Versuch 5: Enzyme (Alkalische Phosphatase)
Werbung
")
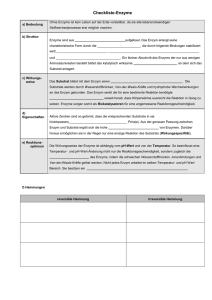
Versuch 5: Enzyme (Alkalische Phosphatase) (V5 20.10.08) Lernziele: 1) Reaktionsgeschwindigkeit, Aktivierungsenergie, chem. Gleichgewicht; 2) Was tun Katalysatoren und Enzyme, 3) Michaelis-Menten Gleichung, Lineweaver-Burk-Diagramm, 4) kompetitive Hemmung, allosterische Regulation, pH-Optimum. Hintergrund: Enzyme sind biologische Katalysatoren. Als solche beschleunigen sie chemische Reaktionen – oftmals um ein Zigtausendfaches, sodaß man Enzyme in solchen Fällen auch als Ein/Aus-Schalter einer Reaktion betrachten kann. Die enzymatische Aktivität wird durch viele Faktoren reguliert, z.B. die chemisch-physikalische Umgebung (pHWert, Temperatur, Ionen, etc.), die Verfügbarkeit von Substrat, die Anwesenheit von bereits gebildetem Produkt, oft auch durch spezifische regulatorische Moleküle, und nicht zuletzt durch die Konzentration des Enzyms selbst. Die Geschwindigkeit der katalysierten Reaktion wird dadurch kontinuierlich regelbar, und das Enzym funktioniert dann wie der Lautstärkeregler an Ihrer Stereoanlage. Damit ein Molekül eine chemische Reaktion eingehen kann, muss es eine bestimmte innere Mindestenergie annehmen, die sog. „Aktivierungsenergie“. Eine Substanz ist umso stabiler, je größer die Differenz ihrer inneren Energie und der Aktivierungsenergie ist (deshalb lagern wir empfindliche Lebensmittel im Kühlschrank). Die chemische Reaktion kann also beschleunigt werden, indem man der Verbindung Energie zuführt, z.B. durch Erhitzen. Dadurch erhöht sich die Chance eines individuellen Moleküls, die Aktivierungsenergie zu erreichen und damit die Reaktion einzugehen. Auch Enzyme beschleunigen die Reaktion, indem sie die Differenz zwischen innerer Energie des Substrats und der Aktivierungsenergie verringern. Die jeweiligen molekularen Mechanismen sind außerordentlich vielfältig und einfallsreich. Manchmal wird durch die Bindung an ein Enzymmolekül ein Teil der inneren Energie des Enzyms auf das Substrat übertragen. In anderen Fällen stabilisiert das Enzym einen Übergangszustand bzw. ein Zwischenprodukt der Reaktion, was formal einer Herabsetzung der Aktivierungsenergie gleichkommt. In Ihrem ersten Experiment werden Sie eine enzymatisch katalysierte Reaktion durchführen und mehrmals, bei immer höheren Substrat-Konzentrationen, wiederholen. Dabei werden Sie feststellen, dass eine Erhöhung des Substratangebots zunächst annähernd proportional zu einer gesteigerten Geschwindigkeit in der Bildung des Reaktionsproduktes führt. Mit zunehmender Substratkonzentration wird dieser Effekt immer bescheidener, bis schließlich eine abermalige Substraterhöhung keine weitere Beschleunigung mehr bewirkt. Der Grund dafür ist, dass Substratmoleküle nur dann reagieren können, wenn sie an ein Enzymmolekül gebunden haben. Da die Anzahl der verfügbaren Enzymmoleküle in allen Ihren Ansätzen die gleiche war, tritt mit steigender Substratkonzentration eine zunehmende Absättigung der Enzymmoleküle ein, sodass für weiteres Substrat immer weniger freie Enzymmoleküle zur Verfügung stehen, bis letztlich alle vorhandenen Enzymmoleküle zu jedem Zeitpunkt ein Substratmolekül gebunden haben. Die Reaktion läuft dann mit maximal möglicher Geschwindigkeit. Michaelis-Menten Theorie: Wenn Sie in einem Diagramm die gemessenen Geschwindigkeiten (v) gegen die jeweiligen Substratkonzentrationen (S) auftragen, erhalten Sie eine nach rechts hin immer flacher verlaufende Kurve, die sich asymptotisch der maximal möglichen Reaktionsgeschwindigkeit (Vmax) annähert. Sie erinnern sich, dass Vmax dann erreicht wird, wenn alle Enzymmoleküle jederzeit Substrat gebunden haben. Demzufolge haben bei halbmaximaler Geschwindigkeit nur 50% aller Enzymmoleküle Substrat gebunden. Wenn Sie in Ihrer Kurve diesen Punkt suchen, können Sie die dafür erforderliche Substratkonzentration auf der X-Achse ablesen. Hätten Sie nun Gelegenheit, mehrere Enzyme zu untersuchen, dann könnten Sie vielleicht feststellen, dass diese Kurve mal steiler, mal flacher ausfallen. Entsprechend würde die Substratkonzentration, die zum Erreichen der halb-maximalen Geschwindigkeit erforderlich ist, mal geringer, mal höher sein. Dieses Verhalten reflektiert die Festigkeit („Affinität“), mit der Enzym und Substrat aneinander binden. Die Substratkonzentration, die zum Erreichen der halb-maximalen Geschwindigkeit, also zur Besetzung von 50% aller vorhandenen Enzymmoleküle erforderlich ist, wird damit zu einer wichtigen Kennzahl eines Enzym-Substrat-Paares. Diese Kennzahl heißt die Michaelis-Menten Konstante („Km“). Maud Menten hat in ihrer Dissertation bei Leonor Michaelis 1913 ein mathematisches Modell für dieses Verhalten formuliert. Versuchen Sie doch einmal, die zugrunde liegenden Überlegungen dieser Theorie nachzuvollziehen. Sie sind in jedem BasisLehrbuch der Biochemie besser und ausführlicher beschrieben, als es in diesem Rahmen hier möglich wäre. Sie werden dann sehen, warum die Kurve, die Sie in Ihren Experimenten erhalten haben eine Parabel der Form v = Vmax * S / (Km+S) ergeben musste. Lineweaver-Burk und andere Linearisierungsverfahren: Wenn Sie nun aus denselben Messwerten die Kehrwerte (1/v) bilden und gegen die Kehrwerte der dazugehörigen Substratkonzentrationen (1/S) auftragen, dann geschieht etwas Sonderbares: anstelle einer gekrümmten Kurve erhalten Sie eine Gerade, die links der Y-Achse die X-Achse schneidet. Dieser Schnittpunkt repräsentiert den negativen Kehrwert von Km. Der Schnittpunkt der Gerade mit der Y-Achse bildet den Kehrwert der Maximalgeschwindigkeit ab (warum?). Wenn Sie die obige Michelis-Menten Gleichung nach 1/v auflösen, erhalten Sie die entsprechende Geradengleichung. Dieses sogenannte Lineweaver-Burk Diagramm hat einige Vorteile, z.B. den, dass eine Gerade bereits durch zwei Punkte definiert ist, und nicht erst unzählige Wiederholungen des Experiments durchgeführt werden müssen, um Vmax zu bestimmen. Außerdem ist aus der asymptotischen Annäherung der Kurve Vmax ohnehin nur näherungsweise zu bestimmen. Der Nachteil der Lineweaver-Burk Darstellung ist, dass kleine (und deshalb ungenaue) Messwerte zu großen Kehrwerten führen und deshalb relativ kleine Fehler zu großen Ungenauigkeiten führen. Später entwickelte Linearisierungsverfahren umgehen dieses Problem. Aber damit wollen wir Sie bei unseren Erstkontakt mit Enzymen nicht belasten. Links das Michaelis-Menten-Diagramm, rechts die Lineweaver-Burk-Auftragung derselben Daten. Kompetitive Inhibitoren: Zu den vielen Verbindungen, die eine Enzymreaktion beeinflussen können zählen auch solche, die eine gewisse strukturelle Ähnlichkeit mit dem Substrat aufweisen. Solche Verbindungen können zwar an das katalytische Zentrum des Enzyms binden, werden aber dort keine (oder eine andere) chemische Reaktion eingehen. Ihre Besetzung des katalytischen Zentrums verhindert natürlich die gleichzeitige Bindung eines Substratmoleküls. Allerdings kann sie durch einen entsprechenden Überschuss an Substrat wieder aus der Bindungsstelle werden. Da beide Verbindungen um die Bindungsstelle am Enzym im Wettstreit liegen, nennt man solche Substanzen „kompetitive Inhibitoren“. Die Stärke des hemmenden Effekts hängt ab von der Konzentration (I) und (verständlicherweise) der Affinität des Inhibitors (Ki) zum Enzym. Einen kompetitiven Inhibitor kann man an einer scheinbaren Erhöhung des Km-Wertes erkennen (warum?). Die erreichbare Maximalgeschwindigkeit bleibt jedoch auch in Anwesenheit eines kompetitiven Inhibitors gleich (warum?). Mathematisch ergibt sich diese Erhöhung aus dem Term Km’ = Km * (1 + I/Ki). Warum das so ist würde hier zu weit führen, aber Sie brauchen diese Beziehung in Ihrem nächsten Experiment, um die Affinität des Inhibitors zu berechnen. Sie werden in Ihren Experimenten Gelegenheit haben, die Funktionsweise von alkalischer Phosphatase (AP) zu untersuchen. AP katalysiert die Hydrolyse von Phosphorsäureestern, also die Abspaltung von anorganischem Phosphat aus einer Vielzahl von organischen Verbindungen, z.B. phosphorylierten Aminosäure-Seitenketten in Proteinen (welchen?), den 5’-Enden von Nukleinsäurefragmenten, ATP, etc., ja sogar von Verbindungen, die in der Natur gar nicht vorkommen. Das dabei anfallende Phosphat, das eben noch Teil des Substratmoleküls war, hat selbst eine gewisse Affinität zum katalytischen Zentrum des Enzyms und wirkt damit als kompetitiver Inhibitor. Wie Sie diese messen können, entnehmen Sie der Arbeitsanleitung in Experiment A weiter unten. Wenn Sie Ihre Messreihe in einem Puffer wiederholen, der Phosphationen enthält, dann verändert sich das Aussehen der Michaelis-Menten Kurve und die Gerade im Lineweaver-Burk Diagramm (wie werden diese im Vergleich zur ungehemmten Reaktion aussehen?) Allosterische Regulation: Alkalische Phosphatase hat zwei Bindungsstellen für Zn++ und eine für Mg++ Ionen. An das Polypeptid gebunden erhöhen diese Ionen die enzymatische Aktivität ganz entscheidend. Sie sind damit gute Beispiele für allosterische Aktivatoren. Zweiwertige Kationen werden durch geeignete Chelatoren (woher kommt dieser Name?) stark gebunden. Die Zugabe von Ethylendiamin-Tetraessigsäure (EDTA) zum Reaktionsansatz wird dazu führen, dass ein Großteil der Ionen vom Enzym abgezogen und an EDTA gebunden wird. In Ihrem Experiment C können Sie beobachten, welchen Einfluss das auf die Reaktionsgeschwindigkeit hat, und dass sich dieser Effekt durch Zugabe von ZnCl2 im Überschuss wieder kompensieren lässt. Es gibt natürlich auch allosterische Inhibitoren. Da diese, wie der Begriff „allosterisch“ nahe legt, außerhalb des katalytischen Zentrums binden, können sie nicht durch erhöhte Substratkonzentration aus dem Enzym verdrängt werden. Deshalb ändert sich bei allosterischen Hemmungen auch der Km-Wert nicht. Weil aber solche Inhibitoren generell die Aktivität des Enzyms beeinträchtigen, wirkt das so als wäre weniger Enzym vorhanden. Dadurch verringert sich die maximal mögliche Reaktionsgeschwindigkeit (wie würden Michaelis-Menten und Lineweaver-Burk Diagramme dann aussehen?). Bevor es los geht noch ein paar Details zum experimentellen System, mit dem Sie arbeiten werden: „Alkalische Phosphatase“ wird so genannt, weil ihr Aktivitätsoptimum im alkalischen pH-Bereich liegt. Als Testsubstrat verwenden wir p-Nitrophenylphosphat. Die Abspaltung des Phosphatrests führt zur Bildung eines p-Nitrophenolat-Anions, das eine charakteristische gelbe Farbe zeigt, anhand derer wir die Konzentration des Reaktionsprodukts photometrisch messen können. Ihre Experimente: Experiment A: Aufgaben: 1) Bestimmung des Km-Wertes von alkalischer Phosphatase. 2) Bestimmung der Wechselzahl 3) Bestimmung von Km’ in Anwesenheit von 0.5mM Phosphat als kompetitivem Inhibitor. 4) Berechnung der Inhibitor-Affinität (Ki). Durchführung: A.1. Bestimmung der Michaelis-Kontante Km und der Wechselzahl von AP 1) Pipettieren Sie alle angegebenen Lösungen, mit Ausnahme des Enzyms (!), in der angegebenen Reihenfolge in saubere, trockene Küvetten. Küvetten # 1 2 3 4 5 Glycin-Puffer, pH 9.6 (µl) 800 800 800 800 800 0.01 M pNPP (µl) 10 20 40 80 160 H2O (µl) 150 140 120 80 - Enzymlösung (µl) 40 40 40 40 40 2) Stellen Sie die Wellenlänge des Photometers auf 405nm. 3) Stellen Sie die Küvette #1 in das Photometer und gleichen Sie es durch Drücken der "Set Ref" Taste auf Null ab. 4) Nehmen Sie die Küvette aus dem Photometer, geben Sie die Enzymlösung zu und verschließen Sie die Küvette mit einem Stück Parafilm. Mischen Sie den Ansatz und starten Sie dabei gleichzeitig die Stoppuhr. 5) Stellen Sie die Küvette in das Photometer zurück und notieren Sie die Absorption bei t1=20sec und bei t2=80sec. 6) Wiederholen Sie dieselbe Vorgangsweise mit den anderen Küvetten. Das gebildete gelbe p-Nitrophenolat ist ein Produkt der enzymatischen Katalyse. Aus der Absorption E bei 405 nm kann man die Konzentration dieses Produktes bestimmen. Man muß dazu nur die Schichtdicke d der Küvette (1 cm) und den molaren Extinktionskoeffizienten ε von p-Nitrophenolat (ε = 18000 L*mol-1*cm-1) kennen, dann kann man mit Hilfe des Lambert-Beer-Gesetzes (E = ε * c * d) die Konzentration c berechnen. Lassen Sie sich durch diese Gleichungen und Koeffizienten nicht einschüchtern. Eigentlich handelt es sich dabei nur um eine Schlussrechnung, und ε gibt nur an, wie hoch die Absorption einer 1M Lösung wäre. Die wenig anschauliche Dimension von ε ergibt sich dabei aus dem Umstand, dass die Absorption eine dimensionslose Größe ist und die Konzentration mol/L (mol * L-1) im Nenner steht, die Schichtdicke d ebenso. Formen Sie also die obige Gleichung nach c um und setzen Sie die gemessene Absorption für E ein, dann erhalten Sie die Konzentration des Reaktionsproduktes. Die Reaktionsgeschwindigkeit wird ausgedrückt als Konzentrationsänderung pro Zeiteinheit, z.B. µmol * L-1 * min-1 oder, was das gleiche ist, µM/min. Ergebnisse der Messungen: Küvetten # 1 2 3 4 5 E (405 nm) t=20sec E (405 nm) t=80sec ∆E/min v (µM/min) 1/v (min/µM) [pNPP] (µM) 1/[pNPP] (L/µmol) E = Absorption (=Extinktion), ∆E = Änderung der Absorption, v = Geschwindigkeit, [pNPP] = Ausgangskonzentration vom Substrat p-Nitrophenylphosphat (zu berechnen aus Konzentration und Volumen der Stammlösung und Volumen des gesamten Ansatzes). Zeichnen Sie nun aus Ihren Ergebnissen ein Michaelis-Menten und ein Lineweaver-Burk Diagramm und ermitteln Sie aus beiden Darstellungen Km und Vmax (welche Diagrammform eignet sich dazu besser?) Vermeiden Sie bei der Erstellung Ihrer Diagramme Zahlen mit allzuvielen Nullen vor oder hinter dem Komma. Dafür gibt es die entsprechenden Vorsätze m (10-3), µ (10-6), n (10-9) etc., bzw. Zusätze wie „x10-3“, etc. Als absoluter Wert ist Vmax wenig aussagekräftig und eignet sich nur für den Vergleich verschiedener Proben untereinander. Wenn man aber die Konzentration der reinen Enzymmoleküle im Ansatz kennt, dann kann man über Vmax berechnen, wie viele Moleküle Substrat ein Molekül Enzym pro Sekunde umsetzen kann. Dieser Wert heißt die „Wechselzahl“ und ist ein weiteres wichtiges Charakteristikum eines Enzym/Substrat-Paares. Vmax ergibt sich dann aus der Wechselzahl und der Konzentration des Enzyms im Ansatz. Alkalische Phosphatase hat ein Molekulargewicht von 69kDa. Nehmen Sie an, die Enzym-Stammlösung, die Sie erhalten haben, hätte eine Konzentration von 33µg/mL, und berechnen Sie damit aus der von Ihnen ermittelten Vmax die Wechselzahl für AP. A.2 Bestimmung von Km' und Vmax in Gegenwart von anorganischem Phosphat und Berechnung der Inhibitorkonstante Ki für Phosphat Der Versuch wird genauso durchgeführt wie A.1., nur enthält diesmal der Glycinpuffer 0.5 mM Phosphat als kompetitiven Inhibitor. Pipettierschema: Küvetten # 1 2 3 4 5 Glycin-Puffer, pH 9.6 mit 0.5 mM 800 800 800 800 800 0.01 M pNPP (µl) 10 20 40 80 160 H2O (µl) 150 140 120 80 - Enzymlösung (µl) 40 40 40 40 40 Phosphat (µl) Ergebnisse der Messungen: Küvetten # 1 2 3 4 5 E (405 nm) t=20sec E (405 nm) t=80sec ∆E/min v (µM/min) 1/v (min/µM) [pNPP] (µM) 1/[pNPP] (L/µmol) Zeichnen Sie Ihre Messwerte in die Diagramme von Experiment A1 ein und bestimmen Sie auch für den neuen Versuch Km und Vmax. Welchen Einfluss hat anorganisches Phosphat auf die Reaktionskinetik im Michaelis-Menten Diagramm, bzw. in der Umformung nach Lineweaver-Burk? Wie würde das mit einem allosterischen Inhibitor aussehen? Ergebnisse: Versuch # 1 Phosphatkonzentration (in der Küvette) (mM) 0 2 Km (µM) Vmax (µM/min) Die Inhibitorkonstante Ki beschreibt die Affinität eines Inhibitors zum Enzym. Ki lässt sich berechnen nach der Gleichung: Km' = Km · ( 1 + I/Ki). [I] ist dabei die Inhibitorkonzentration. Formen Sie diese Gleichung so um, dass Sie I isolieren können. Ergebnis: Ki = ………….. Experiment B: Aufgabe: Bestimmung des pH-Optimums von AP Der pH-Bereich, in dem alkalische Phosphatase ihr maximale Aktivität zeigt wird gemessen, indem man Puffer verschiedener pH-Werte verwendet. Dazu wird unter Substratsättigung des Enzyms die maximale Reaktionsgeschwindigkeit (Vmax) bestimmt. Durchführung: Der Testansatz enthält: 800 µl 0.1 M Glycin-NaOH Puffer (6 Ansätze mit jeweils unterschiedlichem pH-Wert) 80 µl p-Nitrophenylphosphat (0.01 mol/l) 80 µl dest. Wasser 40 µl Enzym Es stehen Glycinpuffer mit sechs verschiedenen pH-Werten zur Verfügung. Durchführung der Messung wie in Experiment A1 beschrieben. pH-Wert E (405nm) 20 s E (405nm) 80 s ∆E/min In einem Diagramm werden die Reaktionsgeschwindigkeiten (als ∆E/min, y-Achse) gegen die pH-Werte (x-Achse) aufgetragen (warum muss in diesem Fall die Geschwindigkeit nicht in µM/min umgerechnet werden? Muss man das für die Bestimmung von Km aus dem Michaelis-Menten oder dem Lineweaver-Burk Digramm?) Ergebnis: pH-Optimum = ………. Experiment C: Die allosterische Regulation der alkalischen Phosphatase wird untersucht, indem zunächst durch EDTA Zink entzogen und dann durch Zugabe von weiterem Zink der EDTA-Effekt wieder aufgehoben und überkompensiert wird. Alkalische Phosphatase benötigt zweiwertige Kationen (Zn, Mg), um aktiv zu sein. EDTA entzieht dem Enzym diese Ionen, was den Verlust eines allosterischen Aktivators bedeutet. Durch Zugabe von Zink kann der EDTA-Effekt wieder aufgehoben werden. Aufgabe: Inaktivierung der AP durch EDTA und Reaktivierung durch Zink. Durchführung: Der Standardansatz enthält: 800 µl 0.1M Glycin-NaOH Puffer, pH 9.6 80 µl 0.01 mol/l p-Nitrophenylphosphat 80 µl dest. Wasser 40 µl Enzym Pipettieren Sie die Lösungen in der angegebenen Reihenfolge in eine Küvette. Nach Zugabe des Enzyms wird sofort gemischt und bei 405 nm im Photometer abgeglichen. Sobald die Geräteanzeige 0.000 erscheint, lesen Sie 3 Minuten lang alle 30 s die Extinktion ab und notieren Sie die Werte. Mit einem zweiten Ansatz wird genauso verfahren, nur dass vor Zugabe des Enzyms 10 µl EDTA-Lösung zugegeben werden. Nach 3 min geben Sie demselben Ansatz 50 µl ZnCl2-Lösung zu. Mischen Sie den Ansatz schnell und lesen Sie die Extinktion für weitere 3 min alle 30 sec ab. Zur Auswertung erstellen Sie ein Diagramm, in dem Sie die Absorptionswerte beider Ansätze gegen die Zeit auftragen. Material-Liste: Geräte: Kolbenhub-Pipetten, einstellbar für Maximal-Volumina von 1000, 200 und 20µL. Reagenzgläser mit Haltern, Photometer (405 nm) Küvetten Stoppuhr Lösungen: Glycin-NaOH Puffer, pH 9.6 (0.1 mol/l) (MW=75,07 g/mol) p-Nitrophenylphosphat (0.01 mol/l) (Na-Salz, MW= 371 g/mol) Alkalische Phosphatase, 1000 U/ml, 10 mg/ml, mit 1x CIAP-Puffer 1:300 verdünnt Glycin-NaOH Puffer, pH 9.6 (0.1 mol/l) mit 0.5 mmol/l Dinatriumhydrogen-Phosphat) Glycin-NaOH Puffer, pH 8.0 (0.1 mol/l) Glycin-NaOH Puffer, pH 8.8 (0.1 mol/l) Glycin-NaOH Puffer, pH 9.2 (0.1 mol/l) Glycin-NaOH Puffer, pH 9.6 (0.1 mol/l) Glycin-NaOH Puffer, pH 10.0 (0.1 mol/l) Glycin-NaOH Puffer, pH 10.4 (0.1 mol/l) EDTA (Ethylendiamintetraacetat) (0.1 mol/l) ZnCl2-Lösung (0.1 mol/l)