Prinzipien der Thermodynamik, Teil I
Werbung

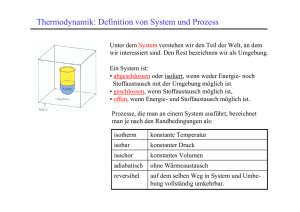
Einführung in die Physikalische Chemie: Inhalt Einführung in die Physikalische Chemie: Inhalt Kapitel 9: Prinzipien der Thermodynamik Inhalt: Literatur: 9.1 Einführung und Definitionen 9.2 Der 0. Hauptsatz und seine mikroskopische Interpretation 9.3 Der 1. Hauptsatz: Zustands- und Transfergrössen, innere Energie, Enthalpie 9.4 Der 2. Hauptsatz: spontane Prozesse und Entropie 9.5 Der 3. Hauptsatz 9.6 Die Freie Enthalpie G und das chemische Potential μ P. Atkins, J. de Paula, “Atkins’ Physical Chemistry”, 8th Ed., Chapters 2,3 I. Tinico et al., “Physical Chemistry, Principles and applications in biological sciences”, 4th Ed., Prentice-Hall 2002 Chapter 2-5 Repetieren Sie den Thermodynamik-Stoff aus Ihrer Physik-Einführungsvorlesung ! 9.1 Einführung und Definitionen Warum Thermodynamik ? Quantenmechanik: Fundamentale Theorie der mikroskopischen Eigenschaften der Materie: - Struktur der Atome und Moleküle - Dynamik der Moleküle - Grundlagen der chemischen Reaktivität Thermodynamik: Fundamentale Theorie der makroskopischen Eigenschaften der Materie: - Energetik makroskopischer Phasen und chemischer Reaktionen - Spontane Prozesse - Konzept der “Temperatur” - Phasenübergänge Thermodynamische Grössen (z.B. die innere Energie U, die Wärmekapazität C,... ) können aus den mikroskopischen Eigenschaften der Moleküle berechnet werden (statistische Thermodynamik: mikroskopische Beschreibung → makroskopische Beschreibung) Einige Definitionen thermodynamischer Begriffe: Thermodynamisches System: Eine Region im Raum, die von der Umgebung durch die Systemgrenzen abgegrenzt wird. Die Systemgrenzen sind frei wählbar, aber nach der Wahl für die weitere Diskussion fixiert. System und Umgebung bilden zusammen das Weltall. dicke Linie=Systemgrenze Arten von Systemen: • Offenes System: Das System kann mit der Umgebung Materie und Energie • • austauschen. Geschlossenes System: Das System kann mit der Umgebung nur Energie austauschen. Abgeschlossenes System: Kein Energie- oder Materieaustausch mit der Umgebung. Zustandsgrössen: physikalische Grössen, die den Zustand des Systems beschreiben (Druck p, Volumen V, Temperatur T, Molzahl n, Innere Energy U, ...). Frei gewählte Zustandsgrössen nennt man Zustandsvariablen, davon abhängige Zustandsgrössen Zustandsfunktionen. Extensive thermodynamische Grössen hängen von der Anzahl Teilchen im System ab (z.B. die Molzahl n, das Volumen V, die innere Energie U...). Intensive thermodynamische Grössen sind unabhängig von der Anzahl Teilchen im System (z.B. der Druck p, die Temperatur T, alle molaren Grössen wie z.B. das Molvolumen Vm, ...) Der Erfahrung nach werden für eine reine Substanz zwei intensive und eine extensive Zustandsvariable benötigt, um ein System zu beschreiben. Alle anderen Zustandsgrössen sind dann Zustandsfunktionen, die von den gewählten Zustandsvariablen abhängen. Beispiel: die Gleichung beschreibt den Zustand eines idealen Gases. • entweder die Molzahl n oder das Volumen V können als extensive Variablen gewählt werden • p, T sind dann die intensiven Zustandsvariablen • wenn zwei intensive und eine extensive Zustandsvariable gewählt sind (z.B. p,T, n), dann hängen alle anderen Zustandsgrössen (V, U, ...) als Zustandsfunktionen von diesen ab. Thermodynamisches Gleichgewicht beschreibt einen Zustand, in dem sich die Zustandsgrössen nicht mit der Zeit ändern. Mikroskopisch wird das thermodynamische Gleichgewicht durch eine Boltzmann-Verteilung der Populationen über alle Energieniveaus des Systems charakterisiert (vgl. Kapitel 5). 9.2 Der 0. Hauptsatz und seine mikroskopische Interpretation Thermodynamik basiert auf vier grundlegenden Gesetzen (die Hauptsätze), aus denen sich die gesamte Theorie entwickeln lässt. Wir werden in den folgenden Abschnitten die vier Hauptsätze und ihre Konsequenzen diskutieren. Der 0. Hauptsatz Befinden sich zwei thermodynamische Systeme im Wärmekontakt, streben sie einem Gleichgewichtszustand zu, der durch eine einheitliche Temperatur charakterisiert ist. Äquivalente Formulierung: Wenn zwei thermodynamische System sich mit einem dritten System im Gleichgewicht befinden, so stehen sie auch untereinander im Gleichgewicht. Der 0. Hauptsatz liefert eine also eine Definition der Temperatur auf Basis des thermodynamischen Gleichgewichts. Auf mikroskopischer Ebene ist das thermodynamische Gleichgewicht durch eine Boltzmann-Verteilung der Populationen Nj der Energieniveaus Ej der Moleküle definiert (vgl. auch Gl. (5.27) in Kapitel 5): (9.1) Die Temperatur erscheint hier als der Parameter, der die Boltzmann-Verteilung und somit das thermodynamische Gleichgewicht definiert. Mikroskopische Interpretation des 0. Hauptsatzes Gemäss dem klassischen Gleichverteilungssatz (s. Kapitel 7) hängt die innere Energie U von der Temperatur ab gemäss (9.2) wobei die Summe über alle Freiheitsgrade i des betreffenden Moleküls läuft, s. Gl. (6.15) in Kapitel 7. Um den 0. Hauptsatz zu illustrieren, zeigen wir im folgenden, dass ein Zustand in welchem die Energie gleichmässig über zwei gekoppelte Systeme verteilt ist, am wahrscheinlichsten ist. Wegen Gl. (9.2) impliziert eine Gleichverteilung der Energie zwischen zwei Systemen eine Angleichung der Temperatur. Wir betrachten beispielhaft zwei identische geschlossene Systeme mit 6 Teilchen und 3 Energieniveaus, die der Boltzmann-Statistik gehorchen: hohe anfängliche Temperatur tiefe anfängliche Temperatur Anfänglich ist die totale Energie Etot=6a im “heissen” System konzentriert. Wir stellen nun thermischen Kontakt zwischen den beiden Systemen her, sodass die Energie ausgetauscht werden kann. Die totale Energie Etot=6a muss dabei erhalten bleiben ! Für jedes System existieren Ω(E) verschiedene Möglichkeiten (=Realisierungen) um einen Zustand mit der Energie E zu erzeugen: → Tafel ... ... ... ... Für jedes System ist die minimal mögliche Energie Emin=0, die maximal mögliche Energie Emax=6a. Die Anzahl Möglichkeiten Etot=6a über beide Systeme zu verteilen beträgt somit: Die wahrscheinlichste Situation entspricht einer Gleichverteilung der Energie: E(1)=E(2)=3a. Gemäss Gl. (9.2) muss daher die Temperatur in beiden Systemen gleich sein: T(1)=T(2). Anders ausgedrückt: eine Situation mit der gleichen Temperatur in beiden Systemen entspricht der grössten Anzahl an Realisierungen und ist damit am wahrscheinlichsten ! (Bem.: Im vorliegenden Beispiel eines sehr kleinen Systems ist die Wahrscheinlichkeit von 31% für die Gleichverteilung der Energie nicht markant höher als die Wahrscheinlichkeit einiger anderer Realisierungen. Es kann gezeigt werden, dass in realistischen Systemen mit ≈NA Teilchen der Zustand mit EnergieGleichverteilung der am weitaus wahrscheinlichste ist !) Somit wird eine anfängliche Temperaturdifferenz immer einen Wärmefluss verursachen, der die Temperatur ausgleicht. Dieser Prozess ist irreversibel. Seine Umkehrung würde einen Zustand mit hoher Wahrscheinlichkeit in einen Zustand mit niedriger Wahrscheinlichkeit überführen und findet daher nicht spontan statt ! 9.3 Der 1. Hautsatz: Zustands- und Transfergrössen, innere Energie, Enthalpie Der 1. Hauptsatz Die Änderung der inneren Energie dU eines Systems ist die Summe der Wärmemenge δq und der Arbeit δw, die mit dem System ausgetauscht werden: (9.3) Dies ist nichts anderes als der thermodynamische Erhaltungssatz der Energie ! 9.3.1 Zustands-und Wegfunktionen Eine Zustandsgrösse ist eine thermodynamische Grösse, deren Wert nur vom Zustand des Systems abhängt und nicht vom Weg, wie das System in diesen Zustand gelangt ist. Bsp.: Druck p, Volumen V, Temperatur T, innere Energie U. Weiters: Enthalpie H, Entropie S, Freie Enthalpie G (s. später) Weggrössen (auch Transfergrössen) sind thermodynamische Grössen deren Wert vom Weg abhängt, in dem das System in den gegenwärtigen Zustand gelangt ist. Wichtige Beispiele: Wärme q und Arbeit w. Notation: (9.4) ein griechisches “δ” bezeichnet eine lateinisches “d” bezeichnet eine infinitesimal kleine Änderung eine infinitesimal kleine Änderung einer einer Weggrösse, d.h. die Änderung Zustandsgrösse, d.h. die Änderung hängt vom Weg ab hängt NICHT vom Weg ab (9.5) ein grischisches “Δ” bezeichnet eine messbare makroskopische Änderung der relevanten thermodynamischen Grösse, die nach der Integration über alle infinitesimal kleinen Änderungen erhalten wird; wir lassen das “Δ” für q und w weg, weil der Transfer von Wärme und Arbeit immer bereits eine Veränderung impliziert. 9.3.2 Zustandsänderungen (“Wege”) Möglichkeiten, einen thermodynamischen Prozess zu führen (“Wege”): • • • • isotherm: die Temperatur bleibt konstant isobar: der Druck bleibt konstant isochor: das Volumen bleibt konstant adiabatisch: kein Wärmefluss δq=0 Alle diese Prozesse können reversibel oder irreversibel geführt werden: • reversibel: Der Prozess kann durch einen infinitesimal kleine • Änderung der relevanten Grösse wieder umgekehrt werden, d.h. das System ist ständig im thermodynamischen Gleichgewicht. irreversibel: Der Prozess kann nicht umgekehrt werden (er passiert spontan) 9.3.4 Expansionsarbeit w und Wärme q Die gängigste Form thermodynamischer Arbeit ist die Expansionsarbeit eines Systems gegen einen äusseren Druck pex: Kraft F=pexA System Gemäss der Figur rechts ist die Arbeit, die geleistet wird, wenn sich das System (schematisch dargestellt als Zylinder) um die Distanz dz ausdehnt, gegeben durch: (9.6) Die gesamte Arbeit w wird durch Integration von Gl. (9.6) erhalten: (9.7) Weil w eine Wegfunktion ist, hängt sie von der Prozessführung ab → Tafel 9.3.5 Die Enthalpie H In der Thermodynamik definiert man eine Reihe von Hilfsgrössen, die in bestimmten Situationen eine spezielle physikalische Bedeutung annehmen. In der Chemie ist eine der wichtigsten Hilfsgrössen die Enthalpie H: (9.8) Physikalische Bedeutung von H ? → Tafel Die Enthalpie H entspricht also der übertragenen Wärme q bei konstantem Druck ! Weil die meisten chemischen Reaktionen unter konstantem Druck durchgeführt werden (offenes Reaktionsgefäss) sind im Labor gemessene Reaktionswärmen immer als Enthalpien zu interpretieren ! ΔH < 0: Wärme wird vom System freigesetzt (exothermer Prozess) ΔH > 0: Wärme wird vom System aufgenommen (endothermer Prozess) 9.3.6 Die Wärmekapazität bei konstantem Druck Cp Pro memoria: die Wärmekapazität C ist definiert als die Ableitung der übertragenen Wärme nach der Temperatur: s. auch Kapitel 7 (9.9) • Wärmekapazität CV bei konstantem Volumen dV=0: δw=-pexdV=0 dU=δq+δw=dq also: (9.10) • Wärmekapazität Cp bei konstantem Druck dp=0: dH=dq also: (9.11) Cp ist also für viele chemische Probleme die relevante Form der Wärmekapazität. In der Regel wird es als molare Wärmekapazität Cp,m tabelliert (s.a. Kapitel 7): (9.12) Es gilt: (Beweis ?) (9.13) Die Temperaturabhängigkeit von Cp,m wird häufig durch die empirische Beziehung (9.14) beschrieben. Die Koeffizienten a,b,c,d können experimentell bestimmt werden: Temperaturabhängigkeit von Cp (Forts.): Als erste Nährung wird oft angenommen, dass Cp temperaturunabhängig ist, d.h., Cp=a und b=c=d=0. Bsp.: Wie gross ist Cp von CO2 bei T=298 K? Cp,m=44.22+8.79.10-23.298 - 8.62.105.298-2 = 37.13 J mol-1 K-1 Temperaturabhängigkeit von Cp (Forts.): Ausgewählte 1-atomige Festkörper Ausgewählte Substanzen Die Temperaturabhängigkeit der Enthalpie errechnet sich aus Gl. (9.11): Integration: (9.15) 9.3.7 Die Adiabatengleichung Beschreibt reversible adiabatische Zustandsänderungen. Herleitung → Tafel Mittels der idealen Gasgleichung pV=nRT kann die Adiabatengleichung in verschiedenen Formen ausgedrückt werden: (9.16) (9.17) (9.18) Vergleiche mit der “Isothermengleichung” (isotherm: T=konst.): (9.19) Adiabate Isotherme