Inhaltsverzeichnis 1. Einleitung 2. Aufgabenstellung 3
Werbung

Enzymkinetik
Inhaltsverzeichnis
1. Einleitung
1.1. Enzyme
1.2. Enzymaktivität
1.3. Cofaktoren
1.4. Reaktionsgeschwindigkeit
1.5. Enzym-Substrat-Affinität
1.6. Michaelis-Menten
1.7. Lineweaver-Burk
1.8. Enzymhemmung
1.9. Extinktion
1.10. Glykolyse, Milchsäuregärung
2. Aufgabenstellung
3. Material, Methoden und Versuchsdurchführung
3.1. Versuch 1
3.2. Versuch 2
3.3. Funktionsprinzip des Spektralphotometers
4. Ergebnisse
4.1. Versuch 1
4.1.1. Berechnung der tatsächlichen Pyruvatkonzentration in der
Küvette
4.1.2. Berechnung der Extinktion ∆E
4.1.3. Berechnung der Reaktionsgeschwindigkeit
4.1.4. Diagramm nach Michaelis-Menten
4.1.5. Diagramm nach Lineweaver-Burk
4.1.6. Berechnung der Enzymaktivität
4.2. Versuch 2
5. Diskussion
5.1. Versuch 1
5.2. Versuch 2
6. Literaturverzeichnis
7. Abbildungsverzeichnis
8. Anhang
1
Enzymkinetik
1. Einleitung
1.1. Enzyme
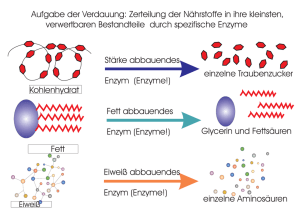
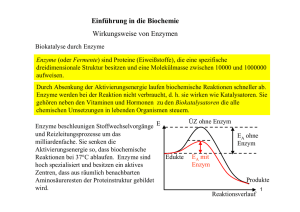
Enzyme gehören zur Klasse der Proteine und erfüllen im Stoffwechsel eine wichtige
biologische Funktion. Sie katalysieren chemische Reaktionen, indem sie die
Aktivierungsenergie EA herabsetzen, d.h. die Energie, die zum Start einer Reaktion
nötig ist. Dadurch können auch Reaktionen mit sehr hoher Aktivierungsenergie bei
Körpertemperatur ablaufen. Die Enzyme werden während der Katalysereaktion
weder verbraucht noch in ihrer chemischen Struktur verändert.
Man kann Enzyme aufgrund ihrer Wirkungsweise klassifizieren:
Oxydoreduktasen:
katalysieren
Redoxreaktionen
(Bsp.:
Lactat-Dehydro-
genase)
Transferasen: Katalyse von Gruppenübertragungen wie Phosphorylierung
oder Methylierung
Hydrolasen: verantwortlich für Hydrolysereaktionen
Lyasen: Bildung von Doppelbindungen durch Hinzufügen oder Entfernen von
einzelnen Atomen oder Molekülgruppen
Isomerasen: katalysieren die Bildung von Konstitutionsisomeren
Ligasen: fügen einzelne Moleküle unter ATP-Verbrauch zusammen
Auch in ihrer chemischen Struktur können Enzyme unterschieden werden:
Reine Proteine: bestehen aus Aminosäuren, die durch Peptidbindungen
miteinander verknüpft sind
Holoenzyme:
zusammengesetzte
Proteine
aus
einem
Apoenzym
(Proteinanteil) und einem Cofaktor
Enzyme sind aufgrund verschiedener Eigenschaften hochspezifisch.
Sie sind in der Lage selektiv Substrate auszuwählen und in ihre entsprechenden
Produkte umzusetzen (Substratspezifität). Dabei müssen die Substrate nach dem
Schlüssel-Schloss-Prinzip zum aktiven Zentrum des Enzyms passen. Einige Enzyme
können sogar Konstitutionsisomere oder andere nah verwandte Verbindungen
unterscheiden.
2
Enzymkinetik
Wenn nun ein spezifisches Substrat am aktiven Zentrum bindet, kann jeder
Enzymtyp
nur
eine
ganz
bestimmte
chemische
Reaktion
katalysieren
(Wirkungsspezifität).
Zum Beispiel kann das Enzym Saccharase nur das Disaccharid Saccharose in
Glukose und Fructose spalten.
Im Folgenden soll nun der Ablauf einer enzymatischen Reaktion erklärt werden.
Voraussetzungen hierfür sind, dass Substrate in ausreichender Menge vorhanden
und die aktiven Zentren der Enzyme nicht besetzt sind.
Ein Substrat bindet unter Bildung einer schwachen Wechselwirkung an das
katalytische Zentrum, sodass ein Enzym-Substrat-Komplex (ES) entsteht. Durch
die
Enzym-Substrat-Wechselwirkung
verändert
das
aktive
Zentrum
seine
Konformation, um sich noch enger an das Substrat anzulagern.
Anschließend wandelt das aktive Zentrum das Substrat in seine Produkte um, welche
anschließend wieder freigesetzt werden.
Nach der Reaktion liegt das Enzym wieder in seiner ursprünglichen Konformation vor
und kann daher mit einem neuen Substratmolekül einen Komplex bilden.
In Abbildung 1 soll dieser Vorgang noch mal verdeutlicht werden. Hierbei dient als
Beispiel die Umsetzung von Saccharose in Glukose und Fructose.
Abb. 1:
Hydrolyse von
Saccharose in
Glukose und
Fructose
durch das
Enzym
Saccharase
(aus:
Campbell,
Biologie,1997)
3
Enzymkinetik
1.2. Enzymaktivität
Unter Enzymaktivität versteht man die katalytische Wirksamkeit eines Enzyms. Sie
wird als Wechselzahl gemessen, d.h. die Anzahl der Substratmoleküle, die pro
Zeiteinheit von einem Mol Enzym umgesetzt werden.
WechselzahlW =
MolSubstratumsatz
MolEnzym × Zeiteinheit
Die Enzymaktivität kann durch verschiedene Faktoren beeinflusst werden:
Temperatur:
Jedes Enzym besitzt ein bestimmtes Temperaturoptimum, bei dem seine Umsatzrate
am höchsten ist. Bei steigender Temperatur nimmt die enzymatische Aktivität stets
zu,
weil
sich
durch
eine
schnellere
Molekülbewegung
(Brownsche
Molekularbewegung) die Wahrscheinlichkeit erhöht, dass Substrat und Enzym
aufeinandertreffen.
Wenn die Temperatur jedoch zu hoch wird, nimmt die Aktivität rapide ab, da Enzyme
ab einer bestimmten Temperatur denaturiert werden. Durch die hohe Wärmeenergie
werden kovalente Bindungen gelöst und so die Tertiärstruktur des Proteins
irreversibel zerstört.
Das Temperaturoptimum menschlicher Enzyme liegt nahe der Körpertemperatur.
Jedes Enzym kann aufgrund einer typischen Reaktionsgeschwindigkeits-TemperaturKurve charakterisiert werden (siehe Abb.2).
Abb. 2: Temperaturoptima am Beispiel zweier Enzyme (aus: Campbell, Biologie, 1997)
pH-Wert:
Neben einem Temperaturoptimum besitzen Enzyme auch einen pH-Bereich, bei dem
sie optimal wirken können. Häufig sind elektrostatische Bindungen an der Bildung
eines Enzym-Substrat-Komplexes beteiligt.
4
Enzymkinetik
Durch eine Zunahme des pH-Wertes wird die Anzahl der negativen aktiven Zentren
erhöht, wobei eine Bindung mit den positiven Gruppen des Substrats erleichtert wird.
Bei einer Abnahme des pH-Wertes erhöht sich die Anzahl der positiven Zentren, die
mit den negativen Substratgruppen reagieren können.
Je nach Wirkungsort unterscheiden sich die pH-Optima verschiedener Enzyme. Der
optimale pH-Wert liegt für die meisten Enzyme zwischen 6 und 8. Im Gegensatz
dazu bevorzugen Verdauungsenzyme im sauren Millieu des Magens einen niedrigen
pH-Wert (Bsp.: Pepsin, pH 2), während Enzyme im Darm an einen alkalischen pHBereich angepasst sind (Bsp.: Trypsin, pH 8).
Abb. 3: pH-Optima der Enzyme Pepsin und Trypsin (aus: Campbell, Biologie, 1997)
Chemische Substanzen:
Die Aktivität von Enzyme kann durch bestimmte Moleküle gehemmt werden. Solche
Inhibitoren können das aktive Zentrum reversibel und sogar irreversibel verändern,
sodass die Enzyme nicht mehr funktionsfähig sind.
Genaueres dazu wird auf Seite 10 erläutert.
1.3. Cofaktoren
Viele Enzyme benötigen für ihre katalytische Aktivität die Mitwirkung kleinerer
Moleküle, sogenannter Cofaktoren.
Diese können permanent und fest mit dem Apoenzym verbunden sein; man nennt sie
dann auch prosthetische Gruppe (wenn organisch).
Wenn sie eine reversible Bindung mit dem Enzym eingehen, heißen sie Cosubstrate
(wenn organisch), da sie zusammen mit dem Substrat an das aktive Zentrum
gebunden werden. Nach der katalytischen Reaktion werden sie mit den Produkten
wieder freigesetzt.
Bei Cofaktoren kann es sich um anorganische Moleküle, wie zum Beispiel die
Metallionen Zink, Eisen und Kupfer, handeln. Aber auch organische Moleküle können
Cofaktoren sein; die meisten davon sind Vitamine.
1.4. Reaktionsgeschwindigkeit
5
Enzymkinetik
Die Geschwindigkeit einer Katalysereaktion hängt von der Konzentration des
Substrats, des Produkts und des aktiven Enzyms ab.
Erhöht man bei konstanter Enzymkonzentration die Konzentration des Substrates,
steigt automatisch die Reaktionsgeschwindigkeit an, da die aktiven Zentren öfter mit
Substratmolekülen besetzt werden.
Die maximale Reaktionsgeschwindigkeit ist erreicht, wenn alle Enzyme mit Substrat
gesättigt
sind.
In
diesem
Zustand
bringt
eine
weitere
Erhöhung
der
Substratkonzentration keine Zunahme der Reaktionsgeschwindigkeit mehr mit sich.
Man spricht dabei von einer Sättigung.
Nur eine Erhöhung der Enzymkonzentration kann jetzt die Geschwindigkeit der
Reaktion noch beeinflussen.
Weitere Faktoren, von der die Katalysegeschwindigkeit abhängt, sind die Temperatur
und der pH-Wert.
1.5. Enzym-Substrat-Affinität
Eine enzymatische Reaktion erreicht ihre größtmögliche Reaktionsgeschwindigkeit,
wenn alle beteiligten Enzym mit Substratmolekülen gesättigt sind. Die Enzymkonzentration muss also einschränkend auf die Geschwindigkeit wirken, während die
Konzentration der Substratmoleküle größer als die der Enzymmoleküle sein muss.
Das Maß für die Affinität eines Enzyms zu seinem Substrat wird als MichaelisKonstante bezeichnet. Sie wird durch die Bestimmung des Substratumsatzes (in v) in
Abhängigkeit von der Substratkonzentration {S} ermittelt. Hat die Konstante einen
hohen Wert, ist die Halbsättigung erst bei einer relativ hohen Substratkonzentration
erreicht (die Hälfte des Enzyms ist gesättigt). Ein Enzym wird bevorzugt das Substrat
umsetzen, mit dem es die kleinste Michaelis-Konstante hat. Mit steigender Affinität
sinkt also die Substratkonzentration, die erforderlich ist, um alle Enzymmoleküle zu
sättigen.
1.6. Michaelis-Menten-Gleichung
Diese Gleichung drückt die Abhängigkeit der Reaktionsgeschwindigkeit v von der
Substratkonzentration {S} aus, wobei hier immer nur ein Substrat katalysiert wird.
6
Enzymkinetik
v = anfängliche Reaktionsgeschwindigkeit bei der Substratkonzentration {S}
vmax = Reaktionsgeschwindigkeit bei einem Überschuss an Substrat
Km = Michaelis-Menten-Konstante
{S} = Substratkonzentration
Abb. 4: die normale Sättigungskurve eines Enzyms nach Michaelis-Menten (durchgezogene Linie)
sowie deren Verlauf bei kompetitiver bzw. nichtkompetitiver Hemmung. (aus: Penzlin, Lehrbuch der
Tierphysiologie, 1989)
1.7. Lineweaver-Burk-Gleichung
Die Lineweaver-Burk-Gleichung ergibt sich aus der algebraischen Umformung der
Michaelis-Menten-Gleichung in die reziproke Form.
Sie wird verwendet, um die enzymatische Reaktion besser zu veranschaulichen, da
diese in einer Geradengleichung linear dargestellt wird.
In Abb. 5 wurde zu dieser besseren Darstellung der Umkehrwert der
Reaktionsgeschwindigkeit 1/v gegen den Umkehrwert der Substratkonzentration
1/{S} aufgetragen. Die Gerade (durchgezogene Linie) hat zwei Schnittpunkte: den
Schnittpunkt mit der x-Achse bei -1/Km und den Schnittpunkt mit der y-Achse bei
1/vmax.
Abb. 5: Lineweaver-Burk-Diagramm (aus: Penzlin, Lehrbuch der Tierphysiologie, 1989)
1.8. Enzymhemmung
Bestimmte chemische Stoffe können selektiv eine hemmende Wirkung auf spezielle
Enzyme ausüben.
7
Enzymkinetik
Dabei kann die Inaktivierung irreversibel sein, wenn sich der Inhibitor stabil an das
Enzym bindet (z.B. Schwermetalle wie Blei).
Bei einer reversiblen Hemmung bindet der Hemmstoff nur über schwache Wechselwirkungen an das Enzym ; dadurch wird das Maß an enzymatisch katalysierten
Reaktionen reguliert.
Viele Hemmstoffe ähneln chemisch den Substraten und konkurrieren mit diesen um
den Eintritt in das aktive Zentrum. Andere Inhibitoren wirken indirekt, da sie nicht an
das aktive Zentrum binden, verhindern aber ebenfalls die Substratbindung.
Ein Sonderfall ist hier die unkompetitive Hemmung, bei der der Inhibitor erst am
Enzym-Substrat Komplex angreift.
Abb. 6:
links: Ein Substrat bindet an das aktive Zentrum eines Enzyms
rechts: Ein kompetitiver Inhibitor konkurriert mit dem Substrat um das aktive Zentrum.
(aus: Campbell, Biologie, 1997)
Kompetitive Inhibitoren
Stoffe, welche die Aktivität von Enzymen herabsetzen, indem sie das Substrat an der
Besetzung des aktiven Zentrums hindern, bezeichnet man als kompetitive Inhibitoren
(Abb.6). Normalerweise ist die kompetitive Hemmung reversibel und kann durch eine
Erhöhung der Substratkonzentration überwunden werden, da auch hier das
Massenwirkungsgesetz gilt. Dieses besagt, dass durch die Erhöhung der
Substratkonzentration das Enzymmolekül viel häufiger mit einem Substrat als mit
einem Inhibitormolekül zusammentrifft. Das heißt, vmax ist ohne und mit Inhibitor
gleich groß, wie aus Abb. 4 ersichtlich ist.
(KM erhöht; Vmax konstant)
Nichtkompetitive Inhibitoren
Anders als oben verhält sich die nichtkompetitive Hemmung. Hier ist vmax bei
Anwesenheit des Hemmstoffs stets kleiner als im Normalfall (Abb.4). Der Inhibitor
konkurriert nicht mit dem Substrat um das Enzym, sondern blockiert die
Katalysereaktion von einer anderen Stelle des Enzymmoleküls, die nicht der
Substratbindung dient. Diese Wechselwirkung führt dazu, dass das Enzymmolekül
seine Gestalt ändert. Dadurch kann das aktive Zentrum so verändert werden, dass
8
Enzymkinetik
das Substrat zwar noch gebunden, aber weniger effektiv in das Produkt umgesetzt
werden kann. Die Substratbindung kann aber auch ganz unmöglich sein.
Diese Hemmung kann nicht durch eine Änderung der Substratkonzentration
beeinflusst werden. Erst die Beseitigung des Hemmstoffes oder eventuell eine
Verdünnung kann der Hemmung entgegenwirken.
(KM konstant; Vmax erniedrigt)
Abb. 7:
Ein nichtkompetitiver Inhibitor bindet sich an einer
vom aktiven Zentrum entfernten Stelle an das
Enzym. Dadurch ändert sich die Struktur des
Enzyms so stark, dass das aktive Zentrum nicht
länger voll funktionsfähig ist.
(aus: Campell, Biologie, 1997)
Im Lineweaver-Burk Diagramm von Abb. 5 wird bei der kompetitiven Hemmung die
Neigung der Geraden bei unverändertem y-Achsenschnittpunkt und bei der
nichtkompetitiven Hemmung bei unverändertem x-Achsenschnittpunkt steiler.
Allosterische Regulation
Die selektive Hemmung und Aktivierung von Enzymen durch in der Zelle natürlich
vorkommende Moleküle (Liganden) ist ein sehr wichtiger Mechanismus der
Stoffwechselkontrolle.
Meistens binden Liganden an einen spezifischen Bereich des Enzymmoleküls, der oft
weit vom aktiven Zentrum entfernten liegt; dieser Teil wird auch das allosterische
Zentrum genannt (Abb.8a). Die meisten Enzyme mit allosterischen Zentren sind
Proteine, die aus zwei oder mehr Polypeptidketten bzw. Untereinheiten bestehen.
Jede Untereinheit besitzt ihr eigenes aktives Zentrum. Die allosterischen Zentren
befinden sich dort, wo die Untereinheiten aneinander stoßen. Der gesamte Komplex
pendelt zwischen zwei Zuständen hin und her; einem katalytisch aktiven und einem
inaktiveren (Abb.8b). Die Bindung eines Aktivators an ein allosterisches Zentrum
stabilisiert die aktive Konformation, während die Bindung eines allosterischen
Inhibitors die inaktivere Form stabilisiert.
Da sich diese allosterischen Effektoren über schwache Bindungen an Enzyme
heften, ändert sich bei wechselnder Regulatorkonzentration auch die Enzymaktivität.
Selten gleichen sich Inhibitor und Aktivator allerdings so sehr in ihrer Gestalt, dass
sie um das selbe allosterische Zentrum konkurrieren.
9
Enzymkinetik
Abb. 8: Allosterische Regulation (aus: Campbell, Biologie, 1997)
a) R-Form von relaxed; T-Form von tense
b) heterotroper Effekt
Kooperativität
Bei der oben besprochenen allosterischen Wechselwirkung unterscheidet sich der
allosterische Effektor chemisch vom Substratmolekül. Man spricht daher auch vom
heterotropen Effekt (Abb.8b). Dagegen beeinflussen beim homotropen Effekt die
Substratmoleküle selbst die katalytische Wirksamkeit ihres Enzyms (Abb.9).
Bei einem Enzym mit zwei oder mehr Untereinheiten kann die Wechselwirkung mit
einem Substratmolekül in allen anderen Untereinheiten des Enzyms dieselbe
günstige Strukturänderung hervorrufen. Dieser Mechanismus, der als Kooperativität
bezeichnet wird, verstärkt die Substrataffinität eines Enzyms. Das erste gebundene
Substratmolekül erleichtert es dem Enzym, weitere Substrate zu binden.
In diesem Fall ergibt sich eine sigmoide v/s Kurve, das Enzym verhält sich nicht
entsprechend der Michaelis-Menten Beziehung.
Abb. 9: Kooperativität zwischen Substrat und Enzym
(aus: Campbell, Biologie, 1997)
1.9. Extinktion
Grundlage für die Berechnung der Absorption ist das Lambert-Beer´sche Gesetz:
∆E = є * ∆C * d
E = Extinktion (Maß der Absorption); hier NADH
є = Extinktionskoeffizient bzw. Stoffkonstante (von NADH 6,22*106 cm²/Mol)
C = Konzentration der absorbierenden Substanz
d = Lichtweg (hier 1cm)
1.10. Glykolyse, Milchsäuregärung
10
Enzymkinetik
Als Glykolyse (Zuckerzerlegung) wird ein Vorgang im Cytoplasma bezeichnet, bei
dem durch die Oxidation von Glucose zu Pyruvat Energie freigesetzt wird. Dabei wird
ein Molekül Glukose (C6) zu zwei Molekülen Pyruvat (C3) umgewandelt. Gleichzeitig
werden zwei Moleküle ATP und zwei Moleküle NADH + H+ synthetisiert. Wenn
Sauerstoff vorhanden ist, kann durch oxidative Phosphorilierung weiteres ATP
entstehen.
Es gibt aber auch Zellen (z.B. Muskelzellen), die trotz Sauerstoffmangel durch
Gärung ATP bilden können. Bei diesem Vorgang wird Pyruvat sofort von NADH + H+
zu Lactat (Anion der Milchsäure) reduziert (anaerobe Glykolyse).
Diesen Vorgang nennt man Milchsäuregärung. Die Milchsäuregärung setzt ein, wenn
bei hoher Belastung der Energiebedarf größer ist als die verfügbare
Sauerstoffmenge. Bei der Umsetzung von einem Molekül Glucose zu zwei Molekülen
Lactat werden nur zwei Moleküle ATP gebildet. Das sich ansammelnde Lactat,
welches Muskelerschöpfung und zum Teil Muskelkater herbeiführt, wird nach und
nach durch das Blut in die Leber transportiert und dort wieder zu Pyruvat umgesetzt.
2. Aufgabenstellung
Die Grundlage der folgenden Versuche bildet die Reaktion von Pyruvat zu Lactat:
Bei der Zellatmung entsteht durch die Glycolyse Pyruvat. Dieses wird bei der
Milchsäuregärung durch das Enzym Lactatdehydrogenase (LDH) Abb.10:
zu Lactat reduziert.
Dabei wird des Co-Substrat NADH/H+ zu NAD+ oxidiert.
Umsetzung des
Pyruvats zu Lactat
In den folgenden Versuchen soll nun die Reaktionsgeschwindigkeit in Abhängigkeit
von der Substratkonzentration und vom pH-Wert gemessen
(aus: Skript
Anfängerpraktikum
werden.
Hierzu macht
Enzymkinetik 2004)
man sich die Eigenschaften des Co-Substrats zu nütze: Wenn man das oxidierte
NAD+ mit einer Wellenlänge von 340nm bestrahlt, absorbiert es nicht. NADH/H+
hingegen absorbiert Licht dieser Wellenlänge, was in einem Absorptionsspektrum
sichtbar durch die Bildung eines Peaks wird. Diese Unterschiede beruhen auf der
Veränderung des Pyridin-Rings des NADH + H+ bzw. NAD+ bei Oxidation und
Reduktion.
11
Enzymkinetik
Mit
dem
Spektralphotometer
kann
man
die
Umsetzung
und
damit
die
Reaktionsgeschwindigkeit des Enzyms LDH messen, da bei der Reaktion, wie oben
beschrieben
NADH/H+
zu
NAD+
umgewandelt
wird
und
somit
die
Absorptionsabnahme des NADH/H+ proportional zur Bildung des Produktes Lactat
ist.
3. Material, Methoden und Versuchsdurchführung
3.1. Versuch 1:
Abhängigkeit der Reaktionsgeschwindigkeit von der
Substratkonzentration (Pyruvat)
Das Enzym Lactatdehydrogenase (LDH) wird aus einem Froschmuskel gewonnen.
Hierfür werden 310,7mg eines Froschmuskels mit 10ml eines 0,05molaren kalten
Kalium-Phosphatpuffers (pH=7) in einem Potter-Elvejhem-Homogenisator unter
Eiskühlung homogenisiert. Hierdurch
wird der Muskel zerkleinert und die
Zellmembranen zerstört.
Das dadurch gewonnene Extrakt wird in einer Beckman-Kühlzentrifuge bei 2°C und
10000 U/min 10 min lang zentrifugiert. Dabei werden die Bestandteile des Extraktes
getrennt. Die schwereren bilden das Pellet (z.B. Mitochondrien, Zellkern...),
wohingegen die leichten Bestandteile, u.a. auch LDH und weitere Elemente des
Cytoplasmas als Überstand bezeichnet werden.
Zu 1ml des Überstandes wird nun 1ml Phosphatpuffer hinzugefügt und anschließend
gekühlt.
Da die Konzentrationsgeschwindigkeit in Abhängigkeit von der Substratkonzentration
untersucht werden soll, werden Pyruvatlösungen verschiedener Konzentrationen
durch Verdünnung mit dem Puffer hergestellt. Hiefür wird zuerst eine 100mmol/l
12
Enzymkinetik
Stammlösung gewonnen, von der ausgehend die anderen Substratkonzentrationen
wie folgt (in Tabelle1) hergestellt werden.
Tabelle 1: Hergestellte Verdünnungen der Stammlösung
Verdünnungen,
hergestellt
die Menge der verwendeten Menge
werden Stammlösung in (µl)
müssen: (mmol/l)
des
verwendeten
Puffers (µl)
100
1000
---
50
500
500
40
400
600
30
300
700
20
200
800
10
100
900
8
80
920
6
60
940
5
50
950
4
40
960
3
30
970
2
20
980
Ablauf des Versuches:
13
Enzymkinetik
Um die Absorption in dem Spektralphotometer bei 340nm messen zu können,
werden als mehrere Küvetten mit jeweils 3ml Phosphatpuffer und 0,05ml LDHExtrakt gefüllt.
Um einen Fehler durch den Leerwert zu vermeiden, wird eine Küvette mit dieser
Mischung in das Photometer eingesetzt. Dieser Vorgang wird als Nullwertabgleich
bezeichnet.
Dann werden die Lösungen in den Küvetten noch mit 0,05ml NADH/H+ versetzt und
vermischt. Es wird eine Küvette in das Photometer eingesetzt, die Absorption ohne
Substrat wird gemessen und von dem daran angeschlossenen Schreiber
aufgezeichnet. Anschließend wird in die Küvette je 0,05ml einer bestimmten
Substratkonzentration pipettiert und erneut die Absorption gemessen.
Dieser Vorgang wird für alle Substratkonzentrationen zweimal durchgeführt
(Doppelbestimmung).
3.2. Versuch 2: Abhängigkeit der Reaktionsgeschwindigkeit vom pH-Wert
Um die Abhängigkeit der Reaktionsgeschwindigkeit vom pH-Wert zu messen, ändert
man den pH-Wert, wobei die Substratkonzentration immer gleich bleibt (30mmol/l).
Um 0,05M Pufferlösungen mit pH-Werte von 4 bis 9 zu bekommen, benötigt man
eine 0,05M Di-Kaliumhydrogenphosphat-Lösung (K2HPO4) (pH=9) und eine 0,05M
Kaliumdihydrogenphosphat-Lösung (KH2PO4) (pH=4,5). Diese beiden Lösungen
werden wie folgt (in Tabelle 2) vermischt um jeweils 200ml einer Lösung mit je
unterschiedlichem pH-Wert herzustellen:
Tabelle 2: Herstellung der Pufferlösungen
pH-Wert
KH2PO4 (in ml)
K2HPO4 (in ml)
Zusätze:
4
pur (200ml)
---
1 Tropfen HCl
5
pur (200ml)
---
1 Tropfen NaOH
6
175,4
24,6
---
7
312*
488*
---
8
10,6
189,4
---
9
---
pur (200ml)
---
*Bei pH=7 werden 800ml Lösung benötigt, da dieser Puffer für Versuch1 verwendet wurde.
14
Enzymkinetik
Der Versuch wird genauso durchgeführt wie der erste Versuch.
3.3. Funktionsprinzip eines Spektralphotometers:
Das Spektralphotometer misst wie viel Licht durch eine Lösung dringt bzw. von ihr
absorbiert wird.
Dazu wird Licht auf ein Prisma fallen gelassen, welches das Licht in unterschiedliche
Wellenlängen, also in monochromatisches Licht aufspaltet. Dieses wird dann durch
die Lösung geschickt. Der durchgelassene Anteil wird von einer Photozelle registriert,
die die Lichtenenergie in elektrischen Strom umwandelt, den im Anschluss ein
Galvanometer misst.
Das Galvanometer zeigt an, welcher Teil des Lichtes absorbiert (Extinktion) wurde
bzw. welcher Teil durch gelassen wurde (Transmission).
Der Schreiber des
Photometers zeichnet dann die Daten auf.
Durch die gemessene Absorption kann man nach Lambert-Beer die Konzentration
des absorbierenden Stoffes messen.
4. Ergebnisse
4.1. Versuch 1
4.1.1. Berechnung der tatsächlichen Pyruvatkonzentration in der Küvette
[SKüvette]
Die Substratkonzentration in der Küvette [SKüvette] wird bestimmt, in dem man die
Substratkonzentration [S] durch den Verdünnungsfaktor teilt.
Der Verdünnungsfaktor VFp wird folgendermaßen berechnet:
VFp =
0,05ml(Pyruvat ) + 0,05ml(LDH) + 0,05ml(NADH) + 3ml(Puffer )
0,05ml(Pyruvat )
VFp=63
Substratkonzentration in der Küvette: [S Küvette ] =
[S ]
63
Wenn nun zum Beispiel eine Substratkonzentration von [S]=100mmol/l gegeben ist,
beträgt die Konzentration des Substrates in der Küvette: [S Küvette ] =
100mmol / l
= 1,587
63
4.1.2. Berechnung der Extinktion ∆E
15
Enzymkinetik
Die Absorption, die der Schreiber des Spektralphotometers aufzeichnet, hat Werte
von 0 bis 100. Dies entspricht nach der Skalierung (Umrechnungsfaktor von 100)
einer Absorption von 0 bis 1. Man muss also um die Graphen auswerten zu können,
folgendes berechnen:
∆E Anfangswert Endwert 2
=
−
*
min
100
100 min
Wobei der Anfangswert, der Wert ist, den der Schreiber als erstes registriert hat.
Der Schreiber läuft nun 30 Sekunden lang, was einer Kästchenanzahl in x-Richtung
von 10 entspricht (dies entspricht 1Inch=2,54cm).
Der Endwert ist somit 10 Kästchen in x-Richtung vom Anfangswert entfernt.
Um die Extinktion pro Minute zu berechnen, muss der Term mit 2 multipliziert
werden.
Da wir für dieselben Substratkonzentrationen immer eine Doppelbestimmung
gemacht haben, bildet man aus beiden noch den Mittelwert.
Z.B. ist bei einer Substratkonzentration von 50mmol/l bei der ersten Bestimmung der
Anfangswert bei 80,3. Der Endwert beträgt 57,0.
80,3 57,0
∆E 1 =
−
* 2 = 0,466
100 100
81,0 51,2
∆E 2 =
−
* 2 = 0,596
100 100
∆E gesamt =
∆E1 + ∆E 2 0,466 + 0,596
=
= 0,531
2
2
4.1.3. Berechnung der Reaktionsgeschwindigkeit v
Die Reaktionsgeschwindigkeit v wird anhand des Lambert-Beerschen Gesetzes
berechnet. Dieses Gesetz besagt, dass die Extinktion ∆E der Konzentration ∆c des
absorbierenden Stoffes und der Schichtdicke d der Lösung proportional ist.
Der Absorptionskoeffizient ist ε, er ist abhängig von der Art der Substanz und der
Wellenlänge.
(1) ∆E = ε * ∆c * d ∆c =
(2) v =
∆E
ε*d
∆c
∆c = v * ∆t
∆t
16
Enzymkinetik
Die Geschwindigkeit der Absorptionszunahme ∆E ist zur Reaktionsgeschwindigkeit v
proportional, deshalb kann man Gleichung (1) und (2) gleichsetzten: v * ∆t =
(3) v =
∆E
ε*d
∆E
ε * d * ∆t
Bei unserem Versuch hatten wir eine Schichtdicke bzw. Lichtweg d der Küvette von
2
1cm, der Absorptionskoeffizient ε von NADH ist 6,22*106 cm Mol . Die Wellenlänge
beträgt 340nm und die Zeitspanne ∆t 1min.
Durch einsetzten dieser Werte in Gleichung (3) ergibt sich:
v=
∆E
6,22
v=
2
* 10 6 cm
mol
* 1cm * 1min
=
∆E * mol
∆E
mol
=
in
6
3
6
6,22 * 10 * cm * min 6,22 * 10
ml * min
∆E
µmol
in
6,22
ml * min
Aus den bei 3.1.1, 3.1.2 und 3.1.3 berechneten Werten ergibt sich die Tabelle 3:
Tabelle 3: Tabelle zur Reaktionsgeschwindigkeit
[S]
in [SKüvette]
in 1/[SKüvette] in ∆E/min
v in
µmol
ml * min
1/v
mmol/l
mmol/l
l/mmol
Mittelwert
100
1,587
0,63
0,57 *1
0,092
10,912
50
0,794
1,26
0,531
0,085
11,714
40
0,635
1,575
0,543
0,087
11,455
30
0,476
2,1
0,56
0,090
11,107
20
0,317
3,15
0,188
0,030
33,085
10
0,159
6,3
0,239
0,038
26, 025
8
0,127
7,5
0,153
0,025
40,654
6
0,095
10,5
0,067
0,011
92,836
5
0,079
12,6
0,03
0,005
207,333
4
0,063
15,75
0,031
0,005
200,654
3
0,048
21
0,012 *2
0,002
518,333
2
0,032
31,5
0,022 *2
0,003
282,727
in
ml*min/µmol
1
* : nur einer der beiden Werte aus der Doppelbestimmung war repräsentativ
2
* : es ergab sich hier kaum eine Absorptionsänderung Werte werden im Diagramm 1 und 2 nicht
17
Enzymkinetik
berücksichtigt
4.1.4. Diagramm nach Michaelis-Menten
Diagramm 1: Michaelis-Menten-Diagramm
Legende: blau= eigene Messwerte
schwarz= an eigene Messwerte angenäherte Kurve
rot= vmax
türkis= 1/2vmax bzw. Km-Wert
Der Kurvenverlauf
zeigt,
dass
bei zunehmender Substratkonzentration
die
Reaktionsgeschwindigkeit ansteigt, bis die maximale Reaktionsgeschwindigkeit, die
bei Sättigung des Enzyms mit Substrat eintritt, erreicht wird. Dies hat zur Folge, dass
trotz zunehmender Substratkonzentration die Maximale Reaktionsgeschwindigkeit
konstant bleibt. In Diagramm 1 wird dies durch ein Plateau dargestellt. Für vmax ergab
sich aus unserem Diagramm der Wert: vmax=0,09.
Bei Halbmaximaler Geschwindigkeit (1/2vmax) ist nach Definition die MichaelisMenten-Konstante Km erreicht. Bei uns hat Km den Wert: Km=0,173.
Mit diesen beiden Werten wurde die angenäherte Kurve berechnet:
f (S ) = 0,09 − 0,09 * e −4*S
4.1.5. Diagramm nach Lineweaver-Burk
18
Enzymkinetik
Diagramm 2: Lineweaver-Burk-Diagramm
Legende: rot= eigene Messwerte
schwarz= angenäherte Gerade
Unsere Messwerte ergaben ungefähr eine Gerade, so dass man gut den Messwerten
eine
Gerade
anpassen
konnte,
die
auf
folgender
Gleichung
basiert:
1
1
f = 11 * + 12,5
[S]
[S]
Der Schnittpunkt der Geraden mit der x-Achse ergibt den Wert:
−1
= −1,136 Km
Km=0,880. Der y-Achsenabschnitt gibt die reziproke maximale Geschwindigkeit an:
1
= 12,5 vmax= 0,08
v max
Aus dem Michaelis-Menten-Diagramm und dem Lineweaver-Burk-Diagramm
ergibt sich ein Mittelwert für: v max =
Km =
0,09 + 0,08
= 0,085 , sowie ein Mittelwert für
2
0.173 + 0,880
= 0,527 .
2
19
Enzymkinetik
4.1.6. Berechnung der Enzymaktivität
Zur Berechnung der Enzymaktivität AE der Lactatdehydrogenase im Froschmuskel
wird zur Vereinfachung angenommen, dass die Dichte des Gewebes 1 beträgt und
dass das Gewicht des Froschmuskels (FG) vor und nach der Zentrifugation gleich
groß ist.
AE =
v max
µmol
in
Muske lg ewicht / ml
mg * min
Vmax wird aus dem Mittelwert der in den Diagrammen 1 und 2 erhaltenen
Maximalgeschwindigkeit ermittelt.
Um die in der Küvette vorhandene Gesamtverdünnung des Froschmuskels zu
berechnen, wird das Gewicht, das der Froschmuskel (FG) bei der Einwaage hatte
dividiert durch: [(das Gewicht des Froschmuskels in ml umgerechnet addiert mit dem
Volumen der Pufferlösung (10ml)) * (dem Verdünnungsfaktor 63) * 2]
Muske lg ewicht / ml =
=
AE =
Einwaage(mg)
=
(Einwaage (ml) + 10) * 2 * 63
310,7(mg)
mg
= 0,239
(0,3107(ml) + 10) * 126
ml
0,085
µmol
= 0.356
0,239
mg * min
4.2. Versuch 2
Berechnung der Extinktion ∆E
Die Berechnung der Substratkonzentration wird nicht benötigt, da jeder Versuch bei
einer Substratkonzentration von 30µmol/l durchgeführt wird, also [SKüvette]=0,467 und
1/[SKüvette]=2,1 ist.
Die Berechnung der Extinktion folgt ebenfalls wie oben. Allerdings wurde die
Messung mehrere Male versehentlich vorzeitig gestoppt. Somit wurden noch weitere
Rechnungen notwendig:
Da der x-Wert nicht mehr 10 Kästchen entsprach, wurde er abgemessen und
entspricht xgem. xgem verhält sich zu der gesuchten Zeit t wie 2,54cm (=1Inch=
10Kästchen) zu Gesamtlaufzeit des Schreibers 30 sek.
20
Enzymkinetik
x gem
t
=
2,54cm
30s
t=
x gem * 30s
2,54cm
Um nun die Extinktion ∆E zu bekommen, berechnet man den Anfangswert/100 minus
den Endwert/100, dies wird multipliziert mit dem Verhältnis der oben berechneten
Zeit t zu der Gesamtzeit 60s.
∆E =
60 Anfangswert Endwert
−
t
100
100
Bei dem pH-Wert von 6 wurde z.B. der Schreiber nur 2,2cm lang laufen gelassen.
Der Anfangswert ist 35, der Endwert 25.
t=
2,2cm * 30s
= 25,984
2,54cm
∆E =
60 35
25
−
= 0,231
25,984 100 100
Die Reaktionsgeschwindigkeit v wird wie unter 4.1.3. berechnet.
Daraus ergeben sich die in Tabelle 4 aufgelisteten Werte.
Tabelle 4: Tabelle zur Berechnung der Extinktion
pH-Wert
∆E/min (Mittelwert)
v in µmol/(ml*min)
4
0,073
0,012
5
0,123
0,020
6
0,203
0,033
7
0,56
0,090
8
0,144 *1
0,023
9
*2
*2
1
* es wurde kein Mittelwert aus der Doppelbestimmung gebildet, da einer der beiden Werte nicht
repräsentativ war
2
* Leider waren beide Messwerte nicht brauchbar.
21
Enzymkinetik
Bei niedrigen pH-Werten war die Reaktionsgeschwindigkeit sehr niedrig. Mit
zunehmendem pH-Wert ist sie gestiegen, bis sie ihr Optimum bei einem pH-Wert von
7 erreicht hat. Danach nahm die Reaktionsgeschwindigkeit bei steigendem pH-Wert
ab.
5.Diskussion
5.1. Versuch 1:
Bereits bei der ersten Messung der Substratkonzentration von 100mmol/l kam eine
unerwartet
hohe
Extinktionsänderung
(∆E=1,01)
heraus,
was
eine
Maximalgeschwindigkeit vmax=0,162 zur Folge hatte. Dieser Fehler könnte durch
ungenaues Pipettieren entstanden sein: wahrscheinlich wurde eine zu große Menge
an
Enzym
hinzugefügt.
Deshalb
wurde
auch
für
die
Ermittlung
der
Reaktionsgeschwindigkeit in der Tabelle 3 und den Diagrammen 1 und 2 nur der
zweite Messwert bei der Substratkonzentration von 100mmol/l berücksichtigt.
Die ermittelte Reaktionsgeschwindigkeit bei Substratkonzentrationen von 50mmol/l
und 40mmol/l entsprachen unseren Erwartungen.
Bei
einer
Substratkonzentration
von
30mmol/l
war
die
berechnete
Reaktionsgeschwindigkeit vmax etwas über dem zu erwartenden Wert, was wiederum
22
Enzymkinetik
auf eine etwas zu hohe Menge am Enzym LDH schließen lässt, was durch einen
Pipettierfehler entstanden sein könnte.
Die Reaktionsgeschwindigkeit vmax war bei der Substratkonzentration von 20mmol/l
zu niedrig. Dieser Fehler kann zurückzuführen sein auf eine Verschmutzung der
Küvette, z.B. durch Fingerabdrücke, was zur Folge hat, dass die durch die Küvette
geschickten Wellen schon durch den Schmutz leicht absorbiert wurden und nicht von
NADH/H+. Außerdem wäre es möglich, dass wir zu langsam gearbeitet haben, so
dass die Reaktion schon eine Weile ablief, bevor wir den Schreiber gestartet haben.
Eine andere Erklärung für die zu geringe Reaktionsgeschwindigkeit wäre, dass
entweder zu wenig Enzym in die Küvette pipettiert wurde oder das Enzym am Rand
der Küvette hängen blieb, da nicht richtig umgerührt wurde. Als weitere Fehlerquelle
kann eine zu geringe Substratmenge oder eine zu niedrige Konzentration des
Substrates angesehen werden.
Die für die Reaktionsgeschwindigkeit ermittelten Werte bei Substratkonzentrationen
von 10mmol/l, 8mmol/l und 6mmol/l liegen in einem zu erwartenden Bereich.
Bei einer Substratkonzentration von 5mmol/l bis zu 2mmol/l wurde nur noch eine
sehr
geringe
Extinktionsänderung
gemessen,
was
bewirkt,
dass
die
Reaktionsgeschwindigkeit sehr niedrig ist. Dies kann durch die oben bereits
genannten Fehler auftreten, also Verschmutzung der Küvette, Pipettierfehler bei der
Substratmenge bzw. –konzentration, sowie zu langsame Arbeitsweise. Am
wahrscheinlichsten ist aber ein Fehler bei der Pipettierung des Enzyms, da es hierbei
zur Bildung einer Luftblase in der Pipette gekommen ist und somit eine zu geringe
Menge an Enzym zugesetzt worden ist. Die Werte für 2mmol/l und 3mmol/l wurden
deshalb in den Diagrammen 1 und 2 nicht berücksichtigt.
Trotz einiger Fehler sind unsere Kurven im Großen und Ganzen wie erwartet
verlaufen.
Die
Graphen
des
Schreiberprotokolls
sind
mit
sinkender
Pyruvatkonzentration immer flacher verlaufen, was dadurch zu erklären ist, dass das
LDH immer weniger Substrat zur Verfügung hatte und somit weniger häufig ein
Enzym-Substart-Komplex gebildet wurde. Dies hat zur Folge, dass weniger NADH/H+
umgesetzt wurde und somit die Absorptionsänderung geringer ist.
23
Enzymkinetik
5.2. Versuch 2:
Dieser Versuch musste wiederholt werden, weil die Absorption sich nicht geändert
hatte, was passierte, da kein NADH/H+ hinzugefügt wurde. Als wir dann NADH/H+
hinzufügten, lief die Reaktion mit doppelter Geschwindigkeit ab, da wir zu Beginn
versehentlich die doppelte Menge an Enzym hinzugefügt hatten. Da wir für pH=8 nur
eine der beiden Lösungen in den Küvetten zur Doppelbestimmung erneuert hatten,
spiegelt sich dieser Fehler bei der ersten pH=8 Bestimmung wider, weshalb nur der
andere Wert berücksichtigt wurde.
Bei sehr niedrigem pH-Wert ist für das Enzym LDH zu erwarten, dass die
Reaktionsgeschwindigkeit sehr gering ist, da es durch zu viele Protonen im
Umgebungsmilieu deformiert wird und somit die Aktivität eingeschränkt ist. Mit
zunehmendem pH-Wert steigt dann die Reaktionsgeschwindigkeit, bis sie ihr
Optimum bei einem pH-Wert von ca. 7 erreicht hat. Danach sinkt die
Reaktionsgeschwindigkeit mit zunehmendem pH-Wert wieder.
Die Ergebnisse bei der zweiten Durchführung des Versuches für die pH-Werte
4,5,6,7 und 8 entsprechen den gerade beschriebenen Erwartungen.
Bei dem pH-Wert 9 ist uns bei beiden Messungen aus versehen ein Fehler
unterlaufen, deshalb wird der pH-Wert von 9 im Diagramm 3 nicht berücksichtigt. Bei
der ersten Messung wäre eine Reaktionsgeschwindigkeit von v=0,081 µmol/(ml*min)
herausgekommen. Diese Reaktionsgeschwindigkeit ist ungefähr so hoch, wie die bei
einem pH-Wert von 7, wo das Enzym sein optimales Umgebungsmilieu hat, in dem
es am Leistungsfähigsten ist. Allerdings sollte dieser Wert doch deutlich unter dem
Optimalen liegen, da mit zunehmendem pH-Wert nach dem Hochpunkt die
Reaktionsgeschwindigkeit stetig sinkt. Der wahrscheinlichste Grund hierfür ist, dass
wie auch bei pH=9 die doppelte Enzymmenge hinzu gegeben wurde. Bei der zweiten
Messung war die Absorptionsänderung ∆E negativ. Dies bedeutet, dass die
Absorption zugenommen hat, anstatt abzunehmen. Dies hat zur Folge, dass auch
die Maximalgeschwindigkeit negativ wäre. Dem liegen wahrscheinlich zwei Fehler
zugrunde. Als erstes kann kein Enzym hinzugefügt worden sein, da ansonsten eine,
wenn auch geringe Absorptionsabnahme stattgefunden hätte. Wenn das NADH
tatsächlich an der Wand hing, kann das Enzym auch nichts damit anfangen, ihr müsst es also nicht
unbedingt vergessen haben!
Zweitens muss das in die Küvette pipettierte NADH/H+ an
den Wänden der Küvette hängen geblieben und im Verlauf des Schreibens in die
Küvette getropft sein. Da nur hierdurch eine Absorptionszunahme möglich ist.
24
Enzymkinetik
Wahrscheinlich habt ihr einfach nur nicht richtig gemischt oder es waren Luftblasen in der Küvette, auf
diese Weisekommt es auch zu Schwankungen der Absorption.
6. Literaturverzeichnis
1.
2.
3.
4.
5.
ECKERT, R.: Tierphysiologie. 4. Auflage, Thieme Verlag
CAMPBELL, Neil A.: Biologie. 2. Auflage, Spektrum Akademischer Verlag
PENZLIN, H.: Lehrbuch der Tierphysiologie, 4.Auflage, Gustav Fischer Verlag
Skript zum Praktikum Enzymkinetik WS 3004/05
Biologie heute, Schroedel Verlag
7. Abbildungsverzeichnis
Abb. 1: Campbell, Neil A.: Biologie, 2. Auflage, Spektrum Akademischer Verlag
Seite 108, 6.12
Abb. 2/3: Campbell, Neil A.: Biologie, 2. Auflage, Spektrum Verlag
Seite 109, 6.13
Abb. 4/5: Penzlin, H.: Lehrbuch der Tierphysiologie, 4.Auflage, Gustav Fischer
Verlag, Seite 27, 1.3
Abb. 6/7: Campbell, Neil A.: Biologie, 2. Auflage, Spektrum Verlag
Seite110, 6.15
Abb. 8/9: Campbell, Neil A.: Biologie, 2. Auflage, Spektrum Verlag
Seite 111, 6.16
Abb 10: Skript zum Praktikum Enzymkinetik, WS 2004/05
8. Anhang
Ausdruck zur Enzymkinetik
25