Synthese und Kristallstruktur von Iodmethyltriphenylphosphonium
Werbung

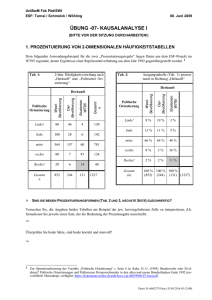
Synthese und Kristallstruktur von Iodmethyltriphenylphosphonium-iodid (C6H5)3PCH2M Synthesis and Crystal Structure of Iodomethyltriphenylphosphonium Iodide (C 6 H 5 )3 PCH 2 M H. Vogt*, K. Lauritsen, L. Riesel, M. von Löwis Institut für Anorganische und Allgemeine Chemie der Humboldt-Universität zu Berlin, Hessische Straße 1/2, D-10115 Berlin G. Reck Bundesanstalt für Materialforschung und -prüfung, Rudower Chaussee, D-12489 Berlin Z. Naturforsch. 48 b, 1760-1766 (1993); eingegangen am 22. März 1993 Iodomethyltriphenylphosphonium Iodide, Synthesis, Crystal Structure Iodomethyltriphenylphosphonium iodide, (C6H 5)3PCH 2I+T, has been prepared by the re­ action of (C6H5)3P with CH2I2 in dichloromethane forming colourless needle like crystals. The crystal and molecular structures have been determined by an X-ray structure analysis. The crystals are orthorhombic, space group Pca2!, Z = 8 ; a = 1478,8(3) pm, b = 1249,3(3) pm, c = 2053,2(3) pm. R = 0.050 for 3219 observed reflections with I > 2cr(I). In the solid state the title compound exists as two discrete monomeric units, (C6H 5)3 PCH2I+T. In both symmetry independent units the I - I distances are surprisingly short with 346,5(1) pm and 356,3(1) pm. For the title compound the results of AM 1, PM 3, and MNDO calculations are in good agree­ ment with corresponding values determined by the X-ray analysis. Einleitung Im Zusam m enhang mit unseren systematischen Untersuchungen im System Phosphan/Halomethan [1] beschäftigten wir uns auch mit der Reaktion von Diiodm ethan mit Triphenylphosphan und erhielten in Analogie zu [2] das erwartete Iodmethyltriphenylphosphoniumiodid. Die Aufklärung der Kri­ stallstruktur der Titelverbindung sollte die Frage nach der A rt und Weise einer möglichen Io d -Io d Wechselwirkung innerhalb des Moleküls beant­ worten, d.h. die Frage klären, ob die Titelverbindung als ein gewöhnliches Phosphoniumsalz oder als ein phosphanstabilisiertes isomeres Methyleniodid aufgefaßt werden muß. Seit dem Nachweis des I3~-Anions und weiterer Polyiodide [3] wurde eine ganze Reihe von V er­ bindungen, in denen Iod-Iod-W echselw irkungen auftreten, synthetisiert und ihre M olekülstruktur aufgeklärt [4 -7]. Charakteristisch für diese V er­ bindungen ist es, daß ein im Kation gebundenes Iodatom mit einem Iodid-Anion oder einem im Anion gebundenem Iodatom (I3_, [A1I4]~, [IW (CO) 4I2] ) wechselwirkt. Die Kationen in den bisher bekannten Beispielen enthalten als Z en­ tralatom immer ein Elem ent der 5. Hauptgruppe und lassen sich durch die allgemeine Formel R 3 E I+ (E = N, P, As) beschreiben. Die in den Verbindun­ gen ermittelten Io d -Io d -A b stän d e überstreichen mit 282,9 pm (Me 3 N I 2 [4]) und 373,1 pm (P 2I 5 +A1I4~ [7]) den recht großen Bereich von 91,2 pm, während die betreffenden E -I -I-W in k e l mit einer A us­ nahme ([terf-Bu 3 PI]+[W(CO)4 I3]- [5]) nur W erte zwischen 170 und 179° aufweisen und somit relativ wenig von einer linearen Anordnung abweichen. Obwohl Weiss et a l [8 ] in den salzartigen V er­ bindungen l,3-Bis(dimethylamino)-l-3-diiodallyliodid und l,3-Bis(dimethylamino)-3-iodpropargyliodid experimentelle Hinweise auf das Vorliegen einer Iod-Iod-W echselw irkung erhielten, ist mit der erfolgreichen Strukturaufklärung des Iodmethyltriphenylphosphoniumiodids der eindeutige Nachweis erbracht, daß auch am Kohlenstoff ge­ bundene Iodatom e mit Iodid-Anionen in Wechsel­ wirkung treten. * Sonderdruckanforderungen an Dr. H. Vogt. Verlag der Zeitschrift für Naturforschung, D-72072 Tübingen 0932-0776/93/1200-1760/$ 01.00/0 Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschung in Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht: Creative Commons Namensnennung-Keine Bearbeitung 3.0 Deutschland Lizenz. This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution-NoDerivs 3.0 Germany License. Zum 01.01.2015 ist eine Anpassung der Lizenzbedingungen (Entfall der Creative Commons Lizenzbedingung „Keine Bearbeitung“) beabsichtigt, um eine Nachnutzung auch im Rahmen zukünftiger wissenschaftlicher Nutzungsformen zu ermöglichen. On 01.01.2015 it is planned to change the License Conditions (the removal of the Creative Commons License condition “no derivative works”). This is to allow reuse in the area of future scientific usage. H. Vogt et al. ■Iodmethyltriphenylphosphonium-iodid (C6HS)3PCHIT~ Tab. I. Kristalldaten und Angaben zur Strukturbestim­ mung von (C6H 5 )3 PCH2I+I". Experimenteller Teil Darstellung des lodm ethyltriphenylphosphonium -iodids Zu einer Lösung von 10 mmol (2,68 g) Diiodm ethan in 20 ml Dichlorm ethan wurde bei -4 0 °C eine Lösung von 10 mmol (2,62 g) Triphenylphosphan in 20 ml Dichlorm ethan unter Rühren lang­ sam zugetropft. Nach dem Erw ärm en auf R.T. wurde noch drei Stunden gerührt. Aus der klaren farblosen Lösung fielen nach m ehreren Tagen na­ delförmige farblose Kristalle aus. Nach 28 Tagen beträgt die A usbeute etwa 80%. D er mittels DTA ermittelte Schmelzpunkt beträgt 240 °C und geht mit einer Zersetzung der Verbindung einher. Elementar analyse CI9H 17PI2 Ber. Gef. C 43,05 C 43,0 H 3,23 H 3,21 1761 P5,84% , P6,06% . 3 1 P-NMR: Singulett bei +23 ppm (CH 2 C12). Massenspektrum: m /z = 51 (71,4%), 77 (31,2%), 107 (49,1%), 108 (77,9%), 127 (84%), 152 (23,7%), 183 (100% ), 184 (23,6% ), 254 (47,1 % ), 262 (49,7% ), 276 (1,8%). Gang der Strukturbestimmung Für die Röntgenkristallstrukturanalyse wurde ein nadelförmiger Kristall mit den Abmessungen 0,35 X 0,2 X 0,2 mm verwendet. Die Kristalldaten und Einzelheiten der Messung der Reflexintensitä­ ten, die auf einem Enraf-Nonius-CAD 4-Diffraktom eter erfolgten, sind Tab. I zu entnehmen. Aufgrund der beobachteten systematisch aus­ gelöschten Reflexe (0 k l, l = 2 n + l und hOl, h = 2n + 1) und einer Intensitätsstatistik konnte auf die Kristallsystem Gitterkonstanten Zellvolumen Formeleinheiten/Zelle Dichte, berechnet Raumgruppe Meßgerät Strahlung orthorhombisch a = 1478,8(3) pm b = 1249,3(3) pm c = 2053,2(3) pm V = 3793,2 106 pm 3 Z Dx= 1,856 g/cm3 Pca21 Enraf-Nonius CAD-4 MoKa (Graphit-Mono­ chromator) Zahl der Reflexe zur Gitter- 25 konstantenbestimmung Meßbereich 3° < 2 6 < 50°; 2 Q-a>-scan Zahl der gemessenen 3430 Reflexe Zahl der unabhängigen 3219 mit I> 2 a (I) Reflexe Korrekturen Lorentz, Polarisation Verfeinerung Einheitsgewichte mit w = 1 R 0,054 für alle Reflexe 0,050 für verwendete Re­ flexe 0,063 Rw 0,5 eÄ - 3 Restelektronendichte Atomformfaktoren International Tables for X-ray Crystallography (1974) Rechenprogramme SDP-Enraf-N onius, MOLGRAF [9]; CELLGRAF [9] Raumgruppe P c a 2 1 mit zwei sym m etrieunabhän­ gigen Formeleinheiten C 19H 17 PI 2 geschlossen wer­ den. Dies wurde durch das Ergebnis der Struktur­ analyse bestätigt. C31 C37 C18 C12 Abb. 1. Darstellung der beiden kristallographisch unterschiedlichen Moleküle des (C6H 5)3 PCH 2I+I . 1762 Atom 11 12 13 14 PI P2 CI C2 C3 C4 C5 C6 C7 C8 C9 CIO C ll C 12 C13 C14 C15 C16 C 17 C 18 C19 C20 C21 C22 C23 C24 C25 C26 C27 C28 C29 C30 C31 C32 C33 C34 C35 C36 C37 C38 H. Vogt et al. • Iodmethyltriphenylphosphonium-iodid (C6H5)3PCHI+T X 0,04406(8) 0,20823(9) 0,23138(9) 0,0288(1) -0,1108(3) 0,3610(3) -0,074(1) 0,325(1) - 0 ,2 1 1 ( 1 ) -0,263(1) -0,340(1) -0,364(1) -0,315(1) -0,236(1) -0,140(1) -0,224(1) -0,245(2) -0,176(1) -0,089(1) -0,067(1) -0,024(1) 0,036(1) 0,103(2) 0,109(2) 0,045(2) -0,024(1) 0,382(1) 0,467(1) 0,479(1) 0,408(2) 0,323(1) 0,308(1) 0,461(1) 0,509(2) 0,585(2) 0,614(2) 0,566(2) 0,487(2) 0,277(1) 0,296(2) 0,227(2) 0,157(2) 0,137(2) 0 ,2 0 0 ( 1 ) 0,03713(9) 0,55047(9) -0,0667(1) 0,4267(1) 0,0542(3) 0,5655(4) 0,113(1) 0,634(1) 0,125(1) 0,185(1) 0,235(2) 0,225(2) 0,169(2) 0,119(2) -0,089(1) -0,114(2) -0,224(2) -0,295(2) -0,269(2) -0,161(1) 0,073(1) 0,156(2) 0,166(2) 0,107(2) 0,027(2) 0,005(2) 0,425(1) 0,390(2) 0,285(2) 0,215(2) 0,248(2) 0,354(2) 0,633(1) 0,594(2) 0,648(2) 0,742(2) 0,780(2) 0,728(2) 0,582(2) 0,518(2) 0,540(2) 0,614(2) 0,667(2) 0,651(2) Tab. II. Atomkoordinaten und ihre Standardab­ weichung. z B/A2 0 ,0 0 0 3,48(2) 3,72(2) 4,13(3) 4,96(3) 2,57(8) 2,94(9) 3,3(3) 3,4(3) 2,6(3)* 3,1(3)* 4,3(4)* 4,3(4)* 4,2(4)* 3,7(4)* 3,3(3)* 4,1(4)* 4,8(4)* 4,3(4)* 3,9(4)* 3,3(3)* 3,3(3)* 4,4(4)* 5,6(5)* 5,4(5)* 6 ,1 (6 )* 4,2(4)* 3,1(3)* 4,2(4)* 4,8(4)* 5,3(5)* 4,6(4)* 4,1(4)* 3,2(3)* 4,9(5)* 4,9(5)* 4,9(5)* 5,5(5)* 4,9(5)* 4,1(4)* 5,3(5)* 6 ,1 (6 )* 6,5(6)* 6 ,2 (6 )* 4,6(4)* 0,00990(7) 0,08903(7) -0,06828(8) -0,1154(2) 0,1288(2) -0,0397(9) 0,0558(9) -0,1407(8) -0,0976(9) -0,118(1) -0,186(1) -0,226(1) -0,207(1) -0,1044(9) -0,089(1) -0,075(1) -0,078(1) -0,094(1) -0,1056(9) -0,1760(9) -0,169(1) -0,219(1) -0,268(1) -0,277(1) -0,230(1) 0,1141(9) 0,094(1) 0,076(1) 0,078(1) 0,095(1) 0,115(1) 0,1556(9) 0 ,2 1 2 ( 1 ) 0,235(1) 0,206(1) 0,151(1) 0,129(1) 0,194(1) 0,253(1) 0,303(1) 0,293(1) 0,236(1) 0,183(1) Falls die auch denkbare zentrosymmetrische R aum gruppe Pcam vorliegen würde, müßten die M oleküle A und B durch eine Spiegelebene inein­ ander überführbar sein. Diese Spiegelebene ist auch in grober N äherung nicht vorhanden. D arüber hin­ aus unterscheiden sich die beiden Moleküle deut­ lich in ihrer Konform ation (vgl. Abb. 1). Ein Versuch, die Struktur mit direkten M etho­ den zu bestimmen, scheiterte. Deshalb wurden die Positionen der vier I-Atome aus einer PattersonSynthese ermittelt. Eine folgende DifferenzFourier-Synthese ergab die Lage beider P-Atome. Nach einer Verfeinerung der Koordinaten und an­ Die mit * gekennzeichneten Atome wurden isotrop verfeinert, die anderen anisotrop ent­ sprechend: (4/3) •[a2*B(l,l) + b 2*B(2,2) + c2*B(3,3) + ab (cosy)*B(1,2) + ac(cos/?)*B(l,3) + bc(cosa)*B(2,3)]. isotropen Tem peraturfaktoren konnten aus einer weiteren Differenz-Fourier-Synthese alle C-Atome lokalisiert werden. Die abschließende full-matrixleast-squares-Verfeinerung der Atom koordinaten, der anisotropen Tem peraturfaktoren der schweren Atome und der M ethylkohlenstoffatome sowie der isotropen Tem peraturfaktoren der anderen CAtome konvergiert unter Verwendung einheit­ licher Gewichte für alle Reflexe mit R = 0,050. Die O rtsparam eter der A tom e - mit Ausnahme der H-Atome - sowie deren Standardabweichung sind in Tab. II zusammengefaßt. Die wichtigstenBindungsabstände und -winkel enthält Tab. III. H. Vogt et al. ■Iodmethyltriphenylphosphonium-iodid (C6H5)3PCHIT 1763 Tab. III. Ausgewählte Bindungslängen und -winkel im (C6H 5)3 PCH 2I2. a) Bindungsabstände in pm P (l)-C (l) 180,3(8) 180,5(7) P (l)-C (3) 184,9(6) P(l)-C (9) 180,6(8) P(l)-C (15) 214,5(8) C (l)-I(l) 356,3(6) 1(1) —1(3) P(2)-C(2) P(2)-C(21) P(2)-C(27) P(2)-C(33) C(2)-I(2) 1(2)-1(4) b) Bindungswinkel in ° C (l)-P (l)-C (3 ) 107,4(4) C (l)-P (l)-C (9 ) 111,0(4) C (l)-P (l)-C (1 5 ) 108,9(4) C (3 )-P (l)-C (9 ) 108,4(4) C (3)-P(l)-C (15) 108,9(4) C (9)-P(l)-C (15) 112,1(4) I ( l) - C ( l) - P ( l) 113,2(4) C (l)-I(l)-I(3 ) 170,9(9) C(2)-P(2)-C(21) C(2) - P(2) - C(27) C(2) - P(2) - C(33) C(21) - P(2) - C(27) C(21) - P(2) - C(33) C(27) - P(2) - C(33) I(2)-C (2)-P(2) C (2)-I(2)-I(4) 180,5(8) 181,2(7) 178,7(7) 183,3(9) 222,0(9) 346,6(6) Strukturbeschreibung und Diskussion Die Kristallstruktur des Iodm ethyltriphenylphosphonium-iodids wird durch zwei kristallographisch unterschiedliche (C 6H 5 ) 3 PCH 2 I2-Einheiten aufgebaut (Abb. 1). Parallel zur (OOl)-Ebene sind alternierend P (l)- und P(2)-M oleküle so ausge­ richtet, daß jeweils eine Phenylgruppe der P (l)-E in ­ heit mit einer Phenylgruppe der P(2)-Einheit wech­ selwirkt. D er Winkel zwischen den wechselwirken­ den Phenylgruppen beträgt nur 8 °. D er kürzeste C -C -A b stan d zwischen diesen Phenylgruppen be­ trägt 326,8 pm (C (8 )-C (37)) und der längste 356,0 pm (C (6)-C (35)). A ußerdem sind die P (l)und P(2)-Einheiten so ausgerichtet, daß die jewei­ ligen CH 2I2-Gruppierungen zueinander orientiert sind, so daß die I-Atom e quasi eine eigene Schicht bilden (Abb. 2). Die CH 2 I2-G ruppen sind parallel zur (OOl)-Ebene gestaffelt angeordnet, so daß die I-Atome sich gegenseitig wenig behindern. Interes­ santerweise sind die I-A tom e 1(3) und 1(4) zu den P-Atom en P(2) und P (l) orientiert. D er Abstand der Iodatom e zu den betrachteten P-Atom en beträgt 504,4 pm (I(3 )-P (2 )) und 518,1pm (I(4 )-P (l)). Die P-Atom e im (C 6H 5 ) 3 PCH 2 I 2 sind durch die vier C-Atome nahezu tetraedrisch koordiniert. Die P -C -A b stän d e von 178,7(7) bis 181,2(7) pm (Tab. III) weisen keine B esonderheiten auf und stimmen mit den P -C -A b stän d en überein, die z. B. für Halogenotriphenylphosphonium -Kationen er­ m ittelt wurden [10]. Interessanter ist der signifi­ kante Unterschied in den C -I-A b stä n d en der bei­ den kristallographisch unterschiedlichen Moleküle 111,7(4) 106,3(4) 110,3(5) 111,6(4) 110,3(5) 106,3(5) 111,4(4) 177,4(9) Die Bindungsabstände und -winkel inner­ halb der Phenylringe weisen keine Beson­ derheiten auf und sind deshalb nicht mit auf­ geführt. von 7,5 pm. W ährend der C -I-A b sta n d im ,,P(1)Molekül“ mit 214,5(8) pm identisch mit dem C - I Abstand im CI 4 (215 pm [11]) ist und in etwa der Summe der Kovalenzradien des Kohlenstoffs und des Iods (210pm [11]) entspricht, ist der C -I- A b ­ stand im „P(2)-M olekül“ mit 222,0(9) pm deutlich vergrößert. Die I-I-A b stän d e von 356,3(6) pm (1(1)-1(3)) und 346,6(6) pm (1(2)-1(4)) sind rela­ tiv lang und liegen damit zwischen der Summe der Kovalenzradien (266 pm [11]) und der Summe der van der Waals-Radien (390 - 424 pm [11]) und stim­ men in etwa mit der Summe aus Kovalenz- und Ionenradius (339 pm [11]) überein. Io d -Io d -A b ­ stände im Bereich von 300 bis 350 pm sind für 12 12 12 Abb. 2. Kristallstruktur des (C6H 5)3 PCH 2I+T. Abstände (pm): P (l)-I(4 ) 518,1; P(2)-I(3) 504,4. 1764 H. Vogt et al. • Iodmethyltriphenylphosphonium-iodid (C6H5)3PCHI+I~ a) Bindungsabstände in pm P -C H 2I2 C -I I-I KSA 180,3a 214,5 356,3 b) Bindungswinkel in ° P -C -I C -I-I 113,2 170,9 c) Torsionswinkel in ° I( l) -C (l)-P (l)-C (9 ) 1(2) - C(2) - P(2) - C(21) C (l)-P (l)-C (9 )-C (1 4 ) C(2)-P(2)-C(21)-C(26) C(l) -P (l) - C(3) - C(4) C(2) - P(2) - C(27) - C(28) C (l)-P (l)-C (1 5)-C (16) C(2) - P(2) - C(33) - C(34) 346,6 AMI 161,5 228,7 257,9 PM3 175,5 203,1 275,2 MNDO 181,3 208,8 257,4 111,3 177,4 126,3 178,2 117,3 173,1 114,4 180,0 -41,6 -52,5 -40,6 -32,8 -54,4 -57,0 -27,5 -34,2 -44,6 -36,3 -28,0 -22,4 1,87 -0,78 0,52 -0,30 0,13 -0,52 180,5b 2 2 2 ,0 -60,2 -52,3 -90,2 -89,2 19,2 -10,7 24,2 5,3 d) berechnete Nettoladungen der P-- C - I - I-Gruppierung P 3,38 C -1,57 I 0,13 I -0,40 Verbindung Ref. ^E-I dw Me3NI+IPh3 PI+IPh3AsI-I(f-Bu)3PI+IPh3PI+I3[(Ph3PP) 2I 3-]I3(r-Bu)3PI+(W(CO)4I3)pi 4+a i i 4p 2i 5+a i i 4 4 4 5 5 227 248,1 264,0 246,1 239,5 240,1 240,1 239,6* 240,9* 282,9 316 300,5 332,6 355,1 350,6 355,1 342,2* 345,5*’a 373,1 *•» 356,3 346,6 Ph3 PCH2I+I^ Ph3PCH 2I+I_d 6 6 5 7 7 Tab. IV. Vergleich der expe­ rimentell erhaltenen (KSA) und der berechneten Bin­ dungsabstände und -winkel, sowie die berechnete La­ dungsverteilung in der P - C - 1- I-Gruppierung. 214,5 2 2 2 ,0 die verschiedenen Polyiodid-Ionen vermessen wor­ den [3]. D arüber hinaus wurden in den anderen Verbindungen mit Iod-Iod-W echselw irkungen Io d -Io d -A b stände von 282,9 bis 373 pm ermittelt (vgl. Tab. V). Vergleicht man die verschiedenen Iodphosphonium-Verbindungen miteinander, so fällt auf, daß die Verkürzung des P -I-A b stan d es mit einer A uf­ weitung der I-I-B in d u n g einhergeht. Insbesonde­ re wird das im Vergleich des (C 6 H 5 ) 3 PI+T mit dem (C 6 H 5 ) 3 PI+I3_ deutlich. W ährend die P -I-B indung im (C 6 H 5)3P T T gegenüber der im (C 6 H 5 )3 PI+I3~ um 8 , 6 pm vergrößert ist, verringert sich der Iod - Iod- 0 ,2 2 -0,79 509,9 564 564,5 578,7 594,6 590,7 595,2 581,8* 586,8*’a 614,0*b 570,8 568,6 179 178,2 174,8 177,6 156,8 177,4 171,1 169-174 170,9 177,4 a „P(l)-Molekül“; b ,,P(2)Molekül“. Tab. V. Elem ent-Iodund Iod - Iod-Bindungsabstände (pm), sowie Element - I o d - Iod-Bindungswinkel (°) in bisher bekann­ ten Verbindungen mit Iod-IodWechselwirkungen. * Durchschnittswerte; a P(III), b P(V); c P(l)-Molekül; d P(2)Molekül. Bindungsabstand in den betreffenden M olekülen um 39,1 pm. Dieser Sachverhalt kann nach [5] auf die geringere Nucleophilie des I3~-Anions gegen­ über dem T-Anion zurückgeführt werden. In die­ sem Zusammenhang ist es bem erkenswert, daß auch beim Vergleich der beiden kristallographisch unabhängigen M oleküle des (C 6H 5 ) 3 PCH 2T T fest­ zustellen ist, daß eine Verkürzung des C -I- A b ­ standes um von 7,5 pm mit einer Verlängerung des I - I-Abstandes um 9,7 pm verbunden ist (vgl. Tab. V). Die unterschiedlichen P - I - und I - I - A b ­ stände in den beiden chemisch äquivalenten M o­ lekülen können in diesem Fall nicht über intram o­ H. Vogt et al. • Iodmethyltriphenylphosphonium-iodid (C6H 5)3PCHIT 1765 lekulare Wechselwirkungen wie im Fall der Iodtriphenylphosphonium-iodide erklärt werden, son­ dern sind auf intermolekulare Wechselwirkungen innerhalb des Kristallgitters zurückzuführen. bis 100 pm kürzer erhalten. Ebenfalls große A b­ weichungen zwischen Rechnungen und Experi­ ment zeigen die aufgeführten Torsions winkel (Tab. IV, Abb. 3). Semiempirische Berechnungen Schlußfolgerungen Die semiempirischen Berechnungen (A M I, PM 3, MNDO) wurden auf einer RS/6000-320 mit dem Programm M OP AC [12] durchgeführt. Die für diese Diskussion wesentlichen M olekülparam eter sind in der Tab. IV zusammen m it denen aus der Kristallstrukturanalyse erhaltenen aufgeführt. Die berechneten Bindungslängen P - C H 2 I2, C - I und I - I differieren innerhalb der drei M ethoden um etwa 20 pm, die Bindungswinkel P - C - I und C - I - I um 12° und 7°. Deutlich größere Abwei­ chungen weisen die aufgeführten Torsionswinkel auf. D er Vergleich der aus der Kristallstrukturana­ lyse erhaltenen M olekülparameter mit den berech­ neten zeigt, daß sowohl die A bstände C - I und P - C H 2 I 2 als auch die Winkel P - C - I und C - I - I durch die MNDO-Rechnung gut reflektiert w er­ den. Die aus den AM 1- und PM 3-Rechnungen er­ haltenen D aten weichen von den experimentell b e­ stimmten deutlich ab. Der I-I-A b sta n d wird im Vergleich zu den experimentell erm ittelten um 80 Zusammenfassend kann festgestellt werden, daß das (C 6 H 5 ) 3 PCH 2 I 2 als ein Ionenpaar (C 6H 5 ) 3 PCH 2 I+T z u betrachten ist. Sowohl der I - I A bstand von 346,5 bzw. 356,3 pm als auch der C -I-I-W in k e l von 170,9° bzw. 177,4° sprechen für diese Betrachtung. Dam it im Einklang steht auch die Ladungsverteilung innerhalb der PCH 2II-Gruppe, die aus den Berechnungen (Tab. IV) folgt, sowie die relativ gute Löslichkeit in polaren Lösungsmit­ teln wie Acetonitril und Methylenchlorid und die sehr geringe Löslichkeit in E ther und Benzen und das 3 1 P-NMR-Signal von + 32 ppm einer Lösung der Titelverbindung in Dichlormethan. Somit wird das Spektrum der ionogenen Verbin­ dungen mit I-I-W echselw irkungen um einen V er­ treter erweitert, bei dem das im Kation gebundene Iodatom an Kohlenstoff und nicht an ein Elem ent der 5. H auptgruppe gebunden ist. Gegen die Auffassung des (C 6H 5 )3 PCH 2 I+T als ein phosphanstabilisiertes isomeres Methyleniodid spricht der große I - I-Abstand, der gegenüber der Rechnung um 80 bzw. 100 pm vergrößert gemessen wurde und der C -I-I-W in k e l von nahe 180°. Für isomere Dihalogenm ethane wurden dagegen C -X -X -W in k e l von 120°berechnet (MNDO- und A M I-B erechnungen für C H ^ [13]; für CH 2Br2, CH 2 BrCl, CH 2 C12 [14-15]). Die Listen der beobachteten F-W erte und der Tem peraturfaktoren können beim Fachinformationszentrum Karlsruhe, Gesellschaft für wissen­ schaftlich-technische Information mbH, D-76344 Eggenstein-Leopoldshafen, unter Angabe der H in­ terlegungsnum m er CSD 57100, der A utoren und des Zeitschriftenzitats angefordert werden. Abb. 3. Vergleich der Molekülstrukturen; starke Linie: KSA „P(l)-Molekül“; normale Linie, große Kreise: AM I; normale Linie, mittlere Kreise: PM3; normale Linie, kleine Kreise: MNDO. Die A utoren danken dem Fonds der Chemischen Industrie für die Bereitstellung der RS/6000-320. 1766 H. Vogt et al. ■Iodmethyltriphenylphosphonium-iodid (C6H5)3PCHI+r [1] K. Lauritsen, H. Vogt, L. Riesel, Phosphorus, Sul­ fur, and Silicon 77,211 (1993). [2] D. Seyferth, J. K. Heeren, G. Singh, S. O. Grim, W. B. Hughes, J. Organometallic Chem. 5, 267 (1966). [3] A. F. Wells, Structural Inorganic Chemistry, 5. ed., S. 398, Clarendon Press, Oxford (1984). [4] S. M. Godfrey, D. G. Kelly, C. A. McAucliffe, A. G. Machie, R. G. Pritchard, S. M. Watson, J. Chem. Soc. Chem. Commun. 1991,1163. [5] M. Bätcher, W.-W. Du Mont, S. Pohl, W. Saak, Phosphorus, Sulfur, and Silicon 49/50, 147 (1990); N. Kuhn, R. Jüschke, W.-W. Du Mont, M. Bätcher, D. Bläser, R. Boese, Z. Naturforsch. 44 b, 9 (1989). [6 ] F. A. Cotton, P. A. Kibala, J. Am. Chem. Soc. 109, 3308 (1987). [7] S. Pohl, Z. Anorg. Allg. Chem. 498, 15 (1983) und 498, 20 (1983). [8 ] R. Weiss, H. Wolf, V. Schubert, T. Clark, J. Am. Chem. Soc. 103, 6142 (1981). [9] G. Reck, R.-G. Kretschmer, Computerprogramme zur Darstellung und Interpretation von Molekülund Kristallstrukturen, Bundesanstalt für Material­ forschung und -prüfung, Berlin. [10] H. Vogt, S. I. Trojanov, V. B. Rybakov, Z. Natur­ forsch. 48b, 160 (1993). [11] J. E. Huheey, Anorganische Chemie/Prinzipien von Struktur und Reaktivität, S. 279 f., Walther de Gruyter, Berlin-New York (1982). [12] MOPAC, a Semiempirical Molecular Orbital Pro­ gram, in der Modifikation von Dr. T. Clark, Institut für Organische Chemie, Friedrich-Alexander-Universität, Erlangen-Nürnberg. [13] M. von Löwis und H. Vogt, unveröffentlicht. [14] G. Maier, H. P. Reisenauer, J. Hu, L. J. Schaad, B. A. Hess (Jr.), J. Am. Chem. Soc. 112,5117 (1990). [15] H.-U. Gremlich, T. K. Ha, G. Zumofen, R. E. Bühler, J. Phys. Chem. 85, 1336 (1981).