Kapitel 10: Grundlagen der Thermodynamik
Werbung

Einführung in die Physikalische Chemie
Teil 2: Makroskopische Phänomene und Thermodynamik
Kapitel 7:
Boltzmann-Verteilung
Zustandsbesetzungen im Teilchenensemble
Maxwell-Boltzmann-Verteilung
Kapitel 8:
Statistische Beschreibung
makroskopischer Grössen
Herleitung thermodynamischer Grössen aus
Eigenschaften des Teilchenensembles
Kapitel 9:
Thermodynamik:
Vorbereitung
Zustandsfunktionen und totales Differential
Homogene Funktionen und mechanische Koeffizienten
Kapitel 10:
Grundlagen der
Thermodynamik
Übergang von
mikroskopischer zu
makroskopischer
Beschreibung
Kapitel 11:
Thermochemie
Klassische
Thermodynamik:
Makroskopische
Chemische Anwendungen der Thermodynamik: Beschreibung der
Materie
thermochemische Grössen, Satz von Hess
Kapitel 12:
Chemisches Gleichgewicht
Reaktionsgleichgewicht, Gleichgewichtskonstanten
Temperaturabhängigkeit
Kapitel 13:
Phasenübergänge
Phasengleichgewichte und -diagramme
Clapeyron-Gleichung und Phasenregel
Kapitel 14:
Transportvorgänge
Materie-, Energie- und Impuls-Transport
in Gasen und Flüssigkeiten
Die vier Hauptsätze der Thermodynamik
Chemisches Potential
Kapitel 10: Grundlagen der Thermodynamik
Übersicht:
10.1 Einführung und Definitionen
10.2 Der 0. Hauptsatz und seine mikroskopische Interpretation
10.3 Der 1. Hauptsatz: Zustands- und Transfergrössen, innere
Energie, Enthalpie
10.4 Der 2. Hauptsatz: spontane Prozesse und Entropie
10.5 Der 3. Hauptsatz
10.6 Die Freie Enthalpie G und das chemische Potential μ
Literatur:
Atkins, de Paula, Physikalische Chemie (4. Aufl.), Kapitel 2,3
Atkins, de Paula, Kurzlehrbuch Physikalische Chemie (4. Aufl.), Kapitel 2
10.1 Einführung und Definitionen
Warum Thermodynamik ?
Quantenmechanik:
Fundamentale Theorie der mikroskopischen Eigenschaften der Materie:
- Struktur der Atome und Moleküle
- Dynamik der Moleküle
- Grundlagen der chemischen Reaktivität
Thermodynamik:
Fundamentale Theorie der makroskopischen Eigenschaften der Materie:
- Energetik makroskopischer Phasen und chemischer Reaktionen
- Spontane Prozesse
- Konzept der “Temperatur”
- Phasenübergänge
Thermodynamische Grössen (z.B. die innere Energie U, die Wärmekapazität
C,... ) können aus den mikroskopischen Eigenschaften der Moleküle berechnet
werden (statistische Thermodynamik: mikroskopische Beschreibung →
makroskopische Beschreibung)
Einige Definitionen thermodynamischer Begriffe:
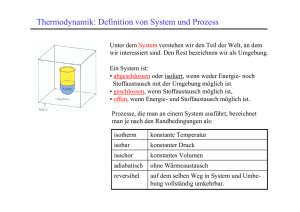
Thermodynamisches System: Eine Region im Raum, die von der Umgebung
durch die Systemgrenzen abgegrenzt wird. Die Systemgrenzen sind frei wählbar,
aber nach der Wahl für die weitere Diskussion fixiert. System und Umgebung
bilden zusammen das Weltall.
dicke Linie=Systemgrenze
Arten von Systemen:
•
•
•
Offenes System: Das System kann mit der Umgebung Materie und Energie
austauschen.
Geschlossenes System: Das System kann mit der Umgebung nur Energie
austauschen.
Abgeschlossenes System: Kein Energie- oder Materieaustausch mit der
Umgebung.
Zustandsgrössen: physikalische Grössen, die den Zustand des Systems
beschreiben (Druck p, Volumen V, Temperatur T, Molzahl n, Innere Energy U, ...).
Frei gewählte Zustandsgrössen nennt man Zustandsvariablen, davon abhängige
Zustandsgrössen Zustandsfunktionen.
Extensive thermodynamische Grössen hängen von der Anzahl Teilchen im
System ab (z.B. die Molzahl n, das Volumen V, die innere Energie U...).
Intensive thermodynamische Grössen sind unabhängig von der Anzahl Teilchen
im System (z.B. der Druck p, die Temperatur T, alle molaren Grössen wie z.B. das
Molvolumen Vm, ...)
Der Erfahrung nach werden für eine reine Substanz zwei intensive und eine
extensive Zustandsvariable benötigt, um ein System zu beschreiben. Alle anderen
Zustandsgrössen sind dann Zustandsfunktionen, die von den gewählten
Zustandsvariablen abhängen.
Beispiel: die Gleichung
beschreibt den Zustand eines idealen Gases.
• entweder die Molzahl n oder das Volumen V können als extensive
Variablen gewählt werden
• p, T sind dann die intensiven Zustandsvariablen
•
wenn zwei intensive und eine extensive Zustandsvariable gewählt sind
(z.B. p,T, n), dann hängen alle anderen Zustandsgrössen (V, U, ...) als
Zustandsfunktionen von diesen ab.
Thermodynamisches Gleichgewicht beschreibt einen Zustand, in dem sich die
Zustandsgrössen nicht mit der Zeit ändern. Mikroskopisch wird das
thermodynamische Gleichgewicht durch eine Boltzmann-Verteilung der
Populationen über alle Energieniveaus des Systems charakterisiert (vgl. Kapitel
7).
10.2 Der 0. Hauptsatz und seine mikroskopische Interpretation
Thermodynamik basiert auf vier grundlegenden Gesetzen (die Hauptsätze), aus
denen sich die gesamte Theorie entwickeln lässt. Wir werden in den folgenden
Abschnitten die vier Hauptsätze und ihre Konsequenzen diskutieren.
Der 0. Hauptsatz
Befinden sich zwei thermodynamische Systeme im Wärmekontakt,
streben sie einem Gleichgewichtszustand zu, der durch eine
einheitliche Temperatur charakterisiert ist.
Äquivalente Formulierung:
Wenn zwei thermodynamische System sich mit einem dritten
System im Gleichgewicht befinden, so stehen sie auch
untereinander im Gleichgewicht.
Der 0. Hauptsatz liefert eine Definition der Temperatur auf Basis des
thermodynamischen Gleichgewichts.
Auf mikroskopischer Ebene ist das thermodynamische Gleichgewicht durch eine
Boltzmann-Verteilung der Besetzungszahlen Ni der Energieniveaus Ei der
Moleküle definiert (s. Gl. (7.3.4) in Kapitel 7):
Ni⇤
exp{ Ei /kB T }
P
=N
j exp{ Ej /kB T }
(7.3.4)
Die Temperatur erscheint hier als der Parameter, der die Boltzmann-Verteilung
und somit das thermodynamische Gleichgewicht definiert.
Mikroskopische Interpretation des 0. Hauptsatzes
Gemäss dem klassischen Gleichverteilungssatz (s. Kapitel 8) hängt die molare
innere Energie U von der Temperatur ab gemäss
X
1
Um = U0,m +
(10.2.1)
2 RT
i
wobei die Summe über alle quadratischen Freiheitsgrade i des betreffenden
Moleküls läuft, s. Gl. (8.4.6) in Kapitel 8 (U0,m ist die Nullpunktsenergie).
Um den 0. Hauptsatz zu illustrieren, zeigen wir im folgenden, dass ein Zustand in
welchem die Energie gleichmässig über zwei gekoppelte Systeme verteilt ist, am
wahrscheinlichsten ist.
Wegen Gl. (10.2.1) impliziert eine Gleichverteilung der Energie zwischen zwei
Systemen eine Angleichung der Temperatur.
Wir betrachten beispielhaft zwei identische geschlossene Systeme mit 6 Teilchen
und 3 Energieniveaus, die der Boltzmann-Statistik gehorchen:
hohe anfängliche
Temperatur
tiefe anfängliche
Temperatur
Anfänglich ist die totale Energie Etot=6a im “heissen” System konzentriert.
Wir stellen nun thermischen Kontakt zwischen den beiden Systemen her, sodass
die Energie ausgetauscht werden kann. Die totale Energie Etot=6a muss dabei
erhalten bleiben !
Für jedes System existieren W(E) verschiedene Möglichkeiten (=Realisierungen)
um einen Zustand mit der Energie E zu erzeugen (s. Gl. (7.2.3) in Kapitel 7)
W
...
W
...
W
...
W
...
W
Für jedes System ist die minimal mögliche
Energie Emin=0, die maximal mögliche Energie
Emax=6a.
W
W
Die Anzahl Möglichkeiten Etot=6a über beide Systeme zu verteilen beträgt somit:
W
W
*W
Die wahrscheinlichste Situation entspricht einer Gleichverteilung der Energie:
E(1)=E(2)=3a. Gemäss Gl. (10.2.1) muss daher die Temperatur in beiden Systemen
gleich sein: T(1)=T(2).
Anders ausgedrückt: eine Situation mit der gleichen Temperatur in beiden
Systemen entspricht der grössten Anzahl an Realisierungen und ist damit am
wahrscheinlichsten !
(Bem.: Im vorliegenden Beispiel eines sehr kleinen Systems ist die Wahrscheinlichkeit von 31% für die
Gleichverteilung der Energie nicht markant höher als die Wahrscheinlichkeit einiger anderer Realisierungen.
Es kann gezeigt werden, dass in realistischen Systemen mit ≈NA Teilchen der Zustand mit EnergieGleichverteilung der am weitaus wahrscheinlichste ist !)
Somit wird eine anfängliche Temperaturdifferenz immer einen Wärmefluss
verursachen, der die Temperatur ausgleicht.
Dieser Prozess ist irreversibel. Seine Umkehrung würde einen Zustand mit hoher
Wahrscheinlichkeit in einen Zustand mit niedriger Wahrscheinlichkeit überführen
und findet daher nicht spontan statt !
10.3 Der 1. Hautsatz:
Zustands- und Transfergrössen, innere Energie, Enthalpie
Der 1. Hauptsatz
Die Änderung der inneren Energie dU eines Systems ist die
Summe der Wärmemenge δq und der Arbeit δw, die mit dem
System ausgetauscht werden:
(10.3.1)
Dies ist nichts anderes als der thermodynamische Erhaltungssatz der Energie !
10.3.1 Zustands-und Wegfunktionen
Eine Zustandsgrösse ist eine thermodynamische Grösse, deren Wert nur vom
Zustand des Systems abhängt und nicht vom Weg, wie das System in diesen
Zustand gelangt ist. Bsp.: Druck p, Volumen V, Temperatur T, innere Energie U.
Weiters: Enthalpie H, Entropie S, Freie Enthalpie G (s. später)
Weggrössen (auch Transfergrössen) sind thermodynamische Grössen deren Wert
vom Weg abhängt, in dem das System in den gegenwärtigen Zustand gelangt ist.
Wichtige Beispiele: Wärme q und Arbeit w.
Notation:
•
Differentielle Formulierung (unendlich kleine Änderungen):
(10.3.2)
ein griechisches “δ” bezeichnet
eine lateinisches “d” bezeichnet
eine infinitesimal kleine Änderung
eine infinitesimal kleine Änderung einer
einer Weggrösse, d.h. die Änderung
Zustandsgrösse, d.h. die Änderung
hängt vom Weg ab
hängt NICHT vom Weg ab
•
Makroskopisch messbare Änderungen thermodynamischer Grössen werden
durch Integration über alle infinitesimalen Änderungsschritte erhalten:
(10.3.3)
ein grischisches “Δ” bezeichnet eine messbare makroskopische Änderung der relevanten
thermodynamischen Grösse, die nach der Integration über alle infinitesimal kleinen Änderungen erhalten
wird; wir lassen das “Δ” für q und w weg, weil der Transfer von Wärme und Arbeit immer bereits eine
Veränderung impliziert.
10.3.2 Zustandsänderungen (“Wege”)
Möglichkeiten, einen thermodynamischen Prozess zu führen (“Wege”):
•
•
•
•
isotherm: die Temperatur bleibt konstant
isobar: der Druck bleibt konstant
isochor: das Volumen bleibt konstant
adiabatisch: kein Wärmefluss δq=0
Alle diese Prozesse können reversibel oder irreversibel geführt werden:
•
•
reversibel: Der Prozess kann durch einen infinitesimal kleine
Änderung der relevanten Grösse wieder umgekehrt werden, d.h. das
System ist ständig im thermodynamischen Gleichgewicht.
irreversibel: Der Prozess kann nicht umgekehrt werden (er passiert
spontan)
10.3.3 Expansionsarbeit w und Wärme q
Die gängigste Form thermodynamischer Arbeit ist die
Expansionsarbeit eines Systems gegen einen äusseren
Druck pex:
Kraft F=pexA
System
Gemäss der Figur rechts ist die Arbeit, die geleistet wird,
wenn sich das System (schematisch dargestellt als
Zylinder) um die Distanz dz ausdehnt, gegeben durch:
(10.3.4)
Die gesamte Arbeit w wird durch Integration von Gl. (10.3.4) erhalten:
(10.3.5)
Weil w eine Wegfunktion ist, hängt sie von der Prozessführung ab → Tafel
10.3.4 Die Enthalpie H
In der Thermodynamik definiert man eine Reihe von Hilfsgrössen, die in
bestimmten Situationen eine spezielle physikalische Bedeutung annehmen. In
der Chemie ist eine der wichtigsten Hilfsgrössen die Enthalpie H:
(10.3.6)
Physikalische Bedeutung von H ? → Tafel
Die Enthalpie H entspricht also der übertragenen Wärme q bei konstantem Druck !
Weil die meisten chemischen Reaktionen unter konstantem Druck durchgeführt
werden (offenes Reaktionsgefäss), sind im Labor gemessene Reaktionswärmen
üblicherweise als Enthalpien zu interpretieren !
ΔH < 0: Wärme wird vom System freigesetzt (exothermer Prozess)
ΔH > 0: Wärme wird vom System aufgenommen (endothermer Prozess)
10.3.5 Die Wärmekapazität bei konstantem Druck Cp
Pro memoria: die Wärmekapazität C ist definiert als die Ableitung der
übertragenen Wärme nach der Temperatur:
s. auch Kapitel 8
(10.3.7)
•
•
Wärmekapazität CV bei konstantem Volumen dV=0:
δw=-pexdV=0
also:
dU=δq+δw=dq
(10.3.8)
Wärmekapazität Cp bei konstantem Druck dp=0:
dH=dq
also:
(10.3.9)
Cp ist also für viele chemische Probleme die relevante Form der Wärmekapazität.
In der Regel wird sie als molare Wärmekapazität Cp,m tabelliert:
(10.3.10)
Es gilt: (Beweis ?)
(10.3.11)
Die Temperaturabhängigkeit von Cp,m wird häufig durch die empirische Beziehung
(10.3.12)
beschrieben. Die Koeffizienten a,b,c,d können experimentell bestimmt werden:
Temperaturabhängigkeit von Cp (Forts.):
Als erste Nährung wird oft angenommen, dass Cp temperaturunabhängig ist,
d.h., Cp=a und b=c=d=0.
Bsp.: Wie gross ist Cp von CO2 bei T=298 K?
Cp,m=44.22+8.79.10-23.298 - 8.62.105.298-2 = 37.13 J mol-1 K-1
= a
+
b
.T
+
c
.T-2
Temperaturabhängigkeit von Cp (Forts.):
Ausgewählte 1-atomige Festkörper
Ausgewählte Substanzen
Die Temperaturabhängigkeit der Enthalpie errechnet sich aus Gl. (9.11):
Integration:
(10.3.13)
10.3.6 Die Adiabatengleichung
Beschreibt reversible adiabatische Zustandsänderungen. Herleitung → Tafel
Mittels der idealen Gasgleichung pV=nRT kann die Adiabatengleichung in
verschiedenen Formen ausgedrückt werden:
✓ ◆ 1
T2
V1
(10.3.14)
=
, T V 1 = konst.
T1
V2
Adiabate
✓ ◆
p2
V1
(10.3.15)
=
, pV = konst.
p1
V2
✓ ◆1
✓ ◆
Isotherme
p2
T1
=
, p 1 T = konst. (10.3.16)
p1
T2
mit
= Cp,m /CV,m
Vergleiche mit der “Isothermengleichung”
(isotherm: T=konst.):
(10.3.17)
10.4 Der 2. Hautsatz: spontane Prozesse und Entropie
Beispiele für spontane Prozesse:
•
•
•
•
•
Ein heisser Körper kühlt sich auf Umgebungstemperatur ab.
Ein kalter Köper erwärmt sich auf Umgebungstemperatur.
Die Moleküle einer offenen Parfumflasche verteilen sich im Raum.
1 l Wasser und 1 l Ethanol vermischen sich.
Ein Gemisch von H2 und O2 reagiert zu H2O.
Spontaneität sagt nichts über die Geschwindigkeit des Prozesses aus.
Ein spontaner Prozess läuft unter den selben Bedingungen nie in die
entgegengesetzte Richtung. Er ist irreversibel.
Spontaneität hat nichts mit der Minimierung von Energie zu tun. Welche der
obigen Prozesse sind nicht exotherm ?
Was ist die Triebkraft hinter der Spontaneität ?
Der 2. Hauptsatz
Ein Prozess läuft spontan ab, wenn sich dabei die totale Entropie
im Weltall erhöht: ΔStot > 0
10.4.1 Die Entropie
Mikroskopische Interpretation des 2. Hauptsatzes: spontane Prozesse gehen
immer mit einer Verteilung von Energie einher.
Illustration:
Bei einem springenden Ball geht
bei jedem Auftreffen auf dem
Boden ein Teil seiner kinetischen
Energie in Wärmebewegung der
Moleküle im Boden über.
Ein Ball liegt auf einer warmen
Oberfläche.
(a) Die Moleküle im Ball üben eine
ungeordnete Wärmebewegung aus.
(b) Damit der Ball spontan wegspringt,
müssen sich die Moleküle im Ball
gleichzeitig in die selbe Richtung
bewegen. Dieser Zustand ist extrem
unwahrscheinlich !
Mikroskopische Interpretation der Entropie:
Boltzmann-Formel:
mit:
•
•
S = kB ln(W )
(10.4.1)
W(E) ... Anzahl der Realisierungen = Anzahl
der Möglichkeiten (=Mikrozustände), die
Energie E über alle Zustände des Systems zu
verteilen (s. Gl. (7.2.3) in Kapitel 7)
kB=1.38.10-23 J K-1 (Boltzmann-Konstante)
E entspricht hier natürlich der inneren Energie U im thermodynamischen Sinn
Illustration: ein Wasserkristall und ein Alkoholkristall aus je 2 Molekülen mischen
sich:
ungemischter
Zustand
gemischter
Zustand
viel wahrscheinlicher
Je grösser die Anzahl Möglichkeiten, die Energie zu verteilen (die Anzahl an
Mikrozuständen), desto grösser die Entropie.
Molare Entropie und physikalische Eigenschaften ([J mol-1 K-1]):
→ mehr räumliche Realisierungsmöglichkeiten
→ mehr räumliche Realisierungsmöglichkeiten
→ weniger räumliche Realisierungsmöglichkeiten
→ je grösser die Masse, desto kleiner der Abstand zwischen zwei Energieniveaus (s. Teilchen im Kasten,
Kap. 3.5.5), desto mehr zur Verfügung stehende Niveaus bei einer gegebenen Energie E
Molare Entropie und physikalische Eigenschaften (Forts.):
→ “weiche” Bindungen → tiefe Vibrationsfrequenzen → mehr Energieniveaus bei gegebener Energie E
→ “weiche” Bindungen → tiefe Vibrationsfrequenzen → mehr Energieniveaus bei gegebener Energie E
→ komplexere Verbindungen → mehr Freiheitsgrade → mehr Energieniveaus
Thermodynamische Definition der Entropie: die Änderung der Entropie im Verlauf
eines Prozesses entspricht der reversibel ausgetauschten Wärme dqrev dividiert
durch die Temperatur:
(10.4.2)
Gl. (10.4.2) beschreibt die Entropieänderung für das System. Die Integrationsgrenzen 1,2 beziehen sich auf den Anfangs- und Endzustand des Systems.
Motivation: es ist einsichtig, dass die Änderung der Entropie zur übertragenen
Wärme proportional sein muss. Die inverse Abhängigkeit zur Temperatur erklärt
sich aus der geringeren Anzahl zur Verfügung stehender Mikrozustände bei tiefen
Temperaturen (vgl. Kapitel 7).
Die Entropie ist eine Zustandsfunktion: die Anzahl der Mikrozustände in einem
thermodynamischen Zustand hängt nicht vom Weg ab, auf dem das System in
diesen Zustand gelangt ist !
Die Entropieänderung des Systems ΔS kann somit für jeden beliebigen Prozess
(reversibel oder irreversibel) gemäss Gl. (10.4.2) berechnet werden, indem man
einen reversiblen Weg findet, der Anfangs- und Endzustand verknüpft.
Da S eine Zustandsfunktion ist, muss die Entropieänderung entlang eines
geschlossenen reversiblen Weges verschwinden:
(10.4.3)
Berechnung von ΔS für verschiedene Prozesse: (→ Tafel)
1. Isotherme Expansion
2. Adiabatische Expansion
3. Entropieänderung bei Phasenübergängen
4. Entropieänderung bei Temperaturveränderungen
10.4.2 Entropieänderungen in System und Umgebung
Die Änderung der totalen Entropie im Weltall dStot ergibt sich aus der Summe der
Entropieänderungen im System dS und in der Umgebung dS’:
(10.4.4)
total
System
Umgebung
Gemäss dem zweiten Hauptsatz muss dStot gilt für einen beliebigen Prozess:
(10.4.5)
Das Gleichheitszeichen in Gl. (10.4.5) gilt nur für einen vollständig reversiblen
Prozess:
(10.4.6)
Illustration: für eine reversible isotherme Expansion wird der Umgebung
reversibel eine Wärmemenge qrev entnommen und dem System zugeführt. Die
Wärmeänderung in der Umgebung ist somit:
und
Die totale Entropieänderung ΔStot beträgt:
Allgemein gilt:
wobei δq die der Umgebung entnommene und dem System zugeführte Wärme
bezeichnet. Aus. Gl. (10.4.5)
folgt die
Clausius’sche Ungleichung:
(10.4.7)
Beispiel: Ein Mol Eis bei 0°C wird in einen See mit einer Temperatur von 10°C
geworfen. Wie gross ist die totale Entropieänderung ΔStot ? → Tafel
10.4.3 Die Temperaturabhängigkeit der Entropie
Aus Gl. (10.4.2)
und
(bei konstantem Druck, s.
Abschnitt 10.3.6) folgt direkt:
(10.4.7)
Analog gilt bei konstantem Volumen:
(10.4.8)
10.5 Der 3. Hauptsatz
Alle Stoffe besitzen am absoluten Temperatur-Nullpunkt T=0
eine einheitliche Entropie. Die Entropie am absoluten Nullpunkt wird
konventionsgemäss S (T=0) = 0 gesetzt.
Ausgehend vom 3. Hauptsatz können mit Gl. (10.4.7) und (10.4.8) die Entropien
bei beliebigen Temperaturen berechnet werden vorausgesetzt, die
Wärmekapazitäten sind bekannt.
Mikroskopische Interpretation: beim absoluten Nullpunkt ist gemäss der
Boltzmann-Statistik nur der quantenmechanische Grundzustand besetzt. Ist
dieser nicht entartet, d.h., es gibt nur einen einzigen Zustand bei der tiefsten
Energie, so gibt es nur genau einen möglichen Mikrozustand (W(T=0)=1) und
somit S=kB.lnW=0.
10.6 Die Freie Enthalpie G und das chemische Potential μ
10.6.1 Definition und Interpretation der Freien Enthalpie
Aus der Clausius’schen Ungleichung (10.4.7)
konstantem Druck folgt:
und dq=dH bei
(10.6.1)
Diese Beziehung legt die Definition einer neuen thermodynamischen Hilfsgrösse
nahe: die Freie Enthalpie G (auch Gibbs-Energie oder Gibbs’sche Freie Enthalpie)
(10.6.2)
Bei konstantem Druck und konstanter Temperatur gilt:
somit mit Ungl. (10.4.7):
(10.6.3)
Bei konstantem Druck und konstanter Temperatur geht ein spontaner Prozess mit
einer Abnahme von G einher !
Im Gleichgewicht gilt folglich:
(10.6.4)
Dies gilt insbesondere auch für das chemische Gleichgewicht. Wir werden später
noch im Detail sehen, dass G die thermodynamische Grösse ist, die das chemische
Gleichgewicht charakterisiert.
Für Prozesse bei konstantem Volumen führt man völlig analog die Helmholtzsche
Freie Energie A ein:
(10.6.5)
Für einen spontanen Prozess bei konstantem Volumen und konstanter Temperatur
gilt dann:
(10.6.6)
A ist jedoch für die Chemie von weit geringerer Bedeutung als G.
Was ist die physikalische Bedeutung von G ? → Tafel
ΔG entspricht also der maximalen Nicht-Volumenarbeit bei konstantem Druck
und konstanter Temperatur.
10.6.2 Fundamentalgleichungen der Thermodynamik
Wir betrachten einen vollständig reversiblen Prozess, bei dem nur Volumenarbeit
geleistet wird:
(10.6.7)
Gemäss dem 2. Hauptsatz gilt dann:
(10.6.8)
Einsetzen von Gl. (10.6.7) und (10.6.8) in den 1. Hauptsatz Gl. (10.3.1)
liefert:
(10.6.9)
Gl. (10.6.9) ist eine allgemeingültige Kombination des 1. und 2. Hauptsatzes und
wird deshalb als Fundamentalgleichung bezeichnet. Da U eine Zustandsfunktion
ist, gilt Gl. (10.6.9) für beliebige (auch irreversible) Prozesse, bei denen ausser
Volumenarbeit keine weitere Arbeit geleistet wird
Für die Freie Enthalpie G lässt sich folgende Fundamentalgleichung herleiten
(→Tafel):
(10.6.10)
10.6.3 Temperatur- und Druckabhängigkeit der Freien Enthalpie
Aus der Fundamentalgleichung (10.6.10)
•
folgt sofort:
Druckabhängigkeit von G:
(10.6.11)
Trennung der Variablen
und Integration:
(10.6.12)
•
Temperaturabhängigkeit von G:
(10.6.13)
Dies kann umgeformt werden zu (→Tafel):
Gibbs-Helmholtz-Gleichung:
(10.6.14)
10.6.4 Das chemische Potential μ
Die freie Enthalpie G ist eng verknüpft mit einer weiteren zentralen Grösse in der
Chemie: dem chemischen Potential μ
(10.6.15)
μ ist die Ableitung von G nach der Stoffmenge n bei konstantem p,T. Mit anderen
Worten: μ beschreibt, wie sich die freie Enthalpie bei einer Veränderung der
Zusammensetzung des Systems verändert.
Für einen reinen Stoff gilt G=nGm (Gm ... molare Freie Enthalpie) und somit:
(10.6.16)
Für einen reinen Stoff ist also das chemische Potential gleich der molaren Freien
Enthalpie.
Für Systeme, die eine Mischung aus mehreren Stoffen 1,2,... mit Molzahlen n1,
n2,.. darstellen, ergibt sich die Variation von G zu:
Fundamentalgleichung (10.6.10)
Definition des chemischen Potentials (10.6.15)
Wir erhalten somit:
(10.6.17)
wobei die Summe über alle Komponenten der Mischung j läuft. Dieser Ausdruck
ersetzt die Fundamentalgleichung (10.6.10) für Systeme von Mischungen mit
mehreren Komponenten.
Bei konstantem Druck und konstanter Temperatur erhalten wir:
(10.6.18)