Autoimmun polyglanduläre Syndrome
Werbung

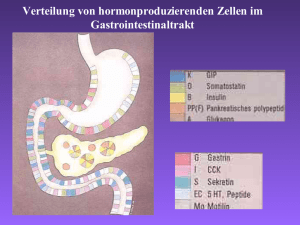
Endokrine Tumoren des Gastrointestinaltraktes = Apudome = GEP-NET (Gastroentero-pankreatische neuroendokrine Tumoren Hormonproduzierenden Zellen im Gastrointestinaltrakt Der Gastrointestinaltrakt als hormonelles System: Peptidhormon Zelltyp Hauptvorkommen Gastrin G-Zelle Antrum, prox. Duodenum, ZNS Sekretin S-Zelle Duodenum, Jejunum Cholezystokinin I-Zelle Duodenum, Jejunum Enteroglukagon EG(A)-Zelle Magenfundus, Duodenum, Pankreasglukagon A-Zelle Pankreas Insulin B-Zelle Pankreas Somatostatin D-Zelle Magen, Pankreas, Jejunum, ZNS Gastric inhibitory Peptide D1(K)-Zelle Duodenum, Jejunum Vasoactive intestinal Peptid H-Zelle Jejunum, Ileum, Colon, ZNS Pankreatisches Polypeptid V-D1-Zelle Pankreas Motilin EC-Zelle Jejunum Histochemische Eigenschaften der APUD-Zellen Amine – Precursor Uptake Decarboxylation Spezifische Hormonproduktion: VIP, Insulin, Serotonin, PP, Gastrin, Somatostatin, alpha-, beta-HCG Methylierung von Tumorsuppressorgenen ( 9p21) Elektronenmikroskopie: sekretorische Vesikel Eigenschaften der APUD-Zellen Argyrophilie, maskierte Metachromasie Charakteristische Enzyme und Marker: Alpha-Glycerophosphatase, Menadion-Reduktase, Esterase, Neuronenspez. Enolase, Sekretogranin I und II, Chromogranin A, Synaptophysin Endokrine GEP-Tumoren GEP_NET Häufigkeit (autoptisch) 0,5-1,5 % Klinische Diagnose <1/100.000 Karzinoid 0,2-0,5/100.000 Klassifikation endokriner GEP-Tumoren Bezeichnung Funktionell inaktiv Insulinom Gastrinom VIPom Glukagonom Somatostatinom Neurotensinom PPom Tumoren mit ektoper Hormonproduktion (Cortisol, ACTH, GH, CRF o.ä.) Malignität Lokalisation (Primär-Tumor) > 75 % < 10 % > 90 % > 75 % > 50 % > 50 % ? ? > 90 % Pankreas, Dünndarm Pankreas, Dünndarm Pankreas, Duodenum Pankreas, Grenzstrang Pankreas Pankreas, Dünndarm Pankreas, Dünndarm Pankreas, Dünndarm Pankreas Carcinoid-Syndrom Definition: Krankheitsbild durch Tumoren, die – Serotonin – Histamin – Kallikrein – Bradykinin produzieren Metastasen • Leber, Lymphknoten, Skelett Klassifikation der Karzinoide • Foregut-Karzinoide – Lunge, Thymus, Magen, Schilddrüse – Ca. 20 % • Midgut-Karzinoide – Dünndarm, Pankreas, Appendix, Coecum – Ca. 65 % • Hindgut-Karzinoide – Colon, Rektum, Ovar – Ca. 15 % Klinik der Karzinoide • Serotoninwirkung • Tryptophan-Mangelerscheinungen – Proteinmalnutrition – Leichte Pellagra (Niacin-Mangel) Carcinoid-Syndrom Klinische Symptomatik FLUSH: Rötung, Hitzegefühl und Brennen im Gesicht mit Ausbreitung auf Hals und Brust. explosionsartig Gewichtsabnahme Eventuell: Ileus, Perforation, Blutung Begleitend: Übelkeit, Kopfschmerzen, Tachykardie Auslöser: Phys. und psych. Belastung, Alkohol, Nahrungsaufnahme Carcinoid-Syndrom Spezielle Diagnostik – Bestimmung von 5-Hydroxyindolessigsäure im Urin (unter tryptophanarmer Kost) – Bestimmung von Serotonin – Bildgebung Tumoren mit ektoper Hormonproduktion (Pat. A.H.) Klinik (Pat. A.H.) Hormonanalytik Beurteilung: Hyperkortisolismus, ACTH-abhängig Keine Supprimierbarkeit nach Dexa Keine Stmulierbarkeit nach CRF Tumoren mit ektoper Hormonproduktion (Cortisol, ACTH, GH, CRF o.ä.) • Klinische Symptomatik – Cushing-Syndrom – Hirsutismus • Spezielle Diagnostik – Bestimmung von Cortisol, ACTH, Testosteron u.a. – CRF-Test – Bildgebung Okkulte ektope ACTH Sekretion: häufige Quellen 1. 2. 3. 4. 5. 6. Bronchuskarzinoid Thymuskarzinoid Medulläres Schilddrüsenkarzinom Phäochromozytom Gastrinom Endokrine Pankreastumoren Insulinom Falldarstellung I.W. *1969 Anamnese am 02.05.90 • Seit 2 Monaten „seltsames Gefühl, wenn nüchtern“. Eltern morgens: • „etwas verwirrt, wieder ins Bett gegangen“ • „nicht erweckbar, bewußtlos“ Notarzt: • Gluco-Stick: < 20 mg/dl • Schlagartige Besserung durch Glukosezufuhr (40 ml G40 i.v.,/ 500 G5) Stationäre Aufnahme: • Innere Abteilung Insulinom Klinische Symptomatik – Whipple‘ Trias • Hypoglykämie nach Fasten oder körp. Belastung • Blutzuckerwerte < 30 mg/dl • Schlagartige Besserung durch Glukosezufuhr – Gewichtszunahme (Adipositas) – Zeichen der Neuroglukopenie oder adrenergen Reaktion: Verwirrtheit, Schwächegefühl, Kopfschmerzen, Doppelbilder, Gedächtnisstörungen, Polyneuropathie Palpitationen, Tachykardie, Schweißausbrüche, Hunger Schwindel, Unruhe Krämpfe, Koma Insulinom Spezielle Diagnostik • Hungerversuch (stationär) Insulin (U/ml)____ Bestimmung von Blutglukose (mg/dl): – Blutzucker Normal < 0,25 Pathologisch > 0,33 – Insulin, C-Peptid – Sulfonylharnstoffen, Gliniden • Glukose-Toleranztest • • • • Glukagontest Insulin-Suppressionstest Euglykämie-Versuch Somatostininduzierte Insulinhemmung Insulinom Wichtige Differentialdiagnosen • Artefizielle Hypoglykämien • Reaktive Hypoglykämien (Spät-Dumping) • Mangel an gegenregulatorischen Hormonen (Nebenniereninsuffizienz) • IgF1 –Produktion in Tumoren – Fibrosarkome, Mesotheliome, Hämangioperizytome, Nebennierenkarzinome, Hepatome u.a. • Multiple endokrine Adenomatose Untersuchungen zur Lokalisation endokriner Pankreastumoren 1. 2. 3. 4. 5. 6. 7. 8. Sonografie Endosonografie Computertomografie mit KM-Bolus-Gabe Kernspintomografie Angiografie Somatostatinrezeptorszintigrafie evtl. selektive Venenblutentnahme Positronen-Emissionstomografie Untersuchungen zur Lokalisation endokriner Pankreastumoren Sonografie Insulinom CT-Apudom Untersuchungen zur Lokalisation endokriner Pankreastumoren Somatostatinrezeptorszintigrafie Vipom Angiografie Insulinom selektive Venenblutentnahme Glukagonom • Klinische Symptomatik – – – – – – – Diabetes mellitus Erythema necretolyticans migrans Normochrome, normozytäre Anämie Atrophische Glossitis, Stomatitis Gewichtsverlust (Diarrhöen) (thromboembolische Komplikationen) Glukagonom Klinische Symptomatik Erythema necretolyticans migrans Glukagonom Klinische Symptomatik Glukagonom • Spezielle Diagnostik – Nachweis mehrfach erhöhter Plasma-Glukagonkonzentrationen – Tumorsuche • • • • • • DD: Diabetes mellitus, Nierenversagen, Leberzirrhose, Bakteriämie, Cushing-Syndrom Gastrinom • Klinische Symptomatik – – – – Magensäurehypersekretion Rezidivierende Ulzera Steatorrhö Diarrhö • Spezielle Diagnostik – Gastrin i.S. – Sekretintest – Kalzium-, Glukagonstimulation Erhöhte Plasma-Gastrinspiegel: • • • • • • Gastrinom Antraler G-Zell-Hyperplasie Belassener Antrumrest nach BII-Op Niereninsuffizienz Ausgeprägte Dünndarmresektion Atrophische Fundusgastritis mit erhaltenem Antrum (z. B. perniziöser Anämie) • Folge einer Vagotomie ohne Antrumresektion • Magen-Ca mit Sub- und Anazidität Somatostatinom • Klinische Symptomatik – Diabetes mellitus – Steatorrhoe – Cholelithiasis – Verminderte Magensäuresekretion – Völlegefühl • Spezielle Diagnostik – Bestimmung von Plasma-Somatostatin (zehnfach erhöht) VIPom Fallbeispiel • G., K.J. 56 Jahre, Landwirt – Plötzlich auftretende wässrige Diarrhöen (bis 12 l/d) – Lethargie, Muskelschwäche – Übelkeit und Erbrechen • Stationäre Aufnahme – Klinisch: ausgeprägte Exsikkose – Laborchemisch: Hypokaliämie, Achlorhydrie, Hyperkalzämie, eingeschränkte Nierenfunktion – Spezielle Diagnostik: Stuhlanalyse auf pathogene Keime und weitere Durchfalldiagnostik negativ VIPom Fallbeispiel G., K.J. 56 Jahre, Landwirt Sonografisch und im CT Nachweis multipler Leberherde Verlegung UKM Münster Spezielle Diagnostik Nachweis erhöhter PlasmaVIP- und SerumCalcitoninKonzentrationen Chemotherapie VIPom (Synonym:Verner-Morrison-Syndrom, WDHA-Syndrom) • Klinische Symptomatik – – – – – – – – Wässrige Diarrhöen (bis 12 l/d) Hypokaliämie Achlorhydrie Dehydratations-Schock Hyperkalzämie Lethargie, Muskelschwäche Übelkeit und Erbrechen Passagere renal-tubuläre Dysfunktion • Spezielle Diagnostik – Stuhlanalyse – Nachweis erhöhter Plasma-VIP-Konzentrationen Therapieprinzipien bei endokrinen Pankreastumoren 1. Chirurgische Tumorentfernung 2. Medikamentöse Kontrolle der hormonellen Symptomatik – präoperativ, – postoperativ bei Nichtauffinden des Tumors, – bei diffuser Metastasierung 3. Kontrolle des Tumorwachstums – Chemotherapie – Metastasenchirurgie, Debulking-Operationen, Transplantation – Embolisation von Ästen der A. hepatica – 90 Y-Somatostatin-Radiotherapie Chemotherapie bei endokrinen Pankreastumoren • • • • • • • • • Streptozotozin Streptozotozin plus 5 FU Adriblastin (Response 40-60 %) L-Asparaginase Dacarbazin Etoposid und Cis-Platin (Response 60 %) DTIC Somatostatin (Response 60-70 %) Interferon (Response 50 %) Symptomatische medikamentöse Therapie bei endokrinen Pankreastumoren Tumor Symptom Therapie Insulinom Hypoglykämie Diazoxid , Somatostatin, Corticosteroide Gastrinom Hyperchlorhydrie, Ulzera, Diarrhöe H2-Rezeptorblocker, Protonenpumpenhemmer Vipom Diarrhöe Somatostatin, Indomethacin, Lithium Glukagonom Erythema necrolyticans migrans Somatostatin, Diphenylhydantoin ACTH- bzw. Cortisolproduzierende Tumoren Cushing-Syndrom Aminogluthelthimid, Mitotane, Somatostatin MEN ( Multiple endokrine Neoplasie) Synonym: Multiple endokrine Adenomatose (MEA) Definition: Syndromenkomplex assoziiert mit Tumoren oder Hyperplasie in zwei oder mehreren endokrinen Organen MEN I MEN II a MEN II b = Wermer`s Syndrom = Sipple`s Syndrom = Mutiple MucosalNeuroma-Syndrom Klinik der MEN I (Wermer-Syndrom) Tumorlokalisation Hormonproduktion Klinik Hypophyse ( ca. 65 %) GH ACTH HPrl Nichtfunktionell Akromegalie Cushing Prolaktinom HVL-Insuffizienz Nebenschilddrüse ( ca. 87 %) PTH Hyperparathyreoidismus Pankreas ( ca. 81 %) Gastrin Insulin VIP Glukagon Calcitonin, PP Zollinger-Ellison-Syndrom Hypoglykämie (Insulinom) WDHA-Syndrom Diabetes, nekrolytische Dermatose (Glukagonom) asymtomatisch Mutation im Chromosom 11 = Menin-Gen Autosomal dominant Klinik der MEN IIa (Sipple`s -Syndrom) Tumorlokalisation Hormonproduktion Klinik Schilddrüse (fast 100 %) Calcitonin Medulläres Schilddrüsenkarzinom Nebenschilddrüse ( ca. 20 %) PTH Hyperparathyreoidismus Nebenniere ( ca. 50 %) Adrenalin, Noradre- Phäochromozytom nalin, Metanephrin, Dopamin Mutation im RET-Protoonkogen Autosomal dominant Klinik der MEN IIb (Multiple mucosal neuroma -Syndrom) Tumorlokalisation Hormonproduktion Klinik Schilddrüse (fast 100 %) Calcitonin Medulläres Schilddrüsenkarzinom Haut Nebenniere Neurome (Augenlid, Lippe, Zunge, Colon) Adrenalin, Noradrenalin, Phäochromozytom Metanephrin, Dopamin Mutation im RET-Protoonkogen Autosomal dominant MEN-IIB Autoimmune polyglanduläre Syndrome Genetische Faktoren Umweltfaktoren HLA-DR, DQ, CTLA-4, T-Zell Defekt Sexualhormone, Jodid, Virusinfektion, Streß Autoimmunität Humorale und zelluläre Immunreaktion Stimulierend Blockierend Destruierend TSH- Rezeptor stimulierende AK Zytokine TSH- Rezeptor blockierende AK IF- , IL-1, TNF- AK verm. Zytotox. Zytotox. T- Lympho. TNF- M. Basedow Hashimoto Hypothyreose Charakteristika der polyglandulären Autoimmunsyndrome Typ I Inzidenz m:w Manifestationsalter Genetik HLA Assoziationen <1:100.000/J 3:4 Kindheit monogenetisch autosomal rezessiv Mutationen im AIRE Gen Chrom. 21 DRB1* 03 (M. Addison) DRB1* 04 DQB1* 0302- (Alopezie) DRB1* 15DQB1* 0602 (D.m.I.) Typ II 1-2: 100.000/J 1:3 30.-40. LJ polygenetisch dominant Gene Chromosom 6 DR3-DQB1* 0,201 multiple Komponenten DR4-DQB1* 0302 (D.m.I.) B8-Allele (M. Basedow) B8/DRw3 Autoimmun polyglanduläre Syndrome • Autoimmun polyglanduläres Syndrom Typ I (APS-1) • Synonyma: Autoimmunes PolyendokrinopathieKandidiasis-Ektodermales-DystrophieSyndrom (APECED) • Autoimmun polyglanduläres Syndrom Typ II (APS-2) • Synonyma: Schmidt-Carpenter Syndrom Autoimmun polyglanduläres Syndrom Typ I Ausfall von Krankheit Kinder, Jugendliche Folgen/Klinik Nebenschilddrüse Hypoparathyreoidismus (Morbus Farr) Hypokalzämie, Hypophosphatämie, Katarakte Basalganglienverkalkung Nebennierenrinde Morbus Addison Schwäche, Hypotonie, Hyperkaliämie, braune Haut Gonaden Gonadeninsuffizienz (Hypogonadismus) Libidoverlust, Impotenz, Amenorrhö Intrinsic factor Perniziosa Atrophische Gastritis, Anämie, (Präcanzerose) ? Autoimmunhepatitis Ikterus, Transaminasenerhöhung ? Candidiasis Alopezie Vitiligo Pilzbefall der Haut Haarausfall, Glatze Weißflecken Häufigkeit der Krankheitskomponenten Autoimmun polyglanduläres Syndrom Typ II (Schmidt-Syndrom) Erwachsene Frauen Ausfall von Krankheit Folgen Schilddrüse Hypothyreose Verlangsamung, Gewichtszunahme, tiefe Stimme, trockene Haut, struppige Haare, Hypothermie Nebennierenrinde Morbus Addison Schwäche, Hypotonie, Hyperkaliämie, braune Haut Endokrines Pankreas Hyperglykämie, Glukosurie, Durst, Müdigkeit, Koma Diabetes mellitus Typ 1 Erstkrankheit bei Patienten mit APS II Häufigste Krankheitskombinationen bei Patienten mit APS II Zeitintervall von Krankheitserstmanifestation und Zweitmanifestation bei Patienten mit APS II Komplikation der Insulintherapie bei Schmidt-Carpenter-Syndrom