DIABETES MELLITUS ["Honigsüsser Durchfluss"]
Werbung

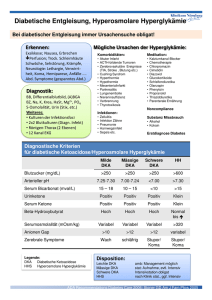
DIABETES MELLITUS ["Honigsüsser Durchfluss"] - ZUCKERHARNRUHR HISTORISCHES: Diabetes → allgemeine Bezeichnung für Erkrankungen, die mit einer überhöhten Ausscheidung von Urin (=Polyurie) einhergehen. Das Wort "Diabetes" kommt aus dem Griechischen und bedeutet eigentlich "die Beine spreizend" (aufgrund des verstärkten Harnflusses). Im allgemeinen wird es jedoch mit "Durchgang" oder auch "Harnruhr" übersetzt. Auch beim Diabetes mellitus, einer speziellen Form des Diabetes, tritt das Symptom der überhöhten Harnausscheidung auf. "Mellitus" ist lateinisch und bedeutet "honigsüß". Der Name DM [also "honigsüßer Durchfluss" o. "Zuckerharnruhr"] bezieht sich auf den süßen Geschmack des Urins von Zuckerkranken, der erstmalig im 17. Jahrhundert von dem engl. Mediziner und Naturphilosophen Thomas Willis beschrieben wurde. Das "Schmecken" des Urins war zu dieser Zeit die einzige Möglichkeit, die Krankheit zu diagnostizieren. DEF: → chron. STW-Erkrankung, die auf einem absoluten oder relativen Insulinmangel beruht und in deren Folge es – zumeist erst nach längerer Krh.dauer - zu Schäden an anderen Organen [v.a. Blutgefässen] kommen kann, d.h. es handelt sich um eine heterogene Gruppe von Erkrankungen daher: SYNDROM DM eine Störung im KH-STW mit dauernder Hyperglykämie [BlutGlucose: > 120 mg/dl] und latenter oder manifester Glukosurie! → später – sekundär - auch Störungen des Protein-STW und Fett-STW; → Unbehandelt völlige STW-Entgleisung COMA DIABETICUM! Blutzucker normal: nüchtern: postprandial: < 110 mg/dl max.140 mg/dl > 126 mg/dl → "diabetischen Stoffwechsellage" > 160-180 mg/dl → Nierenschwelle für Glukose überschritten [Glucosurie] Kurz-Exkurs Physio: KH-Resorption im DüDa in Form von Monosacchariden [Gluc,Gal,Fruc] Leber z.t. Polymerisation zu Glykogen = Glykogenese via G-6-P [Steigerung: Insulin // Hemmung: Glukagon, Adrenalin] resorbierte überschüssige Gluc passiert LE und gelangt ins strömende Blut entweder zur Glucosedeckung der Peripherie [Gehirn, Herz, Muskulatur oder Umwandlung in FS [Depotfett]; REGULATION KH-STW: BLUTZUCKERSPIEGEL Auffüllung der Glykogenreserve in LE und MU und Umwandlung von Gluc zu Fett; BLUTZUCKERSPIEGEL Mobilisierung von Glykogen aus LE und MU und ev. Gluconeogenes aus AS; 1. INSULIN – BLUTZUCKER Bildung reguliert durch Nahrungsaufnahme: reichliche KH-Aufnahme mit Blutzucker Insulin Hunger [KH-Mangel] Insulin LANGERHANS'schen INSELORGAN – Pancreas: B-Zellen [~80%] INSULIN A-Zellen [~10%] GLUKAGON D-Zellen [~10%] Somatostatin, Gastrin D1-Zellen VIP [vasoaktives intestinales Peptid] PP-Zellen [~2%] PP [pankreatisches Polypeptid] 2. ADRENALIN - Blutzucker Glykogenabbau in LE + MU und Freisetzung von GLUC; ANTAGONIST zu INSULIN; 3. GLUCAGON - Blutzucker Glykogenabbau nur in LE ; 4. GLUCOCORTICOIDE + ACTH - Blutzucker Gluconeogenese aus AS ; Oxydation von KH 5. STH [Somatotropin] - Blutzucker Glucoseoxydation durch Proteinsynthese EPI: INSULIN: Biosynthese von Proinsulin u. Speicherung erfolgen in den Beta- = B-Zellen der LANGERHANS* Inseln. Aus diesem ringförmigen, einkettigen Molekül entsteht durch enzymatische Abspaltung eines C-Peptids (»connecting peptide«) das Insulin, das aus zwei durch 2 Disulfidbrücken verknüpften Peptidketten A u. B besteht; das Molekulargewicht beträgt ca. 5000–6000. Die Gesamtmenge im ruhenden Pankreas beträgt ca. 250 IE entspr. 250 U entspr. 10 mg. Die Sekretion (ca. 1–2 mg/d) wird durch verschiedene physiolog. Reize stimuliert oder gehemmt (Tab.). – Abbau erfolgt v.a. in der Leber (Spaltung der Disulfidbrücken durch Insulinase; Proteolyse der A- u. BKette), ferner in Niere, sonstigen Geweben u. Blut (biologische Halbwertszeit ca. 30 Min.); z.T. erfolgt Ausscheidung mit dem Harn die Zahl der Typ 2-Diabetiker steigt mit http://www.8ung.at/prionerl 1 DISCLAIMER ! dem Ausmass an Überernährung; Prävalenz manifester Diabetiker ist altersabhängig: < 50 J. 1-2% / >50 J. im Durchschnitt ca. 5% bei den Inuit in Grönland selten! KLASSIFIKATION nach der ÄTIOLOGIE [WHO und ADA american diabetes association, 1997] PRIMÄRER DM [idiopathisch!] 1. Typ-1-D = Insulinabhängiger DM = früher: JUVENILER DM / IDDM ( 5% ) DEF: Autoimmunerkrankung, die - bei genetischer Disposition und unter Einfluss verschiedener Faktoren – zu einer autoaggressiven lymphozytären Insulitis mit selektiver B-Zelldesktruktion und – daraus folgendem absoluten Insulinmangel führt ! Typ-1-A: immunologisch bedingt Typ-1-B: idiopathisch [in Europa selten!] EPI: v.a. zw. 4.-5. LJ und 12.-14. LJ [Adoleszenz] auftretend; gelegentlich aber auch nach dem 20.-30. LJ [=JODA]! ♀ + ♂ gleich häufig; ÄTIO/PATHO: Ursache nicht eindeutig geklärt! Vermutung: EXOGENE NOXEN, z.B.: Viren [Mumps / Rubella / Myxovirus influenzae, Coxsackie] verändern Proteinstruktur der B-Zellen, wodurch diese sich dann – unter bestimmter genetischer Voraussetzung - als AUTO-AG's präsentieren; Genetische Prädisposition ~ 20 % → geringe positive Familienanamnese [Gene auf Chromosom 6p; HLA II] Konkordanz ["Übereinstimmung bei eineiigen Zwillingen]: nur ~ 30-50% daher exogene Faktoren vermutet; 95% der Typ 1-D exprimieren: HLA-DR3 u./o. HLA-DR4 [aber auch bei ~40% der Nicht-Diabetiker vorhanden!] genetische Basis vorhanden – Vererbungsmodus aber nicht bekannt; Exogene Faktoren: aufgrund nicht eindeutiger genetischer Prädisposition → exogene Faktoren mitverantwortlich → Viren !!! [s.o.] Immunpathologische Reaktion: - Indikatorläsion → autoagressive INSULITIS: Infiltration der Inseln durch zytotoxische T-Lymphos! - diese reagieren mit den B-Zellen Nekrose von Inselzzellen und Bildung von Auto-AK*! *AUTO-ANTIKÖRPER = ICA = islet-cell antibodies [70-90% der Pat.]: haben unterschiedliche Ziel-AG, daher Einteilung in: Insulin-Auto-AK GADA [AK gg. Glutamat-Decarboxylase] AK gg. Thyrosinphosphatase - in etwa 10% Assoziation mit and. Autoimmunerkrankungen [chron. lymphozyt. Thyreoditis, perniziöse Anämie]; MORPH: bei Diagnosestellung viele Inseln keine insulinproduzierend B-Zellen mehr [ Nekrose ] Inseln mit noch funkt., vorhandenen HYPERTROPHEN B-ZELLEN mit meist wenig lymphozyt. Infiltration; ATROPHIE des exokrinen Pancreas [Insulin ist Wachstumsfaktor!] → Gewicht ↓ meist < 50 g [normal: Weisswurst (80-120g/12-15cm/gelblich-weiß)] keine Insel-Amyloidose! [# DM Typ 2] KLIN/PATH.: erst wenn > 80% der B-Zellen zerstört Manifestation des DM! 1. lange, stumme präklinische Phase Monate bis Jahre, jedoch hohe ICA - Titer! 2. klinische Manifestation: meist akut, abrupt infolge von Infekt; ev. mit diabetischer Ketoazidose Coma acidoticum http://www.8ung.at/prionerl 2 DISCLAIMER ! 3. Honey-Moon-Phase kurze vorübergehende Verbesserung unter effektiver Behandlung 4. Vollständige Insulinabhängigkeit exogene Insulinzufuhr notwendig !!! Kr.h. lediglich symptomatisch behandelbar; exokrine Funktionsstörungen!!! durch Atrophie des exokirnen Pancreas Beim Typ-I-Diabetes sind die klassischen Symptome meist vorhanden: Symptome: Polydipsie, Polyurie, Müdigkeit, Abgeschlagenheit, Leistungsminderung, Pruritus genitalis, Gewichtsabnahme, Sehstörungen, Hypoglykämien mit Hungergefühl, Kaltschweißigkeit u. Tachykardie, Muskelkrämpfe, Übelkeit u. Erbrechen. Akute Komplikationen: diabetische Ketoazidose ketoazidotisches Koma! Glucose nicht verwertbar Energie Lipolyse freie FS Ketonkörper metabolische Ketoazidose [Acetongeruch! Kussmaul-Atmung] Coma acidoticum! 2. Typ 2-D = InsulinUNabhängiger DM = früher: ALTERS DM / NIDDM ( 95% ) DEF: Familiär gehäuft auftretende Störung des KH-STW mit Insulinresistenz und Insulinmangel ! 1. gestörte Insulinsekretion [ob in Zusammenhang mit Inselamyloid → unklar!] 2. herabgesetzte Insulinwirkung/Insulinresistenz Ursache ist ein Insulinrezeptordefekt und ein Postrezeptordefekt = Glucoseverwertung in Zelle [Gesunde: nur HIR-A # Typ-2-Diabetiker: HIR-A und HIR-B auf Zellmembran [HIR=Human Insulin-Rezeptor] → B-Zellen lange erhalten werden mit zunehmender Amyloidose weniger; EPI: selten vor dem 40. LJ. auftretend → AUSNAHME: autosomal dominanter MODY [s.u.] ÄTIO/PATHO: Ursache nicht genau bekannt*; Die Mehrzahl der Erkr. jedoch entwickeln sich auf dem Boden eines: METABOLISCHEN SYNDROMS [=WOHLSTANDSSYNDROM]! Gehäuftes Zusammentreffen folgender 4 Faktoren: Stammbetonte Adipositas und Überernährung Dyslipoproteinämie [ Triglyceride ↑ / HDL-Cholesterin ↓ ] essentielle Hypertonie Glucosetoleranzstörung *Vermutung: genetische Defekte sowie Manifestation durch Umweltfaktoren: genetische Prädisposition Konkordanz: > 90% keine spez. HLA-Expressionsmuster periphere Insulinresistenz = Endorganresistenz erhöhte Resistenz von Muskel- und Fettgewebe ggüber Insulin Ursache [genetisch/exogen] + zelluläre Basis [Insulin-/Postrezeptor/Glucosetransporter] ungeklärt; exogene Faktoren Häufigkeit korreliert mit Häufigkeit Adipositas → Wohlstandserkrankung rund 80% der Typ-2-DM → adipös ADIPOSITAS induziert periphere Insulinresistenz → Kompensation durch vorübergehende Hyperinsulinismus möglichweise durch "Überlastung" der B-Zellen bei Adipositas zusammen mit genetischer Prädisposition → Manifestation der Insulinresistenz! B-Zell-Funktionsstörung inadäquate Proinsulin-/Insulinproduktion der B-Zellen auf Glucosereiz! zu viel Insulin wird gebildet!!! Sensibilität der Rezpetoren ↓ "Down-Regulation" → Circulus vitiosus! vermutlich Glucose-Rezeptorstörung der B-Zellen; z.t. aber auch verringerte B-Zellzahl bei einem Teil der Patienten; Inselamyloid = IAPP [Amylin=Inselamyloid-Polypeptid, AEndokrines-Amyoloid] physiolog. Funktion unbekannt! zu 50% ident mit CGP der C-Zellen [Schilddrüse]; wird von B-Zellen synthetisiert und zusammen mit Insulin sezerniert → IAPP → proteolytisch gespalten → Polymerisation zu Amyloidfirillen [ß-Fibrillen mit ßFalltblattstruktur] → in Langerhans Inseln - ausserhalb der B-Zellen - Deponierung als Inselamyloid → dadurch "Erstickung" der Inselfunktion → B-Zellen werden weniger; http://www.8ung.at/prionerl 3 DISCLAIMER ! MORPH: immer eine grosse Anzahl von morhpologisch intakt erscheinend B-Zellen; einzige Abweichung vom Normalbefund: - bei etwa 80 % der Pat → Ablagerung von Amyloid um Inselkapillaren; - bei kleinem Teil der Pat. → 50%ige Reduktion der B-Zellen; KLIN/PATH.: beginnt schleichend, klassische Symptome fehlen häufig; oft erst an drastischen Spätkomplikationen erkannt!! selten Ketoazidose; hyperosmolares Koma [Dehydratation bei zu geringer Flüssigkeitsaufnahme!] zuerst Diät + Sulfonylharnstoff [Insulinsekretion↑], später Insulintherapie [deutl. Reduktion d. B-Zellen] SEKUNDÄRER DM [Ätio bekannt!] durch Zerstörung von Pancreasgewebe durch hormonelle Dysregulationen sekundärer Diabetes mellitus infolge von Pankreaserkrankungen (z.B. chronische Pankreatitis; akute Pancreatitis - Pancreasnekrose, Malnutritionsdiabetes) endokrinen Erkrankungen (z.B. Phäochromozytom, CUSHING* Syndrom, Akromegalie, Hyperthyreose, Glucagonom, VIPom, Somatostatinom) iatrogenen oder toxischen Noxen (z.B. Corticoide, Diuretika, Diazoxid, Ciclosporin, Cyclophosphamid, L-Asparaginase, Pentamidin, Pyriminil [Rattengift]) genetischen Syndromen Diabetes mellitus aufgrund anderer Faktoren z.B. insulinresistenter Diabetes mit Acanthosis nigricans Gruppe A: junge Frauen mit Hirsutismus u. polyzystischen Ovarien mit reduzierter Insulinrezeptorenzahl durch Punktmutation Gruppe B: ältere Patienten mit Antikörpern gegen Insulinrezeptoren, erhöhter BSG u. DNS-Antikörpern Gestationsdiabetes Folge von insulinantagonistischen Wirkungen durch HPL [human placental lactogen], Cortisol, Progesteron, Prolactin u. erhöhter Insulindegradation (meist bei Anlage zu Typ-II-Diabetes) bei allen resp. den meisten dieser Erkrankungen – auch bei langen Kr.h.verlauf → "normale" Insulinsez. B-Zellen und mehr oder weniger normale Zellzahl [Ausnahme: Hämochromatose]; bei chron. Pancreatitis + zystischer Fibrose → Korrelation zw. Schweregrad/Dauer der Erkrankung und Entwicklung eines DM [Vermutung, dass eine Sklerose des exokrinen Pancreas die Inselfunktion direkt [vaskulär?] schädigt, sowie durch Reduktion der Inselzellzahl;] MOD = Maturity-Onset-Diabetes der erst in höherem Lebensalter (gemäß WHO nach dem 65. Lj.) auftretende DiabeDM (»Altersdiabetes«) des Typs II; MODY = Maturity-Onset-Diabetes of the Young der Typ-II-Diabetes bei Jugendlichen; JODA = Juvenile-Onset-Diabetes of the Adult der Typ-I-Diabetes bei Erwachsenen (> 40 J.) gegenüber. LADA = latent autoimmune diabetes with adult onset [frühere Bezeichnung für Typ-1-DM bei > 30Jährigen] KLINIK – ALLGEMEIN: unspezifische Allgemeinsymptome Müdigkeit, Leistungsminderung Symptome durch Hyperinsulinismus und passagere Hypoglykämien [Initialstadium Typ-2-D]: Heisshunger, Schwitzen, Kopfschmerzen etc. Symptome infolge Hyperglykämien und Glukosurie mit osmotischer Diurese: Polyurie, Durst, Polydipsie, Gewichtsverlust Symptome durch Störungen im Elektrolyt und Flüssigkeitshaushalt: nächtliche Wadenkrämfe , Sehstörungen [wechselnder Turgor der Augenlinse] Haut - Pruritus [oft genito-anale Lokalisation] - Bakterielle/mykotische Hautinfektionen [z.B.: Furunkulose! / Candidamykose!] - Rubeosis diabetica [diabetische Gesichtsröte] - Necrobiosis lipoidica [meist an beiden US, bräunlich rote Herde, Ulzerationen möglich] Potenzstörungen, Amenorrhoe; http://www.8ung.at/prionerl 4 DISCLAIMER ! KOMPLIKATIONEN / FOLGEKRANKHEITEN – DM AKUTE KOMPLIKATION → COMA DIABETICUM !!! [siehe Herold S. 614!] KETOAZIDOTISCHES KOMA → typisch für Typ-1-DM HYPEROSMOLARES KOMA → typisch für Typ-2-DM akuter Insulinmangel → Aufnahmestopp von Glucose in Muskel- und Fettgewebszellen und erhöhte Glykolyse in Leber → HYPERGLYKÄMIE und Überschreiten der Nierenschwelle [ Glucoserückresorption ] → GLUKOSURIE und damit POLYURIE aufgrund osmotischer Diurese → Verlust von H2O und Elektrolyten; Glucose im Blut → Hyperosmolarität im Blut [EZR: Dehydratation HypoVOL! // IZR: Dehydratation Bewusstseinsstörung!] + [zusammen mit Polyurie] → POLYDYPSIE; kompletter Insulinmangel [ Glucoseaufnahme absolut ] → Energiegewinnung des Körpers durch → LIPOLYSE mit Erhöhung der freien FS im Blut → in der Leber → Oxidation zu KETONKÖRPERN → METABOLISCHE KETOAZIDOSE → COMA ! unvollständigem Insulinmangel [Typ-2-D] → DEHYDRATION im Vordergrund → bei ungenügender H2Oaufnahme → HYPEROSMOLARES NICHTKETOAZIDOTISCHES KOMA ! anhaltende Hyperglykämie → stimuliert nichtenzymatische Glykosylierung [nichtenzymatisches Ankoppeln von Glukosemolekülen an Glykoproteine] → Struktur- und Funktionsänderung der Glykoproteine → veränderte Glykoproteine in BM → Verdickung und Lumeneinengung → diabetische Mikroangiopathie! in der Intima von Muskeltyp-Arterien → irreversible Verbindung dieser "schlechten" Glykoproteine mit LDL → LDL's werden in Intima "gefangen" gehalten → beschleunigte Ablagerung von Cholesterin [LDL-gebunden!!!] → ATHEROSKLEROSE! weiters übermässige Aufnahme von Glucose in "insulinUNabhängige" Zellen [NZ, Gefässperizyten = Adventitiazellen + Linsenzellen] → durch Aldosereduktase Umwandlung von Glukose → SORBITOL [ein Polyol] und Fruktase → IZR Osmolarität ↑ → H2O-Einstrom → Zellschwellung und Trübung [z.B.: Katarakt]; Sorbitol → ATPase ↓ → Schwann'sche Zellen + Perizyten → möglicherweise dadurch Polyneuropathien und Mikroaneurysmen in Retinakapillaren! MERKE: Diabetesspezifische Krankheitsbilder !!! → diabetische Retinopathie und Zuckerstar → diabetische Nephropathie → diabetische Neuropathie → diabetische (Fuss-)Gangrän diabetische Gangrän: die v.a. an den Zehen (»Diabetesfuß«) vorkommende trockene oder – superinfiziert – feuchte Gangrän (Abb.) als Komplikation des DM, bedingt durch eine Kombination von Arteriosklerose u. diabetischer Mikroangiopathie u. Polyneuropathie. a) mikroangiopathisches Ulkus der Fußsohle; b) mikroangiopathische Zehengangrän 1. Diabetische Makroangiopathie [= Atherosklerose] diabet. Hyperlipidämie + Hypercholesterinämie frühzeitige Veränderung der Arterien aller Kaliber → Sklerose!!! Folgen/Komplikationen: Myokardinfarkt [~ 40% aller Diabetiker sterben an kardiovaskulären Komplikationen]; Hirninfarkt diabetisches Extremitätengangrän [v.a. an Zehen] s.o.; http://www.8ung.at/prionerl 5 DISCLAIMER ! 2. Diabetische Mikroangiopathie [v.a. Typ-1-D, weil früher Beginn] – 50 % aller Amputationen betrifft alle Kapillaren, v.a. aber Niere + Auge! sog. nichtenzymatische Proteinglykosilierung [s.o.] → Sutrukturstörung der mikrovaskulären BM mit Verbreiterungen [ Glomerulum ] → Permeabilitätsstörung!! Komplikationen: Retinopathia diabetica/Diabetische Glomerulosklerose [s.u.] Diabetische Angiopathie: Ausbildung von Mikroaneurysmen der Arteriolen u. Kapillaren der Retina des Auges; Sie entsprechen morphologisch den Veränderungen glomerulärer Gefäße der Niere; 3. DIABETISCHE NEPHROPATHIE – Hälfte aller Dialysepatienten sind Typ-2-Diabetiker! = Überbegriff → völlig unterschiedliche Nierenveränderungen können einzeln oder kombiniert auftreten; Makroskopisch sind Diabetesnieren oft "ziegelrot/gelbrot" → Oxidationsprodukt zw. O2 und den gespeicherten Fett-, Glykogen- und Glucosesubstanzen; DIABETISCHE GLOMERULOPATHIE/-SKLEROSE (S. 794 Böcker) ist eine besondere Manifestation der diabetischen Mikroangiopathie [Verdickung der BM + Vermehrung der Mesangiumzellen, sowie der mesangialen Matrix]; ist 3.häufigste Ursache der chronischen Niereninsuffizienz ! ÄTIO/PATH: nur in Ansätzen bekannt! Bildung von AGE's [advanced glycosilation endproducts] → Glykosylierung von Matrixproteinen der BM → Anhäufung; in vitro - Glucoseerhöhung → Neuexpression von Genen, z.B.: Gene für CTGF [connective tissue growth factor] → stimuliert Matrixbildung + Proliferation von Fibroblasten im Bindegewebe; MORPH: 2 Formen – Unterschied lediglich quantitativ: DIFFUSE GLOMERULOSKLEROSE: - Verdickung der BM der Glomerulusschlingen + vermehrte Ablagerung von BM-Material in mesangialer Matrix; - diffuse Homogenisierung des Mesangiums durch Ablagerung von PAS-positvier Grundsubstanz; NODULÄRE GLOMERULOSKLEROSE = KIMMESTIEL-WILSON-SYNDROM - diabetesspezifische Läsion; in Assoziation mit einer diffusen GS bei ~ 30% der Diabetiker; - Synthese- und Abbaustörung der glomerulären BM → BM-Verdickung → noduläre Ablagerung von PASpositiven BM-Material im Mesangium; [Glomerulusschlingen liegen wie "Kappen" auf nodulären Ablagerungen]; - eosinophile Niederschläge ["Tropfen"] an der Innenseite der Glomeruluskapsel [Proteinverlust!]; - "Fibrinkappen" an der Aussenseite der Glomeruluskapsel [Proteinverlust!] - weiters ausgeprägte Arteriosklerose + Arteriolosklerose KLIN/PATH.: mehr oder weniger stark bei den meisten Diabetikern [nach ~10 J.] bei starker Ausprägung → starker Verlust von glomerulären Proteinen → Proteinurie! in weiterem Verlauf → Verödung der Lumina der Glomerulusschlingen → GFR↓ → renale Hypertension! bei vollständiger Sklerose von mehr als 50% der Glomeruli [makroskopisch deutl. Schrumpfniere] → chronische Niereninsuffizienz! http://www.8ung.at/prionerl 6 DISCLAIMER ! Low power view showing sclerosis of para-glomerular arterioles in kidney High power view of diabetic glomerulopathy "Kimmelstiel-Wilson" diabetische Triopathie das KIMMELSTIEL*-WILSON* Syndrom, kombiniert mit diabetischer Retinopathie u. Polyneuropathie; GLYKOGEN-NEPHROSE schwere, über Stunden anhaltende Hyperglykämie → verstärkte Glucoserückresorption bei Glucosurie aus dem Harn → Deposition von Glykogen in Tubulusepithelzellen → Schwellung der Tubulusepithelzellen → sog. ARMANNI-EBSTEIN-ZELLEN → [feinvakuoläres bis optisch leeres Zytoplasma] weiters in NIERE: Diabetische Mikroangiopathie: Vasa afferentia + Vasa efferentia betroffen → hyaline Arteriolosklerose; Beachte: Vasa efferentia bei NICHT-DIABETISCHEN Formen praktisch nicht befallen; Eitrige Pyelonephritis [Infektionsanfälligkeit ↑] ischämische Papillennekrosen [durch örtliche Zirkulationsstörungen] 4. AUGE – rund 12 % aller Erblindungen ! CATARACTA DIABETICA = ZUCKERSTAR Osmotische, vakuoläre Degeneration des LinsenEP → Linsentrübung; RETINOPATHIA DIABETICA = häufigste Erblindungsursache in den Industrieländern! Kapilläre Mikroaneurysmen + Mikroangiopathie → Mikroinfarkte [=Punkt-Klecks-Hämorrhagien] Retinitis proliferans [Organisation kleiner Blutungen] → Glaskörperschrumpfung durch Bildung von Granulationsgewebe → Amotio/Ablatio retinae [Netzhautablösung] Diabetische Retinopathie Mikroangiopathie mit folg. Veränderungen: 1. 2. 3. 4. 5. 6. Mikroaneurysmen intraretinale multiple flohstichartige o. grösserflächige Blutungen Arteriolen- und Kapillarverschlüsse intraretinale und präretinale Gefäßproliferationen harte Exsudate Cotton-Wool-Herde [retinale Nekrose; kleine, helle, wolk. Exsudatherde am Augenhintergrund, bes. an den Verzweigungsstellen d. Netzhautvenen] Ätio der Mikroangiopathie: Perizytenschaden + Aufsplitterung der kap. BM; Häufige Spätschäden: - http://www.8ung.at/prionerl Rubeosis iridis [vermehrte Füllung u.Schlängelung (z.T. Neubildungen) d. Blutgefäße d. Iris] sek. Winkelblockglaukom [Engwinkelglaukom; durch Einengung des Kammerwinkels] Iridopathia diabetica, Cataracta diabetica diabetische Opticusatrophie 7 DISCLAIMER ! 5. DIABETISCHE LEBER Hyperglykämie → Glykogenablagerung in Hepatozyten [sekundäre Glykogenose*: Gruppe angeb. STW-Krankheiten mit Enzymdefekten] Präp.fixation: sog. Lochkerne; gleichzeitig häufig bei Typ-2-DM → LEBERZELLVERFETTUNG; 6. ZNS DIABETISCHE POLYNEUROPATHIE [Spätkomplikation, nach ca. 25 J bei ca. 50% der Pat.] entwickelt sich v.a. peripher → mehr die sensiblen, weniger die motorischen Funktionen betroffen; Zerstörung der Axone u/o Markscheiden [=Entmarkung] → Hyporeflexie, Störung d. Tiefensensibilität [Stimmgabeltest]; Komplikationen: diabetische Mikroangiopathie + diabet. Polyneuropathie → ZEHENGANGRÄN !!!! MIKROANGIOPATHIE IM ZNS 7. HAUT XANTHOMA DIABETICUM /XANTHELASMEN [an Augenlidern] gutartiger (bis pflaumengroßer) gelbliche Tumoren aus Fibroblasten, Reticulinfasern u. Histiozyten mit schaumigem Zytoplasma (Schaumzellen/Makrophagen; bei entspr. Stoffwechselstörung Lipoidtröpfchen, evtl. auch Cholesterinester enthaltend, nach Lipoidextraktion »hell«). Bei stärkerem Bindegewebsgehalt als »Xanthofibrom« bezeichnet; Xanthelasmen in der Lidhaut rötliche oder strohgelbe, bis fingernagelgroße, z.T. streifenförmige, scharf begrenzte Einlagerungen in der Haut; flach erhaben (= X. planum), ganz selten höher (= X. tuberosum); häufig Symptom einer Fettstoffwechselstörung (Hypercholesterinämie, Diabetes mellitus). Am häufigsten an den Lidern (= X. palpebrarum, Abb.), v.a. am inneren Augenwinkel. NECROBIOSIS LIPOIDICA DIABETICORUM = OPPENHEIM*-URBACH* KRANKHEIT Necrobiosis lipoidica diabeticorum - zentrale Nekrosen mit umgebenden Granulationsgewebe, Schaumzellen, Riesenzellen; - symmetrische, bis handtellergroße, (braun-)rote, evtl. gesprenkelte, flach eingesunkene, evtl. ulzeröse Hautatrophie (s. Abb.) mit großbogiger violetter Begrenzung an den Streckseiten der Unterschenkel (seltener Hände u. Oberschenkel), oft kombiniert mit (Prä-)Diabetes mellitus; - histol: Endangiitis, Kollagennekrose (mittl. Korium), Lipoidspeicherung, lymphohistiozytärer Randwall, Epitheloid- u. Fremdkörperriesenzellen. 8. INFEKTANFÄLLIGKEIT Störung der Erregerabwehr durch "lahme" Granulozyten [Chemotaxisstörung] → eitrig-abszedierende Entzündungen [Furunkulose!!! / Candidamykosis!!!] Infektionen der Harnwege [ v.a. Pyelonephritis + nekrotisierende Papillitis] http://www.8ung.at/prionerl 8 DISCLAIMER ! TODESURSACHEN BEI DM 1. Komplikationen der Arteriosklerose [Myokardinfarkt! Hirninfarkt] 2. Bakterielle Infektionen [Pneumonie, Lungenabszess, Lungengangrän, eitrige Pyelonephritis] 3. Coma diabeticum [Symptome: Hyperglykämie, Azidose, Exszikkose, Elyt-Entgleisung, Bewusstseinsverlust] 4. Urämie [durch diabetische Nephropathie] DIABETISCHE EMBRYO-/FETOPATHIE die Fetopathie bei (Prä-)DM der Schwangeren; [bei schlecht eingestelltem mütterlichen Diabetes]; im letzten Schwangerschaftsdrittel – oft Übergang latenter DM → manifester DM; Folgen: RIESENBABIES mit pastös-adipösem Habitus infolge reaktiver Somatotropin-Sekretion [STH↑] HYALINE MEMBRANKRANKHEIT: - Unreife der Alveolardeckzellen → insuffiziente Surfactant-Bildung → keine Alveolenentfaltung → Alveolen verkleben → Atelektase !!! - Oberflächenspannung ↑ → Ausschwitzung von Fibrin in Alveolen → Hyaline Membranen → IRDS! TRANSITORISCHE HYPOGLYKÄMIEN: - mütterlicher Insulinmangel → Fetus reagiert mit Hyperplasie der B-Zellen → Langerhans Inseln: Makronesie [Inseln vergrössert] und Polynesie [Inselzahl ↑ ] → Gefahr → POSTNATALER HYPOGLYKÄMISCHER SCHOCK ! - Histo: starke lymphozytäre und eosinophile Infiltration des gesamten Pancreas - perinatale Mortalität → 5 x höher! Symptome: Übergröße des Kindes (Riesenkind mit Geburtsgewicht > 4000 g) u. Neigung zu postpartaler Hypoglykämie durch erhöhte eigene Insulinproduktion, Polyglobulie, Atemnotsyndrom, Hypoparathyreoidismus, Hyperbilirubinämie, evtl. – bei Kardiomegalie – Zyanose, Neigung zu Azidose, Elektrolyt-Stoffwechselstörung, häufig auch Hydramnion u. Plazentaanomalien; erhöhte Fehlbildungsrate v.a. des Herzens, hohe Sterblichkeit (prä- u. perinatal). Prophylaxe: gute Stoffwechseleinstellung u. engmaschige Kontrolle der Schwangeren. HYPOGLYKÄMISCHES SYNDROM Hypo|glyk|ämie Absinken des Blutzuckers unter Normalwerte [ < 50 mg/dl ]; i.w.S. die sich daraus ergebenden Krankheitszeichen → voll ausgeprägt ab Schwellenwert von ca. 40 mg/dl; Pathogenese: 1. Mangelhafte Glucosezufuhr - Hungerdystrophie Malabsorptionssyndrom extreme körperlich Belastung mangelhafte Nahrungszufuhr, v.a. wenn kombiniert mit Hyperthyreose während der Schwangerschaft u. in der Stillperiode 2. Mangelhafte Gluconeogenese - schwere LE-Erkrankungen [Hepatitis, Zirrhose, Fettleber u.a.] - chron. Alkoholismus - angeb. hereditäre Enzymopathien [Fruktoseintoleranz, Galaktosämie] 3. Ausfall der hormonalen Insulin-Gegenregulation - Erkrankungen des Hypophysenvorderlappens - Erkrankungen der Nebennierenrinde 4. Hyperinsulinismus - Neoplasmen + Hyperplasien der B-Zellen [z.B. Inselzelladenom] Stimulierung der B-Zellen [z.B. durch Pancreatitis] diabetische Embryopathie Überdosierung von Insulin u. a. Antidiabetika, auch Paracetamol [?] nach Magenresektion [Dumpingsyndrom; verzögerte Passage zu rasche Glucoseresorption übermässige Insulinsekretion] 5. bei extrapankreatioschen Neoplasien - mesenchymale TUen, v.a. des Retroperitoneums [wahrscheinlich vermehrter Glukoseverbrauch] 6. Renale Glukosurie Krankheitszeichen evtl. nur diskret → latente Hyperglykämie mit Gereiztheit, Konzentrationsschwäche, Kopfschmerz, ggf. aber heftig mit Heißhunger, Schwitzen, Zittern, innere Unruhe, psychische Enthemmung, evtl. Somnolenz bis Bewußtlosigkeit = Coma hypoglycaemicum; http://www.8ung.at/prionerl 9 DISCLAIMER !