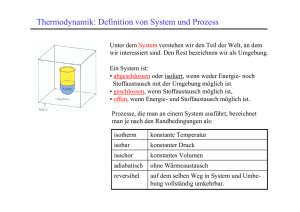
Kapitel 12 - Fakult at f ur Physik
Werbung

Kapitel 12 Thermodynamik Die Thermodynamik behandelt die Umverteilung von Energie in ihren verschiedenen Erscheinungsformen. Historisch entstand die Thermodynamik aus dem Studium der Volumen-, Druck- und Temperaturverhältnisse (der sogenannten Zustandsgrößen) bei Dampfmaschinen. Diese Zustandsgrößen beschreiben makroskopische (im Sinne von auf eine Vielzahl von Einzelteilchen bezogene) physikalische Messgrößen. Nach Einführung der statistischen Mechanik durch Maxwell und Boltzmann entstand ein mikroskopisches Verständnis der bis dahin empirischen Zustandsgleichungen. Zentral ist der Satz der Erhaltung der Energie mit der wichtigen Auflage, dass in Systemen mit vielen Freiheitsgraden irreversible Prozesse die Zeitentwicklung der Zustandsgrößen bestimmen. Eine zentrale Messgröße ist die Temperatur eines Körpers. Sie ist eine intensive Größe wobei intensiv dafür steht, dass sich diese Messgröße nicht ändert, wenn wir den Körper in zwei Hälften teilen. Im Gegensatz dazu ist die innere Energie eines Körpers dU eine extensive Größe, sie halbiert sich wenn wir den Körper in zwei Hälften teilen. 12.1 Temperaturmessung Empirische Temperaturskalen: Celsius, Fahrenheit, später: Anschluss dieser Skalen an die thermodynamische Temperatur welche über die innere Energie definiert ist. Jede messbare Änderung von Materialeigenschaften mit der Temperatur kann zum Messen benutzt werden. • Geometrische Abmessungen (Länge von Stäben, Drähten etc. (Bimetallthermometer), Flüssigkeits- und Gasvolumina (Hg-, Alkohol-, Gasthermometer) • Elektrischer Widerstand (Widerstands-, Halbleiterthermometer) • Elektrische Kontaktspannung (Thermospannung ! Thermoelemente, Thermosäulen) • Emittierte Strahlungsleistung (z.B. Pyrometer, Thermographie, Wärmebildkamera). a) Längenänderungen von Festkörpern Wir messen die Länge eines Metallstabs bei wachsender Temperatur: der Körper dehnt sich proportional zur Temperaturänderung aus: L(T ) = L(T0 ) · (1 + ↵ (T (12.1) T0 )) . 129 130 KAPITEL 12. THERMODYNAMIK Der thermische Längenausdehnungskoeffizient ↵ ist eine Materialeigenschaft. In erster Näherung kann ↵ über kleine Bereiche der Temperaturänderung als konstant angesetzt werden. Seine SI-Einheit ist [↵] = K 1 . (Temperaturänderungen werden in K (Kelvin) gemessen, das ist gleichzeitig die SI-Einheit der thermodynamischen Temperatur, s. später.) Mikroskopisch erklären lässt sich die Ausdehnung durch die Zunahme der Schwingungsenergie der Atome im Festkörper. Da die Schwingung anharmonisch ist, vergrößert sich bei Zunahme der inneren Energie eines Körpers der mittlere Abstand der Atome. Material Aluminium Kupfer Edelstahl Eisen Glas Quarzglas Invar Zerodur vorbehandelter Gummi ↵ / 10 23.8 16.8 16. 12.2 3 ... 9 0.45 1.5 < 0.1 6 K 1 negativ ! Anwendung im Bimetall-Thermometer oder -Schalter: Zwei Metalle mit unterschiedlichen Ausdehnungskoeffizienten sind aneinander gelötet. Durch die unterschiedliche Wärmeausdehnung kommt es beim Erhitzen zu einer Krümmung. ! kann sowohl zum Messen (Übertragung auf eine Anzeigemechanik) als auch zum Schalten benutzt werden (verbogener Stab betätigt Schalter). Experimente: Bimetallstab erwärmen, Ausdehnung einer heissen Kugel b) Volumenänderungen von Festkörpern und Flüssigkeiten Wir beobachten das Volumen einer Flüssigkeit beim Erwärmen. Im Allgemeinen nimmt es proportional zur Temperaturänderung zu: V (T + T ) = V (T ) · (1 + (12.2) T) Der thermische Volumenausdehnungskoeffizient ist eine Materialeigenschaft. Er hängt mit dem Längenausdehnungskoeffizienten zusammen, denn aus der Längenausdehnung ergibt sich für das Volumen V (T + T) = ⇡ ⇡ V (T ) · (1 + ↵ V (T ) · (1 + 3 ↵ T) 3 T ) , d.h. (12.3) 3↵ Seine SI-Einheit ist [ ] = K 1 . Material (20 C) Eis (0 C) Wasser Quecksilber Ethanol Ist das Material anisotrop (z.B. Holz längs und quer zur Faserrichtung, gestreckte Kunststoffe), dann liegen im Allgemeinen drei unterschiedliche Längenausdehnungskoeffizienten ↵x , ↵y , ↵z vor. 3 K 1 1.0000 Wasser relative Dichte Ein wichtiger Fall ist der Volumenausdehnungskoeffizient von Wasser. Zum einen ist die Dichte von Eis rund 10% geringer als die von flüssigem Wasser. Zum anderen besitzt das spezifische Volumen (Volumen / Masse = 1/Dichte) von Wasser ein Minimum bei 4 C. Das ist die Anomalie des Wassers. Sie stellt z.B. sicher, dass sich beim Zufrieren offener Gewässer immer eine etwas wärmere Wasserschicht unterhalb der Eisdecke befindet und so das Überleben von Fische ermöglicht (jedenfalls solange das Wasser nicht bis zum Boden friert). / 10 0.230 0.207 0.182 1.100 0.9995 0.9990 0.9985 0.9980 -10 Eis 0.917 Ø -5 0 5 10 Temperatur 0 C 15 20 12.1. TEMPERATURMESSUNG 131 Die historisch wichtigste Flüssigkeit für Temperaturmessungen ist das Quecksilber (Hg). Damit wurden auch die ersten empirischen Temperaturskalen definiert. Die heute meist gebräuchliche Celsius-Skala verwendet als sog. Fixpunkte schmelzendes Eis (0 C) und siedendes Wasser (100 C), beides bei sog. Normbedingungen, p0 = 1013.25 mb. Eine andere verbreitete Thermometerflüssigkeit ist Alkohol. Sie eignet sich besonders für tiefere Temperaturen, weil Hg schon bei -38,86 C fest wird. Im Vergleich zu Hg hat sie allerdings einen merklich nicht-lineare Ausdehnung (zur Beschreibung braucht man neben dem linearen auch den quadratischen Volumenausdehnungskoeffizienten), was eine nicht-lineare Skala notwendig macht. Vergleicht man den Bereich zwischen 0 und 100 C bei beiden Flüssigkeiten, so zeigt Alkohol bei 50 C auf einer linearen Skala rund 2 Grad zu wenig an. Die Anbindung dieser Temperaturskalen an die absolute Temperaturskala erfolgt durch die Beziehung T [ C] = T [K] 273.15 .Die Anbindung an die Fahrenheitskala erfolgt mit der Beziehung T [ C] = 5(T [ F] 32)/9 . c) Volumen-/Druckänderungen von Gasen Auch Gase dehnen sich aus, wenn man sie erwärmt und dabei den Druck konstant lässt. Diese Nebenbedingung ist hier — im Gegensatz zu Flüssigkeiten und Festkörpern — wichtig, denn Gase besitzen im Gegensatz zu diesen eine hohe Kompressibilität. Bei konstantem Druck erhält man wieder einen linearen Zusammenhang: V (T ) = V (T0 ) · (1 + V (T (12.4) T0 )) , wobei der Index V bedeutet, dass es sich um eine Änderung des Volumens handelt. Es zeigt sich, dass dieser thermische Volumenausdehnungskoeffizient für die meisten Gase gleich ist und bei 273.15 K etwa den Wert 3.7·10 3 K 1 hat. Material (20 C) ideale Gase Helium Argon Sauerstoff CO2 / 10 3.661 3.660 3.671 3.674 3.726 V 3 K 1 Analog findet man bei konstantem Volumen : p(T ) = p(T0 ) · (1 + p (T T0 )) . (12.5) Dieser Zusammenhang wird als das Gay-Lussacsche Gesetz bezeichnet. p bezeichnet man als den linearen thermischen Spannungskoeffizienten. Legt man die Gültigkeit der idealen Gasgleichung pV = N kT , (12.6) zugrunde, so kann man die Änderungen berechnen: Für p = p0 = const gilt p0 V0 = N k T0 und p0 V = N k T , woraus man durch Differenzbildung erhält ✓ ◆ Nk 1 V = V0 + (T T0 ) = V0 · 1 + (T T0 ) . (12.7) p0 T0 Für T0 = 273.15 K ergibt sich daraus V = 1/273.15 K, in Übereinstimmung mit dem obigen Befund. Die gleiche Vorgehensweise mit dem Ansatz V = V0 = const liefert für p das gleiche Ergebnis. Diese Erkenntnis besagt, dass das Volumen (oder der Druck) eines Gases auf Null schrumpft, wenn man T = 273.15 C erreicht. Da es negative Volumina nicht geben kann, hat man seinerzeit argumentiert, dass es einen “absoluten” Temperaturnullpunkt 132 KAPITEL 12. THERMODYNAMIK geben muss, und eine absolute Temperaturskala eingeführt. Durch die kinetische Gastheorie hat sich diese Vorstellung auch als vernünftig erwiesen. Eine Anwendung ist das Gasthermometer. Je nach Variante wird entweder der Druck oder das Volumen konstant gehalten. Diese Thermometer arbeiten über weite Bereiche sehr linear und eignen sich daher gut für Präzisionsmessungen. d) Druckänderungen in Festkörpern Auch in Flüssigkeiten und Festkörpern kann man im Prinzip Experimente bei konstantem Volumen (bzw. konstanter Länge) machen. Im Material baut sich dann, analog zu den Gasen, ein höherer Druck (Druckspannung in Festkörpern) auf. Dabei entstehenden große Kräfte. Eine thermisch bedingte Längenänderung wird durch eine entsprechende Druck- bzw. Zugspannung kompensiert: F =EA Beispiel: l l mit l l =↵ (12.8) T. Elastizitätsmodul Stahl E = 120 GN/m2 , Querschnittsfläche A = 1 cm2 , ↵ = 16·10 6 K 1 , T = 21 K, ! F ⇡ 4000 N. Derartige Kräfte muss man bei Baumaßnahmen einkalkulieren, bei Teilen die dem täglichen Temperaturwechsel unterworfen sind: Ausdehnung von Brücken, Eisenbahnschienen, etc. e) elektrischer Widerstand Der elektrische Widerstand R eines Leiters (messbar als Quotient aus elektrischer Spannung U und elektrischem Strom I) zeigt in vielen Fällen ebenfalls einen linearen Anstieg mit der Temperatur: R(T + T ) = R(T ) (1 + (12.9) T) . ist der lineare Widerstands-Temperaturkoeffizient. Eine derartige Zunahme beobachtet man für praktisch alle Metalle. Für Präzisionsmessungen verwendet man meist Platin-Draht. Der elektrische Widerstand von Halbleitern nimmt dagegen mit der Temperatur stark und nicht-linear ab (Das liegt daran, dass die Ladungsträgerdichte, die die elektrische Leitfähigkeit begrenzende Größe in Halbleitern, mit wachsender Temperatur exponentiell zunimmt. Auch dieser Effekt kann zur Temperaturbestimmung benutzt werden, insbesondere bei sehr tiefen Temperaturen. f ) Thermospannung Verbindet man zwei unterschiedliche Metalldrähte in einer Schleife miteinander so fließt ein geringer Strom durch die Schleife wenn sich die beiden Metallkontakte bei unterschiedlicher Temperatur befinden. Trennt man einen der beiden Metalldrähte auf und verbindet dessen Enden über ein Spannungsmeßgerät, beobachtet man eine von der MaMaterial B Uth Material B terialkombination abhängige sehr kleine Spannung (=Potentialdifferenz). Diese Kontaktspannung ändert sich in guter Näherung proportional zur Temperatur der Kontaktstelle, so dass ein Material A derartiges Thermoelement zum Messen der Temperaturdifferenz genutzt werden kann: Uth = ✏ · (T T0 ) . (12.10) T1 T2 Den Proportionalitätsfaktor ✏ bezeichnet man als “Thermokraft”, ganz im Sinne der elek- 12.2. WÄRMEMENGE, WÄRMEKAPAZITÄT 133 tromotoriaschen Kraft einer Batterie. Als Referenz benutzt man meist eine Mischung aus Eis und Wasser, so dass man eine der Celsius-Temperatur proportionale Spannung erhält. Heute wird anstelle eines aufwendigen Referenzelements diese Kompensationsspannung oft elektronisch erzeugt — leider eine Fehlerquelle in manchen Systemen. Die Größe von ✏ liegt bei -35 µV/K für Pt-Konstantan und bei +22 µV/K für Pt-NiCr. Experiment: Vorzeichenänderung der Thermospannung beim Verdunsten bzw. Erwärmen an einer der beiden Kontaktstellen. g) Änderung der Strahlungsleistung Die von einem Körper als Funktion seiner Temperatur emittierte Wärmestrahlung wird im Abschnitt Wärmetransport eingehender diskutiert. Aus dem bekannten Zusammenhang zwischen Temperatur und Abstrahlung (Plancksches Gesetz) ergibt sich die Möglichkeit eine Temperaturdifferenz zu messen, indem man die Wärmestrahlung von Körpern unterschiedlicher Temperatur vergleicht. Konventionelle Messverfahren die auf der Beobachtung sichtbaren Lichts beruhen sind wegen der steilen Abhängigkeit der sichtbaren Farbe von der Temperatur erst bei hohen Temperaturen (ab etwa 900 C) einsetzbar. Ein solches Gerät, Pyrometer, vergleicht die Helligkeit und Farbe eines glühenden Drahtes mit dem Körper dessen Temperatur bestimmt werden soll. Diese Methodik wird heute von sogenannten Wärmebildkameras übernommen die speziell den Infrarotanteil der Strahlung beobachten und deshalb auch bei Zimmertemperatur einsetzbar sind. 12.2 Wärmemenge, Wärmekapazität Die Temperatur T ist proportional zur im Stoff gespeicherten thermischen Energie. Zufuhr oder -Abfuhr von Energie ist mit einer Temperaturänderung verbunden, Q/ (12.11) T. Den Proportionalitätsfaktor bezeichnet man als Wärmekapazität. Er hängt von der Masse des Stoffes ab und von der spezifischen Wärmekapazität c des Körpers: Q=c·m· T (12.12) Historisch orientierte sich die Einheit der Wärmemenge an der spezifischen Wärmekapazität von Wasser: 1 kcal ist die Wärmeenergie, die benötigt wird, um 1 kg Wasser von 14.5 auf 15.5 C zu erwärmen. Heute benutzt man “normale” Energieeinheiten: 1 J=1 Ws=1 Nm. Den Umrechnungsfaktor bezeichnet man als Wärmeäquivalent: elektrisch: W = Q = U · I · t und Q=c·m· T mechanisch: W durch Reibung erzeugen Q=c·m· T Dabei ist U die elektrische Spannung (Potentialdifferenz),I der elektrische Strom und t die Zeit. Aus beiden Überlegungen ergibt sich 1 kcal = 4186 Nm = 4186 Ws . Mischkalorimeter: Man bringt einen Körper unbekannter Wärmekapazität auf eine bestimmte Temperatur TK und überführt ihn dann in ein (z.B. mit Wasser gefülltes) “Kalorimeter” (Temperatur TW ). Die Temperaturen von Körper und Wasser (und Kalorimeter) gleichen sich zu einer 134 KAPITEL 12. THERMODYNAMIK Mischungstemperatur TM , aus. Aus der Wärmebilanz kann (bei Vernachlässigung von Wärmeverlusten an die Umgebung) die unbekannte spezifische Wärmekapazität des Körpers bestimmt werden: cK mK (TM stoichiometry QK + QW = 0 TK ) + cW mW (TM mW T W cW · · mK T M TW ) TM TK = 0 = cK Molare Größen Für chemische Reaktionen sind nicht so sehr die Massen oder Volumina der Reagenzien maßgebend sondern die Anzahl der reagierenden Teilchen (Stöchiometrie). Es interessieren daher die Energieverluste und -gewinne pro Teilchen. Wir müssen also zählen! Die Zahlen sind allerdings sehr groß, 1 cm3 einer Flüssigkeit enthält größenordnungsmäßig 1022 Teilchen. Als handliches Maß für die Teilchenzahl-Menge eines Stoffs, wurde die sog. Stoffmenge ⌫ eingeführt. Ihre SI-Einheit ist nicht aus anderen Einheiten ableitbar, sie heißt Mol und wird mit dem Symbol “mol” bezeichnet. Sie ist folgendermaßen definiert: 1 mol ist die Stoffmenge mit der gleichen Anzahl von Teilchen (Atome oder Moleküle) wie 12 g Kohlenstoff des Isotops 12 C Diese Teilchenzahl ist gleich der Avogadro-Konstante (auch Loschmidt-Zahl genannt) NA = 6.0221367 ⇥ 1023 mol 1 (12.13) . Die Teilchenzahl in einer Stoffmenge ⌫ ist damit (12.14) N = ⌫ · NA . Wir spezifizieren also anstelle von teilchenzahl-bezogenen Größen stoffmengen-bezogene oder “molare” Größen: m Mm = = molare Masse, [Mm ] = g/mol (12.15) ⌫ m = NA · = NA · MTeilchen . (12.16) N Die Masse eines Teilchens MTeilchen ist abhängig von der jeweiligen Substanz, damit auch die molare Masse. Man führt eine weitere Masseeinheit ein, die, wie auch das Mol, an das Kohlenstoff-Isotop 12 angebunden ist: Die atomare Masseeinheit 1 amu = 1 12 der Masse eines Kohlenstoffatoms des Isotops Das sind 1.6605402 ⇥10 27 kg. 12 C Bezieht man die Massen von Atomen oder Molekülen auf diese Einheit, so nennt man das Resultat die relative Atommasse Ar (oder relative Molekülmasse Mr ): Ar , M r = MTeilchen 1 amu (12.17) Diesen Ausdruck können wir mit NA erweitern und erhalten Ar , M r = Mm 1 amu · NA (12.18) 12.3. INNERE ENERGIE 135 Das Produkt im Nenner ist gleich 1 g/mol, so dass die molare Masse gleich der relativen Atom- bzw. Molekülmasse multipliziert mit der Einheit g/mol ist. Einige Daten zu Atomund molaren Massen: Atom 1 H 12 C 16 O 23 Na Ar = M m 1.007825 12.000000 15.9949 22.989767 g/mol Molekül 1 H2 O 2 H2 O 12 C2 23 Na2 MT eilchen 1.6735 19.92648... 26.5602 38.1754 ⇥10 27 kg Mr = Mm 18.0106 20.026 24.00000 45.9795 g/mol MT eilchen 29.907 33.254 39.853 76.350 ⇥10 27 kg Analog zur molaren Masse definiert man auch das Molvolumen eines Stoffes Vm (12.19) V = Vm ⌫ . Für ideale Gase gilt (12.20) pV = N kT , wobei k = 1.38 ⇥ 10 gilt somit 23 J/K, die Boltzmann-Konstante ist. Für 1 Mol eines idealen Gases (12.21) p Vm = NA k T = R T , wobei wir dem Produkt NA k einen neuen Namen, die sog. allgemeine Gaskonstante, geben R = NA k = 8.31 J K 1 1 mol (12.22) . Die ideale Gasgleichung lässt sich damit schreiben als (12.23) pV = ⌫ RT Das molare Volumen eines idealen Gases hängt von Temperatur und Druck, aber nicht vom Gas selbst ab! Bei sogenannten Normalbedingungen p0 = 1013.15 mb, ist das Molvolumen idealer Gase T0 = 273.15 K = 0 C Vm = 22.4 dm3 . Analog zu den oben genannten molaren Größen definiert man die molare Wärmekapazität C, so dass wir schreiben können Q=c·m· T =C ·⌫· Die Einheiten sind [c] =J K 12.3 1 kg (12.24) T 1 und [C] =J K 1 mol 1 . Innere Energie Generell hängt c bzw. C davon ab, unter welchen Randbedingungen Wärme zu- oder abgeführt wird. Die beiden wichtigsten Spezialfälle sind V = const und p = const. Bei Festkörpern und Flüssigkeiten realisiert man meist nur p = const, weil das die ‘normalen’ Randbedingungen sind; außerdem ist der thermische Volumenausdehnungskoeffizient dort i.d.R. relativ klein, so dass es ohnehin keinen großen Unterschied macht. 136 KAPITEL 12. THERMODYNAMIK Wärmeenergie in idealen Gasen wird in Form von kinetischer Energie ungeordneter translatorischer Bewegung bzw Energie in internen Freiheitsgraden gespeichert: Wkin = N · hEkin i = N f 1 kT . 2 (12.25) Für ideale atomare Gase ist f = 3. Diese Gleichverteilungsprinzip gilt allgemein für beliebige ideale Systeme. Diese gespeicherte Energie bezeichnet man als innere Energie U : U =Nf 1 1 k T = ⌫f R T . 2 2 (12.26) isochore Wärmekapazität: Bei V = const. gilt für die Zu- und Abfuhr von Wärme Q ⌫ CV = T = CV = oder U 1 ⌫f R 2 1 fR 2 T (12.27) und damit (12.28) (12.29) Die Größe f und damit von CV richtet sich nach der Art der Teilchen und der Temperatur 1-atomig 2-atomig 3-atomig (linear) 3-atomig (nicht-linear) N -atomig (nicht-linear) nur Ekin + 2 Rotation + 1 Vibrationa + 2 Rotation + 3 Schwingungen + 3 Rotation + 3 Schwingungen + 3 Rotation + N Schwingungen T T T T T T mittel hoch mittel hoch mittel hoch f f f f f f f CV = 32 R =3 =5 =7 =5 = 11 =6 = 12 CV = 72 R CV = CV = 6 R f = 6N b T hoch 11 R 2 CV = f 2 R a Schwingungen tr zählen doppelt, da kinetische und potentielle Energie beitragen + frot + 2fvib Wegen der Quantelung der Vibrationsenergie werden bei niedrigen Temperaturen Schwingungen unterschiedlicher Schwingungsenergie nicht gleichmäßig angeregt. Typisch setzen verschiedene Schwingungsmoden bei verschiedenen Temperaturen ein (bei N O2 wird zuerst die Biegeschwingung angeregt (f = 8), erst bei höherer Temperatur steigt die Anzahl der aktivierten Freiheitsgrade auf f = 12). Die effektive Anzahl der Freiheitsgrade ist daher zunächst niedriger als der Maximalwert, erst bei hohen T werden alle Vibrationen angeregt. Bei kleinem T werden nicht einmal die Rotationen angeregt. molare Wärmekapazität bf 4R H2 3R vib 2R rot 1R trans 0 10 50100 500 1000 5000 Temperatur HKL Dass linearen Molekülen nur zwei thermische Freiheitsgrade zugeordnet werden hat seinen Ursprung im winzigen Trägheitsmoment I, das mit einer Rotation um die Molekülachse verbunden ist. Gemäß der Quantenmechanik liegen die diskreten Energiestufen für einen starren Rotator bei Erot (`) = `(` + 1)h̄2 /I, wobei der Drehimpuls ` = 0, 1, 2, 3, .... sein muß. 12.3. INNERE ENERGIE 137 Auf Grund des kleinen Trägheitsmomentes um die Molekülachse ist die erste Anregungsstufe energetisch so hoch, dass dieser Freiheitsgrad bei typischen thermischen Bedingungen nicht angeschaltet wird. Bei H2 liegt diese Stufe Erot (1) bei 150 MeV, hingegen für Rotation senkrecht zur Molekülachse bei 15 meV. isobare Wärmekapazität : Bei Zu- und Abfuhr von Wärme bei p = const müssen wir eine Ausdehnung des Gases zulassen, d.h. Arbeit wird vom Gas verrichtet: Q = U+ W U +p V oder 1 ⌫ Cp T = ⌫ f R T + p V und damit 2 1 p V Cp = fR+ mit p V = ⌫ R T 2 ⌫ T f f +2 = R + R = CV + R = R > CV 2 f Den Quotienten beider Wärmekapazitäten bezeichnet man als den Adiabatenkoeffizienten = = Cp f +2 = CV f (12.30) Wärmekapazität von Festkörpern Mit sinkender Temperatur werden alle Stoffe flüssig und (mit Ausnahme von Helium) fest. Freiheitsgrade zur Speicherung innerer Energie gibt es im Festkörper durch die Schwingungen um die Ruhelage gegenüber den nächsten Nachbarn in 3 Dimensionen: d.h. 3 Schwingungsfreiheitsgrade, bzw. 6 Freiheitsgrade der Energiespeicherung (Wkin und Wpot ) für jedes Teilchen. Daraus folgt CV = 3 R = 24.9 J mol 1 K 1 (12.31) Dies ist die Regel von Dulong-Petit, eine Regel, die in vielen Fällen gut erfüllt ist. Schwingungen eines Atoms im Festkörper koppeln in der Regel stark an Nachbaratome ! Wellen; durch Reflexion an Endflächen entstehen Überlagerungen, die im stationären Fall stehende Wellen darstellen. Das Frequenzspektrum dieser Schwingungen ist sehr breit. Bei steigender Temperatur werden mehr und mehr dieser “Moden” angeregt. Die molare Wärmekapazität von Festkörpern steigt bei tiefen Temperaturen / T 3 an, erst bei hohen Temperaturen wird der Wert der Dulong-Petitschen Regel erreicht. Der Unterschied zwischen Cp und CV ist bei festen Körpern in der Regel sehr klein, Cp CV = T Vm ↵2 /, wobei ↵ der lineare Ausdehnungskoeffizient und die Kompressibilität und Vm das Molvolumen ist. Wir verwenden in der Folge Cp = CV für Flüssigkeiten und Festkörper. Einige Daten sind: Cv 24.2 24.3 25.1 25.2 25.5 26.8 5.9 38.1 J mol 1 K 25 Cv J ê K mol Material (20o C) Al Cu Ag Au Fe Pb C (Diamant) Eis (0o C) Kupfer 20 Beryllium 15 Diamant 10 5 0 1 0 200 400 600 Temperatur HKL 800 138 KAPITEL 12. THERMODYNAMIK Latente Wärmen Nicht immer führt die Zufuhr von Wärme Q auch zu einer Temperaturerhöhung T. Experiment: Woodsches Metall (spezielle niedrig schmelzende Legierung) wird von Raumtemperatur (fest) auf rund 100 C (flüssig) erhitzt. Der erwartete Temperaturverlauf (streng monoton ansteigend) wird nicht beobachtet, stattdessen bleibt die Temperatur einige Zeit bei rund 71 C stehen und steigt danach erst weiter. Während dieser Zeit der Temperaturkonstanz wandelt sich das Metall vom festen in den flüssigen Zustand um. Hier wird Wärmemenergie verbraucht, nicht um die Temperatur (und damit die Schwingungsenergie der Teilchen) zu erhöhen, sondern um die Bindungsenergie zu verändern. Das passiert bei allen Materialien beim Schmelzen und beim Sieden, generell bei jeder Art von Phasenumwandlung (Kristallstrukturänderung, Molekülstrukturumwandlungen (z.B. Helix-Umwandlung der DNS). Manchmal wird dabei auch Wärme frei, aber zur Initiierung der Umwandlung muss i.d.R. zunächst eine energetische Schwelle überschritten, und damit Wärmeenergie zugeführt werden, ohne dass sich die Temperatur der Substanz erhöht. Die Wärmeenergie, die für die jeweilige Umwandlung benötigt wird, ist von der Menge des Stoffs und vom Material selbst abhängig, so dass wir schreiben: (12.32) Q = qL · m = Q L · ⌫ qL bzw. QL sind die spezifischen bzw. molaren Umwandlungswärmen. Wasser hat hohe spezifische Wärme und besonders große Umwandlungswärmen. Darin liegt z.B. die Gefahr für Verbrennungen der Haut durch heißen Wasserdampf; große Energieabgabe bei der Wolkenbildung. Eis: Gewässer frieren relativ langsam zu. 12.4 cp 2.1 — 4.2 — 2.0 Jkg 1 K qL Temperatur H o C L Eis Schmelzen von Eis Wasser Verdampfen von Wasser Wasserdampf 333 2256 1 J kg 1 bei konstanter Energiezufuhr 20 10 0 -10 -20 0 100 200 300 400 Zeit HSekundenL 500 Wärmetransport Man kann drei Mechanismen des Wärmetransports unterscheiden: Konvektion Werden in Flüssigkeiten oder Gasen Bereiche unterschiedlicher Dichte erzeugt, so kommt es aufgrund der Schwerkraft zu Ausgleichsströmungen. Ganz leicht erreicht man das z.B., indem man eine Flüssigkeit (Experiment mit lokal eingefärbtem Wasser) im unteren Teil erwärmt; dort sinkt wegen der thermischen Ausdehnung die Dichte. Die Bereiche geringerer Dichte erfahren einen Auftrieb und strömen nach oben, die Bereiche höherer Dichte sinken entsprechend ab. Die Strömungsformen, die sich dabei einstellen, hängen von den speziellen Randbedingungen ab. Kleine Temperaturunterschiede gleichen sich ohne nennenswerte Strömung aus, größere führen sowohl zu laminaren als auch zu turbulenten Strömungen. In besonderen Fällen bilden sich stationäre Strömungen aus, die bestimmten Mustern folgen. In jedem Fall ist mit der Konvektion ein Materietransport verbunden: Volumina mit bestimmten Energieinhalten wandern von einem Ort zum andern und transportieren auf diese Weise Wärmeenergie. 12.4. WÄRMETRANSPORT 139 Wärmeleitung Verbinden wir zwei Wärmereservoire unterschiedlicher Temperaturen T1 und T2 < T1 mit einem homogenen Metallstab mit konstantem Querschnitt A und der Länge l, so zeigt die Erfahrung, dass sich mit der Zeit die beiden Temperaturen angleichen. Wärmeenergie wird demnach vom wärmeren zum kälteren Reservoir transportiert. Der Metallstab leitet Wärme. Stationärer Fall: Wiederholen wir das Experiment, halten aber die beiden Temperaturen T1 und T2 konstant, so findet man, dass die pro Zeiteinheit transportierte Energie proportional zur Temperaturdifferenz und zur Querschnittsfläche des Stabs ist und umgekehrt proportional zu seiner Länge: Q/ t = · A · (T1 l (12.33) T2 ) Der Proportionalitätsfaktor ist eine charakteristische Materialeigenschaft, die man als Wärmeleitfähigkeit bezeichnet. Die SI-Einheit ergibt sich aus den übrigen in die Gleichung eingehenden Größen: [ ]= W K·m (12.34) T1 dQ/dt A T2 l Zusätzlich zum stationäres Gleichgewicht machen wir zwei weitere Voraussetzungen: - Es gibt keine Wärmeverluste zur Seite, der Stab ist gut wärmeisoliert. - Der Wärmekontakt mit den beiden Wärmereservoiren am Ende ist sehr gut, die Stabenden sind bei der Nominaltemperatur. Gleichung (12.33) gilt im stationären Gleichgewicht für jedes Teilstück des Stabs, d.h. wir können für ein infinitesimal kleines Stück der Länge dx mit dem Temperaturgradienten dT /dx dQ/dt = (12.35) A dT /dx schreiben. Bei den hier zugrundegelegten Verhältnissen ist der Gradient negativ: (T1 T2 )/l ! dT /dx, der Wärmefluss findet immer entgegengesetzt zum Temperaturgradienten statt. Dies ist die Wärmeleitgleichung für den stationären Fall. Integriert man sie über den Ort, so ergibt sich bei konstanter Querschnittsfläche A ein linearer Temperaturverlauf: 100 dQêdt = 4l (12.36) Ändert sich die Querschnittfläche mit dem Ort so ist auch der Temperaturverlauf nicht linear und die Rate des Wärmetransports dQ/dt ändert sich. Diesen Umstand vergleicht die Lösung von (12.35) im rechten Bild für drei unterschiedliche Verläufe des Leiterquerschnitts A. dQêdt = 10l 80 dQêdt = 18l T Ho CL T (x) = const x dQ A dt 60 40 20 x 0 0 0 10 2 4 x HcmL 6 8 10 140 KAPITEL 12. THERMODYNAMIK Material Ag Cu Au Al Fe Edelstahl Beton Glas Styropor WK 1 m 1 427 398 315 237 40 ... 85 14.7 2.1 0.7 ... 0,8 0.035 Material Wasser Ethanol WK 1 m 0.60 0.167 H2 He Luft 0.186 0.15 0.026 1 Edelstahl ist ein schlechter metallischer Wärmeleiter, Glas ein sehr schlechter nicht-metallischer. Auch Wasser ist ein schlechter Wärmeleiter, unter den Gasen leitet Luft (wie die meisten schwereren molekularen Gase) ebenfalls schlecht. Experiment: Wärmeleitfähigkeit von Wasser (und Glas): Wasser in Glasrohr: unten einige Eisstücke: Temperatur messen ! wenige C; am oberen Ende heizen ! Wasser siedet oben (100 C), während sich unten die Temperatur nicht ändert, Eis schmilzt nicht. T Nicht-stationärer Fall: Um die Zeitabhängigkeit der Wärmeleitung zu berücksichtigen, müssen wir unseren Ansatz verallgemeinern. Für die Positionen (1) und (2) gilt jeweils wieder Gleichung (12.33) für den Wärmefluss: T1 T2 dQ1/dt A dQ2/dt x dQ1 /dt = A @T1 /@x (12.37) dQ2 /dt = A @T2 /@x (12.38) allerdings müssen wir jetzt die Temperatur partiell nach dem Ort differenzieren, denn T ist jetzt sowohl vom Ort als auch von der Zeit abhängig: T = T (x, t). Ist der Abstand x zwischen den beiden Ebenen (1) und (2) klein, so gilt T2 = T1 + @T @x (12.39) x. Damit ist die Netto-Wärme, die in das durch die beiden Flächen bei (1) und (2) begrenzte Volumen hineinfließt, ✓ ◆ dQ dQ1 dQ2 @ @T = = A T 1 T1 x dt dt dt @x @x @2T = A x. @x2 Lassen wir x gegen Null gehen, so wird daraus d2 Q = dt A @2T dx . @x2 (12.40) Wegen d2 Q = c · dm · dT = c · ⇢ dV · dT = c · ⇢ A dx · dT folgt daraus c ⇢ A dT dx dt dT dt = = @2T dx @x2 @2T , c ⇢ @x2 A oder (12.41) (12.42) die sog. Wärmeleitgleichung. Den Faktor c ⇢ auf der rechten Seite bezeichnet man als Temperaturleitzahl. Er hat die Dimension Fläche pro Zeit, sein Kehrwert stellt damit (bei 12.4. WÄRMETRANSPORT 141 gegebener Fläche) eine Art Zeitkonstante für den Temperaturausgleich dar. Ursachen der Wärmeleitung im Festkörper: • Schwingungen einzelner Atome sind stark an Nachbaratome gekoppelt, wegen Übertragung von Schwingungsenergie zwischen den Atomen kommt es zum Energietransport. • in Metallen: freie Elektronen übertragen zusätzlich kinetische Energie bei Stößen untereinander und mit Atomen; entscheidender Beitrag, d.h. die Wärmeleitfähigkeit ist gekoppelt an die elektrische Leitfähigkeit : = const · T Gesetz von Wiedemann-Franz (12.43) Experiment: Ein Kreuz mit drei verschiedenen Metallen (Cu, Messing, Stahl) wird in der Mitte erhitzt. Streichhölzer, die an den Enden der verschiedenen Materialien liegen, zünden in der Reihenfolge der elektrischen Leitfähigkeit. Ursachen der Wärmeleitung in Flüssigkeiten: • keine Scherkräfte, nur Longitudinalschwingungen koppeln Nachbaratome ! eher geringe Wärmeleitfähigkeit (Beispiel: Wasser). • in elektrisch leitenden Flüssigkeiten (Hg, geschmolzene Metalle) können wieder freie Elektronen kinetische Energie übertragen, hohe Wärmeleitfähigkeit. • In der Brownschen Bewegung wird in Stößen kinetische Energie übertragen. Ursachen der Wärmeleitung in Gasen Austausch von kinetischer Energie der Teilchen duch Stöße zwischen den Teilchen. Wie im Kapitel “Gase” schon behandelt, ist die Wärmeleitfähigkeit bei hohem Druck (normaler atmosphärischer Druck) unabhängig von der Dichte, bei niedrigem Druck, wenn die mittlere freie Weglänge der Teilchen groß wird, ist sie proportional der Teilchendichte n. Im Bereich zwischen 10 3 und 10 mbar kann man daher die Wärmeleitfähigkeit eines Gases als Maß für seinen Druck benutzen (Vakuum-Druckmessung). dW = dt 0.2 0.0 AT4 (12.44) Die Proportionalitätskonstante wHlL HWêm2 mm-1L wHnL H10+5 Wêm2 THz-1L Wärmestrahlung Jede Oberfläche A strahlt Wärme ab, die Abstrahlung hängt stark von der Temperatur T ab. Die abgestrahlte spektrale Leistung ist sowohl eine Funktion der Wellenlänge (oder Frequenz ⌫) als auch der Temperatur T gemäß dem Planckschen Strahlungsgesetz. Integriert man über Raumwin1.2 30 kel, Oberfläche und alle Wellenlängen, erhält man die gesam1.0 25 300 K 5900 K te abgestrahlte Leistung. Sie ist 0.8 20 eine steile Funktion der Tem0.6 15 peratur (Gesetz von Stefan und 2 0.4 68 MWêm 10 460 Wêm2 Boltzmann) 200 400 600 800 10001200 Frequenz n HTHzL 5 0 2 4 6 8 10 12 14 16 18 Wellenlänge l HmmL ist nicht vom Material abhängig, sondern nur von der Art 142 KAPITEL 12. THERMODYNAMIK der Oberfläche. Ihr Maximalwert (schwarzer Strahler) ist = 5.77 · 10 8 Wm 2 K 4 . Die auf eine Oberfläche eingestrahlte Leistung (bei einer bestimmten Wellenlänge) teilt sich auf in absorbierte, reflektierte und transmittierte Leistung, wobei die Koeffizienten A+R+T =1 (12.45) A = f ( ,T) E (12.46) sind. (Der Farbeindruck von Oberflächen spiegelt das spektrale Reflexionsvermögen wieder. Beispiel: Cu: A nimmt im sichtbaren Wellenlängenbereich mit wachsendem ab, R nimmt zu ! rötlicher Farbeindruck). Der Absorptionskoeffizient A hängt mit dem Emissionsvermögen E über eine nur von der Temperatur (nicht vom Material) abhängige Konstante zusammen Ist der Absorptionskoeffizient maximal, so gilt das auch für das Emissionsvermögen; ein ideal “schwarzer Körper” (A = 1) emittiert daher auch die maximale Leistung Experiment: Kugel mit Bohrung, graphitiert (d.h. schon relativ “schwarz”): Raumtemperatur: Bohrung sieht dunkler aus als Kugeloberfläche (d.h. noch schwärzer als Kugeloberfläche); heiße Kugel: Bohrung sieht heller aus als Kugeloberfläche: Hohlraumstrahler ist optimaler schwarzer Körper. Strahlungsgleichgewicht: Ähnlich wie sich durch Wärmeleitung unterschiedliche Temperaturen mit der Zeit angleichen, kann dies auch durch Wärmestrahlung geschehen. Jeder Körper strahlt eine seiner Temperatur entsprechende Leistung ab. Sind andere Körper in der Umgebung, so wird außerdem Strahlungsenergie von diesen Körpern absorbiert. Die Differenz der emittierten und absorbierten Strahlungsleistung wird erst Null, wenn sich die Temperaturen angeglichen haben. Durch Festhalten bestimmter Temperaturen (nur durch Energietransfer mit der Umgebung) können zeitlich stabile Verhältnisse auch bei unterschiedlichen Temperaturen erreicht werden. Aus dem thermischen Gleichgewicht zweier Körper lässt sich auch die Behauptung (12.46) herleiten, dass das Verhältnis emittierter zu absorbierter Leistung nicht vom Material abhängt. Wärmeisolation Zur effektiven Wärmeisolation müssen die Wärmetransportmechanismen (Konvektion, Leitung, Strahlung) unterbunden werden. Typische wärmeisolierende Gefäße sind (Bezeichnung je nach Verwendungszweck) Thermosflasche, Dewar, Kalorimeter. Sie besitzen eine doppelwandige Hülle aus einem schlechten Wärmeleiter (meist Glas oder Edelstahl), deren Zwischenbereich evakuiert ist (Verhinderung von Wärmeleitung oder Konvektion). Die vakuumseitigen Oberflächen der Hülle sind außerdem verspiegelt, so dass auch die Wärmeabstrahlung minimiert wird. Ähnliches gilt für andere Isolationsaufgaben, z.B. im Baubereich: Mauerwerk (Lufteinschlüsse in Mauersteinen, Dämmplatten, isolierende Fenster (doppelwandig, IR-verspiegelt, evtl. Gasfüllung). 12.5 Hauptsätze der Thermodynamik Thermodynamisches System: Eine bestimmte Menge von Atomen / Molekülen befindet sich in Wechselwirkung mit der Umgebung, d.h. Energieaustausch ist erlaubt. Wir wollen unter- 12.6. DER ERSTE HAUPTSATZ 143 suchen, wie sich der Zustand eines solchen Systems mit Energiezu- oder abfuhr ändert. Das System wird durch verschiedene physikalische Größen beschrieben: (12.47) p, T, V, m, ⇢, N, ⌫, ... Die Zusammenhänge zwischen diesen Größen werden durch Zustandsgleichungen und die Hauptsätze der Thermodynamik beschrieben. Zustandsgrößen Gleichgewichtszustand: Wenn sich die Eigenschaften eines Systems zeitlich nicht ändern, sagt man das System ist stationär. Eine sehr langsame Änderung kann man als Durchlaufen einer Folge von Gleichgewichtszuständen darstellen. Chemische Reaktionen laufen i.d.R. sehr weit weg von Gleichgewichtszuständen ab! Drei Zustandsgrößen legen Zustand des Systems eindeutig fest: p, V , T . Zustandsgleichung für ideale Gase (Index m steht für molare Größen): pV = N kT = ⌫ RT (12.48) p Vm = RT (12.49) Auch die innere Energie ist eine Zustandsgröße: U = N f /2 k T (12.50) Um = NA f /2k T = f /2 R T (12.51) wobei für ideale Gase f = 3 ist. 12.6 Der erste Hauptsatz Zufuhr von Wärme (dQ) in ein System kann zum einen die innere Energie U und damit die Temperatur T des Systems erhöhen. Andererseits kann damit das System auch mechanische Arbeit (dW ) leisten, z.B. durch Expansion des Volumens gegen den äußeren Druck. Der erste Hauptsatz ist ein Energieerhaltungssatz dU = dQ + dW Die Summe der einem System von außen zugeführten Wärme (dQ) und Energie (dW ) ist der Zunahme seiner inneren Energie (dU ) gleich. Vorzeichenkonvention: Energie, die in das System hineingeht, hat positives Vorzeichen, Energie, die hinausgeht, hat ein negatives Vorzeichen. Wärmereservoir dQ<0 dQ>0 System p, V, T, U, H, S dW<0 Cv =H ∂U Lv ∂T C p =H ∂H Lp ∂T dW>0 Getriebe Dies ist eine Erfahrungstatsache. Maschinen, von denen man behauptet, dass sie Energie erzeugen könnten (Perpetuum Mobile erster Art) widersprechen dem ersten Hauptsatz. 144 KAPITEL 12. THERMODYNAMIK Die Arbeit, die unser System bei Vergrößerung seines Volumens gegen den äußeren Druck p leistet, ist eine negative Größe: dW = p dV p (12.52) p dV Damit können wir für ein ideales Gas auch schreiben dU = dQ (12.53) p dV Für die folgenden Betrachtungen ist es von Bedeutung, dass zwar U , T , p und T Zustandsgrößen sind, nicht dagegen die Wärmemenge Q. Ein Zustand lässt sich nicht eindeutig durch die Angabe der Wärme, die man ihm zu- oder abgeführt hat, charakterisieren. Wir werden sehen, dass man verschiedene Wärmemengen aufbringen kann, um von einem in einen anderen Zustand zu gelangen. Wärme und Arbeit in speziellen Prozessen Wärme und Arbeit lassen sich mit Hilfe des ersten Hauptsatzes für bestimmte Prozesse relativ einfach berechnen. Dies sind isochor: dV = 0 In diesem Fall wird Wärme vollständig zur Erhöhung der inneren Energie verwendet. Wir betrachten die Änderungen in einem Mol: (12.54) dU = dQ = CV dT CV = ✓ @Q @T ◆ = V ✓ @U @T ◆ . dQ V (12.55) V=const p z p+dp Die Zufuhr von Energie spiegelt sich in eimem Druck und Temperturanstieg wieder. isobar: dp = 0 Bleibt der Druck konstant, dann gilt dU = dQ p dV (12.56) Es ist an dieser Stelle zweckmäßig, eine neue Größe einzuführen, die Enthalpie H. Die Enthalpie ist ein Maß für die gesamte im System enthaltene Energie, sie ist gleich der Summe der inneren Energie U und dem Produkt pV , H = U + pV (12.57) Dann kann man schreiben dH = dU + p dV + V dp = dQ + V dp (12.58) Für dp = 0 gilt somit dH = dQ = Cp dT (12.59) 12.6. DER ERSTE HAUPTSATZ 145 bzw. Cp = ✓ @Q @T ◆ = p ✓ @H @T ◆ (12.60) dQ p=const p V z V+dV Bei Phasenumwandlungen, die meist bei konstantem Druck ablaufen, ist es viel praktischer mit der Enthalpie zu arbeiten als mit der inneren Energie. isotherm: dT = 0 U = f /2 ⌫ R T ist nicht von p oder V abhängig, nur von T . Damit ist dU = 0 und dQ = p dV (Wärme wird vollständig in Arbeit umgewandelt). Für ein ideales Gas gilt dabei das Gesetz von Boyle-Mariotte, p V = const. Diese Zustandsänderungen kann man dann in einem p V - Diagramm als Hyperbel eintragen: Zu jeder Hyperbel gehört eine bestimmte Temperatur. 3 T = 750K 2 p T = 250K 1 0 0 1 2 3 V Die bei isothermer Expansion von V1 auf V2 geleistete Arbeit kann man durch Integration Wisotherm = ZV2 p dV = V1 ZV2 ⌫ RT dV = V V1 ⌫ R T ln V2 V1 (12.61) berechnen, im p V - Diagramm ist sie durch die Fläche unter der für die Temperatur maßgebenden V (p)-Kurve zu erkennen. adiabatisch: dQ = 0 anschaulich: kein Wärmeaustausch mit der Umgebung findet statt; das hat man häufig bei schnell ablaufenden Prozessen (z.B. Schallwellen), da ein Wäremaustausch viel langsamer ablaufen würde als die Zeitskala der Änderungen es zulässt. Hier gilt für 1 Mol dU = p dV = CV dT (Energie wechselt zwischen mechanischer Energie und innerer Energie). Für ideale Gase (p = RT /Vm ) ist dann CV dT dV = R T V (12.62) Diese Differentialgleichung kann man für eine Zustandsänderung von p0 , V0 , T0 nach p, V , T integrieren, wobei wir noch CV /R = CV /(Cp CV ) = 1 1 verwenden, so dass wir erhalten: ln T T0 = T0 T = T ·V p·V 1 = = V ln ( V0 ✓ ◆ V V0 const const 1) damit auch (12.63) 1 oder und damit (12.64) (12.65) (12.66) Diese Gleichungen erscheinen im p V - Diagramm steiler als die Isothermen, da > 1 ist. 146 KAPITEL 12. THERMODYNAMIK 3 3 317K 600K dQ=0 2 2 p 1 200K 378K 153K 0 0 1 1 326K 2 dQ=0 T = 200K 126K 3 V 12.7 T = 600K p 458K 0 0 1 2 3 V Zweiter Hauptsatz Erfahrungstatsachen sind dass Wärme von selbst immer nur vom wärmeren zum kälteren Körper fließt, nie in umgekehrter Richtung. In Kreisprozessen kann mechanische Arbeit vollständig in Wärme umgewandelt werden. In umgekehrter Richtung kann dagegen nur ein Teil der eingebrachten Wärme in mechanische Arbeit umgewandelt werden. Beide Aussagen stellen den wesentlichen Inhalt des zweiten Hauptsatzes der Thermodynamik dar. Der erste Hauptsatz ist ein Satz von der Erhaltung der Energie, bei Umwandlung von Wärme in mechanische Energie bleibt die Gesamtenergie eines Systems erhalten. Im Folgenden soll die Umwandlung von Wärme und mechanischer Arbeit anhand Kreisprozesses von Carnot quantitativ berechnet werden, damit lässt sich die Aussage des Zweiten Hauptsatzes konkretisieren. In einem Kreisprozess durchläuft das System eine Reihe von Zuständen und gelangt zum Ausgangszustand zurück. Reversibler Kreisprozess: kann in beiden Richtungen verlaufen. Kreisprozesse kommen in der Mikrophysik durchaus vor, aber in der Makrophysik sind dies Gedankenexperimente an Vielteilchensystemen, die in Wirklichkeit immer irreversibel verlaufen. Carnot (1842): Erste und immer noch gültige quantitative Aussage über den Bruchteil der Wärme, die maximal in Arbeit umgewandelt werden kann. Das ist ein Gedankenexperiment, bei dem ein ideales Gas durch Expansion und nachfolgende Kompression in zwei isotherme und zwei adiabatische Prozessen einen reversiblen Kreisprozess durchläuft. Reibungsverluste, wie sie in realen Maschinen auftreten, werden vernachlässigt (damit folgt die Aussage, dass eine reale Maschine kann nie effizienter sein kann als der Carnot’sche Prozess). 1 dQ=0 3.0 dQ HzuL 2.5 DW 2 p 2.0 Tk = 375K 1.5 Tw = 500K 4 1.0 0.6 0.8 dQ HabL 1.0 1.2 V 3 1.4 1.6 Vom Startpunkt 1, bei einer Temperatur Tw , erfolgt eine isotherme Expansion zum Zustand 2. Dem System muss eine Wärmemenge Qw zugeführt werden, damit seine Temperatur bei der Expansion konstant bleibt. Wegen T = const. bleibt die Innere Energie des Systems konstant und wir haben Z V2 V2 Qw = p dV = R Tw ln V1 V1 12.7. ZWEITER HAUPTSATZ 147 Danach erfolgt eine adiabatische Expansion zum Zustand 3 mit der Temperatur Tk < Tw . Dabei erfolgt kein Austausch von Energie mit dem äußeren Wärmebad. Wegen dQ = 0 gilt, dass die Ausdehnungsarbeit gleich der Änderung der inneren Energie ist W23 = U2 U3 = p dV Vom Zustand 3 wird das System isotherm komprimiert zum Zustand 4. Dabei muss die Wärmemenge Qk dem System entzogen werden, damit seine Temperatur konstant bleibt. Qk = Z V4 p dV = R Tk ln V3 V4 V3 Schließlich gelangt das System durch adiabatische Kompression wieder in den Ausgangszustand zurück. Wegen dQ = 0 gilt, dass die Ausdehnungsarbeit gleich der Änderung der inneren Energie ist W41 = U4 U1 Gesamtbilanz: Beim Schritt 2 ! 3 wird vom System Arbeit geleistet, beim Schritt 4 ! 1 wird gleich viel Arbeit in das System hineingesteckt, da die Differenz der jeweiligen inneren Energien gleich ist. Bei den isothermen Schritten wird ein Nettobetrag an Arbeit vom System abgegeben. Diese Arbeit ist: W = W12 + W34 = R Tw ln V1 V3 + R Tk ln V2 V4 Unter Verwendung der Adiabatengleichungen T V 1 = const ergibt sich für die Nettoarbeit W = R (Tw Tk ) ln V1 V2 Die Wärmemenge Qk muss an das Kältereservoir abgegeben werden, steht daher nicht zur Umwandlung in mechanische Energie zur Verfügung. Den Wirkungsgrad definiert man als ⌘= W Tw Tk = Q Tw Der Wirkungsgrad steigt mit steigender Temperaturdifferenz. Da aber Tk nicht Null werden kann, bleibt der Wirkungsgrad immer unter 1. Wenn die Carnot-Maschine in umgekehrter Reihenfolge läuft, dann transportiert sie Wärme von einem kälteren Reservoir, Tk , zu einem wärmeren, wozu sie zugeführte Arbeit braucht. Dies wäre der idealisierte Fall einer Wärmepumpe, die auch als Kältemaschine eingesetzt werden kann. Der Arbeitsstoff der Carnot-Maschine ist ein ideales Gas, und bei diesem reversiblen Kreisprozess werden alle Reibungsverluste vernachlässigt. Reale Maschinen haben unvermeidliche Verluste durch Reibung der Kolben, innere Reibung des nicht-idealen Gases, Wärmeleitung, etc, die den Wirkungsgrad weiter verringern. Deshalb gilt: Es gibt keine periodisch arbeitenden Maschinen mit einem Wirkungsgrad der höher ist, als der des Carnot-Prozesses. 148 KAPITEL 12. THERMODYNAMIK Es gibt idealisierte Darstellungen für technische Kreisprozesse, wie sie z.B. im Benzinmotor verwirklicht sind: In thermodynamischen Sinn besteht der Zyklus eines Ottomotors aus den Wegen von 0 ! 1 ! 2 ! 3 ! 4 ! 5 ! 0. In diesem idealen Ottomotor erfolgt kein Wärmeaustausch zwischen Gas und Zylinder und Kolben. 10 Von 0 ! 1 wird bei konstantem Druck frische Luft in den Verbrennungsraum angesaugt. Dabei erhöht 3 8 sich das Volumen um den Faktor V1 /V0 = r. Darauf folgt die adiabatische Kompressionsphase von 1 ! 2 dQ=0 in der sich der Druck um p2 /p1 = r und die Tem6 peratur um T2 /T1 = r 1 erhöhen. In der Verbrenp 2 nungsphase 2 ! 3 erhöhen sich die Temperatur zu 4 W T3 = T2 + f Q cv und der Druck auf p3 = p2 (T3 /T2 ). In der Arbeitsphase 3 ! 4 sinken Temperatur und 4 2 Druck um T4 /T3 = r1 und p4 /p3 = r . Bei Po0 sition 4 öffnen die Auslassventile, der Druck sinkt 1 0 und von 5 ! 0 wird das verbrannte Gemisch in den 0.5 1.0 1.5 2.0 Auspuff geschoben. V In der Phase 1 ! 2 verrichtet der Kolben Arbeit am Gas, in der Phase 3 ! 4 verrichtet das Gas⇥ Arbeit am Kolben.⇤ Der Unterschied zwischen beiden ist die verrichtete Arbeit W = cv (T3 T2 ) (T4 T1 ) . Die Leistung eines Ottomotors ist demnach P = nW f wobei n die Anzahl der Kolben und f die Drehzahl ist. 12.8 Entropie Ein makroskopisches System ist durch die Festlegung der Zustandsgrößen wie p, T, und V bestimmt. Die Mikrozustände sind jedoch unbestimmt. Ein Maß für die Unkenntnis des Mikrozustandes ist die Entropie S. Dem Gas steht nach dem Entfernen der Zwischenwand ein größerer Raum zur Verfügung. Es existieren nach der Expansion also mehr Mikrozustände und das System besitzt eine höhere Entropie. Beim Schmelzen von Eis wird die geordnete Kristallstuktur in eine regellose Bewegung einzelner Wassermoleküle überführt: Die Entropie nimmt dabei zu. Diese Zunahme der Entropie entspricht quantitativ genau der eingebrachten Schmelzwärme, geteilt durch die absolute Temperatur. Die Entropie eines abgeschlossenen Systems ist maximal, wenn sich das System im Gleichgewicht befindet. Einem perfekt geordneten System kann man die Entropie S = 0 zuordnen. In diesem Sinn findet das Konzept Entropie auch in der Informationstheorie Anwendung. Dort ist ein System mit Entropie 0 ein Speicher, der nur mit Nullen besetzt ist. Die Entropie eines Informationssystem kann einen vom System abhängigen Maximalwert nicht überschrei- 12.8. ENTROPIE 149 ten.1 • Entropie kann wohl erzeugt, aber nicht vernichtet werden. • Es ist unmöglich, Wärme von einem kälteren zu einem wärmeren Reservoir zu bringen, ohne in der Umgebung irgendwelche Veränderungen zu hinterlassen. • Es ist unmöglich, eine periodisch arbeitende Maschine zu konstruieren, die nichts weiteres bewirkt als Arbeit zu leisten und ein Wärmereservoir abzukühlen. • Bei einem reversiblen Kreisprozess bleibt die Entropie konstant. Bei irreversiblen Prozessen nimmt die Entropie immer zu. Energie: Fähigkeit des abgeschlossenen Systems zur Arbeitsleistung. Entropie: Fähigkeit des abgeschlossenen Systems sich zu wandeln. (griech. entrepein=drehen, wandeln). Die Entropie wurde ursprünglich als phänomenologisches Maß eingeführt. In der klassischen Thermodynamik wurde die Entropie über die Wärme definiert.2 • Führt man einem System, das schon die Entropie S besitzt, zusätzlich Wärme dQ zu, dann steigt die Entropie des Systems, also dS / dQ. • Die Temperatur ist dabei zu berücksichtigen. Der Grad der Unordnung steigt bei Zufuhr von dQ bei kleiner Temperatur stärker an als bei hoher Temperatur.3 Dies wird in der klassischen Definition der Entropie berücksichtigt: dS = dQ T (12.67) Zwei Beispiele: 1) Zwei sonst gleiche Körper befinden sich auf unterschiedlichen Temperaturen, T1 und T2 . Ihre Wärmeenergien sind: Q1 = m c T1 und Q2 = m c T2 . Man bringt sie in Kontakt und ein Temperaturausgleich findet statt. Wenn keine Wärmeenergie an die Umgebung verloren geht, dann ist die Endtemperatur der beiden Körper Tm = (T1 + T2 )/2. Die Entropieänderung des Körpers 1 ist S1 = Z Tm T1 dQ = mc T Z Tm T1 dT Tm = m c ln T T1 Da Tm < T1 ist folgt S1 < 0. Die Entropieänderung des Körpers 2 ist S2 = Z Tm T2 dQ = mc T Z Tm T2 dT Tm = m c ln T T2 1 Bei einer Festplatte ist dieser Wert durch die Speicherkapazität bestimmt, bei einem physikalischen System durch die Anzahl der Teilchen. 2 Wärme ist eine sehr “unordentliche” Form der Energie mit hoher Entropie 3 Das wird klar aus folgender Überlegung: Führt man einem aufgeräumten (kaltem) Zimmer, 5 Paar Socken willkürlich verteilt zu, dann steigt die Unordnung viel mehr, als wenn schon mal 20 Paar Socken irgendwie herumliegen und wir noch 5 Paar dazulegen. Bei der Berechnung der Entropie über Gl.(12.71) spielt noch eine Rolle inwieweit die Anordnung der Socken im Zimmer und die Socken selbst unterscheidbar sind. 150 KAPITEL 12. THERMODYNAMIK Da Tm > T2 ist folgt S= S1 + S2 > 0. Die Gesamtänderung der Entropie ist S2 = m c ln 2 Tm >0 T1 T2 Dieser Vorgang ist irreversibel, weil nach dem 2. Hauptsatz gilt: der erste Körper erwärmt sich nicht wieder auf Kosten des zweiten Es gelingt immer nur eine Definition der Entropiedifferenz. Die Entropie S(T, p, V ) ist immer nur bis auf eine additive Konstante festgelegt. 2) Die isotherme Diffusion eines idealen Gases aus einem Volumen, V1 , durch ein Öffnung in ein Volumen, V2 . Dieser Vorgang ist nicht reversibel. S = = = Z Q T Z V1 +V2 p dV R dV = T V V1 V1 V1 + V 2 R ln >0 V1 V1 +V2 (12.68) Statistische Deutung der Entropie: Wir betrachten ein Molekül im obigen Experiment. Vorher: Wahrscheinlichkeit das Molekül im Volumen V1 zu finden P1 = 1. 1 Nachher: WSK das Molekül im Volumen V1 zu finden P1 = V1V+V < 1. 2 Bei zwei Molekülen ist die WSK beide Moleküle in V1 zu finden gleich dem Produkt der Einzel-WSK, P22 . Für N Moleküle gilt nachher: ✓ ◆N V1 PN = V 1 + V2 (12.69) Für 1 Mol ist N = NA = R/k = 6 ⇥ 1023 . Damit ist die WSK alle Moleküle in V1 zu finden ✓ ◆N ✓ ◆R/k V1 V1 PN A = = (12.70) V1 + V 2 A V 1 + V2 also praktisch gleich Null. Logarithmieren wir diesen Ausdruck und vergleichen ihn mit (12.68) erhalten wir ✓ ◆ V1 k ln P = R ln V1 + V 2 S = k ln P P war die WSK für einen Mikrozustand. Die Anzahl der Realisierungsmöglichkeiten (Verteilungen der Atome auf beide Volumina): ⌦ = 1/P und demnach S = k ln ⌦ (12.71) Die Entropie ist proportional zur Zahl der Realisierungsmöglichkeiten eines thermodynamischen Systems. Damit ist auch ein Nullpunkt für die Entropie festgelegt! 12.9. REALE GASE UND FLÜSSIGKEITEN 151 Am Beispiel eines Kristalls aus N Atomen untersuchen wir die makroskopische Eigenschaft: Wie viele Leerstellen n gibt es im Kristall? und die Zahl der Realisierungsmöglichkeiten (Mikrozustände): • Zum Makrozustand Alle Atome auf ihren Plätzen, das bedeutet: es gibt keine Leerstellen, (n, N ) = (0, N ) gibt es nur einen Mikrozustand, dieser bedeutet perfekte Ordnung. • Zum Makrozustand Eine Leerstelle irgendwo, also (n, N ) = (1, N ) gibt es gerade N Mikrozustände, dies bedeutet die Leerstelle kann an irgend einem der N Plätze sitzen. • Zum Makrozustand Zwei Leerstellen irgendwo, also (n, N ) = (2, N ) gibt es auf alle Fälle mehr als N Mikrozustände, also Anordnungen, die dadurch unterscheidbar sind, wie die beiden Leerstellen zueinander im Kristall stehen. Die Zahl der möglichen Realisierungen steigt mit der Anzahl der Leerstellen an4 , und die Unordnung im Kristall wächst5 . Daraus folgern wir: Die Zahl der möglichen Mikrozustände zu einem Makrozustand ist ein quantitatives Maßfür den Grad der Unordnung dieses Zustandes. Für ein makroskopisches System kann ein Teil des Systems durchaus eine Entropieabnahme erfahren, aber auf Kosten eines anderen Teiles des Systems. Beispiele dafür sind: • Kristallbildung aus der Schmelze, geordneter Einkristall entsteht. • In Lebewesen entsteht eine geordnete Zellstruktur auf Kosten der Entropiezunahme ihrer Umgebung. In allen Fällen nimmt die Entropie des Gesamtsystems immer zu. • Geordnete Strukturen bilden sich selbst nur in offenen Systemen, fern vom thermodynamischen Gleichgewicht. Nur der Austausch von Energie mit der Umgebung ermöglicht die Erniedrigung der Entropie des Teilsystems. • Die Richtung der Zeit ist mit der Entropiezunahme dS/dt > 0 verknüpft. So gibt es die Möglichkeit zwischen Zukunft und Vergangenheit zu unterscheiden. Eigentlich entsteht der Begriff Zeit zum erstenmal wenn in einem offenen System Wärmeenergie und Arbeit ausgetauscht werden. Ein gutes Konzept zur Messung der Gesamtentropiezunahme fehlt allerdings. Deshalb gibt es auch keine Uhr. Bei herkömmlichen Uhren hat man es immer mit Längenmessungen zu tun. 12.9 12.9.1 Reale Gase und Flüssigkeiten Zustandsgleichung von van-der-Waals Ideales Gas: die Wechselwirkung der Moleküle untereinander und das Volumen der Moleküle werden vernachlässigt. Ein ideales Gas bleibt bei jeder Temperatur gasförmig. Reale Gase können je nach Druck und Temperatur den Aggregatszustand ändern (Kondensation). Bei sehr hohen Drucken ist das Eigenvolumen nicht mehr vernachlässigbar. Wir nehmen die Gasatome als starre Kugeln mit Radius r an. Eigenvolumen: Nächste Annäherung zwischen zwei Atomen ist d = 2r und ein Mindestabstand von der Wand werden berücksichtigt. Aus dieser Überlegung folgt, dass wir für das effektive Volumen eines realen Gases das vierfache Eigenvolumen abziehen müssen. 4 jedenfalls 5 so solange n < N/2 ist wie ein Zimmer mit 25 verstreuten Socken unordentlicher ist als eines mit nur 5 152 KAPITEL 12. THERMODYNAMIK Wechselwirkung : Im Inneren des Gasraumes ist die resultierende Kraft Null. An der Wand bleibt resultierende Kraft, proportional zur Zahl der anziehenden Atome in einer Halbkugel, die von der Wand zum Gas hin gerichtet ist. Die Kraft ist von der Wand nach Innen gerichtet und wirkt wie ein Binnendruck, zusätzlich zum äußeren Druck p. . Diese Modifikationen der allgemeinen Gasgleichung pV = RT führen zur Van-der-Waals’sche Zustandsgleichung ⇣ a ⌘ p + 2 · (V b) = R T (12.72) V 1.0 0.8 kritischer Punkt 0.6 p A 0.4 0.2 B A 0.0 B 0.2 0.4 0.6 320K 315K 310K 305K 300K 0.8 Komprimiert man CO2 bei T = 300 K kontinuierlich, dann folgt die gemessene Kurve p(V ) der Gleichung (12.72) bis zum Punkt B (wie in der Abbildung gezeigt). Dann bleibt der Druck konstant bis zum Punkt A. Bei weiterer Kompression steigt der Druck stark an und folgt dabei wieder der Van-derWaals Kurve. Grund: in Punkt B setzt die Verflüssigung des CO2 Dampfes ein. Der Anteil an Flüssigkeit steigt von B nach A ständig an, bis in A alles verflüssigt ist. Der steile Anstieg bei noch kleineren Volumina ist durch die sehr geringe Kompressibilität der Flüssigkeit bedingt. V 12.9.2 Dampfdruck und Flüssig-Gas-Gleichgewicht Wir betrachten eine Flüssigkeit in einem abgeschlossenem Volumen, teilweise gefüllt. Ein Teil der Flüssigkeit verdampft. Oberhalb der Flüssigkeit bildet sich Dampfphase, die einen Druck auf die Wände und auf die Flüssigkeitsoberfläche ausübt. Bei fester Temperatur stellt sich konstanter Sättigungsdampfdruck pS (T ) ein. Beide Phasen koexisieren nebeneinander (dynamisches Gleichgewicht). ⇤ ist die molare Verdampfungswärme (kJ/Mol). Sie hat zwei Ursachen: Energie muss aufgewand werden, um das Volumen von Vf l auf VD zu erhöhen, und Arbeit wird geleistet, um den mittleren Molekülabstand zu erhöhen. Dieser Teil stellt den größer Anteil dar (92 % bei Wasser). Bei "hoherTemperatur gilt die van’t-Hoff ’sche Gleichung ⇤ RT Die Dampfdruckkurve teilt die Ebene im p T Diagramm in zwei Bereiche: Bei Drucken unterhalb von pS (T ) gibt es nur eine stabile Gasphase, darüber nur die flüssige Phase. Über einer bestimmten kritischen Temperatur, gibt es nur die Dampfphase. An diesem Punkt haben im p V -Diagramm die Vander-Waals Isothermen kein Minimum mehr, sondern nur einen Wendepunkt. (12.73) 220 b Druck pS (T ) / e TK flüssig fest 1b 6.1 mb gasf. TT 0.01 100 374 o Temperatur @ CD 12.9. REALE GASE UND FLÜSSIGKEITEN 12.9.3 153 Joule-Thomson Effekt In einem idealen Gas üben die Teilchen keine Wechselwirkung aus. In diesem Fall hängt der Energieinhalt des Gases nicht vom Volumen ab. Bei einem realen Gas hingegen ändert sich der Energieinhalt bei Entspannung (Druckerniedrigung), wenn diese adiabatisch (ohne Austausch von Wärme) und ohne Arbeitsleistung vor sich geht. Ursache dafür ist, dass dann der mittlere Teilchenabstand größer und die Zahl der Stoßkomplexe die im Gas vorliegen geringer ist. Diesen Effekt verwendet man zur Verflüssigung von Gasen. Ein im Raum 1 beim Druck p1 eingeschlossenes Gas wird durch eine poröse Wand langsam (keine Arbeitsleistung durch Wirbelbildung) in einen zweiten Raum (Druck p2 < p1 ) gedrückt. Mit Luft stellt sich dabei eine Temperatursenkung von einem Grad pro 4 bar Druckdifferenz ein. Damit ein Volumen V1 beim Druck p1 durch die poröse Wand verschwindet verrichtet man von aussen am Gas die Arbeit p1 V1 . Dieselbe Gasmenge erscheint hinter der porösen Wand mit dem Volumen V2 und leistet die Arbeit p2 V2 gegen den Kolben auf der Ausgangsseite. Die Differenz der beiden Arbeiten wurde der inneren Energie des Gases zugeführt, U2 U 1 = p1 V 1 p1 p2 (12.74) p2 V 2 . Dies ist gleichbedeutend mit konstanter Enthalpie, (12.75) H = U1 + p1 V1 = U2 + p2 V2 = const . f 2 RT Ein Mol eines van-der-Waals Gas hat folgende Beiträge zur inneren Energie: und die potentielle Energie a/V , gleichbedeutend mit der Arbeit, die gegen die Kräfte, die den Binnendruck a/V 2 bewirken. Mit RT V b schreibt sich p= a V2 f H = U + pV = RT 2 (12.76) a +V V ✓ RT V b a V2 ◆ = RT ✓ f V + 2 V b ◆ 2a . V (12.77) Wenn die Enthalpie konstant ist und eine Änderung von T über eine Änderung von V erfolgt ergibt sich aus dem totalen Differential ⇣ @H ⌘ ⇣ @H ⌘ dH = 0 = dV + dT (12.78) @V T @T V und nach Berechnung der partiellen Ableitungen von (12.78) ein Ausdruck für die Temperaturänderung dT ⇡ RT b 2a dV . + 1)RV 2 ( f2 (12.79) 2a Für ideale Gase (a = b = 0) ist dT = 0. Unterhalb einer kritischen Temperatur Rb ist dT < 0 und führt zur Kühlung. Bei Helium liegt diese kritische Temperatur bei etwa 45 K, bei H2 bei 180 K, bei N2 und O2 weit über Zimmertemperatur. Je größer der Koeffizient des Binnendrucks a ist, je größer ist die Abkühlung. Dieses Linde Verfahren wird zur Verflüssigung von Luft verwendet ( p1 = 200 bar ! p2 = 20 bar) und und liefert nach Separation des flüssigen Sauerstoffs (verdampft bei 90 K) flüssigen Stickstoff bei 77 K. 154 KAPITEL 12. THERMODYNAMIK 12.9.4 Osmose Ad hoc Annahmen sind notwendig um den osmotischen Druck zu berechnen und zu interpretieren. Formal kann man er aus einer thermodynamischen Betrachtung der Entropie und des Gibbschen Potentials abgeleitet werden. Bildlich vorstellbar ist die Bewegung eines gelösten Moleküls in einem Lösungsmittel: Bei kleiner Konzentration trifft dieses Molekül nie auf einen gleichartigen Partner sondern irrt im Lösungsmittel herum wie ein Brown’sches Teilchen. Es folgt einem Diffusionvorgang als Folge der ungeordneten Molekularbewegung bei der vorliegenden Wärmeenergie. Darauf beruht der Vorschlag von van’t Hoff (1887) : In verdünnten flüssigen Lösungen gelten für die gelösten Moleküle dieselben Gesetze wie für ideale Gase. Der osmotische Druck • ist proportional zu der molaren Konzentration des gelösten Stoffes • ist proportional zur absoluten Temperatur • von Lösungen hängt nur von der Teilchenzahl des gelösten Stoffes ab • einer Lösung von 1 mol in 22,4 l Lösungsmittel beträgt bei 273,15 K (0 C) 101,325 kPa (Standarddruck) Im idealen Gas haben wir die Gleichung p = nkT . (12.80) In der idealen Lösung gilt für das Produkt aus Dichte und und Boltzmann-Konstante nk ) cR , (12.81) wobei c die Konzentration und R die Gaskonstante ist. Damit ist der osmotische Druck ⇧ ⇧ = cRT . (12.82) Wir legen in ein Gefäß eine fiktive Wand, die jetzt zwei Räume mit Lösung gleicher Konzentration gelöster Moleküle beinhaltet. Der osmotische Druck auf diese fiktive Wand ist von beiden Seiten gleich. In anderen Worten: gleich viele gelöste Moleküle bewegen sich von links nach rechts wie von rechts nach links. Den osmotischen Druck verspüren wir erst, wenn eine semipermeable Wand zwei Gebiete trennt mit unterschiedlicher Konzentration. Die semipermeable Wand läßt nur das Lösungsmittel durch, nicht aber die Moleküle. Dann ist ⇧ nicht durch einen entgegengesetzt gleichen Druck kompensiert und ⇧ tritt als osmotischer Druck in Erscheinung auf der Seite in dem sich die gelösten Moleküle befinden. Deren Druck auf die Wand wird nicht durch einen Gegendruck kompensiert.