LAAC1_Teil_II [Kompatibilitätsmodus]
Werbung

Seminar zum Praktikum
LAAC--1
LAAC
„Qualitativer Teil“
Wintersemester 2010/1
Prof. Dr. K. Sünkel
Chemische Analytik
Für den Chemiker (und oft nicht nur für
diesen) sind bei einer unbekannten
Substanz zwei Dinge interessant:
Was ist enthalten? Handelt es sich um ein
Gemisch oder eine Reinsubstanz?
Wieviel ist von welcher Substanz
enthalten?
Chemische AnalytikAnalytik- II
Die erste Frage ist Gegenstand der
„Qualitativen Analyse“ und soll uns in
diesem Semester beschäftigen
Die zweite Frage ist Gegenstand der
„Quantitativen Analyse“ und hat uns in
relativ vereinfachter Form im letzten
Semester beschäftigt
1
Qualitative Analyse
Eine Einführung
Kapitel 1: Einleitung
1.1. Übersicht
1.2. Lösungsversuche und Aufschlüsse
1.3. Trennungsmethoden
1.4. Nachweisreaktionen
1.1. Übersicht
Am Anfang steht die „optische Inspektion“
(charakteristische Farben, Kristallformen)
Dann folgen Lösungsversuche, eventuell
Aufschlüsse
Jetzt geht die eigentliche Chemie los:
Trennungsversuche und
Nachweisreaktionen
Und bei allem:
DENKEN NICHT VERGESSEN!
2
1.2. Lösungsversuche
Im Prinzip kann man hier schon eine Art
VorVor- Trennungsgang durchführen:
Löslichkeit in reinem Wasser
Löslichkeit in Salzsäure
Löslichkeit in konz. Salpetersäure
Löslichkeit in Königswasser
Was jetzt noch unlöslich ist, wird einem
Schmelzaufschluss unterzogen!
Schmelzaufschlüsse
Soda-Pottasche
SodaPottasche--Aufschluss für saure Oxide
und Oxosalze
Kaliumhydrogensulfat-- Aufschluss für
Kaliumhydrogensulfat
basische Oxide und Oxosalze
Oxidationsschmelze mit Kaliumnitrat
Freiberger Aufschluss für Zinnstein
1.3. Trennungsversuche
Um ein Substanzgemisch in seine
Bestandteile aufzutrennen, bedient man
sich im wesentlichen zweier
grundsätzlicher Methoden
1. Fraktionierte Fällung (der „klassische
Trennungsgang“)
2. Chromatographie unter Ausnutzung
unterschiedlichen Adsorptionsverhaltens
3
1.3.1. Der klassische
Trennungsgang der Kationen
Zuerst werden die schwerlöslichen
Chloride abgetrennt
Dann folgen die auch in stark saurer
Lösung schwerlöslichen Sulfide
Im Anschluss hieran fällt man die im
Ammoniakalischen schwerlöslichen Sulfide
und Hydroxide, und zuletzt
Die schwerlöslichen Carbonate
1.3.2. Anionentrennungsgang
Prinzipiell gibt es auch einen
Trennungsgang der Anionen, der sich aber
wegen vieler „Überschneidungen“ nicht
durchgesetzt hat. Man unterscheidet z.B.
Fällung mit SilberSilber- Ionen
Fällung mit CalciumCalcium- Ionen
Fällung mit BariumBarium- Ionen
1.3.3. Chromatographie
Es gibt sehr viele chromatographische
Trennverfahren. In diesem Praktikum soll
uns vor allem die
Dünnschichtchromatographie („DC“) sowie
die Ionenchromatographie („IC“)
beschäftigen.
Die chromatographischen Verfahren
dienen in der Praxis auch zur Aufreinigung
von Substanzen
4
1.4. Nachweisreaktionen
Als Nachweisreaktionen eignen sich vor
allem FarbFarb- und Fällungsreaktionen.
Zur ersten Orientierung können
sogenannte „Vorproben“ direkt an der
„Ursubstanz“ vorgenommen werden.
Um sicher zu gehen ist aber eine
Trennung des erhaltenen
Substanzgemisches unumgänglich
1.4.1. Vorproben
Hierher gehören allgemeine
Untersuchungen:
Säure-- BaseSäure
Base- Verhalten
Redox-- Verhalten
Redox
Flammenfärbungen und andere
Farbreaktionen
Fällungsreaktionen mit „Gruppen„GruppenFällungs--Reagenzien“
Fällungs
Geruchstests und Geschmackstests
1.4.2. Einzelnachweise
Es gibt Reagenzien, die absolut spezifisch
auf eine einzige Substanz reagieren
Viel häufiger sind aber „Stör„Stör- Reaktionen“
anzutreffen, d.h. ein Reagenz reagiert
ähnlich oder gleich mit mehreren
Substanzen, oder die Anwesenheit einer
Substanz verhindert oder verändert die
Nachweisreaktion einer anderen Substanz
5
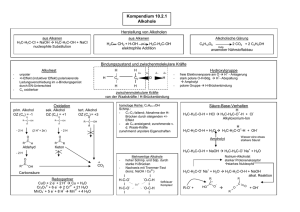
Kapitel 2: Lösungsversuche
Wasser ….trivial!
HCl: Kationen und/ oder Anionen werden
durch Protonen in eine protonierte Form
überführt: Beisp.: CO32- ⇒ HCO3HNO3 und Königswasser: Kationen und/
oder Anionen werden oxidiert: Beisp.: S2⇒ SO42-
… Lösungsversuche
Wichtig:: Sobald wir Substanzgemische
Wichtig
haben, beeinflussen sich die
verschiedenen Bestandteile gegenseitig in
ihrem Lösungsverhalten!!!
…Lösungsversuche
Beispielsweise sind BaCl2 und Na2SO4 für
sich allein gut wasserlöslich. „Löst“ man
ihre feste Mischung in Wasser auf, bildet
sich mehr oder weniger sofort
schwerlösliches BaSO4!
BaCO3 ist in Wasser schwerlöslich, löst
sich hingegen in verdünnter HCl. Ist
gleichzeitig Na2SO4 anwesend, fällt das
gerade in Lösung gebrachte Ba2+ sofort
wieder als BaSO4 aus
6
… Schmelzaufschlüsse
Zur Theorie: Es handelt sich nicht um
Reaktionen in wässriger Lösung! Beim
Aufstellen von Reaktionsgleichungen
haben also „H2O“, „H3O+“ und dergleichen
nichts verloren (außer als verdampfendes
Wasser)!
Zur Praxis: Gutes mechanisches
Vermengen hilft! Naturgemäß hohe
Temperaturen beim Schmelzen bergen
Verbrennungsgefahr!
Der SodaSoda-Pottasche
Pottasche-- Aufschluss
Gemisch von Na2CO3 + K2CO3 zur
Schmelzpunkterniedrigung („Eutektikum“)
Wichtig ist nur das Carbonat, das in der
Schmelze die Rolle von OH- in wässriger
Lösung übernimmt:
CO32- ≡ CO2 + O2OH- ≡ H2O + O2-
Beispiel: Aluminiumoxid
Al2O3 ist amphoter und kann sowohl
alkalisch als auch sauer aufgeschlossen
werden:
Al2O3 + Na2CO3 ⇒ 2 NaAlO2 + CO2
Al2O3 + 6 KHSO4 ⇒
Al2(SO4)3 + 3 K2SO4 + 3 H2O↑
7
Boraxperle- auch eine Art
Boraxperle„Schmelzaufschluss“!
„Borax“ = [Na(H2O)4]2 [B4O5(OH)4]
Na2B4O7*10H2O (T>350°
(T>350°C)
C)→
→ Na2B4O7
T>878°°C
T>878
Na2B4O7 → 2 NaBO2 + B2O3
T>878°°C
T>878
MSO4 + B2O3 → M(BO2)2 + SO3
M = Fe, Co, Ni, Cu, Mn, Cr
Beispiel: CuCu-Borat
Kapitel 3: Trennungsmethoden
Klassischer Kationentrennungsgang
Anionentrennung
Chromatographie
8
3.1. Der „klassische KationenKationenTrennungsgang“
3.1.1. Die HClHCl- Gruppe: Ag+, Pb2+, Hg22+
3.1.2. Die H2S- Gruppe: Hg2+, Pb2+, Bi3+,
Cu2+, Cd2+, As3+, Sb3+, Sn2+
3.1.3. Die AmmoniumsulfidAmmoniumsulfid- UrotropinUrotropinGruppe: Co2+, Ni2+, Mn2+, Zn2+, Fe2+/3+,
Al3+, Cr3+
3.1.4. Die Ammoniumcarbonatgruppe: Ca2+,
Sr2+, Ba2+
3.1.5. Die „lösliche Gruppe“: Li+, Na+, K+,
NH4+, Mg2+
3.1.1. Die HClHCl- Gruppe
Prinzipiell als erster Schritt des
Trennungsgangs durchführbar
Da Chloride aber grundsätzlich schwer
quantitativ ausfällbar sind (Probleme mit
der Stöchiometrie, Bildung löslicher
Chlorido-- Komplexe), lässt sich
Chlorido
„Verschleppung“ in die H2S- Gruppe selten
vermeiden
⇒ Meist direkter Einstieg in die H2S- Gruppe
3.1.2. Die H2S- Gruppe
Hierher gehören alle Sulfide, die im stark
Sauren schwerlöslich sind
Für eine weitere „Vorsortierung“ werden
solche Sulfide, die im Alkalischen im
Sulfidüberschuss lösliche Thiokomplexe
bilden können von denen, die diese
Fähigkeit nicht besitzen, abgetrennt:
Kupfergruppe und Arsengruppe
9
3.1.3. Die AmmoniumsulfidAmmoniumsulfid- Gruppe
Hierher gehören diejenigen Sulfide, die im
NH3/NH4+- Puffer schwerlöslich sind
Aufgrund der hierfür nötigen pHpH- Erhöhung ist
die gleichzeitige Mitfällung solcher Ionen, die
schwerlösliche Hydroxide bilden, unvermeidbar!
Um hier eine Vorsortierung zu erreichen, trennt
man zunächst die Hydroxide ab: UrotropinUrotropinGruppe
3.1.4. Die Ammoniumcarbonat
Ammoniumcarbonat-Gruppe
Hierher gehören die Ionen, die im leicht
alkalischen Bereich schwerlösliche
Carbonate bilden
Da dies im wesentlichen die Erdalkalien
sind, wird der Trennungsgang ab jetzt
recht „farblos“
3.1.5. Die „lösliche“ Gruppe
Hierher gehören die restlichen Ionen, für
die es kein gemeinsames Fällungsreagenz
gibt
Problematisch: Mg2+ (fällt unter
Umständen schon vorher im
Trennungsgang aus, da sowohl Hydroxid
als auch Carbonat relativ schwerlöslich
sind)
10
3.2. Die Fällungsreagenzien
Schwefelwasserstoff und Thioacetamid
Ammoniumsulfid
Urotropin
Ammoniumcarbonat
3.2.1. Schwefelwasserstoff
Farbloses Gas; übelriechend; hochgiftig
Intrinsische Löslichkeit in Wasser unter
Standardbedingungen ca. 0.1 (mol/l)
Sehr schwache zweibasige Säure mit
pKa1 = 6.7 und pKa2 ≈ 17.0
(Fehler im „Jander„Jander-Blasius“ um 4 pKpKEinheiten!) ⇒ Sulfid ist sehr starke Base,
pKb = -3.0!
3.2.2. Thioacetamid
Amid der Thio(n)essigsäure
Hydrolysiert, v.a. beim Erwärmen nach:
CH3C(=S)NH2 + H2O → CH3C(=O)NH2 +
H2S ((→
→OAc- + NH4+ + HS-)
11
3.2.3. Ammoniumsulfid
Salz aus der schwachen Säure NH4+ (pKa
= 9.25) und der starken Base S2- (pKb = 3): das GG: NH4++S2-⇔NH3+HS- liegt
vollständig auf der rechten Seite: K>107
Deshalb ist auf den mit „Ammoniumsulfid“
beschrifteten Flaschen im wesentlichen
eine Mischung aus NH3, NH4+ und SHvorhanden
3.2.4. Urotropin
Kondensationsprodukt aus Ammoniak und
Formaldehyd mit „Adamantan„Adamantan- Struktur“
Beim Erhitzen in Wasser zerfällt U. in
Umkehrung seiner Bildungsgleichung in
NH3 und CH2O :
N4(CH2)6 + 6 H2O ↔ 4 NH3 + 6 CH2O
Urotropin--II
Urotropin
Der gebildete Ammoniak reagiert mit
Wasser gemäß:
NH3 + H2O ↔ NH4+ + OHSowohl NH3 als auch OH- können mit
Metallionen reagieren:
Mn+ + m NH3 ↔ [M(NH3)m]n+
Mn+ + n OH- ↔ M(OH)n ↓
12
3.2.5. Ammoniumcarbonat
Zunächst gilt auch hier, dass wegen der
Acidität von NH4+ und der Basizität von
CO32- (pKb ≈ 3.5) nur HCO3- als Anion
vorliegt
Aufgrund eines parallel erfolgenden
nucleophilen Angriffs von NH3 am
Carbonat-- Kohlenstoff gefolgt von H2OCarbonat
Eliminierung liegt auch
Ammoniumcarbaminat vor:
[NH4+][OC(=O)NH2-]
3.3. Anionentrennungsgang
3.3.1. Die Ca(NO3)2- Gruppe
Gruppe:: F-, CO32-, SiO44-,
BO33-, SO32-, PO43-, C2O42-, SiF62-, SO42-, (AsO43-)
3.3.2. Die Ba(NO3)2- Gruppe: SO42-, SiF62-,
IO3-, (CrO42-)
3.3.3. Die Zn(NO3)2- Gruppe: S2-, CN-,
/4([Fe(CN)6]3-/43.3.4. Die AgNO3- Gruppe: Cl-, Br-, I-, SCN-,
S2O32-, IO3-, BrO3-
Der SodaSoda- Auszug
Zur Anionentrennung wird die Ursubstanz
mit einer konzentrierten Lösung von Soda
(Na2CO3) in großem Überschuss gekocht
Nach Filtration bleibt eine Lösung, die
Sodaauszug („SA“) genannt wird
Grundgedanke ist „Le„Le-ChatelierChatelier-Prinzip“:
MXn + Na2CO3 ↔ „MCO3“ ↓ + n Xx-
13
3.3.1. Die Ca(NO3)2- Gruppe
Hierher gehören alle Anionen, die in
schwach alkalischer Lösung schwerlösliche
Ca
Ca-- Salze bilden
SA wird mit 2m NaOH versetzt und dann
mit Ca(NO3)2-Lösung gekocht
Weitere Trennung dann durch Teillösen
mittels Essigsäure
3.3.2. Die Ba(NO3)2- Gruppe
Hierher gehören alle Ionen, die in
schwach alkalischer Lösung schwerlösliche
BaBa-salze bilden
Weitere Auftrennung mittels neutralem
Wasser und verd. HCl
3.3.3. Die Zn(NO3)2 - Gruppe
Hierher gehören die Ionen, die im
schwach alkalischen (Carbonat(Carbonat-Pufferung)
schwerlösliche Zinksalze bilden
Weitere Auftrennung mittels HCl
14
3.3.4. Die AgNO3- Gruppe
Hierher gehören alle Ionen, die im
schwach HNO3- sauren Milieu
schwerlösliche AgAg- Salze bilden
Filtrat der letzten Gruppe mit NH3versetzen, 5% AgAg-NO3- Lösung zusetzen,
erhitzen, mit verd. HNO3 ansäuern und
aufkochen
Weitere Trennung mittels gesättigter
Ammoniumcarbonat-- Lösung
Ammoniumcarbonat
3.4. Chromatographie
3.4.1. Definition und Historisches
3.4.2. PhysikalischPhysikalisch- chemische Grundlagen
3.4.3. Systematik: Die wichtigsten
Methoden im Überblick
3.4.4. Im Praktikum verwendete Methoden
3.4.1.1. Definition
„Chromatographie“ ist allgemein eine
Bezeichnung für eine Vielzahl physikalischphysikalischchemischer Trennverfahren, bei denen die
zu trennenden Komponenten zwischen
einer ruhenden („stationären“) und einer
sich bewegenden („mobilen“) Phase
verteilt werden
15
3.4.1.2. Historisches
1822: F.J. Runge: Auftrennung von
Farbstoffkomponenten durch Auftragung
eines Tropfens auf Löschpapier:
Historisches--II
Historisches
1903:M. Tswett: Auftrennung
der Farbstoffe des Blattgrüns
auf mit einem Absorbens
gefüllten Trennsäulen (⇒
(⇒“Farb“FarbSchreiben“)
Bildquelle:
http://www.waters.com/waters/nav.htm?locale=de_DE&cid=10048919
16
Historisches--III
Historisches
1942: Martin & Synge: FlüssigFlüssig-FlüssigFlüssigVerteilungschromatographie: Nobelpreis
1954
1951: James & Martin:
Gaschromatographie
1975: Ionenchromatographie
3.4.2. Physikalisch
Physikalisch-- chemische
Grundlagen
Vorbedingung für Auftrennung eines
Gemischs ist, dass die verschiedenen in
der mobilen Phase gelösten Substanzen
eine unterschiedliche Affinität zur
stationären Phase haben: je höher die
Affinität zur festen Phase, um stärker wird
die gelöste Substanz an dieser
festgehalten
Die Wechselwirkungen der verschiedenen
Bestandteile des Analyten beruhen auf
Adsorptions-- und/ oder
Adsorptions
Verteilungsgleichgewichten
Sowohl Thermodynamik als auch Kinetik
dieser Prozesse sind für die Effektivität
einer Trennung von Bedeutung
17
3.4.2.1. Verteilungsgleichgewichte
Beispiel: 2 nicht mischbare Flüssigkeiten
und eine Substanz, die sich in diesen
Flüssigkeiten löst
Das Nernst‘sche Verteilungsgesetz sagt,
dass der Verteilungskoeffizient α = c1 / c2,
wobei ci die Konzentration des gelösten
Stoffes in der Flüssigkeit „i“ ist, bei
gleichen äußeren Bedingungen konstant
ist
Anwendungsbeispiele
Alle Extraktionsverfahren; im
Labor z.B. „Ausschütteln“ im
Scheidetrichter
Entkoffeinierung von Kaffee:
Roselius-- Verfahren (mit
Roselius
Benzol), direktes Verfahren
und indirektes Verfahren (mit
Dichlormethan)
Bildquelle: http://www.restena.lu/ddnuc/COURS/2/220m.htm
3.4.2.2. Adsorptionsgleichgewichte
Grenzflächenreaktion zwischen einem
gelösten Stoff und einer festen Oberfläche
Man unterscheidet Physisorption und
Chemisorption
18
3.4.2.2.1. Physisorption
Schwache Wechselwirkung (van(van-derderWaals--Kräfte, Eads 8- 40 kJ/mol)
Waals
Sehr geringe Aktivierungsenergie
Voll reversibel
Ideal geeignet für chromatographische
Trennungen
3.4.2.2.2. Chemisorption
Starke Wechselwirkung (chem. Reaktion,
evtl. Bindungsbruch, Eads = 80 -600
kJ/mol)
Kinetisch meist stark gehemmt (hohe
Aktivierungsenergie)
Häufig irreversibel
3.4.3. Systematik
Einteilungsmöglichkeiten nach
verschiedenen Gesichtspunkten:
3.4.3.1. Aggregatzustände der beiden
Phasen
3.4.3.2. apparative Trenntechnik
3.4.3.3. grundlegender
Trennmechanismus
3.4.3.4. relative Polarität von mobiler und
stationärer Phase
19
3.4.3.1. Aggregatzustände der
Phasen
Quelle: http://www.analytchem.tugraz.at/institut/download/Instrumentelle_Analytik_Teil_4.pdf
3.4.3.2. Apparative
Trenntechnik
3.4.3.3. Trennmechanismus
Adsorptionschromatographie:
Adsorptionschromatographie:
klassische Form der
Flüssigkeitschromatographie. Verwendung
stationärer Phasen mit polarer Oberfläche
(Kieselgel oder Aluminiumoxid)
Verteilungschromatographie:: heute
Verteilungschromatographie
am häufigsten eingesetzte Methode;
weitere Unterteilung in Flüssig
Flüssig--Flüssig
Flüssig-Chromatographie und Chr. an chemisch
gebundenen Phasen
20
Verteilungschromatographie
Klassische Variante: stationäre flüssige
Phase wird durch Physisorption
festgehalten: Nachteil ist die leichte
Auswaschbarkeit der flüssigen Phase
Stationäre Phase durch chemische
Bindung am Träger fixiert: Normalphasen
Normalphasen-und Umkehrphasen
Umkehrphasen-- Chromatographie;
Spezialfall Ionenaustausch
Ionenaustausch-Chromatographie
3.4.3.4. Relative Polarität
Lösungsmittelpolarität
21
3.4.4. Im Praktikum verwendete
Methoden
3.4.4.1.
3.4.4.2.
3.4.4.3.
3.4.4.4.
Papierchromatographie
Dünnschichtchromatographie
Ionenaustauscher
Ionenchromatographie
3.4.4.1. Papierchromatographie
Zur Einstimmung:
http://www.youtube.com/watch?v=NTDEY
X4TtZg
Im Prinzip dasselbe wie bei den vor 200
Jahren beschriebenen RUNGERUNGE- Bildern
Einfach, schnell, empfindlich (µg(µg-Mengen),
vielseitig (biologische Flüssigkeiten,
Hydrolysate hochmolekularer Verb.,
anorgan. Salzmischungen)
Trennprinzip
Papier nimmt Wasser aufauf- es bildet sich
ein Cellulose
Cellulose-- WasserWasser- Komplex,
Komplex, der
als stationäre Phase wirkt
Durch Kapillarkräfte wird das
Chromatographie-- LM angesaugt
Chromatographie
Trennung der Analysensubstanz entspricht
einem mehrstufigen „Ausschütteln“
zwischen mobiler flüssiger Phase und
diesem Komplex
22
Durchführungsmöglichkeiten
Eindimensionale P.C. : aufsteigende und
absteigende P.C.
Zweidimensionale P.C.
Rundfiltertechnik
3.4.4.2.
Dünnschichtchromatographie (DC)
Weiterentwicklung der
Papierchromatographie mit größerer
Anwendungsbreite, da keine
Beschränkung auf wäßrige
Lösungsmittelsysteme
Wieder was Interessantes aus dem
Internet:
http://www.lehrer--online.de/dc.php
http://www.lehrer
DC: Vorteile
Geringer apparativer Aufwand
Rel. hohe Leistungsfähigkeit bei der
Trennung kleinster Mengen
Sehr niedrige Nachweisgrenzen
Breite Variabilität der stationären Phase
Freie Indikatorwahl
Trennung sowohl hydrophiler als auch
lipophiler Substanzen möglich, auch
präparativ
23
DC: Möglichkeiten
Verwendung von „DC„DC-Platten“ aus Glas
oder „DC„DC-Folien“ aus Aluminium oder
Kunststoff
Für analytisches Arbeiten ist eine ca.
250µm (Glaspl.)/200µm (Alupl.) dicke
Schicht von im Mittel 1010-12 µm großen
Sorbenzien-- Partikeln zusammen mit
Sorbenzien
einem polymeren Trägermaterial
aufgetragen
DC: Sorbenzien
Kieselgel, klassisch oder chemisch
modifiziert (silanisiert, CN, diol , NH etc.)
Aluminiumoxid
Cellulose
Kieselguhr
Durchführung
Ähnlich wie bei Papierchromatographie:
Auftragen mit Kapillare am unteren Ende
der Platte (mind. 1cm Abstand!)
Eintauchen in DCDC- Kammer, die Eluens am
Boden ca 5 mm hoch enthält; Gasraum
muss lösungsmitteldampflösungsmitteldampf- gesättigt seinseinDeckel zu!
24
3.4.4.3. Ionenaustauscher
Kationen oder Anionen gehen eine
schwache ionische Bindung mit der
stationären Phase ein
Kationen benötigen einen
Kationenaustauscher
Anionen einen Anionenaustauscher
In beiden Fällen sind sowohl stationäre als
auch mobile Phase ionischer Natur
http://www.chemieunterricht.de/dc2/iat/
Stationäre Phasen
Auch Carboxylat oder Phosphonat-anker
Mobile Phase
Beim Kationenaustausch z.B. verd. HCl,
HNO3, Weinsäure, Zitronensäure…
Beim Anionenaustausch z.B. verd.
Sodalösung, verd. KOH, Phthalat, Benzoat
25
Trennmechanismus
Eine nette Animation im Internet:
http://www.chemgapedia.de/vsengine/vlu
/vsc/de/ch/9/mac/werkstoff_polystyren/an
wendungen/psx/psx_ionenaustauscher.vlu
/Page/vsc/de/ch/9/mac/werkstoff_polystyr
en/anwendungen/psx/ionenaustausch/ion
enaustauscher.vscml.html
IA: Selektivität
„Größeres Ion“ verdrängt kleineres (also
z.B. K+ verdrängt Na+, I- verdrängt NO3-)
Höhergeladenes Ion verdrängt
niedergeladenes (also z.B.
Al3+>Ca2+>Na+, oder PO43->SO42- )
Schwächer gebundene Ionen in großem
Überschuss können stärker gebundene
verdrängen („Regeneration“)
26
IA: Anwendungen
Wasserenthärtung (Leitungswasser,
Aquarium)
Waschmittel
Präparative Trennung von Lanthanoiden
…
IA: Durchführung
„Frisches“ IAIA-Harz muss mit Wasser
vorgequollen werden
Überführen in die „H„H-Form“ bei KationenKationenA., in die „OH„OH-Form“ bei AnionenAnionen-A.
Aufgeben der Analysenlösung
3.4.4.4. Ionenchromatographie
„High-Tech“ großteils automatisierte
„HighVariante der Chromatographie von
Ionengemischen
Trennprinzipien weitgehend identisch mit
den Vorgängen in den bisher behandelten
„manuellen“ Ionenaustauschersäulen
Detektion der eluierten Ionen mittels
Leitfähigkeitsmessung
27
IC: Prinzipieller Aufbau
Ionenleitfähigkeit
28
Kapitel 4
Einzelnachweise im nasschemischen
Trennungsgang und „Chemie der
Elemente“
Einzelnachweise und Chemie der
Elemente
4.1. Hauptgruppenelemente
4.2. NebengruppenNebengruppen-Elemente
Ionen im Praktikum LAACLAAC-I
Cu2+, Bi3+, Sb3+, Sn2+
Co2+, Zn2+, Mn2+, Fe2+/3+, Al3+
Li+, K+, NH4+, Ca2+, Ba2+ (Mg2+, Sr2+)
F-, Cl-, Br-, IS2-, SO42NO3-, PO43CO32-, C2O42-, SiO2
29
4.1.1. Die Ionen der 1.
Hauptgruppe
Li+, Na+ und K+ gehören alle der „löslichen
Gruppe“ an
Der einfachste Nachweis erfolgt über die
Flammenfärbung, ist aber wegen seiner
großen Empfindlichkeit (Spuren(SpurenVerunreinigungen!) etwas riskant
4.1.1.1 Lithium
Ähnlichkeiten mit Mg („Schrägbeziehung“)
In der Natur hauptsächlich als Silikat oder
Phosphat
Schwerlöslich sind LiF, Li2CO3, Li3PO4
NW durch Fällung von Phosphat im
Alkalischen
4.1.1.2. Natrium
Viele mineralische Vorkommen in der
Natur, jedoch für Nachweisreaktionen
keine geeigneten Vorbilder (entweder
Löslichkeit zu groß –NaCl, NaNO3,
Na2SO4- oder zu komplex für Analytik –
Feldspate)
Als Fällungsform geeignet ist NaSb(OH)6 –
allerdings ist vorherige Abtrennung aller
anderen Ionen außer K+ im
Trennungsgang erforderlich!
30
4.1.1.2. Kalium
Auch beim Kalium lässt sich aus den mineralischen
Vorkommen kein analytisch brauchbarer Hinweis auf
schwerlösliche Verbindungen erhalten
Aufgrund der ähnlichen Größe werden alle
Fällungsreaktionen des K+ von NH4+ gestört!
Am besten geeignet ist die Fällung von KClO4
aus saurer LösungLösung- spezifisch, aber nicht sehr
empfindlich
4.1.2. Die Ionen der 2.
Hauptgruppe
Hier wird –etwas willkürlichwillkürlich- das Mg2+ der
„löslichen Gruppe“, Ca2+, Sr2+ und Ba2+
der „Ammoniumcarbonatgruppe“
zugeordnet
Tatsächlich unterscheidet sich das Mg2+
aufgrund seiner relativ hohen Ladungsdichte
deutlich von seinen höheren Homologen: es ist
z.B. das einzige, das nicht durch
Flammenfärbung erkannt werden kann
4.1.2.1. Magnesium
Liegt in Abwesenheit koordinierender Anionen
und neutraler Donoren in Wasser als
[Mg(H2O)6]2+ mit schwach aciden Eigenschaften
vor (pKa= 11.2)
Bei pHpH- Erhöhung (>10) fällt Mg(OH)2 aus
(pKL= 11), an Luft auch MgCO3,(pKL= 5).
Ersteres findet sich in der Natur als
„Brucit“, letzteres als Mineral „Magnesit“
und Bestandteil von „Dolomit“ (mit
CaCO3)
31
4.1.2.2. Calcium
In der Natur in Form vieler schwerlöslicher
Verbindungen wie CaCO3 (Calcit), CaSO4
(Gips), Ca5(PO4)3(OH|F) (Apatit), CaF2
(Fluorit), die gute Anhaltspunkte für
analytische Nachweise durch
Niederschlagsbildung liefern
In reinem Wasser liegt Ca2+ mit 6
6--8
Wassermolekülen koordiniert vor; der
Aquakomplex ist schwach acid (pKa>12)
Calcium II
Ein relativ empfindlicher Nachweis ist die
Fällung des Oxalats bei pH 55-9 mit
Ammoniumoxalat (Ba2+ stört)
Die Fällung von Gips im HCl sauren ist
insofern charakteristisch, als die
entstehende Niederschlagsform als
Büschel von weißen Kristallnadeln von
keinem anderen Sulfat gebildet wird
4.1.2.3. Strontium
In der Natur als Strontianit (SrCO3)
oder Cölestin (SrSO4)
NW durch karminrote
Flammenfärbung
NW durch Fällung als Iodat (Störung:
Ba2+)
32
4.1.2.4. Barium
Auch Barium kommt in der Natur als
Carbonat und Sulfat vor, die allerdings
beide wesentlich schwerlöslicher als die
entsprechenden CaCa- Salze sind
Daneben ist v.a. die Fällung des gelben
Chromats im HOAc/AcetatHOAc/Acetat-Puffer
charakteristisch; sowohl mit CrO42- als
auch Cr2O72- durchführbar
4.1.3. Die Ionen der 3.
Hauptgruppe
Bor würde bei den Anionen (Borat) behandelt
werden
Aluminium kommt im wässrigen System
nur in der dreiwertigen Form vor und
gehört zur UrotropinUrotropin- Gruppe
4.1.3.1. Aluminium
In der Natur vor allem als „Korund“ Al2O3
in verschiedenen Varietäten, als „Bauxit“
AlO(OH)*aq sowie in zahlreichen
Alumosilikaten (Feldspate, Glimmer, Tone)
In Wasser liegt das acide [Al(H2O)63+] (pKa
= 5.0) vor, das eine hohe
Hydrolysetendenz aufgrund der
Schwerlöslichkeit des HydrolyseHydrolyseEndprodukts Al(OH)3 (pKL = 33) besitzt
33
Aluminium II
Als Nachweis eignet sich v.a. das durch
Zusammenschmelzen mit Co(NO3)2 gebildete
„Thenards Blau“ CoAl2O4, am besten aus dem
Trennungsgang, da Störung durch Silikat und
Phosphat
Daneben ist die mit Morin im Essigsauren
entstehende grüne Fluoreszenz ein empfindlicher
Nachweis (ppb(ppb- Bereich!). Hauptstörung durch
NaOH (fluoresziert ebenfalls)
4.1.4. Die Ionen der 4.
Hauptgruppe
C in Form von Carbonat oder Oxalat und
Si als SiO2 oder Silikat werden bei den
Anionen--Gruppen nachgewiesenAnionen
nachgewiesen- in
Wasser gibt es keine CC- oder SiSi- haltigen
Kationen
Zinn (und Blei) fallen in der H2S- Gruppe
aus
4.1.4.1. Carbonat
Bestandteil vieler Minerale
In Wasser liegt wegen der Basizität des
CO32- immer auch HCO3- vor
Aus festen Carbonaten lässt sich durch
Erhitzen oder durch Versetzen mit HCl
gasförmiges CO2 freisetzen; Einleiten des
freigesetzten Gases in ein „Gärröhrchen“
mit Barytwasser (=Ba(OH)2) führt zu
Ausfällung von weißem BaCO3
34
4.1.4.2. Oxalat
NW durch Fällung von CaC2O4 im schwach
Ammoniakalischen; löslich in starken
Säuren
NW durch Reaktion mit KMnO4 unter
Entfärbung und Gasentwicklung:
2 MnO4- + 5 C2O42- + 16 H+ ⇒
2 Mn2+ + 10 CO2 + 8 H2O
4.1.4.3. SiO2 und Silikate
Silikate sind wohl die größte Gruppe im Bereich
der Minerale und haben eine reichhaltige
Strukturchemie
Beim Erhitzen von Silikaten mit starken Säuren
wird über die Stufe oligomerer Kieselsäuren
letztendlich wasserhaltiges SiO2 abgeschieden
Beim Erhitzen im Sodaauszug bzw. im SodaSodaPottasche--Aufschluss bilden sich Na
Pottasche
Na-- Salze der
Ortho-- und/oder Meta
Ortho
Meta--Kieselsäure
Silikate II
Bester Nachweis ist die „Bleitiegel„Bleitiegel- Probe“
Durch Zugabe von CaF2 und H2SO4 bildet sich
HF: CaF2 + H2SO4 ⇔ CaSO4 + 2 HF
HF reagiert mit SiO2 zu gasförmigem SiF4; das
dabei mitgebildete Wasser wird von der konz.
H2SO4 gebunden: SiO2 +4 HF ⇔ SiF4 + 2H2O
Das SiF4 hydrolysiert am feuchten Filterpapier in
Umkehrung seiner Bildungsgleichung: Weißer
Fleck durch SiO2*aq
35
4.1.4.4. Zinn
In der Natur v.a. als Sn(IV) in Zinnstein
(SnO2)
In wässriger Lösung sind sowohl Sn(II) als
auch Sn(IV) bekannt:
Bei pH=0 liegen [Sn(H2O)3-n(OH)n]2-n mit n= 0
und 1 vor; pHpH- Erhöhung führt zu [Sn3(OH)4]2+;
an Luft leicht Oxidation zu SnO2 (1<pH<11.6)
(E
(E°°Sn(IV)/Sn(II)= 0.15V)
Zinn (II)
In Gegenwart von Halogeniden Bildung
von Komplexen [SnX3]-; mit verd. HCl
(0.01 m) Ausfällung von [SnCl(OH)(H2O)]
Bei hohem pH Bildung von [Sn(OH)3]-
Sn(II) - II
H2S fällt SnS ; auch im Überschuss von
Sulfid und pHpH- Erhöhung keine Bildung
von Thiokomplexen
SnS wird durch Polysulfide zu [SnS3]2oxidiert; LiOHLiOH- Lösung löst es langsam
unter Bildung von [Sn(OH)3]-, bzw. in
Gegenwart von KNO3 B. von [Sn(OH)6]2-
36
Sn(IV)
In „normalen“ pHpH- Bereichen keine
Lösungschemie wg. Schwerlöslichkeit von
SnO2
In verd. Lösungen liegt bei pH=0 [Sn(H2O)6]4+,
bei pH=14 [Sn(OH)6]2- vor; in Gegenwart von
Chlorid [SnCln(H2O)6-n-m(OH)m]4-n-m
Sn(IV)-- II
Sn(IV)
Bei pH=0 fällt H2S SnS2; bei höherem pH
und hohen Sulfidkonzentrationen bilden
sich [SnS3]2- {= [SnS2S2/2]2-} und [SnS4]4-,
die etwas besser löslich sind
Zinn-- Nachweise
Zinn
Unabhängig von der AusgangsAusgangsOxidationsstufe ist der beste Nachweis die
„Leuchtprobe“, die bei Abwesenheit von
Nitrat und Arsen auch aus der Ursubstanz
durchgeführt werden kann
Reduktion mittels Zn zu elementarem Sn
als schwarzer „schwammiger“ Belag
37
4.1.4.5. Blei
In der Natur v.a. als „Bleiglanz“ PbS, als
„Cerussit“ PbCO3, „Anglesit“ PbSO4 sowie
„Rot„Rot-“ und „Gelbbleierz“ (Chromat und
Molybdat) und gediegen
In Wasser ist die zweiwertige Stufe
dominierend, da Pb(IV) (zumindest
thermodynamisch) Wasser oxidieren kann
(E
(E°°Pb(IV/II)=1.46 V)
Blei (II)
In verdünnter Lösung als [Pb(H2O)4] 2+ und [Pb(H2O)6]2+
, sonst schnelle Hydrolyse und Kondensation zu
[Pb4(OH)4] 4+ und [Pb6O(OH)6] 4+ bzw. [Pb(OH)n]2-n
(n=2,3 4)
HX (X=F,Cl,Br,I) fällen PbX2 ,im Überschuss
löslich zu [PbX3]- und [PbCl4]2- (Nachweis!)
H2S fällt auch im stark sauren PbS (schwarz)
Blei (II)(II)-II
PbS kann entweder durch oxidative
Zerstörung des Sulfids mittels HNO3 oder
durch Komplexierung mit großem
Sulfidüberschuss als [Pb(SH)3]- in Lösung
gebracht werden.
In der Kälte fällt verd. H2SO4 weißes
PbSO4; Kaliumchromat fällt im schwach
Essigsauren gelbes PbCrO4 (Störung
durch Ba2+)
38
4.1.5. Die Ionen der 5.
Hauptgruppe
Stickstoff als NH4+ in der löslichen Gruppe,
als Nitrit und Nitrat bei den Anionen
Phosphor als Phosphat bei den Anionen
(Arsen,) Antimon und Bismut gehören zur
H2S- Gruppe
4.1.5.1. NH4+
Aufgrund ähnlicher Größe ähnliche
Fällungsreaktionen wie K+
Bester Nachweis ist Verreiben der
Ursubstanz mit NaOH: Bei Gegenwart von
NH4+ wird Ammoniak freigesetzt:
erkennbar am Geruch, oder durch
Blaufärbung eines angefeuchteten pHpHIndikator-- Papiers
Indikator
4.1.5.2. Nitrit NO2Entsteht in der Natur durch mikrobielle
(Nitrosomonas u.ä.) NH3/ NH4+Oxidation:
(a) NH4+ + 0.5 O2 → NH2OH + H+
(b) NH2OH + O2 → NO2- + H3O+
Nachweis durch die „Ringprobe“
(zahlreiche Störungen!) oder durch
„Lunges Reagenz“ (ebenfalls zahlreiche
Störungen!)
39
Die „Ringprobe“
Störungen durch Halogenide und Sulfid
durch vorheriges Versetzen mit Ag2SO4Lösung beseitigen!
Ansäuern mit verdünnter Schwefelsäure
NO2- + [Fe(H2O)6]2+ + 2H+→
NO + [Fe(H2O)6]3++ H2O
NO + [Fe(H2O)6]2+ ⇔ [FeIII(H2O)5(NO)]2+ +
H2O
Lunges Reagenz
Der Nachweis beruht auf der Azokupplung
eines DiazoniumDiazonium-Salzes und einem
aromatischen Amin; das DiazoniumDiazonium- Salz
entsteht aus einem anderen aromatischen
Amin und salpetriger Säure (pKa= 3.3), :
HOAc + NO2- ⇔ HNO2 + OAcStörung durch SH-, Br-, I- (Entfernung
durch Ag2SO4), CrO42- (Entf. d. BaCl2) und
Fe3+
4.1.5.3. Nitrat
In Abwesenheit von Nitrit ebenfalls mit
„Ringprobe“ bzw. „Lunges Reagenz“
nachweisbar (erstere ohne
Vorbehandlung, da: NO3- + 4 H+ + 3e- →
(E°°= 0.96V) letzteres nach
NO + 2 H2O (E
Reduktion mittels Zn/HOAc )
Reduktion mit Zn und NaOH: Ammoniak!
NO3- +4 Zn +3 OH- +6H2O→4[Zn(OH)3]+NH3↑ (Störung: Nitrit und NH4+)
40
4.1.5.4. Phosphate HnPO43-n
Aufgrund der hohen Basizität von PO43liegen in „normalen“ pHpH-Bereichen nur
HPO42- und H2PO4- vor
Bester Nachweis ist Fällung im stark
Salzsauren mit ZrOCl2 zu weißem gallertgallertartigen Zr3(PO4)2 (Störung nur durch
Silikat)
Fällung der gelben 1212Molybdophosphorsäure muss genau nach
Vorschrift durchgeführt werden!
4.1.5.5. Arsen
In der Natur v.a. sulfidisch als
„Auripigment“ As2S3 und „Realgar“ As4S4
Im wässrigen System sind zwei
Oxidationsstufen beständig, III und V
Bester Nachweis ist die Marsh‘sche Probe,
die unabhängig vom Ausgangsmaterial zu
hochtoxischem AsH3 führt
4.1.5.6. Antimon
In der Natur als „Grauspießglanz“ (Sb2S3)
und „Weißspießglanz“ (Sb2O3)
In Wasser sind sowohl Sb(III) als auch
Sb(V) stabil
Nachweise: Marsh‘sche Probe und
„Eisennagelprobe“ funktionieren
unabhängig von der AnfangsAnfangsOxidationsstufe
41
Sb(III)
Im pHpH-Bereich 22-11 als „Sb(OH)3“
(pKa≈12)
In Salzsäure liegen diverse ChloridoChloridoKomplexe vor, in konz. HCl sogar [SbCl6]3H2S fällt im Sauren Sb2S3,
In schwach salzsaurer Lösung Reduktion
durch Fe zum Element unter Abscheidung
schwarzer Flocken („Eisennagelprobe“)
Sb2S3
Mit Ammoniumpolysulfid bildet sich
lösliches Tetrathioantimonat:
Sb2S3 + 2 Sx2- + 3 S2- → 2 SbS43- + 2 Sx-12Mit einem Gemisch aus LiOH und KNO3
bilden sich gemischte ThioThio-Oxido
Antimonate (III):
Sb2S3 + 6 OH- → 2 SbS3-nOn3- + 3 H2O
Sb(V)
Bei pH>4 (verd.) bzw. >6 (konz.) dominiert
[Sb(OH)6]-, im Sauren [H3O]+[Sb(OH)6]-,
eine einbasige Säure mit pK≈
pK≈ 2.5
In konz. HCl Bildung von [SbCl6]-, auch aus
festem Sb2S5:
Sb2S5 + 12 HCl ⇔ 2 [SbCl6]- + 5 H2S + 2 H+
Reduktion mittels Eisen liefert wieder Sb0:
2 [SbCl6]- + 5 Fe → 2 Sb
Sb↓↓ + 5 Fe2+aq + 12
Cl
42
Die Marsh‘sche Probe
Cu
Cu--Zn + 2 H3O+ → Cu + Zn2+ + 2H +
2H2O
2 H → H2
Sb2O3 + 12 H → 2 SbH3 + 3 H2O
4 SbH3 + 2 H2 + 4 O2 → 4 Sb
Sb↓↓ + 8 H2O
4.1.5.7. Bismut
In der Natur als Oxid („Bismutocker“),
Sulfid („Bi(„Bi-glanz“) und Selenid („Se(„Se-Bi
Bi-glanz“)
In Wasser nur Bi(III) von Bedeutung, da
Bi(V) als starkes Oxidationsmittel Wasser an
Licht zu Sauerstoff oxidiert (E°
(E°= 2.03 V)
3+
Bei pH 0 als [Bi(H2O)9] mit pKa = 1.0;
schnelle Hydrolyse zu [Bi6O4(OH)4]6+
(„Bismutylsalze“) und höherkernigen
Clustern
Bismut-- II
Bismut
Im HClHCl-sauren liegen neben BiCl3 diverse
Chlorido-- Komplexe vor
Chlorido
H2S fällt Bi2S3
Mit Iodid bildet sich schwarzes BiI3, im
Überschuss zu orangem [BiI4]- löslich, das
mit oxin als hellrotes Oxiniumsalz gefällt
werden kann (Nachweis!)
43
4.1.6. Die Ionen der 6.
Hauptgruppe
Im Praktikum kommen nur die schwefelschwefel-haltigen
Anionen Sulfid und Sulfat vor
SO42- - Nachweis mit salzsaurer BaCl2 – Lösung
(Fällung von weißem BaSO4)
Störung durch F- und SiF62S2- - Nachweis durch Ansäuern mit HCl: das
gebildete H2S ist durch seinen typischen Geruch
erkennbar, oder durch Schwärzung von
feuchtem BleiacetatBleiacetat- Papier
4.1.7. Die Ionen der 7.
Hauptgruppe
Im Praktikum kommen alle einatomigen
Halogenide (außer At-) vor
Wie häufig im PSE beobachtet,
unterscheidet sich das leichteste
Homologe, F-, deutlich von seinen
schwereren Homologen, Cl-, Br- und I(Basizität, Oxidierbarkeit,
Fällungsreagenzien)
4.1.7.1. Fluorid
In der Natur als Flußspat, Fluorapatit und
Kryolith (CaF2, Ca5(PO4)3F und Na3AlF6)
Hohes Komplexierungsvermögen für
höherwertige Kationen (wie Al3+ oder
Fe3+), die dadurch „maskiert“ werden
können
Nachweise beruhen meist auf Bildung des
hoch
hoch--aggressiven HFHF- deshalb VORSICHT
44
Fluorid-- Nachweise
Fluorid
„Bleitiegelprobe“
„Kriechprobe“: mit heißer konz. H2SO4
gebildetes HFHF- Gas steigt in der
hochviskosen Säure nur langsam nach
oben
„Ätzprobe“: gebildetes HF löst Silikat aus
dem Glas unter Bildung von SiF4 heraus,
wodurch die Struktur des Glases
oberflächlich zerstört wird: Trübung!
4.1.7.2. Chlorid
In der Natur v.a. in „Salz“„Salz“- Lagerstätten,
also v.a. NaCl , KCl und MgCl2
Bester Nachweis ist die Fällung mit
salpetersaurer AgNO3- Lösung zu weißem
(lichtempfindlichen!) AgCl. Dieses löst sich
beim Kochen in AmmoniumcarbonatAmmoniumcarbonatLösung:
AgCl + (NH4+/NH3/HCO3-) ⇔ [Ag(NH3)2]+
+ Cl- + H2O + CO2
4.1.7.3. Bromid
Leichter als Chlorid oxidierbar: dies nutzt
man durch Oxidation mit Chlorwasser aus:
2Br- + Cl2 → Br2 + 2Cldas gebildete Brom kann an seiner
braunen Farbe erkannt werden, am besten
durch Ausschütteln mit Chloroform
„Überoxidation“ führt zu weitgehender
Entfärbung aufgrund der Bildung von BrCl
45
Bromid--II
Bromid
Das mit HNO3-AgNO3- Lösung gebildete
gelbe AgBr lässt sich durch Kochen mit
Ammoniumcarbonat-- Lösung nicht
Ammoniumcarbonat
auflösen; dies gelingt entweder durch
konz. Ammoniak oder durch Reduktion
mittels Zink und verd. Schwefelsäure:
2 AgBr + Zn ⇔ 2 Ag + Zn2+ + 2 Br-, bzw.
AgBr + Hnasc + H2O ⇔ Ag + H3O+ + Br-
4.1.7.4. Iodid
Bildet mit großen „weichen“ Kationen zahlreiche
schwerlösliche Niederschläge; AgI löst sich auch
nicht in konz. Ammoniak, aber in Natriumthiosulfat
(„Fixiersalz“) oder Zn / H2SO4
Aufgrund seines relativ niedrigen Redoxpotentials
wird es von zahlreichen Oxidationsmitteln (H2SO4,
NO2-, MnO2 etc.) zum Element oxidiert, erkennbar
an der violetten Farbe
4.2. Die Ionen der
Nebengruppen
Im Praktikum kommt aus der 1. Gruppe
nur das Kupfer vor, das der H2S-Gruppe
zugerechnet wird
Aus der 2. Gruppe tritt nur das Zink auf,
das zur AmmoniumsulfidAmmoniumsulfid- Gruppe gehört
Aus der 3.3.-5. Nebengruppe werden im
Praktikum keine Ionen bestimmt
46
Dem Trend der ersten drei Gruppen
folgend, kommen aus diesen Gruppen nur
die jeweils leichtesten Homologen im
Praktikum vor
Alle Elementspezies werden durch die
Vorbehandlung im Lauf des
Trennungsgangs in die 22- oder dreiwertige
Stufe überführt, wodurch erstere in der
Ammoniumsulfid--, letztere in der
Ammoniumsulfid
Urotropingruppe „landen“
4.2.1. Das Kupfer
In der Natur gediegen, sulfidisch
(„Kupferkies“ CuFeS2, „Kupferglanz“ Cu2S,
„Buntkupferkies“ Cu3FeS3) oder als
Carbonat („Malachit“ CuCO3*Cu(OH)2,
„Azurit“ 2CuCO3*Cu(OH)2)
In Wasser kommt Cu(I) nur in Form
einiger weniger Komplexe vor, ansonsten
dominiert Cu(II)
Cu(I)
Hohe thermodynamische
Disproportionierungs-- Tendenz:
Disproportionierungs
2 Cu+ ⇔ Cu2+ + Cu (K= 106),
aber starke kinetische Hemmung
Bei hohem pH wegen Schwerlöslichkeit
von Cu2O umgekehrt Komproportionierung
(K=1023)
47
Cu(I)-- II
Cu(I)
Bei RT werden durch Halogenide (Cl-, Br-,
I-) und Pseudohalogenide (CN-, SCN-)
schwerlösliche Niederschläge CuX
gebildet; diese sind v.a. in der Wärme im
Überschuss von X- löslich, z.B. [CuCl4]3(ß4 = 105.6) oder [Cu(CN)4]3- (ß4 = 1028)
Cu(II)
In Wasser ohne weitere koordinierende
Liganden liegt [Cu(H2O)5]2+ vor, das eine
schwache Säure (pKa= 8.0) darstellt.
Durch pHpH- Erhöhung über 10 fällt Cu(OH)2
aus, das im HydroxidHydroxid- Überschuss unter
Bildung von Hydroxokomplexen löslich ist
Mit nicht allzu konzentriertem Ammoniak
bildet sich der „Tetrammin„Tetrammin- Komplex“,
Cu(II)-- II
Cu(II)
Durch H2S wird
schwarzes CuS gefällt,
auch „Covellit“ genannt.
Entgegen der Erwartung
enthält dieses kein Cu(II),
sondern nur Cu(I), und
sollte besser als „Cu2S2“
formuliert werden; in
Lösung liegt vermutlich
[Cu3S3(H2O)6] vor
G.W.Luther III et al.
48
Quelle: siehe letzte Folie
4.2.2. Zink
In der Natur als ZnS („Zinkblende“ und
„Wurtzit“), ZnCO3 („Zinkspat“, „Galmei“)
und Zn4(OH)2(Si2O7)*H2O („Hemimorphit“)
In Wasser nur als Zn(II), und zwar als
[Zn(H2O)6]2+ in verd. Lösungen und pH<7,
bei hoher Konzentration als [Zn(H2O)4]2+,
bei pHpH- Erhöhung zunächst Ausfällung von
Zn(OH)2 und Wiederauflösung als
[Zn(OH)4]2-
Zink-- Nachweise
Zink
Im Acetatpuffer wird durch H2S weißes
ZnS gefällt
Beim Glühen mit wenig(!) Co(NO3)2 bildet
sich „Rinmanns
„Rinmanns Grün“:
Grün“:
x ZnO + (1(1-x) Co(NO3)2 → ZnxCo1-xO +
(2
(2--2x) NO2 +(1
(1--x)/2 O2
Mit rotem Blutlaugensalz entsteht ein
braungelber Niederschlag:
3Zn2+ + 2[Fe(CN)6]3- → Zn3[Fe(CN)6]2 ↓
49
4.2.3. Chrom
In der Natur hauptsächlich als Chromeisenstein
FeCr2O4 , als farbgebende Komponente im
„Rubin“ sowie im „Krokoit“ PbCrO4
Auch im Wasser sind die beiden
Oxidationsstufen (III) und (VI) stabil, wobei die
höherwertige aufgrund ihrer einerseits größeren
Löslichkeit, andererseits deutlich höheren
Toxizität ein Umweltproblem darstellt
Chrom (III)
Im Sauren violettes [Cr(H2O)6]3+, im stark
Alkalischen grünes [Cr(OH)6]3-,
dazwischen AquaAqua-HydroxoHydroxo- Komplexe und
Ausfällung von grünem Cr(OH)3
In Gegenwart von Cl- reversible Bildung
von oktaedrischen ChloroChloro- Komplexen mit
bis zu drei ChlorChlor- Liganden
Nachweise für Cr(III)
Oxidationsschmelze macht aus grünem
Cr(OH)3 gelbes Chromat:
2Cr(OH)3 + 3NO3- + 2CO32- → 2 CrO42- +
3NO2 + 2CO2 + 3H2O
Die gleiche Oxidation läuft auch
nasschemisch beim „alkalischen Sturz“ mit
NaOH/ H2O2 ab:
2 Cr3+ + 3 H2O2 + 10 OH-→ 2CrO42- +
8H2O
50
Cr(VI)
Als Chromat CrO42-, das mit sinkendem pH über
[Cr2O7]2- in polymeres [CrnO3n+1]2- übergeht
Zugabe von saurem H2O2 führt zu blauem
tetraedrischem [HCrO6]- , das mit
überschüssigem H2O2 zu Cr(III) unter O2Entwicklung reduziert wird; in Anwesenheit von
Ether geht das neutrale CrO5 in die organische
Phase über
Cr(VI) - II
Die Reduktion von schwefelsaurem
Chromat mit Ethanol unter Grünfärbung ist
Grundlage der AlcotestAlcotest- Röhrchen, kann
aber auch als ChromatChromat- Nachweis
verwendet werden.
Die Fällung rot bis orange gefärbter
Schwermetallchromate wie BaCrO4 oder
PbCrO4 kann ebenfalls zum
Chromatnachweis verwendet werden
4.2.4. Mangan
In der Natur vor allem oxidisch (MnO2
„Pyrolusit“, MnO(OH) „Manganit“, Mn3O4
„Hausmannit“) und als Carbonat („Manganspat“
MnCO3
Die wässrige Chemie wird v.a. durch Mn(II)
bestimmt, das das thermodynamische
Energieminimum im Sauren darstellt, und dem
kinetisch stabilen Permanganat, das sich in der
Natur nicht findet
51
Mn(II)
Bei pH<7 als [Mn(H2O)6] 2+ mit geringer
Hydrolysetendenz
Im Alkalischen instabil gegenüber Oxidation zu
Mn(III) und v.a. MnO2 (fällt aus), mit Brom wird
violettes Permanganat gebildet:
2 Mn2++5Br2+16OH- → 2MnO4-+10Br-+8H2O
Im Sauren werden stärkere Oxidationsmittel
benötigt, um zu MnO4- zu oxidieren:
2 Mn2++5PbO2+4H+ → 2MnO4-+5Pb2++2H2O
Mn(VII)
Permanganat wird im Kationentrennungsgang
aufgrund der sauren reduzierenden
Bedingungen (HCl+H2S) zu Mn2+ reduziert. Es
kann allerdings im Sodaauszug auftreten und
muss dort wegen seiner intensiven Farbe durch
Reduktion beseitigt werden (z.B. Oxalat)
VORSICHT bei der Reaktion mit konz. H2SO4:
EXPLOSIONSGEFAHR!
2 MnO4- + 2 H+ → Mn2O7
+ H2O
4.2.5. Eisen
In der Natur sulfidisch als „Pyrit“ FeS2,
„Magnetkies“ FeS, oxidisch als „Hämatit“
Fe2O3, „Magnetit“ Fe3O4 sowie zahlreichen
Silikaten und Carbonaten
In Wasser sind unter Luftausschluss Fe(II)
und Fe(III) stabil, an Luft ist Fe(II) nur in
Gegenwart stabilisierender
Komplexliganden längere Zeit haltbar
(E
(E°°FeII/III= 0.77 V)
52
Fe(II)
Bei pH<7 als schwach blaugrünes
[Fe(H2O)6]2+ mit geringer
Hydrolysetendenz zu ausschließlich
einkernigen AquaAqua-HydroxidoHydroxido- Komplexen;
bei höherem pH Ausfällung von Fe(OH)2
In konz. HCl liegt tetraedrisches [FeCl4]2vor
Hohe Affinität zu Cyanid, Bildung von
[Fe(CN)6]4- mit ß6 = 1036
Fe(II)-- II
Fe(II)
Im pHpH- Bereich um 7 wird mit HS- schwarzes
„FeS“ gefällt
Durch Zugabe von rotem Blutlaugensalz wird
eine kräftig blaue Farbe erzeugt, „Berliner Blau“
mit dem Anion [FeIIFeIII(CN)6]-,
An Luft, v.a. bei höherem pH, wird leicht Fe(III)
gebildet, sodass der sehr empfindliche Nachweis
mit Rhodanid meist auch hier funktioniert
Fe(III)
Aufgrund der hohen Acidität des an Fe3+
koordinierten Wassers liegen in diesem bei
üblichen Konzentrationen zweizwei- oder
dreikernige AquaAqua-HydroxoHydroxo- Komplexe vor
In warmer konz. HCl liegt [FeCl4]- vor,
während bei RT transtrans-[Fe(H2O)4Cl2]+
gebildet wirdwird- beide Ionen sind kräftig
gelb
53
Fe(III)-- II
Fe(III)
Mit Fluorid werden diverse Komplexe gebildet;
die Existenz von [FeF6]3- in wässriger Lösung ist
noch nicht bewiesen
In Abwesenheit von Komplexbildnern fällt bei
„normalem“ pH Fe(OH)3 aus
Als Nachweisreaktionen geeignet sind die
Bildung von Berliner Blau bei Zugabe von
gelbem Blutlaugensalz sowie die Rotfärbung bei
Zugabe von Rhodanid:
Fe(III)-- III
Fe(III)
Fe3+aq + [[Fe(CN)
Fe(CN)6]4-→ [FeIIFeIII(CN)6][Fe(H2O)4Cl2]+ + SCN- + H2O ⇔
[Fe(H2O)5(SCN)]
(SCN)]+ + 2 Cl- (u.a. …)
4.2.6. Cobalt
In der Natur meist mit Nickel
vergesellschaftet; Hauptmineralien sind
„Speiscobalt“ CoAs3, „Cobaltglanz“ CoAsS
und Cobaltkies Co3S4
In Wasser sind nur die Oxidationsstufen II
und III von Bedeutung, wobei letztere in
Abwesenheit stabilisierender Liganden
wegen seiner Oxidationskraft weniger
wichtig ist
54
Co(II)
Bei pH<7 liegen oktaedrisches
[Co(H2O)6]2+ und tetraedrisches
pH- Erhöhung unter
[Co(H2O)4]2+ im GG; pHLuftausschluss führt zu Fällung von
Co(OH)2 bzw. löslichen tetraedrischen
[Co(H2O)4-n(OH)n]2-n mit n=3 -4 bzw. in
Gegenwart von NH3 zu [Co(NH3)6]2+; an
Luft werden stattdessen entweder
Co2O3*aq ausgefällt oder [Co(NH3)6]3+
gebildet
Co(II)-- II
Co(II)
In Gegenwart von Chlorid lässt sich die Bildung
von blauem tetraedrischem [CoCl4]2- nachweisen
(ß4 = 10-2.6)
Mit ammoniakalischer AmmoniumsulfidAmmoniumsulfid- Lösung
bildet sich unter Luftausschluss zunächst
Co(OH,SH)2, das sich langsam in eine Mischung
aus Co1-nS und Co9S8 bzw. unter Luftzutritt in
Co(OH)S bzw. „Co2S3“ umwandelt
Co(III)
In Wasser nur in Gegenwart „starker“
Liganden, die lowlow-spin
spin-- Komplexe bilden,
thermodynamisch stabilisierbar
(Ausnahme: [CoF6]3- ist HighHigh-spin, aber
trotzdem stabil)
Kinetische Stabilisierung durch hohe
Acidität: blaues [Co(H2O)6]3+ bei pH=0
haltbar, bei pHpH- Erhöhung parallel
Hydrolyse (pKa≈2) und Wasseroxidation
(E
(E°° CoII/III = 1.84V)
55
Cobalt-- Nachweise
Cobalt
Im Trennungsgang wird der gealterte SulfidSulfidNiederschlag mittels HOAc/ H2O2 gelöst, wobei
Sulfid zu Sulfat oxidiert und Co(III) zu Co(II)
reduziert werden
Co(II) reagiert im Essigsauren mit NH4SCN zu
blauem Co(SCN)2, das im Reagenzüberschuss
zum blauen Komplex [Co(NCS)4]2weiterreagiert. Beim Schütteln mit Ether nimmt
dieser eine blaue Farbe an (Co(SCN)2 und/oder
H2[Co(NCS)4]. Störung durch Fe(III)
4.2.7. Nickel
In der Natur häufig mit Fe, Cu, Co
vergesellschaftet! Daneben eigene
Mineralien v.a. in Verbindungen mit Sulfid
und Arsenid
In Wasser ist nur Ni(II) wichtig: grünes
Hydrolyse[Ni(H2O)6]2+ mit geringer HydrolyseTendenz zu {[Ni(H2O)3]4(µ3-OH)4}4+; pH
pH-Erhöhung führt entweder zu Ni(OH)2Ausfällung oder Bildung von blauviolettem
[Ni(NH3)6]2+
Nickel-- Nachweise
Nickel
Bester Nachweis ist aus dem Trennungsgang (Auflösung
des HClHCl-unlöslichen Sulfidniederschlags mit essigsaurem
Wasserstoffperoxid) die Fällung des roten
Diacetyldioxim-- Komplexes bei pH 7Diacetyldioxim
7-9
Störung durch Fe(III) und Co(II), die braunrote
Färbungen hervorrufen
HNO3 und H2O2 zerstören den organischen Liganden
und müssen deshalb vorher entfernt werden
56
Kapitel 5
Die organischen Farbreagebzien
bei der Chromatographie
5.1. Dithizon
5.1.1. D. als Komplexligand
57
5.2. PAN
N
N
N
OH
1-(2-Pyridylazo)-naphthol
5.2.1. PAN als Komplexligand
5.3. Oxin
58
5.3.1. Oxin als Komplexligand
5.4. Diphenylcarbazid
Ende
59