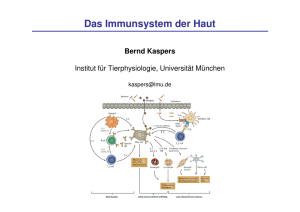
Masterarbeit
Werbung

Masterarbeit Vorgelegt zur Erlangung des Grades eines Master of Science an der Fakultät für Biologie und Biotechnologie der Ruhr-Universität Bochum Charakterisierung des alternativen Korezeptorgebrauchs von HIV-Isolaten Characterization of the coreceptor usage of HIV isolates vorgelegt von Hanna Flamme angefertigt am Robert Koch-Institut in der Abteilung für HIV und Retrovirologie Bochum, im Juli 2012 Betreuer: Dr. Claudia Kücherer Referent: Prof. Dr. Franz Narberhaus Koreferent: Prof. Dr. Klaus Überla Danksagung An erster Stelle möchte ich mich bei Frau Dr. Claudia Kücherer und Herrn Dr. Norbert Bannert für die Ermöglichung meiner Masterarbeit am Robert Koch-Institut bedanken. Zudem danke ich Prof. Dr. Franz Narberhaus und Prof. Dr. Klaus Überla für die Unterstützung an der Ruhr-Universität Bochum. Den Mitarbeitern des Robert KochInstituts danke ich für das herzliche Arbeitsklima, im Besonderen Dr. Claudia Kücherer, Dr. Karolin Meixenberger, Katrin Arndt und Stefan Fiedler für die Einarbeitung und die besondere Unterstützung im Labor. Zu Letzt möchte ich meine Familie und meine Freunde erwähnen, die mir stets unterstützend und motivierend zur Seite standen. II Inhaltsverzeichnis 1 2 Einleitung ........................................................................................................................................ 1 1.1 Die epidemische Verbreitung von humanen Immundefizienzviren........................................ 1 1.2 Das Genom und die Struktur von HIV-1 .................................................................................. 2 1.3 Der Verlauf der HIV-Infektion.................................................................................................. 3 1.4 Der Replikationszyklus von HIV ............................................................................................... 5 1.5 Der Eintritt von HIV-1 in die Zielzelle mittels Korezeptorbindung .......................................... 6 1.6 Mutationen im CCR5 Korezeptor und deren Einfluss auf den HIV Krankheitsverlauf ............ 8 1.7 Antiretrovirale Therapie mit einem CCR5 Korezeptorantagonist ........................................... 9 1.8 Aufgabenstellung................................................................................................................... 10 Material und Methoden ............................................................................................................... 12 2.1 Material ................................................................................................................................. 12 2.1.1 Blutspender ................................................................................................................... 12 2.1.2 Plasmide ........................................................................................................................ 12 2.1.3 Zellen ............................................................................................................................. 12 2.1.4 Virusisolate .................................................................................................................... 13 2.1.5 Chemikalien/ Feinchemikalien/ Reagenzien ................................................................. 14 2.1.6 Puffer ............................................................................................................................. 15 2.1.7 Antikörper...................................................................................................................... 16 2.1.8 Primer ............................................................................................................................ 16 2.1.9 Kits ................................................................................................................................. 16 2.1.10 Kunststoffmaterialien .................................................................................................... 17 2.1.11 Geräte ............................................................................................................................ 17 2.1.12 Software ........................................................................................................................ 18 2.2 Methoden .............................................................................................................................. 18 2.2.1 Kultivierung der 293T-Zellen ......................................................................................... 18 2.2.2 Kultivierung und Wachstum der U87.CD4 Zellen .......................................................... 18 2.2.3 Isolierung von PBMCs aus Buffy Coats .......................................................................... 19 2.2.4 Virusvermehrung ........................................................................................................... 20 2.2.5 Virustitration ................................................................................................................. 21 2.2.6 p24 Antigen Test............................................................................................................ 21 2.2.7 Stabile Transfektion ....................................................................................................... 22 2.2.8 Transduktion der parentalen U87.CD4 Zelllinie mit dem CX3CR1-Gen ........................ 23 2.2.9 FACS-Analyse ................................................................................................................. 25 2.2.10 Infektionskinetik in den U87.CD4-Zelllinien .................................................................. 25 2.2.11 Inhibitionsversuche mit CXCR4 und CX3CR1 spezifischen Liganden ............................. 26 2.2.12 DNA Extraktion aus PBMCs ........................................................................................... 26 III 3 2.2.13 RNA-Extraktion aus Virusstocks .................................................................................... 27 2.2.14 PCR zur Analyse des CCR5-Genotyps............................................................................. 27 2.2.15 env gp120 RT-PCR .......................................................................................................... 27 2.2.16 DNA Agarose-Gelelektrophorese .................................................................................. 28 2.2.17 PCR-Produktreinigung und DNA Sequenzierung ........................................................... 29 2.2.18 Sequenzauswertung ...................................................................................................... 29 Ergebnisse ..................................................................................................................................... 30 3.1 Herstellung einer U87.CD4 Zelllinie mit stabiler CX3CR1 Expression ................................... 30 3.1.1 Kontrolle der Expression des CD4-Rezeptors ................................................................ 31 3.1.2 Kontrolle der CXCR4- und CCR5-Korezeptorexpression ................................................ 33 3.1.3 Überprüfung der Spezifität der Antikörper für die Korezeptoren ................................ 34 3.2 CCR5 Genotyp der PBMC-Spender für die Virusvermehrung und Titration ......................... 35 3.3 Herstellung der Virusstocks und Überprüfung der partiellen viralen Hüllproteinsequenz .. 36 3.3.1 Virusvermehrung auf PBMCs und Titerbestimmung..................................................... 37 3.3.2 Kontrolle der V3-Loop Sequenzen im Hüllprotein der HIV-Virusstocks ........................ 37 3.4 Phänotypische Bestimmungen des Korezeptorgebrauchs der Virusisolate ......................... 39 3.4.1 3.5 4 Infektionskinetik mit und ohne Mediumwechsel.......................................................... 40 Inhibition der HIV-1 Infektion mit Korezeptor-Antagonisten................................................ 47 3.5.1 Inhibition des CXCR4-Korezeptors mit AMD3100 ......................................................... 47 3.5.2 Inhibitionsversuch mit dem natürlichen Liganden von CX3CR1 (Fraktalkin) ................ 48 Diskussion ..................................................................................................................................... 51 4.1 Genotypische Analysen des Korezeptorgebrauchs ............................................................... 52 4.2 Der CX3CR1-Korezeptors ist kein alternativer Korezeptor für das HIV-Isolat ....................... 54 4.3 Inhibitionsversuche mit den Korezeptor-Antagonisten AMD3100 und Fraktalkin ............... 54 5 Zusammenfassung ........................................................................................................................ 58 6 Abstract......................................................................................................................................... 60 7 Literatur ........................................................................................................................................ 62 8 Erklärung ....................................................................................................................................... 65 IV Abbildungsverzeichnis Abb.1 Schematische Darstellung eines HIV-1 Partikels. .................................................................... 2 Abb.2 Die genomische Organisation/ Struktur von HIV-1 ................................................................. 3 Abb.3 Natürlicher Verlauf einer HIV-Infektion .................................................................................. 4 Abb.4 Replikationszyklus von HIV-1................................................................................................... 6 Abb.5 Eintritt von HIV-1 in die Wirtszelle .......................................................................................... 7 Abb.6 Charakteristische Mutationen in der CCR5 Korezeptor Aminosäuresequenz ........................ 9 Abb.7 Transduktion von CX3CR1 mit Pseudotypviren..................................................................... 24 Abb.8 Schematische Darstellung der HIV-1 env Region .................................................................. 28 Abb.9 Nachweis der Expression des CX3CR1-Korezeptors in der U87.CD4.CX3CR1 Zelllinie.......... 31 Abb.10 Expression des CD4-Rezeptors in dem U87-Zellpanel........................................................... 32 Abb.11 Expression des CXCR4-Rezeptors in der U87.CD.CXCR4 Zelllinie .......................................... 33 Abb.12 Expression des CCR5-Korezeptors in der U87.CD.CCR5 Zelllinie ........................................... 34 Abb.13 Überprüfung der Kreuzreaktivität der Antikörper ............................................................... 35 Abb.14 Bestimmung des CCR5 Genotyps der PBMC-Spender........................................................... 36 Abb.15 Sequenzvergleich des V3-Loops im Hüllprotein der HIV-1-Isolate........................................ 38 Abb.16 Infektionskinetik mit und ohne Mediumwechsel ................................................................. 40 Abb.17 A-D. Infektionskinetik U87.CD4-Zellpanel ............................................................................. 43 Abb.18 Phänotypischer Korezeptorgebrauch der Virusisolate im U87.CD4-Rezeptorzellpanel ....... 44 Abb.19 Infektionsversuche ................................................................................................................ 45 Abb.20 Infektionsversuche ................................................................................................................ 46 Abb.21 Inhibition der Infektion mit X4-Viren. ................................................................................... 48 Abb.22 Inhibition mit Fraktalkin ........................................................................................................ 49 V Abkürzungsverzeichnis AIDS Acquired Immunodeficiency Syndrome as antisense ATP Adenosintriphosphat ART antiretrovirale Therapie Bp Basenpaare CA Capsidprotein (p24) cART combination antiretroviral therapy CCR5 CC-Chemokinrezeptor 5 CD4 cluster of differentiation 4 CDC Centers for Disease Control and Prevention cDNA complementary DNA (komplementäre DNA) CDS codierende Sequenz CRF circulating recombinant form CXCR4 CXC-Chemokinrezeptor 4 CX3CR1 CX3C-Chemokinrezeptor CX3CR1 (V28) DMSO Dimethylsulfoxid DNA Desoxyribonukleinsäure dNTP Desoxynukleosidtriphosphat d.p.i. Tage post Infektion dsDNA doppelsträngige DNA DTT Dithiothreitol EDTA Ethylendiamintetraacetat Env envelope (Hüllprotein) et al. et alii FKS fötales Kälberserum FPR false positive rate Gag group specific antigen (gruppenspezifisches Antigen) geq Genomäquivalente VI GFP green fluorescent protein gp Glykoprotein HAART highly active antiretroviral therapy HIV-1 human immunodeficiency virus type 1 (Humanes Immundefizienz Virus) IN Integrase kB Kilobasen LB lysogeny broth LI Linkprotein (p6) LTNP long-term nonprogressor LTR long terminal repeat MA Matrixprotein (p17) MHC II Major Histocompatibility Complex II (Haupthistokompatibilitätskomplex II) MOI multiplicity of infection MoMuLV moloney murine leukemia virus mRNA messenger RNA NC Nukleokapsidprotein (p7) Nef negative factor ORF open reading frame p Protein PBMC peripheral blood mononuclear cells PBS phosphatgepufferte Kochsalzlösung PCR Polymerase-Kettenreaktion PE Phycoerythrin PEG Polyethylenglycol PHA lectin from phaseolus vulgaris typS Pol Polymerase PR Protease rDNA ribosomale DNA Rev regulator of expression VII RCLB red cell lysis buffer RNA Ribonukleinsäure RNaseH RibonukleaseH RNP Ribonukleoproteinkomplex RT Reverse Transkriptase s sense SNP Single-Nukleotid-Polymorphismus SU externes Oberflächen-Glykoprotein (gp120) TAE Tris/Acetat/EDTA Tat Transkriptionsaktivator TBS tris buffered saline TCID50 median tissue culture infective dose TM transmembranes Glykoprotein (gp41) V Versen Vif virion infectivity factor Vpr viral protein Ra Vpu viral protein U VIII Einleitung 1 1.1 Einleitung Die epidemische Verbreitung von humanen Immundefizienzviren Die erste bekannte Infektion mit den für die humanen Immundefizienzviren (HIV) typischen Symptomen wurde bereits 1959 bei einem Mann in Kinshasa, der demokratischen Republik Kongo, festgestellt [1]. Im Jahre 1983 wurde ein Virus aus einem Lymphknoten eines immungeschwächten Individuums gewonnen [2-3], dessen Erreger von der internationalen Kommission für Virus Taxonomie als humanes Immundefizienzvirus Typ 1 (HIV-1) klassifiziert werden konnte [4]. Die Krankheitssymptome wurden anschließend vom Center for Disease Control and Prevention (CDC) als erworbenes Immundefekt Syndrom (englisch: Aquired Immuno Deficieny Syndrome AIDS) zusammengefasst. Noirau postuliert bereits 1987, dass das neue humane Immundefizienz Virus Typ 1 (HIV-1) über einen nicht-humanen Primaten auf den Menschen übertragen wurde und daraufhin die Ausbreitung in Zentralafrika begann [5]. Das humane Immundefizienzvirus ist der Auslöser für mehr als 60 Millionen Infektionen und 25 Millionen Todesfälle weltweit [6]. Global infizierten sich im Jahre 2010 nahezu 2.7 Millionen Menschen mit HIV und mehr als 34 Millionen Menschen leben derzeit mit einer HIV Infektion (UNAIDS/WHO). In Deutschland lag die Neuinfektionsrate im Jahr 2011 bei ca. 3000 Personen, wobei der Subtyp B eine Prävalenz von 90% hat [7]. Dabei dominiert die Übertragung des Virus in der Transmissionsgruppe „Männer, die Sex mit Männern haben“ (MSM) (ca. 2000 von 3000 Neuinfektionen pro Jahr) [7]. HIV ist das am besten untersuchte Virus in der Wissenschaftsgeschichte. Die Struktur und Pathogenese von HIV-1 werden seit der Entdeckung des humanpathogenen Virus intensiv erforscht. Anhand von phylogenetischen Analysen wird zwischen den HIV-Typen 1 und 2 unterschieden. HIV-1 ist epidemisch global verbreitet, wohingegen das Verbreitungsgebiet von HIV-2 hauptsächlich auf Westafrika beschränkt ist [8]. HIV-1 wird in die Gruppen M (major-group), O (outlier), N (non-major, non-outlier) und P (putativ) unterteilt [9]. Innerhalb der Gruppe M sind eine Vielzahl von genetischen Subtypen (A, B, C, D, F, G, H, J, K) und mehr als 50 zirkulierende rekombinante Formen (CRF) definiert. Subtyp C kommt weltweit am häufigsten vor. Die meisten HIV-positiven Menschen leben in Sub-Sahara Afrika (UNAIDS/WHO), wo eine sehr heterogene Verteilung der Subtypen und Gruppen vorliegt. In diesen Regionen 1 Einleitung überwiegt die Übertragung durch heterosexuellen Geschlechtsverkehr. Viren der Gruppe N, O und P wurden nur in Kamerun beobachtet. 1.2 Das Genom und die Struktur von HIV-1 HIV-1 ist ein Lentivirus (Genus), das zu der Familie der Retroviridae und zur Unterfamilie der Orthoretrovirinae gehört [10]. Das reife Virion besteht aus einem konischen Kapsid (Core), welches aus p24 Monomeren aufgebaut und von einer Matrix aus p17-Proteinen umgeben ist (Abb.1). In diesem Kern befinden sich zwei einzelsträngige RNA-Genome (ssRNA), sowie die viralen Enzyme Protease (p12), Reverse Transkriptase/-RNaseH (p51, p66) und Integrase (p32). Der Durchmesser des Virions, das von einer Lipidmembran umgebenen ist, beträgt ca. 120 nm. In der Phospholipiddoppelschicht befinden sich ca. 70 Glykoproteinkomplexe, bestehend aus dem Transmembranprotein gp41 (TM) und dem Oberflächenprotein gp120 (SU). Das gp120 spielt eine entscheidende Rolle in der Erkennung der spezifischen Rezeptoren auf der Oberfläche der Zielzelle. Abb.1 Schematische Darstellung eines HIV-1 Partikels. Hüllmembran mit externem Glykoprotein (gp120, SU) und transmembranem Glykoprotein (gp41, TM), Matrixprotein (p17, MA), Kapsidprotein (p24, CA), Nukleokapsidproteine (p7, NC), Linkprotein (p6, LI), Lateralkörper, Enzyme: Integrase (p32, IN), Protease (p12, PR), Reverse Transkriptase/ RNaseH (p51/ p66 *, RT/ RH) [11]. Das gesamte HIV-1 RNA Genom ist ca. 9,5 kB groß und enthält neun überlappende Leserahmen (open reading frames, ORF). Die Genexpression von HIV-1 wird von der 5´-LTR (long terminal repeat), die als Promoter-/ Enhancer-Region fungiert, gesteuert. Die Transkription wird in der 3´-LTR terminiert. Das HIV-1 Genom beinhaltet die offenen Leserahmen gag, pol und env. Diese offenen Leserahmen sind typisch für alle Retroviren und 2 Einleitung kodieren die Strukturproteine (gruppenspezifische Antigene (gag)), die Enzyme (Polymerase (pol) etc.) und Hüllglykoprotein (env). Der Leserahmen für env beinhaltet sechs konservierte (C1-C6) und fünf hypervariable Regionen (V1-V5). Die V3-Region (V3-Loop) interagiert mit dem Haupt- und Korezeptor auf der Zielzelle und initiiert eine Kaskade von Konformationsveränderungen, die essentiell für das Einschleusen des Viruspartikels in die Zelle ist. Mutationen in dieser Region können zu einem Wechsel in der KorezeptorVerwendung führen. Darüber hinaus kodiert das Genom des komplexen Retrovirus HIV-1 die essentiellen regulatorischen Proteine Tat und Rev, wobei Tat als Transkriptionsaktivator dient und Rev für das Splicing und den Transport der viralen RNA aus dem Zellkern ins Zytoplasma verantwortlich ist. Die akzessorischen Proteine (Vif, Vpr, Vpu, Nef) beeinflussen die Infektiosität und die Pathogenität des Virus [12-13]. Abb.2 Die genomische Organisation/ Struktur von HIV-1 5’LTR-gag-pol-vif-vpr-tat-rev-env-nef-3’LTR; ORF sind teilweise überlappend 1 1.3 Der Verlauf der HIV-Infektion Der natürliche Verlauf einer HIV-Infektion wird in drei unterschiedliche Phasen eingeteilt. Zu Beginn tritt die hochvirämische akute Phase auf, gefolgt von einer klinisch asymptomatischen Phase (klinische Latenz) bis hin zur finalen, klinisch symptomatischen Phase, die AIDS-Stadium genannt wird [14] (Abb. 3). Die erste Phase ist definiert als der Zeitraum zwischen der Infektion und der Antikörperentwicklung (Serokonversion). Nach der erfolgten Ansteckung mit HIV-1 (Infektionszeitpunkt) erleiden ca. 30 % der Patienten ein kurzzeitiges akutes Syndrom, was sich durch eine hohe Anzahl von Viren im Blut (Virämie) und Grippe-ähnlichen Symptomen auszeichnet. Während dieser ersten Phase wird eine HIV1 spezifische Immunantwort induziert. ___________________________________________________________________________ 1 http://www.hiv.lanl.gov/content/sequence/HIV/MAP/landmark.html 3 Einleitung Die Zellzahl der CD4+-T-Zellen/ CD4+-T-Lymphozyten, die Zielzellen von HIV, sinken rapide ab und die Konzentration der Viruspartikel im Plasma nimmt zu (Abb. 3). Nach der Entwicklung der HIV-spezifischen Antikörper im Patienten nimmt die CD4-Zellzahl wieder zu, erreicht jedoch nie mehr den Ausgangswert und es beginnt die klinisch stabile, asymptomatische Phase (Latenzphase). Diese asymptomatische Phase kann bis zu mehreren Jahren andauern, je nachdem wie die virale Fitness (Replikationsrate von HIV) und der genetische Hintergrund des Wirts sind. Diese Phase zeichnet sich durch eine dauerhafte niedrige Viruskonzentration sowie eine relativ stabile Zahl an CD4+-T-Lymphozyten aus. Im Spätstadium der Infektion fällt die Zahl der CD4+-T-Zellen unter 200 Zellen/µl und die Viruslast steigt erneut enorm an. Dieses AIDS-Stadium ist durch die niedrige Anzahl der CD4+-T-Zellen und erneuten Anstieg der Viruslast charakterisiert. Bei unbehandelten Patienten können in diesem klinisch symptomatischen Stadium opportunistische Infektionen, wie Pneumonien, Lymphome und das Kaposi-Sarkom auftreten. Abb.3 Natürlicher Verlauf einer HIV-Infektion Entwicklung der CD4+-T-Zellen ( ) im peripheren Blut (CD4+ Zellen/µl) und Verlauf der Viruslast im Plasma ( RNA Kopien/ml) während des natürlichen Verlaufs einer HIV-1-Infektion. Heutzutage stehen verschiedene Medikamente für die antiretrovirale Therapien zur Verfügung, wodurch die Krankheitsprogression zum AIDS definierenden Stadium verlangsamt wird. Eine hochaktive Kombination von antiretroviralen Medikamenten (HAART, cART) kann die Vermehrung der Viruspartikel erfolgreich verhindern, die Viruslast unter die Nachweisgrenze absenken und die Wahrscheinlichkeit der Transmission der Viruspartikel verringern. Die Reduktion der Viruspartikel im Blut führt zur einer Entlastung des Immunsystems [15]. Eine Heilung der Krankheit ist jedoch nicht möglich. 4 Einleitung 1.4 Der Replikationszyklus von HIV Der Replikationszyklus beginnt mit der Rezeptor-spezifischen Bindung des Viruspartikels an die Wirtszelle. Dabei bindet das virale Hüllglykoprotein gp120 an das monomere zelluläre Protein CD4 auf der Zelle. Mit der dadurch ausgelösten Konformationsänderung im gp120 kommt es im Anschluss zur zusätzlichen Interaktion mit einem Korezeptor auf der Zielzelle. Durch eine weitere Kaskade von Konformationsänderungen folgt die Fusion der Virushülle mit der Zellmembran und das Einschleusen des Viruspartikels in das Zytoplasma (uncoating). Durch die Reverse Transkriptase wird die virale RNA in cDNA umgeschrieben. Die Integrase bindet im Anschluss im Zytoplasma an die virale dsDNA und bildet einen Präintegrationskomplex. Dieser wird in den Zellkern transportiert und die DNA als Provirus in das Genom integriert. Bei der Aktivierung infizierter Immunzellen wird die Transkription des integrierten Provirus aktiviert und die provirale DNA wird in die virale Boten-RNA umgeschrieben (englisch: messenger RNA, mRNA). Die mRNA wird für die folgende Translation der viralen Proteine mithilfe von Rev ins Zytoplasma transportiert [16]. In einem „Selfassembly“-Prozess entstehen neue Viruspartikel an der Zellmembran der infizierten Zelle. Durch Knospung (budding) kommt es zur Freisetzung der Viruspartikel. Mithilfe der Protease werden Vorläuferproteine gespalten; ein Vorgang der zur Reifung der infektiösen Partikel außerhalb der Zelle führt [17]. 5 Einleitung Abb.4 Replikationszyklus von HIV-1 Zu Beginn bindet das Virus an die Rezeptoren auf der Zelloberfläche der T-Zelle, gefolgt von der Fusion der Membranen und Eintritt in das Zytoplasma. Es folgt die Reverse Transkription der viralen RNA und die Integration der dsDNA in die chromsomoale DNA zum Provirus. Die Transkriptionsaktivierung und Translation der Viruspartikel folgen. Es kommt zur Knospung, bei der die Proteine und das konische Core des infektiösen Viruspartikels ausgebildet werden. Die Abbildung wurde aus der Referenz [18] entnommen. 1.5 Der Eintritt von HIV-1 in die Zielzelle mittels Korezeptorbindung Für den Eintritt eines HIV-1 Partikels in die Zielzelle bedarf es zunächst einer spezifischen Bindung des viralen Hüllproteins an die Zelloberfläche der Zielzelle mittels eines zellulären CD4-Rezeptors und eines Chemokinrezeptors. Der CD4-Rezeptor ist der Hauptrezeptor, welcher auf verschiedenen Zellen (CD4+-T-Zellen, Monozyten und Makrophagen) exprimiert wird [19]. Das Virus bindet mit dem immundominanten V3-Loop des externen Glykoproteins gp120 an den monomeren Glykoproteinrezeptor CD4. In nicht infizierten Immunzellen interagiert der CD4-Rezeptor mit dem Haupthistokompatibilitätskomplex II (MHCII) für die Antigenpräsentation. Durch die Bindung von HIV an CD4 wird eine Kaskade an Konformationsveränderungen ausgelöst, die zur Interaktion mit dem Korezeptor und im 6 Einleitung weiteren Verlauf zur Membranfusion durch das transmembrane Glykoprotein gp41 führt. HIV kann verschiedene Korezeptoren benutzen. Die Korezeptoren werden nach den Aminosäuremotiven der natürlichen Liganden in die Gruppen: CXC, CC, CX3C und C unterteilt. Hauptsächlich werden die zwei Korezeptoren CCR5 (CC-Motiv) und CXCR4 (CXCMotiv) verwendet [20]. Viren, die den CCR5-Korezeptor benutzen, was in der frühen Infektionsphase der Fall ist, werden als R5-trope Viren bezeichnet. T-Zell-trope Viren verwenden den CXCR4-Korezeptor für das Anheften, diese Viren treten meist in der späteren Infektionsphase auf (X4-Viren). Während der Infektion findet ein „Shift“ der M-tropen zu den T-tropen HIV-1-Varianten statt. Die R5- und X4-Viren können in einem Patienten auch rekombinieren und dadurch zu einem neuen dualtropen R5X4-Virus mutieren [21]. Weiterhin wurden mehrere Chemokinrezeptoren mit dem CC-Motiv CCR2b, CCR3, CCR6 und CCR8, dem CXC-Motiv CXCR6 (BONZO), dem CX3C-Motiv CX3CR1 (V28) und andere Rezeptoren wie dem GPR15 (BOB) als alternative Korezeptoren identifiziert [22]. Der immundominante V3-Loop des Glykoproteins gp120 ist für die Interaktion zwischen dem viralen Hüllprotein und dem Korezeptor entscheidend. Die Aminosäuren an den Positionen 11, 24 und 25 im V3-Loop bestimmen hauptsächlich die Interaktion mit dem Chemokinrezeptor [23]. Es gibt jedoch Hinweise, dass es weitere Positionen auf dem EnvProtein gibt, die für die Korezeptor Erkennung wichtig sind [24]. Abb.5 Eintritt von HIV-1 in die Wirtszelle Zuerst bindet das virale Glykoprotein gp120 an den CD4 Rezeptor an der Oberfläche der Zielzelle. Danach wird die Korezeptorbindung durch eine Konformationsänderung des Proteins eingeleitet. Nach der Bindung beider Rezeptoren, die für den Eintritt notwendig ist, findet eine weitere Konformationsänderung statt, die die Fusion des gp41 Proteins auslöst. Die virale Membran fusioniert mit der Membran der Zielzelle [25]. 7 Einleitung 1.6 Mutationen im CCR5 Korezeptor und deren Einfluss auf den HIV Krankheitsverlauf Es wurden unterschiedliche genetische Faktoren im menschlichen Genom identifiziert, welche die Empfänglichkeit von HIV-1 reduzieren und zu einer verlangsamten Progression der HIV-1 Infektion führen können [26]. Sehr gut charakterisiert ist eine 32 Basenpaar (Bp) Deletion des CCR5-Korezeptor-Gens, die zu einem verkürzten, nicht funktionellen CCR5Korezeptor führt. Die homozygote Deletion (CCR5Δ32/Δ32) wurde bei 1% der kaukasischen Bevölkerung identifiziert, die heterozygote Deletion (CCR5∆32/wt) tritt bei 5-14% der Bevölkerung auf. Die homozygote Deletion führt zu einem fast vollständigen Schutz gegenüber einer Infektion mit HIV-1, die heterozygote Deletion zu einem verlangsamten Krankheitsverlauf [26-30]. Es sind weltweit nur sehr wenige Individuen bekannt, die trotz der homozygoten Deletion mit HIV-1 infiziert sind [31]. Hütter et al. erreichten im Jahre 2009 eine langfristige Kontrolle, wenn nicht sogar Heilung von HIV-1 durch eine Transplantation von Stammzellen eines homozygoten CCR5Δ32 Spenders auf einen HIV-1 positiven Empfänger mit akuter myeloischer Leukämie [31]. Weitere Mutationen, welche die CCR5-Korezeptor-Funktion beeinflussen, sind bekannt, jedoch hat keine Mutation einen so enormen Effekt wie das CCR5Δ32 Allel [32]. Auch in der deutschen HIV-1 Serokonverterstudie des Robert Koch-Instituts konnten Untersuchungen einen verlangsamenden Effekt der heterozygoten CCR5Δ32 Deletion auf den Krankheitsverlauf zeigen. Bei einem heterozygoten CCR5Δ32 Patienten konnte HIV-1 den CCR5-Korezeptor, allerdings keine alternativen Korezeptoren, für eine Infektion der Wirtszelle nutzen [33-34]. Zudem wurde ein seltener positiver homozygoter CCR5∆32/Δ32 HIV-1 Patienten identifiziert [27]. 8 Einleitung Abb.6 Charakteristische Mutationen in der CCR5 Korezeptor Aminosäuresequenz Das Schema zeigt Mutationen in der CCR5-Aminsoäurensequenz in verschiedenen menschlichen Populationen. Die 32 Bp Deletion führt zu einer Leserasterverschiebung bei Aminosäure 185 und resultiert in einem verkürzten Protein, das nicht mehr auf der Oberfläche exprimiert wird [32]. Mit dem roten Pfeil ist die Position im Wildtyp-Protein markiert, an der die 32 Bp Deletion beginnt. 1.7 Antiretrovirale Therapie mit einem CCR5 Korezeptorantagonist Der Korezeptorgebrauch von HIV-1 ist einer der wichtigen Angriffspunkte der antiretroviralen Therapie. Der CCR5-Antagonist Maraviroc wurde für die Anwendung im Jahr 2007 zugelassen (FDA Antiviral Products Advisory Committee, 2007). Es müssen jedoch vor der Behandlung mit einem Korezeptorantagonisten genotypische oder phänotypische Analysen des Korezeptorgebrauchs durchgeführt werden. Wenn ein Patient mit einem CCR5Inhibitor behandelt wird, jedoch ein X4-Virus vorhanden ist, führt dies zu einem Therapieversagen. Im Internet stehen Analyse-Programme, wie zum Beispiel das Geno2pheno[coreceptor] oder das WebPSSM zur Verfügung, mit deren Hilfe der Korezeptorgebrauch von HIV-1 Isolaten anhand der V3-Loop Sequenz ermittelt werden kann. Dabei wird nur die Verwendung der Korezeptoren CCR5 und CXCR4 analysiert aber nicht die von alternativen Korezeptoren erfasst. Für die Bestimmung eines alternativen Korezeptorgebrauchs müssten phänotypische Analysen mit Wirtszellen durchgeführt werden, die alternative Rezeptoren neben CD4 aufweisen. 9 Einleitung 1.8 Aufgabenstellung Für den Eintritt eines HIV-Partikels bedarf es der Bindung des CD4-Rezeptors und eines spezifischen Korezeptors an der Oberfläche der Zielzelle. Die beiden am häufigsten verwendeten Korezeptoren, die HIV für die Infektion der Zielzellen verwendet, sind der CXCR4- und der CCR5-Korezeptor. HIV ist jedoch auch in der Lage über Interaktion mit alternativen Korezeptoren, wie zum Beispiel dem CX3CR1-Korezeptor, seine Zielzellen zu infizieren. In der Serokonverterstudie des Robert Koch-Instituts wurde 2002 ein HIV-1 positiver Patient mit einer 32 Bp homozygoten Deletion (CCR5∆32/∆32) im CCR5-Gen diagnostiziert. Der Korezeptorgebrauch von einem Erstisolat und einem Verlaufsisolat aus dem CCR5Δ32 homozygot defizienten Serokonverterstudienpatienten wurde in der Arbeitsgruppe untersucht. Hierbei wurde festgestellt, dass das Erstisolat den CXCR4- und den CCR5Korezeptor verwenden konnte. Das Verlaufsisolat konnte den CXCR4-Korezeptor, jedoch nicht den CCR5-Korezeptor anwenden. Es war zudem in der Lage, auch die alternativen Korezeptoren, insbesondere CX3CR1, und möglicherweise auch CCR3, CCR8, BONZO und BOB für den Eintritt in die Zielzelle zu verwenden. Daraus wurde geschlossen, dass das Virus seinen CCR5-Tropismus verlor und die Fähigkeit entwickelte, alternative Korezeptoren, wie den CX3CR1-Korezeptor, neben CXCR4 zur Infektion der Wirtszellen zu nutzen [34]. In Examensarbeiten von Can Demiroglu und Myriam Friedel wurden Infektionsversuche mit zwei Virusisolaten des homozygoten CCR5Δ32 Patienten auf dem GHOST-Zellpanel durchgeführt. Diese Reporter-Zelllinie aus den humanen Osteosarkom-Zellen exprimiert den CD4 Rezeptor und einen von 8 Korezeptoren (CXCR4, CCR5, CX3CR1 etc.). Um den Gebrauch der beiden Korezeptoren genauer zu untersuchen wurden Inhibitionsversuche durchgeführt. Es konnte in den Inhibitionsversuchen mit den CXCR4-Antagonisten AMD3100 und T140 und dem natürlichen Liganden von CX3CR1 (Fraktalkin) keine eindeutige Differenzierung zwischen dem CXCR4- und dem CX3CR1-Gebrauch gezeigt werden [35]. Es wurde vermutet, dass die Infektion mit dem CX3CR1-Korezeptor auf die niedrige Hintergrundexpression von CXCR4 in der Zelllinie zurückzuführen ist. Das Ziel dieser Arbeit bestand darin, die CXCR4-negative Glioma Reporterzelllinie U87.CD4 stabil mit dem alternativen CX3CR1-Korezeptor-Gen zu transduzieren, um mit diesen Zellen den Korezeptorgebrauch der Virusisolate aus dem homozygoten CCR5∆32 HIV-Patienten eindeutig zu charakterisieren. Es wurde in der direkt vorangegangenen Bachelorarbeit ein 10 Einleitung pPABE-puro-CX3CR1 Konstrukt für die Transduktion hergestellt und stand somit für die Produktion von Pseudotypviren zur Verfügung [11]. Das CX3CR1-Expressionsvektor mit dem Transgen CX3CR1 sollte mit zwei weiteren Vektoren, dem Verpackungsvektor und dem Expressionsvektor für das CMV-Hüllprotein, zur Produktion von Pseudotypviren in einer Verpackungszelllinie (293T) verwendet werden. Durch Infektion der U87.CD4 Zelllinie sollte dann das Transgen transduziert und eine stabil CX3CR1 exprimierende Zelllinie selektioniert werden. Es wurden Virusisolate des CCR5∆32/Δ32-Patienten und Referenzviren auf PBMCs vermehrt, um Infektions- und Inhibitionsversuche in dem U87.CD4-Zellpanel (CXCR4, CCR5, CX3CR1) durchzuführen. Als Referenzviren wurde HTLVIIIB (X4-Virus) und BAL (R5-Virus) verwendet. Die env-Sequenzen der vermehrten Virusstocks auf CCR5-Spender-PBMCs wurden kontrolliert, um potentiell auftretende Mutationen bewerten zu können. Das Wachstum der Virusisolate des homozygoten CCR5∆32 HIV-Patienten und der Referenzviren wurde in der phänotypischen Analyse in U87.CD4, -CXCR4, -CX3CR1 und CCR5-Zellen verglichen. Inhibitionsversuche mit dem natürlichen Liganden von CX3CR1 (Fraktalkin) und dem CXCR4-Antagonisten AMD3100 wurden zur spezifischen Analyse des Korezeptorgebrauchs durchgeführt. 11 Material und Methoden 2 2.1 Material und Methoden Material 2.1.1 Blutspender Die in dieser Arbeit verwendeten HIV Isolate wurden in peripheren mononukleären Blutzellen (PBMCs) verschiedener Blutspender vermehrt. Vor Beginn der Infektionskinetiken wurden alle Virusisolate auf PBMCs eines Spenders (D) titriert, um die Infektionsexperimente mit vergleichbaren infektiösen Einheiten durchführen zu können (Tabelle 1). Tabelle 1 Spender Blutspender GRC HIV-1 ELISA CCR5 Genotyp Virusisolat/ Verwendung A 85211776184 negativ wt/∆32 BAL B 85211776190 negativ wt/wt 02-0165, 08-0755 C 85212627300 negativ wt/wt HTLVIIIB D 85212829286 negativ wt/wt Titration 2.1.2 Plasmide Expressionsvektor pBABE-puro-CX3CR1 Addgene, MA, USA pCMV-VSV-G Addgene, MA, USA pUMVC-gag-pol Addgene, MA, USA 2.1.3 Zellen Die humane embryonale Nierenzelllinie HEK 293T Die 293T Zelllinie stammt von humanen, embryonalen Nierenzellen ab und ist mit der temperatursensitiven Mutante des SV40 T-Antigens stabil transformiert. Es handelt sich um eine adhärenten Zelllinie mit einer Generationszeit von 24-30 h. Die Zelllinie wurde vom NRZ für Retrovirologie, Erlangen zur Verfügung gestellt. 12 Material und Methoden U87 abgeleitete Zellen (U87.CD4, U87.CD4.CXCR4, U87.CD4.CCR5) Die U87.CD4 Zelllinie leitet sich von menschlichen epithelialen Glioblastomazellen ab, die stabil mit dem retroviralen Vektor pMV7neo.CD4 transduziert wurden [36]. Die stabile CD4 Expression wurde durch die Selektion mit Geniticin (G418) erreicht. In dieser Arbeit wurden CXCR4 und CCR5 exprimierende U87.CD4 Zellen und auch die parentale Zelllinie U87.CD4 verwendet. Das Zellpanel stammte aus dem NIH AIDS Research and Reference Program und wurde freundlicherweise von Ursula Dietrich, Georg Speyer-Haus, Frankfurt zur Verfügung gestellt. Die parentale U87.CD4 Zelllinie wurde in dieser Arbeit für die Herstellung der U87.CD4.CX3CR1 Zelllinie verwendet. 2.1.4 Virusisolate Die Virusisolate wurden auf den PBMCs vermehrt (Tabelle 2). Die Abnahmen erfolgten an den Tagen 5, 10 bzw. 11, sowie an den Tagen 14 bzw. 15. Die p24 Konzentration der Virusabnahmen wurde ermittelt. Von den Virusstocks mit den höchsten p24 Werten wurde der infektiöse Titer (TCID50/ml) bestimmt. Die Titration wurde mit dem p24-Antigentest ausgewertet. Tabelle 2 Name Virusisolate Virus Korezeptorgebrauch verwendeter Virusstock CCR5 zur Patient Vermehrung Genotyp dpi, Erntedatum HTLVIIIB Referenzlaborstamm CXCR4 d10, 14.06.2010 unbekannt BAL Referenzlaborstamm CCR5 d25, 29.06.2010 unbekannt 02-0165 Primärisolat zu bestimmen d10, 17.07.2008 ∆32/ ∆32 zu bestimmen d10, 17.07.2008 ∆32/ ∆32 (Erstisolat) 08-0755 Primärisolat (Verlaufsisolat) 13 Material und Methoden 2.1.5 Chemikalien/ Feinchemikalien/ Reagenzien Agarose (Ultra-Pur) peQlab, Erlangen, Deutschland AMD 3100, Octahydrochloride Sigma-Aldrich, Schnelldorf, Deutschland CombiZyme™ DNA Polymerase Invitek, Berlin, Deutschland dNTP Invitrogen GmbH, Karlsruhe, Deutschland DMSO Merck, Darmstadt, Deutschland Empigen BB 30% Merck, Darmstadt, Deutschland Ethanol 99,8% Carl Roth GmbH + Co.KG, Karlsruhe, Deutschland Ethidiumbromid Roth, Karlsruhe, Deutschland Expand High Fidelity DNA Polymerase Roche, Grenzach-Wyhlen, Deutschland Fötales Kälberserum Biochrom AG, Berlin, Deutschland Ficoll-Paque Plus GE Healthcare, Uppsala, Schweden Fractalkine (Chemokin Domain) R&D Systems, MN, USA FuGene Roche, Mannheim, Deutschland GeneRuler™ 100bp Ladder Fermentas, St. Leon-Rot, Deutschland GeneRuler™ 1kb Ladder Fermentas, St. Leon-Rot, Deutschland Geneticin (G418-Sulfat) Gibco BRL, Eggenstein, Deutschland IL-2 Sigma-Aldrich, Schnelldorf, Deutschland L-Glutamine PAA Laboratories GmbH, Cölbe Low DNA Mass™ Ladder Invitrogen GmbH, Karlsruhe, Deutschland p24-Antigen Aalto Bio Reagents, Dublin, Irland 14 Material und Methoden p24 Konjugat (Alkalische Phosphatase) Aalto Bio Reagents, Dublin, Irland Penicillin/Streptomycin Gibco BRL, Eggenstein, Deutschland PHA (Phytohämagglutinin) Sigma-Aldrich, Schnelldorf, Deutschland Platinum Taq DNA Polymerase Invitrogen, Karlsruhe, Deutschland Polybren Sigma-Aldrich, Schnelldorf, Deutschland Polyethylenimin PEQLAB, Erlangen, Deutschland Puromycin Sigma-Aldrich, Schnelldorf, Deutschland RNasin (RNase-Inhibitor) Promega GmbH, Mannheim, Deutschland RPMI-1640 Gibco BRL, Eggenstein, Deutschland Schafserum Sigma-Aldrich, Schnelldorf, Deutschland Superscript II Reverse Transkriptase Invitrogen, Karlsruhe, Deutschland TROPIX CSPD Saphire Substrat ABI, Weiterstadt, Deutschland Trypsin 250 Difco, USA Tween®20 Fluka, Schweiz Versen (0,1% EDTA in PBS) Gibco BRL, Eggenstein, Deutschland 2.1.6 Puffer 6x Ladepuffer für Agarose Gelelektrophorese Ficoll 400 25%; Bromphenolblau 0.25%; TAE Puffer 1x PBS “ohne” (pH 7.2) 137 mM NaCl; 2.7 mM KCl; 4.3 mM Na2HPO4; 1.47 mM KH2PO4 RCLB 1.3 mM EDTA; 10 mM KHCO3; 155 mM NH4Cl TBS (10x) 144 mM NaCl; 25 mM Tris 15 Material und Methoden TAE Puffer (10x) 400 mM Tris-HCl; 10 mM EDTA; 500 mM Sodium Acetat Tropix Waschpuffer 200 mM Tris-HCl; 10 mM MgCl2 2.1.7 Antikörper Anti-CCR5-Antikörper (PE) BD Biosciences, CA, USA Anti-CD4-Antikörper (FITC) BD Biosciences, CA, USA Anti-CX3CR1-Antikörper (PE) BioLegend, CA, USA Anti-CXCR4-Antikörper (PE) BD Biosciences, CA, USA Anti-p24-Antikörper Aalto Bio Reagents, Dublin, Irland 2.1.8 Primer Tabelle 3 Primer Alle Oligonukleotide wurden von der Firma Metabion GmbH, 82152 Martinsried, Deutschland synthetisiert. Name Sequenz Verwendung Lokalisation* CCR5Δ32s 5'-CTT CAT CAT CCT CCT GAC AAT CG-3' CCR5 PCR 708-730 CCR5Δ32as 5'-GAC CAG CCC CAA GAT GAC TAT C-3' CCR5 PCR 948-969 6537s 5'-AAT GTC AGC ACA GTA CAA TGT ACA C-3' env gp120 PCR/ Sequenzierung 7254as 5'-TCA TAT CTC CTC CTC CAG GTC TGA A-3' env gp120 PCR 5asc 5'-TCC TTS GAT GGG AGG GGC ATA CAT TGC-3' env gp120 PCR 2as 5'-AGA AAA ATT CCC CTC CAC AAT TAA-3' env gp120 Sequenzierung 2s 5'-TTA ATT GTG GAG GGG AAT TTT TCT-3' env gp120 Sequenzierung 6945-6969 7650-7626 7547-7521 7374-7351 7351-7374 *Lokalisation in HXB2 Acc.No. K03455 2.1.9 Kits BigDye Terminator v 3.1 Cycle Sequencing Kit ABI, Weiterstadt, Deutschland QiAmp DNA Mini and Blood Mini Kit QIAGEN GmbH, Hilden, Deutschland QiAmp Viral RNA Mini Kit QIAGEN GmbH, Hilden, Deutschland 16 Material und Methoden 2.1.10 Kunststoffmaterialien CentriSpin®-10 Columns GENAXXON bioscience, Ulm, Deutschland Corning Costar Polystyrene 96-Loch Platten Aalto Bio Reagents, Dublin, Ireland Cryokonservierungsröhrchen (1,5 ml) Sarstedt, Nümbrecht, Deutschland Zentrifugenröhrchen (15 ml, 50 ml) TPP Techno Plastic Products AG, Trasadingen, Schweiz Mikropipetten (2, 10, 20, 100, 200, 1000 μl) Eppendorf, Hamburg, Deutschland Parafilm M Pechiney, IL, USA Pasteur Pipetten Micro Bio Tec Giessen, Deutschland PCR-Reaktionsgefäße (0,2 ml) Rapidozym, Luckenwalde, Deutschland Polystyrol-Round-Bottom-Tube (5 ml) Becton Dickinson Labware, NJ, USA Pipetten (5 ml, 10 ml, 25 ml) Carl Roth GmbH + Co.KG, Karlsruhe, Deutschland Reaktionsgefäße (1,5 ml, 2 ml) Sarstedt, Numbrecht, Deutschland Zellkulturflasche (25 cm2, 75 cm2, 125 cm2) Thermo Scientific (nunc), Roskilde, Dänemark Zellkulturtestplatten (6-Loch, 24-Loch, 96-Loch) TPP Techno Plastic Products AG, Trasadingen, Schweiz 2.1.11 Geräte Avanti® J-E Zentrifuge Beckman Coulter GmbH, Krefeld, Deutschland CO2-Zellkulturinkubator Thermo Fisher Scientific, MA, USA FACS Calibur® System BD Biosciences, CA, USA Geldokumentationssystem E.A.S.Y. RH-3 Herolab, Wiesloch, Deutschland Gelelektrophorese-Kammer HorizonR58 Gibco BRL, Eggstein, Deutschland Heraus Multifuge X3FR Zentrifuge Thermo Fisher Scientific, MA, USA 17 Material und Methoden Lichtmikroskop Carl Zeiss, Jena , Deutschland Mastercycler® pro S Eppendorf, Hamburg, Deutschland Mikrobiologische Sicherheitswerkbank Karl Bleymehl Reinraumtechnik, Pier, Deutschland Mikrozentrifuge Carl Roth GmbH + Co.KG, Karlsruhe, Deutschland NanoDrop™ND1000 Thermo Fisher Scientific, MA, USA Reinluftwerkbank (Zellen, Virus ) Sicherheitsklasse II Thermo Fisher Scientific, MA, USA 2.1.12 Software E.A.S.Y Win 32 Herolab, Wiesloch, Deutschland Lasergene 8 DNAStar Inc., WI, USA BD CellQuest Pro BD Biosciences, CA, USA FlowJo Ver. 8.6.1 Tree Star Inc., Ashland, OR, USA Magellan 6.3 Tecan Trading AG, Schweiz BioEdit 7.0.9 Ibis Biosciences, CA, USA 2.2 Methoden 2.2.1 Kultivierung der 293T-Zellen Die Kultivierung der 293T-Zellen erfolgte in einem Vollmedium aus D-MEM mit 10% FKS (fötalem Kälberserum) und 200 mM L-Glutamin bei 37°C und einem Kohlenstoffdioxidgehalt von 5%. Die Zellen wurden zweimal in der Woche in einem Verhältnis von 1:10 in 75 cm2 Flaschen passagiert und dabei mit Hilfe einer 0,2% Versen-Lösung vom Boden der Zellkulturflaschen gelöst. 2.2.2 Kultivierung und Wachstum der U87.CD4 Zellen Die adhärente Zelllinie U87.CD4 wurde mit D-MEM und 15% FKS, 200 mM L-Glutamin und 300 µg/ml Geniticin kultiviert. Die Inkubation der Zellen erfolgte bei 37°C in einer 5%-igen CO2 haltigen Atmosphäre. Zum Umsetzen wurden die Zellen mit PBS „ohne“ gewaschen und 18 Material und Methoden mit Versen (5 mM EDTA in PBS) abgelöst. Eine Aufteilung der Zellen 1:5 wurde durchgeführt. Die Zelllinie U87.CD4.CXCR4, U87.CD4.CCR5 und U87.CD4.CX3CR1 wurden mit den Antibiotika Geniticin (300 µg/ml) und Puromycin (1 µg/ml) kultiviert. Bei der Transduktion und den Infektions- bzw. Inhibitionsversuchen wurde das Medium mit 25 mM HEPES versetzt, um den pH-Gehalt im Medium aufrecht zu erhalten (pH-Wert: 7,5). 2.2.3 Isolierung von PBMCs aus Buffy Coats PBMCs wurden für die Virusvermehrungen und die Titrationen verwendet und aus Buffy Coats von gesunden Spendern isoliert. Die Buffy Coats stammen vom Blutspendedienst DRK Heckeshorn. Das Buffy Coat enthält alle zellulären Bestandteile von EDTA-Blut. Das Plasma und ein Teil der Erythrozyten wurden vorab entfernt. Für die Isolierung der PBMCs wurde das Buffy Coat im Verhältnis 1:3 mit PBS (Phosphat gepufferte Kochsalzlösung) versetzt. Falcon Röhrchen (50 ml) wurden bereitgestellt, mit 15 ml Ficoll gefüllt und vorsichtig mit je 30 ml verdünnten Buffy Coat überschichtet. Es folgte ein Zentrifugationsschritt für 45 min, 509 g bei Raumtemperatur (RT) ohne Bremse. Während des Zentrifugationsschrittes trennten sich die PBMCs von den Erythrozyten und Granulozyten und bildeten eine Bande über dem Ficollkissen. Die PBMCs-Bande wurden vorsichtig mit einer sterilen Pipette in neue Falconröhrchen überführt und anschließend mit PBS auf ein Volumen von 50 ml aufgefüllt. Ein Zentrifugationsschritt von 10 min, 509 g bei RT diente zur Reinigung der Zellen vom restlichen Ficoll. Es erfolgte optional eine Behandlung mit RCLB (Red Cell Lysis Buffer), um die restlichen Erythrozyten zu entfernen, falls diese sich im Pellet zeigten. Dazu wurden die PBMCs mit 10 ml RCLB für 15 min bei RT versetzt und zwei weitere Waschschritten mit PBS (+ 1% FKS) folgten. Die gereinigten Zellen wurden in 5 ml RPMI mit 10% FKS und 200 mM L-Glutamin aufgenommen und die Zellzahl pro ml in einer Neubauer Zählkammer bestimmt. Für die Bestimmung der Zellzahl wurde die Zellen zuvor 1 zu 10 in RPMI+-Medium verdünnt und 10 µl der Verdünnung auf die Zählkammer pipettiert. Die Konzentration der Zellen ergibt sich aus der Multiplikation von Zellzahl, Verdünnungsfaktor und Kammerfaktor (104). ℎ: !ℎ ℎ = ∙ 1000 = ! " ü(1100) ∙ ℎöℎ(110) 19 Material und Methoden Für die Virusvermehrung wurden 1x105 PBMCs in eine 25 cm2 Zellkulturflasche überführt und mit 5 µg/ml PHA (Phytohämagglutinin) in RPMI (+ 20% FKS, 1% L-Glu) für 48 Stunden versetzt. PHA ist ein Pflanzenlektin, was einen stark mitotischen und polyklonal stimulierend verklumpenden Effekt auf Leukozyten hat, wodurch die Monozyten ausdifferenzieren. Vor der Virusinfektion erfolgte ein Mediumwechsel zu Interleukin-2 (IL-2) haltigem Medium, das während der gesamten Infektionsdauer verwendet wurde (Anregung zur HIVProduktion). Hierzu musste vorher das PHA von den PBMCs entfernt werden und diese mit IL-2 (1 ng/ml) während der gesamten Infektionsphase/Kultivierung versetzt werden. Die zu diesem Zeitpunkt nicht benötigten Zellen wurden im Freezemix (RPMI +10% FKS, 1% L-Glu und 10% DMSO) nach dem Mr. Frosty Protokoll in Cryoröhrchen eingefroren und nach der kontrollierten Absenkung der Temperatur auf -70°C in flüssigem N2 gelagert (-168°C). 2.2.4 Virusvermehrung Zur Vermehrung der Virusisolate wurden PBMCs in einer 25 cm2-Zellkulturflasche mit 2 bis 3 ml Virusstock infiziert. Das Zellkulturmedium wurde in definierter Menge bis zu einem Restmedium von ca. 2 ml von den Zellen abgenommen. Nach Zugabe von 2 ml Virus (02-0165, 08-0755, HTLVIIIB) und 3 ml (BAL), wurde nach einer Inkubationszeit von 3 h bei 37°C mit Medium (RPMI, + 20% FKS, 1% L-GLu) auf 10 ml aufgefüllt. Die Virusvermehrung dauerte bis zu 15 Tagen. An Tag 4 p.i. („post infectionem“) wurden die Zellen 1:2 aufgeteilt und in eine weitere 25 cm2 Zellkulturflasche überführt. 7 Tage nach der Infektion wurden die Zellen erneut 1:2 aufgeteilt und in 75 cm2 Zellkulturflaschen verbreitet. Bei der Aufteilung der Zellen wurden jeweils neue PBMCs zugegeben, sodass die Zellzahl konstant bei ca. 1x107 Zellen blieb. Zudem wurden an Tag 0, 3, 7, 10 und 14/15 jeweils 500 µl des Virusüberstands entnommen, mit 125 µl Empigen (Endkonzentration 1%) versetzt, um die HIV-Partikel zu inaktivieren, und für den p24-Antigen-Test bei -20°C weggefroren. An Tag 10 wurden jeweils 5 ml aus den 25 cm2 Flaschen Virusüberstand entnommen, in ein Falcon-Röhrchen überführt und bei 161 g für 10 min (4°C) zentrifugiert, um Zelltrümmer zu entfernen. Es wurden von dem Virusüberstand fünf Aliquots à 1 ml auf Eis hergestellt und bei -70°C weggefroren. Die Endabnahme erfolgte an Tag 14 oder 15. Die Virusproduktion wurde mittels p24-Antigen-Test einmal die Woche kontrolliert. Bei einer M.O.I. von 0,01 ergab sich in der Regel eine maximale Virusproduktion an Tag 10-14 p.i.. 20 Material und Methoden 2.2.5 Virustitration Der infektiöse Titer des Virus-Arbeitsstocks wurde gemäß der Spearman-Kärber-Methode (Spearmen C., 1909) bestimmt, um die Infektionsexperimente zu standardisieren. Zunächst wurden unbehandelte, eingefrorene PBMCs in RPMI (+ 10% FKS + 1% L-Glu) aufgenommen und für 24 h in 5 µg/ml PHA stimuliert. Die Zellen wurden bei 161 g und Raumtemperatur für 10 min zentrifugiert, um das PHA-haltige Medium zu entfernen. Es folgte die Aufnahme der Zellen im RPMI+-Medium versetzt mit 1 ng/ml Interleukin (IL-2), die Bestimmung der Zellzahl und die entsprechende Aufteilung der Zellen von 5x105 Zellen in 100 µl pro Loch in einer 96-Loch-Platte für weitere 24 h. Nach diesen 24 h folgte die Infektion der Zellen mit 100 µl der Virusverdünnung. Die Verdünnungen mit RPMI+-Medium des Virusstocks wurden zunächst auf Eis, beginnend mit einer 1:10 Verdünnung und einer seriell folgenden 1:3 Verdünnung, hergestellt. Nach Zugabe der Virusverdünnungen zu den Zellen wurden die Kulturen sieben Tage bei 37°C und 5% CO2 kultiviert. Abschließend wurden 125 µl des Überstands jeder Verdünnungsstufe mit 5% Empigen versetzt, was zur Inaktivierung des Virus führte. Der Überstand wurde für die Messung des p24 Antigen ELISA und des TCID50/ml verwendet. Anhand des p24-Tests wurde bestimmt, bis zu welcher Verdünnungsstufe Virus produziert wurde. Pro Verdünnungsstufe wurden vier Parallelkulturen gemessen. Von den positiv getesteten Kulturen wurden nach der SpearmanKärber-Methode die TCID50/ml berechnet (die Konzentration, bei der die Hälfte aller Testkulturen positiv war). + #$%&'() = #*+ − # -. ℎ/ − 0.53 /2) xk = höchste getestete Verdünnung d = Verdünnungsfaktor ∑+/2) ℎ/ = Anzahl der positiven Wells 2.2.6 p24 Antigen Test Für die Bestimmung des viralen p24 Antigens im Zellkulturüberstand wurde ein In-House-Test mit kommerziellen Bestandteilen von Fa. Aalto verwendet. Der Test basiert auf der Bindung des p24 HIV-1 Antigen an einen Schaf-α-HIV-1-p24-Antikörper. Das 21 Material und Methoden gebundene p24 Antigen wurde durch einen mit alkalischer Phosphatase gekoppelten monoklonalen Antikörper nachgewiesen. Anschließend wurde Substrat dazu gegeben, durch dessen Umsatz die Chemielumineszenz entstand. Die Lumineszenz wurde mit Hilfe eines ELISA-Lesegerätes ermittelt. Zu Beginn wurden 96 Loch-Platten mit polyklonalen Schaf-α-HIV-1-p24-Antikörpern beschichtet. Diese Platten wurden mit 200 µl 1xTBS mit 2% BSA pro Loch beladen und bei -20°C gelagert. Von allen infizierten Zellen wurden 125 µl Überstand mit 1% Empigen in verschiedenen Verdünnungen (1:5, 1:10, 1:100, 1:500, 1:1000, 1:5000) gemessen. Zwei unabhängig voneinander hergestellte Konzentrationsstandardreihen von p24-Protein (0,078 ng/ml bis 10 ng/ml) aus rekombinanten p24 HIV-1 Antigen dienten zur Erstellung der Eichkurve für die Bestimmung des Antigengehaltes in den unbehandelten Proben. Jede Testplatte enthielt mehrere Negativkontrollen (RPMI bzw. D-MEM Medium mit 1% Empigen) sowie Leerwerte. Für die Berechnung der p24-Konzentration in den Zellkulturüberständen wurde eine Eichkurve (Regressionsgerade) aus dem Konzentrationsstandard erstellt und die Konzentration anhand der Geradengleichung durch die Auswertesoftware berechnet (Magellan 6.3). Zur Bestimmung des infektiösen Titers (TCID50/ml) von Virusstocks wurde eine qualitative Analyse des p24 Antigens von den seriellen Virusverdünnungen der infizierten Zellkulturüberstände durchgeführt. 2.2.7 Stabile Transfektion Bei einer stabilen Transfektion liegt das Plasmid extrachromosomal im Zytoplasma der transfizierten Zelle vor und trägt alle regulatorischen Elemente, die zur Transkription und Translation des Fremdgens nötig sind. Für den Versuch wurde das Transfektionsreagenz Polyethylenimine (PEI) verwendet. Die U87.CD4 Zellen wurden 24 h vor der Transfektion mit einer Zellzahl von 4x106 Zellen in eine 75 cm2 Zellkulturfalsche ausgesät und über Nacht bei 37 °C mit 5% CO2 kultiviert. Die Zellen sollten nach 24 h eine Konfluenz von 60–80% erreicht haben. In einem 2 ml Reaktionsgefäß wurde 790 µl NaCl (125 mM) vorgelegt und 24 µl pBABE-puro inklusive CX3CR1-Insert (Klon 15, c=1355,39 ng/µl) und 24 µl PEI (1 µl PEI/µg DNA) zugegeben. Der pBABE-Klon, der verwendet wurde, ist bereits zuvor in der Arbeitsgruppe hergestellt worden und enthält das CX3CR1-Gen (Bachelorarbeit Heller S.; 2011). 22 Material und Methoden Die Zellen wurden mit PBS “ohne“ gewaschen und anschließend mit dem Transfektionsansatz versetzt. Nach 3 h Inkubation wurden die Zellen erneut mit 4 ml PBS “ohne“ gewaschen. Nach der Zugabe von 15 ml Vollmedium erfolgte eine Kultivierung bis zu sieben Tagen. Die Expression wurde mittels Durchflusszytometrie (FACS) nach drei und nach sieben Tagen überprüft. 2.2.8 Transduktion der parentalen U87.CD4 Zelllinie mit dem CX3CR1-Gen 2.2.8.1 Transfektion von drei Vektoren zur Produktion von Pseudotypviren Durch eine Kotransfektion in 293T-Zellen des retroviralen Expressionsvektors pBABE-puroCX3CR1 (A), mit dem Verpackungssignal (Ψ), einem Vesicular Stomatitis Virus env exprimierenden Vektor pCMV-VSV-G (B) und einem Verpackungsvektor pUMVC-gag-pol (C), der die ORFs gag und pol aus dem murinen Leukämievirus (MuLV) exprimiert, wurden Pseudotypviren produziert (Abb. 7 A). Die drei Vektoren wurden zusammen in die humanen 293T-Zellen transfiziert. Für diese Kotransfektion wurde das FuGene-Transfektionsreagenz nach Angaben des Herstellers verwendet. Zu Beginn wurden in Zellkulturplatten mit 6 Vertiefungen (6-Loch-Zellkulturplatte) 5x105 293T-Zellen pro Vertiefung ausgesät. Nach 24 h hatten die Zellen eine Konfluenz von ca. 30 % erreicht, um mit der Vektor-DNA transfiziert zu werden. Es wurde zunächst pro Ansatz 90 µl D-MEM in zwei Eppendorf-Reaktionsgefäße vorgelegt und mit 6 µl FuGene-Transfektionsreagenz versetzt. Zu den Ansätzen wurden 1-2 µg Vektor-DNA pBABE-puro-CX3CR1 (Klon 15) sowie 0,1 µg pCMV-VSV-G und 0,9 µg pUMVC (gag/pol) zugegeben. Nach einer Einwirkzeit von 15 min bei RT wurde der Transfektionsansatz vorsichtig auf die vorbereiteten 293T Zellen getropft. Nach 24 h wurde das Transfektionsmedium entfernt und durch Vollmedium (D-MEM, 15% FKS, 200 nM L -Glu, Geniticin, 25 mM HEPES) ersetzt. Der Transvektor (A) mit dem Transgen (CX3CR1-Gen) enthält ein Verpackungssignal Ψ, sodass durch Einkapselung der viralen Struktur- (B) und Hüllproteine (C) die Verpackung der CX3CR1-RNA erfolgen kann. 24 h und 48 h nach der Transfektion der 293T Zellen wurden 2 ml des Zellkulturüberstandes mit den produzierten Pseudotypviren steril mit einem 45 µm Syringe Filter filtriert. 23 Material und Methoden Expressionsvektor Ψ A Infektion der U87 Zellen (Transduktion von CX3CR1) B Pseudotypvirus im Zellkulturüberstand Abb.7 Transduktion von CX3CR1 mit Pseudotypviren A: Herstellung von Pseudotypviren durch Transfektion A) Transfervektor mit Expressionskassette (CX3CR1-Gen) und Verpackungssignal Ψ, flankiert von viralen LTRs, B) Verpackungsvektor mit MuLV gag-pol-Strukturgenen; C) Hüllprotein-Vektor mit VSV env-Gen. Das Verpackungssignal Ψ im Transfervektor steuert die Verpackung (Strukturproteine) und Einhüllung (Hüllproteine) der CX3CR1-RNA und es erfolgt die Produktion von replikations-defekten Pseudotypviren im Zellkulturüberstand [37]. B: Infektion mit Pseudotypviren Nach der Sterilfiltration der Pseudotypviren werden die Empfängerzellen infiziert. Bei der Infektion der Zielzellen (U87.CD4) wird das Transgen (CX3CR1) übertragen (Transduktion) [37]. 2.2.8.2 Infektion mit Pseudotypvirus Zwei Tage vor der Ernte der Pseudotypviren wurden in einer 6-Loch-Zellkulturplatte 5x105 U87.CD4 Zellen ausgesät, sodass am Tag der Ernte der Pseudotypviren eine Konfluenz der U87.CD4 Zellen von ca. 30% erreicht war. Das Sterilfiltrat der Pseudotypviren wurde zu den ausgesäten U87.CD4-Zellen gegeben; 3 h nach der Infektion erfolgte die Selektion der U87.CD4 Zellen mit dem Antibiotikum Puromycin (1 µg/ml). Im weiteren Verlauf wurden die 24 Material und Methoden Zellen je nach Wachstum auf 12-Loch-Platten bzw. in eine 25 cm2 -Zellkulturflaschen mit dem Selektionsmarker Puromycin kultiviert, sodass das Transgen CX3CR1 zusammen mit dem Gen für die Puromycin-Resistenz selektiert wurde. Die infizierten U87.CD4 Zellen wurden bis zu drei Wochen kultiviert, bis genügend Zellen selektiert waren (ca. 1x106 Z/ml), um diese in Arbeitsstocks à 1 ml einzufrieren (-70°C). Der Erfolg der Transduktion wurde durch Antikörperfärbung von CX3CR1 in der Durchflusszytometrie (FACS) überprüft. 2.2.9 FACS-Analyse Um die Produktion von CD4 und die Korezeptor-Expression zu bestimmen, wurden kommerzielle spezifische Antikörper mit fluoreszierenden Markern verwendet. Die markierten Zellen wurden anschließend in der Durchflusszytometrie (FACS) quantifiziert. Für die FACS-Analyse wurden 2,5x105 Zellen der jeweiligen Zelllinie in 1 ml PBS mit 1% FKS resuspendiert. Endobulin (humanes lgG) wurde für die Inhibition einer unspezifischen FcBindung für 5 min zu den Zellen hinzugefügt. Es folgte die Inkubation der monoklonalen Antikörper (Anti-CX3CR1, Anti-CD4, Anti-CXCR4, Anti-CCR5) für 30 min auf Eis. Es wurde mit Antikörpern in einer 1:5-Verdünnung in PBS mit 1% FKS gearbeitet. Nach der Inkubation wurden die Zellen mit PBS + 1% FKS gewaschen und in 300 µl PBS (1% FKS) aufgenommen. Anschließend wurden die fluoreszierenden Zellen im FACS Calibur System gemessen. Die Größe der Zellen wurde mit dem Vorwärts-Scatter (FSC) ermittelt und die Granularität der Zellen mittels des Seitwärts-Scatters (SSC) bestimmt. Abschließend wurde die Anzahl der fluoreszierenden vitalen Zellen bestimmt. 2x104 Zellen wurden von jeder Probe analysiert. Die Auswertung erfolgte an dem BD FACS-Calibur Durchflusszytometer mit dem Cell-QuestPro-Programm und im Anschluss mit der FlowJo-Software (flow cytometry analysis software). Das Softwareprogramm gibt den Prozentsatz der fluoreszierenden Zellen unter den gemessenen Gesamtzellen wieder. 2.2.10 Infektionskinetik in den U87.CD4-Zelllinien Für die Infektion des U87-Zellpanels wurden von allen vier Zelllinien (U87.CD4 parental, U87.CX3CR1, U87.CXCR4, U87.CCR5) 1x104 Zellen in 250 µl Medium, welches die jeweils erforderlichen Antibiotika (Geniticin, Puromycin) enthielt, in einer 96-Loch-Platte ausgesät. Nach 24 Stunden wurden 200 µl des Medium entnommen. Die Zellen wurden im Anschluss für 3 h mit 100 µl Virussuspension (HTLVIIIB, BAL, 02-0165, 08-0755) mit einer M.O.I. von 0,01 bzw. 0,1 (02-0165, 08-0755) infiziert und im Anschluss wurde die Virussuspension 25 Material und Methoden entnommen und mit 200 µl Mediums aufgefüllt. An den Tagen 5, 8 und 10 wurden jeweils 150 µl Überstand aus den für den jeweiligen Abnahmetag vorgesehen Vertiefungen entnommen und mit 37,5 µl Empigen (1%) versetzt. Die Virusproduktion in den abgenommen Überständen wurden anschließend im p24-Antigen-ELISA quantifiziert. Die p24-Konzentration in Inokulum wurde kontrolliert, war jedoch so gering, im Vergleich zu den tatsächlichen gemessenen Werten an Tag 5, 10, 14 und 24 p.i., sodass sie vernachlässigt wurde. 2.2.11 Inhibitionsversuche mit CXCR4 und CX3CR1 spezifischen Liganden Fraktalkin ist der natürliche Ligand des CX3CR1 Korezeptors und ist in der Lage den CX3CR1 Gebrauch von HIV zu hemmen. AMD3100 ist ein CXCR4 Inhibitor, der CXCR4 vermittelte Infektionen verhindert. Es wurden von den Zelllinie U87.CXCR4 und U87.CCR5 jeweils 1x104 Zellen/Loch in einer 96-Loch-Platte ausgesät und nach 24 h mit 100 µl Medium mit steigender Konzentration von AMD3100 und Fraktalkin (1 nM, 10 nM, 100 nM, 500 nM) für 30-45 min vorinkubiert. Es folgte eine Infektion der Zellen mit den Viren HTLVIIIB, BAL, 02-0165 und 08-0755 mit einer M.O.I. von 0,01 bzw. M.O.I. 0,1 für 3 h. Nach der Inkubationsphase von drei Stunden wurden die Vertiefungen mit frischem Medium, welches die erforderlichen Konzentrationen von AMD3100 bzw. Fraktalkin enthielt, auf ein Volumen von 250 µl aufgefüllt. An den folgenden Tagen 5, 8, 10/11 wurde 150 µl Überstand der infizierten Vertiefungen abgenommen, mit 37,5 µl Empigen (1%) versetzt und mit 150 µl frisches Medium, was die entsprechenden Konzentrationen der Inhibitoren enthielt, aufgefüllt. Die Virusproduktion wurde wiederum mit dem p24-Antigentest quantitativ bestimmt. Der p24-Wert im Kulturansatz an Tag 10 p.i. ohne Inhibitor wurde für die Inhibitionskinetiken als 100% gesetzt. 2.2.12 DNA Extraktion aus PBMCs Zur DNA-Extraktion aus PBMCs wurde das QIAamp Blood Mini Kit entsprechend der Anleitung des Herstellers verwendet. Es wurden 5x106 PBMCs zu Proteinase K gegeben und mit 200 µl Lysispuffer (AVL) für 10 min bei 65°C lysiert. Nach Zugabe von 200 µl Ethanol (99%) wurde das Lysat auf eine Säule überführt. Die an die Säule gebundene DNA wurde mit je 500 µl Waschpuffer AW1 und AW2 gewaschen. Die Elution erfolgte mit 200 µl Elutionspuffer (AVE). Anschließend wurde die Konzentration der extrahierten DNA mit Hilfe 26 Material und Methoden des NanoDrop Spektrometers (260–280 nm) bestimmt. Die Proben wurden im Anschluss bei -70°C gelagert. 2.2.13 RNA-Extraktion aus Virusstocks Zur Extraktion der viralen RNA wurde das QIAamp Viral RNA Mini Kit verwendet. Dazu wurden 140 µl Zellkulturüberstand der infizierten PBMCs mit 560 µl AVL-Puffer inklusive t-RNA (10 µg/ml) versetzt und für 10 min bei Raumtemperatur lysiert. Im Anschluss wurden je 560 µl Ethanol (99%) zu den Proben gegeben und anschließend auf eine Säule überführt. Die weitere Extraktion wurde laut Herstellerprotokoll durchgeführt. Abschließend erfolgte die Elution mit 60 µl Elutionspuffer (AVE), die extrahierte RNA wurde portioniert und bei -70°C gelagert. 2.2.14 PCR zur Analyse des CCR5-Genotyps Der CCR5 Genotyp wurde nach dem PCR-Protokoll von Kristiansen analysiert (Kristiansen et al., 2001). Es wurden 100 ng DNA eingesetzt und die PCR wurde mit 1x PCR-Puffer (Invitrogen), 200 µM dNTPs, 3 mM MgCl2, die Primer CCR5Δ32s und CCR5Δ32as mit einer finalen Konzentration von 0,4 µM und 2 U Platinum Taq DNA Polymerase in einem Volumen von 25 µl durchgeführt. Die Amplifikation erfolgte nach folgendem Zyklusprogramm: einem Denaturierungsschritt von 5 min bei 95°C, 40 Zyklen für 30 Sekunden bei 95°C, 30 s bei 58°C und 30 s bei 74°C und endet mit einer fünf-minütigen finalen Elongation bei 72°C. Die Länge der PCR-Produkte wurde in der Agarose-Gelelektrophorese bestimmt. 2.2.15 env gp120 RT-PCR Für die gp120 RT-PCR wurden 9 µl der viralen RNA aus Virusstocks bei 65°C für 10 min gefolgt von einem 3 min Schritt bei 80 °C denaturiert. Nach der Abkühlung auf 4°C wurde ein Mix aus 1x Puffer (Invitrogen), 3 mM MgCl2, 0,5 mM dNTP, 10 ng/µl Primer env7254asc, 5 mM DTT, 20 U RNasin und 100 U/ml Superscript II (Invitrogen) zugegeben, sodass ein finales Volumen von 21 µl vorlag. Die cDNA-Synthese erfolgte für eine Stunde bei 42°C und die Reverse Transkriptase wurde bei 96°C für 10 min inaktiviert. Für die Amplifikation der gp120 cDNA wurde das Roche High Fidelity-System verwendet. Eine semi-nested PCR wurde durchgeführt, bei der 10 µl des cDNA templates, 1x Roche PCR-Puffer 3, 0,2 mM dNTPs, 2,5 mM MgCl2, die Primer 6537s und 7254as (Tabelle 3), jeweils mit einer Konzentration von 0,25 μM, 0,5 μl DMSO und 1,75 U DNA Polymerase (Roche 1) in einem finalen Volumen von 27 Material und Methoden 50 μl eingesetzt wurde. Für ür die innere PCR wurden 2,5 µl des ersten PCR-Ansatzes PCR mit 1x Roche 3 PCR-Puffer, Puffer, 0,2 mM dNTP, 2,5 mM MgCl2, Primer 6537s und 5as (je 0,25 μM), 0,5μl DMSO und 1,75 U DNA Polymerase (Roche 1) versetzt und amplifiziert. Die Denaturierung erfolgte für 2 Minuten bei 94°C, gefolgt von 30 Zyklen bei 49°C für 1 Minute, 2,5 Minuten bei 65°C und letztlich die Elongation für 10 Minuten bei 68°C. Die qualitative qualitative Analyse erfolgte im anschließenden Agarose-Gel. Gel. In Abb.8 ist eine schematische Darstellung der HIV-1 HIV env-PCR dargestellt. Abb.8 Schematische Darstellung der HIV-1 env Region und den Produkten der envgp120C2V5 PCR 2.2.16 DNA Agarose-Gelelektrophorese Gelelektrophorese Die Agarose-Gelelektrophorese Gelelektrophorese wurde verwendet, um die PCR-Produkte PCR Produkte zu visualisieren und ihre Länge zu bestimmen. Die Auftrennung der DNA-Moleküle DNA Moleküle erfolgte in einem elektrischen Feld, bei dem die DNA-Moleküle Moleküle aufgrund der der negativen Ladung zum Pluspol wandern. Kleinere Moleküle wandern schneller in der Gelmatrix als die großen Moleküle. Die Größe bzw. Konzentration der aufgetrennten Proben wurde anhand eines GrößenGrößen bzw. Konzentrationsstandards ermittelt. Ethidiumbromid diente diente zum Sichtbarmachen der DNADNA Banden im Gel. Es interkaliert in die ds-DNA ds DNA und fluoresziert bei einer Bestrahlung des Gels mit ultraviolettem Licht (260 nm). Der Puffer für die Elektrophorese bestand aus 1x TAE mit 0,5 µg/ml Ethidiumbromid, auch das Gel enthielt 0,5 µg/ml Ethidiumbromid. Eine 1 kBpkBp Leiter (Fermentas) diente als Größenstandard zur qualitativen Analyse. Weiterhin konnte mit einem m Konzentrationsstandard (LML=Low (LML=Low Mass Ladder, Fermentas) eine quantitative Auswertung erfolgen, bei der die Menge der DNA in den DNA-Banden Banden bestimmt wurde. Die Gelelektrophorese erfolgte in einem 1,5% Gel mit 90 V für mindestens 30 Minuten. 28 Material und Methoden 2.2.17 PCR-Produktreinigung und DNA Sequenzierung Die PCR-Produkte wurden mit dem MSB Spin PCRapace Kit nach dem Herstellerprotokoll aufgereinigt. Die PCR-Produkte wurden in 50 µl Elutionspuffer eluiert und bis zur Sequenzierung bei 4°C gelagert. Die Sequenzierung erfolgte nach dem KettenabbruchVerfahren nach Sanger (Zyklussequenzierung mit fluoreszenzmarkierten Terminatoren). Hierzu wurden 10 ng Template-DNA mit jeweils 5 pM Sequenzierungsprimer, 1x Reaktionspuffer und 2 µl Big Dye3.1 in einem finalen Volumen von 10 µl gemischt. Für die Sequenzierung der envC2V5 PCR-Produkte wurden die Primer 6537s, 2s, 2as und 5asc verwendet (Tabelle 3). Das Programm für die Zyklussequenzierung umfasste folgende Schritte: 2 min bei 96°C für die initiale Denaturierung, 40 Zyklen von 10 s bei 96°C, 5 s bei 55°C und 4 min bei 60°C. Die Sequenzanalyse erfolgte im zentralen Sequenzierlabor des Robert Koch-Instituts. Hier wurden die Sequenzprodukte mit Sephadex (G50) aufgereinigt und durch Kapillarelektrophorese aufgetrennt. Die Fluoreszenz der Terminatoren wurde angeregt, detektiert und als Chromatogramm ausgegeben. 2.2.18 Sequenzauswertung Die Auswertung der Einzelsequenzen erfolgte mit SeqMan aus dem Programmpaket der DNAStar Lasergene Software. Aus vier Einzelsequenzen des env-PCR-Produktes wurde eine Konsensussequenz als Contig erzeugt, die später als FASTA-Datei gespeichert wurde. Bei HIV können Variantenmischungen vorliegen, die bei der Sanger-Sequenzierung ab einem Anteil von 20-25% in der Viruspopulation als Basenmischung in der Sequenz detektiert werden. Bei einer Variantenmischung kommt es zu Überlagerung von Fluoreszenzsignalen, da Sequenzmoleküle gleicher Länge mit verschiedenen Basen terminiert werden. Die Varianten wurden nach den entsprechenden IUB/IUPAC Codes (International Union of Biochemists/ Pure and Applied Chemistry) mit definierten Buchstaben benannt. Es musste bei der Auswertung beachtet werden, dass die Mischpositionen in mindestens zwei verschiedenen Sequenzen vorliegen und dass die Einzelsequenzen in dem Bereich kein Hintergrundrauschen aufweisen. Beim Vorliegen einer Mischung setzt das Programm an dieser Stelle ein X für die translatierte Aminosäure ein. 29 Ergebnisse 3 3.1 Ergebnisse Herstellung einer U87.CD4 Zelllinie mit stabiler CX3CR1 Expression Um den Korezeptorgebrauch der Verlaufsisolate des CCR5Δ32/Δ32 homozygot defizienten Patienten zu untersuchen, musste zunächst eine Zelllinie hergestellt werden, die den Korezeptor CX3CR1 stabil exprimiert und keine CXCR4 Hintergrundexpression zeigte. Hierfür wurde zunächst versucht, ob bei einer direkten Transfektion des CX3CR1 Expressionsvektors und (pBABE-puro.CX3CR1, Klon 15, Bachelorarbeit Stephanie Heller) der parentalen U87.CD4 Zelllinie mit dem Transfektionsreagenz Polyethylenimin und anschließender Selektion der transfizierten Zellen mit dem Antibiotikum Puromycin, stabil wachsende Zellen erhalten wurden. In der FACS-Analyse konnten nach Anfärben mit dem Anti-CX3CR1-Antikörper keine CX3CR1+-Zellen identifiziert werden. Nach sieben Tagen Kultivierung waren keine vitalen Zellen mehr vorhanden. Daher wurde wie ursprünglich geplant, eine Infektion mit Pseudotypviren, die das CX3CR1Transgen trugen, vorgenommen. Zur Herstellung der Pseudotypviren wurden zunächst 293TZellen mit dem pBABE-puro-CX3CR1 Expressionsvektor, der das retrovirale Verpackungssignal (ᴪ) kodiert, dem Verpackungsvektor pUMVC-gag-pol, das die HIV-1 gag und pol-Proteine kodiert, und dem Expressionsvektor für das CMV-Hüllprotein (pCMV-VSVG) kotransfiziert. Durch Rekombination in der 293T-Zelle und ausgelöst durch das retrovirale Verpackungssignal (Ψ) entstanden Viruspartikel mit dem CMV-Hüllprotein und dem CX3CR1Gen in der verpackten viralen RNA. Die Pseudotypviren wurde aus dem Zellkulturüberstand geerntet und damit die parentale U87.CD4 Zelllinie infiziert. Bei dieser Infektion mit den Pseudotypviren können die Zielzellen nur einmal infiziert werden (single cycle infection), da aufgrund der fehlenden Leserahmen der HIV-Proteine keine neuen infektiösen Partikel gebildet werden können. Durch den Infektionsprozess wurde das Transgen CX3CR1 in mehr Zielzellen eingeschleust als bei einer Transfektion. Nach der Infektion mit Pseudotypviren wurden CX3CR1-exprimierende Zellen wiederum mit dem Antibiotikum Puromycin selektiert und nach vier Passagen im FACS auf die Expression des CX3CR1-Gens überprüft. Bei erfolgreicher Transduktion liegt das Transgen zusammen mit dem selektiven Antibiotikumsmarker integriert im Genom der Zielzelle vor (stabile Zelllinie). Als Negativkontrolle wurde in einem parallelen Ansatz das pBABE-puro-Plasmid ohne CX3CR1Gen mitgeführt. 30 Ergebnisse Die parentale U87.CD4 Zelllinie wurde als Kontrolle im Vergleich zur U87.CD4.CX3CR1 Zelllinie mit dem Anti-CX3CR1-Antikörper angefärbt (Abb.9 A und B). Es war eine deutliche Expression von CX3CR1 in der transduzierten Zelllinie zu erkennen, 91,5% der Zellen waren positiv für CX3CR1. Während die parentalen Zellen keine CX3CR1 Expression zeigten (93,5% negativ). Die Zellen wurden unter weiterer Puromycin- und Geniticin-Selektion (Geniticin für stabile CD4+-Expression) kultiviert und verbreitert, damit eine Arbeitscharge für die folgenden Infektionsexperimente mit den Referenzviren und Virusisolaten aus dem CCR5Δ32 Patienten zur Verfügung stand (Vital-Einfrierung im Freeze-Mix). Abb.9 Nachweis der Expression des CX3CR1-Korezeptors in der U87.CD4.CX3CR1 Zelllinie Die parentale Zelllinie U87.CD4 und die mit Pseudotypvirus transduzierten Zellen wurden 7 Tage p.i. mit dem CX3CR1-spezifischen Antikörper angefärbt (PE-Konjugat). 2,5x105 Zellen wurden für jedes Experiment gefärbt, 20.000 Zellen wurden im FACS analysiert. A: parentale U87.CD4 Zelllinie; B: U87.CD4.CX3CR1 Zelllinie. 3.1.1 Kontrolle der Expression des CD4-Rezeptors Da HIV den CD4-Rezeptor als Hauptrezeptor vor der Interaktion mit dem Korezeptor für eine erfolgreiche Infektion benötigt, wurde die CD4-Expression in dem U87-Zellpanel überprüft. Nach der Herstellung der Arbeitscharge (U87.CD4 Zellen) und der Zelllinien U87.CD4.CXCR4, U87.CD4.CCR5 und U87.CD4.CX3CR1, wurden die Zellen mit einem spezifischen CD4Antikörper gefärbt, welcher mit dem Fluoreszenzmarker FITC konjugiert war. Nach der Antikörperfärbung der Zellen folgte die Analyse der markierten Zellen im FACS-Calibur. In 31 Ergebnisse allen Zelllinien wurde der CD4-Marker exprimiert, wenn auch zu unterschiedlichen Anteilen. Die Ergebnisse der Färbung mit dem FITC-Anti-CD4-Antikörper zeigten bei der parentalen U87.CD4 Zelllinie 56,1% CD4+-Zellen, bei der U87.CD4.CXCR4 96,6%, bei der U87.CD4.CCR5 93,8% und bei der U87.CD4.CX3CR1 wiederum nur 46,5% CD4+-Zellen. Der Anteil CD4+-Zellen wurde als ausreichend für die Infektionsexperimente bewertet. Abb.10 Expression des CD4-Rezeptors in dem U87-Zellpanel Die vier Zelllinien (parental, CXCR4, CCR5, CX3CR1) wurden mit dem CD4 spezifischen Antikörper (FITC-Anti-CD4-Antikörper) markiert. 2,5x105 Zellen wurden für jedes Experiment gefärbt, 20.000 Zellen wurden analysiert. A: parentale U87.CD4 Zelllinie; B: U87.CD4.CXCR4 Zelllinie, C: U87.CD4.CX3CR1 und D: U87.CD4.CCR5 mit dem FITC-anti-CD4-Antikörper. Auswertung mit FlowJoSoftware. 32 Ergebnisse 3.1.2 Kontrolle der CXCR4- und CCR5-Korezeptorexpression Vor den Infektionsexperimenten wurde in dem U87-Zellpanel auch die Expression der beiden anderen Korezeptoren CXCR4 und CCR5 kontrolliert. Die parentale Zelllinie U87.CD4 diente dabei als Negativkontrolle für die beiden Korezeptoren. Die Korezeptor-Expression wurde durch eine Färbung mit monoklonalen Antikörpern (PE-Anti-CXCR4- und PE-Anti-CCR5Antikörper) und anschließende FACS-Analyse überprüft. Die U87.CD4.CXCR4 Zelllinie zeigt 90,5% CXCR4+-Zellen (Abb.11 B), während in den parentalen Zellen praktisch keine CXCR4+Zellen nachweisbar waren (Abb.11 A, 3,9% Hintergrundfluoreszenz wie sie häufiger bei negativen Zellen beobachtet wurde). Abb.11 Expression des CXCR4-Rezeptors in der U87.CD.CXCR4 Zelllinie Die parentale und die CXCR4 positive U87.CD4 Zelllinie wurden mit dem CXCR4 spezifischen PE markierten Antikörper markiert. 2,5x105 Zellen wurden für jedes Experiment gefärbt, 20.000 Zellen wurden analysiert. A: parentale U87.CD4 Zelllinie; B: U87.CD4.CXCR4 Zelllinie. In Abbildung 12 B ist die CCR5 Expression in der U87.CD4.CCR5 Zelllinie dargestellt. 94,3% der U87.CD4.CCR5 Zellen exprimieren den CCR5.Korezeptor (Abb.12 B), während in der parentalen Zelllinie (Abb.12 A) nur eine geringe Hintergrundfluoreszenz beobachtet wurde (3,4%). 33 Ergebnisse Abb.12 Expression des CCR5-Korezeptors in der U87.CD.CCR5 Zelllinie Die Zelllinien wurden mit dem PE-markierten CCR5 spezifischen Antikörper angefärbt. 2,5x105 Zellen wurde für jedes Experiment gefärbt, 20.000 Zellen wurden analysiert. A: parentale U87.CD4 Zelllinie; U87.CD4.CCR5 Zelllinie. 3.1.3 Überprüfung der Spezifität der Antikörper für die Korezeptoren In vorangegangenen Untersuchungen gab es Hinweise auf eine Kreuzreaktivität des CX3CR1 Antikörpers mit dem CXCR4-Korezeptor [35]. Aufgrund dieser Kreuzreaktivität wurde eine strukturelle Verwandtschaft der beiden Korezeptoren vermutet. Zur Überprüfung, ob auch der CXCR4- und CCR5-Antikörper eine Kreuzreaktion mit dem CX3CR1-Antikörper aufweisen, wurden die drei U87.CD4 Zelllinien mit CXCR4-, CCR5- und CX3CR1-Expression in getrennten Ansätzen mit den jeweils spezifischen Antikörpern PE-Anti-CXCR4, PE-Anti-CCR5 bzw. PEAnti-CX3CR1 angefärbt. Die Ergebnisse der FACS-Analyse sind in Abbildung 13 dargestellt. Es wurde jeweils nur eine geringe Hintergrundfluoreszenz von 4,5% CXCR4 (Abb.13 A) bei der U87.CD4.CX3CR1 Zelllinie bzw. 4,3% CX3CR1 (Abb.13 B) bei der U87.CD4.CXCR4 Zelllinie festgestellt, die in der Größenordnung der Hintergrundfluoreszenz der Negativkontrolle (parentale Zelllinie) für die Korezeptorexpression liegt. Auch bei den U87.CD4.CX3CR1 und U87.CD4.CCR5 Zelllinien wurde jeweils nur eine geringe Hintergrundfluoreszenz von 4,3% CCR5 (Abb.13 C) bei der U87.CD4.CX3CR1 Zelllinie bzw. 4,2% CX3CR1 (Abb.13 D) bei der U87.CD4.CCR5 Zelllinie festgestellt 34 Ergebnisse Abb.13 Überprüfung der Kreuzreaktivität der Antikörper gegen die unterschiedlichen Korezeptoren Die Zelllinien U87.CD4.CXCR4, U87.CD4.CCR5 und U87.CD4.CX3CR1 wurden mit dem CXCR4-, CCR5und dem CX3CR1-spezifischen Antikörper angefärbt. 2,5x105 Zellen wurden für jedes Experiment gefärbt, 20.000 Zellen wurden analysiert. A: U87.CD4.CX3CR1 Zelllinie mit dem PE-Anti-CXCR4 Antikörper angefärbt; B: U87.CD4.CXCR4 Zelllinie mit dem PE-Anti-CXC3CR1 Antikörper angefärbt, C: U87.CD4.CX3CR1 Zelllinie mit dem PE-Anti-CCR5 Antikörper, D: U87.CD4.CCR5 Zelllinie mit dem PEAnti-CX3CR1 Antikörper. 3.2 CCR5 Genotyp der PBMC-Spender für die Virusvermehrung und Titration Die Virusstocks müssen auf PBMCs von gesunden Blutspendern produziert werden. Es konnten für diese Experimente keine PBMCs von homozygoten CCR5∆32/∆32 defizienten Spendern verwendet werden, da die CCR5∆32/∆32 Deletion nur bei weniger als 1% der 35 Ergebnisse kaukasischen Bevölkerung vorkommt [30]. Der CCR5-Genotyp wurde mit einer DNA-PCR analysiert, die ein 32 Bp kleineres PCR Produkt zeigte, wenn die Deletion homozygot vorhanden war [38]. Bei Vorliegen eines homozygoten CCR5-Wildtypallels wurde ein 262 Bp PCR Produkt erhalten, die 32 Bp Deletion führte zu einem 230 Bp großem PCR Produkt. Bei Heterozygotie liegen beide DNA-Banden im Agarosegel vor. Als Kontrollen wurde DNA von Patienten mit bekanntem Genotyp durch eine PCR analysiert (Abb.14). Die Ergebnisse der CCR5-Genotyp PCR zeigten für die Spender B (#190), C (#300) und D (#286), die homozygoten Wildtyp-Allele des CCR5 Korezeptors und für den Spender A die heterozygoten CCR5wt/∆32 Allele. Der Spender B (#190) wurde für die Vermehrung der beiden Primärisolate des homozygoten Patienten verwendet. Spender A (#184) wurde für die Vermehrung von BAL und Spender C (#300) für die Vermehrung von HTLVIIIB eingesetzt. Die Titration aller Virusstocks erfolgte auf dem Spender D (#286). 1 2 3 4 5 6 7 8 400 Bp 300 Bp 262 Bp 230 Bp 200 Bp Abb.14 Bestimmung des CCR5 Genotyps der PBMC-Spender 2%-iges Agarose-Gel mit Ethidiumbromid (0,5 µg/ml). Spurbelegung: Spur 1) MW-Standard, Spur 2) Spender B (#190) CCR5wt; Spur 3) Spender A [19][19](#184) CCR5wt/Δ32; Spur 4) Spender D (#286) CCR5wt/wt; Spur 5) Spender C (#300) CCR5wt; Spur 6) Positivkontrolle für CCR5wt/wt; Spur 7) Positivkontrolle für CCR5wt/Δ32; Spur 8) negative Kontrolle (Aqua Bidest). 3.3 Herstellung der Virusstocks und Überprüfung der partiellen viralen Hüllproteinsequenz In dieser Arbeit wurden Virusstocks von Primärisolaten aus dem CCR5-defizienten Patienten sowie von zwei Referenzviren hergestellt. Das Erstisolat (02-0165), welches innerhalb der ersten Infektionswoche gewonnen wurde, und das Verlaufsisolat (08-0755), das 72 Monate nach der Infektion unter antiretroviraler Therapie aus PBMCs isoliert werden konnte, wurden weiter auf PBMCs CCR5-genotypisierter Spender vermehrt (3.2). Parallel erfolgte die 36 Ergebnisse Anzucht der Referenzviren, um Referenzen für den CXCR4- bzw. den CCR5Korezeptogebrauch für die Infektions- und Inhibitionsversuche zu haben. 3.3.1 Virusvermehrung auf PBMCs und Titerbestimmung Die Primärisolate des homozygoten CCR5Δ32/Δ32 Patienten wurden auf PBMCs vermehrt, um ausreichend Virusstocks für die geplanten Experimente zu erhalten. Als Referenzviren für die Phänotypisierung des Korezeptorgebrauchs wurden HTLVIIIB als CXCR4-gebrauchendes Virus und BAL als CCR5-gebrauchendes Virus ebenfalls auf PBMCs vermehrt. Die Virusmenge in den Virusstocks wurde durch Bestimmung des p24-Gehaltes (Kapsidprotein) als Surrogatmarker HIV-Replikation quantifiziert. Für die Infektionsversuche war es erforderlich, alle Reporter-Zelllinien mit derselben infektiösen Dosis (Multiplizität der Infektion, M.O.I.) zu infizieren, um die Infektionskinetiken in dem U87.CD4-Zellpanel direkt vergleichen zu können (Tabelle 4). Dazu wurden alle Virusstocks auf PBMCs desselben Spenders titriert (Spender D, #286). Tabelle 4 Virusstocks Die p24-Konzentration und TCID50/ml der hergestellten Virusisolate p24 [ng/ml] Virustiter [TCID50/ml] HTLVIIIB 7,7 BAL 6,65 1,3x105 02-0165 10,29 1,8x104 08-0755 6,59 2,4x104 1x10 4 Erntetag, Datum d15 p.i., 10.02.2012 d11 p.i., 23.12.2011 d11 p.i., 23.12.2011 d11 p.i., 23.12.2011 3.3.2 Kontrolle der V3-Loop Sequenzen im Hüllprotein der HIV-Virusstocks Da die Viren zur Virusstockproduktion auf neuen PBMCs Spendern vermehrt werden mussten, bestand die Möglichkeit, dass HIV in diesen neuen Wirtszellen mutiert und somit neue Virusvarianten bei dieser Vermehrung entstehen. Insbesondere Mutationen im Hüllprotein könnten Einfluss auf den phänotypisch messbaren Korezeptorgebrauch haben. Wie in der Einleitung beschrieben, spielt der immundominante V3-Loop des Hüllproteins eine entscheidende Rolle für die Interaktion des Virus mit Haupt- und Korezeptor. Die Positionen 11, 24 und 25 im V3-Loop bestimmen wesentlich den Korezeptorgebrauch mit 37 Ergebnisse [23]. Daher wurde rde nach der Virusvermehrung die V3-Loop Sequenz, die 35-37 35 Aminosäuren lang ist, von PCR-Amplifikat Amplifikaten en der viralen RNA bestimmt und mit den vorliegenden Sequenzen der Primärisolate des Patienten bzw. Datenbanksequenzen der Referenzviren HTLVIIIB und BAL verglichen, um potentielle Mutationen zu identifizieren. Von dem Verlaufsisolat solat des Patienten war keine kei Plasma-RNA-Sequenz Sequenz vorhanden, da die virale Replikation durch die antiretrovirale Therapie gehemmt war. Hier wurde die d provirale DNA-Sequenz, die aus den PBMCs des de Patienten ermittelt worden war, für den Vergleich verwendet. Abb.15 Sequenzvergleich des V3-Loops V3 im Hüllprotein der HIV-1-Isolate Im Referenzvirus HXB2 ist der V3-Loop V3 zwischen den Nukleotiden 7110 und 7214 (Acc.No. K03455) bzw. den Aminosäuren säuren 266 und 301 lokalisiert. A, B) Die Sequenzen der vermehrten Referenzviren HTLVIIIB und BAL (Acc.No.. AB521136) wurden mit den Datenbanksequenzen uenzen und früheren Stockproduktionen verglichen. C) Die vermehrten Patienten-Viren wurden mit der viralen PlasmaRNA-Sequenz und der proviralen DNA-Sequenz DNA sowie mit Sequenzen der Primärisolate verglichen (Spender 300 (HF-diese Arbeit); ); Spender 855 (Can (C Demiroglu); Spender 324 (CD); Spender 184 (HF(HF diese Arbeit); Spender 190 (HF-diese diese Arbeit)). Aus dem Sequenzvergleich lässt sich schließen, schließen dass sich die V3-Loop oop Aminosäuresequenz Aminosäures während der Virusvermehrung ermehrungen überwiegend iegend nicht verändert hat. Im Verlaufsisolat (08-0755) traten jedoch einige Veränderungen im Vergleich zum Erstisolat auf. Die provirale 38 Ergebnisse DNA-Sequenz wies bereits eine Mutation von N zu Y an der Position 7 des V3-Loops auf (Abb.15), mutierte jedoch nicht während der Vermehrung auf PBMCs der Spender #855 (CD) und B (#190), sondern während der Evolution des Virus in dem CCR5 defizienten Wirt. Die Position 7 ist nicht bekannt als relevante Stelle für die Korezeptor Bindung. N wird durch das Codon AAT und Y von TAT codiert. Die P zu L Mutation an Position 16 des Verlaufsisolates beruht auf einer Mutation von CCA (Codon für Prolin) zu CTA (Codon für Leucin). Diese Position im Hüllprotein wird jedoch ebenfalls nicht als relevante Mutation beschrieben. Diese Mutation verschwand während der Vermehrung des Virus auf PBMCs mit CCR5Wildtypen wieder. Weiterhin zeigte das Erstisolat (02-0165) eine Mutation von R zu W an Position 9 (Codon AGA zu Codon TGA), was auch zu einem Stoppcodon führen könnte (TGA). Die Referenzviren wurden mit den Sequenzen der HIV Datenbank verglichen. Die HTLVIIIB Sequenz wurde der HXB2 Sequenz (Acc.No. K03455, molekularer Klon aus HTLVIIIB) gegenübergestellt. Bei der Vermehrung von HTLVIIIB auf dem Spender C zeigten sich Veränderungen an den Positionen 11 und 16. Die Position 11 ist relevant für den Korezeptorgebrauch. An dieser Position wurde eine Mischung aus R und K identifiziert, die in der translatierten Sequenz als X ausgegeben wird (Codon AGA und AAA). Die Sequenz von BAL wurde mit der entsprechenden Datenbanksequenz verglichen, BAL (Acc.No. AB521136). Bei der Auswertung dieser Sequenzen wurde eine Veränderung an der Position 20 analysiert, von der Aminosäure F zu einer Mischung aus F und L, die durch das Codon TTA bzw. TTT codiert werden. Beide Referenzviren sind im Labor schon vielfältig auf verschiedenen Spenderzellen vermehrt worden, so dass die Abweichungen zur Datenbanksequenz nicht überraschend sind. 3.4 Phänotypische Bestimmungen des Korezeptorgebrauchs der Virusisolate Für die Evaluation des möglichen alternativen CX3CR1-Korezeptorgebrauchs der Virusisolate aus dem CCR5 negativen Patienten wurde das U87-Zellpanel aus den vier Zelllinien, die jeweils gesichert keinen Korezeptor oder jeweils CXCR4, CCR5 bzw. CX3CR1 exprimierten, in Infektionsversuchen verwendet. Als Marker für die Infektionskinetiken über 14 Tage wurde wiederum die Konzentration des Kapsidproteins p24 im Zellkulturüberstand bestimmt. 39 Ergebnisse 3.4.1 Infektionskinetik mit und ohne Mediumwechsel Mediumwechse Eine erste Infektionskinetik wurde in zwei Parallelansätzen nsätzen mit der parentalen U87.CD4 Zelllinie und den Zelllinien U87.CD4.CXCR4, -CCR5 und -CX3CR1 und den Referenzviren HTLVIIIB und BAL, sowie den beiden Verlaufsisolaten des CCR5 defizienten Patienten (02-0165, Erstprobe und 08-0755 0755, 7 Jahre nach Infektion) gezeigt.. Dabei wurde überprüft, welchen Einfluss ein Mediumwechsel in den Kulturansätzen an den Tagen 5, 8 und 10 auf die Virusproduktion hatte. Alternativ wurde für die verschiedenen verschiedene Erntetage nach Infektion die entsprechend geplante Anzahl an getrennten Kulturansätzen infiziert und über die geplante Infektionsdauer ohne Mediumwechsel inkubiert (Abb.16).. Das Verfahren mit Mediumwechsel hätte tte den Vorteil, dass weniger Virusstock für die Infektionskinetik verbraucht wurde. 3000 2500 p24 [ng/ml] 2000 mit Mediumwechsel 1500 1000 ohne Mediumwechsel 500 0 d0 d5 d8_1 d8_2 d10_1 d10_2 Abb.16 Infektionskinetik mit und ohne Mediumwechsel am Beispiel von HTLVIIIB in der U87.CD4.CXCR4 Zelllinie linie Für das Experiment wurden 1x10 x104 U87.CD4.CXCR4-Zellen mit einer M.O.I. von 0,01 infiziert infiz und die p24-Produktion wurde an den Tagen Tag 0, 5, 8 und 10 gemessen (96 Lochplatte).. An Tag 5 (d8_2) und Tag 8 (d10_2) wurde parallel in zwei Löchern ein Mediumwechsel durchgeführt und die p24p24 Produktion im Überstand gemessen. Die Infektionskinetiken wurden rden in unabhängigen Doppelbestimmung ermittelt. Jede Abnahme wurde dreifach im p24-Test Test gemessen (drei verschiedene Verdünnungen). Die Abweichungen der Konzentration der Doppelbestimmung wurden als Fehlerbalken alken in der Abbildung markiert. Die Messung der Virusproduktion irusproduktion (p24) zeigte eine effektivere Infektion von HTLVIIIB, HTLVIIIB wenn ein Mediumwechsel an den Erntetagen 5, 8 und 10 durchgeführt wurde. Die Kultivierung ohne Mediumwechsel führte dagegen zu einer geringerern Virusproduktion. Eine weitere 40 Ergebnisse Versuchsreihe, sowie auch die Hemmversuche mit spezifischen Inhibitoren wurden daraufhin nur mit Mediumwechsel durchgeführt. Die Ergebnisse der Infektionskinetik sind in den Abbildungen 17 A-D dargestellt. Der Infektionsversuch mit der U87.CD4.CXCR4 Zelllinie bestätigte den CXCR4- Korezeptorgebrauch des Referenzvirus HTLVIIIB und der beiden homozygoten Isolate (020165, 08-0755, Abb.17 B). Bei der Infektion mit dem Referenzvirus HTLVIIIB war ab Tag 5 ein stetiger Anstieg bis zu einem p24-Wert von 1566 ng/ml zu verzeichnen. Das Erstisolat (020165) zeigte einen p24-Wert von 824 ng/ml an Tag 8 und das Verlaufsisolat (08-0755) ein p24-Wert von 1288 ng/ml an Tag 10. Bei der U87.CD4.CCR5 Zelllinie bestätige sich die Infizierbarkeit mit dem R5-Referenzvirus BAL und dem Erstisolat (02-0165, Abb.17 C). BAL zeigte einen p24-Wert von 48,7 ng/ml an Tag 10 und das Erstisolat (02-0165) einen p24-Wert von 53,21 ng/ml an Tag 10. Auch wenn die p24-Werte wesentlich geringer waren, als die der Infektion der CXCR4+-Zellen, kann von einer eindeutigen Infektion gesprochen werden (unterschiedliche Skalierung in Abb.17 C). Die parentale U87.CD4 Zelllinie zeigte bei allen Viren keine Virusproduktion, es war keine Infektion dieser Zellen möglich (Abb.17 A). Die CX3CR1+-Zellen konnten weder von einem Patientenisolat, noch von den Referenzviren infiziert werden (Abb.17 D). Diese Ergebnisse in dieser Arbeit mit dem U87.CD4-Zellpanel unterscheiden sich somit eindeutig von den Ergebnissen mit dem GHOST-Zellpanel. Zur Kontrolle des CX3CR1 Gebrauchs wurde daher noch eine Infektionskinetik bis zu 14 Tagen p.i. mit einer 10fach höheren M.O.I. von 0,1 durchgeführt, was auch nicht zu einem anderen Ergebnis führte. Die Ausgangswerte von 0,9 ng/ml (HTLVIIIB), 0 ng/ml (BAL), 1,16 ng/ml (020165) und 1,62 ng/ml (08-0755) an Tag 0 der Infektion entsprechen dem Virusinokulum, dessen p24-Gehalt zur Kontrolle gemessen wurde (Methodenteil 2.2.10). 41 Ergebnisse parentale U87.CD4 2 1,8 1,6 p24 [ng/ml] 1,4 HTLVIIIB 1,2 Bal 1 02-1065 0,8 08-0755 0,6 0,4 0,2 0 d0 A) d5 d8 d10 U87.CD4.CXCR4 1600 1400 p24 [ng/ml] 1200 HTLVIIIB 1000 Bal 800 02-1065 600 08-0755 400 200 0 d0 d5 d8 d10 B) U87.CD4.CCR5 1600 1400 p24 [ng/ml] 1200 1000 HTLVIIIB Bal 800 02-0165 600 08-0755 400 200 0 C) d0 d5 d8 d10 42 Ergebnisse U87.CD4.CX3CR1 2 1,8 p24[ng/ml] 1,6 1,4 HTLVIIIB 1,2 Bal 1 02-1065 0,8 08-0755 0,6 0,4 0,2 0 D) d0 d5 d8 d10 Abb.17 A-D. Infektionskinetik U87.CD4-Zellpanel A) Parentale U87.CD4 Zelllinie, B) U87.CD4.CXCR4 Zelllinie, C) U87.CD4.CCR5 Zelllinie und D) U87.CD4.CX3CR1 Zelllinie mit den Viren HTLVIIIB, BAL, dem Erst- (02-0165) und dem Verlaufsisolat (08-0755). Für das Experiment wurden jeweils 1x104 der U87.CD4-Zellen mit einer M.O.I. von 0,01 infiziert und die Produktion von p24 wurde an den Tagen 0, 5, 8 und 11 gemessen. Der Versuch wurde parallel in zwei Ansätzen durchgeführt und jeder Überstand dreifach gemessen. Zur Übersicht wurden die Standardabweichungen nicht in der Abbildung angezeigt. In Abbildung 18 sind die Ergebnisse der zwei Infektionskinetiken der Viren HTLVIIIB, BAL, 020165 und 08-0755 auf den Zelllinien U87.CD4, U87.CD4.CXCR4, U87.CD4.CCR5 und U87.CD4.CX3CR1 für den Tag 10 p.i. gezeigt. Als Kontrolle diente die parentale U87.CD4 Zelllinie, die nur die Expression des CD4-Rezeptors zeigte. 43 Ergebnisse 10000 1000 p24 [ng/ml] 100 10 HTLV3B BAL 02-0165 08-0755 HTLV3B BAL 02-0165 08-0755 Parental CCR5 CXCR4 HTLV3B BAL 02-0165 08-0755 HTLV3B BAL 02-0165 08-0755 1 CX3CR1 Abb.18 Phänotypischer Korezeptorgebrauch der Virusisolate im U87.CD4-Rezeptorzellpanel Die p24-Produktion der verschiedenen Virusisolate wurde gemessen. Für das Experiment wurden jeweils 1x104 Zellen mit einer M.O.I. von 0,01 infiziert und die p24-Produktion wurde an Tag 8, 10 und 14 gemessen. Die Auswertung für Tag 10 ist abgebildet (logarithmische Skalierung). Der Versuch erfolgte in unabhängiger Doppelbestimmung. Jeder Kulturüberstand wurde in drei Verdünnungen im p24-Antigentest quantifiziert. Die Standardabweichung aus allen Messungen ist als Fehlerbalken in der Abbildung angezeigt. Das Experiment bestätigte die Infektion der CCR5+-Zellen durch das R5-Referenzvirus BAL. Trotz der beobachteten Mutation im V3-Loop war das Virus nur in der Lage CCR5+-Zellen zu infizieren. Das X4-Virus HTLVIIIB kann ebenfalls trotz einiger Mutationen im V3-Loop nur CXCR4+-Zellen infizieren. Das Primärisolat (02-0165) kann, wie bereits in früheren Arbeiten der Arbeitsgruppe mit dem GHOST-Zellpanel festgestellt, CXCR4- und CCR5+-Zellen infizieren, die CXCR4+-Zellen sogar 10-fach besser (Abb.18). Das Verlaufsisolat 08-0755 zeigte den bereits bekannten Verlust des CCR5-Gebrauchs und den Gebrauch des CXCR4-Korezeptors. In Abbildung 19 und 20 sind die lichtmikroskopischen Aufnahmen der Infektionskinetiken gezeigt. Die adhärenten, nicht infizierten U87.CD4 Zellen bildeten auf dem Plattenboden netzwerkartige Strukturen aus. Wenn die Zellen zu dicht wachsen, neigte die U87.CD4 zur Ablösung und Clusterbildung (Abb.19 A). Bei erfolgreicher HIV-Infektion war häufig der 44 Ergebnisse zytopathische Effekt von HIV (CP), die Hüllprotein vermittelte Fusion von infizierten Zellen zu vielkernigen Synzytien, zu erkennen (Abb.19 F). HTLVIIIB BAL CXCR4 parentale U87.CD4 B C D E F G H I CX3CR1 CCR5 A Abb.19 Infektionsversuche Die parentale U87.CD4 Zellinie uninfiziert als Negativ Kontrolle (A, D, G). Die U87.CD4.CXCR4 Zelllinie mit HTLVIIIB (B), mit BAL (C), die U87.CD4.CCR5 Zelllinie mit HTLVIIIB (E), mit BAL (F) und die U87.CD4.CX3CR1 Zelllinie mit HTLVIIIB (H) und mit BAL (I). 45 Ergebnisse 02-0165 08-0755 CXCR4 parentale U87.CD4 B C D E F G H I CX3CR1 CCR5 A Abb.20 Infektionsversuche Die parentale U87.CD4 Zellinie uninfiziert als Negativ Kontrolle (A, D, G), die U87.CD4.CXCR4 Zelllinie mit dem Erstisolat (02-0165) (B) und dem Verlaufsisolat (08-0755) (C), die U87.CD4.CCR5 Zelllinie mit dem Erstisolat (02-0165) (E) und dem Verlaufsisolat (08-0755) (F) und die U87.CD4.CX3CR1 Zelllinie mit dem Erstisolat (02-0165) (H) und dem Verlaufsisolat (08-0755) (I). Die Abbildung 19 A, D, G zeigt die nicht infizierte parentale U87.CD4 Zelllinie als Negativ Kontrolle. Weiterhin ist in Abbildung 19 B, C und Abbildung 20 B, C die Zelllinie U87.CD4.CXCR4 infiziert mit HTLVIIIB (Abb.19 B), BAL (Abb.19 C), dem Erstisolat (02-0165) (Abb.20 B) und dem Verlaufsisolat (08-0755) (Abb.20 C) dargestellt. Eine Bildung von Synzytien ist bei HTLVIIIB, dem Erst- und dem Verlaufsisolat auf der CXCR4 Zelllinie zu erkennen. BAL zeigte auf den CXCR4+-Zellen keine Infektion (Abb.19 C). In den Abbildungen 19 E, F und 20 E, F ist die U87.CD4.CCR5 Zelllinie mit HTLVIIIB (Abb.19 E), BAL (Abb.19 F), dem Erstisolat (Abb.20 E) und dem Verlaufsisolat (Abb.20 F) gezeigt. Nur das Erstisolat und BAL waren in der Lage, die CCR5+-Zelle zu infizieren und die Bildung von Synzytien in Gang zu setzen. Weiterhin ist noch die U87.CD4.CX3CR1 Zelllinie mit HTLVIIIB (Abb.19 H), BAL (Abb.19 I), dem Erstisolat (Abb.20 H) und dem Verlaufsisolat (Abb.20 I) dargestellt. Keines der Viren konnte die CX3CR1+-Zellen infizieren. 46 Ergebnisse 3.5 Inhibition der HIV-1 Infektion mit Korezeptor-Antagonisten Um zu zeigen, dass die Infektion der U87.CD4 Zelllinie tatsächlich über die jeweiligen Korezeptoren erfolgte, standen ein Antagonist des CXCR4-Korezeptors (AMD3100) und der natürliche Ligand von CX3CR1 (Fraktalkin) für Inhibitions- bzw. Kompetitionsversuche zur Verfügung. Damit konnte zusätzlich überprüft werden, ob es doch eine schwache Hintergrundexpression des CXCR4-Korezeptors in der CX3CR1-Zelllinie gibt bzw. CX3CR1Expression in der CXCR4 Zelllinie. Wenn eine Inhibition der Infektion erreicht wird, kann damit bestätigt werden, ob HIV einen spezifischen Korezeptor verwendet. 3.5.1 Inhibition des CXCR4-Korezeptors mit AMD3100 AMD3100 ist ein CXCR4 Antagonist, der in der Lage ist, Infektionen mit einem X4Virusstamm zu blockieren. In der Literatur ist der IC50-Wert von AMD3100 mit 2 µM beschrieben [39]. Eine 100%-ige Inhibition wurde in dieser Arbeit bereits ab einem Wert von 1 nM AMD3100 erreicht. Die IC50 entspricht der Inhibitorkonzentration, bei der 50% der Zellen nicht mehr infiziert werden. Es wurden verschiedene Konzentrationen des Antagonisten eingesetzt, so dass man auch den IC50-Wert aus der Dosis-Wirkungskurve bestimmen kann. AMD3100 wurde in den Konzentrationen von 1 nM, 10 nM, 100 nM, 500 nM eingesetzt. Die Zellen wurden bereits vor der Inkubation mit den jeweiligen Viren mit AMD3100 inkubiert, damit der Inhibitor an den CXCR4-Korezeptor binden konnte. CXCR4 AMD3100 120 Infektion [%] 100 80 HTLVIIIB 60 40 BAL 02-0165 IC50 08-0755 20 0 1nM A) 10nM 100nM 500nM AMD3100 [nM] 47 Ergebnisse CCR5 AMD3100 120 Infektion [%] 100 80 HTLVIIIB 60 BAL 02-0165 40 08-0755 20 0 1nM B) 10nM 100nM 500nM AMD3100 [nM] Abb.21 Inhibition der Infektion mit X4-Viren. Konzentrationen von AMD3100 von 0 nM bis 500 nM wurden getestet; die U87.CD4.CXCR4 (A) und U87.CD4.CCR5 Zellen (B) wurden nach Vorinkubation mit AMD3100 mit einer M.O.I. von 0,01 infiziert; das Experiment wurde in zwei parallelen Ansätzen durchgeführt. Die p24-Werte der U87.CD4.CXCR4 und U87.CD4.CCR5 Zelllinie nach Infektion mit den Virusisolaten HTLVIIIB, BAL, 020165 und 08-0755 von Tag 10 p.i. sind dargestellt. Die p24-Konzentration nach Infektion in Abwesenheit des Inhibitors wurde auf 100% gesetzt. Die p24-Werte der Infektion ohne Inhibitor an Tag 10 p.i. dienten zur Normierung und wurden auf 100% gesetzt. Alle Viren, die CXCR4 als Korezeptor verwenden konnten, wurden auch durch AMD3100 inhibiert (Abb.21 A). BAL, das R5-trope Virus dagegen konnte diese Zelllinie nicht infizieren und somit auch nicht inhibiert werden. Die IC50 von AMD3100 lag für die Infektion der U87.CD4.CXCR4 Zelllinie mit allen drei X4-tropen Viren bei 0,5 nM. Ab einer Konzentration von 1 nM AMD3100 wurde die Infektion der U87.CD4.CXCR4 Zellen mit den drei X4-tropen Viren bereits komplett gehemmt (Abb.21 A). Der R5-Stamm BAL war auch in Gegenwart von AMD3100 in der Lage CCR5+-Zellen zu infizieren (Abb.21 B). Die Infektion der CCR5+-Zelllinie durch das Erstisolat 02-0165 des CCR5 defizienten Patienten konnte jedoch teilweise mit dem Antagonisten AMD3100 gehemmt werden. Ab einer AMD3100 Konzentration von 1 nM zeigte sich überraschend eine 20%-ige Hemmung. 3.5.2 Inhibitionsversuch mit dem natürlichen Liganden von CX3CR1 (Fraktalkin) Obwohl keines der Virusisolate die CX3CR1+-Zelllinie infizieren konnte, wurden Inhibitionsversuche mit dem CX3CR1-Antagonisten in den CCR5+- und CXCR4+-Zellen durchgeführt, um eine mögliche Inhibition anderer Korezeptoren durch Kreuzreaktivität der Liganden auszuschließen. Es wurde in der Arbeitsgruppe von Combadiere gezeigt, dass 48 Ergebnisse Fraktalkin, der natürliche Ligand des CX3CR1-Rezeptors, in der Lage ist eine CX3CR1verursachte HIV Infektion ab einer Konzentration von ≥ 100 nM zu inhibieren [40]. Die Infektionen der U87.CD4.CXCR4 und die U87.CD4.CCR5 mit den Viren HTLVIIIB, BAL, 02-0165 und 08-0755 wurden mit einer steigenden Konzentration (0 nm–500 nM) von Fraktalkin gehemmt. In Abbildung 22 A und B sind die Ergebnisse des Inhibitionsversuchs mit Fraktalkin dargestellt. CXCR4 Fraktalkin 120 100 HTLVIIIB Infektion [%] 80 BAL 60 02-0165 IC50 40 08-0755 20 0 1nM 10nM 100nM 500nM Fraktalkin [nM] A) CCR5 Fraktalkin 120 Infektion [%] 100 HTLVIIIB 80 BAL 60 02-0165 40 08-0755 20 0 1nM B) 10nM 100nM 500nM Fraktalkin [nM] Abb.22 Inhibition mit Fraktalkin Konzentrationen von Fraktalkin von 0 nM bis 500 nM wurden getestet; die U87.CD4.CXCR4 (A) und U87.CD4.CCR5 Zellen (B) wurden mit einer M.O.I. von 0,01 infiziert; das Experiment wurde in zwei parallelen Ansätzen durchgeführt. Die p24-Mittelwerte der Infektion von U87.CD4.CXCR4 und U87.CD4.CCR5 Zelllinie mit HTLVIIIB, BAL, 02-0165 und 08-0755 von Tag 10 p.i. sind dargestellt. Die p24-Konzentration ohne Inhibitor wurde auf 100% gesetzt. 49 Ergebnisse Die Infektion der CXCR4+-Zelllinie mit den X4-tropen Virusisolaten HTLVIIIB, 02-0165 und 080755 wurden unerwartet ab einer Konzentration von 1 nM Fraktalkin gehemmt (Abb.22 A). Diese Ergebnisse weisen darauf hin, dass der CX3CR1-Ligand Fraktalkin auch mit dem CXCR4Korezeptor interagiert. BAL vermehrte sich wie erwartet nicht in der CXCR4+-Zelllinie und es konnte somit auch keine Hemmung durch Fraktalkin beobachtet werden. Dagegen erfolgte ein Wachstum von BAL in der CCR5+-Zelllinie, ebenso wie von 02-0165 (Abb.22 B). Fraktalkin zeigte keinen Effekt auf die Infektion mit BAL, während das Erstisolat aus dem CCR5defizienten Patienten eine ca. 30%-ige Hemmbarkeit zeigte (Abb.22 B). 50 Diskussion 4 Diskussion In den letzten Jahren gab es eine Reihe von Studien, die den Effekt von Polymorphismen im CCR5-Gen auf den natürlichen Verlauf einer HIV-1 Infektion beschrieben. Die 32 Bp Deletion im CCR5-Gen ist die relevanteste Mutation, da sie die Empfänglichkeit für HIV-1 und den Verlauf der HIV-1 Infektion stark beeinflusst [30]. Individuen, die eine heterozygote 32 Bp CCR5 Deletion besitzen, zeigen einen langsameren Verlauf der HIV-1 Infektion im Vergleich zu Individuen mit homozygoten CCR5-Wildtyp. Diese weisen eine geringere Viruslast und einen langsameren Abfall der CD4+-T-Zellzahl auf [32]. Liegt die 32 Bp Deletion homozygot vor, bewirkt sie eine Resistenz gegenüber HIV-1. Trotzdem sind einige wenige homozygote Träger beschrieben, die mit HIV-1 infiziert sind [41-50]. Auch in der HIV-1-Serokonverter-Studie des Robert Koch-Instituts wurde ein Patient mit einer homozygoten 32 Bp Deletion identifiziert [27]. Von diesem Patienten wurden in der Arbeitsgruppe ein Virusisolat nahe der Serokonversion des Patienten und ein Verlaufsisolat (6 Jahre nach der Serokonversion) angezüchtet und im Hinblick auf den Korezeptorgebrauch analysiert. Man fand heraus, dass das Erstisolat ein dualtropes Virus war, das die Korezeptoren CXCR4 und CCR5 verwenden konnte. Im Verlauf der Infektion ging die Fähigkeit zum CCR5-Korezeptorgebrauch verloren. In vorherigen Arbeiten der Arbeitsgruppe wurde für das Verlaufsisolat in GHOST-Zellen phänotypisch zusätzlich zu dem CXCR4-Korezeptorgebrauch eine Verwendung des alternativen Korezeptors CX3CR1 festgestellt. Die Infektion der CX3CR1+-Zelllinie verlief im Vergleich zu den CXCR4+- und den CCR5+-Zellen am effizientesten. Der CX3CR1-Korezeptor wird vor allem in Epithelzellen der Darmmucosa exprimiert, daher könnte er eine Rolle während der HIV-Infektion spielen [51-52]. CX3CR1 wurde als Korezeptor für manche HIV-1 Isolate beschrieben und ein Polymorphismus in diesem Korezeptorgen wurde mit der raschen Verbreitung von HIV im Körper in Verbindung gebracht [53-54]. Die Infektion der CX3CR1+-GHOST-Zellen mit dem Verlaufsisolat war jedoch vollständig mit dem CXCR4-Inhibitor AMD3100 hemmbar [35]. Es war beschrieben und auch durch FACSAnalyse bestätigt worden, dass GHOST-Zellen schwache Hintergrundexpression von CXCR4 aufweisen [35]. Dies könnte die Analyse des Gebrauchs des alternativen Korezeptors CX3CR1 in den GHOST-Zellen vorgetäuscht haben. Allerdings konnte das dualtrope Erstisolat nicht 51 Diskussion die CX3CR1+-GHOST-Zelllinie infizieren. Um abzuklären, ob die scheinbar CX3CR1-vermittelte Infektion tatsächlich auf der Hintergrundexpression des CXCR4-Korezeptors beruhte, wurde eine Zelllinie benötigt, die keinen CXCR4-Korezeptor exprimierte. Die Zelllinie U87.CD4 wurde für die Herstellung CX3CR1 exprimierender Wirtszellen für HIV gewählt, da beschrieben war, dass sie kein CXCR4 exprimiert. U87.CD4 stammt von humanen Glioblastomzellen ab, die den natürlichen Wirtszellen von HIV-1 viel ähnlicher sind als die GHOST-Zellen, die von humanen Osteosarkom Zellen abgeleitet werden. Von der parentalen U87.CD4 war von Princen et al. sowohl eine CXCR4+- als auch eine CCR5+-Zelllinie erzeugt worden [36]. Für die Herstellung der CX3CR1-exprimierenden U87.CD4 Zelllinie wurde bereits in einer vorangegangenen Arbeit ein Expressionsvektor mit dem offenen Leserahmen von CX3CR1 hergestellt (pBABE-puro-CX3CR1_Klon15; Bachelor SH [11]). In dieser Arbeit wurde eine CX3CR1+-Zelllinie durch Transduktion erzeugt und es wurden Arbeitsstocks der HIV-1 Isolate des CCR5 defizienten Patienten hergestellt, um deren Korezeptorgebrauch phänotypisch im U87.CD4-Zellpanel zu untersuchen. Die HIV-Isolate wurden genotypisch und phänotypisch im Vergleich zu Referenzviren charakterisiert. Durch den Einsatz eines spezifischen Antagonisten des CXCR4-Korezepors (AMD3100) sowie des natürlichen Liganden von CX3CR1 (Fraktalkin) konnte der jeweilige Korezeptorgebrauch der Isolate eindeutig charakterisiert werden. 4.1 Genotypische Analysen des Korezeptorgebrauchs Das Hüllprotein gp120 spielt eine wichtige Rolle bei der Erkennung des monomeren Glykoproteinrezeptors CD4 auf der Oberfläche der Zielzelle. Für die Interaktion zwischen dem Hüllprotein und einem der möglichen Korezeptoren ist die Ladung des immundominanten V3-Loop (v. a. Pos. 11, 24 und 25) im Glykoproteins gp120 entscheidend [34]. Um die Infektionsversuche vergleichen zu können, sollten möglichst alle Virusisolate auf Spendern mit dem gleichen Genotyp vermehrt werden. Jedoch war dies aus praktischen und zeitlichen Gründen nicht möglich. Für Forschungszwecke kann pro Spender nur ein Buffy Coat erworben werden und es stehen keine Folgespenden desselben Blutspenders (mit demselben CCR5-Genotyp) zur Verfügung, so dass der Spenderwechsel bei Virusvermehrung zur Herstellung einer Arbeitscharge unvermeidlich ist. 52 Diskussion Daher war es notwendig, die Aminosäuresequenz der in PBMCs gesunder Spender produzierten Virusstocks mit den Sequenzen der Primärisolate zu vergleichen, um potentielle Mutationen, die während der Virusvermehrung entstehen können, zu erkennen. Falls Mutationen in der V3-Loop Aminosäuresequenz der verschiedenen Referenzstämme und Virusisolate während der Vermehrung in Zellkultur (Spendern #184, #190 und #300) aufgetreten wären, könnten diese eine Veränderung in dem Korezeptorgebrauch auslösen und damit einen Korezeptorgebrauch aufweisen, der nicht dem originalen Virusstamm in dem Patienten zum Zeitpunkt der Blutabnahme entspricht. Daher wurden die Sequenzen der Virusstocks mit der Sequenz der Plasma-RNA bzw. proviralen DNA der HIV-isolate, sowie mit Sequenzen vorheriger Arbeitschargen verglichen, um potentielle Mutationen im V3-Loop festzustellen [34-35]. Im Verlaufsisolat (08-0755) wurde eine Mutation an der Position 7 des V3-Loops im Vergleich zum Erstisolat festgestellt. Die N (Asparagin) zu Y (Tyrosin) Mutation ist im in vivo Infektionsverlauf aufgetreten und blieb bei allen weiteren in vitro Vermehrungen in PBMCs erhalten. Das in Spenderzellen (#190) vermehrte Erstisolat zeigte an der Position 9 eine Mischung aus den Codons AGA (R) und TGA (Stoppcodon), dass heißt, es liegt eine Mischung mit replikations-defizienten Viren vor. Diese Position ist nicht als relevante Stelle für den Korezeptorgebrauch beschrieben und ging bei der Vermehrung in Spenderzellen (#190) in dieser Arbeit wieder verloren. Auch die P (Prolin) zu L (Leucin) Mutation an Position 16 des Verlaufsisolates ist nicht als relevante Mutation für den Korezeptorgebrauch beschrieben. Die weiter vermehrten Viren zeigten somit im V3-Loop keine Mutationen im Vergleich zu den zuvor hergestellten Virusstocks bzw. den Viren im Patienten (Erstprobe: Plasma-Probe, Verlaufsprobe: DNA-Sequenz), die auf eine Veränderung des Korezeptorgebrauchs hinweisen. Mutationen außerhalb des V3-Loops sind jedoch nicht auszuschließen, aber größere Sequenzabschnitte wurden bisher nicht untersucht. Obwohl der R5-Referenzstamm BAL auf einem PBMCs Spender mit einem heterozygoten CCR5-Genotyp vermehrt wurde (#184), wurden in der BAL-Sequenz keine relevanten Abweichungen zur Referenzsequenz aus der HIV-Datenbank festgestellt (eine Aminosäuremischung an Position 20). In dem X4 Referenzvirus HTLVIIIB wurden zwei Mutationen beobachtet. Obwohl eine davon sogar an der Position 11 auftrat, die für die 53 Diskussion Interaktion mit dem Korezeptor als relevant beschrieben ist, wurde keine Beeinträchtigung des CXCR4-Korezeptorgebrauchs oder Änderung des Korezeptorgebrauchs ermittelt. 4.2 Der CX3CR1-Korezeptors ist kein alternativer Korezeptor für das HIV-Isolat aus dem homozygoten CCR5∆32 Patienten Für die Infektions- und Inhibitionsversuche und vor allem für die Untersuchung der Rolle des Korezeptors CX3CR1 als alternativen Korezeptor, wurde die U87.CD4.CX3CR1 Zelllinie durch eine Transfektion mit Herstellung von Pseudotypviren und anschließender Infektion hergestellt. Nach erfolgreicher Transduktion und Puromycin-Selektion zeigten über 90% der transduzierten Zellen eine stabile CX3CR1-Expression. Und auch die weiter vermehrten Arbeitschargen der Zelllinien U87.CD4, U87.CD4.CXCR4 und U87.CD4.CCR5 exprimierten die jeweiligen Korezeptoren zu mehr als 90% auf der Oberfläche. Sie unterschieden sich jedoch in der CD4-Expression. Nur 56% der parentalen Zellen und 46,5% der CX3CR1+-Zellen exprimierten den Hauptrezeptor CD4 trotz Selektion mit Geniticin. Die Infektionsversuche mit den Referenzviren für den Korezeptorgebrauch zeigten die erwarteten Ergebnisse: der X4-Referenzstamm HTLVIIIB infizierte nur CXCR4+-Zellen und der R5-Referenzstamm BAL war nur in der Lage, CCR5+-Zellen zu infizieren. Die Isolate aus dem homozygoten CCR5-defizienten Patienten infizierten CXCR4+-Zellen und das Erstisolat war zudem noch in der Lage, wie auch in den GHOST-Reporterzellen, CCR5+-Zellen zu infizieren. Das Verlaufsisolat verlor im Verlauf der Infektion die Fähigkeit CCR5+-Zellen zu infizieren. Eine Verwendung des CX3CR1-Korezeptors des Verlaufsisolates für die Infektion von Wirtszellen konnte eindeutig ausgeschlossen werden. Auch bei Infektion mit 10fach höherer M.O.I. (0,1) konnte keinerlei Virusproduktion durch p24-Antigenbestimmung im Zellkulturüberstand nachgewiesen werden. 4.3 Inhibitionsversuche mit den Korezeptor-Antagonisten AMD3100 und Fraktalkin Zur Charakterisierung des Korezeptorgebrauchs wurden Inhibitionsversuche mit dem CXCR4Antagonisten (AMD3100) und dem natürlichen Liganden von CX3CR1 (Fraktalkin) durchgeführt. Die Hemmung mit AMD3100 zeigte die erwartete Wirkung: die Infektionen von CXCR4+-Zellen mit den X4-Viren HTLVIIIB und den beiden Verlaufsisolaten konnten ab einer Konzentration von 1 nM gehemmt werden. Der R5-Stamm BAL dagegen war weiterhin in der Lage CCR5+-Zellen zu infizieren und die CCR5 vermittelte Infektion konnte nicht durch AMD3100 inhibiert werden. Da kein Virus die CX3CR1+-Zellen infizieren konnte, konnte auch 54 Diskussion kein Hemmversuch mit dieser Zelllinie durchgeführt werden. Das im Vergleich zu den GHOST-Zellen völlig andere Ergebnis hinsichtlich des CX3CR1 Korezeptorgebrauchs wird nicht auf Mutationen im gp120 V3-Loop zurückgeführt, da keine Mutationen an den Positionen 11, 24/25 in den Virusstocks beobachtet wurden. Die geringe CD4-Expression auf der Oberfläche könnte zwar zu einer geringeren Infektionsrate in den CX3CR1 Zellen führen, aber da absolut keine Virusvermehrung in dieser Zelllinie nachweisbar war (Nachweisgrenze des p24-Antigentestes ≤7,8 ng/ml), wird ausgeschlossen, dass die Nichtinfizierbarkeit der Zelllinie auf die schwächere CD4-Expression zurückzuführen ist. Um die Korezeptorspezifität von Fraktalkin zu zeigen, wurden Infektionen der CXCR4+- und der CCR5+-Zellen in Gegenwart von Fraktalkin durchgeführt. Die Inhibitionsversuche mit dem CX3CR1-Antagonisten Fraktalkin zeigten auf der U87.CD4.CCR5 Zelllinie unerwartet eine geringe Hemmung des dualtropen R5/X4-Erstisolates (02-0165). Ebenso überraschend war die effiziente Hemmung aller X4-Viren in den U87.CD4.CXCR4 Zelle einschließlich des Verlausisolates durch Fraktalkin. Da der Versuch in einer unabhängigen Doppelbestimmung mit übereinstimmenden Messwerten durchgeführt wurde, wird eine Inhibitorverwechselung ausgeschlossen. Eine mögliche Erklärung für den Hemmeffekt könnte das Vorliegen eines strukturverwandten Epitops auf CXCR4 (und auch CCR5, 30% Hemmung) sein, an das Fraktalkin binden kann und dadurch auch die erfolgreiche Infektion verhindert. Fraktalkin ist der einzige natürliche Ligand des Korezeptors CX3CR1 [40]. CXCR4 wird ebenfalls durch einen spezifischen Liganden zur Chemotaxis aktiviert. Die Chemokinliganden werden wie die Korezeptoren aufgrund ihrer Struktur in vier Gruppen klassifiziert. Man unterscheidet die CC-, CXC-, CX3C- und die XC-Chemokine. Der Unterschied zwischen den CXC- und den CX3C-Chemokine ist, dass bei den CXC-Chemokinen eine Aminosäure und bei den CX3C-Chemokinen drei Aminosäuren zwischen den konservierten Cysteinen liegen [55]. Die nachgewiesene Inhibitionsfunktion von Fraktalkin auf den CXCR4+-Zellen spräche für die mögliche Bindung des CX3C-Chemokins an Rezeptoren, die ein CXC-Motiv aufweisen. Eine Strukturverwandtschaft der Korezeptoren könnte die beobachtete Hemmung durch Fraktalkin erklären. Allerdings sollte dann wiederum auch die Infektion der CCR5+-Zellen mit BAL gehemmt werden, was nicht der Fall war. 55 Diskussion Schwache Hinweise auf eine mögliche Strukturverwandtschaft könnte man aus Kreuzreaktivität der spezifischen Antikörper gegen die jeweiligen verschiedenen Korezeptoren ableiten. Eine geringe Anzahl Zellen von 4,2% zeigte eine Expression des CX3CR1-Korezeptors auf der U87.CD4.CXCR4-Zelllinie sowie 4,2% der CCR5+-Zellen. In derselben Größenordnung reagierten CX3CR1+-Zellen mit dem Antikörper gegen CXCR4 (3,8%), und gegen CCR5 (4,3%). Aufgrund der sauberen Messpeaks der FACS-Analyse (keine Schulter, die eine Verschiebung zum positiven Signal anzeigte) war dies als unspezifische Hintergrundfluoreszenz des Messverfahrens bewertet worden und wird auch als die wahrscheinlichste Interpretation angesehen. In zwei anderen Publikationen wurde eine Ähnlichkeit zwischen dem CX3CR1 Korezeptor und den Korezeptoren mit dem CC-Motiv zu 30–42% nachgewiesen [56]. Weiterhin ist ein viraler Antagonist (vMIPII–virales Makrophagen inflammatorisches Protein II) bekannt, der in der Lage war beide Korezeptoren, CXCR4 und CX3CR1, zu hemmen, was ebenfalls für eine ähnliche Struktur spricht [57]. Eine geringfügige Expression aller Korezeptoren in der parentalen Zelllinie U87.CD4 würde eine alternative Erklärung für den geringen Anteil an CCR5+-, CXCR4+- und CX3CR1+-Zellen in den Kontrollfärbungen für die FACS-Analyse darstellen. Um das abzuklären, sollte man die Korezeptorexpression auf mRNA-Ebene durch eine spezifische RT-PCR überprüfen. Für die CX3CR1-Transkription in U87.CD4 wurde die mRNA bereits überprüft, es konnte keine CX3CR1 spezifische mRNA nachgewiesen werden [35]. Die in dieser Arbeit festgestellte inhibierende Kreuzreaktivität von Fraktalkin gegenüber CXCR4- bzw. CCR5-vermittelter Infektion sollte zudem durch Verwendung eines Produktes eines anderen Herstellers überprüft und reproduziert werden. Die Tatsache, dass die Infektion der CCR5+-U87-Zelllinie mit dem dualtropen Erstisolat im Gegensatz zur Infektion mit BAL hemmbar war, weist auch auf besondere Eigenschaften des viralen Hüllproteins hin. Um die Funktionalität des Hüllproteins aus dem CCR5-defizienten Patienten umfassend zu charakterisieren, sollte eine Amplifikation und Klonierung des vollständigen Hüllproteins durchgeführt werden. Anhand molekularer Expressionsklone und rekombinanter HIV mit dem Hüllprotein aus den Patientenisolaten können mögliche 56 Diskussion Funktionsunterschiede in Infektiösität, Evolution des Korezeptorgebrauchs von X4/R5 zu X4 und Fusionsaktivität detailliert charakterisiert werden [58]. Trotz der überraschenden Kreuzaktivität von Fraktalkin gegenüber CXCR4 und auch CCR5 bleibt festzuhalten, dass kein alternativer CX3CR1-Gebrauch der Virusisolate aus dem CCR5 defizienten Patienten vorliegt. Der Korezeptor, den HIV in dem Patienten mit CCR5Δ32 Homozygotie somit hauptsächlich benutzt, ist der CXCR4-Korezeptor. Dies steht in Übereinstimmung mit früheren Publikationen, in denen gezeigt wurde, dass CCR5 und CXCR4 die hauptsächlich genutzten Korezeptoren von HIV sind [20]. Eine Therapie mit dem CCR5 Antagonisten Maraviroc wäre daher in diesem Patienten nicht zu empfehlen. 57 Zusammenfassung 5 Zusammenfassung Das humane Immundefizienzvirus (HIV) dringt über die Interaktion des viralen Hüllproteins mit dem Hauptrezeptor CD4 und einem Chemokinrezeptor in die Wirtszelle ein. Die hauptsächlich verwendeten Korezeptoren sind der CXCR4- und der CCR5-Chemokinrezeptor. Mutationen im CCR5-Korezeptor haben einen Einfluss auf den Verlauf einer HIV-1 Infektion. Insbesondere eine 32 Bp Deletion im CCR5-Gen vermittelt bei einer homozygoten Mutation einen fast vollständigen Schutz gegenüber einer HIV-1 Infektion. In der HIV-1 Serokonverterstudie des Robert Koch-Instituts wurde ein HIV-1 positiver Patient identifiziert, welcher die homozygote CCR5Δ32 Mutation trägt. In vorangegangen Untersuchungen der Arbeitsgruppe von zwei Verlaufsisolaten zeigte sich, dass HIV nahe am Infektionszeitpunkt den CCR5- und den CXCR4-Korezeptor in GHOST-Reporterzellen für eine Infektion nutzen konnte. Im Infektionsverlauf (6 Jahre nach der Infektion) verlor das Virus die Fähigkeit zum CCR5-Gebrauch und die Infektion wurde verstärkt durch den CXCR4-Korezeptor vermittelt. Zudem wurde der Gebrauch des alternativen Korezeptor CX3CR1 durch das spätere Verlaufsisolat beobachtet. In spezifischen Hemmversuchen mit AMD3100, einem CXCR4Inhibitor, konnte jedoch die CX3CR1-vermittelte Infektion in den GHOST-Zellen verhindert werden. Es wurde vermutet, dass die HIV-Infektion in den CX3CR1-GHOST-Zellen durch die Hintergrundexpression von CXCR4 in den GHOST-Zellen ermöglicht wurde. Ziel der Arbeit war daher, eine CXCR4-freie Glioma-Zelllinie (U87.CD4) mit der CX3CR1-Expression herzustellen, um den möglichen Gebrauch des alternativen Korezeptors CX3CR1 durch die Viren aus dem CCR5-defizienten Patienten abzuklären. Dazu wurden Virusstocks von dem frühen Isolat, sowie dem Verlaufsisolat und von den Referenzviren HTLVIIIB (X4) und BAL (R5) auf humanen PBMCs hergestellt. Der CCR5Genotyp der PBMCs-Spender und die Hüllproteinsequenz (V3-Loop) der produzierten Virusstocks wurden kontrolliert. Eine CX3CR1+-Zelllinie wurde über eine effiziente stabile Transduktion der parentalen U87.CD4 Zellen mit Hilfe von Pseudotypviren mit dem Transgen CX3CR1, die zuvor in einer Verpackungszelllinie produziert wurden, hergestellt. Die Expression der Korezeptoren wurde in FACS-Analysen kontrolliert (91,5 % der transduzierten Zellen waren CX3CR1-positiv). Nach der Virustitration auf PBMCs folgten die phänotypischen Analysen. Es wurden Infektionskinetiken in der parentalen U87.CD4, der U87.CD4.CXCR4, U87.CD4.CCR5 und der neuen U87.CD4.CX3CR1 Zelllinie durchgeführt. Die HIV Produktion 58 Zusammenfassung der verschiedenen U87.CD4 Zellen wurde an verschiedenen Tagen (post Infektion) mit dem p24-Antigentest bestimmt. Zusätzlich wurden Inhibitionsversuche mit dem CXCR4Antagonisten AMD3100 und dem natürlichen CX3CR1-Liganden Fraktalkin durchgeführt. Die Infektion mit den Referenzviren führte zu den erwarteten Ergebnisse: das X4-Virus HTLVIIIB infizierte nur die CXCR4+-Zellen und das R5-Virus BAL infizierte nur die CCR5+-Zellen. Wie auch in den GHOST Zelllinien wies das frühe Isolat einen dualtropen Korezeptorgebrauch auf (R5/X4), während das Verlaufsisolat nur noch U87.CD4.CXCR4+Zellen infizieren konnte. Die phänotypischen Analysen in der U87.CD4.CX3CR1 Zelllinie zeigten jedoch deutlich, dass im Gegensatz zu dem Experiment in GHOST Zellen das Verlaufsisolat nicht den CX3CR1-Korezeptor für eine Infektion nutzen konnte. Die Inhibitionsversuche mit dem CXCR4-Antagonisten zeigten wie erwartet eine Inhibition der Infektion von CXCR4+-Zellen mit den X4-Viren. Der CX3CR1-Ligand konnte überraschend ebenfalls die CXCR4-vermittelte Infektion hemmen. Bei dem Erstisolat des Patienten wurde durch Fraktalkin die CCR5 vermittelte Infektion auch zu 30% gehemmt. Diese Ergebnisse scheinen auf eine Ähnlichkeit in der Struktur der beiden Korezeptoren CXCR4 und CX3CR1 hinzuweisen. Jedoch zeigte die Anfärbung mit den Korezeptor-spezifischen Antikörpern der U87-Zelllinien nur eine geringe Hintergrundfluoreszenz durch Kreuzreaktivität, die deutlich niedriger als in den GHOST-Zellen war. Zusammenfassend lässt sich sagen, dass die nachgewiesene Infektion der CX3CR1+-GHOST Zellen offensichtlich auf der Hintergrundexpression von CXCR4 beruhte und keine Infektion über den CX3CR1-Korezeptor darstellte. Eine funktionale Charakterisierung des vollständigen Hüllproteins von HIV aus dem CCR5-defizienten Patienten kann dazu beitragen, den Einfluss von Proteinstrukturen außerhalb des V3-Loops im Oberflächenprotein auf den Korezeptorgebrauch zu analysieren, die möglicherweise mit der Fraktalkinhemmbarkeit in Zusammenhang stehen. 59 Abstract 6 Abstract The human immunodeficiency virus (HIV) entry into the host cell is initiated by the interaction between the viral envelope protein, the main HIV receptor CD4 and a coreceptor. The main coreceptors used by HIV are the CCR5 and CXCR4 chemokinereceptors. Mutations in the CCR5 gene impact the course of the HIV infection. In particular a homozygous 32 Bp deletion in the CCR5 gene provides a high protective level to HIV. In the German HIV-1 seroconverter cohort a homozygous HIV-1 infected carrier of the highly protective CCR5Δ32 allele was identified. The coreceptor usage of two sequential follow-up isolates of the study patient was analyzed in previous work of the laboratory group. Using the GHOST reporter cell panel it was shown that the first isolate, isolated close the seroconversion used the CXCR4 and the CCR5 coreceptors for cell entry whereas the follow-up isolate, six years after infection, used only the CXCR4 coreceptor more efficiently for infection. Additionally, the alternative CX3CR1 coreceptor was observed. However in inhibition experiments which AMD3100, a CXCR4-Inhibitor, the CX3CR1-mediated infection in the GHOST cells was blocked and it was therefore assumed that the infection of the CX3CR1 positive cells was mediated by the known CXCR4 background expression of the GHOST cells. The aim of this study was therefore to produce a CXCR4 negative cell line (U87.CD4) with express CX3CR1 to clarify the usage of the alternative CX3CR1 coreceptor by the follow-up isolate from the CCR5 deficient patient. Virus stocks of patient’s isolates and the reference viruses HTLVIIIB (X4) and BAL (R5) were propagated and titrated on PBMCs from healthy blood donors. The CCR5 genotypes of the PBMCs from blood donors and the V3 loop sequences of the propagated virus stocks were controlled. The CX3CR1+ cell line was generated by the efficient and stable transduction of U87.CD4 cells with by infection with CX3CR1-pseudotyp viruses first produced in a packaging cell line. The coreceptor expression was controlled by FACS analysis (91,5% positive of the transduced U87.CD4.CX3CR1 cells). Infection kinetics were performed in the different cell lines U87.CD4, U87.CD4.CXCR4, U87.CD4.CCR5 and also in the new cell line U87.CD4.CX3CR1. Successful HIV infection of the different U87.CD4 cells was determinated by p24-protein quantification in the culture supernatant of different days (post infection). Finally the CXCR4 inhibitor AMD3100 and the CX3CR1 ligand fractalkine were used to inhibit 60 Abstract the HIV infection of U87.CD4 cell panel. Infection with the reference viruses showed the expected results: the X4 virus HTLVIIIB infected only the CXCR4+ cells and the R5 virus BAL infected the CCR5+ cells. Ass observed in the GHOST cells the early isolate was dualtropic in contrast to the follow-up isolate which could only infect CXCR4+ cells. However, the phenotypic analyses clearly revealed that the follow-up isolate could not infect the CX3CR1+ cells, in contrast to the results in GHOST cells. The inhibition experiments showed that infection of U87.CD4.CXCR4+ cells by X4-tropic strains could was completely inhibited by the CXCR4 inhibitor AMD3100. Surprisingly, the CX3CR1 ligand fractalkine could also inhibit the CXCR4 mediated infection. During the infection of the CCR5+ cells by the early dualtropic isolate a 30% inhibition by fractalkine was observed. These results would suggest a structural relationship of the two coreceptors CXCR4 and CX3CR1. However, FACS analyses with coreceptor specific antibodies showed only little background fluorescence due to potential cross reactivity, which was clearly lower than observed in the GHOST cell panel. In summary, the results of the phenotypic analyses clearly indicate that infection of the CX3CR1+ GHOST cells was mediated by the known CXCR4 background of the GHOST cells but not by the CX3CR1 coreceptor. A functional characterization of the full-length envelope protein of HIV isolated from the CCR5 deficient patient could contribute analyze the impact of protein structures outside the V3 loop on the coreceptor usage which might cause the fractalkine mediated inhibition of the infection. 61 Literatur 7 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. Literatur Zhu, T., et al., An African HIV-1 sequence from 1959 and implications for the origin of the epidemic. Nature, 1998. 391(6667): p. 594-7. Barre-Sinoussi, F., et al., Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science, 1983. 220(4599): p. 868-71. Gallo, R.C., et al., Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science, 1984. 224(4648): p. 500-3. Coffin, J., et al., What to call the AIDS virus? Nature, 1986. 321(6065): p. 10. Noireau, F., HIV transmission from monkey to man. Lancet, 1987. 1(8548): p. 1498-9. Fauci, A.S., 25 years of HIV. Nature, 2008. 453(7193): p. 289-90. Bulletin, E., Robert Koch-Institut, 46/ 2011. Kanki, P.J., et al., Response: HIV-2 and Natural Protection Against HIV-1 Infection. Science, 1996. 272(5270): p. 1959b-60b. Plantier, J.C., et al., A new human immunodeficiency virus derived from gorillas. Nat Med, 2009. 15(8): p. 871-2. Fauquet, C.M. and D. Fargette, International Committee on Taxonomy of Viruses and the 3,142 unassigned species. Virol J, 2005. 2: p. 64. Heller, S., „Stable Transduction of U87.CD4-Cells with the HIV-coreceptor CX3CR1“. Humboldt Universität Berlin, 2011. Cohen, E.A., R.A. Subbramanian, and H.G. Gottlinger, Role of auxiliary proteins in retroviral morphogenesis. Curr Top Microbiol Immunol, 1996. 214: p. 219-35. Collins, K.L., et al., HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature, 1998. 391(6665): p. 397-401. Weber, J., The pathogenesis of HIV-1 infection. Br Med Bull, 2001. 58: p. 61-72. Hammer, S.M., et al., Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society-USA panel. JAMA, 2006. 296(7): p. 827-43. Fanales-Belasio, E., et al., HIV virology and pathogenetic mechanisms of infection: a brief overview. Ann Ist Super Sanita, 2010. 46(1): p. 5-14. Kaplan, A.H., M. Manchester, and R. Swanstrom, The activity of the protease of human immunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J Virol, 1994. 68(10): p. 6782-6. Furtado, M.R., et al., Persistence of HIV-1 transcription in peripheral-blood mononuclear cells in patients receiving potent antiretroviral therapy. N Engl J Med, 1999. 340(21): p. 1614-22. Bowers, K., C. Pitcher, and M. Marsh, CD4: a co-receptor in the immune response and HIV infection. Int J Biochem Cell Biol, 1997. 29(6): p. 871-5. Bjorndal, A., et al., Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J Virol, 1997. 71(10): p. 7478-87. Mild, M., et al., Frequent intrapatient recombination between human immunodeficiency virus type 1 R5 and X4 envelopes: implications for coreceptor switch. J Virol, 2007. 81(7): p. 336976. Zhang, L., et al., Chemokine coreceptor usage by diverse primary isolates of human immunodeficiency virus type 1. J Virol, 1998. 72(11): p. 9307-12. Cardozo, T., et al., Structural basis for coreceptor selectivity by the HIV type 1 V3 loop. AIDS Res Hum Retroviruses, 2007. 23(3): p. 415-26. Huang, W., et al., Coreceptor tropism can be influenced by amino acid substitutions in the gp41 transmembrane subunit of human immunodeficiency virus type 1 envelope protein. J Virol, 2008. 82(11): p. 5584-93. Moore, J.P. and R.W. Doms, The entry of entry inhibitors: a fusion of science and medicine. Proc Natl Acad Sci U S A, 2003. 100(19): p. 10598-602. 62 Literatur 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. Dean, M., et al., Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science, 1996. 273(5283): p. 1856-62. Oh, D.Y., et al., CCR5Delta32 genotypes in a German HIV-1 seroconverter cohort and report of HIV-1 infection in a CCR5Delta32 homozygous individual. PLoS One, 2008. 3(7): p. e2747. Katzenstein, T.L., et al., HIV-infected individuals with the CCR delta32/CCR5 genotype have lower HIV RNA levels and higher CD4 cell counts in the early years of the infection than do patients with the wild type. Copenhagen AIDS Cohort Study Group. J Acquir Immune Defic Syndr Hum Retrovirol, 1997. 16(1): p. 10-4. Libert, F., et al., The deltaccr5 mutation conferring protection against HIV-1 in Caucasian populations has a single and recent origin in Northeastern Europe. Hum Mol Genet, 1998. 7(3): p. 399-406. Ansari-Lari, M.A., et al., The extent of genetic variation in the CCR5 gene. Nat Genet, 1997. 16(3): p. 221-2. Hutter, G., et al., Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med, 2009. 360(7): p. 692-8. Arenzana-Seisdedos, F. and M. Parmentier, Genetics of resistance to HIV infection: Role of coreceptors and co-receptor ligands. Semin Immunol, 2006. 18(6): p. 387-403. Brett, K., Characterization of the coreceptor usage of HIV-1 isolates of patients with CCR5 mutations. Humboldt Universität Berlin, 2010. Demiroglu, C., Characterization of HIV-1 isolates from patients with CCR5 coreceptor mutations. Technische Universität: Berlin, 2008. Friedel, M., “Co-receptor usage of HIV-1 isolates from a patient with a homozygous Δ32/Δ32 mutation in the CCR5 Co-Receptor”. Humboldt Universität Berlin, 2010. Princen, K., et al., Establishment of a novel CCR5 and CXCR4 expressing CD4+ cell line which is highly sensitive to HIV and suitable for high-throughput evaluation of CCR5 and CXCR4 antagonists. Retrovirology, 2004. 1: p. 2. Stripecke, R., Lentiviral and Retroviral Vector Systems. Cancer Drug Discovery and Development: Gene Therapy for Cancer, 2007. Kristiansen, T.B., et al., A new multiplex PCR strategy for the simultaneous determination of four genetic polymorphisms affecting HIV-1 disease progression. J Immunol Methods, 2001. 252(1-2): p. 147-51. Schols, D., et al., Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J Exp Med, 1997. 186(8): p. 1383-8. Combadiere, C., et al., Identification of CX3CR1. A chemotactic receptor for the human CX3C chemokine fractalkine and a fusion coreceptor for HIV-1. J Biol Chem, 1998. 273(37): p. 23799-804. Balotta, C., et al., Homozygous delta 32 deletion of the CCR-5 chemokine receptor gene in an HIV-1-infected patient. AIDS, 1997. 11(10): p. F67-71. Biti, R., et al., HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nat Med, 1997. 3(3): p. 252-3. Gorry, P.R., et al., Persistence of dual-tropic HIV-1 in an individual homozygous for the CCR5 Delta 32 allele. Lancet, 2002. 359(9320): p. 1832-4. Gray, L., et al., Genetic and functional analysis of R5X4 human immunodeficiency virus type 1 envelope glycoproteins derived from two individuals homozygous for the CCR5delta32 allele. J Virol, 2006. 80(7): p. 3684-91. Heiken, H., et al., HIV-1 infection in a heterosexual man homozygous for CCR-5 delta32. AIDS, 1999. 13(4): p. 529-30. Kuipers, H., et al., An HIV-1-infected individual homozygous for the CCR-5 delta32 allele and the SDF-1 3'A allele. AIDS, 1999. 13(3): p. 433-4. 63 Literatur 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. O'Brien, T.R., et al., HIV-1 infection in a man homozygous for CCR5 delta 32. Lancet, 1997. 349(9060): p. 1219. Sheppard, H.W., et al., HIV-1 infection in individuals with the CCR5-Delta32/Delta32 genotype: acquisition of syncytium-inducing virus at seroconversion. J Acquir Immune Defic Syndr, 2002. 29(3): p. 307-13. Theodorou, I., et al., HIV-1 infection in an individual homozygous for CCR5 delta 32. Seroco Study Group. Lancet, 1997. 349(9060): p. 1219-20. Iversen, A.K., et al., Limited protective effect of the CCR5Delta32/CCR5Delta32 genotype on human immunodeficiency virus infection incidence in a cohort of patients with hemophilia and selection for genotypic X4 virus. J Infect Dis, 2003. 187(2): p. 215-25. Raport, C.J., et al., The orphan G-protein-coupled receptor-encoding gene V28 is closely related to genes for chemokine receptors and is expressed in lymphoid and neural tissues. Gene, 1995. 163(2): p. 295-9. Muehlhoefer, A., et al., Fractalkine is an epithelial and endothelial cell-derived chemoattractant for intraepithelial lymphocytes in the small intestinal mucosa. J Immunol, 2000. 164(6): p. 3368-76. Faure, S., et al., Deleterious genetic influence of CX3CR1 genotypes on HIV-1 disease progression. J Acquir Immune Defic Syndr, 2003. 32(3): p. 335-7. McDermott, D.H., et al., Genetic polymorphism in CX3CR1 and risk of HIV disease. Science, 2000. 290(5499): p. 2031. Schulte, A., Zum Vorkommen und zur Regulation der transmembranen Chemokine CXCL16 und CX3CL1. Christian- Albrechts- Universität zu Kiel, 2006. Murphy, P.M., International Union of Pharmacology. XXX. Update on chemokine receptor nomenclature. Pharmacol Rev, 2002. 54(2): p. 227-9. Fernandez, E.J. and E. Lolis, Structure, function, and inhibition of chemokines. Annu Rev Pharmacol Toxicol, 2002. 42: p. 469-99. Gharu, L., R. Ringe, and J. Bhattacharya, Evidence of extended alternate coreceptor usage by HIV-1 clade C envelope obtained from an Indian patient. Virus Res, 2012. 163(1): p. 410-4. 64 Erklärung 8 Erklärung Hiermit erkläre ich, dass ich die heute eingereichte Masterarbeit selbstständig verfasst und keine anderen als die angegeben Quellen und Hilfsmittel benutzt, sowie Zitate kenntlich gemacht habe. Bei der vorliegenden Masterarbeit handelt es sich um in Wort und Bild völlig übereinstimmende Exemplare. Weiterhin erkläre ich, dass digitale Abbildungen nur die originalen Daten enthalten und in keinem Fall inhaltsveränderte Bildbearbeitungen vorgenommen wurden. Bochum, den ___________________________ 65