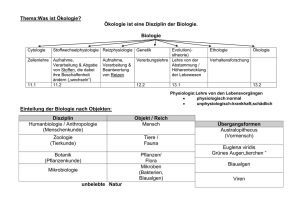
PowerPoint-Präsentation - Goethe
Werbung

Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Baysian Estimation of the Number of Inversions in the History of Two Chromosomes T. L. York, R. Durrett and R. Nielsen Baysian Estimation of Genomic Distance T. L. York, R. Durrett and R. Nielsen Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Allgemein evolvieren Genome durch: Translocations Fusions Fisions Inversions Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Ist es sinnvoll nur Inversionen zu betrachten? Ja, für bestimmte Datensätze: mitochondriale und Chloroplasten DNA Gonosomen DNA von Drosophila spec. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Wie werden die Chromosomen in den Datensätzen repräsentiert? Über sog. „Marker“ werden spezifische Gene, die auf beiden Chromosomen auftauchen, lokalisiert. Marker können 2 Orientierungen besitzen ( + , - ) Ist sowohl die Markerordnung als auch die Orientierung identisch, gelten auch die Chromosomen als identisch. Repräsentation also „nur“ über einen Satz von Markern. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Frühere Ansätze zur Bestimmung der Verwandtschaft über Inversionen: 1. Bestimmung der „Inversions Distanz“: Idee: Kleinst mögliche Zahl an Inversionen, die nötig sind, um Chromosom 1 in Chromosom 2 zu überführen.~ „Sorting by Reversals“ NP- Hard aber branch-and-bound Methode verfügbar. Problem: Keine Garantie, dass Minimum Anzahl = wahre Anzahl an Inversion ist Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Frühere Ansätze zur Bestimmung der Verwandtschaft über Inversionen: 2. Schätzung der wahren Anzahl von Inversionen per "break points“ Break points: Anzahl an adjazenten Marken in einem Chromosom, die in dem anderen nicht adjazent sind Idee: Die Inversions Distanz ist mindestens ½ mal die Anzahl von break points.~ Problem: Diese Grenze ist sehr grob geschätzt. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Der „break point“ – Graph Der Schlüssel zum Ermitteln der Inversions Distanz ist der break point graph von Hannenhalli & Pevzner, 1995 1.Fall: Markerorientierung ist bekannt ( „signed permutation“ ) Jeder Marker kann sich mit „Startpunkt“ und „Endpunkt“ vorgestellt werden. Der Graph einer orientierten Permutation von N Markern, pa, relativ zu einem anderen, pb, hat 2N+2 Knoten, für jedes Ende eines Markers jeweils einen, plus jeweils einen für die Enden des Chromosoms. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Der break point Graph - signed, ein Beispiel: Der Datensatz des ersten Chromosoms (a) ist (+2,-3,+1,+4)~ Der „Startpunkt“ des Markers k wird jetzt zu 2k-1 und der „Endpunkt“ zu 2k 3:4, 6:5,1:2,7:8 Anfügen von 0 links und 2N+1 = 9 rechts führt zu (2,-3,1,4) (0,3:4, 6:5,1:2,7:8,9). Die mit „:“ getrennten Paare sind jeweils die Enden eines Markers => Sie bleiben in jeder Permutation adjazent Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Der break point Graph - signed, ein Beispiel: Der Datensatz des zweiten Chromosoms (b) ist (-1,-4,2,3) ~ (-1,-4,2,3) (0,2:1, 8:7,3:4,5:6,9). Aus beiden Sätzen kann man dann den Break point graph pa relativ zu pb erzeugen: Jeder, in dieser Permutation adjazente Marker aus Pa (Pb) ist durch eine „black Edge“ ( „grey Edge“ ) verbunden. Die verbundenen Marker bilden eine cycle decomposition Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Der break point – Graph - signed Sei die Anzahl an Zykeln in der Zykel-Dekomposition c(pa,pb). Eine Inversion I auf pa verursacht den Bruch zweier „black edges“ und erzeugt zwei neue. 3 mögliche Effekte auf die Zykel Dekomposition: 1. Ein Zykel wird in zwei neue aufgespalten 2. Zwei Zykel werden zu einem vereint 3. Veränderung der Knoten Reihenfolge innerhalb eines Zykels c = c(I pa,pb)- c(pa,pb) = +1, -1 or 0 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Der break point Graph - signed Wenn pa = pb dann ist die Anzahl der Zykel = N+1 pa = pb = (2,-3,1,4) (0,3:4, 6:5,1:2,7:8,9) Da eine Inversion maximal 1 zusätzlichen Zykel erzeugt braucht man mindestens N+1-c Inversionen um pa in pb zu überführen. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Komplikationen im break point Graph - signed 1. Hurdles: Angenommen pa = (3,2,1) und pb = ( 1,2,3 ) keine Inversion kann die Anzahl der Zykel erhöhen => Wenn h(pa, pb) die Anzahl der Hurdles ist, dann ist n + 1- c + h eine untere Grenze für die Inversions Distanz 2. Fortress Eine bestimmte Anordnung von Hurdles erzeugt eine Fortress ein weiterer Schritt notwendig Sei f (pa, pb) =1 wenn der Graph eine Fortress ist und 0 sonst Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Der break point Graph - signed Formel für die untere Grenze der Inversions Distanz im Graph ist also: d (pa, pb) = n+1-c+h+f Hannenhalli und Pevzner haben gezeigt, dass man für orientierte Permutationen, die Distanz in polynomieller Zeit berechnen kann. Algorithmus liefert gute Ergebnisse für signed Permutations. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Der break point Graph - unsigned 2. Fall: Markerorientierung ist nicht bekannt. ( „ unsigned permuation, partially signed permutation „) Realistischer Fall, da genomic Data in Form von comperative Maps meist unsigned oder nur partiell orientiert vorliegt Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Gen Start Kb ANT3 Cyto Cattle order Xp22.32 1 AMELX 8,950 Xp22.31 2 SAT 18,652 Xp22.1 3 Xp21.1 4 CYBB MAOA 38,289 Xp11.4 5 SYN1 42,783 Xp11.23 7 TIMP1 42,792 Xp11.23 8 SYP 44,288 Xp11.22 6 CITED1 64,082 Xq13.1 9 PLP1 97,418 Xq22 11 FACL4 103,471 Xq23 10 HPRT1 128,965 Xq26 14 TNFSF5 130,747 Xq26 13 SLC6A8 148,934 Xq28 12 1,2,3,4,5 +1 7,8 +3 6 9 ?2 ?4 11,10 -5 14,13,12 -6 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Der break point Graph - unsigned +1, +3, ?2, ?4, -5, -6 Mit wenig Aufwand kann man die 4 Möglichkeiten ausprobieren, und kommt zu der signed permutation +1, +3, -2, +4, -5, -6 mit der minimalen Distanz 4. In den meisten Fällen ist durchprobieren nicht möglich. Beim Vergleich von D. melanogaster und D. repleta müsste man 260 > 1018 Möglichkeiten durchprobieren... Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Modell Annahmen • Umordnung geschieht nur infolge von Inversionen • Das Auftreten einer Inversion ist ein Poisson Prozess mit unbekannten Mittelwert ; Die Wahrscheinlichkeit von exakt L aufgetreten Inversionen ist: P(L| )=e-L/L! L = 1, 2, ... • Wir nehmen a priori eine Gleichverteilung für an: P() = 1/ max für 0 < <= max • Die Anzahl der verglichenen Marker auf beiden Chromosomen ist N. Dabei ist es egal ob wir die Orientierung eines Markers kennen oder nicht. Ist die Orientierung bekannt ( nicht bekannt ) repräsentieren wir die Daten D als ein Paar von signed ( unsigned ) Permutationen pa, pb. • Wir unterscheiden N(N+1)/2 mögliche Inversionen. • Jede der N(N+1)/2 möglichen Inversionen hat die gleiche Wahrscheinlichkeit Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Es gibt insgesamt (N(N+1)/2)Lx gleichwahrscheinliche Inversionssequenzen X der Länge Lx +1 +2 +3 +4 4(4+1)/2 = 10 mögliche Inversionen P4 P2 P1 I1 I3 P6 I4 P5 I5 I2 P3 105 mögliche Inversionssequenzen 5X der Länge 5 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Sei die Menge aller möglichen Inversionssequenzen, dann ist die Wahrscheinlichkeit für jedes X geben P(X| ) = (e-Lx/Lx!)(N(N+1)/2)-Lx Ws für das Auftreten von Genau L Inversionen Ws für genau eine Sequenz der Länge L aus allen möglichen Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach- MCMC Ziel ist es die postiori Ws von X und zu erhalten. P(X|D) und P (|D). Erzeugen einer Markov Kette mit dem Zustandsraum: x R+ und stationärer Verteilung P(X, |D), X , R+ Anzahl der Inversionen Rate der Inversionen Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach- MCMC Idee: Samplen der Werte von und X wenn die Markov Kette in ihrer stationären Verteilung ist. Gibt nicht nur den wahrscheinlichsten Zustand aus, sondern eine Menge wahrscheinlicher Zustände, die für die gegeben Daten als „typisch“ bezeichnet werden können ABER: Wie kommt man in die stationäre Verteilung? Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach- Metropolis Hasting Algorithmus min 1, y* Qyx x Qxy x Qxy „proposal step“ Qyx y Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Bayes Theorem: Stationäre Verteilung P(X, |D) = P(D| X, )P(X, )/P(D) = P(D| X, )P(X| )P( )/P(D) = 1 (e-Lx/Lx!)(N(N+1)/2)-Lx P(X|Y) =P(Y|X)*P(X) P(Y) P(X,Y)=P(X|Y)*P(Y) 1 /P(D) max y* Qyx x Qxy Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X Pa I2 X kann man sich als ein „Inversionspfad“ vorstellen, der Sequenzen von Permutationen, p0 = pa, p1,... pL = pb und von Inversionen I1, I2, ... IL mit pi = Iipi-1, i = 1, 2, ... L umfasst. I4 Der neue vorgeschlagene Pfad, Y, wird wie folgt konstruiert: I1 I3 I5 I6 pb I7 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X Pa 1. Wähle einen zu ersetzenden Bereich von X. Wähle mit Ws qL(l,j), eine Länge,l ,(0 < l < L) und eine Startpermutation, pj, ( 0 < j < L-l ). Der Subpfad von p= pj zu p = pj+l wird in Y von einem neuen ersetzt. I1 I2 p I3 I4 I5 I6 pb p I7 2. Generiere einen neuen Subpfad. Verwende den breakpoint Graph von p relative zu p, wähle eine Inversion, I1‘, zufällig, aber c = 1 mit hoher Ws. Dann fahre in gleicher Weise fort, wähle I2‘, unter Betrachtung von I1‘p relativ zu p, und so weiter bis I1‘, I2‘... Il‘‘p = p. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail 1. Wähle einen Bereich von X, der ersetzt werden soll Wähle die Länge l des zu ersetzenden Teilstückes durch das sampeln aus einer Verteilung q(l) Wähle j gleichverteilt zufällig aus 0, 1, ... Li- l Q(l) 1- tanh l -1 N mit = 8 und = 0.65 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail 2. Erzeugung eines neuen Subpfades Start: p = pj End: p = pj+1 Wir suchen eine Sequenz von Inversionen I1‘, I2‘, ... Il‘ und dazwischenliegenden Permutationen p0‘ = p, p1‘, p2‘... pl‘‘= p mit pi‘ = Ii‘pi-1, i = 1,2,...l‘. Wir betrachten dazu den breakpoint Graph von pi-1‘ relativ zu p Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail 2. Erzeugung eines neuen Subpfades Klassifiziere alle Inversionen nach c = +1, 0, -1 Erinnerung: pa = pb wenn Anzahl der Zykel = n+1 Wähle I so, dass mit hoher Ws c = +1 => Ein Schritt „vorwärts“ Man nimmt also an, dass kurze Pfade wahrscheinlicher sind als lange. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail 2. Erzeugung eines neuen Subpfades Wenn N+1, N0 und N-1 die Anzahl der Inversionen ist für c = +1, 0, -1 dann ist die relative Ws für eines davon 1, 1, 2. Nach der Auswahl ziehen wir gleichverteilt eine der N Inversionen => Ws für ein c = +1 = 1/((1+1+ 2)N+1) Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail 2. Erzeugung eines neuen Subpfades Wenn N+1 = N0 = 0 sind die Permutationen gleich ~ Mit Ws 1- 3 stoppen wir hier bzw. mit Ws 3 fahren wir mit c = -1 fort. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail 2. Erzeugung eines neuen Subpfades Die Ws qnew für den neuen Subpfad der Länge l‘ ist das Produkt von l‘ +1 Faktoren Einen pro Inversion + Ws für das Stoppen wenn man pb erreicht Die Länge des vorgeschlagenen Pfades ist L‘ = L+ l‘-l Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail 3. Errechnen der „ forward proposal probabiltiy“ Q(Y|X) = Q(XY) = qL(l,j) qnew 4. Errechnen der „ acceptance probabiltiy“ Q(X|Y) = Q(YX) = qL‘(l‘,j) qold Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail ( unsigned ) Anstatt die Markerorientierung zu berechnen verwendet man einfach orientierte Permutationen Man lässt die Startpermutation über die 2N orientierten Permutationen laufen. Update also sowohl für die Markerorientierung als auch für die Inversionen Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail ( unsigned ) Pa F Fpa 1. Wähle eine Subpfad genau wie im F Signed case. 2. Wende den Flip Operator F an Palpha F Fpalpha Pa an und lasse die Inversionen bis Palpha gleich. 3. Erzeuge den neuen Pfad genau wie Im signed case Wieso kann man das machen? Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating X in Detail ( unsigned ) Pa F Fpa Das Durchführen eines Flips ist eine F Inversion eines einzelnen Markers. Palpha F Fpalpha Inversionen kann man über c bewerten! Führe Flips mit c = -1 mit Ws 4, 0,5 für c = 0 oder c = 1 durch. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating Ein Gibbs Schritt wird verwendet, um zu aktualisieren D.h. wird zufällig gemäß der Ws von gegeben die Anzahl der Inversionen und der Daten aktualisiert P(|X,D) P(X|)P() e- Lx P() Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – convergence monitoring Methode von Gelman und Rubin: Man brauch mindestens 2 Chains für die gleichen Daten Definiere eine „between chain variance“ B und eine „within chain variance“ W. Konvergenz ist erreicht wenn R0,5 = ((n-1)/n+B/W)0,5 sich 1 nähert. Hier verwendet: 5 – 10 Ketten, Burn In Phase bis R0,5 1.1 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – convergence monitoring Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – improving convergence Das Update Schema hat viele Parameter, die die Konvergenz der Ketten beinflussen: und : kontrollieren die Länge des zu ersetzenden Teilstücks 1, 2. 3 : kontrollieren die Erzeugung des neuen Subpfades ( kurze vs. Lange Pfade 4 : kontrolliert die Bevorzugung für c = +1 Markerflips Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – improving convergence Durch Läufe mit simulierten Daten ist man zu folgenden Ergebnissen gekommen: :8 : 0.65 1: 0.03 2 : 1/2 3 : 1 2 4 : 0.025 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Applications to Real Data 1. Human-cattle data 14 unsigned Markers 8 simultane Ketten Startzustände werden mit verschiedenen 1 erzeugt. 815.104 Iterationen Konvergenz nach 8.192 Iterationen Danach über jede Iteration gemittelt Laufzeit: 254 Sekunden Athlon 1,2 GHz Prozessor Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Applications to Real Data 1. Human-cattle data Ergebnisse: Wahrscheinlichster Wert für L = dem parsimony Wert: 4 Aber es ist wahrscheinlich, das die Anzahl der wahren Inversionen höher als 4 ist: 95% credible set für L ( 4 L 9 ) => die Erwartete Anzahl an Inversion ist wesentlich höher als 4 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Applications to Real Data Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Applications to Real Data 1. Human-cattle data Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Applications to Real Data 2. D. melanogaster und D. repleta Daten 79 unsigned Markers 6 simultane Ketten Startzustände werden mit verschiedenen 1 erzeugt. 43 mio Iterationen Konvergenz 1,7 mio Iterationen Danach über jede Iteration gemittelt Laufzeit: 3,456 x105 sec 4 Tage Athlon 1,2 GHz Prozessor Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Applications to Real Data 2. D. melanogaster und D. repleta Daten Ergebnisse: Wahrscheinlichster Wert für L = 87 Erwartungswert: 92 Inversionen 95% credible set für L: ( 71 L 118) Parsemony Wert: 53 95% credible set für : (64.14 125.00 ) Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Applications to Real Data Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Applications to Real Data Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Applications to Real Data Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Conclusion • Lösung des Problems durch einen voll probabilistischen Ansatz machbar • Für große Datensätze versagen die Parsemony Methoden • Im Gegensatz zu den Parsymony Ansätzen ermöglicht der Bayes Ansatz zusätzlich die Beantwortung von folgenden Fragen: Treten alle Inversionen mit der gleichen Rate auf? Sind Inversionsraten über Abstammungslinien konstant • Ziele Einbinden von Translokationen, Chromosomenbrüchen und Fusionen Erweiterung auf komplette Genome i.d „Genomic Distance“ Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Baysian Estimation of Genomic Distance T. L. York, R. Durrett and R. Nielsen Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Motivation für den erweiterten Ansatz „Understanding the relationship between the organization of two genomes is important for transfering information betweens species. For example, for finding animal models of human deseases or locating genes of agricultural importance.“ Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Allgemein evolvieren Genome durch: Translocations Fusions Fisions Inversions Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Allgemein evolvieren Genome durch: Translocations Fusions Fisions Inversions Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Allgemein evolvieren Genome durch: Translocations Fusions Fisions Inversions Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Allgemein evolvieren Genome durch: Translocations Fusions Fisions Inversions Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Parsimony Methods - Hannenhalli, Pevzner and the breakpoint graph Problem der minimalen Chromosomen Distanz gelöst Idee: Die Chromosomen eines Genoms „verketten“, so dass ein langes Chromosom entsteht und erzeugen eines modifizierten break point graphs. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Parsimony Methods - Erzeugung des break points Graphs Zu Untersuchende Daten: Genom der Aubergine vs. Tomate Eggplant: E3: 1 2 3 4 5 6 E4: 7 8 E5: 9 10 E10: 11 12 13 14 15 16 17 18 E11: 19 20 21 22 E12: 23 24 25 26 27 Tomato: T3: 1 -5 2 6 T4: 21 -22 -20 8 T5: -4 14 11 -15 3 9 T10: 7 16 -18 17 T11: -19 24 -26 27 25 T12: -12 23 13 10 Ergänzen von „leeren Chromosomen“ wenn notwendig Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Parsimony Methods - Erzeugung des break points Graphs Eggplant Doubled, Ends Added 1000 , 1 2 , 3 4 , 5 6 , 7 8 , 9 10 , 11 12 , 1001 1002 , 13 14 , 15 16 , 1003 1004 , 17 18 , 19 20 , 1005 1006 , 21 22 , 23 24 , 25 26 , 27 28 , 29 30 , 31 32 , 33 34 , 35 36 , 1007 1008 , 37 38 , 39 40 , 41 42 , 43 44 , 1009 1010 , 45 46 , 47 48 , 49 50 , 51 52 , 53 54 , 1011 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Parsimony Methods - Erzeugung des break points Graphs Tomato Doubled, Ends Added 2000 , 1 2 , 10 9 , 3 4 , 11 12 , 2001 2002 , 41 42 , 44 43 , 40 39 , 15 16 , 2003 2004 , 8 7 , 27 28 , 21 22 , 30 29 , 5 6 , 17 18 , 2005 2006 , 13 14 , 31 32 , 36 35 , 33 34 , 2007 2008 , 38 37 , 47 48 , 52 51 , 53 54 , 49 50 , 2009 2010 , 24 23 , 45 46 , 25 26 , 19 20 , 2011 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Parsimony Methods - Erzeugung des break points Graphs Mögliche enstehende Komponenten im Graph: Pfade die 2 Enden verbinden „kurze Pfade“ ( hier 5 ) „lange Pfade“ ( hier 7 ) Pfade die die Enden eines Genoms verbinden ( hier 0 ) Zykel ( hier 0 ) Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Parsimony Methods - Erzeugung des break points Graphs Die untere Grenze für die Genom Distance berechnet sich aus: M +N ( Anzahl der Kommas, hier 33 ) minus Anzahl der kurzen und langen Pfade ( hier 5 + 7= 12) minus der Anzahl der Zykel Plus Anzahl der Pfade die im gleichen Genom anfangen und Enden ( hier 0 ) Um Tomate in Aubergine zu überführen sind also mindestens 33-12 = 21 Inversionen oder Translokationen notwendig Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Parsimony Methods - Erzeugung des break points Graphs Problem: Wieder nur für „signed“ Marker möglich Im Tomaten- Auberginen Problem waren 5 einzelne Marker: 25 = 32 einfach Mensch - Katze: 221 kompliziert Mensch - Rind: 275 umöglich Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Modell Annahmen • Umordnung geschieht infolge von Inversionen, Translokationen, Fusions and Fissions) • Fusions and Fissions werden als spezial Fall von Translokationen behandelt, bei dem entweder das input oder output Chromosom leer ist • Wenn zu einer Zeit t IT(t) Translokationen und II(t) Inversionen möglich sind dann ist die Rate mit denen beide auftreten jeweils IT(t)T und II(t)I • Die Zeit bis zu einem Ereignis E ist expotential verteilt mit Parameter IT(ti) T+II(ti) I)ti • Wir nehmen a priori eine Gleichverteilung für E an: P(E) = 1/ Emax für 0 < E <= Emax • Die Anzahl der Marker, deren Ordnung auf beiden Genomem bekannt ist, und die verglichen werden soll, ist N. • Die Anzahl der Chromosomen die in beiden Genomen vorhanden ist ( inkl. Leere Chromosome) ist M. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Inversionen und Translokationen im Genom bilden eine Markovkette mit dem Zustandsraum ( 0 ) der durch alle möglichen Anordnungen der N geordneten Marker auf den M geordneten Chromosomen erzeugt wird. (M+N-1)! N |0| = 2 (M-1)! 0(x) = 2-N (M-1)! (M+N-1)! , x 0 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Ordnung der Chromosome egal neue Markovkette mit kollabierten Zustandsraum (U) Zustände in U mit M0 leeren Chromosomen sind Äquivalenzklassen aus 2(M-M0) M!/M0! Elementen aus 0 0 U Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Daraus folgt die stationäre Wahrscheinlichkeit von einem Element aus mit M0 leeren ChromosomenU u(x) = 2(M-N-M0) M!(M-1)! , x U M0!(M+N-1)! Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Genom 1 (x1) , Genom 2 (x2) x1 kann in x2 durch eine Sequenz von Translokationen und Inversionen überführt werden Pr(x1, x2| ) = Pr(x1) Pr(x1 x2| ) wobei Pr(x1 x2| ) die Übergangswahrscheinlichkeit von x1nach x2 ist. ist der Vektor der Parameter T I.. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Die Ws für x1 ist unabhängig von daher: L() = Pr(x1 x2| ) Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Sei die Menge aller möglichen Pfade von x1 zu x2. D.h.: Pr(x1 x2| ) = y Pr(y|) Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Um zu schätzen wird eine neue Markovkette erzeugt mit dem Zustandsraum [0,)2 x und mit stationärer Verteilung geben durch die verbundene Wahrscheinlichkeit von und dem evolutionären Pfad (y,T ,I ) = p(y,T ,I|x1,x2), y , E [0,) Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Verwendung von Metropolis Hasting um Prozess konvergieren zu lassen: min 1, y* Qyx x Qxy Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach Umformung nach Bayes (y,T ,I ) = p(y,T ,I|x1,x2) = p(x1,x2| y,T ,I ) p(y|T ,I) p(T ) p(I) / P( x1,x2) p(y|T ,I) p(T ) p(I) Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach 1 2 . . . E e-(IT(ti) T-II(ti) I)ti E e-(IT(ti) T-II(ti) I)ti p(y|T ,I)= E(i) e-(IT(ti) T-II(ti) I)ti S+1 i=1 E(i) = E e-(IT(ti) T-II(ti) I)ti S T wenn i S und Translokation I wenn i S und Inversion 1 wenn i = S+1 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Proposal Probability Gleiches Vorgehen wie im ersten Paper schlage einen Teil des Pfades vor, der ersetzt werden soll erzeuge neuen Subpfad unter Verwendung des break point Graphen Berechne die Ws für den neuen Pfad Ws für den umgekehrten Weg Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Updating T ,I Y, T und I werden alternierend aktualisiert, wobei Tneu und Ineu unabhängig von den jeweils anderen Werten in einem Fenster um den alten Wert Talt ,Ialt gewählt werden. Die Verteilung der Werte von T und I ist proportional zur deren Likelihood Funktion. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – convergence Verwendung von Metropolis Coupled Markov Chain Monte Carlo Feststellung der Konvergenz über „between chain“ und „within chain variance“ ( Gelman & Rubin ) Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Analysis of Three Data Sets 1. Tomate vs. Aubergine 170 Marker minimale Distanz 28, 23 Inversionen und 5 Translokationen Bayes Ansatz: 6 unheated Chains, 459.000 updates, 20 Stunden Burn in: 14.000 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Analysis of Three Data Sets 1. Tomate vs. Aubergine Ergebnisse des Bayes Ansatz: 95% credible Intervalls Inversionen: [5,7] Transkriptionen[21,31] Number of Events [28,37] T = 0.000219 I = 0.0194 0.000219*30271=6.629 0.0194*1335=25.899 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Analysis of Three Data Sets Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Analysis of Three Data Sets 2. Human vs. Cat 269 Marker parsimony Distanz 78, 64 Inversionen und 14 Translokationen Bayes Ansatz: 6 unheated Chains, 2.2 mio updates, 9 Tage Burn in: 306.000 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Analysis of Three Data Sets 2. Human vs. Cat Ergebnisse des Bayes Ansatz: 95% credible Intervalls Inversionen: [71,89] Transkriptionen[12,15] Number of Events [85,102] T = 0.000161 I = 0.0350 0.000161*79650=12.82 0.0350*2370=82.95 Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Analysis of Three Data Sets 2. Human vs. Cattle 422 Marker parsimony Distanz 155, 135 Inversionen und 20 Translokationen Bayes Ansatz: 4 unheated Chains, 1.3 mio updates, 30 Tage Burn in: Konvergenz konnte nicht erreicht werden Burn in nach 600.000 updates beendet Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Analysis of Three Data Sets 2. Human vs. Cattle Keine sichere Aussage möglich, da die 95% credible sets von Lauf zu Lauf sehr verschieden sind. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Analysis of Three Data Sets Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik A Baysian Approach – Conclusion • Lösung des Problems durch einen voll probabilistischen Ansatz machbar, allerdings entstehen Probleme bei großen Datansätzen wie der Cattle – Mensch Datensatz gezeigt hat • Für große Datensätze versagen die Parsemony Methoden allerdings genauso und früher • Muster erkennbar, das Inversionen 4-7 so häufig sind wie Translokationen Biologen gehen von 2 mal häufigerem Auftreten von Inversionen aus. Der Schluss aus den Parsemony Methoden • Ziele Anpassen der Inversionswahrscheinlichkeiten, so dass sie von der Größe des invertierten Stücks abhängen Erweitern, so dass Genduplikationen erfasst werden. Johann Wolfgang Goethe Universität Frankfurt am Main Fachbereich 15: Biologie und Informatik Bioinformatik Fragen?