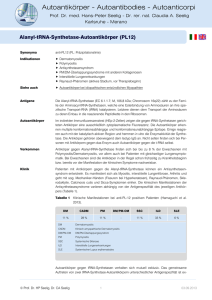
48 Muskelerkrankungen Myositis Netz Darstellung des klinisch-wissenschaftlichen Verbundes und Aktuelles zur Diagnostik und Therapie entzündlicher Muskelerkrankungen J. Zschüntzsch2*; C. Preusse1*; T. Ruck3; S. G. Meuth3; W. Müller-Felber4; W. Stenzel1; J. Schmidt2; für das Myositis Netz# 1Institut für Neuropathologie, Charité-Universitätsmedizin Berlin; 2Klinik für Neurologie, Universitätsmedizin Göttingen; 3Institut für translationale Neurologie und Klinik für Allgemeine Neurologie, Westfälische Wilhelms-Universität Münster; 4Klinik für Kinder- und Jugendmedizin, Klinikum der Universität München Schlüsselwörter Entzündliche Muskelerkrankung, Myositis Netz, myositisassoziierte Autoantikörper Zusammenfassung Entzündliche Muskelerkrankungen stellen eine wichtige Gruppe der seltenen neuromuskulären Erkrankungen dar. Die Diagnose der Myositiden hat einen besonderen Stellenwert, da sie im Gegensatz zu vielen anderen hereditären neuromuskulären Erkrankungen kausal behandelt werden können. Daher ist eine rasche, genaue Diagnosestellung wichtig, um Patienten helfen zu können, bevor irreversible Defizite entstehen. Um Patienten mit der Diagnosestellung einer Myositis und deren Angehörigen bessere Informationen über die Erkrankung zu erKorrespondenzadresse Prof. Dr. Jens Schmidt Klinik für Neurologie, Universitätsmedizin Göttingen Robert-Koch-Str. 40, 37075 Göttingen Tel. 0551/3922355, Fax 0551/398405 [email protected] * gleichberechtigte Erstautoren # Mitglieder des Myositis Netz in alphabetischer Reihenfolge: R. Biesen; H. H. Goebel; S. Glumm; AK. Güttsches; S. Krause; D. Lemmer; S. G. Meuth; F. Montagnese; S. Patschan; D. Pehl; C. Preuße; J. Radke; A. Rosenbohm; T. Ruck; A. Schänzer; U. Schara; K. Schmidt; J. Schmidt; W. Schulz-Schaeffer; C. Seitz; W. Stenzel; J. Wanschitz; J. Zschüntzsch. Das Myositis Netz ist ein deutsch-österreichischer Verbund, in dem sich klinisch und wissenschaftlich tätige Experten mit dem Schwerpunkt für entzündliche Muskelerkrankungen aus den Disziplinen der Neurologie, Neuropathologie, Rheumato- möglichen, hat sich vor einigen Jahren innerhalb der Selbsthilfe der Deutschen Gesellschaft für Muskelkranke e. V. die Diagnosegruppe Myositis gebildet (www.dgm.org). Zum interdisziplinären Austausch von Ärzten und Wissenschaftlern wurde 2015 das Myositis Netz gegründet, welches eine Plattform für wissenschaftliche Kooperationen bietet (www.myositis-netz.de). Hauptziele des Myositis Netz sind es, die Therapie bei Myositis zu optimieren und standardisieren und die Wissenschaft in diesem Bereich zu fördern. In dem vorliegenden Beitrag soll das Myositis Netz mit seinen Aufgaben, Zielen und Teilnehmern vorgestellt werden. Darüber hinaus werden die wesentlichen Fakten zur Diagnostik und Therapie der Myositis kompakt und übersichtlich dargestellt. Keywords logie, Neuropädiatrie und Kinder-Rheumatologie mit den Vertretern der Myositis-Patientenvereinigung zusammengeschlossen haben. Hauptziele des Netzwerkes sind: • Inflammatory muscle disorders, Myositis Netz, myositis-associated auto-antibodies Summary Inflammatory myopathies make up an important group of rare Neuromuscular Disorders. Diagnosis of myositis is important since – contrary to many hereditary Neuromuscular disorder –, myositis can be treated by immunosuppression. Therefore, a quick and precise diagnosis is important in order to help the patient before irreversible deficits occur. In order to better inform patients with myositis and their relatives, several years ago the patient support group “Diagnosegruppe Myositis” was formed within the Deutsche Gesellschaft für Muskelkranke (DGM; www.dgm. org). The Myositis Netz was founded in 2015 to foster the interdisciplinary exchange beMyositis Netz: Presentation of the clinico-scientific tween Clinicians and Researchers (www. network and update on diagnosis and therapy of myositis-netz.de). Main goals of this network inflammatory myopathies Nervenheilkunde 2017; 36: 48–54 are the optimization and standardization of eingegangen am: 16. September 2016 the treatment of myositis and to support reangenommen am: 23. September 2016 search in this area. Hereby we provide an overview of the goals, tasks and members of the Myositis Netz. As second part of this report, we summarize the most important facts for diagnosis and treatment of myositis. • • Verbesserung der interdisziplinäre Patientenversorgung, Förderung von translationaler Forschung der Myositiden, Bildung eines Informationsforums für Betroffene, Wissenschaftler und Ärzte © Schattauer 2017 Nervenheilkunde 1–2/2017 Downloaded from www.nervenheilkunde-online.de on 2017-05-17 | IP: 212.68.80.134 For personal or educational use only. No other uses without permission. All rights reserved. 49 J. Zschüntzsch et al.: Myositis Netz um das Bewusstsein für die Erkrankung bei Förderorganisationen, Firmen und in der Öffentlichkeit zu verbessern. Die Myositiden gehören zu der Gruppe der neuromuskulären Erkrankungen. Es gibt zahlreiche, überwiegend angeborene Muskelerkrankungen im Kindes- und Erwachsenenalter mit sehr unterschiedlicher Ausprägung. Die einzelnen Muskelerkrankungen gehören zwar zu den seltenen Erkrankungen, nimmt man jedoch alle zusammen, so leiden in Deutschland derzeit ca. 100 000 Patienten an einer neuromuskulären Erkrankung. Eine neuromuskuläre Erkrankung wird als solche häufig nicht erkannt. Daher ist die Diagnose meist verzögert, welches insbesondere bei behandelbaren Erkrankungen zu einem raschen Fortschreiten der Symptome führen kann. In Deutschland stehen zur Versorgung der Patienten mit neuromuskulären Erkrankungen von der DGM zertifizierte Zentren zur Verfügung (www.dgm.org). Diese Zentren betreuen meist das gesamte Spektrum der neuromuskulären Erkrankungen, jedoch sind viele Zentren meist für eine Erkrankung speziell ausgewiesen. Myositiden stellen die größte Untergruppe der erworbenen Muskelerkrankungen dar, und man geht von einer autoimmunentzündlichen Genese aus. Die Prävalenz wird aufgrund epidemiologischer Daten aus anderen Ländern extrapoliert. Es ist davon auszugehen, dass in Deutschland mehr als 10 000 Patienten an einer Myositis erkrankt sind. Die Myositiserkrankungen sprechen meist gut auf eine immunsuppressive Therapie an, daher ist eine verlässliche und effiziente Diagnostik notwendig, um den Patienten gut zu therapieren. Eine Hauptaufgabe des Myositis Netz ist daher, die Versorgung der Patienten mit Myositis zu verbessern. Dies beinhaltet Entwicklung von Diagnosestandards, Informationsaustausch zwischen Experten und Vorstellung von Therapien auf dem aktuellsten Stand der Wissenschaft. Darüber hinaus soll Betroffenen eine Plattform zum Austausch und ein Ansprechpartner geboten werden. Dabei ist außerdem hervorzuheben, dass gerade bei seltenen Erkrankungen wie den Myositiden eine Betreuung an spezialisierten Zentren notwendig ist, um eine enge Verzahnung von kli- weitere universitäre Einrichtungen in Deutschland und Österreich (▶Abb. 1). Das Myositis Netz ist eng mit der Selbsthilfegruppe Diagnosegruppe Myositis der DGM assoziiert (www.myositis-netz.de). Interessenten, die sich an der zukünftigen Arbeit des Myositis Netz beteiligen wollen, sind herzlich willkommen. Die Ziele Abb. 1 Übersicht über die Standorte des Myositis Netz nischer Versorgung, Studien und Wissenschaft gewährleisten zu können. Durch die Einführung einheitlicher Diagnosekriterien können Patienten für wissenschaftliche oder Therapiestudien einfacher rekrutiert werden. Das Myositis Netz fördert dadurch neue Forschungsprojekte und die Einwerbung von Forschungsgeldern. Neben der optimalen Versorgung der Patienten soll damit auch die Grundlagenund klinische Forschung an der Erkrankung gefördert werden. Zum Beispiel sind die Identifikation von Biomarkern und ein besseres Verständnis der Pathogenese der Erkrankung wesentlich, um Therapien zu verbessern. Das Myositis Netz möchte neben den klinischen und wissenschaftlichen Aspekten zusätzlich die Wahrnehmung für die Erkrankung in der Öffentlichkeit fördern. Struktur und Historie Das Myositis Netz Deutschland wurde im Juni 2015 gegründet. Neben den beiden Vorsitzenden Prof. Dr. J. Schmidt, Göttingen, und Prof. Dr. W. Stenzel, Berlin, erstreckt sich das Myositis Netz bisher auf 11 Die Hauptziele des Myositis Netz sind: • Verbesserung des Informationsaustausches und Verstärkung gemeinsamer Forschungsprojekte durch stärkere Vernetzung von Myositis-Experten • Verbesserung der Patientenversorgung durch bessere und schnellere Kommunikation zwischen Wissenschaftlern und klinisch tätigen Ärzten, • Intensivierung der finanziellen Förderung zur Myositisforschung durch verstärkte Öffentlichkeitsarbeit, • Förderung der Kommunikation mit Betroffenen durch effektivere Information auf der Webseite und in Broschüren. Die Ziele des Myositis Netz sollen über folgende Projekte realisiert werden: Etablierung eines Diagnosestandards Bei den Myositiden handelt es sich um seltene Erkrankungen, die verschiedene Entitäten umfassen. Dadurch ist es notwendig, definierte Diagnosestandards zu verwenden, die gleichsam klinische, pathologische und laborserologische Kriterien umfassen. Der Fokus der initialen Projektphase des Netzwerkes liegt daher in der Weiterentwicklung nationaler und internationaler Diagnosestandards und Förderung deren Verwendung. Hierbei besteht eine enge Kooperation aller entsprechenden Fachverbände wie der Deutschen Gesellschaft für Muskelkranke (DGM), der Deutschen Gesellschaft für Neurologie (DGN), der Deutschen Gesellschaft für Neuropathologie und Neuroanatomie (DGNN) mit dem Referenzzentrum für neuromuskuläre Erkrankungen, der Deutschen Gesellschaft für Rheumatologie (DGRh), der Deutschen dermatologischen Gesellschaft Nervenheilkunde 1–2/2017 © Schattauer 2017 Downloaded from www.nervenheilkunde-online.de on 2017-05-17 | IP: 212.68.80.134 For personal or educational use only. No other uses without permission. All rights reserved. 50 J. Zschüntzsch et al.: Myositis Netz (DDG), der Gesellschaft für Neuropädiatrie (GNP), der Gesellschaft für Kinderund Jugendrheumatologie (GKJR), der Europäischen Akademie für Neurologie (EAN) und des European Neuromuscular Center (ENMC). Aufbau einer Daten- und Biobank Essenziell für eine gute wissenschaftliche Zusammenarbeit ist der Aufbau einer Biobank, mit der Asservierung von Muskelbiopsien, Blutproben, ggf, Fibroblastenkulturen und Erhebung standardisierter klinischer Daten. Zur Konzeption der Biobank können die Forscher aus Münster auf eine jahrelange Expertise aus dem Kompetenznetz der Multiplen Sklerose zurückgreifen und somit die Infrastruktur für eine zentrale Datenbank liefern. In Ergänzung zu den dezentral und zentral über das Friedrich-Baur-Institut in München vorhandenen Muskelproben sollen Muskel- und Blutproben von Patienten mit Myositiden ergänzt werden. Das Ziel ist die Entwicklung eines Patientenregisters und einer Biobank für Patientenproben – ähnlich dem IBM Register aus München (www.ibm-registry.org), das als wissenschaftliches Projekt von der Firma Novartis gefördert wird. Darüber hinaus arbeitet das Myositis Netz eng mit dem Euromyositis Register zusammen (www. euromyositis.eu). Über die Registerdatenbanken sollen vor allem auch longitudinale Informationen von Patienten zusammengetragen werden, um z. B. die unterschiedlichen klinischen Verläufe und Zusammenhänge mit Tumoren charakterisieren zu können. Förderung wissenschaftlicher Kooperationen Projekte/Kooperationsprojekte im Myositis Netz bauen auf den grundlagenwissenschaftlich orientierten Projekten auf. Derzeit liegt der Schwerpunkt auf der Klassifizierung, Pathogenese und therapeutischen Strategien der verschiedenen Myositiden. Die Methoden der assoziierten Arbeitsgruppen sind sehr umfangreich und umfassen verschiedenste Techniken zur Untersuchung von humanem Gewebe, Zellkulturen und Mausmodellen. Die klinisch orientierten Projekte nut- zen neue diagnostische Methoden wie z. B. Echtzeit-MRT-Bildgebung des Herzens und der Speiseröhre. In den Zentren u. a. in Bochum, Göttingen, Berlin, München, Münster und Ulm werden nationale sowie internationale Studien an Patienten mit entzündlichen Muskelerkrankungen durchgeführt. In-vitroUntersuchungen werden mittels Zellkulturprojekten und In-vivo-Untersuchungen an murinen Modellen durchgeführt. Öffentlichkeitsarbeit Das Myositis Netz hat eine Internetseite, die über aktuelle Projekte und Veranstaltungen informiert. Zusätzlich sollen Workshops, Symposien auf Kongressen und regelmäßige Arbeitstreffen über den aktuellen Stand bei den Myositiden organisiert werden. Darüber hinaus gibt es eine enge Zusammenarbeit mit der Selbsthilfegruppe (Diagnosegruppe Myositis der DGM). Der wissenschaftliche Nachwuchs soll z. B. durch das Angebot von Nachwuchsakademien gefördert werden. Zusammenfassend ist das Hauptziel des Myositis Netz, einen kontinuierlichen und regen Austausch zwischen den verschiedenen Disziplinen aus Grundlagenforschung und Klinik zu der Erforschung von entzündlichen Muskelerkrankungen zu fördern. In dem ersten Jahr wurden bereits erste gemeinsame Projekte erfolgreich publiziert (1, 2). Aktuelle Aspekte zur Diagnostik und Therapie Klassifikation Myositiden sind erworbene entzündliche Erkrankungen der Skelettmuskulatur, die im Kinder- und Erwachsenenalter auftreten können. Es gibt nach der aktuellen Klassifikation verschiedene Unterformen, zu denen u. a. die Dermatomyositis (DM), Polymyositis (PM), immunvermittelte nekrotisierende Myopathie (NM) und Einschlusskörpermyositis (inclusion body myositis, IBM) gehören. Des Weiteren existiert eine Vielzahl von selteneren Myositisformen, zu denen die granulomatösen Myositiden, die eosinophile Fasziitis, die Makrophagen-Myofasziitis, eine Myositis im Rahmen anderer Kollagenosen als Overlap Syndrom und andere gehören. Infektiöse Myositiden müssen von den autoimmun-entzündlichen Myopathien ebenfalls abgegrenzt werden. Eine Übersicht der Myositisformen bietet die ▶Tabelle. Für den deutschsprachigen Raum wurden 2014 die zusammengefassten diagnostischen Kriterien und therapeutischen Empfehlungen zur Myositis von der Deutschen Gesellschaft für Neurologie (DGN) erstmals fachübergreifend mit Beteiligung von Rheumatologen, Dermatologen und Neurologen erstellt und auch von der Fachgesellschaft für Dermatologie als Leitlinie implementiert (3) (in ausführlicher Form frei zugänglich unter www.awmf.org -> Leitlinie Myositissyndrome). Symptome Führende Symptome einer Myositis sind proximal betonte, symmetrische Paresen an Armen und Beinen, die häufig mit Schmerzen einhergehen und zu einer Beeinträchtigung von Alltagsaktivitäten führen. Der Krankheitsverlauf ist meist subakut. Eine Ausnahme bildet die IBM mit einem chronisch-progredienten Verlauf und der Beteiligung distaler Muskelgruppen. In seltenen Fällen, z. B. bei der amyopathischen Dermatomyositis, finden sich definitionsgemäß keine Paresen. Typisch für eine Myositis ist eine zumeist deutliche, z. T. 20- bis 50-fache Erhöhung der Serumcreatinkinase (CK). Eine normale CK schließt jedoch eine Myositis keineswegs aus. Bei der IBM besteht typischerweise eine nur 3- bis 5-fach erhöhte CK. Neben dem Skelettmuskel können bei der Myositis andere Organe mitbetroffen sein. Vor allem eine charakteristische Entzündung der Haut bei DM ist häufig und trägt durch die typische Phänomenologie der Effloreszenzen zur korrekten Diagnosestellung bei. Bei der klassischen DM kommt es zu einem typischen fliederfarbenen Erythem im Dekolletee, Wangen- und Lidbereich sowie den rötlichen GottronPapeln an den Streckseiten der Finger und Hände. Zusätzlich kann es bei einer Myositis zu einer Gelenkbeteiligung im Sinne einer (Poly-)Arthritis kommen. Eine Herzmuskelbeteiligung kommt bei verschiedenen © Schattauer 2017 Nervenheilkunde 1–2/2017 Downloaded from www.nervenheilkunde-online.de on 2017-05-17 | IP: 212.68.80.134 For personal or educational use only. No other uses without permission. All rights reserved. 51 J. Zschüntzsch et al.: Myositis Netz Tab. Übersicht der relevanten klinischen und paraklinischen Befunde bei den wichtigsten und definierten Entitäten der Myositis. ARS, Aminoacyl-t RNA Synthetase; ILD, interstitielle Lungenerkrankung; MHC, Major histocompatibility complex; *Bei Nachweis dieser Autoantikörper sollte ganz besonders eine möglicherweise assoziierte Neoplasie bedacht werden. IBM PM Anfang, Verlauf langsam progredient; immer chronisch akuter/ subakuter akuter/ subakuter Beginn; meist chronisch Beginn; benigne kurze und schwere, chronische Verläufe Paresen, weitere Symptome lange Fingerbeuger, proximale Tetraparese Kniestrecker, Dysphagie ± Dysphagie (z. T. früh bzw. initial) CK-Wert Normal bis ~5-fach erhöht ~10- bis 50-fach erhöht normal oder ~10- bis 50-fach erhöht ~10- bis 50-fach erhöht ~10- bis 50-fach erhöht Autoantikörper Anti-cN1A („Mup44“) unspezifisch Anti-Mi-2 Anti-MDA5 Anti-TIF-1gamma* Anti-NXP2* Anti-SAE Anti-SRP AntiHMGCoAR* 8 Anti-ARS AK: Jo-1, PL-7, PL-12, Ro, Ku, EJ, OJ, PM/Scl) Myopathologie CD8+ T-Zellen, MHC Kl. CD8+ T-Zellen I, Amyloid, Tubulofilaendomysial mente, Vakuolen, Mitochondriale Dysfunktion, (COX, parakr. Einschl.) Entzündung perimysial, perifaszikuläre Atrophie, MHC Kl. I, Komplement auf Kapillaren und/oder Sarkolemm, Kapillarverlust diffus verteilte Nekrosen/Myo-phagozytosen/regenerate, MHC Kl. I milde, Komplement auf Sarkolemm Muskelfasernekrosen perifaszikulär, perimysiale Fragmentierung, Komplement auf Sarkolemm, myonukleäre Aktineinschlüsse Formen der Myositis vor und kann die Prognose ungünstig beeinflussen. Eine Lungenbeteiligung ist vor allem bei dem Anti-Synthetasesyndrom typisch mit einer interstitiellen Lungenerkrankung, die vor allem zu einer Fibrose führt (www.awmf. org -> Leitlinie Myositissyndrome). Eine unterschiedlich stark ausgeprägte Schluckstörung kann Bestandteil jeder Form einer Myositis sein und vor allem während der subakuten Anfangszeit bzw. vor Einleitung einer Therapie vorkommen. Bei der IBM kann eine Schluckstörung auch als Erstsymptom vorkommen. Vor allem bei der IBM stellt eine Dysphagie ein erhöhtes Aspirationsrisiko dar, was einen wesentlichen Faktor für eine erhöhte Mortalität darstellt (4). Besonderheiten im Kindesalter Im Kindesalter kommt fast ausschließlich die DM vor, wobei allerdings die Hauterscheinungen zum Teil nur vorübergehend sein können und bisweilen auch sehr diskret sind. Leitsymptom im Kindesalter ist DM proximale Tetraparese ± Dysphagie; spez. Haut- und Organbeteiligung, Malignome bei Erwachsenen* die Kombination aus deutlich gestörtem Allgemeinbefinden („Malaise“), Muskelschwäche und Hauterscheinungen. Anders als beim Erwachsenen steht beim Kind die Vaskulitis ganz im Vordergrund. Im Verlauf kommt es häufiger als bei Erwachsenen zu teilweise sehr ausgeprägten Verkalkungen, insbesondere wenn die Therapie spät beginnt und nicht konsequent durchgeführt wird. Im Unterschied zum Erwachsenenalter können die CK-Werte auch normal sein. Übersicht zur Diagnostik Essenziell für eine verlässliche Diagnostik ist die gleichrangige Bewertung des klinischen Befundes inklusive Neurophysiologie, der Muskelbiopsie und der Autoantikörperdiagnostik (▶Abb. 2). Nur durch diese Kombination der diagnostischen Verfahren ist eine Unterscheidung der einzelnen Myositissyndrome und zudem eine Abgrenzung zu anderen Muskelerkrankungen wie Muskeldystrophien oder myofibrillären Myopathien möglich (5). Nur im Kindesalter kann im gebotenen Einzelfall bei einer sehr klassischen NM ASS-assoziierte Myositis akuter/ subakuter Beginn, chronische, langsam progrediente Verläufe möglich Akuter/ subakuter Beginn; meist chronisch proximale Tetraparese, selten Herz- und/oder Lungenbeteiligung, Malignome* proximale Tetraparese; interstit. Lungenerkrankung (ILD), RaynaudSyndrom, Arthritis, Mechanikerhände Befundkonstellation (Hauterscheinungen, proximale Schwäche, CK-Erhöhung) auf eine Biopsie verzichtet werden. Die aktuell im deutschsprachigen Raum vorrangig verwendeten und in der Leitlinie Myositissyndrome der DGN enthaltenen Diagnosekriterien für die Myositis wurden in einem ENMC Workshop entwickelt (6; ▶Tab.; www.awmf.org -> Leitlinie Myositissyndrome). International werden die Diagnosekriterien für die Myositis gegenwärtig durch eine interdisziplinäre Expertengruppe unter Einbeziehung der International Myositis Assessment & Clinical Studies Group (IMACS) und unter Federführung des Euromyositis-Konsortiums weiterentwickelt (International Myositis Classification Criteria Project, IMCCP) und wurden den rheumatologischen Fachgesellschaften in Europa und den USA (EULAR und ACR) zur Verabschiedung vorgelegt (7). Für die IBM sind in einem ENMC Workshop 2011 neue Diagnosekriterien von einem internationalen Expertengremium erarbeitet worden (8). Obwohl nicht alle Punkte dieser Kriterien optimal erscheinen, werden sie gegenwärtig als Goldstandard für Nervenheilkunde 1–2/2017 © Schattauer 2017 Downloaded from www.nervenheilkunde-online.de on 2017-05-17 | IP: 212.68.80.134 For personal or educational use only. No other uses without permission. All rights reserved. 52 J. Zschüntzsch et al.: Myositis Netz Abb. 2 Diagnostische Trias für die Myositis: Klinischer Befund, Muskelhistologie, Myositisautoantikörper die IBM angesehen. Wesentliche klinische Charakteristika beinhalten eine Parese der langen Fingerbeuger und der Oberschenkelstrecker (8; ▶Tab.; Leitlinie der DGN: frei zugänglich unter www.awmf.org -> Leitlinie Myositissyndrome). Autoantikörper In den letzten Jahren ist eine zunehmende Zahl an „myositisspezifischen“ und „myositisassoziierten“ Autoantikörpern identifiziert worden (9). Es besteht kein Zweifel daran, dass Autoantikörper einen zunehmend wichtigen Stellenwert bei der Diagnostik der Myositis einnehmen. Wegen der Vielzahl an Antikörpern und der zunehmenden Heterogenität ist eine „Panel-Untersuchung“ durch ein erfahrenes, zuverlässiges Labor unabdingbar (9, 10; ▶Tab.; www.awmf.org -> Leitlinie Myositissyndrome). Der Wert der Antikörperdiagnostik besteht zum einen in der Abgrenzung erblicher Myopathien, indem ein positiver Nachweis die autoimmunentzündliche Genese der Symptome unterstützt. Zum anderen können Autoantikörper direkt zur spezifischen Diagnosestellung beitragen. AntiTIF-1γ und Anti-NXP-2-Autoantikörper sind von wesentlicher prognostischer und therapeutischer Bedeutung, da sie im Erwachsenenalter mit einem sehr hohen Risiko mit Malignomen assoziiert sind. Der anti-NXP-2-Autoantikörpernachweis ist im Kindesalter mit schweren Verläufen und Verkalkungen assoziiert. Der MDA5-Antikörper, der bei amyopathischer DM zu finden ist, ist mit interstitieller Lungenerkrankung assoziiert. Bei der Nekrotisierenden Myopathie finden sich oft die Antikörper anti-SRP und anti-HMGCR (HMG-CoA Reduktase), wobei letztgenannte auch mit Tumoren assoziiert sein können (11). Anti-HMGCR-Antikörper sind u. a. in Zusammenhang mit der Einnahme von Statinen beschrieben worden, kommen aber auch bei Patienten vor, die nie zuvor hiermit behandelt worden sind (12). Im Hinblick auf eine IBM sind bis zu 30–50% der Patienten positiv für anticN1A (13, 14), jedoch zeigen neuere Befunde, dass dieser Antikörper keineswegs spezifisch ist, sondern auch bei PM oder DM und sogar ganz ohne Myositis auch bei anderen Kollagenosen wie z.B. SLE oder Sjögren-Syndrom vorkommt (15, 16). Im Kindesalter spielt die Antikörperdiagnostik vor allem eine Rolle in der Abgrenzung zu Mischkollagenosen. Der im Erwachsenenalter häufig nachweisbare Mi2-Antikörper kommt bei der kindlichen /juvenilen Dermatomyositis nur selten vor. Muskelbiopsie Die Muskelbiopsie stellt einen unverzichtbaren Bestandteil der Diagnostik für die Myositis dar. In den letzten Jahren hat sich gezeigt, dass eine pathognomonische Histomorphologie einem bestimmten klinischen Subtyp zuzuordnen ist. Die Dermatomyositiden weisen eine ausgeprägt heterogene Morphologie auf, eine weitergehende Einordnung erfolgt z. B. durch den Nachweis von myositisassoziierten Autoantikörpern sowie durch typische klinische Charakteristika. Bei der mit dem Autoantikörper-Mi-2-assoziierten DM zeigt sich das klassische klinische Bild der DM. Morphologisch findet man einen schweren Befall der Muskulatur mit ausgeprägter Kalibervarianz und schwerer perifaszikulärer Atrophie, zahlreichen perimysial gelegenen entzündlichen Infiltraten und Komplementablagerungen auf dem Sarkolemm der Fasern. Die mit dem Autoantikörper TIF-1γ-assoziierte DM zeigt ebenfalls eine ausgeprägte perifaszikuläre Atrophie und ein oft erhebliches perimysiales Ödem. Die Dichte entzündlicher Infiltrate ist variabel, auffälligstes Merkmal sind kräftige Komplementablagerungen auf Kapillaren. Bei der mit dem Autoantikörper MDA-5-assoziierten DM (entspricht der klinisch amyopathischen DM: CADM) besteht oft durch eine schwere Lungenbeteiligung eine schlechte Prognose. Histologisch zeigt sich u. a. keine klassische perifaszikuläre Faseratrophie, wenig bis gar keine entzündlichen Infiltrate und wenig MHC Klasse I Färbung auf dem Sarkolemm (17). Bei der immunvermittelten NM, die mit anti-SRP- oder anti-HMGCR-Autoantikörpern assoziiert sind, sieht man Muskelfasernekrosen mit diffuser Verteilung in unterschiedlichen Stadien des Unterganges und Myophagozytosen sowie reichlich Regeneration. MHC-Klasse-I-Moleküle sind wechselnd kräftig, aber diffus verteilt sarkolemmal positiv, lymphozytäre Infiltrate finden sich fokal betont und manchmal nur gering ausgeprägt. Komplementdepots auf dem Sarkolemm und auf Kapillaren kommen vor (18). Die antisynthetaseassoziierte Myositis ist mit einem von acht bekannten Autoantikörpern gegen tRNA-Synthetasen assoziiert. Man sieht Muskelfasernekrosen in perifaszikulärer Lage, perimysiale Fragmentierung des Bindegewebes, dichte lymphozytäre Infiltrate, sowohl MHC-Klasse-I als auch MHC-Klasse-II und myonukleäre Aktininklusionen (19–22). © Schattauer 2017 Nervenheilkunde 1–2/2017 Downloaded from www.nervenheilkunde-online.de on 2017-05-17 | IP: 212.68.80.134 For personal or educational use only. No other uses without permission. All rights reserved. 53 J. Zschüntzsch et al.: Myositis Netz Bildgebung und weitere Zusatzdiagnostik Bei allen Patienten mit Myositis, bei denen eine paraneoplastische Genese möglich oder sogar wahrscheinlich erscheint, ist eine sorgfältige Schnittbilddiagnostik mittels CT oder MRT von Thorax und Abdomen notwendig, um einen Tumor zu belegen oder ihn auszuschließen. Mitunter kann sogar ein PET-CT sinnvoll sein und eine weitere Tumorsuche z. B. mittels Gastroskopie oder Koloskopie muss vom jeweiligen Einzelfall und dessen klinischen Verlauf abhängig gemacht werden. Im Kindesalter ist eine Tumorsuche hingegen nicht notwendig. Eine MRT-Diagnostik der Muskulatur ist in den letzten Jahren zunehmend standardisierter geworden. Zur Diagnostik bei Myositis eignet sich das MRT zum einen initial, um eine Entzündung im Muskel bereits vor einer Biopsie erkennen zu können und zum anderen, um einen möglichst geeigneten Muskel zur Biopsie auszuwählen. Auch als zusätzlicher Verlaufsparameter zur Therapieevaluation ist ein MuskelMRT eine sinnvolle apparative Ergänzung. Weitere Zusatzdiagnostik mittels Echokardiografie und/oder Kardio-MRT zur Diagnostik und Therapiekontrolle kann bei entsprechender Konstellation sinnvoll sein. Bei einer Atemstörung ist eine formale Blutgasanalyse und Lungenfunktionstes- tung essenziell, bei einer Schluckstörung sollte eine FEES (flexible endoskopische Ösophaguskopie) und ggf. eine Videofluoroskopie durchgeführt werden. In Zukunft könnte auch eine Echtzeit-MRT-Untersuchung erfolgen (23). Therapie Aufgrund unzureichender klinischer Studien basieren die meisten Therapieempfehlungen weiterhin auf Expertenmeinungen. Eine detaillierte Darstellung des empfohlenen therapeutischen Vorgehens ist den aktuellen Leitlinien der DGN zu entnehmen (www.awmf.org -> Leitlinie Myositissyndrome) und aktuellen Übersichtsarbeiten (24, 25). Die Basis der Myositisbehandlung besteht in einer Immunsuppression durch Glukokortikoide wie Prednisolon (▶Abb. 3). In der Akutphase der Erkrankung wird üblicherweise eine Dosis von 1 mg/kg pro Tag verwendet und in den ersten 1–3 bzw. 5 Tagen auch eine intravenöse Pulstherapie als Einleitung der Behandlung mit 250 mg bis 1 000 mg Prednisolon pro Tag. Wichtig ist, dass die Immunsuppression nicht alleinig aufgrund eines gebesserten CK-Wertes reduziert wird, sondern dass sich die Dauer und Dosierung der Behandlung am Verlauf des klinischen Befundes orientiert, also den objektiv nachweisbaren Paresen. Üblicherweise ist eine Besserung nach mehreren Wochen zu erwarten, manchmal ist jedoch eine längere Gabe bzw. aggressivere Immunsuppression notwendig. Für eine dauerhafte Immunsuppression und zur Einsparung von Glukokortikoiden aufgrund deren Langzeitnebenwirkungen erfolgt der Einsatz von Immunsuppressiva wie Azathioprin oder Methotrexat. Vor der Eindosierung von Azathioprin sollte die Enzymaktivität der Thiopurinmethyltransferase (TPMT) bestimmt werden, um einer Überdosierung mit Lymphopenie vorzubeugen. Eine Add-on-Therapie mit intravenösen Immunglobulinen ist bei unzureichender Wirksamkeit der Primärtherapie und insbesondere auch bei ausgeprägter Dysphagie Foto: ©Mit Genehmigung des Thieme Verlag KG Die histologischen Charakteristika der IBM umfassen den Nachweis einer spezifischen Entzündung, die überwiegend aus T-Zellen und Makrophagen sowie eine vakuoläre Degeneration mit Nachweis von geränderten Vakuolen, Zeichen der Autophagie und pathologischer amyloidogener Proteinaggregate. Eine mitochondriale Schädigung ist ebenso typisch und oft besteht ein schwer myopathisch geschädigtes, bindegewebig umgebautes Gesamtgewebebild. Eine typische PM ist histologisch durch zytotoxische CD8-immunreaktive T-Lymphozyten charakterisiert, die Skelettmuskelfasern direkt infiltrieren. Da dies ein sehr wenig spezifisches Diagnosekriterium ist, was bei nicht primär immunvermittelten Myopathien vorkommt, sollte die PM nur als Ausschlussdiagnose gestellt werden, wenn keine der Charakteristika zutrifft. Abb. 3 Übersicht der Myositistherapie (aus 24); TPMT, Thiopurinmethyltransferase Nervenheilkunde 1–2/2017 © Schattauer 2017 Downloaded from www.nervenheilkunde-online.de on 2017-05-17 | IP: 212.68.80.134 For personal or educational use only. No other uses without permission. All rights reserved. 54 J. Zschüntzsch et al.: Myositis Netz Interessenkonflikt Fazit für die Praxis Myositiden stellen eine wichtige Untergruppe der neuromuskulären Erkrankungen dar. Der klinische Verlauf und die erforderliche Therapie der einzelnen Syndrome sind z. T. sehr spezifisch, sodass eine akkurate Diagnostik essenziell ist. Diese wird nur durch eine gleichsame Bewertung der Befunde des klinischen Verlaufes, der Myositisautoantikörper, der Muskelbiopsie und der übrigen paraklinischen Befunde möglich. Zur Information über Myositis für Betroffene, Ärzte und Wissenschaftler sowie zum interdisziplinären Austausch zwischen Ärzten und Wissenschaftlern ist vor kurzem das Myositis Netz etabliert worden (www.myositisnetz.de). indiziert. Eine Kostenübernahme dieser Off-label-Behandlung bei schwer verlaufender Myositis ist durch ein GBA-Votum seit 2013 abgedeckt (www.g-ba.de/informa tionen/beschluesse/1701/). Eine frühe Behandlung mit i.v. Immunglobulinen ist bei intolerablen Nebenwirkungen von Glukokortikoiden und bei Kindern zu erwägen. Bei schweren Verläufen aller Formen der Myositis, insbesondere auch bei der NM, ist oft eine frühe Eskalation zur B-Zell-Depletion mittels Rituximab zu erwägen. Sobald eine klinische Stabilisierung eingetreten ist, kann eine vorsichtige, schrittweise Herabdosierung des Glukokortikoids erfolgen. Vor einer Dosisreduktion sollte auch die Langzeitimmunsuppression bzw. ein langfristiges Therapiekonzept etabliert worden sein. Parallel zur medikamentösen Behandlung ist eine intensive und kontinuierliche Physiotherapie ein essenzieller Bestandteil der erfolgreichen Therapie. Je nach Symptomen und Verlauf sind darüber hinaus auch Ergotherapie, Logopädie, neurologische Rehabilitation und eine psychologische Betreuung wichtig. Die gesamte Therapie muss ebenso entsprechend der Symptome interdisziplinär durch ein eingespieltes Team aus Rheumatologe, Neurologe, Dermatologe, Pulmonologe, Kardiologe und Therapeuten erfolgen. S. G. Meuth hat Honoraria für Vorträge, Reisekostenerstattungen oder Projektförderungen erhalten von Bayer, Bayer Schering, Biogen Idec, Genzyme, Merck Serono, Merck Sharp & Dohme, Novartis, Novo Nordisk, Sanofi-Aventis und Teva. J. Schmidt hat Honorare, Forschungsunterstützungen oder Reisekostenunterstützungen erhalten von Bayer, Biomarin, Biotest, Grifols, Novartis, Octapharma und VitalAire. Für die anderen Koautoren bestehen keine Interessenkonflikte. Literatur 1. Afzali AM, Ruck T, Herrmann AM, Iking J, Sommer C, Kleinschnitz C et al. The potassium channels TASK2 and TREK1 regulate functional differentiation of murine skeletal muscle cells. Am J Physiol Cell Physiol 2016 (im Druck). 2. Ruck T, Bittner S, Afzali AM, Gobel K, Glumm S, Kraft P et al. The NKG2D-IL-15 signaling pathway contributes to T-cell mediated pathology in inflammatory myopathies. Oncotarget 2015; 6(41): 43230–43. 3. Sunderkotter C, Nast A, Worm M, Dengler R, Dorner T, Ganter H et al. Guidelines on dermatomyositis – excerpt from the interdisciplinary S2k guidelines on myositis syndromes by the German Society of Neurology. J Dtsch Dermatol Ges 2016; 14(3): 321–38. 4. Price MA, Barghout V, Benveniste O, ChristopherStine L, Corbett A, de Visser M et al. Mortality and causes of death inpatients with sporadic inclusion body myositis: Survey study based on the clinical experience of specialists in Australia, Europe and the USA. J Neuromuscul Dis 2016; 3(1): 67–75. 5. Hilton-Jones D. Myositis mimics: how to recognize them. Curr Opin Rheumatol 2014; 26(6): 663–70. 6. Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004; 14(5): 337–45. 7. Lundberg IE, Miller FW, Tjarnlund A, Bottai M. Diagnosis and classification of idiopathic inflammatory myopathies. J Intern Med 2016; 280(1): 39–51. 8. Rose MR. 188th ENMC International Workshop: Inclusion Body Myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord 2013; 23(12): 1044–55. 9. Betteridge Z, McHugh N. Myositis-specific autoantibodies: an important tool to support diagnosis of myositis. J Intern Med 2016; 280(1): 8–23. 10. Benveniste O, Stenzel W, Allenbach Y. Advances in serological diagnostics of inflammatory myopathies. Curr Opin Neurol 2016; 29(5): 662–73. 11. Allenbach Y, Hervier B, Stenzel W, Benveniste O. Reply: Perifascicular necrosis in anti-synthetase syndrome beyond anti-Jo-1. Brain 2016; 139(Pt 9): e51. 12. Mammen AL, Chung T, Christopher-Stine L, Rosen P, Rosen A, Doering KR et al. Autoantibodies against 3-hydroxy-3-methylglutaryl-coenzyme A reductase in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011; 63(3): 713–21. 13. Larman HB, Salajegheh M, Nazareno R, Lam T, Sauld J, Steen H et al. Cytosolic 5‘-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann Neurol 2013; 73(3): 408–18. 14. Pluk H, van Hoeve BJ, van Dooren SH, Stammen-Vogelzangs J, van der Heijden A, Schelhaas HJ, et al. Autoantibodies to cytosolic 5‘-nucleotidase 1A in inclusion body myositis. Ann Neurol 2013; 73(3): 397–407. 15. Lloyd TE, Christopher-Stine L, Pinal-Fernandez I, Tiniakou E, Petri M, Baer A et al. Cytosolic 5‘-Nucleotidase 1A as a target of circulating autoantibodies in autoimmune diseases. Arthritis Care Res 2016; 68(1): 66–71. 16. Herbert MK, Stammen-Vogelzangs J, Verbeek MM, Rietveld A, Lundberg IE, Chinoy H et al. Disease specificity of autoantibodies to cytosolic 5‘-nucleotidase 1A in sporadic inclusion body myositis versus known autoimmune diseases. Ann Rheum Dis 2016; 75(4): 696–701. 17. Allenbach Y, Leroux G, Suarez-Calvet X, Preusse C, Gallardo E, Hervier B et al. Dermatomyositis with or without anti-melanoma differentiation-associated gene 5 antibodies: Common interferon signature but distinct NOS2 expression. Am J Pathol 2016; 186(3): 691–700. 18. Stenzel W, Goebel HH, Aronica E. Review: immune-mediated necrotizing myopathies – a heterogeneous group of diseases with specific myopathological features. Neuropathol Appl Neurobiol 2012; 38(7): 632–46. 19. Uruha A, Suzuki S, Suzuki N, Nishino I. Perifascicular necrosis in anti-synthetase syndrome beyond anti-Jo-1. Brain 2016; 139(Pt 9): e50. 20. Mescam-Mancini L, Allenbach Y, Hervier B, Devilliers H, Mariampillay K, Dubourg O et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain 2015; 138(Pt 9): 2485–92. 21. Stenzel W, Preusse C, Allenbach Y, Pehl D, Junckerstorff R, Heppner FL et al. Nuclear actin aggregation is a hallmark of anti-synthetase syndromeinduced dysimmune myopathy. Neurology 2015; 84(13): 1346–54. 22. Aouizerate J, De AM, Bassez G, Gherardi RK, Berenbaum F, Guillevin L et al. Myofiber HLA-DR expression is a distinctive biomarker for antisynthetase-associated myopathy. Acta Neuropathol Commun 2014; 2: 154. 23. Olthoff A, Carstens PO, Zhang S, von Fintel E, Friede T, Lotz J et al. Evaluation of dysphagia by novel real-time magnetic resonance imaging. Neurology 2016; 87(20): 2132–2138. 24. Fitzner M, Schmidt J. Entzündliche Muskelerkrankungen – Aktueller Stand zur Diagnostik und Therapie der Myositis. Akt Rheumatol 2016; 41(3): 232–42. 25. Breithaupt M, Schmidt J. Diagnostik und Therapie von Myositiden – Zertifizierte Fortbildung. NeuroTransmitter 2014; 25(12): 46–55. © Schattauer 2017 Nervenheilkunde 1–2/2017 Downloaded from www.nervenheilkunde-online.de on 2017-05-17 | IP: 212.68.80.134 For personal or educational use only. No other uses without permission. All rights reserved.