24.11.2009
Werbung

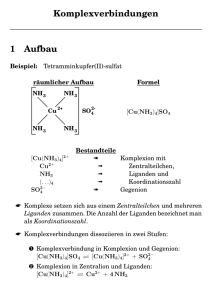
Werners Analysemethoden Die Methoden, die Alfred Werner zur Erkenntnis führten, dass es Komplexe gibt, sind noch heute gebräuchlich, allerdings nicht zur Strukturbestimmung. Die heute üblichen spektroskopischen Untersuchungsmethoden waren damals unbekannt. Er benutzte Leitfähigkeitsmessungen und Titrationstechniken zur Festlegung stöchiometrischer Zusammenhänge, und die elementare Zusammensetzung konnte damals schon recht genau durch Pyrolyse- und Verbrennungstechniken bestimmt werden. Dabei wurden die Zersetzungsgase aufgefangen und quantitativ gemessen, eine Technik, die schon die Lavoisiers perfektioniert hatten (und heute noch benutzt wird). Den Metallgehalt erhielt man meist mit gravimetrischen Methoden, indem man das Metall nach Zersetzung der Verbindung in eine bekannte Form, z.B. ein Oxid oder Sulfat überführte und dessen Masse genau bestimmte. Man muss hier anmerken, dass Werner in erster Linie so genannt kinetisch inerte Komplexe studierte. Das war keine Absicht, aber auch kein Zufall. Die viel zahlreicheren kinetisch labilen Komplexe lassen sich meist nicht in der Form, in der sie in Lösung vorliegen, als Festkörper isolieren. Die kinetisch inerten Komplexe zerfallen ähnlich langsam wie organische Moleküle, also kann man klar zwischen koordinierten Molekülen und Ionen und solchen, die zur Erhaltung der Elektroneutralität vorhanden sind, unterscheiden. Jene dissoziieren in der Lösung und können durch geeignete Reaktionen der qualitativen Analytik direkt nachgewiesen werden, die am Metallion gebundenen hingegen nicht. Ausserdem kann man diese Gegenionen zum Komplex titrieren, sofern eine geeignete Reaktion existiert, die nicht durch den inerten Komplex gestört wird. Durch Messung der Aequivalentleitfähigkeit (Leitfähigkeit pro Konzentrationseinheit) kann man alternativ erkennen, wie viele dissoziierte Ionen vorliegen. Anhand der Feststellung, dass bei gleicher Zusammensetzung verschiedene Farben auftreten können, schloss Werner auf das Vorhandensein von Isomeren. Man könnte also behaupten, Werner habe „Spektroskopie“ im rudimentären Sinn eingesetzt. Vorkommen von Metallkomplexen und Funktion Metallkomplexe kommen an vielen Orten vor: In Mineralien, im Oberflächen- und Grundwasser, in Organismen, und selbstverständlich in Gewerbe und Industrie. Am seltensten sind sie in der Atmosphäre, es gibt wenige flüchtige Verbindungen dieser Art. Ebenso 1 vielfältig ist ihre Funktion: In Mineralien sind sie strukturbestimmende Bestandteile, im Wasser sind sie schlicht die Formen, in denen Metallionen darin überhaupt existieren, in Organismen sind sie Zentren in Katalysatorfunktionen (Enzyme). In der Technik dienen sie ebenfalls als Katalysatoren, als Farbpigmente und vieles Andere. Das Leben benötigt Kohlenstoff, Licht und Wasser. Ohne Metallkomplexe würde jedoch nicht viel laufen: Die essentiellen Prozesse der Photosynthese und Zellatmung beruhen alle auf Metallkomplexen, neben Tausenden von weiteren biochemischen Prozessen. Bindungen in Komplexen Das Auffälligste an den Metallkomplexen ist die in der Regel grosse Zahl von Bindungen an einem zentralen Atom. Alfred Werner selbst hatte noch kein Modell dafür, die Geometrie der Verbindungen bestimmte er nur über die von ihm so bezeichnete Koordinationszahl, das ist die Zahl der unmittelbar am Metall gebundenen Liganden. Er argumentierte völlig korrekt, dass sich die Liganden aus energetischen Gründen in eine Anordnung geringster Raumforderung und Abstossung begeben würden. Für 6 Liganden ist das ein Oktaeder, für 4 ein Tetraeder. Ausserdem kennt man Komplexe mit 2 und 5 Liganden sowie 7-12 Liganden bei sehr grossen Metallatomen. Eher selten ist die Koordinationszahl 3. Bei der Koordinationszahl 4 wurden auch Strukturen gefunden, die quadratisch planar sind. Bei näherer Betrachtung stellt sich bei diesen heraus, dass sie eigentlich Oktaeder sind, die auf einer Achse keine oder nur sehr lose gebundene Liganden tragen. Häufige Komplexformen mit je einem Beispiel: 2 5 [AgCl2]‾ Fe(CO)5 3 [HgI3]‾ [SnCl3]‾ 4 6 [Co(NH3)6]3+ [Zn(NH3)4]2+ [Ni(CN)4]2‾ 2 Die elektronische Natur der Metall-Ligand Bindung wurde bald als Interaktion von nichtbindenden Elektronenpaaren der Liganden mit den an Elektronen defizitären Metallzentren erkannt, i.a. fehlen ihnen zumindest die s- und p-Elektronen der Valenzschale, und oft noch ein Teil der d-Elektronen der nächst tieferen Schale. Es gibt zwar Komplexe mit tiefen Oxidationsstufen von Metallen, aber nur mit sehr speziellen Liganden und nicht in Lösungsmitteln, die selbst als Liganden auftreten können. Eine Besonderheit ist auch das gelegentliche Auftreten von C=C Doppelbindungen als Liganden anstelle von Atomen mit nichtbindenden Elektronenpaaren. Wir kennen solche Bindungen schon von früher, sie entsprechen dem Säure-Base Konzept von G. N. Lewis. Der Unterschied zu damals besteht darin, dass der Elektronenpaar-Akzeptor, das Metall, mehrere Elektronenpaar-Donoren, also Basen bzw. Liganden, binden kann. Tatsächlich binden die meisten Liganden auch H+. Damit lassen sich die Bindungsverhältnisse mit dem VSEPR-Modell meist korrekt behandeln wie bei kovalenten Verbindungen, obwohl die Bindungen selbst oft eher ionischen Charakter besitzen. Je nach Koordinationszahl und Zahl der d-Elektronen sieht die Besetzung etwas anders aus. Auf jeden Fall besetzen Ligand-Elektronenpaare zuerst die 4s- und 4p-Zustände (bis zu 8 e−) im Metall (der 4. Periode), bevor weitere Elektronen in die 3d-Zustände gehen. Sind die 3d-Zustände im Metall schon besetzt, so wird Besetzung der 4d-Zustände angenommen. Das ist eher etwas unrealistisch, solche Komplexe haben ziemlich ionischen Charakter. Ti Ti3+(H2O)6 FeCl4- CoF63- Co(NH3)63+ 3d 4s 4p 4d 3d 4s 4p 4d 3d 4s 4p 4d 3d 4s 4p 4d 3d 4s 4p 4d Metall-Elektronen Donor-Elektronenpaare 3 LCAO-MO-Modell der Bindung in oktaedrischen, tetraedrischen und quadratisch-planaren Komplexen: Wir kombinieren 4 oder 6 Ligandorbitale, i.a. vom s- oder σ-Typ, mit den 4s- und 4p- Orbitalen des Metalls (aus der 4. Periode). Die 3d-Elektronen tragen normalerweise nicht viel zum bindenden Anteil bei, die Bindungselektronen stammen, wie bei der normalen Säure-Base Reaktion, ausschliesslich vom Donor (Base). Es existiert allerdings eine Interaktion zwischen den d-Orbitalen, die auf die Donororbitale ausgerichtet sind. Diese ist antibindender Art, die betreffenden d-Orbitale werden energetisch angehoben. Im Falle von Koordinationszahl 6 sind dies im selben Ausmass das d x2 − y 2 und das d z2 Orbital. Im Falle des quadratisch-planaren Komplexes ist es hauptsächlich das d x2 − y 2 Orbital und weniger das d z2 Orbital. Bei Koordinationszahl 4 und tetraedrischer Geometrie werden die d xy , d xz und d yz Orbitale energetisch ungünstig, dafür d x2 − y 2 und d z2 nicht. Nebenstehend ist das MO- Schema eines rel. Energie 4p symmetrischen oktaedrischen Komplexes dargestellt, die bindenden 4s Orbitale sind energetisch 3d näher den LigandLiganden-MOs Donororbitalen. Die Energie der d-Elektronen wird charakteristisch aufgeteilt in drei nichtbindende und zwei schwach antibindende Zustände, wie vorgehend beschrieben. Das nächste Schema zeigt den quadratisch planaren Fall, wo nur noch 4 rel. Energie 4p Ligandorbitale beteiligt sind, was strenggenommen nicht korrekt ist, weil es 4s sich um gestreckte 3d Liganden-MOs Oktaeder handelt. Eine tiefer gehende Behandlung würde den Rahmen der Basisvorlesung sprengen. 4 Detaillierte Betrachtungen sind späteren Spezialvorlesungen vorbehalten. Mit 4 Liganden und tetraedrischer Geometrie rel. Energie 4p sieht das MO-Schema nochmals anders aus, wie links gezeigt. Hier kehrt 4s sich die energetische 3d Verteilung der dLiganden-MOs Funktionen um, aus den Gründen, die schon weiter oben dargelegt wurden. Die energetische Verschiebung der d- Elektronen hat wenig Einfluss auf die Ligandbindungsstärke, jedoch auf physikalische Eigenschaften des Komplexes und sein reaktionskinetisches Verhalten. Ligandfeld-Aufspaltung Wie schon oben erwähnt, wird unter dem Einfluss der Donor-Elektronen der Liganden die Energie der d-Zustände im Metall verändert. Die Liganden werden einerseits vom Metallzentrum elektrostatisch angezogen, andererseits stossen sie sich mit der Elektronendichte der d-Zustände ab. Besetzung dieser Zustände, wenn die Orbitalgeometrie Vorzugsrichtungen auf die Bindungsachsen der Donoren aufweist, vermindert die Stabilität des Systems. Das Phänomen wird Ligandfeldaufspaltung der d-Energien genannt. In einem symmetrischen oktaedrischen Komplex wird der entstandene Energieunterschied ∆o genannt, in einem symmetrischen tetraedrischen System ∆t. In der Regel ist ∆o > ∆t, weil 6 Liganden ein stärkeres elektrisches Feld erzeugen als 4. Wenn ∆o grösser wird als die Energie, die es kostet, zwei Elektronen mit antiparallelem Spin in einem Energiezustand unterzubringen (Spin-Paarungsenergie), dann werden die d-Elektronen so weit wie möglich gepaart. Dadurch verschwindet das magnetische Moment des Spins dieser Elektronen, und das gesamte magnetische Moment des Metalls nimmt ab. Sind alle d-Elektronen gepaart, ist das Metall diamagnetisch. Komplexe mit maximal gepaarten d-Elektronen nennt man low-spin, solche mit maximal ungepaarten high-spin. Unterschiedliche Aufspaltung ∆o hat noch weitere Folgen: Obwohl die Absorption eines Lichtquants (Photons) durch einen Übergang eines Elektrons von einem d-Zustand zu einem andern d-Zustand quantenmechanisch verboten ist, 5 weil sich die Symmetrie des Elektronenzustands nicht ändert, gibt es eine Restwahrscheinlichkeit dafür. Der Grund dafür ∆o < Ep high spin ∆o sind geringe Abweichungen von der perfekten oktaederischen Form. Deshalb wird dennoch etwas Licht von spezifischer Wellenlänge absorbiert, was den Übergangsmetallkomplexen ihre charakteristischen, aber nicht allzu kräftigen ∆o > Ep low spin ∆o stattfinden, diese Ionen sind farblos. Farben verleiht. Das gilt auch für quadratisch- planare und tetraedrische Komplexe. Ein noch stärkeres Verbot betrifft die Spinumkehr bei der Anregung von Elektronen. Bei einer d5-high spin Konfiguration können deshalb keine Übergänge hν hν d6 high spin d5 high spin Die d3 Konfiguration und die d6 low spin Konfiguration zeigen aufgrund ihrer hohen Symmetrie eine bemerkenswerte Trägheit bei Austauschreaktionen von Liganden. Um einen Liganden austreten und einen neuen eintreten zu lassen, müsste die hohe Symmetrie kurzfristig aufgehoben werden. Dies geschieht nur selten, also ist die Reaktionsrate gering. Diese Art Komplexe nennt man, wie schon erwähnt, kinetisch inert. Alfred Werners erste Studienobjekte waren die d6 low spin Komplexe von CoIII. d3 d6 low spin Die Grösse von ∆o und ∆t hängt wesentlich von der Donorstärke der Liganden ab. Je stärkere Lewisbasen sie sind, desto grösser wird die Aufspaltung der d-Energien. Grosse Aufspaltung bedeutet dann Lichtabsorption bei kurzen Wellenlängen, weil viel Energie zur Anregung 6 benötigt wird. Es bedeutet auch Tendenz zu low spin Systemen, weil die Stabilisierung der Bindung die Paarung der Elektronen erlaubt. Reaktionen von Komplexen Metallkomplexe gehen in erster Linie zwei Arten von Reaktionen ein: Ligandaustauschreaktionen, die man auch als Lewis Säure-Base Reaktionen auffassen kann, und Redoxreaktionen, die vor allem das Metallion betreffen. [Cu(H2O)6]2+ + 4 NH3 ⇀ [Cu(NH3)4]2+ + 6 H2O ↽ 2 [Fe(CN)6]4– + Br2 ⇀ 2 [Fe(CN)6]3– + 2 Br– ↽ Ligandaustausch Redoxreaktion Komplexgleichgewichte Weil es sich bei Lewis Säure-Base Reaktionen immer um Gleichgewichte handelt, können wir im Zusammenhang mit Komplexbildungsreaktionen und damit gekoppelten Vorgängen wieder das Massenwirkungsgesetz anwenden. Die „Komplexbildung“ selbst ist eigentlich eine Ligandaustauschreaktion. Geht ein festes Metallsalz in Lösung, so tritt das Metallkation tatsächlich aus einem Komplex mit den Anionen (oder dem Kristallwasser) im Kristall über in einen Komplex mit Lösungsmittelmolekülen. Gibt man dann zur Lösung einen Donor stärker als das Lösungsmittel, wird dieses ausgetauscht und wir haben die „Komplexbildung“, wie sie üblicherweise gemeint ist. Alle Komplexbildungen sind Stufenreaktionen, sofern mehr als ein Ligand gebunden wird. Beispiel: Ein Metallion M2+ liegt hydratisiert vor und tauscht bei der Zugabe des Liganden L mit steigender Konzentration das H2O gegen den Liganden aus. Wir nehmen Koordinationszahl 6 an. [M(H2O)6]2+ + L [ML(H2O)5]2+ + L [ML2(H2O)4]2+ + L [ML3(H2O)3]2+ + L [ML4(H2O)2]2+ + L [ML5(H2O)]2+ + L ⇀ ↽ ⇀ ↽ ⇀ ↽ ⇀ ↽ ⇀ ↽ ⇀ ↽ [ML(H2O)5]2+ + H2O K1 [ML3(H2O)3]2+ + H2O K3 [ML2(H2O)4]2+ + H2O [ML4(H2O)2]2+ + H2O [ML5(H2O)]2+ + H2O [ML6]2+ + H2O K2 K4 K5 K6 Falls der Ligand L ein Anion ist, ändert sich natürlich fortlaufend die Ladung. Oft kann man die einzelnen K nicht richtig bestimmen, sondern nur Bruttoreaktionen bis zu einer 7 bestimmten Stufe. Die Bruttobildungskonstante bis zu einer Komplexbildungsstufe n berechnet sich zu βn = K1 i K 2 i K 3 …i K n , wie man einfach durch Kombinieren der zu den K- Werten gehörigen Massenwirkungsgesetze herausfindet. Die Konstanten nehmen in der Regel mit zunehmendem Koordinationsgrad ab. Komplexbildungsgleichgewichte können mit andern Gleichgewichten gekoppelt sein. Typisch ist das für die Protonierungsgleichgewichte der Liganden. Mit Säure kann man manchen Komplex wieder zerlegen. Beispiel: ⇀ [Cu(NH3)4]2+ Cu2+ + 4 NH3 ↽ NH4+ ⇀ NH3 + H+ ↽ lg K1 =4.25; lg K2 =3.61; lg K3 =2.98; lg K4=2.24 lg β4 =13.1 lg Ka = -9 Setzt man das MWG für die Säuredissoziation in das Brutto-Komplexbildungsgleichgewicht ein, erhält man β4 = β4 [Cu(NH 3 ) 24+ ][H + ]4 . Das kann man noch zu [Cu 2+ ][NH 4+ ]4 K a4 K a4 [Cu(NH 3 ) 24+ ] = = β 4' umschreiben und erhält das pH-abhängige β4’. Bei pH = 4 [H + ]4 [Cu 2+ ][NH 4+ ]4 z.B. beträgt β4’ = β = 10 ' 4 13.1 10 −36 M = 10 −7.9 M −4 . Das bedeutet, dass bei diesem pH praktisch −16 10 −4 kein Komplex existiert. Bei pH=9 sieht das anders aus, β = 10 ' 4 Komplex ist voll ausgebildet. 13.1 10−36 M = 1013.1 M −4 , der −36 10 −4 Ein weiteres Beispiel demonstriert den Einfluss von Komplexbildungen auf Elektrodenpotentiale. Das Standardelektrodenpotential für Fe3+ + e– ⇀ Fe2+ ↽ E 0Fe3+ / Fe2+ = 0.77V weist Fe3+ als Oxidationsmittel aus. Komplexiert man Fe3+ mit dem Citration, dem Anion der Zitronensäure, so sieht das ganz anders aus. Fe3+ bindet ein Citration mit K1 = 1012. K1 = [Fe(cit ) − ][H + ] [Fe(cit ) − ][H + ] 12 3+ = 10 wird aufgelöst [Fe ] = und in die Nernstgleichung [Fe3+ ][Hcit 3− ] K1[Hcit 3− ] eingesetzt: E = E 0Fe3+ /Fe2+ + E = E 0Fe3+ / Fe2+ + RT [Fe(cit ) − ][H + ] ln Weil K1 eine Konstante ist, kann man F K1[Hcit 3− ][Fe 2+ ] RT 1 RT [Fe(cit ) − ][H + ] ln + ln separieren und ein neues E°’ definieren: F K1 F [Hcit 3− ][Fe 2+ ] E 0' = E 0Fe3+ / Fe2+ + RT 1 ln = 0.77 V − 0.71V = 0.06 V . Das Elektrodenpotential wird nahezu 0! F K1 8 Wenn man dann noch einen hohen pH-Wert einstellt, kann man das Fe3+ in diesem Komplex nur noch schlecht zu Fe2+ reduzieren, was beim Aqua-Ion kein Problem ist. Auch Löslichkeitsprodukte werden beeinflusst. Kso(AgCl) = 10–10 M2 =[Ag+][Cl–], aber β2 (AgCl2− ) = 105 M −2 = [AgCl−2 ] [Ag + ] 1 und damit = . Man sieht, dass [Ag+] − + − 2 [Ag ][Cl ] [AgCl 2 ] β 2 [Cl− ]2 durch β2 und die [Cl–] abgesenkt werden, was bei genügend [Cl–] zur Unterschreitung des Löslichkeitsprodukts führt, weil die Komplexbildung von [Cl–]2 abhängt, Kso aber nur linear. Eine andere Darstellungsform dieses Sachverhalts zeigt die algebraische Umformung des Komplexbildungs-MWG unter Separation des Ionenprodukts: [AgCl −2 ] [Ag ][Cl ] = β2 [Cl− ] + − Falls das Ionenprodukt links Kso überschreitet, findet Fällung statt. Unter hoher [Cl–] wird das aber nicht der Fall sein, weil der Bruch rechts mit steigender [Cl–] immer kleiner wird. [AgCl2–] ist begrenzt durch [Ag+]0, die totale Konzentration an Ag+, auch wenn immer mehr Cl– zugefügt wird. Spezielle Komplexformen Rückbindung Wenn ein Metallzentrum elektronenreich ist, was bei niedrigen Oxidatonszahlen gegeben ist, so kann es bei bestimmten Liganden dazu kommen, dass das das Metall als Donor auftritt. Das Donor-Orbital ist dann ein besetztes d-Orbital, das Akzeptor-Orbital kann ein π*-Orbital oder ein unbesetztes d-Orbital eines Nichtmetalls der 3. oder höheren Periode sein. Es handelt sich im zweiten Fall um ein ähnliches Phänomen wie die zu Beginn beschriebene Hypervalenz in der Valenzbindungstheorie von Atomen in Perioden > 2. Ein typischer Ligand mit π*-Rückbindung ist das Kohlenmonoxid CO, das Komplexe mit Übergangsmetallen der Oxidationszahlen –I, 0, I und II bilden kann (Ni(CO)4, Fe(CO)5, Porphyrin-Fe(II)-CO, etc.). Diese Bindungsart tritt in Ergänzung der σ-Donorbindung des Liganden auf, nicht allein. 9 C CO π-Orbital (p kombiniert mit p) besetzt O Metall d-Orbital besetzt C O C O CO π∗-Orbital (p kombiniert mit p) unbesetzt d - π∗ Rückbindung Mehrzähnige Liganden – Chelate Metalle binden mehrere Liganden, das ist soweit klar. Wenn das Metall aber mehrere Bindungsstellen hat, kann ein Ligand mit mehr als einer Lewis Base-Funktion und geeigneter Struktur auch mehrere dieser Bindungsstellen besetzen, und es entstehen Ringstrukturen. Den Verbindungstyp nennt man Chelat, von griech. chelè, was „Klaue“ oder „Krebsschere“ bedeutet, weil der Ligand das Metall wie mit einer Zange „packt“. Die Basen eines mehrfach bindenden Liganden werden auch Zähne genannt, es wird von mehrzähnigen Liganden gesprochen. Auffällig dabei ist, dass die Stabilität mit der Zahl der Zähne zunimmt. Das scheint zwar intuitiv richtig zu sein, kann aber dennoch auf verschiedene Weise begründet werden. Die der Intuition näher stehende Begründung besagt, dass es sehr unwahrscheinlich ist, dass sich mehrere Zähne zugleich ablösen, wenn der Ligand einmal gebunden ist. Die Komplexbildung verläuft nämlich, experimentell belegt, auch für solche Liganden schrittweise, und deshalb auch die Dissoziation. Weil der Ligand sich nicht auf einmal lösen kann, ist die Wahrscheinlichkeit der erneuten Bindung eines dissoziierten Zahns hoch, also kann sich der gesamte Ligand schlecht aus der Koordinationssphäre lösen. Die zweite Begründung stützt sich auf die Entropie-Änderung ∆S bei der Komplexbildung. Die Entropiefunktion eines molekularen Systems hängt von den so genannten BewegungsFreiheitsgraden ab. Davon existieren translatorische, rotatorische und vibratorische Freiheitsgrade. Jedes Molekül hat 3 translatorische (Bewegung im Raum) sowie 2-3 10 rotatorische Freiheitsgrade (linear oder dreidimensional). Dazu kommen noch 3n-6 vibratorische Freiheitsgrade (3n-5 bei linearen Molekülen), wobei n die Zahl der Atome des Moleküls ist. Diese Bewegungs-Freiheitsgrade machen die Fähigkeit eines Stoffs, Energie zu speichern, aus. Die physikalische Grösse, die dies ausdrückt, ist die Wärmekapazität dS cp = T . Ein normaler 2-zähniger Ligand beispielsweise hat 3 translatorische und 3 dT p rotatorische Freiheitsgrade sowie die vibratorischen. Wenn er koordiniert wird, treten 2 Lösungsmittelmoleküle aus, die bis zu diesem Zeitpunkt keine eigenen translatorischen und rotatorischen Freiheitsgrade hatten. Für den Verlust von 3 rotatorischen und 3 translatorischen Freiheitsgraden des neuen Liganden werden je 2 mal 3 rotatorische und translatorische Freiheitsgrade erzeugt, die Gesamtzahl der Freiheitsgrade des Systems steigt und damit seine Entropie. Die vibratorischen Freiheitsgrade bleiben mehr oder weniger erhalten, weil sie ein intramolekulares Phänomen sind. Weil ∆G = ∆H − T∆S und bei positivem ∆S das ∆G negativ wird, bevorzugt die Entropieänderung den 2-zähnigen Liganden in der gebundenen Form, der Komplex ist thermodynamisch stabiler als mit 2 einzähnigen Liganden. Beispiele für mehrzähnige Liganden und ihre Komplexe: NH2 H2N O O 1,2-Diaminoethan Ethylendiamin (en) O - - O + O NH + NH OH N CH3 O - O CH3 - O O 1,2- Dimethyl-3-hydroxypyrid-4(1H)-on Deferipron 2-[2-(Bis(carboxymethyl)amino)ethyl-(carboxymethyl) amino]acetat Ethylendiamin-Tetraacetat (EDTA) O - O O P P HO Si CH3 P Tris(diphenylphosphanomethyl)-methylsilan Siliphos O O O - - 3-carboxylato-3-hydroxy-pentan-1,5dicarboxylat Citrat 11 H2N O NH2 NH2 O O NH2 O H2N O H2N - N - N O oktaedrischer tris-en Komplex - O - O oktaedrischer EDTA-Komplex O O H2O O 3+ Fe H2O O O H2N NH2 - - H2N Cl O - O Fe(III)-Citrat-Komplex Cl - Cl Cl NH2 - - NH2 - NH2 - H2N H2N Enantiomerenpaar eines oktaedrischen bis-en-dichlorido-Komplexes Lewis Säuren – Basen Affinität Um die Affinität („Vorliebe“) einer Base für Protonen zu charakterisieren, verwenden wir die Dissoziationskonstanten Ka. Für Lewis Säure-Base Kombinationen existiert so etwas nicht. Es lässt sich aber feststellen, dass gewisse Ligandtypen sich stärker an bestimmte Metalltypen binden als an andere, es gibt also auch hier unterschiedliche Affinitäten. In den 1960er Jahren verglich R. G. Pearson die Bildungskonstanten zahlreicher Komplexe mit den verschiedensten Liganden. Dabei kam er zum Schluss, dass kleine, schlecht polarisierbare Ligandatome sich stärker an kleine, stark geladene Metallionen binden als an solche mit niedriger Ladung und grossem Radius. Umgekehrt bilden grosse, gut polarisierbare Ligandatome sehr stabile Komplexe mit grossen, wenig geladenen Metallionen. „Gleich und gleich gesellt sich gern!“ Pearson nannte die grossen, gut polarisierbaren Ionen und Atome „weich“, wegen der Beweglichkeit ihrer Elektronenhülle (entspricht der Polarisierbarkeit). Die kleinen, schlecht polarisierbaren Partikel nannte er analog „hart“. Die Idee ist als „hart-weich Konzept der Lewis Säuren und Basen“ oder HSAB (hard and soft acids and bases) bekannt geworden. Die Regel ist gut brauchbar, z.B. bildet Al3+ (sehr hart) eigentlich nur stabile Komplexe mit F– und O-Liganden (sehr hart). Cd2+ (weich) bildet stabile Komplexe mit R-S– Liganden 12 (Thiolate, weich), ebenso weich ist Ag+. Die Metallionen der Perioden 5 und 6 sind auf Grund ihrer grossen Radien weich, ausser sie tragen Ladungen > +2. Sehr interessant ist der Effekt bei Wechsel der Oxidationszahl des Metalls: Fe3+ ist wie Al3+ sehr hart und komplexiert wirklich gut nur F– und O-Liganden. Nach der Reduktion zu Fe2+ ist die Stabilität mit F– praktisch verschwunden, und O-Liganden werden nur noch schlecht gebunden. Dafür werden jetzt Amin-Liganden angenommen, was Fe3+ nur tut, wenn auch noch O dabei ist, oder das Ligandgerüst so steif wie bei einem Porphyrin. Amine gelten zwar noch als hart, sind das aber schon viel weniger als O. Eine Anomalie findet man bei niedrig geladenen Metallionen der 6. Periode. Au+, Hg2+ und Pb2+ binden weiche Liganden sehr gut, aber merkwürdigerweise auch sehr harte wie OH– oder Carboxylate R-COO–. Au+ reagiert mit H2O unter Freisetzung von H+, der pKa für diese Reaktion ist 4, also saurer als Essigsäure, und das trotz der geringen Ladung von +1! Der Grund dafür ist, dass sich hier Quantenmechanik und spezielle Relativität treffen: Das primäre Akzeptororbital der genannten anomalen Ionen ist das 6s, das wie alle s-Funktionen eine ziemlich hohe Dichte am bei diesen grossen Atomen hoch geladenen Kern besitzt. Die eingespeisten Donorelektronen erfahren also eine sehr grosse elektrostatische Kraft. Berechnet man die scheinbare Geschwindigkeit dieser Elektronen, so liegt der Wert nahe bei der Lichtgeschwindigkeit. Ob sich diese Elektronen nun tatsächlich „bewegen“ oder nicht, der relativistische Effekt der Zeitdilatation in Kernnähe ist vorhanden, damit halten sich die Elektronen von aussen gesehen länger nahe am Kern auf als unter nicht-relativistischer Betrachtung. Der Effekt bewirkt eine stärkere Bindung der Donorelektronen als unter nichtrelativistischen Bedingungen erwartet, das Ion ist eine starke Lewis Säure. 13 Anhang: Komplex-Isomere Konstitutionsisomere Ionisationsisomere [Co(NH3)5(SO4)]Br oder [Co(NH3)5(Br)]SO4 Hydratisomere [Cr(OH2)6]Cl3 oder [Cr(OH2)4Cl2 ]Cl•2H2O oder [Cr(OH2)5Cl ]Cl2•H2O Koordinationsisomere [PtII(NH3)4][PtIVCl6] oder [PtIV(NH3)4Cl2][PtIICl4] Bindungsisomere M − NO2 oder M − ONO dioxidonitrato-κN oder dioxidonitrato-κO M – CN oder M – NC cyanido-κN oder cyanido-κO M – SCN oder M – NCS thiocyanato-κS oder thiocyanato-κN Stereoisomere Diastereoisomere NH3 [PtCl2(NH3)2] cis und trans Pt NH3 [CoCl2(NH3)4] cis und trans Cl 2+ Cl - NH3 - Cl Pt Cl 2+ - - NH3 Enantiomere (Spiegelbild-Isomerie) [CoCl2(en)2]+ und cis-trans H2N 3+ Co NH2 NH2 H2N H2N NH2 Cl - Cl - Cl Cl - - NH2 3+ Co H2N H2N Cl 3+ Co NH2 NH2 Cl - NH2 14