Das Retinoblastom – Diagnose und Therapie
Werbung

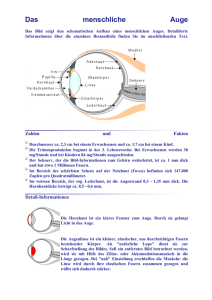
Das Retinoblastom – Diagnose und Therapie 1. Was ist ein Retinoblastom? ........................................................ 2 2. Aufbau und Funktion des Auges ................................................. 2 3. Klinik des Retinoblastoms ........................................................... 3 4. Wie erkennt man ein Retinoblastom (Symptome und Diagnose) ......................................................... 5 5. Stadieneinteilung des Retinoblastoms........................................ 8 6. Ablauf der Erstuntersuchung an der Universitäts-Augenklinik Essen .................................................. 8 7. Die Behandlung des Retinoblastoms .......................................... 9 8. Therapiekontrolle und Prognose ............................................... 13 9. Kontaktadressen ....................................................................... 15 © Zentrum für Augenheilkunde - Universitätsklinikum Essen Hufelandstraße 55 45122 Essen Tel.: 0201 723 2969 E-Mail: [email protected] Seite 1 1. Was ist ein Retinoblastom? Das Retinoblastom ist der häufigste im Auge auftretende Tumor im Kindesalter mit einer Häufigkeit von einem betroffenen Kind auf etwa 18.000 Lebendgeburten. In absoluten Zahlen tritt der Tumor nur selten auf (in Deutschland ca. 60 Fälle/Jahr). Es handelt sich um einen bösartigen Tumor, der von genetisch veränderten, unreifen Netzhautzellen (Retinoblasten) ausgeht und unbehandelt zum Tod führt. Gleichzeitig ist es eine der wenigen heilbaren bösartigen Krebserkrankungen: Frühzeitig erkannt und therapiert überleben mehr als 95 % der Patienten. Wenn nur ein Auge betroffen ist, spricht man von einem unilateralen (einseitigen) Retinoblastom. Dies betrifft etwa zwei Drittel der erkrankten Kinder. Sind beide Augen erkrankt, handelt es sich um ein bilaterales (beidseitiges) Retinoblastom. Im Folgenden möchten wir einen kurzen Überblick über die Symptome, Diagnostik und Behandlung des Retinoblastoms geben sowie auf die Erblichkeit dieser Erkrankung eingehen. Zum besseren Verständnis wird zunächst der Aufbau des Auges erklärt. 2. Aufbau und Funktion des Auges Das Auge ist ein Sinnesorgan und leitet seine Informationen über den Sehnerv direkt zum Gehirn weiter. Abb. 1: Anatomie des Auges Die aus festem Bindegewebe bestehende schützende äußere Hülle des Auges besteht aus der weißen Lederhaut und geht vorne in die durchsichtige Hornhaut über, welche sozusagen das Fenster des Auges bildet. Die Netzhaut kleidet den Augapfel von innen aus und enthält die Sinneszellen, welche über ein komplexes Netz aus Nervenzellen Verbindung zu den Sehnervenfasern haben. Diese laufen in der Sehnervenscheibe zusammen und bilden den Sehnerv. Die Aderhaut bildet mit einem dichten Gefäßnetz die mittlere Schicht und ernährt die äußeren Netzhautschichten. Sie geht vorne in Seite 2 den Strahlenkörper und die Regenbogenhaut über mit einer zentralen Öffnung, der Pupille, die sich je nach Helligkeit verengt und weitet. Hinter der Iris ist die aus durchsichtigen elastischen Fasern bestehende Linse am Ziliarkörper aufgehängt. Die Regenbogenhaut trennt die zwischen Hornhaut und Linse gelegene, mit Kammerwasser gefüllte Augenkammer in eine Vorder- und eine Hinterkammer. Der Glaskörper füllt den Raum hinter der Linse aus, er besteht aus einem Fasergerüst und einer darin eingebetteten transparenten, gallertartigen Masse. Die Funktion des Auges kann am besten mit der einer Kamera verglichen werden. Als Objektiv dienen Hornhaut und Linse, dem Film entspricht die Netzhaut. Das durch Lichtreize entworfene Bild auf der Netzhaut wird über die Nervenfasern als elektrische Impulse dem Gehirn zugeleitet, wo dann der Seheindruck entsteht. 3. Klinik des Retinoblastoms Das Retinoblastom entsteht in der Netzhaut und kann sich in verschiedene Richtungen ausbreiten. In der Frühphase entstehen weißliche Tumoren zunächst am hinteren Augenpol, die unterhalb oder oberhalb der Netzhaut wachsen können (Abb. 2a). Bei einem Wachstum in Richtung des Glaskörpers können sich Tumorzellen von der Oberfläche lösen und im Glaskörperraum schweben. In diesem Fall spricht man von einer Glaskörperaussaat des Tumors (Abb. 2b). Siedeln sich die abgelösten Tumoranteile an einer anderen Stelle der Netzhaut an, so entstehen intraokulare Absiedlungen. Dadurch kann das Bild mehrerer Tumore (multifokal) vorgetäuscht werden, obwohl es sich um einen einzelnen (solitären) Tumor handelt. Infolge der Glaskörperaussaat kann auch der vordere Augenabschnitt mitbeteiligt sein. Sehnerv Abb. 2a: Typisches Retinoblastom am hinteren Augenpol unmittelbar neben dem Sehnervenkopf Seite 3 Abb 2b: Großes Retinoblastom mit diffusem Wachstum in den Glaskörperraum Bei einem Wachstum in Richtung Aderhaut tritt oft eine Netzhautablösung auf. Tumoranteile, die sich ablösen können führen zu einer Absiedlung unter die Netzhaut. Es besteht eine erhöhte Gefahr für einen Einbruch des Tumors in die Aderhaut. In sehr fortgeschrittenen Fällen kann auch die Lederhaut durchwachsen werden und es so zu einer Infiltration der Augenhöhle kommen. Die Ausbreitung des Tumors entlang des Sehnervs führt zu einer Mitbeteiligung der Gehirnstrukturen, der Hirnhaut und der Liquorräume (Rückenmarkflüssigkeit bzw. Hirnwasser). Durch einen Einbruch in das Gefäßsystem der Aderhaut kann es zu Metastasen in Knochenmark, Knochen, Lymphknoten und Leber kommen. Die regionalen Lymphknoten werden über einen Einbruch des Tumors in das vordere Augensegment sowie in die Augenhöhle erreicht. Grundsätzlich kann das Retinoblastom infolge eines kontinuierlichen Wachstums jede Struktur des Auges infiltrieren. Das Auftreten von Gefäßneubildungen in der Regenbogenhaut, Tumorzellen in der Vorderkammer, eine begleitende Entzündung der Augenhöhle oder ein erhöhter Augeninnendruck können dabei in sehr fortgeschrittenen Fällen auftreten. In einem Drittel der Fälle tritt das Retinoblastom in beiden Augen auf. Hierbei handelt es sich nicht um ein Tumorwachstum von dem einen in das andere Auge, sondern um neue Tumoren, die nicht zeitgleich auftreten müssen. Zunächst einseitige Erkrankungen können im weiteren Verlauf beidseitig werden. Es können sich auch in einem Auge mehrere, unabhängig voneinander entstandene Tumoren entwickeln. Seite 4 4. Wie erkennt man ein Retinoblastom? (Symptome und Diagnose) Der Tumor tritt praktisch immer vor dem 5. Lebensjahr auf, da das Wachstum des Retinoblastoms nur von unreifen Netzhautzellen ausgehen kann. Das durchschnittliche Alter bei der Diagnosestellung beträgt bei den einseitigen Retinoblastomen etwa 23 Monate und liegt bei den beidseitigen Retinoblastomen bei ca. 12 Monaten. Etwa 10 % der Retinoblastome sind bereits bei der Geburt nachweisbar, knapp die Hälfte im ersten Lebensjahr. In unserer Klinik werden 90 % der Fälle vor einem Alter von 3 Jahren diagnostiziert. Da der Tumor in einem äußerlich unveränderten Auge bei Säuglingen und Kleinkindern wächst, kann er über längere Zeit symptomlos bleiben. Die einseitige Beeinträchtigung des Sehvermögens der kleinen Kinder wird meist erst durch eine Schielstellung (in 25 %) bemerkt. Das häufigste Erstsymptom ist allerdings eine bei bestimmten Beleuchtungsverhältnissen weiß erscheinende Pupille, die wie ein Katzenauge wirkt und meist auf Fotos auffällt (Leukokorie) im Gegensatz zu einer rot aufleuchtenden oder schwarz erscheinenden Pupille (Abb. 3). Seltener fällt ein erhöhter Augeninnendruck, ein schmerzhaftes rotes Auge, eine Sehverschlechterung, Entzündung der Augenhöhle oder eine Pupillenveränderung auf. Abb. 3 Leukokorie des linken Auges bei fortgeschrittenem Retinoblastom Da in den meisten Fällen die ersten Symptome von den Eltern bemerkt werden, kommt dem zuerst aufgesuchten Kinder- oder Augenarzt eine Schlüsselfunktion zu; er muss an einen seltenen, das Sehvermögen und das Leben bedrohenden Tumor denken, dem er bisher möglicherweise noch nie begegnet ist. Deutliche Warnzeichen liegen vor • wenn eine oder beide Pupillen erweitert oder weißlich-gelb gefärbt sind (Leukokorie), • wenn ein Auge gerötet ist und schmerzt, • bei Schielen oder einer Sehstörung. Zur Diagnosestellung muss immer eine Augenhintergrunduntersuchung in Seite 5 Narkose bei maximal erweiterter Pupille durchgeführt werden sowie eine Ultraschalluntersuchung. Bei ungehindertem Einblick auf den Augenhintergrund kann der erfahrene Augenarzt die Diagnose eines Retinoblastoms anhand der charakteristischen Erscheinung meist ohne Zusatzdiagnostik stellen und von anderen, gutartigen Erkrankungen abgrenzen. Um krankhafte Veränderungen in der Augenhöhle und im Schädel zu erfassen, ist weiterhin ein bildgebendes Verfahren notwendig, wobei eine Kernspintomographie des Kopfes (MRT Schädel) mit einer speziell für die Untersuchung von Augen zur Verfügung stehenden Zusatzeinrichtung durchgeführt wird, um die Tumorausdehnung sicher festzustellen. Zusätzlich muss eine kinderärztliche Untersuchung durchgeführt werden, ggf. müssen weitere Zusatzuntersuchungen eingeleitet werden. In Einzelfällen kann eine Punktion des Knochenmarks und der Rückenmarksflüssigkeit notwendig werden. Retinom Mit dieser Bezeichnung ist ein spontan zurückgebildetes Retinoblastom gemeint, welches meist als Zufallsbefund in 1% bis 2 % bei der Untersuchung nicht erkrankter Verwandter von an Retinoblastom erkrankten Kindern zu finden ist. Es tritt nicht im Kindesalter auf und weist keine bösartige Entartung auf. Klinisch erscheint dieser Tumor wie ein Retinoblastom im Rückbildungsstadium nach Strahlentherapie. Eine Behandlung ist nicht erforderlich, jedoch sind regelmäßige, lebenslange Kontrollen wie bei einem behandelten Retinoblastom notwendig, da vitale (lebende) und potentiell wachstumsfähige Tumorzellen vorliegen. Genetisch verhält sich das Retinom wie ein echtes Retinoblastom mit allen sich daraus ergebenden Konsequenzen in der genetischen Beratung. Trilaterales Retinoblastom Man versteht darunter die sehr seltene Kombination eines erblichen Retinoblastoms mit einem Hirntumor im Bereich der Mittellinie. Es handelt sich hierbei nicht um Metastasen, sondern um einen selbständig wachsenden Tumor, der feingeweblich der Struktur des Retinoblastoms ähnelt. Die Manifestation erfolgt meist gleichzeitig mit dem Retinoblastom, die Prognose ist jedoch sehr viel schlechter. Vererbung/Genetik des Retinoblastoms Es muss zwischen einer erblichen und einer nicht erblichen Form des Retinoblastoms unterschieden werden. Die Mehrzahl der Fälle (90 %) entsteht isoliert neu (sporadisch), d. h. die Erkrankung ist bei keinem der Angehörigen nachzuweisen. In ca. 10 % sind bereits weitere Erkrankungen in der Familie bekannt (familiäres Retinoblastom). Zugrunde liegt eine Veränderung des genetischen Materials (Mutation) im Retinoblastom-Gen (RB1-Gen), welches sich auf dem Chromosom 13 befindet. Zur Tumorentstehung müssen beide Kopien des RB1-Gens inaktiviert werden. Die nicht-erbliche Form geht von zwei Veränderungen in einer einzigen Netzhautzelle aus (somatische Mutation) und kann deshalb nicht an eigene Kinder weitergegeben werden. Sie ist stets einseitig und es findet sich in dem betroffenen Auge nur ein einziger Tumor. Die erbliche Form tritt bei etwa 40% aller Patienten auf und weist bereits Seite 6 eine Veränderung des Retinoblastom-Gens in den Keimbahnzellen auf (germinale Mutation), so dass in allen Körperzellen die genetische Veränderung vorhanden ist. Aus diesem Grund können in den meisten Fällen auch Blutzellen zur Identifikation der für die Veranlagung zum Retinoblastom ursächlichen Mutation verwendet werden. Durch einen Verlust der zweiten Kopie des Retinoblastom-Gens in mindestens einer Netzhautzelle entsteht der Tumor. Das erbliche Retinoblastom betrifft deshalb meist beide Augen. Außerdem finden sich häufig mehrere Tumoren in einem Auge. Die Veranlagung zur Entwicklung eines Retinoblastoms wird als dominant erbliches Merkmal weitergeben. Hat ein Kind mehrere Tumoren oder sind nahe Angehörige ebenfalls erkrankt, kann so von einer erblichen Form ausgegangen werden. Beim familiären Retinoblastom sind meist alle Beteiligten beidseits erkrankt. Aufgrund des Erbgangs wird von dem betroffenen Elternteil die krankheitsverursachende Mutation mit einem Risiko von nahezu 50 % an die Nachkommen vererbt. Wegen einer unvollständigen Penetranz (nicht alle Genträger erkranken) ist das tatsächliche Erkrankungsrisiko jedoch geringer. In seltenen Fällen überwiegen in einer Familie einseitige Erkrankungen (verminderte Expressivität). Die Tumordisposition wird dann auch über nicht erkrankte Angehörige weitergegeben, so dass auch für die Kinder nicht erkrankter Angehöriger ein erhöhtes Risiko besteht. Bei nicht-familiär beidseitig erkrankten Patienten und einem Teil der isoliert einseitigen Erkrankungen beträgt das Risiko für die Entwicklung eines Retinoblastoms bei eigenen Kindern nahezu 50 %. Auch bei Geschwistern besteht ein erhöhtes Risiko von 2 %. Ist die krankheitsverursachende Mutation aus Blut bzw. Tumormaterial bekannt, so kann das Risiko genau bestimmt werden. Bei Kindern mit nur einseitig auftretendem Retinoblastom liegt in 90 % die nicht-erbliche Form des Retinoblastoms vor. In den verbliebenen 10 % der Fälle handelt es sich jedoch trotz einseitiger Erkrankung um ein erbliches Retinoblastom, so dass bei den Nachkommen von einem erhöhten Risiko ausgegangen werden muss (1–7 %). Für Geschwister von einseitig erkrankten Kindern wurde ein Wiederholungsrisiko von 1 % ermittelt. Durch Untersuchung von Tumormaterial können die ursächlichen Mutationen bei den meisten Betroffenen erkannt werden. Wenn diese nicht im Blut nachweisbar sind, kann ein Wiederholungsrisiko für Geschwister ausgeschlossen werden. Durch molekulargenetische Untersuchungen kann die Beurteilung des Erkrankungsrisikos für den Einzelnen entscheidend verbessert werden. Durch Mutationsanalysen kann häufig ein erhöhtes Risiko festgestellt oder mit hoher Wahrscheinlichkeit ausgeschlossen werden. So können die Vorsorge- und Kontrolluntersuchungen auf jene Kinder beschränkt bleiben, welche ein mutiertes RB1-Gen geerbt haben. Dadurch wird auch eine psychische Entlastung für die betroffenen Familien erreicht. Eine wesentliche Voraussetzung für den Erfolg der molekulargenetischen Risikobestimmung ist die Verfügbarkeit der erforderlichen Proben. Es sollte von dem erkrankten Kind, seinen Geschwistern und den Eltern Blut bereitgestellt werden sowie nach Möglichkeit Tumormaterial, insbesondere bei den nicht-familiären Erkrankungen. Bei neugeborenen Kindern aus betroffenen Familien ist es wichtig, Nabelschnurblut zur genetischen Untersuchung abzunehmen. Seite 7 5. Stadieneinteilung des Retinoblastoms Das Retinoblastom wird weltweit nach der sog. ABC-Klassifikation eingestuft. Im Stadium A finden sich umschriebene, kleine Tumoren die auf die Netzhaut beschränkt sind und mindestens 3 mm vom Netzhautzentrum und 1,5 mm vom Sehnerv entfernt sind. Im Stadium B finden sich alle anderen auf die Netzhaut beschränkten Tumoren. Im Studium C finden sich Tumoren mit einer begrenzten, die Netzhaut überschreitenden Aussaat und im Stadium D große, multiple Tumoren mit diffuser Aussaat und Abhebung der Netzhaut. Das Stadium E beschreibt große Tumoren mit Komplikationen wie Erhöhung des Augendrucks, Tumoraussaat in die vordere Augenkammer, Tumorwachstum außerhalb des Auges und beginnende Schrumpfung des Auges. In diesem Stadium ist in der Regel eine Therapie mit dem Ziel, das Auge zu erhalten, nicht mehr möglich. 6. Ablauf der Erstuntersuchung an der UniversitätsAugenklinik Essen Wird ein Kind mit dem Verdacht auf ein Retinoblastom unserer Abteilung zugewiesen, so erfolgt zunächst nach einem ersten Gespräch über die Vorgeschichte (Familie, Schwangerschaft, Geburt, Entwicklung, Erkrankungen) und der Aufklärung durch die Anästhesie am folgenden Tag eine Inspektion beider Augen in Narkose mit weitgetropften Pupillen, verbunden mit einer Ultraschalluntersuchung der Augen und einer Blutentnahme für die Humangenetik (Abb. 4). Wegen der Narkose muss das Kind im ersten Lebensjahr mindestens vier Stunden und nach dem 12. Lebensmonat mindestens sechs Stunden vor der Untersuchung nüchtern bleiben, d. h. es darf nichts essen und trinken. Bis zu zwei Stunden vor der Untersuchung darf das Kind klare Flüssigkeit (z. B. ungesüßter Tee) zu sich nehmen. Nach Absprache kann eine Unterbringung im Elternhaus der Essener Elterninitiative für krebskranke Kinder (s. Kontaktadressen) arrangiert werden. Abb. 4: Untersuchung eines Kinds mit Retinoblastom in Narkose Seite 8 Bestätigt sich durch die klinische Untersuchung die Diagnose eines Retinoblastoms, wird gemeinsam mit den Eltern die Therapie geplant und eingeleitet. Der Therapieplan wird innerhalb einer Tumorkonferenz unter Mitarbeit aller beteiligten Fachabteilungen (Augenklinik, Kinderklinik, Strahlenklinik, Röntgendiagnostik, Humangenetik) am Folgetag intensiv besprochen. Da es klinisch stumme Verlaufsformen der Erkrankung gibt (z. B. Retinome), werden sowohl die Eltern als auch die Geschwister augenärztlich untersucht und eine Blutprobe an die Humangenetik weitergeleitet, um die familiäre Form zu erkennen. Ferner wird im weiteren Verlauf ein Termin zur Beratung in der humangenetischen Sprechstunde vereinbart. Die genetische Beratung von Eltern, die ein erkranktes Kind haben oder selbst erkrankt sind, ist zwingend erforderlich, um eine verlässliche Berechnung des Erkrankungsrisikos der Nachkommen zu ermöglichen. 7. Die Behandlung des Retinoblastoms Grundsätzlich steht bei der Wahl der Therapieform der Erhalt des Lebens über dem Erhalt des Sehvermögens. Entscheidend ist die vollständige Zerstörung bzw. Entfernung des Tumors. In fortgeschrittenen Fällen kann es deshalb notwendig sein, das betroffene Auge zu entfernen, um das Überleben des Kindes zu sichern, insbesondere dann, wenn die Funktion des betroffenen Auges weitgehend verloren ist. In allen anderen Fällen muss sorgfältig geprüft werden, ob eine Zerstörung des Tumors unter Erhalt des betroffenen Auges und einer nutzbaren Funktion möglich ist, ohne die Überlebenschance des betroffenen Kindes zu verschlechtern oder ihm eine nicht angemessene therapiebedingte Belastung zuzumuten. Die Therapieplanung wird sowohl von der Ausdehnung des Tumorleidens und dem Befall eines oder beider Augen als auch vom Alter des Kindes bestimmt und erfordert eine enge Zusammenarbeit zwischen Augenärzten, Radiologen, Humangenetikern, Strahlentherapeuten, Kinderärzten und den betroffenen Eltern. Therapie des einseitigen Retinoblastoms Bei fortgeschrittenen Tumoren mit weitgehendem Verlust des Sehvermögens ist die Entfernung des Auges (Enukleation) die sicherste Behandlungsmethode, da so bei einem normal sehenden Partnerauge die Risiken anderer Behandlungsstrategien vermieden werden können. Durch die vollständige Entfernung des Tumors wird gleichzeitig bei den nicht-erblichen Retinoblastomen das betroffene Kind von seinem Tumorleiden geheilt. In ausgesuchten Fällen kann bei einer frühzeitigen Diagnose eine Behandlung mit dem Ziel, das betroffene Auge zu erhalten, versucht werden. Hier kommt insbesondere die lokale Strahlentherapie, ggfs. in Kombination mit anderen Behandlungsmethoden, in Frage. Da die einseitigen Retinoblastome meistens erst sehr spät erkannt werden und deshalb bereits einen weit fortgeschrittenen Befund aufweisen, sind die betroffenen Augen auch sehr häufig zum Zeitpunkt der Erstdiagnose erblindet. Die Entfernung des meist erblindeten Tumorauges bedeutet für das Kind deshalb keine veränderte Wahrnehmung oder schlechtere Orientierung. Seite 9 Therapie des beidseitigen Retinoblastoms Das Ziel jeder Therapie ist die vollständige Tumorkontrolle und Rückbildung unter Erhalt eines möglichst guten Sehvermögens an zumindest einem Auge durch eine individuelle Kombination der zur Verfügung stehenden Therapiemöglichkeiten. Falls möglich sollten die Tumoren möglichst lokal behandelt werden. Solitäre kleine Retinoblastome können sicher zerstört werden. Kommt eine solche Behandlung nicht mehr in Frage, weil bereits zu große Tumoren oder sogar eine Glaskörperaussaat vorliegt, kann in vielen Fällen durch eine Chemotherapie eine Verkleinerung der Tumoren angestrebt werden. Häufig findet sich in einem Auge ein weit fortgeschrittener Befund, so dass der Erhalt des Augapfels nicht sinnvoll erscheint. Sollte jedoch an dem besseren Auge eine Chemotherapie notwendig sein, kann mit der Enukleation des stärker betroffenen Auges zunächst gewartet werden, da manchmal unter der Therapie eine massive Tumorrückbildung eintritt und eine augenerhaltende Behandlung möglich werden kann. Wenn allerdings das schlechtere Auge bereits erblindet ist, eine Glaskörperaussaat oder Infiltration des vorderen Augensegments oder des Sehnervs besteht, gibt es keine Alternative zur Enukleation. Kommt es am letzten, besseren Auge zu einer Infiltration des Sehnervs oder Glaskörperaussaat, bleibt als einzige augenerhaltende Therapie nur noch die Bestrahlung von außen (perkutane Strahlentherapie). Ist allerdings kein nützliches Sehvermögen zu erwarten, so muss auch das zweite Auge entfernt werden, um das Leben des Kindes nicht zu gefährden. Da eine moderne Chemotherapie mit hoher Wahrscheinlichkeit ein sehr viel geringeres Risiko für die Entstehung weiterer bösartige Tumoren hat als die perkutane Bestrahlung, ist man heute bestrebt, auf die perkutane Strahlentherapie so weit wie möglich zu verzichten, insbesondere wenn das betroffene Kind das erste Lebensjahr noch nicht vollendet hat. Im Folgenden werden die einzelnen therapeutischen Möglichkeiten genauer erläutert: 7.1 Enukleation Darunter versteht man die operative Entfernung des Auges. Sie ist bei funktionslosen Augen oder Augen, die nach einer Therapie erblinden würden, die einzig sinnvolle Option. Das Auge wird dabei in Vollnarkose mit einem möglichst langen Anteil des Sehnervs entfernt. Lider, Tränendrüse und Muskeln werden nicht beeinträchtigt. Als Platzhalter wird in die Tiefe der Augenhöhle ein Kunststoffimplantat eingesetzt. Darüber werden die Muskeln sowie die Bindehaut vernäht. Unmittelbar nach dem Eingriff können Schmerzen auftreten, es kann zu einem Bluterguss in die Augenhöhle kommen sowie in extrem seltenen Fällen zu einer Infektion. Etwa 10 bis 14 Tage nach der Operation wird eine erste Prothese angepasst und in den Bindehautsack eingesetzt, wenn die Wunde geheilt und die Schwellung zurückgegangen ist. Dieses künstliche Auge aus Glas wird dem Partnerauge entsprechend individuell angefertigt und kann oft kaum von einem natürlichen Auge unterschieden werden. Durch die über dem Platzhalter vernähten Muskeln werden die Augenbewegungen des verbleibenden Auges in begrenztem Ausmaß durch die Prothese mitgemacht. Nach der Entfernung des Auges ist noch ein stationärer Aufenthalt von ungefähr 2 Tagen notwendig. Das entnommene Auge wird histologisch feingeweblich aufgearbeitet. Dabei kommt einem möglichen Einwachsen von Tumorzellen in den Sehnerv Seite 10 oder in die Aderhaut besondere Bedeutung zu, da hierdurch ein erhöhtes Risiko für eine Metastasierung des Tumors besteht und ggf. eine Chemotherapie notwendig wird. Bei jeder Enukleation wird zudem Tumorgewebe entnommen, um eine molekulargenetische Untersuchung durchführen zu können und damit eine genauere Aussage über das Vererbungsrisiko treffen zu können. 7.2 Lokale Therapieverfahren 7.2.1 Laserkoagulation Dabei wird in Narkose ein Laserstrahl durch die Pupille auf den Tumor gelenkt und das Tumorgewebe durch Hitze zerstört. Die Laserbehandlung kommt bei kleineren Tumoren am hinteren Pol und in der peripheren Netzhaut in Frage. Meist ist eine wiederholte Behandlung erforderlich. 7.2.2. Kryokoagulation (Vereisung) Der Tumor wird von außen unter Kontrolle des Augenspiegels mit einer Metallsonde lokalisiert und mehrmals durchgefroren. Hierdurch werden die kälteempfindlichen Tumorzellen zerstört. Die Therapie eignet sich für kleine Tumoren der peripheren Netzhaut, größere Tumoren müssen oft wiederholt oder durch andere Therapieverfahren ergänzt behandelt werden. Im Vergleich zu der Laserkoagulation geht allerdings auch ein größerer Teil gesunder Netzhaut verloren. Die Behandlung verursacht zudem eine vorübergehende Schwellung der Lider und der Bindehaut. 7.2.3. Behandlung mit Strahlenträgern (Brachytherapie) Bei dieser sogenannten Brachytherapie wird ein Strahlenträger, der auf seiner Innenseite mit einem strahlenden Material beschichtet ist (meist Ruthenium-106), operativ im Bereich des zu behandelnden Tumors nach Eröffnen der Bindehaut von außen auf die Lederhaut aufgenäht und nach Erreichen einer bestimmten Strahlendosis an der Tumorspitze wieder entfernt. Es sind somit zwei Operationen in Vollnarkose notwendig. Wie lange der Strahlenträger auf dem Auge verbleiben muss, wird von der Höhe des Tumors beeinflusst und genau berechnet. Es wird lokal eine hohe Strahlendosis im Bereich des Tumors erreicht unter Schonung des umliegenden Gewebes. Das strahlensensible Retinoblastom wird dann allmählich in ein inaktives Narbengewebe umgewandelt. Dieses Therapieverfahren eignet sich für einzelne mittelgroße Tumoren. Auch bei einer begrenzten, lokalisierten Glaskörperaussaat kann eine Brachytherapie noch angewandt werden sowie in Kombination mit anderen Therapieverfahren. Die Komplikationen sind abhängig von der Lage des Tumors. Es kann zu einer Linsentrübung kommen, eine strahlenbedingte Schädigung der Netzhaut oder eine Sehnervenschädigung eintreten, die zur Sehverschlechterung führen und auch Blutungen sowie Durchblutungsstörungen im Auge verursachen können. Aus rechtlichen Gründen (Strahlenschutzbestimmungen) muss das Kind während der in der Regel höchstens 2 Tage dauernden Bestrahlung isoliert bleiben, wobei nach entsprechender Aufklärung ein Elternteil mit aufgenommen und bei dem Kind bleiben kann. 7.3. Chemotherapie Die systemische Chemotherapie ist in den letzten Jahren zu der am häufigsten angewandten Methode in der augenerhaltenden Therapie des Retinoblastoms geworden, nachdem die Langzeitkomplikationen der Seite 11 perkutanen Strahlentherapie bekannt wurden (s. unten). Dabei werden Medikamente wie Cyclophosphamid, Carboplatin, Vincristin und Etoposid über eine Vene (intravenös) verabreicht und erreichen so die Tumorgefäße, um hier wirksam zu werden. Moderne Studien haben zeigen können, dass damit eine hohe lokale Tumorkontrollrate erreicht werden kann. Bei der sogenannten Thermochemotherapie wird nach Gabe von Carboplatin eine Erwärmung des Tumorgewebes mittels Laserbehandlung durchgeführt. Jeder einzelne Tumor wird 10 bis 20 Minuten behandelt. Dadurch kann die Wirksamkeit der Chemotherapie soweit verbessert werden, dass eine vollständige Tumorzerstörung erreicht wird. Bei der Chemoreduktion wird die Chemotherapie primär zur Verkleinerung des Tumors eingesetzt, um anschließend den Tumor lokal behandeln zu können (Kryo- oder Laserbehandlung, Brachytherapie). Durch eine ca. 24 Stunden vor der Chemotherapie durchgeführte Kältebehandlung der Netzhaut wird in ausgewählten Fällen bei bestehender, begrenzter Glaskörperaussaat versucht, die Wirkstoffkonzentration der Medikamente im Auge zu erhöhen. Wird nach Enukleation eines an Retinoblastom erkrankten Auges histologisch ein großflächiger Einbruch in die Aderhaut oder eine Infiltration des Sehnervs jenseits des Auges nachgewiesen, ist ebenfalls eine ergänzende (adjuvante) Chemotherapie angezeigt. Bei eingetretener Metastasierung kann die Überlebensrate der Kinder durch eine Chemotherapie gebessert werden. 7.4. Neue Chemotherapie-Protokolle 7.4.1. Intraarterielle Chemotherapie Bei der intraarteriellen Chemotherapie wird ein chemotherapeutisches Medikament (in der Regel Melphalan) über einen über die Arterie eines Beines bis nahe an die Augenhöhle vorgeschobenen Katheter direkt in das Blutgefäßsystem des Auges gegeben. Dies hat den entscheidenden Vorteil, dass Chemotherapeutika nicht im ganzen Körper wirken und lokal eine hohe Konzentration erreicht wird, erfordert aber einen hohen apparativen Aufwand und hat spezifische Risiken, die wesentlich durch die Katheterisierung von wichtigen Blutgefäßen bei in der Regel kleinen Kindern bedingt sind. Diese Methode wird schon seit mehr als 20 Jahren in Japan durchgeführt und hat durch das Engagement der New Yorker Arbeitsgruppe in der letzten Zeit weltweite Aufmerksamkeit erlangt. Der Stellenwert der Methode ist noch unklar. Gesichert ist, dass in einzelnen Fällen Tumorkontrollen erreicht werden, die mit anderen Methoden nicht erreicht werden können. Offensichtlich bestehen aber auch spezifische Risiken durch die Punktion der Arterie; häufig lässt die anatomische Situation aber auch keine gezielte Platzierung des Katheters zu. Im Einzelfall muss über den Einsatz dieser Methode unter Würdigung aller anderen Alternativen entschieden werden. 7.4.2. Intravitreale Chemotherapie Die intravitreale Chemotherapie (Eingabe von Medikamenten in den Glaskörper) galt lange Zeit nicht als mögliche Option, da die Eröffnung eines Auges mit einem Retinoblastom als zu gefährlich galt und möglicherweise ein Wachstum des Retinoblastoms außerhalb des Auges zur Folge haben könnte. Neuere Studien haben gezeigt, dass dieses Risiko unter Berücksichtigung einer Vielzahl von Vorsichtsmaßnahmen sehr gering ist und eine solche Seite 12 direkte Eingabe eines Chemotherapeutikums in das Auge Komplikationen ersparen kann, wie sie z.B. bei der intraarteriellen Chemotherapie auftreten können. Das verwendete Medikament ist in der Regel Melphalan, ähnlich wie bei der intraarteriellen Chemotherapie. Auch mit dieser Methode können Augen behandelt werden, die mit anderen Methoden wie oben beschrieben nicht oder nicht mehr behandelt werden können. Dies gilt insbesondere für Augen mit einem erneuten Tumorwachstum nach Ausschöpfung aller konventionellen Behandlungsmethoden. Auch bei dieser Methode muss im Einzelfall entschieden werden, ob eine Anwendung möglich ist, da weltweit erst begrenzte Erfahrungen bestehen. 7.5. Perkutane Strahlentherapie Sie erlaubt in bis zu 80% der Fälle eine vollständige Tumorkontrolle unter Erhalt des Auges und eines brauchbaren Sehvermögens durch eine Bestrahlung im seitlichen Feld mit Aussparung der Linse. Das Auge wird während der Bestrahlung durch eine Vakuumkontaktlinse in einer festen Position gehalten. Die Gesamtstrahlendosis wird in 25 Sitzungen appliziert. Die Behandlung verläuft somit über 5 Wochen fünfmal wöchentlich und kann bei den kleinen Kindern nur in Narkose durchgeführt werden. Die Fraktionierung hilft, die strahlenbedingten Nebenwirkungen zu reduzieren. Moderne Strahlenquellen mit genau ausrichtbarem Strahlengang vermeiden die Bestrahlung der vorderen Augenabschnitte. Da das Retinoblastom strahlensensibel ist, wird es durch die Behandlung in inaktives Narbengewebe umgewandelt. Als Komplikation können strahlenbedingte Schäden der Netzhaut und des Sehnervs, ein vermindertes Knochenwachstum im Bestrahlungsfeld und die Entwicklung einer Linsentrübung sowie eine Schädigung der Tränendrüse mit nachfolgend Problemen eines trockenen Auges auftreten. Die perkutane Bestrahlung war bis vor wenigen Jahren die Standardtherapie fortgeschrittener beidseitiger Retinoblastome und führte zu einer enormen Steigerung der Heilungsrate. Allerdings haben Langzeituntersuchungen gezeigt, dass ein Drittel der auf diese Weise behandelten Patienten bis zum 30. Lebensjahr an einem bösartigen Zweittumor im Bestrahlungsfeld erkranken. Daher hat diese Therapie ihren Stellenwert verloren, und es wird nach Alternativen gesucht. Die perkutane Strahlentherapie kommt deshalb als Therapiemöglichkeit nur in Frage, wenn alle anderen Therapiemöglichkeiten ausgeschöpft sind und noch ein nützliches Sehvermögen des betroffenen Auges zu erwarten ist. 8. Therapiekontrolle und Prognose Wenn der erste Behandlungszyklus des Retinoblastoms beendet ist, müssen beide Augen bzw. die Augenhöhle regelmäßig kontrolliert werden, um neue Tumoren und ein erneutes Tumorwachstum (Rezidiv) frühzeitig zu erkennen. Der Abstand zwischen den Untersuchungen hängt sowohl vom Alter des Kindes als auch der gewählten Therapieform sowie dem genetischen Befund ab. In den ersten beiden Lebensjahren können relativ kurze Abstände erforderlich sein, da dann das Risiko für neue Tumoren am höchsten ist. Unmittelbar nach einer Therapie sollten etwa alle 4 Wochen Narkoseuntersuchungen durchgeführt werden, so dass mögliche neue Tumoren früh erkannt und lokal behandelt werden können. Zusätzlich sollten ambulante Nachsorgetermine in der Kinderklinik stattfinden, wobei die Seite 13 Untersuchungsabstände im Einzelfall abgestimmt werden müssen. Ist ein Kind Merkmalträger und hat die Veranlagung zur Entwicklung eines Retionoblastoms geerbt, sind erste Tumorherde oft schon bald nach der Geburt erkennbar. Mit engmaschigen augenärztlichen Kontrollen sollte so früh wie möglich begonnen werden, d. h. unmittelbar nach der Geburt. Bis zum 4./5. Lebensjahr müssen die Untersuchungen in Narkose durchgeführt werden, um auch die peripheren, kritischen Areale der Netzhaut sicher einsehen zu können. Mit 5 Jahren ist meistens eine zuverlässige Untersuchung ohne Narkose möglich. Im Zweifelsfall, insbesondere bei einer massiven Abwehr des Kindes, sollte jedoch immer eine Narkoseuntersuchung durchgeführt werden. Aufgrund der deutlichen Fortschritte in der Therapie werden heute 5Jahresüberlebensraten von mehr als 95 % für ein- und beidseitige Retinoblastome in Ländern mit einem entwickelten Gesundheitssystem erreicht. Kinder mit unilateralem Retinoblastom haben ein gesundes Auge ohne Beeinträchtigung des Sehvermögens und können ein ganz normales Leben führen. Auch bei der Mehrzahl der Kinder mit bilateralem Retinoblastom bleibt mindestens ein Auge mit ausreichender Funktion erhalten. Die Langzeitprognose bei Patienten mit erblichem Retinoblastom wird wesentlich durch ein erhöhtes Risiko für die Entwicklung vom Tumoren außerhalb des Auges (Zweittumoren) beeinflusst. Die häufigsten Zweittumoren sind bösartige Knochentumoren und Weichteiltumoren. Dies muss bei der Therapieplanung berücksichtigt werden. Nach einer perkutanen Strahlentherapie ist eine erhöhte Rate von Zweittumoren im Bestrahlungsfeld gesichert. Ob es auch nach einer Chemotherapie zu einem vermehrten Auftreten von Zweittumoren kommt, ist derzeit noch nicht bekannt. Seite 14 9. Kontaktadressen Universitätsklinikum Essen Tumorsprechstunde des Zentrums für Augenheilkunde Herr Prof. Bornfeld, Frau Dr. Biewald, Frau Dr. Metz, Frau Dr. Deike Frau Augusto (Sekretärin) Hufelandstraße 55 45122 Essen Telefon.: 0201-723-2969 Telefax: 0201 723 E-Mail: [email protected] Kinderstation (Station A1) Telefon: 0201 723 2271 Klinik für pädiatrische Onkologie Frau Prof. Fleischhack, Frau Dr. Temming Telefon: 0201 2768 Station K3: 0201-723-2255 Institut für Humangenetik Herr Prof. Lohmann Telefon: 0201 723 4560 Klinik für Strahlentherapie Herr Prof. Sauerwein Telefon: 0201 723 2051 Elternhaus, Selbsthilfegruppen Essener Elterninitiative zur Unterstützung krebskranker Kinder e.V. Kaulbachstr. 8-10 45147 Essen Telefon: 0201 878570 Telefax: 0201 /87857155 E-Mail: [email protected] Internet: http://www.krebskranke-kinder-essen.de/Startseite/37_de_Startseite.html Kinder-Augenkrebs-Stiftung (KAKS) Adenauerallee 13 53113 Bonn Telefon. 0228 688460 Telefax 0228 6884644 E-Mail: [email protected] Internet: http://www.kinderaugenkrebsstiftung.de/ Seite 15