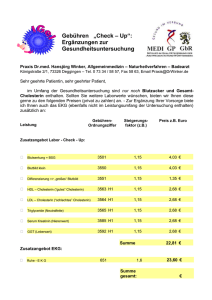
Cholesterin und Schlaganfallrisiko
Werbung

Botenstoff Beispiele für Botenstoffe und ihre Wirkung. Als Botenstoff (Mediator, Signalstoff, Elicitor, Semiochemikalie) bezeichnet man verschiedene chemische Stoffe, die in einem Organismus oder zwischen Spezies der Übertragung von Signalen bzw. Informationen (chemische Kommunikation) dienen. Botenstoffe sind essentiell für das Zusammenspiel der Zellen in einem Organismus (Kommunikation zwischen den Zellen). Bei Pflanzen regulieren Botenstoffe u.a. das Wachstum und die Entwicklung sowie auch den eigenen Schutz, z. B. vor Krankheitserregern oder Fressfeinden.[1][2] Die Kommunikation zwischen den Organismen erfolgt häufig auch über chemische Botenstoffe, die sog. Semiochemikalien. Bei den Semiochemikalien wird generell zwischen Pheromonen und Allelochemikalien unterschieden. Während Pheromone der Kommunikation zwischen Organismen einer Art (intraspezifisch) dienen, vermitteln Allelochemikalien Informationen zwischen verschiedenen Arten (interspezifisch). Beispiele für eine interspezifische Wirkung sind das Vermögen einiger Pflanzen, über bestimmte Stoffe, die sog. Allomone Parasiten von Pflanzenschädlingen anzulocken.[1] das Anlocken von Nachtfaltern als Beute durch eine Spinnenart mittels eines Sexualpheromons.[3] Die Botenstoffe können in verschiedene funktionelle Gruppen oder gemäß ihrer Funktion und Wirkung unterteilt werden, wobei die Einteilung häufig gleitende Übergänge hat bzw. recht willkürlich ist: Gruppen von Botenstoffen[4] Gruppe Bemerkungen, Eigenschaften 1 Beispiel(e) Referenzen Hormone werden im Organismus synthetisiert und übermitteln an Organe, Gewebe oder Zellgruppen, die vom Bildungsort mehr oder weniger weit entfernt liegen können, Signale od. Botschaften, die auf deren Funktion bestimmte physiologische Wirkungen ausüben; dabei wirken Hormone nicht direkt, sondern indirekt, beispielsweise durch Veränderung der Enzymkonzentration [5] Neuropeptide (Cytokine; spezielle Neurotransmitter des Gehirns); regeln die Stärke von bestimmten Botenstoffe des Nervensystems, Reaktionen; Endorphine die die Nervenzellen erregen Neurotransmitter hemmen beispielsweise oder hemmen; eng begrenzte starke Schmerzen, können lokale Wirkung; aber auch Glücksgefühle und Entspannung nach starken körperlichen Anstrengungen vermitteln [6][7] Parahormone Botenstoffe, die in irgendeiner Weise nicht alle Kriterien erfüllen, die für die Definition eines Hormons notwendig sind Kohlendioxid: fungiert im Rahmen der Atmungsregulation als Kommunikationsstoff [8] Pheromone Werden in die Umgebung ausgeschieden und lösen einen bestimmten Effekt oder ein bestimmtes Verhalten aus; Pheromone wirken, im Gegensatz zu den Allomonen zwischen Individuen derselben Spezies (intraspezifisch) Pheromone beeinflussen beispielsweise auch das Zusammenleben der Menschen [9] Botenstoffe in Pflanzen; beeinflussen Wachstums- und Ethylen, Auxine; Auxine stimulieren in geringer Konzentration Wachstums- Phytohormone 2 [10] Differenzierungsprozesse und Entwicklungsprozesse wie Zellteilung und Zellstreckung in der Pflanze. Ethylen ist bei Pflanzen an Wachstumsvorgängen und Stressreaktionen beteiligt Unterteilung von Botenstoffen (Semiochemikalien) nach ihrer Funktion und Wirkung[11] Wirkung Stoffklasse Bezeichnung und Wirkung Intraspezifisch Pheromone Primer: physiologische Veränderung Intraspezifisch Pheromone Releaser: Verhaltensänderung Allomone: Vorteil für produzierenden Interspezifisch Allelochemikalien Organismus bzw. Schaden für Empfänger Beispiel(e) Antibiotika, Toxine, fraßhemmende Geschmacksstoffe bei Pflanzen Kairomone: Vorteil für Interspezifisch Allelochemikalien empfangenden Organismus Synonome: Vorteil für produzierenden und Interspezifisch Allelochemikalien Blütenduft empfangenden Organismus Apneumone: Freisetzung durch Interspezifisch Allelochemikalien abiotische Substrate; können für Empfänger von Vorteil und für das 3 Referenzen Substrat bewohnende Organismen von Nachteil sein Triglyceride Allgemeine Struktur von Triacylglycerinen. Die Reste R stehen für die Kohlenwasserstoffketten meist verschiedener Fettsäuren. Triglyceride, Triglyzeride, auch Glycerol-Triester, sind dreifache Ester des dreiwertigen Alkohols Glycerin mit drei Säuremolekülen. Verbindungen mit Carbonsäuren sollten nach der IUPAC-Empfehlung ausschließlich als Triacylglycerine bezeichnet werden. Die Vorsilbe Tri verweist auf drei Acyl-Säurereste, die mit Glycerin verestert sind. Triacylglycerine mit drei Fettsäuren sind die Verbindungen in Fetten und fetten Ölen. Natürliche Fette bestehen zum überwiegenden Teil aus Triglyceriden mit drei langkettigen Fettsäuren, die meist aus unverzweigten Ketten mit 4 bis 26, typischerweise 12 bis 22 Kohlenstoff-Atomen bestehen. Sind sie bei Raumtemperatur flüssig, werden sie auch als Öle oder, um sie von den Mineralölen zu unterscheiden, fette Öle bezeichnet. Reine Triacylglycerine von Fettsäuren werden auch als Neutralfette bezeichnet. Man kann zwischen MCTs (medium chain triglycerides) (6 bis 12 C-Atome) LCTs (long chain triglycerides) (14 bis 24 C-Atome) unterscheiden, MCTs haben Fettsäuren mittlerer Länge gebunden, LCTs haben Fettsäuren großer Länge gebunden. Phosphoglyceride wie z.B. Lecithine sind Triglyceride aus zwei Fettsäuren und einer organischen Phosphorsäureverbindung. Beide Verbindungstypen zählen zur Klasse der Lipide. Neben pflanzlichen und tierische Triglyceriden gibt es auch synthetische Triglyceride. 4 Cholesterin Strukturformel Allgemeines Name Cholesterin Andere Namen Summenformel C27H46O CAS-Nummer 57-88-5 PubChem 5997 DrugBank EXPT00945 Cholesterol Cholest-5-en-3β-ol 5-Cholesten-3β-ol Kurzbeschreibung weißer, fast geruchloser Feststoff Eigenschaften Molare Masse 386,67 g·mol−1 Aggregatzustand fest Dichte 1,07 g·cm−3 (20 °C) [1] Schmelzpunkt 147–150 °C [1] Siedepunkt Zersetzung bei 360 °C [1] Löslichkeit praktisch unlöslich in Wasser Sicherheitshinweise Gefahrstoffkennzeichnung [1] keine Gefahrensymbole R- und S-Sätze R: keine R-Sätze S: keine S-Sätze 5 Soweit möglich und gebräuchlich, werden SI-Einheiten verwendet. Wenn nicht anders vermerkt, gelten die angegebenen Daten bei Standardbedingungen. Nummerierung der Kohlenstoffringe und Bezeichnung der Ringe im Cholesteringerüst. Das Cholesterin (auch Cholesterol) ist ein in allen tierischen Zellen vorkommender Naturstoff. Der Name leitet sich vom griechischen „chole“ (Galle) und „stereos“ (fest) ab, da es in Gallensteinen bereits im 18. Jahrhundert gefunden wurde. Inhaltsverzeichnis 1 Funktion 2 Chemische Einordnung 3 Biosynthese und Abbau 4 Cholesterinbiosynthese 5 Cholesterintransport (Lipoproteine) 6 Blutspiegel o 6.1 Gesamtcholesterinspiegel o 6.2 LDL-Cholesterinspiegel o 6.3 HDL-Cholesterinspiegel o 6.4 Quotienten o 6.5 Messung und Labor-Referenzwerte o 6.6 Einheiten und Umrechnung 7 Erkrankungen o 7.1 Familiäre Hypercholesterinämie o 7.2 Gallensteine o 7.3 Weitere Krankheitsformen 8 Cholesterin und die Koronare Herzkrankheit (KHK) o 8.1 Bedeutung der Hypothese o 8.2 Empirische Hinweise o 8.3 Die Rolle von High Density Lipoprotein und Low Density Lipoprotein o 8.4 Zielwerte und Richtlinien o 8.5 Kritik 8.5.1 Zweifel an der Kausalkette Ernährung – Cholesterin – KHKErkrankung 8.5.2 Kritische Bewertung von Nutzen und Risiko einer medikamentösen Cholesterin-Senkung 8.5.3 Einfluss wirtschaftlicher Faktoren auf Forschung, Fachgesellschaften und veröffentlichte Meinung 6 9 Cholesterin und Schlaganfallrisiko o 9.1 Serum-Cholesterinspiegel und Schlaganfallrisiko o 9.2 Cholesterinsenkende Medikamente und ihr Einfluss auf das SchlaganfallRisiko 10 Cholesterin und Krebserkrankungen o 10.1 Serum-Cholesterinspiegel und Krebsrisiko o 10.2 Cholesterinsenkende Medikamente und ihr Einfluss auf das Krebsrisiko 10.2.1 Erhöhung des Krebsrisikos 10.2.2 Senkung des Krebsrisikos 10.2.3 Kein Einfluss auf Krebsrisiko 11 Cholesterin und Ernährung o 11.1 Empfehlung bei hohem Cholesterinspiegel o 11.2 Einfluss der Ernährung auf den Cholesterinspiegel o 11.3 Einfluss der Ernährung auf den Cholesterinspiegel durch Prostaglandine o 11.4 Cholesterin und Schwangerschaft o 11.5 Cholesterin und Muttermilch 12 Cholesterin, Psyche und Gedächtnis o 12.1 Cholesterin und Gewaltbereitschaft o 12.2 Cholesterin und Depressionen o 12.3 Cholesterin und Gedächtnis o 12.4 Cholesterinsenkung und Albträume 13 Arzneimittel o 13.1 Fibrate o 13.2 Statine o 13.3 Ezetimib 14 Studien o 14.1 Framingham-Studie o 14.2 Metastudien o 14.3 CARE o 14.4 EXCEL o 14.5 4S o 14.6 PROCAM o 14.7 LIPID o 14.8 HPS o 14.9 4D o 14.10 Hu 1999 15 Zitate 16 Einzelnachweise 17 Literatur 18 Weblinks Funktion Cholesterin ist ein lebenswichtiges Lipid und ein wichtiger Bestandteil der Plasmamembran. Hier erhöht es die Stabilität der Membran und wirkt zusammen mit Proteinen in der Zellmembran an der Ein- und Ausschleusung von Signalstoffen mit. Der Cholesteringehalt des menschlichen Körpers beträgt etwa 140 g. Da es wasserunlöslich ist, befinden sich über 95 % des Cholesterins intrazellulär. Im Blut wird es gebunden an Lipoproteine transportiert, 7 die mit zunehmender Dichte als Chylomikronen, VLDL, IDL, LDL und HDL bezeichnet werden. Cholesterin ist außerdem Vorstufe der Gallensäuren und Steroidhormone. Es wird durch das Cholesterin-Seitenkettentrennungsenzym zu Pregnenolon umgewandelt, welches wiederum die Ausgangsverbindung für die Hormone/Corticoide (Testosteron, Östradiol, Cortisol, Progesteron und Aldosteron) und für die Gallensäuren (Cholanssäure und Cholsäure) ist.[2] Ein Zwischenprodukt der Cholesterinbiosynthese, das 7-Dehydrocholesterin, ist das Provitamin zur Bildung von Vitamin D durch UV-Licht. Neue Forschungen zeigen zudem, dass der Körper Cholesterin zur Biosynthese herzwirksamer Glykoside nutzt. Welche Bedeutung diese endogen synthetisierten Glykoside haben, ist noch weitgehend unbekannt. Beispielsweise aufgrund von Sedimentfunden mit chemischen Cholesterin-Verwandten (Sterolen) wird von einigen Forschern angenommen, dass das Cholesterinmolekül für den Fall, dass es nie anders als in belebter Materie auftrat, auch evolutionsgeschichtlich sehr alt wäre.[3] Die Biosynthese des Moleküls funktioniere allerdings erst, seit Sauerstoff in der Atmosphäre vorhanden sei. In Bakterien und den Membranen von Mitochondrien finde sich aus diesem Grund kaum Cholesterin. Pflanzen und Pilze enthalten ebenfalls kein Cholesterin, dafür aber andere, strukturell ähnliche Sterole. Chemische Einordnung Cholesterin ist ein polyzyklischer Alkohol. Herkömmlich wird es als zur Gruppe der Sterine (Sterole) gehörendes Steroid zu den Lipiden gerechnet. Entgegen einer verbreiteten Verwechslung ist es jedoch kein Fett, selbst die Einordnung als Lipid ist nicht zwingend. Biosynthese und Abbau Cholesterin ist ein für Menschen und Tiere lebenswichtiges Zoosterin. Beim Menschen wird Cholesterin zum Großteil (90 %) im Körper selbst hergestellt (synthetisiert), beim Erwachsenen in einer Menge von 1 bis 2 g pro Tag, und nur zu einem kleinen Teil mit der Nahrung aufgenommen. Die Cholesterinresorption liegt im Durchschnitt bei 0,1 bis 0,3 g pro Tag und kann höchstens auf 0,5 g pro Tag gesteigert werden. Das entspricht 30 bis 60 % des in der Nahrung enthaltenen Cholesterins. Beim Menschen sind die Leber und die Darmschleimhaut die Hauptorte der Cholesterinsynthese. Die Biosynthese des Moleküls erfolgt über viele Zwischenstufen aus der aktivierten Essigsäure, dem Acetyl-Coenzym A. Außer in Leber und Darm kann die Cholesterinbiosynthese mit wenigen Ausnahmen in fast allen Zellen des Körpers ablaufen. Das Gehirn synthetisiert das von ihm benötigte Cholesterin vollständig selbst, da dieses die Blut-Hirn-Schranke nicht passieren kann. Organe mit hohem Cholesterinbedarf sind das Gehirn sowie die Steroidhormone produzierenden Organe (Nebennieren, Eierstöcke und Hoden). Etwa ein Viertel des gesamten Cholesterins ist im Gehirn enthalten, wo es vor allem in den lipidreichen Myelinscheiden der Axone vorkommt.[4] 8 Das Gleichgewicht zwischen benötigtem, selbst produziertem und über die Nahrung aufgenommenem Cholesterin wird über vielfältige Mechanismen aufrechterhalten. Als wichtig kann dabei die Hemmung der HMG-CoA-Reduktase, des wichtigsten Enzyms der Cholesterinbiosynthese, durch Cholesterin gelten (noch stärker wird die HMG-CoAReduktase durch Lanosterol, eine Vorstufe von Cholesterin, gehemmt). Damit hemmen Produkte dieses Stoffwechselwegs (Cholesterinsynthese) „ihr“ Enzym; dies ist ein typisches Beispiel negativer Rückkopplung. Außerdem verkürzt sich die Halbwertszeit der HMG-CoAReduktase bei erhöhtem Lanosterolspiegel stark, da sie dann vermehrt an Insigs (insulininduzierte Gene) bindet, was schließlich zu ihrem Abbau im Proteasom führt. Es gibt noch viele andere, weniger direkte Regulationsmechanismen, die auf transkriptioneller Ebene ablaufen. Hier sind die Proteine SCAP, Insig-1 und -2 wichtig, die in Anwesenheit von Cholesterin, für das sie eine Bindungsstelle besitzen, über die proteolytische Aktivierung von SREBPs die Aktivität einer größeren Anzahl Gene regulieren. Auch Insulin spielt hier eine Rolle, da es u. a. die Transkription von SREBP1c steigert. Die HMG-CoA-Reduktase, das Schlüsselenzym der Cholesterinbiosynthese, kann spezifisch und effektiv durch verschiedene Substanzen gehemmt werden (beispielsweise Statine, die als HMG-CoA-Reduktase-Hemmer eine bestimmte Klasse von Medikamenten darstellen). Über den LDL-Rezeptor wird die Aufnahme in die Zelle aktiviert. Die Höhe des Cholesterinspiegels hängt vor allem von der körpereigenen Produktion ab und erst in zweiter Linie von der Zufuhr über die Nahrung. Daneben gibt es eine Vielzahl genetisch bedingter Hypercholesterinämien. Auch als Folge anderer Erkrankungen kann der Cholesterinspiegel erhöht sein (beispielsweise durch Hypothyreose, Niereninsuffizienz oder metabolisches Syndrom). Cholesterin wird über die Leber ausgeschieden, indem es in Form von Gallensäuren über die Gallenwege in den Darm sezerniert wird. Diese Gallensäuren sind gleichzeitig für die Resorption wasserunlöslicher Nahrungsbestandteile, also auch Cholesterin, erforderlich. Cholesterin wird durch Gallensäuren emulgiert und im Dünndarm resorbiert. Da etwa 90 % der Gallensäuren wieder aufgenommen werden, ist die Ausscheidung von Cholesterin dementsprechend ineffektiv. Durch Einnahme von Medikamenten wie Colestyramin, die Gallensäuren binden und die Wiederaufnahme damit erschweren, kann die Cholesterinausscheidung gesteigert werden. Allerdings wird die Senkung des Cholesterinspiegels durch Zunahme der LDL-Rezeptordichte auf Leberzellen und die damit gesteigerte Cholesterinaufnahme aus dem Blut in die Leber teilweise durch eine vermehrte Neusynthese ausgeglichen. 9 Cholesterinbiosynthese Cholesterinbiosynthese. Aus drei Molekülen aktivierter Essigsäure (Acetyl-CoA) entsteht über das Zwischenprodukt Acetoacetyl-CoA das β-Hydroxymethylglutaryl-CoA (β-HMG-CoA). Mit Hilfe des Enzyms HMG-CoA-Reduktase und NADPH wird das β-HMG-CoA zu Mevalonat reduziert. Letzterer ist der geschwindigkeitsbestimmende Schritt der gesamten Synthese. Die Biosynthese des Cholesterins geht von den Endprodukten des Mevalonatbiosyntheseweges, von Dimethylallylpyrophosphat und von Isopentenylpyrophosphat aus. Das oben genannte Schlüsselenzym, die HMG-CoA-Reduktase, befindet sich im Mevalonatbiosyntheseweg, deshalb wird hier auf die Artikel Dimethylallylpyrophosphat und Terpene verwiesen. Dimethylallylpyrophosphat (DMAPP) und Isopentenylpyrophosphat (IPP) werden durch eine Kopf zu Schwanz Kondensation durch die Farnesylpyrophosphatsynthase zu Geranylpyrophosphat (GPP) verknüpft, die gleiche Farnesylpyrophosphatsynthase verknüpft anschließend durch eine weitere Kopf zu Schwanz Kondensation ein weiteres IPP mit dem GPP zu Farnesylpyrophosphat (FPP). Zwei FPP werden in einer Kopf zu Kopf Kondensation, über die Zwischenverbindung Praesqualenpyrophosphat, durch die Squalensynthase zu Squalen verknüpft. Squalen reagiert durch die Squalenepoxidase zu 2,3-Epoxysqualen, welches durch die Squalenoxidocyclase, über ein Protosterol-Kation, zu Lanosterol reagiert. Lanosterol wird über neunzehn verschiedene Zwischenverbindungen zu Cholesterin umgewandelt, dieser gesamte enzymatische Prozess ist in der Membran des Endoplasmatischen Retikulums lokalisiert. Cholesterin ist wiederum eine Ausgangsverbindung für menschliche Hormone, wie im Abschnitt Funktion beschrieben. 10 Cholesterintransport (Lipoproteine) Da Cholesterin in Wasser unlöslich ist, erfolgt der Transport im Blutplasma zusammen mit anderen lipophilen Substanzen wie Phospholipiden, Triglyceriden oder Fettsäuren, mit Hilfe von Transportvesikeln, den Lipoproteinen. Das über die Nahrung zugeführte Cholesterin sowie Triglyceride werden nach der Resorption aus dem Darm von den Chylomikronen aufgenommen und von dort in die Gewebe transportiert. Lipoproteine verschiedener Dichte (VLDL, IDL und LDL) transportieren selbst hergestelltes und aufgenommenes Cholesterin von der Leber zu den Geweben. HDL nehmen Cholesterin aus den Geweben auf und bringen es zur Leber zurück. Das Cholesterin in den Lipoproteinen ist überwiegend mit Fettsäuren verestert. Das Spektrum dieser Fettsäuren ist in starkem Maße durch die mit der Nahrung aufgenommenen Triglyceride zu beeinflussen. Dies zeigen insbesondere Studien an Bevölkerungsgruppen mit speziellen Ernährungsformen wie z.B. Vegetarier und Veganer.[5] Für den Abbau des LDL-Cholesterins im Blut gibt es im menschlichen Körper zwei voneinander unabhängige Wege, den LDL-Rezeptorweg und den sogenannten ScavengerPathway. Der größte Teil, ca. 65 % des LDL-Cholesterins im Plasma, wird über LDLRezeptoren verstoffwechselt. LDL-Rezeptoren findet man in allen Zelltypen der Arterien und in Hepatozyten (Leberzellen). Neben dem LDL-Rezeptorweg werden circa 15 % des LDLCholesterins im Plasma über den sogenannten Scavenger Pathway in den Blutgefäßen abgebaut. Als Scavenger-Zellen werden die Makrophagen bezeichnet. Sie besitzen sogenannte Scavenger-Rezeptoren, über die chemisch modifizierte (oxidierte) LDL ungehemmt und konzentrationsunabhängig aufgenommen und gespeichert werden können. Zusammenfassend lassen sich drei verschiedene Wege beschreiben, die das Cholesterin (unabhängig ob über die Nahrung oder selbst synthetisiert) im Organismus nimmt: 1. Ausscheidung in die Galle und damit in einen enterohepatischen Kreislauf (Leber → Galle → Darm → Lymphe → Blut → Leber). 2. Umwandlung zu Gallensäuren, die an den Darm abgegeben werden. 3. Abgabe ins Blut in Form von Lipoproteinen (VLDL → LDL → HDL) zur Synthese von Steroiden und Bildung von Membranen in anderen Organen. Blutspiegel Der durchschnittliche Gesamtcholesterinspiegel wie auch die LDL- und HDL-Spiegel der gesunden Normalbevölkerung sind von Land zu Land verschieden und darüber hinaus altersund geschlechtsabhängig. Eine Korrelation zwischen den Blutcholesterin-Werten und dem Body-Mass-Index besteht nicht. 11 Gesamtcholesterinspiegel Generell nimmt der Gesamtcholesterinspiegel mit dem Alter deutlich zu. In der Regel ist er bei jungen Frauen etwas niedriger als bei jungen Männern. Mit zunehmendem Alter gleicht sich dieser Unterschied jedoch aus, und ältere Frauen haben schließlich im Mittel einen höheren Cholesterinspiegel als ältere Männer. Einen Sonderfall stellt die Schwangerschaft dar, in der der Gesamtcholesterinspiegel im Normalfall deutlich erhöht ist. Der durchschnittliche Gesamtcholesterinspiegel der Altersgruppe zwischen 35 und 65 Jahren in Deutschland liegt bei etwa 236 mg/dl (entspricht 6,1 mmol/l), die Standardabweichung bei ±46 mg/dl. Das bedeutet näherungsweise, dass etwa zwei Drittel der deutschen Bevölkerung in dieser Altersgruppe einen Gesamtcholesterinwert im Bereich zwischen 190 mg/dl und 280 mg/dl aufweisen, und jeweils ein Sechstel der Deutschen in dieser Altersgruppe Werte oberhalb beziehungsweise unterhalb dieses Bereichs. LDL-Cholesterinspiegel s. Hauptartikel: Low Density Lipoprotein Der LDL-Cholesterinspiegel unterliegt einer ähnlichen alters- und geschlechtsabhängigen Verteilung. Auch hier ist der altersabhängige Anstieg bei den Frauen deutlich stärker ausgeprägt als bei den Männern. Der Mittelwert der Altersgruppe zwischen 35 und 65 Jahren liegt dabei bei den deutschen Frauen bei 164 mg/dl (Standardabweichung ±44 mg/dl), bei den Männern bei 168 mg/dl (±43 mg/dl). HDL-Cholesterinspiegel s. Hauptartikel: High Density Lipoprotein Der durchschnittliche HDL-Spiegel unterscheidet sich stärker zwischen den beiden Geschlechtern, wobei Frauen im mittleren Alter einen höheren HDL-Spiegel aufweisen als 12 Männer. Die Altersabhängigkeit zeigt sich hier bei beiden Geschlechtern in einem Absinken ab einem Alter von etwa 55 Jahren. Der durchschnittliche HDL-Spiegel bei den deutschen Frauen in der Altersgruppe zwischen 35 und 65 Jahren liegt bei 45 mg/dl (±12 mg/dl), bei den Männern bei 37 mg/dl (±11 mg/dl). Quotienten Auf Grundlage der vorgenannten Parameter werden gelegentlich Quotienten aus diesen Werten bestimmt. Der Mittelwert des Quotienten aus LDL- und HDL-Spiegel liegt für die deutschen Frauen zwischen 35 und 65 Jahren bei 3,9 (±1,6), bei den Männern bei 4,9 (±1,9). Die entsprechenden Durchschnittswerte für den Quotienten aus dem Gesamtcholesterin- und dem HDL-Spiegel liegen für die Frauen bei 5,7 (±2,1), für die Männer bei 7,0 (±2,3). Messung und Labor-Referenzwerte Die Bestimmung der Konzentration von Cholesterin im Blut in medizinischen Routinelabors gehört heute zu den Bestimmungsmethoden, die in Deutschland ringversuchspflichtig sind. Ein Ringversuch ist die externe Qualitätskontrolle von Laborparametern, die von der Bundesärztekammer kontrolliert und zertifiziert wird. An die so genannten „Richtlinien der Bundesärztekammer“ (RiLiBÄK) muss sich jedes Labor in Deutschland halten. Der Referenzbereich (oftmals irreführend als „Normalwert“ bezeichnet) ist vom Messgerät und der Methode abhängig. Für die Bestimmung von Cholesterin werden in Deutschland in den meisten Labors Geräte von Roche Diagnostics (früher Boehringer Mannheim) verwendet. Auf dem Modell Hitachi wird als Referenzwert für das Gesamtcholesterin 110–230 mg/dl angegeben. Beim neueren Gerät Modular wird als Referenzbereich <240 mg/dl angegeben. Die Referenzbereiche wurden in den letzten Jahren mehrfach nach oben korrigiert. Um eine Verfälschung der Ergebnisse auszuschließen, wird die Bestimmung häufig erst 12 bis 16 Stunden nach der letzten Mahlzeit durchgeführt. Lange Zeit wurde im Labor nur das Gesamtcholesterin bestimmt, da die direkte Messung der verschiedenen Lipoproteine nicht möglich bzw. sehr aufwendig war. Das hat sich mittlerweile geändert. Das LDL-Cholesterin wird nicht direkt bestimmt, sondern aus den direkt gemessenen Werten für Gesamtcholesterin, Triglyceride und HDL nach Friedewald et al.[6] abgeschätzt als Gesamtcholesterin minus HDL-Cholesterin minus ein Fünftel des Triglyceridwertes (alle Angaben in mg/dl). Diese Methode kann nicht angewendet werden für Triglyzeridwerte über 400 mg/dl oder bei Vorliegen einer Chylomikronämie. Verschiedene Korrekturfaktoren sind vorgeschlagen worden, um die Präzision dieser Abschätzung zu erhöhen, jedoch sind sie bisher nicht in die klinische Praxis eingegangen. Der Referenzbereich für den LDL-Cholesterinspiegel wird für Frauen und Männer zwischen 70 und 180 mg/dl angegeben. 13 Einheiten und Umrechnung In Westdeutschland wird für die Angabe der Konzentration von Cholesterin im Blut häufig die Einheit „mg/dl“ (Milligramm pro Deziliter) verwendet. In Ostdeutschland wird dagegen – wie im angelsächsischen Sprachraum – überwiegend die Einheit „mmol/l“ (Millimol pro Liter, vergleiche Milli und Mol) benutzt. Für Cholesterin (nicht jedoch für Triglyceride oder andere Stoffe) gilt der folgende Zusammenhang zwischen diesen Maßeinheiten: 1 mg/dl = 0,02586 mmol/l 1 mmol/l = 38,67 mg/dl Beispiel: 236 mg/dl = 236 · 0,02586 mmol/l = 6,10 mmol/l 6,10 mmol/l = 6,10 · 38,67 mg/dl = 236 mg/dl Für Triglyceride gelten die folgenden Umrechnungsformeln: 1 mg/dl = 0,0113 mmol/l 1 mmol/l = 88,57 mg/dl Erkrankungen Zu den bekannten Erkrankungen im Zusammenhang mit Cholesterin gehören die familiäre Hypercholesterinämie und Gallensteine (Gallenkonkrement). Familiäre Hypercholesterinämie Es gibt erbliche Störungen des Cholesterinstoffwechsels (familiäre Hypercholesterinämie), die unabhängig von der Nahrungsaufnahme zu stark erhöhten Cholesterinwerten im Blut führen. Bei einer der bekannten Formen der Hypercholesterinämie sind die LDL-Rezeptoren nur unvollständig ausgebildet oder fehlen ganz. Heterozygote Träger dieser Erbfaktoren sind überdurchschnittlich häufig schon in jüngeren Jahren von Herzinfarkten und anderen Gefäßkrankheiten betroffen. Gemäß einer Untersuchung aus dem Jahre 1991 gilt dies nicht mehr für ältere Personen. Hier geht die Mortalität sogar deutlich zurück und liegt nur bei 44 % gegenüber dem Standard [7]. Die Prävalenz der häufigsten monogenetischen Hypercholesterinämie, der sogenannten autosomal dominanten familiären Hypercholesterinämie, liegt bei ca 1:500. Allerdings scheint es im Verlauf der letzten 200 Jahre eine bedeutende Variabilität in der Häufigkeit von Symptomen bei Betroffenen gegeben zu haben, was auf eine Interaktion einer veränderten Umwelt (beispielsweise Ernährung, Lebensstil) mit dem Genotyp hindeutet [8]. 14 Gallensteine Cholesterin wird mit der Gallensäure im Darm vom Körper aufgenommen. Dabei wird Cholesterin emulgiert und im Dünndarm resorbiert. Die Löslichkeit von Cholesterin in der Gesamtgalle liegt bei 0,26 %. Bei einer Veränderung der Zusammensetzung der Galle kommt es zur Bildung von Cholesterinsteinen. 80 % der Gallensteine sind cholesterinreich und 50 % reine Cholesterinsteine. Die Bildung von Gallensteinen erfolgt nur in der Gallenblase. Weitere Krankheitsformen Weniger bekannte Erkrankungen sind zum Beispiel die Cholesterinspeicherkrankheit (Xanthomatose oder Hand-Schüller-Christian-Syndrom) bei der Cholesterin krankhaft unter anderem in der Haut gespeichert wird. Mit einer Häufigkeit von ca. 1:60.000 kommt in Europa das Smith-Lemli-Opitz-Syndrom (SLO) vor. Grund für die Erkrankung mit SLO-Syndrom ist ein Defekt des letzten Enzyms des Cholesterin-Biosynthesewegs, der 7-Dehydrocholesterin-Reduktase. Das klinische Bild ist gekennzeichnet durch geistige Retardierung, Wachstumsprobleme, Entwicklungsstörungen und Gesichtsveränderungen. Weiterhin ist eine Hypocholesterinämie bekannt, bei der der Cholesterinspiegel unter 130 mg/dl im Blut vorliegt. Dies tritt vor allem bei Leberschädigung oder Behandlung mit Cortison auf. Dabei kann unter anderem das Vitamin E nicht mehr an seine entsprechenden Zielorte transportiert werden. Cholesterin und die Koronare Herzkrankheit (KHK) Herz-Kreislauferkrankungen, dabei insbesondere die koronare Herzerkrankung (KHK), lösten mit steigendem Lebensstandard im 20. Jahrhundert in den westlichen Industrienationen die Infektionskrankheiten als häufigste Todesursache ab. Wesentlich dazu beigetragen hat die steigende Lebenserwartung bei einem gleichzeitigen Rückgang von Infektionskrankheiten, besonders aufgrund verbesserter hygienischer Verhältnisse und der zunehmenden Verbreitung von Antibiotika. In den 50er-Jahren des 20. Jahrhunderts fand die Hypothese des amerikanischen Ernährungsforschers Ancel Keys große Beachtung, diese Entwicklung sei zusätzlich dadurch begünstigt, dass der steigende Wohlstand mit einer falschen, zu fetthaltigen Ernährung einhergehe. Insbesondere führe eine cholesterinreiche Ernährung (in erster Linie Fleisch, Hühnerei, Milch, Butter und andere Milchprodukte) zu einem erhöhten Cholesterinspiegel, und der erhöhte Cholesterinspiegel führe wiederum zu Arteriosklerose. Da die Mehrzahl der Herzinfarkte durch Arteriosklerose ausgelöst wird, sei die Aufnahme von cholesterinhaltiger Nahrung somit eine wesentliche Ursache für Herzinfarkte (Diese These ist immer mehr umstritten. Siehe dazu den Abschnitt Kritik). Bedeutung der Hypothese Die Hypothese, cholesterinreiche Ernährung und ein hoher Blut-Cholesterinspiegel spielen eine ursächliche Rolle bei der Entstehung von Herzinfarkten, hat in den vergangenen Jahrzehnten im wissenschaftlichen Umfeld wie in der öffentlichen Wahrnehmung große Verbreitung gefunden und bildet heute in der medizinischen Praxis ein wesentliches Element der vorbeugenden Behandlung von Herzinfarkten. Sie führte insbesondere in den USA, aber auch in Europa zur Verbreitung künstlich cholesterinreduzierter oder cholesterinfreier 15 Lebensmittel (beispielsweise Margarine), und darüber hinaus zu einer routinemäßigen Verschreibung von Medikamenten zur Senkung des Cholesterinspiegels. Cholesterinsenker stellen heute das weltweit umsatzstärkste Segment des Pharmamarktes dar. Im Jahre 2004 wurden mit Cholesterinsenkern weltweit Umsätze von 27 Milliarden Dollar erzielt, bei einer Wachstumsrate von 10,9 %. Umsatzstärkstes Medikament ist Atorvastatin (Lipitor®, Sortis®) des US-Herstellers Pfizer, welches 2005 einen Umsatz von weltweit 12,2 Milliarden Dollar erzielte.[9] Dieses Medikament spielt allerdings auf dem deutschen Markt heute keine wesentliche Rolle mehr, seit die Krankenkassen eine Festbetragsregelung für Statine eingeführt haben. Weltweit nehmen etwa 25 Millionen Menschen regelmäßig cholesterinsenkende Präparate ein. Empirische Hinweise Die Hypothese stützt sich vor allem auf folgende Beobachtungen: Bei Hasen und anderen überwiegend vegetarisch lebenden Tieren führt im Tierversuch die Verabreichung einer stark cholesterinhaltigen Nahrung (Milch, Eigelb) zur Entwicklung einer Arteriosklerose. Diese Beobachtung wurde erstmals 1908 von dem russischen Wissenschaftler Alexander Ignatowski veröffentlicht. Umstritten ist allerdings die Übertragbarkeit dieser Ergebnisse auf den Menschen, da dessen natürlicher Regelmechanismus für die Höhe des Cholesterinspiegels die Aufnahme von Cholesterin über die Nahrung nahezu vollständig kompensiert. Daher wurden später ähnliche Untersuchungen an Schweinen vorgenommen, welche eine 70 % Homologie zum Menschen aufweisen, beziehungsweise auch an Affen, mit ähnlichen Ergebnissen wie bei den Hasen. Bei einzelnen der untersuchten Affenarten (wie die Schweine meist Allesfresser mit überwiegend vegetarischer Ernährung) fand man allerdings starke individuelle Unterschiede auch innerhalb einer Art. Bei einzelnen Individuen lässt sich der Cholesterinspiegel demnach durch die Ernährung beeinflussen („hyperresponders“), bei anderen nicht („hypo-responders“). Cholesterin ist ein wesentlicher Bestandteil der arteriosklerotischen Plaques. Dies wurde 1910 vom deutschen Chemiker und späteren Nobelpreisträger Adolf Windaus nachgewiesen. Ancel Keys veröffentlichte in den 1950er Jahren aufsehenerregende vergleichende Studien von sechs bzw. sieben Ländern, in denen er für diese Länder länderübergreifend eine Korrelation zwischen der KHK-Rate und dem Anteil tierischer Fette in der Ernährung zeigte. Insbesondere in Japan zeigte sich eine niedrige KHK-Rate bei gleichzeitig geringem Anteil tierischer Fette in der Nahrung, in den USA zeigte sich das Gegenteil. Später wurde ihm allerdings zum Vorwurf gemacht, dass er gezielt nur diejenigen der zu diesem Zeitpunkt veröffentlichten Länder-Datensätze präsentiert hätte, die die von ihm postulierte Korrelation zu unterstützen scheinen. Andere Studien, welche die KHK-Rate von eingewanderten Japanern in den USA untersuchten, konstatierten eine Angleichung der niedrigeren japanischen an die USA-KHK-Rate. Dies könnte wiederum für ernährungsbedingte Faktoren sprechen, wäre aber auch durch andere Faktoren erklärbar, die mit dem Lebensstil zusammenhängen. Kritiker stellen darüber hinaus die in beiden Fällen vorausgesetzte Vergleichbarkeit der von verschiedenen Ländern veröffentlichten Todesursachen in Frage, da bei der Feststellung der Todesursache auch kulturelle Faktoren eine Rolle spielten. 16 Bei jüngeren Männern bis zum Alter von etwa 45 Jahren geht ein hoher Gesamt- bzw. LDL-Cholesterinspiegel mit einem erhöhten Auftreten von KHK-Erkrankungen einher und stellt dabei neben den weiteren bekannten Risikofaktoren einen eigenständigen Risikofaktor dar. Das bedeutet, dass sich diese Korrelation nicht allein durch die Korrelation des Cholesterinspiegels mit anderen bekannten KHK-Risikofaktoren erklären lässt. Weitere bekannte Risikofaktoren sind Lebensalter, Geschlecht, positive Familienanamnese (d. h. Auftreten von Herzinfarkt in der näheren Verwandtschaft), Rauchen, Diabetes mellitus, Bluthochdruck, Übergewicht und Bewegungsmangel. Für jüngere wie ältere Frauen und für ältere Männer stellt ein hoher Cholesterinspiegel allerdings entgegen der weit verbreiteten Meinung keinen Risikofaktor für KHKErkrankungen dar. Patienten mit familiärer Hypercholesterinämie haben aufgrund eines erblichen Gendefekts einen sehr hohen Cholesterinspiegel (oft 400 mg/dl und mehr), und in jungen Jahren ein gegenüber der Normalbevölkerung um ein Vielfaches gesteigertes KHK-Risiko. Durch die Vergabe verschiedener Lipidsenker konnte die Lebenserwartung dieser Patienten erhöht werden. Das KHK-Risiko dieser Patienten normalisiert sich allerdings in einem Alter ab etwa 55 Jahren. In zahlreichen Studien wurde demonstriert, dass die Einnahme von Medikamenten zur Cholesterinsenkung insbesondere bei männlichen KHK-Hochrisikopatienten zu einem Rückgang des Herzinfarktrisikos führen kann, der allerdings in aller Regel durch eine Zunahme anderer Todesursachen kompensiert wurde. In den vergangenen Jahren konnte mit der Medikamentengruppe der Statine in einzelnen Studien erstmals auch ein geringer lebensverlängernder Nutzen der Einnahme eines CholesterinsenkungsPräparats demonstriert werden. Dieser zeigte sich allerdings nur in einem Teil der durchgeführten Studien und nur bei männlichen KHK-Hochrisikopatienten mittleren Alters. Menschen mit einer bestimmten Variante im Gen für den Low-DensityLipoprotein(LDL-)Rezeptor haben ein Leben lang niedrigere Cholesterinspiegel im Blut. Das Herzinfarkt-Risiko ist bei diesen Menschen um 23% vermindert.[10] Die Rolle von High Density Lipoprotein und Low Density Lipoprotein Die ursprüngliche Hypothese, ein erhöhter Cholesterinspiegel sei kausal verantwortlich für die koronare Herzerkrankung, wird in jüngerer Zeit meist in etwas modifizierter Form vertreten. Unterschieden wird nun zwischen HDL- und LDL-Cholesterin, wobei ein hoher HDL-Cholesterinspiegel als günstig, ein hoher LDL-Spiegel dagegen als weniger günstig angesehen wird. Entsprechend dieser Vorstellung wird HDL populärwissenschaftlich als „gutes“ Cholesterin bezeichnet, LDL als „schlechtes“ oder „böses“ Cholesterin. Diese Vorstellung stützt sich im Wesentlichen auf folgende Beobachtungen: HDL dient dem Transport von Cholesterin vom Gewebe zur Leber, LDL dient dem Transport in umgekehrter Richtung. Auf Grundlage dieser Erkenntnis wird vermutet, dass ein hoher HDL-Spiegel und ein niedriger LDL-Spiegel dazu führen, dass im Verhältnis mehr Cholesterin von den Gefäßen zur Leber transportiert wird und sich deshalb weniger arteriosklerotische Plaques bilden können. Das Verhältnis von HDL und LDL korreliert noch stärker als der Gesamtcholesterinspiegel mit den bekannten Risikofaktoren für Arteriosklerose, wie Alter, Geschlecht, Rauchen, Übergewicht und Bewegungsmangel. Betrachtet man also lediglich die HDL- und LDL-Spiegel, ohne eine Normierung bezüglich der bekannten Risikofaktoren vorzunehmen, so zeigt sich der vermutete Zusammenhang sehr 17 deutlich. Allerdings ergibt sich nach dem Herausrechnen dieser Korrelationen keine höhere prognostische Kraft für das KHK-Risiko als beim Gesamtcholesterinspiegel. In wissenschaftlichen Untersuchungen der letzten 20–30 Jahre hat man festgestellt, dass die arteriosklerotischen Plaques überwiegend aus chemisch modifizierten (oxidiertem) LDL-Cholesterin entstehen (siehe auch Lipoprotein-induced atherosclerosis Hypothese unter Arteriosklerose). Zielwerte und Richtlinien [Bearbeiten] Die Hypothese, Cholesterin sei kausal verantwortlich für Herzinfarkte, führte bereits in den 1960er Jahren zu einer breit angelegten öffentlichen Informationskampagne in den USA, um die Bevölkerung vor den möglichen Gefahren eines hohen Cholesterinspiegels zu warnen. Im Jahre 1984 warnte das amerikanische Nachrichtenmagazin Time in einer Titelgeschichte vor dem Verzehr von Eiern und Wurst. Im Jahre 1985 wurde zur Ausweitung dieser Kampagne durch die American Heart Association (AHA, Amerikanischer Kardiologenverband) das National Cholesterol Education Program (NCEP, Nationales CholesterinErziehungsprogramm) ins Leben gerufen. Das NCEP gibt seit seiner Gründung regelmäßig Empfehlungen heraus, an denen sich die Behandlung von Patienten mit hohem Cholesterinspiegel orientieren soll. In Deutschland ist die Deutsche Gesellschaft für Kardiologie (DGK) die entsprechende Fachgesellschaft, die eigene Zielwerte herausgibt, die aber in der Regel den amerikanischen Werten sehr ähnlich sind. Eine vergleichbare Rolle wie das NCEP übernimmt in Deutschland die industrienahe Lipid-Liga. Die grundlegenden Richtlinien der NCEP III, denen sich die europäischen und deutschen Gesellschaften angeschlossen haben, unterscheiden drei gestaffelte Risikogruppen. Zur Gruppe 1 zählen alle Patienten, die bereits eine KHK entwickelt haben oder ein vergleichbares Risiko aufweisen (dazu zählt z. B. auch eine Diabeteserkrankung). Diese Patienten haben ein 10-Jahres-Risiko für ein kardiales Ereignis von >20 %. Zur Gruppe 2 zählen die Patienten, die mindestens zwei Risikofaktoren aufweisen, zur Gruppe 3 die Patienten, die weniger als zwei Risikofaktoren aufweisen.[11] Patienten der Gruppe 1 sollten bei LDL-Werten über 100 mg/dl Lebensstiländerungen vornehmen (Ernährung etc.), bei Werten über 130 mg/dl eine medikamentöse Therapie beginnen. Ziel sollte für sie sein, LDL-Werte unter 100 mg/dl zu erreichen. Patienten der Gruppe 2 sollten bei LDL-Werten über 130 mg/dl Lebensstiländerungen vornehmen, bei Werten über 130 mg/dl oder 160 mg/dl (abhängig von der spezifischen Risikoberechnung) eine medikamentöse Therapie beginnen. Ziel sollte sein, LDL-Werte unter 130 mg/dl zu erreichen. Patienten der Gruppe 3 sollten bei LDL-Werten über 160 mg/dl eine Lebensstiländerung vornehmen und eine medikamentöse Therapie erwägen, ab 190 mg/dl wird eine medikamentöse Therapie dringend empfohlen. Als Risikofaktoren gelten: Rauchen erhöhter Blutdruck (über 140/90 mmHg oder eine aktuelle hypertensive Behandlung) niedriges HDL-Cholesterin (<40 mg/dl) KHK-Erkrankungen in der Familie (bei männlichen Verwandten ersten Grades unter 55 Jahren oder weiblichen Verwandten ersten Grades unter 65 Jahren) 18 Alter (Männer über 45, Frauen über 55 Jahre) Als Lebensstiländerungen werden empfohlen: Reduktion der verzehrten gesättigten Fettsäuren (<7 % der Gesamtkalorien) und Cholesterins Nichtmedikamentöse Therapieoptionen zur LDL-Senkung (z. B. pflanzliche Sterole (2 g/Tag) etc.) Gewichtsreduktion Erhöhte körperliche Betätigung Die Anwendung dieser Zielwerte wird von den deutschen Fachgesellschaften der Kardiologen und Internisten unterstützt und befürwortet.[12] Kritik Die Forderung, ein (LDL-)Cholesterinspiegel oberhalb der publizierten Zielwerte müsse gegebenenfalls durch Ernährungsumstellung und/oder eine medikamentöse Therapie abgesenkt werden, war und ist umstritten. Im Folgenden werden die wichtigsten Kritikpunkte aufgeführt: Zweifel an der Kausalkette Ernährung – Cholesterin – KHK-Erkrankung Auf Basis der umfangreichen Studienlage zu dieser Fragestellung werden Zweifel daran geäußert, dass beim Menschen überhaupt ein relevanter Zusammenhang zwischen Ernährung und Cholesterinspiegel besteht (vgl. Einfluss der Ernährung auf den Cholesterinspiegel). Die Empfehlungen zur Ernährungsumstellung seien daher von vorneherein zum Scheitern verurteilt und führten regelmäßig dazu, dass sich gesunde Menschen anschließend einer meist lebenslangen medikamentösen Therapie unterziehen. Cholesterin ist Bestandteil der Zellwände und eine der häufigsten im Körper vorkommenden Substanzen. Sie spielt, wie die unten aufgeführten Beispiele zeigen, unter anderem für den Gehirnstoffwechsel eine wichtige Rolle - weshalb der Körper sich auch nicht auf die Zufuhr von außen verlässt, sondern den Spiegel selbst reguliert. Welche Nebenfolgen man mit medikamentösen Eingriffen in diesen Mechanismus auslöst, ist kaum abzusehen. Ein hoher (LDL-)Cholesterinspiegel korreliert nur bei Männern bis 45 Jahren mit der Anzahl der KHK-Erkrankungen. Aus einer bloßen Korrelation lasse sich aber noch nicht einmal bei dieser Bevölkerungsgruppe auf eine Kausalität schließen. Wahrscheinlicher sei vielmehr eine gemeinsame Ursache für den Anstieg des Cholesterinspiegels und des KHK-Risikos. Unter anderem wird vermutet, Cholesterinablagerungen in den Arterien könnten eine Reparaturmaßnahme sein, mit der der Körper auf geschädigte Blutgefäße reagiert. Unterdrückt man diese Reparaturmaßnahme durch Reduzierung des freien Cholesterins, dann mag dies in Extremfällen die Infarktgefahr reduzieren, aber nur um den Preis anderer, womöglich größerer Schäden – etwa eines erhöhten Krebsrisikos (s. unten). Dazu passt der nächste Kritikpunkt: Ein hoher (LDL-)Cholesterinspiegel ist statistisch nicht mit einer Verkürzung der Lebenserwartung verknüpft. Das Senken des Infarktrisikos wird also, so es überhaupt stattfindet, durch das Ansteigen anderer tödlicher Krankheiten jedenfalls wieder ausgeglichen. 19 Wäre ein hoher Serum-(LDL)-Cholesterinspiegel ein Auslöser für Arteriosklerose, so müsste sich in pathologischen Untersuchungen an verstorbenen Patienten eine deutliche Korrelation zwischen dem (LDL-)Cholesterinspiegel und dem Grad der arteriosklerotischen Veränderungen der Gefäße zeigen. Dieser Zusammenhang müsste sich sogar weitaus deutlicher zeigen, als eine etwaige Korrelation mit der KHK-Rate, da nur ein Teil der Herzinfarkte durch Arteriosklerose ausgelöst wird. Eine 1998 veröffentlichte Analyse aller vorliegenden Autopsiestudien zu dieser Fragestellung kommt jedoch auch unter Berücksichtigung der methodischen Schwächen der Untersuchungen zur einzig möglichen Schlussfolgerung, dass es keine signifikante Beziehung zwischen Serum-Cholesterin und Atherogenese gibt. Die größte jemals durchgeführte Ernährungs-Interventionsstudie, die Anfang 2006 veröffentlicht wurde, zeigte keinerlei Vorteile einer fettarmen Ernährung. Weder das Risiko für Herz-Kreislauferkrankungen, noch das Risiko für Schlaganfall, noch das Risiko für verschiedene Krebserkrankungen konnte durch die Ernährungsumstellung (weniger Fett, mehr Obst und Gemüse) reduziert werden. An der Studie nahmen fast 50.000 Frauen im Alter zwischen 50 und 79 Jahren teil, die über einen Zeitraum von etwa 8 Jahren beobachtet wurden. Der LDL-Cholesterinspiegel der Studienteilnehmer reduzierte sich durch die Ernährungsumstellung nur marginal um durchschnittlich 2,7 mg/dl (0,07 mmol/l). Kritische Bewertung von Nutzen und Risiko einer medikamentösen CholesterinSenkung Die Ergebnisse von Studien zur medikamentösen Senkung des Cholesterinspiegels rechtfertigen nach Ansicht von Kritikern keineswegs den breiten Einsatz dieser Medikamente. In einer Vielzahl von Studien sei zwar ein Nachweis erbracht worden, dass sich mit diesen Medikamenten effektiv eine Absenkung des Cholesterinspiegels erzielen lasse. Die Erfolge im Hinblick auf einen echten Patientennutzen, insbesondere eine lebensverlängernde Wirkung, seien jedoch gering. Die Fokussierung auf die Höhe des Cholesterinspiegels und auf das KHK-Risiko führe dazu, dass solche Cholesterin-Senkungs-Studien von den Autoren selbst dann noch als Erfolg dargestellt würden, wenn es, wie in einzelnen Studien geschehen, in der Behandlungsgruppe zu einem erheblichen und statistisch signifikanten Anstieg von Krebserkrankungen und Todesfällen gekommen sei. Der Nutzen von Statinen bei der Reduktion des Herzinfarktrisikos insbesondere von männlichen KHK-Hochrisikopatienten sei auch durch andere Wirkmechanismen erklärlich als durch die Absenkung des Cholesterinspiegels. Dafür spreche auch, dass der Ausgangs-Cholesterinspiegel für den Erfolg einer Statin-Behandlung keine Rolle spielt. Statine haben im Vergleich zu anderen Gruppen von Lipid-Senkern relativ seltene, aber u. U. schwerste Nebenwirkungen, teilweise mit tödlichen Folgen. Die medikamentöse Absenkung des Cholesterinspiegels im ersten Schwangerschaftsdrittel, z. B. mit Statinen, führt mit hoher Wahrscheinlichkeit zu schwersten Fehlbildungen in der Ausbildung des zentralen Nervensystems und der Gliedmaßen des Kindes, vergleichbar mit Schädigungen durch Thalidomid. Eine Verschreibung von Cholesterinsenkern in der Schwangerschaft ist deshalb kontraindiziert. Kritiker bemängeln, dass eine Verschreibung an junge Frauen bei Sicherstellung einer „zuverlässigen“ Verhütung dennoch zulässig ist, was dazu geführt hat, dass zahlreiche entsprechende Fälle in der medizinischen Literatur dokumentiert sind. 20 Der LDL-Cholesterinspiegel korreliert mit der Gedächtnisleistung und mit anderen kognitiven Funktionen. Eine Absenkung des LDL-Cholesterinspiegels führt zu einem signifikanten Rückgang von Gedächtnisleistung und Aufmerksamkeit. Als mögliche Erklärung für diesen in verschiedenen Studien beobachteten Effekt kommt die Tatsache in Frage, dass Cholesterin bekanntermaßen bei der Ausbildung von Synapsen im Gehirn eine wesentliche Rolle spielt. Die Ausbildung von Synapsen ist wiederum von wesentlicher Bedeutung beim Lernen und bei der Funktion des Gedächtnisses. Bekannt ist auch, dass sich der Cholesterinspiegel bei Gabe von Statinen nicht nur im Blut, sondern auch im Gehirn deutlich absenkt. In diesem Zusammenhang ist es bemerkenswert, dass in der medizinischen Literatur zahlreiche Fälle von totalem Gedächtnisverlust im direkten Zusammenhang mit der Einnahme von cholesterinsenkenden Präparaten dokumentiert sind. Eine kritische Bewertung der Studien zur Auswirkung von Cholesterin, Cholesterinsenkern und Ernährung finden sich in dem Buch „Mythos Cholesterin“ von Uffe Ravnskov. Er untersucht nicht nur die statistischen Methoden und die Vorgehensweise innerhalb der Studien von 1953 bis 2003, sondern stellt auch die wirtschaftliche Interessenlage der Auftraggeber der Studien vor. Einfluss wirtschaftlicher Faktoren auf Forschung, Fachgesellschaften und veröffentlichte Meinung Folgt man den Richtlinien und Zielwerten, so handelt es sich bei dem überwiegenden Teil der erwachsenen Bevölkerung um behandlungsbedürftige „KHK-Risikopatienten“. So sollte etwa ein gesunder 40-jähriger deutscher Mann mit normalem Blutdruck, der nie geraucht hat und keine KHK-Erkrankungen in der Verwandtschaft hat, mit für seine Altersgruppe durchschnittlichen LDL- und HDL-Werten (168 mg/dl bzw. 37 mg/dl), entsprechend den Richtlinien bereits eine medikamentöse Therapie in Erwägung ziehen. Erreicht er mit diesen durchschnittlichen HDL- und LDL-Werten das Alter von 45 Jahren, so gehört er bereits in die „Risikoklasse 2“, in der er entsprechend den Richtlinien bereits mittels einer medikamentösen Therapie seinen LDL-Spiegel auf 130 mg/dl absenken sollte. Erreicht er ein durchschnittliches Lebensalter, so ist damit zu rechnen, dass er etwa 35 Jahre lang regelmäßig Medikamente zur Cholesterinsenkung einnehmen wird. Demgegenüber liegt bis heute keine einzige Studie vor, die für diesen „Patienten“ auch nur einen geringfügigen Nutzen einer Cholesterinsenkungstherapie zeigen würde. Kritiker sehen in diesen Richtlinien daher in erster Linie ein Instrument zur Steigerung der Umsätze der pharmazeutischen Industrie. Die überwiegende Zahl der Forscher im Bereich Cholesterin und KHK-Erkrankungen, darunter auch die Autoren der NCEP-Richtlinien und die Vorstände der deutschen DGFF (Lipid-Liga), seien in einem hohen Maße finanziell von Fördermitteln der Pharma-Industrie abhängig oder profitierten sogar persönlich von Beratungs- und Vortragshonoraren oder Aktienoptionen dieser Firmen, für die wiederum die Medikamente zur Cholesterinsenkung der größte Umsatzträger sind. Folgen dieser Abhängigkeit seien: o Die Tatsache, dass ein hoher Cholesterinspiegel, anders als vielfach suggeriert, statistisch nicht mit einer Verkürzung der Lebenserwartung verknüpft ist, würde in der kardiologischen Fachwelt weitgehend ignoriert. o Gleiches gelte für Hinweise auf die Bedeutung des Cholesterinspiegels für die Gedächtnisleistung und Aufmerksamkeit sowie für Hinweise darauf, dass 21 o o o niedrige Cholesterinspiegel einen Risikofaktor für verschiedene Krebserkrankungen darstellen. Wissenschaftliche Studien, die einen Zusammenhang zwischen KHKErkrankungen und Cholesterin zu belegen scheinen, werden sechsmal häufiger zitiert als Studien, deren Ergebnisse zu dieser Hypothese eher im Widerspruch stehen, obwohl sich die Gesamtzahl der veröffentlichten Studien insgesamt in der Waage hält. Ergebnisse von Studien, die für die Hersteller der CholesterinsenkungsPräparate ungünstig verlaufen seien, würden zum Teil nicht vollständig veröffentlicht, so etwa im Fall der EXCEL-Studie. Die wissenschaftliche Qualität der fast ausschließlich von Herstellern finanzierten Medikamentenstudien zum Thema Cholesterinsenkung wird in Frage gestellt. So bezeichnete etwa im Jahr 2005 das deutsche Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen die wissenschaftliche Qualität der vorliegenden Statin-Studien generell als „mangelhaft“. Besonders die als einer der wichtigsten Belege für den Nutzen einer Statin-Behandlung angeführte 4S-Studie steht methodisch erheblich in der Kritik. Der hohe Grad der Finanzierung durch Mittel der Pharmaindustrie trifft für den Großteil der gesamten medizinischen Forschung und Entwicklung zu. Gerade universitäre Institute werden von öffentlicher Seite dringend angehalten, Drittmittel für ihre Forschungsarbeit einzutreiben. Letzteres gilt jedoch nicht für die besonders problematischen direkten finanziellen Zuwendungen der Pharmaindustrie an sogenannte „Meinungsbildner“, die in Form von sogenannten Beratungs- und Vortragshonoraren ausgezahlt werden. Nach einer Untersuchung aus dem Jahr 2001 werden etwa 3 % des Marketingbudgets der Pharmaindustrie – im Falle von cholesterinsenkenden Präparaten entspräche dieser Anteil jährlich einem dreistelligen Euro-Millionenbetrag – in Form von substanziellen Zuwendungen an eine relativ kleine Gruppe von meist international, national oder regional bekannten Professoren ausgeschüttet. Diese finanziellen Verflechtungen werden in Deutschland i. d. R. auch dann nicht transparent gemacht, wenn sich solche Professoren etwa in Beiträgen oder Kommentaren für Fachzeitschriften oder in Publikumsmedien für den verstärkten Einsatz von cholesterinsenkenden Präparaten aussprechen. Seit Januar 2006 fordert das „Deutsche Ärzteblatt“ allerdings seine Autoren auf, solche Abhängigkeiten bekanntzugeben und zu veröffentlichen, entsprechend den Gepflogenheiten in internationalen Fachpublikationen. In einer im Jahre 2005 veröffentlichten Studie kritisiert der deutsche Zweig der internationalen Anti-Korruptions-Organisation Transparency International sowohl die Abhängigkeit der medizinischen Forschung von der Pharma-Industrie als auch die nach seiner Ansicht „alltägliche Praxis der Pharmaindustrie“, sich medizinische Meinungsbildner zu „kaufen“, und spricht in diesem Zusammenhang von einer „strukturellen Korruption“. Die Vorsitzende des Verbandes deutscher Medizinjournalisten (VDMJ) hält es für eine gängige Praxis, dass auch Medizinjournalisten von der Pharmaindustrie für ihre Artikel bezahlt werden. Das enorme wirtschaftliche Gewicht von Cholesterinsenkungspräparaten für die Pharma-Industrie führt nach Ansicht von Kritikern gerade aus diesem Grunde zu einer ebenso häufigen wie unkritischen Thematisierung von Cholesterin und Cholesterinsenkern in Publikumsmedien. Die Abhängigkeit der meisten an medizinische Praktiker (niedergelassene Ärzte oder Krankenhausärzte) gerichteten medizinischen Publikationen (z. B. Ärzte-Zeitung, Medical Tribune, Der Internist) vom Werbebudget der großen Pharmakonzerne 22 verhindere eine kritische Auseinandersetzung mit dem Thema in solchen Zeitschriften. Die Redaktion der Fachpublikation „BDI aktuell“ des Berufsverbandes deutscher Internisten (BDI) führt einen kritischen Artikel zum Thema Cholesterinsenkung mit den Worten ein: „... und kommt zu Schlussfolgerungen, die Deutschland-weit kein Anzeigen-finanziertes medizinisches Blatt zu drucken bereit wäre.“[13] Apotheken partizipieren in Deutschland mit einer Gewinnspanne von 3 % und einem Festzuschlag von 5,80 Euro (8,10 Euro abzüglich eines Kassenrabatts von 2,30 Euro) pro Packung an den Medikamentenumsätzen. Wie für die Pharmaindustrie stellen Cholesterinsenker daher auch für Apotheken einen Hauptumsatzträger dar. Gleichzeitig spielen Apothekerfunktionäre und Apotheker eine wesentliche Rolle bei der Aufrechterhaltung der öffentlichen Aufmerksamkeit für das Thema Cholesterinspiegel-Messung und -Senkung. So treten etwa Funktionäre von Apothekerkammern oder Apothekerverbänden in Gesundheitssendungen als „Cholesterin-Experten“ auf oder veröffentlichen Ratgeber zum Thema Cholesterinsenkung. Zahlreiche Apotheken wirken bei dem von der Lipid-Liga veranstalteten „Tag des Cholesterins“ mit. Darüber hinaus wird das Thema in den in Apotheken ausliegenden kostenlosen Publikationen regelmäßig im Sinne der Cholesterin-KHK-Hypothese aufgegriffen. Kritiker stellen angesichts der klaren Interessenkollision in Frage, ob bei diesem Engagement ausschließlich das Patientenwohl im Vordergrund steht.[14][15][16] Auf politischer Ebene spielen industrienahe Denkfabriken wie das „Stockholm Network“ eine große Rolle bei der Verbreitung der Botschaft, dass Cholesterinsenkung notwendig und nützlich sei. So veröffentlichte das „Stockholm Network“ mit seiner Unterorganisation „Centre for the New Europe“ (CNE) im Jahre 2006 eine Studie mit dem Titel „Cholesterin: Die öffentlichen Politik - Implikationen, des nicht genugtuns “ (Cholesterol: The Public Policy Implications of Not Doing Enough) und prophezeit eine „Gesundheitskrise“ bis 2020, falls die gegenwärtige Praxis des „Cholesterin-Managements“ nicht im Sinne einer verstärkten Anwendung von Cholesterinsenkungspräparaten geändert würde. Zu den Gründern des „Stockholm Network“ gehört Pfizer-Vorstand Michael W. Hodin; im Vorstand des CNE sitzt darüber hinaus Catherine Windels, die gleichzeitig als „Director of International Affairs“ für Pfizer tätig ist. Berichten zufolge erhält das CNE über 50 % seiner Finanzierung alleine von Pfizer, dessen Hauptumsatzträger der Cholesterinsenker Lipitor/Sortis ist, daneben soll auch der Pharmakonzern Merck Sharp & Dohme (MSD), Hersteller der Cholesterinsenker Zocor und Ezetrol (Ezetimb), zur Finanzierung der Organisation beitragen. Zocor ist mit 4,4 Milliarden Dollar Jahresumsatz (2005) Hauptumsatzträger von MSD. Der genannte Bericht selbst wurde nach Angaben des Stockholm Network durch die Pharmakonzerne MSD und Schering Plough Corporation finanziert; letzterer vermarktet Ezetrol gemeinsam mit MSD.[17][18][19][20][21][22][23][24] 23 Cholesterin und Schlaganfallrisiko Ein hoher Cholesterinspiegel wird häufig als Risikofaktor für Schlaganfälle dargestellt. Die industrienahe Lipid-Liga bezeichnet Cholesterin als einen der wichtigsten Risikofaktoren für Schlaganfälle[25], und gelegentlich wird Cholesterin sogar als „der wichtigste Risikofaktor“[26] für Schlaganfälle dargestellt, und eine Senkung des Cholesterinspiegels wird als Vorbeugemaßnahme empfohlen. Tatsächlich existiert nach Studienlage kein Zusammenhang zwischen dem Cholesterinspiegel und dem Schlaganfallrisiko, der diese Behauptung rechtfertigen würde, und eine mögliche Rolle des Serum-Cholesterins bei der Entstehung von Schlaganfällen und ein Nutzen von cholesterinsenkenden Medikamenten ist umstritten.[27] Serum-Cholesterinspiegel und Schlaganfallrisiko In der Framingham-Studie, der größten zu dieser Fragestellung vorliegenden Kohortenstudie, findet sich keinerlei Korrelation zwischen dem Cholesterinspiegel und dem Schlaganfallrisiko.[28] Auch eine Metaanalyse von 45 Kohortenstudien mit insgesamt 450.000 beobachteten Individuen und über 13.000 beobachteten Schlaganfällen ergab keinerlei Korrelation zwischen dem Cholesterinspiegel und dem Schlaganfallrisiko. Allenfalls bei unter 45-jährigen Patienten besteht möglicherweise eine leichte positive Korrelation.[29] Andere Studien zeigen bei jungen Frauen ebenfalls eine positive Korrelation zwischen dem (in diesem Alter allerdings absolut gesehen sehr geringen) Schlaganfallrisiko und dem Cholesterinspiegel, während bei älteren Frauen ab dem 50. Lebensjahr das Schlaganfallrisiko mit steigendem Cholesterinspiegel sogar sinkt.[30] Unterscheidet man zwischen den unterschiedlichen Arten von Schlaganfällen, so sind niedrige Cholesterinspiegel mit einem leicht erhöhten Risiko für hämorrhagische Schlaganfälle verbunden, während hohe Cholesterinspiegel mit einem leicht erhöhten Risiko für ischämische Schlaganfälle einhergehen.[31] Cholesterinsenkende Medikamente und ihr Einfluss auf das SchlaganfallRisiko Bis heute liegt keine randomisierte Studie vor, die dafür angelegt war, den Einfluss von Cholesterinsenkung auf das Schlaganfallrisiko zu untersuchen. Allerdings können die vorliegenden Cholesterinsenkungs-Studien zur KHK-Prävention, meist mit KHKHochrisikopatienten (i. d. R. mit vorangegangenem Herzinfarkt oder Diabetes) als Probanden, als Grundlage für entsprechende Auswertungen herangezogen werden. In einer im Jahr 2003 veröffentlichten Meta-Analyse von 38 randomisierten Cholesterinsenkungs-Studien mit unterschiedlichen Präparaten zeigte sich in der Behandlungsgruppe eine zwar geringe aber statistisch signifikante relative Reduzierung der Schlaganfall-Häufigkeit um 17 Prozent, gleichzeitig jedoch eine nicht signifikante Zunahme der tödlichen Schlaganfälle um 9 Prozent.[32] Statine sind dabei die einzige Wirkstoffgruppe, die zu einer statistisch signifikanten Reduzierung des Schlaganfallsrisikos führt. Möglicherweise ist hierfür allerdings die geringe aber statistisch signifikant nachgewiesene 24 blutdrucksenkende Wirkung von Statinen verantwortlich; Bluthochdruck gilt als wichtiger Schlaganfall-Risikofaktor.[33] In der einzigen vorliegenden randomisierten Cholesterinsenkungsstudie mit älteren Patienten (PROSPER) zeigte sich ebenfalls ein Rückgang nichttödlicher ischämischer Schlaganfälle bei einer gleichzeitigen Zunahme tödlicher Schlaganfälle. In der im Jahre 2005 veröffentlichten 4D-Studie mit unter Typ2-Diabetes leidenden Dialysepatienten kam es zu einer statistisch signifikanten Verdopplung der tödlichen Schlaganfälle in der mit Statinen behandelten Gruppe (siehe Abschnitt „Studien“). Cholesterin und Krebserkrankungen Serum-Cholesterinspiegel und Krebsrisiko Bei Krebserkrankung ist der Cholesterinspiegel z.B. von Brustkrebskranken Frauen im Vergleich zu Gesunden erhöht.[34][35] Bei fortschreitendem Leberkrebs wird die Cholesterinbildung eingeschränkt und als Folge sinkt auch der Serum-Cholesterinspiegel. Cholesterinsenkende Medikamente und ihr Einfluss auf das Krebsrisiko Von besonderer Bedeutung ist darüber hinaus die Fragestellung, ob eine Cholesterinsenkung eine präventive Wirkung gegenüber bestimmten Krebserkrankungen hat, oder ob diese die Entstehung von Krebserkrankungen sogar begünstigt. Erhöhung des Krebsrisikos Eine im Juli 2007 veröffentlichte Metaanalyse von prospektiven Cholesterinsenkungsstudien ergab eine signifikante Korrelation des Krebsrisikos mit der Einnahme von Statinen. Je niedriger die erzielten LDL-Cholesterinwerte, desto höher war der Anteil der Patienten, die an Krebs erkrankten. Innerhalb einer Beobachtungsdauer zwischen einem und fünf Jahren wurde in der Gruppe der Patienten mit den niedrigsten erzielten LDL-Cholesterinspiegeln etwa eine zusätzliche Krebserkrankung auf 1000 Patienten beobachtet. [36] In der 1996 veröffentlichten CARE-Studie[37] hatte sich ein hochsignifikanter Anstieg der Brustkrebsfälle in der mit Pravastatin behandelten Gruppe gezeigt (von 1 auf 12). In der 2002 veröffentlichten PROSPER-Studie[38] mit einem im Vergleich zu anderen Statin-Studien vergleichsweise hohen mittleren Alter (und damit Krebsrisiko) der Probanden fand sich ein statistisch signifikanter Anstieg von Krebserkrankungen in der mit Pravastatin behandelten Gruppe. Auch in der 4S- und HPS-Studie zeigte sich jeweils ein (nicht signifikanter) Anstieg von Krebserkrankungen in der mit Simvastatin behandelten Gruppe.[39] Senkung des Krebsrisikos In den letzten Jahren fand auf der Grundlage verschiedener Fall-Kontroll-Studien die gegenteilige Hypothese große Beachtung, Statine hätten möglicherweise gegen verschiedene Krebserkrankungen (u. a. Prostata-Karzinom[40], Kolorektal-Karzinom[41], Brustkrebs[42], Nierenkrebs[43]) sogar eine vorbeugende Wirkung. Grundlage für die zum Teil euphorische Medienberichterstattung[44] war folgende Beobachtung: Unter denjenigen Patienten, die die jeweilige Krebserkrankung entwickelt hatten, war der Anteil der Patienten, die 25 Cholesterinsenker eingenommen hatten niedriger, als in einer Vergleichsgruppe ohne Krebserkrankung. Solche nicht randomisierten Fall-Kontroll-Studien sind allerdings statistisch nur begrenzt aussagekräftig[45], und erlauben insbesondere keinerlei Aussage über UrsacheWirkungsbeziehungen (siehe auch Fall-Kontroll-Studie). Der hier beobachtete Effekt kann beispielsweise auch darauf beruhen, dass Patienten mit hohem Cholesterinspiegel, die bekanntermaßen eine niedrigere Krebsrate haben, häufiger Cholesterinsenker verschrieben bekommen. Bei dieser Verschreibungspraxis würde sich auch bei einem völlig wirkungsfreien Medikament ergeben, dass diejenigen Patienten, die das Medikament einnehmen, eine niedrigere Krebsrate aufweisen. Kein Einfluss auf Krebsrisiko Die Fragestellung, ob Statine eine präventive Wirkung gegen das Kolorektal-Karzinom haben, wurde in einer 2006 veröffentlichten Analyse einer großen Kohortenstudie geprüft. Es fand sich jedoch eine nicht signifikante Erhöhung des Krebsrisikos bei der Patientengruppe, die mit cholesterinsenkenden Mitteln behandelt worden war[46]. Eine im gleichen Jahr erschienene Meta-Analyse der zahlreichen Statin-Studien kommt gleichfalls zu dem Schluss, dass eine Cholesterinsenkung mit Statinen eindeutig keine präventive Wirkung gegenüber Krebserkrankungen hat, weder auf die Gesamtheit aller Krebserkrankungen noch auf einzelne Krebsarten, die Entstehung von Krebs jedoch auch nicht statistisch signifikant begünstigt. [47] Die eindeutig negativen Ergebnisse der beiden letztgenannten Studien lassen weitere Studien zu der erhofften krebspräventiven Wirkung von Cholesterinsenkungspräparaten nach Einschätzung von Experten nicht sinnvoll erscheinen.[48] Cholesterin und Ernährung Das Hühnerei wird wegen des hohen Cholesteringehalts im Eigelb häufig als „Cholesterinbombe“ angeprangert. Nach einer Diagnose eines hohen Cholesterinspiegels wird in der Regel als erste Maßnahme eine fettmodifizierte und cholesterinarme Ernährung empfohlen. Diese Empfehlung ist allerdings umstritten. Empfehlung bei hohem Cholesterinspiegel 26 Gemäß den Empfehlungen der DGFF (Lipid-Liga) sollten hierbei folgende Punkte bei der Nahrungsaufnahme bedacht werden: 1. 2. 3. 4. 5. 6. Weniger fettes Fleisch, Innereien, Wurstwaren, Käse und Eigelb Fettarme Zubereitung Weniger tierische Lebensmittel Mehrmals am Tag frisches Obst und Gemüse Verwendung von Pflanzenöl Verzicht auf Alkohol Einfluss der Ernährung auf den Cholesterinspiegel Kritiker halten dagegen, dass der Einfluss einer kurzfristigen Nahrungsumstellung auf den Cholesterinspiegel nur gering ist, da die Nahrungsaufnahme nur ein geringer Anteil bei der Bildung von Cholesterin ist. Eine prospektive Studie, die Verbundstudie Ernährungserhebung und Risikofaktoren Analytik (VERA, von 1985 bis 1988 mit 25.000 Teilnehmern) ergab, dass auch bei verschiedenen Mengen von gesättigten, aber auch ungesättigten Fettsäuren sowohl die HDL- als auch die LDL-Werte sich, wenn überhaupt, nur minimal änderten. Dagegen wird wiederum eingewendet, die Zusammensetzung des LDLs werde ignoriert: es gebe Hinweise, dass es einen Unterschied machen würde, mit welchen Fettsäuren (überwiegend ungesättigt wie bei Veganern versus überwiegend gesättigte) das Lipoprotein LDL „bestückt“ sei. Die Oxidierbarkeit des LDLs, und damit eine potenzielle Schädigung der Gefäße, hänge von den transportierten Fettsäuren ab, und hier würden die ungesättigten Fettsäuren besser abschneiden - besonders in Verbindung mit Vitamin E, welches ebenfalls in den LDLMolekülen im Blut transportiert wird. Selbst wenn sich diese Zusammenhänge bestätigen sollten, bleibt unbewiesen, dass der Anteil ungesättigter Fette an der Nahrung seinerseits Einfluss auf die Zusammensetzung des LDLs hat, und welche ungesättigten Fettsäuren dabei welche Wirkung haben. Allerdings lässt sich durch eine langfristige drastische Verringerung der Fettzufuhr, z. B. durch einen verlängerten Fastentest, auch der Cholesterinspiegel senken. Bei Vegetariern und Veganern werden verringerte Cholesterinspiegel beobachtet, deren Ursache allerdings nicht geklärt ist: sie können auf ihrer geringen Cholesterinaufnahme mit der Nahrung beruhen, aber auch Nebeneffekt einer insgesamt gesundheitsbewußteren Lebensweise sein. Dafür spricht, dass man (gegenüber US-Amerikanern) deutlich verringerte Cholesterinwerte auch in Gruppen gefunden hat, die sich fast ausschließlich von Milch und Fleisch ernähren (belegt für Massai-Angehörige und von Samburu-Männer). Nicht beobachtet werden konnte umgekehrt eine Erhöhung des Cholesterinspiegels (über als normal geltende Werte hinaus) durch eine besonders cholesterinhaltige Nahrung. Dies belegt etwa eine Studie an der Universität Missouri-Columbia, bei der selbst ein wöchentlicher Verzehr von 24 Eiern den Cholesterinspiegel nicht steigern konnte. Einfluss der Ernährung auf den Cholesterinspiegel durch Prostaglandine Neben der umstrittenen These des Einflusses von direkter Aufnahme von Cholesterin in der Nahrung besteht auch ein Einfluss der Ernährung auf die Cholesterinsynthese durch die Beeinflussung der Synthese von Prostaglandinen. Prostaglandine sind Gewebshormone, die 27 unter Anderem die Synthese von Cholesterin steuern, wobei ein Prostaglandin in die eine Richtung wirkt (etwa das cholesterinsenkende Serie-1 PGE1) und ein anderes gegensätzlich (hier Serie-2 PGE2). Die Bildung von Serie-1 oder Serie-2 Prostaglandinen wiederum wird durch das Verhältnis von mehrfach ungesättigten Fettsäuren (omega-3 zu omega-6) in der Nahrung beeinflusst[49][50]. Prostaglandine steuern außer der Cholesterinsynthese auch andere Faktoren der Entstehung von Arteriosklerose, so z.B. Lipoprotein(A) und Entzündungsparameter. Die Ernährungsempfehlungen, um die Prostaglandine günstig zur Cholesterinsenkung zu beeinflussen, wären etwa: Erhöhung des Anteils an Omega-3-Ölen (Leinöl, Walnussöl) generell, Erhöhung des Verhältnisses von Omega-3-Ölen zu Omega-6-Ölen,[51] direkte Aufnahme von langkettigen Omega-3-Fettsäuren („Fischöl“).[52] Cholesterin und Schwangerschaft Niedrige Cholesterinspiegel der werdenden Mutter (Gesamtcholesterin unter 160 mg/dl) sind ein Risikofaktor für Frühgeburt und niedriges Geburtsgewicht.[53] Cholesterin und Muttermilch Muttermilch enthält einen sehr hohen Anteil an Cholesterin (ca. 25 mg / 100 g, Kuhmilch enthält nur ca. 12 mg / 100 g). Es wird vermutet, dass der höhere Cholesterinanteil der Muttermilch dafür verantwortlich sein könnte, dass gestillte Kinder später im Mittel einen höheren IQ entwickeln, auch weil bekannt ist, dass Cholesterin beim Aufbau des Gehirns und Nervensystems eine wesentliche Rolle spielt. Babynahrungshersteller verzichten auf die Anreicherung von Muttermilch-Ersatz mit Cholesterin, vermutlich weil sie wegen negativer Assoziationen der Verbraucher mit diesem Stoff mit Absatzproblemen rechnen müssten. Cholesterin, Psyche und Gedächtnis Cholesterin und Gewaltbereitschaft In einer im Jahre 2005 veröffentlichten Studie zeigte sich ein statistisch signifikanter Zusammenhang zwischen einem niedrigen Gesamtcholesterinspiegel bei Kindern und Schulverweisen. Kinder und Jugendliche mit einem Gesamtcholesterinspiegel unterhalb des 25-Prozent-Perzentils (<145 mg/dl) hatten eine fast dreifach erhöhte Wahrscheinlichkeit, in ihrer Schullaufbahn von der Schule verwiesen worden zu sein. Dies wird von den Autoren als weiterer Hinweis dafür gewertet, dass niedrige Cholesterinspiegel mit einer erhöhten Aggressivität im Zusammenhang stehen.[54] Cholesterin und Depressionen Niedrige Cholesterinspiegel haben sich in verschiedenen Studien als Risikofaktor für das Auftreten von Depressionen herausgestellt. So zeigte sich beispielsweise bei jungen, gesunden Frauen mit einem Gesamtcholesterinspiegel unterhalb von 4,14mmol/l (160 mg/dl) ein etwa doppelt so hohes Risiko für das Auftreten von Depressionen wie bei Frauen mit mittlerem bis hohem Cholesterinspiegel.[55] Auch die Einnahme von cholesterinsenkenden Medikamenten begünstigt offenbar die Entstehung von Depressionen. So zeigte sich in einer Studie an 234 älteren, depressiven 28 Patienten, dass diejenigen Patienten, die Cholesterinsenker einnahmen, ein statistisch signifikant um fast 80% erhöhtes relatives Risiko für das Auftreten eines Rückfalls hatten als Patienten ohne diese Medikation.[56] In einer placebokontrollierten Studie an über 70-jährigen Patienten zeigte sich, dass die Stimmungslage der Patienten in der mit einem Cholesterinsenker behandelten Patientengruppe statistisch signifikant negativ beeinträchtigt war.[57] Von möglichen positiven Auswirkungen der Statin-Einnahme auf die Psyche berichtet eine Kohortenstudie, in der Patienten, die über Zeitraum von vier Jahren ununterbrochen Statine eingenommen hatten mit solchen Patienten verglichen wurden, die gar nicht oder nur mit Unterbrechungen Statine eingenommen hatten. In der ersten Gruppe zeigte sich eine reduzierte Prävalenz von Depressionen, die jedoch nicht mit dem Maß der Cholesterinsenkung in Zusammenhang stand.[58] Die Aussagekraft dieser Studie ist jedoch dadurch beeinträchtigt, dass Patienten, die z. B. wegen möglicher Nebenwirkungen der Medikamenteneinnahme aus der Studie ausschieden, in der Auswertung nicht berücksichtigt werden konnten. Cholesterin und Gedächtnis In verschiedenen Studien wurde der Einfluss einer Cholesterinsenkung auf die Gedächtnisleistung untersucht. In einer im Jahr 2000 veröffentlichten Studie an 192 gesunden Erwachsenen zeigte sich, dass sowohl die Gedächtnisleistung als auch die Aufmerksamkeit der Probanden in der mit Lovastatin behandelten Gruppe signifikant schlechter ausfiel als in der Kontrollgruppe. Der Leistungsunterschied war signifikant verknüpft mit den absoluten LDL-Cholesterinwerten nach der Behandlung, d. h. niedrigere Cholesterinwerte gingen mit einer schlechteren Gedächtnisleistung einher. Auch in einer an 326 Frauen mittleren Alters durchgeführten und 2003 veröffentlichten Studie zeigte sich eine lineare Korrelation der Gedächtnisleistung mit dem LDL-Cholesterinspiegel. In einem im Jahr 2003 veröffentlichten Übersichtsartikel werden 60 Fälle von totalem Gedächtnisverlust im Zusammenhang mit einer Statin-Behandlung beschrieben. Nach Absetzen der Statin-Behandlung verschwanden in etwas weniger als der Hälfte der dokumentierten Fälle die Gedächtnisstörungen ganz oder teilweise. Cholesterinsenkung und Albträume Niedrige Serum-Cholesterinspiegel stehen offenbar mit dem Auftreten von nächtlichen Albträumen in Verbindung.[59] Darüber hinaus gibt es einzelne Fallberichte, in denen ein direkter Zusammenhang zwischen der Einnahme von Cholesterinsenkern und dem Auftreten von Albträumen beschrieben wurde.[60] Arzneimittel Die ersten Mittel zur Senkung des Cholesterinspiegels waren Gallensäureaustauscherharze (Cholestipol). Später kamen dann Fibrate sowie Nikotinsäurepräparate und deren Derivate auf den Markt. Heute werden in diesem Indikationsbereich fast nur noch Statine und Cholesterinwiederaufnahmehemmer eingesetzt, in Einzelfällen noch Fibrate. 29 Fibrate Derzeit sind die Wirkstoffe Bezafibrat, Fenofibrat und Gemfibrozil im Einsatz. Fibrate zeichnen sich durch eine gute Triglyceridsenkung aus und werden heute deshalb vor allem bei Diabetikern eingesetzt. Statine Als die zur Zeit wirksamsten Medikamente zur Senkung des Cholesterinspiegels gilt die Gruppe der Statine. Sie gehören zur Gruppe der HMG-CoA-Reduktase-Hemmer (CSEHemmer), da sie das Schlüsselenzym der Cholesterinsynthese in der Zelle, die β-Hydroxy-βmethylglutaryl-Coenzym-A-Reduktase hemmen. Als Folge stellt die Zelle benötigtes Cholesterin nicht mehr selbst her, sondern nimmt Cholesterin aus dem Blut, über LDLRezeptoren, auf. Derzeit sind in Deutschland die Wirkstoffe Lovastatin, Simvastatin, Pravastatin, Fluvastatin und Atorvastatin am Markt. Der Wirkstoff Cerivastatin (Zenas, Lipobay) wurde im Jahr 2001 vom Hersteller Bayer vom Markt genommen, nachdem – vor allem, da es trotz Warnhinweisen in einer riskanten Kombination mit Fibraten verordnet wurde – Todesfälle und schwere Gewebeschäden im Zusammenhang mit der Einnahme des Medikaments aufgetreten waren. Der Wirkstoff Rosuvastatin befindet sich derzeit im Zulassungsverfahren und ist in einigen europäischen Ländern bereits im Handel (Crestor). Statine bewirken eine starke LDL-Senkung, aber eine eher schwache Triglycerid-Senkung und HDL-Steigerung. Ezetimib Der relativ neuartige Wirkstoff Ezetimib ist ein im Darm wirkender, selektiver Cholesterinwiederaufnahmehemmer, der gezielt das Niemann-Pick C1-Like 1 (NPC1-L1)Protein blockiert. NPC1-L1 sitzt in der Membran von Entherozyten der Dünndarmwand und ist für die Aufnahme von Cholesterin und Phytosterolen aus dem Darm zuständig. Laut Fachinformation ist Ezetimib für folgende Anwendungsgebiete zugelassen: Primäre Hypercholesterinämie Ezetimib ist zusammen mit einem HMGCoA-Reduktase-Hemmer (Statin) eingenommen begleitend zu Diät angezeigt zur Anwendung bei Patienten mit primärer (heterozygoter familiärer und nicht familiärer) Hypercholesterinämie, bei denen die Therapie mit einem Statin allein nicht ausreicht. Monotherapie Eine Monotherapie mit Ezetimib ist begleitend zu Diät angezeigt zur Anwendung bei Patienten mit primärer (heterozygoter familiärer und nicht familiärer) Hypercholesterinämie, bei denen ein Statin als ungeeignet erachtet oder nicht vertragen wird. Homozygote familiäre Hypercholesterinämie (HoFH) Ezetimib ist zusammen mit einem Statin eingenommen begleitend zu Diät angezeigt zur Anwendung bei Patienten mit homozygoter familiärer Hypercholesterinämie. Die Patienten können weitere begleitende Therapien (wie LDL-Apherese) erhalten. 30 Homozygote Sitosterinämie (Phytosterinämie) Ezetimib ist begleitend zu Diät angezeigt zur Anwendung bei Patienten mit homozygoter familiärer Sitosterinämie. Studien, welche die Wirkung von Ezetimib zur Prävention der Komplikationen von Atherosklerose belegen, sind noch nicht abgeschlossen Studien In zahlreichen Studien wurde die Auswirkung des Cholesterinspiegels auf die Inzidenz von Herz-Kreislauferkrankungen untersucht, aber auch andere Fragestellungen im Zusammenhang mit dem Cholesterinspiegel. Die Vielzahl der Studien macht das Heranziehen einzelner Studien zur Begründung eines Effekts grundsätzlich problematisch, da die Durchführung mehrerer Studien zur Beantwortung der gleichen Fragestellung die vermeintliche statistische Signifikanz einer einzelnen Studie außer Kraft setzen kann. So genügen im Mittel zwanzig für sich betrachtet methodisch korrekt angelegte Studien, um einen nicht vorhandenen Effekt einmal statistisch signifikant „nachzuweisen“. Metastudien gewinnen daher im Zusammenhang mit der Cholesterin-Thematik ein besonderes Gewicht. Auch diese sind jedoch durch den sogenannten „Publikationsbias“ (engl) beeinflusst. Framingham-Studie Eine der wegweisenden Studien auf dem Gebiet der Untersuchung von KHK-Risikofaktoren war die Framingham-Studie, die heute als die wichtigste epidemiologische Studie der USA gilt. Sie untersuchte 6.000 Personen zweier Generationen in Framingham/Massachusetts. Über die Framingham wurden bis zum heutigen Tag über 1.000 wissenschaftliche Publikationen erstellt. Im Rahmen dieser Studie wurden unter anderem nachgewiesen, dass Rauchen und Übergewicht wichtige KHK-Risikofaktoren sind. Es ergab sich darüber hinaus, dass bei Männern im Alter von 30–59 Jahren das Auftreten von KHK entsprechend dem Cholesteringehalt im Blut erhöht ist. Bei Männern in den Dreißigern wiesen die Personen mit dem höchsten Gesamtcholesteringehalt im Blut ein viermal höheres Risiko auf als diejenigen mit dem geringsten Cholesterin. Für Frauen und für Personen über 50 Jahre zeigte sich kein solcher Zusammenhang. Eine Prüfung der Framingham-Studie im Jahre 1987 zeigte, dass eine Absenkung des Cholesterinspiegels um 1 mg/dl tatsächlich zu einer Steigerung der Gesamttodesrate von 11 % und zu einer Steigerung der Todesrate durch Herzkrankheiten um 14 % führt. Die Darstellung in der ursprünglichen Veröffentlichung wurde teilweise kritisiert, da in ihr der gegenteilige Eindruck erweckt worden sein soll. Die Framingham-Studie ist in der Zwischenzeit als eines der Musterbeispiele zum Interpretationsspielraum von Studien in die Lehrbücher eingegangen. Metastudien Das American National Heart, Lung and Blood-Institute führte Metastudien zum gesundheitlichen Nutzen der Cholesterinsenkung durch. 19 Studien wurden analysiert. Untersucht wurden 650.000 Menschen und 70.000 Todesfälle: Geringe Cholesterinspiegel gehen nicht mit einer allgemeinen Erhöhung der Lebenserwartung einher, sondern beziehen sich nur auf Herz-Kreislauf-Erkrankungen, sie erhöhen das Risiko von Schlaganfällen und das 31 Krebsrisiko. Allerdings ist immer noch umstritten, wo hier Ursache und Wirkung liegen; zum Zeitpunkt der Messung könnten niedrige wie auch hohe Cholesterinspiegel auch durch (noch nicht diagnostizierte) Krankheiten im Anfangsstadium verursacht sein. Als gesichert gilt, dass sehr hohe, sehr niedrige und fallende Cholesterinspiegel mit einer erhöhten Mortalität verbunden sind, wobei unklar bleibt, ob das Cholesterin Ursache oder eben nur Indiz eines verschlechterten Naturstoffe (engl. natural products) sind chemische Stoffe, welche aus der belebten oder unbelebten Natur gewonnen werden.[1] Naturstoffe besitzen häufig biologische Aktivität und finden auch als Arzneistoffe Anwendung. Auch wenn der Begriff Naturstoff sich sprachlich und inhaltlich vom Kunststoff absetzt, können auch totalsynthetisch gewonnene Substanzen als Naturstoffe betrachtet werden, nach anderer Definition sind sie naturidentische Stoffe (Beispiele für Unterscheidungen aufgrund gesetzlicher Vorgaben: Ascorbinsäure und Vitamin E). Bekannte Beispiele für Naturstoffe, die als Arzneistoffe verwendet werden, sind Penicilline, Aminoglykoside und andere Antibiotika, Taxol, Morphin, Cyclosporin, Mevastatin und Digitalisglykoside. Naturstoffe zeichnen sich oft durch ihre hohe chemische/strukturelle Komplexität aus, was eine rein synthetische Herstellung erschwert oder verhindert. Außerdem zeichnen sich Naturstoffe häufig dadurch aus, dass sie ein äußerst komplexes Gemisch einer Vielzahl einander sehr ähnlicher Stoffe (z. B. Isomere oder Polymere differierender Kettenlänge bzw. Verzweigung) darstellen. Eben dieser hohe Grad an Komplexität und Diversität macht die Naturstoffchemie ziemlich unübersichtlich, führt allerdings auch zur einmaligen Stellung der Naturstoffe als Therapeutika unterschiedlicher Krankheitsbilder. Die Reindarstellung von Naturstoffen gestaltet sich in der Regel als anspruchsvolles Unterfangen. Die Einteilung von Naturstoffen in verschiedene Klassen ist über ihre Produzenten (Bakterien, Pilze, etc.) sowie ihre chemische Struktur (Polyketide, Nichtribosomale Peptide, Terpene, Glykoside, etc.) möglich. Inhaltsverzeichnis 1 Definition im Chemikalienrecht 2 Quellen von Naturstoffen o 2.1 Mineralische Naturstoffe o 2.2 Naturstoffe aus Pflanzen o 2.3 Naturstoffe aus Bakterien o 2.4 Marine Naturstoffe 3 Verwendung 4 Gefahrstoffe o 4.1 Toxine o 4.2 Reizstoffe o 4.3 Entzündliche Stoffe 5 Siehe auch 6 Einzelnachweise 32 Definition im Chemikalienrecht In der Verordnung EG 1907/2006 („REACH“) wird ein Naturstoff, wie folgt, definiert: natürlich vorkommender Stoff als solcher, unverarbeitet oder lediglich manuell, mechanisch oder durch Gravitationskraft, durch Auflösung in Wasser, durch Flotation, durch Extraktion mit Wasser, durch Dampfdestillation oder durch Erhitzung zum Wasserentzug verarbeitet oder durch beliebige Mittel aus der Luft entnommen. Quellen von Naturstoffen Die Gewinnung von Naturstoffen erfolgt aus unterschiedlichen Mineralien, Pflanzen, Tieren oder Mikroorganismen. Hierzu werden Blätter, Nadeln, tierische Gewebe oder Fermentationsbrühen extrahiert. Ein so hergestellter Rohextrakt besteht aus einem Gemisch unterschiedlicher Substanzen, die durch aufwändige Trennmethoden gereinigt werden müssen um das sogenannte aktive Prinzip zu isolieren. Da Naturstoffe zumeist keine ersichtliche Bedeutung für den Organismus, aus welchem sie gewonnen werden besitzen, handelt sich zumeist um sogenannte Sekundärmetabolite. Mineralische Naturstoffe Hierzu gehören beispielsweise Wasser, Edelsteine, Kalk. Naturstoffe aus Pflanzen Die ältesten bekannten pharmazeutischen Substanzen entstammen der Ethnomedizin. So waren die schmerzlindernden und fiebersenkenden Wirkungen der Weidenrinde (Salix, Salicylat enthaltend) oder die Antimalariaaktivität der Artemisia annua lange vor der Entdeckung ihres aktiven Prinzips bekannt. Beispiele für pflanzliche Wirkstoffe sind Chinin, Kokain, Digitalis oder Opium. Neben diesen altbekannten Naturstoffen sind Pflanzen auch für die moderne pharmazeutische Forschung von immenser Bedeutung. Das aus der pazifischen Eibe (Taxus brevifolia) isolierte Taxol dient unter dem Namen Paclitaxel als Antikrebsmedikament, die Bekämpfung der Malaria ist durch die Anwendung des Artemisinins aus Artemisia annua verbessert worden. Häufig sind die Produzenten der aus Pflanzen isolierten Naturstoffe nicht die Pflanzen selbst sondern in Symbiose (vgl. Endosymbiontentheorie) lebende Bakterien und Pilze. Naturstoffe aus Bakterien Viele der Bakterien, welche Anwendung als Produzenten von Naturstoffen finden, werden aus Erdproben isoliert. Ihre Fermentation in großen Volumina von Nährmedien macht eine Isolierung der aktiven Substanzen möglich. Den größten Bekanntheitsgrad unter den aus Bakterien isolierten Naturstoffen besitzen Antibiotika wie Erythromycin, Vancomycin, Rifamycin oder Tetracyclin. Weitere wichtige bakterielle Naturstoffe sind Immunsuppressiva wie Rapamycin und Tacrolimus und Cytostatika wie Mitomycin und Doxorubicin. Das Suffix der Nomenklatur bakterieller Naturstoffe richtet sich häufig nach der biologischen Aktivität und dem produzierenden Organismus. Die Endungen -mycin und -micin stehen für antibakterielle Aktivität, erstgenannte Substanzen (Bsp. Kanamycin) wurden aus Streptomyces 33 sp., letztgenannte (Bsp. Gentamicin) aus Micromonospora sp. isoliert. Das Suffix -carcin (Bsp. Tetrocarcin) hingegen deutet eine cytotoxische Aktivität an. Marine Naturstoffe Ein in jüngster Zeit stark untersuchtes Gebiet der Naturstoffe beschäftigt sich mit Metaboliten marinen Ursprungs. Als Produzenten dienen hierbei vor allem Korallen, Schwämme oder Algen. Ein bekannter Vertreter dieser Naturstoffklasse ist das Discodermolid. Die Gewinnung solcher Naturstoffe ist aus ökologischer Hinsicht problematisch. Häufig handelt es sich wie bei Pflanzen um Naturstoffe aus Bakterien und Pilzen, welche (vgl. Naturstoffe aus Pflanzen) in Symbiose mit Wirtsorganismen wie Schwämmen leben. Verwendung Naturstoffe finden in allen Produkten Anwendung, zumindest als Rohstoffe: Nahrungsmittel, Arzneimittel, Baustoffen, technischen Produkten (Kautschuk als Rohstoff in Reifen, Quarz als Rohstoff für Glas oder Halbleiter, Erz als Rohstoff im Metallbau), Energieträgern (Mineralöl, Kohle, Uran), Pflanzenfasern als Rohstoff für Papierprodukte … Naturstoffe können vielfältig aufbereitet werden: Sammeln, Isolieren, Waschen, Zerkleinern, mit anderen (Natur-) Stoffen kombinieren … Gefahrstoffe Naturstoffe sind nicht prinzipiell gefahrlos, nur weil sie in der Natur vorkommen. Toxine Sehr viele Akutgifte sind Naturstoffe (Beispiele: Arsen, Digitalistoxin), aber auch viele Gifte chronischer Intoxikationen (z. B. Asbest und die meisten Schwermetalle). Reizstoffe Praktisch alle hydrophilen Naturstoffe sind als lyophylisierte Trockenpulver reizend, besonders für Augen und Lunge. Außerdem können praktisch alle Trockenpulver von Naturstoffen organischen Ursprungs Allergien auslösen (sind allergisierend). Sie werden daher in manchem Sicherheitsdatenblatt als Reizstoffe ausgewiesen (z. B. die meisten Aminosäuren). Nach EU-Richtlinie sind solche Naturstoffe jedoch in aller Regel keine Gefahrstoffe Die Galle (gr. χολή cholé; lat. bilis) ist eine zähe Körperflüssigkeit, die in der Leber produziert wird, um in der Gallenblase gespeichert und zu den Mahlzeiten in den Zwölffingerdarm (Duodenum) ausgeschüttet zu werden. Ihre Färbung wechselt je nach Anteil der hauptsächlichen Farbstoffe Bilirubin und Biliverdin von gelblich bis grünlich. Stark eingedickt nimmt sie einen bräunlichen Ton an. Die Galle dient der Fettverdauung, indem sie Lipide emulgiert, das heißt in kleine, für fettspaltende Enzyme (Lipasen) angreifbare Tröpfchen zersetzt. Weiterhin ist die Galle ein 34 Ausscheidungsmedium für Substanzen, die schwer wasserlöslich sind und in der Leber in eine eliminierbare Form gebracht werden. Die Fortbewegung der Galle in den Gallenwegen einschließlich zugehöriger Bewegungsabläufe der Gallenblase und Gallengänge wird als Cholekinese bezeichnet. Inhaltsverzeichnis 1 Zusammensetzung 2 Physiologie o 2.1 Bildung o 2.2 Transport und Speicherung o 2.3 Bedeutung o 2.4 Kreislauf der Gallensalze 3 Beschwerden 4 Nutzung 5 Kulturgeschichte 6 Quellen o 6.1 Literatur o 6.2 Einzelnachweise 7 Weblinks Zusammensetzung Galle besteht zum größten Teil aus Wasser (82 %) in dem Elektrolytgehalt [1] anorganische Elektrolyte in einer ähnlichen Zusammensetzung Ion Anteil wie im Blutplasma gelöst sind (siehe Tabelle rechts). Galle ist leicht alkalisch. Die wichtigsten funktionellen Bestandteile sind Na+ 130–165 mmol/l jedoch die Gallensalze (12 %), denen eine zentrale Rolle in der Fettverdauung zukommt. Daneben enthält sie auch Alkalische K+ 3–12 mmol/l Phosphatasen, eine Gruppe von Enzymen, die Phosphorsäureester Cl− 90–120 mmol/l hydrolysieren. HCO3− 30 mmol/l Weiterhin findet man in der Galle Lecithin und andere pH-Wert 8,0–8,5 Phospholipide (4 %), nicht verestertes Cholesterin (0,7 %) und Abbauprodukte der Leber, die durch die Galle in den Verdauungstrakt gelangen und von dort mit dem Kot ausgeschieden werden. Zu den letzteren gehören Bilirubin, das Abbauprodukt des Blutfarbstoffs Hämoglobin, sowie einige Hormone und Medikamente. Ihre Farbe erhält die Galle im Wesentlichen durch die Gallenfarbstoffe: das gelbliche Bilirubin und das grünliche Biliverdin. Bilirubin wird im Darm von den dort ansässigen Bakterien unter anderem zu Stercobilin, Bilifuscin und Mesobilifuscin abgebaut, die dem Stuhl seine charakteristische Färbung geben. Der Transport von Cholesterin in der Galle findet in Mizellen statt, die aus Lecithin, Cholesterin und Gallensalzen gebildet werden. Das Mischungsverhältnis dieser drei Stoffe darf nur in sehr engen Grenzen schwanken, damit der Transport des Cholesterins 35 funktionieren kann. Andernfalls kristallisiert das Cholesterin aus und es kommt zur Bildung von Gallensteinen. Physiologie Bildung Der menschliche Körper produziert täglich etwa 700 ml Galle, die interdigestiv, das heißt zwischen den Mahlzeiten, in der Gallenblase gespeichert werden. Galle wird in den Zellen der Leber, den Hepatozyten produziert. Zwischen zwei benachbarten Hepatozyten befinden sich die Gallenkanälchen (Canaliculi), in die die Galle durch Transmembrantransport ausgeschieden wird. Diese Canaliculi vereinigen sich zu größeren Kanälen, die letztendlich die Galle zum Verdauungstrakt befördern (siehe unten). Stoffe, die in die Canaliculi abgesondert werden, sind Lecithin, konjugierte Gallensalze, Cholesterin, mit Glucuronsäure konjugierte Hormone und Bilirubin. Mit Glutathion konjugierte Medikamente können ebenfalls mit der Galle ausgeschieden werden. Die Hepatozyten entnehmen die konjugierten Gallensalze aus den Sinusoiden, mikroskopischen Blutgefäßen, die Blut zu den Hepatozyten transportieren. Die Leberzellen besitzen sowohl in ihrer den Sinusoiden als auch den Canaliculi anliegenden Zellmembranen Transportproteine (Carrier) speziell für Gallensalze. Aus den Sinusoiden werden sie mithilfe eines Natrium-Symport-Transportproteins (NTCP = Na+-taurocholate cotransporting polypeptide) sekundär aktiv aufgenommen, während sie primär aktiv mit Hilfe eines ATP-abhängigen Transporters (hBSEP: human bile salt export pump, auch cBAT: canalicular bile acid transporter) in das Lumen der Canaliculi ausgeschieden werden. Transport und Speicherung Gallenwege und -blase (hier grün dargestellt) Die extrahepatischen (außerhalb der Leber gelegenen) Gallenwege beginnen mit dem Ductus hepaticus communis (gemeinsamer Lebergang), von dem der Ductus cysticus (Verbindung zwischen Gallenblase und Hauptgallengang) zur Gallenblase abzweigt. Der Abschnitt nach dieser Abzweigung heißt Ductus choledochus und mündet schließlich zusammen mit dem Ductus pancreaticus der Bauchspeicheldrüse in den Zwölffingerdarm auf der Papilla duodeni major. In der Gallenblase wird die Galle gespeichert und auf etwa zehn Prozent ihres Volumens[2] eingedickt. Gelangen Lipide mit der Nahrung in den Dünndarm, so regen diese die Produktion 36 des Hormons Cholecystokinin (CCK) in der Dünndarmschleimhaut an. CCK stimuliert die glatte Muskulatur in der Organwand der Gallenblase, so dass diese sich zusammenzieht und ihr Inhalt dem Speisebrei im Duodenum beigemischt wird. Erhöhte Aktivität des parasympathischen Nervus vagus (Vagotonus) hat denselben Effekt. Eine Gallenblase ist jedoch nicht bei allen Wirbeltieren ausgebildet. Bedeutung Die Galle spielt eine wichtige Rolle bei der Aufnahme von Fetten aus der Speise und trägt zur Neutralisierung des nach Magenpassage stark sauren Speisebreis bei. Sie dient auch der Ausscheidung verschiedener Substanzen aus dem Körper wie Cholesterin, Bilirubin sowie vieler Medikamente und ihrer Stoffwechselprodukte. Die Gallebildung ist wesentlich für das Gleichgewicht des Cholesterins im Körper. Die Gallensalze dienen der Fettverdauung, indem sie Mizellen mit den wasserunlöslichen Bestandteilen der Nahrung (Triacylglyceride, freie Fettsäuren, Vitamine und Cholesterin) bilden und damit deren Transport im Blut ermöglichen. Medikamente und ihre Abbauprodukte werden mit Glutathion konjugiert und damit wasserlöslich gemacht, um dann mit der Galle durch den Verdauungstrakt und letztendlich den Kot ausgeschieden zu werden. Dies betrifft ebenso Stoffwechselprodukte wie Bilirubin, das aus dem Abbau von Hämoglobin in den Leberzellen entsteht. Weitere Aufgaben sind die Ausscheidung von Schwermetallen, die Neutralisierung des Zwölffingerdarms nach Magenentleerung und die Aktivierung der Bauchspeicheldrüsenenzyme. Gallensäuren wirken außerdem bakterizid. Kreislauf der Gallensalze Gallensalze werden in primäre und sekundäre Gallensalze unterteilt. Die primären Gallensalze, Cholat und Chenodesoxycholat, werden von der Leber aus Cholesterin synthetisiert. Diese werden von Bakterien im Verdauungstrakt teilweise in sekundäre Gallensalze, Desoxycholat und Lithocholat umgewandelt. Die Gallensalze werden dann im Verdauungstrakt dekonjugiert, von der Schleimhaut absorbiert und in der Pfortader (Vena portae), gebunden an Albumin, wieder zur Leber transportiert. Dort werden sie aufgenommen, wieder mit Taurin und Glycin konjugiert und erneut in die Galle abgesondert. Dieser Kreislauf wird als Enterohepatischer Gallensalzkreislauf bezeichnet und gewährleistet, dass der Gallensalzbestand des Körpers von nur zwei bis vier Gramm den Bedarf der Fettabsorption von 20–30 g decken kann. Dabei zirkuliert er täglich fünf- bis zehnmal. Nur etwa 0,3–0,6 g Gallensalze gehen verloren und müssen in der Leber neu synthetisiert werden. Gallensalze, die nicht mit Taurin oder Glycin konjugiert sind, werden sofort wieder absorbiert, während jene, die konjugiert sind, erst im Ileum (Krummdarm) an der Fettverdauung teilnehmen. 37 Beschwerden Gallensteine in der Gallenblase Bei Störung von Gallebildung oder Gallesekretion beim Menschen auftretende Symptome lassen sich durch ihre Funktionen bei der Fettverdauung und der Ausscheidung von Stoffwechselendprodukten erklären. Eine Verstopfung der Gallenwege mit Rückhaltung von Galle nennt man im medizinischen Sprachgebrauch Cholestase. Bei dieser tritt eine Fettunverträglichkeit auf, da dieses nur noch in geringem Umfang aus dem Darm absorbiert werden kann. Höhere Fettzufuhr in der Nahrung führt zu fettigem Stuhl (Steatorrhoe). Weiterhin tritt der sogenannte posthepatische Ikterus (Gelbsucht) auf, da das Hämoglobinabbauprodukt Bilirubin, ein gelber Farbstoff, nicht mehr ordnungsgemäß ausgeschieden werden kann und eine Gelbfärbung der Haut und Schleimhäute verursacht. Durch das Fehlen der Gallenfarbstoffe nimmt der Stuhl eine lehmartige Färbung an, die als acholisch bezeichnet wird. Diese Verstopfungen können verschiedene Ursachen wie Tumore der Bauchspeicheldrüse, Gallenblase, Gallengänge oder des Zwölffingerdarms haben. Eine andere Ursache können Gallensteine im Ductus hepaticus communis oder im Ductus choledochus sein. Verlegungen des Ductus cysticus führen nur selten zur Blockade der Gallenabgabe (Mirizzi-Syndrom). Gallensteine sind Kristallisationsprodukte, die entstehen, wenn das Mischungsverhältnis zwischen Lecithin, Cholesterin und den Gallensalzen aus dem Gleichgewicht gerät. Symptome treten nur in etwa einem Viertel aller Fälle auf. Dazu gehören Koliken, Druckschmerzen im rechten Oberbauch und die oben erwähnte Gelbsucht. In seltenen Fällen kommt es auch zu Rückenschmerzen. Nutzung Galle-Agar wird aus Rindergalle gewonnen und ist ein in der Mikrobiologie verwendeter Nährboden. Abhängig vom neben der Galle enthaltenen Substrat können zahlreiche Keime wie zum Beispiel Streptokokken, Salmonellen und Shigellen, aber auch Pilze angezüchtet werden. Sollen Salmonellen aus dem Blut eines Patienten angereichert werden, so wird eine Galle-Bouillon, bestehend aus drei Teilen Galle und einem Teil Blut, gemischt. In ihr können sich die Salmonellen vermehren. Für die Herstellung von Gallseife wird ebenfalls Rindergalle verwendet. 38 Kulturgeschichte Das Wort Galle (mhd. galle, ahd. galla) leitet sich von der indogermanischen Wurzel *ghel-, „gelb, grün“ ab; die Galle ist somit nach ihrer Farbe benannt. Aus dieser Wurzel hat sich im Griechischen χολή (cholé) „Galle“ entwickelt; die Cholera (χολέρα, choléra; „Gallendurchfall“) erhielt ihren Namen durch die fälschlichen Annahme, sie würde durch eine Gallenstörung verursacht. In der Humoralpathologie der Hippokratiker, die um 400 v. Chr. entwickelt wurde und die medizinische Lehre für über tausend Jahre bestimmte, bis sie mit Paracelsus an Bedeutung verlor, nimmt die Galle eine zentrale Rolle ein. Es wurde dabei zwischen Gelber Galle und Schwarzer Galle unterschieden. Diese beiden gehören, neben Blut und Schleim, zu den vier so genannten Kardinalsäften. Befinden sich diese im Gleichgewicht (Eukrasie), sei der Mensch gesund. Bei einem Ungleichgewicht (Dyskrasie) komme es zur Krankheit. Gelbe Galle werde in der Leber produziert und mit Cholerikern assoziiert. Als schwarze Galle wurde geronnenes Blut fehlgedeutet. Es werde nach der Humoralpathologie in den Hoden und der Milz produziert und mit Melancholikern (von mélaina cholé, schwarze Galle) in Verbindung gebracht. Sprichwörtliche Ausdrücke wie „mir kommt die Galle hoch“ beziehungsweise „Gift und Galle spucken“, beides Metaphern für Wut, begründen sich in dieser Lehre. Lipide Lipide (von griechisch λίπος lípos „Fett“) ist eine Sammelbezeichnung für ganz oder zumindest größtenteils wasserunlösliche (hydrophobe) Naturstoffe, die sich dagegen aufgrund ihrer geringen Polarität sehr gut in hydrophoben beziehungsweise lipophilen Lösungsmitteln wie Hexan lösen. Ihre Wasserunlöslichkeit rührt vor allem von den langen Kohlenwasserstoff-Resten, welche die allermeisten Lipide besitzen. In lebenden Organismen werden Lipide hauptsächlich als Strukturkomponente in Zellmembranen, als Energiespeicher oder als Signalmoleküle gebraucht. Die meisten biologischen Lipide sind amphiphil, besitzen also einen lipophilen Kohlenwasserstoff-Rest und eine polare hydrophile Kopfgruppe, deshalb bilden sie in polaren Lösungsmitteln wie Wasser Micellen oder Membranen. Oft wird der Begriff „Fett“ als Synonym für Lipide gebraucht, jedoch stellen die Fette (Triglyceride) nur eine Untergruppe der Lipide dar. Die Lipide können in sieben Gruppen eingeteilt werden: Fettsäuren, Triacylglyceride (Fette und Öle), Wachse, Phospholipide, Sphingolipide, Lipopolysaccharide und Isoprenoide (Steroide, Carotinoide etc.) 39 Inhaltsverzeichnis 1 Fettsäuren, Triacylglyceride (Fette und fette Öle) und Wachse o 1.1 Fettsäuren o 1.2 Triacylglyceride o 1.3 Wachse 2 Membranbildende Lipide o 2.1 Phospholipide o 2.2 Sphingolipide o 2.3 Glycolipide 3 Isoprenoide o 3.1 Steroide o 3.2 Carotinoide 4 Biologische Funktionen o 4.1 Essentielle Fettsäuren o 4.2 Fettlösliche Vitamine 5 Literatur 6 Weblinks Fettsäuren, Triacylglyceride (Fette und fette Öle) und Wachse Die Triacylglycerole (Triglyceride) machen mit mehr als 90 Prozent den Hauptanteil der Nahrungslipide aus. Sie sind ein wichtiger Energielieferant (1 g Fett enthält 39 kJ Energie, 1 g Zucker nur 17 kJ). Außerdem bilden Triglyceride den wichtigsten Energiespeicher des Körpers (Zucker, d. h. Glucose, wird dagegen in viel geringerer Menge als Glycogen in der Leber gespeichert), sie sind ein guter Kälteschutz in der Haut und schützen diese auch vor Verletzungen. Alle wichtigen Organe werden durch einen Fettmantel geschützt. Fettsäuren Sowohl Myristinsäure (eine gesättigte Fettsäure) als auch Myristoleinsäure (eine ungesättigte Fettsäure) haben 14 Kohlenstoffatome. Myristoleinsäure weist im Gegensatz zur Myristinsäure eine Doppelbindung auf. Fettsäuren sind meist unverzweigte Monocarbonsäuren, die aus einer Kette aus Kohlenstoffatomen bestehen, an deren einem Ende sich eine Carboxylgruppe befindet (siehe Bild). 40 Unterschieden wird zwischen gesättigten Fettsäuren, in denen keine Doppelbindungen vorkommen, und ungesättigten Fettsäuren, die eine oder mehrere Doppelbindungen besitzen (in der Natur meist in cis-Stellung und nicht in Konjugation miteinander). Die einfachste gesättigte Fettsäure ist die Buttersäure und enthält nur vier Kohlenstoffatome. Wichtige Vertreter der ungesättigten Fettsäuren sind Ölsäure (einfach ungesättigt) und Arachidonsäure (vierfach ungesättigt). Je mehr Doppelbindungen eine Fettsäure enthält, desto niedriger liegt ihr Schmelzpunkt. Ungesättigte Fettsäuren können vom tierischen Organismus nur unter Einschränkung synthetisiert werden. Man bezeichnet daher all jene Fettsäuren, die mit der Nahrung aufgenommen werden müssen, als essenzielle Fettsäuren (s. u.). Triacylglyceride Allgemeine Struktur der Triacylglycerole. Die Seitenketten R1, R2 und R3 stehen für Alkylreste der Fettsäuren. Triacylglyceride (allgem.: Fette und fette Öle) stellen, wie oben erwähnt, die größte Gruppe der Nahrungslipide dar. Sowohl Fette als auch Öle sind Dreifachester des Glycerins und werden als Triacylglyceride bezeichnet. Werden Triacylglyceride durch Verseifung gespalten, entstehen Glycerol und die entsprechenden Salze der Fettsäuren. Es gibt zwei Arten von Triacylglyceriden, einfache und gemischte: Bei einfachen Triacylglyceriden sind die Seitenketten (also die Fettsäurereste) identisch, bei gemischten sind sie verschieden. Die Ursache dafür, dass Fett fest und Öl flüssig ist, liegt im wesentlich höheren Anteil an ungesättigten Fettsäuren in Ölen. Die ungesättigten Fettsäuren besitzen meist cisDoppelbindungen, was die Kristallbildung erschwert und somit den Schmelzpunkt heruntersetzt. Sind in einem Triacylglycerid die Seitenketten R1 und R3 verschieden, so liegt ein chirales Molekül vor und man kann eine optische Aktivität beobachten (d. h. das Spiegelbild des Moleküls ist nicht deckungsgleich mit dem Original und eine Lösung des Moleküles ist in der Lage, einfallendes polarisiertes Licht zu drehen). 41 Wachse Bestandteile von Bienenwachs als Stellvertreter für Wachse Wachse sind Einfachester von Fettsäuren und unterscheiden sich als solche von den DreifachEstern der Fette und Öle. Sowohl der Säuren- als auch der Alkoholteil von Wachsen haben lange gesättigte Alkylreste. Im Gegensatz zu Triglyceriden sind Wachse weniger „ölig“, außerdem härter und poröser. Eine andere Definition (Deutsche Gesellschaft für Fettwissenschaft) sieht Wachse als Stoffklasse, die ausschließlich über ihre mechanisch-physikalischen Eigenschaften definiert wird. Laut dieser Definition sind Wachse bei 20 °C knetbar, fest bis brüchig hart, sie weisen eine grobe bis feinkristalline Struktur auf, farblich sind sie durchscheinend bis opak (undurchsichtig), aber nicht glasartig, über 40 °C schmelzen sie ohne Zersetzung, wenig oberhalb des Schmelzpunktes sind sie leicht flüssig (wenig viskos), weisen eine stark temperaturabhängige Konsistenz und Löslichkeit auf und sind unter leichtem Druck polierbar. Membranbildende Lipide Membranbildende Lipide sind Lipide, die einen hydrophilen und einen hydrophoben Teil besitzen – also amphiphil sind. Dies erlaubt es ihnen, in polaren Lösungsmitteln wie Wasser je nach Beschaffenheit entweder Mizellen (kugelförmige Aggregate aus amphiphilen Molekülen, die sich in einem Dispersionsmedium spontan zusammenlagern) oder Doppellipidschichten zu bilden – wobei immer der hydrophile Teil mit dem polaren Lösungsmittel interagiert. Aus diesen Doppellipidschichten sind alle Biomembranen aufgebaut, welche den Inhalt einer Zelle gegen die Umgebung abgrenzen. Membranbildende Lipide gehören daher zu einer der Grundvoraussetzungen für Leben im Allgemeinen. Phospholipide Allgemeine Struktur der Phosphoglyceride Die Reste R1 und R2 bestimmen die Fettsäuren, der Rest X bestimmt die Klasse. Bei X = H liegt Phosphatidsäure vor Phospholipide bilden den Hauptbestandteil von Biomembranen. Man unterscheidet dabei Phosphoglyceride und Sphingomyeline. Die Struktur der Phosphogylzeride leitet sich von der Phosphatidsäure ab, welche den Triglyceriden ähnelt, mit dem Unterschied, dass sich an der 42 C3-Hydroxylgruppe statt des Acylrestes eine Phosphorylgruppe befindet. Sphingomyeline hingegen unterscheiden sich von Glycerolipiden durch ihr Sphingosin-Grundgerüst. Die Phosphorsäurediestergruppe aller Phospholipide ist hydrophil (d. h. interagiert mit Wasser) und wird „Kopf“ genannt. Die Acylreste beziehungsweise der unpolare Teil des Sphingosins werden als „Schwanz“ bezeichnet und sind hydrophob. Dieser gegensätzliche Charakter führt zur Bildung von Lipid-Doppelschichten, bei denen der hydrophobe Teil der Membranlipide nach innen und der hydrophile Teil nach außen zeigen. Die wichtigsten am Aufbau von Biomembranen beteiligten Phospholipide sind die Phosphoglyceride Phosphatidylcholin (auch Lecithin), Phosphatidylethanolamin, Phosphatidylserin, Phosphatidylcholin und auch Sphingomyeline. Letztere zählen sowohl zu den Phospho- als auch den Sphingolipiden. Phosphatidylethanolamin und Phosphatidylserin werden auch als Kephaline bezeichnet. Eine vor allem in der intrazellulären Weiterleitung extrazelluärer Signale (Signaltransduktion) wichtige Gruppe der Phosphoglyceride sind die in verschiedenen Phosphorylierungsstufen auftretenden Phosphatidylinositole; als Kopfgruppe besitzen sie ein Phosphoinositol. Sphingolipide Allgemeine Struktur der Sphingolipide Verschiedene Reste (R) ergeben unterschiedliche Untergruppen. Wasserstoff – Ceramide Phosphocholin oder Phosphoethanolamin – Sphingomyeline Saccharid – Glycolipide Sphingolipide sind ebenfalls Bestandteile von Zellmembranen. Ihr Grundgerüst besteht aus einer Fettsäure und Sphingosin. Sie werden unterschieden in die Gruppen der Ceramide, der Sphingomyeline und Glycolipide. Sphingolipide finden sich im Nervengewebe, sie spielen eine wichtige Rolle in der Signalübertragung und der Interaktion einzelner Zellen. Glycolipide Glycolipide sind phosphatfreie, sphingosinhaltige Lipide mit einem glycosidisch an die 1Hydroxyl-Gruppe des Sphingosin gebundenen Kohlenhydrat-Anteil. Sie bilden häufig die Außenseite biologischer Membranen, wobei ihr Kohlenhydrat-Anteil auf der Zellmembran präsentiert wird. Es wird vermutet, dass diese eine Rolle in der Kommunikation und Interaktion zwischen einzelnen Zellen spielen. Glycolipide werden in Cerebroside, Ganglioside und Sulfatide unterschieden. Isoprenoide Als Isoprenoide (auch Terpenoide) werden Verbindungen bezeichnet, die auf Isopreneinheiten aufbauen. Zu den Lipiden zählende Verbindungen sind die Steroide und die Carotinoide. Natürlich vorkommende Steroide gehören zu den Triterpenoid-Derivaten (Triterpenoid bedeutet es besteht aus 30 Kohlenstoffatomen), da sie alle ausgehend von Squalen biosynthetisiert werden. Carotinoide werden zu den Tetraterpenoid-Derivaten (40 Kohlenstoffatome) gezählt, sie leiten sich von Lycopen ab. 43 Steroide Grundstruktur aller Steroide, das Steran-Molekül Alle Steroide haben als Grundstruktur ein System aus vier, üblicherweise trans-verbundenen Kohlenstoffringen, drei sechseckigen und einem fünfeckigen. Der bekannteste Vertreter der Steroide ist das zu den Sterinen zählende Cholesterin. Es ist unter anderem auch ein essentieller Bestandteil aller Zellmembranen mit Ausnahme der Innenmembran der Mitochondrien, und kann somit im erweiterten Sinne auch zu den Membranlipiden gezählt werden. Es liegt in der Regel in veresterter Form als Cholesterinester der Fettsäuren vor. Das Spektrum der Fettsäuren der Cholesterinester ist stark ernährungsabhängig. Gallensäuren, die an der Fettverdauung beteiligt sind, besitzen einen hydrophoben und einen hydrophilen Teil, können somit Fette ummanteln und damit deren Absorption im Verdauungstrakt erleichtern. Sexualhormone sind in den Eierstöcken und den Hoden produzierte Steroide, die die Fortpflanzung und die Ausbildung der sekundären Geschlechtsmerkmale steuern. Die weiblichen Geschlechtshormone sind Progesteron und Östrogen, die männlichen Androgene (z. B. Testosteron und Androsteron). Weitere Beispiele sind andere Zoo-, Myco- und Phytosterine und deren Ester wie z.B. Ergosterin, Vitamin D und Herzglycoside (z. B. Digitalis und Strophantin). Phytosterine wie z.B. β-Sitosterin, Stigmasterin und Campesterin und deren Ester treten vermehrt bei vegetarischer Ernährung im Humanserum auf. 44 Carotinoide Carotinoide sind Polymerisationsprodukte von Isopren, die ausschließlich in Pflanzen hergestellt werden und dort als gelb bis rötliche Farbstoffe fungieren. Sie bestehen meist aus ungesättigten Kohlenwasserstoffketten und deren Oxidationsprodukten, und sind aus acht Isopren-Einheiten aufgebaut. Sie werden in Carotine und Xanthophylle unterschieden. Das bekannteste und am häufigsten vorkommende Carotinoid ist das β-Carotin, auch bekannt als Provitamin A. Es wird im Körper in Retinol (Vitamin A) umgewandelt, das eine wichtige Rolle für den Sehvorgang spielt. Biologische Funktionen Die biologischen Funktionen der Lipide sind ebenso vielfältig wie ihre chemische Struktur. Sie dienen als Brennstoff (β-Oxidation der Fettsäuren) Energiespeicher (Triacylglycerole) Membranbausteine (Phospholipide) Signalmoleküle (Diacylglycerol; IP3-Kaskade) Hormone (Eicosanoide; Prostaglandine etc.) Fettlösliche Vitamine (Vitamine A, D, E, K) Cofaktoren (Dolichol) Pigmente (Carotinoide) Während manche Lipide vom menschlichen Körper im Fettstoffwechsel selbst gebildet werden können, müssen andere mit der Nahrung aufgenommen werden. Daher werden diese als essentielle Lipide bezeichnet. Essentielle Fettsäuren Doppelbindungen in der Kohlenwasserstoff-Kette einer Fettsäure, die mehr als neun C-Atome von der Carboxyl-Gruppe entfernt sind, kann der Organismus nicht eigenständig herstellen. Diese sind jedoch von wichtiger Bedeutung und müssen daher über die Nahrung aufgenommen werden, deshalb werden sie als essentiell bezeichnet. Zu den Vertretern der essentiellen Omega-3-Fettsäuren zählen die Linolensäure, Eicosapentaensäure und Docosahexaensäure. Essentielle Omega-6-Fettsäuren sind die Linolsäure und die Arachidonsäure. Aus der Arachidonsäure werden Eikosanoide synthetisiert, diese sind wichtige Gewebshormone und Mediatoren im Körper. Omega-9-Fettsäuren sind nicht essentiell, da sie aus Omega-3- und Omega-6-Fettsäuren synthetisiert werden können. Mögliche Quellen für Omega-3- und Omega-6-Fettsäuren in Nahrungsmitteln sind Fische, Leinsamen, Sojaöl, Hanföl, Kürbiskerne oder Walnüsse. Essentielle Fettsäuren spielen eine wichtige Rolle in vielen Stoffwechselprozessen, und es gibt Hinweise, dass Mängel oder Ungleichgewichte in der Aufnahme der essentiellen Fettsäuren Ursache zahlreicher Krankheiten sind. 45 Fettlösliche Vitamine Die fettlöslichen Vitamine sind: Vitamin A, ein Terpen, das eine wichtige Rolle zum einen beim Sehvorgang, zum anderen für Wachstum, Funktion und Aufbau von Haut und Schleimhäuten spielt, Vitamin D, zuständig für die Regelung der Kalzium- und Phosphor-Konzentrationen im Blut und somit für die Knochenstabilität von entscheidender Bedeutung Vitamin E, ein Terpenoid mit antioxidativer Wirkung und Vitamin K, ein Terpenoid, das bei der Blutgerinnung mitwirkt. Lipoproteine Lipoproteine sind nicht-kovalente Aggregate aus Lipid und Protein und bilden mizellenähnliche Partikel mit einem unpolaren Kern aus Cholesterinestern und Triglyceriden und einer zur wässrigen Phase gerichteten Hülle mit polaren, hydrophilen Anteilen bestehend aus Protein, Phospholipiden und den Hydroxylgruppen von unverestertem Cholesterin. Sie dienen in allen Tierklassen dem Transport der wasserunlöslichen Lipide (Fette) wie Cholesterin im Blut. Nach ihren funktionellen und physikalischen Eigenschaften, z. B. ihres Verhaltens in der Elektrophorese oder die durch Ultrazentrifugation bestimmbare Dichte, werden die Lipoproteine in fünf Klassen eingeteilt: VLDL, IDL, LDL, HDL (Alphalipoprotein) und VHDL, sowie die Chylomikronen. In folgender Tabelle sind die Säugerlipoproteine mit einigen Unterformen aufgeführt. Lipoprotein Durchmesser (nm) Apolipoproteine Konzentration Konzentration bei Männern bei Frauen (g/l) (g/l) ApoB-48, ApoA-I & II, ApoC-II & III, < 0,1 ApoE < 0,1 VLDL - Very Low Density 50 Lipoprotein ApoB-100, ApoC-II, 0,5-2,0 ApoE 0,5-1,5 IDL - Intermediate Density 30 Lipoprotein ApoB-100, ApoC-II, ApoE Chylomikronen LDL - Low Density < 1000 21 ApoB-100 46 2,0-3,5 2,0-3,0 Lipoprotein Lipoprotein a Lp(a) 25 ApoB-100, Apo(a) 0,01-0,5 0,01-0,5 HDL - High Density Lipoprotein HDLE 12 ApoA-I & II <0,05 <0,05 HDL2 10 ApoA-I & II 0,5-1,0 0,5-1,0 HDL3 8 ApoA-I & II 1,0-2,0 1,0-2,0 0,1-0,2 0,1-0,2 VHDL - Very High Density Lipoprotein 7 Über die Nahrung zugeführtes Cholesterin sowie Triglyceride werden vom Dünndarm aufgenommen und dann in Form von Chylomikronen in die Blutbahn abgegeben. Dort werden sie durch die Einwirkung von endothelialen Lipasen zu den so genannten Chylomikronen Remnants, die schließlich von der Leber aufgenommen werden. VLDL, so wie seine Metabolite IDL und vor allem LDL, transportieren vom Körper selbst synthetisiertes Cholesterin und die von den Chylomikronen aufgenommenen Triglyceride von der Leber zu den peripheren Geweben. HDL (genauer HDL3) nehmen Cholesterin unter Vermittlung des Enzyms Lecithin-CholesterinAcyltransferase (LCAT) aus den Geweben, aber auch von anderen Lipoproteinen auf, und transportieren es zur Leber zurück (reverser Cholesterintransport). Das transportierte Cholesterin in den Lipoproteinen ist überwiegend mit Fettsäuren verestert. Über 75 % des Kohlen- und des Wasserstoffes in energieliefernden Substraten im Stoffwechsel sind Lipidbestandteile. Die Transportproteine werden als Apolipoproteine (ApoLp) bezeichnet. Sie geben den Lipidmizellen Stabilität und bestimmen im Stoffwechsel der Lipidoproteine die Umsatzgeschwindigkeit und dirigieren sie zu bestimmten Zielorganen. 47 Chylomikronen Chylomikron-Struktur ApoA, ApoB, ApoC, ApoE (Apolipoproteine); T (Triacylglycerol); C (Cholesterol); grün (Phospholipide) Chylomikronen sind Lipoproteinpartikel von 0,5–1,0 µm Durchmesser und einer Dichte von unter 1,000 g/ml, die vom Dünndarm in die Blutbahn sezerniert werden. Ihr Lipidkern enthält hauptsächlich Triacylglyceride sowie eine im Vergleich geringe Menge an Cholesterinestern. In die Cholesterin-haltige Phospholipidhülle ist Apolipoprotein B-48 als Strukturapolipoprotein eingelagert. Weitere Apolipoproteine, die z. T. zu einem späteren Zeitpunkt auf Chylomikronen übertragen werden, sind Apolipoprotein E (ApoE), die Apolipoprotein-C-Familie sowie Apolipoprotein A-IV. Die Chylomikronen transportieren die im Darm aufgenommenen Nahrungsfette zur Leber und haben in etwa folgende Zusammensetzung: 1 % Eiweiß 4 % Cholesterin und Cholesterinester 5 % Phospholipide 90 % Triglyceride Chylomikronen gelangen über Lymphgefäße und den Milchbrustgang (Ductus thoracicus) in die Blutbahn. In den Kapillaren des Fett- und Muskelgewebes spaltet die Lipoproteinlipase einen Großteil der in den Chylomikronen enthaltenen Triglyceride. Die freigesetzten Fettsäuren werden dann in die umliegenden Fett- und Muskelzellen aufgenommen und gespeichert bzw. verbraucht. Die in der Blutbahn zurückbleibenden Partikel, die nun einen stark erhöhten relativen Cholesteringehalt haben, werden als Chylomikronen-Remnants bezeichnet. An sie lagert sich das Apolipoprotein E an, welches schließlich die rezeptorvermittelte Aufnahme der Chylomikronen-Remnants in die Leber vermittelt (insbesondere über den Membranrezeptor LRP1, in geringerem Maße auch über den LDLRezeptor). Wird ca. zehn Stunden keine Nahrung aufgenommen, sind Chylomikronen nicht mehr nachweisbar. 48 Steroidhormon Steroidhormone sind Steroide, die als Hormone wirken. Zu ihnen gehören die Sexualhormone der Keimdrüsen und die Corticosteroide der Nebennierenrinde. Die Steroidhormone der Säugetiere können in fünf Gruppen eingeteilt werden, nach den Steroidrezeptoren an denen sie binden: Glucocorticoide, Mineralocorticoide, Androgene, Estrogene und Gestagene. Inhaltsverzeichnis 1 Stoffgruppe 2 Metabolismus 3 Regelmechanismus 4 Therapeutische Anwendung 5 Weblinks Stoffgruppe Steroidhormone leiten sich vom Cholesterin ab und sind daher gut fettlöslich und schwer wasserlöslich. Dadurch können sie im Gegensatz zu den anderen Hormongruppen direkt in die Zelle gelangen und brauchen keinen Second Messenger. Dort binden sie an ihre entsprechenden Rezeptoren und können so ihre Wirkung entfalten. Der Steroid-RezeptorKomplex gelangt dann in den Zellkern, wo er auf den Stoffwechsel der Zelle Einfluss nehmen kann. Im Zellkern bewirkt er die Transkription spezifischer DNA-Abschnitte, sodass beispielsweise andere Struktur-Proteine hergestellt werden, welche die Membraneigenschaften der Zelle dauerhaft verändern können. Steroidhormone sind sogar in der Lage, die Blut-Hirn-Schranke zu überwinden. Ihre Wirkungsdauer geht von einigen Stunden bis zu Tagen, wonach sie in der Leber wieder abgebaut werden. Metabolismus Ausgehend vom Cholesterin entsteht Pregnenolon, welches die zentrale Ausgangsverbindung der Steroidhormonbiosythese ist. Regelmechanismus Der Ausstoß der Steroidhormone wird von den Peptidhormonen der Hypophyse kontrolliert, welche wiederum von Neuronen des Hypothalamus gesteuert wird. Beim Transport über die Blutbahn sind sie durch ihre wasserabweisenden Eigenschaften auf Plasmaproteine und spezielle Transportproteine angewiesen. 49 Hormon Epinephrin (Adrenalin), ein Hormon aus der Gruppe der Katecholamine Ein Hormon (griech. ὁρμάω hormáō „antreiben“) ist ein biochemischer Botenstoff. Hormone übermitteln innerhalb eines Lebewesens Informationen von einem Organ zum anderen oder von einem Gewebe zum anderen, ähnlich wie es auch Nerven tun. Im Vergleich erreichen die durch Nerven vermittelten Informationen sehr schnell ihr Zielorgan, während der Informationsfluss durch Hormone vergleichsweise langsam von statten geht. Dabei sind die Zeiträume von der Hormonausschüttung bis zur Hormonwirkung je nach Hormon sehr unterschiedlich, einige Hormone wirken sehr schnell (z. B. Adrenalin), während die Wirkung von anderen Hormonen wie z. B. Steroidhormonen erst nach Stunden einsetzt. Hormone bei Tieren werden durch das Blut oder bei Insekten, Krebsen oder Schnecken mit der vergleichbaren Hämolymphe zu ihren Zielorganen transportiert. Das ist das Wesen der endokrinen Wirkung. Gewebshormone, die im selben Organ gebildet werden und wirken, sind dagegen parakrine Stimulatoren. Inhaltsverzeichnis 1 Allgemeine Einführung 2 Hormonbildende Zellen 3 Endokrine Kaskaden: Hypothalamisch-Hypophysäre Achsen 4 Hormonfreisetzung 5 Hormonähnliche Stoffe 6 Endokrinologie 7 Beispiele für hormonelle Regulation 8 Einteilung nach chemischer Klassifikation 9 Einteilung nach Herkunft 10 Biochemische Eigenschaften 11 Liste von pflanzlichen Hormonen 12 Hormone in der Umwelt 13 Siehe auch 14 Quellen 15 Literatur 16 Weblinks 50 Allgemeine Einführung [Bearbeiten] Hormone wurden seit den frühen Jahrzehnten des 20. Jahrhunderts entdeckt; der Begriff Hormon wurde 1905 von Ernest Starling geprägt. Er entdeckte, dass bei der Stimulation durch Salzsäure aus der Darmwand ein Stoff freigesetzt wurde, der die Pankreas-Sekretion anregte (ein Augenzeugenbericht[1]). Diesen Stoff nannte er Sekretin. Hormone wirken nur auf bestimmte Zielorgane. Nur dort finden sich spezielle Hormonrezeptoren, an welche die Hormonmoleküle binden. Häufig sind diese Rezeptoren Membranproteine, die auf der Zelloberfläche das Hormon binden und auf der Innenseite der Membran nach Hormonbindung Signale auslösen. Einige Hormone (Schilddrüsenhormon, Vitamin D3 und die Steroidhormone, s. u.) erreichen ihre Rezeptoren erst, wenn sie die Zellmembran durch Diffusion durchdrungen haben. Ihre Rezeptoren liegen im Zytoplasma vor oder im Zellkern. Nach der Bindung von Hormon und Rezeptor aggregieren die Rezeptor/Hormon-Komplexe zu Rezeptordimeren, dringen in den Zellkern und steuern dort Genaktivierung. Hormonbildende Zellen Hormone werden von speziellen Hormonproduzierenden Zellen gebildet: Diese finden sich in Drüsen, in der Hirnanhangdrüse (Hypophyse), der Zirbeldrüse, der Schilddrüse, der Nebenniere, den Langerhans’schen Inselzellen der Bauchspeicheldrüse. Einige Hormone werden auch von Nervenzellen gebildet, diese nennt man Neurohormone oder Neuropeptide. Hormone des Magen/Darm-Traktes finden sich verteilt in den Krypten. Zudem werden in der Leber Vorstufen des Angiotensins gebildet. Geschlechtshormone werden von spezialisierten Zellen der weiblichen oder männlichen Geschlechtsorgane gebildet: Theca- und GranulosaZellen bei der Frau und Leydig-Zellen beim Mann. Charakteristisch für die hormonproduzierenden Zellen sind Enzyme, die nur in diesen Zellen vorkommen. Die Freisetzung der Hormone ist individuell für jedes Hormon geregelt. Häufig werden Hormone in der Zelle gespeichert und nach Stimulation durch einen Freisetzungsstimulus freigesetzt. Die Freisetzungsstimuli können Freisetzungshormone (Releasing-Hormone) sein, Liberine, siehe unten. Endokrine Kaskaden: Hypothalamisch-Hypophysäre Achsen Häufig finden wir hormonelle Achsen: die hypothalamisch-hypophysär-gonadotrophe Achse: Das Gonadotropin-Releasing Hormon (GnRH) aus Nervenzellen des Hypothalamus setzt in der Hypophyse die Gonadotropine frei, die wiederum in den Geschlechtsorganen die Bildung von Sexualsteroiden anregen. die hypothalamisch-hypophysär-adrenotrophe Achse: Das Corticotropin-Releasing Hormon (CRH) aus Nervenzellen des Hypothalamus setzt in der Hypophyse das ACTH frei, das in der Nebenniere die Kortisol-Bildung anregt. die hypothalamisch-hypophysär-thyreotrophe Achse: Thyreotropin-Releasing Hormon (TRH) aus Nervenzellen des Hypothalamus setzt in der Hypophyse das Thyrotropin frei, das in der Schilddrüse die Freisetzung des Tyroxin und des Trijodthyronin anregt. 51 Hormonfreisetzung Die Hormonfreisetzung (mit Ausnahme der parakrinen Stimulatoren) erfolgt in der Nähe von Blutgefäßen, die viele kleine Fenster haben, durch die Hormone direkt ins Blut übergehen können. Bei auf die Sekretion von Neuropeptiden spezialisierten Stellen spricht man von Neurohämalorganen. Durch die Bindung eines Stimulus für die Hormonfreisetzung kommt es häufig in der Zelle zu einem Anstieg des intrazellulären Kalziums. Dieser Kalzium-Anstieg erlaubt die Fusion der Zellorganellen, in denen sich die vorgefertigten Hormone befinden, mit der Zellmembran. In dem Moment, in dem die Membran Organelle mit der Zellmembran fusioniert, haben die Hormon freien Zugang zum Raum außerhalb der Zelle und können in die dort benachbarten Blutgefäße durch die gefensterten Blutgefäßwand wandern. Hormonähnliche Stoffe Die bei Pflanzen vorkommenden Hormone werden als Phytohormone bezeichnet. Sie teilen mit den tierischen Hormonen die Eigenschaft, Signalwirkung über eine größere Distanz zu entfalten und in geringen Konzentrationen wirksam zu sein. Die bei Tieren vorkommenden Pheromone sind Botenstoffe zwischen Individuen. Sie sind nicht an den Organismus gebunden, in dem sie gebildet wurden und können über große Distanzen signalisieren. Endokrinologie Die Wissenschaft, die sich mit der Erforschung der Hormone befasst, ist die Endokrinologie. Ein Wissenschaftler oder Arzt, der sich mit der Erforschung der Hormone, ihrer Wirkungsweisen und mit Erkrankungen des hormonalen Geschehens beschäftigt, wird als Endokrinologe bezeichnet. Beispiele für hormonelle Regulation [Bearbeiten] 52 Beispiel für einen Rückkopplungsmechanismus Zuckerstoffwechsel, Fettstoffwechsel, Nahrungsaufnahme Menstruationszyklus der Frau, Sexualentwicklung bei Mann und Frau Knochenwachstum Anpassung an Angst und Stress Thyreotroper Regelkreis Hormone werden selber: 1. durch Regelkreise (Rückkopplung, feedback system; in der hypothalamischhypophysären-thyreotrophen Achse zum Beispiel unterdrückt das Endprodukt Schilddrüsenhormon (Trijodthyronin) die Bildung des TRH im Hypothalamus und des Thyreotropins aus der Hypophyse.), Die Freisetzung der meisten Hormone wird durch negative Rückkopplungen gesteuert, wie beispielsweise die der Glukokortikoide der Nebennierenrinde. Der Hypothalamus setzt corticotropin-releasing-Hormon (CRH) frei, das in der Hypophyse die Freisetzung von adrenocortocotrophes Hormone (ACTH) stimuliert (blauer Pfeil +). Dieses stimuliert in der Nebennierenrinde die Bildung und Freisetzung von Kortisol und anderen Glukokortikoiden (blauer Pfeil +). Über das Blut in das Gehirn und die Hypophyse gebracht unterdrückt Kortisol andererseits die Bildung und Freisetzung von CRH und ACTH (rote Pfeile −), wodurch die Kortisolbildung wieder aussetzt.[2] 2. durch das autonome Nervensystem sowie 3. durch nichthormonelle chemische Botenstoffe wie zum Beispiel die Kalziumkonzentration oder die Glukosekonzentration im Blut reguliert. Einteilung nach chemischer Klassifikation Protein- und Peptidhormone mit charakteristischer Aminosäuresequenz o o o Neuropeptide des Hypothalamus: Freisetzungshormone für LH/FSH, TSH, ACTH, GH Somatostatin Agouti-ähnliches Peptid Neuropeptid Y Leptin Ghrelin Glykoproteinhormone der Adenohypophyse: Follikelstimulierendes Hormon Follitropin (FSH) Luteinisierendes Hormon Luteotropin (LH) Schilddrüsenstimulierendes Hormon Thyreotropin (TSH) Adrenocortikotropin (ACTH) weitere adenohypophysäre Hormone: Wachstumshormone: GH Prolaktin Melanozytenstimulierendes Hormon (MSH) Galanin Kisspeptin 53 o o o o o o o Neuropeptide der Neurohypophyse: Adiuretin (Vasopressin) Oxytocin Hormone der Nebenschilddrüsen Parathormon Kalzitonin Hormon des Herzen Atrial-Natriuretisches Peptid (ANP) Hormone der pankreatischen Inselzellen: Insulin Glucagon Somatostatin Pankreatisches Polypeptid Peptidhormone des Magen- und Darmtraktes Cholezystokinin (CCK) Sekretin Gastrin Ghrelin Vasoaktives intestinales Peptid (VIP) Gastro-inbitorisches Peptid (GIP) Peptid Tyrosyl-Tyrosin (PYY) Peptidhormon der Leber Insulin-like growth factor (IGF) Proteinhormone der Gonaden Inhibin und Aktivin Protein/Peptid-Hormone bei Vertebraten, nicht beim Menschen gefunden o bei Lurchen Caerulein o bei Fischen Stanniokalzin; jetzt auch beim Menschen (und weiteren Vertebraten) zwei Stanniokalzine mit noch unbekannter Funktion gefunden Aminosäurederivate o Katecholamine Adrenalin Noradrenalin Dopamin o Thyroxin (T4) und Triiodthyronin (T3) o Serotonin und Melatonin Isoprenderivate – wie Juvenilhormon und Neotenin bei Insekten Steroidhormone – wie die Nebennierenrinden- und Geschlechtshormone o Mineralocorticoide – wie Aldosteron o Glucocorticoide – wie Cortisol o Estrogene – wie Estradiol o Gestagene – wie Progesteron o Androgene – wie Testosteron 54 Arachidonsäurederivate (Eicosanoide) o Prostaglandine o Leukotriene o Thromboxane Einteilung nach Herkunft Es gibt spezielle Hormondrüsen, in denen hormonbildende Zellen im engen Verbund zusammenhängen. Viele Hormone werden aber von Zellen gebildet, die nicht ausschließlich mit hormonbildenden Zellen im Verbund stehen. So liegen die Zellen, die Gastrin bilden, vereinzelt in den Liebermann’schen Krypten des Magens vor. Ähnlich ist es mit den Zellen für die Hormone Cholezystokinin, Sekretin oder Somatostatin in der Darmwand. Entscheidend für die Hormonproduktion ist nicht die äußere Umgebung einer Zelle, sondern die Ausrüstung innerhalb mit den charakteristischen Enzymen. Spezialisierte Hormondrüsen o Hypophyse Hypophysen-Vorderlappen, die Adenohypophyse): Hier werden LH/FSH, ACTH, Prolaktin, GH und TSH gebildet. Hypophysen-Hinterlappen, Neurohypophyse): Diese ist keine Hormondrüse im eigentlichen Sinne, da hier die Hormone Oxytozin und Vasopressin (Adiuretin) an Nervenenden ausgeschüttet werden, wobei die Nervenzellkerne sich im Hypothalamus befinden und deren Nervenbahnen durch den Hypophysenstiel laufen. o Zirbeldrüse: Bildung des Hormons Melatonin o Schilddrüse: Bildung des Schilddrüsenhormons Nebenschilddrüse (Parathyroidea): Bildung von Parathormon und Calzitonin o Nebenniere: Bildung von Aldosteron (Mineralokortikoid), Androgenen (Androstendion) und Adrenalin (Epinephrin). o Inselzellen in der Bauchspeicheldrüse: Bildung von Insulin, Glukagon, Somatostatin und Pankreatischem Polypeptid Neurohormone, die von Neuronen im ZNS produziert werden. o Hypothalamische Neuropeptide: Bildung von GnRH,CRH,TRH oder GHRH: Speicherung an den Nervenenden in der Eminentia mediana; Bildung von Oxytozin und Vasopressin (Adiuretin), Speicherung an den Nervenenden in der Neurohypophyse; Bildung von NPY, GHrelin, Agouti-ähnlichem Peptid Gewebe mit Hormonbildenden Zellen: o Haut: Bildung von Vitamin D3 durch Bestrahlung von 7-Dehydrocholesterin mit UV-Licht o Herz: Bildung des Atrial-Natriuretischen Peptides durch Muskelzellen (Myozyten) des rechten Herzvorhofes o Leber: Bildung des Angiotensinogen, des Vorläufers des Angiotensin, Bildung von Insulin-ähnlichen Wachstumsfaktoren (IGF) o Magen- und Darmtrakt: Bildung von Cholezystokinin, Gastrin, Sekretin, GHrelin aus einzeln in die Magen- oder Darmwand verteilten endokrinen Zellen. o Gonaden 55 Hoden: Bildung von Testosteron (und Östradiol) durch die LeydigZellen, von Inhibin und Aktivin Ovarien: Bildung von Testosteron durch Theka-Zellen und durch Östradiol durch Granulosa-Zellen, Bildung von Inhibin und Aktivin Weitere Organe mit Steuerungsfunktion bestimmter endokriner Regelkreise o Niere: Die Zellen des juxtaglomerulären Apparates setzen bei erniedrigtem Blutdruck das Enzym Renin frei, das das Angiotensinogen aus der Leber zum Angiotensin I spaltet. o Lunge: Hier wird das Angiotensin I durch das Angiotensin-konvertierende Enzym (ACE) zum wirksamen Angiotensin II verkürzt. Biochemische Eigenschaften Man unterscheidet zwischen zwei Arten von Hormonen: Lipidunlösliche Hormone (Peptide): Diese Substanzen können wegen ihrer Lipidunlöslichkeit die Zellmembran nicht passieren. Stattdessen binden sie sich an spezifische membrangebundene Rezeptoren der Zielzellen. Zusammen mit dem Rezeptor wird ein Hormon-Rezeptor-Komplex gebildet. Der Rezeptor aktiviert ein Enzym, welches ATP in cyclisches AMP (cAMP, ein Second Messenger) umwandeln kann. Das cAMP bindet an einen hemmenden Proteinanteil, welches wiederum an ein anderes Enzym im Cytoplasma gebunden ist und dieses Enzym vorläufig deaktiviert. Durch die Bindung von cAMP an den hemmenden Proteinanteil wird dieser vom Enzym getrennt. Das Enzym wird dadurch aktiviert und bewirkt seinerseits bestimmte Stoffwechselprozesse in der Zelle. Lipidlösliche Hormone (Steroide): Diese Substanzen können aufgrund ihrer Lipidlöslichkeit durch die Zellmembran in die Zelle eindringen. Der Stoff bindet im Cytoplasma an spezifische Rezeptoren und bildet ein Hormon-Protein-Komplex. Dieser Komplex hat, im Gegensatz zum ursprünglichen Hormon, die Fähigkeit, durch die Zellkernmembran zur DNA zu gelangen. Es werden nun spezifische Gene aktiviert, es kommt zur Proteinbiosynthese. Steroidhormone stammen alle vom Cholesterin ab und werden in den Mitochondrien, mittels Enzymen aus Cholesterin gebildet. Hierbei wird immer erst ein Gestagen gebildet aus dem dann ein Glucocorticoid, ein Mineralocorticoid oder ein Androgen gebildet wird. Aus Androgenen werden Estrogene gebildet. Diese Reihenfolge ist starr, d. h. es kann aus einem Gestagen nicht sofort ein Estrogen gebildet werden. Aus diesem Grund nennt man Steroidhormone auch Precurser-Hormone. Aldosteron → Dursthormon, Salzzurückhaltung im Körper Angiotensin I und Angiotensin II → u. a. Stimulierung der Aldosteron und AdiuretinFreisetzung Antidiuretisches Hormon = ADH = Adiuretin = Vasopressin → Wasserzurückhaltung im Körper ANP = atriale natriuretische Peptide Calciferol (Vitamin D3: Cholecalciferol; Vitamin D2: Ergocalciferol), mehr Hormon als Vitamin Calcitonin (Cardionatrin = ANP) Cholecystokinin (CCK) = Pankreozymin 56 Corticotropin-releasing Hormon (CRH) Cortisol Cortison = Stresshormon 2 Dopamin (DA) Erythropoietin (EPO) Hormon, das die Bildung von roten Blutkörperchen anregt Follikel stimulierendes Hormon (FSH) Gastrin Glucagon – Blutzucker steigernd Gonadotropin-releasing Hormon GnRH Humanes Choriongonadotropin (HCG) Insulin – Blutzucker senkend Insulin-like growth factor Leptin Luteinisierendes Hormon LH Melatonin = Beeinflusst den Tag-Nacht-Rhythmus des Körpers Neuropeptid Y Noradrenalin (NA) Östrogen = Weibliches Sexualhormon Oxytocin = Soziales Bindungshormon, Geburtshormon Parathormon (PTH) Phenethylamin (PEA) = Glückshormon POMC = Vorstufe von vielen Hormonen und Regulation der Nahrungsaufnahme Progesteron = Gelbkörperhormon Prolaktin (PRL) Schilddrüsenhormone Thyroxin T4 und Triiodthyronin T3 Secretin Serotonin = 5-Hydroxytryptamin (5-HT) Somatoliberin – Growth-hormone-releasing-hormone – GHRH Somatostatin Somatotropin = Wachstumshormone = HGH (Human Growth Hormon) Testosteron = Männliches Sexualhormon Thrombopoietin Thyroidea stimulierendes Hormon TSH TSH-Releasing Hormon TRH Thyroxin T4 Triiodthyronin T3 Eine spezielle Gruppe von Hormonen sind die trophischen Hormone, die andere Hormondrüsen zur Produktion anregen. So regt das Thyroidea stimulierende Hormon (TSH) das Wachstum und die Aktivität der Schilddrüse an. Hunger-Hormone: Ghrelin PYY-336 Sattheitshormon Leptin Orexine 57 Liste von pflanzlichen Hormonen Auxin Cytokinin Ethylen Abszisinsäure Gibberellinsäure Brassinosteroid Jasmonsäure Salicylsäure Hormone in der Umwelt Besondere Aufmerksamkeit verdient die Tatsache, dass Hormone zunehmend in die Umwelt eingetragen werden und später über die pflanzliche und tierische Nahrungskette in ungünstiger und unkontrollierter Dosierung vom Menschen wieder aufgenommen werden. Ein Beispiel sind die Hormone der Anti-Baby-Pille, die von Kläranlagen nicht erkannt und gefiltert werden. Sie werden mit dem „sauberen“ Wasser in die Flüsse eingeleitet. Da die Kläranlagen auf den Medikamenteneintrag nicht ausgelegt sind, gelangen Medikamente und ihre Rückstände fast ungehindert über die Oberflächengewässer auch wieder ins Trinkwasser. Mehr als 180 der 3000 in Deutschland zugelassenen Wirkstoffe lassen sich in deutschen Gewässern nachweisen: Von Hormonen und Lipidsenkern über Schmerzmittel und Antibiotika bis hin zum Röntgenkontrastmittel.[3] Auch bestimmte Schadstoffe wie beispielsweise DDT, PCB, PBDE oder Phthalate wirken wie Hormone und beeinflussen etwa die immer früher einsetzende erste Monatsperiode bei Mädchen. Gewebshormone Während "klassische" (glanduläre) Hormone in Drüsen gebildet werden, entstehen die Gewebshormone in spezialisierten Einzelzellen, die über ein Gewebe verteilt sein können. Gewebshormone vom parakrinen Typ gelangen durch Diffusion zu ihren Erfolgsorganen, während solche vom endokrinen Typ über den Blutkreislauf dorthin gelangen Beispiele Biogene Amine wie Histamin und Serotonin. Diese entstehen durch Decarboxylierung aus Histidin bzw. Tryptophan. Beide agieren über Rezeptoren entweder als Gewebshormon oder als Neurotransmitter. Die Signaltransduktion kann hierbei über GProteine (cAMP, PLC, 5HT-1,2 und 4) oder über die Öffnung von Kationen-Kanälen (5HT3)erfolgen. Peptidhormone wie die Angiotensine. Kinine. Kininogene sind höhermolekulare Plasmaproteine, aus denen durch verschiedene lokal aktivierte Proteasen (z.B. Kallikrein) Peptidmediatoren freigesetzt werden können. Ein wichtiges Produkt ist Bradykinin, ein Nonapeptid, das 58 gefäßerweiternd, blutdrucksenkend und auf die glatte Muskulatur von Bronchien, Darm und Uterus kontrahierend wirkt. die Eikosanoide, C-20-Fettsäurederivate, die sich in drei Untergruppen aufteilen lassen: Prostaglandine (PG), Leukotriene und Epoxide. Es ist kaum möglich, alle Wirkungen darzustellen, jedoch gilt, dass PG und andere Eikosanoide an fast allen Signalwegen als lokal wirksame second messenger beteiligt sind. Gase mit Signalfunktion: NO (Stickoxid, Stickstoffmonoxid) wird durch NitroxidSynthase aus Arginin hergestellt. Das Gas hat folgende Wirkungen: Gefäßtonusregulierend, Herzkontraktion-fördernd, manchmal toxische Effekte. Es ist Neurotransmitter und beeinflusst die Genexpression. Teils ist es für diese Wirkungen selbst verantwortlich, teils sind es seine Umwandlungsprodukte N2O3, ONOO− (Peroxynitrid), NO− oder NO2. Katecholamine Katecholamine, auch Catecholamine oder Brenzcatechinamine, sind eine Klasse von körpereigenen und künstlichen Stoffen, die an den sympathischen Alpha- und BetaRezeptoren des Herz-Kreislaufsystems eine anregende Wirkung haben. Somit fungieren Katcholamine als Hormone, die pharmazeutisch zu den Sympathomimetika zählen. Sie sind alle chemisch ähnlich aufgebaut (sie alle sind Derivate des 1,2-Dihydroxybenzols, englisch Catechol, ferner Phenylethylamine mit Catechol als Ortho-Diphenolfunktion in 3,4-Position). Im Speziellen fasst man unter dem Begriff Katecholamin die Hormone und Neurotransmitter Adrenalin, Noradrenalin und Dopamin, sowie die Arzneistoffe Isoprenalin, Dobutamin und Dopexamin zusammen. Inhaltsverzeichnis 1 Physiologie o 1.1 Biosynthese o 1.2 Wirkungen o 1.3 Nebenwirkungen 2 Katecholamine in der Diagnostik 3 Katecholamine in der Therapie 4 Weblinks 59 Physiologie Biosynthese Biosynthese der Katecholamine aus L-Tyrosin. 60 Die Biosynthese der Katecholamine findet in den Nebennieren und im Nervensystem statt. Sie geht von der Aminosäure Tyrosin aus (s. Abb.), die zunächst mittels des Enzyms Tyrosinhydroxylase zu Levodopa umgewandelt wird. Im nächsten Schritt entsteht aus Dopa mithilfe der aromatische-L-Aminosäure-Decarboxylase Dopamin. Dopamin kann in einem weiteren Schritt zu Noradrenalin hydroxyliert werden, wozu die Dopaminhydroxylase gebraucht wird. Den optionalen letzten Schritt, die Methylierung von Noradrenalin zu Adrenalin, katalysiert die Noradrenalin-N-Methyltransferase. Wirkungen grobe Einteilung im niedrigen und mittleren Dosisbereich: o Adrenalin steigert Blutdruck und Herzfrequenz. o Noradrenalin. steigert vor allem den Blutdruck, nicht so sehr die Herzfrequenz o Isoprenalin. steigert vor allem die Herzfrequenz, nicht so sehr den Blutdruck. o Dopamin positiv inotrop o Dobutamin positiv inotrop Nebenwirkungen Alle sind in höherer Dosis arrhythmogen; alle sind in hoher Dosis kardiotoxisch; in hoher Dosis Kreislaufzentralisation unter Noradrenalin. Katecholamine in der Diagnostik Die aus dem Nebennierenmark und aus den sympathischen Nerven freigesetzten Katecholamine Noradrenalin und Adrenalin werden zu etwa 1 % im Harn ausgeschieden. 80– 85 % der Katecholaminausscheidung erfolgen als Vanillinmandelsäure und ca. 15 % als Metanephrine. Bei Verdacht auf Phäochromozytom ist die Bestimmung von freien Metanephrinen im Harn notwendig, da diese Tumore meist große Mengen Noradrenalin produzieren. Die Bestimmung der Metanephrine erfolgt dabei im Urin, der über 24 Stunden gesammelt wurde. Zusätzlich wird meist eine Bestimmung im Blutserum vorgenommen. Katecholamine in der Therapie Katecholamine stehen auch als Medikamente zur Verfügung. Sie werden fast ausschließlich in der Intensivmedizin und bei Reanimation verabreicht. Sie sind stark wirksam und werden meist intravenös gegeben. Eine alternative Verabreichungsform bei der Reanimation stellt die endotracheale Gabe von Adrenalin dar. Dies ist dann sinnvoll, wenn ein Endotrachealtubus, aber kein intravenöser Zugang zur Verfügung steht. Die Resorptionsgeschwindigkeit ist vergleichbar, die Dosis muss jedoch angepasst werden. Neuere Studien haben jedoch gezeigt, dass die endotracheale Applikation der intravenösen deutlich unterlegen ist. So lässt sich die 61 Resorptionsgeschwindigkeit eben nicht zuverlässig bestimmen, es kann zu Depotbildungen kommen, die nach der Wiederkehr einer spontanen Blutzirkulation noch zu Herzrhythmusstörungen führen. In den 2005er Richtlinien des ERC wird daher die endobronchiale Gabe nur noch im Ausnahmefall empfohlen und nicht wie früher als Regelmaßnahme. Bei der Katecholaminanwendung zur Kreislaufstabilisierung in der Intensivmedizin ist auf eine sehr gleichmäßige Zufuhr in den Körper zu achten, da sonst erhebliche Druck- und Herzfrequenzspitzen, bzw. (bei Unterbrechung der Zufuhr) Blutdruck- und Herzfrequenzabfälle auftreten können. Eine enge Herzkreislaufüberwachung ist unabdingbar, da diese Stoffe in höherer Dosierung ein erhebliches arrhythmogenes Potential haben (d. h. Herzrhythmusstörungen bis hin zum Kammerflimmern auslösen können). Bei der Anwendung kann es zu Herzinfarkten und Hirnblutungen durch zu hohen Blutdruck kommen. Die Wirksamkeit der Katecholamine beim kardiogenen Schock ist bis jetzt nicht eindeutig belegt. Auch sonst beruht der Einsatz der Katecholamine in der Intensivmedizin meist auf empirischer Erfahrung. Randomisierte Vergleichsstudien sind Mangelware, da die Katecholamine vor der verbreiteten Akzeptanz der Kriterien der „Evidence based medicine“ zur Verfügung standen und ihre Anwendung oft der einzige Weg ist, eine mit dem Überleben vereinbare Kreislaufsituation aufrechtzuerhalten, so dass sich Vergleichsstudien mit Placebos aus ethischen Gründen verbieten. Angiotensin I Angiotensin I Kugelmodell nach PDB 1N9U Größe 10 Aminosäuren Precursor Angiotensinogen Bezeichner Gen-Namen AGT; ANHU; SERPINA8 Externe IDs OMIM: 106150 UniProt: P01019 MGI: 87963 62 Angiotensin I ist ein Dekapeptid und ein Prohormon. Es ist ein wichtiges Bindeglied in dem für die Aufrechterhaltung des Blutdrucks und des Wasserhaushaltes zuständigen ReninAngiotensin-Aldosteron-System (RAAS). Inhaltsverzeichnis 1 Struktur 2 Wirkmechanismus 3 Geschichte 4 Quellen Struktur Die Primärstruktur von Angiotensin I besteht aus zehn Aminosäuren (H2N-Asp–Arg–Val– Tyr–Ile–His–Pro–Phe–His–Leu–COOH) mit einer Molekülmasse von 1296,49 Da.[1] Wirkmechanismus Angiotensin I wird im Organismus enzymatisch durch Renin aus Angiotensinogen gebildet. Es ist selbst weitgehend inaktiv. In Gegenwart des Angiotensin Converting Enzyme (ACE) wird Angiotensin I in das Oktapeptid Angiotensin II gespalten, welches für die blutgefäßkontrahierenden Wirkungen verantwortlich ist. Ein weiterer, von Angiotensin I ausgehender Aktivierungsweg wurde erst kürzlich entdeckt. Angiotensin I kann in Gegenwart des Angiotensin Converting Enzyme Typ 2 und weiterer Peptidasen zum Heptapeptid Angiotensin(1-7) gespalten werden, welches mit einem bisher noch unbekannten Angiotensin-Rezeptor interagiert. Geschichte Angiotensin, ursprünglich Angiotonin bzw. Hypertensin genannt, wurde erstmals 1940 durch I.H. Page beschrieben. Er stellte fest, dass das in der Leber gebildete Angiotensinogen ein Substrat für das der Niere entstammende Enzym Renin ist. Als Resultat einer enzymatischen Umsetzung konnte eine Substanz gefunden werden, die zu einer Vasokonstriktion und einer Blutdrucksteigerung führt. Es benötigte aber noch mehr als ein Jahrzehnt bis durch Leonard T. Skeggs gezeigt werden konnte, dass das Angiotensin ein Gemisch aus mindestens zwei verschiedenen Substanzen ist: dem weitgehend inaktiven Angiotensin I und dem gefäßkontrahierenden Angiotensin II. 63 Angiotensin II Angiotensin II Kalottenmodell nach PDB 1N9V Größe Precursor 8 Aminosäuren Angiotensinogen Bezeichner Gen-Namen AGT; ANHU; SERPINA8 Externe IDs OMIM: 106150 UniProt: P01019 MGI: 87963 Arzneistoffangaben ATC-Code C01CX06 Angiotensin II ist ein zu den Gewebshormonen zählendes Peptidhormon, bestehend aus acht Aminosäuren (Oktapeptid). Es nimmt die Schlüsselposition in dem für die Aufrechterhaltung des Blutdrucks und des Wasserhaushaltes zuständigen Renin-Angiotensin-Aldosteron-System (RAAS) ein. Inhaltsverzeichnis 1 Biochemie o 1.1 Struktur o 1.2 Biosynthese o 1.3 Wirkmechanismus o 1.4 Abbau 2 Geschichte 64 3 Quellenangaben Biochemie Struktur Die Primärstruktur von Angiotensin II besteht aus acht Aminosäuren (H2N-Asp–Arg–Val– Tyr–Ile–His–Pro–Phe–COOH) mit einer Molekülmasse von 1046,19 Da.[1] Biosynthese Angiotensin II wird aus dem Dekapeptid Angiotensin I im Organismus enzymatisch durch das Angiotensin Converting Enzyme (ACE) gespalten. Das Angiotensin Converting Enzyme ist der Angriffspunkt der ACE-Hemmer. Wirkmechanismus Das gebildete Angiotensin II interagiert mit Angiotensin-Rezeptoren (AT-Rezeptoren). Durch eine Aktivierung des AT1-Rezeptors kann primär in Blutgefäßen eine Kontraktion, in der Niere eine erniedrigte glomeruläre Filtration bei gleichzeitig verminderter Na+-Ausscheidung, in der Nebenniere eine Stimulierung der Aldosteron- und Adrenalinfreisetzung und in der Hypophyse eine Freisetzung von Vasopressin beobachtet werden. Auch das Durstgefühl wird auf eine akute Stimulation von AT1-Rezeptoren im Hypothalamus zurückgeführt. Eine chronische Stimulation des AT1-Rezeptors führt hingegen zu einer Stimulation mitogener Effekte und somit beispielsweise zur Hypertrophie des Herzes. Akute und chronische Wirkungen von Angiotensin II auf den AT1-Rezeptor können indirekt durch ACE-Hemmer und direkt durch AT1-Rezeptorantagonisten (Sartane) unterdrückt werden. Angiotensin II zeigt ebenfalls eine hohe Affinität zu AT2-Rezeptoren. Die Bedeutung dieser Rezeptoren an den durch Angiotensin II vermittelten Effekten ist hingegen umstritten. Angiotensinamid, ein Abkömmling des Angiotensin II, ist ein kardiostimulierender und blutdrucksteigernder Arzneistoff. Abbau Angiotensin II wird durch Aminopeptidasen in einem mehrstufigen Prozess zu inaktiven Produkten abgebaut. Anfallende Zwischenprodukte, wie das Angiotensin III und das Angiotensin IV können jedoch durchaus noch eine biologische Aktivität besitzen. Angiotensin III bindet mit mäßiger Potenz an den AT1-Rezeptor, während Angiotensin IV ein Ligand an dem noch wenig erforschten AT4-Rezeptor ist. Ein alternativer Spaltungsweg des Angiotensins II mit Hilfe des Angiotensin Converting Enzyme Typ 2 wurde erst kürzlich entdeckt. 65 Geschichte Angiotensin, ursprünglich Angiotonin bzw. Hypertensin genannt, wurde erstmals 1940 durch I. H. Page beschrieben. Er stellte fest, dass das in der Leber gebildete Angiotensinogen ein Substrat für das der Niere entstammende Enzym Renin ist. Als Resultat einer enzymatischen Umsetzung konnte eine Substanz gefunden werden, die zu einer Vasokonstriktion und einer Blutdrucksteigerung führt. Es benötigte aber noch mehr als ein Jahrzehnt bis durch Leonard T. Skeggs gezeigt werden konnte, dass das Angiotensin ein Gemisch aus mindestens zwei verschiedenen Substanzen ist: dem weitgehend inaktiven Angiotensin I und dem gefäßkontrahierenden Angiotensin II. Releasing-Hormone Releasing-Hormone, auch Liberine, sind Neuropeptide, die in bestimmten Kerngebieten im Hypothalamus gebildet werden. Die Nerven enden in der Eminentia mediana (einem Neurohämalorgan am unteren Rand des Hypothalamus). Hier werden unter der Kontrolle von weiteren Hormonen und Neurotransmittern die Releasing-Hormone in Blutgefäße ausgeschüttet, die direkt zum Hypophysenvorderlappen reichen. Man spricht von einem Portalsystem. In der Hypophyse regen die Releasing-Hormone die Freisetzung anderer Hormone an. Somatostatin und Dopamin sind Release-Inhibiting-Hormone (Statine), also Regulatoren, die die Freisetzung unterdrücken. Die Bildung der Hormone der Hypophyse steht nicht unter der Kontrolle der Releasing-Hormone, nur deren Freisetzung: Inhaltsverzeichnis 1 Die hypothalamischen Releasing-Hormone des Menschen und ihre Wirkung 2 Die hypothalamischen Release-inhibiting-Hormone des Menschen und ihre Wirkung 3 Referenzen 4 Literatur Die hypothalamischen Releasing-Hormone des Menschen und ihre Wirkung TRH (Thyreotropin-Releasing-Hormon), auch Thyreoliberin, bewirkt die Freisetzung von Thyreotropin (TSH). CRH (Corticotropin-Releasing-Hormon), auch Corticoliberin, bewirkt die Freisetzung von Adrenocorticotropin (ACTH). GnRH (Gonadotropin-Releasing-Hormon), auch Gonadoliberin, bewirkt die Freisetzung des Follikelstimulierenden Hormons (FSH) und des Luteinisierenden Hormons (LH). GHRH (Growth-Hormone-Releasing-Hormon), auch Somatoliberin, bewirkt die Freisetzung von Somatotropin (Wachstumshormon, Growth Hormone, GH). PRH (Prolaktin-Releasing-Hormon) gibt es nach dem Stand der Wissenschaft nicht. Die Prolaktin-Releasing Peptide (PrRP) (aus dem Hypophysenzwischenlappen)[1] 66 können zwar in der Zellkultur Prolaktin-Freisetzung erreichen, aber dort, wo die übrigen Releasing-Hormone ins Portalsystem freigesetzt werden, um dann zur Hypophyse transportiert zu werden, in der Eminentia mediana, findet man die PrRPNeuronen nicht. Daher bestehen große Zweifel, ob die hypophysäre ProlaktinFreisetzung durch PrRP stimuliert wird[2]. Die Prolaktin-Freisetzung steht unter negativer Regulation von Dopamin (siehe unten). Die hypothalamischen Release-inhibiting-Hormone des Menschen und ihre Wirkung Somatostatin Gemeinsam mit Somatoliberin (Wachstumshormon-Releasing-Hormon) steuert Somatotostatin die Freisetzung des Wachstumshormon. Darüber hinaus ist es ein Regulator weiterer Hormonfreisetzungen in den Langerhans'schen Inselzellen und im Magen- und Darmtrakt. Dopamin Anders als die vorstehenden Neuropeptide ist Dopamin kein Neuropeptid, sondern als Derivat des Tyrosins ein Katecholamin. Es wirkt sowohl als Neurotransmitter als auch als Hormon. Von der Eminentia mediana freigesetzt und über das Portalsystem zur Hypophyse transportiert, unterdrückt es als Hormon die Prolaktinfreisetzung. Parathormon Parathormon Bänder-/Stäbchenmodell von Aminosäuren 1-34 des PTH nach PDB 1BWX Vorhandene Strukturdaten: 1bwx, 1et1, 1fvy, 1hph, 1hpy, 1zwa, 1zwb, 1zwc, 1zwd, 1zwe, 1zwf, 1zwg Größe Precursor 84 Aminosäuren (115 Aminosäuren) Bezeichner Gen-Namen PTH; Externe IDs OMIM: 168450 UniProt: P01270 MGI: 97799 67 Vorkommen Homologie-Familie Übergeordnetes Taxon Parathormon Amnioten Das Parathormon, auch Parathyrin (PTH), ist ein Peptidhormon, bestehend aus 84 Aminosäuren[1], welches in den Nebenschilddrüsen (Epithelkörperchen) gebildet wird. Die Hauptfunktion des Parathormons ist die Erhöhung der Calcium-Konzentration im Blutplasma. Eine Verminderung dieser Konzentration induziert vermehrte PTH-Sekretion. Verschiedene Ursachen können für einen zu hohen bzw. zu niedrigen Gehalt des Bluts an PTH verantwortlich sein (Hyperparathyreoidismus bzw. Hypoparathyreoidismus). Veränderungen im PTH-Gen, die zu Hypoparathyroidismus führen, sind bekannt. Inhaltsverzeichnis 1 Biosynthese 2 Abbau 3 Mechanismus und Steuerung der Sekretion 4 Wirkungen o 4.1 Induktion der Osteolyse o 4.2 Hemmung der Phosphatresorption o 4.3 Induktion der Biosynthese von Calcitriol 5 Klinik 6 Quellen 7 Literatur Biosynthese Das Parathormon wird als PräPro-Hormon (115 Aminosäuren) an membrangebundenen Ribosomen synthetisiert und cotranslational unter Abspaltung der aminoterminalen Signalsequenz (Prä-Sequenz) prozessiert, so dass Pro-Parathormon (90 Aminosäuren) entsteht. Durch Prozessierung im Golgi-Apparat entsteht das fertige Parathormon (siehe Translation). Abbau Das Parathormon besitzt nur eine Halbwertzeit von wenigen Minuten und wird sowohl in den Epithelkörperchen selbst, als auch in der Leber und der Niere proteolytisch abgebaut. Da nur ein sehr kleiner Anteil des Parathormons für die biologische Aktivität verantwortlich ist, entstehen bei der Proteolyse teilweise Zwischenprodukte, die noch biologische Aktivität aufweisen und im Blut nachweisbar sind. 68 Mechanismus und Steuerung der Sekretion Die PTH-Sekretionsrate wird in Abhängigkeit von der Plasma-Calcium-Konzentration (ionisierte Fraktion) reziprok reguliert, das heißt ein Anstieg über den Normalwert hemmt die PTH-Sekretion (negative Rückkopplung). Bei einer Konzentration von 1 mmol/l ionisiertem Plasma-Calcium wird die maximale PTH-Sekretionsrate erreicht und bei 1,25 mmol/l eine minimale Sekretionsrate von 10 %. Ein Anstieg über 1,25 mmol/l führt also zu keiner weiteren Senkung der PTH-Sekretionsrate (basale Sekretionsaktivität). Für diesen Regulationsmechanismus ist der 1993 entdeckte Calciumsensitive Rezeptor verantwortlich. Dabei handelt es sich um einen G-Protein-gekoppelten Rezeptor, der bei Aktivierung durch den Liganden (hohe Calcium-Konzentration) den Inositolphosphatweg in Gang setzt, der zur Erhöhung der cytoplasmatischen Inositoltriphosphat- und DiacylglycerinKonzentration führt. Wahrscheinlich hemmt Inositoltriphosphat die Adenylylcyclase, so dass die cytoplasmatische cAMP-Konzentration abfällt und dadurch die Sekretion von Parathormon sinkt. Wirkungen Induktion der Osteolyse PTH führt indirekt zur Reifung und Aktivierung der Osteoklasten und damit zu einer Calcium-Phosphat-Mobilisierung aus dem Knochengewebe. Die Osteoklasten und die Stromazellen des Knochens besitzen PTH-Rezeptoren. Bei Bindung von Parathormon an den Rezeptoren wird u. a. der Osteoklasten-differenzierende Faktor (ODF, engl.: Osteoclast differentiating factor) in die Plasmamembran der Osteoblasten eingebaut. ODF interagiert mit RANK (Receptor aktivator of nuclear factor κB), einem Membranrezeptor, der von Osteoklasten exponiert wird und bei Aktivierung die Osteoklastogenese fördert. Eine negative Kalziumbilanz des Knochens tritt jedoch nur bei pathologisch erhöhten PTH-Konzentrationen auf. Hemmung der Phosphatresorption PTH hemmt in der Niere die Phosphat-Resorption (Wiederaufnahme) im proximalen Tubulus und erhöht die Calcium-Resorption (Wiederaufnahme) im distalen Tubulus. Phosphat wird in erster Linie über einen Natrium-Phosphat-Kotransporter (NPT2) aus dem Primärharn zurück in die Nierenepithelzellen des proximalen Tubulus transportiert. Von dort gelangt Phosphat zurück ins Blut. Wenn über die Nahrung ausreichend Phosphat aufgenommen wird, hemmt Parathormon die Phosphatrückaufnahme in der Niere. Die Hemmung findet dadurch statt, dass unter der Parathormonwirkung der NPT2-Kotransporter internalisiert und in Lysosomen abgebaut wird. Dadurch wird die Zahl aktiver Transportmoleküle auf der Zelle stark reduziert. Der Phosphatspiegel im Blut sinkt also, da mehr Phosphat mit dem Urin ausgeschieden wird. Dies ist durchaus sinnvoll, da dadurch im Blut wieder mehr freies ionisiertes Calcium vorliegen kann (das bei zu hoher Phosphatkonzentration mit diesem in der Niere einen schwerlöslichen Komplex bildet, der ausfällt und zur sogenannten Kalkniere führt). 69 Induktion der Biosynthese von Calcitriol Des Weiteren steigert PTH die Aktivität der 1α-Hydroxylase, dem Schlüsselenzym der Calcitriol-Biosynthese (Vitamin D3), die vor allem in der Niere lokalisiert ist, und mit deren Hilfe (Vitamin D3) PTH auch die enterale Calcium-Resorption im terminalen Ileum erhöht. Klinik Blutwert (Referenzbereich): Parathormon im Serum: 12-72 ng/l bzw. 1,5-6,0 pmol/l Erhöhte Blutwerte (Hyperparathyreoidismus): Entartete Epithelkörperchen (Adenome oder selten Karzinome) unterliegen nicht mehr der kalziumabhängigen Sekretionssteuerung. Es kommt zur unkontrollierten Erhöhung des Parathormonspiegels (primärer Hyperparathyreoidismus) und dadurch zur Hyperkalzämie. Auf eine Hypokalzämie infolge von Nieren- Leber- oder Darmerkrankungen reagiert der Körper mit einer verstärkten Sekretion von Parathormon (sekundärer Hyperparathyreoidismus) durch Hyperplasie der Epithelkörperchen. Wird die Ursache für einen sekundären Hyperparathyreoidismus plötzlich therapiert (z.B. Nierentransplantation) bleibt die Basalsekretion vom Parathormon aufgrund der reaktiven Epithelkörperchenhyperplasie erhöht (tertiärer Hyperparathyreoidismus). Folge ist eine Hyperkalzämie. Im Rahmen von malignen Erkrankungen (Bronchialkarzinom, Mammakarzinom, Prostatakarzinom, Plasmozytom) kann es zur paraneoplastischen Hyperkalzämie kommen. Die entarteten Zellen bilden ein dem Parathormon verwandtes Peptid (PTHrP), welches wie Parathormon wirkt (Pseudohyperparathyreoidismus). Erniedrigte Blutwerte (Hypoparathyreoidismus): Nach Schilddrüsenoperationen, Epithelkörperchenadenomentfernung oder autoimmun kann ein Parathormonmangel entstehen. Es kommt zum Absinken des Kalziumspiegels und in der Folge zur hypokalzämischen Tetanie. Die Konstellation Hypokalzämie, Hypomagnesiämie und Hyperphosphatämie bei normaler Nierenfunktion (Kreatininspiegel) sowie Ausschluss einer Malassimilation (Albuminspiegel) deutet auf eine Funktionsstörung der Epithelkörperchen hin. Ein erniedrigter Blutspiegel von Parathormon beweist die Diagnose Hypoparathyreoidismus. 70 Hyperparathyreoidismus Klassifikation nach ICD-10 E21.0Primärer Hyperparathyreoidismus E21.1Sekundärer Hyperparathyreoidismus E21.2Tertiärer, quartärer oder quintärer Hyperparathyreoidismus ICD-10 online (WHO-Version 2006) Hyperparathyreoidismus (HPT) ist eine Regulationsstörung der Epithelkörperchen (Nebenschilddrüsen). Der Hyperparathyreoidismus ist gekennzeichnet durch eine vermehrte Bildung von Nebenschilddrüsenhormon (Parathormon), welches den Calcium-Spiegel im Blut reguliert. Liegt der vermehrten Bildung von Parathormon eine gutartige Geschwulst (Adenom) der Nebenschilddrüse zugrunde, spricht man von einer primären Überfunktion der Nebenschilddrüsen (Primärer Hyperparathyreoidismus). Kennzeichen des primären Hyperparathyreoidismus sind ein erhöhter Parathormon-Spiegel und ein erhöhtes SerumCalcium. Ist die vermehrte Bildung von Parathormon die adäquate Reaktion der Nebenschilddrüsen auf ein vermindertes Serum-Calcium (z. B. bei Vitamin D - Mangel), spricht man von sekundärem Hyperparathyreoidismus. Charakteristisch für den sekundären Hyperparathyreoidismus ist ein erhöhter Parathormon-Spiegel bei niedrigem Serum-Calcium. Eine wichtige Ursache des sekundären Hyperparathyreoidismus ist die verminderte Aktivierung von Vitamin D aufgrund einer chronischen Nierenerkrankung. Ein über lange Zeit bestehender sekundärer Hyperparathyreoidismus kann aufgrund einer chronischen Überstimulierung der Nebenschilddrüsen zu einem inadäquaten Anstieg des Parathormons führen. Parathormon-Spiegel und Serum-Calcium sind erhöht, man spricht von tertiärem Hyperparathyreoidismus. Vom primären Hyperparathyreoidismus kann der tertiäre Hyperparathyreoidismus durch die Krankenvorgeschichte abgegrenzt werden. Folgen des Hyperparathyreoidismus sind Abbau von Knochensubstanz aufgrund einer vermehrten Calcium-Freisetzung aus dem Knochen, Nierensteine aufgrund einer vermehrten Calcium-Ausscheidung in den Urin, Verkalkungen der Blutgefäße durch Ablagerung von Calcium und Phosphat sowie eine Vielzahl weiterer, zum Teil unspezifischer Symptome. Da eine Bestimmung des Serum-Calciums häufig im Rahmen einer Routine-Blutuntersuchung erfolgt, wird die Diagnose meist in einem frühen Stadium gestellt, in dem noch keine oder nur unspezifische Symptome bestehen. Die Therapie des primären Hyperparathyreoidismus erfolgt durch operative Entfernung des Nebenschilddrüsen-Adenoms. Ist eine Operation nicht möglich oder wird diese nicht gewünscht, kann bei geringgradig erhöhtem Serum-Calcium der Krankeitsverlauf durch regelmäßige Kontrollen von Parathormon und Calcium beobachtet werden. Bei stark erhöhtem Calcium kann die Bildung von Parathormon durch das Medikament Cinacalcet gehemmt werden. 71 Der sekundäre Hyperparathyreoidismus wird behandelt mit Vitamin D, Cinacalcet und Phosphatbindern, letztere senken bei chronischer Nierenkrankheit erhöhte Phosphatspiegel. Der tertäre Hyperparathyreoidismus wird durch operative Entfernung der Nebenschildrüsen behandelt. Um eine ausreichende Bildung von Parathormon zu gewährleisten, wird entweder ein Teil eines Epithelkörperches belassen (subtotale Parathyreoidektomie) oder ein Teil eines Epithelkörperchens wird an anderer Stelle in einen Muskel eingepflanzt (autologe Retransplantation). Inhaltsverzeichnis 1 Die Nebenschilddrüsen (Epithelkörperchen) 2 Parathormon und Calcium-Haushalt 3 Primärer Hyperparathyreoidismus o 3.1 Pathogenese o 3.2 Ätiologie 3.2.1 Ionisierende Strahlung 3.2.2 Genetische Veränderungen o 3.3 Klinik 4 Sekundärer Hyperparathyreoidismus 5 Tertiärer Hyperparathyreoidismus 6 Quartärer und quintärer Hyperparathyreoidismus 7 Symptome des Hyperparathyreoidismus o 7.1 Asymptomatischer Hyperparathyreoidismus o 7.2 Symptomatischer Hyperparathyreoidismus 8 Epidemiologie 9 Histologie o 9.1 Nebenschilddrüse o 9.2 Knochen 10 Diagnose o 10.1 Diagnose des primären Hyperparathyreoidismus o 10.2 Differentialdiagnose des primären Hyperparathyreoidismus o 10.3 Diagnose des sekundären Hyperparathyreoidismus o 10.4 Differentialdiagnose des sekundären Hyperparathyreoidismus o 10.5 Klinische Chemie o 10.6 Osteodensitometrie o 10.7 Lokalisationsdiagnostik 11 Therapie des Hyperparathyreoidismus o 11.1 Therapie des primären Hyperparathyreoidismus 11.1.1 Chirurgische Therapie 11.1.2 Nicht chirurgische Therapie o 11.2 Therapie des sekundären Hyperparathyreoidismus 11.2.1 Sekundärer Hyperparathyreoidismus bei chronischem Nierenversagen o 11.3 Therapie des tertiären Hyperparathyreoidismus 12 Siehe auch 13 Quellen o 13.1 Weblinks o 13.2 Leitlinien 72 o 13.3 Einzelnachweise Die Nebenschilddrüsen (Epithelkörperchen) Die Epithelkörperchen sind in etwa linsengroße Organe. Sie haben einen Durchmesser von 5 bis 8 mm und ein Gewicht von etwa 20 bis 50 mg. Sie liegen in der Regel jeweils hinten am oberen und unteren Pol der Schilddrüse. Selten können aber auch Nebenschilddrüsen im Bereich des Thorax gefunden werden. Der Grund findet sich in der embryologischen Entwicklung der Nebenschilddrüsen. Die meisten Menschen haben vier Epithelkörperchen. Sie bilden das Parathormon, ein Hormon, welches den Calciumspiegel im Körper reguliert. Parathormon und Calcium-Haushalt Parathormon und Calcitriol sind die beiden hauptsächlichen Hormone, welche den CalciumPhosphat-Haushalt regulieren. Parathormon hält den Serum-Spiegel von Calcium in einem engen Bereich, kurzfristig durch vermehrte Rückresorption von Calcium im Tubulussystem der Niere und Förderung der Freisetzung von Calcium durch Abbau von Knochensubstanz. Längerfristig fördert Parathormon die Umwandlung von Calcidiol in Calcitriol und stimuliert so die Calcium-Aufnahme im Darm. Die Sekretion von Parathormon wird durch den calciumsensitiven Rezeptor reguliert, welcher an der Oberfläche der Nebenschilddrüsen-Zellen exprimiert wird. Eine Zunahme der Konzentration an ionisiertem Calcium führt über eine Aktivierung des calciumsensitiven Rezeptors zu einer Hemmung der Parathormon-Sekretion, ein Abfall des ionisierten Calciums führt zu einer gesteigerten Sekretion von Parathormon. Primärer Hyperparathyreoidismus Pathogenese Bei Patienten mit primärem Hyperparathyreoidismus ist die Parathormon-Sekretion in Relation zur Calcium-Konzentration im Serum inadäquat hoch, ausgelöst durch ein Adenom oder eine Hyperplasie einer oder mehrerer Nebenschilddrüsen (Epithelkörperchen). Ursache der vermehrten Parathormon-Sekretion ist eine verminderte Sensitivität (Empfindlichkeit) des calciumsensitiven Rezeptors aufgrund einer verminderten Anzahl von calciumsensitiven Rezeptoren pro Nebenschilddrüsen-Zelle und/oder eine vermehrte Masse an Nebenschilddrüsen-Gewebe durch eine vermehrte Anzahl an Nebenschilddrüsen-Zellen. Die Regulation der Parathormon-Sekretion ist dabei nicht vollständig aufgehoben, es ist aber eine höhere Calcium-Konzentration erforderlich, um die Parathormon-Sekretion zu hemmen - der Soll-Wert der Calcium-Konzentration ist nach rechts verschoben.[1][2] Ätiologie Nur in seltenen Fällen kann eine Ursache des primären Hyperparathyreoidismus, ionisierende Strahlung oder genetische Veränderungen gefunden werden. 73 Ionisierende Strahlung Eine erhöhte Inzidenz von primärem Hyperparathyreoidismus fand sich nach Bestrahlung der Halsregion[3], das Risiko, einen primären Hyperparathyreoidismus zu entwickeln steigt mit der Strahlendosis.[4] Auch bei Überlebenden des Atombombenabwurfs auf Hiroshima fand sich eine erhöhte Inzidenz an primärem Hyperparathyreoidismus.[5] Bei Patienten mit Hyperparathyreoidismus,der durch Strahlung ausgelöst wurde, ist die Inzidenz von Schilddrüsenkrebs erhöht, ansonsten unterscheidet sich der Verlauf aber nicht von Patienten mit idiopathischem Hyperparathyreoidismus (d.h. Hyperparathyreoidismus ohne nachweisbare Ursache).[6] Die Daten zum Auftreten eines primären Hyperparathyreoidismus nach Radiojodtherapie sind widersprüchlich, eine Studie fand ein gehäuftes Auftreten [7], in einer weiteren Studie konnte ein Zusammenhang zwischen Radiojodtherapie und Hyperparathyreoidismus dagegen nicht bestätigt werden.[8] Genetische Veränderungen Bei primärem Hyperparathyreoidismus findet sich in der Regel eine monoklonale Vermehrung atypischer Nebenschilddrüsenzellen, d.h. die veränderten Zellen gehen auf eine einzige veränderte Mutterzelle zurück. Im veränderten Nebenschildrüsengewebe wurden Mutationen in einer ganzen Reihe von Genen nachgewiesen: In Genen, welche das Zellwachstum kontrollieren, in Protoonkogenen sowie in Tumorsuppressorgenen. Im einzelnen wurden bisher Veränderungen in folgenden Genen beschrieben: CYCLIN D1 hat multiple Funktionen beim Zellwachstum, bei der Zelldifferenzierung und im Zellstoffwechsel. Eine erhöhte Aktivität von Cyclin D1 spielt eine wichtige Rolle bei der Entstehung von Tumoren einschließlich von Adenomen der Nebenschilddrüse. Multiple endokrine Neoplasie Typ I; MEN1: Inaktivierende Mutationen in MEN1, einem Tumorsuppressorgen führen zu autosomal-dominant vererbten Neoplasien von Nebenschilddrüsen, der Hypophyse und der Inselzellen im Pankreas sowie zu vermehrtem Auftreten von Magengeschwüren. HRPT2 kodiert für das Protein Parafibromin, das im Komples mit weiteren Proteinen (PAF1 und CTR9) an RNA-Polymerase II und an Histon-Methyltransferase bindet. Mutationen in HRPT2 führen zu Adenomen der Nebenschilddrüsen in Verbindung mit Kiefer-Tumoren, sowie zu Nebenschilddrüsenkarzinom. RET kodiert für eine Rezeptor-Tyrosinkinase. Mutationen führen unter anderem zur multiplen endokrinen Neoplasie Typ IIA. Die betroffenen Patienten entwickeln CZell-Karzinome der Schilddrüse, Phäochromozytome und in 15 bis 20 % der Patienten einen primären Hyperparathyreoidismus. Klinik Der erhöhte Parathormonspiegel führt zu einem gesteigerten Knochenabbau und damit zu einer erhöhten Calcium-Konzentration im Blut. Der Knochen wird demineralisiert. Knochenschmerzen können auftreten. In der Niere sorgt das Parathormon für eine verminderte Calciumausscheidung mit dem Urin, so dass die Calciummenge im Blut zusätzlich ansteigt. Durch die Rückresorption von Calcium aus dem Urin, kann das Löslichkeitsprodukt überschritten werden. In der Folge entstehen Nierensteine. Außerdem können Gallensteine und Entzündungen der Bauchspeicheldrüse auftreten. Die Beschwerden 74 des Hyperparathyreoidismus werden gerne mit den drei Worten "Stein-, Bein- und Magenpein" umschrieben. Sekundärer Hyperparathyreoidismus Ursache hier ist eine verstärkte Hormonproduktion als Reaktion auf einen erhöhten Calciumverlust (Hypokalzämie) des Körpers. Durch die kompensatorisch vermehrte Hormonproduktion kann die Calcium-Konzentration im Blut im unteren Normalbereich liegen. Zugrunde liegende Erkrankungen des sekundären Hyperparathyreoidismus: Chronische Niereninsuffizienz (typisch: Calciumspiegel niedrig, Phosphatspiegel zu hoch) Malassimilationssyndrom: Störung der Calcium-Aufnahme im Darm. Leberzirrhose: Gestörte Umwandlung von Vitamin D3 (Cholecalciferol) in 25Hydroxycholecalciferol in der Leber. Durch das fehlende aktive Vitamin D3 wird im Darm weniger Calcium aufgenommen und in der Niere weniger Calcium reabsorbiert. Cholestase: Durch den Mangel an Gallensäuren ist die Resorption von Vitamin D3 aus der Nahrung gestört. Fehlende Sonnenlichtexposition: In der Haut kann Vitamin D3 nicht mehr aus Cholesterin (bzw. 7-Dehydro-Cholesterin) gebildet werden. Tertiärer Hyperparathyreoidismus Wenn über einen längeren Zeitraum ein sekundärer Hyperparathyreoidismus besteht, wird außer der Funktion der Nebenschilddrüsen auch deren Wachstum stimuliert, so dass es schließlich zu einer autonomen Überproduktion in den Epithelkörperchen kommen kann. Wie beim primären Hyperparathyreoidismus ist der Regelkreis an der Stelle der Nebenschilddrüsen aufgetrennt. Quartärer und quintärer Hyperparathyreoidismus Selten sind quartäre und quintäre Formen eines Hyperparathyreoidismus. Als quartärer Hyperparathyreoidismus wird ein sekundärer Hyperparathyreoidismus auf dem Boden einer Nierenschädigung bezeichnet, wenn die Nierenschädigung ihrerseits durch einen primären Hyperparathyreoidismus verursacht worden war. Pathophysiologisch gesehen ist der Regelkreis wie beim sekundären Hyperparathyreoidismus an der Niere aufgetrennt. Als quintär wird ein Hyperparathyreoidismus bezeichnet, wenn die Entkoppelung der Parathormonsekretion aus einem oder mehreren verbliebenen Epithelkörperchen auf einem langjährigen quartären Hyperparathyreoidismus beruht. Symptome des Hyperparathyreoidismus Bei Diagnosestellung eines primären Hyperparathyreoidismus können folgende Krankheitsmanifestationen bestehen: zufällig entdeckte Erhöhung des Calciumspiegels im Blut, ohne dass klinische Krankheitszeichen bestehen (asymptomatische Hyperkalziämie), 75 Erhöhung des Calciumspiegels im Blut mit klinischen Krankheitszeichen (symptomatische Hyperkalziämie), Folgekrankheiten des Hyperparathyreoidismus: verminderte Knochendichte (Osteopenie), Knochenschwund (Osteoporose), Nierensteine (Nephrolithiasis), selten typische Knochenveränderungen (Osteitis fibrosa cystica) oder hyperparathyreotischen Krise. In früheren Jahren standen bei Erstmanifestation meist die klassischen Symptome der fortgeschrittenen Erkrankung im Vordergrund: Stein-Bein-Magen-Pein durch Nierensteine, pathologische Frakturen und Magengeschwüre. Seit der Zunahme von Blutuntersuchungen und Knochendichtemessungen überwiegen symptomlose (asymptomatische) und symptomarme (oligosymptomatische) Verläufe.[9] Asymptomatischer Hyperparathyreoidismus Mindestens 80 % der Patienten mit primärem Hyperparathyreoidismus werden durch den Zufallsbefund eines erhöhten Serum-Calciums diagnostiziert. Diese Patienten haben meist eine milde (im Mittel 2,8 mmol/, Norm 2,1–2,6 mmol/l) und intermittierende Hyperkalziämie. [10] Gelegentlich sind die Calcium-Spiegel normal, und der primäre Hyperparathyreoidismus wird aufgrund einer verminderten Knochendichte festgestellt.[9] Bei genauerer Anamnese geben manche Patienten unspezifische Sypmtome an wie Appetitlosigkeit (Anorexie), milde depressive Verstimmung[11], milde Gedächtnis- oder neuromuskuläre Störungen. [12] Häufig besteht bei primärem Hyperparathyreoidismus ein Vitamin D - Mangel, der die Diagnose verschleiern kann. Nach Gabe von Vitamin D kann es dann zu einem Anstieg des Calciumspiegels kommen, der auf die Diagnose hinweist.[13] Bei Vitamin D Mangel und Hyperparathyreoidismus kann es aber auch zu ausgeprägteren Krankheitsbildern kommen mit größeren Nebenschilddrüsen-Adenomen, höheren Konzentrationen an Parathormon, erhöhtem Knochenumbau und vermehrten Knochenbrüchen.[14] Symptomatischer Hyperparathyreoidismus Durch den erhöhten Parathormonspiegel wird Calcium aus den Knochen abgebaut. Die Knochendichte nimmt ab, Calciumspiegel im Blut und Calciumausscheidung im Urin steigen an. Der erhöhte Calciumspiegel im Blut (Hyperkalzämie) führt zu unspezifischen Beschwerden Appetitlosigkeit (Anorexie), Übelkeit (Nausea), Verstopfung (Obstipation), gesteigerter Durst (Polydipsie) und vermehrte Harnproduktion (Polyurie). Am Knochen kommt es zu Osteoporose und pathologischen Frakturen, insbesondere im Bereich der Wirbelsäule.[15] Bei langjährigem sekundärem renalen Hyperparathyreoidismus können in den Knochen sog. braune Tumore auftreten, die aus knochen-abbauenden Zellen (Osteoklasten) und Bindegewebe bestehen. Die Bezeichnung brauner Tumor bezieht sich auf die braune Färbung, die von Einblutungen hervorgerufen wird.[16] In der Nieren führt die erhöhte Calciumausscheidung zum Ausfällen von Nierensteinen (Nephrolithiais), Weichteilverkalkungen (Nephrokalzinose), Nierenkoliken, chronischem 76 Nierenversagen und Störungen der Tubulusfunktion. Die Nierensteine bestehen vorwiegend aus Calciumoxalat, seltener Calciumphosphat. Bei etwa 15-20% der Patienten mit primärem Hyperparathyreoidismus liegen Nierensteine vor, dem gegenüber findet sich bei etwa 5% der Nierensteinträger ein primärer Hyperparathyreoidismus als Ursache des Steinleidens.[17] Häufig klagen Patienten über neuromuskuläre Beschwerden wie Schwäche, Abgeschlagenheit und Müdgkeit. [18] Zudem treten bei Patienten mit primärem Hyperparathyreoidismus häufiger als in der Normlabevölkerung psychiatrische Erkrankungen auf wie Teilnahmslosigkeit (Lethargie), Depressionen, Verlust des Realitätsbezuges (Psychosen), verminderte soziale Interaktionsfähigkeit und Einschränkung der kognitiven Fähigkeiten (Demenz). Exakte Zahlen hierzu liegen aber nicht vor.[19] Es besteht eine erhöhte Prävalenz an Bluthochdruck[20], Übergewicht [21] und gestörter Glukosetoleranz [22] sowie an rheumatologischen Erkrankungen wie erhöhter Harnsäure (Hyperurikämie), Gicht und Pseudogicht[23]. Es gibt Hinweise darauf, dass die Prävalenz von Krebserkrankungen bei primärem Hyperparathyreoidismus erhöht ist, und das erhöhte Krebsrisiko auch nach operativer Entfernung des Nebenschilddrüsenadenoms fortbesteht. Aufgrund dieser Befunde wird über gemeinsame prädisponierende Faktoren für Hyperparathreoidismus und Krebserkrankungen spekuliert.[24][25][26] Bei Patienten mit primärem Hyperparathyreoidismus besteht eine hohe Inzidenz an kardiovaskuläre Erkrankungen, insbsondere Linksherzhypertrophie sowie Verkalkungen von Herzmuskel (Myokard), Aorten- und Mitralklappe. Das Risiko, an einer Herzerkrankung zu versterben, ist möglicherweise erhöht.[27][28] Bereits bei mildem primärem Hyperparathyreoidismus kommt es zu einer Versteifung der Blutgefäße.[29][30] Insbesondere beim sekundärem renalem Hyperparathyreoidismus kann es zu schweren Verkalkungen der Gefäße kommen, darunter auch der Herzkranzgefäße. Die Folgen sind eine erheblich erhöhte Morbidität und Mortalität an kardiovaskuläre Erkrankungen.[31][32] Die enge Verzahnung zwischen Knochen- und Herzkreislauferkrankung beim sekundären Hyperparathyreoidismushat hat dazu geführt, dass in den neuesten Leitlinien der Begriff Renale Osteopathie durch den Begriff Renale Mineral- und Knochenerkrankung (Chronic kidney disease-mineral and bone disorder) ersetzt wurde, welcher sowohl Knochenstoffwechselstörung als auch Gefäßverkalkungen und deren Folgen beihaltet.[33] Eine besonders schwere Komplikation des sekundären Hyperparathyreoidismus ist die (Calciphylaxie), bei der Calciumablagerungen zu Thrombosen in den Arteriolen der Haut führen. Diese können schwere Durchblutungsstörungen mit ausgedehnten Haut-Nekrosen hervorrufen, die häufig zum Tode führen.[34] Außerdem wird die Gastrinproduktion angeregt, so dass es zu einer Magenschleimhautentzündung (Gastritis) und Zwölffingerdarmgeschwür (Ulcus duodeni) kommen kann. [35] 77 Epidemiologie Die Häufigkeit des primären Hyperparathyreoidismus hat in den letzten Jahren abgenommen. In einem amerikanischen Register betrug die Inzidenz im Zeitraum von 1993 bis 2001 21,6 Fälle pro 100,000 Personen-Jahre, im Zeitraum von 1983 bis 1992 29,1 Fälle und von 1974 bis 1982 82,5 Fälle. Die Gründe für diese Abnahme der Häufigkeit sind nicht bekannt, mögliche Ursachen sind eine verminderte Exposition gegenüber ionisierender Strahlung und eine verbesserte Versorgung mit Vitamin D. Der primäre Hyperparathyreoidismus kann in jedem Lebensalter auftreten, die meisten Erkrankungen manifestieren sich aber erst nach dem 45. Lebensjahr. Frauen sind etwa doppelt so oft betroffen wie Männer, möglicherweise weil der nach der Menopause eintretende Knochenabbau einen latenten Hyperparathyreoidismus demaskieren kann. [36] Histologie Nebenschilddrüse Bei primärem Hyperparathyreoidismus werden bei der feingeweblichen Untersuchung der Nebenschilddrüsen folgende Veränderungen beschrieben:[37] In 89 % der Fälle von primärem Hyperparathyreoidismus wird eine einzelne gutartige Geschwulst (Adenom) der Nebenschilddrüse gefunden, in etwa 5 % der Fälle finden sich zwei Adenome.[38] Die meisten Adenome werden von den Hauptzellen der Nebenschilddrüse gebildet und sind von einer Kapsel umgeben. Gelegentlich werden Adenome gefunden, die aus oxyphilen Zellen bestehen, diese Adenome sind in der Regel größer. Es gibt auch Parathormon-bildende Adenome im Thymus.[39] In 6% der Fälle findet sich eine glanduläre Hyperplasie, d.h. eine diffuse Vergrößerung aller vier Nebenschilddrüsenkörperchen durch Vermehrung der Hauptzellen. Sehr selten wird eine glanduläre Hyperplasie durch eine Vermehrung von Klarzellen hervorgerufen.[40] In ca. 2 % der Fälle findet sich ein Nebenschilddrüsenkarzinom, das gekennzeichnet ist durch Invasion von Kapsel und Gefäßen, Lymphknoten- und Fernmetastasen.[41] Knochen Bei der feingeweblichen Untersuchung des Knochens finden sich Veränderungen, die als Osteitis fibrosa beschrieben werden. Der erhöhte Parathormon-Spiegel aktiviert Osteoklasten. Die vermehrte Osteoklasten-Tätigkeit führt zu einem Abbau von Knochensubstanz. Die feingewebliche Untersuchung zeigt Mikrofrakturen und Einblutungen. Es bilden sich Hohlräume, die gefüllt sind mit Bindegewebe, Osteoklasten und Hämosiderin-beladenen Makrophagen. Zunehmende Auflösung (Resorption) von Knochengewebe und bindegewebiger Umbau (Fibrose) führt zur Bildung von Knochenzysten, die mit bloßem Auge sichtbar sind. Bei weiterem Fortschreiten der Erkrankung verschmelzen die Knochenzysten zu braunen Tumoren, die braune Färbung ist Folge von Einblutungen und Hämosiderinablagerungen (Fallbeispiele und Abb. unter [42][43]) 78 Diagnose Diagnose des primären Hyperparathyreoidismus Erster Hinweis auf das Vorliegen eines primären Hyperparathyreoidismus ist ein erhöhter Calcium-Spiegel (Hyperkalzämie). Andere Ursachen einer Hyperkalzämie können durch Bestimmung der Parathormon-Konzentration abgegrenzt werden. [10] Die Diagnose eines primären Hyperparathyreoidismus wird gestellt, wenn das Parathormon erhöht ist, oder wenn das Parathormon zwar im Normalbereich von 10 bis 60 pg/ml liegt, aber relativ erhöht ist, bezogen auf einen gleichzeitig erhöhten Calcium-Spiegel. [44] Etwa 80-90% der Patienten mit primärem Hyperparathyreoidismus haben ein erhöhtes Parathormon, bei 10-20% der Patienten liegt das Parathormon im Normalbereich.[45] Differentialdiagnose des primären Hyperparathyreoidismus Häufigste Ursache einer Hyperkalzämie sind neben dem Hyperparathyreoidismus bösartige Krebserkrankungen. Meist ist bei Auftreten einer Hyperkalzämie die Krebserkrankung bekannt und weit fortgeschritten, zudem sind die Calciumspiegel bei Krebserkrankungen höher und die damit verbundenen Symptome schwerer. Das Parathormon ist bei Hyperkalzämie aufgrund einer Krebserkrankung meist sehr niedrig. Andere Ursachen einer Hyperkalzämie wie Milch-Alkali-Syndrom, Sarkoidose und Vitamin D-Überdosierung können ebenfalls durch ein erniedrigtes Parathormon vom Hyperparathyreoidismus abgegrenzt werden. Bei der familiären hypokalzurischen Hyperkalzämie (FHH) ist die Calcium-Ausscheidung im Urin vermindert, bei Patienten mit primärem Hyperparathyreoidismus ist die CalciumAusscheidung dagegen normal oder erhöht.[46] Thiaziddiuretika vermindern die Calciumausscheidung über die Nieren und führen zu einer milden Hyperkalzämie. Bei Patienten mit mildem Hyperparathyreoidismus können Thiaziddiuretika die Grunderkrankung demaskieren, in dem sie zu einem deutlichen CalciumAnstieg im Serum führen. Fällt ein erhöhter Calcium-Spiegel nach Absetzen von Thiaziddiuretika nicht ab, spricht dies für das Vorliegen eines primären Hyperparathyreoidismus. Lithium verstärkt die Parathormon-Sekretion und vermindert die Calciumausscheidung über die Nieren, es kommt zu Hyperkalzämie und Hypokalzurie, einige Patienten weisen erhöhte Parathormon-Spiegel auf. Nach Absetzen des Lithium kann sich die Hyperkalzämie normalisieren, nach längerer (über 10 Jahre) dauernder Behandlung mit Lithium ist eine Normalisierung der Calcium-Spiegel nach Beendigung der Therapie weniger wahrscheinlich. Diagnose des sekundären Hyperparathyreoidismus Der sekundäre Hyperparathyreoidismus ist charakterisiert durch einen erniedrigten CalciumSpiegels mit adäquatem Anstieg des Parathormons. Häufigste Ursache des sekundären Hyperparathyreoidismus ist eine verminderte Produktion von aktiviertem Vitamin D (Calcitriol) aufgrund einer chronischen Nierenfunktionseinschränkung. 79 Weitere Ursachen eines sekundären Hyperparathyreoidismus sind: verminderte Calcium-Zufuhr mit der Nahrung verminderte Calcium-Aufnahme über den Darm (Calcium-Malabsorption) erhöhtes Phosphat (Hyperphosphatämie) Vitamin D - Mangel Adipositaschirurgie Zöliakie Exokrine Pankreasinsuffizienz Calciumverluste über die Nieren Idiopathische Hyperkalzurie Schleifendiuretika Verminderter Knochenumbau, z. B. aufgrund von Bisphosphonaten Hungry bone syndrome - Syndrom des hungrigen Knochens Differentialdiagnose des sekundären Hyperparathyreoidismus Einige Patienten mit primärem Hyperparathyreoidismus haben normales Serum-Calcium (Normocalzämischer primärer Hyperparathyreoidismus). In diesen Fällen ist die Abgrenzung zwischen primärem und sekundärem Hyperparathyreoidismus schwierig. Häufig ist eine verminderte Knochendichte erste Manifestation der Erkrankung. Ursachen des normocalzämischen primären Hyperparathyreoidismus sind: Frühstadium oder unvollständige Ausprägung ("forme fruste") der Erkrankung; im Verlauf von 3 Jahren entwickeln etwa 40% der Betroffenen weitere KrankheitsManifestationen (in der Hälfte der Fälle Hyperkalzämie, ansonsten Nierensteine, Knochenbrüche, Hyperkalzurie, Osteoporose). [47] Verlaufskontrollen sind daher angezeigt. Primärer Hyperparathyreoidismus bei gleichzeitigem Vitamin D-Mangel Klinische Chemie Die Diagnose wird durch Laboruntersuchungen gestellt. Ein erhöhtes oder im hohen Normalbereich liegendes Parathormon weist bei gleichzeitig erhöhtem Serum-Calcium auf einen primären Hyperparathyreoidismus hin. Die Calcium-Ausscheidung im 24 Stunden Sammelurin ist bei ca. 40% der Patienten mit primärem Hyperparathyreoidismus erhöht, bei ca. 60% normal. Bei einer CalciumAusscheidung über 400 mg/24 h steigt das Risiko von Nierensteinen. Eine verminderte Calcium-Ausscheidung findet sich bei Familiärer hypokalzurischer Hyperkalzämie oder bei primärem Hyperparathyreoidismus bei gleichzeitig bestehendem mit Vitamin D - Mangel. Durch einen verminderten Blutspiegel an 25(OH)Vitamin D3 kann ein primärer Hyperparathyreoidismus bei gleichzeitig bestehendem mit Vitamin D - Mangel von einer Familiären hypokalzurischen Hyperkalzämie abgegrenzt werden. 80 Bei primärem Hyperparathyreoidismus liegen die Serumspiegel von Calcitriol im hochnormalem Bereich oder sind erhöht, bei sekundärem Hyperparathreoidismus aufgrund eines chronischen Nierenversagens sind die Calcitriol-Spiegel vermindert. Bei primärem Hyperparathyreoidismus ist die Phosphat-Ausscheidung über die Nieren erhöht, der Phosphatspiegel im Serum ist vermindert (Hypophosphatämie) oder liegt im niedrig normalen Bereich. Bei sekundärem renalem Hyperparathyreoidismus ist der Phosphatspíegel typischerweise erhöht. Ein erhöhtes Serum-Kreatinin weist auf eine Nierenfunktionseinschränkung. Diese kann Folge der Hyperkalzämie bei primärem Hyperparathyreoidismus sein aber auch Ursache eines sekundären Hyperparathyreoidismus. Biochemisch Marker des Knochenumsatzes (z. B. die Alkalische Phosphatase) liegen bei primärem und sekundärem Hyperparathyreoidismus im hochnormalen oder erhöhten Bereich. Osteodensitometrie Bei Patienten mit primärem Hyperparathyreoidismus ist der Mineralsalzgehalt des Knochens vermindert. Die Knochendichtemessung (Osteodensitometrie) ist nicht zur Diagnosestellung erforderlich, sondern zur Beurteilung der Schwere der Erkrankung, zur Verlaufsbeobachtung und zur Entscheidung, ob eine konservative oder eine operative Therapie angezeigt ist. Bei Patienten mit sekundärem renalem Hyperparathyreoidismus besteht keine enge Beziehung zwischen Knochenbrüchen und Knochendichte. Eine routinemäßige Knochendichtemessung ist bei renalem Hyperparathyreoidismus nach dem derzeitigen Kenntnisstand nicht sinnvoll.[48][49] Lokalisationsdiagnostik Vor einer geplanten Operation können Lokalisation und Größe der betroffenen Nebenschilddrüsenkörperchen durch Sonografie, Szintigrafie mit Technetium-99m-Sestamibi, Computertomografie oder Kernspintomografie dargestellt werden. Therapie des Hyperparathyreoidismus Therapie des primären Hyperparathyreoidismus Chirurgische Therapie Die chirurgische Entfernung des (oder aller) vergrößerten Epithelkörperchens, führt im Vergleich zur Verlaufsbeobachtung zu einer Verbesserung von Knochendichte und Lebensqualität [50] sowie zu einer Verminderung von Knochenbrüchen, insbesondere von Schenkelhalsfrakturen. [51] In der Mehrzahl der asymptomatischen Patienten finden sich aber auch bei langer Verlaufsbeobachtung keine Hinweise auf ein Fortschreiten der Erkrankung wie Abnahme von Knochendichte, Anstieg von Calcium und Parathormon oder Auftreten von Nierensteinen. Aus diesem Grund wurden Leitlinien entwickelt, deren Ziel ist, diejenigen Patienten einer chirurgischen Therapie zuzuführen, bei denen das Risiko für Organschäden oder ein Fortschreiten der Erkrankung am höchsten ist. 81 Kriterien des 2002 NIH Workshop on Asymptomatic Primary Hyperparathyroidism[52] zur chirurgischen Therapie: Serum Calcium 0,25 mmol/l (1,0 mg/dl) über der oberen Normgrenze. Calcium Ausscheidung im Urin über 10 mmol/Tag (400 mg/Tag) bei normaler Kost. Kreatinin-Clearance 30% des Altersdurchschnitts oder weniger. Knochendichte an Hüfte, Lendenwirbelsäule oder Radius mehr als 2,5 Standardabweichungen unter dem Mittel (T-Wert < -2,5). Alter unter 50 Jahren. Regelmäßige Verlaufskontrollen nicht gewährleistet. Die Operation sollte nur von erfahrenen Chirurgen durchgeführt werden. Nicht chirurgische Therapie Obwohl die chirurgische Therapie zu einer definitiven Heilung des primären Hyperparathyreoidismus führt, werden nicht alle Patienten operiert, insbesondere bei Fehlen von Symptomen, nur geringer Erhöhung des Calciums, bei einem Lebensalter über 50 Jahren, wenn schwere Begleiterkrankungen bestehen oder wenn eine Operation nicht gewünscht wird. In diesen Fällen stehen folgende nicht operative Verfahren zur Verfügung: Bisphosphonate hemmen den Knochenabbau durch Osteoklasten. Alendronat führt bei asymptomatischem primären Hyperparathyreoidismus zu einer Zunahme der Knochenmasse. Risedronat senkt erhöhte Calcium-Spiegel. Raloxifen ist ein selektver Östrogenrezeptor-Modulator. Bei postmenopausalen Frauen mit primärem Hyperparathyreoidismus senkt Raloxifen das Serum-Calcium. Nach sorgfältiger Nutzen-Risiko-Abwägung kann alternativ eine Östrogen-Ersatztherapie in Erwägung gezogen werden. Seit 2004 sind Calcimimetika (z. B. Cinacalcet) verfügbar, die am calciumsensitiven Rezeptor der Nebenschilddrüsenzelle angreifen und somit direkt die Parathormonausscheidung hemmen. Perkutane Alkoholablation: Durch Injektion von Alkohol in das vergrößerte Nebenschilddrüsenkörperchen kann dieses zerstört werden. Nebenwirkungen sind eine Schädigung des Nervus laryngeus recurrens und ein zu starkes Absinken des SerumCalciums (Hypokalzämie) Darüber hinaus werden eine calciumarme Ernährung mit ausreichendem Gehalt an Vitamin D sowie regelmäßige körperliche Betätigung empfohlen[53] Bei Patienten, bei denen keine Operation durchgeführt wird, werden Calcium halbjährlich, Kreatinin und Knochendichte jährlich kontrolliert. Bei Hinweisen auf ein Fortschreiten der Erkrankung wird operiert. Therapie des sekundären Hyperparathyreoidismus [Bearbeiten] Der sekundäre Hyperparathyreoidismus wird im Allgemeinen durch Beseitigung der Grunderkrankung behandelt. Für den sekundäre Hyperparathyreoidismus infolge eines chronischen Nierenversagens existiert eine abgestufte, durch Leitlinien festgelegte medikamentöse Therapie. Sekundärer Hyperparathyreoidismus bei chronischem Nierenversagen [Bearbeiten] 82 Der sekundäre Hyperparathyreoidismus bei chronischem Nierenversagen wird mit phosphatarmer Diät, Vitamin-D-Metaboliten, Phosphatbindern und Cinalcalcet behandelt.[54][55] Liegt der Parathormonspiegel im Stadium 2-3 des chronischen Nierenversagens über 70 pg/ml, (bzw. > 110 pg/ml im Stadium 4 oder > 300 pg/ml im Stadium 5) wird mit einer phosphatreduzierten Ernährung begonnen. Gelingt es nicht, durch eine phosphatarme Diät den Phosphatspiegel ausreichend zu senken, werden Medikamente eingesetzt, welche das mit der Nahrung zugeführte Phosphat im Darm binden (orale Phosphatbinder): Calciumcarbonat, Calciumacetat, Aluminiumhydroxid, Sevelamer oder Lanthancarbonat. Calciumhaltige Phosphatbinder werden wegen der Gefahr der Weichteilverkalkung nicht bei erhöhten Calcium-Werten eingesetzt, die Menge ist begrenzt auf 2,5g Calcium pro Tag. Aluminiumhaltige Phosphatbinder werden wegen der Gefahr der Aluminiumablagerung in Knochen und Gehirn nur bei sehr hohen Phosphatspiegeln und über einen begrenzten Zeitraum angewandt. Ein verminderter Vitamin D - Spiegel im Serum, (25(OH)Vitamin-D3 < 30 ng/ml) wird durch Gabe von Vitamin D ausgeglichen. Sinkt der Parathormonspiegel trotz dieser Maßnahmen nicht in den Zielbereich, wird zusätzlich aktives Vitamin D (Alfacalcidol oder Calcitriol) verabreicht. Nebenwirkung der Therapie mit aktivem Vitamin D ist ein Anstieg des Calciumund Phosphatspiegels mit der Gefahr von Weichteilverkalkungen. Diese Nebenwirkung ist geringer bei Verwendung von pharmakologisch veränderten Vitamin D Metaboliten wie Paricalcitol. Führen phosphatarme Diät, Phosphatbinder und Vitamin D - Metaboliten nicht zum Erfolg, wird Cinacalcet, ein Antagonist des calciumsensitiven Rezeptors eingesetzt. Ohne Therapie ist ein Übergang in einen tertiären Hyperparathyreoidismus wahrscheinlich. Therapie des tertiären Hyperparathyreoidismus [Bearbeiten] Steigt der Parathormonspiegel trotz medikamentöser Therapie auf Werte über 1000 pg/ml und kommt es zu Knochenveränderungen und therapierefraktärer Hyperkalzämie und/oder Hyperphosphatämie müssen alle 4 (bis 8) Epithelkörperchen entfernt (Parathyroidektomie) und anschließend ein Teil eines Epithelkörperchens in Unterarm oder Musculus sternocleidomastoideus autotransplantiert werden, da sonst gar keine Parathormonbildung mehr stattfinden könnte. Alternativ kann auch eine subtotale Parathyroidektomie erfolgen, d.h. ein Nebenschilddrüsenkörperchen wird nicht vollständig entfernt.[56] 83