Kongress über Lungenhochdruck
Werbung

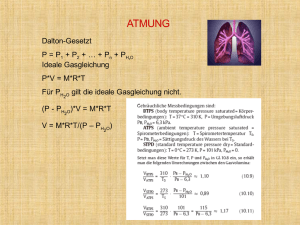
Pressemitteilung Internationaler Kongress über Lungenhochdruck Vom 08. – 10. Dezember 2011 findet im Albert-Fraenkel-Saal der ThoraxklinikHeidelberg wieder eine Internationale Tagung über Lungenhochdruck statt, bei der renommierte Experten aus Deutschland, Österreich und der Schweiz vortragen. Die Veranstaltung steht unter der Schirmherrschaft der Deutschen Gesellschaft für Kardiologie, wird mit 16 Fortbildungspunkten bewertet und bietet neben Vorträgen viel Zeit für Diskussionen und Austausch. Die Tagung behandelt u.a. Themen über das rechte Herz, das häufig "vergessen" wird, aber doch bei vielen Erkrankungen wie Atemwegserkrankungen eine wichtige Rolle spielt. Bei der Tagung werden auch neue Therapieverfahren vorgestellt, wie die Herzunterstützungsmaschine ECMO als Brücke zur Lungentransplantation, neue medikamentöse Therapien oder die ergänzende Atem- und Bewegungstherapie. Die Tagung wurde von der Arbeitsgemeinschaft für Lungenhochdruck der Fachgesellschaften für Kardiologie, Pneumologie und Kinderkardiologie unter der Federführung ihres Ko-Sprechers Professor Ekkehard Grünig organisiert. Er leitet das Lungenhochdruckzentrum der Thoraxklinik Heidelberg. Lungenhochdruckzentrum: Prof. Dr. med. Ekkehard Grünig Leiter des Zentrums für Pulmonale Hypertonie Thoraxklinik Heidelberg Amalienstr. 5 , 69126 Heidelberg Tel: 06221/396-8053 mail: [email protected] Kontakt: Pressestelle: Kirsten Gerlach M.A. Thoraxklinik-Heidelberg gGmbH Amalienstr. 5, 69126 Heidelberg Tel: 06221/3962101, Fax: 06221/3962102 [email protected] Weitergehende Infos: Die pulmonale Hypertonie ist durch eine Erhöhung des Blutdrucks im Lungenkreislauf charakterisiert. Sie ist eine schwerwiegende Erkrankung, bei der die Lunge und das Herz betroffen sind. Beim Gesunden ist der Blutdruck in diesem sog. „Niederdrucksystem“ des Körpers mit ca. 25/10 mmHg relativ niedrig und steigt auch bei körperlichen Belastungen nicht wesentlich an. Demgegenüber weisen Patienten mit pulmonaler Hypertonie bereits in Ruhe einen erhöhten pulmonal arteriellen Druck auf, der bei Belastung weiter ansteigt. Der systolische pulmonal arterielle Druck (PAPs) lässt sich mit Hilfe der Ultraschalluntersuchung (Echokardiographie) des rechten Herzens abschätzen, Werte > 35 mmHg gelten als erhöht. Zur Diagnosesicherung ist die Rechtsherzkatheter-Untersuchung notwendig. Per Definition liegt eine pulmonale Hypertonie vor, wenn der mittels Rechtsherzkatheter gemessene pulmonal arterielle Mitteldruck (PAPm) ≥ 25 mmHg beträgt. In den letzten Jahren konnten bei der PAH neue Erkenntnisse über die Genetik und Pathophysiologie des Lungenkreislaufes und des Gefäßremodellings gewonnen und neue Therapieverfahren entwickelt werden. So wurden bei einigen Unterformen, wie der idiopathischen und familiären PAH Mutationen des BMPR2-Gens gefunden, einem Rezeptor für wachstumshemmende Faktoren. Durch die Mutationen kann das für die PAH typische tumorartige Zuwachsen der kleinen Lungengefäße (plexiforme Läsionen) ausgelöst werden. Zudem wurden PAH-spezifische Medikamente entwickelt, wie die Enthothelinantagonisten Bosentan, Sitaxsentan und Ambrisentan, die Prostacyclinanaloga Ventavis inhalativ, Ilomedin und Epoprostenol intravenös, Treprostenil subcutan sowie der Phosphodiesteraseinhibitor Sildenafil. Trotz der therapeutischen Erfolge kann die PAH nicht kausal behandelt oder gar geheilt werden. In den meisten Fällen kann man aber das Voranschreiten der Erkrankung deutlich verlangsamen. Ein weiteres großes Problem ist die sehr späte Diagnose und der verzögerte Therapiebeginn. In der Regel werden die Patienten mit PAH erst dann diagnostiziert, wenn das rechte Herz bereits deutlich vergrößert und die rechtsventrikuläre Pumpfunktion eingeschränkt ist. Zu diesem Zeitpunkt haben die Patienten bereits massive Beschwerden wie Luftnot bei geringster Anstrengung entsprechend NYHA-Klasse III-IV, einen mittleren pulmonal-arteriellen Druck von etwa 50 mmHg und eine eingeschränkte Prognose. Von Symptombeginn bis zur richtigen Diagnose vergehen immer noch Jahre und die Patienten müssen in der Regel mehrere Ärzte aufsuchen, bis jemand an einen Lungenhochdruck denkt. PH führt zudem zu einer Zunahme von behandlungsbedürftigen Begleiterkrankungen wie Depressionen und Angststörungen sowie zu einer massiven Einschränkung der Lebensqualität. Lungenhochdruck und Atem- und Bewegungstherapie: Als Ergänzung der PHspezifischen medikamentösen Therapie kann unter stationärer Überwachung durchgeführte vorsichtige Atem- und Bewegungstherapie die körperliche Belastbarkeit und die Lebensqualität der Patienten möglicherweise verbessern. Hierfür sprechen die Ergebnisse einer aktuellen prospektiven Studie bei 183 Patienten mit verschiedenen Lungenhochdruckerkrankungen. Die Patienten der Trainingsgruppe wiesen nach 15 Wochen eine signifikante Verbesserung der 6-Minuten Gehstrecke, der Lebensqualität, NYHAKlasse, maximalen Sauerstoffaufnahme und maximalen Belastungsstufe auf dem Fahrradergometer auf.