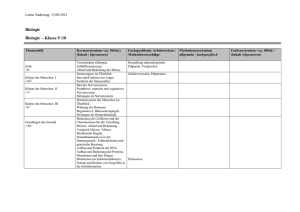
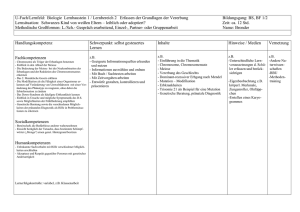
Klinische Humangenetik
Werbung

Klinische Humangenetik Bücher: – jörg schmidtke: Vererbung und Ererbtes , 2. aufl, guc, 2002 – tariverdian, buselmaier, humangenetik, 3 . aufl springer Gentechnologie: – klassische (seit Menschengedenken) Züchtung, Reproduktion... Genmanipulation: veränderung von Menge, Qualität oder Funktion genetischer Information durch menschliche Einwirkung a) direkt b) indirekt indirekt z.B. Agamie Polygamie Homogamie Erb- und Ehegesundheitsgesetzgebung Eheverbote „modern“: Kontrazeption / Abtreibung Reproduktive Kompensation „Germinal choice“ (Eugenikerquark, nobelpreisträgerbesamung) Genetische Beratung Sterilisation „assortative mating“ „Paarungssiebung“ „Homogamie“: gleiche Iqs gesellen sich zueinander Moderne Gentechnologie: 1859 Evolution durch Selektion (Darwin) 1883 Eugenik (genetischer Reduktionismus) 1902 Wiederentdeckung Mendels 1915 Chromosomentheorie der Vererbung 1927 Mutation durch Strahlen (2005 Kreationismus / Intelligent Design) --> www.flying spaghetti monster Überschätzung der Rolle der Genetik um 1900 selektionstheorie Darwins – Sozialdarwinismus Anwendung der Mendel'schen Erbregeln auf komplexe mesnchliche Eigenschaften (Verhalten) Genetik als alleinige Ursache sozialen Elends Eugenik gegen „Degeneration“ und „Dekadenz“ Häufigkeit von genetische (mit-)bedingten Sehstörungen (Kurz- und Weitsichtigkeit) bei Bewohnern Afrikas vgl England grössere Häufigkeit von Sehrstörung wird durch Verringerung der natürlichen Selektion mittels Sehhilfen erklärt 1 / 63 Eugenics (Francis Galton 1883) Die ersten Eugeniker waren Pferdezüchter positive Eugenik: Erhöhung der Geburtenzahl in Familien mit „guten“ Genen und v. v. American breeders association statement 1915: sterilisation genetisch „Defekter“ würde Millionen $ in Zukunft einsparen Zwischen 1910 und 1935: Sterilisationsgesetze in 35 Ländern wie USA, DE, Skandinavien „Eugenic is not a science“ (C.C. Li, 1999) Mutationen in Krankheitsverursachenden (rezessiven) Genen kommen mit einer Häufigkeit von 1/20 bis 1/500 und seltener vor. Um eine Genhäufigkeit von 1/200 durch Verhinderung der Fortpflanzung auf 1/400 zu halbieren, bedarf es 200 Generationen. Dies entspricht etwa 6000 Jahren, Auf grund von immer wieder auftretenden Neumutationen könnte eine solche Halbierung niemals erreicht werden. Auswirkungen Eugenik und Sozialdarwinismus: 1. erhöhte Kriegsberetischaft (natürliche Auslese) 2. Einführung von Sterilisationsgesetzen weltweit, Verschärfung Einwanderungsbestimmungen USA 3. Euthanasie und Holocaust DE berühmt in DE: Plötz... Rassenhygiene Kindergeld, soziale Verbesserung für Familien geht auf eugenische Programme zurück Benno Müller-Hill : Tödliche Wissenschaft: Die Aussonderung von Juden, ... Postmoderne Gentechnologie 1944 DNA als genetisches Material 1953 Doppelhelix 1956 46 Chromosomen ... 2001 Human genome project Gentransfer mittels – viren – physikalisch – zellfusion – chemisch inzwischen über 100 transgene tiermodelle wie z.b. wachstumshormon – minderwuchs insulingene – diabetes amyloidgene – alzheimer bsp pax3gen knockout-maus vgl kind triparentale maus 2 / 63 Genmanipulation Veränderung von Menge, Qualität oder Funktion genetischer Information 3 / 63 Argumente für direkte genetische Manipulation: 1. Bekämpfung von Erbkrankheiten (Gentherapie) 2. Schutz vor Virus- und Krebskrankheiten (genetische Impverfahren) 3. Überwindung biologischer Imperfektion (genetische Verbesserung des Menschen) mögliche Anwendung z.B. bei Frankoniaanämie somatische Gentherapie funktioniert bisher nicht... zahlreiche Problemstellen. Vektoren, an- und abschalten von Genen, Immunantwort... Komplikationen waren auch schon lethal: massive Immunantwort auf Adenoviren, insertionelle Mutagenese -> insertion in Nähe Onkogen Verbesserung der Lebenserwartung für CF-Patienten wahnsinnig gestiegen – durch konventionelle Therapie, hier z.B. damit gute Ergebnisse auch ohne Gentherapie mögliches Feld der Zukunft: Tumortherapie Argumente gegen direkte Genmanipulation am Menschen: 1. Mensch ist nicht nur Produkt der Gene sondern ebenso der Umwelt 2. lange Reproduktionszeit 3. . 4. . Defekte können auch teratogen sein (und nichts mit Genetik zu tun haben) Abnahme der Inzucht / Konsanguinität durch Zunahme der Mobilität -> weniger Autosomalrezessive Erkrankungen Wieviel Gene brauchen wir fürn Body? Gehirn 3195 Ösophagus 70 uswusf Genetische und biochemische Komplexität -> Barriere gegen Genmanipulation Vergleich von direkter und indirekter genetische Manipulation am Menschne kategorie direkte indirekt technik gentechnologie verhalten, einfach zeitdauer sehr lange schnell zielsetzung erb-und krebskrankheiten gesellschaftspolitisch gefahr missbrauch extrem hoch hoch Watson schrieb 1997: Konsumergenetik ist denkbar, wenn schlauere kinder machbar wären würde die leute sie haben wollen... Eugenik? Ich möchte gesunde kinder haben Zielsetzung der eugenik: steuerung der individuellen reproduktion durch gesellschaftliche und gesetzliche massnahmen im interesse der „volksgemeinschaft“ zielsetzung der humangenetik: analyse und information üüber struktur, funktion, weitergabe und 4 / 63 pathologie des menschlichen erbgutes -> grundsätzliche Unterschiede schlussfolgerung u.a. gefahr einer schleichenden eugenik in unserer leistungsgesellschaft die gefahr eines genetisch-reduktionistischen weltbildes darf nicht unterschätzt werden v.a. Kommerzialisierung von genetischen testmethoden, sowie materialistsiche grundeinstellung welche der folgenden manipulationen der reproduktion hat keinen einfluss auf den genpool zukünftiger generationen? A) homogamie b) paarungssiebung c) agamie d) therapeutische klonierung !!!!! e) assortative mating (entspr. Homogamie/ paarungssiebung) warum können erbkrankheiten durch eugenische massnahmen nicht grundsätzlich verhindert werden? a. eugenische massnahmen finden keine generelle akzeptanz b. sind kostenintensiv c. neumutationen treten unabhängig von jedweder eugenik auf !!!!! d. die aufklärung der bevölkerung über eugenik ist unzureichend e. die negative eugenik macht die erfolge der positiven eugenik zunichte welcher der folgenden gründe ist nicht für den fehlenden erfolg der gentherapie von erbkrankheiten verantwortlich? 1. insertionelle mutagenese 2. zu geringer einsatz von forschungsgeldern !!!!!! 3. immunologische reaktion gegen vektor-proteine 4. zu geringe transduktionseffizienz von stammzellen 5. problematik des gentransfers in zellen des zns welches der folgenden argumente kann im hinblick afu die erfahrungen der wissenschaftsgeschichte eine genetische manipulation des menschen wahrscheinlich am wenigsten verhindern? 1. der mensch ist nicht nur produkt seiner gene, sondern ebenso produkt seiner umwelt 2. nicht alles machbare darf auch gemacht werden (ethische argumentation) !!!!!! 3. lange reproduktionszeit des menschen 4. genetische komplexität des menschen 5. wissenschaftliche sinnlosigkeit überschätzung der rolle der genetik ab 1970 annahme der unbeschränkten übertragbarkeit von gentechnischen eingriffen bei e. Coli, drosophila , oder maus auf den menschen „der mensch nach mass“ (bild der wissenschaft) „genetisches hiroshima“ (benda) „molekulargenetische endlösung“ (beck) bsp muskeldystrophie duchenne: pränatales screening kann 100%ig den phänotyp vorhersagen dna-fingerprinting -> eindeutige identitätsfestellung klassischer genbegriff (watson-crick) Ein Gen – ein Polypeptid – linear, statisch und deterministisch 5 / 63 neuer, konditionaler Genbegriff (neumann-held) Gen als Prozess in Abhängigkeit von der Umwelt Bsp: Siamkatze: an-und Abschalten von Gen in Abhängigkeit von Körpertemperatur macht schwarze Akren Steuerung der Gen-Aktivität durch Umweltfaktoren und Proteine Umwelt bestimmt Ablesung der Gene zu Großem Teil Alternatives Splicing (wodurch Mensch nur 35000 Gene braucht vgl Drosophila 13600) 6 / 63 Veränderung der genetischen Information durch Alternatives Splicing, RNA-Editing und posttranslationelle Modifikationen (Phosphorylierung u.a.) -> Ein Gen erlaubt viele Proteinsynthesen Bsp Apo B-100 und Apo B-48 entstehen aus gleicher Sequenz Epigenetik (aktiv acetyliert/inaktiv: methyliert): unterschiedlicher Ausprägung bei gleicher Sequenz... Versuch eineiige Zwillinge: Methylierung divergiert im Laufe des Lebens Der neue, konditionale Genbegriff nach Neumann-Held schließt nicht ein: 1. Linearität, Statik und Determinismus !!!!! 2. rna-editing 3. alternatives splicing 4. epigenetik Welcher der folgenden Faktoren führt nicht zu einer Veränderung der genetisch kodierten information auf proteinebene 1. rna-editing 2. alternatives splicing 3. nicht-translatierter 3'bereich eines gens !!!! 4. methylierung 5. posttranslationelle modifikation (z.b. glykosylierung) Klonen anagefangen hats mit den fröschen dolly ist nur 3 jahre geworden ami-firma klont einem seinen lieblingshund (leider klappts sehr selten und die hunde werden auch nicht alt) Frösche: in frühen entwicklungsstadien Klonierung recht erfolgreich, Forscher gaben jedoch auf bei Versuchen der Klonierung aus reifen Organismus -> Problem bereits aktivierter Gene Durch Kerntransfer geklonte Primatenembryonen zeigen Spindel-Defekte und Chromosomenverluste Gründe für das (überwiegende) Scheitern der Klonierung adulter Säugerzellen – Fehler der Reprogrammierung durch das Zytoplasma der entkernten Eizelle , daher aberrante Methylieruns- und Acetylierungsmuster. Führen z.B. zum „fetal overgrowth syndrome“ – Erhöhtes Risiko für chromosomale Aneupoidien auf Grund von mitotischen Spindel-Problemen führen zu frühen Spontanaborten – Persistenz von DNA-Schäden, welche normalerweise während der meiotischen Paarung eliminiert werden (führen zur vorzeitigen Alterung) Klonierung des Menschen 1. Natürlich bzw. spontan: jeder 200. Mensch hat einen klon (eineiiger Zwilling) . Die natürliche Klonierung (Zwillingsbidlung) kann bis zum Blastula-STadium vorkommen (Embryo-Splitting) 2. Künstlich z.b. durch Kerntransfer in eine entkernte Eizelle. Biologische Gründe sprechen dagegen, dass reproduktive Klonierung gesunde Nachkommen erzeugen kann 3. Umwelt: effektivste und extrem erfolgreiche Methode der Klonierung von Menschen (fundamentalistische Religionen; Medien, Internet) 7 / 63 Die Überschätzung der Genetik geschieht in einer Welt, in der das risiko eines menschen, vorgeburtlich abgetrieben, nach der geburt an unterernährung oder drogenkonsum zu sterben,... noch lange zeit sehr viel realer und höher sein wird als das bisher rein theretische risiko, als produkt einer direkten genaminpulation entstehten und leben zu müssen. Welche der folgenden Gründe ist NICHT für das überwiegende Scheitern der konierung von säugetieren aus adulten somazellen verantworltich? 1. fehler der reprogrammierung durch das zytoplasma der entkernten eizelle 2. erhöhtes risiko für chromosomale aneupoidien auf grund von mittischen spindel-problemen 3. persistenz von dnä-schäden 4. unterschiede der prägung (imprinting) zwischen keimbahn- und soma-zellen 5. zu geringe finanzielle unterstützung der entsprechenden Forschung !!! Bedrohung des Menschen durch Gentechnik ? 1. Gründe Gentechnik genetisch manipulierte Lebensmittel (genfood) kennzeichnungsprflicht ab 18.4. 2004 2. Klonierung des Menschen Therapeutisches Klonen Reproduktives Klonen Vorstellung Gesundheit-Krankheit konventionell: gesund oder krank humangenetik: irgendwo dazwischen Früher z.B. autosomal rezessive Erkrankung wie Albinismus: Problem : am Phänotyp der Eltern keine Heterozygotie für Krankheit ablesbar Heute: Humangenetik kann auf genotypischer Ebene analysieren (Einfaches Beispiel: Chromosomenanalyse) Chromosomnomenklatur anhand von Bandenmuster (p, q, ..) Elektronenmikroskopie bringt Humangenetik nix -> kein Informationsgewinn gegenüber Lichtmikroskopie „chromosome painting“ molekularbiologisch macht Translokationen sichtbar Mutationen beim Menschen Genommutationen Triploidie Chromosomenmutationen Trisomie 21 Genmutationen Substituition Triploidie auf grund von digynie (haploides spermium befruchtete eine diploide eizelle Phänotyp: kleine plazenta) von diandrie:(2 spermien+ haploide Eizelle : symmetrische retardierung kind, plazenta riesig) Welches der folgenden phänomene kann nicht auf impriniting zurückgeführt werden? 1. unterschiede zwischen digynen und diandrischen triploiden foeten 8 / 63 2. unterschiedliche methylierung in der männl und weibl keimbahn 3. gene silencing 4. reprogrammierung in der frühen embryogenese 5. biallelische expresssion von autosomalen genen !!!! Schwangerschaft als „Filter“ – Isolierte Fehlbildungen – Komplexe Fehlbildungen und Anlagestörungen – Chromosomenaberrationen – über 90% der Schwangerschaften enden mit einer spontanen Fehlgeburt Neuralrohdefekt (spina bifida aperta) Neuralrohdefekt: Anenzephalie (Defekt beim Verschluss des oberen Neuralrohrs im 2. Schwangerschaftsmonat) Gaumenspalte jedes 200ste Kind kommt auf die Welt mit Chromosomenveränderung (die meisten werden vorher abortiert) Trisomie 16 (Abort) 13.18.21,Sex-Chromosomen lebensfähig Körpergröße hängt ab vom Vorhandensein 1. eines oder mehrerer y-Chromosomen y-Chromosom besteht überwiegend aus Heterochromatin (welches wurscht is) wichtige y-gene sry= sex determining region = TDF AZF = Azoospermie-Faktor Welches der folgenden Gene spielt KEINE Rolle bei der Geschlechtsdifferenzierung? 1. Hgprt !!!!! 2. sry 3. sox9 4. dmrt1 5. sf1 durch welchen faktor wird das männliche gonadale geschlecht biem menschen determiniert 1. anzahl der urkeimzellen 2. testosteron und dihydrotestosteron 3. sry-gen auf yp !!! 4. anzahl der gonosomen 5. zeitpunkt der inaktivierung des x-chromosoms während der embryonalentwicklung 9 / 63 Genetische Beratung im Olymp oder Götter sind auch nur Menschen Genetische Beratung 1. Genetische Diagnostik 2. Risikoberechnung (Translokationstrisomie 21 bis zu 100% , andere teilweise sehr gering) 3. Informationsmitteilung Nicht-direktive Beratung (der Beratene muss selbst entscheiden) Recht-auf-Nicht-Wissen einzelne Syndrome können zu unterschiedlichen Phänotypen führen (mit gemeinsamem Grund“fehlbildungs“bild z.B. der Extremitäten bei griechischen Göttern) Homeobox: einheitliche, sich wiederholende Sequenz Genfamilien Von zentraler Bedeutung für embryonale Entwicklung beim menschen sind v.a. Drei genfamilien: 1. Homeobox (HOX-Gene) 2. Paired-Box (PAX-Gene) 3. Zinkfingergene PAX-gene bsp mutationen im pax3-gen (2q35 : waardenburg-syndrom typ 1 (Schwerhörigkeit, weiße haarsträhne über stirnmitte, iris unterschiedlich eingefärbt) zinkfingerprotein: mutationen im wt1-gen (Wilms-Tumor (Embryonaler nierentumor)) bei starker Blutsverwandtschaft kann sich auch ein seltenes Krankheitsbild, welches autosomal rezessiv ist, weit verbreiten Risikoberechnung auf der Basis der Theorie der Wahrscheinlichkeit Wahrscheinlichkeit 1. Jemand fragt 2. die Antwort ist ungewiss 3. Es gibt Hinweise darauf, ob eine Antwort zu empfehlen ist Bayessche Formel: Erste Arbeit zur Wahrscheinlichkeitsberechnung wir fangen an mit a-priori-wahrscheinlichkeiten -> daraus folgen konditionale wahrscheinlichkeiten „Durchmultiplikation“ -> a posteriori-Wahrscheinlickeit wenn wahrscheinlichkeiten nicth 1 ergeben -> Normierung -> Wahrscheinlichkeit durch Gesamtwahrscheinlichkeitssumme teilen Bsp Auto-Tür-spiel Folgerung: in der genetischen Beratung richtig rechnen und nicht aus dem Gefühl heraus Risiken 10 / 63 angeben! Nach dem tod ihres ehemannes II/2 geht die frau II/3 eine zweite ehe mit dessen bruder II/1 ein. Der einzige sohn aus der ersten ehe ist ebenso wie II/2 und II/4 von rot-grün-blindheit betroffen (abbildung) wie hoch ist das Wiederholungsrisiko, von rot-grün-blindheit betroffen zu sein, für jedes kind der frau II/3 in der zweiten Ehe? 1. 75% 2. 50% 3. 25%!! 4. 12,5% 5. das wiedehrolungsrisiko entspricht der mutationsrate -> rot-grün-Blindheit x-chromosomal rezessiv söhne 50% , töchter 0% es steht fest, dass die schwester eines an muskeldystrophie duchenne leidenden mannes konduktorin ist. Sie möchte ihren vetter heiraten und fragt nach dem risiko für einen sohn aus dieser ehe, ebefnalls an muskeldytrophie vom typ duchenne zu leiden das risiko beträgt: 1. 12,5% 2. 25% 3. 50% !! 4. 75% 5. 100% es wird nur nach sohn gefragt! Im stammbaum bezeichnen a,b,c und d die urgroßelterlichen allele an einem autosomalen locus . Wie groß i´st die wahrscheinlichkeit , dass ein urenkel homozygot für das urgroßväterliche allel a ist? (ein urenkel der von zwei enkeln aus verchiedenen nachkommen gezeugt wird) 1. 1/80 2. 1/64 !!! 3. 1/32 4. 1/16 5. 1/8 -> wahrscheinlichkeiten werden multipliziert! 11 / 63 Mutationen beim Menschen Genommutationen (Triploidie) Chromosomenmutationen (Trisomie 21) Genmutationen – Substitution (Sichelzellhämoglobin) – Deletion (Muskeldystrophie) – Duplikation (Duchenne und Becker) – Insertion (Neurofibromatose I) – Amplifikation (Fragiles-X Syndrom) Processingmutationen – Splicing-Mutationen; Polyadenylierungs-M. – Initiations-Codon-M; Stop-Codon Mutationen welches der folgenden phänomene kann nicht auf impringting zurückgeführt werden 1. utnerschiede zwischen digynen un ddaidrischen triplodien foteten 2. unterschiedliche methylierrung in männl weibl keimbahn 3. gene sliencing 4. reprogrammierung in embryongenese 5. biallelische expression von autosomalen genen !!! Welche aussage zum neuralrohrdefekt (NTD) trifft nicth zu : 1. multifaktorielle Vererbung 2. Häufigkeit regional unterschiedlich 3. Empirisches Wiederholungsrisiko ca. 5% 4. Prävention durch perikonzeptionelle Folsäuresubstitiution 5. Abhängig von paternalem Alter !!! Die nachfolgenden Geschwister eines Kindes mit einer Spina bifida haben ein gegebnüber der allgemeinbevölkerung erhöhtes wiederholungsrisiko für diese fehlbidlung . Welche der folgendne mass nahmen vermindert wiederholungs risikoberechnungdie perikonzeptionelle behandlung 2. vater mit folsäure 3. der mutter mit folsäure!!! 4. vater hydrocortison 5. mutter hydrocortison 6. mutter ascoribinsäure mit welcher Häufigkeit treten Chromosomenanomalien bei Spontanaborten bis zur 12. SS-Woche auf? 40-60% 0,01%, 0,1 %; 0,5-1%; 10% Welche Genom / Chromosomenaberrationen findet man in der Regel NICHT unter Spontanaborten vor der 12. Schwangerschaftswoche 1. triploidie auf grund von digynie 2. monosomie X 3. trisomie 16 4. 45,XY,rob (13q14q) (Robertson'sche Translokation) !!!!!!!!! 5. Triploidie auf Grund von Diandrie Welche der folgenden Genom- oder Chromosomenstörungen wird unter Spontanaborten am 12 / 63 Seltensten gefunden? 1. Trisomie 21 2. Trisomie 16 3. 69,XXY 4. 47, XXY !!!! 5. 45, X y-Chrosomen-Zahl -> Größe (ein Wachstumsinduktor) wichtige y-Gene sry – sex dtermining region = TDF (testis determierender faktor) Azf- Azoospermie-.Faktor Entwicklung Geschlecht Bipotente Gonadenanlage default-Way --> Weiblich (nur abhängig von SF1) , Müllerscher Gang männlich schwieriger (v.a. SRY,.... WT1, SOX9,SF1) , Wolffscher Gang -> sehr viel häufiger Störungen der männlichen Geschlechtsdifferenzierung Welches der folgenden Gene spielt KEINE Rolle bei der Geschlechtsdifferenzierung? 1. HGPRT !!!! 2. SRY 3. SOX9 4. DMRT1 5. SF1 Durch welchen Faktor wird das männliche gonadale Geschlechtbeim Menschen determiniert? 1. Anzahl urkeimzellen 2. testosteron und dihydrotestosteron 3. sry-gen auf yp !!!!! 4. anzahl der gonosomen 5. zeitpunkt der inaktivierung des x-chromosoms während der embyronalentwicklung Androgen-Insensitivitäts-Syndrom (AIS) auf Grund von Mutationen im X-chromosomalen Androgen-Rezeptor-Gen (früher:testikuläre Feminisierung) vagina endet blind, entwickelt hoden (werden chirurgisch entfernt, Gefahr der malignen Entartung wegen dort höherer Temperatur) Chromosomensatz: 46,XY -> Androgenrezeptordefekt, kein Eintritt von Dihydrotestosteron in die Zelle CAIS: complete PAIS: partial MAIS: mild Bei testikulärer Feminisierung (Androgen-Insesitivitäts-Syndrom ) liegt folgende GonosomenKonstellation vor: 1. del Xq 2. XX 3. XY !!!!!! 4. XXY 5. XYY 13 / 63 Welcher Befund ist für die testikuläre Feminisierung (Androgen-Insensitivitäts-Syndrom) charaktersistisch 1. hypophysentumor 2. klitiorsihypertrophie und viriliseriung 3. fehlender uteruns und fehlnede adnexe!!!!!! 4. streak-gonaden und kleinwuchs 5. karyotyp 47, XXY die REIHENFOLGE DER ANTWORTEN KANN SICH ÄNDERN UI UI UI!!!!! adrenogenitales syndrom (AGS) : Viriliseriung des äusseren Genitales bei weiblichem = 46,XX Chromosomensatz (Pseudohermaphroditismus feminisnus) auf Grund angeborerener Nebennierenrinden-Hyperplasie . Häufigste Ursache: 21-Hydroxylase-efizienz mit oder ohne Salzverlust-Syndrom (autosomal-rezessiver Erbgang) ( bei betroffenem kind pränatale diagnose und eingreifen mittels cortisontherapie vorgeburtlich möglich -> viriliseriungsverhinderung) xx-männer: 1. durch ein crossover zwischen xp und yp gelangt das sry-gen auf xp 2. adrenogenitales syndrom (21-hydroxylase-Def.) Xy-Frauen 1. androgen-insensitivitäts-sndrome (testikuläre feminisierung auf Grund einer utation im Androgen-Rezeptor gen auf dem X-chromosom) 2. Ausfall des sry-gens durch mutationen führen zur xy-gonadendysgenesei (swyer-syndrom) = pseudohermaphroditismus masculinus. 3. Bei 70% der fälle von xy-gonadendysgenesie ursache unbekannt (sry-positive fälle) Welche der folgenden Veränderungen ist KEINE erklärung für Frauen mit männlichem chromsomensatz? 1. durch ein crossover zwischen xp und yp gelangt das sry-gen auf xp !!!! 2. mutaion im androgen-rezeptor-gen auf dem x-chromosom 3. xy-gonadendysgenesie( swyer-syndrom) = pseudohermaphroditismus masculinus 4. sry-positive gonadendysgenesie 5. testikuläre feminisierung Gonosomale Polysomien 1. 47,XYY 2. 47, XXX (Triplo-X Syndrom) 3. 47, XXY (Klinefelter-Syndrom) zeigen alle eine liechte iq-verminderung von ca 10-15 punkten im vergleich zu ihren geschwistern Tony, 47XYY -> normaler laborarbeiter, sehr groß, jähzornig, nicht ganz so schlau wie geschwister Claire, 47XXX -> auch nicht smart genug für studium, jetzt sozialarbeiterin, unauffällig v.a. Phänotypisch 14 / 63 klinefelter-syndrom 47,xxy (angedeutete gynäkomastie, schambehaarung begrenzt, kleiner penis + gonaden) – wird idR entdeckt bei kinderwunsch und infertilität (azoospermie / oligospermie) auch leichte iq-verminderung kevin, 7 jahre, variante des klinefelter-syndroms : 49,xxxxy -> iq unter 80 (steigende beeinträchtigung), kleines geburtsgewicht ursache für iq-beeinträchtiung: x-y homologe gene unterliegen nicht der x-inaktivierung (transkribierung auch aus barr-körperchen), nur ca. 15 gene bekannt der iq von menschen mit xxy- xxx, und xyy liegt ca. 10-15 iq-punkte unter dem ihrer euproloiden geschwister was ist die waqhrscheilichste urssache 1. inaktivierung der überzähligen x-chormosomen 2. gehäuftes auftreten von adhs 3. störungen der gonadenfunktion 4. gen-dosiss effekt von x-y homologen genen !!!! 5. diskriminierung und isolation in schule und familie welche der folgenden aussagen triffauf das phänomen der x-inaktivierung NICHT zu: 1. beruht auf diefferentieller Methylierung der beiden x-choromosomen 2. wird während der oogenese aufgehoben 3. breitet sich von der pseudoautosomalen region über das x-chromosom aus !!!!!! 4. erfolgt bei klinefelter -männern 5. führt zu x-chromosomaler mosaik-konstellation bei frauen abhängigkeit der körpergröße vom kompletten oder partiellen verlust des 2. x-chromosoms elke, 5 jahre , turner-syndrom körpergrösse unter der 3. perzentile 95% aller monosomie-x Schwangerschaften enden mit einer spontanen fehlgeburt – idR fehlbildung der lymphwege v.a. Im nackenbereich turner-syndrom chromosomensatz: 45, X (monosomie X) neugeborenes kind: nacken – und fussrücken-ödeme (ganz charaktersistisch!) fehlende entwicklung von menachre und sekundären geschlechts-merkmalen auf grund von gonaden-dysgenesie idR keine kognitiven Einschränungen bis auf Teilleistungsstörungen v.a. Bereich der sozialen Kompetenz einzige ausnahme mit ernster geistiger behinderung: zentrisches Fragment des 2ten x-chromosoms übriggeblieben, 2% aller turnersyndrome soziale kompetenz nicht bewusstsein der gefühle anderer personen um uns herum keine rücksicht auf andere zeit anderer über gebühr beanspruchen ohne realisieren sturheit (nicht zugänglich rationalen argumenten) einfaches reinplatzen in gespräche 15 / 63 körpersprache nicht verstehen auf vorschläge anderer leute nicht reagieren -> männer haben schlechtere social cognition (bei turner-syndrom jene mit x vom vater ebenbürtig normalem mann, die mit x von der mutter (noch) wesentlich schlechter ) turner syndrom (monosomie x) 60% haben maternales x (xmat) 40% xpat vergleich social cognition s.o. Dies lässt auf einen genort für social cognition auf dem x-chromosom schliessen, der dem imprinting phänomen unterliegt und daher nur von dem väterlichen, nicht aber von dem mütterlichen x-choromsom exprimiert wird. Dies könnte erkären, warum 46 , xy männer ,die immer das xmat haben, eine schlechtere socaial cognition aufweisen als 46, xx frauen, ei denn in der hälfte der zellen das xpat aktiv ist welcher befund ist für eine konstitutionelle monosomie x charakteristisch? 1. hypophysentumor 2. klitorishypertrophie und viriliseriung 3. fehlender uteruns und fehlende adnexe 4. streak-gonaden und kleinwuchs !!!! 5. karyotyp 47, XXY welche der folgenden aussagen über gonosomale aberrationen trifft NICHT zu? 1. menschen mit monosomie X sind in der regel geistig behindert !!!! 2. über 90% der konzeptionen mit 45, x enden als spontane fehlgeburt 3. knaben mit 47, xyy sind im durschchnitt grösser als ihre geschweister 4. bei azoospermie besteht der vérdacht auf ein klinefelter – ysndrom 5. triple-x frauen sind fertil welche aussage über das menschliche x-chorommsom trifft nicht zu ??? 1. kann in weiblichen zellen inaktiv sein 2. enthält überwiegend gene für die geschlechtsentwicklung !!!! 3. enthält sehr viele gene für die kognition 4. kann in weibl . Zellen spät replizieren 5. ist bei säurgern evolutionär hoch konserviert defekte des farben-sehens auf dem x-chromosom beispiel: rot-grün-blindheit (hab nur männer) immun-defizienz-gene auf dem x-chromosom cgd = chronische granulomatose was – wiscott-aldrich syndrome scid = severe combined immune deficiency xla – x-linked agammaglobulinämie 16 / 63 xlhm = x-linked hyper-IgM syndrom ab gewissem alter überproportionale zunahme von autoimmunkrankheiten bei frauen: annahme: xchromosomale inaktivierung funktioniert nicht mehr so gut männer sind 1/3 häufiger geistig behindert; da sie bei heterozygotie in der hälfte der zellen das andere x-chromosom einsetzen, können das also eher ausgleichen iq-verteilung von frauen und männern: bei männern viel größere varianz, aber gleicher mittelwert (mehr extreme) -> sehr große studie warum gibt es mehr superschlaue männer als frauen??? durch meiotische paarung und crossover zwischen den maternalen x-chromosomen entsteht ein „super-hyplotyp“ auf dem x, welches der sohn von seiner mutter erhält. In der nächsten generation wrid dieses super-x an eine frau weitergegeben, in deren meiose es wiederum durch crossover zur auflösung des super-haplotyps kommt -> Genies treten also zufällig in Familien auf, aber nachkommen werden nicht derart hochbegabt sein Evolutionäre Anreicherung von Genen für kognitive Fähigkeiten auf dem menschlichen XChromosom 3,1 fach höhere Frequenz von MR-Genen auf dem X-Chromosom als auf den Autosomen. Erklärung? Female mate choice: im laufe der hominiden-evolution bevorzugten frauen bei ihrer partnerwahl männer mit besonders guten geistigen fähigkeiten. Intelligenz brachte somit einen selktionsvorteil. Zusätzlich beeinflussen vilee MRX-gene auch die fertilität. Intelligentere männer hätten somit mehr nachkommen gehabt. Die rapide evolution des menschlichen gehirns kann als folge der bevorzugtenpaarung von frauen mit intelligenten männern gesehen werdne . Syndromale XLMR Gene (neben geistiger auch körperlich auffällige behinderung) Bsp: Fragiles X-Syndrom (brüchige Stelle am langen arm des x-chromosoms) im kindesalter unauffällig, allerdings verlangsamte entwicklung und verhalten zunahme des testikulären volumens nach der pubertät: makroorchidie große ohren als ausdruck einer generalisierten bindegewebsschwäche langes gesicht 17 / 63 non-random-x-inaktivierung: Hypothese, die kranke Frauen in x-chromosomal rezessiven Erbgängen erklären soll lässt aber nicht alle erklären phänomen „transmitting male“ bei fragilem x-Syndrom. Erklärung CGG repeats (auf FMR1-Gen): er liegt zwischen den gesunden und sicher erkrankten in der repeat-zahl zu viele Repeats inaktivieren das gesamte Gen (obwohl es im Prinzip intakt ist) die repeat-Größe kann auch somatisch (im Individuum) instabil sein Triplett-Repeat Expansion erfolgt nur über die weibliche Linie Bei welcher Expansionsgrösse des CGG-Triplets im 5' Bereich des FMR-1Gens kommt es zur geistigen Behinderung 1. 5-25 2. 25-50 3. 50-100 4. 100-200 5. 200-1000 !!!! Welche der folgenden Aussagen über das fragile X-Syndrom triff NICHT zu: 1. sowohl knaben als auch mädchen können betorffen sein 2. ursache ist eine triplett-expansion im 5'Bereich des FMR1-Gens 3. Männer können Überträger sein 4. Die Expansion einer Prämutation erfolgt in der weiblichen Keimbahn 5. 50% der Nachkommen von männlichen Genträgern sind betroffen !!!!! Ein 15jähriger junge gesunder eltern lässt die folgenden symptome erkennen : iq = 50, vergöerte sprachentwicklung , kg 185cm, hohes und schmaels gesicht, große ohren, makroorchidismus . Diagnose? 1. adrenogenitales syndrom 2. xyy Syndrom 3. martin-bell (Fra-x) Syndrom !!!!! 4. xxy-Syndrom 5. testikuläre Feminisierung Klinische Manifestationen bei Prämutations-Trägern (55-200 CGG repeats) – Frauen: POF-Syndrom (Preamture Ovarian Failure) (kommt nur zum Tragen wenn vom Vater Prämutation vererbt) – überstürztes Altern der Eierstöcker – Männer: FXTAS (fragile -x-associated Tremor / Ataxia Syndrom) Toxische RNA: bei verschiedenen Triplettexpansionskrankheiten beim Menschen: in einem mittleren Expansionsbereich versucht die Zelle, mehr zu transkribieren, es wird aber weniger Protein gebildet. Weiteres Bsp.: Myotone Dystrophie (Typ 1 + 2 mit völlig unterschiedlichen phänotypischen Merkmale ) Antizipation: Repeats vermehren sich in der Erblinie (bei Genen die auf Autosomen liegen v.a.) und damit zu immer früherem Auftreten Krankheitsbild und immer schwererem Verlauf 18 / 63 Wenn ein Mann Prämutationsträger einer cgg-expansion im promotor-bereich des fmr1-gens ist, treffen folgende behauptungen NICHT zu: 1. er ist in der regel geistig behindert !!! 2. seine kinder sind nicht betroffen 3. in seinen keimzellen kommt es zur repeat-expansion !!!!!! 4. im höheren alter kann er von fxtas betroffen sein 5. er ist sohn einer überträger-mutter wenn eine frau prämutationsträgerin einer cgg-expansion im promotor-bereich des fmr1-gens ist, trifft folgende behauptung NICHT zu: 1. sie ist tochter eines prämutationsträgers 2. ihre brüder tragen expansionen von über 200 cgg repeats !!!! 3. sie ist in der regel nicht von geistiger behinderung betroffen 4. ihre söhne können geistig behindert sein 5. sie kann von pof betroffen sein frage müsste eindeutiger sein: müsste gesagt werden dass prämutation von der väterlichen linie kommt, ansonsten könnten söhne über 200 repeats haben wenn gen von mutter Klassen und Konsequenzen von Triplett- Repeat Erkrankungen Erkrankung Triplett Konsequenzen Fragiles X-Syndrom Friedreich'sche ataxie myoklonus epilepsie 1 CGG GAA komplex Loss of function Myotone dystrophie ½ Ctg/cctg Toxische rna (Fra-X Prämutationsträger) Cgg Toxische rna) Chorea huntington spinobulbäre Muskelatrophie spinozerebelläre ataxien typ 1,2,3,6,7,17 Cag Gain of function (PolyglutaminErkrankungen) Welche der folgenden aussagen trifft auf triplett-expansions erkrankungen des menschen nicht zu : 1. können zu loss of function führen 2. können zu gain of function frühren 3. können auf rna-ebene toxisch sein 4. können somatisch instabil sein 5. können durch punktmutationen erklärt werden !!!! Autosomale Chromosomenaberrationen: Kardinalsymptome 1. Intrauteriner Wachstumsrückstand (unter 2500g) 2. Perinatale Probleme (z.B. Immaturität; neonatale Asphyxie; prolongierter Ikterus) 3. Dysmorphien (= Gestaltvarianten ohne Funktionsbeeinträchtigung) 4. Mehrfahce Fehlbildungen 19 / 63 (verbunden mit Dysmorphien und Wachstumsrückstand) 5. Statomotorische Entwicklungsverzögerung und geistige Behinderung Trisomie 21 Sonogramm der 13. Woche; Nackentransparenz > 3mm Dysmorphien z.B. Vierfingerfurche (nicht beweisend!), Hypoplasie des 4. Fingers Dysmrphie= Gestaltvariante, die den Träger funktional nicht belastet Beispiele: Epikanthus, Hypertelorismus, schärgstehende Lidachse,... www.ohrenkuss.de Welche der folgenden Veränderungen / Symptome ist NICHT typische für eine autosomale Chromosomenaberration? 1. niedriges Geburtsgewicht 2. perinatale Probleme 3. Anenzephalie !!! 4. multiple Fehlbildungen 5. Statomotorische entwicklungsverzögerung -> isolierte Fehlbildung nicht typisch... Typische Fehlbildungen bei lebensfähigen autosomalen Trisomien Tris13 (meistens tod kurz nach Tris18 (meistens tod kruz nach Tris21 (lebenserwartung bis 60 geburt) geburt) jahre) Holoprosenzephalie lkg-spalte kolobome (spaltbildungen der iris) herz/urogenitalfehlbildungen Herzfehlbildungen hufeisenniere meckel'sches divertikel fingerfehlstellung Ösophagusatreise/ duodenalatresie megakolon septumdefekt grosse zunge (desewegen klassiches mundaufstehenlassen) Abfall des IQ bei Tris21 mit Frühförderung (schon im 1. LJ) wesentlich geringer! Down-Syndrom-Critical Region 21q22 nur 1/3 der gene zeigen trisomen gendosis-effekt 1/3 der gene sogar niedriger exprimiert als bei disomie Mögliche Gendosis-effekte bei tris21 1. ets2-gen (leukämierisiko) 2. gart-gen (zns-schädigung <- purin-überproduktion) 3. alpha-a-kristallin gen (katrakt/linsenfehler) 4. sod1-gen (superoxid-dismutase, radikalfänger – auffällig wenig arteriosklerose) 5. app-gen (amyloid-precursor protein) bei tris21 ab dem 30 lebensjahr häufung sog. „seniler plaques“ in der grosshirnrinde = aggregate von pathologischen spaltprodukten des app-gens -> alzheimersche erscheinungen (verlust kurzzeitgedächtnis) relativ früh 20 / 63 Frauen mit Trisomie21 sind fertil; die Hälfte der Nachkommen haben wiederum eine Trisomie 21 die nachkommen mit 46 chromosomen zeigen ebenfalls kognitive Einschränkungen, wenn sie überwiegend von der trisomen Mutter betreut wurden. Höchster Risikofaktor für Trisomie21: Mütterliches Alter trotz pränataldiagnostik und folgend erhöhter abtreibungsrate zunahme der tris21-geburten wegen immer späterer schwangerschaften (1980 durchschnittsalter 25, heute fast 30) Welche der folgenden Aussagen trifft auf das Down-Syndrom nicht zu: – alle phänotypischen ... müssen ausgeprägt sein.!! Hypothesen zur Erklärung der Zunahme von Aneuploidien mit dem mütterlichen Alter. 1. Weniger effektive („filterfunktion“ in schwangerschaften älterer Frauchen (ImmunseneszenzHypothese – nicht bewiesen da abortrate mit alter von anfangs 15 auf über 30% ansteigt) 2. diktyotän-hypothese (langes ruhestadium der eizelle in der prophase der meiose I) 3. rekokmbinations-hypothese (erniedrigte crossing-over raten in oozyten älterer Frauen) 4. Oozyten-Selektions Hypothese (Keimzellenmosaik) Diktyotän-Phase: erstmal 22 mitotische Teilungen in weiblicher Keimbahn .. nach befruchtung dann 2 meiotische Teilungen = insgesamt 24 Teilungen... ob jetzt prophase der meiose I 15 oder 40 Jahre besteht macht natürlich einen Unterschied Rekombinations-Hypothese:mit zunehmendem Alter weniger Rekombinationen – Verkürzung der genetischen Länge von Chromosom 21 als Ausdruck davon Welche der folgenden Hypothesen kann die erhöhte aneuploidierate bei schwangerschaften älterer mütter NICHT erklären? 1. immunseneszenztheorie 2. diktyotänhypothese 3. oocytenselektions-theorie 4. rekombinations-hypothese 5. replikations-fehler-hypothese !!!!!!!! die der freien trisomie 21 zugrunde liegende nondisjungciton erfolg überweigend in der 1. paternalen meiose I 2. postzygotisch 3. maternalen meiose II 4. maternalen Meiose I !!!!! 5. paternalen meiose I Genetische Altersrisiken Frauen: Nachkommen mit Chromosomenaberrationen (Aneuplidien) Männer: Nachkommen mit Punktmutationen (DNA-replikations-Hypothese) Zunahme des paternalen Alters... männliche Keimbahn 30 mitotische Teilungen bis zur Pubertät 23 Stammzellmitosen pro Jahr sowie 4 mitotische und 2 meiotische Reifeteilungen im 90 Tage 21 / 63 Zyklus gesamtzahl der Teilungen (DNA-Replikationen) bis 28 Jahre: ca 380 35 Jahre: 540 65 Jahre > 1000 bei jeder DNA-Replikation besteht das (geringe) Risiko eines fehlerhaften Basen-Einbaus = Punktmutation Bsp: Neurofibromatose-Wahrscheinlichkeit nimmt mit väterlichem Alter zu, ebenso Achondroplasie und das Apert-Syndrom (Akrozephalosyndaktylie) Apert-Syndrom: überproportionale Zunahme durch Selektionsvorteil der Spermien mit Mutation (trotz späterer schwer negativer Folgen) welche der folgenden aussagen kann den paternalen alterseffekt (eröhthe zahl von nachkommen mit punktmutationen ) nicht erklären: 1. zunahme der zahl der dna-replikationen in spermien älterer männern 2. selektionsvorteil für spermien mit bestimmten mutationen 3. nachlassen der effektivität und przäsion von dna-reperatur mit zunahmenedem alter 4. nachlassen der selektion gegen mutations-tragende spermien !!! 5. kontinuierliche und alterskorrelierte zunahme der zahl mitotischer zellteilugen während der männlichen spermatogenese – die haben sogar manchmal selektionsvorteil wie oben steht... Zytogenetik des Down-Syndroms 90% feie Trisomie 21 (überwiegend maternale Meiose I Fehler, 5% postzygotische Fehlverteilung) 2-5% Mosaike (klinisch u.U. Leichterer Verlauf) 5% Translokationstrisomie (überwiegend t(14q21q) Robertsonsche Translokationen) welche der folgenden aussagen trifft auf robertson'sche translokationen NICHT zu? 1. beruhen auf einer fusion zwischen akrozentrischen chromosomen 2. bedingen ein höheres risiko für die nachkommen von überträgerfrauen als für die nachkommen von überträger-männnern 3. betreffen am häufigsten das chromosom 15!!!!!! 4. sind die ursache von 3-5% aller individuen mit down-syndrom 5. können familiär auftreten stimmt nüscht sondern 14... alternierte vs adjacent Teilung in Meiose bestimmt ob nachkommen translozierten genabschnitt normal 2x oder 3x haben... Folgen von familiären Chromosomentranslokationen (Häufigkeit von Translokationsträgern ca. 1:500) 1. unauffällige nachkommen (50% translokationsträger, 50% unauffällige chromosomen; alternierendes segregationsmuster in der meiose I) 2. gehäufte spontanaborte (unbalanzierte gameten) auf Grund von „adjacent“ segregationsmuster in der meiose I 3. lebendgeborene mit angeobrenen fehlbildungen, dysmorphien und entwicklungsstörungen (unbalanzierte gameten, adjacent-segregation) 22 / 63 4. sub- bzw. infertilität (bei männlichen translokationsträgern) Duplikations-defiziente Gameten entstehen als Folge folgender Segregation in der Meiose I: 1. adjacent I !!! 2. adjacent II !!!! 3. alternierend 4. asynaptisch 5. trivalent was gehört NICHT zu den folgen familiärer chromosomentranslokationen 1. sub- bzw. infertilität bei männlichen carriern !! 2. sub- bzw. infertilität bei weilbcliehn carriern 3. gehäufte spontanaborte 4. uanuffällige nachkommen 5. geburt von genetisch unbalanzierten nachkommen Spontanaborte – Infertilität genetische Untersuchungen: • Chromosomentranslokationen • klinefelter-syndrom • azf-muattionen • mutationen im CF-Gen Häufigste Art Pränataldiagnostik: Amniozentese, 13.-16. Woche, 10-15 ml Fruchtwasser, Zellkulturdauer: 2 wochen; Risiko: 0,5% invasive Pränataldiagnostik Chorionzottenbioplsie Amniozentese 10. SW 16. SW Gewebsentnahme durch Scheide und Muttermund Transabdominale Punktion durch das Peritoneum in die Fruchtblase Direktpräparation und Kurzzeitkultivierung Mehrere parallele Langzeitkulturen Dauer Chromosomenanalyse 1 tag bis 1 woche 2 ½ bis 3 wochen Fehlgeburtsrate 3,5-4% 0,5-1% Welche der folgenden aussagen trifft auf die amniozentese nicht zu: 1. hat hinsichtlich des chromosomenbefundes ein niedrigeres fehlerrisiko als eine chorionbiopsie 2. wird bei einer 30jährigen mutter empfohlen, die bereits ein kind mit einer lkg-spalte hat !!!!!!! 3. ist die methode der wahl zur pränatalen diagnostik von chromosomenstörungen 4. gestattet die durchführung einer fish-schnelldiagnostik 5. hat ein durchschnittliches fehlgeburtsrisiko von unter 1% da kann man höchstens hochauflösendes ultraschall machen lkg ja chirurgisch korrigierbar welche der folgenden methoden der genetischen pränataldiagnostik gehört zur kategorie der nichtinvasiven methoden: 23 / 63 1. 2. 3. 4. 5. farbkodierte sonografie!!! amniozentese chorionzottenbiopsie plazentazentese nabelschnurpunktion Strukturelle Chromosomenaberrationen: Deletionen Grundsätzlich hat der Verlust von genetischem Material (Monosomie) im menschlichen Genom schwerweigendere Auswirkungen als die Duplikation von gentischem Material (z.B. Trisomie) Partielle Monosomien sind z.T. Lebensfähig, z.B. del 4p, 5p (cri-du-chat syndrom), 7p (mit prämaturer synostose, kurzer turmschädel) Mikrodeletionssyndrome submikrokopische deletionen , welche mit hilfe von in situ hybridisierung (FISH() mit spezifischen dna-sonden erkannt werden können . In den meisten fällen handelt es sich um sogenannte „contiguous gene syndromes“, d.h. Mehrere benachbarte gene in der deletions-region... BLA BLA williams-beuren syndrom: mikrodeletion 7q11 z.B. (u.a. Elastin-Gen) Aortenklappenstenose; häufig auch Pulmonalstenose 100te Mikrodeletionssyndrome bekannt... Prader Willi, MillerDieker (Lissenzephalie), DiGeorge... , Catch 22 (deletion 22q11... cardiac defects, abnormal facies, thymus hypoplasia, cleft palate, hypocalcemia synonyme: DiGeorge, velocardiofaciales syndrom) Welcher der folgenden Befunde ist nicht typisch für das CATCH 22... 1. hypokalzämie 2. thymusaplasie 3. konotrunkale herzfehlbildung 4. gaumenspalte 5. Duodenalatresie !!! die findet man bei kindern mit DOWN-Syndrom welches ist die methode der wahl zur diagnostik von mikrodeletionssyndromen 1. FISH fluoreszen-in-situ-hybridisierung mit spezifischen DNA-Sonden !!! 2. chromosomenanalyse mit GTG-bänderung 3. dna-sequenzierung nach sanger 4. chromosomen painting 5. spektrale karyotypisierung noch ein mikrodeletionssyndrom: prader-willi-syndrom adipositas per magna (bedingt durch hyperphagie, kein sättigungsgefühl) mittelschwere geistige behinderung hypoton (muskeln) (mutter merkt wenig bewegung in schwangerschaft, danach schlecht beim saugen usw) therapie: kühlschrank abschliessen, schild „bitte nicht füttern“ umhängen 70% deletion 15q pat 30% maternale uniparentale disomie (offensichtlich imprinting phänomen! 24 / 63 Angelman-Syndrom: > 90% Deletion 15qmat ; 3% paternale uniparentale disomie Prader-willi-Syndrom: 70% Deletion 15q pat , 30% maternale uniparentale disomie UPD uniparentale disomie 7 : zygote mit tris7; verlust eines chromosoms 7 in der frühen embryogenese ... „trisomie rescue“ bei 1/3 bei „trisomie rescue“ evtl diploide plazenta, triploider embryo -> diese unwägbarkeit reduziert verlässliichkeit der chorionbiopsie im vergleich zur amniozentese wir alle tragen ca. 20 autosomal rezessive krankheitsgene, bei UPDs gefahr der verdopplung derselben -> Krankheit in der nächsten generation was ist die häufigste ursache für die entstehung einer uniparentalen disomie (UPD)? 1. unequal crossing over in meiose I 2. imprinting defekt der männlichen keimzellen 3. dispermie 4. postzygotisch trisomie-reduktion !!!! 5. konsanguinität bei welchem der folgenden syndrome scheidet ein impringting defekt als ursache aus ? 1. transienter neonataler diabetes mellitus 2. angelman-syndrom 3. russel-silver-syndrom 4. klinefelter-syndrom !!!! 5. prader-willi-syndrom mutter: heterozygot für delta f508 vater: keine mutation im cf-gen nachweisbar kind: zystische fibrose (autosomal rezessiv) wie geht daS? Häufigste erklärung: mutter hat quergehauen oder UPD oder Neumutation 90% menschlicher neoplasien: monoklonaler ursprung selektionsvorteil führt zum wachstum eines tumors leiomyome, AML, burkitt-lymphom (atherom) 10%: multizellulär neurofibromatose, trichoepitheliome, verrucae (warzen) Neoplasie: die genetische bzw epigenetische veränderung ist notwendig, aber nicht hinreichend zur entstehung der malignen zelle. sekundär entscheidend ist, ob die genetisch oder epigenetisch veränderte zelle der apoptose entgeht und einen selektionsvorteil (Proliferationsvorteil ) gegenüber unveränderten zellen hat 25 / 63 genetische veränderungen bei krebs 1. gen-mutation genetische aktivierung von proto-onkogenen = wachstumsreguulierende gene, bsp: retprotoonkogen statt sich zu differenzieren und ihre spezischeifschen funktionen auszuüben, teilt sich die zelle immer weiter 2. gen-transloaktion (genomumbau ) vor allem bei leukämien = blutkrebs paradebeispiel: philadelphia translokation bei CML 3. gen-deletion (gen-verlust) vor allem bei soliden tumoren, die in teilungsfähigen geweben entstehen bsp: retionblastom, neuroblastom, nephroblastom, mammakarzinom, kolonkarzinom betrifft idR sogenannte „tumorsuppressorgene“ durch den gen-verlust differenziert sich die betroffene stammzelle nicht, sondern teilt sich immer weiter medulläres schilddrüsenkarzinom (MTC) als leit-manifestation von MEN2 = multiple endokrine neoplasie typ 2 (missense mutationen im RET-gen) genotyp-phänotyp korrelationen beim medullären schilddrüsen ca als leitmanifestation von MEN2A (calcitonin-positives c-zell ca; aktivierende missense mutationen im RET-gen = gain of function!) mutation führt je nach codon zu stark unterschiedlichem erkrankungsalter fämiliäres medulläres schilddrüsenkarzinom (FMTC) 1. 3-4 familienmitglieder von calicitonin-pos-C-zell-karzi8nomen betroffe n 2. autosomal dominanter erbgang 3. penetranz nahezu 100% 4. nachweis von mutationen im RET-proto-onkogen 5. genotyp-phänotyp-korrelationen (3 risikogruppen) 6. möglcihkeit MEN2a-symptom, nach phäochromozyto forschen bei welchen der folgfenden neoplasien kann keine monoklonalität nachgewiesen werden? 1. chronisch myeloische leukämie 2. leiomyom des uterus 3. neurofibrom !!! 4. burkitt-lymphom 5. verruca vulgaris !!! welche der folgenden aussagen gehört nicht zu den klinisch relevanten kriterien bei der multiplen endokrinen neoplasie (MEN ) 2A 1. familiäres auftreten eine papillären schilddrüsenkarzinoms (FPTC) !!!! 2. medulläres schilddrüsenca. Plus phäochromozytom und nebenschilddrüsenadenom 3. gain of function mutationen im RET tyrosin kinase genabschnitt 4. familiäres auftreten eines medullären schilddrüsenkarzinoms (calcitonin-positives c-zellkarzinom (FMTC) 5. lage der mutation im RET-gen für den zeitpunkt des auftretens eines mtc entscheidend ... nur somatische mutatioenn in den tumorzellen selbst 26 / 63 therapeutisches targeting der fusionsproteine bei leukämischen chromosomentranslokationen CML (chronische myeloische Leukämie) translokation : t(9q; 22q) (philadelphia-translokation) fusionsprotein: abl/bcr (aberrante tyrosinkinase) therapie: imatinib (tyrosinkinase-inhibitor) APL (akute promyelozytäre Leukämie) translokation: t(15,17) (q22;q21) fusionsprotein: PML/RARa(defekter retinsäure-rezeptor therapie: überangebot an all-trans-retinsäure (ATRA) Der verlauf der leukämischen erkrankung (prognose) wird entscheidend durch die art der zytogenetischen veränderung bestimmt bsp: MDS (myelodysplastisches syndrom), AML (akute myeloische leukämie) welche aussage trifft NICHT zu ? Das philadelphia-chromosom 1. ist ein deletiertes chromosom 21 !!!!! 2. wurde nach der stadt benannt, in der es erstmalig beschrieben wurde 3. entsteht durch translokationen 4. trägt ein onkogenen 5. findet sich bei der mehrzahl der patienten mit chronischer myeloischer leukämie welcher der folgenden zytogenetischen befunde ist NICHT mit einer schlechten prognose bei MDS oder AML verbunden ? 1. 2. 3. 4. 5. monosomie 7 del 3q rea 11q23 t(6q;9q) trisomie 8 !!!!! monosomie 7 immer SCHEISSE, trisomie 8 hier immer GUT! Welche der folgenden aberrationen ist NICHT mit einer günstigen prognose bei MDS oder AML verbunden ? 1. t(15Q; 17q) 2. trisomie 8 3. monosomie 7 !!!! 4. inv (16) 5. t(8q;21q) translokation 8q;14q beim Burkitt-lymphom c-myc von 8 auf 14 immunglobulin heavy-chain-locus über 1/3 der tumoren im kindesalter sind leukämien – warum ist das unvermeidlich ? Die kinder sind den keimen der ganzen welt ausgesetzt, Ig's der Mutter sind aufgebraucht, kind muss sein eigenes repertoire entwickelt (bis zu 70mio unterschiedliche antikörper über somatische rekombination – das passiert auf chromosom 14 – schwere Ig-Kette) wenn während der rekombination irgendwo anders ein bruch ist, glaubt die rekombinase dass sie das stück auch mit einbauen muss – sozusagen eine translokation als fehler der somatischen rekombination -> potentielle lymphomzelle ! 27 / 63 Welche der folgenden translokationen steht NICHT im ursächlichen zusammenhang mit der entstehung einer leukämie / eines lymphoms = 1. t(9q;22q) 2. t(15q;17q) 3. t(8q;21q) 4. t(14q21q) !!!! 5. t(8q;14q) das ist die robertson'sche translokation: kein strichpunkt zur TRENNUNG!!!! BABY!!!! warum findet sich bei b-zell lymphomen / leukämien assoziierten chromosomen-translokationen häufig eine beteiligung des chromosoms nr. 14 1. chromosom 14 beherbergt das C-myc onkogen !!!! 2. auf chromosom 14 befinden sich mehr als drei fragile stellen 3. der immunglobulin schwere ketten gencluster kartiert auf 14q32 4. auf chromosom 14 befindet sich variables perizentrisches heterochromatin 5. als akrozentrisches bla durch eine chromosomale translokation kann es zur entstehung von fusions-genen und chimären proteinen kommen . Bei welcher der folgenden erkrankungen spielt diser vorgang eine entscheidende rolle? – polyposis coli – retinoblastom – chronisch-myeloische leukämie !!!!! – sichelzellanämie – neurofibromatose mutationen im p53 tumor-suppressor gen in einer familie mit erhöhtem neoplasie-risiko (lifraumeni)... bsp familie mit mammakarzinomen, weichteilsarkomen, gehirntumoren... eine vererbte mutation – wesentlich erhöhtes risiko durch punktimutationen p53 komplett auszuschalten – extreme vorsicht bei strahlen, kohlenwasserstoffe u.a. 28 / 63 Systematik der Ch Chromosomenaberrationen I. Konstituionelle Aberrationen (in allen Körperzellen nachweisbar, bereits in der zygote vorhanden; entstehung in oder übertragung durch elterliche keimzellen) 1. Aberrationen ohne direkte Auswirkungen a) Strukturvarianten b) reziproke Translokationen (=balanziert) 2. Aberrationen mit milden (=segmentalen) Auswirkungen a) gonosomale Aberrationen 3. Aberrationen mit schweren Auswirkungen a) autosomale Aneuploidien (=numerische Aberration) b) unbalanzierte strukturelle Veränderungen II. Somatische Aberrationen 1. Chromosomen-Instabilitäs-Syndrome 2. klonale aberrationen (= leukämien, solide tumoren ) 29 / 63 welche der folgenden aussagen trifft nicht zu tumorsuppressorgene – besitzen einen hemmenden einfluss auf die zellproliferation – kölnnen durhc dletion auf keimbahnebene zur entstehtung herdeditärer tumoren beitragen – entsprechen dem prinzip des two-hit modells der tumorentstehung – entwickleln ihre tumorigenen eigenschaften durch mutation oder deletion – entfalten ihre tumorigene wirkung bereits nach mono-allelischer inaktivierung !!! welche der folgenden neoplasien entsteht nicht auf der grundlage der knudson two-hit hypothese 1. retinoblastom 2. wilms-tumor 3. multiple endokrine neoplasie typ 1 4. neurofibromatose 5. burkitt – lymphom!!! -> translokation, keine punktmutation Mammakarzinom BRCA1/2 wichtig für dna-reperatur (homologe Rekombination) keine Genotyp/Phänotyp Korrelation für Risiko mehr Brustkrebs oder Eierstockkrebs 75% sporadisch, 25% familiär, davon 10% monogen 4% brca 1 (ovarialkarzinom u.a.) 3% brca 2 (ovarialkarzinom, pankreas-ca u.a.., brustkrebs bei männern (1/100 risiko der frau)) 3 % brca x (Atm, chck1 u.a.) laufende studien: taxol – verzögerung des tumors bei mutationsträger-frauen abnahme der eierstöcke führt zur 70%igen Reduktion des Risikos (tumor anscheinend hormonsensitiv welche der folgenden faktoren ist nicht mit einem erhöhten brustkrebsrisiko verbunden ? 1. erstschwangerschaft > 30. lj 2. menopause > 50 lj 3. multiparität !!! 4. orale kontrazeption länger als 10 jahre 5. hormonersatztherapie (kombinationspräparate) das altersbereinigte krebsrisiko ist heute genau so hoch oder etwas niedriger als vor 100 jahren Kolonkarzinom sporadisch 70% familiär 20%, monogen 10% +polyposis -> fap / juvenile polyposis / peutz-jaeger syndrom -polyposis -> HNPCC – lynch syndrom I /II , muir-torre syndrom – – die kolorektalen karzinome gehören zu den häufigsten krebserkrankungen in den westlichen ländern häufigkeit 30 erkrankungen/100k sporadisches kolonkarzinom – 85% CIN (chromosomale instabilität) führt zur erhöhung der mutaitonsrate (initiation : apc-gen defekte) 30 / 63 15% MIN (mikrosatelliten instabilität ) 1.000fache erhöhung der mutationsrate (mutationen in mismatch-reparatur genen) Entstehnung des kolonkarzinoms: adenom-karzinom-sequenz (dauer 20 bis 40 jahre) genetische instabilität cin > min ... – adenomatöse polyposis coli (APC) gen möglicher mutationsbereich: chrpe = congenitale hypertrophie des retinalen pigment epithels -> augenarzt kann diagnose stellen : „hochrisikopatient für dickdarmkrebs“ (untersuchung augenhintergrund -> hypertrophe bereiche der retina ) risiko für extrakolonische tumoren bei FAP-patienten: – hirntumoren 7fach – schilddrüse 8fach – dünndarm 380fach – ampulla vateri 124fach – hepatoblastom 847fach – pankreas 5fach Amsterdam Kriterien 1. drei verwandte mit histologisch gesichertem kolorektalem karzinom. Einer der betroffenen muss verwandeter 1. grades sein 2. es müssen mindestens zwei aufeinanderfolgende generationen betroffen sein 3. bei mindestens einem verwandten muss das kolorektale karzinom vor dem 50. lebensjahr diagnostiziert worden sein erweiterte amsterdam kriterien 1. einbezug von endometrium und dünndarmkarzinom 2. zusätzlich folgendde tumoren: ovarical ca vor 50lj magen ca vor 50lj pankreas- oder hepatobiliäres ca blasen-urothel (jedoch nicht nierenzell-)ca die diagnose von zwei dieser tumoren bei einer person (unabhängig vom alter) bei welcher der folgenden konstellationen ergibt sich kein verdacht auf ein familiäres krebssyndrom 1. zwei doer mehr familienmitglieder von krebs betroffen 2. auftreten eienr krebserkrankung im alter von unter 50 jahren 3. ein fmalilienmitglied an versch tumoren erkrankt 4. ein famlilienmitglied im alter von 75jahren an krebs erkrankt !!! 5. auftreten der gleichen tumorart bei mehreren familienmitgliedern hereditäres nmicht polypöses kolonkarzinom (HNPCC) – keimbahnmutation eines mismatch-reperaturgens – keine polyposis, schnelle entwicklung des karzinoms aus einem polypen – mittleres manifestationsalter des karzinoms : 46 jahrenlokalisation eher rechtsseitig – hnpcc assoziierte tumoren z.b. endometriumkarzinom epigenetische und genetische inaktivierung von mismatch-reperatur genen bei hnpcc hmsh2(31%), hmlh1 (33%), hpms1/2 (2/4%) genetische veränderungen bei krebs 31 / 63 4. loss of imprinting (LOI) (epigenetische genexpressionsstörungen = „gene silencing“ durch veränderungen von dnamethylierung und chromatin-konformation) hypermethylierung von cpg inseln in den promoter-regionen von tumor-suppressor-genen beispiele: mismatch-reperatur gene (hmlh1 und mgmt) Welche der folgenden anamnestischen Angaben weist NICHT auf das vorliegen auf die möglichkeit einer familiären tumoerkrankung hin? 1. ein betroffener ersten grades mit erkrankungsalter über 80 jahre !!!! 2. hyperplasie des retinalen pigmentepithels 3. auftreten der erkrankung im frühen erwachsenenalter 4. mehrere betroffene ersten grades mit frühem erkrankungsalter 5. mehrere organe von tumoren betroffen durch eine chromosomoale translokation kann es zur entstehung von fusionsgenen und chimären proteinen kommen. Bei welcher der folgenden erkrankungen spielt dieser vorgang eine entscheidende rolle ? 1. polyposis coli 2. retinoblastom 3. chronisch-myeloische leukämie !!!! 4. sichelzellanämie 5. neurofibromatose welcher defekt eines DNA-reperatur-systems manifestiert sich als mikrosatelliten-instabilität? 1. defekt in der nukleotid-exzisions-reparatur 2. defekt in der homologen rekombinationsreparatur 3. defekt in dem nicht-homologen end joining 4. defekt in der mismatch reperatur !!! 5. defekte in der basen-exzisions-reparatur umweltfaktoren (ernährung) eindeutig mitverantwortlich für kolorektales karzinom v.a. Bei „amerikanischer diät“ magenkarzinom z.b. in japan sehr hoch bsp: folsäuresubstitutionsversuch: 15 jahre eingenommen (oder gemüse und obst) senkt colon cancer risiko auf 0.3 genetische veränderungen bei krebs 5. gen-amplifikation (gen-vermehrung) vor allem bei fortgeschrittenen soliden tumoren i.d.R. Manifestation der tumorprogression (mehrschritt-tumorentstehung) manche neoplasien reagieren mit genamplifikation auf zytostatika, dadurch werden diese malignome therapie-resistent ionisierende strahlung hat signifikanten einfluss auf tumorgenese natürliche strahlenexposition 2 mSv pro jahr künstliche strahlenexposition 1,5 mSv (fast ausschließlich medizinisch) (unvermeidliche ) endogene dna-schädigung bei 37 grad körpertemperatur und energiegewinnung durch oxidative phosphorylierung purin-verlust 12k / Z / T 32 / 63 einzelstrangbruch 55k /Z / T doppelstrangbruch 9 / Z / T crosslinks 8 / Z / T pyrimidin-verlust 600 / Z / T cytosindesaminierung 200 / Z / T Caretaker genes protect us from exogenous and endogenous dna damage – exogenous dna damage: ionizing radiation , chemicals, viruses – endogenous dna damage thermoinstability , reactive oxygen species (unvermeidlich) dna- reperatur – strategien – BER = Basen exzisions-reparatur (ionisierende strahlen, freie radikale,..) – NER = nukleotid-exzisions-reperatur (UV-Licht, polyzyklische arom. Kohlenw...) – RR = rekombinationsreparatur (ionisierende strahlung ,chemotherapeutika) HR = Homologe rekombination, fehlerfrei NHEJ = nicht-homologes end joining, fehlerhaft – MMR = Mismatch – Reparatur (Replikationsfehler) bsp: xeroderma pigmentosum Genetische instabilitäts-syndrome 1. chromosomale instabilität 2. ... zu 1.: werner syndrom (adulte progerie) life expectancy < 50, vorzeitige alterung (hair loss ,cataracts, ...) multiple translokationen, genomumbau.. gendefekt einer helicase autosomal rezessiv 33 / 63 Achtung Klausur eine Woche vorverlgt auf 30.1. 8:30 16.1.2006 + 1 Stunde extra !!! Welche der folgenden Schädigungsarten unserer DNA sind endogen und damit unvermeidlich? 1. Schädigung durch Alkylantien 2. Schädigung durch reaktive Sauerstoffspezies („freie Radikale“) !!! 3. Schädigung durch ionisierende Strahlung 4. Schädigung auf Grund von Thermoinstailität der DNA !!!! 5. Schädigung durch klastogene Viren Welche der folgenden DNA-Reparaturstrategien findet sich nicht biem menschen? 1. dna-exzisions-reparatur 2. nukleotid-exzisions-reparatur 3. rekombinationsreparatur 4. mismatch-reparatur 5. alles -oder-nichts-reparatur !!! caretaker-gen-syndrome neben werner-syndrom auch bloom-syndrom (extreme lichtempfindlichkeit der haut mit allen arten von tumoren, kleine statur) – ein helicasedefekt; rothmund thompson syndrom (haarverlust, poiklodermie – fleckige hautveränderungen, knochensarkome) – ebenfalls ein helicasedefekt human caretaker gene syndromes (x-ray sensitivity syndromes) atm-gene (reparaturgene für schäden ,die durch ionisierte strahlen verursacht werden) ataxia telangiectasia (ataxie aufgrund cerebellärer atrophie, teleangieektasien (erweiterte kleinste gefäße), erhöhtes AFP – unreife leber, immundefizienz, lymphoretikuläre neoplasien, chromosomtranslokationen 7/14) nbs nijmegen breakage syndrom (kleinwuchs, mikrozephalie, strahlensensitivität, infertilität , lymphome, normales afp fanconi anämie (Auch caretakergen-defekt) „wachsartiges“ gesicht, niedriger hb-wert (anämie eben) – bis zur aplastischen anämie (panzytopenie), ohne passende knochenmarkspende früher tod am MOS (multiorganversagen) 70% extrem kleinwüchsig, multiple angeborene fehlbildungen extrem empfindlich gegen dna-crosslinks (alkylierende subtanzen u.a.) zellen auf niedrige sauerstoffspannung ausgerichtet, besonders empfindlich gegen sauerstoffinduzierte schäden das knochenmark ist „heilbar“, die restlichen bleiben aber sehr tumoranfällig -> früher tumoren 34 / 63 es gibt fälle von „selbstheilung“ durch intragenes crossover bei compound heterozygotie (bildung eines knochenmarkmosaiks) homozygotie: mutation in beiden allelen an der selben stelle compound heterozygotie : mutationen auf jedem allel (allerdings unterschiedliche, sitzen nicht an gleicher stelle), knocken ebenfalls das target aus: Vorteil: Chance auf Selbbstheilung/molekulare Korrektur (durch genkonversion oder intragenes crossover) dafür gibt's auch andere Krankheitsbeispiele wie Tyrosinämie, Wiscott-Aldrich-Syndorm, epidermolysis bullosa... Welches der folgenden Syndrome gehört nicht zu den chromosomalen Instabilitätssyndromen? 1. Werner-Syndrom 2. Ataxia telangiectasia 3. Fanconi Anämie 4. Down-Syndrom !!! 5. Bloom Syndrom sehr wenige sind homozygot, aber viele heterozygot für genetische krankheiten – das sind hypothetischerweise diejenigen, die früh an krebs erkranken Dyskeratosis congenita (aberrante Telomere) – abnormale pigmentierung, mukosale leukoplakie, nageldystrophie, knochenmarksversagen, pulmonale und leber-erkrankungen, hautkrebs (ganz selten) verschiedene erbformen: x-linked, autosomal-dominant TERT: human telomerase reverse transcriptase , dyskerin.... Laminopathien (lamin stabilisiert die Kernhülle) – Dominant Emery-dreifuss musculäre dystrophie limb-girdle ... hutchinson-gilford .. – Rezessiv emery-dreifuss. Charcot.... Emery-Dreifuss muscular dystrophy emd1: x-linked: mutationen im emerin emd2: autosomal-dominant emd3: autosomal-dominant, lmna-mutation (macht u.a. lipodystrophie , fettgewebe nix gut!) lebenserwartung fast um die hälfte reduziert hutchinson-gilford juvenile progerie (HGS) beschleunigte körper-alterung (lebenerwartung <20, fulminatne atherosklerose usw) mutation im lamin-gen... Take-home-message Alterungs-und Krankheitsprozesse werden beschleunigt durch: – Chromosomale Instabilität 35 / 63 Telomer-Instabilität – Chromatin-Instabilität Für die Stabilität von Chromosomen, Telomeren und Chromatin sind u.a. intakte und gut funktionierende Caretaker- und Lamin-Gene (=Anti-Aging-Gene) erforderlich welches der folgenden krankheitsbilder gehört nicht zu den laminopathien? – dyskeratosis congenita !!! – warum sind genetisch (mit)bedingte alterserkrankungen so häufig ???? – die meisten menschen sterben nicht an altersschwäche, sondern an alterskorrelierten erkrankungen – die häufigkeit von geneitsch bedingten und geneitsch mitbedingten alterserkrankungen erklärt sich durch das fehlen von selektion gegen gendefekte, welche sich erst anch abschluss der reproduktion als krankheit manifestieren Antagonistische Pleiotropie – gene, welche alterskrankheiten (mit)bedingen, folgen dem prinzip der antagonistischen pleiotropie – dies bedeutet ,dass sich ein bestimmtes gen in der jugend positiv, im alter jedoch negativ auswirkt – Bsp: Hypercholesterinämie, Hypertonie, Diabetes II, M. Alzheimer Take-home-Message – fehlende Selektion gegen gene, die Alterskrankheiten mitbedingen – sowie eine grosse zahl von genen mit antagonistisch-pleiotroper wirkung erklären die häufigkeit von alterskrankheiten monogene formen von demenzerkrankungen uiii viele z.b. – APP – M. Alzheimer – präsenilin I, II – M. Alzheimer – Alpha-Synuclein – M. Parkinson – Notch 3 – CADASIL – Huntingtin – M. Huntington – Prion-Protein-Gen – CJD, GSS , FFI (auch Demenzerkrankungen) Folien jetzt angeblich alle im Netz!! Prionenkrankheiten – Human: CJD, Kuru, nCJD, GSS (Gerstmann-Sträussler-Scheinker), Fatal Familial Insomnia (FFI) – Tier: Scrapie u.a. entstehen durch eine konformaitons-änderung des normalen zellulären prion-proteins! Die lösliche form besteht hauptsächlich aus alpha-helices, während die unlösliche form überwiegend beta-sheets hat (die die dimerisierung des proteins begünstigen) 36 / 63 Monogene Prionenkrankheiten (autosomal dominante Vererbung) – Menschliches PRNP (Prionprotein)-Gen auf Chromosom 20) mehr als 15 mutationen bekannt, Häufigste E200K (über 50 familien familiäre Creutfeld-jakoberkrankung CJD) – genotyp-phänotypö-korrelation (bei mutation d178n) plus valin : klassische CJD plus methionin: FFI – p102l mutation bei 30 familien mit GSS-Erkrankung Anfälligkeit für Prion-Erkrankungen Rolle des Polymorphismus im Codon 129 des PRP Gens Met/met und Val/val hOmozygotie erhöhen das risiko für die sporadische und iatrogene creutzfeldjakob-erkrankung Umweltfaktor: sie müssen kontaminierte Fleischprodukte zu sich nehmen alle vCJD-patienten („BSE“) sind im Codon 129 Met/met homozygot!!!! (Populationsgenetik in Deutschland „besser“ als in England – weniger/keine Fälle) met/val und val/val individuen sind möglicherweise resistent gegenüber bse oder haben eine sehr lange inkubationszeit M. Huntington: prototyp einer polyglutamin-erkrankung eine triplett-expansions-erkrankung: CAG-repeats > 36´(CAG gibt glutamin, die vermehrung gibt nen conformation - change) CADASIL (cerebrale autosomal dominante arteriopathie mit subcorticalen infarkten und leukoenzephalopathie) kriegen erstmal schwere depression, die gängige medikation hilft aber gar nix, im gegenteil, demenz manifestiert sich auch nocht - > muss an cadasil gedacht werden mutationen im notch3-gen auf chromosom 19 Frontotemporale Demenz: (mutation tau-gen auf chromosom 17) – Tauopathien, aggregation -> neurodegeneration Alzheimer zig genetische und nichtgenetische ursachen (umwelt, alter) – app, presenilin, apolipoprotein e, late-onset gene, down syndrom – toxine, viren, prionen, traumen (boxer) gute Argumente für genetische Determination aller Alzheimer-Fälle (die betroffenen Boxer ham auch alle eine bestimmtes Allel...) über 40 gene damit assoziiert zwillingsstudien über 60% gemeinsamkeit u.a. die monogenen Formen treten früh auf (early onset), die late onset in der regel polygen Amyloid: Aggregation und ablagerung im gehirn early-onset alzheimer – autosomal dominante vererbung – praktisch nur missense-mutationen – krankheitsdauer: je nach betroffenem gen unterschiedlich (6-16 a) 37 / 63 Prävention der Alzheimer-Erkrankung Verringerung der Ablagerungsgeschwindigkeit von Amyloid-Proteinen 1 pro sekunde pro Ablagerungsstelle -> macht in 30 jahren ca 1 Milliarde 0,5 pro sekunde währen's 60 jahre! Bei welcher erkrankung ist eine prädiktive genetische Testung NICHT indiziert? 1. familiäre alzheimer erkrankung !!!! 2. familiäre mamma-ca 3. familiäres colon-ca 4. multiple endokrine neoplasie typ 2 5. famliäre thrombose neigung -> keine testung da keine prävention/therapie verfügbar was gehört nicht zu den risikofaktoren der alzheimer-demenz erkrankung? 1. mehrere betroffene in familie 2. mutationen in app oder präsenilin-genen 3. alter 4. nikotinkonsum !!! 5. apolipoprotein E4 was zählt NICHT zu den risikofaktoren für morbus alzheimer ? 1. missnese mutationen im app-gen 2. männliches geschlecht !!!! 3. lebensalter 4. trisomie 21 5. trägerschaft des apoe4-allels bei welcher demenzerkrankung sind KEINE monogfenen formen bekannt ? 1. Multi-infarkt-demenz 2. frontotemporale demenzerkrankung 3. morbus parkinson 4. CADASIL 5. creutzfeld-jakob erkrankung -> hier könnt man keine ankreuzen! Deswegen wird die frage auch umformuliert welche der folgenden Erkrankungen wird NICHT durch mutationen im prion-protein-gen ausgelöst? 1. Fatale familiäre insomnie 2. creutzfeld-jakob erkrankung 3. m. parkinson !!! 4. variante creutzfeld-jakob-erkrankung 5. gestmann-sträussler-scheinker erkrankung 38 / 63 in der klausur darf man so lange schreiben wie man will.... x-chromosomaler Erbgang x-chromosomal-rezessiv: Bsp-Erbgang mit Frage: wie hoch ist Erkrankungsrisiko Tochter von krankem Vater und gesunder Mutter ? 100% bsp-Krankheit BMD: ist Tochter Konduktorin? CK-Wert im vgl zu Kontrollgruppe erhöht datenbanken im internet http://www3.ncbi.nlm.nih.gov/Omim http://www.bvdh.de punnet-quadrat: Bsp: mutter heterozygot (gesund) und vater hemizygot gesund: 50% söhne erkranken, 50% töchter heterozygot Lyon-Hypothese besagt, dass während der frühen Embryonalentwicklung des Menschen (ca 100zellStadium) ein X-Chromosom der Frau inaktiviert wird und dies in allen Klonen der Zelle beibehalten wird. Welches X-Chromosom abgeschaltet wird ist zufällig. Duchenne-Muskeldystrophie DMD verzögerte Sprachentwicklung zunehmende Gehschwierigkeiten gauersche's zeichen : können sich nicht aus der hocke wieder aufrichten ohne armunterstützung Klinik – verzögerte primärentwicklung besonders sprache, 1/3 geistig retardiert – ab 3. lj vermehrte ermuüdbarkeit, häufiges hinstürzen – watchelder gang, hyperlordose – spitzfusshaltung – abgeschwächte e-rflexe – wadenhypertrophie diagnostische Merkmale – ck über 2000 U/l, auch TA und LDH hoch – vermehrte Echogeniätt im muskelsonogramm – muskelumbau im MRT – kardiomyopathie im Echo – ... W. Erb trennt klar die Atrophien von den Dystrophien Genetik – x-chromosomal rezessiv – genort: xp21.2 – mutationen im dystrophin-gen 60% deletionen 5% duplikationen 35% punktmutationen – hohe Neumutationsrate – inzidenz: DMD 1/3000, BMD (beckersche muskeldystrophie – allelisch im gleichen genort) 39 / 63 1/18000 Problem Neumutationen: selbst bei gesunden Eltern kranke Kinder unterschied meiose – ein kind genträger mitose – keimzellmosaike, mehrere kinder genträger (dann wdhrisiko ~10%) x-chrom rezessiv: wenn papa betroffen alle söhne gesund alle töchter genträgerinnen Becker: drei muskeldystrophien autosomal rezessiv: gliedergürteldystrophie Autosomal dominant: schultergürtel u.a. dystrophie x-chromosomal: DMD (nur Knaben betroffen) dazu : Familie mit x-chromosomalem Erbgang und gutartigem Verlauf: BMD (Beckersche Muskeldystrophie) verändertes Dystrophin (bei DMD: gar kein Dystrophin, normal: normales Dystrophin) Erklärung: bei Duchenne Leserasterverschiebung bei Becker Mutationen, bei denen das Gesamtleseraster erhalten bleibt (damit Restfunktion) Deletionen entstehen vor allem in Oogenese, Punktmutationen in Spermatogenese daher: mutationsrate ist immer geschlechtsspezifisch (bezeichnung: μ im weiblichen,ν im männl. Geschlecht) DMD: Problem verstorbene Elternteile: indirekte Genotypdiagnostik (da Komplettsequenzierung zur Punktmutationssuche sehr Kompliziert) Untersuchung eines Polymorphen Markers aufgrund der Daten, die von Kindern stammen Rückschluss auf Genotyp des Vaters Verkomplizierung : Crossing-over kann Markerproteine verschieben – geringes Restrisiko (10%), aber auch Chance auf gesunde Nachkommen mit „krankem“ Marker Reduktion des Risikos durch Wählen der Marker nahe der vermuteten Mutation Therapieoptionen bei DMD – heilpädagogische frühforderung, ergotherapie, logopädie u.a. – Physiotherapie – spitzfussprävention – kortikoide – kreatin – rideau-operation (rücken) – wirbelsäulenaufrichtung – hilfsmittel – cpap-heimbeatmung – clebuterol, gentamycin, myoblasteninfusionen Autosomal rezessive MD Adhalin-Defekt (alpha-Sarkoglykan) Diagnose anhand Western blot 40 / 63 Emery-Dreifuss-Muskeldystrophie eine neue X-chromosomale Muskeldystrophie, ähnlich wie DMD/BMD, aber frühe Kontrakturen und Herzrhythmusstörungen EMD (Emerin) Genort: Xq28 Inzidenz 1/100000 punktmutationen Diagnose anhand Western blot (Emerin-Fehlen) auch autosomale-dominante Form mit gleichem Krankheitsbild Mutation an anderer Stelle mit gleichem Effekt: Instabilität der Kernmembran über Veränderung im Lamin-A-Gen (7 bekannte Typen) Intelligenzminderung / geistige Retardierung leichte / schwere 50-70 / <50 iq usw häufigste formen – down-syndrom – fragiles x-syndrom – rett-syndrom – prader-willi-syndrom diagnostik der intelligenzminderung .... bsp : Fragiles-x-syndrom mutation im FMR1-gen (cgg-repeat erkrankt ab 200 einheiten, prämutation 50-100) x-chromosomaler erbgang, aber männer mit prämutationen unauffällig heterozygote frauen mit vollmutation oft klinisch betroffen (ca 30%) problem der wiedervererbung ... frau mit prämutation hat 50% vollmutations-söhne therapie Fra(x)-syndrom – ursächlich nicht möglich – sonder-/heilpädagogische förderung Rett-syndrom ab 8. lm auffallende entwicklungsverzögerung (kopf, hände...) – 1:15k – progredienter mikrozephalus – 100 andere sachen – idR mädchen betroffen (knaben letal) – 95% sporadisch (gesunde geschwister normales geschlechtsverhältnis) – x-chromosomal DOMINANTER erbgang – mutationen in MECP2 – reguliert transkription (wichtig embryonalentwicklung) – klinisch variabel (falls nur intelligenzminderung überleben betroffene knaben, aber schwerer betroffen als mädchen) 41 / 63 Fragen wiederholungsrisiko für muskelkrankheit bei sohn von II/1 ( kranker vater gesunde mutter) – entspricht allgemeinbevölkerung welche aussage DMD trifft nicht zu – ursächlich veränderung im dystrophin – übertragung vater auf sohn in regel möglich !!! – krankheit bei 1/3 patienten durch neumutation – krankheitsbild gehört hyperlordose lws dazu – krankheitbild pseudohypertrophie waden dna-analyse mit RFLP wahrscheinlichkeit für konduktorin älteste tochter (die ältesten nachkommen immer links) kranke alle großes allel, gesunde kleines... also 0% x-chromosom inaktivierung im weibl. Geschlecht was ist falsch ? – eines der beiden x-chrom in somatischen zellen größtenteils inaktiviert – mechanismus zur dosiskompensation x-chromosomaler genprodukte – inaktivierung bei feten in 12. SW !!!! – inaktiviertes x-chromosom in versch zellen väterlicher oder mütterlicher herkunft – barr-körperchen als morphologisches substrat häufigste genmutation(en) bei patienten mit BMD oder DMD ist (sind) – instabile trinukleotidsequenz – punktmutaitonen mit vorzeitigem abbruch proteinsynthese – deletionen !!! – inversionen und duplikationen – promotormuationen kleinkind: symptomkombination primordialer disproportionierter minderwuchs, verkürzte extremitäten, fronto-parietales ausladen des schädels, normale geistige entwicklung für – achondroplasie !!!! – katzenschrei-syndrom – tris 13 – tris 18 – tris 21 -> wichtig ist die normale geistige entwicklung (damit scheiden alle anderen aus) erkrankungsrisiko für jedes kind praktisch 100% bei??? – beide eltern taubstumm – beide eltern diabetes I – der vater bluter ,mutter konduktorin für hämophilie A – beide eltern achondroplasie – ein elternteil träger isochromosoms 21 oder robertsonsche translokation (21q21q) !!!! taubstumm: autosomal rezessiv, aber verschiedenste genorte und heterogenes krankheitsbild hämophilie a : bei mutter konduktorin nur 50% söhne achondroplasie autosomal dominant ...nur 75% 42 / 63 bei welcher situation pränataldiagnostik mit chorionzottenbiopsie absolut kontraindiziert? – alter mutter über 40 – mutter trägern balancierte 14q21q robertsonschen translokation – vorausgegangenes kind mit zystischer fibrose (homozygotie für delta f508) – vorausgegangenes kind mit spina bifida !!!!! – mutter konduktorin DMD mit bekannter deletion dystrophin- gen muss AFP bestimmen, fruchtwasser gewinn ich nicht bei chorionzottenbiopsie, brauch ich amniozentese 27jährige frau 8. Ssw gen. Beratung. Verstorbener bruder klinisches downsyndrom, chromosomen wurden nicht untersucht, keine weiteren geschweister, ihr vater verstorben, welche massnahme sollte unter berücksichtigung minimaler invasivität zunächst durchgeführt werden? – chromosomenanalyse bei beiden ehepartnern – chromosomenanalyse bei der schwangeren !!! – amniozentese – chorionzottenbiopsie – nabelschnurpunktion gesunde eltern haben ein kind mit klassischer PKU wie hoch ist das wiederholungsrisiko, wenn die eltern verwandt sind (vetter-base 1. grades ) ? – 25% !!!! – 33% – 50% – 66% – 100% vetter / base = cousin(e) aber blutsverwandschaft spielt keine rolle welche aussage über multifaktorielle vererbung trifft nicht zu ? – bei multifaktorieller vererbung gibt es unterschiede zwischen den geshclehctern in der häufigkit der ausprägung eines merkmals – die ausprägung multifaktoriell bedingeter merkmale kann von umweltfaktoren beeinflusst werden – für multifaktoriell verursachte erkrankungen kann im rahmen einer genteitschen beratung das wiederholungsrirsiko entsprehcne den mendleschen regeln exakt bestimmt weerden !!!! – die vererbung der eizelen, an einem multifaktoriellen systsem beteiligten gene folge den mendelshcne regeln – die widerholungswahscheinlichkeit für ein multifaktoriell.... was falsch – tumorsuppressorgene – besitzen einen hemmenden einfluss auf zellproliferation – können durch deltion auf keimbahnebene zur entstehung ehreditärer tumoren beitragen – .. – .. – entfalten ihre tumroigene wirkung bereits nach mono-allelische inaktivierung !!!! autosomale rezessiv erbliche krankheit hat häufigkeit von 1 /10000 in bevölkerung. wie häufig ist 43 / 63 das krankheitsverursachende abnorme gen? – 1:50 – 1:100 !!! – 1:200 – 1:5000 – 1:10000 welches reproduktive outcome ist nicht typisch für eine familiäre chromosomentranslokation – geburt von kindern mit fehlbildungen – verringerte fruchtbarkeit männl. Individuen – erhöhte fehlgeburtsrate – geburt von kindern mit XXY !!!! – geburt von phänotypisch unauffälligen kindern die der freien trisomie 21 zugreunde ligende nondisjunction erfolgt überwiegend in der – paternalen meiose I – postzygotisch – maternalen meiose II – maternalen meiose I !!! – paternalen meiose II 44 / 63 Abweichungen vom Paradigma der Mendel-Genetik : Imprinting, Epigenetik Autosomal-dominanter Erbgang – ein gen überwiegt stets über sein allel im zustand der heterozygotie – keine bevorzugung eines geschlechts – mermalsträger geben dieses gen an die hälfte ihrere nachkommen weiter – unter den nachkommen merkmalsfreier personen tritt das merkmal nicht auf Paternale Transmission, mütterlich „geprägt“ - mann dazwischengeschaltet -merkmal tritt in nächster generation auf, mutter dazwischengeschaltet – merkmal tritt nicht auf Mechanismen der Prägung – vor der fertilization – reversibel bei passage durch die entgegengesetzte parentale keimbahn (.d.h. Ein mütterlich geprägtes gen muss in väterlichen keimzellen reaktiviert werden) – stabile weitergabe durch die mitose in somatischen zellen – transkription blockiert, dna-methylierung Kerntransferexperimente (Maus) – androgenetische Embryonen (beide kerne vom vater): wachstumsretardiert, throphoblast vergrößert – gynogenetische embryonen (beide kerne von der mutter): kaum extraembryonales gewebe -> abort Komplette Blasenmole – paternal – ovum ohne nucleus – diploid, 90% , 46,XX – Hyperplasie des Trophoblasten, kein fetales Gewebe – kann tumurös in die Uteruswand einwachsen und metastasieren Ovarialteratom – Maternal – Parthenogenese, 46,XX – Produkte aus allen 3 Keimblättern in stark verkümmertem Zustand – rudimentärer _Organismus – kein tumor, keine proliferation, sondern retentionszyste (sekrete) mit neigung zu stieldrehung und vereiterung Triploidie – 2% aller Kontrazeptionen – 20% der chromosomenbedingten Fehlgeburten in der 8.-12. SSW – in 90% stammt der überzählige chromosomensatz vom Vater (2 spermien oder 1 diploides Spermium) : grosse Zystische Plazenta (partielle Blasenmole), Embryo altersentsprechend gross, mikrozephal – in 10% maternal : Plazenta fibrotisch, schwere Wachstumsretardierung und Makrozephalie Fazit – Paternale und maternale Genom sind NICHT funktional äquivalent 45 / 63 – – beide sidn essentiell für eine normale Entwicklung -> eine normale entwicklung erfordert nicht nur einen diploiden chromosomensatz, sondern auch eine biparentale vererbung Epigenetik – reversible, funktionelle Modifikation der Genaktivität im Gegensatz zu konstanten DNAMutationen Prägung (Imprinting) – Elternabhängige Aktivität / Expression einiger Gene durch Methylierungsmuster Epigenetische Modifikationen – führen zu elternabhängiger Inaktivierung bestimmter chromosomaler Regionen: Genomische Imprinting – Funktionale Hemizygotie einer kleinen Anzahl von Genen, da nur das väterliche oder mütterliche Allel exprimiert wird – ist dieses defekt, steht kein gesundes Allel zur Kompensation zur Verfügung da dieses inaktiv ist Prader-Willi-Syndrom 15q11-13 – 1:10k – im säuglingsalter Gedeihstörung – 2-3 Jahre: Hyperphagie und Übergewicht – Minderwuchs – Hypogenitalismus – Lernbehinderung Angelmann-Syndrom 15q11-13 – „Happy Puppet Syndrome“ – Marionettenähnlicher ataktischer Gang – unmotiv. Lachepisoden – Epilepsie – Schwere geistige Behinderung – Fehlende Sprachentwicklung Ursachen: PWS: Fehlen d. Paternalen Allels AS: Fehlen des Maternalen Allels Molekularzytogenetische Labordiagnostik von Deletionen z.B. am UBI3a gen: angelmann-syndrom Molekulare Mechanismen, die zur Entstehung von PWS/AS führen 70% 15q11-q13 – Deletion uniparentale Disomie Imprinting-center-Mutation Single-Gen-Mutation, andere (nur AS) Uniparentale Disomie (UPD) – beide homologen chromosomen eines chromosomenpaares oder einer chromosomalen Region stammen von einem einzigen Elternteil 46 / 63 – – verlujst des normalen biparentalen beitrags in einer region mit geprägten Genen genomisches Imprinting gestört wie kommt es zur Uniparentalen Heterodisomie? – fehler in Meiose I (beide chromosomen in 1 Gameten, diploide Eizelle/spermium) – Trisomic rescue (bei befruchtung Triploide Zelle) Uniparentale Isodisomie – Fehler in Meiose II (1 Chromosom komplett in 1 Eizelle/spermium statt 1 Chromatid) – Gamete complementation (ahja!) DNA-Methylierung – maternale und paternale DNA zeigen differentielle Methylierungsmuster – Methylierung von Genen im Promotorbereich (1-2 kb CpG islands) -> transkriptionelle Inaktivierung – DNA-Methyltransferasen sichern stabile Weitergabe durch Mitosen Familienberatung bei PWS/AS – genaue Diagnostik: Nachweis der Deletion, UPD oder Punktmutaiton – Empirisches Wiederholungsrisiko gering (ca 1%), da in der Regel sporadisch, aber auch Fälle mit 50% – Pränataldiagnostik bei bekanntem Defekt möglich therapie bei PWS/AS – Elternaufklärung – Heilpädagogik / Sonderpädagogik – Schlaf-/Wach-RHythmus – Physiotherapie – Sport – Epilepsie-Behandlung bei AS – konsequente Reduktionsdiät bei PWS (evtl. Wachstumshormonbehandlung) Beckwith-Wiedemann Syndrom 11p15.5 – Gigantismus prä- und postnatal – Makroglossie – Exomphalos – Viszeromegalie – Akzeleriertes knochenwachstum – normale geistige Entwicklung – – – – Paternale UPD 11 Überexpression des väterlich exprimierten embryonale Wachstumsfaktors insulin-like Growth factor 2 (IGF2) Mäuse mit mutiertem väterlichen IGF2 sind nur halb so gross wie normale Mäuse Mäuse mit defektem mütterlichem IGF2 sind normal Geprägte regionen im humanen Genom gibt nicht viele, etwa 30 z.b. am chromosom 15 PWS/AS, 11 BWS.... 47 / 63 Postzygotische Reprogrammierung des Methylierungsmusters – elternspezifische Prägung des Genoms während der Geschlechtszellrefiung – bis auf wenige „geprägte“ Gene kommt es im frühen Embryo zu einer genomweiten Demethylierung (zuerst väterlicher Vorkern, mütterlicher langsamer über erste Teilungen) – neue somatische Methylierungsmusters – Reprogrammierung mütterlich kontrolliert in-vitro-Ferilisation (IVF) – unterdurchschnittliches Geburtsgewicht bei IVF-Kindern – imprintingsyndrome (BWS,AS) gehäuft ?? – für die Abschätzung eines denkbaren epigenetischen Risikos bei Kindern die durch IVF/ICSI entstanden sind fehlen Langzeitstudien Embryoklonierung unverantwortlich – diploide Genom einer Körperzelle in aktivierte entkernte Eizelle eingesetzt – inkomplette Demethylierung des diploiden Spendergenoms in klonierten Rinderembryonen – abnorme somatische Genexpressionsmuster in klonierten Mäusen – nur 0,1 bis „Lampentausch erforderlich“ klone entwickeln sich bis zur Geburt , abnorme Phänotypen Krebsepigenetik – erhöhte DNA-instabilität durch Hypomethylierung rpetitiver Sequenzen – Aktivierung möglicher Krebsgene (Protoonkogene) durch Hypomethylierung – Inaktivierung von Tumorsuppressorgenen durch Hypermethylierung der Promotoren – Hypermethylierungsmuster: Prognose? – Rekativierung genetisch stillgelegter TSG druch demethylierende Substanzen? Welche Aussage trifft nicht zu ? Das prader-willi-syndrom 1. wird in ca 70% durch den verlust des väterlichen 15q11-q13-alles verursacht 2. wird in ca 30% durch eine mütterliche uniparentale disomie hervorgerufen 3. ist durch eine inaktivierung des mütterlichen allels bedingt !!!!! 4. geht mit eienr lernbehinderung einher 5. sollte durch kontrolliertes essverhalten u.a. therapiert werden was ist falsch? das angelmann-syndrom 1. beruht auf einer deletion der maternalen chromosomenregion 15q11-q13 2. väterlichen uniparentalen disomie 15 3. ist durch einen marionettenähnlichen gang un unmotivierte lachepisoden gekennzeichnet 4. tritt nur bei mädchen auf !!!! 5. erfordert in der regel eine antiepileptische therapie beim beckwith – wiedemann-.syndrom wird ein prä- und postnataler gigantismus bei normaler geistiger entwicklung beobachtet weil durch eine väterliche uniparentale disomie 11p15 u.a. väterliche gene wie das igf2 gene im überschuss vorliegen 48 / 63 ja, weil ja!!! 49 / 63 Autosomal dominanter und autosomal rezessiver Erbgang Autosomal dominanter Erbgang ADE siehe schulzeit Dn + nn (krank/gesund) Dn / nn / Dn / nn -> 50% krank Neurofibromatose durch Tumor Bildung beweglicher Fibrome überall am Körper cafe-au-lait-flecken typisch für autosomal dominanter Erbgang ADE: Variable expressivität wenn Erkrankung nach gesunden Eltern -> Neumutation (oder Kuckskind) 40jähriger Mann hat etwa 10fach höheres Neumutationsrisiko gegenüber 20jährigem Mann bei DMD 1:10000, häufig 1:100k; 1:1mio d.h. Das absolute Risiko, ein Kind mit Erbkrankheit zu bekommen, bleibt sehr, sehr niedrig Kasuistik A.B., geb. 1982 – Grundschule – Realschule – 1 Jahr Canada – Gymnasium (Abbruch wg. schlechter Schulleistung) – Praktikum (Kindergarten) – Fachabitur (2004) – Berufsausbildung Ergotherapeutin Symptome – Schulprobleme – antriebsarm – schlaffe Körperhaltung – ermüdet schnell – verwaschene Sprache – – – – – – – – – – – 2000 KJP Wü, V.a. ADS ; Ritalin-Therapie empfohlen (nicht akzeptiert) 2001 Ausschluss Narkolepsie 2002 KJP Köln ADS: Ritalin-Therapie deutliche Besserung Frühjahr 2004 beim Abitur sehr starke Magenschmerzen Labordiagnostik: CK um 1000 IU/l CK-Wewrt bleibt pathologisch hoch Internistisch kann keine Ursache gefunden werden Neurologische Untersuchung Herbst 2004 Muskelschwäche im Gesicht, Schultergürtel, Hände beginnende Spitzfussstellung Faustschlussmyotonie bds. EMG: myotone Serien FA: keine neuromuskulären Erkrankungen Mutter mit 41 J bds Katarakt-OP V.a. Myotone Dystrophie exzessive Tagesmüdigkeit (EDS) bei ca 30% der DM1-Patienten 50 / 63 – Therapie Ritalin – überdeckt Symptome - aber KI bei DM1 wegen kardialer Probleme alternativ z.B. Modafinil bisher als Indikation nur bei Narkolepsie keine negative Beeitrichtigung des Herzen Myotone Dystrophie DM1 (Curschmann-steinert) ADE, 1:20k Klinik: – Muskelschwäche – Myotonie – Katarakt – Herzbeteiligung – Endokrine Störungen – Stirngaltze – Intelligenzminderung – Wesensänderungen Genetik – ADE – Inzidenz 1:20k – CTG-Repeat 5-30 normal, 50-150 leicht betr , 100-1000 klassische DM , über 2000 kong. DM – Antizipation (von Generation zu Generation stärker Betroffene – bei maternaler Übertragung Repeatanstieg) – Chromosom 19q13.3 DMPK-Gen Facio-scapulo-humerale Muskeldystrophie (FSHD) Klinik: – oft Schwäche in der fazialen Muskulatur – zunehmende Schwäche im Schultergürtel – verkrümmung der Wirbelsäule – progrediente Schwerhöhrigkeit – CK nur leicht erhöht – EMG: Myopathiemuster Genetik: – ADE – variable Expressivität – Genort 4q35 – ca 1/3 Neumutationen – Keimzellmosaike – relat. Fertilität verringert (0,6-0,8) – „umgekehrte Repeat-Erkrankung“: FSHD bei <10 D4Z4-Repeats M. Crouzon – Mutation im FGFR2-Gen Apert-Syndrom (Synostosen z.B. der Finger) – ebenfalls Mutation im FGFR2-Gen -> nix mit 1 Gen, 1 Krankheit – es ist mal wieder komplizierter! Chorea Huntington – ADE, 4p16.3 – Huntingtin-Gen normal n=5-34, erkrankt n = 39-121 51 / 63 – Prädiktive Diagnostik nur mit genetischer Beratung , Recht auf Nicht-Wissen (50% nutzen das hier nach Beratung) Kopplung Krankheit zu Allel in Familie – aber dieses Allel ist nicht identisch mit der Mutation die Kopplung sagt nur etwas über den Genort, nichts über die Mutation aus dieses Allel kann auch vom nicht betroffenen Elternteil sein, demzufolge kann Nachkomme mit dem Allel auch die Mutation nicht tragen Kopplungsanalyse Segregation von zwei Loci (A,a und B,b) • die beiden Genorte (2 von papa A,B, 2 von mama a,b) liegen auf unterschiedlichen Chromosomen, werden unabhängig (gemäß Mendelscher Regeln) vererbt möglichkeiten: AB, aB, Ab, ab • liegen die beiden Genorte auf den gleichen Chromosomen dicht beeinander (kein crossover möglich)-> abhängige Vererbung (beides wird gemeinsam vererbt) Möglichkeiten: Aa, Bb • die beiden genorte liegen auf dem gleichen chromosom, gemeinsame vererbgung bis in der meiose ein crossing over eintritt (theta = Rekombinationsrate) LOD-Score: Logarithmus der Wahrscheinlichkeit von Kopplung Autosomal rezessiver Erbgang beide Eltern heterozygot gesund Nd Nd NN Nd Nd dd .... ¼ Kinder gesund , ½ heterozygot gesund, ¼ krank Achtung: gesunde Geschwister beim autosomal rezessiven Erbgang haben eine Wahrscheinlichkeit von 2/3 heterozygote Genträger zu sein! (bei gesundem Phänotyp Ausschluss des kranken ¼ ) bei NN dd homozygot gesund/krank Nd Nd Nd Nd -> alle heterozygot Gesund Weinberg/Hardy und ihre Vererbungsgesetze Weinberg: Merkmal Zwillingsschwangerschaft in Querschnittsuntersuchung Zwillingshäufigkeit ist geographisch/genetisch sehr unterschiedlich (weltweit ca 1,2%) Hardy-Weinberg-Regel p² + 2pq + q² = 1 AA Aa aa Voraussetzungen – Panmixie (random mating, keine Inzucht usw) – keine Auslese, keine Inzucht , keine Migration... – keine Selektion (z.B. verminderte Fertilität) 52 / 63 Versuch: Überprüfung ob ein HWE vorliegt: – 2 Arten von Bonbons a und b – jeder nimmt blind aus der tüte zwei bonbbons aa: 4 3,06 ab: 16 17,87 bb: 27 26,06 geschätzte allelfrequenzen: 0,256 (a); 0,744 (b) chi²=0,51 alpha=5%, FG=2.... tabellenwert 5,99 signifikanzwert darf nicht überschritten werden -> wir unterliegen Hardy-Weinberg-GGW Schätzung von Genfrequenzen – Autosomal rezessiver Erbgang – Kranke=homozygote a a = q² – q² = 1/10k – q = 1/100 p+q =1 (muss 1 sein) p=99/100 ... ≈ 1 – Heterozygotenhäufigkeit 2pq ≈ 2q ≈ 1/50 Spinale Muskelatrophie Werding-Hoffmann • ARE, Inzidenz 1:10k, heterozygoten: 1:50 , auf 5q13.3 • Basisdefekt: Mutationen im SMN, teileweise auch benachbarten NAIP, um 95% deletionen • hypoton • ein Großteil Kinder stirbt im 1. LJ • 3 typen Schwer betroffene: proximale muskelschwäche, adynamie, fehlende eigenreflexe, gutes kontaktverhalten, differenzierte feinmotorik, vermehrtes speicheln, aspiratiosngefahr, skolioseentwicklung • normale ck oder leichte erhöhung • abnorme muskelsono, mrt, emg • normale nlg • ... • verschiedene formen: sma 1-3, sma kennedy (x-chromosomal), sma mit ateminsuffizienz , als (nur in wenigen fällen genetisch) Prader-Willi-Syndrom (hatten wir schon 2x..... GNARF*kopier*) adipositas per magna (bedingt durch hyperphagie, kein sättigungsgefühl) mittelschwere geistige behinderung hypoton (muskeln) (mutter merkt wenig bewegung in schwangerschaft, danach schlecht beim saugen usw) therapie: kühlschrank abschliessen, schild „bitte nicht füttern“ umhängen 70% deletion 15q pat 30% maternale uniparentale disomie (offensichtlich imprinting phänomen! Angelman-Syndrom: > 90% Deletion 15qmat ; 3% paternale uniparentale disomie Prader-willi-Syndrom: 70% Deletion 15q pat , 30% maternale uniparentale disomie in einer Familie kann PWS und AS auftreten Genetik Angelman-Syndrom 65% maternale Deletion häufig Punktmutationen ... 53 / 63 Familienberatung beim PWS / AS genaue diagnostik nachweis der deletion bzw isodisomie oder punktmutation empirisches wdh-risiko sehr gering therapie pws/as: aufklärung / sonderpädagogik / schlafwachrhythmus, physiotherapie, epilepsiebehandlung bei as Inzidenz / Prävalenz inz: häuf erkrankten unter allen leben neugeborenen (down-syndrom: 1/700) präv: häuf erkrankten bezogen auf lebende bevölkerung (zu einem bestimmten zeitpunkt oder periode) z.b. beim down-syndrom deutlich niedriger als inzidenz aufgrund verkürzter lebensspanne Berechnung der Erblichkeit eines quantitativen Merkmals P=G+E (phänotyp, genotyp, umwelteinflüsse(environment) Varianzen: Vp=Vg+Ve Vg=Va +Vd Va = additive Genwirkung der beiden allele Vd = dominanz zwischen den biedne allelen und epistase Heritabilität h² dient zum schätzen genetischer Varianz zu welchem anteil der phänotyp auf den genotyp zurückzuführen ist, sagt jedoch nicht über die anzahl der beteiligten gene, deren eskLAMPENTAUSCH ERFODERLICH... oder genprodukte aus sehr hoch : umweltfaktoren spielen große rolle, niedrig: genetik große rolle 54 / 63 in einer population ist jeder 50. mensch für ein bestimmtes gen heterozygoter genträger wie hoch ist die homozygotenhäufigkeit? 1. 1:100 2. 1:2500 3. 1:5000 4. 1:10000 !!! 5. 1:100000 hardy-weinberg: p²+2pq+q² 2 pq = 1/50 p>>>q (geht gegen 1, also p ≈ 1) 2q ≈ 1/50 q ≈ 1/100 q² = 1/10000 !! ein autosomal-rezessiv erbliche krankheit hat eine häufigkeit von 1/10k in der bevölkerung wie häufig ist das krankheitsverursachende abnorme gen? 1. 2. 3. 4. 5. 1:50 1:100 !!! 1:200 1:5000 1:10000 Es wird nach q gefragt!! die frequenz eiens autosomalen gens ist 0,01 welche der angegebenen schätzungen der häufigkeit der heterozygoten inst bei annahme eines systems von zwei allelen unter den bedingungen unter denen hardyweinberg gilt in bester nährerung zutreffend ? 1. 0,00001 2. 0,0002 3. 0,005 4. 0,02 !!! 5. 0,05 gegeben q heterozygotenhäufigkeit 2pq p geht gegen 1 also 0,02 eine frau hat mit einem mann, der inzw verstorben , ein kind mit seltenen rezessiven erbleiden. Sie will wieder heiraten, den bruder des verstorbenen mannes wie groß risiko für 1. kind aus dieser verbindung mit dem gleichen rezessiven leiden behaftet zu sein ? 1. unter 1/100 2. 1/16 3. 1/8 !!! 4. ¼ 5. ½ 55 / 63 geschwister haben immer 50% gene gemeinsam, wenn bruder 100% heterozygot ist hat der neue 50% 1(mutter)* ½ (neuer ehemann) * ¼ (jeder heterozygote gibt mit 50% wahrscheinlichkeit das gen an nachkommen weiter, also ½ * ½ ) = 1/8 1*1/2*1/4=1/8 eine frau leidet an achondroplasie, ihr mann an vit-d-resistenter hypophosphatämischer rachitis wie groß ist wahrscheinlichkeit für einen sohn, beide krankheiten zu haben ? 1. Nahezu 0 !!! 2. 12,5% 3. 25% 4. 50% 5. 75% Vererbung: achondroplasie ADE vit-d resistent achitis x-chromosomal – der vater vererbt aber das y – also 0 welche aussage über das genomische imprinting trifft nicht zu ? 1. es bezeichnet die wirksamkeit von genen in abhängigkeit von ihrer elterlichen herkunft 2. es wird durch inaktivierung von genen in der keimbahn erreicht 3. es kommt bei beiden geschlechtern vor 4. es wird druch protein-methylierung erreicht !!! 5. es wird durch dna-methylierung erreicht welche häufigkeit ein mensch gleichzeitig heterozygot für die AR Krankheiten a (1:40k ) und b (1:10k) etwa ein mensch unter 1. 400.000.000 menschen 2. 20.000 menschen 3. 10k 4. 5k !!! 5. 2,5k q² gegeben -> q also 1:200, 1:100 2 pq ≈ 2 q ≈ 1:100 bzw 1:50 1:100 * 1:50 -> 1:5000 multiplikation der heterozygoten genträger in der bevölkerung (unabhängig wird multipliziert) bei welchem der folgenden genetisch bedingten krankheitsbilder sind café-au-lait-Flecken ein wichtiges diagnostisches zeichen (kurz : wo cafe-au-lait, mann?) 1. zystische fibrose 2. chorea huntington 3. neurofibromatose typ I !!! 4. adrenogenitales syndrom 5. galaktosämie 56 / 63 das erste und einzige kind eines 52jährigen mannes und einer 31jährigen frau , beide gesund, hat eine achondroplasie welche ursache am wahrscheinlichsten 1. neumutation !!! 2. homozygotie bei autosomal-rezessiver vererbung 3. frühe embryonale noxe 4. phänokopie (es sieht so aus als hätte ich den gendefekt, hab ihn aber nicht – evtl nen anderen) 5. dominante vererbung mit verminderter penetranz neumutationsrisiko steigt bei paps mit dem alter, ist hier recht groß beteiligt bei dem 30jährigen sohn einer an chorea hunt. Verstorb patientin mit genetischen methoden prädiktive diagnostik welche untersuchung ist aussagefähig 1. untersuchung des huntingtin-gens auf deletionen 2. untersuchung des huntingtin-gens auf rasterverschiebung 3. untersuchung des huntingtin-gens auf trinukleotid-repeat-verlängerung !!! 4. untersuchung des huntingtin-gens auf missense-mutation 5. chromosomendarstellung mit anfärbung der huntingtin-bande die bei der zystischen fibrose (mucoviszidose ) vorkommende mutation delta f508 ist eine 1. punktmutation 2. nonsense-mutation 3. frame-shift-mutation 4. splice-site-mutation 5. deletion von drei basenpaaren!!! delta ist abkürzung für deletion !!! die weitaus häufigste(n) genmutation(en) bei patienten mit zF (mucoviszidose) aus mitteleuropa ist / sind del von 3 basenpaaren im codon 508 (dF508) welche der folgenden Aussagen über das fragile x-syndrom trifft NICHT zu 1. sowohl knaben als auch mädlchen können betroffen sein 2. ursache ist eine triplett-expansion im 5'-Bereich des FMR1-Gens 3. männer können überträger sein 4. die expansion einer prämutation erfolgt in der weiblichen keimbahn 5. 50% der nachkommen von männlichen genträgern sind betroffen !!!! 15jähr junge gesunde eltern symptome: iq 50, verzögerte sprachentwicklung, 185cm , hohes shcmales gesicht, große ohren makroorchidismus diagnose? 1. AGS 2. XYY-syndrom 3. martin-bell (Fra-X)Syndrom !!! 4. XXY-Syndrom 5. testikuläre Feminisierung 57 / 63 v.a. Wegen der geistigen entwicklung kleinkind, symptomkombi: primordialer disproportionierter minderwuchs, verkürzte extremitäten, fronto-parietales ausladen des schädels, normale geistige entwicklung 1. achondroplasie !!! 2. katzenschrei-syndrom 3. tris13 4. tris18 5. tris21 knackpunkt geistige entwicklung: chromosomenstörungen immer retardiert weitaus häufigste(n) genmutation(en) bei pat. Mit MD typ duchenne oder becker: 1. instabile trinukleotidsequenz 2. punktmutation , die zum vorzeitigen abbruch proteinsynthese führen (nonsense-mutation) 3. deletionen !!! 4. inversionen und duplikationen 5. promotormutationen x-chromosom-inaktivierung (lyon-hypothese) im weibl geschlecht, was ist falsch? 1. in somatischen zellen eines der beiden x-chromosomen größtenteils inatkviert 2. inaktvierung x-chromosom mechanismus zur dosiskompensation x-chromosomaler genprodukte 3. inaktivierung des x-chromsosoms erfolgt bei fete etwa in 12. SSW !!! 4. inaktiviertes x-chromosom kann in vershciedenne zellen veäterlicher oder mütterlicher herkunft sein 5. das morphologische substrat des inaktivierten x-chromosoms ist das sog. Geschlechtschromatin (syn. Barr-körperchen) -> ausschlussverfahren bei welcher situation pränataldiagnostik mittels chorionzottenbiopsie absolut k.i.? 1. alter mutter > 40 2. mutter trägerin balancierte 14q21q robertsonsche translokation 3. vorausgegangenes kdin mit zF (homozygotie für delta f 508) 4. vorausgegangenes kind mit spina bifida!!! 5. mutter ist konduktorin für MD typ duchenne mit bekannter deletion im dystrophin-gen -> spina bifida multifaktoriell 27jährige Frau, kommt in 8SSW gen beratung verstorbener bruder klinisch diagnostiziertes down-syndrom, keine chromosomenuntersucung keien weiteren geschwister , vater verstorben welche massnahme sollte unter berücksichtigung minimaler invasisivtät zunächst durchgeführt werden? 1. chromosomenanalyse bei beiden ehepartnern 2. chromosomenanalyse bei der schwangeren !!! 3. amniozentese 4. chorionzottenbiopsie 5. nabelschnurpunktion welche aussage über multifaktorielle vererbung trifft nicht zu? 1. dabei utnerschiede zwischen geschlechtern in häufigkeit ausprägung eines merkmals 2. ausprägung kann von umwelteinflüssen beeinflusst werden 58 / 63 3. für erkrankungen kann im rahmen einer gen. Beratung das wdh-risk entsprechen den mendelschen regeln exakt bestimmt werden !!! 4. die vererbung der einzelen an einem multifaktoriellen system beteiligten gene folgt den mendenlschen regeln 5. die wiederholungswahrscheinlichkeit für ein multifaktoriell bedingtes merkmal steigt mit der zahl der betroffenen nahen verwandten tumorsupressorgene, was falsch ? – besitzen hemmenden einfluss auf zellproliferation – beitrag durch deletion auf keimbahneben zur entstehung herdeditärer tumoren – entsprechen prinzip des two-hit-modells der tumorentstehung (nach knudson) – entwickeln ihre tumorigenen eigenschaften durch mutation oder delteion – entfalten ihre tumorigene wirkung bereits nach mono-allelischer inaktivierung !!! welches reproduktive outcome ist NICHT typisch für eine familiäre chromosomentranslokation? 1. geburt von kindern mit fehlbildungen 2. verringerte fruchtbarkeit männlicher individuen 3. erhöhte fehlgeburtsrate 4. geburt von kindern mit XXY !!! 5. geburt von phänotypisch unauffälligen kindern die der freien tris21 zugrunde liegende nondisjunction erfolgt überwiegend in der 1. paternalen meiose I 2. postzygotisch 3. maternalen meiose II 4. maternalen meiose I !!! 5. paternalen meiose II welche aussage zur MD typ duchenne trifft nicht zu? 1. ursächlich veränderung dystrophin-gen 2. übertragung vom vater auf den sohn ist in der regel möglich !! 3. kranhiet wird bei etwa 1/3 patientten neumutation 4. hyperlordose lWS 5. krankheits bild pseudohypertrophie waden -> x-chrom : nö 2. bei welcher konstellation erkrankungsrisiko für kind praktisch 100%? 1. beide eltern taubstumm 2. beide eltern diab m typ I 3. vater bluter , mutter konduktorin hämophilie A 4. beide eltern achondroplasie 5. ein elternteil ist träger eines isochromosoms 21 oder einer robertsonschen translokation (21q21q) !!! -> taubstumm heterogenes krankheitsbild, multifaktoriell; bei 5. entweder monosomie oder tris21 ein isochromosom: langer arm, zentromer, langer arm (oder kurzer arm, zentromer, kurzer arm) frau mit ehemann ein kind mit phenylketonurie der ehemann stirbt, sie heiratet seinen bruder wie groß risiko eine phenylketonurie zu bekommen für das erste kind aus dieser ehe 12,5% -> 1*1/2*1/2*1/2 = 1/8 59 / 63 beckersche MD (x-chromosomal-rezessiv ) fragesetellung . Mit welcher wahrscheinlichkeit ist die tochter heterozygot für BMD ? 100% (erbt von betroffenen vater immer das x-chromosom) geschwister II1/II2 leiden an krankheit mit ARE, die mit einer häufigkeit von 1:40k auftritt elternpaar II3/II4 ratsuchend ist gesund, familie des mannes keine krankheit vorkommen wie hoch wahrscheinlichkeit, dass ein kind des paares II3/II4 an krankheit von tante und onkel leidet? 2/3*1/100*1/4 -> 1/600 II/3 gesunde , die konduktorin sein könnte (2/3 konduktorin) II/4 bevölkerungsrisiko vater I2 des mannes II1 leidet an krankheit mit ARE, die mit einer häufigkeit von 1:2,5k auftritt mit welcher wahrschienlichkeit tritt krankheit bei kind der ratsuchenden (II1/II2) auf ? II/1 mit sichheit heterozygot (gibt weiter) frau: 1/2500: wurzel 1/50 (q) -> 2pq .... 2*1*1/50 -> 1/25 1/25 * ¼ -> 1% die personen mit I/1 und II/1 sind von BMD betroffen wie hoch wdh-risiko für MD bei einem sohn von II/1 ... mutter II/2 gesund risiko entspricht dem der allgemeinbevölkerung -> kriegt ja vom daddy das y RFLP: versch. Banden ... einfach vergleichen ältestes kind steht links 2 molekulare marker markerabstand 10cM (centiMorgan) rekombinationshäufigkeiten zwischen diesen markern und einem krankheit verursachenden gen marker 1: gen 2% rekombination marker 2: gen 8% rekombination welche lage es gens lässt sich aus diesem befund ermitteln? E (gen liegt näher bei marker 1, 8cM vom marker 2 weg) zyst fibrose, kind mit gesunden eltern, wie hoch risiko ? Natürlich der kleinste wert (geringer als 1:2500)ui ist ja normalbevölkerungsrisiko nächstes bsp, sind kranke drin im stammbaum , aber die haen nix mit dem kind zu tun dessen risiko gefragt ist -> also allgemeinbevölkerungsrisiko gesunde eltern haben ein kind mit klassischer phenylketonurie wie hoch ist das wiederholungsrisiko , wenn die eltern verwandt sind (vetter-base 1.Grades)? 25% -> verwandtschaft total wurscht, eltern sind beide eindeutig hetterozygot 60 / 63 Embryonale Entwicklung und Fehlbildungen des Menschen Epigenese (Entwicklung gemäß eines Programms) vs Präformation (alles schon da von Beginn an) Mechanismen : – Induktion – Achsenbildung (durch Induktion und Migration) – Musterbildung isometrisches (alle Körperteile wachsen gleichschnell – Kopf erstmal so groß wie Rumpf) und allometrisches (Proportionierung des Körpers – Kopf nur noch 1/10 der gesamten Körperlänge) Wachstum bsp: Achsenbildung beim Hühnchen... pH-Gradient, Membranpotential li-re-Achsenbildung über Signaltransduktionskaskade Zeittafel der Embryonalentwicklung – erstellt über Geburtsdefekte, ermöglicht wiederum Diagnose selbiger wenn in der pränatalen Periode was schiefläuft, kommt's in der Regel zum Abort – deswegen Besprechungsstart bei der embryonalentwicklung bekannte Teratogene Chemische Substanzen – Alkohol – Heroin – Aminoglykoside – Diphenylhydantoin – Valproat – Thalidomid (heute in Krebstherapie) – Warfarin (Marcumar) Ionisierende Strahlung, Hyperthermie Infektionen – Herpes simplex – Rubella – Toxoplasmose (cave Katzen) – Syphilis Metabolische Entgleisungen der Mutter – Fehlernährung – Diabetes – Phenylketonurie -> bei Gabe teratogener Mittel Verhütung beachten... Alkoholembryopathie – minderwuchs, Mikrozephalie – Kraniofaziale Dysmorphie 61 / 63 – – – Statomotorische Verzögerung ZNS-Störungen Häufig! Rötelnembryopathie – Katarakt mit häufiger Mikrophthalmie – Mikrocephalie, psychomotorische Entwicklungsstörung – Innenohrschwerhörigkeit – persistierender Ductus arteriosus, Pulmonalstenosen – etwa bei ¼ aller Rötel-infizierten Mütter!! Thalidomid-Embryopathie (1958-1963) Symptomatik nach Thalidomideinnahme 34.-50. Tag p.m.: – Gliedmaßenfehlbildungen aller Grade – Gehörmuschelfehlbildungen bis zur Anotie – Mikrophthalmie, Kolobom – Fehlbildungen des Herzens, der großen Gefäße und der Lunge Warfarin-Embryopathie nach Marcumar-Exposition – Hypoplasie des Mittelgesichts – Pränatale Dystophie mit postnatalem dysprop. Minderwuchs – Hemmung der Arylsulfataseaktivität (genetische Phänokopie) Achsenbildung bei den Gliedmaßen – Proximal-distale Achse (Schulter-Hand): FGF-Familie – Anterior-posteriore Achse (Daumen-kl. Finger ): SHH (sonic hedgehog) – dorsale-ventrale Achse (Handrücken-Handfläche ): WNT7A mit FGF8 können wir auch wunderbar beim Hühnchen drei Extremitäten auf einer Seite induzieren Holt-Oram-Syndorm, kardiodigitales Syndrom Mutationen im T-Box5-Gen (TBX5, 12q24), Transkriptionsfaktor Neben der Aktivität in der Extremitätenknospe beeinflusst TBX5 die Differenzierung von Kardiomyozyten, Interaktion mit dem Transkriptionsfaktor GATA 4 – Fehlbildungen der oberen Extremität – Angeborener Herzfehler (VSD, ASD) -> monogenetische Störung früh in der Embryogenese macht diffiziles Krankheitsbild LMX1B-Defekte verursachen des Nagel-Patella Syndrom WNT7A kativiert den LMX1b-Transkriptionsfaktor im dorsalen Mesenchym der Extremitäten Symptomatik – Nagelhypoplasien und Dysplasien – Hypoplasie der Kniescheibe mit häufiger Luxation – Tastbare „Beckenhörner“ Das Problem des Schädelwachstums: Kraniosynostosen – Knochenplatten des Schädeldaches durch membranöse Verknöcherung (ohne Knorpelbildung) – Schädelvolumen muss mit dem Wachstum des Telenzephalons Schritt halten (Ende mit dem Erwachsenwerden) – Nähte und Fontanellen dienen als Wachstumszonen 62 / 63 Genetik der Kraniosynostosen – Inzidenz ist 1:2k bis 1:3k – 10-20% aller haben autosomal dom Erbgang – mehr als 75 Syndrom mit Kraniosynostose als Zusatzsymptom – Vorkommen auch bei chromosomalen Aberrationen , z.B. del(7p) – Neumutationen sind in aller Regel paternalen Ursprungs FGFR-assoziierte Kraniosynostosen: Apert- / Pfeiffer- und Crouzon-Syndrom überlappende Gain of fucnction Mutationen im FGFR2-Gen Membranständige Tyrosinkinase-Rezeptoren (FGFR1 – FGFR4) – Chr. 8P, 10q26, 4p16 in weiteren Domänen von FGFR3 sind Punktmutationen für die Achondroplasie, Hypochondroplasie, Thanatophoren Zwergwuchs Achondroplasie v.a. Wegen alter Väter – diese Punktmutationen entstehen ausschließlich in der männlichen Keimbahn (in sich autosomal dominant, idR aber Neumutationen da die Achondroplastiker selten Partner finden) 63 / 63