Osimertinib
Werbung

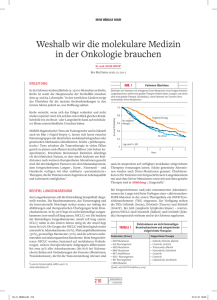
Pharmazeutische Chemie – Osimertinib Osimertinib (Tagrisso®) Die bedeutende Rolle des epidermalen Wachstumsfaktor-Rezeptors (epidermal growth factor receptor = EGFR) beim nichtkleinzelligen Lungenkarzinom (non-smallcell lung cancer = NSCLC) ist weithin bekannt. Onkogene Mutationen am EGFRezeptor führen zu einer Erhöhung der Aktivität des Rezeptors, indem durch diese Mutationen eine intrazelluläre Tyrosinkinase-Aktivität ohne Bindung des Liganden erzeugt und somit eine konstitutive Aktivität des EGF-Rezeptors ermöglicht wird. Tumoren mit solchen aktivierenden Mutationen am EGF-Rezeptor (wie vielfach beim NSCLC) sind in ihrem Wachstum abhängig von einer funktionierenden EGFRSignalkaskade, eine Hemmung des EGFR durch Tyrosinkinase-Inhibitoren (TKIs) führt zu einer Unterbrechung des Signalweges und zu einer Wachstumshemmung des Tumors (Nguyen et al. 2014, Stinchcombe 2014). Abbildung 1 Die EGFR-Inhibitoren der ersten Generation Erlotinib (Tarceva®) und Gefitinib (Iressa®) sowie der Zweitgenerationen-EGFR-Inhibitor Afatinib (Giotrif®) entfalten ihre Wirkung insbesondere dann, wenn aktivierende Mutationen (EGFRm+) in der Tyrosinkinase- und ATP-bindenden Domäne (Exone 18-24) des EGF-Rezeptors vorliegen (s. Tabelle 1) (s. auch Neue Arzneistoffe 2013: Afatinib). Über 90% der der EGFR-Gen-Mutationen betreffen die Exone 18-21, also einen vergleichsweise kleinen Abschnitt des EGFR-Gens. Die beiden häufigsten aktivierenden Mutationen finden sich in den Exonen 19 und 21: im Exon 19 liegt eine Deletion ohne Rasterverschiebung vor (ex19del), im Exon 21 führt eine Punktmutation zur Substitution des Leucins durch Arginin (L858R). Patienten mit diesen Mutationen zeigen ein gutes Ansprechen auf eine Therapie mit den oben genannten Tyrosinkinase-Inhibitoren (Paez et al. 2004, Nguyen et al. 2014, Maione et al. 2015). Dementsprechend sind diese TKIs auch als Erstlinientherapie beim lokal fortgeschrittenen oder metastasierten NSCLC zugelassen sofern aktivierende, onkogene Mutationen vom Typ exdel19 sowie L858R vorliegen. Der Nachweis einer solchen aktivierenden Mutation im EGFR-Gen des Tumorgewebes stellt somit einen wichtigen positiven, prädiktiven Faktor für das Ansprechen einer Therapie mit einem EGFR-Inhibitor (Ji et al. 2006, Sequist et al. 2007, Fachinformation Iressa® 2014, Fachinformation Giotrif® 2015, Fachinformation Tarceva® 2016). Schätzungen zeigen, dass die beiden aktivierenden Mutationen exdel19 und L858R bei 50% der asiatischen und 15% der westlichen NSCLC-Patienten vorliegen (Rosell et al. 2009, Nguyen et al. 2014). 1 CA 18.2.2016 Pharmazeutische Chemie – Osimertinib Typischerweise entwickelt sich bei Patienten mit aktivierenden Mutationen, die anfangs gut auf eine Therapie mit den Erstgenerationen-Inhibitoren Erlotinib und Gefitinib ansprechen, nach 9 bis 14 Monaten eine sekundäre Resistenz. Ausschlaggebend für diese sekundäre Resistenz ist in den meisten Fällen die sogenannte T790M-Mutation, eine Mutation in Exon 20, die zu einer Substitution eines Threonins durch ein Methionin führt. Die T790M-Mutation wird auch als „Gatekeeper“-Mutation bezeichnet, weil die betreffende Aminosäure in der ATPBindungstasche lokalisiert ist. Es wird angenommen, dass durch die voluminöse, räumlich anspruchsvolle Methionin-Seitenkette im Vergleich zum Threonin beim T790M-EGFR die Bindung der Erstgenerationen-TKIs Erlotinib und Gefitinib sterisch verhindert wird (Pao et al. 2005, Sequist et al. 2011, Cross et al. 2014). EGFR-Inhibitor Markteinführung Deutschland Indikation(en) Erlotinib (Tarceva®) 2005 NSCLC mit EGFRm+ Inhibierte EGFRs Dosis/ Art der Anwendung EGFRm+ 1x täglich 1 Filmtablette (150mg), mindstens 1 Stunde vor oder 2 Stunden nach einer Mahlzeit einzunehmen Pankreaskarzinom (+Gemcitabin) Gefitinib (Iressa®) 2009 NSCLC mit EGFRm+ EGFRm+ 1x täglich 1 Filmtablette (250mg), unabhängig von den Mahlzeiten Afatinib (Giotrif®) 2013 NSCLC mit EGFRm+ EGFRm+ (T790M-EGFR) HER2 ErbB4 1x täglich 2 Filmtabletten zu 20mg enstprechend 40mg, mindestens 3 Stunden vor oder eine Stunde nach Einnahme keine Nahrung Osimertinib (Tagrisso®) 2016 NSCLC mit T790M-EGFR EGFRm+, T790M-EGFR 1x täglich 1 Filmtablette (80mg), unabhängig von den Mahlzeiten Tabelle 1: in Deutschland zugelassene EGFR-TK-Inhibitoren zur Behandlung des NSCLC (EGFRm+ = aktivierende Mutationen wie ex19del oder L858R) Therapeutische Optionen für Patienten mit dieser sekundären, erworbenen Resistenz gegen Erlotinib und Gefitinib sind stark limitiert. Der ZweitgenerationenTKI Afatinib ist ebenso wie die Erstgenerationen-Inhibitoren wirksam beim unbehandelten NSCLC mit aktivierenden Mutationen. Zudem wurde bei diesem TKI eine Wirksamkeit bei der T790M-Mutation nachgewiesen. Allerdings scheint es so zu 2 CA 18.2.2016 Pharmazeutische Chemie – Osimertinib sein, dass Afatinib keine durch eine Erlotinib. bzw. Gefitinib-Therapie erworbene und auf die T790M-Mutation zurückzuführende Resistenz durchbrechen kann, da die benötigten Konzentrationen an Afatinib dafür viel zu hoch sind und bei den Patienten aufgrund dosislimitierender Toxizitäten nicht gegeben werden können( s. auch Neue Arzneistoffe 2013: Afatinib) (Miller et al. 2012, Katakami et al. 2013). Nur ein Therapieregime, bei dem Afatinib in Kombination mit dem Anti-EGFR-Antikörper Cetuximab gegeben wurde, zeigte eine Response-Rate von 32% beim NSCLC mit EGFRm+ und einer erworbenen Erlotinib-Resistenz (Janjigian et al. 2011, Cross et al. 2014). Dementsprechend sind die bisher verfügbaren EGFR-Inhibitoren Erlotinib, Gefitinib und Afatinib beim NSCLC nur zugelassen, sofern es sich um einen TKInaiven Tumor mit aktivierenden Mutationen handelt (s. Tabelle 1). Was dringend benötigt wird, ist ein peroral bioverfügbarer TKI, der effektiv sowohl EGFRm+ als auch T790M-EGFR inhibiert. Diese Option steht nun anscheinend mit dem TKI Osimertinib (Tagrisso®), einem EGFR-Inhibitor der sogenannten dritten Generation, zur Verfügung (Strukturformel s. Abbildung 1). Osimertinib ist zugelassen zur Therapie erwachsener Patienten mit lokal fortgeschrittenem oder metastasiertem NSCLC und einer nachgewiesenen T79M-Mutation des EGFR. Die Tagrisso®-Filmtabletten werden einmal täglich im Ganzen mit Wasser unabhängig von den Mahlzeiten und immer zum selben Zeitpunkt eingenommen, ohne dass die Tabletten zerdrückt, geteilt oder zerkaut werden. Gleichzeitige Nahrungsaufnahme beeinflusst die Bioverfügbarkeit des Osimertinibs nicht in klinisch relevantem Ausmaß. Bei Starken Schluckbeschwerden des Patienten kann die Tablette auch in stillem Wasser (50ml) unter Rühren dispergiert werden. Die Dosis beträgt in der Regel 80mg (1 Filmtablette), bei bestimmten Nebenwirkungen/ Toxizitäten kann unter Umständen auf eine Filmtablette mit 40mg einmal täglich reduziert werden. Die Therapie wird fortgesetzt bis zur Krankheitsprogression bzw. bis zum Auftreten einer inakzeptablen Toxizität. (Fachinformation Tagrisso® 2016). Erlotinib, Gefitinib und Afatinib gehören zu den TKIs mit 4-AnilinochinazolinPartialstruktur. Die beiden Erstgenerationen-Inhibitoren Erlotinib und Gefitinib binden reversibel in der ATP-Tasche der Kinase-Domäne und verdrängen ATP kompetitiv aus seiner Bindung. Die Bindungsstelle des Afatinibs liegt auch in der ATP-Tasche, allerdings bildet Afatinib über seine elektrophile α,β-ungesättigte Carbonyl-Gruppe in einer Michael-Addition mit der SH-Gruppe des Cysteins797 in der ATP-Tasche eine kovalente Bindung. Der gebildete Thioether ist sehr stabil, die Bindung zwischen Afatinib und EGFR ist irreversibel (s. Neue Arzneistoffe 2013: Afatinib). Abbildung 2: Vergelich der chemischen Strukturen der beiden irreversiblen EGFR-Inhibitoren Osimertinib und Afatinib mit elektrophiler Acrylamid-Partialstruktur (lila) 3 CA 18.2.2016 Pharmazeutische Chemie – Osimertinib Im Gegensatz zu den drei vorherigen EGFR-Inhibitoren besitzt der neue Inhibitor Osimertinib keine 4-Anilinochinazolin-Partialstruktur. Stattdessen liegt als zentraler Baustein eine Phenylaminopyrimidin-Struktur vor (s. Abbildung 2) (Finlay et al. 2014). Abbildung 3: Bildung der kovalenten, irreversiblen Bindung zwischenm dem Cystein797 im aktiven Zentrum von EGFRm+ und T790M-EGFR über eine Michael-Addition 4 CA 18.2.2016 Pharmazeutische Chemie – Osimertinib Der Pyrimidin-Ring trägt zusätzlich an Position 4 als Substituenten einen Indol-Ring, dessen Stickstoff methyliert ist. Der Phenyl-Ring besitzt eine Methoxy-Gruppe sowie zwei weitere Substituenten. Zum einen einen basischen N,N,N‘Trimethylethylendiamin-Rest, zum anderen ein 2-Propenamid (Acrylamid), das eine elektrophile α,β-ungesättigte Carbonyl-Gruppe enthält, die in Analogie zum Afatinib als Michael-Akzeptor für die kovalente Bindung zum Cystein797 des EGFR verantwortlich ist. Die nukleophile Thiol-Gruppe des Cysteins797 fungiert als Michael-Donor (s. Abbildung 3). Erstaunlich ist, dass die Elektrophilie der Doppelbindung ausreicht, da die Carbonyl-Gruppe als Amid vorliegt und somit die Elektronendichte eher erhöht wird. Wahrscheinlich aber reicht der elektronenziehende Effekt des angrenzenden Phenyl-Ringes aus, um den +M-Effekt der Amino-Gruppe auszugleichen. Das Ergebnis der irreversiblen Michael-Addition des Osimertinibs mit seiner elektrophilen α,β-ungesättigten Carbonyl-Gruppe an die nukleophile SH-Gruppe des Cysteins797 ist ein kovalenter Thioether analog zum Afatinib. Eine Kristallstruktur des Osimertinibs im aktiven Zentrum des T790M-EGFR liegt bislang nicht vor, dafür wurde der Bindungsmodus aber über Molcular Modeling ausgehend von einer Kristallstruktur des T790M-EGFR berechnet (Cross et al. 2014, Finlay et al. 2014). In diesem Strukturmodell ist die kovalente Thioether-Bindung zwischen Cystein797 und der Acrylamid-Struktur des Osimertinibs deutlich zu erkennen. Der Pyrimidin-Ring bildet zwei Wasserstoffbrücken mit der Hinge-Region (Methionin793) aus. Der Indol-Ring scheint für die Aktivität gegenüber T790M-EGFR mitverantwortlich zu sein. In der Modeling-Struktur liegt er direkt angrenzend zum Gatekeeper-Rest Methionin790. Natürlich hemmt Osimertinib zusätzlich zu T790MEGFR auch die aktivierenden, onkogenen Mutationen wie z.B. L858R (Cross et al. 2014, Finlay et al. 2014). Literatur: Cross, A:A:E: Cancer Discov 2014, 4, 1046 Fachinformation Giotrif® 2015, Boehringer Ingelheim International GmbH Fachinformation Iressa® 2014, AstraZeneca AB Fachinformation Tagrisso® 2016, Astra Zeneca AB Fachinformation Tarceva® 2016, Roche Registration Limited Finley, M.R. et al. J Med Chem 2014, 57, 8249 Janjigian, Y.Y. et al. J Clin Oncol 2011, 29(suppl), abstr 7525 Ji, H. et al. Cancer Cell 2006, 9, 485 Katakami, N. et al. J Clin Oncol 2013, 31, 3335 Nguyen, K.S. et al. World J Clin Oncol 2014, 5, 576 Maione, P. et al. Ther Adv Med Oncol 2015, 715, 263 Miller, V.A. et al. Lancet Oncol 2012, 13, 528 Pao, W. et al. PLoS Med 2005, 2, e73 Rosell, R. et al. N Engl J Med 2009, 361, 958 Paez, J.G. et al. Science 2004, 304, 1497 Sequist, L.V. et al. J Clin Oncol 2007, 25, 587 Sequist, L. et al. Sci Transl Med 2011, 3, 75ra26 Stinchcombe, T.E. F1000Prime Reports 2014, 6, 117 5 CA 18.2.2016