1977) und wird durch ein K+-Potential energetisiert (Harvey et al., 1983, Wieczorek et al., 1986). Die in der Gobletzellapikalmembran (GCAM) lokalisierte "K+Pumpe" wurde als ein Zweikomponentensystem aus einer V-ATPase und einem K+/nH+Antiporter identifiziert (Schweikl et al., 1989; Wieczorek et al.; 1989). Das Mitteldarmepithel von M. sexta war damit das erste tierische Epithel, dessen Energetisierung durch eine VATPase nachgewiesen werden konnte (Wieczorek et al., 1991). Die in das Darmlumen transportierten K+-Ionen erzeugen somit zum einen das K+-Potential, das zur Energetisierung von Aminosäure/K+-Symporten in die Columnarzellen benötigt wird (Giordana et al., 1989). Andererseits entzieht der Antiporter wegen seiner Stöchiometrie von 2H+ pro K+ dem Darmlumen Protonen, was letztlich zu einem extrem hohen alkalischer pH-Wert von bis zu 12 im Lumen des Mitteldarms führt (Azuma et al., 1995). Die Alkalinität des Mitteldarmlumens verhindert vermutlich eine Vernetzung der durch die pflanzliche Nahrung aufgenommenen Tannine, welche ansonsten zu einer Inaktivierung von Enzymen führt (Goldstein & Swain, 1965; Berenbaum, 1980). 5 Einleitung Abb. 1: Energetisierung des Mitteldarmepithels von M. sexta durch die V-ATPase. Der aktive elektrogene K+-Transport erfolgt über die Gobletzellapikalmembran (GCAM) durch den von der V-ATPase (V) energetisierten K+/2H+-Antiporter. Durch das Zusammenspiel von V-ATPase und Antiporter entsteht zum einen der alkalische pH-Wert im Mitteldarmlumen und zum anderen eine Spannung von 150 mV über dem Epithel. Durch einen Aminosäure/K+-Symporter (S) in der Columnarzellapikalmembran (CCAM) werden Aminosäuren (AS) aufgenommen (modifiziert nach Wieczorek et al., 2000). Ein besonderer experimenteller Vorteil der V-ATPase von M. sexta liegt in der extrem hohen Dichte in der das Enzym in der GCAM vorliegt (Abb. 2). Schon in ersten elektronenmikroskopischen Aufnahmen von Schnitten des Mitteldarms fielen ca. 10 nm große Partikel auf, welche die Apikalmembran der Gobletzellen bedeckten und als Portasomen bezeichnet und mit dem Ionentransport in Verbindung gebracht wurden (Harvey et al., 1981). Die Isolierung dieser Membran ermöglichte die ersten grundlegenden Charakterisierungen des Ionentransports und die erste Solubilisierung und Reinigung der V-ATPase (Cioffi & Wolfersberger, 1983; Wieczorek et al., 1986; Schweikl et al., 1989). Bis zu Beginn der vorliegenden Arbeit gelang es durch die Kombination von molekularbiologischen und biochemischen Methoden, die Untereinheiten A, B, E, F und G des V1 Komplexes, sowie die Untereinheiten c und d des Vo Komplexes von M. sexta zu identifizieren (Dow et al., 1992; Gräf et al., 1992; Novak et al., 1992; Gräf et al., 1994a; Gräf et al., 1994b; Lepier et al., 1996; Merzendorfer et al., 1997b). Die Untereinheit F konnte dabei erstmals als konstitutive V-ATPase Untereinheit beschrieben werden und ebenso wie die Untereinheit G erstmals dem V1 Komplex zugeordnet werden. Für alle anderen dem V1 Komplex zugehörigen Untereinheiten (C, D und H) gab es, wie aus der SDS-PAGE der gereinigten V-ATPase zu erkennen war, potentielle Kandidaten in Form von Proteinbanden mit der entsprechenden Molekularmasse. Bei den dem Vo Komplex zugehörigen Untereinheiten war dies nicht der 6 Einleitung Fall; so konnte trotz der Verwendung eines ganzen Cocktails von Proteaseinhibitoren die als proteolytisch sehr anfällig bekannte Untereinheit a nicht nachgewiesen werden, was als Besonderheit von Insekten-V-ATPasen interpretiert wurde (Wieczorek, 1992; Wieczorek et al., 2000). Die war insofern interessant, da die gereinigte V-ATPase aus M. sexta sich durch Bafilomycin hemmen läßt und die Untereinheit a eigentlich als Bafilomycin-bindende Untereinheit betrachtet wurde (Wieczorek et al., 1991; Zhang et al., 1994). Abb. 2: Aufnahmen von Vesikeln der Gobletzellapikalmembran von M. sexta. Die Vesikel der gereinigten GCAM aus dem posterioren Mitteldarm wurden von Dr. Michael Radermacher und Dr. Teresa Ruiz (MPI für Biophysik, Frankfurt) wie in Radermacher et al. (1999) beschrieben negativ-kontrastiert und elektronenmikroskopisch abgebildet. In der Aufsicht ist deutlich die dichte Packung der V-ATPase (ca. 10000/µm2) und die hexagonale Struktur des V1 Komplexes mit ca. 10 nm Durchmesser zu sehen (a, b). In der Seitenansicht ist die Unterteilung Kopf und Stiel zusehen, wobei Einheit aus Stiel und Kopf ca. 15 nm aus der Membran herausragt (a). (Radermacher, Ruiz, Huss und Wieczorek unpubliziert). 1.3. Regulation von V-ATPasen Für ein komplexes Enzym wie die V-ATPase, das in verschiedensten Organellen, Organen und Organismen ganz unterschiedliche Aufgaben erfüllt und dabei große Mengen von Stoffwechselenergie in Form von ATP verbraucht, erscheinen Kontroll- und Regulationsmechanismen unbedingt erforderlich. So konsumiert die V-ATPase des Mitteldarmepithels der Raupe von M. sexta 10% des gesamten ATPs des Tieres (Dow, 1984). Deshalb dürfte es von großer Bedeutung sein, dass die V-ATPase-Aktivität den jeweiligen Erfordernissen und Bedingungen angepasst wird, um nicht "unnötig" Energie zu verbrauchen. Für V-ATPasen sind, unter anderem aus Untersuchungen an M. sexta, bereits einige Regulationsmöglichkeiten bekannt. Schon die Biosynthese von V-ATPase Untereinheiten wird auf der Ebene der Transkription reguliert. Ursprünglich wurden V-ATPasen in vielen 7 Einleitung Organismen aufgrund von G+C-reichen Regionen in den Promotoren der Gene für einzelne Untereinheiten als konstitutiv exprimierte house-keeping Proteine betrachtet (Wechser & Bowman, 1995). Allerdings enthalten die Promotoren der Untereinheiten B, G und d von M. sexta verschiedene Elemente, wie TATA-Boxen, CCAAT-Boxen oder Ecdyson-responsiveElemente die auf eine transkriptionelle Regulation hindeuten (Merzendorfer et al., 1997a). In diesem Zusammenhang konnte mit Northern Blots gezeigt werden, dass die Menge der mRNA für fast alle V-ATPase-Untereinheiten während der Häutung und bei Hunger abnimmt und dies mit dem Anstieg des 20-Hydroxyecdyson-Titers (20-HE) in der Hämolymphe korreliert (Reineke et al., 2001). Weiterhin konnte durch Injektion von 20-HE in die Hämolymphe der Raupen und durch Reportergenstudien mit den Promotoren der Gene der Untereinheiten B, G und d ein 20-HE-abhängiges Absinken der Transkriptionsrate für diese Untereinheiten bestätigt werden (Reineke et al., 2001). Auch auf posttranskriptioneller Ebene können die Untereinheiten der V-ATPase reguliert werden. Bei M. sexta wurden erste Hinweise für eine Regulation durch die Transkription von Antisense-RNA für die Untereinheiten d und H gefunden (Merzendorfer et al., 1997a; Merzendorfer et al., 1997b; Merzendorfer, Düvel und Wieczorek unpubliziert). AntisenseRNA kann mit ihrer Sense-mRNA einen Doppelstrang ausbilden, der durch spezifische Doppelstrang-RNAsen (dsRNAsen) abgebaut wird, wodurch die Translationsrate der entsprechenden Untereinheit erniedrigt wird (Nellen & Lichtenstein, 1993). Änderungen des Redoxpotentials in der Zelle könnten ebenfalls eine Möglichkeit darstellen die Aktivität der V-ATPase zu modulieren. Ein Beispiel hierfür ist die redox-abhängige Ausbildung von Disulfidbrücken zwischen zwei konservierten Cysteinresten in der Untereinheit A der V-ATPase aus Clathrinvesikeln mit der daraus resultierenden Inhibierung (Taiz et al., 1994; Merzendorfer et al., 1997a; Stevens & Forgac, 1997). Auch die Aktivität der V-ATPase aus N. crassa wird durch reduzierende bzw. oxidierende Agenzien modifiziert (Dschida & Bowman, 1995). Die Aktivität der V-ATPase lässt sich aber auch über die Menge der zur Verfügung stehenden funktionsfähigen Enzyme in der Membran regulieren. Hierfür wurden bisher zwei verschiedene Mechanismen entdeckt: Zum einen kann die Anzahl der V-ATPasen durch Exozytose bzw. Endozytose von V-ATPase-haltigen Vesikeln gesteuert werden. Im distalen und proximalen Tubulus der Säugerniere oder im Epithel der Krötenharnblase verschmelzen dicht mit V-ATPase bepackte Vesikel nach Bedarf mit der Plasmamembran oder werden wieder endozytiert (Gluck et al., 1982; Schwartz & Al-Awqati, 1985; Brown et al., 1987). Zum anderen kann die Anzahl der aktiven V-ATPasen durch reversible Dissoziation des 8 Einleitung cytosolischen V1 Komplexes vom membranständigen Vo Komplex gesteuert werden (Abb. 3). Dieser Mechanismus wurde erstmals für die V-ATPase im Mitteldarm der Tabakschwärmerraupe von M. sexta entdeckt (Sumner et al., 1995). Nimmt die Raupe keine Nahrung zu sich, z. B. während der Häutung oder bei Futtermangel, dissoziieren die V1 Komplexe von der Membran ab (Sumner et al., 1995; Gräf et al., 1996). Durch diese Trennung in V1 und Vo Komplex wird die V-ATPase sehr effektiv ausgeschaltet, da der cytosolische V1 Komplex in vivo keine Mg-ATPase-Aktivität und der Vo Komplex keine Protonenleitfähigkeit besitzt (Gräf et al., 1996; Beltran & Nelson, 1992). Abb. 3: Die V-ATPase und ihre reversible Dissoziation in die beiden Subkomplexe V 1 und Vo. In diesem Modell wurden alle bekannten Untereinheiten der V-ATPase von M. sexta und die aktuellen Kenntnisse über deren Topologie und Stöchiometrie berücksichtigt. Durch die Dissoziation erhöht sich der Anteil von freien V1 Komplexen an cytosolischem Protein von 1% auf 2% (Gräf et al., 1996). Aus diesem Pool von V1 Komplexen gelang es erstmals einen nativen V1 Komplex aus den Untereinheiten A, B, E, F und G zu reinigen. Dieser Komplex hat im Gegensatz zum V1Vo Holoenzym keine Mg-ATPase-Aktivität, kann aber ebenso wie dieses die Hydrolyse von ATP in Gegenwart von Ca2+ katalysieren (Gräf et al., 1996). Allerdings kann mit Zugabe von Methanol eine Mg-ATP-Hydrolyse-Aktivität des V1 Komplexes induziert werden (Gräf et al., 1996). Wenn die Raupen wieder Nahrung zu sich nehmen, reassoziiert der V1 Komplex wieder an die Membran. Die zur Reassoziation benötigten V1 Komplexe werden offensichtlich aus dem cytosolischen Pool rekrutiert, da hierfür keine Proteinneusynthese erforderlich ist (Jäger & Klein, 1996). Deshalb kann man davon ausgehen, dass die Untereinheiten des Vo Komplexes in der Plasmamembran 9 Einleitung verbleiben und ebenfalls wiederverwendet werden. Ob der Vo Komplex dabei allerdings als ganzer intakter Komplex oder in seine Untereinheiten dissoziiert vorliegt, ist noch nicht geklärt. Auch bei der Hefe S. cerevisiae wurde eine ähnliche reversible Dissoziation der VATPase nachgewiesen (Kane, 1995). Wird den Hefezellen durch einen Wechsel des Nährmediums Glukose als Kohlenstoffquelle entzogen oder erreichen die Zellen die stationäre Wachstumsphase, dissoziiert die V-ATPase, wird den Zellen wieder Glukose im Medium angeboten, reassoziiert die V-ATPase (Kane, 1995; Parra & Kane, 1998). Da die reversible Dissoziation der V-ATPase bei zwei phylogenetisch weit voneinander entfernten Organismen entdeckt wurde, kann man auf einen allgemein gültigen Regulationsmechanismus für V-ATPasen schließen. Die genaue Steuerung der Dissoziation ist weitgehend unbekannt. Möglicherweise spielt die Untereinheit C eine Rolle, da sie beim abdissoziierten V1 Komplex aus Hefe nicht und aus M. sexta nur in substöchiometrischen Mengen vorhanden ist, im reassoziiertem Holoenzym aber wieder auftaucht (Kane, 1995; Gräf et al., 1996). Glukose spielt bei der Hefe offensichtlich eine entscheidende Rolle, da ihre Entfernung aus dem Medium bereits nach fünf Minuten die Dissoziation der V-ATPase bewirkt (Kane, 1995). Auch bei M. sexta sinkt innerhalb von nur 30 min nach Futterentzug die Glukosekonzentration in der Hämolymphe um mehr als 60% ab und beträgt nach 10 h nur noch 5% der Ursprungskonzentration (Gies et al., 1988). Bis jetzt konnte aber kein direkter Zusammenhang zwischen Glukose und der Dissoziation gezeigt werden. Alle Glukosestoffwechsel- und auch Signaltransduktionsmutanten der Hefe, die bisher untersucht wurden, zeigten ein vergleichbares Dissoziationsverhalten wie der Wildtyp (Parra & Kane, 1998). Es ist allerdings bekannt, dass die V-ATPase unter dem Einfluß von chaotropen Ionen und/oder Nukleotiden auch in vitro zu einer Dissoziation in ihren V1 und Vo Komplex neigt (Moriyama & Nelson, 1989), wodurch die Untersuchung von grundlegenden zur Dissoziation der V-ATPase führenden Mechanismen am gereinigten V1Vo Holoenzym möglich sein sollte. 1.4. Die Charakterisierung von ATPasen mit Hilfe von Inhibitoren Wie bei nahezu jeder Proteinfamilie gibt es auch für die V-ATPasen spezifische Inhibitoren. Diese können zum einen zur Klassifizierung herangezogen werden, zum anderen ermöglichen sie es, die Funktion und die Struktur des jeweiligen Proteins zu untersuchen. So konnten beispielsweise mit Hilfe des Phosphatanalogons ortho-Vanadat Teile des Reaktionszyklus der P-ATPasen aufgeklärt werden (Cantley et al., 1978; Pedersen & Carafoli, 1987). Mit dem Inhibitor NEM wurde die Untereinheit A als die katalytisch aktive Untereinheit der V-ATPase bestätigt (Feng & Forgac, 1992). N´,N´-Dicyclohexylcarbodiimid (DCCD), das sowohl F- als 10 Einleitung auch V-ATPasen hemmt, bindet bei beiden ATPasen an einen sauren Aminosäurerest in der membranständigen Untereinheit c des Fo- bzw. Vo-Komplexes, die zueinander homolog sind (Sebald et al., 1980; Arai et al., 1987). Auch Plecomakrolide wie die Bafilomycine und die Concanamycine sind gegen mehrere Klassen von Ionentransport-ATPasen wirksam. Sie hemmen die V-ATPasen in nanomolaren Konzentrationen, sind aber überraschenderweise gegen die verwandten F-ATPasen vollkommen wirkungslos, obwohl Bafilomycin mit dem membranständigen Komplex wechselwirkt, der bei beiden ATPasen zu einem großen Teil aus den zueinander homologen Untereinheiten c besteht (Hanada et al., 1990; Dröse et al., 1993). Dagegen werden PATPasen, wie die Kdp-ATPase aus E. coli und die Na+/K+-ATPase aus Rind, die ganz offensichtlich keinerlei Gemeinsamkeiten mit den V-ATPasen haben, in mikromolaren Konzentrationen gehemmt (Bowman et al., 1988). Auch einige Vertreter der ABCTransporter (ATP-Binding-Cassette) erwiesen sich als sensitiv gegenüber Bafilomycin (Higgins, 1992; Sharom et al., 1995; Hunke et al., 1995). Für keine der drei ATPase-Klassen, die durch Plecomakrolide gehemmt werden, ist bisher die Bindestelle bekannt. Deshalb war es von großem Interesse, zunächst für eine der ATPasen, im speziellem der V-ATPase, welche am sensitivsten auf die Plecomakrolide reagiert, die Bindestelle am Enzym zu bestimmen. Weiterhin interessant sind auch Inhibitoren, die innerhalb einer Klasse von ATPasen eine unterschiedlich starke Wirkung entfalten, da sie zum einen weiteren Aufschluß über den Wirkungsmechanismus bzw. die Interaktion zwischen Inhibitor und Enzym geben und zum anderen wichtige Erkenntnisse für die gezielte Veränderung der Inhibitorstruktur für therapeutische Zwecke liefern können. So sind für Bafilomycin und Concanamycin für VATPasen aus verschiedenen Spezies sehr unterschiedliche IC50-Werte gemessen worden (Dröse et al., 2001). Auch wurde kürzlich für eine neue Gruppe von Makroliden, die Benzolactone-Enamide, entdeckt, dass sie unterschiedlich effizient gegenüber V-ATPasen unterschiedlicher Herkunft sind (Erickson et al.,1997; Reichenbach, 1997; Kunze et al., 1998; Boyd et al., 2001). Besonders auffällig war dabei die hohe Selektivität, mit der die Benzolacton-Enamide V-ATPasen aus Säugetieren hemmten, nicht aber V-ATPasen aus Pilzen (Boyd et al., 2001). 11 Einleitung 1.5. Aufgabenstellung Durch die hohe Dichte der V-ATPase in der Gobletzellapikalmembran bietet der Mitteldarm des Tabakschwärmers M. sexta ideale Vorraussetzungen für biochemische und strukturelle Untersuchungen. Zunächst sollten die Präparationsmethoden für das V1Vo Holoenzym und den V1 Komplex verbessert werden, um eine deutlich größere Ausbeute, Reinheit und Stabilität des Holoenzyms und der V1-ATPase zu erhalten. Weiterhin sollte überprüft werden, ob der Vo Komplex nach Abdissoziation des V1 Komplexes als ganzes in der Membran erhalten bleibt. Gegebenenfalls sollte dieser native Vo Komplex dann gereinigt werden. Diese drei gereinigten Komplexe sollten dann als Grundlage für alle weiteren geplanten Untersuchungen dienen. Ein primäres Ziel war die Aufklärung der Untereinheitenzusammensetzung der V-ATPase und die Identifizierung der Untereinheiten auf Proteinebene, eine Vorraussetzung für spätere hochauflösende Strukturanalysen. Dazu sollten neben etablierten gelelektrophoretischen und immunochemischen Methoden auch die N-terminale Sequenzierung und die MALDI-Massenspektrometrie herangezogen werden. Ein besonderes Augenmerk sollte dabei der weitgehend unbekannten Struktur des V o Komplexes gelten, für den bisher nur die Untereinheiten c und d identifiziert waren. Mit einem semisynthetischen Concanamycin A Derivat sollte die im Vo Komplex lokalisierte Plecomakrolid-bindende Untereinheit der V-ATPase detektiert und wenn möglich die genaue Bindestelle eingegrenzt werden. Auch sollte die Wirkung einiger neuer Makrolide auf die Aktivität der V-ATPase charakterisiert werden. Der abdissoziierte V1 Komplex hat im Gegensatz zum V1Vo Holoenzym keine MgATPase-Aktivität, kann aber ebenso wie dieses die Hydrolyse von ATP in Gegenwart von Ca2+ katalysieren. Durch die Zugabe von Methanol erhält der V1 Komplex die Fähigkeit, Mg-ATP zu hydrolysieren zurück. Die Ursache dieses Phänomens sollte unter anderem durch einen Vergleich der Enzymaktivitäten des V1 Komplexes und des V1Vo Holoenzyms untersucht werden. Da der Vorgang der Dissoziation der V-ATPase noch weitgehend unverstanden ist, sollte er im Rahmen der vorliegenden Arbeit vor allem in Hinblick auf eine mögliche Rolle von Nukleotiden näher betrachtet werden. 12 Material 2. Material 2.1. Chemikalien, Enzyme und sekundäre Antikörper Amersham Pharmacia Biotech: 14C-DCCD Amresco: Acrylamide (40%), Sequenziergelfertigmischung Biomol: Pefabloc SC Fluka: Ammoniummolybdat, Bafilomycin B1, Chloroform-Isoamylalkohol (24:1), Concanamycin A, Dinatrium-Hydrogen-Phosphat, Saccharose, -Aminocapronsäure, NaEGTA, Essigsäure, Ethanol, Glutaraldehyd, Glycin, Kaliumjodid, Salzsäure (37%), Isopropanol, Methanol, n-Butanol, MgCl2, MOPS, NaCl, Natriumhydroxid, Natriumthiosulfat, SDS, Silbernitrat, TCA, TEMED FMC: Sea Plaque (low melting point) Agarose Gibco: Agar, Agarose (ultrapure), Hefeextrakt, LB-Broth, NZY-Broth, TB-Broth, Pepton 140 Grüssing: Formaldehyd (37%), Glycerin (87%) Loewe: IPTG, Tris Merck: Amidoschwarz, Natriummolybdat, Schwefelsäure (95-98%), Tween 20, Coomassie Brilliant-blau R-250 New England Biolabs: Restriktionsenzyme, T4-DNA-Ligase Perkin Elmer: Taq Polymerase Riedel-de Haen: Aceton, CaCl2 x 2 H2O, KCl, Natriumazid, Serva: APS, BCIP, Bromphenolblau, BSA, Na2-EDTA, Fischgelatine (flüssig), Natriumacetat, Natriumcarbonat, NBT, SDS Sigma: ADP (K-Salz), AMP-PNP (Li-Salz), ATP (Na-Salz), ATP (Tris-Salz), alkalische Phosphatase, Alkalische Phosphatase-konjungierte Anti-Maus-IgG, Alkalische Phosphatasekonjungierte Anti-Kaninchen IgG, Alkalische Phosphatase-konjungierte Anti- Meerschweinchen IgG, Ampicillin, Bisacrylamid, -Mercaptoethanol, C12E10, DCCD, DTT, EDTA (freie Säure), GTP (K-Salz), Jodacetamid, Malachitgrün, Mannit, Orthovanadat, PEP, Luciferin/Luciferase, Ponceau S Stratagene: Restriktionsenzyme, T4-DNA-Polymerase, alkalische Phosphatase Alle verwendeten Chemikalien hatten, sofern nicht anders erwähnt, p. a. Qualität. Die nicht aufgeführten Standardchemikalien wurden in der Regel von den Firmen Fluka, Sigma, Merck 13 Material und Riedel de Haen bezogen. Die für die PCR eingesetzten Oligonukleotide wurden von Amersham Pharmacia Biotech synthetisiert. Wurden für die Experimente käufliche Kits eingesetzt ist dies im Text vermerkt. 2.2. Puffer und Medien b-Medium: Einwaage pro Liter Medium: 5 g Hefeextrakt, 20 g Pepton 140, 5 g MgSO4, pH mit KOH auf 7,6 eingestellt a-Medium: entspricht b-Medium plus 14 g Agar APP-Puffer: 100 mM NaCl, 50 mM MgCl2, 100 mM Tris-HCl pH 9,5 APP-Puffer mit NBT/BCIP: 45 µl NBT und 35 µl BCIP pro 10 ml APP-Puffer BCIP: 50 mg/ml 5-Bromo-4-Chloroindoxylphosphat in reinem Dimethylformamid Blockpuffer: 3% Gelatine in TTBSN Blotpuffer 1: 300 mM Tris (nach dem Ansetzen 1/4 Volumenteil Methanol zugeben) Blotpuffer 2: 30 mM Tris (nach dem Ansetzen 1/4 Volumenteil Methanol zugeben) Blotpuffer 3: 30 mM Tris 40 mM -Aminocapronsäure (nach dem Ansetzen 1/4 Volumenteil Methanol zugeben) ET: 5 mM Na2-EDTA, 5 mM Tris-HCl pH 8,1 HI: 1 M NaCl, 20 mM Tris-HCl pH 8,1 HIM: 9,6 mM -Mercaptoethanol in HI HIMC: 0,01% C12E10 in HIM Luciferin/Luciferase-Puffer: 60 mM Tris-Acetat pH 7,75, 10 mM MgCl2, 1 mM KCl, 1,5 mM EDTA, 2,5 mM -Mercaptoethanol, 0,4 mg/ml Luciferin/Luciferase (Sigma) LO: 50 mM NaCl, 20 mM Tris-HCl pH 8,1 LOM: 9,6 mM -Mercaptoethanol in LO LOMC: 0,01% C12E10 in LOM MENT: 300 mM Mannit, 5 mM EDTA, 50 mM NaCl, 17 mM Tris-HCl pH 8,1 MENTP: 5 mM Pefabloc SC in MENT NBT: 75 mg/ml Nitroblau Tetrazolium in 70% Dimethylformamid Pyruvatkinase-Lösung: 70 mM Tris-Acetat pH 7,75, 9 mM MgCl2, 5 mM KCl, 2 mM Phosphoenolpyruvat, 2 mM EDTA, 500 U/ml Pyruvatkinase SET: 250 mM Saccharose in ET 14 Material SETP: 5 mM Pefabloc SC in SET TBS: 500 mM NaCl, 20 mM Tris-HCl pH 7,5 TBSN: 0,02% NaN3 in TBS TE: 0,32 mM EDTA, 16 mM Tris-HCl pH 8,1 TEK: 200 mM KCl in TE TEM: 9,6 mM -Mercaptoethanol in TE TEN: 50 mM NaCl in TE Tfb-1: 30 mM K-Acetat, 100 mM RbCl, 10 mM CaCl2, 15% Glycerin, 50 mM MnCl2, pH 5,8 Tfb-2: 10 mM MOPS, 10 mM RbCl, 75 mM CaCl2, 15% Glycerin, pH 6,5 TTBSN: 0,05% Tween 20 in TBSN Verdünnungspuffer: 1% Gelatine in TTBSN 2.3. Versuchstiere Bei den für die Präparationen verwendeten Tieren handelt es sich um Raupen des Tabakschwärmers Manduca sexta (Lepidoptera, Sphingidae, Abb. 4), die wenn nicht anders erwähnt im 5. Larvalstadium waren und ein Gewicht von ca. 6 g hatten. Die Raupen wurden in einem Laborbrutschrank bei 27°C und 16 h Langtag-Bedingungen gehalten und mit synthetischem Futter ernährt (Bell & Joachim, 1974). Die Falter wurden in Käfigen mit einem Volumen von mindestens 1 m3 gehalten (27°C, 16 h Tageslicht), wobei eine 30%ige Zuckerlösung als Nahrung diente. Abb. 4: Der Tabakschwärmer (Manduca sexta). Raupe im 5. Larvenstadium 15 Methoden 3. Methoden 3.1. Biochemische Methoden 3.1.1. Präparation von angereicherten und hochreinen Gobletzell-Apikalmembranen Zur Anreicherung von Apikalmembranen der Gobletzellen wurde die Methode von Cioffi und Wolfersberger (1983), modifiziert durch Wieczorek et al. (1990), verwendet. Als Ausgangsmaterial dienten posteriore Mitteldärme aus M. sexta Raupen (5. Larvenstadium, 5-7 g). Diese wurden nach Entfernen der Längsmuskulatur entfaltet, in ca. 2 mm große Stücke geschnitten und in eiskaltem SET-Puffer gesammelt. Durch Ultraschall (Branson-Sonifier 250, Mikrospitze, Ausgangsleistung 2, Arbeitspuls 35%/s, 15-25 Pulse) wurde zunächst der Mikrovillisaum der Columnarzellen abgeschert und der Zellinhalt durch dreimaliges Waschen mit SETPuffer entfernt. Die Apikalmembran der Gobletzellen wurde durch Triturieren mit einer Pasteurpipette vom Rest des Zellverbandes gelöst und durch Filtrieren (4 Lagen Gaze) abgetrennt. Die so gewonnenen Membranen wurden abzentrifugiert, in SET resuspendiert und in einem gestuften Saccharosedichtegradienten (3 ml 45%, 3 ml 41% und 3 ml 37% Saccharose in ET w/w) 4 h bzw. über Nacht bei 107.000 x g und 4°C isopyknisch zentrifugiert (SW 41 Ti Rotor, Beckman). Die an der Grenze von 37% und 41% Saccharose angereicherten Gobletzell-Apikalmembranen wurden vom Saccharosegradienten abgenommen, in 4 Schritten (0,5, 1, 1 und 1,5 Volumenteile) langsam mit ET-Puffer 1:5 verdünnt und bei 10.000 x g (30 min, 4°C, Avanti Kühlzentrifuge, JA25.50, Beckman) zentrifugiert. Das Pellet wurde in 1 ml SET-Puffer resuspendiert, bei 48.000 x g (15 min, 4°C, Avanti Kühlzentrifuge, JA25.50, Beckman) erneut zentrifugiert und entweder direkt weiter verwendet oder in flüssigem Stickstoff tiefgefroren und bei –20°C gelagert. Die Präparation von reiner GCAM (Wieczorek et al., 1990) erfolgte bis zur Verdünnung der vom Saccharosedichtegradienten abgenommenen Bande wie oben beschrieben, allerdings enthielt der Gradient kein EDTA. Die verdünnte Suspension wurde dann auf Corexzentrifugengläser verteilt (5-6 ml pro Röhrchen), mit 4 Pulsen ultrabeschallt (BransonSonifier 250, Mikrospitze, Ausgangsleistung 2, Arbeitspuls 35%/s) und für 30 min bei 10.000 x g zentrifugiert (4°C, Avanti Kühlzentrifuge, JA25.50, Beckman). Dabei lösen sich vor allem noch an der GCAM anhaftende Mitochondrien. Die Überstände, die reine GCAM enthielten, wurden in einem Erlenmeyerkolben auf Eis gesammelt, die Pellets in jeweils 3 ml SET 16 Methoden resuspendiert und erneut, wie vorher beschrieben, beschallt und zentrifugiert. Nach zweimaliger Wiederholung dieser Prozedur wurden die gesammelten Überstände aus den 4 Zentrifugationsschritten auf Polycarbonatröhrchen verteilt und für 1 h bei 30.000 x g zentrifugiert. Das durchsichtige Pellet, welches die gereinigte GCAM enthielt, wurde in dem für die Versuche relevanten Puffer resuspendiert und auf Eis bzw. im Kühlschrank gelagert. 3.1.2. Reinigung des V1Vo Holoenzyms aus angereicherter GCAM oder aus Mitteldarmrohhomogenat Zur Reinigung von V1Vo Holoenzyms wurden zunächst tiefgefrorene angereicherte Gobletzell-Apikalmembranen benutzt und direkt wie weiter unten beschrieben solubilisiert und aufgereinigt (Schweikl et al., 1989). Um eine höhere Ausbeute an V1Vo Holoenzym mit weniger Aufwand zu erhalten, wurde eine modifizierte Präparation aus Rohhomogenat von ganzen Mitteldärmen entwickelt. Dazu wurden isolierte Mitteldärme von bis zu 24, für 15 min auf Eis gekühlten Raupen (5. Larvalstadium) in kaltem Puffer SETP (250 mM Saccharose, 5 mM EDTA, 5 mM Tris-HCl, 5 mM Pefabloc SC, pH 8,1) mit einem Ultraturrax (IKA, Deutschland) 30 s bei 20500 rpm homogenisiert. Nach einer Zentrifugation von 5 min bei 10.000 x g und 4°C in einem Festwinkelrotor (Avanti Kühlzentrifuge, JA25.50, Beckman) wurde das Pellet in SETP resuspendiert und erneut unter den gleichen Bedingungen zentrifugiert. Dieses Pellet wurde wieder in SETP aufgenommen und, um das noch vorhandene Fett effektiv abzutrennen, nun für 30 min bei 200.000 x g und 4°C in einem Festwinkelrotor (90 Ti, Beckman) zentrifugiert. Anschließend wurde das Pellet in TEM mit 0,1% C12E10 bei 4°C solubilisiert, wobei im Falle der tiefgefrorenen angereicherten Gobletzell-Apikalmembran das Detergens zu Protein Verhältnis dabei 2:1 betrug. Für das Membranpellet aus dem Rohhomogenat von 24 Raupen wurden standardmäßig 400 ml TEM mit 0,1% C12E10 verwendet. Nach einer 1-stündigen Zentrifugation (200.000 x g, 4°C, 90 Ti, Beckman) wurde der Überstand mit den gelösten Proteinen durch eine Membran mit einer Porengröße von 0,22 µm filtriert und entweder durch Ultrafiltration in einer Rührzelle unter Druck oder per Zentrifugation in Mikrokonzentratoren ankonzentriert (beide Amicon, 10 kDa Ausschlußgröße). Das Konzentrat von höchstens 4 Raupen wurde auf je einen diskontinuierlichen Saccharosegradienten geschichtet (3 ml 40%, 2 ml 30%, 2 ml 20% und 1,6 ml 10% Saccharose gelöst in TEM mit 0,01% C12E10 und 200 mM KCl) und für 90 min bei 310.000 x g und 4°C zentrifugiert (Vertikalrotor VTi 65.1, Beckman). Mit der 30% Fraktion wurde nach einer 4-fachen Verdünnung mit LOMC-Puffer (20 mM Tris-HCl, 9,6 17 Methoden mM 2-Mercaptoethanol, 0,01% C12E10, pH 8,1, mit 50 mM NaCl) eine Anionenaustauscher Chromatographie mittels FPLC auf einer Mono Q bzw. Resource Q (beide Säulen und FPLC, Amersham-Pharmacia Biotechnology Inc.) oder mittels HPLC auf einer Hyper D Q Säule (HPLC Biosys 2000, Beckman; Säule, Biosepra) durchgeführt. Das V1Vo Holoenzym eluierte in einem linearen Salzgradienten (50 – 400 mM NaCl in LOMC) zwischen 230 und 280 mM NaCl. Mit Hilfe eines Zentrifugalkonzentrators (Ausschlußgröße 100 kDa) wurde das Eluat auf 100 µl ankonzentriert. Der letzte Reinigungsschritt erfolgte mittels FPLC über eine Gelpermeationssäule (Superdex 200 Amersham-Pharmacia Biotechnology Inc.) mit dem Puffer LOMC, der 150 mM NaCl enthielt. 3.1.3. Reinigung des Vo Komplexes aus angereicherter GCAM oder aus Mitteldarmrohhomogenat Bei den ersten Versuchen, den Vo Komplex der V-ATPase aus dem Mitteldarm von M. sexta zu reinigen, wurde zunächst die Gobletzell-Apikalmembran aus Raupen im 5. Larvenstadium, die für 16-20 h gehungert hatten, genau wie für das V1Vo Holoenzym (3.1.1-2) beschrieben angereichert, solubilisiert und gereinigt. Der Vo Komplex befand sich in der 20% Saccharose Fraktion des diskontinuierlichen Gradienten, welche dann auch weiter verarbeitet wurde. Auch hier wurde, um eine höhere Ausbeute zu erzielen, auf das Rohhomogenat von ganzen Därmen hungernder Tiere als Startmaterial zurückgegriffen. Als weitere Modifikation des Protokolls wurde das zweite Pellet der Zentrifugation bei 12.000 x g in ETPKI (5 mM EDTA, 5 mM Tris-HCl, 5 mM Pefabloc SC, 0,8 M KI, pH 8,1) resuspendiert und für 30 min auf Eis inkubiert. Anschließend wurde die Probe mit TEM auf eine KI Konzentration von 40 mM verdünnt und bei 200.000 x g für 30 min bei 4°C zentrifugiert. Das Pellet wurde in TEM resuspendiert und nochmals zentrifugiert. Das letzte Pellet wurde dann genau wie das V1Vo Holoenzym (3.1.2.) solubilisiert und gereinigt, nur dass, wie schon erwähnt, hierbei die 20% Saccharose Fraktion nach der zonalen Zentrifugation für die Anionenaustauscher- und die Gelpermeationschromatographie weiter verwendet wurde. 3.1.4. Reinigung des V1 Komplexes aus Mitteldarm Der V1 Komplex aus dem Mitteldarm wurde nach der Methode von Gräf et al. (1996) mit einigen wichtigen Modifikationen durchgeführt. Die Mitteldärme von bis zu 30 eisgekühlten 18 Methoden Raupen (Ernährungs-, bzw. Entwicklungsstatus versuchsabhängig) wurden in eiskaltem MENT-Puffer gesammelt, dreimal mit diesem gewaschen und in 5 ml MENTP mit einem Ultraturrax bei 20500 rpm für 50 s homogenisiert. Das Homogenat wurde bei 10.000 x g und 4°C in einem Festwinkelrotor (JA-25.50, Beckman) für 5 min, und der daraus resultierende Überstand 30 min bei 200.000 x g und 4°C in einem Festwinkelrotor (90 Ti, Beckman) zentrifugiert. Dieser Überstand wurde nach einer Filtration (0,22 µm) langsam mit einem gleich großen Volumen kalter gesättigter Ammoniumsulfatlösung gemischt und unter Rühren für 30 Minuten auf Eis inkubiert. Das Präzipitat wurde durch eine 5-minütige Zentrifugation bei 10.000 x g und 4°C in einem Festwinkelrotor (JA-25.50, Beckman) pelletiert, in TENPuffer mit 9,6 mM -Mercaptoethanol resuspendiert, auf einen diskontinuierlichen Saccharosegradienten (3 ml 40%, 2 ml 30%, 2 ml 20% und 1,6 ml 10% Saccharose in TENPuffer mit 9,6 mM -Mercaptoethanol, ein Gradient pro 4 Raupenausgangsmaterial) geschichtet und für 90 min bei 310.000 x g-Max und 4°C zentrifugiert. Die gesamte 30%Saccharosefraktion (2 ml) und die untere Hälfte (1 ml) der 20%-Saccharosefraktion wurden gepoolt, filtriert (0,22 µm) und mittels eines FPLC- oder HPLC-Systems (Pharmacia oder Beckman) über eine Anionenaustauschersäule (Mono Q, Pharmacia oder Hyper D Q, Biosepra) weiter aufgereinigt. Der V1-Komplex eluiert in einem linearen Salzgradienten (50 – 400 mM NaCl in LOM) bei ca. 220 mM als einzelner Peak. Dieser wurde durch Ultrafiltration (100 kDa Ausschlußgröße) auf 100 µl ankonzentriert und über Gelpermeationschromatographie (FPLC, Superdex 200, Pharmacia) endgültig gereinigt. 3.1.5. SDS-PAGE Die SDS-Polyacrylamidgelelektrophorese (SDS-PAGE) wurde modifiziert nach Lämmli et al. (1970) in vertikalen Gelkammern unterschiedlicher Fabrikate durchgeführt (BIORAD, Hoefer, Biometra). Die Proteinproben wurden mit einer 5-fach Stammlösung Probenauftragspuffer auf eine Endkonzentration von 125 mM Tris-HCl pH 6,8, 5% Saccharose, 2% SDS, 0,005% Bromphenolblau und 2% -Mercaptoethanol gebracht, für 45 s bzw. 3 min bei 95°C erhitzt und auf Eis abgekühlt. Die Proben wurden entweder direkt auf ein Gel geladen oder bei -20°C bis zur Verwendung gelagert. Waren die Proteinkonzentrationen sehr niedrig, wurden die Proben mit Trichloressigsäure (10% Endkonzentration) für 30 min auf Eis gefällt. Zweimal mit eiskaltem Aceton gewaschen und für je 10 min zentrifugiert (20.000 x g 10, 4°C, Festwinkelrotor). Anschließend wurden die 19 Methoden Proben in einem angemessenen Volumen einfach konzentriertem Probenauftragspuffer resuspendiert und wie oben weiter behandelt. Falls nicht anderweitig beschrieben, bestanden die Sammelgele aus 5,4% T/ 2,3% C Acrylamid in 125 mM Tris-HCl pH 6,8 und 0,2% SDS. Für die Trenngele wurde entweder 17% T/ 0,4% C oder 12% T/ 0,4% C in 330 mM Tris-HCl pH 8,7, 01% SDS verwendet. Der Laufpuffer enthielt 100 mM Tris, 100 mM Glycin, 0,1% SDS und pH 8,8. Die Elektophorese wurde in der Regel mit einer konstanten Stromstärke von 15 mA/Gel gestartet. Nachdem die Probe vollständig ins Sammelgel eingelaufen war wurde die Stromstärke auf 30 mA/Gel erhöht. Alternativ wurde außerdem die Tricine-SDS-PAGE nach Schägger & Jagow (1987) in einer Hoefer SE 600 Kammer (Pharmacia) verwendet. Die Gele setzten sich aus einem Trenngel (16,5% T, 3% C), Spacergel (10% T, 3% C) und einem Sammelgel (4% T, 3% C) zusammen. Die Proben wurden mit 3-fach Probenauftragspuffer (Endkonzentration: 4% SDS, 12% Glycerin, 2% -Mercaptoethanol, 0,01% Serva blue G 50 mM Tris-HCl pH 6,8) versetzt und entweder für 45 s bei 95°C erhitzt oder für 30 min bei 40°C inkubiert. Die Elektrophorese wurde mit 35 V gestartet, und, nachdem die gesamte Probe ins Gel eingewandert war (ca. 1h), bei 90 V für 16 h durchgeführt (Anodenpuffer, 200 mM Tris-HCl pH 8,9; Kathodenpuffer, 100 mM Tris, 100 mM Tricine, 0,1% SDS pH 8,25). Für die mit J-Concanolid A markierten Proben wurden teilweise auch Gradientengele mit 1015% T und 3,3% C verwendet (Ratajczak, 1994). 3.1.6. Proteinbestimmung Die photometrische Bestimmung der Proteinkonzentration mit Amidoschwarz erfolgte nach Wieczorek et al. (1990) modifiziert nach Popov et al. (1975). Der Test beruht auf der Bindung des Farbstoffes Amidoschwarz an basische Aminosäuren und hat den Vorteil, dass er bei hoher Sensitivität relativ unempfindlich gegen Detergenzien, Reduktionsmittel und viele bei anderen Proteinnachweisen störende Substanzen ist. Die Proteinproben wurden mit jeweils 300 µl der Amidoschwarzlösung versetzt und 5 min bei RT inkubiert. Anschließend wurde bei 14000 rpm (Eppendorf-Tischzentrifuge) 4 min zentrifugiert, der Überstand abgesaugt, das Pellet zweimal mit 500 µl Essigsäure/Methanol (1:10) gewaschen und für je 3 min zentrifugiert. Dann wurde das Pellet in 350 µl 0,1 N NaOH gelöst, die Extinktion bei 615 20 Methoden nm gemessen und die Proteinmenge mit einer parallel erstellten Eichreihe aus BSAStandardlösungen (2, 4 und 6 µg BSA) bestimmt. 3.1.7. Western Blot Um die aufgetrennten Proteine nach einer Gelelektrophorese auf eine PVDF- oder Nitrocellulosemembran zu übertragen, wurden Western-Blots nach Towbin et al. (1979) im Semidry-Verfahren (Trans-Blot SD, BIO-RAD) durchgeführt. Das benutzte dreiteilige Puffersystem nach Khyse-Anderson (1984) wurde durch Zugabe von 20% Methanol zu den Blotpuffern modifiziert. Der Transfer war in der Regel nach 60 min bei 1 mA/cm2 Gelfläche quantitativ abgeschlossen, was durch reversibles Färben der Blotmembran mit 0,02% Ponceau S und Anfärben des Gels mit Coomassie Blau R-250 überprüft wurde. 3.1.8. Reinigung von Antikörpern Die verwendeten Antikörper aus Blutseren bzw. Hybridomazellkulturüberständen wurden per Affinitätschromatographie über Protein A- bzw. Protein G-gekoppelten Sepharosesäulen (Pharmacia und Sigma) gereinigt. Entsprechend dem für die Produktion von Antikörpern verwendeten Wirtsorganismus wurde das Säulenmaterial mit der jeweils höheren Affinität ausgewählt. Zunächst wurden die Proben mit TBS (500 mM NaCl, 20 mM Tris-HCl pH 7,6) 5-fach verdünnt, sterilfiltriert (0,22 µm) und auf die Säule aufgetragen. Anschließend wurde mit TBS gewaschen und die gebundenen Antikörper mit Elutionspuffer (0,1 M Glycin-HCl pH 2,7) eluiert. Die gesammelten Fraktionen wurden durch vorgelegten Neutralisationspuffer (1 M Tris-HCl pH 8,0) neutralisiert. Mit Hilfe von Dotblots wurden die Fraktionen, welche Antikörper enthielten, bestimmt, gepoolt und gefriergetrocknet. Bei den Antiseren gegen Fusionsproteine mit maltosebindendem Protein (MBP) wurden die spezifischen Antikörper gegen MBP mittels Affinitätschromatographie im Batch-Verfahren entfernt. Hierzu wurde 1 mg MBP (New England Biolabs) an 60 mg Cyanbromid-aktivierte Sepharose 4B (Sigma) gekoppelt, mit 100 mM Glycin-HCl (pH 2,7) gewaschen und mit 2 ml TBS-Puffer äquilibriert. Dann wurden zu 500 µl Aliquots der Immunseren 125 ml 5x TBSPuffer gegeben, filtriert (0,45 µm) und mit der MBP-Sepharose vermischt. Nach 1,5 h Inkubation bei Raumtemperatur wurde die MBP-Sepharose bei 2000 x g (5 min, RT) pelletiert und der Serumüberstand abgenommen. Anschließend wurden die MBP-Antikörper 21 Methoden mit 100 mM Glycin-HCl (pH 2,7) von der MBP-Sepharose eluiert und mit (1 M Tris-HCl pH 8,0) neutralisiert. Die MBP-Sepharose wurde mit TBS-Puffer regeneriert und wiederverwendet. Die Abreicherung der MBP-Antikörper aus dem Serum wurde mit Dotblots kontrolliert und mit dem Serumüberstand wiederholt, bis das Eluat der MBP-Sepharose keine Antikörper gegen MBP mehr enthielt. 3.1.9. Immunfärbung Um unspezifische Markierungen zu verhindern, wurde die Blotmembran zunächst für bis zu 60 min in Blockpuffer und anschließend mit einer Verdünnung des primären Antikörpers in Verdünnungspuffer für eine weitere Stunde inkubiert (siehe Tab. 1). Nachdem die Blotmembran durch 3 x 5 min waschen mit TTBSN von überschüssigen primären Antikörpern befreit war, erfolgte eine einstündige Inkubation mit den entsprechenden sekundären Antikörpern, die mit alkalischer Phosphatase gekoppelt waren (Verdünnungen siehe Tab.1). Nicht gebundene Antikörper wurden durch 3-maliges Waschen mit TTBSN (je 5 min) entfernt, die Membran mit destilliertem Wasser gespült und die spezifisch immunmarkierten Proteinbanden durch Zugabe des APP-Puffers mit NBT/BCIP sichtbar gemacht. Tab.1: Übersicht der verwendeten Antikörper. Die monoklonalen Antikörper in Mäusen und die polyklonalen Antikörper in Kaninchen wurden in unserem Labor hergestellt. Die polyklonalen Antikörper aus Meerschweinchen wurden durch die Firma Charles River (Deutschland) hergestellt. Antikörper Antigen: Hergestellt in: Verdünnung 90-7 E Untereinheit Maus, monoklonal 1:100 47-5 E und G Untereinheit Maus, monoklonal 1:100 221-9 A Untereinheit Maus, monoklonal 1:10-100 224-3 B und e Untereinheit Maus, monoklonal 1:10-100 488-1,2 C Untereinheit Meerschweinchen, polyklonal 1:1000 C23 B Untereinheit (AS 243-428) Meerschweinchen, polyklonal 1:1000 C34 B Untereinheit (AS 475-494) Meerschweinchen, polyklonal 1:100 M40 d Untereinheit Meerschweinchen, polyklonal 1:1000 353-1,2,4 V1 Komplex Meerschweinchen, polyklonal 1:10000 373-3 a Untereinheit aus Rind M115 Kaninchen, polyklonal 1:100 K2 Holoenzym Kaninchen, polyklonal 1:10000 22 Methoden 3.1.10. Coomassie-Färbung Die Gele wurden im Anschluß an die Elektrophorese für mindestens 30 min mit CoomassieLösung (0,25% Coomassie Brilliant-Blue R-250 w/v, 50% Methanol und 10% Essigsäure) fixiert und gefärbt (Bollag & Edelstein, 1991). Zur Entfärbung wurden die Gele entweder für 15 min in 200 ml H2O/Gel in einer Haushaltsmikrowelle (600 W) gekocht oder mit einem zweiteiligen Puffersystem für 30 min in Entfärber I (25% Isopropanol, 10% Essigsäure) und danach für 60 min Entfärber II (7,5% Methanol, 10% Essigsäure) entfärbt. 3.1.11. Silber-Färbung Wenn es nötig war geringe Proteinmengen, besonders membranständiger Untereinheiten auf den Gelen zu visualisieren, wurde die Methode der Silberfärbung nach Heukeshoven & Dernick (1988) benutzt. Dafür wurden die Gele zuerst für 30 min fixiert (40% Ethanol, 10% Essigsäure) und anschließend für 30 min sensitiviert (30% Ethanol, 0,125% Glutaraldehyd (w/v), 0,2% Thiosulfat (w/v) und 6,8% Natriumacetat (w/v)). Nach 3-maligem Waschen mit H2O (5 min) wurde für 20 min die eigentliche Silberreaktion durchgeführt (0,25% Silbernitrat (w/v) und 0,015% Formaldehyd(w/v)). Die Entwicklung, sprich die Reduktion der Silberionen erfolgte in einer Lösung aus 2,5% Natriumcarbonat (w/v) und 0,0075% Formaldehyd (w/v). Durch Waschen mit 1,5% Na-EDTA (w/v) wurde die Entwicklung gestoppt. Um ein Zerspringen der Gele bei der Trocknung zu vermeiden, wurden sie für mindestens 30 min in 30% Ethanol mit 4% Glycerin inkubiert. 3.1.12. Markierung mit 14C-DCCD Die Markierung und Identifizierung des Proteolipids, der Untereinheit c der V-ATPase durch 14 C-N´,N´-Dicyclohexylcarbodiimid (14C-DCCD) erfolgte in Anlehnung an Lepier et al. (1996). Dazu wurde zunächst 6 µg Protein in 500 µl DCCD-Labellingpuffer (50 mm TrisMOPS pH 8,1, 1 mM MgCl2, 1 mM -Mercaptoethanol, 0,1 mM EDTA, 10 µM 14C-DCCD (0,3 µCi)) für 30 min bei 30°C inkubiert. Die Probe wurde, wie unter (3.1.7.) beschrieben, mit TCA gefällt und für die SDS-PAGE vorbereitet, wobei sie allerdings nicht auf 95°C erhitzt, sondern 1 h bei 37°C inkubiert wurde, um ein Aggregieren des Proteolipids zu verhindern. Nach der Gelelektrophorese, dem Färben mit Coomassie Brilliant-Blue 250-R und dem 23 Methoden Entfärben wurde das Gel, vor dem Trocknen auf Whatmanpapier für je 30 min in ENHANCE-Lösung (DuPont) und in kaltem H2O inkubiert. Für die Autoradiographie der mit 14 C-DCCD markierten Banden wurde das getrocknete Gel entweder auf einen Kodak XAR 5 Röntgenfilm ohne Verstärkerscreen oder auf einen Phosphoscreen für mehrere Tage aufgelegt und anschließend entwickelt bzw. mit einem Phosphoimager ausgewertet. 3.1.13. Markierung mit 125J-Concanamycin A 20-30 µg der Proben wurden in einem Volumen von 30-40 µl mit 0,01-100 µM 9-O-[p(Trifluoroethyldiazirinyl)-benzoyl]-21,23-dideoxy-23-epi-[125I]Iodo-concanolid A (J-Con- canolid A) für 1 h auf Eis oder 3 min bei Raumtemperatur inkubiert und anschließend auf Eis für 3-5 min UV bestrahlt (366 nm, 2 x 6 W). Die Proben für die Schutzversuche wurden zusätzlich mit 1 µM, 10 µM, 100 µM und 300 µM Concanamycin A bzw. mit 10 µM und 100 µM der anderen Substanzen für 1 h auf Eis vor Zugabe des J-Concanolid A vorinkubiert. Alle Makrolide wurden in DMSO gelöst, die Konzentration anhand ihres Extinktionskoeffizienten in Methanol photometrisch bestimmt, aliquotiert, bei -70°C gelagert und nur einmal direkt vor dem entsprechenden Versuch aufgetaut. Nach der Bestrahlung wurden 7,5 –10 µl 5-fach SDSProbenpuffer hinzugefügt, die Proben für 45 s auf 95°C erhitzt oder für 30 min bei 40°C inkubiert, auf Eis abgekühlt und auf ein SDS-Gel aufgetragen. Die Gele wurden entweder mit Coomassie gefärbt und nach dem Entfärben auf Whatmanpapier getrocknet oder mit Zn2+gefärbt und in Folie eingeschweißt. Anschließend wurden sie für bis zu 72 h auf einem Phosphoscreen exponiert. Die Auswertung erfolgte mit einem Phosphoimager (Molecular Probes). Nach der Exposition wurden die Zn2+-gefärbten Gele mit Coomassie nachgefärbt und gegebenenfalls die Untereinheit c für MALDI-MS-Analysen bzw. die Messung der gebundenen Radioaktivität in einem Gamma-Counter (Philips, PW 4800) ausgeschnitten. 3.1.14. Isoelektrische Fokussierung mit anschließender 2-D Elektrophorese Für die isoelekrische Fokussierung der Proteinkomplexe wurde das DryStrip-System von Pharmacia benutzt. Dazu wurden die Drystrips in 240 µl Rehydrierungslösung (8 M Harnstoff, 2% Chaps, 2% IPG-Puffer, 0,3% DTT und 0,01% Bromphenolblau) über Nacht rehydriert und dann auf einer Multiphor II in der "Drystripwanne" positioniert. Die Proben wurden mit Probenauftragspuffer versetzt (9,8 M Harnstoff, 2% -Mercaptoethanol, 2% IPGPuffer, 0,5% Triton X-100) und mittels Probenauftragsbechern kathodisch aufgetragen. Die Elektrophorese erfolgte zunächst für 4,5 h bei 300 V und dann für 11,5 h bei 2000 V. 24 Methoden Anschließend wurden die Drystrips entweder sofort weiter verwendet oder bei -70°C gelagert. Für die zweite Dimension wurden die Drystrips zuerst 10 min in Äquilibrierungslösung I (100 mM Tris-HCl pH 6,8, 6 M Harnstoff, 30% Glycerin, 1% SDS, 1% DTT) und dann 10 min in Äquilibrierungslösung II (100 mM Tris-HCl pH 6,8, 6 M Harnstoff, 30% Glycerin, 1% SDS, 200 mM Jodacetamid und 0,01% Bromphenolblau) inkubiert und auf ein vertikales SDS-Gel (Sammelgel ohne Taschen) in einer Biometra-Kammer aufgelegt. Die Elektrophorese erfolgte wie unter (3.1.7.) beschrieben. 3.1.15. Röntgenkleinwinkelstreuung Die Röntgenkleinwinkelstreuungsexperimente wurden zusammen mit Dr. Gerhard Grüber (Osnabrück) und Dr. Michel Koch (EMBL, Hamburg) in der Hamburg Outstation des EMBL wie in Svergun et al. (1998) für den V1 Komplex beschrieben durchgeführt. 3.1.16. Matrix-unterstützte Laserdesorptions Ionisations Massenspektroskopie (MALDI-MS) Die massenspektrometrischen Untersuchungen der Untereinheiten der V-ATPase wurden von Dr. Simone König im Proteomik-Labor der Zentralen Projektgruppe "Integrierte Funktionelle Genomik" des Interdisziplinären Klinischen Forschungszentrum der Medizinischen Fakultät der Universität Münster durchgeführt. Nach der SDS-PAGE wurden die mit Coomassie gefärbten Proteinbanden mit einer modifizierten Methode nach Shevchenko et al. (1996) und Zhang et al. (1998) für die Massenspektrometrie vorbereitet. Die Banden wurden ausgeschnitten und mit 25 mM NH4HCO3 in 50% Methanol entfärbt. Das Gelstück wurde zunächst mit Eisessig/Methanol/Wasser (10/45/45, v/v/v) 30 min und anschließend mit Wasser 30 min gewaschen. Danach wurde es mit Acetonitril geschrumpft, getrocknet und in 20 µl 50 mM NH4HCO3 mit 400 ng Trypsin oder Chymotrypsin rehydriert. Überschüssige Proteaselösung wurde entfernt und das Gelstück mit Puffer überschichtet. Der Verdau (37°C, über Nacht) wurde durch Zugabe von 5-10 ml Eisessig gestoppt und der Überstand in ein neues Eppendorfreaktionsgefäß transferiert. Das Gelstück wurde zweimal mit 70 µl Acetonitril extrahiert und die Überstände vereinigt und lyophilisiert. Das Lyophilisat wurde in 7 µl 0,1% wässriger TFA gelöst, mit ZipTips (Millipore, Bedford, MA, USA) gereinigt und die Peptide mit 7 µl 70% Acetonitril eluiert. Für die Matrix wurden 10 mg α-cyano-4hydroxycinnamic Säure mit Aceton gewaschen und in 1 ml Acetonitril/Ethanol (50/50, v/v) mit 1% 0,1% wässriger TFA aufgenommen. Davon wurden 0,7 µl und 0,7 µl Probe auf den Probenträger aufgebracht und sofort gemischt. Für die MALDI wurde ein TofSpec-2E Gerät 25 Methoden (Micromass Ltd., Manchester, UK) benutzt. Die Proben wurden im positive ion reflectron mode mit einer Matrix-Unterdrückung von 500 gemessen. Die Massen-Kalibrierung erfolgte extern und wurde entweder mit der lock mass Option oder den Trypsin Autolyse Peaks intern korrigiert. 3.1.17. N-terminale Sequenzierung Für die N-terminalen Sequenzierungen wurde das V1Vo Holoenzym, der V1 Komplex oder der Vo Komplex wie beschrieben gereinigt, mittels SDS-PAGE aufgetrennt und auf PVDFMembran geblottet. Anschließend wurden die Proteine mit Amidoschwarz- oder CoomassieFärbung sichtbar gemacht und die entsprechenden Banden ausgeschnitten. Die Sequenzierung der Proben erfolgte durch Dr. Roland Schmid (Universität Osnabrück) bzw. durch Dr. Barbara Schedding (Universität Münster) entweder in einem Applied Biosystems Sequencer Modell 473A oder Modell 492-01. 3.1.18. ATPase-Aktivitätstest und Phosphatbestimmung Um die spezifische ATPase-Aktivität der verschieden Proben zu ermitteln, wurde das bei der Hydrolyse von ATP entstehende anorganische Phosphat photometrisch bestimmt (Wieczorek et al., 1990). Der Vorteil dieser verwendeten Methode gegenüber anderen Pi-Bestimmungen liegt in ihrer hohen Sensitivität (Extinktion 0,3 ≙ 4 ng Pi) und in ihrer Linearität bis zu einer Extinktion von über 2. Der Standardansatz mit einem Endvolumen von 160 µl enthielt 50 mM Tris-MOPS (pH 8,1), 20 mM KCl, 1 mM Tris-ATP, 3 mM -Mercaptoethanol und 1 mM MgCl2 bzw. 3 mM CaCl2. Nach einer 5-10 minütigen Vorinkubation ohne Substrat im Wasserbad bei 30°C wurde die Enzymreaktion durch Zugabe von ATP gestartet, nach Ablauf der gewählten Reaktionszeit durch Tiefgefrieren in flüssigem Stickstoff gestoppt und die Probe danach bei -20°C gelagert. Die Quantifizierung des freigesetzten anorganischen Phosphats erfolgte photometrisch als Malachitgrün-Phosphomolybdat-Komplex. Zu den 160 µl der tiefgefrorenen Probe des Aktivitätstests wurden 50 µl 20% Trichloressigsäure pipettiert und das Ganze im Wasserbad bei 70°C aufgetaut. Das ausgefallene Protein wurde durch Zentrifugation pelletiert (1 min, 13200 rpm, Eppendorf-Tischzentrifuge 5415D). Exakt 3,5 min nach der TCA-Zugabe wurden 60 µl des Überstandes mit 100 µl saurem Molybdat (2,4% H2SO4, 60 mM Na-Molybdat) vermischt. Nach weiteren 15 s wurden 30 µl Malachitgrünlösung (7,4% in gesättigtem, filtriertem Polyvinylalkohol) zupipettiert. Nach insgesamt 5 min 45 s wurde nicht 26 Methoden komplexiertes Malachitgrün mit 200 µl 7,8% H2SO4 zerstört. Die Extinktion der resultierenden Lösung bei 625 nm wurde nach 70-100 min photometrisch bestimmt (SpektraMax Plus, Molecular Devices) und der Phosphatgehalt an Hand von mitgeführten Phosphatstandardwerten (0, 10 und 25 nmol) errechnet. 3.1.19. Bestimmung und Entfernung endogener Nukleotide Der Nachweis und die Quantifizierung der im Enzym fest gebundenen Nukleotide erfolgte mit der leicht modifizierten Luciferin/Luciferase-Methode nach Senior et al. (1992). Die Proben wurden in einem Volumen von 100-200 µl (Proteinkonzentration zwischen 0,5 und 2 µg/µl; Puffer für den V1 Komplex 150 mM NaCl, 9,6 mM -Mercaptoethanol, 20 mM TrisHCl pH 8,1 und für das V1Vo Holoenzym zusätzlich 0,01% C12E10) für 5 min bei 100°C denaturiert, auf Eis abgekühlt und das denaturierte Protein 5 min bei 4°C abzentrifugiert (20.000 x g, Beckman-Tischzentrifuge). Der Überstand wurde abgenommen, bei -20°C eingefroren und dort bis zur Bestimmung des Nukleotidgehalts gelagert. Um das freigesetzte ATP zu messen, wurden zu 10 oder 20 µl des Überstandes zu 500 µl Luciferin/Luciferase-Puffer (60 mM Tris-Acetat pH 7,75, 10 mM MgCl2, 1 mM KCl, 1,5 mM EDTA, 2,5 mM -Mercaptoethanol und 0,4 mg/ml Luciferin/Luciferase (Sigma)) gegeben. Nach 30 s Vorinkubation wurde in einem Luminometer (Lumat LB 9507, EG &G Berthold) die Lichtemission für 60 s gemessen. Zur Quantifizierung des ATP-Gehaltes wurde jeweils parallel zu den Proben eine Eichreihe (0,1 pmol-1 nmol ATP) und eine nur Puffer enthaltende Kontrolle mitgeführt. Die Eichung ergab jeweils eine lineare Beziehung zwischen der Menge an eingesetztem Nukleotid und der gemessenen Lichtemission. Zur Bestimmung des freigesetzten ADPs wurden zu 10 oder 20 µl des Überstandes 90 µl Luciferin/Luciferase-Puffer (ohne Luciferin/Luciferase) und 10 µl Pyruvatkinase-Lösung (70 mM Tris-Acetat pH 7,75, 9 mM MgCl2, 5 mM KCl, 2 mM Phosphoenolpyruvat, 2 mM EDTA und 500 U/ml Pyruvatkinase) gegeben und das Ganze für 40 min bei 30°C inkubiert. Anschließend wurde die Probe mit 400 µl Luciferin/Luciferase-Puffer vermischt und die Lichtemission nach Ablauf von 30 s für 1 min gemessen. Auch hier wurde jeweils parallel eine ADP-Eichreihe (0,1 pmol-1 nmol ADP), welche ebenfalls eine gerade Eichkurve ergab, und eine Pufferkontrolle mitgeführt. Zur Entfernung von Nukleotiden aus dem V1 Komplex wurde im Anschluß an die Standardreinigung (3.1.4.) eine zusätzliche Gelpermeationschromatographie auf einer Superdex 200 Säule (Pharmacia) durchgeführt, wobei dem Laufpuffer (150 mM NaCl, 20 mm 27 Methoden Tris-HCl pH 8,1, 9,6 mM -Mercaptoethanol) entweder a) nichts (Kontrolle), b) 25% Methanol, c) 5 mM EDTA oder d) 0,01% C12E10 zugesetzt wurden. Außerdem wurde noch ein nach Senior et al., (1992) adaptiertes Verfahren mit einer Sephadex G-50 Säule (1,6 cm x 100 cm, Flußrate 0,03 ml/min, RT) und einem Säulenpuffer aus 100 mM Tris-SO4 pH=8,0, 4 mM EDTA, 50% Glycerin verwendet. Zur Kontrolle wurde ein entsprechendes Aliquot parallel bei RT inkubiert. Letztlich wurde auch versucht, Nukleotide durch Behandlung des V1 Komplexes mit alkalischer Phophatase (APP) zu entfernen (Digel et al., 1996). Hierzu wurden 460 µg V1 Komplex in 110 µl Puffer (150 mM NaCl, 20 mm Tris-HCl pH 8,1, 9,6 mM Mercaptoethanol) mit 57 U APP (Stratagene, Nr. 6000015) für 18 h bei 25°C in einem Eppendorfschüttler (300 rpm) inkubiert. Zur Kontrolle wurde parallel je ein Aliquot mit V1 Komplex ohne APP und ein Aliquot ohne V1 Komplex mit APP inkubiert. Anschließend wurde die APP mittels FPLC auf einer Superdex 200 Säule (Pharmacia) quantitativ vom V1 Komplex abgetrennt, was mit Hilfe der NBT/BCIP-Reaktion in einem Titerplattentest der Fraktionen überprüft wurde. 3.1.20. Behandlung des V1 Komplexes mit chaotropen Ionen (KI-Stripping) Der V1 Komplex wurde in 0,8 M KI, 150 mm NaCl, 20 mm Tris-HCl und 9,6 mM 2Mercaptoethanol bei pH 8,1 für 45 min auf Eis inkubiert. Anschließend wurde mit der Probe eine Gelpermeationschromatographie auf einer Superdex 200 HR 10/30 (Pharmacia) mittels FPLC bei einer Flußrate von 0,5 ml/min im gleichen Puffer ohne KI durchgeführt. 3.1.21. Messung des ATP-getriebenen Protonentransports Der ATP-getriebene Protonentransport wurde an GCAM-Vesikeln nach Wieczorek et al. (1991) gemessen. Dazu wurden die Membranpellets in THTCl resuspendiert und ein Standardansatz von 790 µl (pH=8,1) vorbereitet, der sich folgendermaßen zusammensetzte: 20 µl 40 mM Tris-ATP in 200 mM Tris, 10 µl 75 µM Acridinorange, 10 µl THTCl-Vesikel, 8 µl 50 mM NaN3 in THTCl, 8 µl 10 mM Na3VO4 in THTCl und 734 µl THTCl; wenn mehr als 10 µl THTCl-Vesikel verwendet wurden, verringerte sich das THTCl-Volumen entsprechend. Die Reaktion wurde mit 10 µl 80 mM MgCl2 gestartet, so dass sich bei einem Endvolumen von 800 µl die Konzentrationen von 1 mM ATP, 0,9 µM Acridinorange, 0,5 mM NaN3, 0,1 mM Na3VO4 und 1 mM MgCl2 in THTCl ergaben. Die Ansäuerung des Vesikelinneren durch die V-ATPase wurde fluorometrisch gemessen (Fluorometer: LS 50 B, 28 Methoden Perkin Elmer) und aufgezeichnet. Die Rückstellung der Fluoreszenz erfolgte durch Zerstörung des Protonengradienten mit 10 µl 1,6 M KCl bzw. 10 µl 1,6 M NH4Cl (Endkonzentration jeweils 20 mM). 3.1.22. Rekonstitution des V1Vo Holoenzyms in Liposomen Für die Messungen von kapazitiven Strömen am blacklipid Bilayer wurde das V1Vo Holoenzym nach der Methode von Fendler et al. (1996) in Liposomen rekonstituiert. Dazu wurden zunächst 2,5 mg in Chloroform gelöste Phospholipide im Rotationsverdampfer unter Stickstoffbegasung für ca. 90 min getrocknet und anschließend in 250 µl LOM mit 1% C 12E10 resuspendiert. Nach 1 h Inkubation bei Raumtemperatur wurde die Suspension bis zur Opaleszenz im Eisbad beschallt. Für die Rekonstitution des V1Vo Holoenzyms wurde ein Protein-Lipid-Verhältnis von 1:20 (w/w) gewählt und 250 µl der vorgeformten Liposomen mit 0,25 mg Protein (in 275 µl 1% C12E10, 9,6 mM -Mercaptoethanol, 150 mM NaCl und 20 mM Tris-HCl pH 8,1) gemischt und für 15 min auf Eis inkubiert. Das Detergenz wurde durch Zugabe von Biobeads (Biorad) entfernt, zunächst wurde ca. 1/3 Volumen Beads zugegeben und die Suspension unter sanftem Schütteln über Nacht bei 4°C inkubiert. Die Beads wurden mittels Filtration durch Glaswolle entfernt und durch ca. 1/4 Volumen frische Beads ersetzt, welche nach 45 min sanftem Schütteln bei Raumtemperatur ebenfalls abfiltriert wurden. Nach 5 min Zentrifugation bei 13000 x g, um evtl. unlösliche Bestandteile zu entfernen, wurden die Proteoliposomen direkt für die Messungen eingesetzt. 3.1.23. Messung kapazitiver Ströme am blacklipid Bilayer Die Messungen an sogenannten "schwarzen" Lipid-Doppelschichten (Blacklipid Bilayer) erfolgten bei Dr. Klaus Fendler (MPI für Biophysik, Frankfurt). Dazu wurden 0,01-0,02 cm2 große optisch schwarze Lipid-Doppelschichten in einer temperierten Teflonzelle (24°C) hergestellt (Fendler et al., 1985; Borlinghaus et al., 1987). Die Lipid-Doppelschicht wurde aus einer Lösung mit 1,5% (w/v) Diphytanoylphosphatidylcholin und 0,025% (w/v) Octadecylamin gelöst in n-Decan gebildet. Die beiden Kammern der Zelle waren mit je 1,3 ml Puffer (20 mM Tris-HCl pH 7,0, 3 mM DTT, 150 mM NaCl) gefüllt und über Polyacrylamid Salzbrücken und Ag/AgCl Elektroden mit einem externen Meßkreis verbunden, wobei das Meßsignal verstärkt, gefiltert (Cutoff, 500 Hz) und mit einem digitalen Oszilloscop aufgezeichnet wurde. Für eine Meßreihe wurden zwischen 10 und 50 µl Proteoliposomen in eine der Kammern pipettiert und für 30 min gerührt. Die Konzentrationen 29 Methoden von caged ATP (P3-[1-(2-nitrophenyl)ethyl]adenosin 5´-triphosphat, Fendler et al. 1985)), MgCl2, CaCl2 und Concanamycin A konnte durch Zugabe aus diversen Stammlösungen variiert werden. Die Photolyse des caged ATP wurde durch einen 10 ns langen Laserpuls (308 nm, 12 mm dm Blende, Energie zwischen 10,7 und 11,7 mJ) der auf die Lipid-Doppelschicht fokusiert war bewirkt. Dabei wurden zwischen 15% und 30% des caged ATP im Laserfokus freigesetzt. Zwischen den einzelnen Laserblitzen wurde für 10 min gerührt um das freigesetzte ATP zu verdünnen bzw. die Hydrolyse durch die V-ATPase der nicht an die Lipid-Doppelschicht adsorbierten Proteoliposomen zu ermöglichen. 3.1.24. Auftrennung von Subkomplexen des V1Vo Holoenzyms Das V1Vo Holoenzym wurde in 70 µl Aliquots (100 µg Protein, 150 mM NaCl, 9,6 mM Mercaptoethanol, 20 mM Tris-HCl pH 8,1) für 10 min bei 30°C im Wasserbad vorinkubiert und dann mit 10 µl des Test-Nukleotids versetzt und entsprechend der gewählten Zeit bis zu 1 h weiter inkubiert. Im Anschluß an diese Behandlung wurde der V1 und Vo Komplex durch Zentrifugation in einem Saccharosestufengradienten getrennt. Hierfür wurden 920 µl Puffer (5% Saccharose in TEK, 0,01% C12E10, 9,6 mM -Mercaptoethanol, RT) zur fertig inkubierten Probe gemischt und auf den Dichtegradienten geschichtet (2 ml 40%, 1 ml 35%, 1 ml 30%, 1 ml 25%, 1 ml 20%,1 ml 15%,1 ml 10% Saccharose in TEK, 0,01% C12E10, 9,6 mM -Mercaptoethanol). Die Zentrifugation erfolgte für 90 min bei 310.000 x g und 25°C in einem Vertikalrotor (VTi 65.1, Beckman). Die Gradienten wurden anschließend mit einer Peristaltikpumpe und Glaskapillare beginnend mit der 40% Saccharosestufe von unten in 500 µl große Aliquots fraktioniert (Benennung der Fraktionen: 40/1, 40/2, 40/3, 40/4, 35/1, 35/2, 30/1, 30/2, 25/1 usw.). 3.2. Molekularbiologische Methoden 3.2.1. Polymerase-Kettenreaktion (PCR) Für die Polymerase-Kettenreaktionen wurde ein GeneATAQ-Controller (Amersham Pharmacia Biotech) verwendet. Ein Standardansatz (25 µl) enthielt 50 mM KCl, 10 mM TrisHCl pH 8,3, 1,5 mM MgCl2, 0,01% Gelatine, je 200µM dATP, dGTP, dCTP und dTTP, je 0,75 µM der beiden Primer, bis zu 250 ng Template-DNA und 0,625 U Taq Polymerase. Als Verdunstungsschutz wurden die Ansätze mit 2 Tropfen Mineralöl überschichtet. Die Reaktion 30 Methoden wurde wie folgt durchgeführt: 1 x 2 min 93°C, 35 x ( 30 s 93°C, 40 s Annealing-Temperatur, 60 s/1 kb 73°C), wobei die Annealing-Temperatur nach der Formel 4°C x (G + C) + 2° C x (A + T) - 3°C (Lion & Haas, 1990) berechnet und die niedrigere Temperatur des Primerpaars verwendet wurde. 3.2.2. Phenolextraktion und Ethanolfällung Die Aufreinigung von DNA aus den PCR-Ansätzen erfolgte mittels einer Phenolextraktion. Hierzu wurde zu der jeweiligen DNA-Lösung ein Volumenanteil Phenol:Chloroform: Isoamylalkohol (25:24:1) gegeben, durch Vortexen gemischt und 1 min zentrifugiert (13.000 x g). Aus dem wässrigen Überstand wurde das restliche Phenol mit 1 Volumen Chloroform:Isoamylalkohol (24:1) und erneutem Zentrifugieren entfernt. Anschließend wurde die wässrige Phase mit 1/10 Volumenteil 3 M Natriumacetat und 2,5 Volumenteilen -20°C kaltem Ethanol versetzt und für 30 min bei 14000 x g und 4°C zentrifugiert. Nachdem das Pellet noch einmal mit 70% -20°C kaltem Ethanol gewaschen und für 5 min bei 14000 x g zentrifugiert worden war, wurde es in einer Speedvac getrocknet und bei -20°C gelagert. 3.2.3. Plasmid DNA Präparation Für die Plasmidpräparationen wurde entweder die "klassische" Methode der alkalischen Lyse nach Sambrook et al. (1989) oder ein QIAprep Spin Miniprep Kit der Firma Qiagen benutzt. Erstere wurde vor allem für die Sequenzierungsreaktionen benutzt, da sie in der Regel wegen höherer Reinheit bessere Ergebnisse liefert. 3.2.4. Spaltung von DNA durch Restriktionsenzyme Die enzymatische Spaltung der DNA wurde normalerweise in einem Volumen von 20 µl und mit bis zu 3 µg DNA und 3-5 U Restriktionsenzym durchgeführt. Sowohl die Pufferzusammensetzung als auch die Inkubationstemperatur richtete sich nach den Angaben der Hersteller der einzelnen Restriktionsenzyme. Nach einer Inkubation von 2 h wurde der Verdau durch Einfrieren gestoppt und das Ergebnis durch die Elektrophorese in einem 1% Agarosegel überprüft. 31 Methoden 3.2.5. Agarosegelelektrophorese von DNA Für die gelelektrophoretische Auftrennung von DNA-Fragmenten wurden FlachbettMinigelkammern, 1%ige Agarosegele und das TAE-Puffersystem (40 mm Tris-Acetat pH 8,3, 1 mM Na-EDTA) verwendet. Die Elektrophorese wurde bei 7-10 V/cm durchgeführt. Zur Visualisierung der Fragmente wurden die Gele nach dem Lauf für bis zu 60 min mit Ethidiumbromid (1µg/ml in TAE) inkubiert und anschließend nicht gebundenes Ethidiumbromid durch Waschen der Gele in H2O (30 min) entfernt. Mit einem Eagle-EyeVideosystem (Stratagene) wurden die Ergebnisse dokumentiert. Die gewünschten DNAFragmente wurden mit Hilfe des JetSorb-Systems (Genomed) aus dem Gel isoliert. 3.2.6. Ligation und Transformation Die Ligation von DNA erfolgte über mindestens 24 h bei 16°C und wurde durch 10 min Erhitzen auf 68°C gestoppt. Die 25 µl großen Ligationsansätze enthielten 100 ng Vektor, die 10-fache molare Menge an Insert, 50 mM Tris-HCl pH 7,5, 7 mM MgCl2, 1 mM DTT, 0,5 mM ATP und 2 Weiss-Units T4-DNA-Ligase (New England Biolabs). Für die Transformation wurde ein 200 µl Aliquot tiefgefrorener kompetenter Zellen in einem Eppendorfgefäß in der Hand aufgetaut und für 10 min auf Eis temperiert. Anschließend wurden Sie mit 25 µl des Ligationsansatzes vermischt und für weitere 30 min auf Eis inkubiert. Nach einem 90 s "Hitzeschock" von 42°C, gefolgt von 2 min Abkühlen auf Eis wurden 800 µl Medium zugegeben und für 1 h bei 37°C unter sanftem Schütteln inkubiert. Um positiv transformierte Zellen mittels der Blau-Weiß-Selektion erkennen zu können, wurden die transformierten Zellen auf LBamp XGal/IPTG Platten ausplattiert. 3.2.7. Herstellung kompetenter Zellen Zur Herstellung von kompetenten Zellen wurde ein Protokoll der Firma New England Biolabs angewandt, bei dem die Tranformationseffizienz durch Verwendung von RbCl neben CaCl 2 erhöht wird. Zunächst wurde der Empfängerstamm auf a-Platten ausplattiert und über Nacht bei 37°C angezüchtet. Davon wurde eine einzelne Kolonie zum Animpfen von 5 ml bMedium verwendet und bis zu einer OD550 von 0,3 bei 37°C geschüttelt. Mit dieser Vorkultur wurden wiederum 100 ml auf 37°C vorgewärmtes b-Medium angeimpft und für ca. 2 h bei 37°C unter Schütteln bis zu einer OD550 von 0,5 kultiviert. Nachdem die Zellsuspension für 5 32 Methoden min auf Eis abgekühlt war, wurden die Zellen durch Zentrifugation (5 min, 5000 x g, 4°C) geerntet, in 40 ml eiskaltem Tfb-1 resuspendiert und für 5 min auf Eis inkubiert. Anschließend wurde nochmals zentrifugiert (5 min, 5000 x g, 4°C) und diesmal in 4 ml eiskaltem Tfb-2 resuspendiert. Nach 15 min auf Eis wurden die jetzt kompetenten Zellen in 200 µl großen Aliquots in flüssigem Stickstoff tiefgefroren und bei -80°C gelagert. 3.2.8. Sequenzierung von DNA Bei der Sequenzierung von DNA wurde nach der Dideoxynukleotid-Kettenabruchmethode von Sanger et al. (1977) vorgegangen, wobei der Sequenase 2.0-Kit mit modifizierter T7DNA-Polymerase (Tabor & Richardson, 1987) und 35S-dATP verwendet wurde. Als Primer dienten die Standardprimer T3 und T7, sowie insertspezifische in Auftragssynthese hergestellte Primer. Die Auftrennung der Abruchprodukte erfolgte durch eine Elektrophorese in einem denaturierenden Polyacrylamidgel (Sambrock et al., 1989) mit einem BRL Sequencing System S2. Anschließend wurden die Gele fixiert und auf Whatmann 3MM Filterpapier getrocknet. Nach einer 24 h Exposition auf einen Kodak XAR 5 Röntgenfilm konnte das Ergebnis der Autoradiographie ausgewertet werden. 3.2.9. Klonierung, Expression und Reinigung von rekombinanten Teilen der B Untereinheit im pMal-c2-System Als Antigene zur Herstellung von polyklonalen Antikörpern gegen 2 Abschnitte der Untereinheit B, (Aminosäure 243-428 und Aminosäure 475-494) wurden überexprimierte Fusionsproteine verwendet (Novak et al., 1992). Dazu wurden die entsprechenden Teilsequenzen mittels PCR auf einem cDNA Klon der B-Untereinheit von M. sexta in einem Sputnik-Expressionsvektor (Stratagene, Klon von Dr. Ralph Gräph zur Verfügung gestellt) als Template amplifiziert und in den Expressionsvektor pMal c2 (New England Biolabs) kloniert. Als Primerpaar für den bei V-ATPasen stark konservierten Mittelteil der B-Untereinheit (C2; Aminosäuren 243-428) dienten 56KVORW2 (CTC TTC TTG AAC TTG GCC AA) als Vorwärts-Primer und 56KREV2 (TAC TCA GAA TTC TTA GAG CGC CTC CTC ACC CAC TA) als Rückwärts-Primer. Für den C-Terminus (C3; AS 475-494) wurde 56KVOR3 (ATG TTG AAG CGT ATC CCG GC) als Vorwärts-Primer und 56KREV3 (TAC TCA GAA TTC TTA GTG ACG GGA GTC TCT GGG GT) als Rückwärts-Primer eingesetzt. Die beiden Vorwärts-Primer generierten Blunt-Enden, wohingegen die beiden Rückwärts-Primer eine Schnittstelle für EcoRI (unterstrichene Basen) einfügten. Die PCR-Produkte wurden wie 33 Methoden beschrieben (3.2.1-5) über ein Agarosegel gereinigt, eluiert und nach 2 h Inkubation mit EcoR1 bei 37° C eingefroren. Parallel dazu wurde das Plasmid pMAL-c2 gereinigt und ebenfalls für 2 h mit EcoR1 und XMN1 inkubiert und anschließend eingefroren. Die Ligation erfolgte über 72 h bei 16 ° C (3.2.6.). Anschließend wurden die Plasmide in kompetente E. coli XL1 Blue-Zellen transformiert (3.2.6-7) und auf LBamp-Platten ausgestrichen. Je 5 positive Kolonien aus der Blau-Weiß-Selektion wurden abgenommen (C2 1-5 und C3 1-5) und als Glycerindauerkulturen bei -70°C gelagert. Der korrekte Einbau des Inserts in das Plasmid der Klone wurde später mittels DNA-Sequenzierung überprüft (3.2.8.). Für die Expression der beiden Teilstücke C2 und C3 wurden jeweils zweimal 0,5 l TB-Amp Medium mit je 5 ml einer Übernachtkultur der Klone C23 und C34 angeimpft. Nachdem die Bakterien bis zu einer OD600 von 0,5 herangewachsen waren wurden sie mit 0,3 M IPTG induziert, um das entsprechende rekombinante Protein herzustellen. 2 h später wurden die Zellen durch eine Zentrifugation (20 min, 4000 x g, 4°C) pelletiert, in 50 ml Säulenpuffer (200 mM NaCl, 1 mM Na-EDTA, 20 mM Tris-HCl, pH 7,4) resuspendiert und bei -20°C eingefroren. Am nächsten Tag wurden die Zellen in kaltem Wasser langsam aufgetaut und in 5 ml großen Aliquots auf Eis für 2 min ultrabeschallt (Branson Sonifier, Mikrotip, Stufe 6, 50% Impulsdauer), um die Zellen zu lysieren. Anschließend wurde die Suspension für 30 min bei 9000 x g und 4°C zentrifugiert und der Überstand mit Säulenpuffer auf eine Proteinmenge von ca. 3 mg/ml eingestellt. Die Reinigung des Fusionsproteins aus dem Überstand erfolgte über Affinitätschromatographie, wobei das maltosebindende Protein an die kovalent mit der Agarosematrix der Säule (Volumen ca. 10 ml) verbundene Amylose bindet. Der gefilterte Überstand (Spritzenvorsatzfilter, 0,45 µm) wurde auf die Säule aufgetragen und diese danach mit dem 8fachen Säulenvolumen an Säulenpuffer durchgespült. Das gebundene Protein wurde mit 10 mM Maltose im Säulenpuffer eluiert, gesammelt und auf einem SDS-Gel mit reinem maltosebindendem Protein verglichen. Die Proben wurden ankonzentriert und zur Immunisierung von Meerschweinchen zu Charles River, Deutschland geschickt (8,5 mg C23 bzw. 1 mg C34). 34 Methoden 3.2.10. Herstellung der rekombinanten Untereinheit C im pET System Die rekombinante C Untereinheit, wurde für die Herstellung von polyklonalen Antikörpern und für Reassoziationsversuche mit dem pET Expressionssystem (Novagen) hergestellt. Hierzu wurde der komplette kodierende Abschnitt der cDNA mittels PCR amplifiziert, in den Vektor pET-16b eingefügt und in E. coli BL21 Zellen transformiert (Merzendorfer et al., 2000). Die Zellen wurden mit 0,4 mM IPTG induziert und das rekombinante Protein mit Hilfe eine N-terminal angefügten His-Tags über NTA-Säulen gereinigt. Dies erfolgte nach dem vom Hersteller (Novagen) vorgegeben Protokoll, wobei der Elution des rekombinanten Proteins mit 300 mM Imidazol, 0,5 M NaCl, 20 mM Tris-HCl (pH 7,9) ein Waschschritt mit 100 mM Imidazol, 0,5 M NaCl, 20 mM Tris-HCl (pH 7,9) vorraus ging. Um eine Aggregation des eluierten Proteins durch von der Säule abgelöstes Ni2+ zu unterbinden, wurde beim Fraktionieren eine 200 mM Na-EDTA-Lösung vorgelegt (Endkonzentration 10 mM in den Fraktionen). Das so gewonnene Protein war frei von Verunreinigungen und konnte direkt für die Immunisierung bzw. die Reassoziationsversuche eingesetzt werden. 35 Ergebnisse 4. Ergebnisse 4.1. Die V-ATPase aus M. Sexta 4.1.1. Optimierung der V1Vo Holoenzym Präparation Ein Manko aller bisher beschriebenen Reinigungsprozeduren für V-ATPasen ist die im Verhältnis zu F- und P-Typ-ATPasen relativ geringe Ausbeute pro Präparationsansatz, die aufwendigere biochemische und strukturelle Untersuchungen verhindert. Dies gilt auch für die Reinigung der V-ATPase aus dem Mitteldarm von M. sexta, bei der zudem bisher nur der posteriore Abschnitt des Mitteldarms benutzt wurde, der nur etwa ein Drittel der gesamten Länge ausmacht (Abb. 5). Abb. 5: Unterschiede in der Morphologie des anterioren, medianen und posterioren Mitteldarmepithels von Manduca sexta. (A) longitudinal geöffneter Mitteldarm. Faltungsmuster des (B) anterioren Teils, (C) medianen Teils, und (D) posterioren Teils. (E) Querschnitt durch das anteriore und mediane Mitteldarmepithel. (F) Querschnitt durch das posteriore Mitteldarmepithel. (G) Schnitt durch die Apikalmembran einer Gobletzelle des anterioren und medianen Mitteldarmepithels. (H) Schnitt durch die Apikalmembran einer Gobletzelle des posterioren Mitteldarms. CC, Columnarzelle; GC, Kavität der Gobletzelle; NC, Kern der Columnarzelle; NG, Kern der Gobletzelle; M, Mitochondrium. Die Abbildung wurde übernommen aus Cioffi & Harvey (1981). 36 Ergebnisse Allerdings befinden sich auch in seinem anterioren und medianen Abschnitt Goblet-Zellen, deren Apikalmembranen mit sogenannten Portasomen (V-ATPase) besetzt sind (Cioffi & Harvey, 1981) und in denen die V-ATPase auch durch immuncytochemische Untersuchungen nachgewiesen wurde (Russell et al., 1992). Weiterhin hatten Cioffi & Harvey (1981) gezeigt, dass es zwischen den drei Darmabschnitten keinen Unterschied im aktiven K+-Transport gibt. Um die Präparationsausbeute an V-ATPase zu erhöhen, wurde deshalb versucht, diese auch aus der Gobletzellapikalmembran des anterioren Mitteldarms zu reinigen. Bei der angereicherten GCAM (Bande 2, 3.1.1.) und bei der aus ihr solubilisierten und gereinigten VATPase (3.1.2.) aus dem anterioren Mitteldarm zeigte sich, dass sie in vergleichbarer Menge gewonnen werden konnte und dass die Reinheit und Aktivität der gereinigten V-ATPase mit der aus dem posterioren Abschnitt vergleichbar war (Tab. 2, Abb. 6). Tab. 2: Ausbeute und Aktivität der V-ATPase aus Präparationen des anterioren und posterioren Mitteldarms. Angegeben ist die Ausbeute in µg Protein pro eingesetzter Raupe und die spezifische Aktivität in U/mg aus einem typischen Versuch. Mitteldarmabschnitt Ausbeute an angereicherter GCAM (Bande 2) Ausbeute an gereinigter V-ATPase aus Bande 2 ATPase-Aktivität der gereinigten V-ATPase ATPase-Aktivität der gereinigten V-ATPase mit 0,5 mM NaN3 ATPase-Aktivität der gereinigten V-ATPase mit 0,5 mM NaN3 und 0,1 mM Na3VO4 96 kD 67 kD Posterior 60 µg/Raupe 10 µg/Raupe 1,8 U/mg 1,9 U/mg Anterior 120 µg/Raupe 14 µg/Raupe 1,9 U/mg 1,8 U/mg 1,7 U/mg 1,9 U/mg A B 43 kD C 30 kD D E 20 kD c G F 14 kD 1 2 3 Abb. 6: Gereinigte V-ATPase aus angereicherter GCAM. Coomassie gefärbtes SDS-Gel, Bahn 1, 5 µg Standardprotein, Bahn 2, 10 µg V-ATPase aus anteriorem Mitteldarm, Bahn 3, 10 µg V-ATPase aus posteriorem Mitteldarm. 37 Ergebnisse Ein viel wichtigerer Befund war allerdings, dass das verwendete Detergenz C12E10 die VATPase sehr selektiv solubilisiert, so dass keine Verunreinigungen mit anderen ATPasen vom F-Typ (Hemmung durch Azid) oder P-Typ (Hemmung durch Vanadat) vorhanden waren, welche sich in der gereinigten GCAM aus anteriorem Mitteldarm noch mit bis zu 50% Fremd-ATPase-Aktivität bemerkbar machten (Tab. 3). Tab. 3: Ausbeute und ATPase-Aktivität der reinen GCAM aus anteriorem und posteriorem Mitteldarm. Die Ausbeute ist in µg Protein pro verwendeter Raupe, die ATPase-Aktivität in U pro mg Protein und in Prozent der gesamten ATPase-Aktivität angegeben (n=3, S. D.). Ausbeute ATPase-Aktivität ATPase-Aktivität mit 0,5 mM NaN3 ATPase-Aktivität mit 0,5 mM NaN3 und 0,1 mM Na3VO4 Posteriorer Mitteldarm 6 µg/Raupe 0,65 0,24 U/mg 100% 0,52 0,29 U/mg 80% 0,48 0,11 U/mg 74% Anteriorer Mitteldarm 12 µg/Raupe 0,72 0,28 U/mg 100% 0,41 0,13 U/mg 56% 0,3 0,11 U/mg 50% Diese Selektivität war eine wichtige Voraussetzung für die Entwicklung eines neuen Reinigungsprotokolls für das V1Vo Holoenzym und den Vo Komplex aus dem Mitteldarmrohhomogenat (3.1.2., 3.1.3.), das zu stark erhöhten Ausbeuten führte. Bei diesem neuen Reinigungsprotokoll konnte zum einen auf die Anreicherung der GCAM, welche die zeitaufwendige Entfernung der Längsmuskulaturstreifen vom Darmepithel beinhaltet, verzichtet werden (3.1.1.). Zum anderem wurde nicht nur der posteriore Mitteldarmabschnitt, sondern der ganze Mitteldarm als Ausgangsmaterial verwendet. Der komplette Darm wurde homogenisiert und das daraus gewonnene Membranpellet mit C12E10 solubilisiert. Um Verunreinigungen durch unspezifische elektrostatische Wechselwirkungen zwischen der VATPase und anderen Proteinen zu minimieren, wurde dem Saccharosedichtegradienten, in welchem der konzentrierte Solubilisierungsüberstand aufgetrennt wurde, 200 mM KCl zugesetzt. Dies führte nicht nur zu einer höheren Reinheit, sondern auch zur Entdeckung der bis dato nicht darstellbaren Untereinheit a (siehe 4.1.2.). Um die V-ATPase in einer noch höheren Reinheit zu erhalten, wurden im Anschluß an die Dichtegradientenzentrifugation noch 2 Chromatographieschritte eingeführt. Zunächst wurde eine Anionenaustauscherchromatographie auf einer Mono Q-, Resource Q- (beide Pharmacia) oder Hyper D Q-Säule (Biosepra) durchgeführt, bei der das V1Vo Holoenzym in einem linearen Salzgradienten bei einer NaCl-Konzentration zwischen 230 mM und 280 mM als relativ homogener Peak eluierte (Abb. 7a). Bei einer anschließenden Gelpermeationschromatographie auf einer Superdex 200-Säule (Pharmacia) eluierte das V1Vo Holoenzym dann in einem einzigen Peak mit einer Schulter (Abb. 7b), auf die später noch näher eingegangen wird. Bei einer SDS38 Ergebnisse PAGE des Elutionspeaks der Gelfiltration zeigte sich nach Silberfärbung (Abb. 7c), dass alle Untereinheiten des V1Vo Holoenzyms vorhanden und keine nennenswerten Verunreinigungen enthalten waren. Die Ausbeuten der neu entwickelten Präparation beliefen sich auf im Durchschnitt 100 µg reines V1Vo Holoenzym/Raupe, was gemessen an der bisherigen Ausbeute von ca. 10 µg V1Vo Holoenzym/Raupe mit geringerer Reinheit (Schweikl et al., 1989) eine deutliche Steigerung darstellt. Außerdem ist noch zu erwähnen, dass bei der herkömmlichen Art der Reinigung durch den hohen Zeitaufwand für die längsmuskelfreie Präparation im Schnitt nur ca. 16 Raupen verwendet werden konnten. Da bei der Reinigung aus dem Rohhomogenat von ganzen Mitteldärmen bis zu 32 Raupen verwendet werden können, steigt somit die Gesamtausbeute noch zusätzlich. Abb. 7: Reinigung des V1Vo Holoenzyms. a) Elutionsprofile des V1Vo Holoenzyms der Anionenaustauscherchromatographie (Hyper D Q, Biosepra); b) Elutionsprofile des V 1Vo Holoenzyms Gelpermeationschromatographie (Superdex 200, Pharmacia); c) Silbergefärbtes SDS-Gel des Elutionspeaks der Gelpermeationschromatographie (1 µg Protein). 39 Ergebnisse 4.1.2. Entdeckung der Untereinheit a bei M. sexta Bei den Versuchen, das V1Vo Holoenzym durch direktes Solubilisieren des Membranpellets eines Mitteldarmrohhomogenats zu reinigen, wurde den Saccharoselösungen für die zonale Dichtegradientenzentrifugation 200 mM KCl zugesetzt, um Verunreinigungen mit Proteinen, die durch unspezifische elektrostatische Wechselwirkungen am V1Vo Holoenzym haften zu verhindern (siehe Abb. 6). Bei einer anschließenden Auftrennung der Probe auf einem SDSGel zeigte sich dabei erstmals eine deutliche Bande mit einer apparenten Molekularmasse von ca. 100 kDa (Abb. 8). Im weiteren wurde dann überprüft, ob es sich bei dieser Bande um die Untereinheit a handelt, die bei den meisten anderen V-ATPasen beschrieben wurde, bei M. sexta bis jetzt allerdings nachgewiesen werden konnte (Wieczorek et al., 2000). Abb. 8: Entdeckung der Untereinheit a bei M. sexta. Ein mit Coomassie gefärbtes SDS-Gel (Bahn 1-3) und ein immungefärbter Westernblot (Bahn 4) des aus der GCAM des posterioren Mitteldarms gereinigten Holoenzyms, mit und ohne KCl im Dichtegradienten. Bahn 1, 5 µg Standardproteine (Molekularmassen in kDa), Bahn 2, 5 µg V1Vo Holoenzym aus GCAM, Bahn 3 und 4, 5 µg V1Vo Holoenzym aus GCAM mit 200 mM KCl im Saccharosedichtegradienten, Bahn 4, gefärbt mit Serum gegen die 116 kDa Untereinheit a von chromaffinen Granula aus Rind (Gillespie et al., 1991). Mit Hilfe von Antikörpern gegen die 116 kDa Untereinheit a von chromaffinen Granula aus Rind (Gillespie et al., 1991) konnte die Vermutung, dass es sich bei dem 100 kDa Protein um die Untereinheit a handelt bestätigt werden (Abb. 8). Inzwischen wurde cDNA für die Untereinheit a aus dem Mitteldarm von M. sexta in unserer Arbeitsgruppe kloniert und sequenziert (Merzendorfer et al., 2000; GenBank Acc.-Nr. AJ249390). Deshalb konnte auch durch MALDI-MS-Analysen von tryptischen Peptiden der aus einem SDS-PAGE ausgeschnittenen 100 kDa Bande eine eindeutige Identifizierung dieser Bande als Untereinheit a mit einer Sequenzabdeckung von 36% erreicht werden (Abb. 9). 40 Ergebnisse a) m/z gemessene Peptide 705.411 732.384 763.380 891.470 938.380 954.382 1009.492 1017.501 1025.444 1044.621 1122.564 1126.592 1166.568 1173.552 1260.588 1271.643 1278.637 1288.596 1419.673 1490.661 1465.637 1481.659 1542.697 1558.685 1575.715 1646.750 1670.791 1686.863 1738.841 1821.852 1837.879 2048.957 2064.900 2119.987 2079.948 2121.960 2126.023 2264.173 2370.229 2550.350 2573.259 erwartet m/z 705.405 732.404 763.410 891.505 938.382 954.377 1009.420 1017.548 1025.458 1044.657 1122.605 1126.622 1166.596 1173.606 1260.659 1271.671 1278.706 1288.629 1419.658 1490.695 1465.663 1481.658 1542.690 1558.685 1575.759 1646.796 1670.785 1686.780 1738.872 1821.844 1837.839 2048.978 2064.973 2120.016 2080.027 2121.983 2126.071 2264.153 2370.256 2550.402 2573.267 erste AS 195 182 51 50 57 letzte AS 200 187 56 56 63 180 258 170 122 346 473 804 827 111 121 39 244 187 266 179 130 356 482 812 838 121 130 49 256 357 368 136 148 244 257 135 148 267 357 281 371 75 93 94 201 372 611 320 740 346 110 219 389 630 340 761 368 Sequenz (R)GNVFLR(Q) (R)IPAFER(M) (K)FVNEVR(R) (R)KFVNEVR(R) (R)RCDEMER(K) methionine oxidized cysteine acrylamide (R)ERIPAFER(M) (R)EMAMGVMTR(I) (K)LGFVAGVILR(E) (R)NYLELTELK(H) (R)SGSSVPPILNR(M) (K)SLNIFGSSWR(Q) (R)LHWVEFQSK(F) (R)FEVILDSAGPGR(G) (R)EVNQNAEALKR(N) (K)RNYLELTELK(H) (R)DLNPDVNAFQR(K) (R)ATLYPCPESPADR(R) C acrylamide (R)METLEDPPTYNR(N) M ox. (K)TQVFFDEMADPSR(E) M ox. (R)ATLYPCPESPADRR(E) C acrylamide (R)KTQVFFDEMADPSR(E) M ox. (R)IEDLNTVLGQTQDHR(H) (R)METLEDPPTYNRNNK(F) M ox. (R)DGIPMLEIPGECPEAPQPR(E) M ox. C acrylamide (R)EMIDLEATFEKLENELR(E) (R)QAEIDTPLEDPSSSDQVYK(S) (K)FTQAFQNLIYAYGVATYR(E) (K)TSAYCAPSILITFINMMLFK(T) (K)CLIAECWVPALDLETIQLALR(R) M ox. (R)LWALSLAHAQLAEVAWNMLLRK(G) (R)SGSSVPPILNRMETLEDPPTYNR(N) b) 1 61 121 181 241 301 361 421 481 541 601 661 721 781 841 MGSLFRSEEMTLCQLFLQSEAAYACVSELGELGLVQFRDLNPDVNAFQRKFVNEVRRCDE MERKLRYLEKEIRRDGIPMLEIPGECPEAPQPREMIDLEATFEKLENELREVNQNAEALK RNYLELTELKHILRKTQVFFDEMADPSREEEQVTLLGEEGLMAGGQALKLGFVAGVILRE RIPAFERMLWRACRGNVFLRQAEIDTPLEDPSSSDQVYKSVFIIFFQGDQLKTRVKKICE GFRATLYPCPESPADRREMAMGVMTRIEDLNTVLGQTQDHRHRVLVAAAKNIKNWFVKVR KIKAIYHTLNLFNLDVTQKCLIAECWVPALDLETIQLALRRGTERSGSSVPPILNRMETL EDPPTYNRNNKFTQAFQNLIYAYGVATYREVNPAPYTIITFPFLFAVMFGDLGHGALMAA FGFWMCYKEKPLQAKKIDSEIWNIFFGGRYIILLMGLFSMYTGLIYNDIFSKSLNIFGSS WRQNYNASTLTENKLLQLNPDSPDYLQYPYPFGIDPVWQLAEANKIIFMNAYKMKISIII GVFHMLFGVCLSLWNHLYFKRRISVYVEFIPQILFLTLLFFYMVLLMFIKWTSYGPTPGA FGSQDPAIVKTSAYCAPSILITFINMMLFKTDANTRPQCDDTMYAGQLQLQKFFVIVALL CVPVMLFGKPYFIMKEQKQRARQGHQPVEGAAENGTAGGAPVPSSGHHDDDITEVFIHQG IHTIEYVLGSVSHTASYLRLWALSLAHAQLAEVAWNMLLRKGLMSTDFQGGIFLYIVFAG WAAISVSILVLMEGLSAFLHTLRLHWVEFQSKFYAGEGLSLPTGSRFEVILDSAGPGRGV N 60 120 180 240 300 360 420 480 540 600 660 720 780 840 Abb. 9: MALDI-MS Karte der Untereinheit a. Die durch MALDI-MS identifizierten tryptischen Peptide der 100 kDa Bande (a) und ihre Sequenzabdeckung (b) mit der Untereinheit a der V-ATPase aus M. sexta (GenBank Acc.-Nr. AJ249390). Die gefundenen Fragmente der MALDI-MS sind in der Aminosäuresequenz unterstrichen und fett gedruckt. 41 Ergebnisse 4.1.3. Die Untereinheit B und die Identifizierung ihrer putativen Isoform B´ als Hsp60 Da die bisher zu Immunfärbungen verwendeten Antiseren gegen das V1Vo Holoenzym so gut wie keine Antikörper gegen die Untereinheit B enthielten und gegen diese auch keine monoklonalen Antikörper zur Verfügung standen, sollten polyklonale Antikörper gegen eine rekombinant hergestellte Untereinheit B produziert werden. Um die Möglichkeiten der Anwendung zu erhöhen, wurde nicht die ganze Untereinheit, sondern zwei Teilsequenzen ausgewählt: zum einen der Bereich von Aminosäure 243-428, welcher bei allen V-ATPasen hoch konserviert ist, so dass die Antikörper auch gegen V-ATPasen aus anderen Organismen zu verwenden sind, zum anderen der Bereich von Aminosäure 475-494 aus dem C-Terminus der für M. sexta spezifisch ist und den die Antikörper deshalb nur in der Insekten V-ATPase erkennen sollten (Novak et al., 1992). Beide Teilsequenzen der B Untereinheit wurden mit spezifischen Primern in einer PCR vervielfältigt, die Produkte gereinigt, in den Expressionsvektor pMalc2 (New England Biolabs) kloniert und dieser in E. coli Zellen (XL1 blue) transformiert. Die Expression des rekombinanten Fusionsproteins wurde induziert und das Protein per Affinitätschromatographie auf einer Amylosesäule soweit als möglich gereinigt. Bei einer Kontrolle in der SDS-PAGE waren dann auch Banden mit der für die Fusionsproteine erwarteten Größe zu sehen (Abb. 10). Abb. 10: Fusionsprotein aus Teilsequenzen der B Untereinheit und MBP. Coomassie-gefärbte SDS-PAGE der gereinigten Fusionsproteine. Bahn 1, Standardproteine (5 µg), Bahn 2, MBP (1µg), Bahn 3, MBP-C34 (1µg) und Bahn 4, MBP-C23 (1µg). Die Fusionsproteine wurden direkt zur Immunisierung von zwei Meerschweinchen verwendet und die erhaltenen Antiseren auf gereinigtem V1Vo Holoenzym getestet. Dabei kam es zu einem überraschenden Ergebnis. Das Antiserum C23 detektierte die Untereinheit B und ein weiteres Protein mit ca. 60 kDa im folgenden B´ genannt (Abb. 11b). Das Antiserum C34 erkannte die Untereinheit B überhaupt nicht, dafür aber ebenfalls das 60 kDa Protein B´. Das 42 Ergebnisse Protein B´ wurde bisher häufig als schwache Bande bei der Reinigung der V-ATPase beobachtet und ist auch in der neuen Präparationsmethode als Bande in der bereits erwähnten mal mehr mal weniger ausgeprägten Schulter des Elutionspeaks der Gelfiltration zu sehen (Abb. 11a). Abb. 11: Entdeckung des Proteins B´. a) Elutionsprofil des V1Vo Holoenzyms von einer Superdex 200 Gelfiltrationssäule (Pharmacia). b) SDS-PAGE der Peaks I und II der Gelfiltration. Bahn 1-3 Coomassie gefärbt, Bahn 4-7, Westernblot. Bahn 1, Proteinstandard, Bahn 2, 4 und 6, Peak I; Bahn 3, 5 und 7, Peak II. Bahn 4 und 5, Immunfärbung mit Antiserum C23 und Bahn 6 und 7, Immunfärbung mit Antiserum C34. Abb. 12: Vorkommen von B` in unterschiedlichen Präparationen. Silbergefärbtes SDS-PAGE-Gel. Bahn 1, Standardproteine, Bahn 2, V1Vo Holoenzym aus angereicherter GCAM aus anteriorem Mitteldarm,. Bahn 3, V1Vo Holoenzym aus angereicherter GCAM aus posteriorem Mitteldarm. Bahn 4, V1Vo Holoenzym aus gereinigter GCAM aus anteriorem Mitteldarm. Bahn 5, V1Vo Holoenzym aus gereinigter GCAM aus posteriorem Mitteldarm. Bahn 6, V1 Komplex. 43 Ergebnisse Es stellte sich nun die Frage, ob es sich bei diesem Protein um eine Isoform der Untereinheit B handelt, und wenn ja, wo diese Isoform vorkommt. Zunächst wurde untersucht, in welchen Präparationen die B´-Bande auftaucht. Das V1Vo Holoenzym wurde aus der angereicherten GCAM (3.1.1.) und der gereinigten GCAM des anterioren und posterioren Mitteldarms solubilisiert und isoliert und mit dem V1 Komplex verglichen (Abb. 12). Es zeigte sich, dass beim V1Vo Holoenzym aus der angereicherten GCAM beider Darmabschnitte deutlich die Bande B´ zu sehen ist. Hingegen ist B´ weder im V1Vo Holoenzym aus gereinigter GCAM noch im V1 Komplex enthalten. MALDI-MS-Analysen der beiden Proteinbanden ermöglichten eine eindeutige Identifizierung der Bande B als Untereinheit B entsprechend der von der cDNA abgeleiteten Proteinsequenz (Abb. 15). Die Bande B´ konnte auf diese Weise nicht identifiziert werden, da keine der gemessenen Fragmentmassen ein Pendant in der von der cDNA abgeleiteten Proteinsequenz hatte (nicht gezeigt). Eine eindeutige Identifizierung von B` erfolgte erst durch eine N-terminale Sequenzierung der entsprechenden Proteinbande durch Dr. Barbara Schedding (Universität Münster). Die ersten 30 Aminosäuren konnten eindeutig gelesen werden und ergaben bei der Suche in der Datenbank eine hohe Übereinstimmung mit dem Hitzeschockprotein 60 (Hsp63) von Heliothis vireszens, dem Tobacco Budworm (Abb. 13). Außerdem stimmten die 30 Aminosäuren mit der Sequenz eines, bei einem sogenannten "Shotgun-Screening" für V-ATPase Untereinheiten von Ralph Gräf in unserem Labor gefundenen Klons (Klon10) vollständig überein. Sowohl das Hsp63 aus H. vireszens als auch der Klon 10 beinhalteten eine vorgeschaltete Signalsequenz für den mitochondrialen Import. Für Klon 10 wurde die mitochondriale Lokalisation mit einer Wahrscheinlichkeit größer 90% vorhergesagt (PSORT II server). Hsp60 M. sexta Klon 10 M. sexta Hsp60 H. vireszens 1 10 20 30 40 50 60 +--------+---------+---------+---------+---------+---------+AKDVRFGADVRALMLQGVDILADAVAVTMG SIRTKMLRLPRVVRQSISLHKSQQLT--RFYAKDVRFGADVRALMLQGVDILADAVAVTMG MFKMYRSPHITRNSFKYLKATNINSCRFYAKEVRFGPDVRSLMLQGVDILADADDVTMG 70 80 90 100 110 --------+---------+---------+---------+---------+Hsp60 M. sexta Klon 10 M. sexta Hsp60 H. vireszens PKGRNVILEQSWGSPKITKDGVTVAKGVELKDKFQNIGAKLVQNVANNT PKGVNVILAKNLGPPKITKDGVTVAKGIDLKDKFQNIGARLVQNVANKTN Abb. 13: Sequenzvergleich Hsp60. Vergleich zwischen der durch N-terminale Sequenzierung des Protein B´ erhaltenen Sequenz (Hsp60 M. sexta), des durch ein shotgun screening erhaltenen Klons aus M. sexta (Klon 10 M. sexta) und des Hsp60 aus H. vireszens. Bei allen drei Sequenzen identische Aminosäuren (rot), bei zwei Sequenzen identische Aminosäuren (blau), andere (schwarz). 44 Ergebnisse 4.1.4. Identifizierung der Untereinheit D Zu Beginn der Arbeit lag keinerlei Sequenzinformation zur bis dahin als D Untereinheit bezeichneten SDS-Bande des V1Vo Holoenzyms und des V1 Komplexes vor. Die Bande wurde nur auf Grund ihrer apparenten Molekularmasse von 32 kDa und wegen des Vorkommens einer D Untereinheit bei anderen V-ATPasen als solche benannt. Um die Identität dieser Bande aufzuklären, wurde sie aus einem SDS-PAGE Gel ausgeschnitten. Bei der N-terminalen Sequenzierung eines Cyanbromid-Fragments durch Dr. Roland Schmid (Abt. Mikrobiologie, Universität Osnabrück) konnte die Sequenz GRLAGAQKGHGLL detektiert werden. Bei einer Datenbanksuche ergaben sich eindeutige Übereinstimmungen mit D Untereinheiten anderer V-ATPasen (Beispiele in Abb. 14). Mittlerweile wurde diese Sequenzinformation zur Synthese von degenerierten Primern benutzt, die zu einer erfolgreichen Klonierung und Sequenzierung der cDNA der Untereinheit D von M. sexta führte (Merzendorfer et al., 2000). B. taurus C. albicans S. cerevisiae M. sexta 1 10 20 30 40 +--------+---------+---------+---------+-MSGKDRIEIFPSRMAQTIMKARLKGAQTGRNLLKKKSDALT MSGAGNREQVFPTRMTLGVMKSKLKGAQQGHSLLKRKSEALT MSGNREQVFPTRMTLGLMKTKLKGANQGYSLLKRKSEALT GRLAGAQKGHGLL Abb. 14: Teilsequenz der D Untereinheit. Vergleich der Teilsequenz der D Untereinheit von M. sexta mit den entsprechenden Teilen der D Untereinheiten aus B. taurus, C. albicans und S. cerevisiae. Identische Aminosäuren (rot), andere (schwarz). 4.1.5. Analyse der Untereinheiten der V-ATPase durch MALDI-MS Für weitergehende funktionelle und strukturelle Untersuchungen der V-ATPase war es nicht nur notwendig, das Enzym in ausreichender Quantität und Qualität zu reinigen, sondern auch alle seine Untereinheiten zu identifizieren. Zwar sind inzwischen alle Untereinheiten der VATPase von M. sexta auf der Basis von cDNA-Sequenzen nachgewiesen (Dow et al., 1992; Gräf et al., 1992; Novak et al., 1992; Gräf et al., 1994a; Gräf et al., 1994b; Lepier et al., 1996; Merzendorfer et al., 1997b; Merzendorfer et al., 1999; Merzendorfer et al., 2000), doch stand die eindeutige Identifizierung der Untereinheiten auf Proteinebene noch aus, zumal für eine Reihe von Untereinheiten Isogene bzw. Isoformen existieren. Deshalb wurden alle Banden eines SDS-PAGE Geles des V1Vo Holoenzyms bzw. des V1 Komplexes ausgeschnitten und im Gel tryptisch verdaut (3.1.16.). Die extrahierten Peptide wurden mittels 45 Ergebnisse MALDI-MS analysiert und den entsprechenden Abschnitten der sich aus ihrer cDNA ergebenden Aminosäuresequenz zugeordnet (Abb. 15). Trypsin erwies sich für die Untereinheit c und e als ungeeignet, da die meisten der entstehenden Fragmente nicht im Meßbereich lagen. Bei der Untereinheit c, welche im Zusammenhang mit der Untersuchung zur Makrolidbindestelle wichtig war (4.3.3.), wurde deshalb Chymotrypsin eingesetzt. Mit Ausnahme der Untereinheit e konnten alle Untereinheiten durch eine deutliche Übereinstimmung zwischen den analysierten Fragmenten und den cDNA-Sequenzen identifiziert werden (Sequenzabdeckungen: A = 85%, B = 62 %, H = 58 %, C = 71%, D = 56%, E = 55%, G = 79%, F = 66%, a = 36%, d = 47% und c = 76%). Bei der Untereinheit c konnten zwei auffällige Massenfragmente von 1523,9 und 1739,9 m/z nachgewiesen werden, die nicht durch die cDNA-Sequenz zu erklären sind (siehe auch 5.1.2.). Möglicherweise handelt es sich hierbei um das Fragment einer Isoform der Untereinheit. Weiterhin ergaben sich aus der MALDI-MS keine Hinweise auf Isoformen für andere Untereinheiten. Diese können allerdings nicht vollständig ausgeschlossen werden, da sich Sequenzänderungen auch in nicht repräsentierten oder nachweisbaren Fragmenten befinden können. Abb. 15: MALDI-MS-Analyse aller Untereinheiten des V1Vo Holoenzyms. Für jede Untereinheit wurden die in der MALDI-MS gefundenen Massen ihren potentiellen Peptidfragmenten zugeordnet und in der Aminosäuresequenz fettgedruckt und unterstrichen hervorgehoben. Untereinheit A: 1 51 101 151 201 251 301 351 401 451 501 551 601 MASKGGLKTIANEENEERFGYVFAVSGPVVTAEKMSGSAMYELVRVGYNE LVGEIIRLEGDMATIQVYEETSGVTVGDPVLRTGKPLSVELGPGILGSIF DGIQRPLKDINELTQSIYIPKGVNVPSLAREVDWEFNPLNVKVGSHITGG DLYGIVHENTLVKHKMLMPPRAKGTVTYIAPAGNYKVTDVVLETEFDGEK AQYTMLQVWPVRQPRPVTEKLPANHPLLTGQRVLDSLFPCVQGGTTAIPG AFGCGKTVISQALSKYSNSDVIIYVGCGERGNEMSEVLRDFPELTVEIEG VTESIMKRTALVANTSNMPVAAREASIYTGITLSEYFRDMGYNVSMMADS TSRWAEALREISGRLAEMPADSGYPAYLGARLASFYERAGRVKCLGNPDR EGSVSIVGAVSPPGGDFSDPVTAATLGIVQVFWGLDKKLAQRKHFPSINW LISYSKYMRALDDFYEKNYPEFVPLRTKVKEILQEEEDLSEIVQLVGKAS LAETDKITLEVAKLLKDDFLQQNSYSSYDRFCPFYKTVGMLKNIISFYDM SRHAVESTAQSDNKVTWNVIRDAMGNVLYQLSSMKFKDPVKDGEAKIKAD FDQLLEDMSAAFRNLED 50 10 150 200 250 300 350 400 450 500 550 600 650 Untereinheit B: 1 51 101 151 201 251 301 351 401 451 46 MAKTLSAAQANKEHVLAVSRDFISQPRLTYKTVSGVNGPLVILDEVKFPK FSEIVQLKLADGTHRSGQVLEVSGSKAVVQVFEGTSGIDAKNTLCEFTGD ILRTPVSEDMLGRVFNGSGKPIDKGPPILAEDFLDIQGQPINPWSRIYPE EMIQTGISAIDVMNSIARGQKIPIFSAAGLPHNEIAAQICRQAGLVKIPG KSVLDDHEDNFAIVFAAMGVNMETARFFKQDFEENGSMENVCLFLNLAND PTIERIITPRLALTAAEFLAYQCEKHVLVILTDMSSYAEALREVSAAREE VPGRRGFPGYMYTDLATIYERAGRVEGRNGSITQIPILTMPNDDITHPIP DLTGYITEGQIYVDRQLHNRQIYPPVNVLPSLSRLMKSAIGEGMTRKDHS DVSNQLYACYAIGKDVQAMKAVVGEEALTPDDLLYLEFLTKFEKNFITQG NYENRTVFESLDIGWQLLRIFPKEMLKRIPASILAEFYPRDSRH 50 10 150 200 250 300 350 400 450 500 Ergebnisse Untereinheit H: 1 51 101 151 201 251 301 351 401 451 MANIGDEKVSQLIPTLGDDKIDMIAATSVLQIRASEIRQTQINWQSYLQG QMITQRDHDFIVNLDQRGQKDLPDKNPDACADVFLNLLTHISKDHTIQYI LVLIDDILSEDKSRVKIFRETKYSGNIWQPFLNLLNRQDEFVQHMTARII AKLACWHPQLMDKSDLHFYLSWLKDQLKMNNNDYIQSVARCLQMMLRVDE YRFAFLSVDGISTLLSILASRVNFQVQYQLVFCLWVLTFNPLLAEKMNKF NAIPILADILSDSVKEKVTRIVLAVFRNLIEKPQDQQVAKEHCIAMVQCK VLKQLSILEQKRSDDEDIMNDVEFLNERLQASVQDLSSFDQYATEVKSGR LEWSPVHKSAKFWRENAARLNERGQELLRTLVHLLEKSHDPVVLAVACYD VGEYVRHYPRGKHIIEQLGGKQRVMYLLSHDDPNVRYEALLAVQKLMVHN WEYLGKQLEKEQIDKQAGTVVGAKA 50 10 150 200 250 300 350 400 450 500 Untereinheit C: 1 51 101 151 201 251 301 351 MSEYWLISAPGDKTCQQTWEALNQATKANNLSLNYKFPIPDLKVGTLDQL VGLSDDLGKLDTFVEGVTRKVAQYLGEVLEDQRDKLHENLTANNDDLPHY LTRFQWDMAKYPIKQSLRNIADIISKQVGQIDADLKVKSSAYNALKGNLQ NLEKKQTGSLLTRNLADLVKKEHFILDSEYLTTLLVIVPKSMFNDWNANY EKITDMIVPRSTQLIHQDGDYGLFTVTLFKKVVDEFKLHARERKFVVREF AYNEADLVAGKNEITKLLTDKKKQFGPLVRWLKVNFSECFCAWIHVKALR VFVESVLRYGLPVNFQAALLVPSRRSARRLRDTLHALYAHLDHSAHHHAN AQQDSVELAGLGFGQSEYYPYVFYKINIDMIEKAA 50 10 150 200 250 300 350 Untereinheit D: 1 51 101 151 201 MSGKDRLAIFPSRGAQMLMKGRLAGAQKGHGLLKKKADALQVRFRLILSK IIETKTLMGEVMKEAAFSLAEAKFTTGDFNQVVLQNVTKAQIKIRSKKDN VAGVTLPIFESYQDGSDTYELAGLARGGQQLAKLKKNFQSAVKLLVELAS LQTSFVTLDEVIKITNRRVNAIEHVIIPRLERTLAYIISELDELEREEFY RLKKIQDKKKIIKDKAEAKKAALRAAGQDLRDSANLLDEGDEDLLF 50 10 150 200 250 Untereinheit E: 1 51 101 151 201 MALSDADVQKQIKHMMAFIEQEANEKAEEIDAKAEEEFNIEKGRLVQQQR LKIMEYYEKKEKQVELQKKIQSSNMLNQARLKVLKVREDHVRNVLDEARK RLAEVPKDIKLYSDLLVTLIVQALFQLVEPTVTLRVRQADKALVESLLGR AQQDYKAKIKKDVVLKIDNENFLPPDTCGGIELIAAKGRIKISNTLESRL ELIAQQLLPEIRNALFGRNPNRKFTD 50 10 150 200 Untereinheit G: 1 51 101 MASQTHGIQQLLAAEKRAAEKVSEARKRKAKRLKQAKEEAQDEVEKYRQE RERQFKEFEAKHMGTREGVAAKIDAETRIKIDEMNKMVQTQKEAVIKDVL NLVYDIKPELHINYRVV 50 10 Untereinheit F: 1 51 101 MALHAAVKGKLISVIGDEDTCVGFLLGGIGEINKNRHPNFMVVDKNTPVS EIEECFKRFVKRDDIDIILINQNVAELVRHVIDAHTAPVPSVLEIPSKDH PYDASKDSILRRAKGMFNPEDLVR 50 10 Untereinheit a: (siehe Abb. 9) 47 Ergebnisse Untereinheit d: 1 51 101 151 201 251 301 MKGCIFNIDAGYLEGLCRGFKCGILKQSDYLNLVQCETLEDLKLHLQGTD YGTFLANEPSPLSVSTIDDKLREKLVIEFQHLRNHSVEPLSTFLDFITYS YMIDNIILLITGTLHQRPISELIPKCHPLGSFEQMEAIHVAATPAELYNA VLVDTPLAPFFVDCISEQDLDEMNIEIIRNTLYKAYLEAFYDFCKQIGGT TADVMCEILAFEADRRAIIITINSFGTELSKDDRAKLYPRCGKLNPDGLA ALARADDYEQVKAVAEYYAEYSALFEGAGNNVGDKTLEDKFFEHEVNLNV HAFLQQFHFGVFYSYLKLKEQECRNIVWISECVAQKHRAKIDNYIPIF 50 100 150 200 250 300 350 Untereinheit c; Trypsin und Chymotrypsin: 1 51 101 151 MAENPIYGPFFGVMGAASAIIFSALGAAYGTAKSGTGIAAMSVMRPELIM KSIIPVVMAGIIAIYGLVVAVLIAGSLDSPSNNYTLYRGFIHLGAGLAVG FSGLAAGFAIGIVGDAGVRGTAQQPRLFVGMILILIFAEVLGLYGLIVAI YLYTKQ 50 100 150 Untereinheit e: 1 51 48 MGASFVPITVFTILWGVVGIVCPIFAPKGPNRGIIQVVLMLTAATCWLFW LCAYMAQMNPLIGPRLNNETLIWISRTWGNPISNHTGA 50 Ergebnisse 4.2. Der V1 Komplex 4.2.1. Modifikation der Präparation des V1 Komplexes Um die Struktur und die biochemischen Eigenschaften des V1 Komplexes intensiver untersuchen zu können, war es erforderlich, die Stabilität und Aktivität des Komplexes über längere Zeit zu erhalten. Bei Gräf et al. (1996) wurden alle Experimente direkt im Anschluß an die Präparation durchgeführt, da schon bei einer Lagerung über Nacht die Aktivität des V1 Komplexes sich im Vergleich zur frischen Präparation stark verringerte. Bei Vortests einiger Lagerungsbedingungen zeigte sich, dass sowohl die Zugabe von -Mercaptoethanol als auch ein Aufbewahren bei 4°C im Kühlschrank sich positiv auf die Aktivität des V1 Komplexes auswirken (Tab. 4). Tab. 4: Lagerungsbedingungen für den V1 Komplex und deren Auswirkung auf die Aktivität. Bei der gemessenen Aktivität handelt es sich um die Mg-ATPase-Aktivität in Gegenwart von 25% Methanol. Lagerungsbedingungen Kontrolle: 150 mM NaCl, 20 mM Tris/HCl pH 8,1 Relative Aktivität 100% (2,4 U) Nach 96 h in 150 mM NaCl, 20 mM Tris/HCl pH 8,1, 4°C 43% Nach 96 h in 150 mM NaCl, 20 mM Tris/HCl, 25% Methanol pH 8,1, 4°C 42% Nach 96 h in 150 mM NaCl, 20 mM Tris/HCl, 3 mM -Mercaptoethanol pH 8,1, 4°C 62,5% Einfrieren und/oder Lagern im flüssigem Stickstoff bewirkten einen kompletten Verlust der Aktivität. Wurde der Komplex bei Raumtemperatur gelagert, zeigte sich bei einer anschließenden Anionenaustauscherchromatographie, warum die Aktivität des V1 Komplexes im Vergleich zur Lagerung bei 4°C schlechter wurde. Der V1 Komplex (II) mit hoher Aktivität (Gräf et al., 1996), welcher im zweiten Peak eluiert, wandelt sich im Laufe der Zeit komplett in die inaktivere Form des ersten Elutionspeaks (I) um (siehe Abb. 16). 49 Ergebnisse Abb. 16: Denaturierung des V1 Komplexes bei Raumtemperatur. Zu sehen sind Elutionsprofile von einer Anionenaustauscherchromatographie (Mono Q, Pharmacia). Der 2. Peak (II) einer V 1 Komplex Präparation wurde in 20 mM Tris-HCl pH 8,1, 150 mM NaCl bei RT gelagert und am folgenden Tag erneut auf der Anionenaustauschersäule aufgetrennt a). Dies wurde mit jeweils dem 2. Elutionspeak (II) wiederholt b) und c). Da die einzelnen Untereinheiten des V1 Komplexes viele Cysteine enthalten und diese sowohl membrangebunden im V1Vo Holoenzym als auch im freien V1 Komplex vom reduzierenden Milieu des Cytosols umgeben sind, lag es nahe, schon bei der Präparation des V1 Komplexes ein Reduktionsmittel zur Stabilisierung zu verwenden. Da -Mercaptoethanol bereits seinen positiven Effekt bei der Lagerung des V1 Komplexes gezeigt hatte, wurde es mit einer Endkonzentration von 9,6 mM den Laufpuffern für die Chromatographie zugegeben (Merzendorfer et al., 1997a). Dadurch verschwand der Anteil des V1 Komplexes mit geringerer Aktivität bei der Anionenaustauscherchromatographie, also der 1. Elutionspeak fast vollständig und der aktivere 2. Peak nahm entsprechend zu (Abb. 17). Deshalb wurde auch in der anschließenden Gelpermeationschromatographie -Mercaptoethanol als Reduktionsmittel verwendet. 50 Ergebnisse Abb. 17: Der Einfluß von -Mercaptoethanol auf die Reinigung des V1 Komplexes. Vergleich zweier V1 Komplex Präparationen ohne a) und mit b) 9,6 mM -Mercaptoethanol im Laufpuffer der Anionenaustauscherchromatographie (Mono Q, Pharmacia). Somit waren durch die Zugabe von 9,6 mM -Mercaptoethanol in alle Chromatographiepuffer und die anschließende Lagerung bei 4°C im Kühlschrank die Vorraussetzungen geschaffen, größere Mengen des V1 Komplexes zu sammeln und zeitaufwendigere Experimente, für die ein stabiles Enzym notwendig ist, durchzuführen. Bei Proben, die unter diesen Bedingungen gereinigt und gelagert wurden, konnte nach 4 Wochen noch eine Aktivität von bis zu 95% im Vergleich zur frischen Probe gemessen werden (nicht gezeigt). 51 Ergebnisse 4.2.2. Die Untereinheitenzusammensetztung des V1 Komplexes in der 2-D Gelelektrophorese Da es unklar war, ob zwei verschiedene Proteine wegen gleicher Eigenschaften in der SDSPAGE als eine einzige Proteinbande zu sehen sind, wurde der V1 Komplex mit der Methode der 2-dimensionalen Gelelektrophorese analysiert. Dabei wurde in der ersten Dimension nach dem Isoelektrischen Punkt getrennt, in der zweiten Dimension in einem SDS-PAGE-Gel nach der apparenten Molekularmasse (Abb. 18). Abb. 18: 2-D Gelelektrophorese des V1 Komplexes. Gezeigt ist die 2. Dimension, des Coomassie-gefärbten SDS-PAGE-Gels. Auf dem Gel wurde als Referenz links 5 µg V1 Komplex und rechts 5 µg Proteinstandard aufgegeben. In der ersten Dimension wurden 20 µg V1 Komplex auf einem IEF-Dry Strip (Pharmacia, pH 3-10) aufgetrennt und anschließend auf das SDS-Gel aufgelegt. Die in der 2. Dimension wiedergefundenen Untereinheiten sind im Gel markiert. Ein unbekannter Spot wurde mit einem Pfeil markiert. Bei dieser Auftrennung konnten alle Untereinheiten des V1 Komplexes wiedergefunden werden und soweit spezifische Antikörper (A, B, E G Untereinheit) vorhanden waren, durch Immunfärbung eines Westernblots der 2. Dimension identifiziert werden (nicht gezeigt). Die Position der Untereinheiten im Gel entsprach zum einen ihrer apparenten Molekularmasse, zum anderen aber auch dem von ihrer abgeleiteten Aminosäuresequenz berechneten Isoelektrischen Punkt (Tab. 5). 52 Ergebnisse Tab. 5: Isoelektrische Punkte der Untereinheiten des V1 Komplexes. Der Isoelektrische Punkt wurde aus den abgeleiteten Aminosäuresequenzen ermittelt. Untereinheit A B C D E F G H Isoelektrischer Punkt 4,99 5,2 8,2 9,6 9,4 5,9 9,4 6,3 Einzig eine kleine Bande (Abb. 18, Pfeil), die etwas links und leicht höher als die Untereinheit F lief, könnte auf eine mögliche Isoform der Untereinheit G hindeuten, von der es mehr als ein Gen gibt (Lepier et al., 1996; Merzendorfer et al., 2000). Ein Westernblot mit anschließender Immunfärbung mit dem monoklonalen Antikörper 47-5, welcher gegen ein gemeinsames Epitop der E und G Untereinheit gerichtet ist, erkennt diese Bande allerdings nicht. Inzwischen wird die Problematik der Isoformen von Untereinheiten von Stephan Reineke in unserem Labor genauer untersucht, so dass mit neuen Erkenntnissen zu rechnen ist. Außer diesem einen Punkt gab es keine weiteren auffälligen neuen Banden, die auf eine Isoform oder eine bis dato unbekannte neue Untereinheit hindeuten würden. 4.2. 3. Untersuchung der Untereinheit C des V1 Komplexes Obwohl die Untereinheit C dem katalytischen V1 Komplex des Holoenzyms zu zuordnen ist, kommt sie im gereinigten V1 Komplex im Vergleich zum V1Vo Holoenzym allenfalls in substöchiometrischen Mengen vor (Gräf et al., 1996; Svergun et al., 1998). Bei den Untersuchungen zur Stimulierung der Mg-ATPase-Aktivität des V1 Komplexes durch Alkohole (4.4.1.) zeigte sich, dass die Untereinheit C bei einer Gelpermeationschromatographie in Gegenwart von 25% Methanol, 15% Ethanol, 10% Isopropanol oder 5% 1Propanol vom eigentlichen V1 Komplex abgetrennt werden kann (Abb. 19, nur für Methanol gezeigt). Wird der V1 Komplex vor dem Auftragen auf die Gelfiltrationssäule mit dem Alkohol gemischt, findet man die Untereinheit C in einem V1-Aggregat wieder, welches deutlich größer als der ursprüngliche V1 Komplex ist und nahe dem Ausschlußvolumen eluiert. Derselbe Effekt einer Aggregatbildung mit höherer Molekularmasse war auch bei einer Saccharosedichtegradientenzentrifugation des V1 Komplexes in Gegenwart von 53 Ergebnisse Methanol beobachten, wenn die Probe vorher auf 25% Methanol gebracht wurde. Das Aggregat war dann bei einer wesentlich höheren Dichte wieder zu finden (Abb. 20). Abb. 19: Einfluß von Methanol auf den V1 Komplex (Gelchromatographie). Auftrennung des V1 Komplexes durch Gelchromatographie mit Methanol im Laufpuffer. 2,5 mg (25 µg/µl) V 1 Komplex wurde mit Methanol versetzt [25%] und in einer Superdex 200 Säule (Pharmacia) 150 mM NaCl, 20 mM Tris-HCl pH 8,1, 9,6 mM Mercaptoethanol, 25% Methanol) aufgetrennt (Fraktion I und II). Coomassie-gefärbtes SDS-PAGE Gel von unbehandeltem V1 Komplex (1); Fraktion I (2) und Fraktion II (3). Abb. 20: Einfluß von Methanol auf den V1 Komplex (Dichtegradientenzentrifugation). Auftrennung des V1 Komplexes in einem methanolhaltigen linearen Saccharosedichtegradienten. 2,5 mg (25 µg/µl) V 1 Komplex wurde mit Methanol versetzt [25%] und auf einem linearem Saccharosedichtegradienten (10%-50% Saccharose in 150 mM NaCl, 20 mM Tris-HCl pH 8,1, 9,6 mM -Mercaptoethanol, 25% Methanol) für 90 min bei 310.000 x g und 4°C (VTi 65.1, Beckman) zentrifugiert. Der Proteingehalt der 500 µl Fraktionen (1, hohe Dichte; 20, niedrige Dichte) ist in µg angegeben. Inset, Silbergefärbte SDS-PAGE-Gele gleich großer Aliquots der gepoolten Fraktionen 4-7 (a) und der gepoolten Fraktionen 12-15 (b). 54 Ergebnisse Beim Auftragen des V1 Komplexes ohne vorherige Zugabe von Alkohol auf eine mit Alkohol äquilibrierte Superdexsäule wurde die Untereinheit C ebenfalls vom V1 Komplex abgetrennt, allerdings entstand dabei kein Aggregat (Abb. 21). Die Untereinheit C konnte in diesem Fall wegen der starken Verdünnung in der Säule nicht als Peak detektiert werden. Offensichtlich ist die Bildung von Aggregaten bei hohen verwendeten Proteinkonzentrationen (25 µg/µl) eine Folge der Zugabe von Alkohol. Wird die Probe hingegen langsam auf den entsprechenden Alkoholanteil bei gleichzeitiger Erniedrigung der Proteinkonzentration gebracht, wie dies im letzten Experiment der Fall war, kommt es zu keiner Aggregatbildung, die Untereinheit C wird allerdings auch hier abgelöst. Die Aggregation des V1 Komplexes bei hohen Proteinkonzentrationen in Gegenwart von Alkohol wurde auch durch die Streukurven aus der Röntgenkleinwinkelstreuung bestätigt (Abb. 22). Die Streukurve des V1 Komplexes in methanolhaltigem Puffer (Abb. 22b) zeigte im Gegensatz zur Kontrolle ohne Alkohol (Abb. 22a) im oberen Bereich einen geraden Verlauf, was auf eine Aggregation des Proteins schließen lässt. Abb. 21: Abtrennung der Untereinheit C vom V1 Komplex (Gelchromatographie). Die Auftrennung des V1 Komplexes (2,5 mg, 25 µg/µl) durch Gelchromatographie erfolgte in einer Superdex 200 Säule (Pharmacia; Laufpuffer: 150 mM NaCl, 20 mM Tris-HCl pH 8,1, 9,6 mM -Mercaptoethanol, 25% Methanol). a) Elutionsprofil. b) SDS-PAGE-Gel gefärbt mit Coomassie Blau (je 10 µg Protein). Bahn 1, Kontrolle, Bahn 2, Peak aus a). 55 Ergebnisse Abb. 22: Streukurve des V1 Komplexes im Röntgenkleinwinkelstreuungsexperiment. a) ohne und b) mit 25% Methanol im Puffer (Protein,10 mg/ml). Um das Phänomen der Ablösung der C Untereinheit vom V1 Komplex genauer untersuchen zu können, wurde diese, nachdem die sie codierende cDNA von Xiao-Fan Zhao in unserem Labor erfolgreich kloniert und sequenziert worden war (Merzendorfer et al., 2000) mit Hilfe des PET-Systems in E. coli überexprimiert (siehe 3.2.9.). Bei Reassoziationsexperimenten wurde zunächst der V1 Komplex durch Mischen mit Methanol [25%] und anschließender Gelchromatographie vollständig von der Untereinheit C befreit (Abb. 23a). Dieser C-freie Komplex wurde nach Überführung in einen methanolfreien Puffer mit 10-fachem Überschuß an rekombinanter Untereinheit C inkubiert. Bei der anschließenden Gelchromatographie zeigte sich, dass das rekombinante Protein an den V1 Komplex assoziiert und nicht gebundenes Protein später eluiert (Abb. 23c). In einer Kontrolle ließ sich die rekombinante Untereinheit genau wie die native Untereinheit durch Zugabe von Methanol wieder entfernen (Abb. 23d). Wurde der durch Alkohol von der C Untereinheit befreite Komplex für einen Aktivitätstest eingesetzt, gab es keinen meßbaren Unterschied zu seinem C-haltigem Ursprungskomplex, weder bezüglich der Mg-ATPase-Aktivität in 25% Methanol noch bezüglich der Ca-ATPase-Aktivität in wässrigem Milieu (nicht gezeigt). Auch eine Zugabe von einem Überschuß von rekombinanter Untereinheit C hatte praktisch keinen Einfluß auf 56 Ergebnisse die Eigenschaften des V1 Komplexes. Daraus läßt sich schließen, dass die C Untereinheit offensichtlich keinen Einfluß auf die Aktivität des V1 Komplexes hat. Abb. 23: Reassoziation der rekombinanten Untereinheit C an den V 1 Komplex. Zu sehen sind jeweils die Elutionsprofile der Gelfiltration auf der Superdex 200 Säule (Pharmacia) und die mit Coomassie gefärbten SDSPAGE Gele der entsprechenden Peakfraktionen. a) Entfernung der nativen Untereinheit C vom V 1 Komplex durch 25% Methanol im Laufpuffer (150 mM NaCl, 20 mM Tris-HCl pH 8,1, 9,6 mM -Mercaptoethanol). b) Der C-freie V1 Komplex (110 µl, 1 µg/µl) wurde im Eppendorfschüttler bei 4°C über Nacht im Laufpuffer inkubiert. c) Der C-freie V1 Komplex (110 µl, 1 µg/µl) wurde mit 100 µg rekombinanter Untereinheit C im Eppendorfschüttler bei 4°C über Nacht im Laufpuffer inkubiert. d) Der C-freie V1 Komplex (110 µl, 1 µg/µl) wurde mit 100 µg rekombinanter Untereinheit C im Eppendorfschüttler bei 4°C über Nacht im Laufpuffer inkubiert und vor der Gelfiltration mit 25% Methanol versetzt. Der Laufpuffer enthielt hier ebenfalls 25% Methanol. Es stellte sich nun die Frage, ob sich die Untereinheit C auch beim V1Vo Holoenzym mit Hilfe von Methanol ablösen läßt. Aus diesem Grunde wurde in einem Experiment eine Gelchromatographie des V1Vo Holoenzyms in Gegenwart von 25% Methanol durchgeführt, die erhaltenen Fraktionen in einem SDS-PAGE Gel aufgetrennt und nach Western Blotting mit Antiserum gegen die rekombinante Untereinheit C analysiert (Abb. 24). Dabei zeigte sich, dass sich beim Holoenzym im Gegensatz zum V1 Komplex die Untereinheit C durch den Alkohol nicht ablösen ließ. Überraschenderweise löste sich im Kontrollexperiment in Gegenwart von 0,01% des von uns standardmäßig zur Reinigung des V1Vo Holoenzyms verwendeten Detergenz C12E10 die Untereinheit C vom V1 Komplex ab. Diese schon durch 57 Ergebnisse das milde Detergenz hervorgerufene Dissoziation deutet darauf hin, dass die Untereinheit C im Holoenzym stärker geschützt ist als im V1 Komplex. Der Schutz könnte eine Folge der Lage der Untereinheit C und/oder stärkerer Wechselwirkungen mit dem Holoenzym sein. Abb. 24: Entfernung der Untereinheit C vom V1 Komplex und vom V1Vo Holoenzym. a) Coomassiegefärbtes SDS-PAGE Gel. b) Westernblot eines SDS-PAGE Gels, immungefärbt mit polyklonalen Antikörpern gegen die rekombinante Untereinheit C. St, Standardproteine. 1, 10 µg V 1 Komplex. 2, 10 µg V1 Komplex nach Gelfiltration mit 0,01% C12E10 im Laufpuffer. 3, 10 µg V1 Komplex nach Gelfiltration mit 0,01% C12E10 und 25% Methanol im Laufpuffer. 4, 10 µg V 1Vo Holoenzym nach Gelfiltration mit 0,01% C12E10 im Laufpuffer. 5, 10 µg V1Vo Holoenzym nach Gelfiltration mit 0,01% C12E10 und 25% Methanol im Laufpuffer. 58 Ergebnisse 4.2.4. Behandlung des V1 Komplexes mit chaotropen Ionen Chaotrope Ionen, wie das hier verwendete Iodid, stören die Ordnung von Wasser und machen es lipophiler (Hatefi & Hanstein, 1969). Dies führt unter anderem dazu, dass hydrophobe Wechselwirkungen zwischen Proteinen abgeschwächt oder aufgehoben werden. Wird der V 1 Komplex mit 0,8 M KI 30 min auf Eis inkubiert, zerfällt er in zwei Subkomplexe, die bei einer anschließenden Gelfiltration voneinander getrennt werden können (Subkomplexe I und II, Abb. 25a). Der Subkomplex I besteht aus den Untereinheiten A, B, H, C, E und G und der Subkomplex II aus A, B, E und G (Abb. 25b). Beide Subkomplexe wiesen keinerlei ATPaseAktivität auf (nicht gezeigt). Mit Hilfe von Eichproteinen (Thyroglobulin 669 kDa, Ferritin 440 kDa, Catalase 232 kDa und Aldolase 158 kDa) wurden die apparenten Molekularmassen des V1 Komplexes und der Subkomplexe mittels Gelfiltration bestimmt (Abb. 26, Tab. 6). Für den V1 Komplex ergab sich dabei eine Masse von 484 kDa, für Subkomplex I 275 kDa und für Subkomplex II 190 kDa. Geht man von der mit Hilfe der Röntgenkleinwinkelstreuung ermittelten Molekularmasse des V1 Komplexes von 550 kDa aus (Svergun et al., 1998), ergibt sich ein Korrekturfaktor von 1,14, welcher bei Subkomplex I zu einer Molekularmasse von 314 kDa und bei Subkomplex II zu einer Masse von 217 kDa führt. Diese beiden Massen entsprechen in etwa den aus der cDNA abgeleiteten Massen der Subkomplexe (Tab. 6), wenn man auch die unterschiedliche Stöchiometrie von A und B in den beiden Subkomplexen berücksichtigt. Weiterhin ist bemerkenswert, dass die Untereinheiten D und F in keinem der beiden Komplexe enthalten sind und auch nicht separat gefunden werden. Ersteres deutet auf eine eher periphere Lokalisation dieser beiden Untereinheiten im V1 Komplex hin, letzteres dass diese beiden Untereinheiten als einzelne und kleine Proteine bei der verwendeten Gelfiltration bis unter die Nachweisgrenze verdünnt werden. 59 Ergebnisse Abb. 25: Behandlung des V1 Komplexes mit chaotropen Ionen. Der V1 Komplex zerfällt bei Behandlung mit 0,8 M KI in zwei Subkomplexe. a) Elutionsprofil der beiden Subkomplexe I und II bei einer Gelfiltration (Superdex 200, Pharmacia). b) Silbergefärbtes SDS-PAGE-Gel des unbehandelten V1 Komplexes V und der beiden Subkomplexe I und II. Molekularmassenbestimmung mittels Gelfiltration 0,45 0,4 0,35 Kav 0,3 0,25 y = -0,2622Ln(x) + 3,5342 0,2 0,15 0,1 0,05 0 100000 1000000 Molkularmassen (Da) Abb. 26: Eichung der Superdex 200 Säule. Eichgerade der Proteine, Thyroglobulin 669 kDa, Ferritin 440 kDa, Catalase 232 kDa und Aldolase 158 kDa für die Molekularmassenbestimmung mittels Gelfiltration. Tab. 6: Die Molekularmassen des V1 Komplexes und seiner durch KI-Behandlung entstehenden Subkomplexe. Die Massen wurden entweder mit Hilfe von Eichproteinen in der Gelfiltration oder als Summe der aus den cDNAs abgeleiteten Molekularmassen berechnet, wobei die Stöchiometrie von A und B berücksichtigt wurde. 0,102 Molekularmasse nach Gelfiltration 484 kDa Molekularmasse nach SDS-PAGE (Summe der Untereinheiten) A3B3HCDEFG = 550 kDa Subkomplex I 0,25 275 kDa AB2HCEG = 317 kDa Subkomplex II 0,346 190 kDa A2BEG = 231 kDa 60 Molekül Kav V1 Komplex Ergebnisse 4.3. Der Vo Komplex 4.3.1. Reinigung des Vo Komplexes Nachdem es gelungen war, die während der Häutung bzw. während der Hungerphasen von der GCAM abdissoziierten V1 Untereinheiten als intakten V1 Komplex aus dem Cytosol zu isolieren, stellte sich die Frage, ob die Vo Untereinheiten als ganzer Komplex in der Membran erhalten bleiben. Würde dies der Fall sein, dann sollten sie sich bei der Solubilisierung mit C12E10 ähnlich wie das V1Vo Holoenzym verhalten und genauso wie dieses isoliert werden können. Dazu wurde zuerst die GCAM von hungernden Raupen, genau wie für das V1Vo Holoenzym beschrieben, als Bande 2 angereichert, anschließend mit C12E10 solubilisiert und der Überstand nach Zentrifugation bei 100000 x g über einen Saccharosedichtegradienten aufgetrennt. Dabei zeigte sich schon bei der Proteinverteilung innerhalb der Gradientenfraktionen ein anderes Bild als bei fressenden Raupen (Abb. 27). Proteinverteilung im Saccharosedichtegradienten 35 Prozentuale Proteinverteilung 30 25 20 15 10 5 0 1 2 3 4 5 6 Fraktionen Abb. 27: Proteinverteilung im Saccharosedichtegradienten bei fressenden und hungernden Raupen. Verglichen wird der prozentuale Proteingehalt der einzelnen Fraktionen (Fraktion 1 hohe, Fraktion 6 niedrige Saccharosedichte) bezogen auf die gesamte Proteinmenge. Weiße Balken, fressende Raupen (n=3, Mittelwert S. D.) Schwarze Balken, hungernde Raupen (n=3, Mittelwert S. D.). 61 Ergebnisse Es kam zu einer Umverteilung des Proteins von Fraktionen mit hoher Saccharosedichte, welche normalerweise das V1Vo-Holoenzym enthalten (Fraktion 1 und 2) zu Fraktionen mit geringerer Dichte (Fraktion 3 und 4). Im folgenden wurde dann, mittels gelelektrophoretischer Analyse der Fraktionen und Westernblot mit spezifischen Antikörpern für die Untereinheit d und die Untereinheit e gezeigt, dass es sich bei dieser Verschiebung tatsächlich um die Untereinheiten des Vo Komplexes handelt (Abb. 28). Damit lässt sich feststellen, dass bei hungernden Raupen der Vo Komplex als solcher in der Membran erhalten bleibt und sich als Ganzes solubilisieren und anreichern lässt, da einzelne Untereinheiten nicht soweit in den Saccharosegradienten einwandern würden. Abb. 28: Verteilung der Untereinheiten d und e im Saccharosedichtegradienten. Gezeigt sind immungefärbte Westernblots der Saccharosedichtegradientenfraktionen von fressenden (oben) und hungernden Raupen (unten). Jede Fraktion wurde einmal mit einem Antiserum gegen das V 1Vo Holoenzym (jeweils linker Blotstreifen), einmal mit dem Antiserum M40 gegen die Untereinheit d (jeweils mittlerer Blotstreifen) und einmal mit dem monoklonalen Antikörper 224-3 gegen die Untereinheit e (jeweils rechter Blotstreifen) immungefärbt. Als nächster Reinigungsschritt wurde eine Anionenaustauscherchromatographie (Mono Q, Pharmacia oder Hyper D Q, Biosepra) mit der Vo Komplex-haltigen Fraktion des 62 Ergebnisse Saccharosedichtegradienten durchgeführt, wobei der Vo Komplex relativ breit zwischen 230 und 350 mM NaCl eluiert (Abb. 29). Abb. 29: Reinigung des Vo Komplexes durch Anionenaustauscherchromatographie. a) Der Vo Komplex eluiert bei der Anionenaustauscherchromatographie (Mono Q, Pharmacia) zwischen 230 mM und 350 mM NaCl (Pfeile). b) Silbergefärbtes SDS-PAGE-Gel der Fraktionen der Anionenaustauscherchromatographie. Bahn 1 und 11, 5 µg Standardproteine. Bahn 2-10 und 12-19, je 10 µl der Fraktionen der Anionenaustauscherchromatographie. Die Pfeile markieren den Bereich von 230 mM bis 350 mm NaCl. Die entsprechenden Fraktionen wurden gepoolt, ankonzentriert und einer Gelpermeationschromatographie (Superdex 200, Pharmacia) unterzogen. Der Vo Komplex eluierte als ein einzelner Peak und enthielt alle bekannten, ihm zugeordneten Untereinheiten a, d, e und c, welche entweder mit Antikörpern oder radioaktivem 14 C-DCCD identifiziert wurden, sowie eine zusätzliche Bande mit einer apparenten Molekularmasse von 26 kDa (Abb. 30). Später wurde von der doch sehr aufwendigen Präparation der angereicherten GCAM, genau wie bei der Reinigung des V1Vo Holoenzyms, zur Solubilisierung eines Rohhomogenats aus Mitteldärmen von hungernden Tieren übergegangen, wobei als zusätzlicher Schritt zur Erhöhung des Anteils an Vo Komplexen in der Membran die nicht abdissoziierten V1-Untereinheiten mit Hilfe von Kaliumiodid als chaotropem Ion abgelöst wurden. Ausserdem wurde ebenfalls, wie bei der Reinigung des Holoenzyms, der 63 Ergebnisse Saccharosedichtegradient mit 200 mM KCl versetzt, um lose assoziierte Proteine zu entfernen und die Untereinheit a zu stabilisieren. Abb. 30: Reinigung des Vo Komplexes mittels Gelpermeationschromatographie. a) Elutionsprofil des Vo Komplexes aus angereicherter GCAM mit 200 mM KCl im Saccharosegradienten von einer Superdex 200-Säule (Pharmacia). Der grau unterlegte Bereich entspricht dem V o Komplex. b) SDS-PAGE-Gel dieses Vo Komplexes. Bahn 1, 1 µg Standardproteine, Silberfärbung. Bahn 2, 1 µg Vo Komplex; Silberfärbung. Bahn 3, 5 µg Vo Komplex, Autoradiographie mit radioaktiven 14C-DCCD, das spezifisch an die c Untereinheit bindet. Bahn 4, 1 µg Vo Komplex, Immunfärbung mit dem monoklonalen Antikörper 224-3 gegen die e Untereinheit. Bahn 5, 5 µg Vo Komplex, Immunfärbung mit Antiserum M40 gegen die d Untereinheit. Bahn 6, 5 µg Vo Komplex, Immunfärbung mit Serum gegen die 116 kDa Untereinheit a von chromaffinen Granula aus Rind (Gillespie et al., 1991). 64 Ergebnisse 4.3.2. Identifizierung der 26 kDa Bande des Vo Komplexes Zur Identifizierung der unbekannten 26 kDa Bande im gereinigten Vo Komplex wurde diese nach SDS-PAGE mit einem Westernblots auf eine PVDF-Membran transferiert. Diese Blotmembran wurde mit Coomassie Blau angefärbt, wobei als Besonderheit anzumerken ist, dass sich die 26 kDa-Bande wie auch die Untereinheit c nicht anfärben ließen, sondern im Gegenteil die Blotmembran an dieser Stelle weiß blieb (Abb. 31). Die 26 kDa-Bande wurde ausgeschnitten und von Frau Dr. Schedding (Universität Münster) N-terminal ansequenziert. Abb. 31: Blot der 26 kDa Bande aus dem V o Komplex. Der Vo Komplex wurde nach SDS-PAGE auf PVDFMembran geblottet und mit Coomassie Blau angefärbt. Dabei wurde die Aminosäuresequenz [A R S] E N [P F] [I V] Y G P ermittelt, wobei die Aminosäuren in Klammern eine gleich hohe Wahrscheinlichkeit haben. Bei einer BLASTSuche fand sich diese Teilsequenz ausschließlich als N-terminale Sequenz für c Untereinheiten von V-ATPasen wieder. Die Variante M A E N P I Y G P um das N-terminale Methionin erweitert entspricht genau dem N-Terminus der Untereinheit c von M. sexta (Abb. 32). Alle bisher gefundenen Isoformen c´´ unterscheiden sich von ihren c und c´ Varianten durch die Verlängerung der N-Termini (Abb. 32), deshalb kann man mit hoher Sicherheit ausschließen, dass es sich bei der 26 kDa Bande um die c´´ Isoform von M. sexta handelt. Da die Untereinheit c typischerweise zur Bildung von Aggregaten neigt (Lepier et al., 1996), handelt es sich bei der 26 kDa Bande sehr wahrscheinlich um ein Dimer von c. 65 Ergebnisse Abb. 32: Sequenzvergleich Untereinheit c bzw. die n-terminale Sequenz der 26 kDa Bande mit anderen c Untereinheiten. Die Variante A E N P I Y G P der Teilsequenz die bei der n-terminalen Sequenzierung der 26 kDa Bande erhalten wurde, deckt sich mit dem n-Terminus der Untereinheit c aus M. sexta. Hier wurde diese Sequenz mit den N-Termini von V-ATPase Untereinheiten c aus anderen Organismen verglichen, für die neben der Isoform c auch eine Isoform c´ oder c´´ bekannt ist (S. cerevisiae, C. elegans und H. sapiens). 4.3.3. Markierung der plecomakrolidbindenden Untereinheit der V-ATPase Plecomakrolide sind seit längerem als sehr spezifische Inhibitoren von V-ATPasen bekannt (Bowman et al., 1988; Dröse et al., 1993), insbesondere die Concanamycine und Bafilomycine (Abb. 41). Die Zusammenarbeit der Arbeitsgruppen von Prof. Altendorf in Osnabrück und von Prof. Zeeck in Göttingen führte zu einer Analyse der für die Hemmung entscheidenden Komponenten der Plecomakrolide (Bindseil & Zeeck, 1994; Boddien, 1995; Dröse et al., 1997; Dröse et al., 2001). Im Rahmen dieser Zusammenarbeit gelang es Dr. Gudrun Ingenhorst (Göttingen), ein semisynthetisches Derivat des Concanamycin A herzustellen (Ingenhorst, 2000). Dieses Derivat, das im folgenden als J-Concanolid A bezeichnete 9-O-[p-(Trifluoroethyldiazirinyl)-benzoyl]-21,23-dideoxy-23-epi-[125I]Iodo- concanolid A, besitzt gegenüber der Ausgangssubstanz zwei wichtige Modifikationen. Zum einen hat es eine UV-aktivierbare Diazirinyl-Gruppe an der Position C9, zum anderen ein 125 Jod an Position C23 des Hemiketalrings (Abb. 33). Mit Hilfe der Diazirinyl-Gruppe, die nach UV-Belichtung ein hochreaktives Carben bildet, welches sofort eine kovalente Bindung mit dem in unmittelbarer Nähe zur Verfügung stehenden Partner eingeht. Das 125 I als radioaktiver Marker sollte dann die mit dem Makrolid interagierende Untereinheit der VATPase in der Autoradiographie nach SDS-PAGE sichtbar machen. 66 Ergebnisse Abb. 33: Struktur des 9-O-[p-(Trifluoroethyldiazirinyl)-benzoyl]-21,23-dideoxy-23-epi-[125I]Iodoconcanolid A. Die gegenüber dem Ausgangsstoff Concanamycin A veränderten Positionen sind durch Nummern gekennzeichnet und die Reaktion bei UV-Belichtung dargestellt. Zunächst wurde die Wirkung des J-Concanolids A auf die Aktivität der gereinigten V-ATPase genauer untersucht und mit der Ausgangssubstanz Concanamycin A verglichen (Abb. 34), wobei eine Probe mit nicht radioaktivem Jod verwendet wurde. Bei diesen Versuchen zeigte es sich, wie entscheidend der richtige Umgang mit den Plecomakroliden für die Messung reproduzierbarer Werte ist. Die Plecomakrolide erwiesen sich als äußerst empfindlich gegenüber "Gefrier-Auftau-Zyklen" bzw. Lagerung in gelöster Form im Kühlschrank bei 4°C. Die besten Ergebnisse wurden mit frisch in DMSO gelösten Substanzen erzielt, deren echte Konzentration mit Hilfe ihres Extinktionskoeffizienten spektralphotometrisch bestimmt wurde. Die Proben wurden anschließend entweder direkt verwendet oder aliquotiert, bei -70°C gelagert und nur einmal direkt vor dem jeweiligen Versuch aufgetaut. Für das JConcanolid A ergab sich ein IC50 von 10-20 µM, welcher deutlich schlechter war als für das parallel getestete Concanamycin A (IC50 10 nM), jedoch durchaus als noch ausreichend spezifisch zu betrachten ist. 67 Ergebnisse Abb. 34: Hemmung des V1Vo Holoenzyms durch Plecomakrolide. Jede der Kurven ist das Ergebnis eines Aktivitätstests mit einer unabhängigen V1Vo Holoenzym Präparation, wobei jeder Meßwert dem Mittelwert von 3 Versuchsansätzen entspricht. Die spezifische Aktivität des V1Vo Holoenzyms war im Standardansatz ohne Inhibitor 2,9 0,5 U/mg (S. D.). Concanamycin (▲), J-Concanolid A (■). Abb. 35: Konzentrationsabhängige Photoaffinitätsmarkierung des gereinigten V 1Vo Holoenzyms mit JConcanolid A. Tricine SDS-PAGE ( Sammelgel 4% T, 3% C; Distanzgel 10% T, 3% C; Trenngel 16,5% T, 3% C, Schägger & Jagow 1987). Bahn 1 mit Coomassie gefärbt, Bahn 2-9 Autoradiogramm des Gels nach 48 h Exposition auf einem Phosphoscreen. Es wurden je 20 µg V 1Vo Holoenzym mit 100 µM (Bahn 2,3), 30 µM (Bahn 4), 10 µM (Bahn 5), 3 µM (Bahn 6), 1 µM (Bahn 7), 0,3 µM (Bahn 8) und 0,1 µM (Bahn 9) J-Concanolid A für 1 h auf Eis inkubiert und anschließend für 5 min bei 366 nm bestrahlt (ausser Bahn 2, Dunkelkontrolle). Die Markierung der gereinigten V-ATPase wurde wie unter 3.1.13. beschrieben durchgeführt. In der Autoradiographie ist deutlich zu erkennen, dass nur die Untereinheit c und selbst bei der hohen Konzentration von 100 µM J-Concanolid A keine der anderen Untereinheiten 68 Ergebnisse markiert wurde (Abb. 35). Auch bei der unbelichteten Probe wurde keine der Untereinheiten markiert; die Radioaktivität wanderte unspezifisch ins Gel ein und verteilte sich über die ganze Bahn 2. Bei der belichteten Probe mit 100 µM J-Concanolid A ohne Protein verblieb die gesamte Sonde direkt in der Auftragstasche des Geles, das gleiche geschah auch mit dem nicht an Protein gebundenen Anteil des J-Concanolid A in den Ansätzen mit Protein (nicht gezeigt). Die mit Coomassie Blau gefärbten Banden der Untereinheit c wurden im Anschluß an die Autoradiographie aus dem Gel ausgeschnitten und die gebundene Radioaktivität in einem Gamma-Counter gemessen (Abb. 36). Dabei ergab sich eine eindeutige Korrelation der an die Untereinheit c gebundenen Radioaktivität mit der bei gleichen eingesetzten Konzentrationen an J-Concanolid A gemessenen Hemmung der V-ATPase Aktivität. Abb. 36: Die relative Hemmung des V1Vo Holoenzyms im Vergleich mit der gemessenen Radioaktivität. Die Banden der Untereinheit c aus dem SDS-Gel (Abb. 35) wurden ausgeschnitten und ihre Radioaktivität in einem Gamma-Counter bestimmt (68,3 cpm (100 µM-Bande), 69,9 cpm (30 µM-Bande), 63,6 cpm ( 10 µMBande), 59,1 cpm (3 µM-Bande), 57,4 cpm (1 µM-Bande), 55,2 cpm (0.3 µM-Bande), 56,6 cpm (0.1 µM-Bande) und 51,5 cpm (0 µM-Bande). Die prozentuale Hemmung der V-ATPase durch das J-Concanolid A wurde aus dem parallel durchgeführten Test mit nicht radioaktivem J-Concanolid A entnommen. Um die Identität der als Untereinheit c bezeichneten Bande zweifelsfrei zu sichern, wurden in Zusammenarbeit mit Simone König (Münster) MALDI-MS-Analysen der ausgeschnittenen SDS-Gel Banden durchgeführt. Die nach einem chymotryptischen Verdau erhaltenen Fragmente zeigen zum einen eindeutig, dass es sich bei der ausgeschnittenen Bande tatsächlich um die Untereinheit c handelt und zum andern, dass durch die kovalente Bindung des J-Concanolid A an die Untereinheit neue zusätzliche Fragmente entstehen (Abb. 37, Abb. 69 Ergebnisse 38). Sechs dieser Fragmente konnten ihren möglichen Peptiden zugeordnet werden. Es handelt sich um die Fragmente der Aminosäuren 8-22, 12-29 und 85-87 deren Masse um 842 kDa erhöht wurde und Fragmente der Aminosäuren 1-7, 85-87 und 138-151 deren Masse um 972 kDa erhöht wurde. In Abb. 38 ist ein Ausschnitt aus dem Massenspektrum der mit JConcanolid A modifizierten Untereinheit c zusehen, der drei dieser Fragmente enthält. Abb. 37: MALDI-MS-Karte der Untereinheit c nach einem Chymotrypsinverdau. Die mit J-Concanolid A markierte Proteinbande (oben) und die entsprechende Bande ohne J-Concanolid A Behandlung (unten) wurden aus einem SDS-GEL ausgeschnitten, mit Chymotrypsin verdaut und eine MALDI-MS durchgeführt. Den identifizierten Massen wurden jeweils die entsprechenden chymotryptischen Fragmente mit Angabe der ersten und letzten Aminosäure zugeordnet. Die angegebenen Fragmente sind in beiden Spektren zu finden. Bei der mit J-Concanolid A behandelten Probe wurden einige zusätzliche neue Massen gefunden, die wahrscheinlich auf eine Modifizierung durch J-Concanolid A zurückzuführen sind. Bei diesen Massen handelt es sich um die Fragmente der Aminosäuren 8-22, 12-29 und 85-87 deren Masse um 842 kDa erhöht wurde und Fragmente der Aminosäuren 1-7, 85-87 und 138-151 deren Masse um 972 kDa erhöht wurde. 70 Ergebnisse Abb. 38: Bei Behandlung der Untereinheit c mit J-Concanolid A entstehen Fragmente mit erhöhter Masse. Ein Ausschnitt eines Massenspektrums der Untereinheit c, welche mit 10 µM J-Concanolid A behandelt wurde. Die Pfeile weisen auf drei Fragmente hin die möglicherweise durch J-Concanolid A modifiziert wurden. a) Mögliches Fragment Aminosäuren 8-22 plus 842 kDa (Sonde, die HJ verloren hat). b) Mögliches Fragment Aminosäuren 12-29 plus 842 kDa. c) Mögliches Fragment Aminosäuren 138-151 plus 972 kDa. Um die Spezifität der Bindung von J-Concanolid A an die Untereinheit c zu überprüfen, wurde versucht, die Markierung durch Vorinkubation Concanamycin A zu unterbinden (Abb. 39). 1 µM Concanamycin A führte schon zu einer deutlich schwächeren Markierung der Untereinheit c, und bei Konzentrationen von 10 µm, 100 µm und 300 µM war überhaupt keine Markierung mehr zu sehen. Concanamycin A konkurriert also mit seinem Derivat um die gleiche Bindungsstelle in der Untereinheit c der V-ATPase. Über die bisher geschilderten Markierungsversuche hinaus wurden auch solche mit GCAMVesikeln und mit gereinigtem Vo Komplex durchgeführt (Abb. 40). Auch bei der membrangebundenen V-ATPase, bei der sich alle Untereinheiten in ihrer natürlichen Lipidumgebung befinden, wurde ebenfalls nur die Untereinheit c markiert. Genauso wurde beim gereinigten Vo Komplex ebenfalls nur die Untereinheit c markiert, wobei die für den Vo Komplex typische breite Bande der Untereinheit c im SDS-Gel auffällt. 71 Ergebnisse Abb. 39: Die J-Concanolid A-Markierung der Untereinheit c kann durch Concanamycin A verhindert werden. Autoradiogramm eines Tricine SDS-PAGE Gels ( Sammelgel 4% T, 3% C; Distanzgel 10% T, 3% C; Trenngel 16,5% T, 3% C, Schägger & Jagow 1987) nach 48 h Exposition auf einem Phosphoscreen. Es wurden jeweils 20 µg gereinigtes V1Vo Holoenzym mit 10 µM J-Concanolid A inkubiert (Bahn 1-5). Vorinkubation der Proben mit 1 µM (Bahn 2), 10 µM(Bahn 3), 100 µM (Bahn 4) und 300 µM (Bahn 5) Concanamycin A. Abb. 40: J-Concanolid markiert die Untereinheit c im membrangebundenem V 1Vo Holoenzym und im gereinigtem Vo Komplex. SDS-PAGE Gel (10-15% T, 3,3% C) mit Coomassie gefärbt (Bahn 1, 3 und 5). Autoradiogramme des Gels nach 72 h Exposition auf einem Phosphoscreen (Bahn 2, 4 und 6). Bahn 1,2, 20 µg V1Vo Holoenzym, Bahn 3,4 20 µg gereinigter Vo Komplex, Bahn 5,6, 20 µg gereinigte GCAM. Alle Proben wurden mit 0,1 µM J-Concanolid A inkubiert und für 3 min mit UV-Licht (366 nm) behandelt. 72 Ergebnisse 4.3.4. Die Wirkung neuer Makrolide Nun blieb noch die Frage zu beantworten, ob es sich bei der Bindestelle in der Untereinheit c nur um die Concanamycin A Bindestelle, um die universelle Plecomakrolidbindestelle oder sogar um die Bindestelle für eine noch größere Gruppe von Makrolidantibiotika handelt. Hierzu wurden zunächst die bekannten Plecomakrolide Bafilomycin A1 und Bafilomycin B1 sowie eine Reihe neuerer Makrolide (Apicularen, Archazolid, Salicylihalamid und Cruentaren) auf ihre hemmende Wirkung auf das gereinigte V1Vo Holoenzym getestet (Abb. 41, Abb. 42). Dabei zeigte sich, dass Bafilomycin A1 und B1 (IC50 10-20 nM) in ihrer Hemmwirkung auf die V-ATPase im Gegensatz zu vielen Literaturdaten keine Unterschiede im Vergleich zu Concanamycin A (IC50 10 nM) aufweisen. Abb. 41: Die Strukturen der verwendeten Makrolide. Die Struktur von Cruentaren C33H51NO8 ist bis jetzt noch nicht gelöst. 73 Ergebnisse Abb. 42: Die Hemmung des gereinigten V1Vo Holoenzyms durch Makrolide. Alle Substanzen wurden parallel in einem Versuchsansatz und mit einer V1Vo Holoenzym Präparation getestet. Jeder Wert entspricht dem Mittel aus 3 Versuchsansätzen. Auch für Apikularen, Archazolid und Salicylihalamid ergaben sich gleich niedrige IC 50 Werte von 10-20 nM, lediglich Cruentaren und J-Concanolid A (Abb. 42) wichen mit ihren relativ hohen IC50 Werten von 60 µM bzw. 10-20 µM etwas davon ab. Im Anschluß an die Charakterisierung der Hemmeigenschaften wurden die Substanzen in einem Kompetitionsversuch auf ihre Fähigkeit, mit J-Concanolid A um die Bindestelle in der Untereinheit c zu konkurrieren, getestet (Abb. 43). Als weiterer Inhibitor der V-ATPase, von dem die Bindung an die Untereinheit c bekannt ist, wurde in diesem Versuch auch noch DCCD eingesetzt. Die beiden Bafilomycine und das Archazolid konnten die Markierung der Untereinheit c in gleicher Weise verhindern wie Concanamycin A. Die anderen Makrolide, Apikularen, Salicylihalamid und Cruentaren, waren nicht in der Lage, mit dem J-Concanolid A um die Bindestelle zu konkurrieren, was eventuell auf eine andere Bindungsstelle bzw. Wirkungsweise der Substanzen schließen läßt. Auch die Vorinkubation des V1Vo Holoenzyms mit DCCD, welches kovalent an das Glutamat 139 bindet, bewirkte keine Verdrängung der Markierung. 74 Ergebnisse Abb. 43: Kompetitionsversuche mit anderen Makroliden und DCCD. Tricine SDS-PAGE Gel (Sammelgel 4% T, 3% C; Distanzgel 10% T, 3% C; Trenngel 16,5% T, 3% C, Schägger & Jagow 1987). Bahn 1, Coomassie Blau gefärbt. Bahn 2-16, Autoradiogramm des Gels nach 48 h Exposition auf einem Phosphoscreen. Es wurden jeweils 20 µg gereinigtes V1Vo Holoenzym mit der entsprechenden Substanz vorinkubiert und anschließend mit 10 µM J-Concanolid A versetzt. Bahn 1, 20 µg V1Vo Holoenzym ; Bahn 2/3, 100/10 µM Bafilomycin A1; Bahn 4/11, Kontrolle ohne zusätzliche Substanz; Bahn 5/6, 100/10 µM Bafilomycin B1; Bahn 7/8, 100/10 µM Apicularen; Bahn 9/10, 100/10 µM Archazolid; Bahn 12/13, 100/10 µM Salicylihalamid; Bahn 14, 100 µM Cruentaren; Bahn 15/16, 100/10 µM DCCD. 75 Ergebnisse 4.4. Funktion und Regulation des V1Vo Holoenzyms und der V1 ATPase 4.4.1. Einfluß von Alkoholen und Detergenzien auf die Mg-ATPase-Aktivität des V1 Komplexes Aus der Arbeit von Gräf et al. (1996) war einiges über die enzymatischen Eigenschaften des V1 Komplexes bekannt wie z. B., die Stimulierung seiner Mg-ATPase-Aktivität durch Methanol, ähnlich wie bei den F1 Komplexen aus Bakterien, Chloroplasten und Mitochondrien (Hicks et al., 1986; Sakurai et al., 1981; Schuster, 1979; Ortiz Flores et al., 1982). Die Wirkungsweise des Methanols wird zum einen auf eine Konformationsänderung der Komplexe in der durch die Lösungsmittel bedingten hydrophoberen und somit membranähnlicheren Umgebung und zum anderen durch die mögliche Freisetzung von festgebundenem ADP erklärt (Anthon & Jagendorf, 1983). Eine hydrophobere Umgebung sollten auch Detergenzien herstellen, aber weder C12E10 (0,03% und 0,003%) und Octylglycosid (1,5% und 0,15%) konnten eine Mg-abhängige ATPase-Aktivität des V1 Komplexes induzieren (nicht gezeigt). In der Anwesenheit von Alkoholen zeigte sich jedoch für jeden der verwendeten Alkohole ein Optimum für die Aktivierung der Mg-ATPase, das bei 1-Propanol bei 5%, bei Isopropanol bei 10-15%, bei Ethanol bei 15% und bei Methanol bei 25% lag (Abb. 44). Bei einer Gelchromatographie (Superdex 200, Pharmacia) zeigte sich für alle bei ihrer Optimumskonzentration verwendeten Alkohole, das bereits beschriebene Phänomen der Aggregation und des Ablösens der C Untereinheit. Relative Aktivität Methanol (n=5) 140 Ethanol (n=3) 120 Isopropanol (n=2) 100 1-Propanol (n=3) 80 60 40 20 0 0 5 10 15 20 25 30 35 Alkoholanteil in % (v/v) Abb. 44: Einfluß von Alkoholen auf die Mg-ATPase-Aktivität des V1 Komplexes. Die Aktivitäten wurden auf den Wert bei 25% Methanol normiert, welcher bei jedem Versuch als Kontrolle mitgeführt wurde (Mittelwert S.D.). 76 Ergebnisse In einem weiteren Versuch wurde überprüft, ob die durch Methanol induzierte Mg-ATPaseAktivität über die Zeit linear verläuft. Dazu wurde die Aktivität des V1 Komplexes in Gegenwart von 15%, 20% und 25% Methanol nach 1, 2 und 3 min bestimmt. Wie Abb. 45 zeigt, war die Hydrolyserate von ATP zeitunabhängig für die einzelnen Alkoholkonzentrationen gleich groß. Da sich eine Methanolkonzentration von 25% als optimal erwies und es für diese Bedingung bereits einige Daten gab, wurde diese standardmäßig verwendet, um die Mg-ATPase Eigenschaften des V1 Komplexes zu untersuchen. Weiterhin wurde, um eine einheitliche Population von V1 Komplexen ohne verschieden große Mengen an assoziierter C Untereinheit als Ausgangsmaterial zur Verfügung zu haben, der V1 Komplex durch eine Gelchromatographie mit 25% Methanol im Laufpuffer von Resten der C Untereinheit befreit U/mg und anschließend wieder in lösungsmittelfreien Puffer gebracht. 1,6 1,4 1,2 1 0,8 0,6 0,4 0,2 0 1 min 2 min 3min 15 20 25 Methanolanteil (%) Abb. 45: Die methanolabhängige Mg-ATPase-Aktivität des V1 Komplexes hat einen linearen zeitlichen Verlauf. Gezeigt ist die ATPase-Aktivität in Gegenwart von 15%, 20% und 25% Methanol nach 1, 2 und 3 min eines typischen Experimentes. 77 Ergebnisse 4.4.2. Einfluß von Methanol auf das V1Vo Holoenzym Die Wirkung von Methanol auf die Mg-ATPase-Aktivität wurde auch für das V1Vo Holoenzym untersucht, wobei sich zeigte, dass es bei einer Konzentration von 20% zu einer 20-40%igen Stimulierung durch den Alkohol kommt (Abb. 46). Als naheliegende Ursache für diese Erhöhung der Aktivität wurde ein Ablösen von V1 Komplexen vom V1Vo Holoenzyms durch Methanol vermutet, welche dann einen Beitrag zur Gesamtaktivität leisten. Diese Hypothese wurde durch eine Gelchromatographie (Superdex 200, Pharmacia) des V1Vo Holoenzyms in Gegenwart von 20% Methanol überprüft und widerlegt (siehe Abb. 24). Wahrscheinlich ist die Erhöhung der Aktivität auf die durch Mg-ATP-Hydrolyse bedingte Dissoziation des Holoenzyms in V1 und Vo Komplex zurückzuführen. Betrachtet man den Verlauf der Phosphatproduktion durch das Holoenzym ohne Methanol, so verringert sie sich mit zunehmender Inkubationszeit, denn die abdissoziierten V1 Komplexe haben keinerlei MgATPase-Aktivität. Dagegen nimmt die Phosphatproduktion in Gegenwart von Methanol deutlich weniger ab, da sich hier die freien V1 Komplexe mit ihrer durch Methanol induzierten Mg-ATPase-Aktivität an der Gesamtaktivität beteiligen. a) b) 18 16 14 3 2,5 12 10 8 6 nmol Pi Aktivität (U/mg) 3,5 2 1,5 1 0 % Methanol 4 2 0 0,5 0 0 5 10 15 Methanolanteil (%) 20 25 20 % Methanol 0 1 2 3 Zeit (min) 4 5 Abb. 46: Stimulierung der Mg-ATPase-Aktivität des V1Vo Holoenzyms durch Methanol. (a) Die Aktivität des V1Vo Holoenzyms wurde in Gegenwart von 0% - 25% Methanol bestimmt (2 µg Protein, 2 min Inkubationszeit, n=3, Mittelwerte S. D.). (b) Vergleich der Hydrolyse Aktivität ohne und mit 20% Methanol über die Zeit (2 µg Protein). 78 Ergebnisse 4.4.3. Abhängigkeit der Ca-abhängigen Hydrolyse-Aktivität des V1 Komplexes vom verwendeten Nukleosidtriphosphat Für den V1 Komplex ist eine Ca-ATPase-Aktivität beschrieben, die mit zunehmender Reaktionszeit abnimmt, wobei eine Produkthemmung durch ADP vermutet wurde (Gräf et al., 1996 und Abb. 47). Wird hingegen Ca-GTP als Substrat angeboten, sinkt zwar die Hydrolyserate um ca. 1/3 ab, ändert sich aber mit zunehmender Reaktionszeit nicht. Eine direkte Überprüfung, ob ADP als Endprodukt den V1 Komplex hemmt, konnte in diesem Fall nicht mit Hilfe eines regenerierenden Systems aus Pyruvatkinase und Phosphoenolpyruvat durchgeführt werden, da die Pyruvatkinase ein Mg2+-abhängiges Enzym ist und schon Mg2+Konzentrationen von 0,1 mM die Ca2+-Aktivität des V1 Komplexes inhibieren. Alternativ wurde Kreatinkinase und Phosphokreatin als Mg2+-unabhängiges regenerierendes System getestet, dabei zeigte sich jedoch, dass auch dies nicht geeignet war, da Phosphokreatin sehr säureinstabil ist und bei der Quantifizierung des freigesetzten Pi mit saurem Molybdat zerfällt. Deshalb wurde der Einfluß von ADP auf die Aktivität des V1 Komplexes durch Vorinkubation mit verschiedenen ADP Konzentrationen untersucht. 25 Ca-ATP 4 µg Protein Ca-ATP 8 µg Protein Ca-GTP 4 µg Protein Ca-GTP 8 µg Protein nmol Pi 20 15 10 5 0 0 5 10 15 Zeit (min) Abb. 47: Die Hydrolyse von Ca-ATP und Ca-GTP durch den V1 Komplex. Die Hydrolyse-Aktivität von 4 µg und 8 µg V1 Komplex für Ca-ATP und Ca-GTP nach 0,25, 0,5, 1, 3, 5, 10 und 15 min wurde gemessen. Da mehr als 25 nmol produziertes Pi den Meßbereich des verwendeten Tests überschreiten konnten die 10 min Werte für 8 mg Protein und die 15 min Werte für 4 und 8 µg Protein mit Ca-GTP nicht ausgewertet werden. 79 Ergebnisse 4.4.4. Einfluß von Ca-ADP, Mg-ADP und Pi auf die Hydrolyse von ATP durch das V1Vo Holoenzym und den V1 Komplex Die bisherigen Versuche zur Hydrolyse-Aktivität des V1 Komplexes legen eine Endprodukthemmung der Ca-ATPase-Aktivität nahe, welche unter Einbeziehung des nach der neuen Methode präparierten V1Vo Holoenzyms systematisch untersucht werden sollte. Von diesem war schon durch die Versuche von Schweikl et al. (1989) bekannt, dass die MgAktivitäten des solubilisierten V1Vo Holoenzyms eine starke Endprodukthemmung aufweisen, die durch ein ADP-regenerierendes System aus Pyruvatkinase und Phosphoenolpyruvat teilweise aufgehoben werden kann. Aus diesem Grund wurde zunächst die Aktivität des gereinigten V1Vo Holoenzyms mit und ohne Regeneration von ADP getestet. Es zeigte sich im wesentlichen das gleiche Ergebnis wie bei Schweikl (1988), wobei die Inaktivierung des Holoenzyms bei kleinen Proteinmengen und kurzen Inkubationszeiten nicht ganz so schnell verlief. Ab einer Konzentration von ca. 50 µM produziertem ADP war allerdings ebenfalls eine deutliche Hemmung zu beobachten (Abb. 48). a) b) 14 30 12 25 8 0 U/ml PK 3 U/ml PK 10 U/ml PK 30 U/ml PK 6 4 2 0 nmol Pi nmol Pi 10 20 15 0 U/ml PK 3 U/ml PK 10 U/ml PK 30 U/ml PK 10 5 0 0 0,2 0,4 0,6 0,8 1 Zeit (min) 0 5 10 15 Zeit (min) Abb. 48: Einfluß eines ADP-regenerierenden Systems. Es wurde die Mg-ATPase-Aktivität von 4 µg/160 µl (a) und 1 µg/160 µl (b) gereinigtem V1Vo Holoenzym in Gegenwart von 0, 3, 10 und 30 U/ml Pyruvatkinase und 10 mM Phosphoenolpyruvat (PEP) bestimmt. Um unterscheiden zu können, ob schon die Anwesenheit der Hydrolyseendprodukte ADP oder Pi einen Einfluß auf die Aktivität haben oder ob der Vorgang der Hydrolyse selbst zu einer Inaktivierung notwendig ist, wurden einige Experimente durchgeführt, bei denen der V1 Komplex und das V1Vo Holoenzym mit ADP oder Pi und jeweils Ca2+ oder Mg2+ vorinkubiert wurde. Beim V1 Komplex war die Mg-abhängige Aktivität in Gegenwart von 25% Methanol vollkommen unabhängig von der vorgelegten ADP- oder Pi Konzentration, die in mehreren Versuchen zwischen 0 und 30 µM bzw. 0 und 50 µM variiert wurde (Abb. 49, Abb. 52). Bei 80 Ergebnisse den Vorversuchen für die Ca-ATPase-Aktivität zeigte sich dagegen schon eine deutliche Hemmung bei der Verwendung von 30 µM ADP. In einem weiteren Test wurde dann die ADP Konzentration auf bis zu 1 mM erhöht, was eine Hemmung der Anfangs-Hydrolyserate der Ca-Aktivität um mehr als 70% zur Folge hatte (Abb. 49). Eine Vorinkubation mit Pi hatte keine Auswirkung auf die Aktivität (Abb. 52). 10 0 µM ADP 1 µM ADP 3 µM ADP 10 µM ADP 30 µM ADP nmol Pi 8 6 4 2 0 0 2 3 Zeit (min) 4 5 0 µM ADP 30 µM ADP 100 µM ADP 300 µM ADP 1000 µM ADP 10 8 nmol Pi 1 6 4 2 0 0 1 2 3 Zeit (min) 4 5 Abb. 49: Einfluß von ADP auf die Aktivität des V1 Komplexes. Methanol induzierte Mg-ATPase-Aktivität des V1 Komplexes (1,5 µg Protein/160 µl) nach 5 min Vorinkubation mit den angegebenen Mg-ADPKonzentrationen (oben). Ca-ATPase-Aktivität des V1 Komplexes (4 µg Protein/160 µl) nach 5 min Vorinkubation mit den angegebenen Ca-ADP-Konzentrationen (unten). Die Messung erfolgte in je zwei parallelen Ansätzen, wobei nach den entsprechenden Zeiten je 160 µl Aliquots zur Phosphatbestimmung entnommen wurden. Die Mg- und Ca-ATPase-Aktivität des V1Vo Holoenzyms wurde ebenfalls nicht durch Vorinkubation mit Pi beeinflußt (Abb. 53). Auch die Ca-ATP-Hydrolyserate blieb weitgehend unbeeinflußt von der vorgelegten Menge an Ca-ATP (Abb. 50) Dagegen zeigte hier eine Vorinkubation mit Mg-ADP eine deutliche Hemmung (Abb. 50), die sich vor allem bei kurzen Inkubationszeiten andeutete. Deshalb wurde der Einfluß von Mg-ADP auf die Hydrolyse bei einer Inkubationszeit von 15 s untersucht (Abb. 51). Die Hemmung war konzentrationsabhängig, wobei 100 µM vorgelegtes Mg-ADP zu 85% inhibierte. 81 Ergebnisse Als weitere Parameter wurden noch eine Erhöhung der im Test verwendeten MgKonzentration von 1 mM auf 3 mM bzw. eine Verdopplung der Proteinmenge bei Mg- und Ca- Aktivität untersucht, was jeweils zu gleichen Ergebnissen führte (nicht gezeigt). 18 0 µM ADP 0,1 µM ADP 0,3 µM ADP 1 µM ADP 3 µM ADP 10 µM ADP 30 µM ADP 16 14 nmol Pi 12 10 8 6 4 2 0 0 1 2 3 4 5 Zeit (min) 18 16 0 µM ADP 0,1 µM ADP 0,3 µM ADP 1 µM ADP 3 µM ADP 10 µM ADP 30 µM ADP 14 nmol Pi 12 10 8 6 4 2 0 0 1 2 3 4 5 Zeit (min) Abb. 50: Einfluß von ADP auf die Aktivität des V1Vo Holoenzyms. Mg-ATPase-Aktivität des V1Vo Holoenzyms (1,3 µg/160 µl) nach 5 min Vorinkubation mit den angegebenen Mg-ADP-Konzentrationen (oben). Ca-ATPase-Aktivität des V1Vo Holoenzyms (2,6 µg/160 µl) nach 5 min Vorinkubation mit den angegebenen CaADP-Konzentrationen (unten). Die Messung erfolgte in je zwei parallelen Ansätzen, wobei nach den entsprechenden Zeiten je 160 µl Aliquots zur Phosphatbestimmung entnommen wurden. 82 Ergebnisse 3 noml Pi nach 15 s 2,5 2 1,5 1 0,5 0 0 µM 0,1 µM 0,3 µM 1 µM 3 µM 10 µM 30 µM 100 µM Vorinkubation mit µM [ADP] Abb. 51: Hemmung der Mg-ATPase-Aktivität des V1Vo Holoenzyms durch Mg-ADP bei einer Inkubationszeit von 15 s. Je 4 µg V1Vo Holoenzym wurden für 5 min mit den entsprechenden Mengen MgADP vorinkubiert (je 6 Proben). 20 0 µM Pi 5 µM Pi 20µM Pi 50 µM Pi nmol Pi 15 10 5 0 0 1 2 3 Zeit (min) 4 5 20 0 µM Pi 5 µM Pi 20µM Pi 50 µM Pi nmol pi 15 10 5 0 0 1 2 3 Zeit (min) 4 5 Abb. 52: Einfluß von anorganischem Phosphat auf die Aktivität des V 1 Komplexes. Methanol induzierte Mg-ATPase-Aktivität des V1 Komplexes (1 µg/160 µl) nach 5 min Vorinkubation mit den angegebenen PiKonzentrationen (oben). Ca-ATPase-Aktivität des V1 Komplexes (4 µg/160 µl) nach 5 min Vorinkubation mit den angegebenen Pi-Konzentrationen (unten). Die Messung erfolgte in je zwei parallelen Ansätzen, wobei nach den entsprechenden Zeiten je 160 µl Aliquots zur Phosphatbestimmung entnommen wurden. Das vorgelegte Pi wurde über die Meßwerte für 0 min korrigiert. 83 Ergebnisse 20 18 16 14 12 10 8 6 4 2 0 nmol Pi 0 µM Pi 5 µM Pi 20 µM Pi 50 µM Pi 0 20 18 16 14 12 10 8 6 4 2 0 1 2 3 Zeit (min) 4 5 nmol Pi 0 µM Pi 5 µM Pi 20 µM Pi 50 µM Pi 0 1 2 3 Zeit (min) 4 5 Abb. 53: Einfluß von anorganischem Phosphat auf die Aktivität des V 1Vo Holoenzyms. Mg-ATPaseAktivität des V1Vo Holoenzyms (1,8 µg/ 160 µl) nach 5 min Vorinkubation mit den angegebenen PiKonzentrationen (oben). Ca-ATPase-Aktivität des V1Vo Holoenzyms (3,6 µg/160 µl) nach 5 min Vorinkubation mit den angegebenen Pi-Konzentrationen (unten). Die Messung erfolgte in je zwei parallelen Ansätzen, wobei nach den entsprechenden Zeiten je 160 µl Aliquots zur Phosphatbestimmung entnommen wurden. Das vorgelegte Pi wurde über die Meßwerte für 0 min korrigiert. 84 Ergebnisse 4.4.5. Bestimmung der endogen gebundenen Nukleotide des V1Vo Holoenzyms und des V1 Komplexes Um die Ergebnisse der Experimente zum Einfluss von Nukleotiden auf die V-ATPase besser interpretieren zu können, wurde zunächst der bis dato noch nicht untersuchte Gehalt von endogen gebundenen Nukleotiden im V1Vo Holoenzym und im V1 Komplex aus hungernden Raupen mit Hilfe der Luciferin/Luciferase-Methode bestimmt (3.1.19.). Für die Berechnung des Verhältnisses von Nukleotide zu Protein wurde für das V1Vo Holoenzym eine Molekularmasse von 800 kDa und für den V1 Komplex von 550 kDa angenommen. Dabei ergab sich für das V1Vo Holoenzym ein ATP-Gehalt von 0,005 mol/mol Enzym und für den V1 Komplex ein ATP-Gehalt von 0,0015 mol/mol Enzym (Tab. 7). Höhere Werte ergaben sich für ADP mit einem Gehalt von 0,28 mol/mol Holoenzym und 1,7 mol/mol V1 Komplex (Tab. 7). Der ADP-Gehalt des V1 Komplexes war unabhängig vom physiologischen Zustand der Raupen: fressende, hungernde und sich häutende Tiere unterschieden sich nicht. Tab. 7: Der Nukleotidgehalt des V1Vo Holoenzyms und des V1 Komplexes. Die Messung des Nukleotidgehaltes von mehreren unabhängigen Präparationen (n=3-14) erfolgte mit der Luciferin/LuciferaseMethode (Mittelwerte S. D., n. b., nicht bestimmt). V1Vo Holoenzym ATP-Gehalt ADP-Gehalt 0,005 0,002 mol/mol (n=3) 0,28 0,05 mol/mol (n=6) V1 Komplex V1 Komplex V1 Komplex (hungernde Raupen) (fressende Raupen) (häutende Raupen) 0,0015 0,001 mol/mol (n=3) 1,7 0,57 mol/mol (n=14) n. b. n. b. 1,63 0,42 mol/mol (n=8) 1,75 0,42 mol/mol (n=4) Im weiteren wurde versucht, das im V1 Komplex gebundene ADP zu entfernen (Abb. 54). Neben mehreren Variationen des für die Standardgelchromatographie (Superdex 200, Pharmacia) verwendeten Puffers (150 mM NaCl, 20 mm Tris-HCl pH 8,1, 9,6 mM Mercaptoethanol) durch Zugabe von 25% Methanol, 0,01% C12E10 oder 5 mM EDTA, wurde auch eine Säulenchromatographie nach Senior et al. (1992) und eine Inkubation mit alkalischer Phosphatase (Digel et al., 1996) versucht (Details, siehe 3.1.19.). Alle Proben wurden nach der entsprechenden Behandlung zusammen mit der entsprechenden Ausgangsprobe auf einem SDS-PAGE Gel analysiert, wobei keine Veränderung festgestellt werden konnte (nicht gezeigt). Obwohl Methanol die Mg-ATPase-Aktivität des V1-Komplexes stimuliert und linearisiert, löst es kein ADP aus dem Komplex. Auch 5 mM EDTA, welches durch die Komplexierung 85 Ergebnisse von divalenten Kationen das ADP aus seiner Bindestelle, die möglicherweise von Mg-ADP besetzt ist herauslösen könnte, hatte ebenso wie das für die Reinigung des Holoenzyms verwendete C12E10, welches sehr wenig ADP enthält, keine Wirkung. Die für F-ATPasen erfolgreich zur Entfernung von Nukleotiden eingesetzte Chromatographie mittels einer langen Sephadex G-50 Säule war ebenfalls uneffektiv. Lediglich die Inkubation mit alkalischer Phosphatase bewirkte eine deutliche Reduktion des ADP-Gehaltes auf 1/3 des Ausgangswertes. Ein Aktivitätstest dieses V1 Komplexes unter Standardbedingungen zeigte allerdings keinen Unterschied zur Kontrolle im Hinblick auf die Ca- und die Mg-abhängige Aktivität; dies könnte möglicherweise auf ein sofortiges wiederbesetzten der Bindestelle mit ADP zurückzuführen sein. Für weitere Untersuchungen über den Einfluß von Nukleotiden bzw. die Bindestelle des endogenen ADPs sollte deshalb darüber nachgedacht werden, diese Methode in die Vorbereitung des V1 Komplexes mit einzubinden. 100 relativer ADP-Gehalt (%) 90 80 70 60 50 40 30 20 10 0 Kontrolle Methanol EDTA C12E10 Senior APP Abb. 54: ADP-Gehalt unterschiedlich behandelter V1 Komplexe. Gelchromatographie in Superdex 200 mit 25% Methanol, mit 5 mM EDTA, mit 0,01% C12E10 im Laufpuffer; Gelchromatographie in Sephadex G-50 nach Senior et al. (1992); Inkubation mit alkalischer Phosphatase (APP) nach Digel et al. (1996). Vergleich mit jeweils parallel durchgeführten Kontrollen (100%). 86 Ergebnisse 4.4.6. Dissoziation des V1Vo Holoenzyms in vitro Da gezeigt werden konnte, dass ADP einen starken Einfluß auf die Aktivität der V-ATPase hat, sollte überprüft werden, ob diese Hemmung durch eine Dissoziation des V1Vo Holoenzyms in seine beiden Subkomplexe bewirkt wird. Aus diesem Grunde wurde das V1Vo Holoenzym mit den entsprechenden Nukleotiden vorinkubiert, gefolgt von einer Zentrifugation im Saccharosedichtegradienten (siehe 3.1.24.). Die deutlichste Zunahme an Vo-Untereinheiten in Fraktionen niedriger Dichte, verbunden mit deren Abnahme in Fraktionen hoher Dichte, zeigte sich bei Inkubation des Holoenzyms mit 2 mM Mg-ATP (Abb. 55). Dies bedeutet, dass es unter diesen Inkubationsbedingungen zu einer Dissoziation von V1 Komplex und Vo Komplex kam. Ein schwächerer Effekt wurde durch Inkubation mit 2 mM Mg-ADP sowie mit 2 mM Mg-ADP und 2 mM Pi erzielt, wobei sich zwischen diesen beiden Inkubationsvarianten kein signifikanter Unterschied zeigte. Hingegen ergab sich bei Inkubation des Holoenzyms mit 2 mM Ca-ATP, mit 2 mM Ca-ADP, mit 0,2 mM Mg-ADP, sowie mit 0,2 mM Mg-ADP und 0,2 mM Pi kein deutlich sichtbarer Effekt, der auf eine größere Dissoziationsrate des Holoenzyms schließen lassen könnte. Abb. 55: Einfluß von Mg- und Ca-ADP, Mg- und Ca-ATP und von Pi mit Mg-ADP auf die Dissoziation des V1Vo Holoenzyms. a) und b) Silbergefärbte SDS-PAGE Gele der Saccharosedichtegradientenfraktionen (a) hohe Dichte (b) niedrige Dichte. 1, Kontrolle. 2, 2 mM Mg-ADP. 3, 2 mM Mg-ATP. 4, 2 mM Ca-ADP. 5, 2 mM Ca-ATP. c) und d) Silbergefärbte SDS-PAGE Gele der Saccharosedichtegradientenfraktionen (c) hohe Dichte (d) niedrige Dichte. 1, Kontrolle. 2, 0,2 mM Mg-ADP. 3, 2 mM Mg-ADP. 4, 0,2 mM Mg-ADP, 0,2 mM Pi. 5, 2 mM Mg-ADP, 2 mM Pi. 87 Ergebnisse Bei den vorausgegangenen Experimenten war die Konzentration der Nukleotide so groß, dass jedes Holoenzymmolekül mehr als 1000 ATP hydrolysierte. Daher war es von Interesse, wie sich die V-ATPase bei Inkubation mit äquimolaren Nukleotidkonzentrationen verhält. Bei einer ATP Konzentration von 1 µM steht jedem eingesetzten V1Vo Holoenzym (100 µg/80µl) theoretisch ca. 1 ATP zur Verfügung. Wie Abb. 56 zeigt, reichte dies tatsächlich aus, um den ungefähr gleichen Dissoziationseffekt zu erzeugen. Ein Gemisch von 1 µM ATP und 0,2 mM ADP führte allerdings zu keiner spürbaren Dissoziation von V1 und Vo Komplex. Mit Hilfe des nicht hydrolysierbaren ATP Analogons AMP-PNP sollte getestet werden, ob die Besetzung der ATP-Bindestelle alleine schon eine Dissoziation bewirken kann. Dies ist nicht der Fall, da die Probe mit AMP-PNP keinerlei Unterschied zur Kontrolle aufwies (Abb. 56). Möglicherweise bietet AMP-PNP aber einen Schutz vor der durch ATP bewirkten Dissoziation, da diese in Kombination mit dem Analogon bei einer ATP Konzentration von 2 mM geringer ausfällt als ohne. AMP-PNP konkurriert möglicherweise sowohl mit ATP als auch mit dem entstehenden ADP um die Bindestelle im Enzym. Zur Verdeutlichung dieser Ergebnisse wurde zusätzlich zu den SDS-PAGE Gelen auch noch die Proteinverteilung in den Saccharosegradienten analysiert, wodurch sich der Dissoziationsgrad ebenfalls sehr schön darstellen läßt (Abb. 57). Zusätzlich wurde auch der Nukleotidgehalt der Fraktionen dieses Versuches bestimmt, wozu jeweils die Fraktionen 2-4 und 7-9 zusammengefasst wurden (Abb. 58). In keinem der beiden Pools konnte ATP in nennenswerter Menge nachgewiesen werden. Der ATP-Gehalt der Fraktion 2-4, welche jeweils das komplette V1Vo Holoenzym enthalten, entsprach mit 0,001 mol/mol V-ATPase in etwa dem bereits bestimmten Wert (siehe auch 4.4.5.). In der Fraktion 7-9 in der sich neben dem Vo Komplex auch V1-ATPase befindet konnte, kein ATP nachgewiesen werden. Anders sah es hingegen mit dem ADP-Gehalt der Proben aus. Bei der Inkubation mit 2 mM Mg-ATP erhöhte sich der ADP-Gehalt der Fraktion 2-4 im Vergleich zur Kontrolle um das Dreifache. Auch bei der Mischung aus 2 mM AMP-PNP und 2 mM ATP erhöhte sich der ADP-Gehalt leicht. Bei der Verwendung von 2 mM AMP-PNP alleine konnte eine deutliche Verringerung des ADP-Wertes registriert werden. In der Fraktion 7-9 war der ADP-Gehalt bei allen Versuchsbedingungen ca. gleich groß und pendelte um 0,2 mol ADP/mol Enzym (da V1 und Vo Untereinheiten in der Fraktion 7-9 enthalten sind wurde auch hier mit einer Molekularmasse von 800 kDa gerechnet). Es konnte also keine Erhöhung des ADP-Gehaltes in den abdissoziierten V1 Komplexen festgestellt werden. Hieraus lässt sich zunächst schließen, dass vermutlich nicht die feste Bindung eines ADP-Moleküls an das Enzym zu dessen Dissoziation führt. 88 Ergebnisse Abb. 56: Einfluß von geringen ATP-Konzentrationen und von AMP-PNP auf die Dissoziation des V1Vo Holoenzyms. a)- h) Silbergefärbte SDS-PAGE Gele der Saccharosedichtegradienten (1) Standardproteine, (210) entspricht Fraktion 40/2-25/2 (siehe 3.1.24.). (a) Kontrolle, (b) 2 mM Mg-ATP, (c) 1 µM Mg-ATP, (d) 1 µM Mg-ATP, 0,2 mM Mg-ADP, (e) 0,2 mM Mg-ADP, (f) 2 mM Mg-AMP-PNP, (g) 2 mM Mg-AMP-PNP, 0,2 mM Mg-ATP, (h) 2 mM Mg-AMP-PNP, 2 mM Mg-ATP. 89 Ergebnisse 60 Kontrolle 2 mM ATP 1 µM ATP 1 µM ATP 0,2 mM ADP 0,2 mM ADP 2 mM AMP-PNP 0,2 mM ATP 2 mM AMP-PNP 2 mM ATP 2 mM AMP-PNP Proteinverteilung (%) 50 40 30 20 10 0 1 2 3 4 5 6 7 8 9 Fraktion Abb. 57: Proteinverteilung in den Saccharosedichtegradienten. Gezeigt ist die Proteinverteilung in den Gradienten der Dissoziationversuche mit geringer ATP-Konzentration und AMP-PNP (1-9 entspricht Fraktion 40/2-25/2, siehe auch Abb. 56). 2 1,8 Fraktion 2-4 mol ADP/mol Enzym 1,6 Fraktion 7-9 1,4 1,2 1 0,8 0,6 0,4 0,2 0 M M m m + DP A M A M TP A TP A TP P AT M m 2 TP + A P M PN m P2 M 0, A + M NP m 2 -P P M A P PN P2 2 m µM µM A lle tro M m 2 0, 1 1 2 on K 2 0, M m D A P Abb. 58: ADP-Gehalt der Saccharosedichtegradientenfraktionen. Die Fraktionen 2-4 und 7-9 des Dissoziationversuche mit geringer ATP-Konzentration und AMP-PNP (1-9 entspricht Fraktion 40/2-25/2, siehe auch Abb. 56 und 57) wurden gepoolt, ankonzentriert und ihr Gehalt an gebundenem ADP mit wie unter 3.1.22 beschrieben bestimmt. Bei der Berechnung des Verhältnisses wurde jeweils ein Molekulargewicht des V1Vo Holoenzyms von 800 kDa benutzt. Zur Kontrolle wurde auch der ATP-Gehalt bestimmt, welcher für die Fraktionen 2-4 bei 0,001 mol/mol Enzym lag und sich für die Fraktionen 7-9 unter der Nachweisgrenze befand. 90 Ergebnisse 4.4.7. Einfluß von DCCD und Concanamycin A auf die Mg2+ und Ca2+-ATPase-Aktivität des V1Vo Holoenzyms Das Carbodiimid DCCD und das Plecomakrolid Concanamycin A sind Inhibitoren, die ihre Bindestelle und somit ihren Wirkungsort im Vo Komplex des Enzyms haben, obwohl die Hydrolyse von ATP im V1 Komplex erfolgt. Die Mg-abhängige Hydrolyse von ATP wurde in diesem Versuchsansatz durch 0,01 µM Concanamycin A stark und durch 0,1 µM komplett gehemmt (Abb. 59 , siehe auch 4.3.3.). DCCD hatte nur in höheren Konzentration von 100 µM einen hemmenden Einfluss auf das Enzym. Ganz anders sah das Ergebnis für die CaATPase-Aktivität aus, welche weder durch Concanamycin noch durch DCCD wesentlich beeinflußt wurde (Abb. 60). Hieraus lässt sich schließen, dass die Ca-ATPase-Aktivität des V1Vo Holoenzyms keine Protonentranslokation durch den Vo Komplex induziert. Die Entkopplung von ATP-Hydrolyse und Protonentransport könnte auf eine Uni-site Katalyse in der Gegenwart von Ca2+ zurückzuführen sein. Ob es sich hierbei um ein Phänomen des detergenssolubilisierten und gereinigten V1Vo Holoenzyms handelt oder dies auch in der nativen Membranumgebung erfolgt, wurde in der Folge an gereinigten GCAM-Vesikeln getestet. 91 Ergebnisse Kontrolle 1,5 µM DCCD 100 µM DCCD 0,01 µM Con A 0,1 µM Con A 35 30 nmol Pi 25 20 15 10 5 0 0 1 2 Zeit (min) 3 4 Abb. 59: Einfluß von DCCD und Concanamycin A Mg-ATPase-Aktivität des V1Vo Holoenzyms. Die Proben (840 µl, 25 µg/ml, n=2) wurden mit den entsprechenden Hemmstoffen für 10 min vorinkubiert, die Hydrolyse mit 120 µl ATP (Endkonzentration 1 mM) gestartet und zu den angegebenen Zeiten 160 µl Aliquots zur Phosphatbestimmung entnommen und in N2 eingefroren. 35 Kontrolle 1,5 µM DCCD 100 µM DCCD 0,01 µM Con A 0,1 µM Con A 30 nmol Pi 25 20 15 10 5 0 0 1 2 Zeit (min) 3 4 Abb. 60: Einfluß von DCCD und Concanamycin A Ca-ATPase-Aktivität des V1Vo Holoenzyms. Vorgehen wie Abb. 59. 92 Ergebnisse 4.4.8. Messung des Protonentranports an GCAM-Vesikeln mit Mg2+- und Ca2+-ATP als Substrat Die vorhergehenden Versuche zur Inhibierung der Mg- und Ca-ATPase-Aktivität des V1Vo Holoenzyms durch die Vo Komplex-spezifischen Inhibitoren DCCD und Concanamycin A legten nahe, dass es einen Unterschied zwischen der ATP-Hydrolyse mit Mg2+ oder Ca2+ als divalentem Kation gibt, der sich auf die Protonentranslokation auswirkt. Diese Vermutung wurde durch fluorometrische Messungen des Protonentransports in gereinigten GCAMVesikeln verifiziert. Die Anwesenheit von 1 mM Mg2+ induzierte einen ATP-getriebenen Protonentransport der durch Quenchen der Acridinorange-Fluoreszenz gemessen wurde (Abb. 61a). Durch die Aktivierung des K+/H+-Antiporters in der GCAM mit KCl konnte diese Ansäuerung wieder aufgehoben werden (Wieczorek et al., 1991). Dagegen konnte die Protonentranslokation durch Ca2+-Konzentrationen von 1-15 mM nicht induziert werden (Abb. 61c-d). Ca2+-Konzentrationen bis zu 15 mM beeinflussten die MgATP-induzierte Ansäuerung der Vesikel nicht. Auch der Zusatz von Ca2+, nachdem mit Mg2+ eine Ansäuerung erreicht wurde, hatte keine Wirkung (Abb. 61c). Abb. 61: Fluorometrische Messung des Protonentransports in GCAM-Vesikeln. In den Versuchen (a-d) wurden jeweils 10 µl GCAM-Vesikel (ca. 10 µg Protein) eingesetzt und die Änderung der Acridinorangefluoreszenz nach Zugabe von 1 mM MgCl2 (1), 20 mM KCl (2), 1 mM CaCl2 (3), 3 mM CaCl2 (4) und 15 mM CaCl2 (5) gemessen (es sind jeweils die Endkonzentrationen in der Küvette angegeben). Bei Versuch (c) war bereits 4 mM CaCl2 vorgelegt. 93 Ergebnisse 4.4.9. Messungen kapazitiver Ströme des rekonstituierten V1Vo Holoenzyms Um den elektrogenen Schritt bei der Protonentranslokation über die Membran elektrisch erfassen zu können wurde das V1Vo Holoenzym zunächst in Liposomen rekonstituiert. Diese Proteoliposomen wurden dann an einen Blacklipid Bilayer adsorbiert und die kapazitiven Ströme bei der Hydrolyse von ATP durch das V1Vo Holoenzym gemessen. Der Strom war abhängig von der eingesetzten Konzentration an ATP und die Werte reproduzierbar zu messen (Abb. 62a-c). Da der Strom durch Concanamycin A inhibierbar war, kann er als spezifisch für das rekonstituierte V1Vo Holoenzym betrachtet werden (Abb. 62d.). Auch zeigte sich wie bereits bei der GCAM auch im rekonstituierten System, dass die Hydrolyse von Ca-ATP keinen Protonentransport antreibt (Abb. 62e) und dass Ca2+ die Mg-ATPabhängige Protonentranslokation nicht beeinflusst (Abb. 62f-h). Abb. 62: Blacklipidbilayer: Messungen der Stromstärke von Bilayeradsorbaten. Für die hier gezeigten Versuche wurden jeweils 43 µl Proteoliposomen eingesetzt. Zum Zeitpunkt 0 s wurde mit einem Laserblitz das caged ATP freigesetzt. a) 300 µM caged ATP, 1 mM MgCl2. b) und c) 1 mM caged ATP, 1 mM MgCl2. d) 1 mM caged ATP, 1 mM MgCl2, 10 µM Concanamycin A (10 min Vorinkubation). e) 1 mM caged ATP, 3 mM CaCl2. f) und g) 1 mM caged ATP, 3 mM CaCl2, 1 mM MgCl2. h) 1 mM caged ATP, 3 mM CaCl2, 3 mM MgCl2. 94 Diskussion 5. Diskussion 5.1. Die Struktur der V-ATPase aus M. sexta 5.1.1. Das V1Vo Holoenzym Die Isolierung der V-ATPase aus dem Mitteldarmepithel von Tabakschwärmern (Schweikl et al., 1989) war mit dem Manko behaftet, dass die Präparation im Verhältnis zur Ausbeute sehr aufwendig war und dass deswegen nur eine vergleichsweise geringe Anzahl von Experimenten mit einer Präparation durchgeführt werden konnte. Ausserdem enthielten die Präparationen nicht alle konstitutiven V-ATPase-Untereinheiten (Wieczorek et al., 2000). In dieser Arbeit wurden bestehende Reinigungsprotokolle optimiert und neue Reinigungsprotokolle etabliert. Erst dadurch war es möglich, für das V1Vo Holoenzym sowie seine beiden Teilkomplexe V1 und Vo die genaue Untereinheitenzusammensetzung zu bestimmen und darüber hinaus Strukturdaten, besonders über den katalytischen V1 Komplex, zu gewinnen. Für die Erhöhung der Ausbeute und die wesentlich einfachere und schnellere Präparation des V1Vo Holoenzyms sind vor allem zwei Ergebnisse ausschlaggebend. Zum einen hat die VATPase aus dem anterioren Mitteldarmabschnitt die gleichen enzymatischen Eigenschaften und die gleiche Untereinheitenzusammensetzung wie die V-ATPase aus dem posterioren Mitteldarm, so dass der ganze Mitteldarm als Ausgangsmaterial für die Reinigung der VATPase verwendet werden kann. Zum anderen solubilisiert das Detergenz C 12E10 die VATPase so selektiv aus dem Mitteldarmepithel, dass sich die aufwendige längsmuskelfreie Präparation des Mitteldarmepithels und die Anreicherung der GCAM auf einem Saccharosestufengradienten erübrigt; vielmehr kann ein Rohhomogenat des Mitteldarmepithels als Ausgangsmaterial für die Reinigung der V-ATPase verwendet werden. Die zusätzlichen Chromatographieschritte mit einem Anionenaustauscher und einer Gelfiltration ermöglichten die Reinigung eines V1Vo Holoenzyms aus M. sexta, dass mit den Untereinheiten A, B, C, D, E, F, G und H alle bekannten Untereinheiten des V1 Komplexes und mit den Untereinheiten a, c, d und e auch die des Vo Komplexes enthält. Die Ausbeute an V1Vo Holoenzym liegt mit 100 µg pro Raupe mehr als 10 mal höher als bei der bisherigen Präparationsmethode. Die Identifikation aller Untereinheiten erfolgte über MALDI-MSAnalyse von tryptischen bzw. chymotryptischen Fragmenten ihrer aus einem SDS-PAGE Gel 95 Diskussion ausgeschnittenen Proteinbanden und deren Abgleich mit den Fragmenten eines theoretischen Verdaus ihrer, aus der cDNA abgeleiteten, Aminosäuresequenzen (Dow et al., 1992; Gräf et al., 1992; Novak et al., 1992; Gräf et al., 1994a; Gräf et al., 1994b; Lepier et al., 1996; Merzendorfer et al., 1997; Merzendorfer et al., 1999; Merzendorfer et al., 2000). Bis auf die Untereinheit e, deren tryptische Fragmente außerhalb des Meßbereichs lagen, konnten alle Proteinbanden eindeutig den jeweiligen Untereinheiten zugeordnet werden. Letztere war allerdings bereits durch monoklonale Antikörper und N-terminale Sequenzierung eindeutig identifiziert worden (Merzendorfer et al., 1999). 5.1.2. Der Vo Komplex Bei der neuen Präparationsmethode des V1Vo Holoenzyms ist besonders die Entdeckung der Untereinheit a hervorzuheben, die für lange Zeit bei Insekten nicht dargestellt werden konnte (Wieczorek et al., 2000). Die Abwesenheit dieser Untereinheit stellte ein Problem dar, weil bei Mutageneseexperimenten an der V-ATPase der Hefe gezeigt wurde, dass sie essentiell für die Assemblierung und/oder die Protonentranslokation der V-ATPase ist (Manolson et al., 1994; Leng et al., 1996; Leng et al., 1998). Weiterhin gilt die Untereinheit a als Kandidat für einen wichtigen Bestandteil des sogenannten Stators in Modellen, welche die Arbeitsweise der V-ATPase in Analogie zur F-ATPase erklären (Kagawa & Hamamoto, 1996; Junge et al., 1997). Tatsächlich wurde dieser second stalk erstmalig bei V-ATPasen elektronenmikroskopisch dokumentiert (Boekema et al., 1997), was bald danach zu seiner Entdeckung bei F-ATPasen führte (Wilkens & Capaldi, 1998). Des weiteren wurde die Untereinheit a bisher als Favorit für die Bindung von Plecomakroliden gehandelt (Zhang et al., 1994), wobei dies in dieser Arbeit allerdings widerlegt werden konnte. Die besondere Proteolyse-Empfindlichkeit der Untereinheit a war zwar bekannt (Adachi et al., 1990a), so dass ein Abbau des Proteins während der Reinigung nach Schweikl et al. (1989) durchaus möglich gewesen wäre, allerdings konnte selbst die Verwendung eines ganzen Cocktails von Proteaseinhibitoren (Wieczorek et al., 2000) die scheinbare "Degradation" nicht verhindern. Mit der Erhöhung der Ionenstärke im Saccharosedichtegradienten durch 200 mM KCl gelang es nun erstmals, das V1Vo Holoenzym mit seiner Untereinheit a zu reinigen. Offensichtlich bewirkt die höhere Ionenstärke der Lösung durch verstärkte hydrophobe Wechselwirkungen eine festere Bindung der Untereinheit a an den Rest des V1Vo Holoenzyms. An welche der Untereinheiten der V-ATPase die Untereinheit a fester bindet bzw. mit welcher der Untereinheiten sie überhaupt interaggiert, ist für M. sexta bisher noch nicht untersucht. Auch 96 Diskussion bei anderen V-ATPasen sind die Wechselwirkungen mit den anderen Untereinheiten nicht vollständig geklärt. Bei Hefe sind allerdings Interaktionen der Untereinheit a mit der Untereinheit d, c, A und H beschrieben (Tomashek et al., 1996; Landolt-Marticorena et al., 2000). Mittlerweile ist die Untereinheit a aus dem Mitteldarm kloniert und sequenziert (Merzendorfer et al., 2000), eine weitere Isoform wurde in der cDNA von Malpighischen Gefäßen gefunden und teilweise kloniert und sequenziert (Reineke, Merzendorfer, Vor der Brüggen und Wieczorek, nicht publiziert). Bei Hafer konnte die Untereinheit a bisher ebenfalls nicht als Bestandteil des gereinigten V1Vo Holoenzyms nachgewiesen werden, obwohl sie im Vo Komplex, welcher nativ in der Membran vorkommt, enthalten ist (Li & Sze, 1999). Da das Reinigungsprotokoll für die VATPase aus Hafer keine Salze enthält wäre es interessant zu überprüfen, ob sich bei Erhöhung der Ionenstärke auch bei dieser V-ATPase der gleiche Effekt einstellt. Neben der modifizierten Reinigung des ganzen V1Vo Holoenzyms wurde auch ein Protokoll zur Reinigung des nativen Vo Komplexes etabliert. Dabei wurde die Anreicherung des freien Vo Komplexes in der GCAM durch die Abdissoziation des V1 Komplexes bei hungernden Raupen ausgenutzt (Gräf et al. 1996) und gezeigt, dass neben dem cytosolischen V1 Komplex auch der membranständige Vo Komplex als ganzes erhalten bleibt und mit einer hohen Ausbeute 50 µg/Raupe isoliert werden kann. Durch die hohe Reinheit der Präparation war es möglich, die genaue Untereinheitenzusammensetzung des Vo Komplexes, aus den Untereinheiten a, c, d und e zu bestimmen. Wie bereits für das V1Vo Holoenzym ausgeführt, gelang es auch hier erstmalig die Untereinheit a als konstitutiven Bestandteil des Vo Komplexes einer Insekten V-ATPase darzustellen. Die Untereinheit d, die bereits als Bestandteil des Vo Komplexes aus Clathrinvesikeln von Rindern etabliert war (Zhang et al., 1992; Zhang et al., 1994) konnte auch für die Insekten VATPase nachgewiesen werden (Merzendorfer et al., 1997b). Diese Untereinheit besitzt im Gegensatz zu den anderen Vo Untereinheiten keinen Membrandurchgang (Merzendorfer et al., 1997b) und wird nur durch ihre Wechselwirkungen mit den anderen Untereinheiten an den Vo Komplex gebunden, wo sie jedoch eine wichtige Rolle bei der Assemblierung des V1Vo Holoenzyms spielt (Bauerle et al., 1993). Als neuer konstitutiver Bestandteil des nativen Vo Komplexes konnte die Untereinheit e erstmalig bei M. sexta nachgewiesen werden (Merzendorfer et al., 1999); ihre Entdeckung sicherte ab, dass ein kurz zuvor durch Co-Reinigung mit der Rinder V-ATPase gefundenes Protein auch dort als V-ATPase-Untereinheit angesehen werden kann (Ludwig et al., 1998). Die homologe Untereinheit in Hefe (VMA21) gehört nicht zur reifen V-ATPase, sondern ist 97 Diskussion nur während der Assemblierung im Endoplasmatischen Retikulum mit dem Vo Komplex assoziiert. Im Gegensatz zur Untereinheit e von M. sexta wird sie dort durch ein Retentionssignal zurückgehalten und weist auch keinerlei Glycosylierungen auf, die bei M. sexta immerhin 10 kDa zur apparenten Molekularmasse von 20 kDa beitragen. Die Untereinheit c konnte durch die Autoradiographie nach Inkubation mit 14 C-DCCD und durch die MALDI-MS-Analysen ebenfalls eindeutig dem Vo Komplex zugeordnet werden. Bei vielen anderen Spezies, z. B. S. cerevisiae, C. elegans und H. sapiens konnten inzwischen neben c auch dessen Isoformen c´ und c´´ gefunden werden, wobei die Isoform c´ gleich groß wie c ist, die Isoform c´´ größer ist (19-23 kDa) und wahrscheinlich einen Membrandurchgang mehr besitzt (Hirata et al., 1997; Oka et al., 1997; Nishigori et al., 1998). Für S. cerevisiae konnte gezeigt werden, dass zum einen jede der 3 Isoformen für die richtige Funktion eines V-ATPase-Moleküls exprimiert werden muß (Hirata et al., 1997) und dass zum anderen in jedem Vo Komplex jeweils eine Untereinheit c´ und c´´, sowie mehrere Untereinheiten c vorhanden sind (Powell et al., 2000). Bei M. sexta wurden diese Isoformen bisher vergeblich gesucht, obwohl es zumindest nach Ergebnissen von Northern-BlotHybridisierungen zwei Transskripte unterschiedlicher Größe für das Proteolipid gibt (Dow et al., 1992). Die Bande mit der apparenten Molekularmasse von 26 kDa, die als einziges unbekanntes Protein im gereinigten Vo Komplex zu erkennen ist erschien aus naheliegenden Gründen als guter Kandidat für die Isoform c´´. Dies erschien insbesondere plausibel, da bei früheren Versuchen, die Untereinheit c in der GCAM mit 14 C-DCCD zu markieren, auch ein Signal bei 26 kDa zu finden war (Sumner et al., 1995). Bei der N-terminalen Sequenzierung dieser Bande stellte es sich dann aber heraus, dass die erhaltene Sequenzinformation genau dem N-Terminus der Untereinheit c entspricht. Bei einem Vergleich mit mehreren Isoformen aus anderen Organismen zeigte sich, dass alle Isoformen der Kategorie c´´ ihren Größenunterschied zu c und c´ hauptsächlich durch eine Verlängerung des n-Terminus erhalten. Deshalb ist es sehr unwahrscheinlich, dass es sich bei der 26 kDa Bande um c´´ handelt. Für die Untereinheit c ist andererseits bekannt, dass sie beim Erhitzen der Proben für die SDS-PAGE sehr leicht Aggregate bildet, die sehr groß werden und dadurch eine Einwanderung ins Gel verhindern können (Finbow & Pitts, 1993; Lepier et al., 1996). Um eine Aggregation zu vermeiden, werden die Proben des V1Vo Holoenzyms und des Vo Komplexes in unserem Labor standardmäßig entweder nur 45 s auf 95°C erhitzt oder für 30 min bei 37°C erwärmt. Da das Vermeiden der Aggregation trotzdem nicht vollständig zu gelingen scheint, dürfte es sich bei der 26 kDa Bande mit ziemlicher Sicherheit um eine Dimer der Untereinheit c handeln. Nichtsdestotrotz gibt es, neben den bereits erwähnten zwei 98 Diskussion Produkten aus der Northern-Blot-Hybridisierung, einen weiteren Hinweis auf die Existenz einer Isoform c´ der Untereinheit c. Bei allen MALDI-MS-Analysen der Untereinheit tauchten reproduzierbar zwei markante chymotryptische Massenfragmente auf (1523,9 und 1739,9 m/z), die nicht in der Sequenz von c wiederzufinden sind. Dabei könnte es sich um Fragmente der Isoform c´ handeln, welche normalerweise ca. die gleiche Molekularmasse wie c hat und deshalb in der normalen SDS-PAGE nicht von dieser zu unterscheiden ist. 2-D Gelelektrophoresen, wie für den löslichen V1 Komplex, waren sowohl mit dem V1Vo Holoenzym, als auch mit dem Vo Komplex bisher nicht erfolgreich. Einen neuen Ansatz bietet die trägerfreie isoelektrische Fokussierung mit dem Octopussystem der Firma Dr. Weber, welches gerade für Membranproteine besonders geeignet ist. 5.1.3. Der V1 Komplex Auch die Reinigung des cytosolischen V1 Komplexes wurde im Vergleich zu Gräf et al. (1996) modifiziert, so dass zum einen die Stabilität und Aktivität des Komplexes über mehrere Wochen konserviert werden kann, und zum anderen eine größere Menge an Raupen bei einer Präparation verarbeitet werden konnte, was die Menge an gereinigtem V1 Komplex pro Präparationstag erhöht. Als entscheidend für die Erhaltung der Aktivität erwies sich die Zugabe eines Reduktionsmittels schon früh während der Präparation, um bei der großen Anzahl an Cysteinen im V1 Komplex die Bildung von Disulfidbrücken zu verhindern, die zu einem deutlichen Aktivitätsverlust der V1-ATPase führen (diese Arbeit; Gräf et al., 1996). Auch konnte inzwischen gezeigt werden, dass die Oxidation des V1 Komplexes zu Änderungen seiner 3D-Struktur führt (Grüber et al., 2000b). Einen weiteren wichtigen Einfluß hatte die Lagerungstemperatur von 4°C, bei der sich die V1-ATPase am stabilsten erwies. Mit Hilfe der 2-D Gelelektrophorese konnte die genaue Zusammensetzung des V1 Komplexes aus den Untereinheiten A, B, C, D, E, F, G und H ermittelt werden, welche auch durch die Ergebnisse von Immunfärbungen mit spezifischen Antikörpern und durch MALDIMS-Analysen bestätigt wurde. Die 2-D Gellektrophorese zeigte weiterhin, dass keine Isoformen von Untereinheiten, die sich in ihren isoelektrischen Punkten unterscheiden, im V1 Komplex enthalten sind. 99 Diskussion 5.1.4. Die putative Isoform der Untereinheit B gehört zu den Hsp60-Proteinen Für B Untereinheiten von V-ATPasen sind in mehreren Organismen Isoformen, die in unterschiedlichen Organen bzw. Zellkompartimenten exprimiert werden, beschrieben (Wang & Gluck, 1990; Van Hille et al., 1994; Bartkiewicz et al., 1995). Bei der Verwendung der zwei monospezifischen Antiseren C23 und C34, welche gegen die Aminosäuren 243-428 bzw. gegen die Aminosäuren 475-494 der Untereinheit B gerichtet waren, wurde überraschenderweise eine Bande B´ zwischen der A und der B Untereinheit mit einer Molekularmasse von 60 kDa erkannt. C23 markierte dabei die Banden B und B´, C34 hingegen nur die Bande B´. Die B´ Bande ist sowohl in der gereinigten V-ATPase aus Mitteldarmrohhomogenat als auch in der gereinigten V-ATPase aus angereicherter GCAM von anteriorem und posteriorem Mitteldarm enthalten, nicht aber in der V-ATPase aus gereinigter GCAM des anterioren und posterioren Mitteldarms und im V1 Komplex. Da die letzteren Präparationen entweder aus reinen Membranen erfolgten oder direkt aus dem Cytosol, lag die Vermutung nahe, dass es sich möglicherweise um eine endosomale Isoform der Untereinheit B handeln könnte, die nicht in der Plasmamembran V-ATPase vorkommt. Eine andere Möglichkeit wäre, dass es sich bei der Untereinheit B in der Plasmamembran VATPase um ein posttranslational C-terminal verkürztes Protein handeln könnte, da es nicht durch das Antiserum C34 gegen diesen Bereich erkannt wird. Bei einer MALDSI-MS Analyse zeigte sich allerdings, dass es sich bei der Bande B um das vollständige Protein einschließlich des C-Terminus handelt. Eine N-terminale Sequenzierung von B´ klärte schließlich dessen Identität als Mitglied der Hsp60 Familie, für welches bereits zusätzliche Sequenzinformation durch ein "shotgun screening" vorhanden war. Dieser Klon 10 aus M. sexta enthält eine mitochondriale Lokalisationsequenz, die im Fall von B´ nicht mehr vorhanden ist; dies spricht dafür, dass es sich tatsächlich in Mitochondrien befindet. Durch die Zerstörung der Mitochondrien bei der Homogenisation des Gewebes wird es freigesetzt und bindet eventuell als Chaperon an beschädigte Teile der V-ATPase und wird deshalb mitgereinigt. Aus der späteren Elution in der Gelchromatographie im Vergleich zum reinen Holoenzym lässt sich schließen, dass es nicht an die intakte bzw. vollständige V-ATPase bindet, sondern an Teilkomplexe die möglicherweise "beschädigt" sind. Es stellt sich nun die Frage, warum Antikörper gegen zwei verschiedene Teilbereiche der B-Untereinheit dieses Hsp60 erkennen. Hierfür gibt es zwei mögliche Erklärungen. 1. Bei der Reinigung der Fusionsproteine für die Herstellung der Antiseren wurde aus den zur Expression verwendeten 100 Diskussion E. coli Zellen GroEL, ebenfalls ein Mitglied der Hsp60 Familie, mitgereinigt. Deshalb produzierten die zur Immunisierung verwendeten Meerschweinchen Antikörper gegen GroEL, welche das Hsp60 aus M. sexta erkennen. 2. Es ist bekannt, dass es zwischen der Untereinheit der F-ATPasen und Chaperonen konservierte Regionen gibt, welche auch in der B-Untereinheit von V-ATPasen vorhanden sind und dass Antikörper gegen zwei synthetische Peptide dieser Regionen der Untereinheit von F-ATPasen sehr stark mit GroEL kreuzreagieren (Luis et al., 1990, Alconada et al., 1994). Einige dieser konservierten Motive kommen sowohl bei der B-Untereinheit von M. sexta als auch bei Hsp60 Proteinen der Insekten vor (nicht gezeigt, hier wurde mit Hsp63 von H. vireszens verglichen da nur ein Teil der M. sexta Sequenz bekannt ist), so dass auch hier eine Kreuzreaktion auftreten könnte. 5.1.5. Gehört die Untereinheit C zum V1 Komplex? Bei Gräf et al. (1996) enthielten die gereinigten cytosolischen V1 Komplexe ebensowenig eine Untereinheit C, wie die V1 Komplexe aus S. cerevisiae (Kane, 1995) und die gestrippten V1 Komplexe aus chromaffinen Granula von Rind (Moriyama & Nelson, 1989). Allerdings ist die Untereinheit C in den nach der modifizierten Methode gereinigten V1 Komplexen, wenn auch in substöchiometrischen Mengen, enthalten. Sie läßt sich aber durch die Zugabe von Alkoholen sehr leicht vom V1 Komplex abtrennen, weshalb zunächst vermutet wurde, dass dadurch die Mg-ATPase-Aktivität des V1 Komplexes hervorgerufen wird. Der C-freie Komplex zeigt aber in wässriger Lösung ebenfalls nur Ca-ATPase- und keine Mg-ATPaseAktivität. Die rekombinante Untereinheit C reassoziiert problemlos mit dem C-freien Komplex und dieser hat die gleichen enzymatischen Eigenschaften wie der C-freie oder der native C-haltige V1 Komplex. Dies scheint dem Ergebnis von Peng et al. (1993) zu widersprechen, welche bei Clathrinvesikeln feststellten, dass durch Zugabe der rekombinanten Untereinheit C zu einem Subkomplex aus A, B und D dessen Ca-ATPaseAktivität um ein 15-faches auf ein Drittel der Aktivität des intakten V1 Komplexes anstieg. Dieser Subkomplex wurde allerdings unter sehr harschen Bedingungen hergestellt, so dass die Aktivitätssteigerung möglicherweise durch die Wirkung der Untereinheit C als Stabilisator oder auch Chaperon für diesen Subkomplex zurückzuführen ist und er dadurch erst in eine aktivere Konformation gebracht wird. Der V1 Komplex aus M. sexta scheint hingegen mit und ohne die Untereinheit C bereits in seiner richtigen Konformation zu sein. Eine mögliche Antwort auf die Frage der Rolle der Untereinheit C ergab sich aus dem Befund, dass sie sich mit Alkoholen nicht vom V1Vo Holoenzym entfernen läßt. Dagegen 101 Diskussion reichen bereits 0,01% des Detergens C12E10, welches standardmäßig zur Solubilisierung und Reinigung des V1Vo Holoenzyms benutzt wird, aus, um sie vom V1 Komplex abzulösen. Dies zeigt, dass es einen großen Unterschied in der Wechselwirkung von Untereinheit C mit dem V1Vo Holoenzym und dem V1 Komplex gibt. Es wurde bereits gezeigt, dass die Untereinheit C zwar nicht für die Assemblierung der Komplexe V1 und Vo notwendig ist, wohl aber für die Assemblierung der Beiden zum intakten Holoenzym (Beltran & Nelson, 1992; Ho et al., 1993; Doherty & Kane, 1993). Auch bei der glukoseabhängigen Dissoziation der V-ATPase in Hefe ist die Untereinheit C weder im V1- noch im Vo-Komplex zu finden, bei einer Reassoziation ist sie allerdings wieder im V1Vo Holoenzym zu finden. Deshalb ist es sehr wahrscheinlich, dass bedingt durch die einzige bisher nachgewiesene Aufgabe der Untereinheit C, nämlich bei der Assemblierung des V1Vo Holoenzyms mitzuwirken, ihre Lokalisation am Interface zwischen den beiden Komplexen zu finden ist, wo sie möglicherweise durch die Untereinheit a "eingeklemmt" wird (siehe Modell Abb. 3). Die Anwesenheit im gereinigten V1-Komplex von M. sexta ist vielleicht nur auf eine präparationsbedingte hohe Affinität der Untereinheit C zum V1 Komplex zurückzuführen, was auch die leichte Reassoziation der rekombinanten Untereinheit belegt. 5.1.6. Ist die Untereinheit E bei V-ATPasen das funktionelle Homolog zur Untereinheit der F-ATPasen? Bei der Behandlung des V1 Komplexes mit chaotropen Ionen zerfiel dieser in zwei Subkomplexe bestehend aus den Untereinheiten AB2HCEG und A2BEG mit einem Molekulargewicht von ca. 310 bzw. 230 kDa. Keiner der beiden Komplexe zeigte eine Caoder Mg-ATPase-Aktivität und in keinem der beiden Komplexe waren die Untereinheiten D und F enthalten. Es sieht so aus, als ob der V1 Komplex durch KI in zwei asymmetrische Hälften gespalten würde, die zusammengesetzt fast wieder einen ganzen Komplex von ca. 540 kDa ergeben. Für die F Untereinheit ist bekannt, dass sie leicht vom V1 Komplex abzulösen und auch wieder zu reassozieren ist (Gräf et al., 1994b; Gräf et al., 1996). Ausserdem gibt es Hinweise darauf, dass die beiden Untereinheiten D und F miteinander interagieren (Thomashek et al., 1997). Interessant war jedoch das Verschwinden der D Untereinheit bei gleichzeitigen Verbleiben der E Untereinheit an den Komplexen, da bei den V-ATPasen immer noch sehr kontrovers diskutiert wird, welche dieser beiden Untereinheiten das Homologe für die Untereinheit der F-ATPasen darstellt (Bowman et al., 1995; Nelson et al., 1995; Xu & Forgac, 2000; Grüber et al., 2000a). Im aktuellen Struktur-Funktions-Modell für die F-ATPasen (Review in Stock et al., 2000), das zum einen durch hochauflösenden 102 Diskussion Strukturdaten (Abrahams et al., 1994; Bianchet et al. 1998; Stock et al., 1999; Hausrath et al., 1999; Gibbons et al., 2000) und zum anderen durch Experimente, bei denen die Rotation der Untereinheit relativ zur und Untereinheit gezeigt wurde (Duncan et al., 1995; Sabbert et al., 1996; Noji et al., 1999), gestützt wird, übernimmt die langgestreckte Untereinheit der FATPase die wichtige Aufgabe, die bei der Katalyse von ATP entstehende Bewegung im hexagonalen Kopfteil der F1-ATPase zum für die Protonentranslokation verantwortlichen FoTeil zu vermitteln. Beide Untereinheiten der V-ATPase haben keinerlei auffällige Sequenzhomologie zur Untereinheit der F-ATPasen, aber für beide wird eine langgestreckte Struktur aus -Helices vorhergesagt, wodurch zumindest diese Anforderung erfüllt wäre. Für die Untereinheit E spricht weiterhin ihre Stabilität gegenüber der Trypsinisierung, des V1 Komplexes und des V1Vo Holoenzyms und der Behandlung durch chaotrope Ionen, bei denen die D Untereinheit als eine der ersten verschwindet; dabei bleibt die durch Methanol induzierte Mg-ATPase-Aktivität des V1 Komplexes nichtsdestotrotz erhalten (Grüber et al., 2000a; Reineke, Hunke, Huss und Wieczorek, unpublizierte Ergebnisse). Auch ein Komplex aus den Untereinheiten A, B, C und E der Clathrinvesikel aus Rind zeigt ohne die Untereinheit D eine Ca-ATPase-Aktivität (Peng et al., 1993). Der schnelle Abbau der D Untereinheit deutet, genau wie deren herauslösen aus dem V1 Komplex durch chaotrope Ionen, auf eine eher exponierte Position hin, die sich eigentlich nicht mit einer für ein Homolog a priori zu erwartenden geschützten Lage im Inneren des Enzyms vereinbaren läßt. Weiterhin konnten für die E Untereinheit in Abhängigkeit von zugesetzten Nukleotiden verschiedene Nulllängen-Vernetzungsprodukte entdeckt werden, was auf eine Bewegung der E Untereinheit bei der Hydrolyse, evtl. eine Rotation, hindeutet (Grüber et al., 2000a). Auch ist bei einer Pflanzen-V-ATPase die Entstehung einer Disulfidbrücke innerhalb der E Untereinheit, durch Zugabe eines ATP-Analogons und einer dadurch verursachten Konformationsänderung beobachtet worden (Kawamura et al. 2001). Eine Studie allerdings, bei der die Untereinheit D entweder zufällig oder gerichtet mutiert wurde, zeigte bei vollständig assemblierten V-ATPasen einen eindeutigen Zusammenhang zwischen der Untereinheit D, der ATPase-Aktivität und dem Protonentransport, was wiederum diese als Homolog favorisiert (Xu & Forgac, 2000). Unter diesem Gesichtspunkt können die Ergebnisse der KI-Behandlung auch folgendermaßen interpretiert werden: Die Untereinheit D muß als rotierendes Element leicht drehbar sein und sollte deshalb keine allzu starken Wechselwirkungen, z. B. hydrophober Natur zu anderen Untereinheiten eingehen können. Da der V1 Komplex durch KI "in der Mitte" gespalten wird, was aus den entstehenden 103 Diskussion Fragmenten AB2HCEG und A2BEG ersichtlich ist, könnte in diesem Fall die Untereinheit D einfach aus dem Komplex "herausfallen" (Abb. 63). Ausserdem ist die Untereinheit E in beiden Subkomplexen der KI-Spaltung enthalten und somit möglicherweise mehr als einmal pro Enzym vorhanden. Dies spricht ebenfalls gegen ihre Funktion als Pendant zur Untereinheit, welche in der F-ATPase als einzelne Kopie vorliegt. Auch die ATPase-Aktivität des V1 Komplexes ohne D darf nicht überbewertet werden, da die Ca-ATPase-Aktivität im V1Vo Holoenzym als wahrscheinliche uni-site Katalyse sowieso keinen Protonentransport bewirkt und deshalb auch kein -Homolog erforderlich ist. Möglicherweise handelt es sich auch bei der Methanol stimulierten Mg-ATPase-Aktivität um eine solche uni-site Katalyse. Eine vollständige Klärung, welche der V-ATPase Untereinheiten nun wirklich das Homolog zur Untereinheit der F-ATPasen ist, wird erst durch eine Struktur der V-ATPase in atomarer Auflösung möglich sein. Abb. 63: Modell der Spaltung des V1 Komplexes durch KI. 5.1.7. Die räumliche Struktur des V1 Komplexes Bislang kamen vor allem zwei bildgebende Verfahren, die Röntgenkleinwinkelstreuung und die Elektronenmikroskopie, für die Analyse der räumlichen Struktur des V1 Komplexes zum Einsatz (Svergun et al., 1998; Radermacher et al., 1999; Grüber et al., 2000a; Radermacher et al., 2001). Die erste Methode hat, trotz der relativ geringen Auflösung, den entscheidenden Vorteil, dass sie mit einem unfixierten Objekt arbeitet und damit die erhaltenen Bilder die Struktur unter physiologischen Bedingungen widerspiegeln. In diesen Röntgenkleinwinkel104 Diskussion streuungsexperimenten wurde die Struktur des V1 Komplexes mit einer Auflösung von 3,6 nm bestimmt. Der V1 Komplex hat die Form eines Pilzes, wobei der Durchmesser des Kopfes ca. 14,5 nm beträgt und der Stiel eine Länge von 11 nm hat. Der Kopfteil zeichnet sich ausserdem durch eine dreifache Symmetrie aus, die sich mit der alternierenden Anordnung der jeweils drei Kopien der A und der B Untereinheit erklären läßt, wie sie auch für die und -Untereinheiten der F1-ATPase beobachtet wurde (Abrahams et al., 1994). Die Messung von Konformationsänderungen im V1 Komplex mit der Röntgenkleinwinkelstreuung ist möglich, wie bereits durch eine Veränderung der Struktur in Abhängigkeit vom RedoxZustand gezeigt werden konnte (Grüber et al., 2000b). Um mehr Details über den löslichen V1 Komplex und im besonderen über die Feinstruktur des Stiels zu erhalten, wäre auf jeden Fall eine Untersuchung des vollständig C-freien V1 Komplexes und eine Analyse der durch KIBehandlung entstehenden asymmetrischen Hälften des V1 Komplexes sehr interessant. Die mit Hilfe der Röntgenkleinwinkelstreuung gewonnenen Daten decken sich sehr gut mit den für die V1-ATPase (Radermacher et al., 1999; Grüber et al., 2000a; Radermacher et al., 2001) und das Holoenzym (Abb. 2) aus M. sexta durch Elektronenmikroskopie erhaltenen. Auch sie bestätigen die bereits aus anderen Organismen, wie N. Crassa und C. fervidus bekannten Strukturmerkmale der V-ATPasen (Dschida & Bowman, 1992; Boekema et al., 1997, 1998 und 1999). In der Aufsicht zeigen sie wiederum eine hexagonale Anordnung der Untereinheiten A und B, und in der Seitenansicht die Unterteilung in Kopf und Stiel. Auch die zusätzlichen Massen auf beiden Seiten des Stiels sind zu erkennen, welche in elektronenmikroskopischen Bildern mit höherer Auflösung als zweiter bzw. dritter Stiel der V-ATPase interpretiert wurden (Boekema et al., 1999). Zumindest einer dieser zusätzlichen Stiele ist nach dem vorgeschlagenen Rotor- und Stator-Modell für die Funktionsweise der VATPase in Analogie zur F-ATPase notwendig (Junge et al., 1997; Junge, 1999). Eine Besonderheit der V-ATPase von M. sexta gegenüber V-ATPasen aus anderen Organismen ist die hohe Dichte in der sie in die GCAM integriert ist, wie elektronenmikroskopische Bilder eindrucksvoll zeigen (Abb. 2). In der chromaffinen Granula aus Rindern sind es nur ca. 20 V-ATPasen pro Organelle und in den Clathrinvesikeln vermutlich nur 2 pro Vesikel und auch in der Vakuolenmembran von N. crassa sind es deutlich weniger als in der GCAM von M. sexta (Schmidt et al., 1982; Forgac et al., 1984; Bowman et al., 1989). Die hohe, quasi kristalline Dichte der V-ATPase in der GCAM soll in einer Zusammenarbeit mit der Arbeitsgruppe von Prof. Engel (Biozentrum Basel) dazu genutzt werden, mit Hilfe der Kraftmikroskopie und der Elektronenkristallographie die Struktur der V-ATPase in ihrer nativen Membran weiter zu untersuchen. Vor allem soll dabei 105 Diskussion auch die Struktur des Vo Komplexes, von dem es bis jetzt so gut wie keine Daten gibt, mit einbezogen werden. Zum einen können dafür sogenannte right-side-out Vesikel und zum anderen mit Hilfe von KI vom V1 Komplex befreite Vesikel verwendet werden. 5.2. Die Aktivität der V-ATPase 5.2.1. Ca2+ ist kein vollwertiger Ersatz für Mg2+ In der vorliegenden Arbeit konnte gezeigt werden, dass die Aktivität der V-ATPase von M. sexta deutliche Unterschiede in Hinblick auf ihre Wechselwirkung mit Mg2+ bzw. Ca2+ aufweist. Messungen der Enzymaktivität sowie Protonentransport- und Spannungsmessungen ergaben, dass die Hydrolyse von Ca-ATP im V1 Komplex, im Gegensatz zur Hydrolyse von Mg-ATP, nicht zu einer Kommunikation mit dem Vo Komplex führt, die in eine Protonentranslokation mündet. Auch für die V-ATPase aus Clathrinvesikeln war gezeigt worden, dass Ca2+ nicht als Ersatz für Mg2+ verwendet werden kann, da die Hydrolyse von Ca-ATP dort ebenfalls keinen Protonentransport antreibt (Xie & Stone, 1988). In dieser Arbeit wurde, neben der Aufgabe des Mg2+ als Co-Faktor für die ATP-Hydrolyse, eine weitere Funktion des Mg2+ als Koppler zwischen Hydrolyse und Protonentransport und eine zweite Bindestelle für Mg2+ diskutiert. Begründet wurde dies durch den 500-fachen Konzentrationsunterschied zwischen der geringen, zur Hemmung der Ca-ATPase-Aktivität und der hohen, für eine optimale Mg-ATPase-Aktivität benötigten Mg2+-Konzentration. Auch in zwei weiteren Untersuchungen wurde eine mögliche zusätzliche Mg2+-Bindestelle bei VATPasen postuliert. Zum einen bei Markierungsversuchen mit -[32P]ATP bei der V-ATPase aus chromaffinen Granula deutlich mehr Mg2+ benötigt als für die Bildung von Mg-ATP als Substrat erforderlich war (Moriyama & Nelson, 1987). Zum anderen erhielten David & Baron (1994) sowohl für die Osteoklasten als auch für die Nieren V-ATPase einen jeweils 2-fach höheren Km-Wert für Mg2+, als nach der Bestimmung des Km-Wertes für ATP zu erwarten gewesen wäre. Auch bei F-ATPasen konnte ein ähnlich unterschiedlicher Einfluß der beiden divalenten Kationen auf die Aktivität des Enzyms festgestellt werden. Bei mitochondrialen FATPasen bewirkt Ca2+ zwar ebenso wie Mg2+ eine ATP-Hydrolyse, nicht aber einen Protonentransport (Pullman et al., 1960). Desweiteren konnte gezeigt werden, dass Mg2+ vermutlich bei der Dissoziation und Reassoziation von F1- und F0 Komplex bei E. coli eine Rolle spielt (Vogel & Steinhart, 1976). 106 Diskussion Obwohl es diesen entscheidenden Unterschied zwischen den beiden Kationen gibt, kann die genauere Betrachtung der Ca-ATPase-Aktivität wichtige Erkenntnisse über Teilschritte der ATP-Hydrolyse liefern. Dies gilt im besonderen für Untersuchungen am isolierten V1 Komplex, der in wässriger Lösung nur als Ca-ATPase aktiv ist. Hier konnten bereits mit Hilfe von CuCl2-Vernetzungsexperimenten wechselnde Interaktionen zwischen einzelnen Untereinheiten, in Abhängigkeit von Ca-ATP und Ca-ADP, gefunden werden (Grüber et al., 2000a). 5.2.2. Warum ist der isolierte V1 Komplex inaktiv? Der gereinigte cytosolische V1 Komplex hat eine Ca-abhängige ATPase-Aktivität und keinerlei Mg-ATPase Aktivität. Die Ca-ATPase-Aktivität wird sogar schon durch 0,1 mM Mg2+ vollständig inhibiert (Gräf et al., 1996). Durch die Zugabe von Methanol läßt sich allerdings eine Mg-abhängige Hydrolyse von ATP induzieren (Gräf et al., 1996). Diese methanolabhängige Mg-ATPase-Aktivität des V1 Komplexes ist vollkommen linear und unabhängig vom produzierten oder vorgelegten ADP, ähnlich wie es z. B. für den mitochondrialen F1-Komplex aus Rinderherz beschrieben ist (Ortiz Flores et al., 1982). Bei der Überprüfung, ob dieser Effekt auch mit anderen Alkoholen erreicht werden kann, zeigte sich, dass mit zunehmender Kettenlänge weniger Volumenanteil der Alkohole zugesetzt werden muß, um eine optimale Aktivität zu erhalten. Diese Reihenfolge entspricht der Aktivierung der Mg-ATPase-Aktivität der CF1-ATPase durch Alkohole (Kneusel et al., 1982; Mochimaru & Sakurai, 1998), was auf einen vergleichbaren Aktivierungsmechanismus bei den CF1- und den V1 Komplexen schließen läßt. Allerdings bewirkt Methanol bei der CF1ATPase die Freisetzung von festgebundenen Nukleotiden (Anthon & Jagendorf., 1984), während beim V1 Komplex keine Veränderung des Gehaltes an festgebundenem ADP gemessen werden konnte. Aus diesem Grunde kann die Stimulierung der Mg-abhängigen ATPase-Aktivität durch die Freisetzung eines endogenen inhibitorischen ADPs aus dem Komplex ausgeschlossen werden. Bis auf die Restablösung, der nur leicht assoziierten und substöchiometrisch vorhandenen Untereinheit C, deren An- oder Abwesenheit keinerlei Einfluß auf die Aktivität hat, wurde keine weitere Untereinheit durch die Inkubation mit Alkoholen aus dem Komplex entfernt. Auch die Untereinheit H verbleibt im V1 Komplex, was insofern von Bedeutung ist, da für den V1 Komplex einer Vma13p Mutante der Hefe eine geringe, Methanol-unabhängige Mg-ATPase-Aktivität (0,265 U/mg) beschrieben wurde, die sich durch Zugabe von Methanol verdoppeln läßt (Parra et al., 2000). Eine Aktivierung 107 Diskussion des Komplexes durch die Entfernung einer inhibitorischen Untereinheit, analog zur Entfernung der Untereinheit der CF1-ATPasen (McCarty, 1992), ist deshalb auszuschließen. Für die vollständige Inhibierung der Mg-ATPase-Aktivität des V1 Komplexes in wässriger Lösung, d. h. in Abwesenheit von Methanol, bieten sich zwei Alternativen an. Zum einen könnte bei einer Inkubation mit Mg2+ und ATP genau ein Hydrolyseschritt stattfinden, wobei das entstandene Mg-ADP das Enzym sofort blockiert. Dies bedeutet, dass nach der Inkubation mit Mg2+ und ATP zusätzliche Nukleotide im V1 Komplex enthalten sind. Falls sich diese durch Methanol wieder entfernen lassen, wäre die Wirkungsweise geklärt. Zum anderen könnte sich bei Zugabe von Mg2+ ein inhibierender Komplex aus Mg2+ und bereits gebundenem ADP bilden, der die Hydrolyse des beim Reaktionsstarts zugegebenen TrisATPs verhindert. Bei einem von endogenen Nukleotiden befreiten V1 Komplex sollte sich dann Hydrolyse-Aktivität nachweisen lassen. Eine vergleichbare Wirkung von Mg2+ wurde bei der F1-ATPase und bei der CF1-ATPase nachgewiesen (Drobinskaya et al., 1985; Carmeli et al., 1981; Digel et al., 1996). Bei der CF1-ATPase wird die aus der Entfernung von endogenem Nukleotid resultierende schnellere Startgeschwindigkeit der Hydrolyse durch Wiederbeladen des Enzyms mit Mg-ADP rückgängig gemacht (Digel et al., 1998). Im Gegensatz zur linearen Mg-ATP-Hydrolyse-Aktivität in Gegenwart von Methanol ist die Ca-ATPase-Aktivität des V1 Komplexes einer Endprodukthemmung durch Ca-ADP unterworfen, was durch die starke Hemmung der Hydrolyse nach Vorinkubation mit ADP und die lineare Ca-GTPase-Aktivität bestätigt wird. Auch für den isolierten V1 Komplex aus Hefe ist eine Hemmung der Ca-ATP-Hydrolyse durch ADP und eine lineare Ca-GTPaseAktivität beschrieben (Parra & Kane, 1998). Der verwendete V1 Komplex aus Hefe enthält, im Gegensatz zum V1 Komplex aus M. sexta, keinerlei gebundene Nukleotide. Es handelt sich aber nicht um einen nativ abdissoziierten, sondern einen durch Deletion von Vma3p (Untereinheit c) gewonnenen V1 Komplex. Dieser besteht zwar aus allen Untereinheiten (A, B, D, E, F, G und H), kann aber wegen des defekten Vo Komplexes nicht an die Membran andocken. Somit hat er weder native ATPase-Aktivität, noch eine native Dissoziation hinter sich und enthält möglicherweise deswegen kein Nukleotid. Die V1 Komplexe aus beiden Organismen zeigen aber eine ähnliche Hemmung durch Ca-ADP, weshalb diese sehr wahrscheinlich nicht durch das bei M. sexta bereits endogen vorhandene ADP vermittelt wird, sondern durch ein neu gebundenes Ca-ADP. Dieses besetzt möglicherweise eine der katalytischen Bindestellen und verhindert so weitere Hydrolyse-Aktivität. Für die V1-ATPase aus Thermus thermophilus ist eine derartige Inaktivierung durch gebundenes ADP beschrieben (Yokoyama et al., 1998). Dort liegt der ADP-Gehalt des Enzyms zunächst bei 108 Diskussion weniger als 0,1 mol/mol und steigt während der Hydrolyse bis zur vollständigen Inaktivierung auf 1,5 mol/mol an. 5.2.3. Die V-ATPase-Aktivität wird durch ADP gehemmt Die Hydrolyse-Aktivität des V1Vo Holoenzyms aus M. sexta wird durch Mg-ADP deutlich erniedrigt, wobei man zwischen zwei verschiedenen Arten der Hemmung unterscheiden muß; die Hemmung durch das während der ATP-Hydrolyse gebildete ADP (siehe 5.2.4.) und die Hemmung der Startgeschwindigkeit durch Vorinkubation mit Mg-ADP. Letztere scheinen alle V-ATPasen gemeinsam zu haben, da sie bereits für Vertreter aus verschiedenen Spezies publiziert wurde. So wird die ATP-Hydrolyse der V-ATPase aus chromaffinen Granula durch 100 µM ADP allosterisch gehemmt (Moriyama & Nelson, 1987). Auch für den ATPabhängigen Protonentransport durch die V-ATPasen in der Niere und in den Osteoklasten konnte eine inhibierende Wirkung von ADP auf die Startgeschwindigkeit, mit Ki-Werten von 37 bzw. 17 µM, gezeigt werden (David & Baron, 1994). Weiterhin konnten Webster et al. (1995) einen inhibitorischen Effekt von ADP bei Konzentrationen von 25 bis 200 µM auf die Protonentransportaktivität der V-ATPase in chromaffinen Granula feststellen. In einer späteren Arbeit dieser Autoren gelang es, mit Hilfe von [3H]ADP dessen Bindung an eine hochaffine Bindestelle (Kd=70 nM) nachzuweisen (Webster & Apps, 1996). Für Clathrinvesikel wurde eine bereits bei ADP-Konzentrationen von 0,2 µM beginnende Hemmung des Protonentransport entdeckt (Vasilyeva & Forgac, 1998). Nicht zuletzt hat die V-ATPase aus M. sexta mit einer 50%igen Hemmung der Hydrolyse-Aktivität bei ca. 2 µM ADP eine vergleichbar hohe Empfindlichkeit. Alle diese Ergebnisse sind weitgehend in Einklang mit dem von Webster et al. (1995) aufgrund von kinetischen Daten vorgeschlagenen Modell für die Funktion der V-ATPase. Demnach gibt es für die V-ATPase zwei Konformationszustände, eine katalytisch aktive R-Konformation und einen inaktive TKonformation, mit jeweils drei katalytischen und einer regulatorischen Bindestelle für Nukleotide. ADP bindet dabei bevorzugt an die T-Konformation der V-ATPase und reguliert dadurch die Aktivität des Enzyms. Eine vergleichbare Art der Verzögerung der katalytischen Aktivität gibt es auch bei den FATPasen. Als Ursache für deren Hemmung nach Vorinkubation mit Mg-ADP geht man von der Besetzung der hochaffinen katalytischen Bindestelle durch ein Mg-ADP aus, die ähnlich auch bei der normalen Hydrolyse gemäß dem binding change Mechanismus erfolgt (Vasilyeva et al., 1982; Feldman & Boyer, 1985; Boyer, 1989; Milgrom & Cross, 1993). 109 Diskussion 5.2.4. Mg-ATP spielt eine wichtige Rolle bei der Dissoziation der V-ATPase in den V1 und den Vo Komplex Schon seit geraumer Zeit ist bekannt, dass in vitro bei der membrangebundenen bzw. rekonstituierten V-ATPase der katalytische V1 Komplex, unter dem Einfluß von chaotropen Ionen und/oder von Nukleotiden vom Vo Komplex abgetrennt werden kann, wobei eine Inkubation auf Eis (cold inactivation) die Dissoziation zusätzlich unterstützt (Moriyama & Nelson, 1989; Bowman et al., 1989; Arai et al., 1989; Kane et al., 1989; Adachi et al., 1990b). Dabei wurden die stärksten Dissoziationsraten in Gegenwart von Mg-ATP erreicht. Die Vorinkubation mit NEM, welches die Bindung und Hydrolyse von ATP an die Untereinheit A verhindert, unterbindet auch eine Dissoziation des Holoenzyms (Bowman et al., 1989; Moriyama & Nelson 1987 und 1989). Ausserdem wurde mit Hilfe von nicht hydrolysierbaren ATP-Analoga gezeigt, dass die Bindung von ATP alleine nicht ausreicht, um eine Dissoziation zu induzieren. Deshalb wird vermutet, dass die V-ATPase während der ATP-Hydrolyse einen Konformationszustand durchläuft, der sie anfällig für eine Dissoziation durch chaotrope Ionen macht. Nachdem die Dissoziation der V-ATPase unter in vivo Bedingungen in der Hefe nachgewiesen werden konnte, gibt es einige wichtige Hinweise darauf, dass auch in vivo die ATP-Hydrolyse eine entscheidende Rolle für die Dissoziationsfähigkeit des Enzyms spielen könnte. So sind inzwischen mehrere Hefe-Mutanten bekannt, die nicht mehr in der Lage sind, ATP zu hydrolysieren und offensichtlich als Folge davon auch keine Dissoziation der VATPase nach Glukoseentzug aufweisen (Parra & Kane, 1998; MacLeod et al., 1999). Eine dieser Mutanten ist zwar in der Lage, ATP zu binden, dissoziiert aber trotzdem nicht (MacLeod et al., 1999). Andererseits reagierte ein Hefestamm mit einer Mutation in der Untereinheit D (Vma8), was einen mehr als 90%igen Verlust der V-ATPase-Aktivität und des Protonentransports zur Folge hat genauso wie der Wildtyp mit Dissoziation des Enzyms auf die Entfernung von Glukose aus dem Nährmedium (Xu & Forgac, 2000). Es reicht also schon eine relativ geringe Aktivität der V-ATPase aus, um in vivo die Möglichkeit der Regulation der V-ATPase durch Dissoziation zu erhalten. Die indirekte Vermutung, dass nur wenige Hydrolyseschritte für eine Dissoziation der V-ATPase erforderlich sind, konnte in der vorliegenden Arbeit experimentell bestätigt werden. Allein die Hydrolyse von einem einzigen Mg-ATP pro Holoenzymmolekül scheint vollkommen ausreichend zu sein, die V-ATPase soweit zu destabilisieren, dass eine Dissoziation in V1 und Vo Komplex erfolgt. Auch die 110 Diskussion Inkubation mit hohen Mg-ADP-Konzentrationen löst eine teilweise Dissoziation der VATPase aus (Moriyama & Nelson 1989; Bowman et al., 1989). Für die Dissoziation könnte man sich folgendes Szenario vorstellen. Mg-ATP bindet an die V-ATPase, wird hydrolysiert und das entstehende Mg-ADP wird sofort durch ein neues Mg-ATP ersetzt. Ist aufgrund eines ungünstigen ADP/ATP-Verhältnisses nicht ausreichend ATP vorhanden, verbleibt das MgADP in der Bindetasche. Diese Konformation der V-ATPase ist offensichtlich relativ instabil; das Enzym tendiert damit zur Dissoziation, die umso wahrscheinlicher ist, je länger die instabile Konformation beibehalten wird. Möglicherweise erklärt dies auch den Effekt der cold inactivation bei dem die V-ATPase bei Inkubation mit Mg-ATP auf Eis deutlich schneller dissoziiert als unter gleichen Bedingungen bei Raumtemperatur (Moriyama & Nelson, 1989). Da die ATP-Hydrolyse bei niedrigen Temperaturen langsamer erfolgt ist das Enzym möglicherweise bei jedem Reaktionsschritt länger in einer instabilen Mg-ADPKonformation als bei Raumtemperatur und dissoziiert deshalb leichter. Widersprüchlich sind in diesem Zusammenhang die Befunde in Hinblick auf den ADP-Gehalt der abdissoziierten V1 Komplexe. In den in vitro erhaltenen Komplexen konnte kein ADP nachgewiesen werden, dagegen haben alle nativ von der Membran abdissoziierten V1 Komplexe einen ADP-Gehalt von ca. 1,7 mol/mol. Der ADP-Gehalt im intakten Holoenzym stieg bei den in vitro Versuchen allerdings auf einen ähnlichen Wert von 1,8 mol/mol an. Um die Rolle des ADPs in diesem Zusammenhang zu verstehen, ist es notwendig zu klären, ob 1) ADP in beiden Fällen an der gleichen Stelle gebunden ist, was auf eine gleiche Funktion hinweisen würde oder ob 2) verschiedene Bindestellen vorhanden sind, die unterschiedliche Funktionen implizieren. Beim Holoenzym könnte es sich um einen Sensor für die Energieladung der Zelle handeln, welcher die Dissoziation des V1 Komplexes vom Vo Komplex induziert. Dass die V-ATPase selbst auf wechselnde ATP/ADP-Verhältnisse in der Zelle reagiert, zeigen die Ergebnisse von Dietz et al. (1998). Hier wurde die V-ATPase aus Gerste mit unter verschiedenen Bedingungen in vivo gemessenen ATP/ADP-Verhältnissen inkubiert und die Aktivität bestimmt. Diese korrelierte mit der in vivo gemessenen Ansäuerung der Vakuole unter den selben Bedingungen. Beim V1 Komplex könnte das endogene ADP die Mg-ATPase-Aktivität abschalten, wie es bei T. thermophilus der Fall ist (Yokoyama et al., 1998). Für die Bindung des ADPs bieten sich zunächst natürlich die 3 katalytischen Untereinheiten A an. Nach kinetischen Untersuchungen hat die V-ATPase allerdings neben drei katalytischen Bindestellen auch noch mindestens eine regulatorische Bindestelle (Webster et al., 1995). Es gibt bereits eine ganze Reihe von Befunden, welche belegen dass auch andere Untereinheiten mit Nukleotiden 111 Diskussion interagieren. Die Untereinheit B der V-ATPase aus Hefe kann sowohl mit 2-Azido-ATP als auch mit BzATP markiert werden (Zhang et al., 1995; Vasilyeva & Forgac, 1996). Weiterhin konnte in chromaffinen Granula die Untereinheit D mit radioaktivem ATP markiert werden (Moriyama & Nelson, 1987). Auch konnte kürzlich die Untereinheit E mit 8-N3-3´-biotinylATP markiert werden, wobei diese Markierung allerdings als Folge der engen Nachbarschaft der Untereinheit E, als putatives -Homolog, mit der katalytischen Untereinheit A interpretiert wurde (Schäfer et al., 2001). Da es in einem Vorversuch bereits durch die Verwendung von alkalischer Phophatase gelang, den nativen V1 Komplex deutlich von seinem endogen gebundenen ADP zu befreien und das native Holoenzym quasi frei von Nukleotiden ist sollte es mit den entsprechenden Nukleotid-Sonden möglich sein die Bindestelle des ADPs zu identifizieren. 5.3. Makrolid Antibiotika als Inhibitoren der V-ATPase 5.3.1. Die Identifizierung der plecomakrolidbindenden Untereinheit der V-ATPase Die Plecomakrolide Bafilomycin und Concanamycin, welche von Werner et al. (1983, 1984) erstmals beschrieben wurden, sind seit mehreren Jahren für ihre sehr spezifische und reversible Hemmung von V-Typ ATPasen in nanomolaren Konzentrationen, aber auch für die Hemmung von P-Typ ATPasen in mikromolaren Konzentrationen bekannt (Bowman et al., 1988; Dröse et al., 1993; Dröse & Altendorf, 1997). Über die Art der Inhibierung bzw. die Bindungsstelle der Antibiotika in den Enzymen ist allerdings sehr wenig bekannt. Hanada et al. (1990) berichteten, dass Bafilomycin A1 bei V-ATPasen aus chromaffinen Granula des Rindes mit dem Vo Komplex interagiert, da durch Zugabe von Vo Komplexen zu gereinigtem V1Vo Holoenzym, dessen Hemmung durch Bafilomycin A1 unterdrückt werden konnte, zugesetzte V1 Komplexe dagegen waren wirkungslos. Der Vo Komplex als Zielort der Plecomakrolidhemmung wurde durch zwei weitere Publikationen zur V-ATPase aus Clathrinvesikeln von Rindern bestätigt (Crider et al., 1994; Zhang et al., 1994). Zum einen konnte durch 1 nM Bafilomycin A1 der passive Protonenfluß durch das säureaktivierte rekonstituierte V1Vo Holoenzym und durch den säureaktivierten rekonstituierten Vo Komplex unterbunden werden. Zum anderen wurde die Hemmung des ATP-getriebenen Protonentransports von rekonstituiertem V1Vo Holoenzym durch Bafilomycin A1 untersucht. Durch Zugabe von rekonstituiertem Vo Komplex, rekonstituierter Untereinheit a oder 112 Diskussion gemeinsam rekonstituierten Untereinheiten a und d konnte diese vermindert werden, nicht jedoch durch Untereinheit d alleine oder der Kombination aus c und c´. Daraus schlossen die Autoren auf eine Bindungsstelle für Bafilomycin A1 in der Untereinheit a. Ein Widerspruch ergab sich allerdings aus der Beobachtung, dass Proteoliposomen mit den gemeinsam rekonstituierten Vo-Untereinheiten c, c´ und a, bzw. c, c´, d und a keinerlei Wirkung hatten. Dies läßt auf eine Veränderung der Eigenschaften der Untereinheiten während der Reinigung bzw. der Rekonstitution schließen. Die Ergebnisse aus drei weiteren Untersuchungen deuten darauf hin, dass die Wirkung von Bafilomycinen auf die V-ATPase unabhängig von der Untereinheit a stattfindet. So zeigten Li & Sze (1999), dass bei der gereinigten V-ATPase aus Hafer, welche keine Untereinheit a enthält, die ATP-Hydrolyse durch Bafilomycin inhibiert wird. Ähnliches konnte in Hinblick auf die Enzymaktivität bereits für die gereinigte V-ATPase aus M. sexta gezeigt werden, deren Präparation früher ebenfalls keine Untereinheit a enthielt, die aber trotzdem sensitiv für Bafilomycin war (Wieczorek et al., 1991). Weiterhin beobachten Landolt-Marticorena et al. (2000) eine Hemmung der ATP-Hydrolyse durch Bafilomycin auch nachdem der Protonentransport durch die Trypsinisierung der Untereinheit a von der ATP-Hydrolyse entkoppelt wurde. Einen Hinweis auf die Untereinheit c des Vo Komplexes als möglicher Bindungspartner für Plecomakrolide lieferte eine Affinitätschromatographie mit kovalent gebundenem Bafilomycin C1, welche zur Reinigung der V-ATPase aus Hühnchenosteoklasten eingesetzt wurde (Rautiala et al., 1993). Dabei zeigte die Untereinheit c eine sehr hohe Affinität zur Bafilomycin C1 Säule und eine Vorinkubation mit dem Untereinheit c spezifischen Inhibitor DCCD in einer Konzentration von 1 mM verhinderte die Bindung der V-ATPase an die Säule, woraus auf eine Bindestelle des Bafilomycins in der Untereinheit c nahe der DCCDBindestelle geschlossen wurde. Allerdings bindet DCCD bei dieser hohen Konzentration nicht nur an Carboxylgruppen wie die von Glutamat und Aspartat, sondern auch an Sulfhydrylgruppen und Tyrosin-Seitenketten (Azzi et al., 1984). Deshalb konnte eine Modifikation von Makrolidbindestellen in anderen Untereinheiten bzw. an einer anderen Stelle der Untereinheit c nicht ausgeschlossen werden. Außerdem war zum Zeitpunkt der Publikation die Anwesenheit bzw. Identität der Untereinheit a in der V-ATPase aus Hühnchenosteoklasten noch nicht geklärt, so dass diese möglicherweise nur nicht detektiert werden konnte. Durch ein mit 3H markiertes Derivat des Bafilomycins A1 konnte bei der VATPase aus Hühnchenosteoklasten eine nicht kovalente Bindung an den Vo Komplex nachgewiesen werden (Mattsson & Keeling, 1996), wodurch die direkte Interaktion von 113 Diskussion Plecomakroliden mit dem Vo Komplex endgültig bestätigt wurde. Weiterhin unklar war jedoch, in welcher der Vo Untereinheiten die Plecomakrolidbindestelle sitzt. Diese Frage konnte nun durch die Experimente der vorliegenden Arbeit eindeutig geklärt werden. Wie bereits erwähnt, wurde in einer langjährigen Zusammenarbeit der Arbeitsgruppen von Prof. Altendorf (Universität Osnabrück) und Prof. Zeeck (Universität Göttingen) versucht, die für die Hemmung entscheidenden Strukturelemente der Plecomakrolide zu finden und diejenigen Positionen zu identifizieren, die für eine Modifikation ohne großen Verlust an Spezifität und Hemmung geeignet sind (Dröse et al., 1993; Dröse et al., 2001). Für die biologische Wirksamkeit erwies sich der Makrolactonring als essentiell und der Hemiketalring für eine maximale Hemmung als entscheidend. Die Position C23, an der bei Concanamycin A ein Zuckerrest gebunden ist, der lediglich zu einer höheren chemischen Stabilität der Verbindung beiträgt, konnte ohne Aktivitätsverlust entfernt und modifiziert werden. Ähnliche Untersuchungen an Bafilomycin A1 von Gagliardi et al. (1998, 1999) zeigten ebenfalls, dass diese Position, bei Bafilomycinen handelt es sich um C21, ohne Einfluß auf die Aktivität modifiziert werden kann. Deshalb wurde diese Position mit einem 125 I besetzt, welches zu Detektion mittels Autoradiographie genutzt wurde. Als weitere Stelle, bei der eine Modifikation keinen Einfluß auf die Hemmungseigenschaften hat, bot sich die Position C9 an, deren OH-Gruppe gegen eine UV-aktivierbare Gruppe, ein Trifluormethylaryldiazirin (Brunner et al., 1980) ausgetauscht wurde. Diese Gruppe bildet nach UV-Bestrahlung bei 360 nm ein hochreaktives, aber sehr kurzlebiges Carben, welches mit allen in Proteinen vorkommenden Resten kovalente Bindungen eingehen kann, wobei die Reaktion mit Nucleophilen die bevorzugte ist. Außerdem wurde noch die Position C21 deoxyliert, was sich positiv auf die Stabilität der Verbindung auswirkte. Die daraus resultierende Verbindung 9-O-[p-(Trifluoroethyldiazirinyl)-benzoyl]-21,23-dideoxy-23-epi[125I]Iodo-concanolid A, (J-Concanolid A, Abb. 33) wurde zunächst als nicht radioaktive Verbindung für Hemmstudien des gereinigten V1Vo Holoenzyms eingesetzt und hatte mit einem IC50 von 10-20 µM im Vergleich zu seiner Ausgangssubstanz Concanamycin A mit einem IC50 10 nM zwar eine schlechtere Hemmung, die aber immer noch als ausreichend erachtet wurde. Bei der konzentrationsabhängigen Markierung des V1Vo Holoenzyms mit der Sonde konnte nur die Markierung der Untereinheit c beobachtet werden. Selbst mit einer Konzentration von 100 µM, bei der die ATPase-Aktivität des V1Vo Holoenzyms bis zu 90% gehemmt ist, wurde keine andere Untereinheit markiert. Die direkte Bindung der Sonde an die Untereinheit c wird durch vier weitere Befunde gestützt. Erstens konnte bei Radioaktivitätsmessungen der an Hand der Proteinfärbung identifizierten und aus dem SDS114 Diskussion PAGE Gel ausgeschnittenen Bande eine Zunahme der daran gebundenen Radioaktivität in Abhängigkeit von der eingesetzten Sondenkonzentration gemessen werden. Diese korreliert auch mit dem Grad der Inhibierung des V1Vo Holoenzyms bei den gleichen Konzentrationen. Zweitens wird auch im gereinigten Vo Komplex nur die Bande der Untereinheit c markiert wobei diese Markierung ein, für den Vo Komplex typisches Bild der Untereinheit c, mit einer relative breiten Bande aufweist, die auch in der Proteinfärbung wie in der Autoradiographie mit 14 C-DCCD zu bemerken ist. Drittens wird in den Vesikeln der GCAM, in der sich das V1Vo Holoenzym in seiner nativen Membran mit der originären Lipidzusammensetzung befindet, ebenfalls nur die Untereinheit c markiert. Letztlich konnte die Untereinheit c per MALDI-MS-Analyse der aus dem SDS-PAGE-Gel ausgeschnittenen Banden, die auch Radioaktivität aufwiesen, eindeutig identifiziert werden. In Gegenwart der Sonde konnten kovalente Modifikationen an bestimmten Fragmenten festgestellt werden, die ohne Sonde nicht vorhanden waren. Die Massenzunahme der Fragmente von Aminosäure 8-22, 12-29 und 85-87 um 842 kDa entspricht der Masse des J-Concanolids A, welches seine HI-Gruppe verloren hat. Dieser Verlust ist wahrscheinlich auf die Säureempfindlichkeit der Sonde und die doch sehr niedrigen pH-Werte bei der Gelfixierung und der Vorbereitung für die MALDIMS zurückzuführen. Bei den drei anderen gefundenen Fragmenten, Aminosäure 1-7, 85-87 und 138-151 erhöhte sich die Masse um 972 kDa. Diese Erhöhung ist etwas kritischer zu betrachten, da bei Bindung der ganzen Sonde nur eine Massenzunahme von 970 kDa zu erwarten ist. Möglicherweise ereignet sich eine zusätzliche Modifikation der Sonde, z. B. das Öffnen einer der C-C Doppelbindungen und Anlagerung zweier Protonen. Die Untersuchung einer möglichen Modifikation der Sonde und die sehr zeitaufwendige, genauere Analyse und Sequenzierung der erhaltenen modifizierten Fragmente dauert noch an. Allerdings sprechen mehrere Hinweise dafür, dass es sich bei den Fragmenten tatsächlich um die Umgebung der Bindestelle handelt. Erstens taucht das Fragment 85-87 mit beiden Modifikationen auf. Zweitens überlappen sich die Fragmente 8-22 und 12-29, und das Fragment 1-7 liegt in ihrer direkten Nachbarschaft. Drittens befinden sich die Fragmente 1-7 und 85-87 nach Vorhersagen zur Topologie der Untereinheit c auf der luminalen Seite der Membran und auch die anderen Fragmente sind in der luminalen Hälfte der Membran zu finden (Tab. 9; Findlay et al., 1997). Dagegen war keines der gefundenen Fragmente cytosolisch orientiert. Deshalb kann man vermuten, dass die Bindetasche für J-Concanolid A und somit auch für Concanamycin A auf der luminalen Seite der Membran liegt oder von dort zugänglich ist. 115 Diskussion Tab. 9: Topologie der Untereinheit c. Die Daten für die Untereinheit c von M. sexta wurden aus NiceProt view of SWISS-PROT für Acc. Nr. P31403 übernommen. Aminosäurenummer 1-7 8-30 31-52 53-73 74-92 93-114 115-126 127-152 153-156 Orientierung Lumen Membran Cytosol Membran Lumen Membran Cytosol Membran Lumen Auch die Spezifität der Sonde konnte untermauert werden, da sich die Markierung durch 10 µM J-Concanolid A mit 1 µM vorgelegtem Concanamycin A deutlich abschwächen und durch höhere Konzentrationen (10 µM, 100 µM und 300 µM) ganz verhindern läßt, was eine gemeinsame Bindestelle für Concanamycin A als Ausgangssubstanz und J-Concanolid A als dessen Derivat nahelegt. Für die vollständige Verdrängung der Markierung muß demnach mehr Concanamycin A eingesetzt werden als für die vollständige Hemmung der ATPHydrolyse. Die Ursache dafür ist möglicherweise, dass für eine vollständige Hemmung, ähnlich wie für DCCD gezeigt (Forgac, 1998), nur ein einziges gebundenes Inhibitormolekül pro V-ATPase benötigt wird; im optimalen Fall stünden dem J-Concanolid A noch die 5 weiteren Bindestellen der anderen Untereinheiten c zur Verfügung. Es müssen demgemäß alle sechs Untereinheiten c mit Concanamycin A besetzt sein, bis keine Markierung mehr möglich ist. Neben den Concanamycinen gehören auch die bereits mehrfach erwähnten Bafilomycine zu den Plecomakroliden (Bindseil & Zeeck, 1994). Zwei Vertreter dieser Gruppe, Bafilomycin A1 und Bafilomycin B1, wurden ebenfalls auf ihre Fähigkeit getestet, die V-ATPase zu hemmen bzw. mit J-Concanolid A um die Bindestelle in der Untereinheit c zu konkurrieren. Mit einem IC50 zwischen 10-20 nM lagen sie exakt im Bereich des parallel getesteten Concanamycin A. Dies war zunächst etwas überraschend, da Concanamycin A eigentlich als der potentere Inhibitor für V-ATPasen gilt und der bisher bekannte IC50-Wert für Bafilomycin A1 bei M. sexta auch etwas höher lag (Bowman et al., 1988; Dröse et al., 1993; Wieczorek et al., 1991). Der niedrigere Wert für Bafilomycin A1 liegt wahrscheinlich zum einen an der großen Reinheit der verwendeten Plecomakrolide, die wir direkt von der Arbeitsgruppe von Prof. Zeeck (Göttingen) erhalten hatten und zum anderen an der schon beschriebenen Lagerung der in DMSO gelösten Makrolide bei -70°C und der strikten Vermeidung von 116 Diskussion Gefrier-Auftau-Zyklen, besonders bei den instabileren Bafilomycinen. Gleiche Empfindlichkeit der V-ATPase gegenüber allen drei verwendeten Plecomakroliden kommt nicht nur bei M. sexta vor sondern ist auch für Mesembryanthemum crystallinum beschrieben worden (Dröse et al., 2001). Die Unterschiede zur V-ATPase von N. crassa könnten sich durch unterschiedliche Affinitäten erklären lassen; die höhere Flexibilität des mit 18 CAtomen größeren Lactonrings der Concanamycine erlaubt gegenüber dem steiferen 16 CRing der Bafilomycine eine leichtere Anpassung an eine von Spezies zu Spezies möglicherweise verschieden gestalteten Bindungstasche (Kinashi et al., 1984; Dröse & Altendorf, 1997). Auf jeden Fall konnten beide verwendeten Bafilomycine die Markierung der Untereinheit c durch J-Concanolid A unterbinden, was verallgemeinernd einen Schluss auf eine gemeinsame Bindestelle in der Untereinheit c für Plecomakrolide zulassen dürfte. Leider gibt es außer bei den drei erwähnten Organismen offensichtlich keine vergleichenden Untersuchungen zur Hemmung von Bafilomycinen und Concanamycinen. Bei einem Sequenzvergleich der Untereinheiten c dieser drei Spezies, zeigen sich jedoch gemeinsame Unterschiede von M. sexta und M. crystallinum zu N. crassa, die evtl. zu einer veränderten Bindetasche führen könnten (Abb. 64). Von den 10 gefundenen Aminosäureaustauschen sind 5 neutral, bei den anderen 5 handelt es sich um Wechsel von polaren zu hydrophoben Resten oder umgekehrt (LN, TA, SA, SA), sowie einen Austausch von einem polaren Glutamin (Q91) zu einem positiv geladenem Histidin (H92 bzw. H95). Diese zusätzliche Ladung befindet sich in der unmittelbaren Nachbarschaft des Fragmentes 85-87, welches vermutlich mit J-Concanolid A markiert wird. Durch gerichtete Mutationen an diesen Positionen, unterstützt von vergleichenden Analysen anderer V-ATPasen, die den Austausch QH haben wie z. B. die Pflanzen-V-ATPasen oder nicht haben wie z. B. die Säuger-VATPasen (siehe auch Abb. 65) könnte untersucht werden, ob dieser Zusammenhang wirklich besteht. Abb. 64: Vergleich der Untereinheiten c von N. crassa, M. sexta und M. crystallinum. Die Aminosäuren aus der Sequenz von N. crassa, welche sich von den beiden anderen Sequenzen unterscheiden, diese aber gemeinsam haben, sind hervorgehoben. (grau unterlegt, neutrale Austausche; schwarz unterlegt, nicht neutrale Austausche) 117 Diskussion 5.3.2. Gibt es eine gemeinsame Bindestelle für Makrolide in V-ATPasen? Die Plecomakrolide gehören zur Familie der Makrolide, in deren Reihen einige neue Substanzen mit ganz unterschiedlichen Wirkungen auf V-ATPasen aus verschiedenen Spezies beschrieben sind. Eine dieser neuen Gruppen bei den Makroliden sind die BenzolactonEnamide (Erickson et al., 1997). Zwei Vertreter dieser Gruppe wurden hier bezüglich ihres Einflusses auf die V-ATPase aus M. sexta untersucht, das Salicylihalamid und das Apikularen A. Salicylihalamid wurde erstmals aus dem marinen Schwamm Haliclona sp. isoliert und hat eine stark cytotoxische Wirkung. Sehr wahrscheinlich wird Salicylihalamid allerdings nicht von Haliclona sp. selbst, sondern von einem symbiontischen Mikroorganismus dieses Schwammes produziert (Erickson et al., 1997). Bei Untersuchungen mit V-ATPasen aus verschiedenen Spezies ergab sich für Salicylihalamid ein sehr differenziertes Bild; so wurde die V-ATPase von S. cervisae und N. crassa nicht gehemmt, wohl aber Säuger-V-ATPasen aus Leber, Niere und Osteoklasten (Boyd et al., 2001). Für den zweiten Vertreter der Benzolacton-Enamide, das Apikularen A, isoliert aus dem Myxobakterium Chondromyces robustus ist ähnliches bekannt; es hat keinerlei Effekt auf Hefen und filamentöse Pilze, wirkt aber stark toxisch auf Säugetierzellen (Kunze et al., 1998; Jansen et al., 2000; Boyd et al., 2001). Archazolid und Cruentaren gehören ebenfalls zur Gruppe der Makrolide und werden auch aus Myxobakterien isoliert (Prof. Reichenbach & Dr. Sasse, GBF Braunschweig, persönliche Mitteilung). Archazolid führt bei Wachstumstests in Säugetierzelllinien zu den gleichen cytotoxischen Wirkungen und den gleichen morphologischen Veränderungen wie Salicylihalamid, Concanamycin und Bafilomycin z. B. die Erweiterung des Endoplasmatischen Retikulums oder die Bildung von Streßfaserbündeln (Reichenbach, 1997 und Dr. Sasse, persönliche Mitteilungen). Für Cruentaren werden dagegen wesentlich höhere Konzentrationen für eine Hemmung des Zellwachstums benötigt, und die morphologischen Veränderungen der Zellen haben andere Ausprägungen, was auf eine andere Wirkungsart der Cytotoxizität hinweist (Dr. Sasse, persönliche Mitteilung). Diese letzten drei Vertreter der Makrolide wurden in dieser Arbeit erstmals auf ihre Wirkung auf V-ATPasen getestet, Salicylihalamid zum ersten Mal bei einer Insekten V-ATPase. Für Salicylihalamid, Apikularen und Archazolid wies die V-ATPase die gleiche Empfindlichkeit auf wie für die Plecomakrolide mit einem IC50 von 10-20 nM. Mit der Insekten V-ATPase wurde somit neben den Säuger-V-ATPasen eine zweite Gruppe von V-ATPasen mit hoher Sensitivität 118 Diskussion gegenüber Benzolacton-Enamiden etabliert und Archazolid A als neue V-ATPase hemmende Substanz in die Makrolidfamilie eingeführt. Warum die V-ATPase von M. sexta und die V-ATPasen von Säugern im Gegensatz zu VATPasen aus S. cervisae und N. crassa auf die Benzolacton-Enamide reagieren, ist nicht klar. Geht man allerdings von der Annahme aus, dass die Benzolacton-Enamide, ähnlich wie die Plecomakrolide an die Untereinheit c binden, ergeben sich bei einem Vergleich der Aminosäuresequenzen dieser Untereinheit, welche bei allen Spezies hoch konserviert ist, fünf Positionen, die nur S. cervisae und N. crassa gemeinsam haben (Abb. 65). Ob einer dieser Aminosäureaustausche allerdings die Unempfindlichkeit der V-ATPase aus diesen beiden Organismen gegenüber Benzolacton-Enamiden bewirkt, müßte wiederum durch gezielte Mutageneseexperimente überprüft werden. Auch wurden bisher noch keine Tests an pflanzlichen V-ATPasen durchgeführt, welche im Vergleich (Abb. 65) ebenfalls berücksichtigt sind. Abb. 65: Sequenzvergleich der V-ATPase Untereinheiten c. Es sind die Sequenzen folgender Spezies miteinander verglichen: S. cerevisiae, N. Crassa, R. norvegicus, H. sapiens, B. taurus, A. aegyptie, N. norvegicus, N. tabacum, G. hirsutum, M. crystallinum und O. sativa. Die fünf Aminosäuren die nur S. cerevisiae und N. crassa im Vergleich zu allen anderen Spezies gemeinsam haben, sind hervorgehoben. Besonders interessant waren in diesem Zusammenhang auch die Versuche, die Markierung durch J-Concanolid A durch die vier neuen Makrolide zu kompetitieren. Mit 10 µM Archazolid A gelang es, ähnlich wie mit Concanamycin A, Bafilomycin A1 bzw. B1 die Markierung vollständig zu unterdrücken, mit Salicylihalamid und Apikularen gelang dies, obwohl sie die gleiche Effizienz in der Hemmung der V-ATPase zeigen, nicht. 119 Diskussion Wie nach den Beobachtungen von Dr. Sasse zu erwarten war, hatte Cruentaren einen deutlich höheren IC50 von 60 µM bezüglich der Hemmung der V-ATPase, und es konnte ebenfalls keine Verdrängung der Markierung durch J-Concanolid A nachgewiesen werden. Damit gibt es einen entscheidenden Unterschied in der Wirkungsweise von Plecomakroliden und Archazolid A gegenüber den Benzolacton-Enamiden, da sie bei gleichen IC50 Werten einen verschiedenen Einfluß auf die Markierung mit J-Concanolid A haben. Hierfür bieten sich zwei Erklärungsmöglichkeiten an. Erstens, die beiden Gruppen haben verschiedene Bindungsstellen an die V-ATPase und binden dort unabhängig voneinander an das Enzym, möglicherweise sogar an unterschiedlichen Untereinheiten. Bisher gibt es keine Daten über die Wirkungsweise der Benzolacton-Amide-Hemmung auf die V-ATPase. Es ist nur der Effekt auf Zellkulturen bzw. die V-ATPase-Aktivität, nicht aber ein Einfluß auf die Protonenleitfähigkeit des Vo Komplexes gezeigt, wie dies für Plecomakrolide geschehen ist (Crider et al., 1994; Zhang et al., 1994), so dass durchaus eine andere inhibitorische Wirkung auf die V-ATPase-Aktivität denkbar wäre. Die zweite Möglichkeit könnte in der Größe der Bindetasche der V-ATPase für die Makrolide begründet liegen. Diese muß relativ groß sein, da sowohl der Makrolidring als auch der Hemiketalring einen entscheidenden Einfluß auf die Hemmung der V-ATPase haben (Dröse et al., 1993; Gagliardi et al., 1999; Dröse et al., 2001). Vielleicht ist sie bei einer Vorinkubation mit Plecomakroliden und Archazolid A blockiert, weshalb sich das J-Concanolid A nicht einlagern kann. Dagegen füllen die kleineren Benzolacton-Amide diese Tasche nicht vollständig aus und das J-Concanolid A hat zusätzlich Platz um zu binden oder es verdrängt durch seine Anlagerung das BenzolactonAmid. 5.3.3. Die V-ATPase von M. sexta: Ein Modellsystem für die Osteoporoseforschung Die Untersuchung der V-ATPase und die Wirkungsweise ihrer spezifischen Inhibitoren, vor allem aus der Gruppe der Makrolide, wird im Zusammenhang mit der Osteoporoseforschung immer wichtiger. Die zum Knochen hin orientierten Membranen der Osteoklasten sind dicht mit V-ATPasen bestückt (Blair et al., 1989), und es gibt einen direkten Zusammenhang zwischen der V-ATPase und dem Abbau von Knochenmaterial durch die Osteoklasten (Chatterjee et al., 1992; Blair, 1998). Eine spezifische Hemmung der V-ATPase in Osteoklasten könnte also den Knochenabbau verhindern oder verlangsamen. Es konnte schon gezeigt werden, dass Bafilomycin A1 in vitro und in vivo in der Lage ist, genau dies zu bewirken (Sundquist et al., 1990; Farina et al., 1995). Allerdings wirkt Bafilomycin auch auf 120 Diskussion alle anderen V-ATPasen im Organismus und seine Letaldosis liegt bei 1 mg/kg Lebendgewicht. Bekannt sind aber auch osteoklastenspezifische Isoformen von V-ATPase Untereinheiten (Van Hille et al., 1993, 1995; Bartkiewicz et al., 1995; Lee et al., 1996; Li et al., 1996) und eine Regulation der mRNA für V-ATPase Untereinheiten während der Osteoklastenreifung (Laitala-Leinonen et al., 1996). Deshalb wird versucht, ausgehend von der bekannten Struktur von Bafilomycin, durch Modifikation einen Inhibitor zu finden, der nur die V-ATPase in den Osteoklasten hemmt (Farina & Gagliardi, 1999; Gagliardi et al., 1998; Keeling et al., 1998). Durch die Entdeckung der neuen Mitglieder der Makrolidfamilie, die zwischen V-ATPasen aus verschiedenen Spezies unterscheiden können, bietet sich eine neue Ansatzmöglichkeit, um gewebespezifische Inhibitoren der V-ATPasen zu finden (Boyd et al., 2001; Farina & Gagliardi, 1999). Da die V-ATPase aus M. sexta ähnlich wie die VATPase aus Osteoklasten durch die Benzolacton-Amide gehemmt wird, eignet sie sich auch aufgrund ihrer hohen Reinheit und Verfügbarkeit hervorragend für eine Struktur-FunktionsAnalyse von neuen Inhibitoren. Eine weitere wichtige Gemeinsamkeit zwischen der V-ATPase aus M. sexta und der VATPase aus Osteoklasten spiegelt sich in einer für beide beobachteten Interaktion mit dem Cytoskelett der Zelle wieder. So konnte gezeigt werden, dass die V-ATPase während der Aktivierung der Osteoklasten mit dem Aktincytoskelett direkt interagiert und in vitro an FAktin bindet (Lee et al., 1999; Holliday et al., 2000). Auch die V-ATPase aus M. sexta kann in vitro direkt an Aktin binden, und in der Zelle colokalisiert sie mit Aktinfilamenten (Merzendorfer et al., 2001). Mit ihrer Empfindlichkeit gegenüber Plecomakroliden und der neuen Gruppe der Benzolacton-Amide, sowie ihrer Interaktion mit dem Aktincytoskelett stellt die V-ATPase von M. sexta damit möglicherweise ein ideales Modell dar, mit dem grundlegende Fragen in der Osteoporose-Forschung geklärt werden können. 121 Zusammenfassung 6. Zusammenfassung Die V-ATPase im larvalen Mitteldarm des Tabakschwärmers Manduca sexta energetisiert die gesamten sekundär aktiven Transportprozesse in diesem Epithel. Sie ist einer der best untersuchten Vertreter dieser Klasse von Ionentransport-ATPasen und hat sich als ideales Objekt für die Untersuchung von Struktur, Funktion und Regulation dieser Proteinfamilie erwiesen. In der vorliegenden Dissertation wurde die Struktur des V1Vo Holoenzyms, des V1 Komplexes und des Vo Komplexes, die Regulation der V-ATPase-Aktivität und die Hemmung der V-ATPase durch Makrolide untersucht. Mit der Modifikation von bestehenden und der Etablierung von neuen Protokollen gelang es das V1Vo Holoenzym, den V1 Komplex und den Vo Komplex in Milligramm-Mengen zu reinigen und damit die Vorraussetzung für neue Erkenntnisse über die Struktur der V-ATPase zu schaffen. Durch N-terminale Sequenzierung, Immunfärbung und/oder MALDI-MS konnten neben den bereits bei M. sexta bekannten Untereinheiten A, B, E, F, G, c und d auch die restlichen Untereinheiten C, D, H, a und e identifiziert werden. Das als Kontamination der V-ATPase häufig auftauchende Protein B´ wurde als ein mitochondriales Hsp60 identifiziert. Bei dem sowohl im Holoenzym als auch im Vo Komplex vorhandenen 26 kDa großen Protein handelt es sich sehr wahrscheinlich um ein Dimer der Untereinheit c und nicht, wie bislang vermutet um deren Isoform c´´. Für die im V1 Komplex nur substöchiometrisch vorhandene Untereinheit C konnte gezeigt werden, dass sie im Gegensatz zur Situation beim V 1Vo Holoenzym nur sehr schwach gebunden ist. Sie lässt sich schon unter relativ milden Bedingungen (0,01% C12E10) vom V1 Komplex abtrennen, reassoziiert aber andererseits auch sehr leicht wieder an den V1 Komplex, wie mit einer rekombinanten Untereinheit gezeigt werden konnte. Durch die Behandlung des V1 Komplexes mit chaotropen Ionen ergaben sich Hinweise, die eher auf die Untereinheit D als auf die Untereinheit E als Homolog der VATPasen zur -Untereinheit der F-ATPasen schließen lassen. Durch die Erhöhung der Ionenstärke während eines Reinigungsschrittes gelang es erstmals, die Untereinheit a bei einer Insekten V-ATPase darzustellen. Dies gelang sowohl für das V1Vo Holoenzym als auch für den Vo Komplex und war besonders wichtig, da die Untereinheit als der Favorit für die Bindung der V-ATPase spezifischen inhibitorischen Plecomakrolide angesehen wurde. Wie sich allerdings herausstellte, ist die Untereinheit c des Vo Komplexes die einzige Untereinheit die durch das semisynthetische Concanamycin-Derivat, 9-O-[p-(Trifluoroethyldiazirinyl)122 Zusammenfassung benzoyl]-21,23-dideoxy-23-epi-[125I]Iodo-concanolid A (J-Concanolid A) spezifisch markiert wird. Durch MALDI-MS konnten einige Bereiche der Untereinheit c bestimmt werden, die potentiell mit der Sonde interagieren. Diese Abschnitte befinden sich alle auf der luminalen Seite der Membran. Beim Testen einer Reihe neuer Makrolide zeigte sich erstaunlicherweise, dass Salicylihalamid, Apikularen und Archazolid ähnlich wie Concanamycin A, Bafilomycin A1 und B1, mit einem IC50 von ca. 20 nM die V-ATPase schon bei sehr niedrigen Konzentrationen hemmen. Das ebenfalls getestete Cruentaren lag mit einem IC 50 von 60 µM hingegen deutlich höher. Neben Concanamycin und den Bafilomycinen ist auch Archazolid in der Lage, die Markierung durch J-Concanolid A zu unterbinden, was auf eine gemeinsame Bindestelle dieser drei Makrolide hinweist. Die Untersuchung der Enzymaktivität der V-ATPase zeigte zum einen eine deutliche Endprodukthemmung durch das entstehende ADP und zum anderen dass Mg2+ nicht durch Ca2+ zu ersetzten ist, da Ca2+-ATP im Gegensatz zu Mg2+-ATP keinen Protonentransport unterstützt. Für den Mechanismus der Dissoziation des V1Vo Holoenzyms in seinen V1 und Vo Komplex scheinen Nukleotide von entscheidender Bedeutung zu sein. Während das V1Vo Holoenzym praktisch nukleotidfrei ist, sind im abdissoziierten V1 Komplex ein bis zwei Moleküle ADP enthalten. Die Bindung von ADP oder AMP-PNP an das Holoenzym bewirkt keine Dissoziation der V-ATPase. Allerdings genügt bereits die Hydrolyse von nur einem Molekül Mg2+-ATP pro Enzym, um eine Dissoziation zu induzieren. Aus diesem Befund kann geschlossen werden, dass die V-ATPase während der Hydrolyse einen Konformationszustand durchläuft, in dem sie instabil ist und zur Dissoziation neigt. 123 Literaturverzeichnis 7. Literaturverzeichnis Abrahams, J. P., Leslie, A. G., Lutter, R. and Walker, J. E. (1994). Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria. Nature 370, 621-8. Adachi, I., Arai, H., Pimental, R. and Forgac, M. (1990a). Proteolysis and orientation on reconstitution of the coated vesicle proton pump. J Biol Chem 265, 960-66. Adachi, I., Puopolo, K., Marquez-Sterling, N., Arai, H. and Forgac, M. (1990b). Dissociation, cross-linking, and glycosylation of the coated vesicle proton pump. J Biol Chem 265, 967-73. Alconada, A., Flores, A. I., Blanco, L. and Cuezva, J. M. (1994). Antibodies against F1-ATPase alpha-subunit recognize mitochondrial chaperones. Evidence for an evolutionary relationship between chaperonin and ATPase protein families. J Biol Chem 269, 13670-9. Anraku, Y., Hirata, R., Wada, Y. and Ohya, Y. (1992). Molecular Genetics of the Yeast Vacuolar H+-ATPase. J Exp Biol 172, 67-81. Anthon, G. E. and Jagendorf, A. T. (1984). Methanol-induced release of tightly bound adenine nucleotides from thylakoid-associated CF1. Biochim Biophys Acta 766, 354-62. Arai, H., Berne, M. and Forgac, M. (1987). Inhibition of the Coated Vesicle Proton Pump and Labeling of a 17,000 Dalton Polypeptide by N,N´-dicyclohexylcarbodiimide. J Biol Chem 262, 11006-11. Arai, H., Pink, S. and Forgac, M. (1989). Interaction of anions and ATP with the coated vesicle proton pump. Biochemistry 28, 3075-82. Arai, H., Terres, G., Pink, S. and Forgac, M. (1988). Topography and Subunit Stoichiometry of the Coated Vesicle Proton Pump. J Biol Chem 263, 8796-02. Azuma, M., Harvey, W. R. and Wieczorek, H. (1995). Stoichiometry of K+/H+ antiport helps to explain extracellular pH 11 in a model epithelium. FEBS Lett 361, 153-6. Azzi, A., Casey, R. P. and Nalecz, M. (1984). The effect of N, N'-Dicyclohexylcarbodiimide of enzymes of bioenergetic relevance. Biochim Biophys Acta 768, 209-26. Bartkiewicz, M., Hernando, N., Reddy, S. V., Roodman, G. D. and Baron, R. (1995). Characterization of the osteoclast vacuolar H+-ATPase B-subunit. Gene 160, 157-164. Bauerle, C., Ho, M. N., Lindorfer, M. A. and Stevens, T. H. (1993). The Saccharomyces-Cerevisiae VMA6 Gene Encodes the 36-kDa Subunit of the Vacuolar H+-ATPase Membrane Sector. J Biol Chem 268, 12749-57. Bell, R. A. and Joachim, F. G. (1974). Techniques for rearing laboratory colonies of tobacco hornworms and pink bollworms. Ann Entomol Soc Am 69, 365-73. Beltran, C. and Nelson, N. (1992). The Membrane Sector of Vacuolar H+-ATPases by Itself Is Impermeable to Protons. Acta Physiol Scand 146, 41-47. Berenbaum, M. (1980). Adaptive significance of midgut pH in larval lepidoptera. Am Nat 115, 138-46. Bianchet, M. A., Hullihen, J., Pedersen, P. L. and Amzel, L. M. (1998). The 2.8-Angstrom Structure of Rat Liver F1-Atpase - Configuration of a Critical Intermediate in Atp Synthesis/Hydrolysis. Proc Nat Acad Sci USA 95, 11065-70. Bindseil, K. U. and Zeeck, A. (1994). The Chemistry of Unusual Macrolides .2. Spectroscopic and Biosynthetic Investigations of the V-Type ATPase Inhibitor Concanamycin a. Liebigs Ann Chem 1994, 305-12. Blair, H. C. (1998). How the osteoclast degrades bone. Bioessays 20, 837-46. Blair, H. C., Teitelbaum, S. L., Ghiselli, R. and Gluck, S. (1989). Osteoclastic bone resorption by a polarized vacuolar proton pump. Science 245, 855-57. Boddien, C. (1995). Neue Concanamycin-Derivate als ATPase-Inhibitoren und zur Markierung des aktiven Zentrums der Enzyme sowie Fermentations- und Biosynthesestudien an Streptomyces sp. Dissertation, Universität Göttingen. Boekema, E. J., Ubbink-Kok, T., Lolkema, J. S., Brisson, A. and Konings, W. N. (1998). Structure of V-type ATPase from Clostridium fervidus by electron microscopy. Photosynthesis Research 57, 267-273. Boekema, E. J., Ubbinkkok, T., Lolkema, J. S., Brisson, A. and Konings, W. N. (1997). Visualization of a Peripheral Stalk in V-Type Atpase - Evidence For the Stator Structure Essential to Rotational Catalysis. Proc Natl Acad Sci U S A 94, 14291-93. Boekema, E. J., van Breemen, J. F. L., Brisson, A., Ubbink-Kok, T., Konings, W. N. and Lolkema, J. S. (1999). Biological motors - Connecting stalks in V-type ATPase. Nature 401, 37-38. Bollag and Edelstein (1991) Protein methods. Wiley-Liss Inc., New York 124 Literaturverzeichnis Borlinghaus, R., Apell, H. J. and Läuger, P. (1987). Fast Charge Translocation Associated with Partial Reactions of the Na,K-Pump: I. Current and Voltage Transients after Photochemical release of ATP. J Membrane Biol 97, 161-78. Bowman, B. J., Dschida, W. J., Harris, T. and Bowman, E. J. (1989). The vacuolar ATPase of Neurospora crassa contains an F1-like structure. J Biol Chem 264, 15606-12. Bowman, E. J., Siebers, A. and Altendorf, K. (1988). Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci USA 85, 7972-76. Bowman, E. J., Steinhardt, A. and Bowman, B. J. (1995). Isolation of the vma-4 gene encoding the 26 kDa subunit of the Neurospora crassa vacuolar ATPase. Biochim Biophys Acta 1237, 95-98. Boyd, M. R., Farina, C., Belfiore, P., Gagliardi, S., Kim, J. W., Hayakawa, Y., Beutler, J. A., McKee, T. C., Bowman, B. J. and Bowman, E. J. (2001). Discovery of a novel antitumor benzolactone enamide class that selectively inhibits mammalian vacuolar-type (H+)-atpases. J Pharmacol Exp Ther 297, 114-20. Boyer, P. D. (1989). A perspective of the binding change mechanism for ATP synthesis. FASEB J 3, 2164-78. Brown, D., Gluck, S. and Hartwig, J. (1987). Structure of the Novel Membrane-coating Material in Proton-secreting Epithelial Cells and Identification as an H+ ATPase. J Cell Biol 105, 1637-48. Brunner, J., Senn, H. and Richards, F. M. (1980). 3-Trifluoromethyl-3-phenyldiazirine. A new carbene generating group for photolabeling reagents. J Biol Chem 255, 3313-17. Cantley, L. C., Cantley, L. G. and Josephson, L. (1978). A characterization of vanadate interactions with the Na/KATPase. Mechanistic and regulatory implications. J Biol Chem 253, 7361-68. Carmeli, C., Lifshitz, Y. and Gutman, M. (1981). Modulation by divalent metal ions of the autocatalytic reactivity of adenosinetriphosphatase from chloroplasts. Biochemistry 20, 3940-44. Chatterjee, D., Chakraborty, M., Leit, M., Neff, L., Jamsakellokumpu, S., Fuchs, R., Bartkiewicz, M., Hernando, N. and Baron, R. (1992). The Osteoclast Proton Pump Differs in Its Pharmacology and Catalytic Subunits from Other Vacuolar H+-ATPases. J Exp Biol 172, 193-04. Cioffi, M. and Harvey, W. R. (1981). Comparison of potassium transport in three structurally distinct regions of the insect midgut. J Exp Biol 91, 103-16. Cioffi, M. and Wolfersberger, M. G. (1983). Isolation of separate apical, lateral and basal plasma membrane from cells of an insect epithelium. A procedure based on tissue organization and ultrastructure. Tissue & Cell 15, 781-03. Crider, B. P., Xie, X. S. and Stone, D. K. (1994). Bafilomycin inhibits proton flow through the H+ channel of vacuolar proton pumps. J Biol Chem 269, 17379-81. David, P. and Baron, R. (1994). The catalytic cycle of the vacuolar H(+)-ATPase. Comparison of proton transport in kidney- and osteoclast-derived vesicles. J Biol Chem 269, 30158-63. Deckers-Hebestreit, G. and Altendorf, K. (1996). The F1Fo-type ATP synthases of bacteria: structure and function of the Fo complex. Annu Rev Microbiol 50, 791-24. Dietz, K. J., Heber, U. and Mimura, T. (1998). Modulation of the Vacuolar H+-Atpase By Adenylates As Basis For the Transient Co2-Dependent Acidification of the Leaf Vacuole Upon Illumination. Biochim Biophys Acta 1373, 87-92. Digel, J. G., Kishinevsky, A., Ong, A. M. and McCarty, R. E. (1996). Differences between two tight ADP binding sites of the chloroplast coupling factor 1 and their effects on ATPase activity. J Biol Chem 271, 19976-82. Digel, J. G., Moore, N. D. and McCarty, R. E. (1998). Influence of divalent cations on nucleotide exchange and ATPase activity of chloroplast coupling factor 1. Biochemistry 37, 17209-15. Doherty, R. D. and Kane, P. M. (1993). Partial assembly of the yeast vacuolar H(+)-ATPase in mutants lacking one subunit of the enzyme. J Biol Chem 268, 16845-51. Dow, J. A. T. (1984). Extremely high pH in biological systems: a model for carbonate transport. Am J Physiol 246, 633-36. Dow, J. A. T. (1992). pH Gradients in Lepidopteran Midgut. J Exp Biol 172, 355-75. Dow, J. A., Goodwin, S. F. and Kaiser, K. (1992). Analysis of the gene encoding a 16-kDa proteolipid subunit of the vacuolar H(+)-ATPase from Manduca sexta midgut and tubules. Gene 122, 355-60. Drobinskaya, I. Y., Kozlov, I. A., Muratliev, M. B. and Vulfson, E. N. (1985). Tightly bound adenosine diphosphate, which inhibits the activity of mitochondrial F1-ATPase, is located at the catalytic site of the enzyme. FEBS Lett 182, 419-24. Dröse, S. and Altendorf, K. (1997). Bafilomycins and concanamycins as inhibitors of V-ATPases and PATPases. J Exp Biol 200, 1-8. Dröse, S., Bindseil, K. U., Bowman, E. J., Siebers, A., Zeeck, A. and Altendorf, K. (1993). Inhibitory effect of modified bafilomycins and concanamycins on P- and V-type adenosinetriphosphatases. Biochemistry 32, 3902-6. 125 Literaturverzeichnis Dröse, S., Boddien, C., Gassel, M., Ingenhorst, G., Zeeck, A. and Altendorf, K. (2001). Semisynthetic derivatives of concanamycin a and c, as inhibitors of v- and p-type atpases: structure-activity investigations and developments of photoaffinity probes. Biochemistry 40, 2816-25. Dschida, W. J. and Bowman, B. J. (1992). Structure of the Vacuolar ATPase from Neurospora-Crassa as Determined by Electron Microscopy. J Biol Chem 267, 18783-89. Dschida, W. J. and Bowman, B. J. (1995). The vacuolar ATPase: sulfite stabilization and the mechanism of nitrate inactivation. J Biol Chem 270, 1557-63. Duncan, T. M., Bulygin, V. V., Zhou, Y., Hutcheon, M. L. and Cross, R. L. (1995). Rotation of subunits during catalysis by Escherichia coli F1-ATPase. Proc Natl Acad Sci U S A 92, 10964-8. Erickson, K. L., Beutler, J. A., Cardellina, J. H. And Boyd, M. R. (1997). Salicylihalamides A and B, novel cytotoxic macrolides from marine sponge Haliclona sp. J. org. Chem. 62, 8188-92 Farina, C., Belfiore, P. and Clarke, G. D. (1995). Bafilomycin A1 inhibits bone resorbtion in vivo after systemic administration. J Bone Min Res 10 (Sup.), 296. Farina, C. and Gagliardi, S. (1999). Selective inhibitors of the osteoclast vacuolar proton ATPase as novel bone antiresorptive agents. Drug Discovery Today 4, 163-72. Feldman, R. I. and Boyer, P. D. (1985). The role of tightly bound ADP on chloroplast ATPase. J Biol Chem 260, 13088-94. Fendler, K., Dröse, S., Altendorf, K. and Bamberg, E. (1996). Electrogenic K+ transport by the Kdp-ATPase of escherichia coli. Biochemistry 35, 8000-17. Fendler, K., Grell, E., Haubs, M. and Bamberg, E. (1985). Pump currents generated by the purified Na+K+ ATPase from kidney on black lipid membranes. EMBO J 12, 3079-85. Feng, Y. and Forgac, M. (1992). Cysteine 254 of the 73-kDa a Subunit Is Responsible for Inhibition of the Coated Vesicle (H+)-ATPase upon Modification by Sulfhydryl Reagents. J Biol Chem 267, 5817-22. Fillingame, R. H., Jones, P. C., Jiang, W., Valiyaveetil, F. I. and Dmitriev, O. Y. (1998). Subunit Organization and Structure in the F-0 Sector of Escherichia Coli F1f0 Atp Synthase. Biochim Biophys Acta - Bioenergetics 1365, 135-42. Finbow, M. E. and Pitts, J. D. (1993). Is the gap junction channel--the connexon--made of connexin or ductin? J Cell Sci 106, 463-71. Findlay, J. B., Finbow, M. E., Jones, P. C., Kim, Y. I., Harrison, M. A. and Hughes, G. (1997). A structure-based model for the 16 kDa membrane sector of the vacuolar H(+)-ATPase. Biochem Soc Trans 25, 1107-13. Forgac, M. (1998). Structure, function and regulation of the vacuolar (H+)-ATPases. FEBS Lett 440, 258-63. Forgac, M. and Cantley, L. (1984). Characterization of the ATP-dependent proton pump of clathrin-coated vesicles. J Biol Chem 259, 8101-5. Gagliardi, S., Gatti, P. A., Belfiore, P., Zocchetti, A., Clarke, G. D. and Farina, C. (1998). Synthesis and Structure-Activity Relationships of Bafilomycin a(1) Derivatives As Inhibitors of Vacuolar H+-Atpase. J Med Chem 41, 1883-93. Gagliardi, S., Rees, M. and Farina, C. (1999). Chemistry and structure activity relationships of bafilomycin A(1), a potent and selective inhibitor of the vacuolar H+-ATPase. Current Medicinal Chemistry 6, 1197-12. Ganti, T. (1971). The principle of life. Gondolat, Budapest. Geering, K. (2000). Topogenic motifs in P-type ATPases. J Membrane Biol 174, 181-90. Gibbons, C., Montgomery, M. G., Leslie, A. G. W. and Walker, J. E. (2000). The structure of the central stalk in bovine F-1-ATPase at 2.4 angstrom resolution. Nature Structural Biology 7, 1055-61. Gies, A., Fromm, T. and Ziegler, R. (1988). Energy metabolism in starving larvae of Manduca sexta. Comp. Biochem. Physiol. 91A, 549-55. Gillespie, J., Ozanne, S., Tugal, B., Percy, J., Warren, M., Haywood, J. and Apps, D. (1991). The vacuolar H(+)translocating ATPase of renal tubules contains a 115- kDa glycosylated subunit. FEBS Lett 282, 69-72. Giordana B, S. V., Parenti P, Hanozet GM. (1989). Amino acid transport systems in intestinal brush-border membranes. Amer J Physiol 257, 494-00. Gluck, S. (1992). V-ATPases of the Plasma Membrane. J Exp Biol 172, 29-37. Gluck, S. L., Nelson, R. D., Lee, B. S., Wang, Z. Q., Guo, X. L., Fu, J. Y. and Zhang, K. (1992). Biochemistry of the Renal V-ATPase. J Exp Biol 172, 219-29. Gluck, S., Kelly, S. and Al-Awqati, Q. (1982). The Proton Translocating ATPase Responsible for Urinary Acidification. J Biol Chem 257, 9230-33. Gogarten, J. P., Starke, T., Kibak, H., Fishman, J. and Taiz, L. (1992). Evolution and isoforms of V-ATPase subunits. J Exp Biol 172, 137-47. Goldstein, J. L. and Swain, T. (1965). The inhibition of enzyms by tannins. Phytochemistry 4, 185-92. Gräf, R., Harvey, W. R. and Wieczorek, H. (1994a). Cloning, sequencing and expression of cDNA encoding an insect V-ATPase subunit E. Biochim Biophys Acta 1190, 193-6. 126 Literaturverzeichnis Gräf, R., Harvey, W. R. and Wieczorek, H. (1996). Purification and properties of a cytosolic V1-ATPase. J Biol Chem 271, 20908-13. Gräf, R., Lepier, A., Harvey, W. R. and Wieczorek, H. (1994b). A novel 14-kDa V-ATPase subunit in the tobacco hornworm midgut. J Biol Chem 269, 3767-74. Gräf, R., Novak, F. J., Harvey, W. R. and Wieczorek, H. (1992). Cloning and sequencing of cDNA encoding the putative insect plasma membrane V-ATPase subunit A. FEBS Lett 300, 119-22. Grüber, G., Radermacher, M., Ruiz, T., Godovac-Zimmermann, J., Canas, B., Kleine-Kohlbrecher, D., Huss, M., Harvey, W. R. and Wieczorek, H. (2000a). Three-dimensional structure and subunit topology of the V-1 ATPase from Manduca sexta midgut. Biochemistry 39, 8609-16. Grüber, G., Svergun, D. I., Godovac-Zimmermann, J., Harvey, W. R., Wieczorek, H. and Koch, M. H. (2000b). Evidence for major structural changes in the manduca sexta midgut V1 ATPase due to redox-modulation: A small-angle X-ray scattering study. J Biol Chem 275, 30082-7 Hanada, H., Moriyama, Y., Maeda, M. and Futai, M. (1990). Kinetic studies of chromaffin granule H+-ATPase and effects of bafilomycin A1. Biochem Biophys Res Commun 170, 873-78. Harold, F. (1986). The vital force. A Study of Bioenergetics. Freeman, New York. Harvey WR, Cioffi, M. and Wolfersberger MG. (1983). Chemiosmotic potassium ion pump of insect epithelia. Am J Physiol 244, 163-75. Harvey, B. J. (1992). Energization of Sodium Absorption by the H+-ATPase Pump in Mitochondria-Rich Cells of Frog Skin. J Exp Biol 172, 289-09. Harvey, W. R. (1992). Physiology of V-ATPases. J Exp Biol 172, 1-17. Harvey, W. R., Cioffi, M. and Wolfersberger, M. G. (1981). Portasomes as Coupling Factors in Active Ion Transport and Oxidative Phosphorylation. Amer Zool 21, 775-91. Hatefi, Y. and Hanstein, W. G. (1969). Proc Nat Acad Sci USA 62, 1129-36. Hausrath, A. C., Grüber, G., Matthews, B. W. and Capaldi, R. A. (1999). Structural features of the gamma subunit of the Escherichia coli F-1 ATPase revealed by a 4.4-angstrom resolution map obtained by x-ray crystallography. Proc Nat Acad Sci USA 96, 13697-02. Heukeshoven, J. and Dernick, R. (1988). Improved silver staining procedure for fast staining in PhastSystem Development Unit. I. staining of sodium dodecyl sulfate gels. Electrophoresis 9, 28-32. Hicks, D. B. and Krulwich, T. A. (1986). The Membrane ATPase of Alkalophilic Bacillus firmus RAB is an F1type ATPase. J Biol Chem 261, 12896-02. Higgins, C. F. (1992). ABC Transporters - From Microorganisms to Man. Annu Rev Cell Biol 8, 67-113. Hirata, R., Graham, L. A., Takatsuki, A., Stevens, T. H. and Anraku, Y. (1997). VMA11 and VMA16 encode second and third proteolipid subunits of the Saccharomyces cerevisiae vacuolar membrane H+-ATPase. J Biol Chem 272, 4795-803. Ho, M. N., Hill, K. J., Lindorfer, M. A. and Stevens, T. H. (1993). Isolation of vacuolar membrane H(+)ATPase-deficient yeast mutants; the VMA5 and VMA4 genes are essential for assembly and activity of the vacuolar H(+)-ATPase. J Biol Chem 268, 221-7. Holliday, L. S., Lu, M., Lee, B. S., Nelson, R. D., Solivan, S., Zhang, L. and Gluck, S. L. (2000). The amino-terminal domain of the B subunit of vacuolar H+-ATPase contains a filamentous actin binding site. J Biol Chem 275, 32331-37. Hunke, S., Dröse, S. and Schneider, E. (1995). Vanadate and bafilomycin A1 are potent inhibitorsof the ATPase activity of the recostituted bacterial ATP-binding cassette transporter for maltose (MalFGK2). Biochem Biophys Res Commun 216, 589-94. Ingenhorst, G. (2000). Madumycine aus Actinoplanes sp. A1372 und deren Derivatisierung sowie Semisynthetische Naturstoffderivate für biochemische Untersuchungen. Dissertation, Universität Göttingen. Jäger, D. Klein, U. (1996). Biogenesis and regulation of V-ATPase in the midgut of the tobacco hornworm. Proc Germ zool Soc 89.1, 327. Jansen R., Kunze B., Reichenbach H. and Höfle G. (2000) Antibiotics from gliding bacteria LXXXVI. Apikularens A and B, cytotoxic 10-membered lactones with a novel mechanism of action from Chondromyces species (myxobacteria): isolation, structure elucidation, and biosythesis. Eur J Org Chem 913-19 Junge, W. (1999). ATP synthase and other motor proteins. Proc Nat Acad Sci USA 96, 4735-37. Junge, W., Lill, H. and Engelbrecht, S. (1997). ATP synthase: an electrochemical transducer with rotatory mechanics. Trends Biochem Sci 22, 420-23. Jungreis A M, V. G. L. (1977). Insensitivity of Lepidopteran tissues to ouabain: Absence of ouabain binding and Na+ - K+ ATPases in Larval and adult Midgut. J Insect Physiol 23, 503-09. 127 Literaturverzeichnis Kagawa, Y. and Hamamoto, T. (1996). The energy transmission in ATP synthase: from the gamma-c rotor to the alpha 3 beta 3 oligomer fixed by OSCP-b stator via the beta DELSEED sequence. J Bioenerg Biomembr 28, 421-31. Kane, P. M. (1995). Disassembly and reassembly of the yeast vacuolar H+-ATPase in vivo. J Biol Chem 270, 17025-32. Kane, P. M., Yamashiro, C. T. and Stevens, T. H. (1989). Biochemical characterization of the yeast vacuolar H+ATPase. J Biol Chem 264, 19236-44. Kawamura, Y., Arakawa, K., Maeshima, M. and Yoshida, S. (2001). ATP analogue binding to the A subunit induces conformational changes in the E subunit that involves a disulfide bond formation in plant VATPase. Eur J Biochem 268, 2801-9. Keeling, D. J., Herslof, M., Mattsson, J. P. and Ryberg, B. (1998). Tissue-Selective Inhibition of Vacuolar Acid Pumps. Acta Physiologica Scandinavica 163, 195-01. Kinashi, H., Someno, K. and Sakaguchi, K. (1984). Isolation and characterization of concanamycins A, B and C. J Antibiot (Tokyo) 37, 1333-43. Kneusel, R. E., Merchant, S. and Selman, B. R. (1982). Properties of the solvent-stimulated ATPase activity of chloroplat coupling factor 1 from chlamydomonas reinhardii. Biochim Biophys Acta 681, 337-44. Kunze, B. Jansen, R. Sasse, F. Höfle, G and Reichenbach, H. (1998): Apicularens A and B, new cytostatic macrolides from Chondromyces species (Myxobacteria): production, physico-chemical and biological properties. Journal of Antibiotics, Japan 51, 1075-80 Kyhse-Anderson, J. (1984). Electroblotting of multiple gels: a simple appararus without buffer tank for rapid transfer of proteins from polyacrylamide to nitrocellulose. J biochem biophy Meth 10, 203-09. Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-5. Laitala-Leinonen, T., Howell, M. L., Dean, G. E. and Vaananen, H. K. (1996). Resorption-cycle-dependent polarization of mRNAs for different subunits of V-ATPase in bone-resorbing osteoclasts. Mol Biol Cell 7, 129-42. Landolt-Marticorena, C., Williams, K. M., Correa, J., Chen, W. M. and Manolson, M. F. (2000). Evidence that the NH2 terminus of Vph1p, an integral subunit of the V-0 sector of the yeast V-ATPase, interacts directly with the Vma1p and Vma13p subunits of the V-1 sector. J Biol Chem 275, 15449-57. Lee, B. S., Holliday, L. S., Krits, I. and Gluck, S. L. (1999). Vacuolar H+-ATPase activity and expression in mouse bone marrow cultures. Journal of Bone & Mineral Research 14, 2127-36. Lee, B. S., Holliday, S., Ojikutu, B., Krits, I. and Gluck, S. L. (1996). Osteoclasts express the B2 isoform of vacuolar H+-ATPase intracellularly and on their plasma membranes. Amer J Physiol Cell Physiol 39, 382-88. Leng, X. H., Manolson, M. F. and Forgac, M. (1998). Function of the Cooh-Terminal Domain of Vph1p in Activity and Assembly of the Yeast V-Atpase. J Biol Chem 273, 6717-23. Leng, X. H., Manolson, M. F., Liu, Q. and Forgac, M. (1996). Site-directed mutagenesis of the 100-kDa subunit (Vph1p) of the yeast vacuolar (H+)-ATPase. J Biol Chem 271, 22487-93. Lepier, A., Gräf, R., Azuma, M., Merzendorfer, H., Harvey, W. R. and Wieczorek, H. (1996). The peripheral complex of the tobacco hornworm V-ATPase contains a novel 13-kDa subunit G. J Biol Chem 271, 8502-8. Li, X. and Sze, H. (1999). A 100 kDa polypeptide associates with the V0 membrane sector but not with the active oat vacuolar H(+)-ATPase, suggesting a role in assembly. Plant J 17, 19-30. Li, Y. P., Chen, W. M. and Stashenko, P. (1996). Molecular cloning and characterization of a putative novel human osteoclast-specific 116-kDa vacuolar proton pump subunit. Biochem Biophys Res Commun 218, 813-21. Lion, T. and Haas, O. A. (1990). Nonradioactive Labeling of Probe with Digoxigenin by Polymerase Chain Reaction. Analytical Biochem 188, 335-37. Ludwig, J., Kerscher, S., Brandt, U., Pfeiffer, K., Getlawi, F., Apps, D. K. and Schagger, H. (1998). Identification and Characterization of a Novel 9.2-Kda Membrane Sector-Associated Protein of Vacuolar Proton-Atpase From Chromaffin Granules. J Biol Chem 273, 10939-47. Luis, A. M., Alconada, A. and Cuezva, J. M. (1990). The alpha regulatory subunit of the mitochondrial F1-ATPase complex is a heat-shock protein. Identification of two highly conserved amino acid sequences among the alpha-subunits and molecular chaperones. J Biol Chem 265, 7713-6. MacLeod, K. J., Vasilyeva, E., Merdek, K., Vogel, P. D. and Forgac, M. (1999). Photoaffinity labeling of wild-type and mutant forms of the yeast V-ATPase A subunit by 2-azido-[P-32]ADP. J Biol Chem 274, 32869-74. Mandel M, M. Y., Hulmes, J. D., Pan, Y.C.E., Nelson, H. and Nelson, N. (1988). cDNA sequence encoding the 16kDa proteolipid of chromaffin granules implies gene duplication in the evolution of H+-ATPases. Proc Nat Acad Sci USA 85, 5521-24. 128 Literaturverzeichnis Manolson M. F., Rea P. A. Poole R. J. (1985). Identification of 3-O-(4-Benzoyl)benzoyladenosine 5´Triphosphate-and N, N´-Dicyclohexylcarbodiimide-binding Subunits of a Higher Plant H+translocating Tonoplast ATPase. J Biol Chem 260, 12273-79. Manolson, M. F., Wu, B., Proteau, D., Taillon, B. E., Roberts, B. T., Hoyt, M. A. and Jones, E. W. (1994). STV1 gene encodes functional homologue of 95-kDa yeast vacuolar H(+)- ATPase subunit Vph1p. J Biol Chem 269, 14064-74. Mattsson, J. P. and Keeling, D. J. (1996). [H-3]Bafilomycin as a probe for the transmembrane proton channel of the osteoclast vacuolar H+-ATPase. Biochim Biophys Acta-Biomembranes 1280, 98-106. McCarty, R. (1992). A Plant Biochemists View of H+-ATPases and ATP Synthases. J Exp Biol 172, 431-41. Mellman, I. (1992). The Importance of Being Acid - The Role of Acidification in Intracellular Membrane Traffic. J Exp Biol 172, 39-45. Merzendorfer H., Düvel M., Hunke C., Mädler U. and Wieczorek H. (2001) Interaction between the plasma membrane V-ATPase and the actin cytoskeleton in the midgut goblet cells of the tobacco hornworm. Zoology (Sup. IV, DZG 94.1) 104, 62 Merzendorfer, H., Gräf, R., Huss, M., Harvey, W. R. and Wieczorek, H. (1997a). Regulation of proton-translocating V-ATPases. J Exp Biol 200, 225-35. Merzendorfer, H., Harvey, W. R. and Wieczorek, H. (1997b). Sense and antisense RNA for the membrane associated 40 kDa subunit M40 of the insect V-ATPase. FEBS Lett 411, 239-44. Merzendorfer, H., Huss, M., Schmid, R., Harvey, W. R. and Wieczorek, H. (1999). A novel insect V-ATPase subunit M9.7 is glycosylated extensively. J Biol Chem 274, 17372-8. Merzendorfer, H., Reineke, S., Zhao, X. F., Jacobmeier, B., Harvey, W. R. and Wieczorek, H. (2000). The multigene family of the tobacco hornworm V-ATPase: novel subunits a, C, D, H, and putative isoforms. Biochim Biophys Acta 25, 369-79. Milgrom, Y. M. and Cross, R. L. (1993). Nucleotide binding sites on beef heart mitochondrial F1-ATPase. Cooperative interactions between sites and specificity of noncatalytic sites. J Biol Chem 268, 23179-85. Mitchell, P. (1966). Chemiosmotic coupling in oxidative and photosynthetic phophorylation. Biol Rev Camb Philos Soc 41, 445-02. Mochimaru, M. and Sakurai, H. (1998). Effects of organic solvents and tentoxin on enzyme-bound ATP synthesis in isolated chloroplast coupling factor 1. Photosynthesis Research 57, 305-315. Moriyama, Y. and Nelson, N. (1987). Nucleotide Binding Sites and Chemical Modification of the Chromaffin Granule Proton ATPase. J Biol Chem 262, 14723-14729. Moriyama, Y. and Nelson, N. (1989). Cold inactivation of vacuolar proton-ATPases. J Biol Chem 264, 3577-82. Nellen, W. and Lichtenstein, C. (1993). What Makes an Messenger RNA Anti-sense-itive? Trends Biochem Sci 18, 419-23. Nelson, H., Mandiyan, S. and Nelson, N. (1995). A bovine cDNA and a yeast gene (VMA8) encoding the subunit D of the vacuolar H(+)-ATPase. Proc Natl Acad Sci USA 92, 497-01. Nelson, N. (1992). Structural conservation and functional diversity of V-ATPases. J Bioenerg Biomembr 24, 407-14. Nelson, N. and Taiz, L. (1989). The evolution of H+ -ATPases. Trends Biochem Sci 14, 113-16. Nishigori, H., Yamada, S., Tomura, H., Fernald, A. A., Lebeau, M. M., Takeuchi, T. and Takeda, J. (1998). Identification and Characterization of the Gene Encoding a Second Proteolipid Subunit of Human Vacuolar H+-Atpase (Atp6f). Genomics 50, 222-28. Noji, H., Häsler, K., Junge, W., Kinosita, K., Yoshida, M. and Engelbrecht, S. (1999). Rotation of Escherichia coli F-1-ATPase. Biochem Biophys Res Commun 260, 597-99. Noji, H., Yasuda, R., Yoshida, M. and Kinosita, K., Jr. (1997). Direct observation of the rotation of F1-ATPase. Nature 386, 299-02. Noumi T, B. C., Nelson H, Nelson N. (1991). Mutational Analysis of Yeast Vacuolar H+ -ATPase. Proc Nat Acad Sci USA 88, 1938-42. Novak, F. J., Graf, R., Waring, R. B., Wolfersberger, M. G., Wieczorek, H. and Harvey, W. R. (1992). Primary structure of V-ATPase subunit B from Manduca sexta midgut. Biochim Biophys Acta 1132, 67-71. Oka, T., Yamamoto, R. and Futai, M. (1997). Three vha genes encode proteolipids of Caenorhabditis elegans vacuolar-type ATPase - Gene structures and preferential expression in an H-shaped excretory cell and rectal cells. J Biol Chem 272, 24387-92. Ortiz Flores, G., Acosta, A. and Gomez Puyou, A. (1982). Characteristics of adenylyl imidodiphosphate- and ADP-binding sites insoluble and particulate mitochondrial ATPase. Studies with methanol. Biochim Biophys Acta 679, 466-73. Pänke, O., Gumbiowski, K., Junge, W. and Engelbrecht, S. (2000). F-ATPase: specific observation of the rotating c subunit oligomer of EfoEF1. FEBS Lett 472, 34-38. 129 Literaturverzeichnis Parra, K. J. and Kane, P. M. (1998). Reversible association between the V-1 and V-0 domains of yeast vacuolar H+ATPase is an unconventional glucose-induced effect. Molecular & Cellular Biology 18, 7064-74. Parra, K. J., Keenan, K. L. and Kane, P. M. (2000). The H subunit (Vma13p) of the yeast V-ATPase inhibits the ATPase activity of cytosolic V-1 complexes. J Biol Chem 275, 21761-67. Pedersen, P. L. and Carafoli, E. (1987). Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem Sci 12, 146-150. Peng, S. B., Stone, D. K. and Xie, X. S. (1993). Reconstitution of recombinant 40-kDa subunit of the clathrincoated vesicle H(+)-ATPase. J Biol Chem 268, 23519-23. Popov, N., Schmitt, M., Schulzeck, S. and Matthies, H. (1975). Eine störungsfreie Mikromethode zur Bestimmung des Proteingehaltes in Gewebehomogenaten. Acta biol med germ 34, 1441-46. Powell, B., Graham, L. A. and Stevens, T. H. (2000). Molecular characterization of the yeast vacuolar H+-ATPase proton pore. J Biol Chem 275, 23654-60. Pullman, M. E., Penfsky, H. S., Datta, A. and Racker, E. (1960). J Biol Chem 235, 3322-28. Radermacher, M., Ruiz, T., Harvey, W. R., Wieczorek, H. and Grüber, G. (1999). Molecular architecture of Manduca sexta midgut V1 ATPase visualized by electron microscopy. FEBS Lett 453, 383-6. Radermacher, M., Ruiz, T., Wieczorek, H. and Gruber, G. (2001). The structure of the v(1)-atpase determined by three-dimensional electron microscopy of single particles. J Struct Biol 135, 26-37. Ratajczak, R. (1994). Non ionic-detergent Brij 58 conserves the structure of the tonoplast H+-ATPase of Mesembryanthenum crystallinium L. during solubilization and partial purification. Bot Acta 107, 201-9. Rautiala, T. J., Koskinen, A. M. and Vaananen, H. K. (1993). Purification of vacuolar ATPase with bafilomycin C1 affinity chromatography. Biochem Biophys Res Commun 194, 50-6. Reichenbach, H. (1997): 3.1. Biologisch aktive Sekundärstoffe aus Mikroorganismen: Biologie neuer Naturstoffe und Produzenten. GBF Ergebnisbericht, http://bib.gbf.de/ergebnis bericht/1997; Januar 2001 Reineke, S., Wieczorek, H. and Merzendorfer, H. (2001). Expression of Manduca sexta V-ATPase genes mvB, mvG and mvd is regulated by ecdysteroids. (In Vorbereitung). Russell, V. E., Klein, U., Reuveni, M., Spaeth, D. D., Wolfersberger, M. G. and Harvey, W. R. (1992). Antibodies to mammalian and plant V-ATPases cross react with the V- ATPase of insect cation-transporting plasma membranes. J Exp Biol 166, 131-43. Sabbert, D., Engelbrecht, S. and Junge, W. (1996). Intersubunit rotation in active F-ATPase. Nature 381, 623-5. Sakurai, H., Shinohara, K., Hisabori, T. and Shinohara, K. (1981). Enhancement of adenosine triphosphatase activity of purified chloroplast coupling factor 1 in an aqueous organic solvent. J Biochem 90, 95-102. Sambrook, J., Fritsch, E. F. and Maniatis, T. (1989). Molecular cloning: A Laboratory Manual 2nd Edition. Cold Spring Harbor Laboratory Press, New York. Sanger, F., Niklen, S. and Coulson, A. R. (1977). DNA sequencing with chin-terminating inhibitors. Proc Nat Acad Sci USA 74, 5463-67. Schäfer, H. J., Coskun, U., Eger, O., Godovac-Zimmermann, J., Wieczorek, H., Kagawa, Y. and Grüber, G. (2001). 8-N(3)-3'-biotinyl-ATP, a novel monofunctional reagent: differences in the F(1)- and V(1)ATPases by means of the ATP analogue. Biochem Biophys Res Commun 286, 1218-27. Schägger H. and von Jagow G. (1987). Tricine-Sodium Dodecyl-Polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Analyt Biochem 166, 368-79. Schemidt, R. A., Qu, J., Williams, J. R. and Brusilow, W. S. A. (1998). Effects of Carbon Source On Expression of F-0 Genes and On the Stoichiometry of the C Subunit in the F1f0 Atpase of Escherichia Coli. Journal of Bacteriology 180, 3205-3208. Schmidt, W., Winkler, H. and Plattner, H. (1982). Adrenal chromaffin granules: evidence for an ultrastructural equivalent of the proton-pumping ATPase. Eur J Cell Biol 27, 96-104. Schuster, S. M. (1979). Effect of organic solvents on the beef heart mitochondrial adenosine triphosphatase. Biochemistry 18, 1162-67. Schwartz, G. J. and Al-Awqati, Q. A. (1985). Carbon dioxide causes exocytosis of vesicels containing H+ pumps in isolated perfused proximal and collecting tubules. Journal of clinical investigation 75, 1638-44. Schweikl, H. (1988). Die Kalium-Pumpe der Insekten: Versuche zur Solubilisierung und Reinigung einer VakuolenTyp-ATPase aus K+-tranportierenden Membranen des Mitteldarmepithels von Manduca sexta. Dissertation, Universität München. Schweikl, H., Klein, U., Schindlbeck, M. and Wieczorek, H. (1989). A vacuolar-type ATPase, partially purified from potassium transporting plasma membranes of tobacco hornworm midgut. J Biol Chem 264, 11136-42. Sebald, W., Machleidt, W. and Wachter, E. (1980). N,N´-Dicyclohexylcarbodiimide binds specifically to a single glutamyl residue of the proteolipid subunit of the mitochondrial adenosintriphosphatases from Neurospora crassa and Saccharomyces cerevisiae. Biochemistry 77, 785-789. 130 Literaturverzeichnis Seelert, H., Poetsch, A., Dencher, N. A., Engel, A., Stahlberg, H. and Muller, D. J. (2000). Structural biology. Proton-powered turbine of a plant motor. Nature 405, 418-9. Senior, A. E., Lee, R. S., al-Shawi, M. K. and Weber, J. (1992). Catalytic properties of Escherichia coli F1-ATPase depleted of endogenous nucleotides. Arch Biochem Biophys 297, 340-4. Sharom, J. F., Yu, X., Chu, J. W. K. and Doige, C. A. (1995). Characterization of the ATPase activity of pglycoprotein from multidurg-resistant chinese hamster ovary cells. Biochemical Journal 308, 381-90. Shevchenko, A., Wilm, M., Vorm, O. and Mann, M. (1996). Mass spectrometric sequencing of proteins silverstained polyacrylamide gels. Anal Chem 68, 850-8. Stevens, T. H. and Forgac, M. (1997). Structure, function and regulation of the vacuolar (H+)-ATPase. Annu Rev Cell Dev Biol 13, 779-08. Stock, D., Gibbons, C., Arechaga, I., Leslie, A. G. W. and Walker, J. E. (2000). The rotary mechanism sf ATP synthase. Curr Op Struct Biol 10, 672-79. Stock, D., Leslie, A. G. W. and Walker, J. E. (1999). Molecular architecture of the rotary motor in ATP synthase. Science 286, 1700-05. Sumner, J. P., Dow, J. A. T., Earley, F. G. P., Klein, U., Jäger, D. and Wieczorek, H. (1995). Regulation of plasma membrane V-ATPase activity by dissociation of peripheral subunits. J Biol Chem 270, 5649-53. Sundquist, K., Lakkakorpi, P., Wallmark, B. and Väänänen, K. (1990). Inhibition of Osteoclast Proton Transport by Bafilomycin A1 Abolishes Bone Resorption. Biochem Biophys Res Commun 168, 309-13. Supekova, L., Supek, F. and Nelson, N. (1995). The Saccharomyces cerevisiae VMA10 is an intron-containing gene encoding a novel 13-kDa subunit of vacuolar H(+)-ATPase. J Biol Chem 270, 13726-32. Svergun, D. I., Konrad, S., Huss, M., Koch, M. H. J., Wieczorek, H., Altendorf, K., Volkov, V. V. and Grüber, G. (1998). Quaternary structure of V-1 and F-1 ATPase: Significance of structural homologies and diversities. Biochemistry 37, 17659-63. Tabor, S. and Richardson, C. C. (1987). DNA sequence analysis with a modified bacteriophage T7 DNA polymerase. Proc Nat Acad Sci USA 84, 4767-71. Taiz, L., Nelson, H., Maggert, K., Morgan, L., Yatabe, B., Taiz, S. L., Rubinstein, B. and Nelson, N. (1994). Functional analysis of conserved cysteine residues in the catalytic subunit of the yeast vacuolar H+-ATPase. Biochim Biophys Acta 1194, 329-34. Takase, K., Yamato, I. and Kakinuma, Y. (1993). Cloning and Sequencing of the Genes Coding for the A-Subunit and B-Subunit of Vacuolar-Type Na+-ATPase from Enterococcus-Hirae - Coexistence of Vacuolar-Type and F0F1-Type ATPases in One Bacterial Cell. J Biol Chem 268, 11610-16. Tomashek, J. J., Graham, L. A., Hutchins, M. U., Stevens, T. H. and Klionsky, D. J. (1997). V-1-situated stalk subunits of the yeast vacuolar proton-translocating ATPase. J Biol Chem 272, 26787-93. Tomashek, J. J., Sonnenburg, J. L., Artimovich, J. M. and Klionsky, D. J. (1996). Resolution of subunit interactions and cytoplasmic subcomplexes of the yeast vacuolar proton-translocating ATPase. J Biol Chem 271, 10397404. Towbin, H., Staehhelin, T. and Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76, 4350-54. Tsunoda, S. P., Aggeler, R., Noji, H., Kinosita, K., Yoshida, M. and Capaldi, R. (2000). Observation of rotation within the FoF1-ATP synthase: deciding between rotation of the F(o)c subunit ring and artifact. FEBS Lett 470, 244-48. Van Hille, B., Richener, H., Green, J. R. and Bilbe, G. (1995). The ubiquitous VA68 isoform of subunit A of the vacuolar H+-ATPase is highly expressed in human osteoclasts. Biochem Biophys Res Commun 214, 110813. Van Hille, B., Richener, H., Schmid, P., Puettner, I., Green, J. and Bilbe, G. (1994). Heterogeneity of vacuolar H+ATPase: Differential expression of two human subunit B isoforms. Biochem J 303, 191-98. Van Hille, B., Vanek, M., Richener, H., Green, J. R. and Bilbe, G. (1993). Cloning and tissue distribution of subunits C, D, and E of the human vacuolar H(+)-ATPase. Biochem Biophys Res Commun 197, 15-21. Vasilyeva, E. A., Minkov, I. B., Fitin, A. F. and Vinogradov, A. D. (1982) Kinetic mechanism of mitochondrial adenosine triphosphatase. ADP-specific inhibition as revealed by the steady-state kinetics. Biochem. J. 202, 9-14. Vasilyeva, E. and Forgac, M. (1996). 3'-O-(4-Benzoyl)benzoyladenosine 5'-triphosphate inhibits activity of the vacuolar (H+)-ATPase from bovine brain clathrin-coated vesicles by modification of a rapidly exchangeable, noncatalytic nucleotide binding site on the B subunit. J Biol Chem 271, 12775-82. Vasilyeva, E. and Forgac, M. (1998). Interaction of the clathrin-coated vesicle V-ATPase with ADP and sodium azide. J Biol Chem 273, 23823-9. Vogel, G. and Steinhart, R. (1976). ATPase of Escherichia coli: Purification, Dissociation, and Reconstitution of the Active Complex from the isolated Subunits. Biochemistry 15, 208-16. 131 Literaturverzeichnis Walker, J.E., Saraste, M., Runswick, M. J. and Gay, N. J. (1982). Distantly related sequences in the a- and bsubunits of ATP synthase, myosin, kinases and other ATP-requiring enymes and a common nucleotide binding fold. EMBO J 1, 945-51. Wang, Z. Q. and Gluck, S. (1990). Isolation and properties of bovine kidney brush border vacuolar H(+)-ATPase. A proton pump with enzymatic and structural differences from kidney microsomal H(+)-ATPase. J Biol Chem 265, 21957-65. Webster, L. C. and Apps, D. K. (1996). Analysis of nucleotide binding by a vacuolar proton-translocating adenosine triphosphatase. Eur J Biochem 240, 156-64. Webster, L. C., Perezcastineira, J. R., Atkins, G. L. and Apps, D. K. (1995). Allosteric regulation of proton translocation by a vacuolar adenosinetriphosphatase. Eur J Biochem 232, 586-95. Wechser, M. A. and Bowman, B. J. (1995). Regulation of the expression of three housekeeping genes encoding subunits of the Neurospora crassa vacuolar ATPase. Mol Gen Genet 249, 317-27. Werner, G., Hagenmaier, H., Drautz, H., Baumgartner, A. and Zahner, H. (1984). Metabolic products of microorganisms. 224. Bafilomycins, a new group of macrolide antibiotics. Production, isolation, chemical structure and biological activity. J Antibiot (Tokyo) 37, 110-7. Werner, G., Hagenmaier, H., Drautz, H., Baumgartner, A. and Zähner, H. (1983). Metabolic products of microorganisms. 224. Bafilomycins, a new group of macrolide antibiotics. Production, isolation, chemical structure and biological activity. J Antibiot 37, 100 -117. Wieczorek, H. (1992). The insect V-ATPase, a plasma membrane proton pump energizing secondary active transport: molecular analysis of electrogenic potassium transport in the tobacco hornworm midgut. J Exp Biol 172, 335-43. Wieczorek, H., Brown, D., Grinstein, S., Ehrenfeld, J. and Harvey, W. R. (1999). Animal plasma membrane energization by proton motive V-ATPases [Review]. Bioessays 21, 637-48. Wieczorek, H., Cioffi, M., Klein, U., Harvey, W. R., Schweikl, H. and Wolfersberger, M. G. (1990). Isolation of goblet cell apical membrane from tobacco hornworm midgut and purification of its vacuolar-type ATPase. Methods Enzymol 192, 608-16. Wieczorek, H., Grüber, G., Harvey, W. R., Huss, M., Merzendorfer, H. and Zeiske, W. (2000). Structure and regulation of insect plasma membrane H+V-ATPase. J Exp Biol 203, 127-35. Wieczorek, H., Putzenlechner, M., Zeiske, W. and Klein, U. (1991). A vacuolar-type proton pump energizes K+/H+ antiport in an animal plasma membrane. J Biol Chem 266, 15340-7. Wieczorek, H., Weerth, S., Schindlbeck, M. and Klein, U. (1989). A vacuolar-type proton pump in a vesicle fraction enriched with potassium transporting plasma membranes from tobacco hornworm midgut. J Biol Chem 264, 11143-8. Wieczorek, H., Wolfersberger, M. G., Cioffi, M. and Harvey, W. R. (1986). Cation-stimulated ATPase activity in purified plasma membranes from tobacco hornworm midgut. Biochim Biophys Acta 857, 271-81. Wilkens, S. and Capaldi, R. A. (1998). Electron Microscopic Evidence of Two Stalks Linking the F-1 and F-0 Parts of the Escherichia Coli Atp Synthase. Biochimica et Biophysica Acta - Bioenergetics 1365, 93-97. Xie, X. S. (1996). Reconstitution of ATPase activity from individual subunits of the clathrin-coated vesicle proton pump - The requirement and effect of three small subunits. J Biol Chem 271, 30980-85. Xie, X. S. and Stone, D. K. (1988). Partial Resolution and Reconstitution of the Subunits of the Clathrin-coated Vesicle Proton ATPase Responsible for Ca2+ -activated ATP Hydrolysis. J Biol Chem 263, 9859-67. Xu, T. and Forgac, M. (2000). Subunit D (Vma8p) of the yeast vacuolar H+-ATPase plays a pole in coupling of proton transport and ATP hydrolysis. J Biol Chem275, 22075-81. Yokoyama, K., Muneyuki, E., Amano, T., Mizutani, S., Yoshida, M., Ishida, M. and Ohkuma, S. (1998). V-ATPase of Thermus thermophilus is inactivated during ATP hydrolysis but can synthesize ATP. J Biol Chem 273, 20504-10. Zhang, J. M., Feng, Y. and Forgac, M. (1994). Proton conduction and bafilomycin binding by the V-0 domain of the coated vesicle V-ATPase. J Biol Chem 269, 23518-23. Zhang, J. M., Myers, M. and Forgac, M. (1992). Characterization of the V0 Domain of the Coated Vesicle (H+)ATPase. J Biol Chem 267, 9773-78. Zhang, J. M., Vasilyeva, E., Feng, Y. and Forgac, M. (1995). Inhibition and labeling of the coated vesicle V-ATPase by 2-azido-[P-32]ATP. J Biol Chem 270, 15494-500. Zhang, X., Herring, C. J., Romano, P. R., Szczepanowska, J., Brzeska, H., Hinnebusch, A. G. and Qin, J. (1998). Identification of phosphorylation sites in proteins separated by polyacrylamide gel electrophoresis. Anal Chem 70, 2050-9 132 Publikationen 8. Publikationen Orginalpublikationen: Grüber, G., Radermacher, M., Ruiz, T., Godovac-Zimmermann, J., Canas, B., KleineKohlbrecher, D., Huss, M., Harvey, W. R. and Wieczorek, H. (2000). ThreeDimensional Structure and Subunit Topology of the V(1) ATPase from Manduca sexta Midgut. Biochemistry 39, 8609-16. Huss, M., Gaßel, M., Dröse, S., Ingenhorst, G., Koenig, S., Zeeck, A., Altendorf, K. and Wieczorek, H. (in Revision). Plecomacrolides bind to subunit c of the insect VATPase. Biochemistry. Merzendorfer, H., Graf, R., Huss, M., Harvey, W. R. and Wieczorek, H. (1997). Regulation of proton-translocating V-ATPases. J Exp Biol 200, 225-35. Merzendorfer, H., Huss, M., Schmid, R., Harvey, W. R. and Wieczorek, H. (1999). A novel insect V-ATPase subunit M9.7 is glycosylated extensively. J Biol Chem 274, 17372-8. Svergun, D. I., Konrad, S., Huss, M., Koch, M. H., Wieczorek, H., Altendorf, K., Volkov, V. V. and Grüber, G. (1998). Quaternary structure of V1 and F1 ATPase: significance of structural homologies and diversities. Biochemistry 37, 17659-63. Wieczorek, H., Grüber, G., Harvey, W. R., Huss, M. and Merzendorfer, H. (1999). The plasma membrane H+-V-ATPase from tobacco hornworm midgut. J Bioenerg Biomembr 31, 67-74. Wieczorek, H., Grüber, G., Harvey, W. R., Huss, M., Merzendorfer, H. and Zeiske, W. (2000). Structure and regulation of insect plasma membrane H(+)V-ATPase. J Exp Biol 203, 127-35. Abstracts und Kurzvorträge: Huss, M., Merzendorfer, H., Dietlein, T., Harvey, W. R. and Wieczorek, H. (1997). Regulation of the insect V-ATPases. Verhandlungen der Deutschen Zoologischen Gesellschaft, 107 Huss, M., Grüber, G., Harvey, W. R., Merzendorfer, H., Schmid, R. and Wieczorek, H. (1999). Structural features of the insect V-ATPase: the catalytic V1 complex. Zoology 102, 66 Merzendorfer, H., Huss, M., Reineke, S., Harvey, W. R., Schmid, R. and Wieczorek, H. (1999). Structural features of the insect V-ATPase: the membrane bound Vo complex. Zoology 102, 69 Merzendorfer, H., Huss, M., Reinike, S., Harvey, W. R., Schmid, R. and Wieczorek, H. (1999). Structural features of the insect V-ATPase: the Vo complex. Comparative Biochemistry and Physiology 124A, S81 Kurzvortrag 5th Inter. Congress of Comparative Physiology and Biochemistry, Calgary 1999: Huss, M., Grüber, G., Harvey, W. R., Merzendorfer, H., Schmid, R. and Wieczorek, H. (1999). Structural features of the insect V-ATPase: the V1 complex. Comparative Biochemistry and Physiology 124A, S6 133 Abkürzungsverzeichnis 9. Abkürzungsverzeichnis ADP: Adenosin-Diphosphat AMP-PNP: Adenylyl-Imino-Diphosphat APP: Alkalische Phosphatase APS: Ammoniumpersulfat ATP: Adenosin-Triphosphat BCIP: 5-Bromo-4-Chloro-3-Indolylphosphat BSA: Bovine serum albumin BzATP: 3-O-(4-Benzoyl)Benzoyladenosin-5´-triphosphat C12E10: Polyoxyethylen-10-Laurylether DCCD: N´,N´-Dicyclohexylcarbodiimid DMSO: Dimethylsulfoxid DTT: Dithiothreitol EDTA: Ethylendiamintetraessigsäure GCAM: Goblet cell apical membrane GTP: Guanosin-Triphosphat J-Concanolid A: 9-O-[p-(Trifluoroethyl-diazirinyl)-benzoyl]-21,23-dideoxy-23-epi[125I]Iodo-concanolid A kDa: Kilodalton MALDI-MS: Matrix unterstützte Laserdesorptions-Ionisations-Massenspektroskopie MBP: Maltose bindendes Protein MOPS: N-Morpholinopropansulfonsäure NBT: 4-Nitroblautetrazoliumchlorid-Hydrat NEM: N-Ethylmaleimid PAGE: Polyacrylamid-Gelelektrophorese SDS: Sodium dodecyl sulfate TCA: Trichloressigsäure TEMED: N,N,N´,N´-Tetramethylethylendiamin Tris: 2-amino-2-(hydroxymethyl)-1,3-propandiol Tween 20: Polyoxyethylensorbitanmonolaureat 134 Lebenslauf 10. Lebenslauf Markus Huß Persönliche Angaben Geburtstag: Geburtsort: Staatsangehörigkeit: Familienstand: Eltern: 13. März 1968 Augsburg deutsch seit 25. Juli 1997 verheiratet mit Tanja Huß, geb. Beutmüller Huß Manfred und Renate, geb. Rößler Schulbildung 1974-1978 Besuch der Hans Adlhoch Grundschule in Augsburg 1978-1987 Besuch des Holbein Gymnasiums in Augsburg (mathematischnaturwissenschaftlicher Zweig) Juni 1987 Erlangung der allgemeinen Hochschulreife Wehr/Ersatzdienst Juli 1987-Feb. 1989 Zivildienst beim Malteser Hilfsdienst Augsburg Hochschulbildung Okt. 1989-Sept. 1995 Studium an der Ludwig-Maximillians-Universität München, Diplomstudiengang Biologie Mai 1992 Vordiplom Hauptstudium mit der Fächerbelegung Zoologie (Hauptfach), Biochemie, Pflanzenphysiologie und Anthropologie/Humangenetik Diplomarbeit im Labor von Prof. Dr. Wieczorek, Zoologisches Institut der Universität München Sept. 1995 Abschluß als Diplom-Biologe univ. 1996 Wissenschaftlicher Mitarbeiter von Prof. W. R. Harvey (Temple University, PA, USA) 1997 Promotionsstudium Doktorarbeit im Labor von Prof. Dr. Wieczorek (zunächst Zoologisches Institut der Universität München, seit 1998 Arbeitsgruppe Tierphysiologie Universität Osnabrück) 135 Eidesstattliche Erklärung 11. Danksagung An dieser Stelle möchte ich mich bei allen recht herzlich bedanken, die mich während meiner Studien und der vorliegenden Doktorarbeit unterstützt und zum erfolgreichen Abschluß beigetragen haben: Zunächst bedanke ich mich bei M. sexta ohne deren Mitteldarmepithel es die ganze VATPase Forschung wesentlich schwerer hätte. Prof. Dr. Helmut Wieczorek danke ich für die Betreuung, die guten Forschungsbedingungen in einem modernen Labor und die vielen intensiven, nicht nur forschungsrelevanten Diskussionen. Prof. Dr. Karlheinz Altendorf danke ich für die Übernahme des Koreferates und die Zusammenarbeit im Rahmen der Untersuchungen der Makrolide. Bei allen Mitgliedern unserer ehemaligen Arbeitsgruppe aus München und unserer jetzigen Arbeitsgruppe hier in Osnabrück bedanke ich mich für die gute Zusammenarbeit und das angenehme Arbeitsklima, besonders bei Bruni Förg-Brey, Gundula Key, Daniela KleineKohlbrecher, Thomas Krüppel, Harald Mikoleit, Hans Merzendorfer, Stephan Reineke, Tobias Schafmeier und Wolfgang Zeiske. Weiterhin vielen Dank an die Arbeitsgruppe Mikrobiologie für die stets freundliche Hilfe und Unterstützung bei Problemen aller Art, hier vor allem an Michael Gaßel und Stefanie Konrad. Bei Simone König, Klaus Fendler, Eva Grabsch, Barbara Schedding, Roland Schmid, Gudrun Ingenhorst, Prof. Zeeck, Michel Koch und Gerhard Grüber bedanke ich mich für die erfolgreiche Zusammenarbeit. Weiterhin möchte ich mich bei Prof. Reichenbach und Dr. Sasse von der GBF (Braunschweig) und Dr. Boyd vom NCI (Maryland) für die Bereitstellung von Makroliden bedanken. Nicht zuletzt möchte ich mich bei meiner Familie bedanken, vor allem bei meiner Mutter und meiner Ehefrau Tanja, die mich immer unterstützt und mir den Rücken freigehalten haben. 136