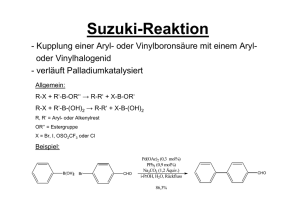
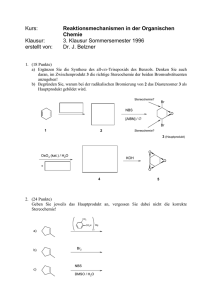
Aromaten und Heteroaromaten WS 2011
Werbung

Aromaten und Heteroaromaten 1. Aromatizität und Antiaromatizität 1.1. Benzol • Im späten 18. Jahrhundert und zu Beginn des 19. Jahrhunderts wurden tierische und pflanzliche Materialien pyrolysiert und aus dem Pyrolysat Substanzen isoliert. • Aus Walöl wurde seit dem Ende des 18. Jahrhunderts in England durch Pyrolyse Leuchtgas für Straßenlaternen hergestellt. Daraus hat 1825 Michael Faraday eine Substanz isoliert, die er bicarburet of hydrogen nannte. Faraday fand bereits aus der Elementaranalyse ein Verhältnis von C : H = 1 : 1 • 1833 isolierte Mitscherlich aus dem Calziumsalz der Benzoesäure dieselbe Substanz, die er Benzin nannte. Seine Ergebnisse veröffentlichte Mitscherlich in Liebigs Annalen der Chemie. Liebig fügte dieser Publikation eine Fußnote hinzu, in der er vorschlug, diese Substanz Benzol zu nennen. Die Endung –ol sollte darauf hinweisen, dass aus der festen Benzoesäure eine Flüssigkeit entstanden war (von lat. Oleum = Öl) • 1835 führte Viktor Mayer eine Molmassenbestimmung von Benzol durch und berechnet daraus die Summenformel C6H6. • In der Folgezeit wurden weitere Benzolderivate entdeckt. Viele wurden aus aromatisch riechenden pflanzlichen Materialien isoliert, z.B. aus Nelken, Zimt, Lorbeer, Anis, bitteren Mandeln, Vanille usw. Deshalb wurden diese Substanzen zur Klasse der Aromatischen Verbindungen (kurz: Aromaten) zusammengefasst. • Im Laufe der Zeit stellte sich heraus, daß diese Aromatischen Verbindungen mehrere Eigenschaften gemeinsam haben, die man unter dem Begriff „Aromatizität“ zusammenfasst. 1.1.1. Das Benzol-Problem • Benzol ist eine hoch ungesättigte Verbindung, ähnlich wie Olefine oder Acetylene. • ABER: Benzol zeigt ganz andere chemische Eigenschaften als Alkene und Alkine. • Benzol reagiert im Dunklen nicht mit Brom unter Entfärbung, nur bei Bestrahlung mit Licht. • Bayer-Probe (Reaktion mit schwach alkalischer KMnO4-Lösung) negativ. • Benzol polymerisiert nicht. • Benzol läßt sich katalytisch hydrieren und geht unter Aufnahme von 3 Equivalenten Wasserstoff in Cyclohexan über. • Benzol reagiert mit Ozon zu Glyoxal. • Benzol zeigt auch ganz andere physikalische Eigenschaften als Alkene und Alkine. • Schmelzpunkt: +6 °C; zum Vergleich 1,3,5-Hexatrien: -12 °C • Siedepunkt: +80 °C; zum Vergleich 1,3,5-Hexatrien: +76 °C 1.1.2. Strukturvorschläge für Benzol • von 1862 bis 1865 schlug August Kekulé verschiedene cyclische Strukturen für Benzol vor. H H H H H H H H H H H H H • 1867 veröffentlichte Dewar seinen Strukturvorschlag. H H H H H • 1869 schlug Ladenburg eine alternative Struktur für Benzol vor. H H H H H H • 1872 verfeinerte Kekulé seine Benzolstruktur. Um zu erklären, dass es nur ein orthoDisubstituiertes Benzol gibt, führte er oszillierende Strukturen ein. H H H H H H H H H H H H • • 1899 schlug Thiele eine Benzolformel vor, die ohne oszillierende Strukturen auskam. Thiele formulierte ganz allgemein Doppelbindungen mit Hilfe sogenannter Partialvalenzen. 1925 übertrugen Robinson und Ingold ihre Vorstellung von abgeschlossenen Elektronenschalen (“Edelgaskonfiguration”) auf Benzol und führten den Begriff “aromatisches Elektronensextett” ein. H H H H H H 1.1.3. VB-Beschreibung von Benzol • Die VB-Theorie fasst die beiden Kekulé-Strukturen nicht mehr als nebeneinander existierende Moleküle auf, sondern als mesomere Grenzformeln zur Beschreibung eines einzigen Moleküls. D. h. die 6 πElektronen sind cyclisch delokalisiert. Energie • Reales Benzol ist energetisch günstiger als beide mesomere Grenzformeln. • Reales Benzol ist also gegenüber den Kekulé-Strukturen stabilisiert. Man nennt diese Stabilisierung Mesomerieenergie oder Resonanzenergie oder Delokalisationsenergie. • Man kann die Mesomerieenergie aus den Hydrierwärmen abschätzen. Energie fiktives Cyclohexatrien Mesomerieenergie 151 kJ/mol 360 kJ/mol 232 kJ/mol 209 kJ/mol 120 kJ/mol 1.1.4. MO-Beschreibung von Benzol • Für konjugierte π-Systeme kann man mit guter Näherung die HMOTheorie anwenden (Hückel-MO-Theorie). Die HMO-Theorie gilt streng nur unter folgenden Bedingungen: • Cyclische Systeme, die nur sp2-hybridisierte C-Atome enthalten. • Alle σ-Bindungen sollen gleich lang sein. • σ-π-Trennung • Für Überlappungsintegrale Sik zwischen einzelnen C-Atomen soll gelten: Sik = 1 für i = k und Sik = 0 für i = k+1. • Die Energien der einzelnen p-Orbitale, die das π-System bilden, sollen gleich sein und dem Coulom-Integral α entsprechen. • Die Wechselwirkungsenergie zweier benachbarter p-Orbitale ist durch das Resonanzintegral β gegeben. • Aus diesen Annahmen lassen sich die Energien der einzelnen πMolekülorbitale berechnen. • MO-Schema von Benzol: α-2β α-β α α+β α+2β _ + _ + _ + + _ _ + _ _ + + ___________________________________________ _ + _ + + + + + + + + _ + _ • Frost-Musulin-Schema für cyclisch konjugierte π-Syszeme: Man zeichnet einen Kreis mit Radius 2β und zeichnet darin ein n-Eck mit der Spitze nach unten so ein, dass die Ecken den Kreis berühren. Dann liegen an den Berührungspunkten die Energieniveaus der MOs. E E 2ß 2ß E 2ß E 2ß Anmerkung: α und β ist für jeden Ring ein anderer Wert. E 2π 2π C3H3+ C4H42+ 6π C5H5- E 6π C6H6 6π C7H7+ 6π C8H82+ E 2π 6π 6π C3H3+ C4H42- C5H5- E 6π C6H6 6π C7H7+ 10π C8H82- Fazit: Bindende und nichtbindende MOs können mit 2, 6, 10,… πElektronen doppelt besetzt werden. Will man 4, 8, 12,… π-Elektronen einfüllen, dann muss man auch antibindende MOs besetzen oder nichtbindende MOs einfach. Dies hat Hückel in einer Regel („Hückel-Regel“) formuliert: Cyclisch konjugierte π-Systeme mit 2, 6, 10,…, (4n+2)π-Elektronen sind stabilisierte aromatische Systeme. Cyclisch konjugierte π-Systeme mit 4, 8, 12,…, (4n)π-Elektronen sind destabilisierte antiaromatische Systeme. aromatisch: C3H3+, C4H42+, C5H5-, C6H6, C7H7+, C8H82+, C8H82antiaromatisch: C4H4, C7H7-, C8H8 1.1.5. Elektronenbeugung und Röntgenstrukturanalyse von Benzol • 1931 hat Wierl Elektronenbeugungsexperimente mit Benzoldampf durchgeführt und dabei C-C- und C-H-Abstände bestimmt. C-C 139,3 pm C-H 108,4 pm • 1958 hat Cox eine Röntgenstrukturanalyse von Benzol angefertigt und den C-C-Abstand zu 139,2 pm bestimmt. 1.1.6. Ringstromeffekt und NMR • Chemische Verschiebung der Benzol-Hs: 7,25 ppm Chemische Verschiebung olefinischer Hs: 4-7 ppm • Erklärung: Ringstromeffekt. Das Magnetfeld induziert im cyclischkonjugierten π-System einen Ringstrom, der wiederum nach der Lenz´schen Regel ein Magnetfeld induziert, das im Innern des Ringstroms zum äusseren Magnetfeld entgegengesetzt gerichtet ist. Dadurch wird im Bereich der H-Atome das äussere Magnetfeld verstärkt. Oberhalb und unterhalb des Rings wird das äussere Magnetfeld abgeschwächt. Nach Larmor führt das zu höheren Resonanzfrequenzen, als auf Grund vom äußeren Magnetfeld erwartet. • Modellvorstellung B0 B0 Ringstrom H H H H H Induziertes Magnetfeld H Die H-Atome sind im Bereich des verstärken Magnetfeldes. Die CAtome bleiben unbeeinflusst. • Experimenteller Beweis bei Ansa-Verbindungen -1,6 ppm bis -4,0 ppm H2C B0 CH2 CH2 B0 H2C CH2 H H H2C 1,3 ppm bis 2,2 ppm CH2 H H 1.1.7. Kriterien aromatischer Zustände Verbindungen sind als „aromatisch“ zu bezeichnen, wenn sie • ein ebenes, cyclisch-konjugiertes π-Elektronensystem mit (4n+2) πElektronen besitzen • Idealen Bindungslängenausgleich zeigen • Im NMR einen Ringstromeffekt zeigen • Chemisches Kriterium: aromatische Verbindungen gehen eher Substitutionsreaktionen als Additionsreaktionen ein. 1.1.8. Valenzisomere von Benzol • 271 theoretisch mögliche Konstitutionsisomere von Benzol: • Valenzisomere sind Verbindungen, die sich nur durch Verschieben von C-C- und C=C-Bindungen ineinander umwandeln lasssen. Valenzisomere von Benzol sind: • Tectadien H H H = H • Benzvalen = • Prisman = • Bicyclopropenyl 1) Synthese von Tectadien COOH COOH Na/Hg + Ac2O O O COOH COOH - AcOH O [2+2] O O + H2O COOH COOH hν O Pb(OAc)4 oder anodische Oxidation Van Tamelen et al., J. Am. Chem. Soc. 1971, 6092. COOMe + Cl2 ∆ H Cl H Cl Cl ∆ COOMe — COOMe Na/Hg Ts MeOOC COOMe MeOOC Cl HH CAN Fe2(CO)9 Fe Cl OC OC CO Ts Cl 2) Synthese von Benzvalen nBuLi NaNH2 Li H CH Katz et al., Org. Synth. 53, 157 (1973). CH2Cl2 – LiCl CH2Cl H 3) Synthese von Prisman O N N N Ph H O H O N [2+2] N O N N Ph N KOH – CO2 – Ph-NH2 hν Cu2+ NH NH O Ph O N – N2 N Katz et al., J. Am. Chem. Soc. 1973, 2738. explodiert beim Kratzen mit dem Spatel 4) Synthese von Bicyclopropenyl SiMe3 H 2 C Me3Si Cl nBu4NF Cl 0 °C, 0,02 Torr SiMe3 Me3Si Cl Billups et al., Angew. Chem. 101, 1735 (1989). 1.2. Cyclobutadien und Cyclooctatetraen 1.2.1. Cyclobutadien • Cyclobutadien ist extrem instabil • Herstellung erstmals 1965 von Pettit et al. In einer Ar-Matrix bei 10 K O hν O O O 10 K – CO2 COOMe + Cl2 Cl COOMe — COOMe Cl H Cl H Cl MeOOC COOMe MeOOC Cl HH Fe2(CO)9 Cl CAN Fe OC CO OC Nachweis durch Abfangreaktionen • Abfangreaktionen für Cyclobutadien. H Dim. H • Nach Hückel sollte Ccylobutadien antiaromatisch und ein Biradikal sein. + E – + – + + – + – • Experimentelle Befunde: kein ESR-Spektrum ⇒ kein Biradikal • Aus IR-Spektrum: Cyclobutadien ist rechteckig, zwei Bindungen sind 137 pm lang (Doppelbindung), zwei sind 154 pm lang (Einfachbindung). + – + – E + – Verzerrung + + – • Aus temperaturabhängigen 13C-NMR-Spektren von substituierten Cyclobutadienen folgt: Cyclobutadien ist ein fluktuierendes Molekül H 2,5 kJ/mol H 2,5 kJ/mol H • Also: Cyclobutadien weicht dem instabilen antiaromatischen Zustand teilweise durch Verzerrung aus. • Bildungsenthalpie: + 114 kcal/mol. Rechnungen ergeben eine Destabilisierung von 87 kcal/mol gegenüber 1,3-Butadien (32 kcal/mol wegen Ringspannung, 55 kcal/mol wegen „Antiaromatizität“). • Substituierte Cyclobutadiene sind deutlich stabiler als der Grundkörper, so dass sie auch isoliert werden können. O O hν O H O O [2+2] H O – CO2 H – CO 5,38 ppm G. Maier et al., Angew. Chem. 85, 1056 (1973). S S S [2+2] 160 pm Krebs et al., Angew. Chem. 84, 952 (1972). 134,4 pm Gelbe Substanz, m.p. 240 °C 1.2.2 Cyclooctatetraen • Cyclooctatetraen COT ist eine im Kühlschrank haltbare Flüssigkeit mit einem Schmelzpunkt von -5 °C und einem Siedepunkt von 142 °C. • Nach Hückel sollte COT ein Antiaromat sein (8 π-Elektronen). • Aber: COT zeigt typische Olefinreaktionen. • Grund: COT ist nicht eben! 126 ° 146 pm • 133 pm Also: HMO-Theorie darf nicht angewendet werden! • Aus Ab-initio-Rechnungen ergibt sich eine Destabilisierung von ebenem antiaromatischen COT gegenüber dem nicht ebenen olefinischen COT von ca. 15 kcal/mol. 15 kcal/mol • Sowohl beim Umklappen der COT-Wanne als auch bei der Bindungsverschiebung wird das ebene, antiaromatische COT als Übergangszustand durchlaufen. • Erste Synthese von COT durch Willstätter und Mitarbeitern (1905) O Na N OH H+ 1) MeI N N EtOH 2) AgOH – H2O 3) N Br 1) MeI Me2NH Br2 2) AgOH 3) N 1) MeI 2) AgOH N 3) Br • Heute Synthese von COT nach Reppe H H H H H NC NC H Ni H H Ni(CN)2 200 bar 90% NC NC Ni 1.3. Weitere Moleküle mit cyclisch-konjugierten π-Elektronen 1.3.1. Monocyclische Teilchen 1) Cyclopropenyl-Kation Cl CCl2 Ph Cl Cl nBu3SnH Cl Cl Ph Cl “ N N – N2 CN ” Ph Cl H Cl SbCl5 Cl Ph CN Ph Ph BF3 • OEt2 Cl Cl “ Cl = Ph “ Ph Cl Cl Cl Ph “ Ph = “ Ph Ph • Das Cyclopropenyl-Kation ist stabiler als das Allyl-Kation • Tri-tert.Butylcyclopropenylium-perchlorat kann man aus Wasser umkristallisieren • Cyclopropenon enthält ein Cyclopropenyl-Kation • Cylopropenyl-Kationen sind aromatisch 2) Cyclobutenyl-Dikation O HO O OH ” O “ O” “ HO OH ” O = O” 2“ HO OH • Das Cyclobutenyl-Dikation kommt z.B. in der Quadratsäure vor • Auf Grund der zwei positiven Ladungen im Ring ist diese Substanz stark sauer: pKs1 = 1,0; pKs2 = 3,5 • Cylobutenyl-Dikationen sind aromatisch 3) Cyclopentadienyl-Anion • Das Cyclopentadienyl-Anion kann leicht aus Cyclopentadien durch Deprotonieren hergestellt werden (pKs = 16) nBuLi HH ” H = ” • Das Cyclopentadinyl-Anion wird häufig als Ligand für Metallkomplexe verwendet und zur Synthese substituierter Cyclopentadiene (z.B. als Dienkomponente für Diels-Alder-Reaktionen) • Das Cyclopentadienyl-Anion ist aromatisch 4) Cyclopentadienyl-Kation • Das Cyclopentadienyl-Anion ist nur unterhalb von -200 °C stabil. 1) nBuLi HH 2) Cl2 SbF5 “ H H Cl = “ • Es zeigt ein ESR-Spektrum, wie es für ein Diradikal erwartet wird • Das Cyclopentadienyl-Kation ist antiaromatisch • Ein Cyclopentadienyl-Kation versteckt sich auch im Cyclopentadienon O H H DIPEA Br O • O “ O ” Cyclopentadienon ist ebenfalls antiaromatisch O 5) Tropylium-Kation • Das Tropylium-Kation kann man durch Hydrid-Abspaltung aus Cycloheptatrien herstellen Ph3C+ BF4– H H • H = “ Bromcycloheptatrien liegt als Salz vor H Br2 H H H • “ Br H H Br DIPEA Br H – HBr “ Tropylium-Kationen treten auch in Tropolonen auf OH O OH O HO OH O HO MeO H NHAc MeO MeO O Tropon Tropolon Purpurogallin Colchicin OH Br– • Herstellung von Tropon Br Br Br Br CH2N2 Br2 DIPEA – HBr Br ” “ Br Br Br Br • H2O O Herstellung von Tropolon Cl ClCl C O Cl HH ” O O H KOH O ” O [2+2] Cl ClCl H Cl OH O OH ” O 6) Cyclooctatetraen-Dianion • COT kann leicht durch Alkalimetalle zweifach reduziert werden. ” + Na + Na • - Na+ - Na+ Disproportionierung • Es bildet sich ein ebenes Dianion mit 10 π-Elektronen • Röntgenstruktur: alle Bindungen gleich lang: 141 pm • COT-Dianion ist aromatisch 2” 7) [10]-Annulen • n-gliedrige Ringe mit cyclisch konjugierten Doppelbindungen werden als [n]-Annulene bezeichnet. 5,6-6,0 ppm H H hν + H + HH Winkelspannung nimmt ab Transannulare Wechselwirkung nimmt zu • [10]-Annulen wird durch Photolyse von Dihydronaphthalin als Isomerenmischung erhalten • 1H-NMR • [10]-Annulen ist nicht eben und damit olefinisch liegt im olefinischen Bereich • [10]-Annulen nach E. Vogel Cl Na / NH3 fl. H H Br Br2 H H KOH H – HBr – 0,5 ppm H • H Br Br H Na / NH3 fl. CCl2 Br H Cl + 7,6 ppm 1,6-Methano- [10]-Annulen zeigt einen Ringstromeffekt, ist also aromatisch. hν • Weitere Vogel-Annulene N O 8) [12]-Annulen hν hν + cis/trans-Isomere • [12]-Annulen ist extrem instabil, weicht aber durch Verdrillung (zumindest teilweise) dem antiaromatischen Zustand aus. 9) [14]-Annulen • [14]-Annulen wurde von Sondheimer et al. hergestellt und untersucht. H H H+ H H Cu(OAc)2 Py – H+ H H + H+ H+ H H H H H • + H H H H – 0,6 ppm H H H H H H H + 7,6 ppm H+ H • H H – H+ H2 / Pd + H+ BaCO3 H H H H H – H+ + H+ H H H H H O H H Ringstromeffekt ⇒ aromatisch • Es gibt auch die entsprechenden überbrückten Vogel-Annulene: eben, aromatisch O H H nicht eben, olefinisch O H H O H H O H H H H 10) [16]-Annulen • [16]-Annulen zeigt alternierende Bindungslängen und ist damit olefinisch. 134 pm 146 pm 11) [18]-Annulen • [18]-Annulen wurde ebenfalls von Sondheimer et al. hergestellt und untersucht. H H H H H H H H – 6 bis – 8 ppm H H H H H H –1,8 ppm H H H H H + 8,9 ppm H H H H H H H H H 139 - 140 pm H H H H H H H H + 9 bis + 10 ppm • Synthese von [18]-Annulen Cu(OAc)2 KOtBu Py / Luft tBuOH H2 / Pd BaCO3 1.3.2. Polycyclische Teilchen • Die Hückel-Regel ist hier nicht mehr anwendbar. Hückel fordert monocyclische Moleküle! • Für Polycyclische Aromaten gilt die Clar´sche Regel: Eine Verbindung ist um so stabiler, je mehr autonome πElektronensextetts sich formulieren lassen. • Clar´sche Regel ist rein empirisch und hat nichts mit der realen Elektronenverteilung zu tun! • Polycyclische Aromaten werden unterteilt in Acene, Phene und Helicene. • Man erhält polycyclische Aromaten formal durch Anhängen von einem oder mehreren Benzolringen an Benzol. 1) Naphthalin und Azulen • Naphthalin und Azulen sind Isomere, beide haben die Summenformel C10H8. • Naphthalin ist farblos (absorbiert also im UV-Bereich), Azulen ist dunkelblau (absorbiert also bei 560-600 nm = grün bis gelb). 139 bis 140 pm 137 pm 141 pm Naphthalin 151 pm Azulen • Naphthalin und Azulen sind Isomere, beide haben die Summenformel C10H8. • Naphthalin ist farblos (absorbiert also im UV-Bereich), Azulen ist dunkelblau (absorbiert also bei 560-600 nm = grün bis gelb). • Naphthalin zeigt alternierende Bindungen, Azulen zeigt bis auf die gemeinsame Bindung einen idealen Bindungslängenausgleich. • Napthalin besitzt kein Diplomoment, Azulen besitzt ein Dipolmoment! ” “ E 0 ∆E = UV Napthalin ∆E = gelbgrün Azulen .... ” “ • Ziegler-Hafner-Azulen-Synthese H N ” N Cl “ H H3C N“ H CH3 NO2 N NO2 N – H+ NO2 NO2 N H CH3 NO2 + H+ H “ N N H CH3 NO2 NO2 NO2 Zincke-Salz H “ N H N H3C H H H N “ N H CH3 NO2 N H3C N H CH3 NO2 N H N H3C H N H CH3 NO2 – H+ NO2 NO2 NO2 +H + H N N H H N“ H CH3 H3C NO2 N – NO2 “ N Cl ” NaOH N H O O2N H2N NO2 König´sches Salz Zincke-Aldehyd Ph H3C N N H H3C H N Ph O H NaOH – H3C Zincke-Aldehyd • N Die „Kunst“ bestand darin, einen Ersatz für den instabilen Glutacondialdehyd zu finden. Der Zincke-Aldehyd erfüllt diese Bedingung. HO H O Glutacondialdehyd instabil N H O Zincke-Aldehyd stabil WISSENSCHAFTS-POSSE Ahnungslose Chemiker entdecken Verbindung zum zweiten Mal Von Jens Lubbadeh (Spiegel online 6. 12. 2007) Ein japanischer Chemiker glaubte, ein geniales neues Ringmolekül hergestellt zu haben. Das aber kam einem emeritierten Professor aus Würzburg so bekannt vor, dass er nachblätterte - und herausfand: Die Entdeckung wurde vor 102 Jahren schon mal gemacht. Isao Yamaguchi war so stolz: Als erster hatte er mit seinen Kollegen einen Weg gefunden, ein Ringmolekül aus 10 Kohlenstoff- und zwei Stickstoffatomen herzustellen. 12-Annulen nennen die Chemiker diese Verbindung und Yamaguchi hatte zudem noch eine spezielle Unterart kreiert. Annulene sind im Prinzip Seifen und senken die Oberflächenspannung des Wassers. Diese Eigenschaft macht man sich unter anderem bei Seifenblasen zunutze - oder wenn man Wasserläufer im Teich ärgern will: Ein Paar Tropfen Spüli ins Wasser, und der Läufer wird zum Säufer, weil ihn das Wasser plötzlich nicht mehr trägt. Der japanische Chemiker reichte seine Entdeckung bei dem Fachmagazin "Organic Letters" ein. Im August 2006 wurde das Papier publiziert. Im August 2007 erschien - auf Yamaguchis Arbeit aufbauend - im Magazin "Angewandte Chemie International Edition" eine weitere Publikation zur Herstellung des besonderen 12-Annulens, diesmal von einem US-Chemikerteam um Frederic Menger. Yamaguchis und Mengers Arbeiten schienen ganz normale Entdeckungen im Wissenschaftsbetrieb zu sein. Doch dann kam Manfred Christl, emeritierter Professor am Institut für Organische Chemie in Würzburg. Trotz Ruhestand verfolgt er noch immer eifrig die Fachpresse. Als Christl das Paper von Frederic Menger in "Angewandte Chemie" las, wurde er stutzig. Ihm kam die Entdeckung ziemlich bekannt vor. Daraufhin las er noch Yamaguchis Veröffentlichung. Plötzlich dämmerte es ihm: Diesen Reaktionstyp zur Gewinnung des besonderen 12-Annulens mit den beiden Stickstoffatomen war keineswegs neu. Christl hatte ihn den Studenten schon vor Jahren in seiner Vorlesung erklärt. Es handelte sich um die so genannte Zincke-Reaktion - benannt nach dem deutschen Chemiker Theodor Zincke, der von 1843 bis 1928 lebte. Er hatte diesen Reaktionstyp 1904 in "Justus Liebigs Annalen der Chemie" veröffentlicht. Ganze 102 Jahre vor Yamaguchi. Falsch interpretierte Reaktion So wurde der Emeritus zum Detektiv: "Ich habe nachgeschaut, ob Zinckes Arbeit von Yamaguchi und Menger zitiert wurde", sagt Christl im Gespräch mit SPIEGEL ONLINE. Aber Fehlanzeige Zincke tauchte weder in Yamaguchis noch in Mengers Papier auf. "Ueber Dinitrophenylpyridiniumchlorid und dessen Umwandlungsproducte", erschienen 1904 in "Justus Liebigs Annalen der Chemie" Zincke war Direktor des Chemischen Instituts in Marburg und hatte einen berühmten Schüler: Otto Hahn, Entdecker der Kernspaltung, promovierte 1901 bei ihm. "Liebigs Annalen" war eine der bedeutendsten Chemie-Fachzeitschriften, 1832 von dem berühmten Chemiker Justus Liebig gegründet. Ihm verdankt die Welt unter anderem die Begründung der organischen Chemie, jener Fachrichtung, in der Yamaguchi und Menger forschen. Außerdem entwickelte Liebig den Dünger und legte damit die Grundlage für die moderne Landwirtschaft. Bis 1997 erschienen die "Annalen", danach wurden sie mit einigen anderen Magazinen zum "European Journal of Organic Chemistry" vereint. Christl schrieb seinen Kollegen Yamaguchi und Menger, dass ihre vermeintliche Neuentdeckung altbekannt war. Noch schlimmer: Yamaguchi und Menger hätten die Reaktion falsch interpretiert. Nicht etwa ein 12-Annulen hatten sie laut Christl hergestellt, sondern nur ein 6-Annulen - also eine ringförmige Verbindung aus sechs statt zwölf Atomen. Zincke dagegen hatte trotz der beschränkten Möglichkeiten seiner Zeit den Reaktionsmechanismus sorgfältig und richtig aufgeklärt. Yamaguchi hatte offenbar schon vor Christl von anderer Seite einen Hinweis bekommen, dass er in seiner Arbeit unvollständig zitiert hatte. Dennoch beharrte der Japaner zunächst darauf, ein 12Annulen hergestellt zu haben. Vier Wochen später zog er allerdings seine Publikation zurück "wegen Ungewissheiten, welche Produkte in der beschriebenen Reaktion entstanden sind", heißt es auf der Homepage von "Organic Letters". Menger reagierte einsichtiger, erzählt Christl. "Eine Viertelstunde nach meiner E-Mail schrieb er mir, er werde das sofort überprüfen." Auch der Amerikaner wähnte sich zunächst im Recht: Er habe tatsächlich ein neues 12-Annulen hergestellt, "I hate to disappoint you", schrieb er Christl selbstsicher - und berief sich auf eine massenspektroskopische Analyse seines Produkts. Die habe nunmal eine höhere Molekularmasse ergeben als bei einem 6-Annulen. Doch Menger übersah ein wichtiges Detail. "Ich bin kein Spezialist in analytischen Methoden - aber ich wusste, dass sich bei der massenspektroskopischen Methode, die Menger verwendet hatte, Teilchen zusammenlagern", sagt Christl. Aus dem 6-Annulen war also während der massenspektroskopischen Analyse ein 12Annulen geworden. Menger kannte diesen Effekt der Analysemethode nicht. Der Zeitschrift "Nature" sagte er zerknirscht: "Ein Umstand, den wir zu der Zeit nicht beachtet hatten." Christl wirft Menger ungenaues Arbeiten vor: "Er hätte seinen Spezialisten für Massenspektroskopie fragen müssen." Menger musste eine Korrektur seiner Arbeit nachreichen - und Zincke kam durch den Einsatz Christls doch noch zu seiner Anerkennung. Dabei waren Yamaguchi und Menger eigentlich schon ganz dicht auf Zinckes Spur gewesen, sagt Christl amüsiert. Beide hätten in ihren Experimenten ein bestimmtes Salz zur Herstellung ihres vermeintlichen 12-Annulens verwendet, das sie Zincke-Salz nannten - nur waren sie offenbar völlig ahnungslos ob der Namensherkunft. "Junge Forscher verlassen den Computer gar nicht mehr" Eine Frage bleibt allerdings: Wie konnte eine 102 Jahre alte Entdeckung einfach übersehen werden - obwohl jeder Artikel von externen Gutachtern der Fachmagazine geprüft wurde? Natürlich - Zincke hatte seine Arbeit damals auf Deutsch veröffentlicht. Damals war Deutschland ein Zentrum der Forschung, Englisch noch nicht die Amtssprache der Wissenschaft. Aber war es wirklich nur die Sprachbarriere, die Yamaguchi und Menger den Zugang zu Zinckes Arbeit erschwert hatte? Christl sagt: "Diese Literatur ist so wichtig, die gibt es auch auf Englisch." Vielleicht nicht Zinckes gesamten Originalartikel aus den "Annalen", aber zumindest eine Beschreibung der Reaktion in den chemischen Handbüchern. Die stehen in den Bibliotheken. Und hier liege das Problem, denn in die Bücher schaue doch kaum noch jemand rein, klagt Christl: "Junge Wissenschaftler verlassen doch gar nicht mehr ihren Computer. Die wissen meist gar nicht, dass es solche Handbücher überhaupt gibt." Zeitmangel und Überlastung der Gutachter seien aber sicher auch schuld. "Die müssen teilweise eine Publikation pro Tag prüfen. Wie soll das gehen?" Es komme immer wieder vor, dass alte Entdeckungen unwissentlich neu entdeckt werden - weil Wissenschaftler einfach ihre Hausaufgaben nicht mehr machen, sagt Christl: "Das Lesen der Literatur ist für angesehene Wissenschaftler zum Luxus geworden, den sie sich kaum noch gönnen können." 2) Acene und Phene • Acene leiten sich vom Naphtalin durch lineare Anellierung von einem oder mehreren Benzolringen ab. • Phene leiten sich vom Naphtalin durch anguläre Anellierung von 7 8b einem oder mehreren Benzolringen ab. 8 6 1 2 3 Aromatischer Charakter und Stabilität nimmt ab 4 9a 9 8a 8 4a 7 2 6 3 10a 10 5 Tetracen, gelb Pentacen, orange Hexacen, blaugrün 1 8a 4 4a 9 5 10a 10 Tetraphen, farblos Pentaphen, gelb Hexaphen, orange • Anthracen und auch die höheren Acene reagieren überwiegend entsprechend der Clar´schen Regel. HH H2 / Pd / C HH Aber: HH 2H2 / Pd / C HH 4H2 / Pd / C H H H H HH HH HH HH H H H H H H H H O + CrO3 / H O Aber: OH OsO4 OH OH OH O O O O O O H H Trypticen • Analoges gilt für Phenanthren und höhere Phene. Br2 Br Br H2 / Pd / C HH H H CrO3 O O • Wichtige Tetracenderivate sind die Tetracycline und die Anthracycline. Beide Substanzklassen können aus StreptomycesKulturen isoliert werden und wirken antibiotisch bzw. cytostatisch. • Tetracycline hemmen die Protein-Biosynthese von Bakterien und werden bei Infektionen als Breitbandantibiotika eingesetzt. • Anthracycline wirken als Topoisomerasehemmer und werden als Cytostatika bei Krebserkrankungen eingesetzt. • Biosynthetisch machen die Streptomyceten solche Substanzen aus vollständig ungesättigten Tetracenderivaten, die aus aktivierten Malonsäurederivaten erzeugt werden. 3) Helicene • Helicene leiten sich vom Phenanthren ab durch anguläre Annellierung von Benzolringen. Helicene besitzen eine Helicale Struktur. HH H H H H H H Tetrahelicen Pentahelicen Hexahelicen • Helicene sind auf Grund ihrer Helix-Struktur chiral (P- und M-Helix). • Tetrahelicen und Pentahelicen lassen sich nicht in Enantiomere trennen. Sie racemisieren bei Raumtemperatur sehr schnell. • Hexahelicen kann in Enantiomere getrennt werden. • Synthese von Hexahelicen O Br H KOH-Schmelze Br NaOAc / Ac2O COOH • – CO2 COOH rac. Racematspaltung von Hexahelicen mit (-)-TAPA = Tetranitroflourenylidenaminooxypropionicacid O2N HNO3 H2SO4 O • – HBr NO2 O2N N O NO2 + H O2N COOH NO2 O2N O TAPA bildet Charge-Transfer-Komplexe mit elektronenreichen Aromaten NO2 N O COOH (-)-TAPA • (-)-TAPA bildet nur mit (+)-Hexahelicen einen Charge-TransferKomplex, der in Benzol/Ethanol in Lösung bleibt, während (-)Hexahelicen keinen Charge-Transfer-Komplex bildet und unlöslich ist. • Aus 2,27 g racemischem Hexahelicen konnte man auf diese Weise durch 8-maliges Umkristallisieren 11,9 mg (!) enantiomerenreines (-)Hexahelicen erhalten. (-)-Hexahelicen (-)-TAPA (+)-Hexahelicen • (-)-TAPA (+)-Hexahelicen + Benzol Ethanol • Schmelzpunkt: 264-267 °C • Spezifische optische Rotation [α]D24 = - 3640 (CHCl3) (-)-Hexahelicen Ÿ 1.4. Heteroaromaten 1.4.1. Elektronenmangel-Heteroaromaten • Pyryllium-Kationen und Derivate, Pyridin und Derivate sowie 6gliedrige Heterocyclen mit mehreren Heteroatomen zählen zu den Elektronenmagel-Aromaten, da die Heteroatome auf Grund der höheren Elektronegativität Ladungsdichte aus dem πElektronensystem abziehen. “ O “ “ .... O O “ “ N 7,4 ppm H H H H N H 7,0 ppm 8,5 ppm H H N N” N” + 0,1 136 ppm H H .... H H H + 0,05 H 124 ppm 149 ppm H N H + 0,15 – 0,5 139 pm H H H H N H 139 pm 134 pm • Kein idealer Bindungslängenausgleich mehr im Pyridin ⇒ HMOTheorie streng genommen nicht mehr anwendbar. • Weitere wichtige Elektronenmangel-Heterocyclen N N N Pyrazin N N N Pyrimidin Pyridazin N N N 1,3,5-Triazin N N N N Chinolin Isochinolin N N Pteridin • Pyridin entspricht von der Elektronenverteilung her dem Nitrobenzol (ähnliche Reaktivität) • Diazine verhalten sich ähnlich wie die entsprechenden Dinitrobenzole, Triazine wie die Trinitrobenzole usw. 1.4.2. Elektronenüberschuss-Heteroaromaten • Furan, Pyrrol und Thiophen sowie deren benzokondensierte Derivate als auch 5-gliedrige Heterocyclen mit mehreren Heteroatomen sind Elektronenüberschuss-Aromaten, da das freie Elektronenpaar des Heteroatoms mit den beiden Doppelbindungen zusammen das aromatische π-Elektronensextett. Dann kommen aber 6 Elektronen auf 5 Ringatome ⇒ Elektronenüberschuss (entspricht dem Cyclopentadienid-Anion) ” N H S ” S“ .... O“ O“ ” .... N“ H N“ H ” O ” ” .... S“ 143 pm 6,2 ppm; 107 ppm 137 pm 138 pm 6,7 ppm; 116 ppm N H 6,3 ppm; 110 ppm 136 pm O 136 pm 142 pm 7,4 ppm; 142 ppm 7,0 ppm; 126 ppm 137 pm 171 pm 142 pm 7,2 ppm; 124 ppm S • Kein idealer Bindungslängenausgleich mehr in den Elektronenüberschussheterocyclen ⇒ HMO-Theorie streng genommen nicht mehr anwendbar. • Weitere wichtige Elektronenmangel-Heterocyclen N N H Imidazol N N O S Oxazol Thiazol N N H Pyrazol O N Isoxazol S N Isothiazol 2. Reaktionen von Aromaten • Die wichtigsten klassischen Reaktionen von Aromaten sind elektrophile und nucleophile Substitutionen, wobei das aromatische System erhalten bleibt. • Durch Deprotonierung von substituierten Aromaten in ortho-Position zu bestimmten Substituenten (ortho-Lithiierung) können elektrophile aromatische Substitutionen leicht durchgeführt werden, die sonst schwierig zu bewerkstelligen sind. • Substitutionsreaktionen an Aromaten, die unter klassischen Bedingungen schwierig oder gar unmöglich sind, können übergangsmetallkatalysiert durchgeführt werden. • Moderne Methoden nutzen Aromaten als 6-gliedrigen cyclischen Baustein für nicht-aromatische Moleküle. Solche Reaktionen fasst man unter dem Begriff Dearomatisierungsreaktionen zusammen. 2.1. Elektrophile Aromatische Substitution 2.1.1. Allgemeiner Mechanismus • Das Elektrophil E+ nähert sich der π-Elektronenwolke des Aromaten und bildet dabei zunächst reversibel einen sogenannten π-Komplex. • π-Komplexe können spektroskopisch nachgewiesen werden, sind aber nur kurzlebig und i.d.R. nicht stabil. • Erst 2001 gelang es, π-Komplexe zu kristallisieren und durch Röngenstrukturanalyse zu untersuchen. • Bei der Ausbildung des π-Komplexes lagert sich das Elektrophil an die π-Elektronenwolke zwischen zwei C-Atome an. Bei Toluol bilden sich zwei π-Komplexe, bei denen die Brommoleküle bereits über der 1,2Bindung (Angriff in ortho-Position) oder über der 3,4-Bindung (Angriff in para-Position) sitzen. • Der π-Komplex geht anschließend in den σ-Komplex (WhelandKomplex) über. • Von einem σ-Komplex aus Brom und Hexamthylbenzol existiert ebenfalls eine Röntgenstruktur. • Im σ-Komplex wird die cyclische Konjugation der π-Elektronen unterbrochen. • Durch Abspalten von H+ geht der σ-Komplex in das Produkt über. • Auf Grund des Prinzips der mikroskopischen Reversibilität muss man annehmen, dass bei der H+-Abspaltung auch ein πKomplex durchlaufen wird. • Reaktionsprofil einer elektrophilen aromatischen Substitution (schematisch) E σ π1 π2 Ar + E+ Ar-E + H+ E+ E + H H H H H H H H H H H H H H H E H “H H H+ H H E H H H H H E H H H H+ 2.1.2. Reaktivität • Elektronenschiebende Substituenten am Aromaten erhöhen die Elektronendichte im aromatischen π-System und damit auch die Reaktivität gegenüber Elektrophilen („der Aromat wird nucleophiler als Benzol“) +I, +M O ” S ” O –I, +M OH OPh OR O R SH SR O NH2 NHR NR2 O +I R O ” indifferent H Ph H H NH R • Elektronenziehende Substituenten am Aromaten erniedrigen die Elektronendichte im aromatischen π-System und damit auch die Reaktivität gegenüber Elektrophilen („der Aromat wird weniger nucleophil als Benzol“) –I –I, –M + + SR2+ CF3 O O O R OH OR CN SO3H SO2R NR3 PR3 O H O NR2 NO2 –I, +M F Cl Br I • Nicht alle Elektrophile können mit allen Aromaten reagieren. • Elektrophile, die mit aktivierten und desaktivierten Aromaten reagieren: O H “ O N O H2SO4 O N“ O ” "Br+" Br Br "Cl+" Cl Cl SO3 O + HOSO2 HO S O Cl MXn MXn O H “ O N“ O ” H NO2+ – H2O Br Br MXn Br+ BrMXn– Cl Cl MXn Cl+ ClMXn– • Elektrophile, die mit aktivierten Aromaten und Benzol reagieren: R3C + R3C RCH2+ RCO Cl RCH2 Cl O + R MXn MXn MXn X O R R3C+ ClMXn– RCH2 R “ “ MXn R X RCOH2+ O R R2COH O R R O ” O MXn “ + H O H R H “ + H O H R R MXn MXn+1– X H + Cl + RCH2 ClMXn– • Elektrophile, die nur mit stark aktivierten Aromaten reagieren: “ N H R + NO “ Ar N N + R N H O N O Ar NH2 Ar NH2 H H+ NaNO2 HCl iAmONO NaOH R “ N H “ H O N O H “ Ar N N “ Ar N N NO+ – H2O 2.1.3. Regioselektivität bei Mehrfachsubstitutionen • Substituenten 1. Ordnung am Aromaten dirigieren Zweitsubstituenten in ortho- und para-Position. +I, +M O ” S ” O –I, +M OH OR OPh O R SH SR O NH2 NHR F Cl O +I R O ” NR2 Br NH R I • Substituenten 2. Ordnung am Aromaten dirigieren Zweitsubstituenten in meta-Position. –I –I, –M NR3+ + + PR3 SR2 CF3 O O O O O H R OH OR NR2 NO2 CN SO3H SO2R • Erklärung mit Hilfe des Hammond-Postulats: Frühe Übergangszustände sind eduktähnlich, späte Übergangszustände sind produktähnlich. • Bei elektrophilen aromatischen Substitutionen liegen überwiegend späte Übergangszustände vor, nur bei sehr reaktiven Elektrophilen treten frühe Übergangszustände auf. • Substituenten 1. Ordnung stabilisieren die Übergangszustände, die zu o- und p-Produkt führen stärker, als die, die zu m-Produkt führen. • Substituenten 2. Ordnung destabilisieren die Übergangszustände, die zum m-Produkt führen, weniger stark als die, die zu o- und p-Produkt führen. • E Reaktionsprofil einer SEAr mit Substituenten 1. Ordnung Benzol meta ortho para Ar + E+ Ar-E + H+ • Die Stabilität der Übergangszustände kann aus den mesomeren Grenzformen für die jeweiligen Übergangszustände abgeschätzt werden • Reaktionsprofil einer SEAr mit Substituenten 2. Ordnung E Benzol meta ortho para Ar + E+ Ar-E + H+ • Die Stabilität der Übergangszustände kann aus den mesomeren Grenzformen für die jeweiligen Übergangszustände abgeschätzt werden • Experimentelle Daten 2.2. • Nucleophile Aromatische Substitution Nucleophile aromatische Substitutionen können nach zwei verschiedenen Mechanismen ablaufen, a) Additions-EliminierungsMechanismus und b) Eliminierungs-Additions-Mechanismus (ArinMechanismus). 2.2.1. Additions-Eliminierungs-Mechanismus • Elektronenarme Aromaten reagieren nach dem AdditionsEliminierungs-Mechanismus. Der geschwindigkeitsbestimmende Schritt ist die Addition des Nucleophils. • Der Aromat muss eine geeignete Abgangsgruppe besitzen, meist ein Halogen, aber auch Methoxy-Gruppen, Nitro-Gruppen oder Sulfonylgruppen können als Abgangsgruppen fungieren. • Hydrid-Ionen sind extrem schlechte Abgangsgruppen und werden nur ganz selten abgespalten. • Als Reaktionspartner können die_ üblichen Nucleophile verwendet _ _ _ _ _ werden, OH , RO , ArO , RS , F , RNH2, RNH , C-Nucleophile sind eher selten. • Nucleophile aromatische Substitutionen nach dem AdditionsEliminierungs-Mechanismus verlaufen besonders leicht, wenn in ortho- oder para-Position zur Abgangsgruppe eine elektronenziehende Gruppe vorhanden ist. X X X Nu ” – Nu ” Nu Z Z Z Meisenheimer-Komplex Nu Nu ” X – X– Z • Z Eine Abgangsgruppe in m-Position zu einem elektronenziehenden Substituenten wird weniger leicht substituiert als eine Abgangsgruppe in o- und/oder p-Position. • Dirigierende Effekte X Nu X + Nu– X – X– Z Z X + Nu– X X – X– Nu Z Z X Nu + Nu– X Z – X– X Z 2.2.2. Eliminierungs-Additions-Mechanismus • Unter drastischen Bedingungen können auch elektronenreiche Aromaten, die eine Abgangsgruppe X besitzen, mit Nucleophilen umgesetzt werden. • Dabei wird zunächst die Abgangsgruppe als HX eliminiert, erst dann addiert das Nucleophil. • Als Zwischenverbindung tritt ein nicht isolierbares Arin auf. Nu X H Base HNu H H Nu + – HX R • R R R Die Regioselektivität ist in der Regel nur schwach ausgeprägt. • Dirigierende Effekte Z Z X Z Z Base Nu H NuH + H H Nu Hauptprodukt Nebenprodukt Z Z Base Z Z NuH + H Nu H X H Nu Hauptprodukt Nebenprodukt Z H NuH Base H Nu H + + + H Nu X viel Z Z Z Z Z Nu wenig Hauptprodukt Nebenprodukte H D D X D D Base H Nu NuH + H Nu H Hauptprodukt Nebenprodukt D D Base D D NuH + H H Nu X Nu H Hauptprodukt Nebenprodukt D D H + + + H Nu X H Nu H NuH Base viel wenig D D D D H Hauptprodukt Nu Nebenprodukte H • Bindungsverhältnisse in Arinen schwache Bindung = • Auf Grund der schwachen Bindung aus 2 sp2-Hybridorbitalen sind Arine extrem reaktiv und können nur durch Abfangreaktionen nachgewiesen werden. • Durch photochemische Abspaltung von CO2 aus Phthaloylperoxid kann Arin in einer Ar-Matrix erzeugt und spektroskopisch untersucht werden. O O O O H2O2 O O O hν – 2 CO2 • Herstellungsmöglichkeiten für Arine F Li F Cl oder Li – LiF nBuLi COOH iAmONO COO– NH2 NaOH N2+ – CO2 – N2 N Pb(OAc)4 N N N NH2 • N N – 2 N2 Wenn kein geeigneter Reaktionspartner vorhanden ist, dann dimerisieren Arine zu Biphenylenen 2 2.2.3. Radikal-Mechanismus • Aromatische Substitutionen lassen sich auch photochemisch bei tiefen Temperaturen (in flüssigem Ammoniak, Sdp. – 33 °C) durchführen. • Die Reaktionen unterscheiden sich von anderen nucleophilen aromatischen Substitutionen dadurch, dass Radikalanionen als Intermediate auftreten. • Weil die angreifenden Teilchen Nucleophile sind, spricht man von einem SRN1-Mechanismus. X X + e– – X– ” • – X– X X Nu + • ” • Nu + Nu– • ” • Es gibt keine dirigierenden Effekte. • Elektronenziehende oder elektronenschiebende Substituenten wirken sich nicht auf die Reaktionsgeschwindigkeit aus. • Der geschwindigkeitsbestimmende Schritt ist die Bildung des Radikalanions des Edukts, unterschiedliche Nucleophile zeigen alle praktisch die gleiche Reaktivität. • Radikalfänger bringen die Reaktion zum Stillstand. • Nucleophile, die nach einem SRN1-Mechanismus reagieren, sind Enolate von Ketonen, 1,3-Diketonen, Estern und Amiden, Phenolate, Thiophenolate, Phosphit-Anionen und Phosphidanionen. 2.3. Radikale Aromatische Substitution • Einige wenige Reaktionen von Aromaten sind radikalische Substitutionsreaktionen. • Aromaten zeigen nur mittelmäßige Reaktivität gegenüber Radikalen. • Von Bedeutung ist die Umsetzung von Aromaten mit Arylradikalen zu Biphenylen. • Arylradikale können aus Aryldiazoniumsalzen oder aus NNitrosoacetaniliden erzeugt werden. • Die Ausbeuten liegen selten über 50%. “ N N – – + OH N N O H + OH N N O – H2O ” Diazohydroxid “ N N 2 N N O N N – N2O Diazoniumoxid Diazoanhydrid • N N 1) AcCl / Py NH2 – AcO – O 2) NaNO2 / HCl O N N O O • • H • – H• • Weil keine dirigierenden Effekte auftreten, können als aromatische Reaktionspartner für Arylradikale nur symmetrisch substituierte Benzole eingesetzt werden, weil man sonst Mischungen aus verschiedenen Regioisomeren erhält. • Diese Reaktion heisst Gomberg-Bachmann-Reaktion. 2.4. Orto-Lithiierung von Aromaten • Der pKS-Wert von Benzol liegt bei ca. 43, der von nBuLi bei ca. 50. • Prinzipiell könnte Benzol also von nBuLi deprotoniert werden, allerdings ist die Reaktion extrem langsam. nBuLi Li H 3 h / 80 °C nur 5 % • Setzt man der Lösung TMEDA (Tetramethylethylendiamin) zu, dann bildet sich nach 3 h bei RT Ph-Li in 76% Ausbeute. nBuLi N N N Li H N 3 h / RT 76 % • Grund: das PhLi wird durch Komplexbildung mit TMEDA stabilisiert, ausserdem wird nBuLi durch denselben Prozess reaktiver gemacht. • Erkenntnis: Aromaten lassen sich nur gut deprotonieren, wenn das entstehende Phenylmetall (hier PhLi) durch Komplexierung stabilisiert wird. • Dies wird bei der ortho-Lithiierung dadurch ausgenutzt, dass sich ein stabilisierender Substituent bereits im Aromaten befindet. O nBuLi O Li H Et2O / T < 0 °C • Lithiierte (= deprotonierte) Aromaten sind sehr gute Nucleophile, d.h. sie können von Elektrophilen sehr leicht angegriffen werden (viel leichter als bei der elektrophilen aromatischen Substitution). • Mechanismus: nBuLi (oder sec. BuLi oder tertBuLi) komplexiert erst an den stabilisierenden Substituenten und wird dadurch so orientiert, dass eine Deprotonierung in ortho-Stellung zu diesem Substituent leicht möglich wird. –I-Effekte erleichtern die Deprotonierung. O O Li Li H • + H Im Laufe der Zeit hat man viele Substituenten entdeckt, die die orthoLithiierung begünstigen. O O O NR2 NR2 N O O S NR2 O NR2 O O Ortho-dirigierende Wirkung nimmt ab F NR2 O O O O • Wenn ein Aromat mehrere ortho-dirigierende Substituenten enthält, dann kann man mit der sehr starken Base nBuLi + KOtBu in orthoPosition zum elektronegativeren Substituenten deprotonieren. • Mit der schwächeren Base nBuLi deprotoniert man neben dem besseren Ligand für Lithium. Li Li nBuLi F nBuLi F OMe OMe F OMe KOtBu • Als Elektrophile sind alle denkbaren Elektrophile einsetzbar, z. B. O O R I R R R' H Br2 CO2 NBS O O Cl H NMe2 NMe2 • Es lassen sich auch mehrere ortho-Lithiierungen nacheinander ausführen, wenn der nach der ersten ortho-Lithiierung eingeführte Substituent selbst wieder ein Substituent ist, der die ortho-Lithiierung begünstigt. MeO O NEt2 O MeO O NEt2 O nBuLi + ClCONEt2 O MeO Li O MeO O MeO O nBuLi NEt2 NEt2 O MeI NEt2 O Li O Me O O NEt2 – LiCl NEt2 NEt2 • Um reversibel eine orto-Position zu blockieren, kann man eine TMSSchutzgruppe einführen. H NEt2 O H O H O NEt2 + TMSCl O nBuLi O H NEt2 O Li TMS – LiCl O NEt2 Li O O + ClCONEt2 nBuLi Et2N O Et2N O NEt2 O H O Et2N O TBAF O TMS TMS O NEt2 NEt2 O nBuLi Li O Et2N OH O NEt2 • Trick, um in ortho-Position zu einer Aldehyd-Gruppe ein Elektrophil einzuführen: in situ-Schutzgruppe O H N nBuLi N Li N Li H N O − 78 °C − 78 °C N N Li O nBuLi − 78 °C O 1) DMF N Li 2) H3O+ N H H O • Kann Benzylmethylether auch ortho-lithiiert werden? Nein! Benzylmethylether geht die 1,2-Wittig-Umlagerung ein!!! • R = H, Alkyl, Aryl, Alkenyl, Alkinyl, COOR, COOM, R‘ = Alkyl, Allyl, Benzyl, Aryl R O R' nBuLi ” R O R' R' (1) R O ” (2) ” R O• • R' • R' • R O ” • (1) beschreibt einen konzertierten Mechanismus, (2) einen Radikalmechanismus. Beide Mechanismen laufen nebeneinander ab, wobei (2) aber deutlich überwiegt. • Der Radikal-Mechanismus läuft in einem Lösungsmittelkäfig ab. • Wittig hat solche Umlagerungen lange vor der Wittig-Olefinierung beschrieben. • Ausserdem gibt es noch die 2,3-Wittig-Umlagerung. R' R' R' nBuLi R • O ” R O R O ” Dabei handelt es sich um eine Reaktion, die nach einem konzertierten Mechanismus abläuft (sigmatrope Umlagerung). 2.5. Übergangsmetallkatalysierte Reaktionen 2.5.1. Essentielle Grundlagen der Metallorganik • Die Metallorganische Chemie befasst sich mit Metallkomplexen, die neben anorganischen Liganden auch organische Liganden besitzen. allgemeine Formel [MXaLb] c • M = Metall (meistens ein Nebengruppenmetall) • X und L sind Liganden (mindestens ein organisches Molekül). • c ist die Ladung bei geladenen Komplexen. • In metallorganischen Komplexen liegt mindestens eine MetallKohlenstoff-Bindung vor. • Ganz allgemein: das Metall beeinflusst den organischen Liganden durch elektronische und sterische Effekte und verändert so die Reaktivität des komplexierten organischen Moleküls im Vergleich zum unkomplexierten Zustand. 1) Metall M • Die für die Organische Chemie wichtigsten metallorganischen Komplexe sind solche mit Fe, Co, Ni, Cu, Rh, Pd, Pt. • Hier in dieser Vorlesung: Pd • Wichtig: Elektronenkonfiguration der Metalle. Achtung! Die Elektronenkonfiguration der Metalle in Komplexen sind anders als die der elementaren Metalle! • Bei den elementaren Metallen liegt das n s-Orbital (n = Hauptquantenzahl) energetisch unter den (n-1) d-Orbitalen, wird also vor den d-Orbitalen besetzt. • Bei den Metallen in Komplexen ist das n s-Orbital energiereicher als die (n-1) d-Orbitale, wird also nicht besetzt. 2) Liganden X und L • Übliche Liganden: H, Alkylreste, Arylreste, CO, CN−, H2O, OH−, CH3-COO−, NH3, NR3, Halogene, PPh3, Alkene, Diene, R-CN, R-NC, usw. • Nach Crabtree und Greene werden Liganden in zwei Klassen unterteilt: X und L • Liganden des L-Typs sind neutrale 2-Elektronen-Donoren, wie z. B. CO, PPh3, Ethen, H2O usw. • Liganden des X-Typs sind einfach negativ geladene 2-ElektronenDonoren, wie z. B. CN−, Cl−, CH3− usw. Als Neutralteilchen sind XLiganden 1-Elektron-Donoren. • Liganden können auch gemischte Typen sein, wie z. B. das Allylanion CH2=CH−CH2−. Insgesamt liegt ein 4-Elektronen-Donor vor, der als LX zu klassifizieren ist. • Wenn man alle Liganden eines Komplexes entsprechend klassifiziert hat, dann kann man relativ einfach verschiedene Kenngrößen berechnen, die den Komplex charakterisieren. 3) Charakteristische Größen für Komplexe • Zahl der Elektronen in der Valenzschale des Metalls in einem Komplex [MXaLb]c, wobei M in der Gruppe N des Periodensystems steht: VE = N + a + 2b – c • Koordinationszahl: CN = a + b • Oxidationsstufe des Metalls: OS = a + c • Zahl der d-Elektronen des Metalls: dn = d(N – a – c) • Beispiele: RhCl(PPh3)3 = MXL3 N=9 Pd(PPh3)2Br(C6H5) = ML2X2 N = 10 VE = 9 + 1 + 2·3 - 0 = 16 VE = 10 + 2 + 2·2 - 0 = 16 CN = 1 + 3 = 4 CN = 2 + 2 = 4 OS = 1 + 0 = 1 OS = 2 + 0 = 2 dn = 9 - 1 = 8 dn = 10 - 2 = 8 4) 18-Elektronen-Regel • Hauptgruppenverbindungen gehorchen (mehr oder weniger) der 8Elektronen-Regel (Oktett-Regel). • Analog zur Oktett-Regel existiert für Übergangsmetallkomplexe die 18-Elektronen-Regel, die besagt, dass das Metall im Komplex bestrebt ist, in seinen Verbindungen die Elektronenkonfiguration des nächsten Edelgases einzunehmen. Diese umfasst 10 d-Elektronen, 2 s-Elektronen und 6 p-Elektronen, also zusammen 18 Valenzelektronen. • Es gibt jedoch (wie bei der Oktett-Regel auch) zahlreiche Ausnahmen der 18-Elektronen-Regel. • Beispiele: Cr(CO)6 = MX0L6 N = 6 Rh(H)(H)(PPh3)2(CO)Cl = MX3L3 N = 9 VE = 6 + 2·6 - 0 = 18 VE = 9 + 3 + 2·3 - 0 = 18 OS = 0 + 0 = 0 OS = 3 + 0 = 3 5) Strukturen von Metallkomplexen • Die Struktur von Metallkomplexen hängt von der Koordinationszahl, von der Art des Liganden und von der Zahl der d-Elektronen ab. • Man kann die Struktur von Metallkomplexen mit Modellen zu Bindungsverhältnissen erklären (vgl. nächstes Kapitel), aber nicht ohne weiteres vorhersagen. • Allgemein beobachtet man folgende Strukturen: Koordinationszahl 2 3 Struktur L M L L L M L 4 L L M L L 5 L L M L 6 L L L M L L L M L L L M L L L L L L L M L L L L M L L 6) Bindungsverhältnisse in ÜM-Komplexen • In der Organischen Chemie verwendet man zur Beschreibung der Struktur von organischen Molekülen Formeln, die auf der ValenceBond-Theorie von Pauling basieren. Prinzipielle Vorgehensweise: 1) Hybridisierung der Valenzorbitale des Zentralatoms zu geeignet ausgerichteten Hybridorbitalen. 2) Überlappung dieser Hybridorbitale mit geeigneten Orbitalen der Substituenten zu lokalisierten Bindungsorbitalen. Vorteile: 1) Anschaulich. 2) Die so erhaltenen Formeln lassen sich durch Lewis-Formeln darstellen. 3) In Fällen, in denen 2) nicht möglich ist, wird der Begriff „Mesomerie“ eingeführt. 4) Reaktionsmechanismen lassen sich durch „Elektronenschieben“ verdeutlichen. • Nachteil: Aus heutiger Sicht zu einfach und nicht genau genug, wenn man Molekülstrukturen und Moleküleigenschaften berechnen will. • Im Vergleich dazu sind die Bindungsverhältnisse in Übergangsmetallkomplexen extrem (!) kompliziert. • Es gibt prinzipiell drei verschiedene Möglichkeiten, die Bindungsverhältnisse in ÜM-Komplexen zu beschreiben: VB-Theorie, Ligandenfeldtheorie und MO-Theorie. VB-Theorie Ligandenfeldtheorie MO-Theorie Struktur Struktur Struktur Hybridisierung Aufspaltung der Aufspaltung der des Metalls d-Orbitale des Metalls d-Orbitale des Metalls Überlappung Hinzufügen von Überlappung mit mit Ligandenorbitalen Ligandenorbitalen Ligandenorbitalen geeigneter Symmetrie lokalisierte lokalisierte Bindungsorbitale Bindungsorbitale delokalisierte (Summe aus Metall- Bindungsorbitale und Ligandenorbitalen) (erstrecken sich über mehrere Atome) • Dilemma: egal, durch welche Theorie man die Bindungsverhältnisse beschreibt, man hat immer Probleme, die Verhältnisse mit LewisFormeln zu veranschaulichen. • Ausweg: man beschränkt sich auf die Veranschaulichung einzelner Bindungen und betrachtet in den seltensten Fällen den gesamten Komplex. • Beispiel: Cr(CO)6 oktaedrisch, 18 Valenzelektronen, Koordinationszahl 6, Oxidationsstufe 0, d6-Konfiguration. CO OC OC Cr CO C O C O CO CO übliche Darstellung (die Details muss man sich denken) irgend ein leeres d-Orbital von Cr nichtbindendes Elektronenpaar von CO M-C-σ-Donor-Bindung irgend ein volles d-Orbital von Cr leeres antibinden- des π-MO von CO M-C-π-Akzeptor-Bindung • Beispiel: [Pt(CH2=CH2)Cl3]− quadratisch, [MX3L]−-Typ, N = 10, 16 Valenzelektronen, Koordinationszahl 4, Oxidationsstufe +2, d8Konfiguration. Cl Cl CH2 Pt − CH2 Cl übliche Darstellung (die Details muss man sich denken) irgend ein leeres d-Orbital von Pt CH2 CH2 CH2 CH2 bindendes π-MO von Ethylen M-C-σ-Donor-Bindung irgend ein volles leeres antibindendes d-Orbital von Pt π-MO von Ethylen M-C-π-Akzeptor-Bindung • Fazit: in der Metallorganischen Chemie bedeutet ein Bindungsstrich oft viel mehr als nur eine Einfachbindung. • Dadurch wird das bekannte (und bewährte!) Elektronenschieben der Organischen Chemie nicht unbedingt auf die Metallorganik übertragbar. • Auf Grund der möglichen Bindungsverhältnisse kann man die Liganden genauer klassifizieren. • σ-Donor-Liganden: NR3, OR2, σ-Alkyl, σ-Alkenyl, σ-Aryl, H; erhöhen die Elektronendichte am Metall. • σ-Donor-π-Akzeptor-Liganden: CO, CN−, PPh3, Alkene, Alkine, Aromaten; erniedrigen die Elektronendichte am Metall. • σ-Donor-π-Donor-Liganden: OH− , OR− , F−, Cl−, RCOO− ; erhöhen die Elektronendichte am Metall. • Ob ein Ligand im konkreten Fall als σ-Donor, als π-Donor oder/und als πAkzeptor fungiert, hängt auch vom Metall und dessen Oxidationsstufe ab. • Durch die Koordination eines organischen Liganden an ein Metall wird nicht nur die Elektronendichte im Metall verändert, sondern auch die Elektronendichte im Ligand selbst. • Dadurch kann ein komplexiertes organisches Molekül ganz andere Reaktionen eingehen als das nicht-gebundene Molekül. Dies wird bei Übergangsmetall-katalysierten Reaktionen ausgenutzt. 2.5.2. Reaktionsmechanismen in der Metallorganik 1) Ligandenaustauschreaktionen • Formal wird ein Ligand durch einen anderen ersetzt, z. B. Br− durch OH− Ph3P Br Ph3P + NaOH Pd OH Pd Ph3P – NaBr Ph3P • Ligandenaustauschprozesse können dissoziativ oder assoziativ ablaufen. • Koordinativ gesättigte 18-Elektronenkomplexe reagieren (überwiegend) nach dem dissoziativen Mechanismus: CO OC OC Cr CO CO CO CO 130 °C – CO OC OC Cr CO CO PPh3 CO OC OC Cr CO PPh3 CO • Koordinativ ungesättigte 16-Elektronenkomplexe können nach dem assoziativen Mechanismus reagieren: + N Et3P Cl • Pt N Cl Et3P PEt3 Cl – Cl – Et3P Cl PEt3 Pt N PEt3 Es sind aber auch Fälle bekannt, in denen 16- Elektronenkomplexe nach einem dissoziativen Mechanismus reagieren. Br Ph3P Ph3P Pd Ph3P • Pt Cl – Br – Pd Ph3P + OH– OH Ph3P Pd Ph3P Bisher gibt es noch keine überzeugenden Argumente für einen assoziativen Mechanismus bei quadratisch-planaren Pd(II)Komplexen. 2) Oxidative Addition und Reduktive Eliminierung • Bei der Oxidativen Addition wird eine C-X-Einfachbindung durch einen koordinativ ungesättigten Metallkomplex (oder ein Metall) in niedriger Oxidationsstufe gespalten und beide Komponenten werden koordiniert. Dabei erhöht sich die Oxidationsstufe des Metalls um 2 Einheiten. • Auch die Oxidative Addition kann nach mehreren Mechanismen ablaufen: konzertiert, SN2-artig und nach einem Radikalmechanismus. • Nach einem konzertierten Mechanismus werden unpolare Verbindungen, wie H2, wenig polare C-H-Bindungen und Csp2−X-Bindungen oxidativ addiert. X = Abgangsgruppe Cl, Br, I, OTf, OTs usw. # Br Ph3P R Ph3P Pd Pd PPh3 Br Ph3P Br Pd Ph3P R Ph3P R • Bei einem konzertierten Mechanismus sind die addierten Liganden zunächst immer cis-ständig angeordnet. Anschließende (schnelle) Isomerisierungen sind möglich, müssen aber nicht stattfinden. • Csp3−X-Bindungen werden nach einem SN2-Mechanismus und COXBindungen nach einem SN2t-Mechanismus oxidativ addiert. Dabei fungiert das Metall als Nucleophil. Ph3P OC Ir CH3 Cl Cl PPh3 CH3 Ph3P OC Ir Cl + Cl CH3 ” Ph3P OC PPh3 Ir Cl PPh3 Cl • Die Reduktive Eliminierung ist die Umkehrung der Oxidativen Addition. Dabei müssen die zu eliminierenden Liganden immer cis-ständig sein. • Entscheidend für den Nutzen in der Organischen Synthese: bei der Reduktiven Eliminierung werden andere Liganden entfernt als vorher oxidativ addiert wurden. R2 Ph3P Ph3P Pd 2 + Pd Ph3P PPh3 R1 1 R R 3) Transmetallierung • Bei der Transmetallierung wird ein ÜM-Komplex mit einer Hauptgruppenmetallverbindung (B-Verbindung, Al-Verbindungen, Si-Verbindung, SnVerbindung, Grignard-Verbindung u.ä.) oder mit einer anderen ÜMVerbindung (Cu-organische Verbindung oder Zn-organische Verbindung, Zr-organische Verbindungen u.a.) umgesetzt. • Dabei überträgt die zweite Verbindung ihren organischen Substituenten auf den ÜM-Komplex. Aus der Sicht des übertragenen Substituenten findet eine Transmetallierung („Ummetallierung“) statt. • Die Reaktion funktioniert gut, wenn die Elektronegativität des ÜM größer ist als die des Hauptgruppenmetalls oder des zweiten ÜM. • Es liegt sehr wahrscheinlich ein konzertierter Mechanismus mit einem 4gliedrigen Übergangszustand vor. ” Ph3P OH Pd OH B OH OH Ph3P OH Pd Ph3P Ph3P R1 R1 OH HO B” OH Ph3P Pd – B(OH)4 – Ph3P R1 4) Nucleophile Addition an Liganden • An ÜM koordinierte Liganden verhalten sich chemisch anders als ungebundene Liganden. • Donor-Liganden werden in der Regel durch Koordination an ein ÜM positiver als in freier Form. Dadurch können Nucleophile addiert werden. • π-gebundene Liganden gehen dabei meist in σ-gebundene Liganden über. Ph3P OH Ph3P Pd Ph3P OC Cr CO H + Ph3P OH Pd OC OC Cr CO “ C CO ” O OH Ph3P R CO CO O –H Ph3P “ R CO H Pd CO OC OH R CH3 Li CO OC OC Cr CO O C CO ” CH3 2.6. • Knüpfung von C-C-Bindungen Wichtigste Reaktionen in der modernen synthetischen Chemie: Knüpfung von C-C-Bindungen zum Aufbau von Kohlenstoffgerüsten. 2.6.1. Synthese von aromatischen Carbonsäuren und Derivaten 1) durch Kolbe-Schmidt-Reaktion • Reaktion von Natriumphenolat mit CO2 zu Salicylsäure (als Ausgangsmaterial für Aspirin) ” OH NaOH “ Na O O O ” “ Na O C O ” “ Na H2O / H+ HO O” “ Na OH OH O O COOH • Reaktion von Kaliumphenolat mit CO2 zu p-Hydroxybenzoesäure (als Ausgangsmaterial für Parabene) ” OH KOH O K“ O O O ” OH K C O “ O OH H O ” K“ H2O / H+ O • O COOH ” “ K Grund für die unterschiedlichen Reaktionsverläufe: Komplexierungsverhalten von Na+ und K+. Na ” O “ ” O C O O K“ O C O 2) aus Aryl-Grignard-Verbindungen und CO2 • Arylhalogenide lassen sich mit Mg in Grignard-Verbindungen überführen, die dann mit CO2 zu den entsprechenden Carbonsäuresalzen reagieren. Wässerig-saure Aufarbeitung liefert Benzoesäurederivate. Mg O O C O H+ / H2O O Mg–Br Br OH OMgBr • Der Arylring kann funktionelle Gruppen enthalten, die mit GrignardReagenzien bei RT oder höherer Temperatur kompatibel sind. • Man kann Arylgrignard-Reagenzien auch durch Umgrignardisierung mit Isopropylmagnesiumbromid bei -40 °C herstellen. Dann können Arylhalogenide auch Estergruppen, Nitrilgruppen und Nitrogruppen enthalten (!). Mg FG Br Br FG 1) O C O Mg Br THF / – 40 °C + 2) H / H2O FG O OH • Mit Chlorameisensäureestern kommt man direkt zu Benzoesäureestern. O FG 1) Mg Br Cl OR FG 2) H+ / H2O O OR 3) durch Ortholithiierung und Reaktion mit CO2 • Durch ortho-Lithiierung erzeugte Aryl-Lithium-Verbindungen können ebenfalls mit CO2 in substituierte Benzoesäuren und mit Chlorameisensäureestern in Benzoesäureester überführt werden. O O tBuLi O THF / -90 °C Li O O O tBuLi O THF / -90 °C Li O 1) CO2 + O O 2) H / H2O COOH 1) Cl-CO-OR + 2) H / H2O O O COOR 4) aus Arylhalogeniden und CO • Häufig eingesetzter Katalysator: Pd(PPh3)4 ML4 PPh3 Ph3P • Pd PPh 3 PPh3 N = 10 VE = N + 2·4 = 18 OS = 0 CN = 4 d =d n 10 )=d Pd(PPh3)4 geht durch Verlust von 2 PPh3-Liganden in die katalytisch aktive Spezies über. PPh3 Ph3P Pd PPh3 PPh3 PPh3 • (N-0-0 Pd – 2 PPh3 PPh3 Arylhalogenide lassen sich sehr leicht an Übergangsmetallkomplexe oxidativ addieren. PPh3 Pd PPh3 Ph3P IsomeriPd Br Ph3P Br sierung Ph3P Pd Br PPh3 • Nun folgt ein Ligandenaustausch mit CO Ph3P Pd Br • PPh3 – PPh3 “ O Ph3P Pd Pd Br Br CO Der Arylring greift nun intramolekular das CO nucleophil an („COInsertion“. Ph3P Ph3P Pd Br + PPh3 Pd C O • ” C Ph3P Br C O PPh3 Ph3P Pd Br C O Durch Reaktion mit Wasser bildet sich die Carbonsäure und ein Hydridokomplex. Mit Alkoholen als Nucleophil erhält man Benzoesäureester und mit primären oder secundären Aminen erhält man Carbonsäureamide. PPh3 Ph3P H Pd Br • C O H O PPh3 Ph3P Pd Br + COOH H Eine von aussen zugesetzte Base (K2CO3 oder NEt3) spaltet HBr ab und regeneriert die katalytisch aktive Spezies. PPh3 Ph3P Pd Br • PPh3 H “ Pd O H Br C O ” Ph3P NEt3 Ph3P Pd PPh3 H Üblicherweise werden solche Reaktionssequenzen in einem Katalysecyclus dargestellt (wobei man oft Details wie z.B. Isomerisierungen einfach weglässt). • Katalysecyclus PPh3 Ph3P Pd PPh3 PPh3 – 2 PPh3 HBr PPh3 Br Pd PPh3 Ph3P PPh3 Ph3P Pd Pd Br Ph3P H Br COOH PPh3 Ph3P Ph3P Pd Br Pd C O Br PPh3 Ph3P CO Pd Br CO 5) aus aktivierten Aromaten und Trichloracetonitril • Trichloracetonitril ist in der Lage, als Elektrophil aktivierte (elektronenreiche) Aromaten anzugreifen. • Mechanismus: elektrophile aromatische Substitution. EDG Cl3C C N H+ EDG “ Cl3C C N H – H+ EDG C N – CHCl3 N H CCl3 6) durch Gattermann-Amid-Synthese • Mit Carbamoylchlorid und AlCl3 läßt sich in Aromaten eine Amidgruppe einführen. • Man benötigt äquimolare Mengen an AlCl3. • Mechanismus: elektrophile aromatische Substitution. O H2N AlCl3 Cl H+ / H2O “ H2N O AlCl3– Cl O AlCl3– Cl NH2 O NH2 • Aromatische Amide kann man ebenfalls aus Isocyanaten in Gegenwart von H+ erhalten. 2.6.2. Synthese von Arylketonen und Arylaldehyden durch C-C-Knüpfung 1) durch Friedel-Crafts-Acylierung • Umsetzung von Benzol und aktivierten Aromaten mit Carbonsäuren und Derivaten (Säurechloride, Anhydride) in Gegenwart von Lewis-Säuren oder starken Brönstedt-Säuren zu aromatischen Ketonen. • Mechanismus: Elektrophile aromatische Substitution SEAr • Das Produkt bindet 1 Äquivalent Lewis-Säure, daher benötigt man >1 Äquivalent Lewis-Säure als „Katalysator“. Ausnahme: Sc(OTf)3 als Lewis-Säure! • Angreifendes Elektrophil ist der Komplex aus Säurederivat und LewisSäure oder das freie Acyliumion (hängt von der Stabilität des Acyliumions ab). • Wenn sich durch CO-Abspaltung aus freien Acyliumionen stabile Carbeniumionen bilden können, dann beobachtet man Friedel-CraftsAlkylierungen als Nebenreaktionen oder auch als Hauptreaktionen (hängt auch von der Stabilität des Acyliumions ab). • Beispiel: Haworth-Naphthalin-Synthese O O O O AlCl3 1) Na / EtOH KOH HOOC 200 °C HOOC O + S / 200 °C 2) H+ • H+ N2H4 – H2S Fries-Umlagerung O OH R-COCl O R OH O AlCl3 OH R AlCl3 R O 2) durch Vilsmeier-Formylierung • Es gibt keine Friedel-Crafts-Formylierung weil Ameisensäurechlorid nicht stabil ist. H-CO-Cl zerfällt in CO und HCl !!! Das Anhydrid der Ameisensäure H-CO-O-CO-H ist auch nicht stabil ! Gemischte Anhydride aus Ameisensäure und Essigsäure oder Trifluoressigsäure führen zum acetylierten bzw. trifluoracetylierten Aromaten!!! • Deshalb die Entwicklung der Vilsmeier-Formylierung = Übertragung der Formyl-Gruppe aus Dimethylformamid auf den Aromaten. • Benzol und aktivierte Aromaten reagieren mit DMF/POCl3 zu den entsprechenden aromatischen Aldehyden. Cl • Das reaktive Teilchen ist das Vilsmeier-Salz • Die Vilsmeyer-Reaktion funktioniert auch gut bei elektronenreichen Heterocyclen wie Pyrrol, Thiophen und Furan und deren benzokondensierten Derivaten sowie Imidazol. • Alternativ zu DMF wird oft auch N-Methyl-Formanilid verwendet. “ N H • Mechanismus O O ” O H H O P Cl Cl Cl O O P Cl Cl Cl H “N N “ N ” O P “N Cl H Cl Cl ” O O P N Cl Cl H Cl Cl – PO2Cl2 H • – Cl– N H H N Cl “ N – + H2O / H N “ Cl “ H – H+ H O Beispiele: O H N H 2) TBAF POCl3 / DMF TIPS-Cl 1) POCl3 / DMF N Si DIPEA N H O N H H 3) durch Gattermann-Reaktion • Es gibt verschiedene Varianten der Gattermann-Reaktion • Gattermann-Koch-Reaktion: Einleiten von wasserfreiem HCl und CO zu einem Gemisch aus aktivierten Aromaten und AlCl3 und CuCl Cl “ “ O C Cu2” H Cl HCl CuCl ” “ C O HCl AlCl3 “ ” H C O AlCl4 • Das Reaktionsgemisch verhält sich so, als ob Formylchlorid eingesetzt worden wäre. • Gattermann-Reaktion: Einleiten von Wasserfreier Blausäure und wasserfreie HCl zu einem Gemisch aus aktiviertem Aromaten und Lewis-Säure. H C N • HCl “ H C N H ” Cl Gattermann-Adams-Reaktion: Statt gasförmiger Blausäure wird Zinkcyanid verwendet (besseres Handling). Zn(CN)2 HCl – ZnCl2 H C N HCl “ H C N H ” Cl 4) durch Reimer-Tiemann-Reaktion • Bei der Reimer-Tiemann-Reaktion werden Phenol oder Phenolderivate mit NaOH/CHCl3 zu Salicylaldehyd oder Derivaten davon umgesetzt. • Es entsteht überwiegend ortho-Produkt • Mechanismus: ” Cl Cl Cl ” O H H – H2O ” O Cl Cl H Cl ” Cl Cl Cl – – Cl ” ” O H – Cl– O O O Cl O ” H Cl H ” O H Cl ” Cl O O H OH O H + H2O / H – – Cl – H2O • Ausbeuten klassisch: o-Hydroxybenzaldehyd: 35% p-Hydroxybenzaldehyd: 12% • Verbesserung der Ausbeuten und verkürzen der Reaktionszeit durch Microwellenheizung! H • Mit Pyrrol bildet sich zu 31% der Pyrrol-2-aldehyd und zu 13% 3-Chlorpyridin. CHCl3 / NaOH Cl O N H N H + N H 31% • 13% Mechanismus der Nebenreaktion (wird oft auch als „anormale“ ReimerTiemann-Reaktion bezeichnet): Cl N H Cl Cl Cl N H ” Cl O H – Cl– – H2O N 5) durch Houben-Hösch-Reaktion • Bei der Houben-Hösch-Reaktion werden Phenol oder Phenolderivate mit Nitrilen in Gegenwart von wasserfreier HCl zu aromatischen Ketonen umgesetzt. • Es entsteht überwiegend para-Produkt • Mechanismus: R HCl R C N R H “ N H – Cl– “ R C N H –H R H 2 O / H+ O + N H + H+ 6) durch Ortho-Lithiierung • Die durch ortho-Lithiierung erhaltene Lithiumorganische Verbindung kann mit Dimethylformamid formyliert werden. O O tBuLi O THF / -90 °C Li O O 1) DMF O + 2) H / H2O H O • Als Formylierungsreagenz kann auch ein gemischtes Anhydrid aus Ameisensäure und Essigsäure verwendet werden. O O O tBuLi O THF / -90 °C Li O 1) H O O O O + 2) H / H2O H O • Um bessere Ausbeuten zu erhalten, setzt man der Ar-Li-Verbindung gelegentlich MgBr2 zu (Transmetallierung!). 7) durch übergangsmetallkatalysierte Carbonylierung • Analog zur übergangsmetallkatalysierten Carboxylierung können auch Carbonylierungen (Formylierungen) ausgeführt werden. PPh3 Ph3P Pd PPh3 Pd PPh3 – 2 PPh3 PPh3 PPh3 Ph3P IsomeriPd Pd Br Ph3P PPh3 Ph3P Br Pd Br PPh3 Ph3P Pd sierung ” C Ph3P – PPh3 PPh3 “ O Br Ph3P Pd Br PPh3 Pd Br CO • Unterschied zur Carboxylierung: der entstandene Acyl-Komplex wird durch Wasserstoff zum Aldehyd und zum Hydrido-Komplex reduziert. • Wie diese Reduktion mechanistisch funktioniert, ist ungeklärt. • Gezeigt ist eine hypothetische Möglichkeit. Ph3P H Ph3P Pd Br Br O H Ph3P Pd Br H Pd C PPh3 PPh3 Ph3P Pd H Pd C + Br O C Br + H H H H Ph3P NEt3 Ph3P Pd PPh3 O • PPh3 Katalysecyclus Ph3P Pd PPh3 PPh3 – 2 PPh3 HBr PPh3 Br Pd PPh3 H O Ph3P PPh3 Ph3P Pd Pd Br Ph3P H Br H2 PPh3 Ph3P Ph3P Pd Br Pd C O Br Ph3P CO Pd Br PPh3 PPh3 CO PPh3 2.6.3. Synthese von Benzylalkoholderivaten 1) durch Hydroxyalkylierung • Man setzt Benzol oder aktivierte Aromaten mit Aldehyden oder Ketonen in Gegenwart von katalytischen Mengen Säure um und erhät Benzylakoholderivate. + H O R • O R' H R “ R' R –H OH + R' Beispiel: Cl O Cl3C Cl H + cat. H Cl OH H CCl3 H Cl cat. H + Cl CCl3 DDT 2) durch Blanc-Reaktion • Man setzt Benzol oder aktivierte Aromaten mit Formaldehyd in Gegenwart von HCl um, dann wird der gebildete Benzylalkohol anschließend noch in einer SN1-Reaktion in das Chlorid überführt. H O H • H + O H H “ H H –H + OH H HCl H Cl H Funktioniert auch mit anderen Aldehyden, wird aber selten angewendet. 3) durch Ortho-Lithiierung • Die durch ortho-Lithiierung erhaltene Lithiumorganische Verbindung kann mit Aldehyden und Ketonen in Benzylalkoholderivate überführt werden. O O O tBuLi O THF / -90 °C Li O 1) R O R' O + 2) H / H2O HO R R' • Manchmal findet auch vor der Umsetzung mit Aldehyden oder Ketonen eine Ummetallierung mit MgBr2, mit ZnBr2 oder mit CeCl3 zu den entsprechenden Reagenzien statt. • Durch Ummetallierung erhaltene Zn- und Ce-metallorganische Verbindungen tolerieren viele funktionelle Gruppen im Rest R. • Wichtig: Zn-Organische Reagenzien reagieren nur noch mit Aldehyden. 2.6.4. Synthese von Biarylen, Arylalkenen und Arylalkinen durch C-C-Knüpfung • Die Übergangsmetall-katalysierte Verknüpfung zweier sp2- oder sphybridisierter C-Atome nennt man Kreuzkupplungsreaktionen. • Als Übergangsmetallkatalysatoren werden überwiegend Pd(0)Komplexe eingesetzt. • Alternativ können auch Pd(II)-Verbindungen eingesetzt werden, die in situ zu Pd(0) reduziert werden. • Kombinationsmöglichkeiten: X R R Y R R R' X R' R' R Y Noch mehr Kombinationen unter Einbeziehung von sphybridisierten C-Atomen R' R R' R' 1) durch Heck-Reaktion • Die Heck-Reaktion ist eine der wichtigsten Pd-katalysierten Kreuzkupplungsreaktionen. • Ein Ausgangsmaterial enthält eine C(sp2)-X-Bindung, wobei X = Cl, Br, I oder OTf ist. Das zweite Ausgangsmaterial hat üblicherweise die Struktur CH2=CH-Z, wobei Z eine elektronenziehende Gruppe oder ein Aromat ist. Auch höher substituierte Alkene wurden erfolgreich umgesetzt. • Bruttoreaktionen: R X Pd(0)-Kat. / PAr3 + Z R X Z • Z LM / Base Pd(0)-Kat. / PAr3 + R LM / Base Z ist meist Estergruppe, Keton, Nitril, Aromat. R Z • Für die Heck-Reaktion wurden viele unterschiedliche Reaktionsbedingungen beschrieben, z.B. a) Pd(OAc)2 / PPh3 / NEt3 / DMF (Heck) b) Pd(OAc)2 / nBu4NCl / NaHCO3 / DMF (Jeffrey) c) Pd(OAc)2 / (oTol)3P / nBu4NBr / DMA (Beller) • Der Mechanismus ist ziemlich komplex und noch nicht bis in alle Einzelheiten geklärt. Möglicherweise liegen bei unterschiedlichen Reaktionsbedingungen auch unterschiedliche Mechanismen vor. • Oxidative Addition: PPh3 Ph3P Pd PPh3 PPh3 PPh3 PPh3 Pd PPh3 Pd – 2 PPh3 PPh3 Ph3P IsomeriPd Br Ph3P Br sierung Ph3P Pd Br PPh3 • Coordinierung vom Alken: Ph3P Ph3P Pd Br – PPh3 PPh3 Z Ph3P Pd Ph3P Pd Br Pd Br Br Z • Z Nucleophiler Angriff vom Arylring auf das coordinierte Alken: dies ist die eigentliche Kreuzkupplung der beiden Liganden. Ph3P Ph3P Pd Pd HH Br Br Z H Z Dadurch wird ein π-gebundenes Alken zum σ-gebundenen Alken, wobei eine neue C-C-Bindung zwischen den beiden coordinierten Liganden geknüpft wurde. • Der neu entstandene Ligand kann verschiedene Konformationen annehmen und durch β-Eliminierung weiter reagieren. Ph3P Pd Ph3P HH H Pd Ph3P H Pd Br Br H Br Z H Z H H Z H β-Eliminierung Ph3P H Pd Ph3P H Br Pd H Z Nebenprodukt H Ph3P Pd H Br H Z H Br H Z Ph3P H Hauptprodukt H Pd Br H Z H trans-Alken • Anschließend dissoziiert das neu entstandene Alken ab, die freiwerdende Koordinationsstelle wird durch ein PPh3 besetzt. Durch die eingesetzte Base wird schließlich HX abgespalten. Ph3P H Pd Br H Ph3P Br H H Pd Z H + H Br Z H Pd Ph3P PPh3 PPh3 Ph3P Pd Br H NEt3 – HBr Ph3P Pd PPh3 • PPh3 Katalysecyclus Ph3P Pd PPh3 PPh3 – 2 PPh3 HBr PPh3 Br Pd PPh3 Ph3P PPh3 Ph3P Pd Pd Br Ph3P H Br Z PPh3 Ph3P Ph3P Pd Pd Br Br Z PPh3 Ph3P Pd Z Br Z PPh3 • Beispiel 1) Teilschritt aus der Synthese von FR-900482 OBu H I MeOOC N OBu H Pd(PPh3)4 / Et3N H OMOM O CH3CN / 90 °C / 18 h N OH Pd MeOOC N N O H H OMOM COOMe N COOMe N H H H Pd MeOOC N Pd OBu H H O OMOM MeOOC N N O FR-900482 OBu H O N OCONH2 OH OBu MeOOC 93% COOMe OHC OMOM COOMe S. J. Danishefsky et al. JACS 117, 4722-4723 (1995). N O OMOM N COOMe • Beispiel 2) Teilschritt aus der Synthese von Morphin OMe OH OMe Pd(OCOCF3)2(PPh3)2 OH I PG Pentamethylpiperidin N PG Toluol / 110 °C / 10 h H 60% OMe O CH3 OH Morphin N H L. E. Overman et al., THL 33, 4859-4862 (1992). N H 2) durch Suzuki-Kupplung • Die Suzuki-Kupplung ist eine der wichtigsten Pd-katalysierten Kreuzkupplungsreaktionen. • Ein Ausgangsmaterial enthält eine C(sp2)-X-Bindung, wobei X = Cl, Br, I, OTs, OPO(OR)2 oder OTf ist. Das zweite Ausgangsmaterial ist eine Alkenyl- oder Aryl-Boronsäure oder ein Boronsäurederivat. • Bruttoreaktionen: OH 1 R HO B X + R2 R1 X B + HO • R2 LM / NaOH oder NaOR R2 HO 1 R Pd(0)-Kat. / PAr3 Pd(0)-Kat. / PAr3 R1 LM / NaOH oder NaOR Statt Ar-X kann man auch Vinyl-X oder Allyl-X einsetzen. 2 R • Der Mechanismus der Suzuki-Kupplung ist ziemlich komplex und noch nicht bis in alle Einzelheiten geklärt. • Oxidative Addition: PPh3 Ph3P Pd PPh3 PPh3 PPh3 PPh3 Pd PPh3 • Pd – 2 PPh3 PPh3 Ph3P Ph3P Pd Br Ph3P Br – Br– Pd Ph3P OH – Ph3P Pd Ph3P OH Nach der oxidativen Addition von Ar-X findet ein Ligandenaustausch von X− durch OH− statt. OH− erhöht die Elektronendichte am Metall und macht es nucleophiler für den nachfolgenden Schritt. • Transmetallierung: OH HO Ph3P ” OH B R Pd Pd Ph3P Ph3P Ph3P Ph3P OH HO B OH OH ” OH Pd – B(OH)4– Ph3P R • Reduktive Eliminierung: PPh3 Ph3P Pd Pd R Ph3P R • + PPh3 Wichtig! Die Geometrie der Doppelbindung der Vinylboronsäure bleibt erhalten. • PPh3 Katalysecyclus Ph3P Pd PPh3 PPh3 R – 2 PPh3 Br PPh3 Pd PPh3 Ph3P Pd Ph3P Ph3P Pd Br Ph3P R OH B(OH)4 – Ph3P Pd R B OH HO ” HO Ph3P Br– OH – • Beispiel 1) Teilschritt aus der Synthese von Milbemycin β3 OTBDMS OTBDMS COOMe Pd(OAc)2 / PPh3 I + TBDMSO B O OMe O CH3CN / Tl2CO3 TBDMSO COOMe 80 °C / 68% OMe O O O O OH I. E. Marko et al. THL 37, 25072510 (1996). Die Reaktion geht nur gut mit Tl2CO3! Um geeignete Reaktionsbedingungen zu finden, haben die Autoren Katalysator, Lösungsmittel und Base variiert: Lösungsmittel Katalysator Base Dioxan Pd(PPh3)4 K3PO4 THF PdCl2 NaHCO3 DMF Pd(OAc)2 NaOMe Benzol Pd(dba)2 NaOEt Toluol PdCl2dppf Na2CO3 Acetonitril PdCl2dppe TlNO3 TlOH • Beispiel 2) Teilschritt aus der Synthese von Michellamin A OBn Bn N OBn OBn B(OH)2 Bn N OTf OBn Me OAc OMe MeO OAc Pd(PPh3)4 / DME / H2O Ba(OH)2 / 80 °C / 8h Me OTf 74% Me OAc OMe MeO OAc Me BnO B(OH)2 N BnO N Bn OBn Bn OBn OH H N OH Me OH OH HO OH Michellamine A, B, C Me HO N OH H G. Bringmann et al., THL 35, 76217624 (1994). 3) durch Stille-Reaktion • Die Stille-Reaktion ist in der universitären Forschung eine der wichtigsten Kreuzkupplungsreaktionen. In der Industrie ist die StilleKupplung tabu!!! (Warum???) • Ein Ausgangsmaterial enthält eine C(sp2)-X-Bindung, wobei X = Cl, Br, I, OAc, OPO(OR)2 oder OTf ist. Das zweite Ausgangsmaterial ist ein Alkenyl- oder Aryl-Stannan. • Bruttoreaktionen: 1 R X + R3Sn Pd(0)-Kat. / PAr3 R2 R X + 2 R LM 2 R1 R1 R3Sn Pd(0)-Kat. / PAr3 R2 R1 LM 2 R R1 X + R3Sn Pd(0)-Kat. / PAr3 LM R1 R2 • Als Pd-Katalysatoren verwendet man oft Pd(PPh3)4, PdCl2(CH3CN)2, Pd(dba)2 und Pd(OAc)2. Als Lösungsmittel haben sich THF, Dioxan, DMF, DMA und NMP bewährt. • Wichtig für Syntheseplanung: Der „Dummy“-Rest R am Zinn darf nicht übertragen werden, deshalb wählt man oft R = Me oder R = Bu (bessere Handhabung der Zinn-Verbindung). Übertragbarkeit von Resten am Zinn: Alkinyl > Alkenyl > Aryl > Allyl > Benzyl > Alkyl • Oxidative Addition: PPh3 Ph3P Pd PPh3 PPh3 PPh3 PPh3 Pd PPh3 Pd – 2 PPh3 PPh3 Ph3P Isomeri- Pd Br Ph3P Br sierung Ph3P Ph3P Pd Br PPh3 – PPh3 Pd Br • Transmetallierung und reduktive Eliminierung: R3Sn Ph3P R Ph3P Ph3P Pd Pd – R3SnBr Br R3Sn Br Pd R Ph3P R PPh3 Ph3P Pd Pd R Ph3P R • + PPh3 Wenn Stille-Reaktionen schlecht funktionieren, dann setzt man Ph3As oder (2-Furyl)3P zu. In obigen Reaktionen tauschen dann diese Liganden den PPh3-Liganden aus. Mit Ph3As oder mit (2-Furyl)3P laufen die Transmetallierungen bis zu 1000 mal schneller. • Katalysecyclus PPh3 Ph3P Pd PPh3 PPh3 R – 2 PPh3 Br PPh3 Pd PPh3 Ph3P Pd Ph3P Ph3P Pd Br Ph3P R R3SnBr Ph3P Pd Br R SnR3 PPh3 • Beispiel 1) Teilschritt aus der Synthese von Zearalenon MEMO MEMO O I MEMO O Pd(PPh3)4 O nBu3Sn Toluol O MEMO 54% HO O O HO O Zearalenon O J. K. Stille et al., JOC 56, 2883-2887 (1991). O • Beispiel 2) Teilschritt aus der Synthese von Himastatin I COOtBu TBDMSO N N H Me3Sn Cbz COOtBu TBDMSO N + N H Cbz Cbz Cbz Pd2dba3 / Ph3As DMF / 45 °C 83 % Cbz Cbz H N N tBuOOC COOtBu TBDMSO OTBDMS Himastatin N N H Cbz Cbz S. J. Danishefsky et al., Angew. Chem. 110, 3164-3166 (1998). 4) durch Sonogashira-Kupplung • Die Sonogashira-Kupplung verknüpft sp2-hybridisierte C-Atome mit sphybridisierten C-Atomen. • Als Katalysatoren werden Pd(0)-Komplexe und Cu(I)-Salze verwendet. Zusätzlich werden tertiäre Amine als Basen benötigt. Üblicherweise gibt man noch Triarylphosphin als Ligand zu. • Bruttoreaktionen: R1 + X H Pd(0)-Kat. / PAr3 2 R R1 R2 Base / LM 1 R X + H 2 R Pd(0)-Kat. / PAr3 1 R Base / LM • 2 R Die Sonogashira-Kupplung ist zur Zeit die beste Reaktion zur Herstellung von Arylalkinen. • Der Mechanismus der Sonogashira-Kupplung besteht aus den Teilschritten Oxidative Addition, Transmetallierung und Reduktive Eliminierung. PPh3 Pd Ph3P PPh3 PPh3 PPh3 – 2 PPh3 Ph3P Trans- Pd Br Ph3P PPh3 Ph3P – CuBr H –H + ” R PPh3 PPh3 Pd R Pd Base Br reduktive Pd metallierung Eliminierung R R Pd Ph3P Br Cu PPh3 Pd Ph3P Ph3P PPh3 + R + Cu+ R Cu • Katalysecyclus PPh3 Ph3P Pd PPh3 PPh3 – 2 PPh3 Br PPh3 Pd PPh3 Ph3P R Pd Ph3P Br R Ph3P Pd Ph3P R Cu • Beispiel 1) Teilschritt aus der Synthese von Hellianuol E H MeO Me OH I PdCl2(PPh3)2 / CuI OMe Et2NH / Benzol / RT 99% HO Me O OH MeO OH K. Shishido et al., JOC 66, 309-314 (2001). Me OMe • Beispiel 2) Teilschritt aus der Synthese von Mappicin OH N Cl H TMS PdCl2(PPh3)2 / CuI Et3N / DMF / RT / 1h 98% N O N OH M. Ihara et al., JOC 65, 7110-7113 (2000). OH N TMS 2.6.4. Synthese von Arylalkanen durch C-CKnüpfung 1) durch Friedel-Crafts-Alkylierung • Bei der Friedel-Crafts-Alkylierung werden Alkylhalogenide, Alkene oder Alkohole in Gegenwart von Lewis- oder Brönstedt-Säuren zu Alkylaromaten umgesetzt. Als Alkylhalogenide können primäre, secundäre und tertiäre Alkylhalogenide sowie Benzyl und Allylhalogenide eingesetzt werden. • Reaktivität der Halogenide: I > Br > Cl > F. Letzteres wird selten eingesetzt. • Als Elektrophile treten polarisierte C-Hal-Bindungen bis hin zu Carbeniumionen auf, je nach Stabilität des Carbeniumions. • Wenn freie Carbeniumionen auftreten, die sich durch Umlagerung stabilisieren können, dann laufen diese Umlagerungen ab und man erhält ein umgelagertes Produkt. • Allylkationen können unter SN2‘-Reaktion reagieren (Allylverschiebung). • Problem bei Friedel-Crafts-Alkylierung: Mehrfachalkylierung weil Produkt reaktiver (elektronenreicher!) als Edukt ist. Ausweg: großen Überschuss Edukt verwenden! • Wichtig: Friedel-Crafts-Reaktionen sind reversibel! D.h. man kann sie unter kinetischer Kontrolle (niedrige Temperatur, kurze Reaktionszeit) oder unter thermodynamischer Kontrolle (hohe Temperatur, lange Reaktionszeit) durchführen. • Unter kinetischer Kontrolle beobachtet man die üblichen dirigierenden Effekte (o,p > m). • Unter thermodynamischer Kontrolle erhält man bevorzugt das metaProdukt (Minimierung von sterischer Hinderung). • Friedel-Crafts-Alkylierungen können intermolekular und intramolekular durchgeführt werden. 6-Ringe werden bei intramolekularer Reaktion bevorzugt vor 5-Ringen gebildet. Oft findet dann vor der Cyclisierung noch ein Hydrid-Shift statt. • Beispiel 1) Teilschritt aus der Synthese von Brasilichinon B O O MeO OMe Br OMe SnCl4 / CH2Cl2 0 °C / 1h / 84% O OH O OH V. H. Deshpande et al., THL 40, 4437-4438 (1999). O OMe O • Beispiel 2) Teilschritt aus der Synthese von α-Tocopherol (Vitamin E) OH Me ( HO Me OH Me Sc(OTf)3 )3 H Me HO Me O ( Me 96% H. Yamamoto et al., Bull. Chem. Soc. Jpn. 68, 3569 (1995). )3 H 2) durch Ziegler-Alkylierung • Die Ziegler-Alkylierung ist eine nucleophile aromatische Substitution, die bevorzugt mit Elektronenmangel-Heteroaromaten abläuft, wenn man sie mit Alkyllithiumverbindungen umsetzt. • Die Reaktion verläuft nach einem Additions-Eliminierungs-Mechanismus unter Abspaltung von LiH. • Beispiel: Synthese von Coniin (Schierlingsgift). Li N – LiH H2 / Pd / C N N H 3) durch Suzuki-Kupplung • Durch Suzuki-Kupplung lassen sich auch Alkylgruppen an Arylhalogenide (oder Vinylhalogenide) kuppeln, wenn man 9-Alkyl-BBN als Borankomponente einsetzt. R B R Br Pd(PPh3)4 / NaOH • Das benötigte 9-Alkyl-BBN läßt sich einfach durch Hydroborierung von terminalen Alkenen mit 9-BBN herstellen. 2.7. Synthese von Aryl-Heteroatom-Bindungen 2.7.1. Halogenierung • Aromaten können durch Chlor oder Brom in Gegenwart von LewisSäuren halogeniert werden. Mechanismus: Elektrophile aromatische Substitution. • Elektronenreiche Aromaten wie z.B. Phenole, Aniline, mehrfach alkylierte Aromaten, Naphthalin u.a. reagieren auch ohne Katalysator. Aniline werden oft mehrfach chloriert oder bromiert. Ausweg: Überführen in N-Acetylanilide. • Iod reagiert nur gut mit elektronenreichen Aromaten wie z.B. mit Phenol und mit mehrfach hydroxylierten Benzolen. • Bessere Reaktion zur Iodierung: Sandmeyer-Reaktion. • Direkte Fluorierungen funktionieren sehr schlecht auf Grund der hohen Reaktivität von F2. • Bessere Reaktion zur Fluorierung: Schiemann-Reaktion. 2.7.2. Nitrierung • Reagenzien zur Nitrierung: HNO3/H2SO4, NO2+ BF4−, NO2+ PF6−, Ac2O/HNO3, N2O5/CCl4 u.a. • Reaktive Aromaten (Phenole, Aniline, Pyrrole) werden schon von HNO3 allein nitriert. • Eigentliches Elektrophil: NO2+ • Wichtig: Aniline werden in konz. HNO3 im m-Position nitriert (warum?) Ausweg: N-Acetylierung. • Weitere Nitrierungsreaktion: Sandmeyer-Reaktion mit NaNO2/CuCl. • Weitere Möglichkeiten zur Knüpfung von Ar-N-Bindungen: a) Nitrosierung (nur reaktive Aromaten wie z.B. Phenole oder tert. Aniline). b) Azokupplung 2.7.3. Buchwald-Hartwig-Reaktion (Aminierung) • Durch Buchwald-Hartwig-Reaktion werden Arylhalogenide oder Aryltriflate mit primären oder sekundären Aminen in Gegenwart eines Pd(0)-Katalysators und einer starken Base in Arylamine überführt. • Bruttoreaktion: R R1 X + Pd(0) / Ligand H N R R1 N R2 Base R2 • Als Pd(0)-Quelle verwendet man oft Pd(OAc)2, als Ligand bevorzugt Chelatliganden wie BINAP oder dppf oder dppe, aber auch P(o-Tol)3. Als Base werden üblicherweise NaOtBu, LiHMDS, K2CO3 oder Cs2CO3 in stöchiometrischer Menge eingesetzt. • Der Mechanismus besteht aus den Schritten Oxidative Addition, Ligandenaustausch und reduktive Eliminierung des Produkts unter Rückbildung der katalytisch aktiven Pd(0)-Spezies. • L Katalysecyclus L Pd L L R1 N 2 R L Br L Pd L L Pd L L Pd L Br 1 N R 2 R NaOtBu tBuOH L Pd L 1 R H N R2 NaBr OtBu • Beispiel 1) Teilschritt aus der Synthese von Cyclazocin-Derivaten N N CH3 TfO CH3 Ph-NH2 Pd2dba3 / dppf NaOtBu / Toluol CH3 CH3 Ph N H 80 °C / 57% M. P. Wentland et al., Bioorg. Med. Chem. Lett. 10, 183-187 (2000). • Beispiel 2) Teilschritt aus der Synthese von Mitomycin TBDMSO Br I N3 N H Pd2dba3 / BINAP NaOtBu / Toluol 80 °C / 66% Br Mitomycin N OTBDMS N3 J. Sulikowski et al., JOC 64, 4224-4225 (1999). 2.7.4. Hydroxylierung • Halogenierte oder sulfonierte Aromaten können durch nucleophile aromatische Substitution nach dem Arin-Mechanismus in Phenolderivate überführt werden (Dow-Phenol-Synthese, Alternative: Phenolverkochung von aromatischen Diazoniumsalzen). • Dakin-Reaktion: Elektronenreiche aromatische Ketone oder Aldehyde können mit NaOH/H2O2 oder auch mit Persäuren analog zur BaeyerVilliger-Oxidation in Phenolester überführt werden, die nach Hydrolyse die entsprechenden Phenole liefern. Wichtig: die Aromaten müssen stark elektronenschiebende Substituenten in o- oder/und p-Stellung besitzen. Statt R‘O- kann auch R2‘N- eingesetzt werden. O H2O2 R'O R'O R • O NaOH O OH– R'O OH R Analog zur Buchwald-Hartwig-Aminierung kann auch eine entsprechende Hydroxylierung durchgeführt werden. Wenn man Buchwald-Hartwig-Hydroxylierungen durchführen will, darf man kein Alkoholat als Base verwenden (warum?). • Halogenierte Aromaten können durch Magnesium in Grignardverbindungen überführt werden, die mit B(OMe)3 zu aromatischen Boronsäureestern reagieren. Diese können mit H2O2/NaOH zu Phenolen oxidiert werden. R Mg R X R OMe O B OMe MgX OH– B(OMe)3 R OH R OMe B OMe H2O2 NaOH 2.7.5. Sulfonierung • Klassisch werden Sulfonierungen mit H2SO4 konz., mit Oleum oder mit Chlorsulfonsäure HSO3Cl durchgeführt. Sulfonierungen sind bei hohen Temperaturen thermodynamisch kontrolliert. In der medizinischen Chemie werden Sulfonierungen von Aromaten oft durchgeführt, um Wirkstoffe besser wasserlöslich zu machen. • Die Umsetzung von aromatischen Grignard-Verbindungen mit elementarem Schwefel führt zu Thiophenolen. Die Umsetzung mit elementarem Selen ergibt Selenophenole. • Setzt man Diazoniumsalze mit NaSH oder mit H2S um, dann entstehen analog zur Phenolverkochung Thiophenole. Wichtig: Thiophenole werden an Luft leicht oxidativ zu Disulfiden dimerisiert. Gleiches gilt für Selenophenole. 2.8. Dearomatisierungsreaktionen 2.8.1. Birch-Hückel-Reduktion • Aromaten werden durch Alkalimetalle (Li, Na, K) in flüssigem Ammoniak oder in Aminen zu Cyclohexadienen reduziert. • Birch-Variante: Alkalimetall + Aromat + tert.BuOH in fl. NH3 (Eintopf) Hückel-Variante: Alkalimetall + Aromat in fl. NH3, erst wenn der Aromat vollständig zum Dianion reduziert ist, wird ges. NH4Cl-Lösung zugegeben. • Reaktivität: elektronenreiche Aromaten reagieren langsamer als Benzol, elektronenarme Aromaten reagieren schneller als Benzol. Naphthalinderivate und Heterocyclen werden ebenfalls reduziert. Wichtig: isolierte Doppelbindungen werden nicht reduziert, isolierte innere Dreifachbindungen werden zu Doppelbindungen reduziert. Konjugierte Diene und α,β-ungesättigte Carbonylverbindungen werden ebenfalls reduziert. • Regioselektivität: Z Z Na / NH3 fl. – 37 °C D D Na / NH3 fl. – 37 °C 2 eq. Na 1 eq. Na NH3 fl. / EtOH NH3 fl. / EtOH Beispiel: Teilschritt einer Synthese von Aeruginosin 298-A Li / NH3 fl. COOH NH2 MeO COOH EtOH NH2 MeO HO O HO N OH H H O H NH N O N H CH2OH J. Bonjoch et al., Chem. Eur. J. 7, 3446-3462 (2001) N H NH2 2.8.2. Wesseley-Oxidation und Pelter-Oxidation • Substituierte Phenole werden durch Blei(VI)acetat in o-Chinonmonoacetale oder in o-Chinole überführt (Wesseley). O OH Pb(OAc)4 O OAc OAc OAc O OH Pb(OAc)4 • OAc AcO AcO O Analoge Reaktionen lassen sich mit Diacetoxyiodbenzol PhI(OAc)2, mit Bis-(trifluoracetoxy)-iodbenzol PhI(OOC-CF3)2 (Pelter) und mit Oxon (Mischung aus KHSO5, KHSO4 und K2SO4) in Gegenwart von verschiedenen Nucleophilen durchführen. • Mechanismus: O H O Pb(OAc)4 Pb(OAc)3 O O ” O H O OAc Pb(OAc)4 O CH3 H OAc O O Pb(OAc)3 OAc O ” O CH3 OAc I O Ph H Ph–I(OAc)2 OAc OAc O Ac–O–H - AcOH OAc Beispiel 1): Teilschritt einer Synthese von Aranorosin OH O Ph–I(OAc)2 H Cbz N COOH MeOH / 0 °C O 40% Cbz N H H O O O O O O N H O P. Wipf et al., J. Org. Chem. 58, 7195-7203 (1993) Beispiel 2): Teilschritt einer Synthese von Halleridon Oxon OH O O PPh3 NaHCO3 CH3CN / H2O OH 25 min / RT 80 % HOO OH oder Me2S oder Na2S2O3 O H HO O A. Urbano et al., Angew. Chem. 118, 2803-2807 (2006) HO OH 3. 3.1. • Synthese von Aromaten Reppe-Synthese Reppe entdeckte bei der BASF, dass Acetylen in Gegenwart bestimmter Ni-Katalysatoren entweder trimerisiert oder tetramerisiert. H H Ni (acac)2 150 °C / 150 bar Ph3PNi(CO)3 H H 120 °C / 120 bar • Weitere Reppe-Reaktionen: a) Acetylide + Aldehyde oder Ketone zu Propargylalkoholen, b) Acetylen + Alkohole oder Carbonsäuren zu Vinylethern oder Vinylestern und c) Acetylen + CO + Wasser oder Alkoholen zu Acrylsäure oder Acrylsäureestern. • Mechanismus der Acetylen-Trimerisierung: PPh3 Ni OC CO H CO H PPh3 a b PPh3 Ni - 3 CO Ni a b b PPh3 H H Ni a PPh3 b Ni H H 3.2. • Vollhardt-Synthese Vollhardt wollte analog zu Arbeiten von McComber unsubstituiertes Cyclopentadienon herstellen. tBu McComber tBu tBu CpCo(CO)2 O tBu Co O Vollhardt H H CpCo(CO)2 (geplant) • Reaktion läuft aber nicht wie geplant! Co • Ergebnis: H H CpCo(CO)2 • Erklärung (formal): • Ausführliche Untersuchungen zeigten: R H H R • CpCo(CO)2 R R Hexadiin-1,5 und ein symmetrisches Acetylen liefert Benzocyclobutane. • Mechanismus: CpCo(CO)2 – CO TMS CO TMS CpCo CpCo(CO) – CO TMS TMS TMS CpCo CpCo TMS CpCo TMS TMS TMS TMS TMS TMS CpCo TMS TMS TMS TMS TMS TMS Beispiel: Synthese von Östron 1) 3 nBuLi 2) O 1) OH 1) TsCl / Py O I 2) NaI OTMS MgBr ” LiNH2 2) TMSCl O O NH3 fl. TMS TMS TMS TMS O O O TMS TFA H TMS H H –30 °C TMS TMS O O H H TMS Pb(CF3COO)4 H H H H H H HO K. P. C. Vollhardt et al., J. Am. Chem. Soc. 102, 5253-5261 (1980). 3.3. Sato-Synthese • Sato und Mitarbeiter haben die Reppe-Synthese weiterentwickelt. Ein Nachteil der Reppe-Synthese mit Ni-Katalysatoren war die fehlende Regioselektivität bei der Verwendung von substituierten Acetylenen. • Mit einem Katalysator, der aus Ti(OiPr)4 und iPrMgCl hergestellt wird, lassen sich regioselektiv hoch substituierte Aromaten aufbauen. COOtBu R Ts COOtBu 1) Ti(OiPr)4 + iPrMgCl R 1 2) H 1 + 2 R 2 R + H / H2O Ti(OiPr)2 R 1 COOtBu OiPr Ti OiPr R 2 iPrO OiPr Ti Ts 1 R Ts R iPrO OiPr tBuOOC Ti 1 R Ts 1 R – Ts 2 R 2 COOtBu Ti(OiPr)2Ts ” 2 R 3.4. • Witulski-Synthese Witulski und Mitarbeiter fanden, dass sich mit dem Wilkinson-Katalysator drei Doppelbindungen, die sich in geeignetem Abstand innerhalb einer Kette befinden, zu Aromaten trimerisieren lassen. Die Regioselektivität ist durch die Anordnung der Dreifachbindungen bereits festgelegt. R RhCl(PPh3)3 R CH2Cl2 / RT R L L Rh R RhL2 Beispiel: Synthese von Alcyopterosin E RhCl(PPh3)3 TsO O CH2Cl2 / RT O 71% NBu4NO3 TsO O O O2N O O O B. Witulski et al., Angew. Chem. 114, 3415-3418 (2002). 3.5. Synthese von Aromaten durch Diels-AlderReaktion • Durch die Kombination von Diels-Alder-Reaktion und Retro-Diels-AlderReaktion kann man ebenfalls Aromaten herstellen. • Das Dien ist dabei ein 1,3-Cyclohexadien-Derivat oder ein α-PyronDerivat, als Dienophile verwendet man elektronenarme Acetylene oder auch Arine. R 1 1 R R R 3 COOR COOR 2 R1 R 2 ∆ R2 – 3 R COOR R3 O R1 O O R2 O R 3 1 R COOR COOR 2 R 3 R R1 ∆ – CO2 COOR R2 R3