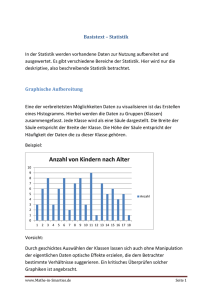
Dokument_38.
Werbung

Aus der Neurochirurgischen Klinik der Friedrich-Alexander-Universität Erlangen-Nürnberg Direktor: Prof. Dr. M. Buchfelder Einfluss präoperativer medikamentöser Therapien auf die Proliferationsaktivität von Hypophysenadenomen bei Akromegalie Inauguraldissertation zur Erlangung der Doktorwürde der Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg vorgelegt von Sven-Martin Schlaffer aus Schwetzingen II Gedruckt mit Erlaubnis der Medizinischen Fakultät der Universität Erlangen-Nürnberg Dekan: Professor Dr. med. Dr. h.c. J. Schüttler Referent: Professor Dr. med. M. Buchfelder Korreferent: Professor Dr. med. O. Ganslandt Tag der mündlichen Prüfung: 30. Juli 2012 III Meiner Frau und meiner wachsenden Familie gewidmet 1 Inhaltsverzeichnis 1. Zusammenfassung 03 1.1 Hintergrund und Ziele 03 1.2 Methoden 03 1.3 Ergebnisse und Beobachtungen 03 1.4 Praktische Schlussfolgerungen 04 2. Summary 05 2.1 Aim and background 05 2.2 Methods 05 2.3 Results 05 2.4 Conclusion 06 3. Einleitung und Problemstellung 17 4. Material und Methoden 13 4.1 Patientenkollektiv 13 4.2 Vorbehandlung 14 4.3 Operationen 17 4.4 Histologische Untersuchungen 17 4.5 Bestimmung der Proliferationsparameter 20 4.6 Statistische Analyse 21 5. Ergebnisse 22 5.1 Proliferationsaktivität ohne medikamentöse Vorbehandlung 22 5.2 Wirkung der Vorbehandlung mit Dopamin-Agonisten 25 5.3 Wirkung der Vorbehandlung mit Somatostatin-Analoga 28 5.4 Wirkung der Kombination aus Dopamin-Agonisten 5.5 5.6 und Somatostatin-Analoga 30 Wirkung der Vorbehandlung mit Pegvisomant 31 5.5.1 Monotherapie mit Pegvisomant 32 5.5.2 Kombinationstherapien mit Pegvisomant 34 Einfluss der medikamentösen Monotherapie in Abhängigkeit von der Histologie 35 5.7 Mehrfach operierte Patienten 37 5.8 Einfluss einer Radiotherapie 40 6. Diskussion 6.1 Proliferationsaktivität von Hypophysenadenomen 41 41 2 6.2 Einfluss einer Vorbehandlung mit Dopamin-Agonisten 44 6.3 Einfluss einer Vorbehandlung mit Somatostatin-Analoga 45 6.4 Einfluss einer Vorbehandlung mit Pegvisomant 46 6.5 Bedeutung mehrfach operierter Patienten 48 6.6 Wirkung der Radiotherapie 50 7. Literaturverzeichnis 52 8. Abbildungsverzeichnis 67 9. Abkürzungsverzeichnis 68 10. Danksagung 69 3 1. Zusammenfassung 1.1 Hintergrund und Ziele Die Akromegalie geht unbehandelt mit einer verkürzten Lebenserwartung einher. Daher ist das Ziel jeglicher Therapie die Normalisierung des Wachstumshormonexzesses. Neben der kausalen, operativen Therapie durch die Resektion des Wachstumshormon-produzierenden Tumorgewebes, stehen verschiedene Medikamente zur Behandlung zur Verfügung. Die Aufgabe dieser Arbeit war es, den Einfluss dieser Medikamente bzw. den einer Radiotherapie auf die immunhistochemische Expression der Proliferationsmarker Ki-67 und Topoisomerase-IIα von Wachstumshormon-sezernierenden vorbehandelten, primär operierten Hypophysenadenomen im Vergleich zu von nicht entsprechend behandelten Patienten zu untersuchen. Von besonderem Interesse war der Einfluss des Wachstumshormon-Rezeptor-Antagonisten Pegvisomant. Unter dieser Therapie kam es in einigen Fällen zu einem deutlichen Tumorprogress mit einmalig nachgewiesenem Anstieg der oben genannten Proliferationsparameter, die dem Wirkungsmechanismus des Medikaments zugeordnet wurden. 1.2 Methoden Insgesamt standen 385 in Formalin fixierte Tumorproben von 368 Patienten zur retrospektiven Analyse der in den Jahren 2002 bis 2010 operierten Patienten zur Verfügung. Die Präparate wurden für die Proliferationsmarker Topoisomerase-IIα und Ki-67 gefärbt und die Proliferationsrate an allen Tumorproben mikroskopisch anhand der positiv gefärbten Zellen bestimmt. Diese Ergebnisse wurden mit den klinischen Parametern sowie der Art, Dauer und Dosis der medikamentösen Vorbehandlung in Beziehung gesetzt. 1.3 Ergebnisse und Beobachtungen Grundsätzlich verhielten sich die untersuchten Proliferationsparameter kongruent zu einander. Es fanden sich statistisch signifikant erhöhte Proliferationsraten bei größeren und invasiven Hypophysenadenomen bzw. jüngeren Patienten. Die Gabe von Dopamin-Agonisten und SomatostatinAnaloga führten zu einer Minderung beider Proliferationsparameter, wobei 4 die Applikationsdauer bzw. die Dosierung der Medikamente keinen signifikanten Einfluss zeigten. Einzig Oktreotid führte aber auch nur in der Gruppe der dicht-granulierten Hypophysenadenome zu einer statistisch signifikanten Minderung der Proliferationsindices. Einen Anstieg der Proliferationsparameter unter der Therapie mit Pegvisomant fand sich dagegen nicht. Bei der Untersuchung von Tumorproben der im Intervall mehrfach operierten Patienten konnte im Einzelfall eine Änderung der Proliferationsindices unter veränderter medikamentöser oder strahlentherapeutischer Therapie nachgewiesen werden. Es fand sich aber auch hier keine Zunahme der individuellen Proliferationsaktivität unter der Therapie mit Pegvisomant. 1.4 Praktische Schlussfolgerungen Es bestätigten sich die teilweise in der Literatur beschriebenen Effekte der Behandlung mit Somatostatin-Analoga bei Patienten mit einer klinisch floriden Akromegalie Hypophysenadenom. und einem Wachstumshormon-sezernierenden Interessanterweise scheinen die proliferations- hemmenden Effekte der Therapie mit Somatostatin-Analoga auch nach deren Beendigung fortzubestehen. Eine Erhöhung der Proliferationsaktivität durch den Einsatz von Pegvisomant, ließ sich anhand der vorliegenden Daten aus der zulassungsgerechten klinischen Anwendung dieser Substanz nicht nachvollziehen. Der bisher unbekannte und erstmalig nachgewiesene, von der Dosis und Dauer abhängige Effekt einer Therapie mit Pegvisomant auf die Expression von Topoisomerase-IIα bedarf weiterer klinischer und experimenteller Untersuchungen. 5 2. Summary 2.1 Aim and background Untreated acromegaly is associated with a shortened life expectancy. Therefore, the goal of any therapy is normalization of growth hormone secretion. In addition to the causal, surgical treatment by resection of the growth hormone-producing tumour tissue, different drugs are available for medical treatment. The purpose of this study was to compare the influence of the last applied drug or radiotherapy on the immunhistochemical expression of the proliferation markers Ki-67 and topoisomerase-IIα in patients with growth hormone-secreting pituitary adenomas. Previously untreated tumours were compared with medically pretreated or irradiated ones. Of particular interest was the influence of the growth hormone receptor antagonist Pegvisomant. In some cases a significant tumour progression occurred during Pegvisomant therapy and in one patient an increase of the above mentioned proliferation parameters was reported. This effect has been assigned to the mechanism of action of the drug itself. 2.2 Methods A total of 385 formalin-fixed tumour samples from 368 patients, operated in the years between 2002 and 2010 were available for retrospective analysis. Each tumour sample topoisomerase-IIα and was Ki-67 stained and the for the proliferation proliferation was markers determined microscopically by counting positively stained cells. These results were correlated with clinical parameters and the nature, duration and dosage of the last applied drug. 2.3 Results Basically, the proliferation parameters studied behaved congruent to each other. There were statistically significantly increased proliferation rates in larger and invasive adenomas or younger patients. The administration of dopamine agonists and somatostatin analogues resulted in a reduction of both proliferation parameters, whereas neither the duration of treatment nor the dosage of the drugs showed a definite influence. Only octreotide led in the group of densely-granulated pituitary adenomas to a statistically 6 significant reduction in the proliferation indices. An increase of proliferation parameters in patients treated with Pegvisomant was not detectable. In addition, investigations of tumour samples of repeatedly operated patients who had been exposed to different drugs or radiation therapy occasionally led to a change of the proliferation indices, but an increase of proliferation during Pegvisomant treatment was never observed. 2.4 Conclusion This study confirmed some of the published effects of treatment with somatostatin analogues in patients with acromegaly and a growth hormonesecreting pituitary adenoma. Interestingly, the antiproliferative effects of a therapy with somatostatin analogues continued even after cessation of the application. An increase in the proliferative activity caused by the currently recommended clinical application of Pegvisomant was to not detected. The up to date unknown and herein demonstrated dose and duration-dependent effect of treatment with Pegvisomant on the expression of topoisomerase-IIα warrants further clinical and experimental studies. 7 3. Einleitung und Problemstellung Hypophysenadenome beim Krankheitsbild der Akromegalie und bei Gigantismus sind im Allgemeinen gutartige und langsam wachsende Tumore. Die vollständige chirurgische Entfernung stellt daher oft eine kurative Behandlung dar [69, 85]. Allerdings können die Geschwülste auch invasiv in die umgebenden Strukturen einwachsen und sind dann nicht mehr komplett resezierbar [91]. Bei den hormonproduzierenden Adenomen bestimmt die hormonelle Übersekretion und damit die Ko-Morbidität sowohl bei der Akromegalie als auch beim Morbus Cushing die Prognose und begründet auch die reduzierte Lebenserwartung der Patienten [5, 35, 96]. Mit einer Normalisierung der Hormonsekretion verringert sich die Ko-Morbidität ganz erheblich. Dies entspricht der Korrektur der Lebenserwartung nach einer erfolgreichen Operation in den Normalbereich hinein [5, 55, 120]. Deshalb ist es für das Schicksal des Patienten mit einer Akromegalie oder einem Gigantismus entscheidend, die Spiegel des Wachstumshormons (somatotropes Hormon, STH) bzw. des nach Stimulation von Wachstumshormon vornehmlich in der Leber synthetisierten Insulinähnlichem Wachstumsfaktor 1 (insulin-like growth factor 1; IGF-1) zu normalisieren [10, 48]. Oft gelingt dies nur mit einer Kombinationstherapie aus Operation, medikamentösen Maßnahmen und ggf. auch einer Radiotherapie [47, 85, 114]. Jedenfalls ist die prämature Mortalität in kaum einem Fall durch das aggressive Wachstum des Hypophysentumors und seine raumfordernde Wirkung bedingt, sondern durch die Ko-Morbidität der Akromegalie [35, 96, 120]. Von Wachstumshormon-produzierenden, aggressiven Hypophysenkarzinomen sind bisher nur in wenigen Kasuistiken berichtet [34, 51, 70]. Die spontane Progression des Hypophysenadenoms ohne Behandlung ließe sich zwar im Prinzip durch serielle Untersuchungen mit bildgebenden Verfahren bestimmen, doch wird wegen der Ko-Morbidität der Akromegalie heute jede diagnostizierte Erkrankung auch unmittelbar behandelt [85]. Obwohl die bildgebende Methode der Wahl, die Magnetresonanztomographie, seit nunmehr über 20 Jahren verfügbar ist, existieren kaum Untersuchungen, aus denen sich die Proliferationstendenz unbehandelter Tumoren über einen längeren Zeitraum ableiten ließe [57, 98]. Eine bessere Datenlage findet sich zum Verlauf von Tumorresten nach einer 8 Operation [24, 37, 52, 117], denn die mikrochirurgische Tumorentfernung ist aufgrund ihrer Effektivität und Sicherheit als Primärtherapie etabliert und bis heute allgemein akzeptiert. Sofern keine Kontraindikationen bestehen, wird dem Patienten daher in aller Regel als erster Schritt der Therapie zur Operation geraten [69, 85]. Oft reicht aber die alleinige Operation nicht aus, um eine vollständige Remission der Akromegalie zu erreichen [83] . Die Kriterien für eine Normalisierung der Wachstumshormon- und IGF-1Spiegel unterliegen wechselnden Definitionen, die in Konsensuskonferenzen ausgehandelt werden Vorbehandlung mit [10, 46, 48]. Der Somatostatin-Analoga Wert wird einer dabei präoperativen unterschiedlich diskutiert [7-9, 12, 22, 27, 29, 75, 86]. In einigen Arbeiten wird eine höhere Normalisierungsquote der Wachstumshormon-Sekretion angegeben, wenn die Patienten präoperativ über Zeiträume von zwei Monaten bis zu einem Jahr mit einem Somatostatin-Analogon, vornehmlich Oktreotid vorbehandelt wurden [4, 22, 28, 118]. Andere Autoren finden weder eine Verbesserung der chirurgischen Ergebnisse, was die Hormonübersekretion betrifft, noch eine Verringerung der perioperativen Komplikationen [1, 11, 66]. Die meisten der heute verwendeten Therapien der Akromegalie sind gegen den Hypophysentumor gerichtet. Die Operation zielt auf seine Entfernung, die Radiotherapie auf seine Inaktivierung. Medikamente, wie Dopamin-Agonisten [45, 90] oder Somatostatin-Analoga [82, 95] interferieren mit intrazellulären Signalübertragungsmechanismen im Hypophysenadenom. Die parallel zur Senkung der Wachstumshormonspiegel zu beobachtende Abnahme der Tumorgröße eines Wachstumshormon-sezernierenden Hypophysenadenoms unter einer Behandlung mit Oktreotid oder Lanreotid dokumentiert diese direkten Effekte [12, 27, 29, 31, 32, 49, 67, 80, 103]. Immerhin kommt es bei der Mehrzahl der Patienten so zu einer Tumorschrumpfung, wenngleich das Ausmaß der Volumenreduktion geringer ist, als der Effekt von DopaminAgonisten bei Prolaktinomen [12]. Lediglich die periphere Blockade des Wachstumshormon-Rezeptors durch Antagonisten, wie Pegvisomant, berücksichtigt den Tumor nicht und verhindert mit der Verminderung der Produktion von IGF-1 ausschließlich die klinischen Manifestationen der Akromegalie [54, 64, 123, 128]. Individuelle Berichte über die 9 Größenzunahme von einigen Hypophysenadenomen unter der Behandlung mit dem Wachstumshormon-Rezeptor-Antagonisten stammen schon aus der Zulassungsstudie und aus anekdotischen Fallbeschreibungen [3, 30, 42, 60, 112, 128]. Diese Angaben beunruhigten viele Kliniker, auch weil die Annahme eines wirkungsvollen Feedback-Mechanismus zwischen Wachstumshormon und IGF-1 und damit die Möglichkeit der Entwicklung besonders aggressiver Hypophysentumoren bei einer Störung dieses Regelkreises befürchtet wurde. Man hat Analogien zum ACTH- sezernierenden Tumor und zu Thyreotropinomen nach Therapien, die gegen die Nebenniere bzw. Schilddrüse gerichtet waren, hypothetisiert [2, 15, 33, 110]. So gelten ACTH-sezernierende Hypophysenadenome nach der Behandlung eines Morbus Cushing durch bilaterale Adrenalektomie und TSH-sezernierende Hypophysenadenome, bei denen die Ersttherapie gegen die Schilddrüse gerichtet war, als besonders aggressiv, proliferationsaktiv und schwierig zu behandeln [2]. Klinische Verlaufsuntersuchungen durch die serielle Analyse von Kernspintomogrammen bei Patienten mit Akromegalie konnten, trotz aller dabei bestehenden methodischen Schwierigkeiten, keinen Wachstums-fördernden Einfluss von Pegvisomant finden [18, 19, 59]. Ein ganz anderer Ansatz dagegen ist die Analyse der Proliferationsaktivität durch Untersuchungen am, bei einer Operation gewonnenen Tumorgewebe. Hier ist die medizinische Literatur schon seit Jahren reich an Daten. Dabei sind unterschiedliche methodische Ansätze gewählt worden. Die früher als Gold-Standard angesehene Flow-Zytometrie liefert zwar Angaben über die prozentualen Anteile der Zellzyklusphasen, benötigt aber Frischgewebe [41, 79]. Ähnlich umständlich ist die Bestimmung der Inkorporation von Bromdesoxyuridin oder Tritium-markierten Thymidin in das Adenomgewebe [21, 26, 87, 115]. Weniger schwierig zu handhaben sind dagegen immunhistochemische Untersuchungen mit Hilfe von Proliferations- assoziierten Antikörpern, die gegen in proliferationsaktiven Zellzyklusphasen exprimierte nukleäre Antigene gerichtet sind und daher auch bei langsam wachsenden Tumoren helfen, die Proliferationsaktivität abzuschätzen und damit das Wachstumspotenzial vorherzusagen [104, 105, 113]. Man verwendet dabei das bei einer Operation gewonnene Gewebe, um prognostische Aussagen treffen zu können. Als Proliferationsmarker hat sich 10 dabei in erster Linie der von Gerdes et al. [43, 44] vorgeschlagene Antikörper Ki-67 bewährt, der in der kommerziell erhältlichen MIB-1 Variante auch für Untersuchungen am paraffinfixierten Gewebe tauglich ist und so auch retrospektive Untersuchungen erlaubt. Im Prinzip vergleichbar, aber weniger häufig angewendet sind die Markierungen von Topoisomerase-IIα [53, 78, 107, 129, 131], PCNA [105, 113], p53 [122] und Galectin [101, 108]. Für jeden der angeführten Marker gibt es gewisse methodische Probleme. Die in der Literatur bisher überwiegend verwendete Methode ist die immunhistochemische Untersuchung der MIB-1 Expression. Hierbei ist die Inhomogenität des Tumors ein erhebliches Problem, so dass sich die Untersuchung eines Tumors kaum prognostisch für das jeweilige Individuum verwerten lässt [127]. Allerdings ist die Bildung von Kohorten aufschlussreich. Man hat den Eindruck, dass Dopamin-Agonisten bei Prolaktinomen und Somatostatin-Analoga bei Wachstumshormon- sezernierenden Hypophysenadenomen die mittlere Proliferationsaktivität vermindern [39, 58, 75, 121, 133, 134]. Der Radiotherapie wird ebenfalls ein Proliferations-reduzierender Einfluss zugeschrieben [124]. Untersuchungen über den Einfluss von Pegvisomant gibt es bisher kaum. Interessant ist die Fallbeschreibung von Drake et al. [38], der von zwei Operationen zu unterschiedlichen Zeitpunkten bei einem Patienten mit einem ungewöhnlich aggressiven und letztlich auch nicht zu beherrschenden Hypophysentumor Gewebeuntersuchungen durchführen konnte. Dabei fand sich nach der Exposition von Pegvisomant eine signifikant erhöhte Proliferation, gemessen an der immunhistochemischen Expression von Ki-67 und Topoisomerase-IIα (siehe Abbildung 1). 11 A B C D Abbildung 1: Mikroskopische Abbildungen der immunhistochemischen Untersuchungen des in der Originalpublikation von Drake et al. beschriebenen Patienten (Überlassung der Abbildung im Original mit freundlicher Genehmigung des Erstautors). In der oberen Reihe (A und B, 400fache Vergrößerung) befinden sich die Färbung für Ki-67, in der unteren Reihe die für die Topoisomerase-IIα (C und D, 1000-fache Vergrößerung). Die Abbildung A und C entsprechen den 1986 entnommenen Tumorproben, B und D denen der Operation im Jahre 2003. Für beide Proliferationsmarker war eine deutliche Zunahme nach der Gabe des Wachstumshormon-Rezeptor-Anatgonisten Pegvisomant in den Jahren 1997 bis 2000 und 2002 bis 2003 zu beobachten (Ki-67: 0 – 0,5 % (1986) auf 1 – 3 % (2003), TopoisomeraseIIα: 2 – 10 % (1986) auf 15 – 80 % (2003)). Ob die Steigerung der Proliferationsindices der spontanen Entwicklung oder sogar Entdifferenzierung dieses riesigen und invasiven Tumors zuzuschreiben war oder Folge der Einwirkung von Pegvisomant, bleibt unklar. Jedenfalls hat diese Arbeit die Ängste vor einer Steigerung des Wachstumspotentials nach Gabe des Wachstumshormon-Rezeptor- Antagonisten genährt. Systematische vergleichende Untersuchungen gibt es in der medizinischen Literatur bisher nicht. Die in der Fallbeschreibung von Drake et al. [38] gemachte Annahme, dass nämlich die immunhistochemisch 12 bestimmte Proliferationsaktivität in dem bei einer Operation gewonnenen Tumorgewebe Rückschlüsse auf die Wirkung von Substanzen zulässt, denen der Patient vor der Operation ausgesetzt war, erscheint schlüssig und für eine systematische Untersuchung geeignet. Jedenfalls kann man hypothetisch annehmen, dass der Einfluss des letzten, vor der Operation verwendeten Medikaments dominierend war. Dabei ist zu bedenken, dass manche Patienten mit unterschiedlichen Pharmaka behandelt wurden und gelegentlich auch eine Kombination medikamentöser Therapien bzw. eine Strahlenbehandlung erfolgte. Die medikamentösen Therapien werden mit unterschiedlichen Präparaten und Dosen durchgeführt. Auch sind keine einheitliche Dauer der Therapie und natürlich auch kein standardisierter zeitlicher Abstand der letzten Gabe des Medikaments vor der Operation zu erwarten gewesen. Die Aufgabe dieser Arbeit war es somit die immunhistochemisch nachweisbare Expression der Proliferationsmarker Ki-67 und Topoisomerase-IIα an einer ausreichend großen Kohorte von Patienten mit Wachstumshormon-sezernierenden Hypophysenadenomen zu untersuchen und dabei den Einfluss von Dopamin-Agonisten, Somatostatin-Analoga, Pegvisomant und einer Radiotherapie durch den Vergleich mit primär operierten, nicht medikamentös behandelten Tumoren herauszufinden. 13 4. Material und Methoden 4.1 Patientenkollektiv Zur retrospektiven Auswertung standen die zwischen dem 01. April 2002 und 31.12.2010 gewonnen Tumorproben von insgesamt 385 Operationen bei 368 Patienten mit einer Akromegalie von zwei deutschen Neurochirurgischen Universitätskliniken (Göttingen und Erlangen) zur Verfügung. Alle Patienten litten an einer floriden Akromegalie mit einem erhöhten basalen Wachstumshormonspiegel, einer unzureichenden Suppression dessen im oralen Glukose-Suppressionstest sowie einem erhöhten IGF-1-Spiegel [46, 85]. Insgesamt konnte in 19 Fällen kein ausreichendes Tumormaterial für die erneute immunhistochemische Färbung aufgearbeitet werden, so dass diese Fälle von der weiteren Analyse ausgeschlossen wurden. Die verbleibenden 366 Tumorproben wurden von insgesamt 352 Patienten gewonnen, von denen 186 (52,8 %) weiblichen (193 Tumorproben; 52,7 %) bzw. 166 (47,2 %) männlichen (173 Tumorproben; 47,3 %) Geschlechts waren. Das Lebensalter zum Zeitpunkt der Operation lag zwischen 14 und 87 Jahren, im Mittel bei 44,96 mit einer Standardabweichung von ± 13,12 Jahren (Anmerkung: im weiteren Verlauf werden alle Mittelwerte ± einer Standardabweichung angegeben). In 81 Fällen (22,1 %) war bereits mindestens eine Operation vorangegangen. Zum Zeitpunkt der Operation erfüllten 85 Tumore (23,2 %) die Kriterien eines Hypophysenmikroadenoms mit einem Tumordurchmesser unter 10 mm, 273 Tumore (74,6 %) die Kriterien eines Hypophysenmakroadenoms mit einem Tumordurchmesser zwischen 10 und 40 mm bzw. 8 Tumore (2,2 %) die Kriterien eines Giant-Hypophysenadenoms mit einer Durchmesser von über 40 mm. Von den 366 untersuchten Adenomen respektierte die überwiegende Zahl (n=229; 62,6 %) zum Zeitpunkt der Operation die anatomischen Grenzen und waren somit im Gegensatz zu den invasiven Adenomen (n=137; 37,4 %) nicht in die umliegenden Strukturen (wie z.B. den Sinus cavernosus) eingewachsen. 14 4.2 Vorbehandlung Von den insgesamt 366 Fällen, war in 216 (59,0 %) keine medikamentöse Therapie zum Zeitpunkt der Operation erfolgt. Eine medikamentöse Monotherapie erfolgte in insgesamt 118 (32,2 %), eine Kombinationstherapie aus zwei verschiedenen Medikamenten in 23 (6,3 %) Fällen bzw. von drei Medikamenten in 1 (0,3 %) Fall. In insgesamt 8 (2,2 %) Fällen war eine Strahlentherapie vorausgegangen (siehe Tab.1). Art der medikamentösen Therapie n= % Keine 216 59,0 Monotherapie mit Somatostatin-Analoga 88 24,0 Dopamin-agonistische Monotherapie 21 5,7 Kombinationstherapie aus DopaminAgonisten und Somatostatin-Analoga 18 4,9 Monotherapie mit Pegvisomant 9 2,5 Kombinationstherapien mit Pegvisomant 6 1,6 stereotaktische Einzeldosiskonvergenzbestrahlung („Radiochirurgie“) 3 0,8 konventionelle Strahlentherapie 5 1,4 andere präoperative Therapien Tabelle 1: Übersicht über die Verteilung der einzelnen Gruppen in Abhängigkeit der präoperativ zuletzt applizierten Medikation bzw. der bisher erfolgten Strahlentherapie. Zum Zeitpunkt der Operation war in 109 (29,8 %) Fällen eine Monotherapie erfolgt, die entweder die Wachstumshormon- oder Prolaktin-Sekretion hemmen. In 21 Fällen erhielten die Patienten eine dopamin-agonistische Monotherapie, die in 13 Fällen mit Cabergolin und in 5 Fällen mit Bromocriptin erfolgte. In 3 weiteren Fällen litten die Patienten an einem Morbus Parkinson und erhielten hierfür Levodopa. 15 Die Therapiedauer der mit Cabergolin behandelten Patienten lag zwischen 1 und 36 Monaten (Mittelwert: 9,5 ± 10,9 Monate; Median: 3 Monate), die der mit Bromocriptin behandelten Patienten zwischen 4 und 120 Monaten (Mittelwert: 47,2 ± 39,6 Monate; Median: 36 Monate). Die kumulative Gesamtdosis des Cabergolins lag zwischen 4 und 2700 mg (Mittelwert: 238 ± 711,43 mg; Median 16 mg), die des Bromocriptins zwischen 0,3 und 16,2 g (Mittelwert: 7,2 ± 5,7 g; Median 7,8 g). Weder die Gesamtdosis noch die Dauer der Levodopa-Medikation konnten hinreichend genau zurückverfolgt werden. Im Falle der Monotherapie mit einem der beiden verwendeten SomatostatinAnaloga Oktreotid (n=80) bzw. Lanreotid (n=8) lag die Gesamtdauer der Oktreotid-Therapie zwischen 0,5 und 89 Monaten (Mittelwert: 11,6 ± 14,5 Monate, Median: 6 Monate), die des Lanreotid zwischen 1 und 31 Monaten (Mittelwert: 12,5 ± 9,2 Monate; Median: 11 Monate). Die applizierte, kumulative Gesamtdosis des Oktreotid lag zwischen 3 und 2544 mg (Mittelwert: 285,8 ± 408,9 mg; Median 120 mg), die des Lanreotid zwischen 120 und 3720 mg (Mittelwert: 1350 ± 1132,0 mg; Median: 1020 mg). In 9 (2,5 %) Fällen wurde zum Zeitpunkt der Operation eine Monotherapie der peripheren Rezeptorblockade des Wachstumshormons durchgeführt. Die Dauer der Therapie lag zwischen 5 und 68 Monaten (Mittelwert: 20,1 ± 18,8 Monate; Median: 15 Monate), die der applizierten Gesamtdosis zwischen 1,5 und 39,6 g (Mittelwert: 10,8 ± 10,8 g; Median 7,65 g). In 18 (4,9 %) Fällen erfolgte bis zur Operation die kombinierte Hemmung der Wachstumshormon- und Prolaktin-Sekretion bestehend aus Oktreotid und verschiedenen Dopamin-Agonisten. In 14 Fällen (3,8 %) erfolgte die Kombinationstherapie mit Cabergolin, in 3 (0,8 %) Fällen mit Bromocriptin und in 1 (0,3 %) Fall mit Quinagolid. In der Kombination aus Oktreotid und Cabergolin lag die Gesamtdauer der Oktreotid-Therapie zwischen 3 und 34 Monaten (Mittelwert: 10,4 ± 9,2 Monate; Median: 7 Monate), die des Cabergolins zwischen 3 und 29 Monaten (Mittelwert: 8,6 ± 6,7 Monate; Median: 6 Monate). Die kumulative Gesamtdosis des Oktreotid lag in dieser Kombinationstherapie zwischen 50 und 1020 mg (Mittelwert: 280,2 ± 285,4 mg; Median: 180 mg), die des 16 Cabergolins zwischen 10 und 540 mg (Mittelwert: 95,1 ± 138,1 mg; Median: 38 mg). In der Kombination Oktreotid und Bromocriptin lag die Gesamtdauer der Oktreotid-Therapie zwischen 1,8 und 24 Monaten (Mittelwert: 16,6 ± 10,5 Monate; Median: 24 Monate), die des Bromocriptin zwischen 1,8 und 24 Monaten (Mittelwert: 16,6 ± 10,5 Monate; Median: 24 Monate). Die kumulative Gesamtdosis des Oktreotid lag in dieser Kombination zwischen 2,1 und 720 mg (Mittelwert: 480,7 ± 338,4 mg; Median: 720 mg), die des Bromocriptin zwischen 105 und 18000 mg (Mittelwert: 12035,0 ± 8435,8 mg; Median: 18000 mg). In der Kombination Oktreotid und Quinagolid lag die Gesamtdauer der Oktreotid-Therapie bei 86 Monaten, die des Quinagolid bei 24 Monaten. Die kumulative Gesamtdosis des Oktreotid lag bei 12520 mg, die des Quinagolids bei 109,5 mg. In 3 (0,8 %) Fällen erfolgte eine Kombination aus der peripheren Rezeptorblockade des Wachstumshormons und dem Somatostatin-Analogon Oktreotid. Die Gesamtdauer der Pegvisomant-Therapie lag zwischen 3 und 6 Monaten (Mittelwert: 5 ± 1,4 Monate; Median: 6 Monate), die des Oktreotid zwischen 6 und 27 Monaten (Mittelwert: 13,3 ± 9,7 Monate; Median: 7 Monate). Die kumulative Gesamtdosis des Pegvisomant lag zwischen 1,8 und 3,6 g (Mittelwert: 2,3 ± 0,5 g; Median: 2,7 g), die des Oktreotid zwischen 180 und 770 mg (Mittelwert: 386,7 ± 271,3 mg; Median: 210 mg). In 2 (0,5 %) Fällen erfolgte eine Kombinationstherapie aus Pegvisomant mit einem Dopamin-Agonisten. In dem Falle des Quinagolid lag die Dauer der Kombinationstherapie bei 23 Monaten. Die kumulative Gesamtdosis der Pegvisomant-Therapie lag bei 10,35 g, die des Quinagolid bei 103,5 mg. Im Falle der Kombination des Pegvisomant mit Cabergolin lag die Dauer der Kombinationstherapie bei 9 Monaten. Die kumulative Gesamtdosis der Pegvisomant-Therapie betrug 8,1 g, die des Cabergolin 36 mg. In einem einzigen Fall (0,3 %) erfolgte die Dreifach-Kombination aus Pegvisomant, Oktreotid und Bromocriptin. Die kumulative Therapiedauer des Pegvisomant betrug 8, die des Oktreotid 67 und die des Bromocriptin 11 Monate. Die kumulative Gesamtdosis des Pegvisomant lag bei 4,8 g, die des Oktreotid bei 2010 mg, die des Bromocriptin bei 1650 mg. 17 4.3 Operationen Von den 366 durchgeführten Operationen wurden 352 (96,2 %) über einen transsphenoidalen und 14 (3,8 %) über einen transkraniellen Zugangsweg durchgeführt. Die jeweiligen standardisierten Operationstechniken sind an anderen Stellen im Detail beschrieben [13, 16, 17]. Insgesamt erfolgten 285 (77,9 %) primäre und 81 (22,1 %) Rezidiv-Operationen. Von den 14 transkraniellen Eingriffen ging in 13 (92,9 %) Fällen bereits eine transsphenoidale (n=12) oder eine transkranielle (n=1) Operation voraus. Von den 352 transsphenoidalen Eingriffen waren 272 (77,3 %) primäre und 80 (22,7 %) Rezidiveingriffe. Eine Besonderheit in der Analyse stellen die 13 sequentiell operierten Patienten dar, bei denen zu unterschiedlichen Zeitpunkten Tumorproben gewonnen werden konnten. Diese flossen sowohl in die Gruppenanalyse ein, werden aber auch gesondert als individuelle Langzeitverläufe betrachtet. 4.4 Histologische Untersuchungen Das während der Operation entfernte Tumorgewebe wurde in physiologischer Kochsalzlösung gesammelt. Repräsentative Tumoranteile wurden für die histologische Untersuchung ausgesucht, in Formalin fixiert, nachfolgend in Paraffin eingebettet, geschnitten und auf Objektträgern aufgebracht. Die histo-pathologische Diagnose eines Hypophysenadenoms mit der Überexpression von Wachstumshormon im Sinne einer Akromegalie wurde von den lokalen Neuropathologischen Instituten in der Universität Göttingen bzw. Erlangen-Nürnberg sowie im Referenzzentrum für Hypophysentumore der Deutschen Gesellschaft für Endokrinologie (Institut für Pathologie, Marienkrankenhaus Hamburg) für alle Tumorproben bestätigt. Routinemäßig wurden Periodic-Acid-Schiff eine Haematoxylin-Eosin-Färbung (PAS)-Reaktion durchgeführt. sowie eine Für die immunhistochemischen Untersuchungen wurden Antikörper gegen die vom Hypophysenvorderlappen sezernierten Hormone genutzt: Prolaktin (PRL), Wachstumshormon (STH), Thyroidea-stimulierendes Hormon (TSH), 18 lutenisierendes und follikel-stimulierendes Hormon (LH und FSH), adrenokortikotrophes Hormon (ACTH) [106]: Antigen Antikörper anti-STH STH (monoklonal) PRL TSH FSH LH Hersteller Sigma Immunochemicals 1:200 St. Louis, USA anti-Prolaktin Zytomed (monoklonal) Berlin, Deutschland anti-TSH Immunotech (monoklonal) Marseille, Frankreich anti-FSH Immunotech (monoklonal) Marseille, Frankreich anti-LH Immunotech (monoklonal) Marseille, Frankreich anti-ACTH Zytomed (polyklonal) Berlin, Deutschland ACTH Verdünnung 1:12000 1:5000 1:20000 1:2500 1:30 Tabelle 2: Übersicht über die im Pathologischen Institut am Marienkrankenhaus Hamburg von Prof. Saeger verwendeten Antikörper in der immunhistologischen Diagnostik. Entsprechend den histologischen Kriterien für Hypophysenadenome wurden diese in: dicht-granulierte, gering-granulierte, gemischt Prolaktin (PRL)- und Wachstumshormon (STH)- sezernierende, mamma-somatotrope bzw. plurihormonelle Adenome unterteilt [72, 106]. Histologisch fanden sich insgesamt 156 dicht-granulierte (42,6 %), 119 gemischt Prolaktin- und Wachstumshormon-sezernierende (32,5 %), 83 19 gering-granulierte (22,7 %), 5 (1,4 %) mamma-somatotrope und 3 (0,6 %) plurihormonelle Adenome. Von den 156 dicht-granulierten Adenomen befanden sich 89 (57,1 %) in der Gruppe ohne eine medikamentöse Therapie, 44 (28,2 %) in der Gruppe der Monotherapie mit einem Somatostatin-Analogon, 9 (5,8 %) in der Gruppe einer medikamentösen Kombinationstherapie, 7 (4,5 %) in der Gruppe der dopamin-agonistischen Monotherapie, 2 (1,3%) in der Gruppe der Monotherapie mit einem Wachstumshormon-Rezeptor-Antagonisten und 5 (3,2 %) in der Gruppe der zuvor radiotherapeutisch behandelten Patienten. Von den 119 gemischt Prolaktin- und Wachstumshormon-sezernierenden Adenomen befanden sich 77 (64,7 %) in der Gruppe ohne eine medikamentöse Therapie, 23 (19,3 %) in der Gruppe der Monotherapie mit einem Somatostatin-Analogon, 7 (5,9 %) in der Gruppe der dopaminagonistischen Monotherapie, 5 (4,2 %) in der Gruppe einer medikamentösen Kombinationstherapie, 5 (4,2 %) in der Gruppe der Monotherapie mit einem Wachstumshormon-Rezeptor-Antagonisten und 2 (1,7 %) in der Gruppe der zuvor radiotherapeutisch behandelten Patienten. Von den 83 gering-granulierten Adenomen befanden sich 45 (54,2 %) in der Gruppe ohne eine medikamentöse Therapie, 20 (24,1 %) in der Gruppe der Monotherapie mit einem Somatostatin-Analogon, 10 (12,0 %) in der Gruppe einer medikamentösen Kombinationstherapie, 7 (8,4 %) in der Gruppe der dopamin-agonistischen Monotherapie und 1 (1,2 %) in der Gruppe der zuvor radiotherapeutisch behandelten Patienten. Von den 5 mamma-somatotropen Adenomen befanden sich 3 (60 %) in der Gruppe ohne eine medikamentöse Therapie und je einer (20 %) in der Gruppe der Monotherapie mit einem Somatostatin-Analogon bzw. einem Wachstumshormon-Rezeptor-Antagonisten. Von den drei plurihormonellen Adenomen befanden sich 2 in der Gruppe ohne eine medikamentöse Therapie und eines in der Gruppe der Monotherapie mit einem Wachstumshormon-Rezeptor-Antagonisten. 20 4.5 Bestimmung der Proliferationsparameter Zusätzlich zu den in 4.4 genutzten Antikörpern wurden folgende Antikörper für die Antigene Ki-67 bzw. Topoisomerase-IIα verwendet: Antigen Antikörper Hersteller Verdünnung Ki-67 Mib-1 (monoklonal) Loxo, Dossenheim, Deuschland 1:100 Topoisomerase-IIα KiS1 (monoklonal) Dako, Glostrup, Dänemark 1:100 Tabelle 3: Übersicht über die verwendeten Antikörper für die Proliferationsmarker Ki-67 und Topoisomerase-IIα. Die gefärbten Präparate wurden unter einem Durchlichtmikroskop (Leica DM1000) systematisch und bei 400-facher Vergrößerung betrachtet. Sofern es das Gesamtgewebe zuließ, wurden mindestens an 10 verschiedenen Stellen des Präparats gelegene Gesichtsfelder und insgesamt mindestens 1000 Zellen gezählt. Nekrotische bzw. fibrotische Areale wurden ebenso wie die Randbereiche des Präparats ausgelassen. Als „positiv“ wurden nur die Zellen gewertet, bei denen die nukleare Färbung homogen nachzuweisen war. Der Proliferationsindex wurde als Quotient der positiv gewerteten Zellen und der Gesamtzellzahl der gezählten Zellen berechnet (sog. Labeling-Index) [63, 109]. 21 4.6 Statistische Analyse Alle statistischen Berechnungen wurden mit der der 18. Version des Statistical Package for the Social Sciences (SPSS) von IBM durchgeführt. In der Annahme, dass parametrische Tests für die Proliferationsindices von Ki-67 und Topoisomerase-IIα nicht valide waren (Kolmogorov-Smirnov p<0,05), wurden die Daten mittels der nicht-parametrischen Kruskal-Wallis-, Mann-Whitney U- und Wilcoxon-Tests verglichen. Für den Vergleich von drei oder mehr Gruppen wurde zunächst der KruskalWallis-Test durchgeführt um zu prüfen, ob ein signifikanter Unterschied bestand. Bei dem Nachweis der Signifikanz wurde eine post hoc Analyse mit dem Mann-Whitney-U-Test und der Bonferroni-Korrektur (p<0,05 /(k(k-1)/2)) durchgeführt (k entsprach der Anzahl der verglichenen Gruppen). Die Korrelationen zwischen den Proliferationsindices Ki-67 bzw. Topoisomerase-IIα und Dosis bzw. Dauer verschiedener medikamentöser Therapien und anderer Variablen wurde der Spearmans- Rangkorrelationskoeffizient berechnet. Unterschiede mit einem p<0,05 wurden als statistisch signifikant angesehen. 22 5. Ergebnisse 5.1 Proliferationsaktivität ohne medikamentöse Vorbehandlung In der Gruppe der nicht behandelten Tumoren (n=216; 59,0 %) fanden sich 56 (25,9 %) Mikro-, 156 (72,2 %) Makro- und 4 (1,9 %) Giant– Hypophysenadenome. In bereits 20 Fällen (9,3 %) war eine Operation vorausgegangen. Das Alter zum Zeitpunkt der Operation lag zwischen 14 und 78 Jahren, im Durchschnitt bei 45,84 ± 12,65 Jahren. Eine Invasion in den Sinus cavernosus fand sich bei 66 Tumoren (30,5 %). Histologisch überwogen die dicht-granulierten (n=89; 41,2 %), gefolgt von gemischt STH/ PRL-exprimierenden (n=77; 35,6 %) und gering-granulierten Adenomen (n=45; 20,8 %). Ferner fanden sich 3 (1,4 %) mamma-somatotrope und zwei (0,9 %) plurihormonelle Adenome. Der mittlere Proliferationsindex aller nicht vorbehandelten Tumore von Ki-67 lag bei 0,70 ± 1,05 %, der Median bei 0,3 % (0,0 – 7,3 %). Der mittlere Proliferationsindex der Topoisomerase-IIα lag bei 0,31 ± 0,55%, der Median bei 0,1 % (0,0 – 4,2 %). Zwischen den Geschlechtern sowie der Unterscheidung zwischen Erst- oder Re-Operation fand sich kein signifikanter Unterschied oder Trend in Hinblick auf die Proliferationsindices (siehe Tab. 4). Mit zunehmender Größe des Adenoms steigen beide Proliferationsindices (Ki-67: von 0,37 ± 0,56 % bei Mikro- über 0,81 ± 1,16 % bei Makro- auf 1,03 ± 0,95 % bei Giant-Adenomen; Topoisomerase-IIα: von 0,20 ± 0,45 % bei Mikro- über 0,35 ± 0,59 % bei Makro- auf 0,45 ± 0,51 % bei GiantAdenomen) stetig an. Die Zunahme der Proliferation war von Mikro- zu Makroadenomen für beide Indices statistisch signifikant (p<0,05), bei dem Vergleich von Mikro- zu Giant-Adenomen nur für Ki-67, aber nicht für die Topoisomerase-IIα. Die Zunahme der Proliferation bei dem Vergleich von Makro- und Giant-Adenomen war für beide Parameter nicht signifikant. Ebenso fanden sich statistisch signifikant höhere Proliferationindices bei invasiven, als bei nicht-invasiven Hypophysenadenomen (Ki-67 nicht-invasiv: 0,60 ± 0,96 %, invasiv: 0,95 ± 1,20 %, p<0,05; Topoisomerase-IIα nichtinvasiv 0,28 ± 0,53 %, invasiv: 0,38 ± 0,61 %, p<0,05). 23 Klinische Parameter Ki-67 – Index Mittelwert (%) Gesamt 0,70 ± 1,05 männlich 0,68 ± 1,11 weiblich 0,72 ± 0,99 Mikroadenom (1) 0,37 ± 0,56 Makroadenom (2) 0,81 ± 1,16 Giantadenom (3) 1,03 ± 0,95 nichtinvasiv 0,60 ± 0,96 invasiv 0,95 ± 1,20 ErstOperation 0,72 ± 1,07 ReOperation 0,52 ± 0,75 Median (%) 0,30 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00 – 7,30) 0,20 (0,00 6,80) 0,40 0,31 ± 0,58 p=0,12 0,31 ± 0,53 (0,00 7,30) 0,10 (0,00 2,30) 0,40 (0,00 7,30) 0,20 ± 0,45 p1,2<0,05 p1,3<0,05 p2,3=0,32 0,70 0,20 0,50 0,28 ± 0,53 p<0,05 0,38 ± 0,61 (0,00 7,30) 0,35 (0,00 7,30) 0,35 ± 0,59 0,45 ± 0,51 (0,30 2,40) (0,00 6,80) Mittelwert (%) 0,31 ± 0,56 p=0,15 0,10 0,30 ± 0,43 (0,00 2,40) Median (%) 0,10 U-Test (0,00 – 4,20) 0,10 (0,00 3,50) 0,10 p=0,99 (0,00 4,20) 0,10 (0,00 3,10) 0,20 (0,00 4,20) p1,2<0,05 p1,3=0,26 p2,3=0,62 0,35 (0,00 1,10) 0,10 (0,00 3,50) 0,20 p<0,05 (0,00 4,20) 0,10 (0,00 4,20) p=0,83 0,10 (0,00 1,40) Tabelle 4: Übersicht über die Ergebnisse des Mann-Whitney U-Tests innerhalb der Gruppe der nicht medikamentös vorbehandelten Tumore der Abhängigkeit verschiedener klinischer Parameter . untersuchten Proliferationsindices in 24 Hinsichtlich der histologischen Klassifikation fand sich eine Zunahme der Proliferation für Ki-67 von dicht- über gering-granulierte und gemischte STH/ PRL-Adenome (Ki-67: dicht-granuliert 0,50 ± 0,78 %, gering-granuliert 0,76 ± 0,93 %, gemischt-STH/ PRL 0,94 ± 1,33 %) ohne aber die Kriterien der Signifikanz zu erfüllen (siehe Tab. 5a). Ki-67 – Index Histologie n= Mittelwert (%) dichtgranuliert (1) 89 0,50 ± 0,78 geringgranuliert (2) 45 0,76 ± 0,93 gemischt PRL/ STH (3) 77 0,94 ± 1,33 Median (%) KruskalWallis-Test U-Test 0,30 (0,00 - 5,80) 0,50 (0,00 - 3,30) p=0,068 p1,2>0,008 p2,3>0,008 0,40 (0,00 - 7,30) Topoisomerase-IIα – Index Histologie n= Mittelwert (%) dichtgranuliert (1) 89 0,23 ± 0,54 geringgranuliert (2) 45 0,44 ± 0,48 gemischt PRL/ STH (3) 77 0,35 ± 0,60 Median (%) KruskalWallis-Test U-Test 0,10 (0,00 - 4,20) 0,20 (0,00 - 1,80) p<0,05 p1,2<0,008 p2,3>0,008 0,10 (0,00 – 3,50) Tabellen 5a und 5b: Übersicht über die Gruppe der nicht medikamentös vorbehandelten Tumoren und den Proliferationsindices (Tab. 5a: Ki-67, Tab. 5b: Topoisomerase-IIα) der verschiedenen histologischen Befunde. Der Vergleich der Parameter zueinander wurde mit dem KruskalWallis-Test sowie einer post-hoc-Analyse durchgeführt (Mann-Whitney U-Test mit Bonferroni-Korrekur; p<0,0083 wurde als signifikant angesehen). Für die Topoisomerase-IIα zeigte sich ein ähnlicher Trend, der für den Unterschied zwischen dicht- und gering-granulierten Adenomen aber statistisch signifikant war (Topoisomerase-IIα: dicht-granuliert 0,23 ± 0,54 %, gering-granuliert 0,44 ± 0,48 %, gemischt-STH/ PRL 0,35 ± 0,60 %, siehe Tab. 5b) 25 In der Korrelationsanalyse nach Spearman fand sich eine negative Korrelation zwischen dem Alter der Patienten und beiden Proliferationsindices Ki-67 (R= - 0,226; p<0.001) bzw. Topoisomerase-IIα (R= - 0,264; p<0,001). Die beiden untersuchten Parameter verhielten sich konkordant zueinander, wobei die absoluten Werte der Topoisomerase-IIα niedriger waren, als die des Ki-67. Die hier erhobenen Parameter der mittleren Proliferationsindices stellten den Ausgangswert für den Vergleich der nachfolgenden, medikamentös vorbehandelten Gruppen dar. 5.2 Wirkung der Vorbehandlung mit Dopamin-Agonisten Wie schon in der Gruppe der nicht vorbehandelten Tumoren fanden sich überwiegend Makroadenome (n= 16; 76,2 %), aber kein Giant- Hypophysenadenom. Fünf Adenome (23,8 %) waren bereits invasiv in die umgebenden Strukturen eingewachsen und in anderen 5 Fällen (23,8 %) wurde schon eine Operation durchgeführt. Das Alter zum Zeitpunkt der Operation lag zwischen 26 und 72 Jahren, das Durchschnittsalter bei 43,29 ± 12,62 Jahren. Histologisch fanden sich in dieser Gruppe je 7 (33,3 %) gemischt STH/ PRLexprimierende, dicht- bzw. gering granulierte Adenome. Insbesondere der Proliferationsindex für Ki-67 lag bei allen verwendeten Dopamin-Agonisten (Cabergolin 0,31 ± 0,55 %; Bromocriptin 0,28 ± 0,47 %; Levopoda 0,23 ± 0,25 %) unterhalb der bei Adenomen ohne Vorbehandlung. Statistisch signifikant war der Unterschied aber nicht (siehe Tab. 6). Der Proliferationsindex der Topoisomerase-IIα verhielt sich ähnlich dem des Ki-67 (Cabergolin 0,25 ± 0,42 %; Bromocriptin 0,28 ± 0,57 %; Levopoda 0,20 ± 0,35 %), aber auch diese waren nicht statistisch signifikant (siehe Tab. 6). 26 Ki-67 – Index n= Mittelwert (%) Median (%) keine Therapie 216 0,70 ± 1,05 0,30 Cabergolin 13 0,31 ± 0,55 Bromocriptin 5 0,28 ± 0,47 Levodopa 3 0,23 ± 0,25 Topoisomerase-IIα – Index U-Test (0,00 - 7,30) 0,10 (0,00 - 1,80) 0,20 (0,00 - 0,50) Median (%) 0,31 ± 0,55 0,10 p=0,07 0,25 ± 0,42 p=0,24 0,28 ± 0,57 p=0,46 0,20 ± 0,35 0,10 (0,00 - 1,10) Mittelwert (%) U-Test (0,00 - 4,20) 0,05 (0,00 - 1,30) p=0,46 0,00 (0,00 - 1,30) 0,00 (0,00 - 0,60) p=0,35 p=0,52 Tabelle 6: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase-IIα in Abhängigkeit der verwendeten dopamin-agonistischen Therapie in Relation zur Gruppe der nicht-vorbehandelten Tumore. Im Vergleich mit der Gruppe der nicht medikamentös vorbehandelten Adenome fand sich keine statistisch signifikante Abhängigkeit von der Gesamtdauer (siehe Tab. 7) bzw. kumulativen Gesamtdosis (siehe Tab. 8) des Cabergolins oder eindeutiger Trend in Hinblick auf die untersuchten Proliferationsindices. Einschränkend ist sicherlich der ohnehin kleine Stichprobenumfang (n=21), so dass die Berechnungen auch nur für die größte Gruppe der dopamin-agonistischen Monotherapie (Cabergolin; n=13) durchgeführt wurden. 27 Cabergolin Therapie (Monate) Ki-67 – Index n= keine Therapie 216 Mittelwert (%) 0,70 ± 1,05 ≤6 7 0,11 ± 0,09 >6 6 0,53 ± 0,78 ≤ 12 10 0,39 ± 0,61 > 12 3 0,03 ± 0,06 Median (%) 0,30 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00 - 7,30) 0,10 (0,00 - 0,20) 0,10 (0,00 - 1,80) 0,15 (0,00 - 1,80) 0,00 (0,00 - 0,10) Mittelwert (%) Median (%) 0,10 U-Test (0,00 - 4,20) 0,11 p=0,06 0,11 ± 0,12 p=0,51 0,44 ± 0,62 p=0,31 0,30 ± 0,44 (0,00 - 1,30) p=0,05 0,00 0,00 (0,00 - 0,30) 0,00 (0,00 - 1,30) 0,15 p=0,41 p=0,84 p=0,93 p=0,10 Tabelle 7: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase-IIα in Abhängigkeit von der kumulativen Dauer der Cabergolin-Gabe in Relation zur Gruppe der nicht-vorbehandelten Tumore. Cabergolin Dosis (mg) Ki-67 – Index n= keine Therapie 216 Mittelwert (%) 0,70 ± 1,05 ≤ 50 7 0,30 ± 0,56 > 50 6 0,33 ± 0,59 Median (%) 0,30 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00 - 7,30) 0,10 (0,00 - 1,80) 0,05 (0,00 - 1,20) Mittelwert (%) p=0,15 0,19 ± 0,29 p=0,26 0,43 ± 0,75 Median (%) 0,10 U-Test (0,00 - 4,20) 0,10 (0,00 - 0,90) 0,00 (0,00 - 1,30) p=0,53 p=0,67 Tabelle 8: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase-IIα in Abhängigkeit von der kumulativen Gesamtdosis der Cabergolin-Gabe in Relation zur Gruppe der nicht-vorbehandelten Tumore. 28 5.3 Wirkung der Vorbehandlung mit Somatostatin-Analoga Die verwendeten Somatostatin-Analoga waren Lanreotid (n=8) und Oktreotid (n=80), wobei beim Letztgenannten sowohl kurzwirksame (tägliche s.c. Injektion), als auch Depotpräparate des Oktreotid (i.m. Injektion alle 4 bzw. 6 Wochen) zum Einsatz kamen. Das Alter zum Zeitpunkt der Operation lag zwischen 19 und 75 Jahren, im Durchschnitt bei 44,88 ± 12,79 Jahre und es fanden sich ähnlich viele weibliche (n=43) wie männliche Patienten (n=45). Der überwiegende Anteil der therapierten Adenome waren Makrodadenome (n=66; 75 %). Mikroadenome fanden sich in 23,9 % der Fälle (n=21) sowie ein GiantHypophysenadenom (1,1 %). Eine Invasion in die umliegenden Strukturen fand sich in 43,2 % (n=38) der Fälle. Histologisch fanden sich 44 dicht- (50 %) bzw. 23 gering-granulierte (26,1 %), 20 gemischte STH/ PRL-exprimierende (22,7 %) Adenome und ein mamma-somatotropes (1,1 %) Adenom. In dieser Gruppe waren bereits 29 Patienten (33,0 %) schon einmal operiert. Im Vergleich der Gruppe der mit Somatostatin-Analoga vorbehandelten Patienten mit der der nicht medikamentös behandelten, waren beide untersuchten Proliferationsindices statistisch signifikant vermindert (Ki-67 0,38 ± 0,97 %; Topoisomerase-IIα 0,18 ± 0,53 %; jeweils p<0,05). In der Subgruppenanalyse der verwendeten Somatostatin-Analoga führen zwar beide im Vergleich mit der Gruppe nicht vorbehandelter Tumoren zu einer Minderung der Proliferation (Oktreotid: Ki-67 0,38 ± 1,01 %, TopoisomeraseIIα 0,18 ± 0,55 %, Lanreotid: Ki-67 0,35 ± 0,42 %, Topoisomerase-IIα 0,18 ± 0,32 %), die aber nur für die Oktreotid-Gruppe statistisch signifikant war (siehe Tab. 9). Betrachtet man die kumulative Gesamtdosis (siehe Tab. 10) bzw. die –dauer (siehe Tab. 11) der Anwendung von Oktreotid, so führt dieses, bereits in einer niedrigen Dosierung (≤ 100 mg) bzw. kurzfristigen Gabe (≤ 6 Monate) zu einer statistisch signifikanten Reduktion beider Proliferationsindices. Eine Steigerung der Gesamtdosis oder Dauer der Anwendung führt dagegen zu keiner weiteren Reduktion der Proliferationsindices. 29 Ki-67 – Index n= keine Therapie 216 alle Mittelwert (%) 0,70 ± 1,05 88 0,38 ± 0,97 Oktreotid 80 0,38 ± 1,01 Lanreotid 8 0,35 ± 0,42 SSA Median (%) 0,30 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00 - 7,30) 0,10 (0,00 - 5,80) 0,00 (0,00 - 5,80) 0,25 (0,00 - 1,00) Mittelwert (%) p<0,05 0,18 ± 0,53 p<0,05 0,18 ± 0,55 p=0,37 0,18 ± 0,32 Median (%) 0,10 U-Test (0,00 - 4,20) 0,11 (0,00 - 4,20) 0,00 (0,00 - 4,20) 0,15 (0,00 - 0,90) p<0,05 p<0,05 p=0,26 Tabelle 9: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase-IIα in Abhängigkeit der verwendeten Somatostatin-Analoga im Vergleich zur Gruppe der nicht-vorbehandelten Tumore. OktreotidTherapie (Monate) Ki-67 – Index n= keine Therapie 216 Mittelwert (%) 0,70 ± 1,05 ≤6 45 0,31 ± 0,95 >6 35 0,46 ± 1,10 ≤ 12 60 0,37 ± 1,01 > 12 20 0,40 ± 1,10 Median (%) 0,30 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00 - 7,30) 0,00 (0,00 - 5,80) 0,00 (0,00 - 4,80) 0,00 (0,00 - 5,80) 0,00 (0,00 - 4,80) Mittelwert (%) p<0,05 0,09 ± 0,18 p<0,05 0,30 ± 0,81 p<0,05 0,18 ± 0,60 p<0,05 0,18 ± 0,37 Median (%) 0,10 U-Test (0,00 - 4,20) 0,00 (0,00 - 0,80) 0,00 (0,00 - 4,20) 0,00 (0,00 - 4,20) 0,00 (0,00 - 1,40) p<0,05 p<0,05 p<0,05 p=0,32 Tabelle 10: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase-IIα in Abhängigkeit von der Dauer der Anwendung von Oktreotid im Vergleich zur Gruppe der nicht-vorbehandelten Tumore. 30 Oktreotid Dosis (mg) Ki-67 – Index Mittelwert (%) n= 0,70 ± 1,05 keine Therapie 216 ≤ 100 38 0,19 ± 0,42 > 100 42 0,53 ± 1,30 Median (%) 0,30 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00 - 7,30) 0,00 (0,00 - 2,00) 0,00 (0,00 - 5,80) Mittelwert (%) p<0,05 0,19 ± 0,29 p<0,05 0,26 ± 0,74 Median (%) 0,10 U-Test (0,00 - 4,20) 0,10 (0,00 - 0,50) 0,00 (0,00 - 4,20) p<0,05 p<0,05 Tabelle 11: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase-IIα in Abhängigkeit von der applizierten Gesamtdosis von Oktreotid im Vergleich zur Gruppe der nicht-vorbehandelten Tumore. 5.4 Wirkung der Kombination aus Dopamin-Agonisten und Somatostatin-Analoga In insgesamt 18 Fällen wurden Somatostatin-Analoga mit einer dopaminagonistisch wirksamen Therapie kombiniert. In 14 Fällen mit Cabergolin, in drei Fällen mit Bromocriptin und in einem Fall mit Quinagolid, welches in der vorliegenden Serie nie als alleiniges Therapeutikum eingesetzt wurde. Das Durchschnittsalter zum Zeitpunkt der Operation lag bei 39,60 ± 14,37 Jahren (14 – 76 Jahre) und es fanden sich neun männliche bzw. weibliche Patienten. In dieser Gruppe überwogen erstmalig die invasiven (n=12; 66,7 %) gegenüber den nicht-invasiven Adenomen (n=8; 33,2 %). Wiederum dominierend waren Makroadenome (n=15; 83,3%), bei zwei Mikro- (11,1 %) und einem Giant-Adenom (5,6 %). Histologisch fanden sich 9 gemischt STH/ PRL-exprimierende (50 %), 6 dicht- (33,3 %) bzw. 3 gering-granulierte Adenome (16,7 %). 31 Ki-67 – Index n= keine Therapie 216 DA + SSA (gesamt) Oktreotid + Cabergolin Oktreotid + Bromocriptin Oktreotid + Quinagolid Mittelwert (%) 0,70 ± 1,05 Median (%) 0,30 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00-7,30) 0,50 18 0,77 ± 0,87 14 0,69 ± 0,94 3 1,30 ± 0,53 (0,90-1,90) 1 0,30 0,30 (0,00-3,40) 0,25 (0,00-3,40) 1,10 Mittelwert (%) Median (%) 0,10 U-Test (0,00-4,20) 0,30 p=0,44 0,36 ± 0,40 p=0,95 0,31 ± 0,44 p=0,06 0,60 ± 0,10 (0,50-0,70) p=0,96 0,30 0,30 (0,00-1,20) 0,10 (0,00-1,20) 0,60 p=0,45 p=0,75 p<0,05 p=0,56 Tabelle 12: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase II-α in Abhängigkeit von der Kombination von Oktreotid und verschiedenen Dopamin-Agonisten in Relation zur Gruppe der nicht-vorbehandelten Tumore Entgegen der vorherigen Ergebnisse fand sich sowohl in der Gesamtheit der Gruppe, als auch in der Subgruppenanalyse der verwendeten Kombinationsbehandlungen von Oktreotid und einem Dopamin-Agonisten keine verminderte Proliferationsaktivität im Vergleich mit der Gruppe der nicht behandelten Tumoren (siehe Tab. 12). 5.5 Wirkung der Vorbehandlung mit Pegvisomant Insgesamt wurden 15 Patientinnen und Patienten vor der Operation mit Pegvisomant behandelt. Neun erhielten eine Monotherapie, drei eine Kombination aus Pegvisomant und Oktreotid, zwei eine Kombination aus Pegvisomant und einem Dopamin-Agonisten (Quinagolid bzw. Cabergolin) und eine Patientin erhielt eine Kombination aus Pegvisomant, Oktreotid und Bromocriptin. Das Alter zum Zeitpunkt der Operation lag zwischen 22 und 87 Jahren, im Durchschnitt bei 46,67 ± 16,67 Jahren und es überwogen Tumore weiblicher Patientinnen (n=12; 80 %). 32 Es fanden sich überwiegend Makroadenome (n=14; 93,3 %), aber kein Giant-Adenom. Ähnlich wie in der Gruppe der kombiniert mit einem Somatostatin-Analogon und einem Dopamin-Agonisten behandelten Patienten überwogen die invasiven Adenome (n=12; 80 %) und die der bereits einmal operierten Patienten (n=9; 60 %). Histologisch fanden sich 7 gering- (46,7 %). 5 dicht-granulierte (33,3 %) und je ein (6,7 %) gemischt STH/ PRL-exprimierendes bzw. mammasomatotropes und plurhormonelles Adenom. Für alle Patienten mit einer Pegvisomant Monotherapie galt, wie oben beschrieben, daß diese zuvor Oktreotid in unterschiedlichen Dosierungen (kumulativ zwischen 20 und 900 mg) bzw. Dauer (kumulativ zwischen 3 und 45 Monaten) erhielten. 5.5.1 Monotherapie mit Pegvisomant In der Gruppe der Patienten, die unmittelbar vor der Operation mit dem Wachstumshormon-Rezeptor-Antagonisten Pegvisomant behandelt wurden, lag die Gesamtdosis zwischen 1,5 und 39,6 g, die Behandlungsdauer zwischen 4 und 68 Monaten. Unter der Therapie mit Pegvisomant zeigte sich eine Reduktion beider Proliferationsindices (Ki-67: 0,27 ± 0,28 %; Topoisomerase-IIα: 0,11 ± 0,16 %) im Vergleich mit der Gruppe der nicht-vorbehandelten Tumoren. Diese war aber nicht statistisch signifikant (siehe Tab. 13). Ki-67 – Index n= Mittelwert (%) keine Therapie 216 Pegvisomant 9 0,70 ± 1,05 0,27 ± 0,28 Median (%) 0,30 (0,00-7,30) 0,30 (0,00-0,80) Topoisomerase-IIα – Index U-Test p=0,26 Mittelwert (%) 0,31 ± 0,55 0,11 ± 0,16 Median (%) 0,10 (0,00-4,20) 0,10 U-Test p=0,25 (0,00-0,50) Tabelle 13: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase II-α in Abhängigkeit von Pegvisomant im Vergleich zur Gruppe der nichtvorbehandelten Tumore. 33 Interessanterweise führte eine längere Therapiedauer zu einer deutlichen Minderung beider Proliferationsindices verglichen mit der Gruppe der nichtvorbehandelten Adenome. Dieser Trend war nicht nur nach 6 Monaten (Ki-67 ≤ 6 Monate: 0,40 ± 0,36 %, > 6 Monate: 0,22 ± 0,24 %; Topoisomerase-IIα ≤ 6 Monate 0,30 ± 0,28 %, > 6 Monate 0,05 ± 0,05%), sondern auch nach 12 Monaten Behandlungsdauer zu beobachten (Ki-67 ≤ 12 Monate: 0,40 ± 0,30 %, > 12 Monate: 0,18 ± 0,25 %; Topoisomerase-IIα ≤ 12 Monate 0,23 ± 0,23 %, > 12 Monate 0,04 ± 0,05 %). Statistisch signifikant waren diese Unterschiede aber nicht (siehe Tab. 14). Eine kumulative Gesamtdosis von über 5 g führte zu einer weiteren, wenn auch minimalen Reduktion beider Proliferationsindices, welche aber wiederum nicht statistisch signifikant waren (Ki-67 ≤ 5 g: 0,30 ± 0,36 %; > 5 g 0, 26 ± 0,24 %; Topoisomerase-IIα ≤ 5 g: 0,23 ±0,23 %; < 5 g 0,04 ± 0,05) (siehe Tab. 15). Pegvisomant Therapie Monate Ki-67 – Index n= keine Therapie 216 Mittelwert (%) 0,70 ± 1,05 ≤6 3 0,40 ± 0,36 >6 6 0,22 ± 0,24 ≤ 12 4 0,40 ± 0,30 > 12 5 0,18 ± 0,25 Median (%) 0,30 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00-7,30) 0,30 (0,10-0,80) 0,20 (0,00-0,50) 0,35 (0,10-0,80) 0,00 (0,00-0,50) Mittelwert (%) p=0,98 0,30 ± 0,28 p=0,17 0,05 ± 0,05 p=0,96 0,23 ± 0,23 p=0,12 0,04 ± 0,05 Median (%) 0,10 U-Test (0,00-4,20) 0,30 (0,10-0,50) 0,05 (0,00-0,10) 0,10 (0,10-0,50) 0,00 (0,00-0,10) p=0,52 p=0,09 p=0,69 p=0,08 Tabelle 14: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase II-α in Abhängigkeit der Dauer der Anwendung von Pegvisomant im Vergleich zur Gruppe der nicht-vorbehandelten Tumore. 34 Pegvisomant Dosis (g) Ki-67 – Index n= keine Therapie 216 Mittelwert (%) 0,70 ± 1,05 ≤5 4 0,30 ± 0,36 >5 5 0,26 ± 0,24 Median (%) 0,30 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00-7,30) 0,20 (0,00-0,80) 0,40 (0,00-0,50) Mittelwert (%) p=0,37 0,23 ± 0,23 p=0,49 0,04 ± 0,05 Median (%) 0,10 U-Test (0,00-4,20) 0,10 (0,10-0,50) 0,00 (0,00-0,10) p=0,69 p=0,08 Tabelle 15: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase II-α in Abhängigkeit von der applizierten Gesamtdosis von Pegvisomant im Vergleich zur Gruppe der nicht-vorbehandelten Tumore. 5.5.2 Kombinationstherapie mit Pegvisomant Insgesamt waren die einzelnen Gruppen für aussagekräftige statistische Betrachtungen zu gering. Zu beobachten aber war eine weitere Abnahme beider Proliferationsindices (siehe Tab. 16) Ki-67 – Index keine Therapie Pegvisomant Monotherapie Pegvisomant + DA Pegvisomant + SSA Pegvisomant + SSA + DA n= Mittelwert (%) Median (%) 0,30 216 0,70 ± 1,05 9 0,27 ± 0,28 (0,00-0,80) 2 0,00 3 1 Topoisomerase-IIα – Index U-Test 0,31 ± 0,55 (0,00-7,30) 0,30 Mittelwert (%) Median (%) 0,10 U-Test (0,00-4,20) 0,10 p=0,26 0,11 ± 0,16 0,00 p=0,06 0,25 ± 0,35 (0,00-0,50) 0,00 0,00 p<0,05 0,00 0,00 p<0,05 0,00 0,00 p=0,23 0,00 0,00 p=0,34 (0,00-0,50) 0,25 p=0,25 p=0,97 Tabelle 16: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase II-α in Abhängigkeit von den verschiedenen Therapien von Pegvisomant im Vergleich zur Gruppe der nicht-vorbehandelten Tumore. 35 5.6 Einfluss der medikamentösen Monotherapie in Abhängigkeit von der Histologie Bei dem Vergleich der Proliferationsindices der drei größten histologischen Gruppen (dicht- bzw. gering-granulierte und gemischt Prolaktin- und Wachstumshormon-sezernierende Hypophysenadenome) gegenüber der medikamentösen Monotherapien (Cabergolin, Pegvisomant und Oktreotid), führte nur die Therapie mit Oktreotid in der Gruppe der dicht-granulierten Hypophysenadenome, die in der Gruppe der nicht medikamentös behandelten Adenome die niedrigste Proliferationsrate aufwiesen, zu einer statistisch signifikanten Reduktion beider untersuchten Proliferationsindices (Ki-67 ohne medikamentöse Therapie: 0,49 ± 0,78 % bzw. Ki-67 unter Oktreotid: 0,14 ± 0,36 %; p=0,000; siehe Tab. 17a; Topoisomerase IIα ohne medikamentöse Therapie: 0,23 ± 0,54 % bzw. Topoisomerase IIα unter Oktreotid: 0,06 ± 0,14 %; p=0,001; siehe Tab. 17b). Auch unter der Therapie mit Cabergolin war eine Reduktion der Proliferationsindices in der Gruppe der dicht-granulierten Hypophysenadenome am stärksten zu verzeichnen. Dieser war aber statistisch nicht signifikant (p>0,083). Unter der Monotherapie mit Pegvisomant war keine statistisch signifikante Änderung, insbesondere kein Anstieg verzeichnen (siehe Tab. 17a und 17b). der Proliferationsindices zu 0,35 ± 0,60 0,44 ± 0,48 0,23 ± 0,54 Ki-67-Index (%) Therapie 5 3 5 n= 5 3 5 n= 0,28 ± 0,43 0,53 ± 0,68 0,06 ± 0,09 IIα-Index (%) Topoisomerase Cabergolin 0,40 ± 0,79 0,53 ± 0,58 0,08 ± 0,05 Ki-67-Index (%) Cabergolin p>0,016 p>0,0083 p>0,0083 U-Test p>0,016 p>0,0083 p>0,0083 U-Test 0 5 2 n= 0 5 2 n= 0,14 ± 0,21 0,10 ± 0,07 IIα-Index (%) Topoisomerase Pegvisomant 0,32 ± 0,34 0,25 ± 0,21 Ki-67-Index (%) Pegvisomant p>0,0083 p>0,0083 U-Test p>0,0083 p>0,0083 U-Test 19 21 39 n= 19 21 39 n= 0,13 ± 0,23 0,46 ± 1,02 0,06 ± 0,14 IIα-Index (%) Topoisomerase Oktreotid 0,61 ± 1,35 0,62 ± 1,40 0,14 ± 0,36 Ki-67-Index (%) Oktreotid p>0,016 p>0,0083 p=0,001 U-Test p>0,016 p>0,0083 p=0,000 U-Test Wallis-Test sowie einer post-hoc-Analyse durchgeführt (Mann-Whitney U-Test mit Bonferroni-Korrekur. p<0,05/(k(k-1)/2) wurde als signifikant angesehen). verschiedenen Gruppen der medikamentösen Monotherapien mit Cabergolin, Pegvisomant und Oktreotid. Der Vergleich der Parameter zueinander wurde mit dem Kruskal- Übersichten über die Verteilung der Proliferationsindices Ki-67 (Tab. 17a) bzw. Topoisomerase IIα (Tab. 17b) gegenüber den histologischen Ergebnissen in den 77 45 p>0,05 p>0,05 89 n= p=0,027 Test Wallis- Kruskal- 0,94 ± 1,33 0,76 ± 0,93 0,49 ± 0,78 Ki-67-Index (%) Therapie keine medikamentöse 77 45 p>0,05 p>0,05 89 n= p=0,002 Test Wallis- Tabellen 17a und 17b : PRL/ STH gemischt granuliert granuliert gering- dicht Histologie PRL/ STH gemischt granuliert granuliert gering- dicht Histologie Kruskal- keine medikamentöse 36 37 5.7 Mehrfach operierte Patienten Im Untersuchungsintervall wurden insgesamt 12 Patienten (8 männliche, 4 weibliche) zweimal, Hypophysenadenom eine bei Patientin sogar persistierender dreimal an einem Akromegalie oder größenprogredientem Adenom operiert und entsprechendes Gewebe zur Analyse gesichert. Bei zwei Patienten (Nr. 1 & 2) wurde keine medikamentöse Therapie, bei drei Patienten (Nr. 3 – 5) keine veränderte medikamentöse Therapie zwischen den Operationen durchgeführt. Bei insgesamt sieben Patienten (Nr. 6 – 12) wurde nach der ersten Operation eine medikamentöse Therapie (1 x Cabergolin, 5 x Oktreotid, 1 x Kombination Cabergolin und Oktreotid) initiiert. Bei einer Patientin (Nr. 13) wurde die Therapie von Oktreotid auf ansteigende Dosierungen von Pegvisomant verändert. Unter der unverändert fortgeführten medikamentösen Therapie (Nr. 3 – 5) findet sich bei allen drei Fällen eine Minderung beider Proliferationsindices im Verlauf. Führte die initiierte medikamentöse Therapie bei (Nr. 6; 9; 11) zu einer Minderung der Proliferationsindices, zeigte sich bei einem Patienten (Nr. 12) keine Änderung und bei drei Patienten sogar ein Anstieg der Proliferationsindices im Verlauf (Nr. 7; 8; 10). Im letzten Fall (Nr. 13, siehe Abbildung 2) bot sich die Gelegenheit, das Verhalten der Proliferationsindices bei einer Patientin sehr differenziert zu beurteilen, die insgesamt viermal an ihrem Hypophysenadenom bei Akromegalie operiert wurde. Es standen insgesamt drei Gewebeproben zur Verfügung. Die erste Operation erfolgte nach mehrmonatiger Exposition mit Oktreotid. Vor einem weiteren Eingriff wurde (in dieser Sequenz) Oktreotid LAR, Cabergolin und Pegvisomant verabreicht. Die Monotherapie mit Pegvisomant wurde dann fortgeführt und es bot sich wegen der Tumorprogredienz eine weitere Möglichkeit Tumorproben zu gewinnen. Die Operationen erfolgten in den Jahren 2003, 2006 und 2010. Zu allen drei Untersuchungszeitpunkten findet sich eine homogene, fast identische Expression der Proliferationsparameter Ki-67 und Topoisomerase-IIα (siehe Tab. 18). 38 PatientGeschlecht Nr. #1 weiblich #2 männlich #3 männlich #4 männlich OPDatum Alter Mai 10 15 Topoisomerase-IIα – Index (%) Therapien vor Operation 0,00 - 0,40 0,10 - 0,10 0,10 - 0,10 0,10 - 1,40 0,30 Oktreotid 0,80 0,10 Oktreotid 2,00 0,10 Oktreotid 0,30 0,30 Oktreotid 0,40 0,10 Oktreotid + Pegvisomant 0,00 0,00 Oktreotid + Pegvisomant 0,70 0,30 - 0,10 0,10 Cabergolin 0,90 0,50 - 1,90 0,60 Oktreotid 0,00 0,00 - 0,10 0,00 Oktreotid 0,30 0,10 - 0,10 0,00 Oktreotid 1,80 1,40 - 2,50 1,60 Oktreotid 3,50 2,20 RTX 2,30 1,50 RTX + Oktreotid 0,10 0,05 - 0,10 0,05 Cabergolin + Oktreotid 0,50 0,05 Oktreotid Nov. 06 0,50 0,05 Pegvisomant Jan. 10 0,50 0,00 Pegvisomant Aug. 10 Okt. 07 53 Okt. 07 Sep. 04 34 Jul. 06 Sep. 07 29 Apr. 08 Mai 04 #5 30 männlich Sep. 04 #6 männlich #7 weiblich #8 männlich #9 weiblich # 10 männlich # 11 weiblich Feb. 05 männlich Okt. 04 weiblich 37 Apr. 05 Jul. 09 33 Apr. 10 Apr. 10 48 Aug. 10 Jul. 03 57 Mai 05 Sep. 07 14 Jan. 08 23 Mai 04 Mai 03 # 13 38 Okt. 05 Mai 03 # 12 Ki-67 – Index (%) 0,00 40 Tabelle 18: Übersicht über die mindestens zweimal operierten Fälle im Untersuchungszeitraum inklusive der Proliferationsindices und der initiierten Therapien vor den jeweiligen Operationen. 39 A B C D E F Abbildung 2: Mikroskopische Abbildungen der immunhistochemischen Untersuchungen für die Proliferationsmarker Ki-67 (A, C, E) und Topoisomerase-IIα (B, D, F) der seriell aus den Jahren 2003 (A und B), 2006 (C und D) und 2010 (D und E) gewonnen Tumorproben der Patientin #13; 400-fache Vergrößerung. Bis zur Operation 2003 erhielt die Patientin Oktreotid über 15 Monate mit einer Gesamtdosis von 135 mg. Im Jahre 2006 und 2010 hatte die Patientin Pegvisomant über 33 bzw. 71 Monate mit einer Gesamtdosis von 13,3 bzw. 39,5 appliziert bekommen. Änderungen der Proliferationsindices fanden sich nicht, welche im Mittel bei jeweils 0,5 % lagen. 40 5.8 Einfluss einer Radiotherapie Insgesamt wurden acht Hypophysenadenome vor der Operation bestrahlt (3 x stereotaktische Einzeldosiskonvergenzbestrahlung, 5 x konventionelle Strahlentherapie). In einem Fall wurde keine medikamentöse Therapie durchgeführt, bei zwei Patienten eine dopamin-agonistische Monotherapie (1 x Cabergolin, 1 x Bromocriptin), in drei Fällen eine Monotherapie mit Oktreotid, in einem Fall eine Kombinationstherapie aus Oktreotid und Cabergolin und in einem Fall eine Kombinationstherapie aus Pegvisomant und Oktreotid bis zur Operation durchgeführt. Das Durchschnittsalter zum Zeitpunkt der Operation betrug 36,0 ± 16,17 Jahre (14 – 63 Jahre) und es fanden sich fast ausschließlich weibliche Patientinnen (n=7: 87,5 %). Ein Mikroadenom fand sich nicht, dafür zwei Giant-Hypophysenadenome (25 %) und sechs Makroadenome (75 %). Die überwiegende Anzahl der Tumore war invasiv entwickelt (n=6; 75 %) und schon einmal operiert (n=7; 87,5 %). Histologisch fanden sich fünf dicht- (62,5 %), zwei gering-granulierte (25 %) sowie ein gemischt STH/ PRL-exprimierendes Adenom. In der Gruppe der bestrahlten Adenome lagen beide Proliferationsindices Ki67 und Topoisomerase-IIα trotz der unterschiedlichen medikamentösen Therapien bis zur Operation höher als in der Gruppe der nicht vorbehandelten Adenome (Ki-67: 1,33 ± 1,39 %, Topoisomerase-IIα 0,59 ± 0,89 %). Eine statistische Signifikanz fand sich aber nicht (siehe Tab. 19). Ki-67 – Index n= Mittel (%) keine Therapie 216 0,70 ± 1,05 Radiotherapie 1,33 ± 1,39 8 Median (%) 0,30 Topoisomerase-IIα – Index U-Test Mittel (%) 0,31 ± 0,55 (0,00 - 7,30) 0,95 (0,10 - 3,50) p=0,26 0,59 ± 0,89 Median (%) 0,10 U-Test (0,00 - 4,20) 0,20 (0,00 - 2,20) p=0,82 Tabelle 19: Übersicht über die Ergebnisse des Mann-Whitney U-Test der Proliferationsindices Ki-67 und Topoisomerase-IIα der radiotherapeutisch behandelten Fälle in Relation zur Gruppe der nicht-vorbehandelten Tumore. 41 6. Diskussion 6.1 Proliferationaktivität von Hypophysenadenomen Hypophysenadenome sind üblicherweise langsam wachsende Tumore, die als gutartig klassifiziert werden [72, 106]. Allerdings unterliegt ihr Wachstumsverhalten ganz erheblichen interindividuellen Schwankungen [57, 98]. Während viele Mikroadenome der Hypophyse über Jahre hinweg auch ohne Therapie in ihrer Größe konstant bleiben können, gibt es auch rasch proliferierende Tumoren, die innerhalb von Monaten an Größe zunehmen und auch nach radiologisch dokumentierter, (anscheinend) vollständiger Resektion immer wieder rasch Rezidive entwickeln [56, 57, 68, 132]. Eine Möglichkeit, die Proliferationsaktivität von Hypophysentumoren radiologisch zu dokumentieren, sind kernspintomographische Verlaufsuntersuchungen mit volumetrischer Auswertung. Diese sind aber für die Akromegalie nicht sinnvoll, da die mit der Erkrankung verbundene Mortalität eine abwartende Haltung verbietet [82, 85]. Überall auf der Welt, wo die Möglichkeit von kernspintomographischen seriellen Untersuchungen besteht, werden die Vorschläge der Fachgesellschaften zur Therapie auch in der klinischen Praxis umgesetzt. So muss, um überhaupt einen Eindruck über das Wachstumsverhalten von Hypophysenadenomen zu bekommen, auf die Gruppe der hormonell inaktiven Hypophysenadenome zurückgriffen werden. Bei einer retrospektiven, volumetrischen Analyse von 15 hormoninaktiven Hypophysenadenomen, haben Honegger et al. [57] mathematische Modelle über das Wachstum erstellt. Sie fanden eine logistische Wachstumskurve bei 5 Adenomen und ein konstantes Tumorvolumen bei einem Patienten. In der Mehrzahl der Fälle (n=9) fand sich ein exponentielles Wachstumsverhalten der Adenome. Daraus lässt sich ableiten, dass bei kurzen Beobachtungszeiträumen nur sehr geringe Volumenveränderungen auftreten können und man bei der Mehrzahl der Patienten nach Zeiträumen von wenigen Monaten bis wenigen Jahren noch keine definitive Aussage über eine Größenzunahme machen kann, die sich bei einer exponentiellen Charakteristik erst nach längeren Beobachtungszeiträumen erkennen lässt [57, 68]. Die operative Therapie ist nach den gängigen Algorhythmen oft die Erstbehandlung der Wahl bei Hypophysenadenomen [13]. Dies gilt insbesondere für die Akromegalie [85, 91]. Durch die Operationen wird 42 Gewebe gewonnen Untersuchungen in und seit langer Zeit den Tumorbiopsien versuchte Informationen man über durch das Wachstumsverhalten des individuellen Tumors zu finden. Mitosen lassen sich bei Hypophysenadenomen kaum nachweisen, so dass man auf Zellzyklusanalysen zurückgreifen musste. Die Zellzyklusanalyse durch FlowZytometrie wird heute nicht verwendet. Hoffnungen hat man in den von Gerdes und Mitarbeitern [43, 44] entwickelten monoklonalen Antikörper Ki-67 gesetzt, der die nukleären Proteine von 345 und 395 kDa in proliferierenden Zellen markiert, wenn Sie nicht in der G0-Phase des Zellzyklus sind. Die Anwendung war zunächst auf Gefrierschnitte beschränkt. Die ersten Anwendungen der Methode in der Neuropathologie sind von Burger et al. [20] beschrieben. Ein Klon des ersten Antikörpers Ki-67, namens MiB-1 erlaubte auch die retrospektive Untersuchung von in Paraffin eingebettetem Gewebe. Bereits frühe Untersuchungen [14, 127] ließen aber Hoffnungen enttäuschen, dass man prognostische Aussagen aus dem Operationsmaterial für das weitere Schicksal des individuellen Patienten ziehen könnte. Innerhalb eines einzigen Tumors kann der Anteil Ki-67positiver Zellen erheblich variieren [14, 127]. Allerdings hatte man dann den Eindruck, dass die Expression des nukleären Antigens Ki-67 in Kohorten von Patienten mit invasiven Hypophysenadenomen, gemessen am Anteil der Zellen, die sich mit dem Antikörper anfärben lassen, bezogen auf alle gezählten Zellen (also dem sogenannten Labeling-Index LI), mit einer Invasion des Tumors korreliert [58, 68, 93, 121, 130, 131]. Dies würde bedeuten, dass aggressiver wachsende Tumoren auch rascher proliferieren. Auch meinte man, die Größe des Tumors, zumindest wenn sie in Mikro-, Makro- und Giant-Adenome kategorisiert wurde, mit der Ki-67-Expression korrelieren zu können. Größere Tumoren hatten in der Regel höhere Proliferationsaktivitäten [58, 74, 125]. In den meisten Untersuchungen aber wurden alle Hypophysenadenome zusammen betrachtet und nicht nach der hormonellen Sekretionsaktivität bzw. den endokrinologischen Krankheitsbildern differenziert. Ähnlich verhält es sich mit der Topoisomerase-IIα, einem Enzym, das in der Kontrolle der DNA-Topologie, beim Aufspleissen und Replikation der DNA beteiligt ist. Auch dieses ist ausschließlich in proliferierenden Zellen 43 außerhalb der G0-Phase exprimiert. In einer Korrelationsanalyse bestätigte sich, dass die beiden Parameter bei Hypophysenadenomen sehr gut miteinander korrelieren [131]. Auch nach einer Ausweitung der Untersuchungen mit den Proliferationsparameter Ki-67 auf Paraffin-fixierte Gewebeproben findet sich kein einheitliches Bild [109]. Während die Mehrzahl der Untersucher eine Korrelation zwischen der invasiven Wachstumstendenz des Hypophysentumors und der Expression des Proliferationsparameters Ki-67 fand [58, 68, 93, 121, 130, 131] konnten andere keine entsprechende Korrelation herstellten [36, 56, 126, 132]. Ähnlich verhält es sich mit der Korrelation zwischen rezidivierenden und nicht-rezidivierenden Tumoren, wobei aber überwiegend keine Korrelation zwischen der Expression von Ki-67 und dem Auftreten eines Rezidivs zu beobachten war [58, 93, 111]. Die Mehrzahl der Untersucher fand zumindest eine positive Korrelation mit der Größe eines Hypophysentumors [14, 58, 76, 126], aber auch hier gibt es gegensätzliche Angaben [36]. Wiederum ähnlich verhält es sich mit dem Geschlecht des Patienten, dem Alter und der sekretorischen Aktivität [109]. Einschränkend muss festgestellt werden, dass sich die genannten Untersuchungen auf die gepoolten Ergebnisse sämtlicher Arten von Hypophysenadenomen beziehen. In allen angefertigten Arbeiten liegt die Zahl der Wachstumshormon-sezernierenden Hypophysenadenome unter 50, mit der Ausnahme von Delgrange et al. [36], die 96 Präparate untersucht haben. Demgegenüber sind die hier vorliegenden statistischen Analysen von insgesamt 366 Tumorproben viel aussagekräftiger. Während in der vorliegenden Arbeit keine Korrelation zwischen dem Geschlecht und der Expression von Ki-67 bei den hier untersuchten Wachstumshormon-sezernierenden Hypophysenadenomen nachzuweisen war, fand sich eine Korrelation zwischen dem Invasionsverhalten und der Größe des Adenoms. Bei invasiv wachsenden Adenomen sowie Makroadenomen waren beide untersuchten Proliferationsparameter Ki-67 und Topoisomerase-IIα statistisch signifikant erhöht. Ferner bestätigte sich, 44 dass eine negative Korrelation von Alter sowohl mit der Ki-67 und Topoisomerase-IIα–Expression (R = -0,226, p<0,001; R = -0,264, p<0,001) besteht. Dies bedeutet, dass das Lebensalter der Patienten mit den untersuchten Proliferationsmarkern invers korreliert. 6.2 Einfluss einer Vorbehandlung mit Dopamin-Agonisten Die Therapie mit Dopamin-Agonisten ist heute bei Prolaktinomen allgemein akzeptiert [62, 84]. Bei der Akromegalie sind Dopamin-Agonisten aber nicht so effektiv wie Somatostatin-Analoga, was die Normalisierung der erhöhten WachsumshormonNormalisierung und dieser IGF-1-Spiegel Parameter betrifft [12, entsprechend 83, der in 114]. Eine Konsensus- Konferenzen geforderten strikten Normalisierungskriterien tritt eher selten auf [83]. Allerdings gibt es auch kasuistische Berichte von eindrucksvoll radiologisch dokumentierten Tumorschrumpfungen bei Patienten mit Akromegalie, allein nach dopamin-agonistischer Therapie mit Cabergolin [100]. Wenn eine Volumenverminderung eines Tumors auftritt, ist eine Hemmung der Proliferationsaktivität zu vermuten. So ist bei Prolaktinomen auch eine antiproliferative Wirkung dokumentiert [45]. In Gewebeproben von Patienten, die zuvor mit Dopamin-Agonisten behandelt wurden, fand sich eine verminderte Expression des Proliferationsparameters DNA Polymerase und ein signifikant niedriger Ki-67 Index [14]. Allerdings sind diese bisher in der Literatur niedergelegten Einflüsse auf die Proliferationsmarker auf Prolaktinome beschränkt. Angaben über den Einfluss einer Vorbehandlung mit Dopamin-Agonisten auf Proliferationsparameter bei akromegalen Patienten finden sich bisher in der medizinischen Literatur nicht. Die dopamin-agonistische Therapie führte in dieser Untersuchung zwar zu einer nachweislichen Minderung der beiden untersuchten Proliferationsparameter, signifikant war diese Reduzierung jedoch nicht und spiegelt auch den klinischen Eindruck des Effekts einer dopaminagonistischen Monotherapie bei Akromegalie wider. Ähnlich verhält es sich bei der Kombinationstherapie aus Somatostatin-Analoga und DopaminAgonisten. Auch hier war in dem untersuchten Material eine Minderung, aber keine signifikante Absenkung der durchschnittlichen Proliferationsindices nachzuweisen. Ursächlich hierfür auch die Therapieresistenz dieser 45 vorwiegend invasiven Adenome sein, da die Monotherapie mit SomatostatinAnaloga nicht zu einer Normalisierung des Hormonexzesses führte und somit der Therapieversuch aus der Kombination initiiert wurde. 6.3 Einfluss einer Vorbehandlung mit Somatostatin-Analoga Somatostatin-Analoga sind heute die, allgemein akzeptiertem primäre medikamentöse Therapie bei der Akromegalie [82, 83, 85, 114]. Bei der Mehrzahl der Patienten kommt es schon nach wenigen Monaten zu einer deutlichen Verminderung der IGF-1-Spiegel. Häufig ist eine Volumenreduktion des Tumors häufig zu beobachten [12]. Sowohl für Oktreotid und Oktreotid LAR ist dieser positive Effekt dokumentiert [12, 29, 31, 32, 49, 67]. Eine signifikante Tumorschrumpfung ist nach 12 Monaten einer Therapie mit Lanreotid als Primärtherapie der Akromegalie auch für Lanreotid beschrieben [27]. Auch Parsireotide, ein neues SomatostatinAnalogon, welches gerade die klinische Prüfung bestanden hat, schrumpft etwa 1/3 der behandelten Tumoren bei akromegalen Patienten nach drei Monaten Behandlungsdauer in einem signifikanten Ausmaß [92]. Allerdings können die antiproliferativen und andere sekretorische Effekte auch divergieren und müssen daher nicht immer konkordant auftreten [99]. Dass Somatostatin-Analoga, parallel zu ihren anderen sekretorischen und Tumorvolumen-reduzierenden Effekten, auch die Proliferationsaktivität der Tumoren (gemessen an im Gewebe bestimmten Proliferationsparametern) vermindern können, ist seit langem bekannt. So haben Buchfelder et al. [14] eine reduzierte Aktivität der DNA-Polymerase gefunden, wenn die Patienten mit Somatostatin-Analoga vorbehandelt wurden. Thapar et al. [121] beschreiben, dass die Wachstumsfraktionen im Adenom bei 16 akromegalen Patienten, die präoperativ mit Oktreotid behandelt worden waren, im Vergleich zu 16 akromegalen, präoperativ nicht medikamentös behandelten Patienten um 83 % vermindert waren. Losa et al. [74] zeigen, dass die Vorbehandlung mit Oktreotid zwar den Ki-67-LI signifikant reduziert, den apoptotischen Index aber nicht verändert. Während die Lebensdauer der Tumorzellen offensichtlich nicht verkürzt war, fand sich eine eindeutige Wirkung auf die Proliferationsaktivität. In ihrer Untersuchung gingen insgesamt 78 Patienten ein, von denen 39 Patienten präoperativ Oktreotid 46 erhielten. Die Kontrollgruppe bestand aus ebenfalls 39 Patienten ohne Medikamentenexposition. Konkordant zu der bisherigen Literatur fand sich anhand der hier verwendeten Tumorproben eine verminderte Proliferationsrate unter der Therapie mit Somatostatin-Analoga. Hierbei zeigte sich, dass der in der Literatur vorbeschriebe Effekt des Oktreotid [40] auf die Proliferationsindices bei dicht-granulierten Hypophysenadenomen statistisch signifikant über denen der anderen histologischen Gruppen lag. Ursächlich hierfür könnte die vermehrte Expression des Somatostatin-Rezeptor-Subtyp-2 in dicht- granulierten, im Vergleich zu gering-granulierten Hypophysenadenomen sein [61], insbesondere da der Verlust dieses Subtyps auch mit einer OktreotidResistenz in Hinblick auf die Normalisierung der Wachstumshormon-Spiegel beobachtet wird [94]. Dieser Effekt könnte insbesondere als prognostischer Faktor in Hinblick auf das Ansprechen einer postoperativen Therapie mit Oktreotid angesehen werden. Interessanterweise war es jedoch unerheblich, wie hoch die kumulative Gesamtdosis oder die Gesamtanwendungsdauer war. Eine Erklärung, warum keine weitere Reduktion der Proliferationsparameter mehr stattfindet, wäre die ohnehin nur sehr niedrige Proliferationsrate der Hypophysenadenome. Da schon eine kumulative Gesamtdosis von bis zu 100 mg (was etwa einer dreimonatigen hochdosierten Therapie mit Oktreotid entspricht) zu einer Minderung der durchschnittlichen Proliferationsaktivität (Ki-67 und Topoisomerase-IIα) auf 0,0 % führt, kann nur spekuliert werden, inwieweit diese bei langfristiger Anwendung noch weiter gesenkt werden kann. Die in anderen Studien beobachtete Schrumpfung der Adenomgröße, auch nach längerer Anwendung als drei Monate, könnte ein entsprechendes Indiz für diese Annahme sein, wenngleich hierfür eine histologische Verifizierung noch fehlt. 6.4 Einfluss einer Vorbehandlung mit Pegvisomant Die periphere Rezeptorblockade des Wachstumshormon-Rezeptors durch Pegvisomant stellt eine der wirksamsten therapeutischen Optionen dar, um eine biochemische Normalisierung der IGF-1-Sekretion und somit den klinischen Manifestationen zu ermöglichen. In den Zulassungsstudien wurde 47 eine Normalisierung von IGF-1 bei fast allen Patienten erreicht [123, 128]. Zwar haben sich die Ergebnisse in Bezug auf die Kontrolle der Akromegalie in Anwendungsbeobachtungen, die die klinische Praxis besser repräsentieren, als nicht ganz so gut erwiesen [112, 119], doch handelt es sich bei den Patienten, die mit Pegvisomant behandelt werden auch um die negative Selektion von Patienten mit einer Akromegalie. Die Zulassung des Medikamentes setzt voraus, dass eine vorhergehende Therapie mit Somatostatin-Analoga nicht zu einer Normalisierung der Akromegalie geführt hat [112]. Schon aufgrund des Wirkungsmechanismus ist keine Minderung der Proliferationsaktivität erwarten. Bereits in des der ursächlichen Hypophysenadenoms Zulassungsstudie fanden sich zu einzelne größenprogrediente Adenome während der Behandlung mit Pegvisomant [128]. Weitere Berichte von Tumorvolumenexpansionen folgten und alarmierten die Therapeuten von Hypophysenadenomen weltweit [3, 30, 42, 60, 112]. Demgegenüber war in volumetrischen, nach strengen Protokollen ausgerichteten, prospektiven kernspintomographischen Untersuchungen des Tumorvolumens über Behandlungsdauern von 12 [30] und 24 Monaten [19] keine Veränderung des mittleren Tumorvolumens nachweisbar. Drake et al. [38] konnten in einem Fall eines invasiven Makroadenoms beeindruckend eine erhöhte Proliferationsaktivität nach der Behandlung mit Pegvisomant nachweisen. In der hier vorliegenden Studie waren die von Drake et al. untersuchten Proliferationsparameter nicht nur nicht-erhöht, sondern auch signifikant im Vergleich zu nicht behandelten Adenomen vermindert. Ursächlich hierfür wird die schon oben beschriebene klinische Anwendung von Pegvisomant diskutiert. In der hier vorgelegten Untersuchung wird davon ausgegangen, dass das letzte Pharmakon, dem der Patient ausgesetzt war, bevor die Tumorprobe dann bei der Operation gewonnen wird, den stärksten Effekt hat. In der medizinischen Literatur ist bis auf die von Drake et al [38] publizierten Fall, bisher nicht über den Einfluss von Pegvisomant auf die Proliferationsaktivität in einem Wachstumshormon-sezernierenden Hypophysenadenom berichtet. Da die Verschreibung von Pegvisomant ein Versagen der Therapie mit Somatostatin-Analoga oder deren Unverträglichkeit voraussetzt, sind die 48 untersuchten Adenome zuvor mit eben auch diesen Präparaten behandelt worden. Die hier nachweisbaren Effekte der Minderung der Proliferationsindices sind somit möglicherweise gar nicht dem Pegvisomant, sondern dem vorausgehenden Einfluss der Somatostatin-Analoga zuzuordnen. Dies würde bedeuten, dass die Effekte der SomatostatinAnaloga-Medikation auf die Proliferationsaktivität im Gewebe auch nach deren Absetzen bestehen bleiben. Demgegenüber steht aber die klinische Beobachtung, dass es nach dem Absetzen von Somatostatin-Analoga klinisch rasch zu einem „rebound“ der pathologischen Sekretion von Wachstumshormon und IGF-1 und zu einer Expansion des Tumors kommt [19, 25, 73, 97, 116]. Interessanterweise aber fand sich eine nahezu signifikant verminderte Expression von Topoisomerase-IIα, wenn die Patienten länger mit Pegvisomant behandelt wurden bzw. wenn die Gesamtdosis 5 g überschritt. Da dieser Effekt nicht in der Gruppe derer auftrat, die kürzer behandelt wurden bzw. eine geringere Gesamtdosis erhielten, könnte dieses ein bisher nicht beobachteter Effekt der Therapie mit Pegvisomant sein. Die Minderung der Expression von Ki-67 folgt dem Verlauf der Topoisomerase-IIα, wenn aber auch nicht in so umfangreichem Ausmaß. Ob dieser Effekt direkt oder indirekt zu Stande kommt, müssen weitere Studien untersuchen. In der hier vorliegenden Untersuchung während der Therapie mit Pegvisomant lässt sich die Beobachtung von Drake et al. jedenfalls nicht nachvollziehen [38]. Eine Exposition von Pegvisomant führte in diesem Material nicht zu einer erhöhten Expression der in den Gewebeproben untersuchten Proliferationsparameter. 6.5 Bedeutung mehrfach operierter Patienten Eine besondere Erkenntnis-theoretische Bedeutung kommt Einzelfällen zu, bei denen man die Gelegenheit hatte, zu mehreren Zeitpunkten die zellulären Mechanismen zu analysieren. Der Anlass zur vorliegenden Untersuchung ist der Fallbericht von Drake et al. [38], die über einen aggressiven und zu keinem Zeitpunkt zufrieden stellend behandelten Patienten berichten. Dieser wurde in einem Intervall von 17 Jahren zweimal operiert, so dass zwei Gewebeproben zur Untersuchung zur Verfügung standen. Der Patient wurde 49 neben den Operationen auch bestrahlt und erhielt eine ganze Reihe medikamentöser Therapien, unter anderem auch über einen Gesamtzeitraum von vier Jahren zur Kontrolle der immer erhöhten IGF-1Spiegel Pegvisomant. In den gewonnenen Tumorproben 1986 bzw. 2003 wurden die Proliferationsindices von Ki-67 und Topoisomerase-IIα bestimmt. Bei der ersten Operation lagen diese für Ki-67 zwischen 0 – 0,5 % und für Topoisomerase-IIα zwischen 2 – 10 %. Bei der Re-Operation im Jahr 2003 ist es dann zu einem erheblichen Anstieg der Proliferationsindices für Ki-67 auf 1 – 3 % gekommen. Parallel dazu stieg die Expression von Topoisomerase-IIα im Jahr 2003 auf 15 – 80 % der Zellen an (siehe Abbildung 1). Keine der Therapien konnte diesen Tumor kontrollieren, an dem der Patient dann letztlich auch verstorben ist, so dass ein ungünstiger Einfluss von Pegvisomant zwar diskutiert werden könnte, aber nicht ursächlich wahrscheinlich ist. Die Besonderheit liegt darin, dass die Gewebeuntersuchung nach Pegvisomant bisher nur in diesem einen kasuistischen Bericht in der medizinischen Literatur dokumentiert ist. Bei ihr bot sich die Gelegenheit eine weitere Patientin, die insgesamt viermal an ihrem Hypophysenadenom bei Akromegalie operiert wurde, zu betrachten. Es standen insgesamt drei Gewebeproben von unterschiedlichen Operationen zur Verfügung. Eine Zunahme der Proliferationsaktivität wurde nicht beobachtet.. Eine Exposition von Pegvisomant ist also nicht notwendigerweise mit einer Erhöhung der Expression dieser Proliferationsparameter vereinbar. Die Patientin weist auch eine bemerkenswerte, im MR überzeugend dokumentierte Tumorprogression auf, zu einem Zeitpunkt, als die Akromegalie noch nicht erkannt und sie mit keiner Therapie behandelt wurde. Wenngleich diese Einzelfallbeobachtung natürlich keine Beweiskraft hat, so dokumentiert sie immerhin, dass eine Exposition mit Pegvisomant nicht notwendigerweise zu einer nachweisbaren Steigerung der im Gewebe nachweisbaren Proliferationsaktivität führen muss und ist in diesem Zusammenhang sicherlich interessant. 50 Insgesamt bot sich die Möglichkeit, 13 Patienten zu verschiedenen Zeitpunkten zu untersuchen. Die Veränderung der Proliferationsaktivität war dabei kaum jemals dramatisch. 6.6 Wirkung der Radiotherapie Die Radiotherapie ist ein sehr probates und zuverlässiges Mittel zur Verminderung der Rezidivquote von Hypophysenadenomen [6, 77]. Wenn es sich technisch realisieren lässt, versucht man heute den Tumor soweit mit chirurgischen Methoden zu verkleinern, dass eine stereotaktische Einzeldosiskonvergenzbestrahlung möglich ist [17]. Bei dieser wird die zur Inaktivierung des Tumors notwendige Dosis in einer einzigen Sitzung mit stereotaktischer Therapieplanung auf den Tumor appliziert [81, 89]. Gerade die Wirkung der stereotaktischen Einzeldosiskonvergenzbestrahlung auf das Tumorvolumen von hormoninaktiven bzw. Hypophysenadenomen bei einer Akromegalie sowie der damit verbundenen antiproliferative Effekt ist in kernspintomographischen Verlaufsuntersuchungen gut dokumentiert [23, 50, 65, 71, 88, 102]. Zum Effekt auf die Proliferationsaktivität im Gewebe finden sich nur wenige Angaben. In der Mehrzahl der Situationen bedingt die Radiotherapie eine Tumorkontrolle und die entsprechenden Patienten werden dann nicht noch einmal operiert, so dass keine weiteren Tumorproben zur Analyse von Proliferationsparametern gewonnen werden können. Zwar fand sich in der hier dokumentierten Gruppe der radiotherapierten Patienten eine höhere Proliferationsrate als in der Gruppe ohne Medikamente. Abzuleiten, dass eine Strahlentherapie zu einer erhöhten Proliferationsaktivität in Hypophysenadenomen führt, wäre aber nicht zulässig. Zum einen ist der Stichprobenumfang von acht Fällen des untersuchten Kollektivs von insgesamt 366 Tumorproben zu klein, zum anderen handelt es sich bei den bestrahlten Patienten um diejenigen, bei denen vorangegangene Operationen und eine medikamentöse Therapie keine Normalisierung der Sekretionsdynamik erreichen konnte oder bei dem der Tumor weiter progredient war. Diese wenigen Fälle entsprechen also der „negativ“-Selektion der nur schwer zu therapierenden Adenome, die mit einer 51 ausgesprochenen Aggressivität trotz erfolgter Strahlentherapie weiter wachsen und somit in keinster Weise mit einem „normalen“ Hypophysenadenom verglichen werden dürfen. Auffällig ist zudem das niedrigere Durchschnittsalter der bestrahlten Patienten. 52 7. Literaturverzeichnis 1. Abe T, Ludecke DK: Effects of preoperative octreotide treatment on different subtypes of 90 GH-secreting pituitary adenomas and outcome in one surgical centre. Eur J Endocrinol. 2001 (145): 137-45. 2. Barber TM, Adams E, Ansorge O, Byrne JV, Karavitaki N, Wass JA: Nelson's syndrome. Eur J Endocrinol. 2010 (163): 495-507. 3. Barkan AL, Burman P, Clemmons DR, Drake WM, Gagel RF, Harris PE, Trainer PJ, van der Lely AJ, Vance ML: Glucose homeostasis and safety in patients with acromegaly converted from long-acting octreotide to pegvisomant. J Clin Endocrinol Metab. 2005 (90): 568491. 4. Barkan AL, Lloyd RV, Chandler WF, Hatfield MK, Gebarski SS, Kelch RP, Beitins IZ: Preoperative treatment of acromegaly with long-acting somatostatin analog SMS 201-995: shrinkage of invasive pituitary macroadenomas and improved surgical remission rate. J Clin Endocrinol Metab. 1988 (67): 1040-8. 5. Beauregard C, Truong U, Hardy J, Serri O: Long-term outcome and mortality after transsphenoidal adenomectomy for acromegaly. Clin Endocrinol (Oxf). 2003 (58): 86-91. 6. Becker G, Kocher M, Kortmann RD, Paulsen F, Jeremic B, Muller RP, Bamberg M: Radiation therapy in the multimodal treatment approach of pituitary adenoma. Strahlenther Onkol. 2002 (178): 173-86. 7. Beckers A: Does preoperative somatostatin analog treatment improve surgical cure rates in acromegaly? A new look at an old question. J Clin Endocrinol Metab. 2008 (93): 2975-7. 8. Beckers A, Kovacs K, Horvath E, Abs R, Reznik M, Stevenaert A: Effect of treatment with octreotide on the morphology of growth hormone—secreting pituitary adenomas: Study of 24 cases. Endocrine Pathology. 1992 (2): 123-131. 9. Bevan JS: Clinical review: The antitumoral effects of somatostatin analog therapy in acromegaly. J Clin Endocrinol Metab. 2005 (90): 1856-63. 10. Biermasz NR: Pituitary gland: New consensus in acromegaly: criteria for cure and control. Nat Rev Endocrinol. 2010 (6): 480-1. 53 11. Biermasz NR, van Dulken H, Roelfsema F: Direct postoperative and follow-up results of transsphenoidal surgery in 19 acromegalic patients pretreated with octreotide compared to those in untreated matched controls. J Clin Endocrinol Metab. 1999 (84): 3551-5. 12. Biller BM, Colao A, Petersenn S, Bonert VS, Boscaro M: Prolactinomas, Cushing's disease and acromegaly: debating the role of medical therapy for secretory pituitary adenomas. BMC Endocr Disord. 2010 (10): 10. 13. Buchfelder M: Treatment of pituitary tumors: surgery. Endocrine. 2005 (28): 67-75. 14. Buchfelder M, Fahlbusch R, Adams EF, Kiesewetter F, Thierauf P: Proliferation parameters for pituitary adenomas. Acta Neurochir Suppl. 1996 (65): 18-21. 15. Buchfelder M, Fahlbusch R, Thierauf P, Muller OA: Observations on the pathophysiology of Nelson's syndrome: a report of three cases. Neurosurgery. 1990 (27): 961-8. 16. Buchfelder M, Kreutzer J: Transcranial surgery for pituitary adenomas. Pituitary. 2008 (11): 375-84. 17. Buchfelder M, Schlaffer S: Surgical treatment of pituitary tumours. Best Pract Res Clin Endocrinol Metab. 2009 (23): 677-92. 18. Buchfelder M, Weigel D, Droste M, Mann K, Saller B, Brubach K, Stalla GK, Bidlingmaier M, Strasburger CJ: Pituitary tumor size in acromegaly during pegvisomant treatment: experience from MR reevaluations of the German Pegvisomant Observational Study. Eur J Endocrinol. 2009 (161): 27-35. 19. Buhk JH, Jung S, Psychogios MN, Goricke S, Hartz S, Schulz-Heise S, Klingebiel R, Forsting M, Bruckmann H, Dorfler A, Jordan M, Buchfelder M, Knauth M: Tumor volume of growth hormone-secreting pituitary adenomas during treatment with pegvisomant: a prospective multicenter study. J Clin Endocrinol Metab. 2010 (95): 552-8. 20. Burger PC, Shibata T, Kleihues P: The use of the monoclonal antibody Ki-67 in the identification of proliferating cells: application to surgical neuropathology. Am J Surg Pathol. 1986 (10): 611-7. 54 21. Carboni P, Jr., Detta A, Hitchcock ER, Postans R: Pituitary adenoma proliferative indices and risk of recurrence. Br J Neurosurg. 1992 (6): 33-40. 22. Carlsen SM, Lund-Johansen M, Schreiner T, Aanderud S, Johannesen O, Svartberg J, Cooper JG, Hald JK, Fougner SL, Bollerslev J: Preoperative octreotide treatment in newly diagnosed acromegalic patients with macroadenomas increases cure short-term postoperative rates: a prospective, randomized trial. J Clin Endocrinol Metab. 2008 (93): 2984-90. 23. Castinetti F, Morange I, Dufour H, Regis J, Brue T: Radiotherapy and radiosurgery in acromegaly. Pituitary. 2009 (12): 3-10. 24. Chakrabortty S, Oi S, Yamaguchi M, Tamaki N, Matsumoto S: Growth hormone-producing pituitary adenomas: MR characteristics and preand postoperative evaluation. Neurol Med Chir (Tokyo). 1993 (33): 815. 25. Charest L, Comtois R, Beauregard H, Serri O: Growth hormone rebound after cessation of sms 201-995 treatment in acromegaly. Can J Neurol Sci. 1989 (16): 442-5. 26. Chatterjee S, May PL, Forster G, Spiller D, Jeffreys RV: Prediction of recurrence in pituitary tumours: a flow cytometric study using in vivo bromodeoxyuridine. Br J Neurosurg. 1993 (7): 165-9. 27. Colao A, Auriemma RS, Rebora A, Galdiero M, Resmini E, Minuto F, Lombardi G, Pivonello R, Ferone D: Significant tumour shrinkage after 12 months of lanreotide Autogel-120 mg treatment given first-line in acromegaly. Clin Endocrinol (Oxf). 2009 (71): 237-45. 28. Colao A, Ferone D, Cappabianca P, del Basso De Caro ML, Marzullo P, Monticelli A, Alfieri A, Merola B, Cali A, de Divitiis E, Lombardi G: Effect of octreotide pretreatment on surgical outcome in acromegaly. J Clin Endocrinol Metab. 1997 (82): 3308-14. 29. Colao A, Pivonello R, Auriemma RS, Briganti F, Galdiero M, Tortora F, Caranci F, Cirillo S, Lombardi G: Predictors of tumor shrinkage after primary therapy with somatostatin analogs in acromegaly: a prospective study in 99 patients. J Clin Endocrinol Metab. 2006 (91): 2112-8. 55 30. Colao A, Pivonello R, Auriemma RS, De Martino MC, Bidlingmaier M, Briganti F, Tortora F, Burman P, Kourides IA, Strasburger CJ, Lombardi G: Efficacy of 12-month treatment with the GH receptor antagonist pegvisomant in patients with acromegaly resistant to longterm, high-dose somatostatin analog treatment: effect on IGF-I levels, tumor mass, hypertension and glucose tolerance. Eur J Endocrinol. 2006 (154): 467-77. 31. Colao A, Pivonello R, Auriemma RS, Galdiero M, Savastano S, Grasso LF, Lombardi G: Growth hormone-secreting tumor shrinkage after 3 months of octreotide-long-acting release therapy predicts the response at 12 months. J Clin Endocrinol Metab. 2008 (93): 3436-42. 32. Cozzi R, Attanasio R, Montini M, Pagani G, Lasio G, Lodrini S, Barausse M, Albizzi M, Dallabonzana D, Pedroncelli AM: Four-year treatment with octreotide-long-acting repeatable in 110 acromegalic patients: predictive value of short-term results? J Clin Endocrinol Metab. 2003 (88): 3090-8. 33. Daousi C, Foy PM, MacFarlane IA: Ablative thyroid treatment for thyrotoxicosis due to thyrotropin-producing pituitary tumours. J Neurol Neurosurg Psychiatry. 2007 (78): 93-5. 34. Dayan C, Guilding T, Hearing S, Thomas P, Nelson R, Moss T, Bradshaw J, Levy A, Lightman S: Biochemical cure of recurrent acromegaly by resection of cervical spinal canal metastases. Clin Endocrinol (Oxf). 1996 (44): 597-602. 35. Dekkers OM, Biermasz NR, Pereira AM, Romijn JA, Vandenbroucke JP: Mortality in acromegaly: a metaanalysis. J Clin Endocrinol Metab. 2008 (93): 61-7. 36. Delgrange E, Donckier J: Proliferation activity in somatotroph pituitary adenomas. Mayo Clin Proc. 1998 (73): 1027-8. 37. Dina TS, Feaster SH, Laws ER, Jr., Davis DO: MR of the pituitary gland postsurgery: serial MR studies following transsphenoidal resection. AJNR Am J Neuroradiol. 1993 (14): 763-9. 38. Drake WM, Berney DM, Kovacs K, Monson JP: Markers of cell proliferation in a GH-producing adenoma of a patient treated with pegvisomant. Eur J Endocrinol. 2005 (153): 203-5. 56 39. Ekramullah SM, Saitoh Y, Ohnishi T, Arita N, Taki T, Hayakawa T: Effects of bromocriptine on staining indices of Ki-67 and proliferating cell nuclear antigen, and nucleolar organizer region number in pituitary adenomas. Neurol Med Chir (Tokyo). 1995 (35): 221-6. 40. Ezzat S, Kontogeorgos G, Redelmeier DA, Horvath E, Harris AG, Kovacs K: In vivo responsiveness of morphological variants of growth hormone-producing pituitary adenomas to octreotide. Eur J Endocrinol. 1995 (133): 686-90. 41. Fitzgibbons PL, Appley AJ, Turner RR, Bishop PC, Parker JW, Breeze RE, Weiss MH, Apuzzo ML: Flow cytometric analysis of pituitary tumors. Correlation of nuclear antigen p105 and DNA content with clinical behavior. Cancer. 1988 (62): 1556-60. 42. Frohman LA, Bonert V: Pituitary tumor enlargement in two patients with acromegaly during pegvisomant therapy. Pituitary. 2007 (10): 283-9. 43. Gerdes J, Becker MH, Key G, Cattoretti G: Immunohistological detection of tumour growth fraction (Ki-67 antigen) in formalin-fixed and routinely processed tissues. J Pathol. 1992 (168): 85-6. 44. Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H: Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984 (133): 1710-5. 45. Gillam MP, Molitch ME, Lombardi G, Colao A: Advances in the treatment of prolactinomas. Endocr Rev. 2006 (27): 485-534. 46. Giustina A, Barkan A, Casanueva FF, Cavagnini F, Frohman L, Ho K, Veldhuis J, Wass J, Von Werder K, Melmed S: Criteria for cure of acromegaly: a consensus statement. J Clin Endocrinol Metab. 2000 (85): 526-9. 47. Giustina A, Bronstein MD, Casanueva FF, Chanson P, Ghigo E, Ho KK, Klibanski A, Lamberts S, Trainer P, Melmed S: Current management practices for acromegaly: an international survey. Pituitary. 2010 (14): 125-33. 48. Giustina A, Chanson P, Bronstein MD, Klibanski A, Lamberts S, Casanueva FF, Trainer P, Ghigo E, Ho K, Melmed S: A consensus on 57 criteria for cure of acromegaly. J Clin Endocrinol Metab. 2010 (95): 3141-8. 49. Giustina A, Mazziotti G, Torri V, Spinello M, Floriani I, Melmed S: Meta-analysis on the effects of octreotide on tumor mass in acromegaly. PLoS One. 2012 (7): e36411. 50. Gopalan R, Schlesinger D, Vance ML, Laws E, Sheehan J: Long-term outcomes after Gamma Knife radiosurgery for patients with a nonfunctioning pituitary adenoma. Neurosurgery. 2011 (69): 284-93. 51. Greenman Y, Woolf P, Coniglio J, O'Mara R, Pei L, Said JW, Melmed S: Remission of acromegaly caused by pituitary carcinoma after surgical excision of growth hormone-secreting metastasis detected by 111-indium pentetreotide scan. J Clin Endocrinol Metab. 1996 (81): 1628-33. 52. Hald JK, Nakstad PH, Kollevold T, Bakke SJ, Skalpe IO: MR imaging of pituitary macroadenomas before and after transsphenoidal surgery. Acta Radiol. 1992 (33): 396-9. 53. Heck MM, Earnshaw WC: Topoisomerase II: A specific marker for cell proliferation. J Cell Biol. 1986 (103): 2569-81. 54. Hodish I, Barkan A: Long-term effects of pegvisomant in patients with acromegaly. Nat Clin Pract Endocrinol Metab. 2008 (4): 324-32. 55. Holdaway IM, Bolland MJ, Gamble GD: A meta-analysis of the effect of lowering serum levels of GH and IGF-I on mortality in acromegaly. Eur J Endocrinol. 2008 (159): 89-95. 56. Honegger J, Prettin C, Feuerhake F, Petrick M, Schulte-Monting J, Reincke M: Expression of Ki-67 antigen in nonfunctioning pituitary adenomas: correlation with growth velocity and invasiveness. J Neurosurg. 2003 (99): 674-9. 57. Honegger J, Zimmermann S, Psaras T, Petrick M, Mittelbronn M, Ernemann U, Reincke M, Dietz K: Growth modelling of non-functioning pituitary adenomas in patients referred for surgery. Eur J Endocrinol. 2008 (158): 287-94. 58. Jaffrain-Rea ML, Di Stefano D, Minniti G, Esposito V, Bultrini A, Ferretti E, Santoro A, Faticanti Scucchi L, Gulino A, Cantore G: A critical reappraisal of MIB-1 labelling index significance in a large 58 series of pituitary tumours: secreting versus non-secreting adenomas. Endocr Relat Cancer. 2002 (9): 103-13. 59. Jimenez C, Burman P, Abs R, Clemmons DR, Drake WM, Hutson KR, Messig M, Thorner MO, Trainer PJ, Gagel RF: Follow-up of pituitary tumor volume in patients with acromegaly treated with pegvisomant in clinical trials. Eur J Endocrinol. 2008 (159): 517-23. 60. Jorgensen JO, Feldt-Rasmussen U, Frystyk J, Chen JW, Kristensen LO, Hagen C, Orskov H: Cotreatment of acromegaly with a somatostatin analog and a growth hormone receptor antagonist. J Clin Endocrinol Metab. 2005 (90): 5627-31. 61. Kato M, Inoshita N, Sugiyama T, Tani Y, Shichiri M, Sano T, Yamada S, Hirata Y: Differential expression of genes related to drug responsiveness between sparsely and densely granulated somatotroph adenomas. Endocr J. 2012 (59): 221-8. 62. Klibanski A: Clinical practice. Prolactinomas. N Engl J Med. 2010 (362): 1219-26. 63. Kontogeorgos G: Classification and pathology of pituitary tumors. Endocrine. 2005 (28): 27-35. 64. Kopchick JJ, Parkinson C, Stevens EC, Trainer PJ: Growth hormone receptor antagonists: discovery, development, and use in patients with acromegaly. Endocr Rev. 2002 (23): 623-46. 65. Kopp C, Theodorou M, Poullos N, Jacob V, Astner ST, Molls M, Grosu AL: Tumor shrinkage assessed by volumetric MRI in long-term followup after fractionated stereotactic radiotherapy of nonfunctioning pituitary adenoma. Int J Radiat Oncol Biol Phys. 2012 (82): 1262-7. 66. Kristof RA, Stoffel-Wagner B, Klingmuller D, Schramm J: Does octreotide treatment improve the surgical results of macro-adenomas in acromegaly? A randomized study. Acta Neurochir (Wien). 1999 (141): 399-405. 67. Lancranjan I, Atkinson AB: Results of a European multicentre study with Sandostatin LAR in acromegalic patients. Sandostatin LAR Group. Pituitary. 1999 (1): 105-14. 68. Landolt AM, Shibata T, Kleihues P: Growth rate of human pituitary adenomas. J Neurosurg. 1987 (67): 803-6. 59 69. Laws ER, Vance ML, Thapar K: Pituitary surgery for the management of acromegaly. Horm Res. 2000 (53 Suppl 3): 71-5. 70. le Roux CW, Mulla A, Meeran K: Pituitary carcinoma as a cause of acromegaly. N Engl J Med. 2001 (345): 1645-6. 71. Lim YL, Leem W, Kim TS, Rhee BA, Kim GK: Four years' experiences in the treatment of pituitary adenomas with gamma knife radiosurgery. Stereotact Funct Neurosurg. 1998 (70 Suppl 1): 95-109. 72. Lloyd R, Kovacs K, Young W, Farrell W, Asa S, Trouillas J, Kontogeorgos G, Sano T, Scheithauer B, Horvath E, Watson R, Lindell E, Barkan A, Saeger W, Nose V, Osamura R, Ezzat S, Yamada S, Roncaroli F, Lopes M, Vidal Ruibal S: Tumours of the pituitary gland. R. DeLellis, R. Lloyd and H. Heitz. Pathology and Genetics of Tumours of Endocrine Organs. (Vol 8). International Agency for Research and Cancer (IARC). Lyon. 2004. 9 - 48. 73. Lorcy Y, Dejager S, Chanson P: Time course of GH and IGF-1 levels following withdrawal of long-acting octreotide in acromegaly. Pituitary. 2000 (3): 193-7. 74. Losa M, Barzaghi RL, Mortini P, Franzin A, Mangili F, Terreni MR, Giovanelli M: Determination of the proliferation and apoptotic index in adrenocorticotropin-secreting pituitary tumors : comparison between micro- and macroadenomas. Am J Pathol. 2000 (156): 245-51. 75. Losa M, Ciccarelli E, Mortini P, Barzaghi R, Gaia D, Faccani G, Papotti M, Mangili F, Terreni MR, Camanni F, Giovanelli M: Effects of octreotide treatment on the proliferation and apoptotic index of GHsecreting pituitary adenomas. J Clin Endocrinol Metab. 2001 (86): 5194-200. 76. Losa M, Franzin A, Mangili F, Terreni MR, Barzaghi R, Veglia F, Mortini P, Giovanelli M: Proliferation index of nonfunctioning pituitary adenomas: correlations with clinical characteristics and long-term follow-up results. Neurosurgery. 2000 (47): 1313-8; discussion 1318-9. 77. Losa M, Picozzi P, Motta M, Valle M, Franzin A, Mortini P: The role of radiation therapy in the management of non-functioning pituitary adenomas. J Endocrinol Invest. 2011 (34): 623-9. 60 78. Lynch BJ, Guinee DG, Jr., Holden JA: Human DNA topoisomerase IIalpha: a new marker of cell proliferation in invasive breast cancer. Hum Pathol. 1997 (28): 1180-8. 79. Machado AL, Nomikos P, Kiesewetter F, Fahlbusch R, Buchfelder M: DNA-flow cytometry of 207 pituitary adenomas: ploidy, proliferation, and prognosis. J Endocrinol Invest. 2005 (28): 795-801. 80. Maiza JC, Vezzosi D, Matta M, Donadille F, Loubes-Lacroix F, Cournot M, Bennet A, Caron P: Long-term (up to 18 years) effects on GH/IGF-1 hypersecretion and tumour size of primary somatostatin analogue (SSTa) therapy in patients with GH-secreting pituitary adenoma responsive to SSTa. Clin Endocrinol (Oxf). 2007 (67): 282-9. 81. Mayberg M, Vermeulen S: Advances in stereotactic radiosurgery in the treatment of pituitary adenomas. Curr Opin Endocrinol Diabetes Obes. 2007 (14): 296-300. 82. Melmed S: Medical progress: Acromegaly. N Engl J Med. 2006 (355): 2558-73. 83. Melmed S, Casanueva F, Cavagnini F, Chanson P, Frohman LA, Gaillard R, Ghigo E, Ho K, Jaquet P, Kleinberg D, Lamberts S, Laws E, Lombardi G, Sheppard MC, Thorner M, Vance ML, Wass JA, Giustina A: Consensus statement: medical management of acromegaly. Eur J Endocrinol. 2005 (153): 737-40. 84. Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, Wass JA: Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011 (96): 273-88. 85. Melmed S, Colao A, Barkan A, Molitch M, Grossman AB, Kleinberg D, Clemmons D, Chanson P, Laws E, Schlechte J, Vance ML, Ho K, Giustina A: Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. 2009 (94): 1509-17. 86. Melmed S, Sternberg R, Cook D, Klibanski A, Chanson P, Bonert V, Vance ML, Rhew D, Kleinberg D, Barkan A: A critical analysis of pituitary tumor shrinkage during primary medical acromegaly. J Clin Endocrinol Metab. 2005 (90): 4405-10. therapy in 61 87. Meyer JS, Marchosky JA, Hickey WF: Cell kinetic classification of tumors of the nervous system by DNA precursor labeling in vitro. Hum Pathol. 1993 (24): 1357-64. 88. Minniti G, Scaringi C, Amelio D, Maurizi Enrici R: Stereotactic Irradiation of GH-Secreting Pituitary Adenomas. Int J Endocrinol. 2012 (2012): 482861. 89. Niranjan A, Flickinger JC: Radiobiology, principle and technique of radiosurgery. Prog Neurol Surg. 2008 (21): 32-42. 90. Nishina Y, Takano K, Yasufuku-Takano J, Teramoto A, Fujita T: Mechanism of D(2) agonist-induced inhibition of GH secretion from human GH-secreting adenoma cells. Endocr J. 2005 (52): 775-9. 91. Nomikos P, Buchfelder M, Fahlbusch R: The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical 'cure'. Eur J Endocrinol. 2005 (152): 379-87. 92. Petersenn S, Schopohl J, Barkan A, Mohideen P, Colao A, Abs R, Buchelt A, Ho YY, Hu K, Farrall AJ, Melmed S, Biller BM: Pasireotide (SOM230) demonstrates efficacy and safety in patients with acromegaly: a randomized, multicenter, phase II trial. J Clin Endocrinol Metab. 2010 (95): 2781-9. 93. Pizarro CB, Oliveira MC, Coutinho LB, Ferreira NP: Measurement of Ki-67 antigen in 159 pituitary adenomas using the MIB-1 monoclonal antibody. Braz J Med Biol Res. 2004 (37): 235-43. 94. Plockinger U, Albrecht S, Mawrin C, Saeger W, Buchfelder M, Petersenn S, Schulz S: Selective loss of somatostatin receptor 2 in octreotide-resistant growth hormone-secreting adenomas. J Clin Endocrinol Metab. 2008 (93): 1203-10. 95. Pyronnet S, Bousquet C, Najib S, Azar R, Laklai H, Susini C: Antitumor effects of somatostatin. Mol Cell Endocrinol. 2008 (286): 230-7. 96. Rajasoorya C, Holdaway IM, Wrightson P, Scott DJ, Ibbertson HK: Determinants of clinical outcome and survival in acromegaly. Clin Endocrinol (Oxf). 1994 (41): 95-102. 97. Ramirez C, Vargas G, Gonzalez B, Grossman A, Rabago J, Sosa E, Espinosa-de-Los-Monteros AL, Mercado M: Discontinuation of 62 octreotide LAR after long term, successful treatment of patients with acromegaly: is it worth trying? Eur J Endocrinol. 2012 (166): 21-6. 98. Reincke M, Allolio B, Saeger W, Menzel J, Winkelmann W: The 'incidentaloma' of the pituitary gland. Is neurosurgery required? Jama. 1990 (263): 2772-6. 99. Resmini E, Dadati P, Ravetti JL, Zona G, Spaziante R, Saveanu A, Jaquet P, Culler MD, Bianchi F, Rebora A, Minuto F, Ferone D: Rapid pituitary tumor shrinkage with dissociation between antiproliferative and antisecretory effects of a long-acting octreotide in an acromegalic patient. J Clin Endocrinol Metab. 2007 (92): 1592-9. 100. Rickels MR, Snyder PJ: Cabergoline decreases somatotroph adenoma size: a case report. Pituitary. 2004 (7): 107-10. 101. Riss D, Jin L, Qian X, Bayliss J, Scheithauer BW, Young WF, Jr., Vidal S, Kovacs K, Raz A, Lloyd RV: Differential expression of galectin-3 in pituitary tumors. Cancer Res. 2003 (63): 2251-5. 102. Ronchi CL, Attanasio R, Verrua E, Cozzi R, Ferrante E, Loli P, Montefusco L, Motti E, Ferrari DI, Giugni E, Beck-Peccoz P, Arosio M: Efficacy and tolerability of gamma knife radiosurgery in acromegaly: a 10-year follow-up study. Clin Endocrinol (Oxf). 2009 (71): 846-52. 103. Saeger W: Effect of drugs on pituitary ultrastructure. Microsc Res Tech. 1992 (20): 162-76. 104. Saeger W: Pituitary tumors: prognostic indicators. Endocrine. 2005 (28): 57-66. 105. Saeger W: Proliferation markers and cell cycle inhibitors in pituitary adenomas. Front Horm Res. 2004 (32): 110-26. 106. Saeger W, Ludecke DK, Buchfelder M, Fahlbusch R, Quabbe HJ, Petersenn S: Pathohistological classification of pituitary tumors: 10 years of experience with the German Pituitary Tumor Registry. Eur J Endocrinol. 2007 (156): 203-16. 107. Saeger W, Schreiber S, Ludecke DK: Cyclins D1 and D3 and topoisomerase II alpha in inactive pituitary adenomas. Endocr Pathol. 2001 (12): 39-47. 63 108. Salehi F, Agur A, Scheithauer BW, Kovacs K, Lloyd RV, Cusimano M: Biomarkers of pituitary neoplasms: a review (Part II). Neurosurgery. 2010 (67): 1790-8; discussion 1798. 109. Salehi F, Agur A, Scheithauer BW, Kovacs K, Lloyd RV, Cusimano M: Ki-67 in pituitary neoplasms: a review--part I. Neurosurgery. 2009 (65): 429-37; discussion 437. 110. Sanno N, Teramoto A, Osamura RY: Thyrotropin-secreting pituitary adenomas. Clinical and biological heterogeneity and current treatment. J Neurooncol. 2001 (54): 179-86. 111. Scheithauer BW, Gaffey TA, Lloyd RV, Sebo TJ, Kovacs KT, Horvath E, Yapicier O, Young WF, Jr., Meyer FB, Kuroki T, Riehle DL, Laws ER, Jr.: Pathobiology of pituitary adenomas and carcinomas. Neurosurgery. 2006 (59): 341-53; discussion 341-53. 112. Schreiber I, Buchfelder M, Droste M, Forssmann K, Mann K, Saller B, Strasburger CJ: Treatment of acromegaly with the GH receptor antagonist pegvisomant in clinical practice: safety and efficacy evaluation from the German Pegvisomant Observational Study. Eur J Endocrinol. 2007 (156): 75-82. 113. Schreiber S, Saeger W, Ludecke DK: Proliferation markers in different types of clinically non-secreting pituitary adenomas. Pituitary. 1999 (1): 213-20. 114. Sherlock M, Woods C, Sheppard MC: Medical therapy in acromegaly. Nat Rev Endocrinol. 2011 (7): 291-300. 115. Shibuya M, Saito F, Miwa T, Davis RL, Wilson CB, Hoshino T: Histochemical study of pituitary adenomas with Ki-67 and anti-DNA polymerase alpha monoclonal antibodies, bromodeoxyuridine labeling, and nucleolar organizer region counts. Acta Neuropathol. 1992 (84): 178-83. 116. Shimatsu A, Murabe H, Nakamura Y, Usui T: Rebound hypersecretion of GH following octreotide withdrawal due to liver dysfunction in an acromegalic patient. Endocr J. 2000 (47): 635-8. 117. Steiner E, Knosp E, Herold CJ, Kramer J, Stiglbauer R, Staniszewski K, Imhof H: Pituitary adenomas: findings of postoperative MR imaging. Radiology. 1992 (185): 521-7. 64 118. Stevenaert A, Harris AG, Kovacs K, Beckers A: Presurgical octreotide treatment in acromegaly. Metabolism. 1992 (41): 51-8. 119. Strasburger CJ, Buchfelder M, Droste M, Mann K, Stalla GK, Saller B: Experience from the German pegvisomant observational study. Horm Res. 2007 (68 Suppl 5): 70-3. 120. Swearingen B, Barker FG, 2nd, Katznelson L, Biller BM, Grinspoon S, Klibanski A, Moayeri N, Black PM, Zervas NT: Long-term mortality after transsphenoidal surgery and adjunctive therapy for acromegaly. J Clin Endocrinol Metab. 1998 (83): 3419-26. 121. Thapar K, Kovacs K, Scheithauer BW, Stefaneanu L, Horvath E, Pernicone PJ, Murray D, Laws ER, Jr.: Proliferative activity and invasiveness among pituitary adenomas and carcinomas: an analysis using the MIB-1 antibody. Neurosurgery. 1996 (38): 99-106; discussion 106-7. 122. Thapar K, Scheithauer BW, Kovacs K, Pernicone PJ, Laws ER, Jr.: p53 expression in pituitary adenomas and carcinomas: correlation with invasiveness and tumor growth fractions. Neurosurgery. 1996 (38): 765-70; discussion 770-1. 123. Trainer PJ, Drake WM, Katznelson L, Freda PU, Herman-Bonert V, van der Lely AJ, Dimaraki EV, Stewart PM, Friend KE, Vance ML, Besser GM, Scarlett JA, Thorner MO, Parkinson C, Klibanski A, Powell JS, Barkan AL, Sheppard MC, Malsonado M, Rose DR, Clemmons DR, Johannsson G, Bengtsson BA, Stavrou S, Kleinberg DL, Cook DM, Phillips LS, Bidlingmaier M, Strasburger CJ, Hackett S, Zib K, Bennett WF, Davis RJ: Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N Engl J Med. 2000 (342): 1171-7. 124. Tsuzuki T, Tsunoda S, Sakaki T, Konishi N, Hiasa Y, Nakamura M, Yoshino E: Tumor cell proliferation and apoptosis associated with the Gamma Knife effect. Stereotact Funct Neurosurg. 1996 (66 Suppl 1): 39-48. 125. Turner HE, Nagy Z, Gatter KC, Esiri MM, Wass JA, Harris AL: Proliferation, bcl-2 expression and angiogenesis in pituitary 65 adenomas: relationship to tumour behaviour. Br J Cancer. 2000 (82): 1441-5. 126. Turner HE, Nagy Z, Sullivan N, Esiri MM, Wass JA: Expression analysis of cyclins in pituitary adenomas and the normal pituitary gland. Clin Endocrinol (Oxf). 2000 (53): 337-44. 127. Turner HE, Wass JA: Are markers of proliferation valuable in the histological assessment of pituitary tumours? Pituitary. 1999 (1): 14751. 128. van der Lely AJ, Hutson RK, Trainer PJ, Besser GM, Barkan AL, Katznelson L, Klibanski A, Herman-Bonert V, Melmed S, Vance ML, Freda PU, Stewart PM, Friend KE, Clemmons DR, Johannsson G, Stavrou S, Cook DM, Phillips LS, Strasburger CJ, Hackett S, Zib KA, Davis RJ, Scarlett JA, Thorner MO: Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet. 2001 (358): 1754-9. 129. Vidal S, Kovacs K, Horvath E, Rotondo F, Kuroki T, Lloyd RV, Scheithauer BW: Topoisomerase IIalpha expression in pituitary adenomas and carcinomas: relationship to tumor behavior. Mod Pathol. 2002 (15): 1205-12. 130. Wierinckx A, Auger C, Devauchelle P, Reynaud A, Chevallier P, Jan M, Perrin G, Fevre-Montange M, Rey C, Figarella-Branger D, Raverot G, Belin MF, Lachuer J, Trouillas J: A diagnostic marker set for invasion, proliferation, and aggressiveness of prolactin pituitary tumors. Endocr Relat Cancer. 2007 (14): 887-900. 131. Wolfsberger S, Wunderer J, Zachenhofer I, Czech T, Bocher-Schwarz HG, Hainfellner J, Knosp E: Expression of cell proliferation markers in pituitary adenomas--correlation and clinical relevance of MIB-1 and anti-topoisomerase-IIalpha. Acta Neurochir (Wien). 2004 (146): 831-9. 132. Yamada S, Ohyama K, Taguchi M, Takeshita A, Morita K, Takano K, Sano T: A study of the correlation between morphological findings and biological activities in clinically nonfunctioning pituitary adenomas. Neurosurgery. 2007 (61): 580-4; discussion 584-5. 133. Zatelli MC, Ambrosio MR, Bondanelli M, Uberti EC: Control of pituitary adenoma cell proliferation by somatostatin analogs, dopamine 66 agonists and novel chimeric compounds. Eur J Endocrinol. 2007 (156 Suppl 1): S29-35. 134. Zatelli MC, Piccin D, Ambrosio MR, Bondanelli M, Uberti EC: Antiproliferative effects of somatostatin analogs in pituitary adenomas. Pituitary. 2006 (9): 27-34. 67 8. Abbildungsverzeichnis Abbildung 1: Mikroskopische Abbildungen der immunhistochmischen Untersuchungen des in der Originalpublikation von Drake et al. beschriebenen Patienten (Überlassung der Abbildung im Original mit Genehmigung des Erstautors). Abbildung 2: Mikroskopische Abbildungen der immunhistochmischen Untersuchungen für die Proliferationsmarker Ki-67 und Topoisomerase-IIα der seriell aus den Jahren 2003, 2006 und 2010 gewonnen Tumorproben der Patientin #13. 68 9. Abkürzungsverzeichnis ACTH Adrenocorticotropes Hormon bzw. beziehungsweise CT Computertomographie DA dopamin-agonistisch g Gramm i.m. intramuskulär IGF-1 insulin-like growth factor-1; Insulinähnlicher Wachstumsfaktor 1 LAR longer-acting slow-release; Depotpräparat LI Labeling-Index mg Milligramm MRT Magnetresonanztomographie Nr. Nummer Pat. Patient Peg. Pegvisomant PRL Prolaktin RTX Radiotherapie s.c. subcutan SSA Somatostatin-Analoga STH Somatotropes Hormon, Wachstumshormon Tab. Tabelle U-Test Mann-Whitney U-Test µg Mikrogramm 69 10. Danksagung Mein besonderer Dank gilt Herrn Professor Dr. med. Michael Buchfelder, dem Direktor der Neurochirurgischen Klinik der Universität ErlangenNürnberg, der für die Diskussion der Problemstellung und für seine fortwährende Unterstützung in allen Stadien dieser Inauguraldissertation zur Verfügung stand. Ich danke Herrn Prof. Dr. med. Wolfgang Saeger, dem Leiter des Pathologischen Instituts Bereitstellung des immunhistochemischen am Marienkrankenhaus Tumormaterials, Färbungen sowie der der Hamburg für die Durchführung der Anleitung der bei mikroskopischen Bewertung der immunhistochemischen Untersuchungen. Ich danke Herrn Dr. William M. Drake, Endokrinologe am St. Bartholomew’s Hospital (West Smithfield, London, England) für die freundliche Überlassung nicht-publizierter Originalbilder der immunhistochemischen Untersuchungen seiner Originalpublikation. Ich danke Frau Silke Speck für die Hilfe bei der Überarbeitung und Korrektur der Arbeit. Ich bedanke mich bei meinen Kollegen, Frau Yu Xi und Herrn Fuhua Lin, die mich bei der Zählkontrolle der Färbeindices sowie den statistischen Auswertung unterstützt und beraten haben. Zuletzt danke ich meiner Frau Jana Schlaffer die mir in allen Phasen der Arbeit mir die Kraft und den Rückhalt zur Erstellung der Arbeit gegeben hat.