Dynamik der Proteine - Lehrstuhl für Biophysik - Ruhr
Werbung

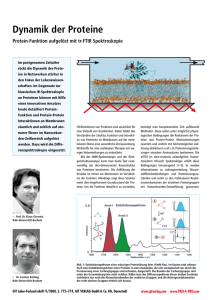
Dynamik der Proteine Protein-Funktion aufgelöst mit tr-FTIR Spektroskopie (time-resolved Fourier-transform Infrared Spectroscopy) Im postgenomen Zeitalter rückt die Dynamik der Proteine in Netzwerken stärker in den Fokus der Lebenswissenschaften. Im Gegensatz zur klassischen IR-Spektroskopie an Proteinen können mit Hilfe eines innovativen Ansatzes heute detailliert Protein-Funktion und Protein-Protein Interaktionen an Membranen räumlich und zeitlich auf atomarer Ebene im NanosekundenZeitbereich aufgelöst werden. Dazu wird die Differenzspektroskopie eingesetzt. Fehlfunktionen von Proteinen sind ursächlich für eine Vielzahl von Krankheiten. Daher bildet das Verständnis der Struktur, Funktion und Interaktion von Proteinen an Membranen auf der molekularen Ebene eine wesentliche Vorraussetzung, Wirkstoffe für eine molekulare Therapie mit weniger Nebenwirkungen zu entwickeln. Mit der NMR-Spektroskopie und der Röntgenstrukturanalyse kann man heute fast routinemäßig die drei-dimensionale Raumstruktur von Proteinen bestimmen. Die Aufklärung der Struktur ist immer ein Meilenstein im Verständnis der Funktion. Allerdings zeigt diese Struktur meist den eingefrorenen Grundzustand der Proteine. Um die Funktion detailliert zu verstehen, benötigt man komplementäre Zeit auflösende Methoden. Diese sollen unter möglichst physiologischen Bedingungen die Reaktionen der Proteine und Protein-Protein Wechselwirkungen räumlich und zeitlich mit höchstmöglicher Auflösung detektieren und z. B. Protonierungsänderungen einzelner Aminosäuren bestimmen. Die trFTIR (tr: time-resolved, zeitaufgelöst; Fourier-transform Infrarot) Spektroskopie erfüllt diese Bedingungen hervorragend [1–3]. Es werden Informationen zu Ladungsverteilung, Wasserstoffbrückenbindungen und Protonierungszuständen und somit zu den molekularen Reaktionsmechanismen der einzelnen Proteingruppen mit Nanosekunden-Zeitauflösung gewonnen. Man beobachtet sozusagen mit einem IR-Nanoskop das Geschehen im aktiven Zentrum des Proteins. Absorptionsspektrum eines Proteins Prof. Dr. Klaus Gerwert, Ruhr-Universität Bochum Dr. Carsten Kötting, Ruhr-Universität Bochum In Abbildung 1 ist ein typisches Absorptionsspektrum eines Proteins in Wasser dargestellt. Aufgrund der Vielzahl von Absorptionen können im Spektrum aber noch keine Absorptionsbanden einzelner Gruppen erkannt werden. Dominierend sind die Absorptionen der Peptidbindung, die Amid-I (C=O) und die Amid-II-Bande (NH+CN). Anhand des Absorptionsspektrums lassen sich Aussagen zum Gesamtprotein, wie z.B. zur Sekundärstruktur treffen. Differenzspektroskopie Um Aussagen über einzelne funktionelle Gruppen und den Reaktionsmechanismus zu erhalten, benötigt man Differenzspekt- BIOforum 6/2008, S. 36–38, GIT VERLAG GmbH & Co. KG, Darmstadt ren. Im einfachen Fall einer Reaktion A→B betrachtet man die Differenz der beiden Absorptionsspektren (B–A). Wichtig ist es hierbei, A und B unter genau den gleichen Bedingungen zu messen, da die Hintergrundabsorption um drei bis fünf Größenordnungen stärker ist als die der wenigen reaktiven Gruppen. Dieses stellt sehr hohe Anforderungen an die Messaufbauten. Unter anderem ist es wichtig, die zu untersuchende Reaktion präzise innerhalb des Spektrometers zu starten. Dies kann für photobiologisch aktive Proteine direkt über einen Laserblitz geschehen. In anderen Fällen ist der Einsatz von „caged“Substanzen möglich, welche nach einem Laserblitz eine biologisch aktive Substanz freisetzen. Auch der Einsatz von nanotechnologisch hergestellten Mikro-Mischzellen ist möglich [4]. Bei der ATR (attenuated total reflection)-Technik wird ein Protein auf einer Oberfläche des ATR Kristalls fixiert (siehe Abb. oben). Mit Hilfe der evanescenten Welle des IR Stahls, der durch den Kristall geführt wird, kann das Spektrum aufgenommen werden. Interaktionspartner, wie Liganden oder andere Proteine können in der Lösung ausgetauscht werden. Ein Schlüsselschritt in der Bestimmung der Proteinfunktion ist die Zuordnung der IR-Banden zu molekularen Gruppen des Proteins. Dies kann entweder durch ortsspezifische Mutagenese (eine Aminosäure wird molekularbiologisch durch eine andere ersetzt, so dass die Bande im Vergleich zum Wildtyp im Spektrum der Mutante fehlt [3]) oder durch Isotopenmarkierung (eine Aminosäure oder ein Ligand wird mit einem www.gitverlag.com www.pro-4-pro.com Abb. 1: Extinktionsspektrum einer wässrigen Proteinlösung (hier 10mM Ras). Im Kasten sind schematisch zwei Extinktionsspektren eines Proteins in zwei Zuständen, die sich exemplarisch nur durch die Protonierung einer Carboxylgruppe unterscheiden, dargestellt. Die Banden der Carboxylgruppe sind unter der Gesamtabsorption nicht sichtbar. Bildet man das Differenzspektrum dieser beiden Zustände, verbleiben nur noch die Absorptionsbanden der reaktiven Gruppen, und die Hintergrundabsorption der nicht aktiven Gruppen mittelt sich heraus. Isotop (z.B. 13C) markiert, wodurch sich die entsprechende Bande spektral verschiebt [5]) geschehen. Wie Proteine Protonen pumpen Die Methode der trFTIR-Differenz-Spektroskopie wurde bei der Untersuchung der lichtgetriebenen Protonenpumpe Bakteriorhodopsin (bR) etabliert. Die entscheidenden Schritte der Protonenpumpe, der Protonentransfer von der zentralen Protonenbindestelle, der Schiffschen Base, zu Asp85 und die Reprotonierung von Asp96 wurden mit Hilfe der trFTIR aufgeklärt [3]. Darüber hinaus konnten die trFTIRMessungen zeigen, dass einzelne Wassermoleküle wesentlich am Protonentransport beteiligt und nicht nur passive Elemente im Protein sind (Abb. 2) [6]. Dieses ist ein Paradigmenwechsel in der Vorstellung über die Rolle der proteingebundenen Wassermoleküle. Schaltmechanismus von GTPasen Das Ras-Protein ist ein molekularer Schalter, welcher unter anderem ein Wachstumssignal für die Zelle kontrolliert [7]. In 30 % aller menschlichen Tumoren findet sich onkogen mutiertes Ras. Uns ist es gelungen, das Abschalten dieses Wachstumssignal, welches durch die Hydrolyse des Nukleotids GTP hervorgerufen wird, infrarotspektroskopisch zu verfolgen (Abb. 3). Für die Katalyse der GTP-Hydrolyse sind sowohl enthalpische als auch entropische [8] Effekte verantwortlich: Durch die ungewöhnliche Verschiebung der Bande des β-Phosphats, konnte ein Ladungstransfer zum β-Phosphat nachgewiesen werden, Abb. 2: A) Die zeitabhängigen Änderungen der Infrarotabsorptionen während des bR Photozyklus von 30ns bis 200ms bei 4 cm–1 Auflösung. Die Absorptionen von protoniertem Asp85 (1762 cm–1) und protonierter Schiffscher Base (PSB) (1190 cm–1) sind markiert. In der nach 60µs abgeschlossenen L → M Reaktion wird ein Proton von der PSB zu Asp85 transferiert, was im Spektrum durch den Abfall der 1190cm–1 Absorption und den Anstieg der Absorption bei 1762 cm–1 deutlich wird. B) Durch eine Licht-induzierte Isomerisierung des Chromophors Retinal wird die starke H-Brücke des Wassers 402 aufgebrochen und etwa die Hälfte der Energie im Protein gespeichert. Da die freie OH-Gruppe von Wasser 401 (dangling water) nach Isomerisierung wasserstoffbrückengebunden wird, stabilisiert Wasser 401 nicht länger die negative Ladung von Asp85. Das Proton wandert von der zentralen Protonen-Bindestelle PSB zum Gegenion Asp85. Durch die Neutralisierung von Asp85 wird eine Bewegung des Arg82 induziert. Die Bewegung der positiv geladenen Guanidinium Gruppe destabilisiert den protonierten Wasserkomplex nahe der Proteinoberfäche. Der protonierte Wassercluster (blau) speichert ein Proton, wahrscheinlich in einem so genannten Eigen-Komplex (H+(H2O)3). Im Gegensatz zum zufälligen Grotthuss-Protonentransfer in Wasser wird im Protein der protonierte Wasserkomplex durch eine gezielte Bewegung des Arg82 deprotoniert. Das Proton wird in der zweiten Hydratschale über Aminosäuren, statt über Wasser stabilisiert. Das Protein hat die physiko-chemischen Eigenschaften der Wasser genutzt, um schnell und gezielt das Proton zu transferieren. lektiv durch kleine Moleküle (Wirkstoffe) angegriffen werden soll. Referenzen [1] Gerwert K.: Berichte der Bunsen-Gesellschaft 92, 978 (1988) [2] Kötting C. und Gerwert K.: ChemPhysChem 6, 881 (2005) [3] Gerwert K. et al.: P Natl Acad Sci USA 86, 4943 (1989) [4] Kauffmann E. et al.: P Natl Acad Sci USA 98, 6646 (2001) [5] Warscheid B., Brucker S., Kallenbach A., Meyer H. E., Gerwert K., Kötting C.: in press 2008, doi: 10.1016/j.vibspec.2007.11.003 [6] Garczarek F. und Gerwert K.: Nature 439, 109 (2006) Abb. 3: Zeitabhängige Veränderungen des Differenzspektrums während der GTPase Reaktionen von Ras und dem Protein-Protein Komplex Ras·GAP. Die Änderungen können direkt die intrinsische GTPase Reaktion und die GAP katalysierte Reaktion charakterisieren. Dies kann als Assay genutzt werden, um z.B. direkt den Einfluss von Wirkstoffen auf onkogene Mutanten zu untersuchen [9]. Bei Ras verschwindet die GTP-Bande bei 1143cm-1 mit der gleichen Rate mit der die GDP Bande bei 1104cm-1 entsteht. Bei Ras·GAP ist zusätzlich ein Intermediat (1116cm-1) zu erkennen. Die trFTIR-Spektroskopie konnte zeigen, dass es sich hierbei um H2PO4-, das über Wasserstoffbrücken und Coulomb-Wechselwirkungen nicht-kovalent im Protein gebunden ist handelt [10]. Entscheidend für die Katalyse ist der „Argininfinger“ vom GAP, dessen Bewegung ebenfalls mit trFTIR aufgelöst werden konnte [8]. [7] Vetter I.R. und Wittinghofer A.: Science 294, welcher die Aktivierungsenthalpie herabsetzt. Beim Hereindrehen des sog. „Argininfingers“ (Abb. 3) wird weiterhin die Aktivierungsentropie erhöht, vermutlich weil geordnete Wassermoleküle verdrängt werden. In einer eukaryontischen Zelle Kontakt: Dr. Carsten Kötting Prof. Dr. Klaus Gerwert Lehrstuhl für Biophysik Ruhr-Universität Bochum [email protected] [email protected] befinden sich etwa 150 verschiedene ­GT‑Pasen, die alle einen ähnlichen Mechanismus aufweisen. Das Verständnis des Mechanismus ist für die Entwicklung von molekularen Therapien sehr wichtig, weil das onkogene Protein direkt und se- 1299 (2001) [8] Kötting C., Kallenbach A., Suveyzdis Y., Wittinghofer A., Gerwert K.: Proc Natl Acad Sci U S A, 105, 6260 (2008) [9] Kötting C. et al.: ChemBioChem 8, 781 (2007) [10]Kötting C. et al.: P Natl Acad Sci USA 103, 13911 (2006)