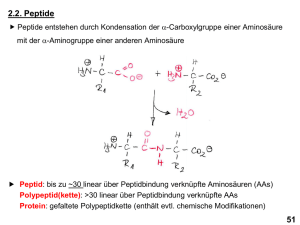
In vitro Testmethoden zur Überprüfung der Aktivität natürlicher und
Werbung

Aus der Abteilung für Plastische Chirurgie und Schwerbrandverletzte, Handchirurgie-Zentrum, im Berufsgenossenschaftlichen Universitätsklinikum Bergmannsheil der Ruhr – Universität Bochum Direktor: Prof. Dr. med. H. U. Steinau In vitro Testmethoden zur Überprüfung der Aktivität natürlicher und synthetischer Host Defense Peptide gegen Bakterien und Pilze mit Vergleich zu klinisch eingesetzten Antibiotika und Antimykotika Inaugural-Dissertation zur Erlangung des Doktorgrades der Medizin einer Hohen Medizinischen Fakultät der Ruhr – Universität Bochum vorgelegt von Gottlieb Pazdzierny aus Kedzierzyn-Kozle 2010 Dekan: Prof. Dr. med. G. Muhr Referent: Prof. Dr. med. L. Steinsträßer Korreferent: Prof. Dr. med. S. Gatermann Tag der mündlichen Prüfung: 05.04.2011 Inhaltsverzeichnis: Seite 1. Einleitung 2 1.1. Antibiotika 2 1.1.1. Eine Welt ohne Antibiotika 2 1.1.2. Die Entdeckung der Antibiotika 3 1.1.3. Resistenzentwicklung innerhalb der Antibiotika 5 1.1.4. Lösungsstrategien zur Minimierung der Resistenzentwicklung 11 1.2. 11 Das Angeborene Immunsystem 1.2.1. Host Defense Peptide 13 1.2.2. Cathelicidine 17 1.3. Aufgaben und Ziele der Studie 20 2. Material und Methoden 22 2.1. Radial Diffusion Assay 22 2.2. Modified CLSI Microbroth Dilution Assay 27 2.3. CLSI Microbroth Dilution Assay für Pilze 30 3. Ergebnisse 33 3.1. Radial Diffusion Assay 33 3.1.1 Grampositive Bakterien 33 3.1.2. Gramnegative Bakterien 34 3.1.3. Pilz 35 3.2. 36 Modified CLSI Microbroth Dilution Assay 3.2.1. Grampositive Bakterien 37 3.2.2. Gramnegative Bakterien 39 3.2.3. Vergleich mit Standardantibiotika 41 3.3. Modified CLSI Microbroth Dilution Assay für Pilze 44 4. Diskussion 45 5. Zusammenfassung 53 6. Literaturverzeichnis 54 7. Danksagung 72 8. Lebenslauf 73 1 1. Einleitung 1.1. Antibiotika 1.1.1. Eine Welt ohne Antibiotika „Although the Soldiery retreated from the Field of Death and encamped out of the City, the Contagion followed, and vanish´d them; many in their Old Age, and others in their Prime, sunk under its cruelties; of the Female Sex most died; and hardly any children escaped; and it was not uncommon to see an Inheritance pass successively to three or four Heirs in as many days; the Number of Sextons were not sufficient to bury the Dead.” - Nathanial Hodges; Loimologia: An account of the 1665 London Plague - Die Bedrohung der Menschheit durch tödliche Infektionskrankheiten, die sich mittels Übertragung zu verheerenden Pandemien ausweiten, ist so alt wie die Menschheit selbst. Dazu zählen nicht nur die oben angesprochene Pest, sondern auch Cholera, Influenza, Typhus, Tuberkulose und viele andere, deren Verbreitung letztlich dafür sorgte, dass die Lebenserwartung der Bevölkerung in früheren Jahrhunderten deutlich eingeschränkt blieb. Aus anderen Erkrankungen, wie Ohr-, Haut- oder Halsentzündungen resultierten oft Taubheit, Entstellung und/oder Tod infolge von Sepsis und anderen Komplikationen [116]. Bis ins 19. Jahrhundert hinein, betrug die durchschnittliche Lebenserwartung in Europa und Nordamerika ca. 50 Jahre und war bestimmt durch – wenn vorhersagbar – den steten Verlust von Familienmitgliedern, Freunden, Gatten und Kollegen. Die Angewohnheit sich nach der Gesundheit des Gegenübers zu erkundigen stammt aus dieser Zeit und war eine Freundlichkeit mit Hintergrund. Sie basierte auf der immerwährenden Bedrohung durch einen plötzlichen Tod, sei es durch eine Seuche, einen Unfall oder eine zufällige Infektion, verursacht durch einen der zahlreichen umweltbezogenen Erreger. Es war eine Welt, in der die Wahrscheinlichkeit durch eine infektiöse Seuche vorzeitig zu versterben bei 40% lag und in der Frauen regelmäßig an den Folgen von perinatalen Infektionen 2 verstarben, deren Heilung in heutigen Standards nahezu selbstverständlich ist [116]. 1.1.2. Die Entdeckung der Antibiotika Zu Beginn des 20. Jahrhunderts änderte sich die Situation mit der Entdeckung neuer Mittel im Kampf gegen die Infektionen des Alltags. 1928 beobachtete der britische Arzt Sir Alexander Flemming, wie ein bestimmter Schimmelpilz namens Penicillium, der die Bakterienkultur des Wissenschaftlers kontaminiert hatte, diese abtötete. Die Entdeckung des Antibiotikums Penicillin sollte der erste entscheidende Schritt, auf dem Weg zur einer veränderten Strategie bei der Behandlung von Infektionen werden. Die Entwicklung vieler weiterer Antibiotika folgte, darunter die der Cephalosporine aus Pilzen sowie einer Vielzahl anderer aus den verschiedenen Stämmen des Filamentbakteriums Streptomyces, wie z. B. Streptomycin, Erythromycin, Tetracyclin und Vancomycin. Dann begannen die semi-synthetischen Modifikationen der Klassen von Penicillinen und Cephalosporinen und damit die Produktion von ß-Laktamen der zweiten und dritten Generation. Die sog. totale Synthese führte dann zur Entwicklung von Zweitgenerations-Erythromycinen wie Klarithromycin und Azithromycin (Abb. 1). Abbildung 1: Übersicht über den zeitlichen Verlauf der Entwicklung neuer Antibiotika [85] 3 Die Einführung dieser Medikamente in den medizinischen Alltag sollte in der Folgezeit Millionen von Leben retten und die Entwicklung von komplexen chirurgischen Eingriffen erlauben, solchen die zuvor wegen des Risikos von postoperativen Infektionen noch als zu risikoreich abgelehnt worden waren. Der herausragende Erfolg der neuen Medikamente verleitete einige Kliniker und Wissenschaftler zur Voraussage, der Planet sei in naher Zukunft von allen pathogenen Bakterien befreit. Alsbald wurde dieser Optimismus durch das Aufkommen antibiotikaresistenter Bakterien ad absurdum geführt. Zwei Jahre nachdem Penicillin Mitte der 40er Jahre in der breiten Masse verfügbar geworden war, registrierten Wissenschaftler das in Erscheinung Treten der ersten Stämme von penicillin-resistenten Stämmen des Staphylococcus aureus. Von diesen ersten Fallbeschreibungen aus, eskalierte das Problem in eine ernste Sorge des öffentlichen Gesundheitswesens mit einer Bedeutung auf der globalen ökonomischen, sozialen und politischen Ebene [104]. Als man immer vertrauter wurde mit der Epidemiologie und den Mechanismen der Resistenz gegenüber antimikrobiellen Medikamenten, sah man sich mit einer beachtliche Fülle von Möglichkeiten konfrontiert, die Bakterien zur Verfügung stand, um sich der Wirkung von Antibiotika zu entziehen. Eine einzelne Mutation kann bereits zur Resistenz eines Stammes führen, ohne dabei seine Pathogenität oder Lebensfähigkeit zu verändern. In der wissenschaftlichen Theorie kann das Problem der Mutationsresistenz überwunden werden durch das Verabreichen einer Kombination von Medikamenten in ausreichender Dosis und Dauer zur Auslöschung der Infektion. In der klinischen Praxis ist das Gegenteil geläufig. Immer dann, wenn Falschanwendung und Missbrauch von Antibiotika stattfindet, entstehen Resistenzen. Zusätzlich töten die so angewendeten Antibiotika die nonresistenten Organismen, den wichtige Status quo des Organismus zerstörend, schaffen sie Platz für die Vermehrung der resistenten Gegenstücke, die dann ihre Antibiotika feindlichen Gene verbreiten können. Einige wichtige Beispiele für solche Organismen sind der methicillin-resistente S. aureus (MRSA), der penicillin-resistente Streptococcus pneumoniea (PRSP) und der vancomycinresistente Enterococcus spp. (VRE). So sind in der ersten Welt schätzungsweise 60% von nosokomialen Infektionen durch antibiotikaresistente Mikroben verursacht. Da in den vergangenen Jahren immer mehr Menschen Träger von 4 multiresistenten Keimen wurden, blieben die Infektionen mit solchen Keimen nicht nur auf das Krankenhaus beschränkt [82, 20]. So bilden diese multiresistenten Stämme eine immer ernster werdende Bedrohung der Arbeit des plastischen Chirurgen, weil die Infektion einen großen Beitrag am Versagen der operativen Behandlung einnimmt. 1.1.3. Resistenzentwicklung innerhalb der Antibiotika Nach einer Lawine von Entdeckungen zwischen 1930 und 1980 hatte es in den letzten 20 Jahren deutlich weniger Innovationen im Kampf gegen infektiöse Organismen gegeben. Pharmazeutische Unternehmen glaubten, es gebe bereits genug Antibiotika, und begannen Forschungsinvestitionen zur Entwicklung neuer Mittel zu reduzieren oder sie aus dem wenig profitablen Antibiotikamarkt umzuleiten [23]. So war es nicht verwunderlich, dass der großen Häufigkeit von Multiresistenzen nur die begrenzte Einführung neuer Mittel zur Behandlung bakterieller Infektionen gegenüberstand. Die üblichen Kosten für ein Antibiotikum von seiner Entdeckung bis zu seiner Markteinführung liegen ca. zwischen 85 Mio. € und 300 Mio. €. Ist einmal die effektive Wirksamkeit des Antibiotikums nachgewiesen und es betritt den klinischen Gebrauch, ist es nur eine Frage der Zeit bis zum Auftreten der ersten Resistenzen. Klinisch signifikante Resistenzen entwickeln sich nahezu ständig im Laufe von Monaten bis Jahren [24]. Nimmt man die riesige Anzahl von Bakterien, die in einem Infektionszyklus involviert ist, dazu noch die schnelle Generationszeit und die intrinsische Rate von Mutationen von ungefähr 1 zu 107, so hätte ein Pool von 1010 Bakterien Mutationen in ca. 1000 Loci zur Folge. Trägt nur eine dieser Mutationen zu einer Resistenz gegenüber dem angewandten Antibiotikum, so werden nur die sensiblen Bakterien sterben, während der resistente Stamm wächst, den Platz der toten Nachbarn einnimmt und schließlich die dominante Variante in dieser Population wird. Diese Organismen können nun entweder ihre Resistenz-Gene an ihr Nachkommen durch Replikation weitergeben oder sie verwandten Bakterien mittels Konjugation weiterreichen, wobei genübertragende Plasmide von einem Organismus zum anderen überbracht werden. Trotz des häufigen Gebrauchs von Antibiotika im Krankenhaus, bleiben Infektionen ein alltägliches Problem der Klinik. Während die Verteilung der pathogenen Keime, die aus chirurgischen Infektionen isoliert werden, sich 5 innerhalb der vergangenen Jahre nicht wesentlich verändert hat [80, 83], stellt die steigende Frequenz von antimikrobiell resistenten Stämmen ein wachsendes Problem dar [81]. Bis 2001 hat man über sechs klinische Infektionen mit Vancomycin-intermediärem S. aureus (VISA) aus den USA berichtet [35], seitdem ist ihre Zahl nicht nur in den USA weiter gestiegen, es wurde auch ein Anstieg an Infektionen in Europa und Asien beobachtet [15, 53, 70, 88]. Heutzutage sind nahezu alle S. aureus Stämme resistent gegenüber Penicillin und das Verhältnis zu S. aureus Isolaten, die gegenüber Methicillin, Oxacillin oder Naficillin (sog. MRSA) resistent sind, steigt kontinuierlich, so dass in einigen Regionen Asiens und der USA eine Prävalenz bis über 50% erreicht wird [56]. Staphylococcus aureus ist und bleibt Wundinfektionen [81]. dabei die häufigste Stämme dieses Ursache von chirurgischen Bakteriums verursachen Hautentzündungen sowie Abszesse und können sich hämatogen ausbreiten, um so zu schweren Organschäden oder gar zum Tod zu führen [54]. Die hohe Anzahl von S. aureus Infektionen in der Chirurgie ist teilweise auf die hohe Rate der Kolonisation von Haut- und Schleimhautoberfläche der Menschen innerhalb der Bevölkerung durch das Bakterium zurückzuführen. Ungefähr 50% der Bevölkerung sind intermittierende Träger der Mikrobe, in Nasenvorhof oder auf der Haut und ca. 30 % der Menschen sind sogar prolongierte Träger des Bakteriums in loco typico [17, 86]. Neuere Beweise machen deutlich, dass die präoperative nasale Eradikation des Keimes mit Mupirocin eine effektive Maßnahme ist, um postoperative Infektionen bei chirurgischen Patienten zu verhindern [118]. Darüber hinaus verlassen sich Kliniker wegen der steigenden Anzahl von methicillinresistenten Koagulase-negativen Staphylokokken immer mehr auf das Vancomycin, welches bei diesem resistenten Keim den letzten Ausweg für beides, sowohl die chirurgische Prophylaxe als auch die Behandlung von Katheter (intravaskulär) oder Biomaterial (alloplastisch, e. g. Brustimplantate) assoziierten Infektionen darstellt [87]. Ein anderer Problemkeim ist der Vancomycin-resistente Enterococcus. Die Rate von Infektionen mit diesen Bakterien hat sich nicht verkleinert, dabei ist das Bakterium bei ein Viertel aller Infektionen, die es verursacht, resistent gegenüber allen von der FDA zugelassenen Antibiotika, einschließlich des Reservemittels Vancomycin [66]. Enterokokken verursachen eine Vielzahl von sowohl mono- als auch polymikrobiellen Infektionen, hauptsächlich bei immunkompromittierten Patienten. Das Spektrum dieser 6 Infektionen reicht dabei von Bakteriämie über Dekubitus bis hin zu diabetischen Fußulzera. Erworben werden sie entweder endogen aus der patienteneigenen intestinalen Flora oder exogen aus einer mit dem Keim kontaminierten Umgebung. Enterokokken sind von Natur aus gegenüber vielen antimikrobiellen Substanzen resistent und erwerben bereitwillig zusätzliche Resistenzen, diese Eigenschaften haben sie zu prominenten nosokomialen Pathogenen werden lassen. Wegen dieser Resistenzen war Vancomycin das praktisch einzige Medikament, auf das man verlässlich bei der Bekämpfung von durch multi-resistente Enterokokken verursachten Infektionen zurückgreifen konnte. Jedoch wurden auch hier 1987 erstmalig Resistenzen beobachtet, die sich dann in den folgenden 4-6 Jahren sogar dramatisch ausgeweitet haben [78]. Die VRE (Vancomycin resistente Enterokokken) besitzen in ihrem Erbgut fünf Gene, die für ihre High-Level Resistenz notwendig und ausreichend sind [2, 114]. Zum einen kodieren diese Gene Peptide der Zellwand und sind so für die Funktion der Zellwand essentiell, zum anderen verschaffen sie bei Vancomycinmedikation eine entsprechenden Überlebensvorteil und führen zur Selektion der Resistenz [2]. Da die Bedingungen, die das Auftreten von Multi-Resistenzen bestärken, auch im neuen Jahrtausend andauern, ist es von entscheidender Bedeutung flexible Strategien zur Detektion und Antwort solcher Probleme schnellstmöglich zu entwickeln. Tabelle 1: Resistenzraten der häufigsten Bakterien auf Intensivstationen gegenüber gebräuchlichen Antibiotika von 2000 bis 5/2005 auf der Basis der Daten von SARI (Surveillance der Antibiotika-Anwendung und der bakteriellen Resistenzen auf Intensivstationen). Dargestellt wurden die Resistenzraten (RR) und Resistenzdichte (RD) zwischen 2000 und Mai 2005. [76] 7 Das wichtigste Ziel der postoperativen chirurgischen Behandlung ist die früh einsetzende Heilung ohne Komplikationen. Komplikationen wie postoperative Wundinfektion, traumatische Wundinfektion, schwere Karbunkel oder das Erysipel kommen dennoch im klinischen Alltag ständig vor, so ist auch die systemische prä- und postoperative antimikrobielle Therapie ein integraler Bestandteil der Therapie neben der chirurgischen Intervention. Die permanent hohe Rate multiresistenter Keime, wie es die Tabelle 1 (Resistenzrate = Anzahl resistenter Isolate einer Spezies pro 100 Isolate dieser Spezies; Resistenzdichte = Anzahl resistenter Erreger pro 1.000 Patiententage, wobei pro Patient nur einmal ein resistentes Isolat gezählt werden darf) darstellt, seien es Staphylokokken (MRSA), Pneumokokken, E. coli oder Enterokokken (VRE) lässt die Notwendigkeit nach einer besseren Prävention der postoperativen Infektion erkennen. Im Zeitalter von Resistenzen gilt es dabei drei entscheidende strategische Ziele zu verfolgen: das Aufdecken der Resistenz, die Berichterstattung darüber und das Verhindern einer weiteren Streuung oder Übertragung des Keimes auf andere Personen. Der wichtigste und einfachste Schritt der Prävention ist dabei die Reinhaltung der Umgebung des Patienten. Wenn multiresistente Keime schon zum Milieu des Patienten gehören, dann muss ihre Streuung und Möglichkeit zur Ausbreitung in diesem Milieu so gering wie möglich gehalten werden. Deshalb sind die Eckpfeiler in der Minimierung der postoperativen Infektion das Händewaschen und die Desinfektion der Haut durch das Personal. Denn das Risiko einer Übertragung von Person zu Person Personalhände ist Versorgungseinheiten, von multiresistenten nirgends auf größer denen Pathogenen als man in über überfüllten, auch noch kontaminierte unterbesetzten schwerstkranke, immuneingeschränkte Patienten vorfindet [115]. Neben den chirurgischen Wundinfektionen, sind es vor allem Infektionen über den Blutweg, die ein ernstes Problem im klinischen Alltag darstellen. Katheter und Gefäßprothesen sind für Bakterien ein willkommene Örtlichkeit zur Ansiedlung und in der Folgezeit zur Aufrechterhaltung einer kontinuierlichen Infektion. So liegen die Letalitätsraten für nosokomiale Blutweginfektionen für S. aureus bei 25%, für Koagulase-negative Staphylokokken bei 21% und für Enterokokken bei 32% [27]. Obwohl der genaue Beitrag der Resistenz zu dem Ausmaß dieser Raten noch unklar ist, suggerieren mehrere Studien, dass nosokomiale Infektionen über den Blutweg die acht höchste Todesursache in den Vereinigten Staaten darstellen 8 [27]. Auch in Europa bedeutet der in den meisten Regionen stattfindende Anstieg der Resistenzen (Abb. 2) eine Limitierung der Therapiemöglichkeiten. Der Gebrauch von Antibiotika ist die zentrale Determinante bei der Entwicklung von Resistenz. Das komplexe Beziehungsgeflecht zwischen Patient, Mikroorganismus und dem Antibiotikum hat eine große Bedeutung für viele Ärzte. Die antimikrobielle Therapie des schwer erkrankten Patienten beginnt nahezu immer in der Unsicherheit über den verursachenden Keim und seiner Empfindlichkeit. So besteht die sog. blinde empirische Initialtherapie oft aus einer hochdosierten, aufs breite Erregerspektrum ausgerichteten Gabe eines oder mehrerer Antibiotika. In dieser Situation kann eine vernünftige Auswahl von Antibiotika nur dann erwartet werden, wenn der behandelnde Arzt über die in seiner Region vorherrschenden Empfindlichkeitsmuster der für die Erkrankung relevanter Stämme Kenntnis besitzt. Wie eminent dies ist, wird anhand von ein paar Zahlen deutlich: zu irgendeinem gegebenen Zeitpunkt erhalten 25 bis 35 Prozent der hospitalisierten Patienten eine systemische Antibiotikatherapie, um eine aktive Infektion zu behandeln oder ihr Ausbrechen zu verhindern. In der Chirurgie beträgt der Anteil der unangemessenen Behandlung dabei 40-75 %, wie aus den USA über die letzten 15 Jahre berichtet wurde [42]. In den Vereinigten Staaten werden dabei 30 Verschreibungen auf 100 Personen pro Jahr geschrieben, so dass ein Verschreibungsvolumen von 4,1 kg Antibiotika auf 100 Personen pro Jahr resultiert [68]. Dieser enorme Verbrauch von Antibiotika birgt ein ebenso großes Potential zur Selektierung oder Beschleunigung des Wachstums resistenter Stämme. Bezogen auf oben erwähnte Zahlen sind vermutlich bis zur Hälfte dieser Anwendungen unangemessen, so wie z. B. bei dem nutzlosen Behandlung von viralen Infektionen des oberen Atemtraktes, der Bronchitis und der Pharyngitis. Neben dem Antibiotikagebrauch selbst spielt die Compliance des Patienten eine wichtige Rolle. Eine sporadische Compliance ihrerseits ruft gleichsam das Äquivalent einer wiederholten Monotherapie hervor, mit dazwischenliegenden für die Resistenzentstehung so entscheidenden Perioden ungehemmten bakteriellen Wachstums. Um sich der Compliance des Patienten zu vergewissern, wurde viel Anstrengung in die sog. DOT (directly observed therapy, Patient wird bei Medikamenteinnahme beobachtet) gelegt, mit dem Ergebnis, dass die Inzidenz von Resistenz assoziiertem Therapieversagen signifikant minimiert werden konnte [62]. 9 Abbildung 2: Resistenzentwicklung für S. aureus in Europa aus den Daten des EARSS (European Antimicrobial Surveillence System) vom Jahr der Gründung 1999 bis zum Jahr 2006. Die Pfeile zeigen Anstieg bzw. Rückgang der Resistenzraten. 10 1.1.4. Lösungsstrategien zur Minimierung der Resistenzentwicklung Das letzte Jahrzehnt kam es zu einem rapiden Fortschritt auf dem Feld der molekularen Biologie und ihrer Anwendung im Bereich der infektiösen Erkrankungen [28]. Im 21. Jahrhundert werden molekulare Werkzeuge wie bakterielles Schnüffeln [43], Nanobiotechnologie [16] sowie Hochdurchsatz-Gen- und Proteinexpressionsanalyse (Genomics, Proteomics) nicht nur die frühe Diagnose der Infektion, sondern auch die Erforschung von Angriffspunkten, Inhibition und Manipulation der Pathogene erleichtern [65]. Einige dieser neuen antimikrobiellen Strategien zielen in ihrem Design gegen Resistenz- und Virulenzmechanismen und werden auf diesem Wege eine interessante Herausforderung für den mikrobiellen Keim darstellen [19]. Angesichts der erodierten Effizienz der sogar neuesten antimikrobiellen Stoffe haben sich Ärzte im zunehmenden Masse auf die Fähigkeit der pharmazeutischen Industrie verlassen, auch immer neuere Agenzien entwickeln zu können. Diese sieht sich ihrerseits einer Eskalation von Kosten gegenüber, sei es für Entdeckung, Entwicklung, Testung oder Genehmigungsverfahren für Antibiotika konfrontiert, während die Risiken einer unerwarteten Toxizität oder des schlichten klinischen Scheiterns einer neuen Substanz gleich geblieben sind [9]. Im Angesicht der Probleme der gesteigerten bakteriellen Resistenzen gegenüber konventionellen Antibiotika rückt das so genannte angeborene Immunsystem immer stärker in den Blickpunkt der Forschung. Einen bedeutenden Teil dieser angeborenen Abwehr stellen die Host Defense Peptide, früher auch als Antimikrobielle Peptide (AP) bezeichnet, dar. Isoliert aus höheren Eukaryoten, weisen sie ein weites Feld struktureller Unterschiede auf und eignen sich gerade deshalb als neuartige Vorlagen für die Entwicklung pharmazeutischer Komponenten. In diesem Bereich liegt die Aufgabe der Entdeckung von neuen potentiellen Therapien gegen die aufkommende Multiresistenz vieler Keime. 1.2. Das Angeborene Immunsystem Warum gerade in den Regionen des Körpers, die sich in höchsten Masse mit eindringenden Mikroorganismen auseinandersetzen müssen, eine derart niedrige Inzidenz von Infektion und entzündlichen Komplikationen vorherrscht, ist darauf zurückzuführen, dass gerade auf den Oberflächen ein lokaler, hoch effektiver und auf ein breites antimikrobielles Spektrum ausgerichteter Verteidi11 gungsmechanismus existiert. Entdeckt wurden diese antimikrobiellen Eigenschaften der Epithelien im letzten Jahrhundert durch Metchnikoff. Er beobachtete, wie Mikroben, die die natürliche Barriere übertreten hatten, von mobilen Zellen (Phagozyten) gestellt, sowie anschließend regelrecht gefressen (phagozytiert) und abgetötet wurden. Als er dies Phänomen der Phagozytose und des Abtötens der Mikroben zum ersten Mal beschrieb, suggerierte er zugleich, dass in den Phagozyten auch mikrozide Substanzen vorhanden sein müssen, mittels derer der Tötungsvorgang stattfinde. Diese Substanzen nannte er damals „Fermente“ (spätere Enzyme) [112]. Diese Verteidigungslinie des Wirtsorganismus, die in den frühen Phasen der Interaktion zwischen eindringenden Mikroorganismen und dem Wirt tätig ist, beruht nicht auf einer spezifischen Antigen- Erkennung, im Gegensatz zum so genannten adaptiven Immunsystem. Dieses System, welches auf Erkennung und gezielter Vernichtung fremdartiger Epitope mittels gezielter Antikörperherstellung beruht, kann im oben genannten Fall nicht schnell genug reagieren, um den Organismus vor der einsetzenden Infektion zu bewahren. Die klonale Expansion von B- und T-Lymphozyten nach einer erstmaligen oder wiederholten Begegnung mit eindringenden Pathogenen benötigt Tage bis Wochen, um die maximale Aktivität zu erreichen [32]. Das adaptive Immunsystem (Antikörperbildung und Antigenerkennung mittels B- und T-Lymphozyten) ist eine späte evolutionäre Entwicklung, zu finden vor allem in höher entwickelten Vertebraten. Die spezifische Antigenerkennung durch Lymphozyten spielt natürlich auch während der initialen Begegnung mit dem Pathogen eine Rolle, wird aber erst beim Eindringen der Mikroorganismen in den Körper entscheidend, weil dann von einer Persistenz der Keime ausgegangen wird, die eine langfristige Reaktion erfordert. Im Gegensatz zu der Spezifität der erworbenen Immunantwort, ist die angeborene Immunantwort unspezifisch und funktioniert auch ohne Berücksichtigung des Antigencharakters der Mikrobe. Die wirksamen Komponenten des Angeborenen Immunsystems unterscheiden sich deswegen auch entscheidend von dem Erworbenen in derlei Hinsicht, als dass sie relativ unspezifisch und schnell induzierbar sind. Ihr Wirkungseintritt beträgt Minuten bis Stunden. Sie sind auf Ziele ausgerichtet, die konservierte Muster der Erkennung darstellen, solche wie die Lipopolysaccharide (LPS), ein natürlicher Wandbestandteil von gramnegativen Bakterien, oder die Lipo12 teichonsäure (LTA), Äquivalent der LPS bei grampositiven Bakterien. Die angeborene Antwort ist eine Wirtsleistung, die auf einem integrierten Multiorgansystem beruht: im Laufe der Entwicklung vom Organismus gestaltet, nicht nur, um mikrobielle Invasionen zu bekämpfen, sondern auch um Gewebeverletzung und Zellzerstörung zu minimieren, Reparationsvorgänge zu fördern sowie die Möglichkeit von sekundären oder opportunistischen Infektionen zu reduzieren [31]. Dieses System richtet sich auch gegen bereits eingedrungene Keime, und füllt so die zeitliche Lücke bis zur Entwicklung einer adäquaten Antwort der erworbenen Immunität. Neue Studien zeigen ferner, dass die angeborene Immunantwort nicht nur die effektive erste Linie der Wirtsverteidigung gegen pathogene Keime bildet, sondern auch durch Interaktionen die Funktionen des erworbenen Systems definiert und beeinflusst [14, 31]. 1.2.1. Host Defense Peptide Im letzten Jahrzehnt erkannte man zunehmend die Bedeutung von Host Defense Peptiden, auch Antimikrobielle Peptide bezeichnet, als die der ersten Verteidigungslinie des Angeborenen Immunsystems. Es existiert eine wachsende Anzahl von Beweisen dafür, dass ihre Rolle in der Abwehr von Pathogenen ebenso wichtig für den Wirtsorganismus ist wie Antikörper, Immunzellen und Phagozyten. Das sich daraus ergebende Konzept ihrer Funktion besteht daraus, dass die Peptide zur protektiven Wirkung der Barriere-Funktion von Epithelien durch das Abtöten von eindringenden Mikroorganismen beitragen [34, 69, 89, 109]. Zellen verschiedener Spezies produzieren eine Vielzahl unterschiedlicher antimikrobieller Substanzen, die sowohl als endogenen Antibiotika als auch als desinfizierende Agenzien fungieren. Die Host Defense Peptide (HDP) als ein Bestandteil des Angeborenen Immunsystems sind üblicherweise definiert als Polypetidverbindungen, bestehend aus weniger als 100 Aminosäuren, mit antimikrobiellen Eigenschaften, kodiert durch Gene und synthetisiert durch Ribosomen. Diese Definition unterscheidet sie von den meisten (keinesfalls allen) Peptidantibiotika der Bakterien und Pilze, die ihrerseits typischerweise auf speziellen metabolischen Wegen hergestellt werden und oft auch exotische Aminosäuren beinhalten [39]. Die meisten der HDP sind amphipathisch, kationisch 13 (sie tragen eine positive Nettoladung), und weisen eine definierte Alpha-Helix oder Beta-Faltblatt-Struktur in membranähnlicher Umgebung (Abbildung 3, 4) auf. Abbildung 3 (links): Protegrin gehört zu der Gruppe der Cathelicidine und besteht aus 18 Aminosäuren. Die wesentliche Funktionsstruktur ist determiniert durch eine beta-Faltblattstruktur, hier blau untermalt. [25] Abbildung 4 (rechts): Ovispirin-1 auch ein Vertreter der Cathelicidine, ist im Gegensatz zum Protegrin jedoch mit einer Alpha-Helix in der Sekundärstruktur ausgestattet. [113] Sie können in nahezu allen Lebensformen gefunden werden, von Pflanzen ausgehend über Insekten bis hin zu Tieren, dabei Amphibien, Vögel, Fische und letztlich auch Säugetiere wie Menschen einschließend [42, 43]. Mehr als 500 dieser Peptide sind bis dato entdeckt worden. In Tabelle 2 sind einige Beispiel von kationischen Peptiden aus Säugern dargestellt. Die Haut ist die epitheliale Hauptbarriere zwischen dem Organismus und der feindlichen Umgebung. Sie übernimmt in dieser Funktion auch die Aufgaben eines aktiven Abwehrorgans [12]. So ist es auch zu erklären, dass man Host Defense Peptide als Teil des Angeborenen Immunsystems häufig auf Oberflächen wie Haut und Schleimhaut findet. Epithelzellen aus gesundem Gewebe demonstrierten die Expression von beta-Defensinen auf niedrigem Niveau. Manche Defensine können aber auch durch Behandlung von pro-inflammatorischen Zytokinen, Bakterien oder ihren Abbauprodukten in ihrer Erzeugung induziert werden. So wird zum Beispiel die Produktion von humanen betaDefensin-2 (HBD-2) in Keratinozyten deutlich heraufreguliert durch die Anwesenheit von gramnegativen Bakterien oder pro-inflammatorischen Zytokinen wie dem Tumor-Nekrose-Faktor-alpha (TNF-alpha) oder Interleukin-1-beta (IL-1-beta) [96]. Die Induzierbarkeit der Peptide in der Immunantwort resultiert in der Fähigkeit dieser Barriere gegen ein breites Spektrum von pathogenen Keimen innerhalb von 14 Minuten in einer unspezifischen Art und Weise reagieren zu können. Die Kinetik der Induktion ist dabei überaus vielsagend über die Rolle der frühen Immunantwort gegen eine Infektion. Tabelle 2: Zusammenfassung ausgewählter Host Defense Peptide Peptid Cathelicidin Name/n LL-37/ Ursprung Mensch Biochemischen Hauptzellen und Eigenschaften Gewebsquellen Kationisch, alpha-helikal hCAP18 Textreferenz Neutrophile, Mastzellen, 4-6, 36, 39, Epithelien 44, 47, 52, 61, 63, 84, 90, 101, 106,108, 119 Cathelicidin CRAMP Maus Amphipathisch, alpha-helikal Cathelicidin Protegrine Schwein Kationisch, Disulfidbrücken Neutrophile, 9, 38, 110, Knochenmark 119 Neutrophile 33, 39, 45, 49, 1-5 50, 52, 55, 57, 73, 98, 102, 108, 105, 119, 122-3 Cathelicidin Ovispirin Schaf Kationisch, alpha-helikal Neutrophile 3, 13, 93, 100, 107, 110, 119 Cathelicidin Cathelicidin CAP18 Dodeca- Hase Kationisch, alpha-helikal Neutrophile, Mastzellen, 18, 39, 60, Epithelien 119, Kuh Kationisch, alpha-helikal Neutrophile 47, 119 Mensch Kationisch, Disulfidbrücken, Neutrophile, Epithelien 12, 40, 52, 63, peptide Alpha- Alpha- Defensine Defensine beta-Faltblatt 74, 96, 1-4 Beta- Beta- Defensine Defensine Mensch Kationisch, Disulfidbrücken, Neutrophile, Epithelien beta-Faltblatt 52, 63, 74, 77, 96 1-4 ThetaDefensine Plectasin Pilz Kationisch, Disulfidbrücken Pilzbestandteil 8, 77, 79 Beta-Faltblatt Schließlich werden die Peptide als antimikrobiell bezeichnet, weil sie ein ungewöhnlich breites Spektrum der Aktivität besitzen. Sie veranschaulichen eine beeindruckende Fähigkeit, nicht nur grampositive oder gramnegative Bakterien, sondern auch Pilze [8], einschließlich Hefearten [29], Parasiten [71], Krebszellen [52] und sogar behüllte Viren wie das HIV [21, 58] und das Herpes-simplex-Virus [58] abzutöten oder zu neutralisieren. Zusätzlich weisen diese Eiweiße auch einige antiendotoxische Eigenschaften auf [92], neben den Fähigkeiten immunmodulatorisch einzuwirken z. B. durch die Induktion von TNF-alpha [67]. Besonders wichtig neben den antimikrobiellen Fähigkeiten der Peptide erscheint gerade für den späteren klinischen Einsatz ihre angiogenetische Potenz [51, 106]. 15 In vivo Studien mit HDP zeigten fernen einen systemischen Schutz gegen Streptococcus pneumoniea in Mäusen [41], lokale Protektion gegen polymikrobische orale Mukositis in Hamstern [72], sowie Schutz gegen Pseudomonas aeruginosa von Verbrennungswunden in Mäusen und Ratten [105]. Gemeinsames Kennzeichen dieser in vivo Studien war die demonstrierte Effizienz der unterschiedlichsten auch synthetischen HDP bei der Clearence von Bakterien, die entweder systemisch oder topisch appliziert wurden. In ihrer Eigenschaft als Peptide ist auch eine weitere Erweiterung der HDPTherapie möglich, nämlich die des Einsatzes in der Gentherapie. Anstelle diese auf einen Langzeit-Gentransfektion auszurichten, wie zum Beispiel bei der Korrektur vererbter genetisch bedingter Erkrankungen, wäre eine transiente Übertragung der Gene zur verbesserten oberflächlichen Wundheilung absolut ausreichend. Diese wurde zuletzt von Bals et al. demonstriert. Sie transfizierten Mäuse mit einem adenoviralen Konstrukt, der das Genom eines menschlichen Host Defense Peptides enthielt. Die Mäuse wiesen hohe Level transgener Expression des Eiweißes in Serum und Lunge auf. Nach einer subletalen bakteriellen Infektion zeigten die Tiere signifikant niedrigere Bakterienraten und eine unterdrückte inflammatorische Antwort. Ebenso demonstrierten die Mäuse eine deutliche Resistenz gegenüber Endotoxin [4]. Neben der Rolle der HDP in der Abwehr von Infektionen sind es auch andere Eigenschaften die ausgewählten kationischen Peptiden opsonierender zugeschrieben Phagozytose [94], werden, wie Chemoattraktion Stimulation von von non- IL-8-stimulierten Neutrophilen [1, 94] und Induktion von Wundheilung [18]. Unter den verschiedenen Klassen von Peptidantibiotika bei den Säugetieren wurden einigen eine zentrale Rolle bei der angeborenen Immunität zugeschrieben. Hierzu zählt man verschiedene Cystein-reiche Peptide wie die Defensine und Protegrine [40, 64, 74] und die strukturell stärker abweichende Cathelicidine [110, 119, 120]. Hergestellt als Präcursoren, benötigen sie ein proteolytisches Processing, um zur reifen, antimikrobiellen Funktion des Peptides zu gelangen. Cathelicidine enthalten eine konservierte N-terminale Domain namens Cathelin und eine strukturell davon unterschiedliche C-terminale Domain, die die antimikrobielle Aktivität des Peptides trägt. Das CAP18 des Hasen war der erste 16 entdeckte Cathelicidin-Präkursor und sein reifes Peptid weist eine antimikrobielle Aktivität mit breitem Spektrum [60] auf. In der Folgezeit sind Cathelicidine in vielen anderen Species identifiziert worden, einschließlich des hCAP18/LL37 im Menschen [1], CRAMP in Mäusen [38, 99] und dem SMAP29 in Schafen [3, 100]. Mögliche Einschränkungen in Bezug auf den therapeutischen Gebrauch natürlich vorkommender Host Defense Peptide beim Menschen beinhalten ihre Zytotoxizität und Faktoren, die sich auf Kosten bei einer groß kalibrierten Produktion beziehen. Bemühungen, diese Hindernisse zu überwinden, führten zur Entwicklung eines neuen alpha-helikalen Host Defense Peptides, Ovispirin-1, dessen Struktur sich an den 18 N-terminalen Resten des Schafscathelicidins SMAP29 orientierte [13, 91, 117]. Bei erhaltener antimikrobieller Effektivität gegenüber gramnegativen und -positiven Keimen zeigte das neue Eiweiß deutlich geringere Zytotoxizität. Kleinere strukturelle Modifikationen von Ovispirin-1 führten dann zur Entstehung von Novispirin G10 und seinen Derivaten. Dieses demonstrierte ähnliche antimikrobielle Aktivität bei geringer Zytotoxizität und Hämolyse unter Einfluss von humanem Blutserum sowie die in vivo vorhandene Fähigkeit Pseudomonas aeruginosa auch in Verbrennungswunden abzutöten [107]. 1.2.2. Cathelicidine Das erste menschliche Cathelicidin, LL37/hCAP-18 wurde zunächst aus einer aus dem menschlichen Knochenmark isolierten cDNA kloniert [1, 22, 61], bevor dann seine Gene gemappt wurden [44, 45]. Die folgenden Studien machten deutlich, dass LL-37/hCAP-18 primär in myeloiden Zellen exprimiert wird, wo es dann in Granula verbleibt [101]. Die Ableitung seines Gens konnte aber auch im Hoden [1] und entzündeter Haut [36] nachgewiesen werden. Bals et al. zeigten dann, dass LL-37/hCAP-18 diffus auch in vielen anderen Epithelien menschlicher Organe exprimiert wird, einschließlich des Oberflächenepithels der Atemwege sowie serösen und mukösen Zellen der submukösen Drüsen des oberen Atemtraktes [6]. Dabei wird das Peptid mit der breiten antimikrobiellen Aktivität gegen grampositive und –negative Organismen in den Oberflächenfilm der Atemwege sezerniert. Eine weitere Studie der Forscher mit einem Xenograft Modell der Zystischen Fibrose demonstrierte eindrucksvoll, wie die Expression und Sekretion dieses Peptides vor bakterieller Kolonisation und Infektion schützt [5]. 17 Die Protegrin (PG) Familie unter den Cathelicidinen besteht aus einer Reihe von HDP (PG-1 bis PG-12), die zuerst in Schweinen identifiziert wurden [57, 122, 123]. Protegrine sind kleine (bis 2 kDa) HDP mit breitem Spektrum, die man in Neutrophilen von Schweinen findet, wo sie als Cathilin-enthaltende Präkursoren gelagert werden. Die reifen, also voll prozessierten, Protegrine enthalten dann 1618 Aminosäureresiduen, werden aber als inaktive Präkursoren freigegeben, die dann extrazellulär durch die neutrophile Elastase aktiviert werden [98]. Wie die gut charakterisierten Defensine, sind auch Protegrine in hohem Maße homolog und teilen gemeinsame Merkmale wie kationische Struktur und Cystein-Reichtum. In Lösung bilden sie eine amphipathische Beta-Faltblatt Struktur, die durch 2 Disulfidbrücken stabilisiert wird. Diese Brücken sind von eminenter Bedeutung für die antimikrobielle Aktivität, weil sie das Peptid mindestens zehnmal so effektiv machen wie in der linearisierten Form. Frühere Studien zeigten, dass PG-1 im Gegensatz zu üblich gebräuchlichen Antibiotika sowohl gramnegative als auch positive Bakterien der log- und Plateau-Phase auf extrem schnelle Art und Weise abtötet [102]. Der Mechanismus des Tötens ist dabei noch nicht vollständig untersucht. Bisher vorliegende Studien sprechen dafür, dass durch die Peptide ein spannungsabhängiger Ionenkanal in der bakteriellen Wand geschaffen wird [49, 50, 73, 79]. Protegrine weisen nämlich eine hohe Affinität zu integralen Bestand-teilen der Bakterienwand auf, solchen wie Lipopolysacchariden in gramnegativen oder Lipoteichonsäuren in grampositiven. Wenn eine Schwellenkonzentration des Protegrins erreicht wird, werden die spannungsabhängigen Ionenkanäle geformt und die Mikrobe rapide abgetötet [33, 49, 55, 103] (Abbildung 5). Bei niedrigeren Konzentrationen wird dieser Effekt nicht beobachtet, und die Peptide möglicherweise durch extrazelluläre Proteasen gespalten. PG-1 ist besonders attraktiv für die klinische Anwendung, weil es, im Gegensatz zu menschlichen Defensinen, eine breite antimikrobielle Aktivität auch bei physiologischer Salzkonzentration und in Anwesenheit von Serum besitzt. 18 Abbildung 5: Abgebildet ist der Wirkmechanismus der Host Defense Peptide am Beispiel des Protegrin (rot-blau). Normalerweise wasserlöslich (blau), vermag es über seine aliphatische Seite, rot dargestellt, sich an die bakterielle Membran zu lagern, um dann durch Interaktion mit benachbarten Peptiden eine Pore in der Membran zu bilden. Diese Poren zerstören die Homöostase des Bakteriums und führen zu seiner Lyse. [110] Abbildung 6: Die Abbildung demonstriert am Beispiel des LL-37, einem alphahelikalem Cathelicidin, den amphipathischen Charakter der Peptide. Im elektrostatischen Oberflächenplot stellt die Blaufärbung den hydrophilen Anteil dar. In dem Schema darunter die Darstellung der hydrophilen (dunkel) und hydrophoben Anteile (hell) des Peptides, typisch im Übrigen als Strukturmerkmal vieler Peptide. [113] 19 Letztere werden beide in Wunden und anderen Entzündungsorten gefunden, die mit einer Störung der kapillären Funktion einhergehen. Steinstraesser et al. haben dann dank dieser einzigartigen Eigenschaften des Peptides seine auch in vivo herausragende Fähigkeit zur bakteriellen Clearance in Verbrennungswunden bestätigen können [105]. 1.3. Aufgaben und Ziele der Studie Vor diesem Hintergrund bestand die Motivation dieser Studien im Vergleich der antimikrobiellen Fähigkeiten ausgewählter sowohl natürlicher (PG-1, LL-37/hCAP18, Plectasin) als auch synthetischer Peptide (Novispirin G10, RIR G10, R2 G10, WS 22, WS 22 N). Das in porkinen Neutrophilen vorkommende Protegrin-1 als Repräsentant der Protegrine ist relativ klein (18 Aminosäuren), weist eine betaFaltblattstruktur auf und besitzt breite antimikrobielle Aktivität [77]. LL-37/h-CAP kommt als menschliches Cathlicidin u. a. auf Oberflächenepithelien des Atemtraktes vor, wo es Teil der angeborenen Barriere gegen eine mikrobielle Invasion ist [5, 6]. Plectasin ist ein aus dem Pilz Pseudoplectania nigrella isoliertes Defensin, welches vor allem im grampositiven Bereich deutliche Wirksamkeit besitzt [79]. Die synthetischen Peptide Novispirin G10, RIR G10 und R2 G10 sind Weiterentwicklungen des Ovispirins (an den Stellen 5 bis 7 der Aminosäuresequenz folgt bei Novispirin G10 ein zweifaches Isoleucin und Arginin (RII); beim RIR G10 folgt dem Isoleucin ein Arginin; beim R 2 G10 ist das erste Isoleucin durch Arginin ersetzt) und fürs Novispirin G10 konnte deutliche Wirksamkeit gegen Pseudomonas aeruginosa bei Verbrennungswunden nachgewiesen werden [107]. WS 22 und WS 22 N sind synthetische Abkömmlinge des LL-37. Sie bestehen nur aus 22 Aminosäuren, wobei bei beiden die Aminosäure Nr. 8 der Sequenz von LL-37 (Lysin) durch Tryptophan ersetzt ist. Beim WS 22 N findet man zusätzlich an der letzten Stelle anstatt des ursprünglichen Glutamin ein Asparagin. Da die Peptide über so ein breites Spektrum interessanter Fähigkeiten verfügen und um die Resultate der Tests miteinander vergleichbar zu machen, entschieden wir uns bei der Überprüfung der antimikrobiellen Potenz für die zehn am häufigsten Wundinfektionen 20 verursachenden Keime sowie multi-resistente Isolate aus der Klinik. Unter den Bakterienstämmen wurden bis auf das klinische Isolat eines MRSA sowie multiresistente Pseudomonaden vornehmlich Qualitätskontrollstämme verwendet, um eine Interassay-Reproduzierbarkeit im Hinblick auf die Austestung zu gewährleisten. Schließlich wählten wir zum einen ein derzeitiges Standardverfahren für einen Vergleich der Peptide untereinander, nämlich das Radial Diffusion Assay. Dieser Versuch dient der Bestimmung der minimalen effektiven Konzentration (MEK) (in µg/ml) der Host Defense Peptide. Die Ermittlung dieser erlaubt es, die Aktivität einzelner Peptide oder Peptidgruppen miteinander zu vergleichen. Eine MEK unter 5 µg/ml wird dabei als klinisch hochwirksam betrachtet [103]. Daneben wurde das CLSI Modified Microbroth Dilution Assay durchgeführt. Der Versuch ist das Standardwerkzeug zur Bestimmung der Aktivität einer antimikrobiellen Substanz, sei sie ein Antibiotikum, die im Versuch als positive Kontrollen eingesetzt wurden, oder wie hier ein Host Defense Peptid. Die Methode ist zur Bestimmung der MHK und MBK unter Mikrobiologen weltweit anerkannt, im Gegensatz zum RDA, bei dem eine MEK bestimmt wird, die noch nicht überall gleichermaßen als standardisierte Größe gilt. So ist in der Durchführung beider Methoden die Absicht zu sehen, sowohl ein Verfahren zu wählen, dessen Ergebnis standardisiert und mit den Resultaten herkömmlicher antimikrobieller Substanzen stets verglichen werden kann, als auch ein Verfahren das im besonderen Maße auf die Host Defense Peptide modelliert wurde. CLSI Microbroth Dilution Assay for Fungi diente der standardisierten Bestimmung einer MHK bei Pilzen. Im eigentlichen Sinne ist er eine Abwandlung des oben beschriebenen Modified CLSI Microbroth Dilution Assay, auf mykotische Kulturen. Verbunden sind die Veränderung mit einer Anpassung an die Unterschiede der Pilze im Wachstum und Ernährung. Im Vergleich zu Bakterien äußert sich dies im Wahl anderer Medien und anderen Kultivierungsbedingungen (Temperatur, Brutzeit). 21 2. Material und Methoden 2.1. Radial Diffusion Assay (RDA) 2.1.1 Chemikalien Monobasischer Natriumphosphatpuffer NaH2PO4 * 2 H2O (J.T.Baker, Deventer, Niederlande), Dibasischer Natriumphosphatpuffer Na2HPO4 * 7 H2O (J.T.Baker, Deventer, Niederlande), Agarose (Agarose NEEO Ultra- Qualität, Carl Roth GmbH, Karlsruhe, Deutschland), Essigsäure (0,01%; J.T.Baker, Deventer, Niederlande), Deionisiertes Wasser. 100 mM monobasischen Natriumphosphatpuffer mit einem pH von 4,5 erhielt man aus der Lösung 15,6 g monobasischen Puffers in 1 L destillierten und deionisierten Wasser. 100 mM dibasischen Natriumphosphatpuffer mit einem pH von 9,5 erhielt man aus der Lösung 26,8 g dibasischen Puffers in 1 L destillierten und deionisierten Wasser. Aus 725 ml des 100 mM dibasischen Puffers und 400 ml des monobasischen Puffers wurden 1,125 L Phosphatpuffer mit einem pH von 7,4 gemischt. Dieser wurde im Folgenenden verwendet. 2.1.2. Antibiotika Ampicillin (Ampicillin K029.1, Carl Roth GmbH, Karlsruhe, Deutschland), Vancomycin (Vial Vancomycin Hydrochlorid, Lilly, Eli Lilly and Co, Indianapolis, USA), Imipenem (Imipenem Powder No. 3540, Merck & Co Inc., West Point, PA, USA), Cefotaxim (Cefotaxim+Na, Aventis Pharma GmbH, Deutschland). Die Antibiotika werden gemäß Gebrauchsanleitung in Lösung gebracht. Dabei werden folgende Konzentrationen aus Stammlösungen frisch am Versuchstag angefertigt, die im Versuch als Positiv- oder Negativkontrolle Verwendung finden: Ampicillin 2 mg/ml; Vancomycin 6 mg/ml; Imipenem 2 mg/ml; Cefotaxim 6 mg/ml. 2.1.3. Host Defense Peptide: Novispirin G10 (Novozymes, Bagsvaerd, Dänemark), R2 G10 a (Novozymes, Bagsvaerd, Dänemark), RIR G10 a (Novozymes, Bagsvaerd, Dänemark), LL-37 (Novozymes, Bagsvaerd, Dänemark), LL-37 (Novozymes, Bagsvaerd, Dänemark), WS 22 (Novozymes, Bagsvaerd, Dänemark), WS 22 N (Novozymes, Bagsvaerd, Dänemark), Plectasin (Novozymes, Bagsvaerd, Dänemark), Protegrin-1 (Bob 22 Lehrer, UCLA, USA). Alle von Novozymes gelieferten Peptide wurden rekombinant in E. coli BL21 (DE3) hergestellt. Nach Extrahierung aus den Zellüberständen der Bakterien erfolgte die Purifikation mit Hilfe der Umkehrphasen – HPLC (high performance liquid chromotography). Die erreichte Homogenität betrug 98 %. Deutschland. Das von Bob Lehrer bereitgestellte Protegrin-1 wurde mittels Festphasensynthese hergestellt und ebenfalls mittels Umkehrphasen – HPLC gereinigt (Purifikationsgrad >98%). Die Peptide werden alle zunächst in 1 mg/ml Aliqouts in 0,01% Essigsäure gelöst und bei -80°C gelagert. Jeweils frisch am Versuchstag werden folgende Konzentrationen, angesetzt: 250 µg/ml, 79,1 µg/ml, 25 µg/ml, 7,91 µg/ml, 2,5 µg/ml, 0,79 µg/ml und 0,25 µg/ml. 2.1.4. Bakterien und Pilze Bakterien: E. coli (DSMZ 1103, ATCC 25922), Pseudomonas aeruginosa (DSMZ 1117, ATCC 27853), Acinetobacter baumannii (DSMZ 30008, ATCC 19606), Klebsiella pneumoniae (DSMZ 681, ATCC 10031), Proteus mirabilis (DSMZ 4479, ATCC 29906), Staphylococcus. epidermidis (DSMZ 1798, ATCC 12228), Enterococcus faecalis (DSMZ 2570, ATCC 29212), Staphylococcus aureus (DSMZ 1104, ATCC 25923), C-MRSA (klinisches Isolat, Prof. Sören Gatermann, Institut der Mikrobiologie, Ruhr - Universität, Bochum, Deutschland), Pseudomonas aeruginosa VA 53383 (klinisches Isolat, Universitätsklinik Plastischen Chirurgie, BG Kliniken, Bergmannsheil, der Ruhr - Universität, Bochum, Deutschland), Pseudomonas aeruginosa VA 53476 (klinisches Isolat, Universitätsklinik der Plastischen Chirurgie, BG Kliniken, Bergmannsheil, Ruhr – Universität, Bochum, Deutschland), Pseudomonas aeruginosa VA 52519 (klinisches Isolat, Universitätsklinik der Plastischen Chirurgie, BG Kliniken, Bergmannsheil, Ruhr – Universität, Bochum, Deutschland). Bei den Stämmen, die DSMZ gelistet sind, handelt es sich um Qualitätskontrollstämme, die in der Antibiotikatestung üblich verwendet werden. Pilze: Candida albicans (klinisches Isolat , Prof. Sören Gatermann, Institut der Mikrobiologie, Ruhr - Universität, Bochum, Deutschland), Candida albicans (DSMZ 11948, ATCC 24433), Candida albicans (DSMZ 11225, ATCC 90028), Candida parapsilosis (DSMZ 5784, ATCC 22019), Issatchenkia orientalis 23 (Saccharomyces krusei; DSMZ 6128, ATCC 6258). Leztgenannter Stamm ist ein Qualitätskontrollstamm für die Austestung von Antimykotika. 2.1.5 Gebrauchsmaterialien Quadratische Petrischalen (10x10 cm; Greiner-Bio-One, Kremsmünster, Österreich); Petrischalen (rund; 8,5 cm Durchmesser; Greiner-Bio-One, Krems- münster, Österreich), Pipetten (Renner GmbH, Dannstadt, Deutschland), Sterile Pipettenspitzen (Renner GmbH, Dannstadt, Deutschland), Konisch zulaufende Mikrozentrifugenröhrchen (1,5 ml; Greiner, Kremsmünster, Österreich), Sterile Impfösen (Renner GmbH, Dannstadt, Deutschland), Zentrifugenröhrchen (15 ml; Greiner, Kremsmünster, Österreich), Serologische Pipetten (50 ml; Greiner, Kremsmünster, Österreich), Falcon-Röhrchen (50 ml; Greiner Kremsmünster, Österreich), 5 ml Reagenzgläser. 2.1.6. Geräte Spektrophotometer (UV – V – IS - Spektrometer, Perkin Elmer 555, Boston, Massachusetts, USA), Nivelliertisch (Greiner, Kremsmünster, Österreich), Gelstanze (3 mm Durchmesser; 9 mm Durchmesser, Universitätsklinik Plastischen Chirurgie, BG Kliniken, Bergmannsheil, Bochum, Deutschland), Schüttelwasserbad (B. Melsungen, Ruhr - Universität, Braun Deutschland), Dampfwasserbad (B. der Braun Melsungen AG, Melsungen AG, Melsungen, Deutschland), Inkubator (37°C; Heraeus, Heraeus Holding GmbH, Hanau, Deutschland), Millimeterlineal (Aristo, Rotring GmbH, Hamburg, Deutschland), Vortex-Schüttler (IKA, Staufen, Deutschland), Schablone (Universitätsklinik der Plastischen Chirurgie, BG Kliniken, Bergmannsheil, Ruhr - Universität, Bochum, Deutschland), Erlmeyer-Kolben (20 ml, 50 ml, 100 ml, 125 ml, 500 ml, 1000 ml, Greiner, Kremsmünster, Österreich), Zentrifuge (Hereaus Megafuge 3.0 R, Hereaus International GmbH, Hanau, Magnetrührer (IKA, Staufen, Deutschland), Autoklav (H+P Deutschland)), Labortechnik, Oberschleißheim, Deutschland) 2.1.7. Medien und Platten Trypticase Soy Broth (TSB; Soja-Bouillon; Difco, Detroit, Michigan, USA) 24 Underlay-Agar: 50 ml des 100 mM Natriumphosphatpuffers werden 5 ml SojaBouillon (Difco, Detroit, Michigan, USA) und 5 g Agarose in einem 1 L Erlenmeyerkolben zugesetzt, anschließend bis 500 ml mit deionisiertem Wasser aufgefüllt. Der pH-Wert wird auf 7,4 gehalten, falls nötig. Die Suspension wird in einem Dampfwasserbad zur Lösung gebracht und in 100 ml Erlenmeyerkolben zu 50 ml Portionen aliquotiert. Diese werden bei 121°C für 20 min autoklaviert und bis zum Gebrauch bei 4°C gelagert. Vor Gebrauch ist der Agar in einem Dampfwasserbad erneut zur Lösung zu bringen. Overlay-Agar: Dieser Agar enthält 60 g TSB Pulvermedium und 10 g Agarose pro 1 L deionisiertes Wasser. Die Suspension wird analog zum Underlay-Agar im Dampfwasserbad zu Lösung gebracht, anschließend in Reagenzgläsern zu 7,5 ml Portionen aliquotiert. Nach Autoklavierung bei 121°C für 20 min erfolgt Lagerung bei 4°C. Vor Gebrauch ist der Agar im Dampfwasserbad in Lösung zu bringen. 2.1.8. Computerprogramme StatView 5.0 (SAS Institute, Cary, North Carolina, USA) Microsoft Excel XP (Microsoft Corporation, USA) 2.1.9. Anzucht von Bakterien Eine einzelne Kolonie wird mit einer Impföse aufgenommen, in ein Reagenzglas mit 3-5 ml Soja-Bouillon transferiert und bei 37°C in einem Schüttelwasserbad für 18-24 h inkubiert. Ein Aliquot (500 µl) der resultierenden stationären Phase der Kultur wird in 50 ml frischen Soja-Bouillons eingebracht und für 2,5 h bei 37°C und 200 rpm inkubiert. 2.1.10. Versuchsdurchführung Die gezüchtete Subkultur wird in ein 50 ml Falconröhrchen transferiert und für 10 min bei 4°C und 880 g / 4000 U zentrifugiert. Das so entstehende bakterielle (o. fungale) Pellet wird einmalig mit 10 ml des kalten 10 mM Natriumphosphatpuffers gewaschen. Nach erneutem Zentrifugieren wird es in 5 ml desselben Puffers gelöst. Nun erfolgt die Messung der optischen Dichte (OD) der Probe. 1 ml wird in eine Messküvette eingebracht und die OD bei 620 nm unter Abgleich mit 1 ml des 25 Puffers gemessen. Die Konzentration der Bakterien (CFU/ml) in den verbleibenden 4 ml wird aus folgender Formel ermittelt: CFU/ml = OD620nm x 2,5 x 108 Für Pilze: CFU/ml = OD620nm x 0,5 x 108 Nun kann das Volumen der gewaschenen Bakteriensuspension berechnet werden, welches 2 x 107 CFU enthält (das Inoculum für 50 ml Underlay-Agar): (2 x 107 CFU) (CFU/ml) = gesuchtes Volumen Dieses Volumen der Bakterien wird in 50 ml geschmolzenen, im Wasserbad bei ca. 45-50°C gehaltenen Underlay-Agar eingebracht und sorgfältig gemischt. Nun wird ein 15 ml Aliquot in ein 50 ml Falconröhrchen transferiert, auf dem vorgewärmten Nivelliertisch auf die quadratische Petrischale (10x10 cm; Greiner), die vorher mittels der Schablone entsprechend eingeteilt wurde, gegeben und dort gleichmäßig verteilt. Nachdem sich der Agar in der Petrischale ein wenig gesetzt hat und transportfähig ist, wird er für 30 min bei 4°C gelagert, wo er sich endgültig verfestigt. Nun werden mittels der Metallstanze, die zur Sterilisation abgeflammt wird, Zylinder (3 mm im Durchmesser) in den Agar gestanzt. Es entstehen 9 Zylinder in einer 3 x 3 Anordnung. Die verschiedenen Proben werden nun in 5 µl Aliqouts, wie folgt, in die Zylinder eingebracht (Abb. 7): zu testendes Antimikrobielles Peptid: 7 Konzentrationen in die Probenfelder 1-7; Positivkontrolle: Antibiotikum Probefeld 8; Negativkontrolle: 0,01% Essigsäure in Probefeld 9. Nach dem Einfüllen werden die Platten zugedeckt und für 3 h bei 37°C inkubiert. Dabei wird jedes Peptid pro Mikroorganismus im Triplett, also dreifach untersucht. Bei dieser Zwischeninkubation kann die Testsubstanz in den Agar diffundieren. Nach 3 Stunden wird jeder Underlay-Agar mit 15 ml geschmolzenem Overlay-Agar begossen. Dieser beinhaltet die für das Bakterien/Pilzwachstum erforderlichen Nährstoffe in ausreichender Menge. Sobald sich der Agar verfestigt hat, wird er zugedeckt umgedreht und bei 37°C über Nacht inkubiert. Am nächsten Tag wird der Durchmesser der entstandenen Hemmzonen mit einem Millimeterlineal gemessen (auf 0,1 mm genau). Es werden drei Wiederholungen von jedem Peptid 26 (Platte) durchgeführt. Zur Ermittlung der MEK wird von dem Durchmesser der Hemmzone der Durchmesser des Loches (3 mm) subtrahiert und diese Differenz über eine Multiplikation mit dem Faktor 10 in so genannte Units überführt (10 U = 1 mm). Diese Units werden bezogen auf die jeweilige Konzentration in StatView eingegeben. Es wird eine logarithmische Regressionsanalyse durchgeführt, mit der Konzentration als unabhängiger und den Units als abhängiger Variable. Es wird nur ein Nullwert mit in die Analyse aufgenommen. Die Regressionsanalyse liefert eine Gleichung zur Beschreibung der Wachstumskurve dieser Art. Da die minimale effektive Konzentration äquivalent mit dem X-Achsenschnittpunkt der Kurve ist, kann die Gleichung zur Bestimmung der MEK mit Y=0 (da XAchsenschnittpunkt) nach X aufgelöst werden und die MEK so ermittelt werden. Abbildung 7: Beispiel für RDA-Testplatte: Zylinder 1-7 Konzentrationen des Host Defense Peptides, Zylinder S Negativkontrolle 0,01% Essigsäure, Zylinder A Positivkontrolle Antibiotikum Der Versuch dient der Bestimmung der minimalen effektiven Konzentration (MEK) (in µg/ml) Host Defense Peptide. Die Ermittlung dieser erlaubt es, die Aktivität 27 einzelner Peptide oder Peptidgruppen miteinander zu vergleichen. Eine MEK unter 5 µg/ml wird dabei als klinisch hochwirksam betrachtet. 2.2. Modified CLSI Microbroth Dilution Assay (MDA) In der folgenden Auflistung fanden nur zusätzliche oder vom RDA abweichende Gebrauchsmedien oder Methoden Erwähnung. 2.2.1. Chemikalien 1% Humanes Serum-Albumin (HSA Fraktion V, SERVA Electrophoresis GmbH, Heidelberg, Deutschland) 2.2.2. Host Defense Peptide Es wurden die gleichen Peptide verwendet wie beim Radial Diffusion Assay. Hier werden Stammlösungen von 1 mg/ml in 0,01% Essigsäure einzelner Peptide angefertigt. Diese werden dann zu 100 µl Proben aliquotiert und bei -80°C bis zum Versuchstag gelagert. 2.2.3. Gebrauchsmaterialien Neben den im RDA erwähnten Geräten fanden hier zusätzlich Verwendung 96Loch Gewebskulturplatten (U-Form; Greiner, Kremsmünster, Österreich), MultiKanal Pipette (10-100 µl, BRAND GMBH + CO KG, Wertheim, Deutschland). 2.2.4. Medien und Platten Müller-Hinton-Bouillon (MHB; Merck GmbH, Hohenbrunn, Deutschland) 2.2.5. Anzucht von Bakterien Eine einzelne Kolonie wird in 3-5 ml Müller-Hinton-Bouillon (MHB) in einem Reagenzglas transferiert. Dieses wird bei 37°C über Nacht inkubiert. Diese Kultur wird am nächsten Morgen 1:20 verdünnt, und zwar 50 µl Kultur zu 950 µl MHB in einer Messküvette zur Bestimmung der optischen Dichte (OD) bei 600 nm. Um die für den Versuch erforderliche Volumen mit einer Konzentration von 4 x 105 CFU/ml zu bestimmen, ist ein Bakterienabhängiger Umrechnungsfaktor (siehe Tabelle 3) notwendig. Dabei wird wie folgt gerechnet: 28 Volumen mit definierter CFU/ml = OD600 x (n x CFU/ml) 0,2 Tabelle 3: Beziehung zwischen CFU und Absorbenz [103] Bakterium E. coli, K. pneumoniae, Proteus mirabilis P. aeruginosa, A. baumannii MRSA, S. aureus, S. epidermidis, E. faecalis (n CFUs/ml) 0,2 A 600 nm 8 x 107 7,8 x 107 2 x 107 Im Folgenden wird ein Verdünnungsfaktor berechnet, anhand dessen die Bakteriensuspension soweit mit MHB verdünnt wird, bis sie die erforderlichen 4 * 105 CFU/ml enthält. Berücksichtigt werden muss, dass man pro Platte ca. 8 ml Suspension braucht. 2.2.6. Versuchsdurchführung Zunächst werden 100 µl Peptiddiluent in die Löcher A2 bis A12 einer 96Lochzylinder Gewebskulturplatten (U-Form; Greiner, Solingen, Deutschland) eingefüllt, dann 200 µl der Peptid- oder Antibiotika-Lösung in Loch A1. Nun transferiert man 100 µl aus A1 in das Loch A2, mischt gut, indem man die Flüssigkeit mit der Pipette auf- und abzieht, und transferiert 100 µl in A3, mischt erneut usw. Das ganze Prozedere führt man bis A11 fort, die 100 µl, die hier entnommen werden, werden verworfen. So dient A12 als Negativkontrolle, weil er nur den Peptiddiluenten beinhaltet und keine aktive Substanz. Mit einer MultiKanal-Pipette werden nun jeweils 100 µl Bakteriensuspension in jedes Loch der Reihe B-D eingebracht. Ein anderes Bakterium wird in die Löcher der Reihen F-H gefüllt. Nun werden mit der Multi-Kanal-Pipette 11 µl aus den Löchern der obersten Reihe mit der Peptid/Antibiotika-Lösung in das jeweils korrespondierende Loch der Bakteriensuspension enthaltenden Reihen eingebracht (A1 in B1, A2 in B2 etc. auch A1 in C1 etc. und A1 in D1 etc.). Die Gewebskulturplatten werden nach Ende dieser Prozedur zugedeckt und über Nacht bei 37° inkubiert. Am nächsten Morgen wird dann die minimale Hemmkonzentration (MHK) abgelesen. Es ist das letzte Loch, das eine klare Flüssigkeit enthält, (von 1 nach 12 lesend), in dem also kein bakterielles Wachstum (= keine Trübung) sichtbar ist. Die 29 Konzentration, die hier ausschlaggebend ist, wird von jeder der drei Reihen abgelesen. Zur Bestimmung der minimalen bakteriziden Konzentration (MBK) werden 10 µl aus den Löchern mit der MHK, der zweifachen, vierfachen und achtfachen MHK entnommen und in die mit 200 µl Soja-Bouillon gefüllten, korrespondierenden Löcher einer neuen Gewebsplatte transferiert, anschließend wird die Platte zugedeckt und erneut über Nacht bei 37°C inkubiert. Das letzte Loch, das eine klare Flüssigkeit enthält, (von 1-12 lesend) zeigt die Konzentration der MBK an. Sowohl die MHK als auch MBK werden dreifach bestimmt sowie unter Verwendung des Student´s T-Tests mittels StatView ausgewertet. Der Versuch ist das Standardwerkzeug zur Bestimmung der Aktivität einer antimikrobiellen Substanz, sei sie ein Antibiotikum, die im Versuch als positive Kontrollen eingesetzt wurden, oder wie hier ein Host Defense Peptid. Die Methode ist zur Bestimmung der MHK und MBK weltweit anerkannt, im Gegensatz zum RDA, bei dem eine MEK bestimmt wird, die noch nicht überall gleichermaßen als standardisierte Größe gilt. So ist in der Durchführung beider Methoden die Absicht zu sehen, sowohl ein Verfahren zu wählen, dessen Ergebnis standardisiert und mit den Resultaten herkömmlicher antimikrobieller Substanzen stets verglichen werden kann, als auch ein Verfahren, das im besonderen Maße auf die Host Defense Peptide modelliert wurde. 2.3. CLSI Microbroth Dilution Assay für Pilze In der folgenden Auflistung fanden nur zusätzliche oder vom RDA oder MDA abweichende Gebrauchsmedien oder Methoden Erwähnung. 2.3.1. Chemikalien Destilliertes Wasser; BaCl2 (0,048mol/L; 1,175% Gewicht/Volumen BaCl2 * 2 H2O; J.T.Baker, Deventer, Niederlande); H2SO4 (0,36 N; 1% Volumen/Volumen; J.T.Baker, Deventer, Niederlande) 2.3.2. Antimykotika Antimykotika: Clotrimazol mikrofein® (Caelo, Caesar&Lorentz GmbH, Hilden, Deutschland), Amphotericin B (Bristol-Myers, Squibb GmbH, Regensburg, Deutschland). Stammlösung des Clotrimazol wird angefertigt, indem 25 mg in 5 ml 30 Ethanol (J.T.Baker, Deventer, Niederlande) zu einer Konzentration von 5 mg/ml aufgelöst werden. Diese wird aliqoutiert und bei -80°C gelagert. 2.3.3. Medien 10,4 g des pulverförmige RPMI-1640-Mediums (Sigma-Aldrich Co., St. Louis, USA) werden in 900 ml destillierten Wassers aufgelöst. 34,53 g MOPS Puffer (Sigma-Aldrich Co., St. Louis, USA) werden hinzugefügt und verrührt, so dass eine finale Konzentration von 0,165 mol/L erreicht wird. Der pH-Wert wird auf 7,0 mit 1 M NaOH (J.T.Baker, Deventer, Niederlande) berichtigt. Destilliertes Wasser wird so zugefügt, dass das Volumen abschließend 1 L beträgt, dann steril filtriert und bei 4°C gelagert. 2.3.4. Puffer und Lösungen McFarland 0,5 Bariumsulfat Mischung Standard wird benutzt, um die Dichte des Inokulums zu standardisieren. 0,5 ml von 0,048 mol/L BaCl 2 (1,175% Gewicht/Volumen BaCl2*2H2O) werden zu 99,5 ml einer 0,18 mol/L (0,36 N) H2SO4 (1% Volumen/Volumen) hinzugefügt. Die korrekte Dichte des Mischungsstandards wird am Spektrophotometer überprüft. Mit einem Gerät, das einen 1 cm Lichtpfad besitzt und bei Gebrauch einer geeigneten Küvette, sollte die Auslöschung bei 625 nm zwischen 0,08 und 0,10 liegen, um die richtige Trübung des 0,5 McFarland Standards zu kennzeichnen. Diese Standard-Mischung wird anschließend in verschraubbare Falconröhrchen aliquotiert zu Portionen à 4-6 ml und bei Raumtemperatur im Dunklen gelagert. Vor dem Gebrauch wird der Standard auf einem Vortex Gerät geschüttelt. Die Trübung wird 3 Monate nach Herstellung überprüft. 2.3.5. Anzucht von Pilzen Das Inokulum besteht aus einigen Kolonien, die größer als 1 mm sind und von 24 h alten Kulturen der oben erwähnten Candida Spezies stammen. Die Kolonien werden in 3 ml sterilem, 0,145 mol/L Salzwasser (8,5 g NaCl in 915 ml H2O; 0,85% NaCl-Lösung) suspendiert. Die resultierende Suspension wird für 15 sec auf einem Vortex geschüttelt und anschließend die Dichte auf einen 0,5 McFarland Standard abgeglichen und nötigenfalls durch Lösung weiterer Kolonien 31 korrigiert. Weiterhin wird zunächst 1:50, und dann 1:20 mit Salzwasser verdünnt, um das erforderliche zweifache Test-Inokulum zu erhalten. 2.3.6. CLSI Microbroth Dilution Assay für Pilze Es wird zunächst über Verdünnung der Antimykotikastammlösung die für den Versuch erforderlichen Antimykotikalösungen hergestellt. Hierzu werden 25,6 µl der Stammlösung in 974,4 µl RPMI-1640 Medium gegeben. So wird eine Konzentration von 128 µg/ml erreicht, die dann über eine Verdünnung durch einen gleichvolumigen Anteil an Pilz-Inokulum auf 64 µg/ml dilutiert wird. Entsprechend stellt man fürs Host Defense Peptide eine Startkonzentration von 128 µg/ml her. Die 96-Loch-Gewebskulturplatte (U-Form; Greiner, Solingen, Deutschland) wird nun bei B1, C1 und D1 mit entweder 100 µl der Antimykotikalösung (128 µg/ml) oder der Peptidlösung (128 µg/ml) befüllt. 50 µl des RPMI-1640 Mediums werden in die Löcher 2-12 der Reihen B-D eingefüllt. 50 µl werden aus B1 entnommen und in B2 transferiert. Es wird viermal gemischt und wieder 50 µl von B2 in B3 transferiert, erneut viermal gemischt und erneut transferiert etc. Die 50 µl aus Loch 11 werden verworfen, weil die letzte Probe als Negativkontrolle dient. Entsprechend wird in Reihe C und D verfahren. Die Löcher 1-12 der Reihen B-D werden nun mit 50 µl des Pilz-Inokulums befüllt, so dass das endgültige Volumen in jeder Probe 100 µl beträgt. Die Platten werden zugedeckt und bei 35°C für 48 h inkubiert. Die minimale Hemmkonzentration (MHK) der Azol-Gruppe der Antimykotika ist definiert als die niedrigste Konzentration, bei der eine deutliche Abnahme der Trübung (Stufe 2) beobachtet wird. Entsprechendes Kriterium wird auch bei den Peptiden in der Auswertung zugrunde gelegt. Hier wird die Platte dreifach wiederholt. Dieser Versuch dient der standardisierten Bestimmung einer MHK bei Pilzen. Im eigentlichen Sinne ist er eine Abwandlung des oben beschriebenen Modified CLSI Microbroth Dilution Assay, der allerdings in erster Linie auf Bakterien modelliert ist, für Pilzkulturen. 32 3. Ergebnisse 3.1. Radial Diffusion Assay Dieser Versuch diente der Bestimmung der minimalen effektiven Konzentration (MEK) (in µg/ml) der Host Defense Peptide. Die Ermittlung dieser erlaubte es, die Aktivität einzelner Peptide oder Peptidgruppen miteinander zu vergleichen. Eine MEK unter 5 µg/ml wurde dabei als klinisch hochwirksam betrachtet [11]. Er wurde benutzt, um die antibiotische und antimykotische Aktivität der Host Defense Peptide, und zwar sowohl der natürlichen als auch ihrer synthetischen Veränderungen, gegen die Mikroorganismen, die für die meisten Infektionen menschlicher Verbrennungswunden verantwortlich sind, miteinander zu vergleichen. Hierbei fanden folgende Bakterien Verwendung: als gramnegative, P. aeruginosa, E. mirabilis und coli, K. drei pneumoniae, klinische Isolate Acinetobacter baumannii, (P. aeruginosa) Proteus aus menschlichen Verbrennungs-wunden, als grampositive, S. aureus, Enterococcus faecalis, S. epidermidis sowie der multiresistente C-MRSA. Die ATCC bzw. DSMZ gelisteten Bakterienstämme wurden speziell ausgesucht, denn es handelte sich dabei um vornehmlich bei Qualitätskontrollen der Antibiotikatestung verwendete Keime. Bei der Überprüfung der antimykotischen Aktivität gebrauchte man folgenden Pilz: Candida albicans. Insgesamt wurden dabei folgende Peptide in ihrer antimikrobiellen Aktivität verglichen, zum einen natürliche wie das Protegrin und das LL-37, zum anderen synthetische Peptide, wie Plectasin oder Novispirin G10, wie auch dessen Weiterentwicklungen RIR G10, R2 G10, oder auf Veränderungen des LL-37 basierende synthetische Peptide wie WS 22 N, WS 22. 3.1.1. Grampositive Bakterien Gegen die wichtigsten grampositiven Bakterienstämme (Tab. 4), die zu Wundinfektionen bei Brandverletzten führen, nämlich S. aureus, S. epidermidis und Enterococcus feacalis sowie den C-MRSA zeigte das synthetische Novispirin G10 die beste antimikrobielle Aktivität. Die MEK (minimale effektive Konzentration, in µg/ml) lag für alle erwähnten Stämme unter 1 µg/ml. Die MEK der synthetischen Modifikation des G10, R 2 G10, waren höher als die des G10, jedoch niedriger als die der weiteren Modifikation, nämlich des RIR G10. Dessen MEK lagen um 5 33 µg/ml für alle Keime. Im Vergleich der beiden natürlichen Peptide, LL-37 und Protegrin-1 waren die MEK des LL-37 höher als die des Protegrin-1 für alle getesteten Stämme. Auch die MEK der beiden synthetischen Modifikationen von LL-37, WS 22 N und WS 22, waren bei Testung aller Bakterien niedriger als die des natürlichen Stammpeptides. Das synthetische Peptid Plectasin wies signifikant niedrigere MEK-Werte als LL-37, jedoch signifikant höhere als die Modifikation WS 22 N. Tabelle 4: Im RDA demonstriert Novispirin G10 beste antimikrobielle Aktivität gegen grampositive Bakterien. (+ p<0,05 G10 vs. Rest) MEK in µg/ml Novispirin G10 C-MRSA + 0,38 ± 0,15 S. aureus S. epidermidis + 0,89 ± 0,40 0,37 ± 0,07 E. faecalis + 0,99 ± 0,20 RIR G10 5,08 ± 0,20 4,56 ± 0,04 4,98 ± 0,15 4,73 ± 0,06 R 2 G10 3,25 ± 0,78 1,52 ± 0,07 2,06 ± 0,12 2,33 ± 0,55 13,42 ± 0,25 14,46 ± 0,27 13,11 ± 0,70 14,29 ± 0,47 WS 22 N 0,93 ± 0,28 0,56 ± 0,03 0,62 ± 0,02 2,98 ± 0,65 WS 22 4,11 ± 0,14 1,03 ± 0,22 1,55 ± 0,05 3,83 ± 0,58 Plectasin 3,37 ± 0,00 2,49 ± 0,88 8,20 ± 2,20 10,60 ± 0,19 Protegrin-1 2,01 ± 0,06 0,92 ± 0,04 4,48 ± 0,23 4,46 ± 0,42 LL-37 3.1.2. Gramnegative Bakterien Gegen die wichtigsten gramnegativen Bakterienstämme (Tab. 5) demonstrierten mehrere Antimikrobielle Peptide ein breites Wirkspektrum (MEK < 5 µg/ml), darunter Novispirin G10, R2 G10, RIR G10, WS 22 N, WS 22 und Protegrin-1. So blieb die MEK von Novispirin bei vier von fünf getesteten Keimen unter 3 µg/ml, für E. coli 1,327 µg/ml, für K. pneumoniae 1,407 µg/ml, für P. aeruginosa 0,845 µg/ml, für Acinetobacter baumannii 2,411 µg/ml und lediglich für Proteus mirabilis 6,573 µg/ml. Damit war Novispirin G10 signifikant (p<0,05) antimikrobieller als die synthetische Modifikation RIR G10, deren Konzentrationen um 4 µg/ml lagen. Die andere synthetische Erweiterung der Novispirin Familie, R2 G10, wies bei E. coli und K. pneumoniae ähnliche Aktivitäten auf (1,547 µg/ml und 1,032 µg/ml) wie das Stammpeptid, war aber bei den Pseudomonaden, P. aeruginosa und Acinetobacter baumannii, sowie bei Proteus mirabilis signifikant (p<0,05) schlechter. Von den natürlichen Peptiden im Test überzeugte lediglich Protegrin-1 in seinem 34 antimikrobiellen Spektrum. LL-37 wies die signifikant schlechtesten Werte der MEK auf. Ganz anders stellten sich die Ergebnisse der synthetischen Modifikationen des LL-37, des WS 22 N und des WS 22, dar. Beide demonstrierten nicht nur eine signifikant (p<0,05) bessere Aktivität, sondern wiesen mit der MEK von 0,52 µg/ml des WS 22 N gegen P. aeruginosa auch die signifikant niedrigste MEK im Test auf. WS 22 zeigte dabei ein ausgeglichenes Spektrum. Die MEK für alle Bakterien, außer Proteus mirabilis (MEK von 13,353 µg/ml), lagen deutlich unter 5 µg/ml. Plectasin zeigte für P. aeruginosa, Acinetobacter baumannii und Proteus mirabilis keine antimikrobielle Aktivität, und für E. coli und K. pneumoniae mit einer MEK von jeweils 79,102 µg/ml eine sehr eingeschränkte. Tabelle 5: Gegen gramnegative Bakterien weist LL-37 im Vergleich zu den restlichen Peptiden (außer Plectasin) eine schwächere antimikrobielle Aktivität auf. (# p<0,05) MEK in µg/ml E. coli K. pneumoniea P. aeruginosa A. baumannii P. mirabilis Novispirin G10 1,32 ± 0,22 1,41 ± 0,51 0,84 ± 0,35 2,41 ± 0,43 6,57 ± 1,98 RIR G10 4,14 ± 0,12 3,65 ± 0,21 4,23 ± 0,10 3,93 ± 0,16 12,47 ± 0,89 R 2 G10 1,55 ± 0,36 1,03 ± 0,30 4,03 ± 0,24 3,46 ± 0,88 13,86 ± 0,22 # # # # LL-37 12,78 ± 0,65 13,74 ± 0,78 21,47 ± 6,70 14,59 ± 0,34 # 33,72 ± 1,13 WS 22 N 4,14 ± 0,18 4,283 ± 0,11 0,52 ± 0,04 2,43 ± 0,71 5,51 ± 1,97 WS 22 2,49 ± 0,51 1,24 ± 0,19 2,66 ± 0,59 2,69 ± 0,60 13,35 ± 0,51 79,10 ± 0,00 79,10 ± 0,00 0 0 0 2,00 ± 0,14 2,34 ± 0,10 3,26 ± 1,07 5,02 ± 0,19 7,03 ± 1,76 Plectasin Protegrin-1 In einem weiteren Test (Tab. 6) des Novispirin G10 gegen multiresistente, klinische Isolate des P. aeruginosa zeigte das Peptid sehr gute Aktivität für alle Isolate (VA 53476, VA 53383, VA 52519), denn die MEK blieb bei allen unter 4 µg/ml. Tabelle 6: Novispirin G10 beweist gegen multiresistente klinische Isolate des P. aeruginosa im Radial Diffusion Assay hocheffektive antimikrobielle Eigenschaften. MEK in µg/ml Novispirin G10 Pseudomonas Pseudomonas Pseudomonas Pseudomonas aeruginosa VA 53383 VA 53476 VA 52519 3,63 ± 0,19 1,97 ± 0,59 2,81 ± 0,06 1,97 ± 0,60 35 3.1.3. Radial Diffusion Assay gegen Pilze Bei diesem Test (Abb. 11) gegen Candida albicans demonstrierte Novispirin G10 die signifikant (p<0,05) höchste antimykotische Aktivität (0,783 µg/ml). Auch die synthetischen Modifikationen zeigten gute Wirksamkeit, wobei jedoch die MEK des R 2 G10 signifikant niedriger war als die des RIR G10. Sowohl das natürliche Peptid LL-37 als auch seine synthetischen Abkömmlinge WS 22 N und WS 22 wiesen im Vergleich zu den anderen Peptiden signifikant (p<0,05) schlechtere antimikrobielle Aktivitäten auf. Hier stellten jedoch die Modifikationen die bessere antimykotische Wirksamkeit gegenüber ihrem Stammpeptid. Tabelle 7: Besonders wirksam gegen Candida albicans ist Novispirin G10 im Vergleich der Peptide im Radial Diffusion Assay. (* p<0,05 G10 vs. Rest) MEK in µg/ml Candida albicans Novispirin G10 0,78 ± 0,02* RIR G10 4,88 ± 0,11 R 2 G10 1,90 ± 0,34 LL-37 31,28 ± 1,62 WS 22 N 6,92 ± 2,26 WS 22 6,53 ± 2,63 Zusammenfassend demonstrierten in dieser Testreihe des RDA mehrere Peptide ein breites Spektrum mit hoher Wirksamkeit (MEK < 5 µg/ml) in grampositivem wie –negativem Bereich. Hierzu zählten die Novispirin Gruppe mit Novispirin G10, RIR G10 und R 2 G10, die beiden synthetischen Modifikationen des LL-37, WS 22 N und WS 22, sowie Protegrin-1. Bis auf den Test gegen Proteus mirabilis blieben die MEK dieser Peptide zumeist deutlich unter 5 µg/ml, und damit in einem hocheffektiven Wirksamkeitsbereich. Die MEK des Plectasin war nur gegen die beiden S. aureus-Stämme so effektiv, LL-37 nur gegen den ATCC gelisteten S. aureus. Gegen Candida albicans bleiben nur die Peptide der Novispirin-Gruppe mit ihrer MEK unter 5 µg/ml. 3.2. Modified CLSI Microbroth Dilution Assay Dieser Versuch ist das Standardwerkzeug zur Bestimmung der Aktivität einer antimikrobiellen Substanz, sei sie ein Antibiotikum, die im Versuch als positive 36 Kontrollen eingesetzt wurden, oder wie hier ein Host Defense Peptid. Die Methode ist zur Bestimmung der MHK und MBK unter Mikrobiologen allgemein anerkannt, im Gegensatz zum RDA, bei dem eine MEK bestimmt wird, die noch nicht überall gleichermaßen als standardisierte Größe gilt. CLSI (Clinical and Laboratory Standards Institute) Microbroth Dilution Assay für Pilze diente der standardisierten Bestimmung einer MHK bei Pilzen. Er stellt eine Abwandlung des oben beschriebenen Modified CLSI Microbroth Dilution Assay für mykotische Kulturen dar. Beide Versuchsverfahren wurden gemäß den Richtlinien des CLSI durchgeführt. Die antimikrobiellen Aktivitäten der Peptide wurden gegenüber den gleichen Mikroorganismen, die schon im Radial Diffusion Assay Verwendung fanden, getestet. Ausnahme hiervon stellte der Dilution Assay für Pilze, bei dem 4 neue (ATCC und DSMZ gelistete) Pilzstämme aus der Familie des Candida albicans herangezogen wurden. Der MDA (Microbroth Dilution Assay) liefert als Ergebnisgröße die MHK (minimale Hemmkonzentration, in µg/ml) und die MBK (minimale bakterizide Konzentration, µg/ml), also per defintionem standardisierte Größen. Denn die MHK ist definiert als die niedrigste Konzentration einer antibakteriellen Substanz, die die Vermehrung eines Bakterienstammes unter definierten Bedingungen verhindert. Die MBK ist wiederum die niedrigste Konzentration einer antibakteriellen Substanz, die einen Bakterienstamm (99,9 % der Population unter definierten Bedingungen) abtötet. 3.2.1. Grampositive Bakterien In der Novispirin-Gruppe (Tab. 8), bestehend aus Novispirin G10, RIR G10 und R2 G10, waren es vor allem die Modifikationen des Stammpeptides Novispirin, also RIR G10 und R2 G10, die ein gutes Wirkprofil hinsichtlich der MHK und MBK für die getesteten Bakterien (S. aureus, C-MRSA, S. epidermidis und E. faecalis) vor allem bei den Staphylokkoken demonstrierten. Beim S. aureus wiesen erwähnte Peptide signifikant (p<0,05) niedrigere MHK auf als Novispirin G10. Die signifikant niedrigste MHK beim C-MRSA zeigte RIR G10. Sehr niedrige MHK wiesen sowohl Novispirin als auch RIR G10 und R2 G10 beim S. epidermidis auf. Die signifikant beste Bakterizidie stellte R 2 G10 sowohl beim S. aureus als auch beim S. epidermidis. Die MBK von RIR G10 (8,333 µg/ml) und 37 R 2 G10 (10,417 µg/ml) für MRSA lagen signifikant (p<0,05) niedriger als die des Novispirin G10 (50 µg/ml). Schwächere Wirksamkeit wurde im grampositiven Bereich für die gesamte Novispirin-Gruppe beim E. faecalis festgestellt, denn die MHK aller Peptide lag bei 50 µg/ml. Nur Novispirin G10 erreichte eine MBK (66,667 µg/ml). Tabelle 8: Im Modified CLSI Microbroth Dilution Assay demonstrieren besonders die RIR G10 und R2 G10 niedrige MHK- und MBK-Werte bei grampositiven Bakterien. (+ p<0,05 RIR G10 vs. Rest, # p<0,05 R2 G10 vs. Rest). In µg/ml Novispirin G10 C-MRSA S. aureus S. epidermidis E. faecalis 50,00 ± 0,00 6,25 ± 0,00 3,13 ± 0,00 50,00 ± 0,00 50,00 ± 0,00 16,67 ± 4,16 6,25 ± 0,00 66,67 ± 33,33 + 1,56 ± 0,00 0,78 ± 0,00 50,00 ± 0,00 8,33 ± 2,08 12,50 ± 0,00 1,56 ± 0,00 0 # 50,00 ± 0,00 MHK Novispirin G10 MBK RIR G10 MHK RIR G10 MBK 3,13 ± 0,00 R 2 G10 MHK 6,25 ± 0,00 1,56 ± 0,00 R 2 G10 MBK 10,41 ± 2,08 4,17 ± 1,04 0,65 ± 0,13 0 LL-37 MHK 0 50,00 ± 0,00 0 100,00 ± 0,00 LL-37 MBK 0 0 0 0 WS 22 N MHK 6,25 ± 0,00 1,56 ± 0,00 3,13 ± 0,00 12,50 ± 0,00 WS 22 N MBK 10,41 ± 2,08 9,38 ± 3,13 7,29 ± 2,75 25,00 ± 0,00 WS 22 MHK 6,25 ± 0,00 3,13 ± 0,00 6,25 ± 0,00 12,50, ± 0,00 WS 22 MBK 12,50 ± 0,00 10,42 ± 2,08 20,83 ± 4,17 29,17 ± 11,02 Plectasin MHK 6,25 ± 0,00 6,25 ± 0,00 25,00 ± 0,00 0 Plectasin MBK 50,00 ± 0,00 25,00 ± 0,00 100,00 ± 0,00 0 0,39 ± 0,00 Ampicillin MHK 0,39 ± 0,00 1,56 ± 0,00 Ampicillin MBK 1,30 ± 0,26 4,16 ± 1,04 Vancomycin MHK 0,19 ± 0,00 0,39 ± 0,00 Vancomycin MBK 0,26 ± 0,06 0,39 ± 0,00 In der LL-37-Gruppe (Tab. 8), die sich aus dem Stammpeptid LL-37 sowie seinen synthetischen Modifikationen WS 22 und WS 22 N zusammensetzte, waren es in erster Linie die synthetischen Peptide, die im grampositiven Bereich für alle Bakterien ein gutes Wirkungsspektrum hinsichtlich der MHK aufzeigten. 38 Beim S. aureus wies WS 22 N die signifikant (p<0,05) niedrigste MHK auf. Die MBK beider synthetischer Modifikationen lagen um 10 µg/ml. Für C-MRSA bot sich im Testergebnis ein ähnliches Bild, während die MHK für beide Veränderungen gleich bei 6,25 µg/ml war, zeigte WS 22 N etwas niedrigere MBK (10,417 µg/ml) als WS 22 (12,5 µg/ml). Beim S. epidermidis wartete erneut WS 22 N mit der signifikant (p<0,05) niedrigeren MHK und diesmal auch MBK gegenüber WS 22 auf. Auch für E. faecalis wiesen sowohl WS 22 N als auch WS 22 gute Wirksamkeit dar. LL-37 zeigte eingeschränkte Wirksamkeit für S. aureus (MHK: 50 µg/ml) und E. faecalis (MHK 100 µg/ml). Im Vergleich mit den Novispirinen zeigten die synthetischen Modifikationen des LL-37 wie dort die Modifikationen des Novispirins ein ähnliches Wirkungsspektrum, mit dem Unterschied, dass die WS 22 N und WS 22 signifikant (p<0,05) besser gegen E. faecalis wirksam sind. Plectasin (Tab. 8) zeigte gute Wirksamkeit im grampositiven Bereich vor allem gegen Staphylokokken, auch bei multiresistenten (C-MRSA). Die MBK war bei CMRSA und S. epidermidis eingeschränkt. 3.2.2. Gramnegative Bakterien Die Novispirin-Gruppe (Novispirin G10, RIR G10 und R 2 G10) (Tab. 9) zeigte geschlossen eine sehr gute Wirksamkeit im gramnegativen Bereich hinsichtlich der MHK und MBK für die getesteten Keime außer Proteus mirabilis. Für E. coli lagen alle MHK- und MBK- Werte der Novispirine unter 5,5 µg/ml. Für K. pneumoniae befanden sich alle gemessenen MHK und MBK sogar unter 1,5 µg/ml. Bei P. aeruginosa wies R 2 G10 die signifikant (p<0,05) niedrigste MHK und MBK auf. Die MHK und MBK von RIR G10 und R 2 G10 lagen beim Acinetobacter baumannii deutlich unter 2 µg/ml. Gegen Proteus mirabilis zeigte keines der drei Peptide Wirksamkeit. Das Wirkspektrum der LL-37-Gruppe (Tab. 9) (das natürliche Peptid LL-37 sowie die beiden synthetischen Modifikationen WS 22 N und WS 22) stellte sich im gramnegativen Bereich, wie folgt dar: sehr gute Wirksamkeit von WS 22 N und WS 22 gegen Acinetobacter baumannii, E. coli und K. pneumoniae sowie eingeschränkte Wirkung gegen P. aeruginosa und fehlende gegen Proteus mirabilis. LL-37 war eher eingeschränkt wirksam gegen die drei ersterwähnten 39 Bakterienstämme. So demonstrierten WS 22 N und WS 22 jeweils niedrige MHKund MBK-Werte bei E. coli. Bei K. pneumoniae wiesen WS 22 und WS 22 N MHK und MBK von unter 5,5 µg/ml auf. Bei Acinetobacter baumannii blieben die MHK und MBK beider Peptide sogar unter 2,2 µg/ml. Tabelle 9: Bis auf Proteus mirabilis zeigen Novispirin G10, RIR G10 und R2 G10 hocheffektive Aktivität gegen gramnegative Bakterien. (Schattiert die MBK; + p<0,05 R 2 G10 vs. Rest) E. coli K. pneumoniea P. aeruginosa A. baumannii P. mirabilis 4,17 ± 1,04 1,30 ± 0,26 6,25 ± 0,00 1,17 ± 0,39 0 G10 MBK 5,21 ± 1,04 1,30 ± 0,26 6,25 ± 0,00 3,12 ± 1,56 0 RIR G10 MHK 2,60 ± 0,52 0,39 ± 0,00 6,25 ± 0,00 1,30 ± 0,26 0 RIR G10 MBK 2,60 ± 0,52 1,04 ± 0,26 6,25 ± 0,00 1,30 ± 0,26 0 + 0,91 ± 0,34 3,13 ± 1,56 + 0 4,17 ± 1,04 0,91 ± 0,34 3,13 ± 1,56 0,65 ± 0,13 0 LL-37 MHK 33,33 ± 8,33 100,00 ± 0,00 66,67 ± 16,67 12,50 ± 0,00 0 LL-37 MBK 58,33 ± 22,05 0 100,00 ± 0,000 20,83 ± 4,17 0 WS 22 N MHK 3,13 ± 0,00 3,13 ± 0,00 50,00 ± 0,000 1,56 ± 0,00 0 WS 22 N MBK 8,33 ± 2,08 3,13 ± 0,00 66,67 ± 16,67 2,08 ± 0,52 0 WS 22 MHK 5,21 ± 1,04 3,13 ± 0,00 100,00 ± 0,000 1,56 ± 0,00 0 WS 22 MBK 5,21 ± 1,04 5,21 ± 1,04 166,67 ± 33,33 1,56 ± 0,00 0 3,13 ± 0,0 0,39 ± 0,00 12,50 ± 0,0 6,25 ± 3,13 3,13 ± 0,00 3,13 ± 0,00 MHK/MBK in µg/ml Novispirin G10 MHK Novispirin R 2 G10 MHK R 2 G10 MBK 2,08 ± 0,52 0,39 ± 0,00 Ampicillin MHK Ampicillin MBK Imipenem MHK Imipenem MBK 16,66 ± 2,08 6,25 ± 0,00 6,25 ± 0,00 33,33 ± 16,66 40 Die Wirkung der LL-37-Gruppe gegen P. aeruginosa war eingeschränkt sowohl in der MHK der Peptide als auch in der MBK (kein Wert unter 50 µg/ml). Wie schon in der Novispirin-Gruppe äußerte sich auch hier die fehlende Wirksamkeit gegen Proteus mirabilis. Plectasin zeigte keinerlei Wirksamkeit im gramnegativen Bereich für die getesteten Keime. Im Vergleich der beiden Peptidgruppen, Novispirine und LL-37, zeigte sich deutlich das breitere Wirkspektrum der Novispirine im gramnegativen Bereich. Bei der LL- 37 Gruppen zeigten zum einen nicht alle Peptide, LL-37 wies eher eingeschränkte Wirksamkeit auf für E. coli, K. pneumoniae und P. aeruginosa, die gleiche Wirksamkeitskonstanz. Für WS 22 N und WS 22, die ein gutes Wirkspektrum kennzeichneten, lagen die VergleichsMHK und MBK auch signifikant (p<0,05) höher als bei den Novispirinen. Zusammenfassend zeigte die MDA-Testung der Peptide gegen die Keime aus dem grampositiven wie –negativen Bereich in erster Linie das breite antimikrobielle Spektrum dieser Peptide, hier seien vor allem die NovispirinGruppe und die synthetischen Abkömmlinge des LL-37 erwähnt. Das Beispiel des Plectasins verdeutlicht, dass die Entwicklung spezifischer Peptide mit einem engen Wirkspektrum durch aus möglich ist, lag doch die Hauptwirksamkeit dieses Peptides vor allem in der Aktivität gegen S. aureus. Zugleich weist die Testung auch darauf hin, sei es durch die fehlende Wirksamkeit der Peptide gegen Proteus mirabilis oder die eingeschränkte Aktivität der Novispirine gegen E. faecalis, dass auch in einem sehr breiten Spektrum Lücken vorhanden sein können. 3.2.3. Vergleich mit Standardantibiotika Die MHK und MBK von Ampicillin wurden beim S. aureus, E. faecalis und E. coli mit der MHK und MBK der beiden Peptidgruppen sowie des Plectasin verglichen. Beim S. aureus (DSMZ 1104, ATCC 25923; Tab. 8) wies Ampicillin zwar die signifikant (p<0,05) niedrigste MHK und MBK auf, jedoch waren die MHK von RIR G10, R 2 G10 und WS 22 N in einem ähnlichen als hocheffektiv anzusehendem Wirkungsbereich. Beim E. faecalis (DSMZ 2570, ATCC 29212; Tab. 8) zeigte Ampicillin erneut die im Vergleich signifikant niedrigste (p<0,05) MHK und MBK auf. Hier waren die 41 MHK der Peptide deutlich höher, die besten im Test waren die MHK der Modifikationen von LL-37, WS 22 N und WS 22, mit jeweils 12,5 µg/ml. Beim E. coli (DSMZ 1103, ATCC 25922; Tab. 9) lag die MHK des Ampicillins signifikant (p<0,05) höher als die der synthetischen Novispirine, RIR G10 und R2 G10. Die MHK der restlichen Peptide, bis auf die von LL-37 und Plectasin, waren darüber hinaus in einem sehr ähnlichen Effektivitätsbereich wie dem des Antibiotikums (3,125 – 5,208 µg/ml). Die MBK aller Peptide (erneut mit Ausnahme des LL-37 und Plectasin) waren signifikant (p<0,05) niedriger als die des Ampicillins. Die Peptide bewiesen bei besserer Hemmung des Wachstums auch die bessere Bakterizidie gegenüber dem Bakterium als das Antibiotikum. Die MHK und MBK von Vancomycin wurden beim C-MRSA und S. epidermidis (DSMZ 1798, ATCC 12228) mit der getesteter Peptiden verglichen. Beim MRSA (Tab. 8) wies Vancomycin sowohl die signifikant (p<0,05) niedrigste MHK als auch MBK auf. Das beste Peptid war hierbei RIR G10, eine synthetische Variante des Novispirin G10, mit einer MHK von 3,125 µg/ml und einer MBK von 8,333 µg/ml. Beim S. epidermidis (DSMZ 1798, ATCC 12228; Tab. 8) konnten die synthetischen Novispirine R 2 G10 (0,391 µg/ml) und RIR G10 (0,781 µg/ml) eine MHK in ähnlichen Bereich wie das Antibiotikum (0,39 µg/ml) erreichen. Auch bei der MBK, obwohl das Antibiotikum hier mit 0,39 µg/ml den signifikant (p<0,05) niedrigsten Wert stellte, lagen die beiden Peptide der Novispirin-Gruppe mit ihren Werten (0,651 µg/ml und 1,563 µg/ml) ebenfalls in diesem hocheffektiven Bereich. Schließlich wurden auch die MHK und MBK von Imipenem bei K. pneumoniae, P. aeruginosa und Acinetobacter baumannii mit den MHK und MBK der getesteten Peptide verglichen. Bei K. pneumoniae (DSMZ 681, ATCC 10031; Tab. 9) erreichte Imipenem eine MHK von 0,391 µg/ml. In diesem hocheffektiven Bereich der MHK bleiben auch die MHK der Novispirin-Gruppe (Novispirin G10, RIR G10 und R 2 G10) mit Werten zwischen 0,391 und 1,303 µg/ml. Bei der MBK lagen die Werte der Peptide sogar signifikant (p<0,05) niedriger als die des Antibiotikums (Ausnahmen hiervon: LL-37 keine MBK und WS 22 mit einer MBK von 5,208 µg/ml). 42 Ein ähnliches Ergebnisbild brachte der Vergleich bei der Testung gegen P. aeruginosa (DSMZ 1117, ATCC 27853; Tab. 9) hervor. Die MHK von Imipenem wies den gleichen Wert auf, wie die von R 2 G10 (beide 3,125 µg/ml). Die MHK der restlichen Novispirine lag bei 6,25 µg/ml. Bei der MBK lag das Antibiotikum dann signifikant (p<0,05) höher als das Peptid R 2 G10. Auch die restliche MBK der Novispirin-Gruppe waren bei 6,25 µg/ml. Bei Testung gegen Acinetobacter baumannii (DSMZ 30008, ATCC 19606; Tab. 9) lag die MHK des Imipenem signifikant (p<0,05) höher als die MHK der Novispirine und auf gleichem Level wie der synthetischen LL-37 Modifikationen. Die MBK (6,25 µg/ml) des Imipenems lag sogar signifikant (p<0,05) höher als sowohl die der gesamten Novispirine als auch der synthetischen LL-37-Peptide. Damit demonstrierten die Peptide hier deutlich ihre hervorragende Fähigkeit zur Hemmung des bakteriellen Wachstums als auch die eigentlich ihrer Wirksamkeit zugrunde liegende bakterizide Aktivität, die in diesen Fällen sogar die Möglichkeiten des Standardantibiotikums Imipenem überstieg. Insgesamt zeigte der Vergleich der MHK- und MBK-Werte von Standardantibiotika und der MHK und MBK von Host Defense Peptiden deutlich die Vorteile der Peptide. Zum einen kam hier deutlich das weite Spektrum der Peptide zutage. Während sich die Wirksamkeit der einzelnen Antibiotika auf zumeist 2 Keime beschränkte, auch aus gleichem Grambereich, reichte das Spektrum von Peptiden wie den Novispirinen oder den synthetischen Abkömmlingen der LL-37-Gruppe über den größten Teil des grampositiven wie gramnegativen Bereiches. Ferner lag die Fähigkeit zur Wirksamkeit der Peptide im Gegensatz zu den verwendeten Antibiotika häufig in der gleichen MHK und MBK. Dies lässt schlussfolgern, dass hier die Hemmung des Wachstums auf einer tatsächlichen Abtötung der Bakterien beruhte. Hier schnitten die Antibiotika wie Imipenem oder Ampicillin schlechter ab, sie bewirkten eine sehr gute Hemmung des bakteriellen Wachstums durch Mechanismen, die nicht auf einer Abtötung des Bakteriums gründeten. 43 3.3. Modified CLSI Microbroth Dilution Assay für Pilze Dieser Versuch sei als Analogon zum Modified CLSI (Clinical and Laboratory Standards Institute) Microbroth Dilution Assay zu betrachten. Er berücksichtigte in besonderer Weise die Erfordernisse der Kultivierung von Pilzen zum Zwecke einer antimikrobiellen Testung. Im Test fanden folgende Pilzkulturen Verwendung Candida albicans ATCC 24433, Candida albicans ATCC 90028, Candida parapsilosis ATCC 22019, Issatchenkia orientalis ATCC 6258. Als Peptid wurde Novispirin G10 getestet, Als Antimykotikum benutzte man Clotrimazol. Die Ergebnisse (Tab. 10) demonstrierten die im Vergleich zum Clotrimazol eingeschränkte antimykotische Aktivität des G10, lediglich gegen Candida parapsilosis ATCC 22019 lag sie bei 3 µg/ml und erreichte damit ein effektives Niveau. Im Vergleich dazu lag die MHK von Clotrimazol bei 0,25 µg/ml. Tabelle 10: Nur gegen Candida parapsilosis demonstriert Novispirin G10 eine effektive Hemmkonzentration im Microbroth Dilution Assay für Pilze. (+ p<0,05 Clotrimazol vs. Novispirin G10) Candida Candida Candida Issatchenkia albicans ATCC albicans ATCC parapsilosis orientalis 24433 90028 ATCC 22019 ATCC 6258 Novispirin G10 64,00 ± 29,0 43,00 ± 21,0 6,00 ± 3,00 53,00 ± 26,0 Clotrimazol 0,25 ± 0,00 MHK in µg/ml + 0,25 ± 0,00 + 0,25 ± 0,00 + + 0,25 ± 0,00 44 4. Diskussion Bakterielle Wundinfektionen in der Chirurgie verzögern oder kehren gar den Heilungsprozess im Krankenhausstation Mikrokosmos können alle der Wunde um. erdenklichen Im Alltag der Einschränkungen der Abwehrmechanismen zu solchen Wundinfektionen führen. Besonders betroffen sind dabei ältere Menschen, sowie Patienten die chirurgischen Eingriffen unterzogen werden. Im Zeitalter steigender Kosten und der Budgetierung des Krankenhausaufenthaltes, die zu einem immer höherem Druck auf den behandelnden Arzt führen, Infektionen, vor allem Wundinfektionen in der Chirurgie, zu verhindern, um so die Heilung und damit den Aufenthalt des Krankenhauspatienten nicht zu verzögern, bleibt es vor dem Hintergrund des zunehmenden Aufkommens an virulenten und der Antibiotikatherapie gegenüber resistenten Bakterien zugleich immer weniger aus, dass Wundinfektionen vorkommen, deren Verlauf durch den Mangel an Therapiemöglichkeiten prolongiert wird. In diesem Lichte existiert schon seit längerer Zeit ein dringender Bedarf an alternativen Therapien von Infektionen. Im Hinblick darauf eröffnet sich mit den Antimikrobiellen Peptiden ein viel versprechendes Forschungsfeld. Die derzeitige Studienlage spricht dafür,, dass die so genannten endogenen Antibiotika eine Schlüsselrolle in den Immunsystemen nahezu aller tierischen Spezies einnehmen. Diese Proteine stellen essentielle Komponenten des angeborenen Teils der Körperabwehr dar, weil sie als Effektorsubstanzen agieren, die die Fähigkeit besitzen ein breites Spektrum von Mikroorganismen zu zerstören [11, 64, 66]. Im Gegensatz zur hohen Spezifität der adaptiven Immunität, die es diesem System unmöglich macht, schnell genug zu reagieren um einen geschwächten Organismus gegen eine akute Invasion verschiedener Pathogene zu verteidigen. Der angeborene Teil des Immunsystems hat eben diese Kapazitäten die es ihm ermöglichen schnell und unspezifisch auf Angriffe zu antworten, durch diese Fähigkeiten übernimmt es die Funktion der ersten Verteidigungslinie gegenüber allen Arten von Infektionen. Einen zentralen Bestandteil dieser Verteidigungsstrategie stellen die Antimikrobiellen Peptide dar. Viele solcher 45 natürlich vorkommender Peptide sind bereits charakterisiert worden, solche wie die Defensine, Protegrine, Lysozyme oder die Magainine [121]. Mit der Aufschlüsselung der genauen Aminosäurensequenz, der Konformation und der räumlichen Ausfaltung, also der Dekodierung von primärer, sekundärer und tertiärer Struktur erlangte man die Möglichkeit zur Modifikation dieser und damit auch zur Veränderung der Funktion des natürlichen Peptides [107]. Was folgte, war die eigenständige Herstellung neuer Peptide auf der Basis der entweder natürlichen oder bereits veränderten synthetischen Peptide, z. B. die hier verwendeten Novispirine G10, RIR G10 sowie R2 G10 oder auch die Abkömmlinge des LL-37 WS 22 und WS 22 N. In der Folgezeit galt es nun in vitro Testmethoden zu finden, die einer sinnvolle Überprüfung vor allem der antimikrobiellen Fähigkeiten gewährleisteten. Mit dem Radial Diffusion Assay (RDA) und dem CLSI Microbroth Dilution Assay (MDA für Bakterien und für Pilze) stellten wir zwei solcher Testmethoden zur Überprüfung der antimikrobiellen Fähigkeiten der Peptide vor [103]. Im Gegensatz zu vielen anderen Studien zuvor [6, 90, 97, 99, 100, 102, 105, 107], die sich vor allem mit der Demonstration der in vitro Fähigkeiten auf einzelne Bakteriengruppen oder gar Bakterien beschränkten, wurde in dieser Arbeit ein systematischer Vergleich der antimikrobiellen Fähigkeiten vollzogen. Hierzu wurde ein breite Auswahl aus dem Spektrum der Wundinfektionen verursachenden Keimen herangezogen. Die Testung der Agenzien erfolgte unter den definierten Standardbedingungen. Dies bedeutete, dass es in der jeweiligen Testreihe möglich war eine Effektivitätsvariable für jedes Peptid zu ermitteln, die quantitativ in der Lage war, die antimikrobielle Aktivität des Peptides wiederzugeben. Im Falle des RDA ermittelte man so die MEK (minimale effektive Konzentration; in µg/ml). Beim MDA erlangte man sogar Kenntnis über die Fähigkeit der Peptide hinsichtlich ihrer Möglichkeiten zur Hemmung des bakteriellen Wachstums oder zur potenziellen direkten Abtötung der Keime. Die MHK spiegelte dabei die Hemmung dar (minimale Hemmkonzentration; in µg/ml), während die MBK das unmittelbare Abtöten der Keime im Test anzeigte (minimale bakterizide Konzentration; in µg/ml). Durch die Aufrechterhaltung identischer definierter Standardbedingungen, 46 die auch bei der Testung von Antibiotika beim Microbroth Dilution Assay bestehen, war es sogar möglich beim MDA die antimikrobielle Fähigkeiten von Peptiden und klinisch eingesetzten Antibiotika gegenüberzustellen. Unterstützt wurde dies Vorhaben durch die Verwendung so genannter Quality Control Strains, also bakteriellen Stämme, die speziell für die antimikrobielle Testung von z. B. Antibiotika Verwendung finden. Hinsichtlich des Spektrums dieser Stämme griff man im Test neben solchen Kontrollstämmen auch auf klinische Isolate zurück, die wiederum besondere Fähigkeiten wie erhöhte Pathogenität oder Multiresistenz aufwiesen. Insgesamt umfasste die Testbreite unter den Mikroben die zehn häufigsten Wundinfektionen verursachenden Keime. Auch wenn die oben genannten Verfahren einzeln oder zum Teil gemeinsam bereits vorher schon in zahlreichen Studien eingesetzt wurden [6, 90, 97, 99, 100, 102, 105], so war es diese Arbeit, die sich zum Ziel nahm, Ergebnisse beider Methoden einander gegenüber zu stellen. Auch wenn es in der Konsequenz bedeutete, dass hinsichtlich der Wirkung einzelner Peptide deutliche Unterschiede in diesem Vergleich gesehen wurden. In den Ergebnissen auffällig sind Unterschiede zwischen den Ergebnissen für die MEK und MHK der Host Defense Peptide, hier zum Beispiel das Novispirin G10. Für eine definitive Erklärung reichen die hier erbrachten Ergebnisse nicht aus. Vermutlich liegen diese Unterschiede aber in der Rolle von Proteinasen, die als Virulenzfaktoren der benutzten Bakterien bekannt sind sowie zur Hemmung der Peptide führen und im Müller-Hinton-Bouillon (MDA) eher eine Wirkung als im Agargel (RDA) entfalten [95]. Daneben könnte auch die Zusammensetzung der Medien hier entscheidend sein. So ist bekannt, dass Glykosaminglykane Host Defense Peptide in ihrer antimikrobiellen Wirkung hemmen [7]. Hier können die Stärke oder der als Fleischinfus benannte Teil des Müller-Hinton-Bouillon eine hemmende Rolle spielen. Neben den Unterschieden zwischen den beiden Methoden gibt es noch Unterschiede, de man als stammspezifisch bezeichnen kann, weil sie den Staphylococcus aureus und den C-MRSA betreffen. So demonstriert erneut die MDA-Versuchsreihe deutliche Unterschiede in der MHK z. b. für Novispirin G10 bei beiden Bakterien. Eine mögliche Erklärung für diese Beobachtung liegt sicherlich in veränderten Virulenzfaktoren der Bakterien. So sieht man bei einigen 47 Spezies von Staphylococcus aureus ein neues Membranprotein MprF, welches die negativ geladenen Phospholipide der bakteriellen Zytoplasmamembran mit LLysin modifiziert. Dies führt zu einer Änderung der Ladung und damit zu Abschwächung der antimikrobiellen Wirkung des Host Defense Peptides [59]. Hinsichtlich der einzelnen Testverfahren wurde in dieser Arbeit bezogen auf die bereits bekannten Peptide (Novispirin G10, Protegrin-1) die in vitro Ergebnisse, was die antimikrobielle Fähigkeiten anbetrifft, früherer Studien bestätigt [6, 90, 102, 105, 107]. Daneben konnte anhand der Auswahl der verschiedenen Bakterienstämme auch ein Stamm mit einer natürlichen Resistenz gegenüber den HDP (Proteus mirabilis) bestätigt werden. Diese Resistenz ist auf veränderte Lipopolysaccharide des Bakteriums zurückzuführen [75], so wird das anionische Lipid A durch die kationische Aminoarabinose ausgetauscht. Es kommt zu einer Veränderung der Ladung auf der Oberfläche der Bakterienwand und damit quasi zu einer Wirkungsminderung des über die anionische Ladung bindenden kationischen Peptides. [30] Weiterhin demonstrierte sich auch in dieser Studie gegenüber zahlreichen anderen [93, 97, 107, 110], die die Wirksamkeit einer AP-Familie bzw. synthetischen Modifikationen von AP im Vergleich zu ihrem Ursprungspeptiden untersuchten, dass aus einer solchen Modifikation heraus die potenzielle Erweiterung des antimikrobiellen Spektrums resultiert. Wie die Erweiterung und Steigerung der antimikrobiellen Fähigkeiten in der LL-37 Gruppe infolge einer veränderten Aminosäurenstruktur des Stammpeptides durch den Austausch einer Aminosäure eindrucksvoll zeigte, können durch die Weiterentwicklung der Peptide, und diese liegt in der synthetischen Modifikation, nicht nur bestehende Lücken im Spektrum geschlossen, sondern auch bestehende antimikrobielle Fähigkeiten potenziert werden. Es muss allerdings in Zukunft weiterhin untersucht werden, ob solche Peptidresistenzen wie oben beschrieben auch mit einer gezielten Veränderung des Peptides umgangen werden können. Die Testung der fungiziden Eigenschaften von Host Defense Peptiden mittels MDA ist zunächst einmalig in der Literatur, offenbarte jedoch einige Diskrepanzen. So fand sich im Versuchsfeld des RDA eine deutliche antimikrobielle Aktivität, die 48 jedoch unter den Bedingungen des Modified Assay for Fungi nicht nachvollzogen werden konnte. Hier stellten sich ähnliche Vermutungen, die schon zur Erklärung der Unterschiede von Ergebnissen des RDA- und des MDA-Testreihen herangezogen wurden. Aus früheren Versuchen mit Host Defense Peptiden weiß man um ihre Empfindlichkeit, vor allem hinsichtlich ihrer antimikrobiellen Aktivität, gegenüber den unterschiedlichen Medien und z. B. dem Salzgehalt des Mediums [5]. Das Problem, was sich hieraus stellte, ist das der definierten Bedingungen, unter den Testreihen wie der MDA durchgeführt werden. Diese definierte Bedingungen zu verändern, und hierzu gehörte auch das Medium, bedeutete den Vergleich aufgeben. Erzielten die Peptide im Falle eines anderen Mediums entsprechend andere MHK-Werte, so könnte man diese nicht mit den Ergebnissen von Antimykotika vergleichen, weil die erforderlichen definierten Bedingungen in der Testung der Peptide nicht vorgeherrscht hätten. Hier ist es weiteren Projekten vorbehalten, das Standardverfahren einer für die HDP geeigneter Modifikation zu unterziehen. Auch besonders in dieser Arbeit ist der gesuchte Vergleich der Fähigkeiten der Peptide mit Antibiotika, der hier in NMDA-Testung vollzogen wurde. Es konnte nun nicht wie früher bei einzelnen Peptiden, sondern in der gesamten Gruppe (Novispirine, die Veränderungen des LL-37) die auch in früheren Studien [5, 6] bereits angedeutete Erkenntnis untermauert werden, die Fähigkeit zur Wirksamkeit der Peptide liege im Gegensatz zu den verwendeten Antibiotika häufig in der gleichen MHK und MBK. Dies lässt schlussfolgern, dass hier die Hemmung des Wachstums auf einer tatsächlichen Abtötung der Bakterien beruhte [50, 57, 63, 117]. Die Host Defense Peptide agieren auf einem anderen Mechanismus aufbauend, sie sind als erste Linie der Verteidigungsstrategie des angeborenen Immunsystems in ihrer Funktion vor allem auf das Abtöten der Mirkoorganismen ausgelegt. Im Gegensatz zu den oben aufgeführten Studien [6, 90, 97] wurde in dieser Versuchsreihe nicht auf die Ermittlung der MHK verzichtet. Erst der Vergleich beider, der MHK und der MBK erlaubt nämlich o. g. Schlüsse zu ziehen. Mit den hier dargestellten in vitro Testsystem bestehend aus dem Radial Diffusion Assay und dem CLSI Microbroth Dilution Assay stehen also zwei standardisierte 49 Verfahren zur Verfügung, die es zweifelsohne erlauben, die antimikrobiellen Fähigkeiten der so genannten endogenen Antibiotika richtig einzuschätzen. Wie die Ergebnisse zeigen, ist die Potenz der Host Defense Peptide, hier sind es vor allem die synthetischen Modifikationen entweder natürlicher Peptide wie WS 22 und WS 22 N, die vom LL-37 abstammen, oder Weiterentwicklungen synthetischer Peptide, wie der Novispirin-Gruppe, bei der aus dem synthetischen Novispirin G10 mit RIR G10 und R 2 G10 weitere synthetische Abkömmlinge geschaffen wurden, beachtlich. So demonstrieren sie nicht nur ein bisher bei den Antibiotika nicht möglich geglaubtes breites antimikrobielles Spektrum. Weitere Vorteile ergeben sich aus der Struktur der Antimikrobiellen Peptide. Sie sind im Gegensatz zu Antibiotika, die entweder Derivate von Pilzprodukten sind oder den menschlichen Zellen ganz fremde Substanzen, aus in der Natur und im menschlichen Körper ubiquitär vorkommenden Aminosäuren aufgebaut. Da LL-37 sogar menschlichen Ursprungs ist, stellt sich nicht nur für das Peptid selbst, sondern auch für seine Derivate WS 22 und WS 22 N die Frage, ob sie in dem Masse allergen wirken können wie Antibiotika [6]. Diese sind aufgrund ihrer dem menschlichen Körper fremden Struktur häufig in der Lage nicht nur allergische Reaktionen, aber auch gastrointestinale Beschwerden, Kreuzreaktionen und andere Komplikationen hervorzurufen. Daraus ergibt sich die Folgefrage nach der Zytotoxizität der Peptide, die in nachfolgenden Untersuchungen (Zytotoxizitätstest, Hämolysetest) geklärt werden muss. Für einige Peptide ist dies bereits geschehen, so für Novispirin G10 [107] und Protegrin-1 [105] sowie für LL-37 [6], für deren Modifikationen wie z. B. die neuen Novispirine oder die synthetischen Derivate des LL-37 noch nicht. Hier ist es Aufgabe zukünftiger Projekte auf der Basis dieser Ergebnisse Peptide gezielt für in vivo Forschung auszuwählen. Dennoch beantworten diese Testreihen nicht nur Fragen hinsichtlich der Fähigkeiten der Host Defense Peptide, sie stellen viele neue, und diese betreffen vor allem den Funktionsmechanismus der Moleküle. Zentraler Punkt hierbei bleibt die Diskrepanz in den Ergebnissen des RDA und MDA. Letztlich weiß man bereits, um Sensibilität der Aktivität der Peptide gegenüber dem pH oder der Salzkonzentration der Umgebung [5, 6]. Veränderungen des äußeren Milieus, hier der Ionen und folglich der Osmolalität, beeinflussen die Struktur der Peptide und damit ihre antimikrobielle Funktion [49]. Doch sind viele andere Faktoren, die 50 möglichen Einfluss auf die antimikrobiellen Eigenschaften der Peptide haben, noch unklar. Hier sind weitere Versuchsreihen mit vermehrter Modifikation der Testbedingungen notwendig, um verantwortlichen Faktoren zu identifizieren. Der Mechanismus wie Host Defense Peptide Bakterien oder Pilze abzutöten vermögen, ist noch nicht bis ins Detail erforscht worden. Bei den meisten bekannten antimikrobiellen Peptiden handelt es sich um positiv geladene polykationische Peptide, die eine amphipathische Struktur besitzen. Diese Struktur macht es den Eiweißen möglich an die negativ geladene bakterielle Membran zu binden. Sie docken förmlich an mit dem kationischen, hydrophilen Part ihrer Struktur an die negativ geladene mikrobielle Membran an [49, 50, 73]. Zugleich können sie mit ihrem apolaren ungeladenen Teil transmembranäre Kanäle bilden, diese sind für Ionen durchlässig. Die Kanäle führen durch ihre Ionenpermeabilität zu einem Zusammenbruch des Membranpotenzials infolge des Anstiegs der Membrandurchlässigkeit und dadurch zum Verlust von Ionen und damit letztlich zum Kollaps des gesamten Mikroorganismus [33] (Abbildung 26). Allerdings ist auch möglich, dass allein die Bindung des positiv geladenen Peptides zu einem Verlust der Membranfluidität führt und damit zu einem Bruch in der Struktur der mikrobiellen Membran. Abbildung 8: Elektronenmikroskopische Aufnahme eines E. coli vor (A und B) und nach Inkubation (C mit Cecropin und D mit LL-37) mit Host Defense Peptiden. Während in beiden Fällen letztlich die Lyse des Bakteriums folgt, ist der Mechanismus bis dahin, nicht nur was die Zeitebene anbetrifft, unterschiedlich und im letzten Detail noch ungeklärt. Die Resultate dieser Studie verschaffen der Behauptung nachhaltig Geltung, dass Host Defense Peptide seit Millionen von Jahren einen immensen Beitrag an der 51 Abwehr von Infektionen in verschiedenen Organismen unterschiedlicher Spezies leisten. In Zukunft könnten aus ihnen die Alternativen zu Antibiotika in der klinischen Therapie bakterieller Infektionen, vornehmlich von Wundinfektionen, die durch unterschiedliche Mikroorganismen herbeigeführt werden. Zum einen ihr breites Spektrum und die hervorragenden bakteriziden und fungiziden Eigenschaften, zum anderen ihre Struktur als Peptide und die Möglichkeit zur Modifikation ihrer Eigenschaften lassen sie zu potenziellen Substraten in der Behandlung topischer Infektionen werden, auch wenn hierzu noch weitere Studien notwendig sind. Aus dem dargelegten Vergleich der Peptide stechen zweifelsohne einige hervor, wie die Novispirin-Gruppe und die synthetischen Derivate des LL37, jedoch sind gerade mit diesen Peptiden weitere Versuche notwendig, die ihre möglichen zellschädigende Eigenschaften aufdecken, wie Kontrollen der Zytotoxizität und Überprüfung potentieller Hämolyse. Darüber hinaus steht dann die Erhebung der Fähigkeit zur Behandlung topischer Infektionen in einem Tiermodell aus (Bochumer Verbrennungsmodell). Der finale Schritt wäre dann die Demonstration der Effizienz ausgewählter Host Defense Peptide gegenüber systemischen Infektionen sowie Detektion möglicher Nebenwirkungen in Tierstudien und klinischen Versuchen. Darüber hinaus steckt gerade wegen der Struktur als Peptide die Möglichkeit zu einer Anwendung in der Gentherapie. Letztlich sind weitere Studien notwendig, die sich auch mit den Funktionsmechanismen auseinandersetzen, um das therapeutische Potenzial der Peptide besser definieren zu können. 52 5. Zusammenfassung In vitro Testmethoden zur Überprüfung der Aktivität natürlicher und synthetischer Host Defense Peptide gegen Bakterien und Pilze mit Vergleich zu klinisch eingesetzten Antibiotika und Antimykotika Problem: Die dramatisch steigende Inzidenz von Wundinfektionen mit multiresistenten Keimen führte bei der Behandlung von Patienten mit gleichzeitig bestehender erhöhter Morbidität und Letalität zu schwerwiegenden Komplikationen. Die Suche nach potentiellen Auswegen fokussiert sich in den letzten Jahren auf das angeborene Immunsystem und dessen Antimikrobiellen Peptide. Ziel dieser Studie war die in vitro Untersuchung der Wirksamkeit von Host Defense Peptiden gegenüber klinisch eingesetzten Antibiotika und Antimykotika. Methode: Getestet wurden das humane Cathelicidin LL-37, LL37-Modifikationen (WS22 und WS22N), Novispirin G10 ein synthetischer Abkömmling des natürlich beim Schaf vorkommenden Ovispirins, Novispirin Modifikationen (R 2 G10, RIR G10), das porkine Peptid Protegrin-1 und das vom Pilz exprimierte Plectasin gegen die häufigsten Infektionen verursachenden Mikroorganismen sowie multiresistente Isolate, wie MRSA und P. aeruginosa, mittels dem Bilayer Radial Diffusion Assay und dem CLSI Microbroth Assay. Verglichen wurde die Aktivität der AP mit klinisch eingesetzten Antibiotika (Ampicillin, Imipenem, Vancomycin). Ergebnisse: Diese Studie zeigte nur für das humane LL-37 eine eingeschränkte antimikrobielle Aktivität. Dagegen zeigten die Protegrin-1, LL37 Modifikationen und die Novispirine ausgesprochen breite und potente Aktivitätsspektra gegen grampositive und -negative Bakterien sowie gegen Candidia albicans. Bei E. coli, K. pneumoniae, A. baumannii zeigten WS22 und die Novispirine signifikant (p < 0.05) niedrigere Hemmkonzentrationen (MHK) und antibakteriellen Konzentrationen (MBK) als Standardantibiotika. Diese Studie veranschaulicht damit, dass Host Defense Peptide gleichzeitig gegen grampositive und -negative Bakterien wie auch Pilze wirksam sind. Diskussion: Host Defense Peptide wie die Novispirine stellen so potentielle Alternativen oder Ergänzungen zu den gegenwärtig klinisch eingesetzten Antibiotika zur Therapie von Wundinfektionen mit multi-resistenten Keimen. 53 6. Literatur/Bildnachweis [1] Agerberth, B., Gunne, H., Odeberg, J., Kogner, P., Boman, H. G., Gudmundsson, G. H. (1995). FALL-39, a putative human peptide antibiotic, is cysteine-free and expressed in bone marrow and testis. Proc Natl Acad Sci USA 92, 195-199 [2] Arthur, M., Courvalin, P. (1993). Genetics and mechanisms of glycopeptide resistance in enterococci. Antimicrob Agents Chemother 37,1563-71 [3] Bagella, L., Scocchi, M., Zanetti, M. (1995). cDNA sequences of three sheep myeloid cathelicidins. FEBS 376, 225-228 [4] Bals, R., Wang, X., Zasloff, M., Wilson, J. M. (1998). The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci USA 95, 9541-9546 [5] Bals, R., Weiner, D. J., Meegalla, R. L., Wilson, J. M. (1999). Transfer of a cathelicidin peptide antibiotic gene restores bacterial killing in a cystic fibrosis xenograft model. J Clin Invest 103, 11131117 [6] Bals, R., Weiner, D. J., Moscioni, A. D., Meegalla, R. L., Wilson, J. M. (1999). Augmentation of innate host defense by expression of a cathelicidin antimicrobial peptide. Infect Immun 67, 6084-6089 [7] Barańska-Rybak, W., Sonesson, A., Nowicki, R., Schmidtchen, A. (2006). Glycosaminoglycans inhibit the antibacterial activity of LL-37 in biological fluids. J Antimicrob Chemother 57, 260-5 [8] Beckloff, N., Laube, D., Castro, T., Furgang, D., Park, S.., Perlin, D., Clements, D., Tang, H., Scott, R. W., Tew, G. N.., Diamond, G. 54 (2007). Activity of an antimicrobial peptide mimetic against planktonic and biofilm cultures of oral pathogens. Antimicrob Agents Chemother 51, 4125-4132 [9] Billstein, S. A. (1994). How the pharmaceutical industry brings an antibiotic drug to market in the United States. Antimicrob Agents Chemother 38, 2679-2682 [10] Boman, H. G. (1995). Peptide antibiotics and their role in innate immunity. Annu Rev Immunol 13, 61-9 [11] Boman, H. G. (1998). Gene-encoded peptide antibiotics and the concept of innate immunity: an update review. Scand J Immunol 48, 15-25 [12] Bos, J. D., Kapsenberg, M. L. (1993). The skin immune system: progress in cutaneous biology. Immunol Today 14, 75-78 [13] Brogden, K. A., Kalfa, V. C., Ackermann, M. R., Palmquist, D. E., McCray, P. B. Jr., Tack, B. F. (2001). The ovine cathelicidin SMAP29 kills ovine respiratory pathogens in vitro and in an ovine model of pulmonary infection. Antimicrob Agents Chemother 45, 331-334 [14] Brown, E., Atkinson, J. P., Fearon, D. T. (1994). Innate immunity: 50 ways to kill a microbe. Curr Opin Immunol 6, 73-74 [15] Bulchandani, D., Nachnani, J., Fitzsimmons, C., Jost, P., Brewer, J. (2008). Beyond MRSA: the growing menace of hVISA and VISA. South Med J 101, 663-4 [16] Cargill, J. F., Lebl, M. (1997). New methods in combinatorial chemistry-robotics and parallel synthesis. Curr Opin Chem Biol 1, 6771 55 [17] Casewell, M. W. (1998). The nose: an underestimated source of Staphylococcus aureus causing wound infection. J Hosp Infect 40, 311 [18] Chan, Y. R., Gallo, R. L. (1998). PR-39, a syndecan-inducing antimicrobial peptide, binds and affects p130(Cas). J Biol Chem 273, 28978-28985 [19] Chopra, I., Hodgson, J., Metcalf, B., Poste, G. (1997). The search for antimicrobial agents effective against bacteria resistant to multiple antibiotics. Antimicrob Agents Chemother 41, 497-503 [20] Cohen, M. L. (1992). Epidemiology of drug resistance: implications for a post-antimicrobial era. Science 257, 1050-1055 [21] Cole, A. M., Cole, A. L. (2008). Antimicrobial polypeptides are key anti-HIV-1 effector molecules of cervicovaginal host defense. Am J Reprod Immunol 59, 27-34 [22] Cowland, J. B., Johnsen, A. H., Borregaard, N. (1995). hCAP-18, a cathelin/pro-bactenecin-like protein of human neutrophil specific granules. FEBS Lett 368, 173-176 [23] Culotta, E. (1994). Funding crunch hobbles antibiotic resistance research. Science 264, 362-363 [24] Davies, J. (1996). Bacteria on the rampage. Nature 383, 219-220 [25] Department of Chemical Engineering and Materials Science, University of Minnesota. (2004). (Zugriff vom 24.02.2004) http://www.cems.umn.edu/biolog/Images/anti-1%20copy.jpg 56 [26] Dürr, U. H. N., Sudheendra, U. S., Ramamoorthy, A. (2006). LL-37, the only human member of the cathelicidin family of antimicrobial peptides, Biochem. Biophys. Acta 1758 1408-1425 [27] Edmond, M. B., Wallace, S. E., McClish, D. K., Pfaller, M. A., Jones, R.. N., Wenzel, R. P. (1999). Nosocomial bloodstream infections in United States hospitals: a three-year analysis. Clin Infect Dis 29, 239-244 [28] Emmanuel, P. J. (1993). Polymerase chain reaction from bench to bedside. Applications for infectious disease. J Fla Med Assoc 80, 627-630 [29] Enrique, M., Marcos, J. F., Yuste, M., Martínez, M., Vallés, S., Manzanares, P. (2007). Antimicrobial action of synthetic peptides towards wine spoilage yeasts. Int J Food Microbiol 118, 318-325 [30] Ernst, R. K., Yi, E. C., Guo, L., Lim, K. B., Burns, J. L., Hackett, M., Miller, S. I. (1999). Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science 286, 1561–1565 [31] Fearon, D. T, Locksley, R. M. (1996). The instructive role of innate immunity in the acquired immune response. Science 272, 50-3 [32] Fearon, D. T. (1997). Seeking wisdom in innate immunity. Nature 388, 323-324 [33] Fortney, K., Totten, P. A., Lehrer, R. I., Spinola, S. M. (1998). Haemophilus ducreyi is susceptible to protegrin. Antimicrob Agents Chemother 42, 2690-2693 [34] Fox, C. L. Jr., Rappole, B. W., Stanford, W. (1969). Control of pseudomonas infection in burns by silver sulfadiazine. Surg Gynecol Obstet 128, 1021-1026 57 [35] Fridkin, S. K. (2001). Vancomycin-intermediate and -resistant Staphylococcus aureus: what the infectious disease specialist needs to know. Clin Infect Dis 32, 108-115 [36] Frohm, M., Agerberth, B., Ahangari, G., Stâhle-Bäckdahl, M., Lidén, S., Wigzell, H., Gudmundsson, G. H. (1997). The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J Biol Chem 272, 15258-15263 [37] Gallo, R. L., Kim, K. J., Bernfield, M., Kozak, C. A., Zanetti, M., Merluzzi, L., Gennaro, R. (1997). Identification of CRAMP, a cathelinrelated antimicrobial peptide expressed in the embryonic and adult mouse. J Biol Chem 272, 13088-13093 [38] Gallo, R. L., Murakami, M., Ohtake, T., Zaiou, M. (2002). Biology and clinical relevance of naturally occurring antimicrobial peptides. J Allergy Clin Immunol 110, 823-831 [39] Ganz, T., Lehrer, R. I. (1995). Defensins. Pharmacol Ther 66, 191205 [40] Ganz, T., Lehrer, R. I. (1999). Antibiotic peptides from higher eukaryotes: biology and applications. Mol Med Today 5, 292-297 [41] Goldstein, B. P., Wei, J., Greenberg, K., Novick, R. (1998). Activity of nisin against Streptococcus pneumoniae, in vitro, and in a mouse infection model. J Antimicrob Chemother 42, 277-278 [42] Gorecki, P., Schein, M., Rucinski, J. C., Wise, L. (1999). Antibiotic administration in patients undergoing common surgical procedures in a community teaching hospital: the chaos continues. World J Surg 23, 429-432 58 [43] Greenwood, J. E., Crawley, B. A., Clark, S. L., Chadwick, P. R., Ellison, D. A., Oppenheim, B. A., McCollum, C. N. (1997). Monitoring wound healing by odour. J Wound Care 6, 219-221 [44] Gudmundsson, G. H., Agerberth, B., Odeberg, J., Bergman, T., Olsson, B., Salcedo, R. (1996). The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. Eur J Biochem 238, 325-332 [45] Gudmundsson, G. H., Magnusson, K. P., Chowdhary, B. P., Johansson, M., Andersson, L., Boman, H. G. (1995). Structure of the gene for porcine peptide antibiotic PR-39, a cathelin gene family member: comparative mapping of the locus for the human peptide antibiotic FALL-39. Proc Natl Acad Sci USA 92, 7085-7089 [46] Hancock, R. E., Chapple, D. S. (1999). Peptide antibiotics. Antimicrob Agents Chemother 43, 1317-1323 [47] Hancock, R. E., Lehrer, R. I. (1998). Cationic peptides: a new source of antibiotics. Trends Biotechnol 16, 82-88 [48] Hancock, R. E., Scott, M. G. (2000). The role of antimicrobial peptides in animal defenses. Proc Natl Acad Sci USA 97, 8856-8861 [49] Harwig, S. S., Waring, A., Yang, H. J., Cho, Y., Tan, L. Lehrer, R. I. (1996). Intramolecular disulfide bonds enhance the antimicrobial and lytic activities of protegrins at physiological sodium chloride concentrations. Eur J Biochem 240, 352-357 [50] Heller, W. T., Waring, A. J., Lehrer, R. I., Harroun, T. A., Weiss, T. M., Yang, L., Huang, H. W. (2000). Membrane thinning effect of the beta-sheet antimicrobial protegrin. Biochemistry 39, 139-145 59 [51] Hirsch, T., Metzig, M., Niederbichler, A., Steinau, H. U., Eriksson, E., Steinstraesser, L. (2008). Role of host defense peptides of the innate immune response in sepsis. Shock 30(2), 117-26 [52] Hoskin, D. W., Ramamoorthy, A. (2008). Studies on anticancer activities of antimicrobial peptides. Biochem Biophys Acta 1778, 357375 [53] Howden, B. P., Davies, J. K., Johnson, P. D., Stinear, T. P., Grayson, M. L. (2010). Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev 23, 99-139 [54] Iwatsuki, K., Yamasaki, O., Morizane, S., Oono, T. (2006). Staphylococcal cutaneous infections: invasion, evasion and aggression. J Dermatol Sci 42, 203-214 [55] Jang, H., Ma, B., Nussinov, R. (2007). Conformational study of the protegrin-1 (PG-1) dimer interaction with lipid bilayers and its effect. BMC Struct Biol 7, 21 [56] Kipp, F., Friedrich, A. W., Becker, K., Eiff, C. (2004). Bedrohliche Zunahme Methicillin-resistenter Staphylococcus-aureus-Stämme: Strategien zur Kontrolle und Prävention in Deutschland. Dtsch Arztebl 101, 2045-50 [57] Kokryakov, V. N., Harwig, S. S., Panyutich, E. A., Shevchenko, A. A., Aleshina, G. M., Shamova, O. V., Korneva, H. A., Lehrer, R. I. (1993). Protegrins: leukocyte antimicrobial peptides that combine features of corticostatic defensins and tachyplesins. FEBS Lett 327, 231-236 60 [58] Kovalchuk, L. V., Gankovskaya, L. V., Gankovskaya, O. A., Lavrov, V. F. (2007). Herpes simplex virus: treatment with antimicrobial peptides. Adv Exp Med Biol 601, 369-376 [59] Kraus, D., Peschel, A. (2008). Staphylococcus aureus evasion of innate antimicrobial defense. Future Microbiol 3, 437-51 [60] Larrick, J. W., Hirata, M., Balint, R. F., Lee, J., Zhong, J., Wright, S. C. (1995). Human CAP18: a novel antimicrobial lipopolysaccharidebinding protein. Infect Immun 63, 1291-1297 [61] Larrick, J. W., Morgan, J. G., Palings, I., Hirata, M., Yen, M. H. (1991). Complementary DNA sequence of rabbit CAP18--a unique lipopolysaccharide binding protein. Biochem Biophys Res Commun 179, 170-175 [62] Lazarus, A., Sanders, J. (2000). Management of tuberculosis. Choosing an effective regimen and ensuring compliance. Postgrad Med 108, 71-74, 77-79, 83-84 [63] Lehrer, R. I., Ganz, T. (1990). Antimicrobial polypeptides of human neutrophils. Blood 76, 2169-2181 [64] Lehrer, R. I., Ganz, T. (1999). Antimicrobial peptides in mammalian and insect host defence. Curr Opin Immunol 11, 23-27 [65] Lennon, G. G. (2000). High-throughput gene expression analysis for drug discovery. Drug Discov Today 5, 59-66 [66] Levison, M. E., Mallela, S. (2000). Increasing Antimicrobial Resistance: Therapeutic Implications for Enterococcal Infections. Curr Infect Dis Rep 2, 417-423 61 [67] Levy, O., Sisson, R. B., Kenyon, J., Eichenwald, E., Macone, A. B., Goldmann, D. (2000). Enhancement of neonatal innate defense: effects of adding an N-terminal recombinant fragment of bactericidal/permeability-increasing protein on growth and tumor necrosis factor-inducing activity of gram-negative bacteria tested in neonatal cord blood ex vivo. Infect Immun 68, 5120-5125 [68] Levy, S. B. (1997). Antibiotic resistance: an ecological imbalance. Ciba Found Symp 207, 1-9; discussion 9-14 [69] Lindberg, R. B., Moncrief, J. A., Switzer, W. E., Order, S. E., Mills, W. Jr. (1965). The successful control of burn wound sepsis. J Trauma 5, 601-616 [70] Liu, C., Chambers, H. F. (2003) Staphylococcus aureus with heterogeneous resistance to vancomycin: epidemiology, clinical significance, and critical assessment of diagnostic methods. Antimicrob Agents Chemother 47, 3040-5 [71] Löfgren, S. E., Miletti, L. C., Steindel, M., Bachère, E., Barracco, M. A. (2008). Trypanocidal and leishmanicidal activities of different antimicrobial peptides (AMPs) isolated from aquatic animals. Exp Parasitol 118, 197-202 [72] Loury, D., Embree, J. R., Steinberg, D. A., Sonis, S. T., Fiddes, J. C. (1999). Effect of local application of the antimicrobial peptide IB-367 on the incidence and severity of oral mucositis in hamsters. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 87, 544-551 [73] Mangoni, M. E., Aumelas, A., Charnet, P., Roumestand, C., Chiche, L., Despaux, E., Grassy, G., Calas, B., Chavanieu, A. (1996). Change in membrane permeability induced by protegrin 1: implication of disulphide bridges for pore formation. FEBS Lett 383, 93-98 62 [74] Martin, E., Ganz, T., Lehrer, R. I. (1995). Defensins and other endogenous peptide antibiotics of vertebrates. J Leukoc Biol 58, 128136 [75] McCoy, A. J., Liu, H., Falla, T. J., Gunn, J. S. (2001). Identification of Proteus mirabilis mutants with increased sensitivity to antimicrobial peptides. Antimicrob Agents Chemother 45, 2030-7 [76] Meyer, E., Schwab, F., Gastmeier, P., Rueden, H., Daschner, F. D. (2006). Surveillance of antimicrobial use and antimicrobial resistance in German intensive care units (SARI): a summary of the data from 2001 through 2004. Infection 34, 303-309 [77] Miyasaki, K. T., Lehrer, R. I. (1998). Beta-sheet antibiotic peptides as potential dental therapeutics. Int J Antimicrob Agents 9, 269-280 [78] Murray, B. E. (1997). Vancomycin-resistant enterococci. Am J Med 102, 284-293 [79] Mygind, P. H., Fischer, R. L., Schnorr, K. M., Hansen, M. T., Sönksen, C. P., Ludvigsen, S., Raventós, D., Buskov, S., Christensen, B., De Maria, L., Taboureau, O., Yaver, D., ElvigJørgensen, S. G., Sørensen, M. V., Christensen, B. E., Kjaerulff, S., Frimodt-Moller, N., Lehrer, R. I., Zasloff, M., Kristensen, H. H. (2005). Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 437, 975-980 [80] National Nosocomial Infections Surveillance (NNIS). (1996). A report from the National Nosocomial Infections Surveillance (NNIS) System, data summary from October 1986 – April 1996.; Am J Infect Control 24, 380-388 63 [81] National Nosocomial Infections Surveillance (NNIS). (1999). System report, data summary from January 1990 – May 1999. Am J Infect Control 27, 520-532 [82] Neu, H. C. (1992). The crisis in antibiotic resistance. Science 257, 1064-1073 [83] Nooyen, S. M., Overbeek, B. P., Brutel de la Rivière, A., Storm, A. J., Langemeyer, J. J. (1994). Prospective randomised comparison of single-dose versus multiple-dose cefuroxime for prophylaxis in coronary artery bypass grafting. Eur J Clin Microbiol Infect Dis 13, 1033-1037 [84] Oren, Z., Lerman, J. C., Gudmundsson, G. H., Agerberth, B., Shai, Y. (1999). Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem J 341, 501-13 [85] Paul-Ehrlich-Gesellschaft für Chemotherapie e.V. (2003). (Zugriff vom 09.10.07) http://www.zuendstoff-antibiotika- resistenz.de/seite11.html [86] Perl, T. M., Golub, J. E. (1998). New approaches to reduce Staphylococcus aureus nosocomial infection rates: treating S. aureus nasal carriage. Ann Pharmacother 32, 7-16 [87] Ranson, M. R., Oppenheim, B. A., Jackson, A., Kamthan, A. G., Scarffe, J. H. (1990). Double-blind placebo controlled study of vancomycin prophylaxis for central venous catheter insertion in cancer patients. J Hosp Infect 15, 95-102 64 [88] Rong, S. L., Leonard, S. N. (2010). Heterogeneous vancomycin resistance in Staphylococcus aureus: a review of epidemiology, diagnosis, and clinical significance. Ann Pharmacother 44, 844-50 [89] Russell, J. P., Diamond, G., Tarver, A. P., Scanlin, T. F., Bevins, C. L. (1996). Coordinate induction of two antibiotic genes in tracheal epithelial cells exposed to the inflammatory mediators lipopolysaccharide and tumor necrosis factor alpha. Infect Immun 64, 1565-8 [90] Saiman, L. (1999). Drug resistant organisms from CF patients are inhibited by cathelicidin peptides. Pulmonology 19, 320 [91] Saiman, L., Tabibi, S., Starner, T. D., San Gabriel, P., Winokur, P. L., Jia, H. P., McCray, P. B. Jr., Tack, B. F. (2001). Cathelicidin peptides inhibit multiply antibiotic-resistant pathogens from patients with cystic fibrosis. Antimicrob Agents Chemother 45, 2838-2844 [92] Santamaría, C., Larios, S., Quirós, S., Pizarro-Cerda, J., Gorvel, J. P., Lomonte, B., Moreno, E. (2005). Bactericidal and antiendotoxic properties of short cationic peptides derived from a snake venom Lys49 phospholipase A2. Antimicrob Agents Chemother 49, 13401345 [93] Sawai, M. V., Waring, A. J., Kearney, W. R., McCray, P. B. Jr., Forsyth, W. R., Lehrer, R. I., Tack, B. F. (2002). Impact of singleresidue mutations on the structure and function of ovispirin/novispirin antimicrobial peptides. Protein Eng 15, 225-232 [94] Sawyer, J. G., Martin, N. L., Hancock, R. E. (1988). Interaction of macrophage cationic proteins with the outer membrane of Pseudomonas aeruginosa. Infect Immun 56, 693-698 65 [95] Schmidtchen, A., Frick, I. M., Andersson, E., Tapper, H., Björck, L. (2002). Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol Microbiol 46, 157-68 [96] Schröder, J. M., Harder, J. (1999). Human beta-defensin-2. Int J Biochem Cell Biol 31, 645-651 [97] Schwab, U., Gilligan, P., Jaynes, J., Henke, D. (1999). In vitro activities of designed antimicrobial peptides against multidrugresistant cystic fibrosis pathogens. Antimicrob Agents Chemother 43, 1435-1440 [98] Shi, J., Ganz, T. (1998). The role of protegrins and other elastaseactivated polypeptides in the bactericidal properties of porcine inflammatory fluids. Infect Immun 66, 3611-3617 [99] Shin, S. Y., Kang, S. W., Lee, D. G., Eom, S. H., Song, W. K., Kim, J. I. (2000). CRAMP analogues having potent antibiotic activity against bacterial, fungal, and tumor cells without hemolytic activity. Biochem Biophys Res Commun 275, 904-909 [100] Skerlavaj, B., Benincasa, M., Risso, A., Zanetti, M., Gennaro, R. (1999). SMAP-29: a potent antibacterial and antifungal peptide from sheep leukocytes. FEBS Lett 463, 58-62 [101] Sørensen, O., Arnljots, K., Cowland, J. B., Bainton, D. F., Borregaard, N. (1997). The human antibacterial cathelicidin, hCAP18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood 90, 2796-2803 [102] Steinberg, D. A., Hurst, M. A., Fujii, C. A., Kung, A. H., Ho, J. F., Cheng, F. C., Loury, D. J., Fiddes, J. C. (1997). Protegrin-1: a broadspectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob Agents Chemother 41, 1738-1742 66 [103] Steinberg, D. A., Lehrer, R. I. (1997). Designer assays for antimicrobial peptides. Disputing the "one-size-fits-all" theory. Methods Mol Biol 78, 169-186 [104] Steinstraesser, L. (2004). Sepsis - new strategies with host defense peptides? Crit Care Med 32, 2555-2556 [105] Steinstraesser, L., Burghard, O., Nemzek, J., Fan, M. H., Merry, A., Remick, D. I., Su, G. L., Steinau, H. U., Wang, S. C. (2003). Protegrin-1 increases bacterial clearance in sepsis but decreases survival. Crit Care Med 31, 221-226 [106] Steinstraesser, L., Ring, A., Bals, R., Steinau, H. U., Langer, S. (2006). The human host defense peptide LL37/hCAP accelerates angiogenesis in PEGT/PBT biopolymers. Ann Plast Surg 56, 93-98 [107] Steinstraesser, L., Tack, B. F., Waring, A. J., Hong, T., Boo, L. M., Fan, M. H., Remick, D. I., Su, G. L., Lehrer, R. I., Wang, S. C. (2002). Activity of novispirin G10 against Pseudomonas aeruginosa in vitro and in infected burns. Antimicrob Agents Chemother 46, 1837-1844 [108] Steinstraesser, L., Tippler, B., Mertens, J., Lamme, E., Homann, H. H., Lehnhardt, M., Wildner, O., Steinau, H. U., Uberla, K. (2005). Inhibition of early steps in the lentiviral replication cycle by cathelicidin host defense peptides. Retrovirology 2, 278-284 [109] Stone, H. H. (1966). Review of pseudomonas sepsis in thermal burns: verdoglobin determination and gentamicin therapy. Ann Surg 163, 297-305 [110] The Olson Laboratory, The Molecular Graphics Laboratory (2003). (Zugriff vom 03.05.04) 67 http://mgl.scripps.edu/people/goodsell/imges_art/mgs_art2 /palay2.html [111] Travis, S. M., Anderson, N. N., Forsyth, W. R., Espiritu, C., Conway, B. D., Greenberg, E. P., McCray, P. B. Jr., Lehrer, R. I., Welsh, M. J., Tack, B. F. (2000). Bactericidal activity of mammalian cathelicidinderived peptides. Infect Immun 68, 2748-2755 [112] Unbekannter Autor. Classics in infectious diseases. Concerning the relationship between phagocytes and anthrax bacilli by Elie Metchnikoff 1884. (1984). Rev Infect Dis. Sep-Oct 6(5), 761-770 [113] University of Minnesota, Chemical Engineering, Kaznessis Group (2004). (Zugriff vom 07.08.2005) http://www.cems.umn.edu/research/ kaznessis/Images/anti-3%20copy.jpg [114] Walsh, C. T., Fisher, S. L., Park, I. S., Prahalad, M., Wu, Z. (1996). Bacterial resistance to vancomycin: five genes and one missing hydrogen bond tell the story. Chem Biol 3, 21-28 [115] Weinstein, R. A. (1991). Epidemiology and control of nosocomial infections in adult intensive care units. Am J Med 16, 179-184 [116] World Health Organization Report on Infectious Diseases (2000). Overcoming Antimicrobial Resistance, Chapter One: A World without Antibiotics; World Health Report on Infectious Diseases. (Zugriff vom 12.07.06). http://www.who.int/infectious-disease-report/2000/ [117] Yamaguchi, S., Huster, D., Waring, A., Lehrer, R. I., Kearney, W., Tack, B. F., Hong, M. (2001). Orientation and dynamics of an antimicrobial peptide in the lipid bilayer by solid-state NMR spectroscopy. Biophys J 81, 2203-2214 68 [118] Yano, M., Doki, Y., Inoue, M., Tsujinaka, T., Shiozaki, H., Monden, M. (2000). Preoperative intranasal mupirocin ointment significantly reduces postoperative infection with Staphylococcus aureus in patients undergoing upper gastrointestinal surgery. Surg Today 30, 16-21 [119] Zanetti, M., Gennaro, R., Romeo, D. (1995). Cathelicidins: a novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett 374, 1-5 [120] Zanetti, M., Gennaro, R., Romeo, D. (1997). The cathelicidin family of antimicrobial peptide precursors: a component of the oxygenindependent defense mechanisms of neutrophils. Ann N Y Acad Sci 832, 147-162 [121] Zasloff, M. (1987). Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci USA 84, 5449-5453 [122] Zhao, C., Ganz, T., Lehrer, R. I. (1995). The structure of porcine protegrin genes. FEBS Lett 368, 197-202 [123] Zhao, C., Liu, L., Lehrer, R. I. (1994). Identification of a new member of the protegrin family by cDNA cloning. FEBS Lett. 346, 285-288 69 /. Danksagung An dieser Stelle möchte ich ganz besonders Herrn Professor Dr. med. Lars Steinsträsser, Juniorprofessor für molekulare Wundheilung und Gentherapie danken, der die vorliegende Arbeit sowohl im praktischen als auch im schriftlichen Teil hervorragend betreut und unterstützt hat. Ein Dank gilt Herrn Prof. Dr. Hans - Ulrich Steinau und Herrn Prof. Dr. Sören Gatermann für die Rahmenbedingungen, die diese Promotionsarbeit ermöglicht haben. Außerdem möchte ich für die Hilfe bei der Durchführung der Doktorarbeit Dr. rer. nat. Frank Jacobsen, Janine Mertens, Andrea Gerhards, Jürgen Beller, Alexander Baraniskin, Kristina Drewermann, Tobias Hirsch und Dominik Mittler danken. Ein besonderer Dank gilt der Medizinischen Mikrobiologie der Ruhr – Universität – Bochum, besonders Frau Susanne Friedrich für die Hilfe und Unterstützung bei der Durchführung der Experimente. 8. Lebenslauf Name: Gottlieb Pazdzierny Geburtsdatum: 16. 12. 1977 Geburtsort: Kedzierzyn-Kozle, Polen Staatsangehörigkeit: deutsch Familienstand: ledig Schulische Ausbildung: 09. 1984 – 02. 1988 Grundschule in Kedzierzyn-Kozle (Polen) 02. 1988 Ausreise nach Deutschland 02. 1988 – 06. 1988 Grundschule in Unna 08- 1988 – 07. 1997 Gymnasium in Menden (Sauerland) 07.1997 Abitur Hochschulausbildung: 10. 1998 – 04. 2005 Studium der Medizin an der Ruhr – Universität Bochum 04. 2004 – 04. 2005 PJ in den Augusta-Krankenanstalten Bochum Wahlfach Neurologie im EVK Hattingen 07. 2001 Erstes Staatsexamen 06. 2004 Zweites Staatsexamen 04. 2005 Drittes Staatsexamen 05. 2005 Approbation Beruflicher Werdegang: 10. 2005 Assistenzarzt in der Abteilung für Allgemeine Medizin, Endokrinologie und Diabetologie in der Med. Klinik I, Prof. Dr. med. H. H. Klein, BG Kliniken Bergmannsheil Bochum Publikationen: Medline gelistete Abstracts 1. Steinstraesser, L., Beller, J., Hirsch, T., Pazdzierny, G., Mittler, D., Lehnhardt, M., Druecke, D., Steinau, H. U.: Transient cutaneous gene transfer in burns with liposomes or „naked“ DNA, Langenbeck’s Archives of Surgery Vol 387 (5-6) Oct 2002, S265, A-148 2. May, P., Steinstraesser, L., Hermann, S., Lehnhardt, M., Hirsch, T., Pazdzierny, G., Steinau, H. U.: Dermisersatzstoffe in porcinen Vollhautwunden. Konressband 2003, Springer Verlag Berlin, Heidelberg, New York, ISBN 3-540-20002-9), 441-442. 3. Schneider, S., Pazdzierny, G., Klein, H. H. Oral antidiabetic therapy. Dtsch Med Wochenschr. 2006 Dec;131 Suppl 8:S264-7. Review. German 4. Steinstraesser, L., Hirsch, T., Beller, J., Mittler, D., Sorkin, M., Pazdzierny, G., Jacobsen, F., Eriksson, E., Steinau, H. U. Transient non-viral cutaneous gene delivery in burn wounds. J Gene Med. 2007 Nov;9(11):949-55. Kongressbeiträge 5. Steinstraesser L., Baraniskin, A., Pazdzierny, G., Lehnhardt, M., Steinau, H. U.: Antimikrobielle Verbrennungsmodel. 22. Aktivität von Jahrestagung humanen der Histonen im Deutschsprachigen Arbeitsgemeinschaft für Verbrennungsbehandlung (DAV), Rottach-Egern, 06.-09. Januar 2004 6. Steinstraesser, L., Baraniskin, A., Pazdzierny, G., Lehnhardt, M., Steinau, H. U.: Antimikrobielle Verbrennungsmodel. 22. Aktivität von Jahrestagung humanen der Histonen im Deutschsprachigen Arbeitsgemeinschaft für Verbrennungsbehandlung (DAV), Rottach-Egern, 06.-09. Januar 2004 7. Pazdzierny, G., Hirsch, T., Lehnhardt, M., Gatermann, S., Steinau, H. U, Steinstraesser, L.: In vitro comparision of naturally occuring and „designer“ antimicrobial peptides with common antibiotics and antimycotics. Plastic Surgery Research Council 48th Annual Meeting 23-26.4.2003, Las Vegas, USA 8. Steinstraesser, L., Beller, J., Hirsch, T., Pazdzierny, G., Mittler, D., Lehnhardt, M., Druecke, D., Steinau, H. U.: Kutaner Gentransfer in Verbrennungswunden mit Liposomen oder “Nackter” DNA 9. Jahrestagung der Deutschen Arbeitsgemeinschaft für Gentherapie, 7-9.11.2002 Schloss Reisensburg bei Günzburg 9. Jacobsen, F., Steinstraesser, L., Beller, J., Mertens, J., Hirsch, T., Pazdzierny, G., von Peter, S., Druecke, D., Steinau, H. U.: Transienter kutaner Gentransfer mit Transfektionsreagenzien oder nackter DNA. 120 Kongress Deutsche Gesellschaft für Chirurgie 29.4.-2.5.2003 in München. 10. Pazdzierny, G., Steinstraesser, L., von Peter, S., Lehnhardt, M., Hirsch, T., Lehrer, R. I., Steinau, H. U.: In vitro Vergleich der Aktivität von natürlichen und designer Antimikrobiellen Peptiden mit klinisch eingesetzten Antibiotika und Antimykotika. 120 Kongress Deutsche Gesellschaft für Chirurgie 29.4.2.5.2003 in München. 11. Pazdzierny, G., Steinstraesser, L., May, P., Beller, J., Mittler, D., Hirsch, T., Steinau, H. U.: Biologisch abbaubare Implantate zur Auffüllung von Weichgewebsdefekten. 120 Kongress Deutsche Gesellschaft für Chirurgie 29.4.-2.5.2003 in München. 12. May, P., Steinstraesser, L., Hermann, S., Lehnhardt, M., Hirsch, T., Pazdzierny, G., Steinau, H. U.: Dermisersatzstoffe in porcinen Vollhautwunden. 120 Kongress Deutsche Gesellschaft für Chirurgie 29.4.2.5.2003 in München. 13. Steinstraesser, L., Beller, J., Hirsch, T., Pazdzierny, G., Mittler, D., Lehnhardt, M., Druecke, D., Steinau H. U.: Transient cutaneous gene transfer in burns with liposomes or „naked“ DNA, 11. Chirurgische Forschungstage, 27-29.11.2002 Universität Köln 14. Steinstraesser, L., Beller, J., Hirsch, T., Pazdzierny, G., Mittler, D., Lehnhardt, M., Druecke, D., Steinau, H. U.: Kutaner Gentransfer in Verbrennungswunden mit Liposomen oder “Nackter” DNA 9. Jahrestagung der Deutschen Arbeitsgemeinschaft für Gentherapie, 7-9.11.2002 Schloss Reisensburg bei Günzburg. 15. Pazdzierny, G., Steinstraesser, L., von Peter, S., Überla, K., Steinau, H. U.: In vitro Vergleich der Wirksamkeit und Zytotoxizität von natürlich vorkommenden und “designer” antimikrobiellen Peptiden gegen HIV. VDPC, 18-21.9.2002 in Heidelberg. 16. Pazdzierny, G., Steinstraesser, L., Hirsch, T., Beller, J., Steinau, H. U.: In vitro Vergleich der Aktivität von Antimikrobiellen Peptiden mit klinisch eingesetzten Heidelberg. Antibiotika und Antimykotika. VDPC, 18-21.9.2002 in