Buch 4.indb - AkadMed.com
Werbung

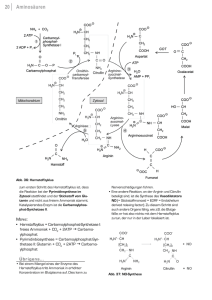
23.7 Harnstoffzyklusstörungen E. Mönch 1. Einleitung Zur Ammoniakentgiftung durch den Harnstoffzyklus sind insgesamt 6 cytosomal bzw. intramitochondrial gelegene Enzyme notwendig. Von allen diesen Enzymen sind angeborene Defekte mit vielen Mutationen bekannt, die zwar in der Mehrzahl schon in der Neonatalzeit zu klinischen Symptomen führen, jedoch in Varianten auch das Kindes- und Erwachachsenalter (late onset) betreffen können. Mit Ausnahme des Arginase-Mangels (Hyperargininämie) sind alle Harnstoffzyklusdefekte gekennzeichnet durch lebensbedrohliche Ammoniakvermehrungen im Blut – auch die late onset Formen! Bei den Harnstoffzyklusstörungen handelt es sich um folgende Enzymdefekte: " – N-Acetylglutamatsynthetase-Mangel (NAGS) (OMIM 237310) # – Carbamylphosphatsynthetase I-Mangel (CPS I) (OMIM 237300) C – Ornithintranscarbamylase-Mangel (OTC) (OMIM 311250) D – Citrullinämie (CIT I) (OMIM 238970) E – Argininbernsteinsäure-Krankheit (Argininosuccinaturie) (OMIM 207900) F – Hyperargininämie (OMIM 207800) Die Inzidenz der einzelnen Harnstoffzyklusdefekte ist sehr unterschiedlich, am häufigsten ist der OTC-Mangel. Insgesamt treten sie mit einer geschätzten Häufigkeit von 1: 8.000 auf. Die Harnstoffzyklusdefekte folgen einem autosomal rezessiven Erbgang, bis auf den OTC-Mangel, der X-chromosomal vererbt wird. 2. Mechanismus des Harnstoffzyklus Der mit der Nahrung aufgenommene Stickstoff wird zu einem Großteil als Harnstoff mit dem Urin ausgeschieden. Das aus den Aminogruppen der Aminosäuren stammende Ammoniak wird mit CO2 verbunden und das entstehende Carbamylphosphat (enthält nur ein Stickstoffmolekül) in den Mitochondrien der Leberzellen an Ornithin gekoppelt, wobei Citrullin entsteht. Bei der Verbindung von Citrullin mit Asparaginsäure im Cytoplasma entsteht Argininbernsteinsäure, die dann in Fumarsäure (Fumarat) und Arginin gespalten wird. Nach Abtrennung des – 2 Stickstoffmoleküle enthaltenden – Harnstoffs entsteht wieder Ornithin, das, nach Transport zurück in die Mitochondrien, dem Zyklus erneut zur Verfügung steht. Über die Ursache der schweren Enzephalopathie bei Hyperammonämie sind nur einige Details bekannt. So findet im Gehirn eine Ammoniakentgiftung durch Bildung von Glutamat und Glutamin statt, was einerseits zur Verarmung von D-Ketoglutarat sowie anderen Citratzyklusmetaboliten (Energiemangel) und andererseits zur massiven intrazellulären Anreicherung an Glutamin führt. Die Entwicklung eines Hirnödems ist die Folge. Nachgewiesen sind auch Störungen der Synthese von Neurotransmittern und deren Rezeptoren. 3. Klinik der Harnstoffzyklusdefekte Die klinische Symptomatik der Harnstoffzyklusstörungen (mit Ausnahme der Hyperargininämie) basiert auf der Wirkung des Ammoniaks. Die Hyperammonämien treten häufig schon am ersten bis dritten Lebenstag auf. Die late onset Formen können bei Stoffwechselentgleisung ebenso dramatisch verlaufen, wie die schweren (klassischen) Formen bei Erstmanifestationen im Neugeborenenalter. Ammoniak sollte aus venösem Blutplasma mit einer enzymatischen Methode gemessen werden. Im Neugeborenenalter sind Werte unter 827 Buch 4.indb 827 15.10.2009 14:20:02 Kap. 23.7 E. Mönch Abb. 1. Schema der Harnstoffsynthese 110 μmol/l (187 μg/dl), im späteren Alter unter 80 μmol/l (136 μg/dl) normal. Ammoniakkonzentration über 150 μmol/l (255 μg/dl) bei Neugeborenen oder über 100 μmol/l (170 μg/dl) bei Kindern entsprechen einer Hyperammonämie. Hinweise auf das Vorliegen einer Hyperammonämie geben die Erhöhungen der Konzentrationen von Glutamin, Glutamat, Alanin und Asparagin im Blut. 3.1. N-Acetylglutamatsynthetase-Mangel Mehrheitlich wird diese Störung des intramitochondrial gelegenen Enzyms zwischen dem ersten bis sechsten Lebenstag in Abhängigkeit von der Eiweißaufnahme klinisch manifest (neonatale Form). Die Symptome sind Nahrungsverweigerung, Somnolenz, Lethargie, Koma, Ataxie, Tachypnoe und Krampfanfälle. Das Produkt der Enzymreaktion, N-Acetylglutamat, ist Coenzym der Carbamylphosphatsynthetase-I, dem ersten Enzym in der Harnstoffsynthese. Wird die initiale Hyperammonämie erfolgreich behandelt, besteht ständig die Gefahr weiterer hyperammonämischer Krisen. Dauern diese mehrere Tage, resultieren nicht nur permanente Ernährungsschwierigkeiten, sondern infolge der eingetretenen Hirnschäden ausgeprägte neurologischer Symptomatiken. Eine mildere Variante ist gekennzeichnet durch neurologische Symptome, wie Ataxien, Verzögerung der statomotorischen Entwicklung, Sehstörungen und auffälliger Aversionen gegen proteinreiche Nahrung. Bei Verdacht auf das Vorliegen eines N-Acetylglutamatsynthetase-Mangels sollte die Hyperammonämie wie unten beschrieben energisch therapiert, zusätzlich die Blockierung der Carbamylphosphatsynthetase-I mangels N-Acetylglutamat durch die orale Substitution eines zellwandpermeablen alternativen Coenzyms, Carbamylglutamat (Carbaglu®, Swedish ORPHAN) in der initialen Dosierung von 100 – 250 (570) mg/kg KG/Tag, beseitigt werden. 3.2. Carbamylphosphatsynthetase IMangel Ist das Enzym des ersten Schrittes der Harnstoffsynthese, die mitochondriale Carbamylphosphatsynthetase I (CPS I), defekt, treten am ersten bis dritten Lebenstag (in Abhängigkeit von der Aufnahme von Eiweiß) Erbrechen, Lethargie, Koma und Krämpfe, seltener Hyperven- 828 Buch 4.indb 828 15.10.2009 14:20:04 Harnstoffzyklusstörungen tilation, Hirnödem, Hypotonie und Hepatomegalie auf. In dieser Phase ist der Carbamylphosphatsynthetase-Mangel vom N-Acetylglutamatsynthetase-Mangel weder klinisch, noch durch Blut- und/oder Urinanalysen zu unterscheiden. Die Gabe von Carbamyglutamat kann bei der differentialdiagnostischen Klärung hilfreich sein (Ammoniakkonzentration fällt bei Vorliegen eines N-Accteylglutamatsynthetase-Mangels nach Gabe von Carbamylglutamat in der Regel ab). Letztendlich muss eine Enzymdiagnostik aus Lebergewebe einen der beiden infrage kommenden Defekte ausschließen. Gelingt es, die neonatale hyperammonämische Phase erfolgreich zu überwinden (Therapie siehe unten), folgen meist Episoden mit Erbrechen und Ernährungsproblemen. Je nach Schwere der durchgemachten Hyperammonämie und der damit verbundenen Hirnschädigung resultieren außerdem statomotorische Entwicklungsrückstände, Hirnatrophie, Ataxie und weitere neurologische Symptome. Die seltenen milden Varianten manifestieren sich in Form von Entwicklungsrückständen, Hirninfarkten, können aber durch ebenso schwere Hyperammonämien wie die neonatele Form auffallen. 3.3. Ornithintranscarbamylase-Mangel Der einzige X-chromosomal vererbte Defekt des Harnstoffzyklus, der Ornithintranscarbamylase-Mangel (OTC-Mangel, mitochondrial), manifestiert sich bei den betroffenen Jungen meist schon bis zum dritten Lebenstag (abhängig von der Eiweißzufuhr) mit Erbrechen, Lethargie, Koma und Krampfanfällen, seltener Hyperventilation, Hypotonie und Hepatomegalie. Von diagnostischer Wichtigkeit beim OTC-Mangel, der unter den Harnstoffzyklusdefekten die höchste Inzidenz hat, ist die Messung der Ausscheidung von Orotsäure mit dem Urin (oft über 1000 μmol/ mol Kreatinin; normal 1–11 μmol/mol Kreatinin). Außerdem ist die Blutkonzentration von Citrullin sehr niedrig (unter 15 μmol/l). Wird die erste hyperammonämische Krise überlebt (Behandlung siehe unten; eventuell Gabe von L-Citrullin anstatt L-Arginin), treten häufiges Erbrechen und andere Ernährungsschwierigkeiten, nicht selten auch Ataxien und muskuläre Hypotonie auf. Bei den selteneren milderen Varianten fallen die Jungen in der Regel durch Verzögerungen der geistigen Entwicklung auf. Aber auch heterozygote Mädchen können klinische Symptome entwickeln (mindestens 10 % der Betroffenen), die von Kopfschmerzen, Ataxien und Lethargie bis zu schweren Hyperammonämien mit Hirnschädigung sogar mit Todesfolge reichen, die z. B. während fieberhafter Erkrankungen (Hyperammonämie bei kataboler Stoffwechsellage) oder nach eiweißreicher Kost auftreten. Mittels eines Allopurinol-Belastungstests und/oder molekulargenetischen Untersuchungen lassen sich die Mehrzahl der Überträgerinnen des OTC-Defektes erfassen. 3.4. Citrullinämie I Die schwere, neonatale Form des Argininbernsteinsäuresynthetase-Mangels (Citrullinämie) ist seltener, als die milderen oder sogar ohne klinische Symptome einhergehenden Varianten. Die neonatale Form dieses cytosomalen Defektes ist klinisch identisch mit dem des CPS- oder OTC-Mangels. Die Hyperammonämie sollte wie unten beschrieben behandelt werden. Wie beim OTC-Mangel, ist auch bei der Citrullinämie die Ausscheidung von Orotsäure mit dem Urin vermehrt, jedoch in geringerem Ausmaß. Bei einem Großteil der im Neugeborenenscreening mittels Tandem-Massenspektrometrie gefundenen Citrullinämien handelt es sich um milde Varianten, die ev. nie klinische Symptome entwickeln werden, oder durch Erbrechen, Durchfall, Lethargie, cerebrale Krämpfe, gelegentlich Echolalie bzw. Dyslalie, Halluzinationen und manische Episoden nach eiweißreicher Mahlzeit auffallen. Zur differentialdiagnosti829 Buch 4.indb 829 15.10.2009 14:20:05 Kap. 23.7 E. Mönch schen Klärung werden heutzutage vorwiegen molekulargenetische Untersuchungen herangezogen. 3.5. Argininbernsteinsäure-Krankheit Bei der Argininbernsteinsäure-Krankeit (Argininosuccinaturie; Mangel der cytoplasmatischen Argininbernsteinsäurelyase) lassen sich 3 klinische Verläufe unterscheiden: r r r neonatale oder maligne Form infantile Form chronische Form Nach einem symptomfreiem Intervall von einigen Tagen treten bei der OFPOBUBMFO 'PSN Trinkschwäche, Apathie, muskuläre Hypotonie und Krampfanfälle auf, die durch die sich gebildete Hyperammonämie zu erklären sind. Die Behandlung (siehe unten) entspricht der der bisher genannten Harnstoffzyklusdefekte. Die JOGBOUJMF'PSN beginnt im Säuglingsalter mit Erbrechen, Tremor, Koma und Krampfanfällen. Zusätzlich sind Hepatomegalie und trockene, schuppige Haut festzustellen. Ohne Behandlung bilden sich körperliche und geistige Retardierung aus. Entwicklungsverzögerung nach dem ersten Lebensjahr ist meist das erste Symptom der DISPOJTDIFO'PSN der Erkrankung. Später kann man nach eiweißreichen Mahlzeiten und anderen Situationen mit Stoffwechselstress (fieberhafte Erkrankungen) Kopfschmerzen, Lethargie, Ataxien und Krampfanfälle beobachten. Hepatomegalie mit Leberfibrose und sprödes, struppiges Haar (trichorrhexis nodosa) sind weitere Symptome. Bei der Argininbernsteinsäure-Krankheit empfiehlt sich eine Substitution von Natriumcitrat (oral, bis zu 650 mg/kg KG/Tag) zur Aufrechterhaltung des Harnstoffzyklus, wenn weniger als 0,3 mol Citrat/mol Kreatinin mit dem Urin ausgeschieden wird. 3.6. Hyperargininämie Ein völlig anderes klinisches Bild als bei den übrigen angeborenen Defekten des Harnstoffzyklus zeigt sich bei dem Arginase-Mangel (Hyperargininämie), der relativ leicht zu verifizieren ist, da das Enzym in Erythrozyten expremiert ist. Hyperammonämien sind bei dieser Erkrankung selten. Der Verlauf ist chronisch, obwohl auch episodenhafte Zustände mit Erbrechen, Hirnödem, stato- und psychomotorischen Entwicklungsrückständen, Mikrozephalie, Chorea und Athetose, spastischer Tetraplegie, Krampfanfällen sowie Hyperaktivität oder Schläfrigkeit nach der Nahrungsaufnahme beobachtet wurden. Die Therapie außerhalb der akuten Zustände besteht in einer reduzierten Eiweißzufuhr. 4. Screening auf Harnstoffzyklusstörungen Das Neugeborenenscreening auf angeborene Aminosäurenstoffwechselstörungen wird heutzutage in der Regel mittels Tandem-Massenspektrometrie durchgeführt. Keine der Harnstoffzyklusdefekte stehen aber auf der Liste der in Deutschland zu screenenden angeborenen Stoffwechselstörungen. Mit den Routineanalysen lassen sich aber durchaus Konzentrationsveränderungen von Citrullin, Glutamin und Alanin feststellen. Prinzipiell ist auch die Messung von Argininbernsteinsäure mittels Tandem-MS möglich. Bei dem klinischen Verdacht auf das Vorliegen eines Harnstoffzyklusdefektes sollte man die kompletten Ergebnisse der Aminosäurenmessungen im Rahmen des Neugeborenenscreenings im Screeninglabor erfragen! 5. Differentialdiagnosen der Harnstoffzyklusstörungen Bei jeder Hyperammonämie, deren Ursache zunächst unbekannt ist, ist unabhängig vom Alter und Geschlecht an einen Harnstoffzyklusdefekt 830 Buch 4.indb 830 15.10.2009 14:20:05 Harnstoffzyklusstörungen zu denken. In dieser Situation sollten die freien Aminosäuren im Blut bestimmt werden, mit dem besonderen Augenmerk auf Glutamat und Glutamin sowie auf Alanin und Citrullin. Quantitative Analysen von Argininbernsteinsäure und Orotsäure (Orotat) im Urin sind notwendig. Andere angeborene Stoffwechseldefekte oder Störungen der Leberfunktion können zu Hyperammonämien führen, z. B.: legentlich eine ausgeprägte Aversion gegen eiweißreiche Nahrungsmittel. r r 7.1. Behandlung einer akuten Hyperammonämie r r r r r r r r r r alpha-1-Antitrypsin-Mangel (OMIM 107410) Galactosämie (Galactose-1-Phosphat-Uridyltransferase-Mangel) (OMIM 230400) Glutamatdehydrogenase Defekt mit Hyperammonämie und Hyperinsulinismus (mit Hypoglykämien) (OMIM 138130) Hepatitis HHH-Syndrom (Hyperammonämie-Hyperornithinämie-Homocitrullinämie) (OMIM 238970) Leberbypass Lysinurische Proteinintoleranz (OMIM 222700) Mitochondropathien Organoacidämien (z. B. Propionacidurie (OMIM 232000), Methylmalonacidurie (OMIM 251000)) Pyrrolin-5c-Carboxylatsynthetase Mangel (OMIM 138250) Synthesestörungen der Gallensäuren Tyrosinose Typ I (OMIM 276700) 7. Therapie der Hyperammonämie Im Vordergrund bei der Therapie der Harnstoffzyklusdefekte steht im Allgemeinen die Beseitigung einer Hyperammonämie bzw. deren Vermeidung. Die Beseitigung einer Hyperammonämie muss unverzüglich und mit allen zur Verfügung stehenden Mitteln in der Regie von stoffwechselversierten Ärzten unter intensivmedizinischen Bedingungen betrieben werden: r r 6. Harnstoffzyklusstörungen assoziierte Veränderungen Überleben die Betroffenen mit den akuten Formen der Harnstoffzyklusstörungen die schweren hyperammonämischen Krisen, bleiben als klinische Symptome häufig Ernährungsschwierigkeiten mit Erbrechen. Aber auch Hypotonie, Ataxie, Kopfschmerzen, Lethargie und psychische Auffälligkeiten sind nicht selten (bes. nach eiweißreicher Nahrung). Patienten mit milderen Formen von Harnstoffzyklusdefekten oder auch Überträgerinnen des OTC-Mangels haben ge- r Reduktion/Stopp der Proteinzufuhr (für maximal 2 Tage) Hochkalorische Ernährung (> 100 kcal/kg KG Tag): Beginn mit mindestens 10 g/kg KG Glucose zusammen mit Elektrolyten für 24 Stunden. Die Glucosemenge kann bis auf 20 – 30 g/kg KG erhöht werden. Falls notwendig Zugabe von Insulin (0,01– 0,5 I. E./kg KG/Stunde), um den Glucoseblutspiegel zwischen 80 und 200 mg/dl zu halten. Das Ziel der hohen Kaloriengabe ist die Vermeidung von Eiweißkatabolismus. Zusätzlich kann Fett infundiert werden (am Anfang 0,5 –1 g/ kg KG/Tag und wenn möglich Steigerung auf 2 – 3 g/kg KG/Tag; unter Kontrolle der Triglyceridkonzentrationen im Blut) Verabreichung von Medikamenten (oral oder i. v.): – Natriumbenzoat 250 mg/kg KG in 10 %iger Glucoselösung, über 2 Stunden – Natriumphenylbutyrat (Ammonaps®, Swedish ORPHAN) 250 mg/kg KG in 10 %iger Glucoselösung, über 1– 2 Stunden 831 Buch 4.indb 831 15.10.2009 14:20:05 Kap. 23.7 E. Mönch – r r Argininhydrochlorid 21%: (1 ml = 1 mmol) 1– 2 mmol/kg KG in 10 %iger Glukoselösung, über 2 Stunden Forcierte Diurese mit Furosemid (Lasix®) (1– 2 mg oral oder 0,5 –1 mg/kg KG i. v., alle 6 –12 Stunden). Hämodiafiltration, ersatzweise Hämodialyse bei Ammoniakspiegeln über 400 μmol/l (680 μg/dl) Erwachsenenalter sinnvoll. Bei Kindern ist die Dosierung so zu wählen, dass weiche, aber nicht wässrige Stühle und keine Bauchschmerzen auftreten. Bei akuter Entgleisung besteht in Extremfällen die Behandlungsmöglichkeit mittels intraportaler Infusion von blutgruppenidentischen gesunden Hepatozyten, mit deren Hilfe eine schnellere Stoffwechselnormalisierung ermöglicht wird. 7.2. Langzeitbehandlung Ziel der Langzeitbehandlung der Harnstoffzyklusdefekte ist die Vermeidung von Hyperammonämien, hervorgerufen durch zu hohe Eiweißzufuhr oder katabole Stoffwechselzustände. Eine Kombination von Reduzierung der Zufuhr von natürlichem Eiweiß, in der Regel mit Substitution von essentiellen Aminosäuren, und der Verabreichung von Medikamenten (Natriumbenzoat, Natriumphenylbutyrat, Argininhydrochlorid) ist empfohlen. Die Eiweißzufuhr soll bis auf den minimalen Bedarf gesenkt werden (Verzicht auf Fleisch, Fisch, Milch, Eier und Getreideprodukte). Bei der Erstbehandlung sollte nach einer etwa 2tägigen Eiweißkarenz mit der Gabe sehr geringer Mengen an natürlichem Protein (0,3 g/kg KG/Tag) begonnen werden, und nachfolgender langsamer Steigerung bis auf etwa 1 g/kg KG und Tag). Meist muss zur Deckung des Tagesbedarfs an Protein zusätzlich ein Gemisch mit essentiellen Aminosäuren verabreicht werden. Eventuell wird außerdem die Gabe von Citrullin (zusammen mit oder anstatt Arginin) notwendig. Für die Eiweißzufuhr insgesamt (natürliches Eiweiß plus Aminosäurengemisch) wird empfohlen: g/kg KG/Tag Säuglinge Kleinkinder Schulkinder Jugendliche/Erwachsene 1,8 – 2,0 1,2 –1,5 1,0 < 0,5 Zur Vermeidung von übermäßigen Ammoniakproduktionen durch untypische Darmbakterien ist eine Gabe von Lactulose (3 u 4 – 20 g/Tag) im Treten gehäuft Stoffwechselkrisen auf und gestaltet sich die Behandlung insgesamt sehr schwierig, ist auch eine Lebertransplantation als Therapieoption in Erwägung zu ziehen. Literaturverzeichnis Mönch E, Link R (2006) Diagnostik und Therapie bei angeborenen Stoffwechselstörungen. SPS Publ., Heilbronn Brusilow SW, Horwich AL (2001) Urea Cycle Enzymes. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Vogelstein B, Childs B, Kinzler KW (online eds) The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, New York, S 1909 –1963 Bachmann C (2003) Inherited Hyperammonemias. In: Blau N, Duran M, Blaskovics ME, Gibson KM (eds) Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases. Springer, Berlin, Heidelberg, New York, pp 261 ff Leonard JV (2006) Disorders of the urea cycle and related enzymes. In: Fernandes J, Saudubray J-M, Van den Berghe G (eds) Inborn Metabolic Diseases, Springer, Berlin, Heidelberg, New York, pp 263 ff Müller E (2003) Harnstoffzyklusstörungen. In: Müller E (Hsg) Praktische Diätetik in der Pädiatrie. Grundlagen für die Ernährungstherapie. SPS Verlagsgesellschaft, Heilbronn, S 89 – 94 Berry GT, Steiner RD (2001) Long-term management of patients with urea cycle disorders. J Pediatr 138: 56 – 61 Bachmann C (2003) Long-term outcome of patients with urea cycle disorders and the question of neonatal screening. Eur J Pediatr 162 (Suppl 1): 29 – 33 Hauser ER, Finkelstein JE, Valle D, Brusilow SW (1990) Allopurinol induced orotidinuria. A test for mutations at the ornithine carbamoyltransferase locus in women. N Engl J Med 322: 1641–1645 Grünewald S, Fairbanks L, Genet S, Cranston T, Hüsing J, Leonard JV, Champion MP (2004) How reliable is the 832 Buch 4.indb 832 15.10.2009 14:20:06 Harnstoffzyklusstörungen allopurinol load in detecting carriers for ornithine transcarbamylase deficiency? J Inher Metab Dis 27: 179 –186 Horslen SP, McCowan TC, Goertzen TC, Warkentin PI, Cai HB, Strom SC, Fox IJ (2003) Isolated hepatocyte transplantation in an infant with a severe urea cycle disorder. Pediatrics 111: 1262 –1267 McBridge KL, Miller G, Carter S, Goss J, Lee B (2003) Development outcome in early liver transplantation for urea cycle disorders. J Inher Metab Dis 26 (Suppl. 2): 77 Bachmann C, Häberle J, Leonard J (eds) (2007) Pathophysiology and Management of Hyperammonaemia, Symposia Proceedings, SPS Verlagsgesellschaft, Heilbronn 833 Buch 4.indb 833 15.10.2009 14:20:06