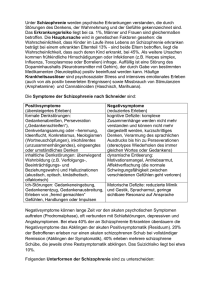
Dokument_1. - OPUS Würzburg
Werbung

Aus der Klinik und Poliklinik für Psychiatrie, Psychosomatik und Psychotherapie der Universität Würzburg Direktor: Prof. Dr. Jürgen Deckert NOS1AP als Kandidatengen für endogene Psychosen Inaugural - Dissertation zur Erlangung der Doktorwürde der Medizinischen Fakultät der Julius-Maximilians-Universität Würzburg vorgelegt von Alexandra Kramer aus Göttingen Würzburg, Dezember 2010 Referent: Prof. Dr. Andreas Reif Korreferent: Priv.-Doz. Dr. Christoph Kleinschnitz Dekan: Prof. Dr. Matthias Frosch Tag der mündlichen Prüfung: 18.07.2011 Die Promovendin ist Ärztin Für meine Eltern Inhaltsverzeichnis Inhaltsverzeichnis Abkürzungsverzeichnis 1 Einleitung ................................................................................................................ 1 1.1 Schizophrenie ................................................................................................... 1 1.1.1 Diagnostik und Klinik .............................................................................. 1 1.1.2 Epidemiologie........................................................................................... 3 1.1.3 Ätiologie ................................................................................................... 3 1.1.4 Neuropathologie ....................................................................................... 4 1.2 Bipolar-affektive Erkrankung ........................................................................... 5 1.2.1 Diagnostik und Klinik .............................................................................. 5 1.2.2 Epidemiologie........................................................................................... 7 1.2.3 Ätiologie ................................................................................................... 8 1.2.4 Neuropathologie ....................................................................................... 8 1.3 Pathophysiologie endogener Psychosen ........................................................... 9 1.3.1 Schizophrenie ........................................................................................... 9 1.3.1.1 Theorie der neuronalen Entwicklungsstörung ...................................... 9 1.3.1.2 Neurotransmitterhypothesen ............................................................... 10 1.3.2 Bipolar-affektive Erkrankung ................................................................. 15 1.3.2.1 Monoaminhypothese .......................................................................... 15 1.3.2.2 Dysfunktion der Hypothalamus-Hypophysen-Nebennieren-Achse ... 16 1.3.2.3 Veränderungen der synaptischen Plastizität ....................................... 16 1.3.2.4 Hinweise auf Beteiligung des Glutamatsystems ................................ 17 1.4 Genetik ........................................................................................................... 18 1.4.1 Vererbung ............................................................................................... 18 1.4.2 Kopplungsanalysen ................................................................................. 19 1.4.3 Assoziationsstudien ................................................................................ 20 1.4.4 Vulnerabilitätsgene endogener Psychosen ............................................. 21 1.4.4.1 Suszeptibilitätsregionen - Ergebnisse aus Kopplungsanalysen .......... 21 1.4.4.2 Suszeptibilitätsgene - Schizophrenie .................................................. 22 1.4.4.3 Suszeptibilitätsgene - Bipolar-affektive Störung................................ 24 1.4.4.4 Hinweise auf Überlappung der Ätiologie ........................................... 24 Inhaltsverzeichnis 1.5 Das Kandidatengen NOS1AP......................................................................... 25 1.5.1 Definition und Funktion ......................................................................... 25 1.5.2 Neuronale NO-Synthase als Zielstruktur von NOS1AP......................... 28 1.5.3 NOS1AP als Kandidatengen: bisherige Studienergebnisse.................... 30 2 Fragestellung ......................................................................................................... 33 3 Material und Methoden ......................................................................................... 34 3.1 Probanden ....................................................................................................... 34 3.2 Material ........................................................................................................... 35 3.2.1 Reagenzien.............................................................................................. 35 3.2.2 Geräte und Verbrauchsmaterialien ......................................................... 37 3.3 Methoden ........................................................................................................ 38 3.3.1 Methodischer Überblick ......................................................................... 38 3.3.2 DNA-Extraktion ..................................................................................... 39 3.3.3 Polymerase-Kettenreaktion .................................................................... 39 3.3.4 Agarose-Gelelektrophorese .................................................................... 41 3.3.5 Amplifizierung nach dem iPlex®-Protokoll............................................ 42 3.3.6 Verdau mit Alkalischer Phosphatase ...................................................... 42 3.3.7 iPlex® Reaktion ...................................................................................... 42 3.3.8 Reinigung durch Ionenaustauschharz ..................................................... 43 3.3.9 MALDI-ToF Massenspektrometrie ........................................................ 43 3.3.10 Statistische Analyse ................................................................................ 44 3.3.11 Qualitätssicherung .................................................................................. 44 4 Ergebnisse.............................................................................................................. 46 4.1 Schizophrenie ................................................................................................. 46 4.1.1 Kopplungsungleichgewicht .................................................................... 46 4.1.2 Assoziationsanalyse ................................................................................ 47 4.2 Bipolar-affektive Störung ............................................................................... 48 4.2.1 Kopplungsungleichgewicht .................................................................... 48 4.2.2 Assoziationsanalyse ................................................................................ 48 4.3 5 Tabellarische Übersicht der Ergebnisse ......................................................... 49 Diskussion ............................................................................................................. 60 5.1 Stärken und Schwächen von Assoziationsstudien .......................................... 60 Inhaltsverzeichnis 5.2 Vergleich mit anderen Studien ....................................................................... 62 5.3 Ursachen für Unterschiede zu anderen Assoziationsstudien .......................... 65 5.4 Andere Kandidatengene auf 1q22 .................................................................. 67 5.5 Bedeutung des nitrinergen Systems ................................................................ 69 5.6 Funktionelle Studien ....................................................................................... 70 5.7 Neue Studienansätze ....................................................................................... 73 5.8 Sozioökonomische Folgen und Ausblick ....................................................... 76 6 Zusammenfassung ................................................................................................. 78 7 Literaturverzeichnis ............................................................................................... 80 8 Abbildungsverzeichnis .......................................................................................... 91 9 Tabellenverzeichnis ............................................................................................... 92 10 Anhang .................................................................................................................. 93 Abkürzungsverzeichnis Abkürzungsverzeichnis Abb. AMDP AMP AMPA AMPAR BDNF bp bzw. C ca. Ca2+ CAPON cGMP COMT CREB CRH CYP d. h. D2-Rezeptor DAAO DAOA DAT dATP dCTP ddNTP dex Dexras dGTP DISC DLPFC DNA dNTP DSM dTTP DTNBP1 EAAT EDTA g GABA GDP ges. Glx GTP Abbildung Arbeitsgemeinschaft für Methodik und Dokumentation in der Psychiatrie Adenosinmonophosphat Alpha-Amino-3-hydroxy-5-methyl-4-isoxazol-Propionat Alpha-Amino-3-hydroxy-5-methyl-4-isoxazol-PropionatRezeptor brain derived neurotrophic factor Basenpaare beziehungsweise Celsius circa Calcium carboxy-terminal PDZ ligand of neuronal NO synthase cyclisches Guanosinmonophosphat Catechol-O-Methyltransferase cyclic AMP response element-binding protein Corticotropin-releasing hormone Cytochrom P450 das heißt Dopamin-2-Rezeptor D-amino acid oxidase D-amino acid oxidase activator Dopamintransporter Desoxyadenosintriphosphat Desoxycytidintriphosphat Didesoxyribonukleosidtriphosphat Dexamethason dexamethasone-induced Ras-related protein Desoxyguanosintriphosphat disrupted in schizophrenia dorso-lateraler präfrontaler Cortex deoxyribonucleic acid Desoxyribonukleosidtriphosphat Diagnostic and Statistical Manual of Mental Disorders Desoxythymidintriphosphat Dysbindin excitatory amino acid transporter Ethylendiamintetraacetat Gramm gamma-aminobutyric acid Guanosindiphosphat gesamt Glutamat/Glutamin Guanosintriphosphat Abkürzungsverzeichnis GTPase HCl HDAC HHN-Achse HLA-DQB1 H-MRS H2O 5-HTT ICD K+ kb KCl kDa KHCO3 l LD LOD LTD LTP M MAGUK MALDI-ToF MAP-Kinase Mb Met mg Mg2+ MgCl2 min ml mM mRNA MRT MS µg µl Na+ NaCl NANC NET neg. NF-L NH4Cl nl nm NMDA NMDAR Guanosintriphosphatase Chlorwasserstoff Histondeacetylase Hypothalamus-Hypophysen-Nebennieren-Achse major histocompatibility complex, class II, DQ beta 1 Protonen-Magnetresonanz-Spektroskopie Wasser 5-Hydroxytryptamintransporter International Statistical Classification of Diseases and Related Health Problems Kalium Kilobase Kaliumchlorid Kilodalton Kaliumhydrogencarbonat Liter linkage disequilibrium logarithm of odds long-term depression long-term potentiation molar membrane-associated guanylate kinase matrix-assisted laser disorption ionisation-time of flight mitogenaktivierte Proteinkinase Megabase Methionin Milligramm Magnesium Magnesiumchlorid Minute Milliliter millimolar messenger ribonucleic acid Magnetresonanztomographie Massenspektrometer Mikrogramm Mikroliter Natrium Natriumchlorid nonadrenerg noncholinerg norepinephrine transporter negativ light neurofilament Ammoniumchlorid Nanoliter Nanometer N-Methyl-D-Aspartat N-Methyl-D-Aspartat Rezeptor Abkürzungsverzeichnis NO nom. NOS NOS1AP Nr. NR1 NR2A-D NR3 p PCR PDZ Perm. PET PFC pH pmol pos. P-p PRODH PSD PTB-Domäne Ras RGS4 RNA rpm SADS-L SAP SAP102 SDS sec sGC SNP sog. Tab. TAE U u. a. UHMK1 UTR UV v. a. Val vs. WHO z. B. ZNS ZO-1 Stickstoffmonoxid nominell NO-Synthase NOS1 adaptor protein Nummer N-Methyl-D-Aspartat Rezeptor Untereinheit 1 N-Methyl-D-Aspartat Rezeptor Untereinheit 2A-D N-Methyl-D-Aspartat Rezeptor Untereinheit 3 p-Wert polymerase chain reaction Postsynaptic Density 95/Disc-Large/Zona Occludens Permutation Positronenemissionstomographie präfrontaler Cortex potentia hydrogenii Pikomol positiv p-Wert im Permutationstest Prolindehydrogenase post synaptic density Phosphotyrosin-bindende Domäne rat sarcoma regulator of G-protein 4 ribonucleic acid revolutions per minute Schedule for Affective Disorders and Schizophrenia-lifetime Shrimp alkalische Phosphatase synapse-associated protein of 102 kDa sodium dodecyl sulphate Sekunde soluble guanylyl cyclase single nucleotide polymorphism sogenannt Tabelle Tris-Acetat-EDTA unit unter anderem U2AF homology motif kinase 1 untranslated region ultraviolett vor allem Valin versus World Health Organization zum Beispiel zentrales Nervensystem Zonula occludens Protein 1 1 Einleitung 1 Einleitung 1.1 Schizophrenie 1.1.1 Diagnostik und Klinik Schizophrenie ist eine schwerwiegende psychiatrische Erkrankung, die weltweit zu den zehn häufigsten Gründen für eine krankheitsbedingte Beeinträchtigung der Lebensqualität gehört und hohe ökonomische und soziale Kosten verursacht (Mueser und McGurk 2004; Tandon et al. 2008b). Es existieren mehrere wissenschaftliche Theorien darüber, wie lange es die Krankheit Schizophrenie schon gibt. Traditionell geht man jedoch davon aus, dass sie so alt ist wie der Homo sapiens selbst, was durch über 1000 Jahre alte Beschreibungen von Psychosen unterstützt wird (wiedergegeben in Adityanjee et al. 1999; Tandon et al. 2009). Im ausgehenden 19. Jahrhundert teilte Emil Kraepelin die funktionellen Psychosen in zwei Kategorien ein, die sich bezüglich ihrer Prognose unterschieden. Er grenzte das „manisch-depressive Irresein“ mit fluktuierendem Verlauf und kompletter Genesung zwischen den Episoden von der Dementia praecox ab. Unter diesem Überbegriff vereinte er die Katatonie, die Hebephrenie, zuvor beschrieben von Karl Ludwig Kahlbaum bzw. Ewald Hecker, und die von ihm beschriebene paranoide Demenz. Kraepelin bemerkte Unterschiede hinsichtlich Ersterkrankungsalter, Familiengeschichte und prämorbider Persönlichkeit zwischen beiden Krankheiten und fand Hinweise auf Vererblichkeit der Dementia praecox (Adityanjee et al. 1999; Naqvi 2008). Am Anfang des 20. Jahrhunderts wurde dann der Begriff Schizophrenie (altgriechisch: schizein, „abspalten“ und phrēn, „Zwerchfell, Seele“) von Eugen Bleuler eingeführt. Er betrachtete die Symptomatik im Querschnitt und formulierte sogenannte Grund- (Assoziationslockerung, Affektstörung, Autismus, Ambivalenz) und Nebensymptome (Wahrnehmungsstörungen, inhaltliche Denkstörungen, Katatonie), wobei er die Grundsymptome allein als charakteristisch für die Schizophrenie ansah (Adityanjee et al. 1999; Naqvi 2008). Später formulierte Kurt Schneider die Symptome ersten und zweiten Ranges, die Wahrnehmungsstörungen, inhaltliche Denkstörungen und Ich-Störungen beschreiben. Seiner Meinung nach wiesen die Symptome ersten Ranges eindeutig auf Schizophrenie hin, allerdings weiß man heute, dass sie auch bei 1 1 Einleitung anderen Zuständen auftreten können. Schneiders Kriterien wurden zum Inhalt vieler strukturierter Interviews und trugen entscheidend zu den heutigen diagnostischen Klassifikationssystemen bei. Heute wird die Diagnose entweder nach ICD-10 oder DSM-IV gestellt. Beide Systeme definieren Symptome und Beeinträchtigungen auf ähnliche Art und Weise und haben die Diagnostik zuverlässiger gemacht (Mueser und McGurk 2004). Bei ICD-10 müssen die notwendigen Diagnosekriterien für den Zeitraum von mindestens einem Monat bestehen, während bei DSM-IV ein Auftreten der charakteristischen Merkmale von mindestens sechs Monaten gefordert wird. Schizoaffektive und andere transiente psychotische Störungen werden ebenfalls in der Gruppe der Schizophrenie klassifiziert (Adityanjee et al. 1999; Mueser und McGurk 2004). Nach ICD-10 erfolgt eine Subtypisierung in die paranoide, hebephrene, katatone und undifferenzierte Schizophrenie, die postpsychotische Depression, das schizophrene Residuum und die Schizophrenia simplex, welche jeweils unterschiedliche Prognosen haben (Brunnhuber et al. 2005). Die Schizophrenie zeichnet sich durch zwei große Symptomenkomplexe aus: positive bzw. psychotische Symptome und negative Symptome. Zudem spielen kognitive Beeinträchtigungen eine wichtige Rolle. Zu den psychotischen Symptomen gehören der Verlust des Realitätsbezuges, Wahnvorstellungen, Halluzinationen und absonderliches Verhalten. Typischerweise kommt es zu auditorischen Halluzinationen, es können jedoch auch visuelle, olfaktorische, gustatorische und taktile Halluzinationen auftreten. Wahnvorstellungen zeigen sich besonders in Form von Verfolgungswahn, Kontrollwahn, Größenwahn und somatischem Wahn und haben häufig einen bizarren Charakter. Unter negativen Symptomen versteht man defizitäre Zustände, in denen es zu einer Einschränkung oder einem Verlust grundlegender Emotionen und Verhaltensprozesse kommt. Dazu gehören Affektverflachung, Anhedonie, Apathie, Verarmung von Sprache, Mimik und Gestik sowie sozialer Rückzug. Während die psychotischen Symptome meist episodisch auftreten, sind Negativsymptome überwiegend konstant vorhanden und beeinträchtigen die psychosoziale Funktion des Betroffenen schwer. Zu den kognitiven Beeinträchtigungen werden Einschränkungen von Aufmerksamkeit und Konzentration, psychomotorischer Geschwindigkeit, Lernen, 2 1 Einleitung Gedächtnis und exekutiven Funktionen wie abstraktem Denken und Problemlösung gezählt (Mueser und McGurk 2004). Erkrankte leiden oft unter Komorbiditäten, darunter besonders kardiovaskuläre Krankheiten. Das Risiko für Alkohol- und Drogenprobleme, Nikotinabusus, infektiöse Krankheiten oder Obdachlosigkeit ist groß und führt zu einer erhöhten Mortalität durch Suizid, Unfälle und Krankheitsfolgen (Mueser und McGurk 2004). 1.1.2 Epidemiologie Schizophrenie hat eine Lebenszeitprävalenz von ca. 1%, die jährliche Inzidenz liegt bei 0,2-0,4/1000 (Brzustowicz et al. 2004; Mueser und McGurk 2004; Tandon et al. 2008b). Bisher ist man davon ausgegangen, dass die Krankheit bei beiden Geschlechtern ungefähr gleich häufig auftritt, allerdings wurde in neueren Metaanalysen ein häufigeres Auftreten beim männlichen Geschlecht festgestellt, das Verhältnis betrug 1,4:1 (Tandon et al. 2008a). Das Ersterkrankungsalter liegt bei Frauen zwischen 25 und 30 Jahren, bei Männern tritt die Krankheit im Durchschnitt fünf Jahre früher auf. Dieser Unterschied ist möglicherweise auf die protektive Wirkung von Östrogen und die reduzierte Sensitivität von D2-Rezeptoren im ZNS von Frauen zurückzuführen. Frauen zeigen zudem meistens einen milderen Verlauf (Mueser und McGurk 2004). Die Inzidenz und die klinischen Symptome sind laut einer WHO-Studie in verschiedenen Kulturen und Ländern, unabhängig vom Entwicklungsgrad, relativ konstant (Mueser und McGurk 2004; Naqvi 2008). 1.1.3 Ätiologie Familien-, Zwillings- und Adoptionsstudien geben deutliche Hinweise auf einen genetischen Hintergrund, die Heritabilität, also der genetisch bedingte Anteil für die Krankheitsanfälligkeit, ist sehr hoch und wird auf bis zu 80% geschätzt (Brzustowicz et al. 2004; Tandon et al. 2008a; Tandon et al. 2008b; Zheng et al. 2005). Die Konkordanzrate bei monozygoten Zwillingen liegt bei 31-78%, bei dizygoten nur bei 628%. Aus Adoptionsstudien weiß man, dass das Erkrankungsrisiko eines adoptierten 3 1 Einleitung Kindes, dessen biologische Mutter an Schizophrenie erkrankt ist, so hoch bleibt wie das eines Verwandten ersten Grades und nicht das allgemeine Risiko seiner Adoptionsfamilie annimmt (Lewis und Levitt 2002). Dies lässt den Einfluss des familiären Umfeldes auf die Krankheitsentstehung eher gering erscheinen. Während das Erkrankungsrisiko in der Allgemeinbevölkerung bei ca. 1% liegt, haben Angehörige ersten Grades eines Betroffenen ein ca. zehnfach erhöhtes Risiko, welches mit der Anzahl an betroffenen Verwandten steigt (Lewis und Levitt 2002; Mueser und McGurk 2004). Sind beide Elternteile betroffen, beträgt das Risiko für das Kind 40% (Tsuang 2000). Allerdings können Schizophrenien auch sporadisch auftreten. Obwohl die Genetik einen großen Beitrag zur Entstehung der Schizophrenie liefert, spielen auch andere Faktoren eine wichtige Rolle. Hinweise dafür sind die erwähnte unvollständige Konkordanz bei eineiigen Zwillingen und die Tatsache, dass das Risiko der Kinder eines betroffenen und eines nicht betroffenen Zwillings an Schizophrenie zu erkranken jeweils gleich groß ist. Dies bedeutet, dass die genetische Prädisposition bei beiden Zwillingen vorhanden ist, die Erkrankung jedoch nur bei einem, möglicherweise durch Umweltfaktoren oder aber auch durch epigenetische Mechanismen, ausgelöst wird (Craddock und Jones 1999; Tsuang 2000). Zu den bekannten Risikofaktoren gehören prä- und perinatale Probleme wie maternale Infektionen, Unterernährung und Rauchen während der Schwangerschaft sowie Entbindungskomplikationen, besonders im Zusammenhang mit fetaler Hypoxie. Geburt im späten Winter oder frühen Frühling ist mit einem 5-10% höheren Erkrankungsrisiko assoziiert. Auch soziodemographische Faktoren wie Armut oder Geburt in städtischer Gegend spielen statistisch gesehen eine Rolle (Mueser und McGurk 2004; Tandon et al. 2008a; Tsuang 2000). 1.1.4 Neuropathologie Auffällig ist ein erweitertes Ventrikelsystem mit einer Reduktion von Gehirnvolumen und grauer Substanz sowohl bei Erkrankten als auch bei gesunden Angehörigen. Zu den Regionen mit vermindertem Volumen gehören Frontallappen, Amygdala, Hippokampus, Parahippokampus, Thalamus, Gyrus temporalis superior und medius sowie Gyrus cinguli. In PET-Studien zeigte sich bei Erkrankten, die Aufgaben 4 1 Einleitung bezüglich exekutiver Funktion, Gedächtnis und Aufmerksamkeit durchführten, außerdem ein abnormaler Blutfluss in frontalem Kortex, Kleinhirn und Thalamus (Mueser und McGurk 2004). Ein weiterer wichtiger neuropathologischer Befund ist das Fehlen einer Gliose, die typisch für neurodegenerative oder inflammatorische Prozesse ist. Generell lassen MRT-Studien eher eine Unterbrechung funktionaler Schaltkreise als eine Dysfunktion einzelner Gehirnregionen vermuten (Mueser und McGurk 2004). 1.2 Bipolar-affektive Erkrankung 1.2.1 Diagnostik und Klinik Die bipolar-affektive Erkrankung wurde in der Geschichte in vielen verschiedenen Kulturen beschrieben (Belmaker 2004). Kraepelin vereinigte Ende des 19. Jahrhunderts verschiedene Formen der Manie und Depression unter dem Begriff „Manischdepressives Irresein“ und grenzte sie im Sinne der von ihm propagierten Dichotomie von der Dementia praecox ab (Angst und Gamma 2008; Cookson 2005; Erfurth und Arolt 2003). Karl Leonhard und Jules Angst etablierten später aufgrund epidemiologischer, klinischer und prognostischer Unterschiede die „bipolar vs. unipolar Dichotomie“ (Brieger 2007; Cookson 2005; Oswald et al. 2007). Im amerikanischen Klassifikationssystem wurde in den 70er Jahren die bipolar-affektive Störung, gestützt durch genetisch-epidemiologische Untersuchungen, wiederum in Bipolar I und Bipolar II untergliedert (Brieger 2007; Cookson 2005; Erfurth und Arolt 2003). Als klassische bipolar-affektive Erkrankung gilt die Bipolar-I-Störung (Belmaker 2004). Nach DSMIV bezeichnet die Bipolar-I-Störung Episoden der Manie mit oder ohne Depression, wobei hier bereits eine Episode der Manie ausreichend für die Diagnose ist, wohingegen bei ICD-10 zwei Episoden notwendig sind, von denen eine manisch, hypomanisch oder gemischt sein muss (Belmaker 2004; Cookson 2005). Als Bipolar-II-Störung bezeichnet man rekurrente depressive Phasen mit hypomanen Episoden, welche vom Betroffenen nicht unbedingt als krankhaft erkannt werden und ohne psychotische und selbst- bzw. fremdgefährdende Symptome einhergehen. Sie fällt bei ICD-10 unter „sonstige bipolaraffektive Störungen“ (Cookson 2005; Belmaker 2004; Brieger 2007; Erfurth und Arolt 2003). Die Dauer der einzelnen Episoden ist individuell oft stabil (Cookson 2005). 5 1 Einleitung Zwischen den Episoden kommt es in der Regel zu einer Remission, ca. 20% der Erkrankten leiden jedoch auch im Intervall unter Stimmungslabilität (Angst 2007; Belmaker 2004; Oswald et al. 2007). Eine besondere Form ist das rapid cycling, bei der mindestens vier Episoden innerhalb eines Jahres auftreten. Diese Form ist besonders schwer zu behandeln und hat eine schlechte Prognose (Cookson 2005; Belmaker 2004; Erfurth und Arolt 2003; Oswald et al. 2007). Klinisch ist die Störung durch zwei entgegengesetzte Pole gekennzeichnet. In der depressiven Episode sind eine Störung der Affektivität im Sinne eines Nichtfühlenkönnens und ein massiv gehemmter Antrieb, dessen maximale Form der depressive Stupor darstellt, charakteristisch. Zudem kommt es zu eingeschränktem und unproduktivem Denken. Oft besteht in der Depression auch ein Wahnerleben, typisch sind Schuldwahn, Verarmungswahn, Krankheitswahn und nihilistischer Wahn. Zudem treten vegetative Störungen mit verändertem Schlaf-Wach-Rhythmus und Schlafmangel auf. Charakteristisch sind auch tageszeitliche Schwankungen mit dem sogenannten Morgentief. In der manischen Phase ist eine Hochstimmung, oft verbunden mit Begeisterung oder Reizbarkeiz ein essentielles Kriterium. Charakteristischerweise kommt es zu Ideenflucht, Gedankenrasen und einem Rededrang, in abgeschwächter Form fällt eine weitschweifige Sprache auf. Außerdem hat der Betroffene eine exzessive Energie und ein deutlich überhöhtes Selbstbewusstsein, was zu einem Gefühl der Grandiosität mit überhöhter Einschätzung der eigenen Fähigkeiten und übersteigertem Ehrgeiz führen kann. Daraus können auch psychotische Wahnvorstellungen und Halluzinationen resultieren, wie z. B. die Überzeugung von großem Reichtum, einem speziellen Auftrag, Verfolgungswahn oder unterstützenden und bestärkenden Stimmen. Auf kognitiver Ebene ist der Betroffene leicht ablenkbar und hat Konzentrationsschwierigkeiten, bleibt aber stets zeitlich und örtlich orientiert. Auch in der manischen Phase treten vegetative Symptome in Form von vermindertem Schlafbedürfnis, verstärktem Appetit und vermehrtem Interesse an Sexualität auf (Cookson 2005; Tölle und Windgassen 2006). Generell kommt es oft zu einem Hochrisikoverhalten durch Verlust der Urteilskraft. Charakteristisch für die manische Episode sind eine fehlende Krankheitseinsicht und ein 6 1 Einleitung nicht mit der normalen Persönlichkeit kongruentes Verhalten des Patienten (Belmaker 2004; Cookson 2005; Shastry 2005). Erschwert wird die Krankheit durch eine hohe Rate an Komorbiditäten wie Alkohol-, Drogen- oder Medikamentenmissbrauch sowie Angst- und Persönlichkeitsstörungen, die zu einem schlechteren Verlauf und einem erhöhten Selbstmordrisiko führen (Angst 2007; Angst und Gamma 2008; Cookson 2005; Oswald et al. 2007). Die bipolaraffektive Störung ist außerdem mit somatischen Krankheiten wie Diabetes mellitus, Hypertonie und anderen kardiovaskulären Erkrankungen assoziiert, woraus auch die höhere Mortalitätsrate im Vergleich zur Allgemeinbevölkerung resultiert, die nicht alleine auf Suizid zurückzuführen ist (Angst 2007; Brieger 2007; Shastry 2005). 1.2.2 Epidemiologie Die weltweite Lebenszeitprävalenz der bipolar-affektiven Störung wird auf 1,0-1,5% geschätzt, nach einer neueren Studie liegt sie mit 3,3% sogar noch höher (Bauer und Pfennig 2005; Brieger 2007). Zur Prävalenz der Bipolar-II-Störung gibt es nur wenige valide Studien, sie wird aber mit 2-6% höher eingeschätzt (Bauer und Pfennig 2005; Belmaker 2004; Brieger 2007; Oswald et al. 2007). Die verschiedenen Prävalenzzahlen zur bipolaren Störung schwanken im Wesentlichen nach den jeweils verwendeten Kriterien zur Einordnung einer Erkrankung in das sog. „bipolare Spektrum“. Die bipolar-affektive Störung generell tritt, anders als die unipolare Depression, bei Männern und Frauen gleich häufig auf (Angst und Gamma 2008; Shastry 2005). Dagegen kommt die Bipolar-II-Störung bei Frauen öfter vor (Brieger 2007). Die Erkrankung tritt meistens erstmals in der Adoleszenz auf, der erste Gipfel des Ersterkrankungsalters liegt im Durchschnitt zwischen 16 und 20 Jahren (Oswald et al. 2007; Sanacora et al. 2008; Shastry 2005). Es versterben 5-20% aller bipolar-affektiv Erkrankten an einem Suizid, was einem ca. 30-fach erhöhten Risiko gegenüber der Allgemeinbevölkerung entspricht (Brieger 2007; Guze und Robins 1970; Jamison 1986; McIntyre et al. 2008). 7 1 Einleitung 1.2.3 Ätiologie Auf die hauptsächlich genetische Komponente der bipolar-affektiven Störung weisen diverse Zwillings- und Familienstudien hin (Le Hellard et al. 2007; Segurado et al. 2003). In der Hälfte der Fälle kommen affektive Krankheiten in der Familie des Erkrankten vor (Belmaker 2004). Die Erkrankungswahrscheinlichkeit liegt bei Verwandten ersten Grades von bipolar-affektiv Erkrankten um ein Zehnfaches höher als in der Allgemeinbevölkerung (Bauer und Pfennig 2005). Das Erkrankungsrisiko eines Kindes mit einem betroffenen Elternteil beträgt 20%, wenn beide erkrankt sind, steigt es auf 50-60% (Tölle und Windgassen 2006). Zwillingsstudien zeigen bei monozygoten Zwillingen eine Konkordanz von 40-80%, bei dizygoten liegt sie bei 10-20% (Belmaker 2004; Kato 2001; Shastry 2005). In Adoptionsstudien ist das Erkrankungsrisiko für die biologischen Eltern eines betroffenen Patienten signifikant höher als für die Adoptiveltern (Craddock und Jones 1999). Wie schon für Schizophrenie beschrieben, weist die unvollständige Konkordanz monozygoter Zwillinge auf die Bedeutung nicht-genetischer Faktoren hin. Bei einer statistischen Heritabilität von 85% bleiben demnach ca. 15% Varianz für individuelle Umweltfaktoren (Bauer und Pfennig 2005; Craddock und Jones 1999). Dabei spielen unter anderem life-events und das soziale Umfeld eine Rolle (Alloy et al. 2005). 1.2.4 Neuropathologie Neuropathologische Abnormalitäten betreffen vor allem den präfrontalen Kortex, hier finden sich ein reduzierter Blutfluss und ein vermindertes Volumen der grauen Substanz. Morphometrisch fällt eine reduzierte Größe und Dichte von Neuronen und Gliazellen sowohl präfrontal als auch temporal auf (Belmaker 2004; Duman 2002; Harrison 2002; Shastry 2005; Zarate et al. 2003). Auch der Hippokampus ist größengemindert und in seiner Formation verändert, übereinstimmend mit der hippokampalen Stress-Atrophie-Hypothese, ebenso besteht eine Atrophie in limbischen Strukturen (Duman 2002; Harrison 2002; Shastry 2005). Des Weiteren wurden Veränderungen in subkortikalen Strukturen, synaptischen Endigungen und Dendriten beobachtet (Harrison 2002). Neben den oben beschriebenen Pathologien wurden in 8 1 Einleitung bildgebenden und postmortalen Studien Abnormalitäten in Striatum, Kleinhirn, anteriorem Cingulum, Basalganglien, subgenualen Raphekernen und Amygdala festgestellt (Bauer und Pfennig 2005; Zarate et al. 2003). Wie bei der Schizophrenie kommt es bei der bipolar-affektiven Störung nicht zu einer Gliose. Es ist nicht zweifelsfrei geklärt, ob die neuropathologischen Veränderungen auf eine Störung der neuronalen Entwicklung zurückgehen, kompensatorische Veränderungen anderer pathogener Prozesse sind oder Folgekomplikationen von affektiven Episoden darstellen (Zarate et al. 2003). Es gibt ähnliche neuropathologische Veränderungen bei Schizophrenie und bipolaraffektiver Erkrankung. So sind beispielsweise die neuronale Größe im präfrontalen Kortex und die neuronale Dichte im anterioren Cingulum vermindert sowie synaptische und dendritische Marker im PFC und Hippokampus reduziert. Zudem besteht bei beiden Krankheiten ein Gliadefizit. Trotzdem gibt es auch spezifische Veränderungen (Harrison 2002). 1.3 Pathophysiologie endogener Psychosen 1.3.1 Schizophrenie 1.3.1.1 Theorie der neuronalen Entwicklungsstörung Wahrscheinlich liegen dem klinischen Bild der Schizophrenie verschiedene pathogenetische Ursachen zu Grunde. So gibt es zum einen die Theorie der neuronalen Entwicklungsstörung. Sie basiert darauf, dass eine zeitliche Differenz zwischen ätiologisch relevanten Umgebungsfaktoren und dem klinischen Auftreten der Krankheit besteht. Viele Faktoren, die im Verdacht stehen, einen Einfluss auf die Krankheitsentstehung zu haben, sind Ereignisse des frühen Lebens, wie beispielsweise Geburt im Winter oder frühen Frühling, Geburt und Aufwachsen in urbanen Gegenden oder prä- und perinatale Komplikationen. Auch fallen bei Individuen, die später eine Schizophrenie entwickeln, in Kindheit und früher Jugend motorische Abnormalitäten und Störungen des Sozialverhaltens auf. Damit übereinstimmend sind strukturelle neuropathologische Veränderungen schon in den frühesten Phasen der Schizophrenie 9 1 Einleitung vorhanden und im Verlauf nicht progredient. Sie könnten also Folgeschäden von frühen Störungen der neuronalen Entwicklung sein (Murray und Lewis 1987; Tsuang 2000). Außerdem ist das Fehlen einer Gliose ebenfalls hinweisend auf eine neuronale Entwicklungsstörung. Eventuell liegt eine fehlerhafte Migration der Neurone in der Entwicklung vor, welche in abnormalen neuronalen Verbindungen und Schaltkreisen resultiert (Mueser und McGurk 2004; Naqvi 2008). Insgesamt gibt es diverse Hinweise darauf, dass Störungen der Gehirnfunktion, zumindest bei einigen Betroffenen, schon früh im Leben vorhanden sind, bevor es später zu ersten Symptomen der Krankheit kommt (Lewis und Levitt 2002). 1.3.1.2 Neurotransmitterhypothesen Es wird außerdem die Beteiligung verschiedenster Neurotransmittersysteme an der Ätiologie der Schizophrenie angenommen, darunter neben Dopamin und Glutamat, welche im Folgenden ausführlicher beschrieben werden, auch GABA, Serotonin sowie das cholinerge und das opioide System (Laruelle et al. 2003). Die älteste und am längsten überdauernde Neurotransmitterhypothese ist die sog. Dopaminhypothese. Sie gründet auf der Feststellung, dass dopaminfreisetzende Stimulantien wie Amphetamin Psychosen auslösen können und antipsychotische Medikamente wie Chlorpromazin dopaminerge D2-Rezeptoren blockieren und ihre Affinität zum D2-Rezeptor mit ihrer klinischen Potenz korreliert (Burt et al. 1976; Coyle et al. 2003; Creese et al. 1976; Seeman et al. 1975; Seeman und Lee 1975). Im Rahmen dieser Theorie wird angenommen, dass negative und kognitive Symptome durch eine verminderte Dopaminfunktion im präfrontalen Kortex und Positivsymptome durch eine subkortikale und mesolimbische dopaminerge Überfunktion verursacht werden (Abi-Dargham und Moore 2003; Andreasen 1999; Belsham 2001; Knable und Weinberger 1997). Die Grenzen dieser Hypothese bestehen darin, dass negative und kognitive Symptome nicht durch antipsychotische Medikation gebessert werden und Amphetamine nur eine Positivsymptomatik auslösen (Belsham 2001; Coyle et al. 2003). Trotzdem geht man heute davon aus, dass Veränderungen der dopaminergen Aktivität wahrscheinlich in Kombination mit anderen Transmittersystemen - eine wichtige pathogenetische Rolle spielen (Benes 2009). 10 1 Einleitung Die Glutamathypothese ist ein alternatives Erklärungsmodell, welches auch die wichtigen Negativsymptome integriert (Stone et al. 2007). 1980 wurde erstmals von Kim et al. vorgeschlagen, dass eine verminderte Glutamataktivität ursächlich an Schizophrenie beteiligt sein könnte, als sie eine reduzierte Glutamatkonzentration im Liquor schizophrener Patienten entdeckten (Kim et al. 1980). Glutamat ist im Gehirn ubiquitär vorhanden und der wichtigste exzitatorische Neurotransmitter. Glutamaterge Neurone projizieren innerhalb des Kortex sowie in subkortikale Regionen wie Locus coeruleus, Raphekerne und Substantia nigra, wo monoaminerge Signalwege moduliert werden (Kugaya und Sanacora 2005). Grundlage der Hypothese ist die Feststellung, dass Phencyclidin und Ketamin, zwei nicht-kompetitive Antagonisten am NMDARezeptor, die den Ionenkanal blockieren, bei Gesunden zu schizophrenietypischen Symptomen wie Halluzinationen, Wahnvorstellungen, kognitiven Störungen und vor allem auch zu Negativsymptomen führen (Goff und Coyle 2001; Javitt 2004; Schosser und Aschauer 2004; Stone et al. 2007; Toro und Deakin 2005). Bei Schizophrenen, die sich in einem stabilen Zustand befinden, können diese Antagonisten einen Rückfall auslösen (Stone et al. 2007). Die Glutamathypothese wird auch als NMDA-RezeptorHypofunktionshypothese bezeichnet. Es gibt insgesamt acht verschiedene Glutamatrezeptoren, darunter die ionotropen NMDA-, AMPA- und Kainatrezeptoren sowie verschiedene metabotrope Rezeptoren (Belsham 2001). Sie sind auf Synapsen und Gliazellen lokalisiert (Kugaya und Sanacora 2005). Der NMDA-Rezeptor ist eine heteromere Struktur aus vier bzw. fünf Untereinheiten, darunter mindestens eine Kopie der obligatorischen NR1-Untereinheit und verschiedene NR2A-D- und NR3-Untereinheiten, die unterschiedliche Kanäle in Bezug auf physiologische und pharmakologische Eigenschaften bilden (Kristiansen und Meador-Woodruff 2005; Lau und Zukin 2007). Die Anzahl und Zusammensetzung der Rezeptoren ist dynamisch und hängt von der neuronalen Aktivität ab (Lau und Zukin 2007). Bei Ruhemembranpotential ist der NMDAR durch Mg2+-Ionen blockiert. Um aktiviert zu werden, muss Glutamat nicht nur an den NMDAR, sondern auch an den AMPAR binden, so dass es zu einer Depolarisation durch Na+-Einstrom kommt, welche zur Freigabe des NMDARIonenkanals führt (Javitt 2004; Lau und Zukin 2007). Der Rezeptor wird also dual reguliert, einerseits über das Potential, andererseits über Ligandenbindung (Kugaya und 11 1 Einleitung Sanacora 2005). Am NMDAR gibt es zudem eine modulierende Glycin-Bindestelle, welche mit den endogenen Liganden Glycin oder D-Serin besetzt sein muss, damit Glutamat den Kanal öffnen kann (Coyle et al. 2003; Javitt 2004). Der Kanal ist hochpermeabel für Ca2+- und zu einem geringeren Grad auch für Na+- und K+-Ionen, wobei der Calciumeinstrom zu einer Aktivierung von Proteinkinasen und Phosphatasen führt (Belsham 2001; Coyle et al. 2003; Lau und Zukin 2007). Der Vorgang wird primär durch die Aufnahme des Transmitters aus der Synapse durch EAATs (excitatory amino acid transporters) auf Neuronen und Gliazellen beendet (Javitt 2004; Sanacora et al. 2008). Abb. 1.1: Schematische Darstellung des NMDA-Rezeptors (nach Albensi 2007) Synaptische NMDAR sind an den postsynaptischen Verdichtungen lokalisiert, wo sie Teil eines makromolekularen Komplexes von Glutamatrezeptoren, Gerüst-, Signal- und Adaptorproteinen sind (Lau und Zukin 2007; Sanacora et al. 2008). Die Adaptorproteine verankern die Rezeptoren an der postsynaptischen Membran, indem sie direkt an PDZ-Erkennungsmotive der NR2-Untereinheit binden (Lau und Zukin 2007). Zu diesen sogenannten MAGUKs (membrane-associated guanylate kinases) gehören PSD95, PSD93, SAP102 und NF-L (Kristiansen und Meador-Woodruff 2005; Toro und Deakin 2005). Neben ihrer Funktion als Ankerproteine modulieren sie die Aktivität des Rezeptors, koppeln ihn an Kinasen, Phosphatasen und andere nachgeschaltete intrazelluläre Signalwege und beeinflussen das Targeting zur Membran (Clinton und Meador-Woodruff 2004; Lau und Zukin 2007). PSD-Proteine stellen somit eine Brücke 12 1 Einleitung zwischen extrazellulären synaptischen Signalen und intrazellulärer Signalübermittlung dar (Toro und Deakin 2005). Der NMDAR hat eine spezielle Bedeutung bei der Langzeitpotenzierung (LTP) und Langzeitdepression (LTD), welche eine wichtige Rolle bei der Bildung des Gedächtnisses und beim Lernen spielen (Kugaya und Sanacora 2005; Lau und Zukin 2007). Entscheidend für die Langzeitpotenzierung ist eine hohe präsynaptische Glutamataktivität, die zu einer Rekrutierung von NMDA-Rezeptoren und konsekutivem Calciumeinstrom führt (Coyle et al. 2003; Lau und Zukin 2007). Des Weiteren führt die Aktivierung des NMDAR zu erhöhter Expression von BDNF (brain derived neurotrophic factor) und, vermittelt durch CREB (cyclic AMP response element binding protein), zu verstärkter Genexpression (Kugaya und Sanacora 2005). NMDARAktivierung hat dementsprechend trophische Effekte, während Inaktivierung zu Apoptose führt (Coyle et al. 2003). Die chronische Einnahme von NMDARAntagonisten induziert beispielsweise die Degeneration von Pyramidenneuronen (Javitt 2004). Bei Überaktivität des NMDAR kommt es zu einer Exzitotoxizität, die Folge einer zu starken Aktivierung calciumabhängiger Enzyme und der Generierung freier Radikale ist. Daher ist eine exakte Kontrolle des Glutamatsystems besonders wichtig (Sanacora et al. 2008; Zarate et al. 2003). Prädiktive Validität erhielt die NMDAR-Hypofunktionshypothese durch die Entdeckung, dass die Co-Agonisten Glycin und D-Cycloserin in Kombination mit konventionellen Antipsychotika zu einer Verbesserung der Negativsymptomatik führen (Belsham 2001; Coyle et al. 2003; Javitt 2004; Laruelle et al. 2003). In einigen Studien konnte dabei eine Korrelation der Höhe des basalen Glycinspiegels mit einer Verbesserung der Negativsymptomatik festgestellt werden (Javitt 2004). Doch auch die vielfältigen Funktionen des NMDAR, insbesondere sein Einfluss auf Lern- und Gedächtnisprozesse, die bei Schizophrenie betroffen sind, und seine Rolle bei synaptischer und neuronaler Plastizität, sprechen für seine Beteiligung an der Pathophysiologie (Javitt 2004; Sanacora et al. 2008). Die Hypofunktion des NMDAR ist vermutlich mit einer exzessiven Glutamatausschüttung und konsekutiver Überstimulation von postsynaptischen Neuronen assoziiert (Belsham 2001). In Übereinstimmung mit dieser Theorie stimulieren NMDAR-Antagonisten akut die präfrontale Glutamatausschüttung (Javitt 2004). Eine enthemmte Glutamataktivität 13 1 Einleitung würde auch ein aberrantes LTP auslösen, was in veränderten Gedächtnisleistungen und möglicherweise auch Wahnvorstellungen resultieren könnte (Belsham 2001). Davon ausgehend, dass Veränderungen in der Funktion des gesamten NMDARKomplexes eine Rolle in der Pathophysiologie spielen könnten, wurden funktionelle Studien durchgeführt, die die Expression einzelner NMDAR-Untereinheiten und assoziierter Moleküle an der Synapse untersuchten (Toro und Deakin 2005). Dabei fanden sich eine verminderte Expression von PSD95 im hippokampalen Gyrus dentatus und eine reduzierte Expression von SAP102 im Striatum (Kristiansen und MeadorWoodruff 2005; Toro und Deakin 2005). Im Thalamus zeigte sich eine signifikante Erhöhung von NR2B- und eine verminderte Expression von NF-L-, PSD95- und SAP102- Transkripten (Clinton und Meador-Woodruff 2004). Allerdings gibt es auch Studien, die diesen Ergebnissen widersprechen und von reduzierter Expression von NR1- und 2C-Untereinheiten sowie erhöhter Expression von NF-L-, PSD95- und SAP102-Transkripten im Thalamus berichten (Clinton und Meador-Woodruff 2004). Obwohl sich aus diesen Befunden keine eindeutige Schlussfolgerung ziehen lässt, kann man davon ausgehen, dass eine veränderte Expression der mit dem NMDAR assoziierten Proteine, insbesondere PSD95, zu einer abnormen Lieferung und Verankerung des Rezeptors in der Synapse sowie veränderter intrazellulärer Signaltransduktion führen könnte. Dies würde im weiteren Sinne mit einer gestörten NMDAR-Funktion übereinstimmen (Clinton und Meador-Woodruff 2004; Kristiansen und Meador-Woodruff 2005; Toro und Deakin 2005). Die NMDAR-Hypofunktionshypothese wäre auch kongruent mit der Theorie der neuronalen Entwicklungsstörung, da eine eingeschränkte Funktion des NMDAR aus gestörten Abläufen in der neuronalen Entwicklung resultieren könnte (Belsham 2001). Eventuell sind Abnormalitäten der NMDAR-Funktion schon früh vorhanden, führen aber erst nach Ausreifung der neuronalen Verbindungen zu psychotischen Symptomen (Stone et al. 2007). Wahrscheinlich ist, dass die Veränderungen nicht nur ein Transmittersystem betreffen, sondern dass es zu Interaktionen kommt, besonders mit Dopamin und GABA (Belsham 2001; Howes et al. 2009). Eine veränderte Funktion des NMDAR könnte z. B. eine verstärkte Dopaminausschüttung zur Folge haben (Coyle et al. 2003). Vorstellbar ist, dass eine NMDAR-Hypofunktion negative und kognitive Symptome auslöst, während 14 1 Einleitung eine verstärkte dopaminerge Neurotransmission die Positivsymptomatik hervorruft (Coyle et al. 2003; Stone et al. 2007). 1.3.2 Bipolar-affektive Erkrankung 1.3.2.1 Monoaminhypothese Die Monoaminhypothese basiert auf Erkenntnissen über die neurobiologische Wirkungsweise von Antidepressiva, welche die Verfügbarkeit von monoaminergen Neurotransmittern im synaptischen Spalt erhöhen (Baumann et al. 2003). Auch die Beobachtung, dass das Antihypertensivum Reserpin, welches die Konzentration von Monoaminen in den synaptischen Vesikeln vermindert, gelegentlich eine depressive Symptomatik auslöst, trug zu dieser Theorie bei (Ackenheil 2001). Heute gibt es diverse Hinweise, die für eine ätiologische Beteiligung von Dopamin, Noradrenalin und Serotonin nicht nur an der Depression, sondern auch an der bipolar-affektiven Erkrankung sprechen. Die Entdeckung, dass Patienten mit bipolar-affektiven Erkrankungen durch direkte oder indirekte Dopaminagonisten eine Manie entwickeln können, deutet auf eine ursächliche Beteiligung des dopaminergen Systems hin (Minton et al. 2009). Möglicherweise liegt in depressiven Episoden ein reduzierter und in manischen Episoden ein normaler oder sogar erhöhter dopaminerger Tonus vor (Brugue und Vieta 2007). Damit übereinstimmend weist der Dopaminmetabolit Homovanillinsäure im Liquor von Erkrankten phasenspezifische Veränderungen auf, er ist während depressiver Episoden vermindert und während manischer Phasen erhöht (Brugue und Vieta 2007). Weiterhin gibt es Hinweise auf funktionelle Defizite im noradrenergen und serotonergen System. Beispielsweise finden sich ein erhöhter Noradrenalinumsatz in verschiedenen kortikalen Regionen und im Thalamus sowie eine Verminderung des Serotoninmetabolits 5-Hydroxyindolessigsäure und des SerotoninTransporters im Kortex (Vawter et al. 2000). 15 1 Einleitung 1.3.2.2 Dysfunktion der Hypothalamus-Hypophysen-Nebennieren-Achse Auch eine gestörte Funktion der Hypothalamus-Hypophysen-Nebennieren (HHN)Achse, welche eine maßgebliche Rolle bei der Stressantwort spielt, steht im Verdacht, an der Entstehung affektiver Störungen beteiligt zu sein. Man geht davon aus, dass eine Überaktivität der HHN-Achse zu einer Hyperkortisolämie führt, was toxische Effekte auf hippokampale Neuronen hat (McEwen 1999; Sapolsky 2000; Vawter et al. 2000). Während nicht eindeutig geklärt ist, ob bei bipolar-affektiv Erkrankten die basalen Kortisolwerte verändert sind, konnte wiederholt festgestellt werden, dass sowohl in depressiven als auch in manischen Phasen eine gesteigerte Antwort auf den dex/CRHTest besteht. Uneinheitliche Ergebnisse gibt es hinsichtlich der Frage, ob eine abnormale Antwort auf den dex/CRH-Test auch bei Patienten in Remission bestehen bleibt (Rybakowski und Twardowska 1999; Schmider et al. 1995; Watson et al. 2004). 1.3.2.3 Veränderungen der synaptischen Plastizität Eine weitere Theorie geht davon aus, dass Beeinträchtigungen der neuronalen Plastizität der Pathophysiologie affektiver Störungen zugrunde liegen könnten und dass Antidepressiva und Stimmungsstabilisatoren hauptsächlich auf Signalwege wirken, die Neuroplastizität und Zellüberleben regulieren (Shastry 2005; Zarate et al. 2003). Wichtig für die Vermittlung langfristiger Neuroplastizität ist die MAP-KinaseSignalkaskade, über die viele endogene Wachstumsfaktoren wie BDNF ihre Effekte vermitteln (Zarate et al. 2003). BDNF ist wichtig für die Differenzierung von Neuronen während der Entwicklung, hat auch im adulten Gehirn neuroprotektive Wirkungen und trägt zur synaptischen Plastizität bei (Duman 2002). Ein nachgeschalteter Effekt der MAP-Kinase ist die Phosphorylierung von CREB, was zu vermehrter Genexpression führt (Zarate et al. 2003). Diese beiden Faktoren tragen eventuell zur Störung der synaptischen Plastizität bei affektiven Störungen bei. Man geht davon aus, dass ihre Expression und Funktion bei Erkrankten vermindert sind und Medikamente wie Noradrenalin- und Serotonin-Wiederaufnahmehemmer dem entgegenwirken (Duman 2002). 16 1 Einleitung 1.3.2.4 Hinweise auf Beteiligung des Glutamatsystems Eine Beteiligung von Glutamat an der Ätiologie affektiver Störungen wurde schon in den 60er Jahren postuliert, als man beobachtete, dass Tuberkulosepatienten, die mit DCycloserin, einem Antibiotikum mit partialagonistischer Wirkung am NMDAR, behandelt wurden, eine klinische Verbesserung ihrer depressiven Symptomatik zeigten (Crane 1961). Bei Personen mit affektiven Störungen wurden außerdem Abnormalitäten des Glutamatspiegels in Plasma, Serum, Liquor und Gehirngewebe gefunden, wobei diese Ergebnisse teilweise nicht repliziert werden konnten (Sanacora et al. 2008; Zarate et al. 2003). Auch pharmakologische Ergebnisse stützen die Hypothese einer Beteiligung des Glutamatsystems. Klinischen Studien zufolge haben NMDARAntagonisten antidepressive Effekte, die möglicherweise auf eine Erhöhung des extrazellulären Glutamatlevels zurückgehen. Ketamin z. B. hat als Einzeldosis eine ca. 72 Stunden andauernde antidepressive Wirkung, die wahrscheinlich durch eine erhöhte präsynaptische Ausschüttung von Glutamat, eventuell vermittelt über GABAerge Interneurone, zustande kommt (Javitt 2004; Kugaya und Sanacora 2005; Sanacora et al. 2008). Dieser erhöhte Glutamatausstrom wirkt vermutlich auf glutamaterge AMPARezeptoren (Kugaya und Sanacora 2005; Sanacora et al. 2008). Ähnlich wie bei Schizophrenie könnten auch bei Depression Zusammenhänge mit Störungen des hippokampalen LTP und der Neurogenese bestehen, die durch eine NMDAR-Blockade oder einen nachfolgenden Reboundeffekt verändert werden könnten (Javitt 2004). Auch Antidepressiva haben Auswirkungen auf das Glutamatsystem. Ihre chronische Applikation wirkt modifizierend und regulierend auf die Aktivität glutamaterger Neurone sowie die NMDAR-Expression und -Funktion (Sanacora et al. 2008; Zarate et al. 2003). Einige stimmungsstabilisierende Medikamente, z. B. das Antikonvulsivum Lamotrigin, wirken wahrscheinlich inhibierend auf die Glutamataktivität (Belsham 2001; Sanacora et al. 2008). Auch Valproat und Lithium reduzieren den synaptischen Glutamatspiegel (Sanacora et al. 2008; Zarate et al. 2003). Lithium schützt außerdem in Nagerneuronzellen vor glutamatinduzierter Exzitotoxizität, wahrscheinlich indem es glutamaterge Überstimulation und den damit verbundenen NMDAR-vermittelten Calciumeinstrom verhindert (Sanacora et al. 2008; Zarate et al. 2003). 17 1 Einleitung H-MRS Studien geben ebenfalls Hinweise auf Abnormalitäten des glutamatergen Systems, so ließen sich Veränderungen des Glutamat/Glutamin (Glx)-Levels in verschiedenen Hirnarealen nachweisen (Michael et al. 2003; Zarate et al. 2003). Hinweise auf eine Beteiligung des NMDAR stammen auch aus Studien, die Untereinheiten des NMDAR und assoziierte Proteine an der Synapse funktionell untersucht haben (Toro und Deakin 2005). Teilweise ergaben sich dabei ähnliche Ergebnisse für die bipolar-affektive Störung wie für Schizophrenie. So war auch hier die Expression von PSD95 in speziellen Regionen des Hippokampus vermindert, während die Expression von NR1 unverändert war (Toro und Deakin 2005). Ebenso war wie bei Schizophrenie die SAP102-Expression im Striatum vermindert, zusätzlich die von PSD95 (Kristiansen und Meador-Woodruff 2005). Die Ergebnisse einer Studie im Thalamus zeigten, wiederum übereinstimmend mit Schizophrenie, eine Verminderung von NF-L-, PSD95- und SAP102-Transkripten (Clinton und MeadorWoodruff 2004). Dies könnten Hinweise auf eine Beteiligung des NMDAR-Komplexes und seinen assoziierten Proteinen an der Pathophysiologie von affektiven Störungen sein. Ob die teilweise überschneidenden Befunde auf einen gemeinsamen Pathomechanismus hindeuten, ist allerdings unklar (Clinton und Meador-Woodruff 2004; Kristiansen und Meador-Woodruff 2005; Toro und Deakin 2005). Wie oben beschrieben besteht der Verdacht, dass Störungen der synaptischen Plastizität eine Rolle bei affektiven Störungen spielen. Da das Glutamatsystem die synaptische Plastizität, Zellwachstum und –atrophie beeinflusst, könnte hier ein Zusammenhang zwischen beiden Theorien bestehen (Sanacora et al. 2008). Auch die Reduktion von Gliazellen könnte über eine verminderte Aufnahme aus der Synapse zu einem erhöhten extrazellulären Glutamatlevel führen und sich so auf das glutamaterge System auswirken (Kugaya und Sanacora 2005). 1.4 Genetik 1.4.1 Vererbung Bei vielen neuropsychiatrischen Erkrankungen, die sich erst im Erwachsenenalter manifestieren, tragen sowohl genetische als auch nicht-genetische Faktoren zur 18 1 Einleitung Entstehung bei. Aus Familien-, Zwillings- und Adoptionsstudien weiß man, dass die genetischen Faktoren bei endogenen Psychosen die wichtigere Rolle spielen (Kennedy et al. 2003). Der genaue molekulargenetische Mechanismus der Vererbung ist heute noch immer nicht geklärt, allerdings stimmt man darin überein, dass kein mendelsches Vererbungsmuster vorliegt (Belmaker 2004; Schosser und Aschauer 2004). Für eine komplexe Genetik spricht die große klinische Heterogenität und das Fehlen pathognomonischer Phänotypen (Brzustowicz et al. 2004). Sowohl für Schizophrenie als auch für die bipolar-affektive Störung geht man heute von einem multifaktoriellen Modell aus, also von einer Interaktion mehrerer Gene und nicht-genetischer Umweltfaktoren (Berrettini 2000; Craddock und Jones 1999; Pulver 2000; Schosser und Aschauer 2004; Shastry 2005). Bei einer solchen Vererbung kommen die Suszeptibilitätsallele in der Population häufig vor, also auch bei gesunden Individuen, wobei der Beitrag eines einzelnen Gens zur Anfälligkeit relativ klein ist (Brzustowicz 2008; Gershon 2000; Kennedy et al. 2003; Schosser und Aschauer 2004). Die Suszeptibilitätsallele erhöhen also das Risiko zu erkranken, sind für eine Manifestation der Störung jedoch weder notwendig noch können sie die Krankheit alleine auslösen (Berrettini 2000; Schosser und Aschauer 2004). Die Krankheit entwickelt sich nur, wenn genetische und umweltbedingte Risikofaktoren in Kombination einen gewissen Schwellenwert überschreiten (Brzustowicz 2008). Alternativ hierzu kann auch eine Vielzahl seltener Risikoallele zur Erkrankung beitragen; vermutlich sind beide Vererbungsmuster von Bedeutung. Auch epigenetische Faktoren spielen wahrscheinlich eine wichtige Rolle (Petronis 2004; Shastry 2005). 1.4.2 Kopplungsanalysen Genkopplung bedeutet, dass Gene, die auf einem Chromosom liegen, häufiger gemeinsam vererbt werden, als es zu erwarten wäre, wenn sie unabhängig voneinander vererbt werden würden (Buselmaier und Tariverdian 2007). Man beschreibt damit also eine Beziehung zwischen verschiedenen Loci (Strachan und Read 2005). Je weiter die Gene voneinander entfernt liegen, desto höher ist die Wahrscheinlichkeit, dass sie durch Crossing over in der Meiose getrennt werden. Als Maß für die Entfernung zweier Gene dient die Rekombinationshäufigkeit, die angibt, wie häufig zwei Gene gemeinsam 19 1 Einleitung vererbt werden. Die statistische Wahrscheinlichkeit für Genkopplung wird mit dem LOD-Score ausgedrückt (Lander und Kruglyak 1995). In Kopplungsanalysen werden genetische Marker bekannter genomischer Position bei betroffenen und nicht betroffenen Familienmitgliedern untersucht. Ist nun ein Marker mit dem krankheitsauslösenden Polymorphismus gekoppelt, kann dieser häufiger bei den erkrankten Familienmitgliedern nachgewiesen werden. Auf diese Weise kann die Lage eines krankheitsverursachenden Gens oder Polymorphismus bestimmt werden. Typischerweise werden dabei Regionen von 10-20 Mb identifiziert, in denen durchschnittlich 75 Gene lokalisiert sind (Berrettini 2000; Brzustowicz 2008; Tandon et al. 2008a). 1.4.3 Assoziationsstudien Assoziationsanalysen testen die Hypothese, dass eine bestimmte Sequenzvariante das Risiko erhöht, eine Krankheit zu entwickeln. Ist diese Hypothese wahr, sollte die Sequenzvariante in einer Gruppe nicht verwandter betroffener Erkrankter häufiger vorkommen als in einer gesunden Kontrollgruppe (Berrettini 2000; Brzustowicz 2008). Es handelt sich hierbei also nicht um ein spezifisches genetisches Phänomen, sondern um eine statistische Feststellung über die Häufigkeit des gemeinsamen Auftretens von Allel und Phänotyp. Ein wichtiger Vorteil dieser Methode ist die Möglichkeit, auch Gene mit kleinem Effekt zu entdecken (Craddock und Jones 1999; Pulver 2000). Neben dem direkten Zusammenhang kann eine Assoziation jedoch auch auf Populationsstratifizierung, Typ-I-Fehler oder Kopplungsungleichgewicht zurückgehen (Buselmaier und Tariverdian 2007). Assoziationsstudien basieren auf der Voraussetzung, dass die für die Anfälligkeit verantwortliche DNA-Veränderung erstmals vor langer Zeit in einem gemeinsamen Vorfahren aufgetreten ist und sich im Laufe der Zeit unter den Betroffenen ausbreiten konnte (Brzustowicz 2008). Voraussetzung für ein valides Ergebnis ist daher, dass Fälle und Kontrollen aus derselben Subpopulation stammen (Buselmaier und Tariverdian 2007). Das HapMap-Projekt hat eine neue Generation genetischer Assoziationsstudien möglich gemacht. In der Genomsequenz gibt es vermutlich ca. 10 Millionen single nucleotide polymorphisms, kurz SNPs. Dies sind Orte, an denen die interindividuelle 20 1 Einleitung Basensequenz differiert. Die meisten SNPs treten in biallelischer Form auf. Es gibt ca. einen SNP pro 100 – 300 Basen, die Frequenz für jedes Allel beträgt per Definition mindestens 1%. In den letzten Jahren wurde herausgefunden, dass nahe beieinander gelegene SNPs häufiger, als es der Zufall vorgeben würde, miteinander vererbt werden. Dies nennt man Kopplungsungleichgewicht. Eine Region stark miteinander assoziierter SNPs nennt man Haploblock (Manolio et al. 2008). Diese Blöcke variieren in ihrer Größe von wenigen bis zu mehr als hundert Kilobasen (Slatkin 2008). Zwischen den Haploblöcken liegen Rekombinationshotspots, an denen fast die gesamte meiotische Rekombination geschieht. Bei hohem Kopplungsungleichgewicht ist die Kenntnis einiger bestimmter SNP-Allele (tagSNPs) ausreichend, um auf die Varianten der restlichen SNPs eines Blocks schließen zu können (Manolio et al. 2008). Auf diese Weise können genomweite Assoziationsstudien, in denen 500.000 bis 1 Million SNPs erfasst werden, mit relativ geringem Aufwand durchgeführt werden (Slatkin 2008). 1.4.4 Vulnerabilitätsgene endogener Psychosen 1.4.4.1 Suszeptibilitätsregionen - Ergebnisse aus Kopplungsanalysen In diversen Kopplungsanalysen wurden verschiedene Suszeptibilitätsloci für endogene Psychosen identifiziert, wobei eine Replikation der Ergebnisse selten gelang (Berrettini 2001). In einer Metaanalyse genomweiter Kopplungsstudien wurden deutliche Hinweise auf die Regionen 13q und 22q als Suszeptibilitätsorte für Schizophrenie und bipolaraffektive Störung gefunden, zudem wurde für Schizophrenie 8p als Suszeptibilitätsregion identifiziert. Weniger starke Hinweise gab es für 1q, 2q, 6q und 15q als Suszeptibilitätsloci für Schizophrenie, eventuell hervorgerufen durch Ergebnisse einzelner Analysen (Badner und Gershon 2002). Zwei andere Metaanalysen genomweiter Kopplungsstudien konnten oben genannte Regionen nicht bestätigen. So zeigte sich in der Metaanalyse für die bipolar-affektive Störung für keine Region eine genomweite Signifikanz, die signifikantesten p-Werte lagen in den Regionen 9p, 10q und 14q und auf Chromosom 18 (Segurado et al. 2003). Die Metaanalyse für Schizophrenie ergab genomweite Signifikanz für Kopplung auf Chromosom 2q. Weitere Regionen konnten als Kandidatenregionen in Betracht gezogen werden, insbesondere 5q, 3p, 11q, 6p, 1q, 22q, 8p, 6p, 20q und 14p (Lewis et al. 2003). 21 1 Einleitung 1.4.4.2 Suszeptibilitätsgene - Schizophrenie In den letzten Jahren wurden durch Assoziationsstudien in chromosomalen Regionen, die in Kopplungsanalysen identifiziert wurden, die ersten Suszeptibilitätsgene für Schizophrenie entdeckt. Ein vielversprechendes Vulnerabilitätsgen ist Neuregulin 1 auf Chromosom 8p21-22, das wiederholt signifikante Assoziation mit Schizophrenie zeigte (Harrison und Owen 2003; Kennedy et al. 2003). Obwohl ein assoziierter Haplotyp, der sogenannte HapICE, in verschiedenen Populationen nachgewiesen werden konnte, ist bisher noch keine ursächliche funktionelle Variante identifiziert (Stefansson et al. 2002; Tosato et al. 2005). Neuregulin 1 fördert die Proliferation von Oligodendrozyten sowie neuronale Migration und Zelldifferenzierung und ist auf diese Weise an neuronaler Entwicklung beteiligt (Harrison und Owen 2003; Schosser und Aschauer 2004). Außerdem wird es im ZNS in Synapsen exprimiert und ist an Expression und Aktivierung von Neurotransmitterrezeptoren, z. B. dem NMDAR, beteiligt (Harrison und Owen 2003; Schosser und Aschauer 2004). Damit übereinstimmend exprimieren Mäuse, die eine Mutation im Neuregulingen haben, weniger funktionale NMDA-Rezeptoren (Schosser und Aschauer 2004; Stefansson et al. 2002). Aufgrund seiner Funktionen in Synaptogenese, Gliadifferenzierung, Myelinisierung und Neurotransmission ist es ein interessantes Kandidatengen (Craddock et al. 2005). Auf 6p22.3 wurde DTNBP1 (Dysbindin) als Suszeptibilitätsgen für Schizophrenie identifiziert. Dysbindin ist Bestandteil synaptischer Systeme und vor allem postsynaptisch im Zytoskelett vorhanden. Man nimmt an, dass es zu einer verminderten Glutamataktivität führt (Kennedy et al. 2003; Schosser und Aschauer 2004). Weitere Suszeptibilitätsgene sind G72, auch DAOA (D-Aminosäure-OxidaseAktivator) genannt, auf 13q34 und DAAO (D-Aminosäure-Oxidase) auf 12q24 (Schosser und Aschauer 2004), in denen jeweils SNPs bzw. Haplotypen mit Schizophrenie assoziiert sind (Craddock et al. 2005; Harrison und Owen 2003). Die Interaktion von G72 und DAAO führt zu einer Aktivierung von DAAO (Craddock et al. 2005), welches dann D-Serin, einen allosterischen Aktivator des NMDAR, oxidiert und konsekutiv zu herabgesetzter Aktivität des NMDAR führt. Auf diese Weise kann die 22 1 Einleitung NMDAR-vermittelte glutamaterge Wirkung reguliert werden (Schosser und Aschauer 2004). Auf 22q11 führt eine Deletion zum velokardiofazialen Syndrom, bei dem eine hohe Inzidenz an Psychosen besteht, die manchmal nicht von Schizophrenie unterschieden werden können (Kennedy et al. 2003). Hier liegt das Gen PRODH (Prolindehydrogenase), in dem mehrere SNPs mit Schizophrenie assoziiert sind. Das Enzym ist an der Bildung von Glutamat aus Prolin beteiligt (Craddock et al. 2005; Schosser und Aschauer 2004). Ein anderes Kandidatengen auf 22q11 ist COMT (Catechol-O-Methyltransferase), welches ebenfalls mit Schizophrenie assoziiert ist. COMT ist für den Abbau von Katecholaminen wie Dopamin verantwortlich und hat besonders im präfrontalen Kortex eine wichtige Funktion, da die Katecholaminwirkung an anderen Orten über Aufnahme aus dem synaptischen Spalt in Nervenendigungen beendet wird (Harrison und Owen 2003; Kennedy et al. 2003; Schosser und Aschauer 2004). Der Val158Met-SNP führt zu einer veränderten Aminosäuresequenz, was Auswirkungen auf die Enzymaktivität hat und Frontallappenfunktion sowie präsynaptische Dopaminaktivität beeinflusst (Craddock et al. 2005; Harrison und Owen 2003). Auch das RGS4-Gen auf 1q21-22 ist mit Schizophrenie assoziiert. Seine Funktion besteht darin, dass es als negativer Regulator die Effekte von Agonisten an G-Proteingekoppelten Rezeptoren abschwächt, indem es die GTPase-Aktivität und damit die Umwandlung in die inaktive GDP-bindende G-Proteinform beschleunigt (Craddock et al. 2005). Somit wird die G-Protein-gekoppelte synaptische Signaltransduktion verkürzt (Schosser und Aschauer 2004). Im Gehirn von Schizophrenen hat man post mortem eine verminderte Expression von RGS4 festgestellt (Craddock et al. 2005; Schosser und Aschauer 2004). In einer Familie mit häufigen Fällen von Schizophrenie und affektiven Störungen wurde eine balancierte Translokation auf Chromosom 1 entdeckt, durch die das Kandidatengen DISC1 (disrupted in schizophrenia) unterbrochen wird. Es gibt Hinweise, dass DISC1 an Neuritenarchitektur und neuronaler Migration beteiligt ist und die Anzahl an NMDAR an der postsynaptischen Membran beeinflusst. Zudem ist es an der Ausbildung des LTP an der glutamatergen Synapse beteiligt (Craddock et al. 2005; Stahl 2007). 23 1 Einleitung Die Gemeinsamkeit dieser Vulnerabilitätsgene ist ein Zusammenhang mit der Glutamathypothese, da sie in Verbindung zur glutamatergen Synapse stehen, an der sie Formation, Plastizität und Signalvermittlung beeinflussen (Harrison und Owen 2003; Schosser und Aschauer 2004). 1.4.4.3 Suszeptibilitätsgene - Bipolar-affektive Störung Für bipolar-affektive Störungen wurden bisher deutlich weniger Suszeptibilitätsgene identifiziert. Im Folgenden werden zwei Kandidatengene, für die replizierte Assoziation besteht, beschrieben. In mehreren Fall-Kontroll-Studien zeigten sich Assoziationen von SNPs und Haplotypen im DAOA-Gen auf 13q34, jedoch wurden nicht immer dieselben SNPs reproduziert. Eine pathologische Variante wurde dementsprechend ebenfalls noch nicht identifiziert (Craddock und Forty 2006; Kennedy et al. 2003). Ein weiteres Kandidatengen ist BDNF aus der Neurotrophinfamilie auf 11p13. Die Funktionen von BDNF wurden oben bereits beschrieben. Durch seine Beteiligung an neuronaler Plastizität ist es mit der Hypothese der synaptischen Plastizität affektiver Störungen vereinbar. In zwei Studien, die dieses Gen untersuchten, zeigte sich ein krankheitsassoziierter SNP, welcher mit einer veränderten Aminosäuresequenz einhergeht. Auch in einer kürzlich veröffentlichen Metaanalyse konnte eine Assoziation des Risikoallels mit der bipolaren Erkrankung, v. a. in asiatischen Populationen, belegt werden. Allerdings gibt es auch hier Studien, die diese Assoziation nicht bestätigten (Craddock et al. 2005; Craddock und Forty 2006; Kennedy et al. 2003). 1.4.4.4 Hinweise auf Überlappung der Ätiologie Die Frage, ob es eine Überlappung in der Ätiologie von Schizophrenie und bipolaraffektiver Erkrankung gibt, wird kontrovers diskutiert. Hinweise dafür liefern ähnliches Ersterkrankungsalter, Lebenszeitrisiko, Suizidrisiko, Geschlechterverhältnis und die geschätzte Heritabilität (Berrettini 2001). Klinisch werden Individuen, die Merkmale beider Krankheiten zeigen, häufig unter der Diagnose „schizoaffektive Störung“ klassifiziert. Auch Familienstudien geben Anhaltspunkte für die Theorie einer 24 1 Einleitung überlappenden Ätiologie (Craddock und Forty 2006). So haben Verwandte ersten Grades von Schizophrenen und bipolar-affektiv Erkrankten ein erhöhtes Risiko, an schizoaffektiven Störungen oder unipolar affektiven Störungen zu erkranken (Berrettini 2000; Berrettini 2003; Gershon 2000). Schizophrenie und bipolar-affektive Erkrankung treten wiederum gehäuft in Familien von Patienten mit schizoaffektiver Störung auf (Craddock und Forty 2006). Allerdings erhöht Schizophrenie nicht das Risiko für erstgradige Verwandte, an bipolar affektiver Störung zu erkranken und umgekehrt (Berrettini 2003). In Kopplungsanalysen wurden 13q, 22q, 8p, 18q, 10p sowie 6p als überlappende Suszeptibilitätsregionen beschrieben, wobei in diesen Regionen einige Suszeptibilitätsgene für Schizophrenie liegen (Berrettini 2003; Maier 2008). In einem Genomscan, in dem Familien von schizoaffektiv Erkrankten untersucht wurden, ergaben sich genomweite Signifikanz für 1q24 und weniger eindeutige Ergebnisse für 22q11 (Craddock und Forty 2006). Der beste Kandidat für ein gemeinsames Vulnerabilitätsgen ist G72 (DAOA), welches sowohl mit bipolar-affektiver Erkrankung als auch mit Schizophrenie assoziiert ist. Auch im Neuregulingen sind einige Marker mit bipolar-affektiver Störung assoziiert (Maier 2008) und es wurde Assoziation eines COMT-Allels mit dem Endophänotyp „rapid cycling“ festgestellt (Kato 2001). Ferner gibt es auch für DISC1 aus Kopplungsund Assoziationsanalysen Hinweise für eine Verbindung zu Schizophrenie, bipolaraffektiver und schizoaffektiver Störung (Craddock und Forty 2006). Diese Daten lassen eine gemeinsame genetische Grundlage möglich erscheinen. Auf der anderen Seite gibt es auch viele Allele mit krankheitsspezifischer Assoziation (Maier 2008). 1.5 Das Kandidatengen NOS1AP 1.5.1 Definition und Funktion NOS1AP (NOS1 adaptor protein), früher als CAPON (carboxy-terminal PDZ ligand of neuronal NOS) bezeichnet, ist auf Chromosom 1q22 lokalisiert (Eastwood 2005). Das Gen hat eine Länge von 30 Kilobasen, von denen nur 1,5 Kilobasen kodieren (Xu et al. 25 1 Einleitung 2005). Das Protein wurde erstmals im Gehirn von Ratten entdeckt, wo die höchste Expression im Kortex und in der Medulla oblongata vorlag und Kolokalisierungen mit der neuronalen NO-Synthase festgestellt wurden (Jaffrey et al. 1998). NOS1AP ist ein 55 kDa großes zytoplasmatisches Protein, dessen C-Terminus an die PDZ-Domäne der NOS-I binden kann, dabei konkurriert es mit PSD95 und PSD93 um diese Bindestelle. Außerdem besitzt es eine N-terminale Phosphotyrosin-bindene Region (Fang et al. 2000; Jaffrey et al. 2002). Es gibt beim Menschen mindestens zwei Isoformen von NOS1AP. Die lange Isoform ist ein Protein aus 501 Aminosäuren, welches aus allen zehn Exons gebildet wird. Das kürzere Protein besteht nur aus 211 Aminosäuren und entsteht aus den letzten zwei Exons (Xu et al. 2005). Es beinhaltet nicht den N-Terminus, sondern nur die PDZbindende Domäne (Brzustowicz 2008). Abb. 1.2: Genomische Konfiguration der langen (oben) bzw. kurzen Isoform (unten) von NOS1AP. Darstellung der Exons durch nummerierte Balken (nach Xu et al. 2005) NOS1AP bindet die NOS-I hochspezifisch, nicht hingegen die anderen Isoformen der NO-Synthase NOS-II und NOS-III (Jaffrey et al. 1998). Die NOS-I ist normalerweise über PDZ-PDZ-Interaktionen mit PSD95 an den NMDA-Rezeptor der postsynaptischen Membran gebunden und somit unmittelbar dem Calciumeinstrom in die Zelle durch den NMDAR-assoziierten Kationenkanal ausgesetzt (Jaffrey et al. 1998). Dieser Calciumeinstrom aktiviert die NOS-I, welche dann Stickoxid (NO) bildet. Durch Bindung von NOS1AP an die NOS-I wird diese ins Zytosol transloziert, sodass es weniger NMDAR/PSD95/NOS-I-Komplexe gibt und die Verfügbarkeit von Calcium für die NOS-I reduziert ist. Folglich ist ein größerer Anteil des Enzyms katalytisch inaktiv und es entsteht weniger Stickoxid. Somit ist NOS1AP ein negativer Regulator der NO-Produktion (Boehning und Snyder 2003; Jaffrey et al. 1998; Nedvetsky et al. 2002). 26 1 Einleitung Die letzten 125 Aminosäuren von NOS1AP binden neben der PDZ-Domäne von NOS-I auch direkt eine PDZ-Domäne von PSD95 und blockieren so die Bindestelle für NOS-I (Brzustowicz 2008). Neben der NOS-I bindet NOS1AP auch weitere Proteine, dabei fungiert es als Adaptorprotein für NOS-I und bindet diese an spezifische physiologische Zielproteine (Jaffrey et al. 2002). Da diese Bindung durch den N-Terminus, welcher der kurzen Isoform fehlt, vermittelt wird, kann die kurze Isoform von NOS1AP nicht als Adaptorprotein fungieren (Brzustowicz 2008). Bindungspartner von NOS1AP sind die Synapsine I, II und III. NOS1AP, NOS-I und Synapsin I bilden einen ternären Komplex, bei dem die C-terminale PDZ-bindende Domäne von NOS1AP an NOS-I und die N-terminale PTB-Domäne an Synapsin I bindet (Jaffrey et al. 2002). Da Synapsine nur präsynaptisch vorkommen, ist diese Interaktion eventuell ein entscheidender Faktor beim präsynaptischen Targeting der NOS-I (Jaffrey et al. 2002). Abb. 1.3: Interaktion der NOS-I mit NOS1AP und weiteren Adaptorproteinen (nach Nedvetsky et al. 2002) Ein anderer Bindungspartner ist das Protein Dexras1, ein Mitglied der Ras-Familie kleiner monomerer G-Proteine. Es interagiert ebenfalls mit der N-terminalen PTBDomäne von NOS1AP und bildet zusammen mit NOS-I einen ternären Komplex. Auf diese Weise kann Dexras1 durch Stickoxid S-nitrosyliert und aktiviert werden. 27 1 Einleitung Wahrscheinlich moduliert Dexras1 in der aktivierten Form Signaltransduktionen (Fang et al. 2000). NOS1AP wird auch im Herzen exprimiert und interagiert dort ebenfalls mit NOS-I, eine physiologische Funktion ist allerdings nicht bekannt. In einer großen Assoziationsstudie in einem deutschen Sample wurde entdeckt, dass Polymorphismen im NOS1AP-Gen signifikant mit Variationen des QT-Intervalls im EKG assoziiert sind. Dieses Ergebnis wurde in verschiedenen Populationen repliziert (Aarnoudse et al. 2007; Arking et al. 2006; Lehtinen et al. 2008; Post et al. 2007). Der Mechanismus, durch den NOS1AP die Länge des QT-Intervalls beeinflusst, ist noch nicht zweifelsfrei geklärt, man geht allerdings von einer Veränderung des basalen Expressionslevels aus, die zu einer beschleunigten kardialen Repolarisation durch Inhibition eines L-Typ Calciumkanals führt (Chang et al. 2008). In einer weiteren Assoziationsstudie stellte sich zudem heraus, dass ein SNP die Wirksamkeit von Sulfonylharnstoffen beeinflusst und zu veränderten Mortalitätsraten bei damit behandelten Patienten führt (Becker et al. 2008). 1.5.2 Neuronale NO-Synthase als Zielstruktur von NOS1AP Es gibt drei verschiedene NO-Synthase-Isoformen, die endotheliale NOS (NOS-III), die induzierbare oder mitochondriale NOS (NOS-II) und die neuronale NOS, auch NOS-I genannt. Die Isoformen sind auf drei verschiedenen Genen kodiert (Bernstein et al. 2005), die neuronale und endotheliale NOS werden konstitutiv exprimiert (Calabrese et al. 2007). Die NOS-I ist mit 160 kDa größer als die anderen Isoformen, da sie eine N-terminale PDZ-Domäne besitzt, mit der sie Proteininteraktionen eingeht (Bernstein et al. 2005; Fang et al. 2000). Im ZNS ist das Enzym für über 90% der NO-Synthese verantwortlich. Die NOS-I kommt hauptsächlich in Kleinhirnrinde, Nucleus accumbens, Hippocampus, Präfrontalcortex, Hypothalamus und Subthalamus vor, dabei ist die Dichte von NOS-Iexprimierenden Neuronen von der Region abhängig (Bernstein et al. 2005; Blum-Degen et al. 1999). Peripher wird sie in nichtadrenergen und nichtcholinergen (NANC) Neuronen, welche glatte Muskelzellen in Gastrointestinaltrakt, Corpora cavernosa, Uterus und Prostata innervieren, exprimiert (Calabrese et al. 2007). 28 1 Einleitung Die NOS-I wird durch Erhöhung des Calciumspiegels und anschließende Komplexierung mit Calmodulin aktiviert (Calabrese et al. 2007; Jaffrey et al. 1998). Die Regulation erfolgt außerdem über Phosphorylierung und Veränderung der Expression, auf subzellulärer Ebene ist die NO-Produktion auch von Bindung an Proteine abhängig (Nedvetsky et al. 2002). So können Proteininteraktionen zu einer Destabilisierung der Homodimere der NOS-I führen und sie so inhibieren (Nedvetsky et al. 2002). Die Hauptfunktion von NOS-I ist die Bildung von NO durch die Konversion von LArginin zu L-Citrullin in Anwesenheit von Sauerstoff (Bernstein et al. 2005; Calabrese et al. 2007). Wie oben beschrieben ist die NOS-I über PSD-Proteine an den NMDAR gekoppelt und beeinflusst über Proteininteraktionen zytosolische Signaltransduktionswege (Bernstein et al. 2005). Derartige Proteininteraktionen führen auch zu einer Kopplung der Stickoxidsynthese durch NOS-I an die Zielstruktur lösliche Guanylatcyclase (sGC) (Nedvetsky et al. 2002). Des Weiteren kann die NOS-I durch SNitrosylierung den NMDA-Rezeptor direkt negativ regulieren (Bernstein et al. 2005; Hata und Takai 1999). Stickoxid (NO), das Produkt der NOS-I, ist ein wichtiger autokriner und parakriner Botenstoff mit diversen Funktionen in Immunsystem, kardiovaskulärem und neuronalem System (Calabrese et al. 2007; Jaffrey et al. 2002). NO ist ein flüchtiger, hochreaktiver Stoff, der Membranen passieren und intrazellulär von Metallionen, Glutathion und Superoxid schnell abgebaut werden kann (Bernstein et al. 2005; Jaffrey et al. 2002). Daher ist eine durch Proteininteraktionen vermittelte räumliche Nähe der NOS-I zu den Zielstrukturen von NO wichtig (Jaffrey et al. 2002). Die wichtigste Zielstruktur von NO ist die sGC, die bei Aktivierung den second messenger cGMP bildet, welcher wiederum Kinasen aktiviert (Bernstein et al. 2005; Boehning und Snyder 2003; Calabrese et al. 2007). Ein weiterer Wirkmechanismus ist die S-Nitrosylierung durch kovalente Bindung an Thiolgruppen (Boehning und Snyder 2003; Calabrese et al. 2007). Mittels dieser nachgeschalteten Reaktionen beeinflusst NO synaptische Plastizität, Relaxation des glatten Muskels, Zellsignalisierung sowie Kontrolle von Schlaf, Appetit und Körpertemperatur (Calabrese et al. 2007). Des Weiteren führt NO zu Gefäßdilatation und Zerstörung von Tumorzellen und Bakterien durch Makrophagenaktivierung (Boehning und Snyder 2003; Fang et al. 2000). NO 29 1 Einleitung beeinflusst außerdem die Ausschüttung der „klassischen“ Transmitter wie GABA, Dopamin, Serotonin, Glutamat und Noradrenalin in vielen Gebieten des Gehirns und ist an der Regulation der neurosekretorischen Aktivität des Hypothalamus beteiligt (Calabrese et al. 2007). Ein neuroprotektiver Effekt besteht in der Beendigung einer verlängerten Stimulation des NMDA-Rezeptors, die zu exzitotoxischem Zelltod führen kann, durch Nitrosylierung der NR1- und NR2-Untereinheit. Auf diese Weise wird der Calciumeinstrom, welcher den Zelltod verursacht, vermindert. Auch eine verlängerte NOS-Aktivierung, die zur Produktion von toxischen Superoxidradikalen führt, wird durch NO-vermittelte Nitrosylierung beendet. Darüber hinaus reguliert NO einige Faktoren, die neuroprotektive Effekte haben, darunter CREB (Calabrese et al. 2007). Allerdings kann NO als freies Radikal in höheren Konzentrationen auch zytotoxische Effekte haben, vor allem durch das aus der Konjugation mit Superoxid entstehende Peroxynitrit (ONNO¯). Dieses wirkt oxidierend und nitrierend und kann zu Lipidperoxidation und Zerstörung von Proteinen, Aminosäuren und Nukleinsäuren führen (Bernstein et al. 2005). Eine überschießende NO-Bildung ist möglicherweise auch an neurodegenerativen Krankheiten wie Alzheimer und Parkinson beteiligt (Calabrese et al. 2007; Guix et al. 2005). 1.5.3 NOS1AP als Kandidatengen: bisherige Studienergebnisse Es gibt sowohl aus Kopplungs- und Assoziationsanalysen als auch aus funktionellen Studien diverse Anhaltspunkte, die NOS1AP zu einem interessanten Kandidatengen für endogene Psychosen machen. Dabei haben sich die Studien bisher auf Schizophrenie konzentriert. Der stärkste Hinweis auf Kopplung im Bereich 1q21-22 stammt von Brzustowicz et al. aus dem Jahr 2000. Diese parametrische Kopplungsstudie in einem kanadischen Sample von Familien, in denen gehäuft Schizophrenie und schizoaffektive Störung vorkommen, zeigte eine signifikante Kopplung mit einem maximalen LOD-Score von 6,5 (Brzustowicz et al. 2000). In einer nachfolgenden fine-mapping Analyse wurde die Region auf eine Länge von ca. 1 Mb eingegrenzt (Brzustowicz et al. 2002). Unterstützt wurde dieses Ergebnis durch weitere Studien. So wurden in einer genomweiten Kopplungsanalyse schwach positive LOD-Scores für verschiedene Regionen, darunter 30 1 Einleitung 1q22-23 gefunden (Shaw et al. 1998). In einer anderen genomweiten Kopplungsstudie fand sich ein signifikanter Wert für den Marker D1S196 im Bereich 1q22.1-23 (Gurling et al. 2001). Aus einer Kopplungsstudie in einem taiwanesischen Sample gibt es ebenfalls Hinweise auf eine Kopplung für Marker D1S1679 im Bereich 1q21-31 (Hwu et al. 2003). Auch eine Metaanalyse weist auf einen Suszeptibilitätsort auf 1q hin (Lewis et al. 2003). Allerdings gibt es auch diverse Studien, die diese Ergebnisse nicht bestätigen (Faraone et al. 2006; Suarez et al. 2006). Dies ist nicht verwunderlich, da die Replikation bei einer genetisch komplexen Krankheit sehr schwierig ist (Brzustowicz 2008). Auch Assoziationsstudien liefern Hinweise auf ein Suszeptibilitätsgen für Schizophrenie im Bereich von NOS1AP. In der ersten Assoziationsstudie in dieser genomischen Region wurden sechs Mikrosatellitenmarker in einem Sample spanischer Kernfamilien untersucht. Dort zeigte sich signifikante allelische Assoziation zu dem Marker D1S1679, welcher 25 kb von NOS1AP entfernt liegt. Um klinische, pathophysiologische und ätiologische Heterogenität zu vermindern, wurde die gleiche Analyse mit dem Endophänotyp „Realitätsdistortionssyndrom“ durchgeführt, dabei zeigte sich wiederum eine bevorzugte Transmission einiger Allele des D1S1679Polymorphismus. Hieraus lässt sich schließen, dass ein Affinitätsgen an dieser Stelle an der Pathophysiologie dieses Syndroms beteiligt sein könnte (Rosa et al. 2002). Auch in einem Sample kolumbianischer Trios wurde signifikante Assoziation zu dem Marker D1S1679 gefunden (Miranda et al. 2006). In einer Studie des oben genannten kanadischen Samples waren sechs SNPs und zwei Mikrosatellitenmarker signifikant mit Schizophrenie assoziiert, diese Marker lagen alle im genomischen Bereich von NOS1AP (Brzustowicz et al. 2004). Eine Studie in der chinesischen Han-Bevölkerung fand signifikante Assoziation zu einem SNP und daraus hervorgehenden Haplotypen, wobei dieser SNP in der kanadischen Studie unauffällig gewesen war. Die Ergebnisse der kanadischen Studie konnten hingegen nicht reproduziert werden (Zheng et al. 2005). Auch in einem südamerikanischen Patientenkollektiv konnte eine Assoziation von acht SNPs innerhalb des NOS1AP-Gens mit Schizophrenie festgestellt werden (Kremeyer et al. 2009). Kürzlich wurde außerdem herausgefunden, dass eine Deletion von 1,38 Mb im Bereich 1q21.1 signifikant mit Schizophrenie assoziiert ist, allerdings ist diese sehr 31 1 Einleitung selten und wahrscheinlich nicht für die positiven Kopplungsbefunde verantwortlich. Es waren keine SNPs in diesem Bereich signifikant mit Schizophrenie assoziiert (Stefansson et al. 2008). Der Versuch einer Reproduktion der Ergebnisse von Brzustowicz et al. in einem britischen Sample gelang nicht. Keiner der im kanadischen Sample assoziierten Marker zeigte hier Assoziation mit Schizophrenie (Puri et al. 2006). Auch eine familienbasierte Assoziationsstudie in der chinesischen Hanbevölkerung fand keine Assoziation von Einzelmarkern oder Haplotypen mit Schizophrenie (Fang et al. 2008). Die Rolle von NOS1AP als Kandidatengen wird auch von funktionellen Studien unterstützt. Eine Sequenzierung der kodierenden Regionen von NOS1AP deckte keine Mutationen auf, die mit der Krankheit assoziiert sind, jedoch gibt es Hinweise auf veränderte Expression bei Schizophrenie und bipolar-affektiver Erkrankung (Xu et al. 2005). 32 2 Fragestellung 2 Fragestellung NOS1AP ist ein interessantes Kandidatengen für endogene Psychosen. Diverse Kopplungs- und Assoziationsstudien unterstreichen vor allem seine Bedeutung bei Schizophrenie. Insbesondere bei Betrachtung seiner physiologischen Funktion zeigt sich eine mögliche Beteiligung an der Krankeitsentstehung. NOS1AP steht im Zusammenhang mit dem NMDAR und spielt durch seine Verbindung zur NOS-I eine wichtige Rolle bei der NO-Synthese. Somit ist es im weiteren Sinne mit neuronaler Signaltransduktion und synaptischer Plastizität verbunden (Brzustowicz et al. 2004; Hata und Takai 1999). Wie oben beschrieben, gibt es auch diverse Hinweise auf eine Beteiligung des Glutamatsystems und des NMDAR-Komplexes an der Ätiologie der bipolar-affektiven Störung. Daher ist es möglich, dass NOS1AP auch bei dieser Erkrankung eine Rolle spielt. Für komplexe genetische Krankheiten wie Schizophrenie und die bipolar-affektive Erkrankung sind Assoziationsstudien mit Fall-Kontroll-Samples besonders geeignet, um Suszeptibilitätsgene mit geringem Effekt zu identifizieren (Brzustowicz 2008). Ziel dieser Arbeit war es, zu untersuchen, ob eine Assoziation von 14 SNPs und den daraus entstehenden Haplotypen im NOS1AP-Gen mit Schizophrenie und der bipolaraffektiven Störung besteht. Dazu wurde eine Fall-Kontroll-Studie in einem unterfränkischen Kollektiv durchgeführt. 33 3 Material und Methoden 3 Material und Methoden 3.1 Probanden Für die Studie wurden 335 nicht miteinander verwandte Patienten aus Unterfranken rekrutiert, darunter 174 Frauen und 161 Männer. Das mittlere Alter betrug 42,6 ± 13,9 Jahre. Im Verlauf ihrer Erkrankung waren alle Patienten mindestens einmal in der Klinik und Poliklinik für Psychiatrie, Psychosomatik und Psychotherapie des Universitätsklinikums Würzburg hospitalisiert. Zur Diagnosestellung wurde ein ausführliches, semi-strukturiertes klinisches Interview analog dem AMDP-Interview (Arbeitsgemeinschaft für Methodik und Dokumentation in der Psychiatrie 2000) durch einen erfahrenen Psychiater vorgenommen, außerdem erfolgte eine Durchsicht der Krankenakte und auswärtiger Arztbriefe und es wurden Angehörigeninterviews durchgeführt. Erfasst wurden zudem Familienanamnese, Suchtmittelabusus und Ersterkrankungsalter. Die Diagnosestellung erfolgte nach den ICD-10-Kriterien. Dementsprechend litten 245 Patienten an einer schizophrenen Störung, darunter 135 Männer und 110 Frauen. Alle Patienten wiesen einen chronischen Verlauf der Erkrankung ohne dauerhafte Remissionen auf. Außerdem nahmen 90 Patienten mit einer bipolar-affektiven Störung an der Studie teil, darunter waren 64 Frauen und 26 Männer. Es wurden nur Patienten mit mindestens einer klinisch manifesten manischen und einer depressiven Episode, die zu einer Behandlung führte, eingeschlossen, das heißt es wurden strenge Bipolar-I-Kriterien angewandt. Keiner der Patienten hatte signifikante neurologische Komorbiditäten wie beispielweise Epilepsie, Retardierung oder somatische Störungen, die zu einer organischen Psychose führen könnten. Patienten mit substanzinduzierter Psychose wurden nicht in die Studie aufgenommen. Die Kontrollproben wurden von 360 gesunden Blutspendern (188 Männer, 172 Frauen) gewonnen, die aus derselben Gegend stammten wie die Patienten und den gleichen genetischen Hintergrund wie die Patienten aufwiesen. Die Kontrollpersonen wurden nicht hinsichtlich psychiatrischer Störungen überprüft, jedoch nahm keine der Kontrollpersonen Medikamente ein und die Studie wurde ausführlich erklärt, so dass 34 3 Material und Methoden die Wahrscheinlichkeit einer schweren psychiatrischen Krankheit unwahrscheinlich ist (Reif et al. 2006). Das mittlere Alter der Kontrollgruppe lag bei 33,0 ± 10,5 Jahren. Patienten und Kontrollen waren kaukasischer Abstammung. Entsprechend den Regeln der Deklaration von Helsinki wurden nur Patienten und Freiwillige, die nach ausführlicher Information schriftlich ihre Zustimmung gaben, in die Studie aufgenommen. Vor Beginn der Studie wurde die Untersuchung durch die Ethik-Kommission der Universität Würzburg genehmigt. 3.2 Material 3.2.1 Reagenzien Primer: SNP 4 (rs1415263) Primer-forward 5’-TGT CAA CTG CTT CTG GGT TG- 3’ Primer-reverse 5’-GTT TGG TTG ATA CCA CGC TC- 3’ Produktgröße: 150 bp SNP 7 (rs4145621) Primer-forward 5’- AGT CTT CGG GGA CCC ATC- 3’ Primer-reverse 5’- CAA GGG GTG ATG GAC AAT AGA- 3’ Produktgröße: 126 bp SNP 13 (rs348624) Primer-forward 5’- ACC AGT TGG CTG CTG AGG- 3’ Primer-reverse 5’- GCA AGA GCT GGA ACT GAA GC- 3’ Produktgröße: 132 bp Hersteller sämtlicher Primer: MWG-Biotech AG, Ebersberg 35 3 Material und Methoden Enzyme und Oligonukleotide: dNTP (100 mM dGTP, dATP, dCTP, dTTP) Promega GmbH, Mannheim Hotstar-Taq-Polymerase Qiagen, Hilden iPlex® Enzym Sequenom, San Diego, USA Pronase E AppliChem, Darmstadt Shrimp Alkalische Phosphatase (1U/l) Sequenom, San Diego, USA Taq-DNA-Polymerase Biorad, München Chemikalien: Agarose Biozym, Rockland, USA Bromphenolblau Merck, Darmstadt 100 bp DNA-Leiter Peqlab Biotechnologie GmbH, Erlangen Ethidiumbromid 10 mg/ml Sigma-Aldrich Chemie GmbH, Steinheim Glycerol Sigma-Aldrich Chemie GmbH, Steinheim H2O Nanopure, Klagenfurt Isopropylalkohol AppliChem, Darmstadt iPlex®-Puffer 10x Sequenom, San Diego, USA iPlex®-Extension Primer Mix Sequenom, San Diego, USA iPlex®-Termination Mix Sequenom, San Diego, USA ® iPlex -Ionenaustauschharz Sequenom, San Diego, USA KCl AppliChem, Darmstadt MgCl2 AppliChem, Darmstadt MgCl2, 25mM Qiagen, Hilden NaCl 6M AppliChem, Darmstadt Na2EDTA AppliChem, Darmstadt SAP-Reaktionspuffer Sequenom, San Diego, USA SDS 10% Sigma, Steinheim 36 3 Material und Methoden Tris-Acetat AppliChem, Darmstadt Tris-HCl AppliChem, Darmstadt Tween-20 AppliChem, Darmstadt Lichrosolv Merck, Darmstadt Xylen Cyanol FF Sigma-Aldrich Chemie GmbH, Steinheim Puffer aus eigener Herstellung: 1x TAE-Puffer 40 mM Tris-Acetat 1 mM EDTA (pH = 8,0) Blaupuffer 0,25% Bromphenolblau 0,25% Xylen Cyanol FF 30% Glycerol in Wasser TE-Puffer (pH = 8,0) Tris-EDTA in wässriger Lösung SE-Puffer (pH = 8,0) 75 mM NaCl 0,5 M EDTA 155 mM NH4Cl Lysispuffer (pH = 7,4) 10 mM KHCO3 0,1 mM EDTA PCR-Puffer mit MgCl2 500 mM KCl 100 mM Tris-HCl (pH = 8,3) 0,25% Tween-20 0,25 mg/ml BSA 15 mM MgCl2 3.2.2 Geräte und Verbrauchsmaterialien Biometra T-Gradient Thermocycler Biometra, Göttingen Biometra UNOII Thermoblock Biometra, Göttingen Chemie Doc Biorad, München Chip Sequenom, San Diego, USA 37 3 Material und Methoden Filterspitzen Sarstedt, Nümbrecht Gelkammern, Kämme, Spacer, Spatel Peqlab Biotechnologie GmbH, Erlangen Glaskolben Schott-AG, Mainz MALDI-ToF Autoflex 1-Massenspektrometer Bruker Daltonik, Bremen Nano Dispenser Sequenom, San Diego, USA Personal Spin-Vortex, Microspin FV-2400 lab-4-you GmbH, Berlin Pipetten Eppendorf AG, Hamburg Reaktionsgefäße: 8er Ketten 0,2 ml PCR-Gefäße Brand GmbH & Co. KG, Wertheim Safe Lock 1,5 ml Eppendorf AG, Hamburg Software: TYPER 3,4 Sequenom, San Diego, USA Spannungsgerät: Power Supply LKB, Bromma, Schweden Thermo fast 384er-Platten ABgene, Hamburg Tischzentrifuge Mikroliter Hettich GmbH& Co. KG, Tuttlingen Ultraviolet Transilluminator UVP, Upland CA, USA Waage Toledo Mettler, Gießen 3.3 Methoden 3.3.1 Methodischer Überblick In dieser Arbeit wurde die Genotypisierung der SNPs mittels einer Primerextension und anschließender Analyse in der MALDI-ToF (matrix-assisted laser desorption ionization-time of flight) Massenspektrometrie durchgeführt. Bei SNP 4 (rs1415263), 7 (rs4145621) und 13 (rs348624) wurde zunächst eine PCR und anschließende Gelelektrophorese durchgeführt, während SNP 1 (rs1572495), 2 (rs1538018), 3 (rs945713), 5 (rs4306106), 6 (rs3924139), 8 (rs2661818), 9 (rs1508263), 10 (rs3751284), 11 (rs7521206), 12 (rs905721) und 14 (rs1964052) mittels des iPlex®Protokolls amplifiziert wurden. Danach folgten jeweils die Primerextension und die Analyse mit MALDI-ToF. Die Primerextension ist eine häufig angewandte Methode um die Allele von SNPs zu 38 3 Material und Methoden genotypisieren. Nach Amplifikation der DNA-Zielfrequenz mittels PCR dient das PCRProdukt als Vorlage für die Primerverlängerung. Übrige Nukleotide und Primerrückstände werden entfernt. Der Primer für die Primerextension bindet mit seiner 3’-terminalen Base direkt oberhalb des SNPs an die Zielsequenz. Die Primerverlängerung mit einem Nukleotid (ddNTP) führt nun zu Produkten unterschiedlicher Masse, die sich in der Spektrometrie unterscheiden lassen (Tost und Gut 2002; Tost und Gut 2005). 3.3.2 DNA-Extraktion Es wurde eine DNA-Extraktion aus dem Vollblut der Probanden entsprechend der Methode nach Miller durchgeführt (Miller et al. 1988). Hierfür wurden die Leukozyten durch Zentrifugieren abgetrennt und anschließend durch hypotone Lösung, Pronase E und SDS-Detergenz lysiert. Peptide und andere Zellbestandteile wurden durch Erhöhung der Salzkonzentration ausgefällt und abzentrifugiert. Die DNA konnte dann mit Isopropanol aus dem wässrigen Überstand präzipitiert werden. Danach erfolgte eine photometrische Konzentrationsbestimmung bei einer Wellenlänge von 260 nm. Hierbei entsprach eine Konzentration von 50 µg/ml doppelsträngiger DNA einer optischen Dichte von 1. 3.3.3 Polymerase-Kettenreaktion Die Polymerase-Kettenreaktion, kurz PCR, wurde 1985 von Kary B. Mullis erfunden (Garcia und Ma 2005). Sie ist eine in vitro Methode, die die Produktion großer Mengen spezifischer DNA-Fragmente einer definierten Länge und Frequenz aus einer kleinen Menge Vorlage (Template) erlaubt (White et al. 1989). Die PCR beruht auf enzymatischer Amplifikation eines DNA-Fragments, welches von zwei Oligonukleotidprimern flankiert wird. Zunächst erfolgt die Denaturierung, das heißt die DNA-Vorlage wird auf 90-95°C erhitzt, damit sich die Einzelstränge voneinander trennen. Im zweiten Schritt, dem Annealing, wird das Gemisch auf eine primerabhängige Temperatur von 50-60°C 39 heruntergekühlt, sodass sich die 3 Material und Methoden Oligonukleotidprimer an die Einzelhelices binden können. Im dritten Schritt, der Extension, wird die Temperatur auf ca. 73°C erhöht, sodass die DNA-Polymerase aus den Desoxynukleotiden einen komplementären DNA-Strang aufbauen kann. Das Extensionsprodukt jedes Primers fungiert dann wiederum als Vorlage für eine weitere Amplifikation. Die drei Schritte - Denaturierung, Annealing und Extension - werden ca. 30-40-mal wiederholt. Dabei entsteht eine exponentielle Menge des DNA-Segments, ungefähr 2n, wobei n für die Anzahl der Zyklen steht. Als DNA-Polymerase wird die thermostabile Taq-Polymerase des Thermus aquaticus benutzt. Zur Amplifizierung der SNPs 4, 7 und 13 wurden folgende PCR-Ansätze angewendet: SNP 4 (rs1415263): DNA-Template 1,5 µl Nukleotide 2,5 mM 1,5 µl Primer forward (10 pmol/µl) 1,5 µl Primer reverse (10 pmol/µl) 1,5 µl Taq-Polymerase 0,15 µl PCR-Puffer mit 15 mM MgCl2 3,75 µl Destilliertes Wasser 27,6 µl Es wurden 45 Zyklen mit einer Annealing-Temperatur von 56,4°C durchgeführt. SNP 7 (rs4145621): DNA-Template 1,5 µl Nukleotide 2,5 mM 1,5 µl Primer forward (10 pmol/µl) 1,5 µl Primer reverse (10 pmol/µl) 1,5 µl Taq-Polymerase 0,15 µl PCR-Puffer mit 15 mM MgCl2 3,75 µl Destilliertes Wasser 27,6 µl Es wurden 45 Zyklen bei einer Annealing-Temperatur von 62,2°C durchgeführt. 40 3 Material und Methoden SNP 13 (rs348624): DNA-Template 1,5 µl Nukleotide 2,5 mM 1,5 µl Primer forward (10 pmol/µl) 1,5 µl Primer reverse (10 pmol/µl) 1,5 µl Taq-Polymerase 0,1 µl PCR-Puffer mit 15 mM MgCl2 3,75 µl Destilliertes Wasser 27,65 µl Es wurden 45 Zyklen bei einer Annealing-Temperatur von 59,8°C durchgeführt. Um eine Verunreinigung ausschließen zu können, wurde bei jeder PCR eine Negativkontrolle, in der alle Reagenzien außer der DNA enthalten waren, mitgeführt. 3.3.4 Agarose-Gelelektrophorese Die Agarose-Gelelektrophorese ist der gängigste Weg, um das PCR-Produkt zu analysieren. Durch Variation des Agaroseanteils kann der Trennbereich der Matrices verändert werden. Je höher das Agarosegel konzentriert ist, desto weiter wird der optimale Trennbereich in Richtung kleinerer DNA-Fragmente verschoben. Mithilfe von Ethidiumbromid, einem Stoff, der fest an die DNA bindet, indem er zwischen den Basen interkaliert, kann das amplifizierte Produkt durch Fluoreszenz sichtbar gemacht werden, da Ethidiumbromid unsichtbares ultraviolettes Licht in sichtbares Licht konvertieren kann. Auf diese Weise lässt sich sofort feststellen, ob das PCR-Produkt die erwartete Größe hat und ob ebenfalls amplifizierte Fremd-DNA enthalten ist. Bei der Herstellung der Gele und der Gelelektrophorese wurde folgendermaßen vorgegangen: Die Gele wurden aus 100 ml TAE-Puffer und 2 g Agarose hergestellt. Das Gemisch wurde in der Mikrowelle ca. drei Minuten erwärmt. Nach Abkühlen auf 50-60°C wurden 3 µl Ethidiumbromid hinzugefügt und das Gel wurde in eine mit Kämmen versehene Gelkammer gegossen. Nach Erkalten wurden jeweils 10 µl PCRProdukt, versetzt mit 5 µl Blaupuffer, in die Kammern pipettiert und eine Spannung von 41 3 Material und Methoden 110 Volt für 30 Minuten angelegt. Dadurch wanderten die negativ geladenen DNAFragmente zur Anode, je nach Länge unterschiedlich schnell. Eine 100 bp Leiter wurde mitgeführt, um später die Größe der PCR-Produkte bewerten zu können. Die Negativkontrolle der PCR wurde ebenfalls untersucht. Unter UV-Licht wurden die Größe des PCR-Produkts und die Negativkontrolle begutachtet. 3.3.5 Amplifizierung nach dem iPlex®-Protokoll Die Amplifikationen von SNP 1, 2, 3, 5, 6, 8, 9, 10, 11, 12 und 14 wurden nach dem iPlex®-Protokoll angefertigt. Es erfolgte eine initiale Denaturierung bei 94°C für 15 min. Danach schlossen sich 44 Zyklen mit einer Denaturierung bei 95°C für 20 sec, einem Annealing bei 56°C für 30 sec und einer Extension bei 72°C für 1 min an. Nach dem letzten Zyklus erfolgte eine finale Elongation bei 72°C für 3 min. 3.3.6 Verdau mit Alkalischer Phosphatase Nach der PCR erfolgte ein enzymatischer Verdau mit Shrimp Alkalischer Phosphatase (SAP), um Reste von Desoxynukleotidtriphosphaten durch Dephosphorylierung zu entfernen. Je Probe wurden 10x SAP-Puffer (0,17 µl), 1,53 µl H2O und 0,30 µl SAP (1U/µl) verwendet. Die Reaktion wurde im Thermocycler auf 384 well Platten für 40 min bei 37°C durchgeführt. Um die SAP zu inaktivieren folgten 5 min bei 85°C. 3.3.7 iPlex® Reaktion Für die auf den SAP-Verdau folgende Einzelbasenextension wurde folgender iPlex® Reaktionsmix erstellt: 412 µl Nanopure H2O wurden mit jeweils 109 µl iPlex®-Puffer Plus (10x) und iPlex®-Termination Mix, außerdem 438 µl Extension Primer Mix (mit steigender Konzentration und Masse) und 21,8 µl iPlex®-Enzym gemischt. Zu den gereinigten PCR-Produkten wurden jeweils 2 µl Mix pipettiert. Daraufhin erfolgten nachstehende Schritte der Primer-Extensionsreaktion im Thermocycler: Nach einer initialen Denaturierung für 30 sec bei 94°C erfolgten 40 Zyklen mit jeweils 5 sec bei 42 3 Material und Methoden 94°C, dann ein Annealing bei 52°C für 5 sec. Die Elongation, d. h. die Verlängerung des Primers um eine Base, erfolgte bei 80°C. In jedem der 40 Zyklen wurden letztere zwei Schritte fünfmal wiederholt. Abschließend folgten 3 min bei 72°C. 3.3.8 Reinigung durch Ionenaustauschharz Um die MALDI-Analyse nicht zu beeinflussen, wurden Salze aus dem iPlex® Reaktionsprodukt folgendermaßen entfernt: In jeden Ansatz wurden 16 µl H2O pipettiert. Daraufhin wurden 6 mg Ionenaustauschharz hinzugefügt, die Platten wurden verschlossen und 20 min gewendet. Als nächster Schritt folgte eine Zentrifugation (3000 rpm) von 5 min. Danach wurden 25 nl des gereinigten iPlex® Ansatzes mittels eines Nano Dispensers auf einen SpectroChip® aufgebracht, es folgte die weitergehende Analyse mittels eines Bruker Autoflex MALDI-ToF Massenspektrometers. 3.3.9 MALDI-ToF Massenspektrometrie Die MALDI-Methode wurde 1988 von Tanaka, Karas und Hillenkamp entwickelt, um große Biomoleküle zu ionisieren und ihre Masse zu analysieren (Griffin und Smith 2000; Tost und Gut 2002). Sie kann zur Analyse von Proteinen, Peptiden und Nukleinsäuren benutzt werden. Die MALDI-ToF Massenspektrometrie ist mittlerweile eine häufig angewandte Methode, um SNPs zu genotypisieren. Große Vorteile sind die extrem kurze Analysezeit sowie die absoluten Ergebnisse, welche als Verhältnis zwischen Masse und Ladung der jeweiligen Moleküle dargestellt werden können (Griffin und Smith 2000). Bei der MALDI-ToF wird der zu analysierende Stoff (z. B. DNA) mit einer Matrix vermischt, dieses Gemisch kristallisiert dann aus. Durch Laserbeschuss mit einer Wellenlänge von ca. 337 nm wird das Analysat ionisiert und gasförmig. Es liegt eine gewisse Spannung an, welche die Ionen beschleunigt. Dabei erreichen Ionen mit kleinerem Verhältnis von Masse zu Ladung den Detektor schneller als solche mit größerem Quotienten. Die jeweilige Flugzeit wird gemessen und darüber das MasseLadungs-Verhältnis berechnet (Griffin und Smith 2000). 43 3 Material und Methoden Abb. 3.1: Schematische Darstellung eines MALDI-ToF Massenspektrometers (nach Sauer 2007) 3.3.10 Statistische Analyse Die Auswertung der Ergebnisse erfolgte mit dem Programm Haploview (Barrett et al. 2005). Zunächst wurde das Kopplungsungleichgewicht zwischen den untersuchten SNPs ermittelt. Eine mögliche Assoziation einzelner Allele bzw. Haplotypen mit Schizophrenie und bipolar-affektiver Erkrankung wurde mittels Chi2-Test berechnet. Zur genaueren Beurteilung signifikanter Ergebnisse wurde weiterhin ein Permutationstest mit 10.000 Permutationen durchgeführt. Die Signifikanzgrenze wurde bei einem p-Wert von 0,05 festgelegt. 3.3.11 Qualitätssicherung Es wurden 14 verschiedene SNPs im NOS1AP-Gen im Bereich von Intron 1, 2, 3, 4, 8 und 9 sowie Exon 6 und 9 untersucht. Für SNP 8 (rs2661818) wurde in allen Fällen nur das Allel C gefunden, es handelt sich daher für das vorliegende Patienten- und Kontrollkollektiv nicht um einen polymorphe Variante, so dass er von den weiteren Analysen ausgeschlossen wurde. Bei Schizophrenie betrug die beobachtete Heterozygotenrate 16,3% bis 52,5%. Die minore Allelfrequenz lag zwischen 9,6% und 45,4%. Die call rate der 13 untersuchten SNPs lag zwischen 87,1% und 99,6%. SNP 13 befand sich nicht im Hardy-Weinberg- 44 3 Material und Methoden Gleichgewicht (p = 0,0065), was am ehesten auf einen Genotypisierungsfehler zurückzuführen ist, weshalb er ebenfalls von weiteren Analysen ausgeschlossen wurde. Bei der bipolar-affektiven Erkrankung lag die beobachtete Heterozygotenrate zwischen 15,1% und 54,9%. Die minore Allelfrequenz betrug 9,4% bis 45,1%. Die call rate lag bei 87,0% bis 99,5%. Außer SNP 13 befanden sich alle SNPs im Hardy-WeinbergGleichgewicht. 45 4 Ergebnisse 4 Ergebnisse 4.1 Schizophrenie 4.1.1 Kopplungsungleichgewicht Das Kopplungsungleichgewicht wurde nach der solid spine-Methode berechnet. Dabei ergab sich in der Gesamtstichprobe für die 13 untersuchten SNPs eine Struktur mit zwei Haploblöcken. Block 1 bestand aus SNP 4-7, die sich mit D’-Werten zwischen 0,87 und 1,0 im Kopplungsungleichgewicht befanden. Der zweite Block bestand aus den SNPs 11-14 mit D’-Werten von 0,98 bis 1,0. Abb. 4.1: LD Struktur Schizophrenie Gesamtstichprobe (D’) 46 4 Ergebnisse 4.1.2 Assoziationsanalyse Acht Individuen wurden von der Analyse ausgeschlossen, weil weniger als 85% der Allele bestimmt werden konnten. Zunächst wurde eine Assoziation der einzelnen Allele untersucht (Tab. 4.1), dabei ergab sich für SNP 2 (rs1538018) ein p-Wert von 0,0085, für SNP 3 (rs945713) ein p-Wert von 0,0072 und für SNP 5 (rs4306106) ein p-Wert von 0,0466 im Chi²-Test. Somit zeigte sich bei diesen drei SNPs eine signifikante Assoziation zu Schizophrenie. Darüber hinaus wurde eine mögliche Assoziation der korrespondierenden Haplotypen untersucht (Tab. 4.2). Dabei zeigte sich jedoch kein signifikant mit Schizophrenie assoziierter Haplotyp. Nach Vergrößerung des Haplotypblocks 1 auf SNP 3-7 ergab sich allerdings eine signifikante nominelle Assoziation des Haplotyps CTAGC mit einem p-Wert von 0,0172 (Tab. 4.3). Dieser Haplotyp kam bei 18% der Erkrankten und 13% der Kontrollen vor. Zur weiteren Analyse der signifikant assoziierten Allele bzw. des Haplotyps wurde ein Permutationstest mit 10.000 Permutationen durchgeführt. Danach fand sich weder für die Allele noch für den Haplotyp eine signifikante Assoziation. Allerdings zeigte sich für SNP 2 mit einem p-Wert von 0,0924 und für SNP 3 mit einem p-Wert von 0,0788 immerhin noch ein statistischer Trend. Anschließend wurde die Analyse geschlechtsspezifisch durchgeführt. Bei den Männern wurden drei Individuen ausgeschlossen. In der Assoziationsanalyse der einzelnen SNPAllele mittels Chi²-Test zeigten SNP 2 (p = 0,0059) und SNP 3 (p = 0,005) signifikante Assoziationen mit Schizophrenie (Tab. 4.4). In der Haplotypanalyse zeigten in Block 1 die Haplotypen TCGAT mit einem p-Wert von 0,0367 und CTAGC mit einem p-Wert von 0,0467 nominelle Assoziation mit Schizophrenie (Tab. 4.5). Der Haplotyp TCGAT kam bei 41% der Patienten und 50% der Kontrollen vor, CTAGC hatten 20% der Patienten und 14% der Kontrollen. Im Permutationstest ergab sich eine grenzwertige Signifikanz für SNP 3 mit einem p-Wert von 0,0588 und ein Trend für SNP 2 mit einem p-Wert von 0,0706. Die Haplotypen waren im Permutationstest nicht mehr signifikant. Bei den Frauen wurden fünf Individuen ausgeschlossen. In der Analyse der SNPs zeigte sich keine signifikante allelische Assoziation (Tab. 4.6). In keinem der Haploblöcke zeigte sich ein signifikant mit Schizophrenie assoziierter Haplotyp (Tab. 4.7). 47 4 Ergebnisse 4.2 Bipolar-affektive Störung 4.2.1 Kopplungsungleichgewicht Nach Berechnung der Daten mit solid spine ergab sich eine Blockstruktur aus zwei Haploblöcken (Abb. 4.2). Haploblock 1 beinhaltete SNP 4-7, es zeigte sich ein Kopplungsungleichgewicht mit D’-Werten von 0,86 bis 1,0. Block 2 bestand aus SNP 11-14 mit D’-Werten von 0,98 bis 1,0. Abb. 4.2: LD Struktur bipolar-affektive Störung Gesamtstichprobe (D’) 4.2.2 Assoziationsanalyse Zwei Individuen wurden von der Assoziationsanalyse ausgeschlossen, da weniger als 85% der Allele bestimmt werden konnten. In der Assoziationsanalyse der SNPs mit dem Chi²-Test zeigte sich keine signifikante allelische Assoziation mit der bipolar- 48 4 Ergebnisse affektiven Störung (Tab. 4.8). Die Haplotypenanalyse ergab ebenfalls keine signifikante Assoziation (Tab. 4.9). Auf eine nach Geschlechtern getrennte Assoziationsanalyse wurde bei unzureichender Subgruppengröße verzichtet. 4.3 Tabellarische Übersicht der Ergebnisse Tab. 4.1: Schizophrenie Gesamtstichprobe - Einzelmarker SNP Nr. 1 Marker rs1572495 LD-Block 2 rs1538018 3 rs945713 4 rs1415263 1 5 rs4306106 1 6 rs3924139 1 7 rs4145621 1 Allele T C χ2 = 0,165 p = 0,6847 P-p = 0,9999 C G χ2 = 6,919 p = 0,0085 P-p = 0,0924 C T χ2 = 7,228 p = 0,0072 P-p= 0,0788 T C χ2 = 3,282 p = 0,07 P-p = 0,5074 A G χ2 = 3,96 p = 0,0466 P-p = 0,3762 G A χ2 = 3,624 p = 0,057 P-p = 0,4378 C T χ2 = 1,598 p = 0,2062 P-p = 0,8946 49 Fälle 45 (10%) 403 (90%) Kontrollen 59 (9%) 575 (91%) 109 (32%) 233 (68%) 152 (24%) 480 (76%) 206 (46%) 244 (54%) 248 (38%) 410 (62%) 190 (43%) 256 (57%) 240 (37%) 406 (63%) 109 (24%) 339 (76%) 126 (19%) 526 (81%) 174 (39%) 270 (61%) 205 (33%) 407 (67%) 199 (48%) 217 (52%) 279 (44%) 357 (56%) 4 Ergebnisse 9 rs1508263 10 rs3751284 11 rs7521206 2 12 rs905721 2 13 rs348624 2 14 rs1964052 2 A G χ2 = 0,03 p = 0,8627 P-p = 1,0 G A χ2 = 0,52 p = 0,4707 P-p = 0,9976 G C χ2 = 0,072 p = 0,7887 P-p = 1,0 T C χ2 = 0,073 p = 0,7869 P-p = 1,0 T C χ2 = 0,407 p = 0,5235 P-p = 0,9988 T C χ2 = 0,002 p = 0,9623 P-p = 1,0 205 (46%) 245 (54%) 299 (45%) 365 (55%) 289 (66%) 151 (34%) 399 (64%) 229 (36%) 146 (35%) 274 (65%) 216 (34%) 420 (66%) 146 (35%) 272 (65%) 217 (34%) 419 (66%) 47 (11%) 385 (89%) 55 (10%) 515 (90%) 56 (12%) 394 (88%) 82 (12%) 582 (88%) Tab. 4.2: Schizophrenie Gesamtstichprobe - Haplotypen Haplotyp LD-Block CGAT 1 Frequenz (Gesamtkollektiv) 0,513 TAGC 1 0,212 50 Fälle pos.:neg. (ges.) 219:231 (450) (49%) χ2 = 2,091 p = 0,1482 P-p = 0,9242 107:343 (450) (24%) χ2 = 3,065 p = 0,08 P-p = 0,7561 Kontrollen pos.:neg. (ges.) 352:312 (664) (53%) 129:535 (664) (19%) 4 Ergebnisse TGGC 1 0,114 CGAC 1 0,074 TGAC 1 0,044 TGGT 1 0,017 CGGC 1 0,012 CCCC 2 0,531 GTCC 2 0,338 CCTT 2 0,121 53:397 (450) (12%) χ2 = 0,057 p = 0,8107 31:419 (450) (7%) χ2 = 0,406 p = 0,5242 22:428 (450) (5%) χ2 = 0,431 p = 0,5114 8:442 (450) (2%) χ2 = 0,0 p = 0,9975 6:444 (450) (1%) χ2 = 0,045 p = 0,8313 236:214 (450) (52%) χ2 = 0,19 p = 0,6631 153:297 (450) (34%) χ2 = 0,019 p = 0,8918 54:396 (450) (12%) χ2 = 0,008 p = 0,9297 75:589 (664) (11%) 52:612 (664) (8%) 27:637 (664) (4%) 11:653 (664) (2%) 8:656 (664) (1%) 355:307 (662) (54%) 223:439 (662) (34%) 81:581 (662) (12%) Tab. 4.3: Schizophrenie Gesamtstichprobe – Haplotypen (vergrößerter Block 1) Haplotyp LD-Block TCGAT 1 Frequenz (Gesamtkollektiv) 0,451 CTAGC 1 0,148 51 Fälle pos.:neg. (ges.) 188:262 (450) (42%) χ2 = 3,592 p = 0,0581 80:370 (450) (18%) χ2 = 5,677 p = 0,0172 Kontrollen pos.:neg. (ges.) 314:348 (662) (47%) 84:578 (662) (13%) 4 Ergebnisse CTGGC 1 0,113 TTAGC 1 0,065 CCGAT 1 0,062 TCGAC 1 0,058 CTGAC 1 0,044 CCGAC 1 0,016 CTGGT 1 0,015 TCGGC 1 0,013 53:397 (450) (12%) χ2 = 0,156 p = 0,6932 27:423 (450) (6%) χ2 = 0,419 p = 0,5174 31:419 (450) (7%) χ2 = 0,72 p = 0,396 24:426 (450) (5%) χ2 = 0,476 p = 0,4903 22:428 (450) (5%) χ2 = 0,449 p = 0,5027 7:443 (450) (2%) χ2 = 0,004 p = 0,951 7:443 (450) (2%) χ2 = 0,065 p = 0,7985 6:444 (450) (1%) χ2 = 0,0 p = 0,9995 73:589 (662) (11%) 46:616 (662) (7%) 38:624 (662) (6%) 41:621 (662) (6%) 27:635 (662) (4%) 10:652 (662) (2%) 10:652 (662) (2%) 9:653 (662) (1%) Tab. 4.4: Schizophrenie Männer - Einzelmarker SNP Nr. 1 Marker rs1572495 2 rs1538018 LD-Block Allele T C χ2 = 0,392 p = 0,5313 P-p = 0,9996 C G χ2 = 7,581 p = 0,0059 P-p = 0,0706 52 Fälle 25 (10%) 223 (90%) Kontrollen 27 (9%) 289 (91%) 65 (35%) 121 (65%) 77 (24%) 249 (76%) 4 Ergebnisse 3 rs945713 1 4 rs1415263 1 5 rs4306106 1 6 rs3924139 1 7 rs4145621 1 9 rs1508263 10 rs3751284 11 rs7521206 2 12 rs905721 2 C T χ2 = 7,874 p = 0,005 P-p = 0,0588 T C χ2 = 2,749 p = 0,0973 P-p = 0,639 A G χ2 = 1,554 p = 0,2126 P-p = 0,9017 G A χ2 = 2,257 p = 0,133 P-p = 0,7624 C T χ2 = 2,83 p = 0,0925 P-p = 0,6256 G A χ2 = 0,317 p = 0,5734 P-p = 1,0 G A χ2 = 0,986 p = 0,3206 P-p = 0,979 C G χ2 = 1,35 p = 0,2453 P-p = 0,9335 C T χ2 = 1,113 p = 0,2915 P-p = 0,9653 53 121 (48%) 129 (52%) 123 (37%) 211 (63%) 111 (44%) 139 (56%) 124 (38%) 206 (62%) 60 (24%) 188 (76%) 66 (20%) 266 (80%) 102 (41%) 146 (59%) 109 (35%) 203 (65%) 113 (50%) 115 (50%) 138 (42%) 188 (58%) 136 (54%) 114 (46%) 177 (52%) 163 (48%) 159 (65%) 87 (35%) 195 (61%) 127 (39%) 157 (69%) 71 (31%) 209 (64%) 117 (36%) 154 (68%) 72 (32%) 208 (64%) 118 (36%) 4 Ergebnisse 13 rs348624 2 14 rs1964052 2 T C χ2 = 1,575 p = 0,2095 P-p = 0,8967 T C χ2 = 1,005 p = 0,3161 P-p = 0,9778 26 (11%) 212 (89%) 23 (8%) 273 (92%) 34 (14%) 216 (86%) 37 (11%) 303 (89%) Tab. 4.5: Schizophrenie Männer - Haplotypen Haplotyp LD-Block TCGAT 1 CTAGC 1 CTGGC 1 TTAGC 1 CCGAT 1 TCGAC 1 CTGAC 1 Frequenz Fälle (Gesamtkollektiv) pos.:neg. (ges.) 0,46 102:148 (250) (41%) χ2 = 4,364 p = 0,0367 P-p = 0,4407 0,162 49:201 (250) (20%) χ2 = 3,957 p = 0,0467 P-p = 0,5411 0,111 32:218 (250) (13%) χ2 = 1,268 p = 0,2602 P-p = 0,9958 0,059 11:239 (250) (4%) χ2 = 1,452 p = 0,2282 P-p = 0,9883 0,049 15:235 (250) (6%) χ2 = 0,892 p = 0,3451 0,046 12:238 (250) (5%) χ2 = 0,048 p = 0,826 0,046 13:237 (250) (5%) χ2 = 0,294 p = 0,5877 54 Kontrollen pos.:neg. (ges.) 168:170 (338) (50%) 46:292 (338) (14%) 33:305 (338) (10%) 23:315 (338) (7%) 14:324 (338) (4%) 15:323 (338) (4%) 14:324 (338) (4%) 4 Ergebnisse CCGAC 1 0,022 CTGGT 1 0,018 TCGGC 1 0,011 CCCC 2 0,539 GTCC 2 0,337 CCTT 2 0,115 6:244 (250) (2%) χ2 = 0,015 p = 0,902 5:245 (250) (2%) χ2 = 0,184 p = 0,6676 3:247 (250) (1%) χ2 = 0,002 p = 0,9659 136:114 (250) (54%) χ2 = 0,083 p = 0,7737 78:172 (250) (31%) χ2 = 1,391 p = 0,2383 P-p = 0,992 32:218 (250) (13%) χ2 = 0,635 p = 0,4255 7:331 (338) (2%) 5:333 (338) (1%) 4:334 (338) (1%) 181:159 (340) (53%) 121:219 (340) (36%) 36:304 (340) (11%) Tab. 4.6: Schizophrenie Frauen - Einzelmarker SNP Nr. 1 Marker rs1572495 LD-Block 2 rs1538018 3 rs945713 1 4 rs1415263 1 Allele C T χ2 = 0,001 p = 0,9815 C G χ2 = 0,738 p = 0,3904 C T χ2 = 0,791 p = 0,3737 T C χ2 = 0,664 p = 0,4152 55 Fälle 180 (90%) 20 (10%) Kontrollen 286 (90%) 32 (10%) 44 (28%) 112 (72%) 75 (25%) 231 (75%) 85 (43%) 115 (57%) 125 (39%) 199 (61%) 79 (40%) 117 (60%) 116 (37%) 200 (63%) 4 Ergebnisse 5 rs4306106 2 6 rs3924139 2 7 rs4145621 2 9 rs1508263 10 rs3751284 11 rs7521206 3 12 rs905721 3 13 rs348624 3 14 rs1964052 3 A G χ2 = 2,456 p = 0,1171 G A χ2 = 1,186 p = 0,276 C T χ2 = 0,003 p = 0,9548 A G χ2 = 0,626 p = 0,4289 G A χ2 = 0,006 p = 0,9366 G C χ2 = 2,659 p = 0,1029 T C χ2 = 2,291 p = 0,1301 C T χ2 = 0,083 p = 0,7739 C T χ2 = 0,926 p = 0,336 49 (25%) 151 (75%) 60 (19%) 260 (81%) 72 (37%) 124 (63%) 96 (32%) 204 (68%) 86 (46%) 102 (54%) 141 (45%) 169 (55%) 91 (46%) 109 (54%) 136 (42%) 188 (58%) 130 (67%) 64 (33%) 204 (67%) 102 (33%) 75 (39%) 117 (61%) 99 (32%) 211 (68%) 74 (39%) 118 (61%) 99 (32%) 211 (68%) 173 (89%) 21 (11%) 242 (88%) 32 (12%) 178 (89%) 22 (11%) 279 (86%) 45 (14%) Tab. 4.7: Schizophrenie Frauen - Haplotypen Haplotyp LD Block TC 1 Frequenz Fälle (Gesamtkollektiv) pos.:neg. (ges.) 0,529 100:100 (200) (50%) χ2 = 1,182 p = 0,277 56 Kontrollen pos.:neg. (ges.) 177:147 (324) (55%) 4 Ergebnisse CT 1 0,312 CC 1 0,089 TT 1 0,07 GAT 2 0,529 AGC 2 0,203 GGC 2 0,132 GAC 2 0,121 CCCC 3 0,523 GTCC 3 0,339 CCTT 3 0,125 66:134 (200) (33%) χ2 = 0,54 p = 0,4623 19:181 (200) (10%) χ2 = 0,112 p = 0,7373 15:185 (200) (8%) χ2 = 0,174 p = 0,6763 103:97 (200) (52%) χ2 = 0,208 p = 0,648 46:154 (200) (23%) χ2 = 1,627 p = 0,2021 24:176 (200) (12%) χ2 = 0,27 p = 0,6032 22:178 (200) (11%) χ2 = 0,479 p = 0,4889 99:101 (200) (50%) χ2 = 1,125 p = 0,2888 76:124 (200) (38%) χ2 = 2,473 p = 0,1158 22:178 (200) (11%) χ2 = 0,696 p = 0,4043 97:227 (324) (30%) 28:296 (324) (9%) 22:302 (324) (7%) 175:151 (326) (54%) 60:266 (326) (18%) 45:281 (326) (14%) 42:284 (326) (13%) 176:148 (324) (54%) 102:222 (324) (31%) 44:280 (324) (14%) Tab. 4.8: Bipolar-affektive Erkrankung Gesamtstichprobe - Einzelmarker SNP Nr. 1 Marker rs1572495 LD-Block Allele T C χ2 = 0,011 p = 0,9157 57 Fälle 14 (10%) 132 (90%) Kontrollen 59 (9%) 575 (91%) 4 Ergebnisse 2 rs1538018 3 rs945713 4 rs1415263 1 5 rs4306106 1 6 rs3924139 1 7 rs4145621 1 9 rs1508263 10 rs3751284 11 rs7521206 2 12 rs905721 2 13 rs348624 2 C G χ2 = 0,785 p = 0,3755 C T χ2 = 0,21 p = 0,6467 T C χ2 = 0,181 p = 0,6702 A G χ2 = 2,096 p = 0,1476 G A χ2 = 0,049 p = 0,824 C T χ2 = 0,171 p = 0,6791 A G χ2 = 0,003 p = 0,9577 G A χ2 = 1,655 p = 0,1983 G C χ2 = 1,443 p = 0,2297 T C χ2 = 1,264 p = 0,261 T C χ2 = 0,016 p = 0,9001 58 35 (28%) 91 (72%) 152 (24%) 480 (76%) 58 (40%) 88 (60%) 248 (38%) 410 (62%) 57 (39%) 89 (61%) 240 (37%) 406 (63%) 36 (25%) 110 (75%) 126 (19%) 526 (81%) 51 (34%) 97 (66%) 205 (33%) 407 (67%) 65 (46%) 77 (54%) 279 (44%) 357 (56%) 67 (45%) 81 (55%) 299 (45%) 365 (55%) 97 (69%) 43 (31%) 399 (64%) 229 (36%) 58 (39%) 90 (61%) 216 (34%) 420 (66%) 57 (39%) 89 (61%) 217 (34%) 419 (66%) 14 (10%) 126 (90%) 55 (10%) 515 (90%) 4 Ergebnisse 14 rs1964052 2 T C χ2 = 0,686 p = 0,4076 22 (15%) 126 (85%) 82 (12%) 582 (88%) Tab. 4.9: Bipolar-affektive Erkrankung Gesamtstichprobe - Haplotypen Haplotyp LD Bock CGAT 1 TAGC 1 TGGC 1 CGAC 1 TGAC 1 TGGT 1 CGGC 1 CCCC 2 GTCC 2 CCTT 2 Frequenz Fälle (Gesamtkollektiv) pos.:neg. (ges.) 0,527 76:72 (148) (51%) χ2 = 0,144 p = 0,7043 0,203 36:112 (148) (24%) χ2 = 1,566 p = 0,2109 0,102 9:139 (148) (6%) χ2 = 3,3 p = 0,0693 0,079 12:136 (148) (8%) χ2 = 0,004 p = 0,9478 0,045 9:139 (148) (6%) χ2 = 1,056 p = 0,304 0,018 3:145 (148) (2%) χ2 = 0,118 p = 0,7312 0,012 2:146 (148) (1%) χ2 = 0,04 p = 0,8417 0,523 68:80 (148) (46%) χ2 = 2,97 p = 0,0848 0,346 58:90 (148) (39%) χ2 = 1,674 p = 0,1957 0,122 20:128 (148) (14%) χ2 = 0,257 p = 0,6121 59 Kontrollen pos.:neg. (ges.) 352:312 (664) (53%) 129:535 (664) (19%) 74:590 (664) (11%) 53:611 (664) (8%) 27:637 (664) (4%) 12:652 (664) (2%) 8:656 (664) (1%) 357:307 (664) (54%) 223:441 (664) (34%) 79:585 (664) (12%) 5 Diskussion 5 Diskussion 5.1 Stärken und Schwächen von Assoziationsstudien Das Ziel von genetischen Assoziationsstudien ist es, statistische Assoziation zwischen genetischen Polymorphismen und einem Phänotyp nachzuweisen und so genetische Risikofaktoren zu identifizieren, die später umfassender analysiert werden können (Lunetta 2008). Ein großer Vorteil von populationsbasierten Assoziationsstudien ist die einfache Rekrutierung von Fall-Kontroll-Samples im Vergleich zu familienbasierten Daten. Auf diese Weise kann man große Samples aufbauen, in denen die Frequenz des Krankheitsallels und das ihm zuschreibbare Risiko gleichzeitig geschätzt werden können. Auch wird es durch diese Art von Studien möglich, Krankheiten mit einem späten Ersterkrankungsalter zu analysieren, bei denen familienbasierte Studien nicht mehr möglich wären, da ältere Angehörige eventuell schon verstorben sind. Man kann in Assoziationsstudien Krankheitsallele, vor allem auch häufig vorkommende Allele mit geringem Einfluss auf die Krankheitsentstehung identifizieren. Durch den technologischen Fortschritt sind mittlerweile kostengünstige Methoden mit hohem Durchsatz verfügbar, sodass viele SNPs im Genom untersucht werden können (Rodriguez-Murillo und Greenberg 2008). Inzwischen können Assoziationsstudien einige große Erfolge nachweisen, die nicht nur psychiatrische Krankheiten betreffen. So konnten beispielsweise SNPs identifiziert werden, die mit Multipler Sklerose, Mammakarzinom und altersabhängiger Makuladegeneration assoziiert sind (Easton et al. 2007; Hafler et al. 2007; Klein et al. 2005). Assoziationsstudien haben jedoch auch einige Nachteile. Besonders schwierig ist es, Assoziation eines bestimmten Gens in ethnisch verschiedenen Samples zu replizieren, da diese aufgrund unterschiedlicher Herkunft genetisch differieren. Falls im Laufe der Zeit in verschiedenen Populationen unterschiedliche anfälligkeitsvermittelnde Veränderungen innerhalb eines Gens auftreten, ist es extrem schwer bis unmöglich, Assoziation zu entdecken. Selbst wenn die gleichen Veränderungen geschehen sind, jedoch zu unterschiedlichen Zeitpunkten, kann es kompliziert sein, Assoziation nachzuweisen. Da sich in einem solchen Fall die flankierenden Regionen 60 5 Diskussion wahrscheinlich unterscheiden, besteht unter Umständen kein Kopplungsungleichgewicht zur kausalen Variante mehr, sodass man exakt die kausale Variante untersuchen müsste, um Assoziation feststellen zu können (Brzustowicz 2008; Rodriguez-Murillo und Greenberg 2008). Es hat sich gezeigt, dass der Grad einer Assoziation in der ersten Studie meist viel höher eingeschätzt wird als in nachfolgenden Replikationsstudien, ein Phänomen, das als „winner’s curse“ bezeichnet wird. Dies kann zum einen darauf beruhen, dass die originale Studie ein falsch positives Ergebnis präsentiert oder die Assoziation überschätzt, andererseits ist es möglich, dass der Geneffekt in einer Subpopulation größer ist als in anderen (Ioannidis et al. 2001). Selbst in scheinbar ähnlichen Subpopulationen herrscht teilweise große Variabilität in der Stärke einer Assoziation, was auf Differenzen in unbekannten Parametern wie ethnischen Unterschieden und verschiedenen Gen-Umwelt-Interaktionen zurückzuführen ist. Die Bewertung solcher Subgruppeneffekte ist schwierig und erfordert große Stichproben (Ioannidis et al. 2001). Ein weiteres Problem ist, dass genetische Assoziation oft von geringem Ausmaß ist und einzelne Studien nicht genug Power haben, um sie zu entdecken (Ioannidis et al. 2001). Zudem haben einzelne assoziierte Allele im Vergleich zu Haplotypen eher eine geringe Aussagekraft (Petronis 2004; Zheng et al. 2005). Durch die Analyse von Haplotypen kann man die Datenmenge verkleinern, wodurch weniger Mehrfachtests nötig sind. Auf diese Weise vergrößert sich die Power, seltene Allele zu entdecken (Malhotra et al. 2004). Was die Interpretation eines positiven Ergebnisses erschwert, ist die schon oben beschriebene Tatsache, dass Assoziation keine genetische Beziehung, sondern lediglich ein statistisches Phänomen beschreibt. Dementsprechend ist auch trotz eines statistisch negativen Ergebnisses eine Assoziation nicht auszuschließen (Ioannidis et al. 2001). Es ergeben sich bei vielen Assoziationsstudien Probleme mit dem Studiendesign, insbesondere durch die fehlende Vergleichbarkeit von Fällen und Kontrollen. Ein solches schlechtes Matchen von Fällen und Kontrollen erhöht die Gefahr eines Typ-1Fehlers und kann beispielsweise durch familienbasierte Studiendesigns verhindert werden, die weniger anfällig für Stratifikation sind und den Einfluss nicht genetischer Risikofaktoren stärker berücksichtigen. Nachteilig ist dabei wiederum die geringere statistische Power und die eingeschränkte Verfügbarkeit ausreichend großer Familiensamples im Vergleich zu Fall-Kontroll-Studien nicht verwandter Individuen 61 5 Diskussion (Abou-Sleiman et al. 2006; Chanock et al. 2007; Craddock und Jones 1999; Kennedy et al. 2003; Malhotra et al. 2004; Petronis 2004). Trotz der genannten Probleme macht die Vielzahl von fraglichen Genotyp-PhänotypAssoziationen gute Replikationsstudien notwendig, um die Aussagekraft von Ergebnissen zu überprüfen (Chanock et al. 2007). 5.2 Vergleich mit anderen Studien In dieser Arbeit wurde die Bedeutung von NOS1AP auf 1q22 als Kandidatengen bei Schizophrenie und bipolar-affektiver Erkrankung untersucht. Der funktionelle Zusammenhang zwischen NOS1AP und Schizophrenie wurde in der Einleitung ausführlich beschrieben und basiert in erster Linie auf der Verbindung zu NMDARezeptor und NO-Synthese. Die Rolle von NOS1AP in der Pathogenese der bipolaraffektiven Erkrankung ist hingegen weniger gut erforscht, es ist jedoch vorstellbar, dass auch affektive Störungen auf Abnormalitäten des Glutamatsystems beruhen und eine Beziehung zum NMDAR besteht. Es gibt verschiedene Kopplungsstudien, die Hinweise auf den Genort 1q22 als Kandidatenregion für Schizophrenie geben (Brzustowicz et al. 2000; Brzustowicz et al. 2002; Gurling et al. 2001; Hwu et al. 2003; Shaw et al. 1998). In insgesamt sechs Assoziationsstudien wurden Marker innerhalb des NOS1AP-Gens bei Schizophrenen untersucht. Ihre Ergebnisse sind allerdings eher widersprüchlich. Während in vier Studien Assoziation von SNPs mit Schizophrenie nachgewiesen werden konnte (Brzustowicz et al. 2004; Kremeyer et al. 2009; Wratten et al. 2009; Zheng et al. 2005), zeigten die anderen zwei Studien negative Ergebnisse (Fang et al. 2008; Puri et al. 2006). Es gibt bisher also keine sicheren Hinweise auf eine Bedeutung von Polymorphismen in diesem Gen für eine Krankheitsanfälligkeit. Die vorliegende Arbeit stellt eine weitere Fall-Kontroll-Studie dar. Ursprünglich wurden 14 SNPs im NOS1APGen untersucht, die auch Gegenstand vorheriger Studien waren. Ein SNP (rs2661818) wurde von der Analyse ausgeschlossen, da es sich in diesem Patienten- und Kontrollkollektiv nicht um einen Polymorphismus handelte. Ein weiterer SNP (rs348624) befand sich nicht im Hardy-Weinberg-Gleichgewicht, sodass dieser SNP ebenfalls von weiteren Analysen ausgeschlossen wurde. 62 5 Diskussion Für die SNPs rs1538018 (p = 0,0085), rs945713 (p = 0,0072) und rs4306106 (p = 0,0466) ergab sich eine nominelle Assoziation mit Schizophrenie. Da es bei FallKontroll-Studien durch die Kombination vieler Einzeltests zu einem erhöhten Risiko für falsch positive Ergebnisse kommt, wurde anschließend ein Permutationstest durchgeführt (Lunetta 2008). Hierbei verfehlten die zuvor nominell assoziierten SNPs rs1538018 (p = 0,0924) und rs945713 (p = 0,0788) das Signifikanzniveau von p = 0,05 knapp, was jedoch nicht ungewöhnlich ist und eine Assoziation dieser Varianten mit Schizophrenie nicht ausschließt. Der Haplotyp CTAGC in Block 1, der das assoziierte C-Allel von SNP rs945713 beinhaltet, war ebenfalls nominell assoziiert (p = 0,0172), wobei sich auch hier kein signifikanter Wert im Permutationstest ergab. Dies unterstreicht trotzdem eine mögliche Relevanz des C-Allels in SNP rs945713 als Risikofaktor für Schizophrenie. In der nach Geschlechtern unterteilten Analyse zeigten bei den Männern SNP rs1538018 (p = 0,0059) und rs945713 (p = 0,005) eine nominelle Assoziation. Nach dem Permutationstest wies SNP rs945713 noch eine grenzwertige Signifikanz auf (p = 0,0588) und auch rs1538018 verfehlte die Signifikanzgrenze nur sehr knapp (p = 0,0706). In Block 1 ergaben sich für zwei Haplotypen, TCGAT (p = 0,0367) und CTAGC (p = 0,0467), nominell signifikante Ergebnisse, wobei der Haplotyp TCGAT häufiger bei Gesunden vorkam, während CTAGC, welcher wiederum das assoziierte CAllel aus SNP rs945713 enthält, mit Schizophrenie assoziiert war. Allerdings hielten erneut beide Haplotypen dem Permutationstest nicht stand. SNP rs1538018 steht nicht im Kopplungsungleichgewicht mit den anderen untersuchten SNPs, sodass es keine resultierenden Haploblöcke gibt. In der Analyse für Frauen ließen sich keine signifikanten Assoziationen nachweisen. Beim Vergleich der vorliegenden Ergebnisse mit den anderen Assoziationsstudien fällt zunächst auf, dass die SNPs rs1538018 und rs945713, welche in der vorliegenden Studie nominell mit Schizophrenie assoziiert waren, in der ursprünglichen Studie von Brzustowicz et al. ebenfalls signifikant assoziiert waren, wobei sie nicht zu den drei Markern gehörten, die genomweite Signifikanz erreichten (Brzustowicz et al. 2004). Eine aktuelle Assoziationsstudie konnte ebenfalls eine signifikante Assoziation des Markers rs945713 in einer kolumbianischen Population feststellen, der SNP rs1538018 war hingegen nicht assoziiert (Kremeyer et al. 2009). Der dritte nominell assoziierte 63 5 Diskussion SNP dieser Arbeit, rs4306106, war lediglich Gegenstand einer weiteren Studie (Wratten et al. 2009). In dieser aktuellen, mit einer neuen Methode durchgeführten Untersuchung des kanadischen Samples, in welchem ursprünglich Assoziation festgestellt worden war, zeigten sich für rs4306106, wie auch für rs1538018 und rs945713, keine deutlichen Hinweise auf eine Assoziation mit Schizophrenie. In der Studie von Puri et al. wiesen rs1538018 und rs945713 ebenfalls kein signifikantes Ergebnis auf, bei Zheng et al. wurde nur SNP rs945713 untersucht, der ebenso negativ war. Keiner dieser beiden SNPs wurde in der Assoziationsstudie von Fang et al. analysiert. Abb. 5.1: Assoziation der untersuchten SNPs (rot) und SNPs anderer, ausgewählter Studien mit Schizophrenie. Umgekehrt logarithmische Auftragung (signifikant: -log(p;10) > 1,3) Unterstützung der vorliegenden Ergebnisse findet man also im kanadischen und kolumbianischen Sample, allerdings muss man erwähnen, dass bei Brzustowicz et al. drei weitere Marker signifikante Ergebnisse aufwiesen, zwei davon sogar genomweite Signifikanz (rs1415263 und rs4145621), die in dieser Untersuchung negativ waren. Auch die Marker rs348624 bzw. rs1964052, die in der Studie von Zheng et al. im 64 5 Diskussion kompletten Kopplungsungleichgewicht waren und signifikante Assoziation mit Schizophrenie aufwiesen, waren in der vorliegenden Studie nicht signifikant assoziiert bzw. bei fehlendem Hardy-Weinberg-Gleichgewicht nicht auswertbar. Die Assoziationsstudie im kolumbianischen Sample zeigte für acht SNPs nominell signifikante Ergebnisse, darunter wiederum rs1415263 und rs4145621 sowie zwei weitere SNPs (rs3924139, rs3751284), die in der vorliegenden Untersuchung ebenfalls negativ waren (Kremeyer et al. 2009). Die Studie von Wratten et al. lieferte mit einer anderen Methode erneut ein positives Ergebnis für die Marker rs1415263 und rs4145621. Zudem wurde für den signifikant assoziierten SNP rs12742393, der nicht Gegenstand der vorliegenden Arbeit war, eine funktionelle Variante identifiziert (Wratten et al. 2009). Insgesamt lassen sich aus den vorliegenden Ergebnissen keine eindeutigen Schlüsse ziehen, jedoch weisen die nominell signifikanten Resultate für die SNPs rs1538018 und rs945713 insbesondere bei Männern auf eine Assoziation von NOS1AP mit Schizophrenie hin. Allerdings ist es möglich, dass die geschlechtergetrennten Untergruppen zu klein sind, um ein aussagekräftiges Ergebnis zu liefern, sodass das Ergebnis der Gesamtstichprobe im Vordergrund steht. Im bipolar-affektiven Kollektiv wurden dieselben SNPs untersucht, jedoch ergab die Assoziationsanalyse der einzelnen SNPs keine signifikanten Werte. Einschränkend muss allerdings gesagt werden, dass das Kollektiv der Patienten relativ klein war. Da es bisher keine Assoziationsstudien gibt, die NOS1AP bei einem Kollektiv bipolar-affektiv Erkrankter untersucht haben, ist ein Vergleich nicht möglich. 5.3 Ursachen für Unterschiede zu anderen Assoziationsstudien Beim Vergleich der Ergebnisse für Schizophrenie mit anderen Fall-Kontroll-Studien fällt auf, dass die Studien nicht zu einem einheitlichen Ergebnis kommen und selbst in den Studien, die Assoziation nachweisen konnten, unterschiedliche SNPs assoziiert waren. Die Ursachen hierfür können vielfältig sein. Einen Hauptgrund stellen ethnische Unterschiede in den verschiedenen Samples dar. Verschiedene Populationen weisen auch unterschiedliche Verteilungen von Geno- und Phänotypen auf (Abou-Sleiman et 65 5 Diskussion al. 2006; Lunetta 2008). Obwohl beispielsweise generell davon ausgegangen wird, dass Europa und der mittlere Osten einen relativ homogenen Genpool darstellen, wurde festgestellt, dass sogar innerhalb Europas erhebliche genetische Variation besteht (Thomas und Witte 2002). In dieser Studie wurde ein Kollektiv kaukasischen Ursprungs aus Unterfranken in Deutschland untersucht. Die bisherigen Assoziationsstudien arbeiteten mit Samples Han-chinesischen, kolumbianischen, keltischen und britischen (d. h. englischen, schottischen, walisischen und irischen) Ursprungs und differierten somit zum Teil deutlich. Einen Hinweis auf unterschiedliche Allelfrequenzen zwischen diesen Populationen liefert beispielsweise der im kolumbianischen und Hanchinesischen Sample biallelische (C/G) SNP rs2661818 (Fang et al. 2008; Kremeyer et al. 2009), der in der vorliegenden Studie auch untersucht wurde, jedoch nicht polymorph war, weil in allen Fällen nur das Allel C gefunden wurde. Auch die unterschiedliche Blockstruktur in verschiedenen Populationen kann differierende Ergebnisse zur Folge haben, wenn man davon ausgeht, dass die Assoziation eines Markers auf Kopplungsungleichgewicht zu der „echten“ Suszeptibilitäts-Variante beruht. Falls dieses Kopplungsungleichgewicht in einer anderen Population nicht auftritt, ist auch keine Assoziation des Markers zur Krankheit nachweisbar. Ein weiteres Phänomen, welches den Vergleich unterschiedlicher Fall-Kontroll-Studien bei so komplexen Krankheiten wie Schizophrenie und bipolar-affektiver Störung erschwert, stellt die genetische Heterogenität dar. Da ein multifaktorielles, polygenetisches Vererbungsmuster vorliegt, können in den betroffenen Individuen verschiedene Gene für die Krankheitsentstehung ursächlich sein. Außerdem können selbst im gleichen Gen unterschiedliche Varianten die Anfälligkeit vermitteln (Alaerts und Del-Favero 2009; McClellan et al. 2007). Dies könnte erklären, warum in den einzelnen Studien verschiedene Marker von NOS1AP mit der Krankheit assoziiert sind. Allerdings sind die assoziierten SNPs überwiegend in einer Region, nämlich im Intron 2 lokalisiert. Sie stehen teilweise im Kopplungsungleichgewicht miteinander und möglicherweise auch mit einem weiteren, bisher unbekannten Suszeptibilitätslocus (Kremeyer et al. 2009). Somit ist es wahrscheinlicher, dass die differierenden Haploblockstrukturen in den verschiedenen unterschiedlichen Ergebnisse verantwortlich sind. 66 Studienpopulationen für die 5 Diskussion Des Weiteren besteht auch eine Heterogenität hinsichtlich des Phänotyps. Es ist vorstellbar, dass unterschiedlichen Phänotypen auch unterschiedliche Gene bzw. Polymorphismen zu Grunde liegen und viele der untersuchten Patientenkollektive zu heterogen sind (Abou-Sleiman et al. 2006). So könnten auch die verschiedenen Diagnosekriterien eine Ursache für die unterschiedlichen Ergebnisse sein. Die heutigen Standard-Diagnosekriterien sind ICD-10 und DSM-IV. In der vorliegenden Studie wurden die Diagnosen entsprechend ICD-10 gestellt, dabei wurden auch schizoaffektiv Erkrankte in die Fall-Gruppe mit einbezogen. Alle Erkrankten wiesen jedoch einen chronischen Verlauf auf. In der Assoziationsstudie von Brzustowicz war DSM-III-R Grundlage der Diagnose, auch in dieser Studie beinhaltete die Patientengruppe Schizophrenie und chronisch schizoaffektive Erkrankung. In den Studien von Zheng et al., Fang et al. und Kremeyer et al. wurden DSM-III-R bzw. DSM-IV als Diagnosekriterien verwendet. Puri et al. wiederum stellten die Diagnosen mit Hilfe der Research Diagnostic Criteria und SADS-L Interviews. Letztere vier Studien schlossen schizoaffektiv Erkrankte aus. Die Diagnosekriterien unterscheiden sich demnach nicht erheblich und folgen den Standardkriterien. Dennoch ist es denkbar, dass die Beschränkung einiger Studien auf die Diagnose „Schizophrenie“ im Gegensatz zur Ausweitung der Fallgruppe mit Einbeziehung der schizoaffektiven Störung, wie in der vorliegenden Studie, zu verschiedenen Ergebnissen führt. Ein weiterer Faktor, der potentiell zu unterschiedlichen Ergebnissen führen kann, ist das Studiendesign. In dieser Studie wurde eine populationsbasierte Fall-KontrollUntersuchung nicht verwandter Individuen durchgeführt, dieselbe Methode wurde bei Puri et al. gewählt. Hingegen waren die Studien von Brzustowicz et al., Wratten et al., Kremeyer et al. und Fang et al. familienbasiert. Somit hatte die vorliegende Studie eine größere statistische Power, seltene Allele zu entdecken, gleichzeitig aber auch ein erhöhtes Risiko für Populationsstratifikation (Malhotra et al. 2004). 5.4 Andere Kandidatengene auf 1q22 Es ist möglich, dass trotz der nominellen Assoziation von Markern innerhalb des NOS1AP-Gens keine kausale Beziehung zu Schizophrenie besteht. Das positive Kopplungssignal der Region 1q22 könnte demzufolge auf ein anderes Kandidatengen in 67 5 Diskussion diesem Bereich hindeuten, insbesondere weil die Kopplungsregion nach dem finemapping auf einen Bereich von 3 Mb eingegrenzt wurde, in welchem ca. 50 Gene enthalten sind (Brzustowicz et al. 2002; Brzustowicz 2008). Selbst die positiven Assoziationsergebnisse wären durch ein anderes Kandidatengen zu erklären, da sich Assoziation über größere genomische Segmente erstrecken kann, die teilweise mehrere Gene enthalten (Brzustowicz 2008). Sogar ein über 40 kb entfernt liegender SNP kann im Kopplungsungleichgewicht mit einer funktionellen Variante stehen (Shinkai et al. 2002). Zudem können regulatorische DNA-Regionen über Distanzen von bis zu 1 Mb wirken und sogar mehrere Gene beeinflussen. Dies zeigt die Möglichkeit auf, dass krankheitsassoziierte Sequenzen in einem Gen die Expression eines oder mehrerer anderer Gene regulieren, wodurch diverse Gene zur Krankheitsanfälligkeit beitragen könnten (Brzustowicz 2008). Tatsächlich wurden bisher zwei andere Kandidatengene in der Nähe von NOS1AP entdeckt. Das eine Kandidatengen ist RGS4, welches 700 kb von NOS1AP entfernt liegt und bereits in mehreren Assoziations- und Funktionsstudien untersucht wurde (Erdely et al. 2006; Levitt et al. 2006; Mirnics et al. 2001). Seine Funktion wurde oben bereits beschrieben. Da die Effekte vieler Neurotransmitter, die potentiell für die Pathophysiologie von Schizophrenie relevant sind, über G-Protein-gekoppelte Rezeptoren vermittelt werden, ist die Rolle von RGS4 interessant und eine Bedeutung für die Krankheitsentstehung gut vorstellbar (Talkowski et al. 2006). Dementsprechend zeigte sich in einer Studie, in der mehrere Populationen untersucht wurden (u. a. kaukasischer, indischer und afroamerikanischer Herkunft), signifikante Assoziation von verschiedenen SNPs mit Schizophrenie (Chowdari et al. 2002). Obwohl auch einige weitere Studien Assoziation feststellten, waren die assoziierten SNPs und Haplotypen in den verschiedenen Samples unterschiedlich. Bisher sind keine Polymorphismen in kodierenden Regionen bekannt, sodass man auch bei RGS4 davon ausgehen kann, dass die assoziierten SNPs zu veränderter Transkription oder Translation und weniger zu strukturell veränderten Proteinen führen und so die Pathophysiologie beeinflussen (Talkowski et al. 2006). So wurde beispielsweise festgestellt, dass eine signifikant verminderte Expression von mRNA und Protein in betroffenen Individuen besteht (Mirnics et al. 2001; Erdely et al. 2006). Einige SNPs sind darüber hinaus mit 68 5 Diskussion funktionellen Veränderungen, wie dem veränderten Volumen des dorsolateralen präfrontalen Kortex, assoziiert (Prasad et al. 2005). Auch das Gen UHMK1 auf 1q23.3 ist ein potentielles Kandidatengen für Schizophrenie und liegt 129 kb von NOS1AP entfernt. Es zeigte in zwei Studien allelische und haplotypische Assoziation mit Schizophrenie (Puri et al. 2007; Puri et al. 2008). UHMK1 kodiert für eine Proteinkinase, die in sich entwickelnden und adulten Neuronen exprimiert wird und verschiedene Substrate wie beispielsweise Stathmin phosphoryliert. Stathmin reguliert die Dynamik von Mikrotubuli und eine Ausschaltung führt zu Verhaltensabnormalitäten. Beim Menschen wurde eine Assoziation von Stathmin mit emotionsregulierter Schreckreaktion, Neurotizismus und Panikstörung gefunden (Reif et al., in Vorbereitung). Es ist vorstellbar, dass ein Knock-out von UHMK1 bei Mäusen ebenfalls zu derartigen Auffälligkeiten führen könnte (Puri et al. 2008). Die positiven Assoziationsergebnisse für SNPs in NOS1AP und RGS4 sowie für den Marker D1S1679, der zwischen NOS1AP und UHMK1 liegt, könnten daher auch auf ein Kopplungsungleichgewicht mit einer anfälligkeitsvermittelnden Variante im UHMK1-Gen zurückgehen. Dies wird unterstützt durch die Entdeckung, dass D1S1679 ein starkes Kopplungsungleichgewicht zum Promotorbereich von UHMK1 aufweist, was Auswirkungen auf die Expression des Gens haben könnte (Puri et al. 2007). Die Erklärungen sind aber keinesfalls exklusiv: so kann es gut sein, dass Varianten in allen drei Risikogenen zu dem Linkagepeak auf Chromosom 1 beitragen; generell könnten solche Phänomene häufiger sein als gedacht und auch bei anderen psychiatrischen Erkrankungen eine Rolle spielen. 5.5 Bedeutung des nitrinergen Systems Das nitrinerge System ist an mehreren Signalkaskaden beteiligt, die im Verdacht stehen, eine Rolle bei der Entstehung endogener Psychosen zu spielen. So agiert das NOgenerierende Enzym NOS-I als nachgeschaltetes Signalmolekül des NMDAR und ist daher Bestandteil der glutamatvermittelten Neurotransmission. Möglicherweise führt eine Unterfunktion des Glutamatsystems zu einer nachfolgend eingeschränkten NOS-IFunktion. Des Weiteren steht die NOS-I nicht nur mit dem Glutamatsystem, sondern auch mit dem serotonergen und dopaminergen System in Verbindung. So inhibiert NO 69 5 Diskussion über S-Nitrosylierung monoaminerge Transporter (5HTT, DAT, NET) und bedingt dadurch eine größere Verfügbarkeit der Transmitter (Kiss und Vizi 2001). Es wurde zudem festgestellt, dass ein NOS1-Knock-out in einer veränderten Serotoninfunktion resultiert (Chiavegatto et al. 2001). Auch funktionelle Studien haben gezeigt, dass nitrinerge Neurone bei Schizophrenie verändert sind. Darüber hinaus wurde Assoziation von Schizophrenie mit einem funktionellen SNP in der Promotorregion von NOS1 festgestellt, der mit verschlechterter präfrontaler Gehirnfunktion in Verbindung steht (Reif et al. 2006). Insgesamt scheint das nitrinerge System eine Verbindung zwischen glutamatergem und monoaminergem System darzustellen und könnte so eine Schlüsselfunktion in der Pathogenese sowohl von schizophrenen als auch affektiven Störungen innehaben. NOS1AP als Adaptorprotein der NOS-I mit einer direkten Auswirkung auf die Produktion von NO steht damit im Mittelpunkt dieser Theorie (Reif et al. 2006). 5.6 Funktionelle Studien Prinzipiell gibt es zwei Möglichkeiten, wie genetische Variation Krankheitsanfälligkeit vermitteln kann. Zum einen kann die Struktur des kodierten Proteins durch den Einbau einer falschen Aminosäure verändert werden und zum anderen kann die Expression und somit die verfügbare Menge des betroffenen Proteins beeinflusst werden. Im Endeffekt bewirken beide Mechanismen eine veränderte Funktion des Proteins und führen zu einem veränderten Phänotyp (Harrison und Weinberger 2005; Kleinjan und van Heyningen 2005). In dieser Arbeit wurde keine Assoziation zu kodierenden SNPs in Exons festgestellt. Eine veränderte Aminosäurefrequenz, die zu einem Protein mit eingeschränkter Funktion führt, ist also eher unwahrscheinlich, obwohl theoretisch Kopplungsungleichgewicht zu einer kodierenden Variante bestehen könnte (Harrison und Weinberger 2005). Dies ist jedoch keine überraschende Entdeckung, da es viele potentielle Mechanismen gibt, die Auswirkungen auf die normale Genfunktion und somit pathologische Folgen haben (Kleinjan und van Heyningen 2005). Auch bei den meisten anderen Kandidatengenen sind die Auswirkungen der assoziierten SNPs noch nicht bekannt, man geht jedoch davon aus, dass eine veränderte Genexpression der 70 5 Diskussion genetischen Assoziation unterliegt (Harrison und Owen 2003; Harrison und Weinberger 2005). Um zu verstehen, auf welche Art und Weise sich Polymorphismen in der DNA auf ein Protein auswirken, sind funktionelle Studien wichtig. Diese untersuchen z. B. eine veränderte Expression, welche auf unterschiedliche mRNA- und Proteinspiegel, strukturelle Veränderungen der primären RNA bzw. Proteinsequenz oder abnorme Proteinmodifikationen zurückgehen kann (Brzustowicz 2008). Ein wichtiger Bestandteil der Regulationsmechanismen ist die transkriptionale Kontrolle, bei der neben dem Promotor noch diverse andere regulatorische Elemente komplex miteinander interagieren. Viele Gene benötigen für die korrekte Expression diese sogenannten cis-regulatorischen Elemente, welche man auch als „enhancer“ oder „repressor“ bezeichnet und die Bindestellen für Transkriptionsfaktoren sind (de Laat und Grosveld 2003; Kleinjan und van Heyningen 2005). Sie können bis zu 1 Mb in 5’oder 3’-Richtung entfernt liegen und sich z. B. in Introns benachbarter Gene befinden (Kleinjan und van Heyningen 2005). Varianten im Promotorbereich oder aber auch SNPs in Introns können also solche regulatorischen Elemente betreffen und die Transkriptionsaktivität beeinflussen (Duan et al. 2003a; Greenwood und Kelsoe 2003; Harrison und Weinberger 2005). Synonyme, d. h. „stille“ SNPs in kodierenden Regionen und SNPs in Introns können darüber hinaus Splicing und mRNA-Stabilität, zwei weitere wichtige Regulatoren der Genexpression, verändern und so die Expression eines Gens beeinflussen. Auch SNPs in UTRs wirken sich auf mRNA-Stabilität aus und können zu eingeschränkter Translationsinitiation führen (Duan et al. 2003b). Zudem zeigen sich mitunter die Effekte einzelner Allele erst in Kombination miteinander und unter Berücksichtigung der Haplotypen (Duan et al. 2003a; Duan et al. 2003b; Harrison und Weinberger 2005). Der Nachteil an funktionellen Studien ist, dass es wahrscheinlich nicht ausreicht, sie nur in vitro oder als Tiermodelle durchzuführen, weil möglicherweise die Genregulation im menschlichen Gehirn einzigartig ist und somit weitaus aufwändigere post mortemStudien erfordert. Ein weiteres Problem ist, dass veränderte Expression eines Anfälligkeitsgens nicht nur auf eine genetische Variante im kodierenden Gen zurückgehen kann, sondern auch Veränderungen in anderen Genen beteiligt sein 71 5 Diskussion können, welche für Proteine kodieren, die am selben Signalweg bzw. Pathomechanismus beteiligt sind. Mehrere funktionelle Studien bestätigen die mögliche Bedeutung von NOS1AP insbesondere für die Krankheitsentstehung von Schizophrenie und analysieren den Mechanismus, über den sich genetische Polymorphismen auswirken könnten. In einer post mortem-Studie wurde von signifikant erhöhter mRNA-Expression der NOS1AP-Kurzform im dorsolateralen präfrontalen Kortex bei Patienten mit Schizophrenie und bipolar-affektiver Störung berichtet. Darüber hinaus fand sich bei den Varianten der SNPs, die im kanadischen Sample mit Schizophrenie assoziiert waren, eine höhere Expression der Kurzform von NOS1AP. Für die lange Isoform zeigten sich keine signifikanten Unterschiede. Interessanterweise war die Expression der Kurzform-mRNA nicht veränderbar durch antipsychotische Medikation, die Expression der langen Isoform hingegen schon, zumindest bei der bipolar-affektiven Störung. Die Überexpression von NOS1AP und die damit verbundene vermehrte Entkopplung der NOS-I vom NMDAR-Komplex wäre mit der NMDAR- Hypofunktionshypothese vereinbar (Xu et al. 2005). In einer weiterführenden Studie wurden bildgebende Untersuchungen an gesunden Probanden, die entsprechend der DNA-Sequenz in einem NOS1AP-SNP unterteilt wurden, durchgeführt. Dabei zeigten gesunde Träger des Risikoallels für Schizophrenie eine stärkere Aktivierung des DLPFC während der Bearbeitung von Aufgaben, die das Arbeitsgedächtnis involvierten. Möglicherweise deutet dies auf eine ineffektive Informationsverarbeitung bei Trägern des krankheitsassoziierten Allels hin (Callicott et al. 2007, wiedergegeben in Brzustowicz 2008). Wratten et al. analysierten in einer weiteren Studie eine Vielzahl an SNPs und evaluierten die drei am stärksten assoziierten SNPs, die miteinander in nahezu vollständigem Kopplungsungleichgewicht standen, hinsichtlich ihrer Funktion. Dabei war ein SNP (rs12742393) mit einer erhöhten Expression von NOS1AP im DLPFC und in neuronalen Zelllinien assoziiert, vermutlich vermittelt durch eine erhöhte Bindungsaffinität von Transkriptionsfaktoren. Dieser spezielle SNP wurde Erstaunlicherweise bisher führten die noch nicht beiden in anderen anderen SNPs Studien trotz untersucht. des hohen Kopplungsungleichgewichts nicht zu einer veränderten Genexpression (Wratten et al. 72 5 Diskussion 2009). Hinweise auf die Bedeutung einer solchen Überexpression liefert eine aktuelle Studie, die darstellen konnte, dass eine erhöhte Expression von NOS1AP zu einer reduzierten Anzahl und Verzweigung von Dendriten in kultivierten hippokampalen Neuronen führt. Während der langen Isoform hierbei vermutlich eine langfristige Bedeutung für Bildung, Wachstum und Erhalt der Dendriten zukommt, hat die kurze Isoform wahrscheinlich eher eine regulative Funktion bezüglich der Dendritenverzweigungen. Diese Entdeckung steht mit der Beobachtung verminderter dendritischer Ausbreitung bei Schizophrenie in Einklang (Carrel et al. 2009). Ein mögliches Modell wäre also eine Beeinflussung der dendritischen Verzweigungen durch NOS1AP während der neuronalen Entwicklung. 5.7 Neue Studienansätze Möglicherweise sind die klassischen, symptombasierten Krankheitsphänotypen zu komplex und heterogen und die Diagnose nach standardisierten Methoden in binärer Form, also betroffen vs. nicht-betroffen, zu simpel und mit zu großem Informationsverlust verbunden, um Kandidatengene zu identifizieren. Bei komplexen Krankheiten stellen Phänotypen die Zusammenfassung verschiedener Merkmale dar, die wiederum die Effekte multipler kausaler und modifizierender Faktoren sind. Studien, die mit sogenannten Endophänotypen arbeiten, versuchen, diesem Problem zu begegnen. Zu Grunde liegt die Annahme, dass die Dysfunktion in einem bestimmten neuronalen System weniger komplex ist als der Krankheitsphänotyp insgesamt. Endophänotypen stehen also unmittelbarer mit den genetisch basierten neurobiologischen Defiziten (Genexpression, Störung neuronaler Kreisläufe) in Verbindung als die Krankheit selber, da diese dem Einfluss vieler Kandidatengene und Dysfunktionen in verschiedenen neuronalen Systemen unterliegt (Braff et al. 2008; Cannon und Keller 2006). Da bestimmte Endophänotypen in mehreren Krankheiten vorkommen können, ist es außerdem möglich, pleiotrope Gene mit Auswirkungen auf mehrere Störungen zu identifizieren (Cannon und Keller 2006). Endophänotypen müssen gewisse Kriterien erfüllen: das Merkmal muss in der Population kausal mit der Krankheit assoziiert und nicht Folge einer Therapie sein, es muss erblich sein, darf nicht vom aktuellen 73 5 Diskussion Krankheitszustand abhängig sein, muss in Familien mit der Krankheit kosegregieren und bei nicht-betroffenen Familienmitgliedern häufiger vorkommen als in der übrigen Bevölkerung (Kennedy et al. 2003; Naqvi 2008; Petronis 2004). Als Endophänotyen für Schizophrenie eignen sich spezifische bildgebende sowie neurophysiologische, -kognitive oder -chemische Merkmale, die zuverlässig und objektiv messbar sind (Cannon und Keller 2006). Potentielle Kandidaten sind unter anderem neurophysiologische Parameter wie zentral inhibitorische Defizite, z. B. Abnormalitäten der Okulomotorik. Auf neurokognitiver Ebene kommen Einschränkungen der Aufmerksamkeit und Vigilanz sowie des deklarativen und verbal episodischen Gedächtnisses in Frage. Im Bereich der neuronalen Bildgebung sind Abnormalitäten in polaren und dorsolateralen präfrontalen Regionen sowie die Reduktion von temporalem kortikalen und hippokampalen Volumen interessante Parameter (Braff et al. 2008; Cannon und Keller 2006; Naqvi 2008). Einer der ersten Erfolge von Endophänotypenstudien in der Schizophrenieforschung ist die Identifikation eines SNPs in der Promotorregion des alpha-7-nikotinergen Rezeptors durch die Analyse der P50-Suppression, welche die Entwicklung spezifischer Rezeptoragonisten ermöglichte (Braff et al. 2008). Allerdings bringen Analysen von Endophänotypen auch Probleme mit sich. So muss unbedingt vermieden werden, dass generell akzeptierte Diagnosekriterien umgangen werden. Des Weiteren finden sich in vielen Studien unzureichende Beschreibungen der Endophänotypen, was an ihrer Zuverlässigkeit zweifeln lässt (Chanock et al. 2007). Ein anderer Forschungsschwerpunkt, der weitere wichtige Erkenntnisse verspricht, ist die Epigenetik. Mit diesem Begriff bezeichnet man Veränderungen der Genexpression, die nicht in der DNA selbst kodiert sind und dennoch sowohl meiotisch als auch mitotisch vererbt werden. Drei verschiedene Mechanismen bewirken epigenetische Regulation von Genen, nämlich DNA-Methylierung, RNA-vermitteltes „Ausschalten“ von Genen und Histonmodifikation (Egger et al. 2004). Sowohl für Schizophrenie als auch für die bipolar-affektive Erkrankung hat dieser Ansatz viel Beachtung gefunden (Petronis 2004). Epigenetische Faktoren im Zusammenspiel mit genetischer Anfälligkeit wären eine Erklärung für einige Merkmale der Krankheiten, die bisher nicht komplett verstanden sind. So ist die Rolle von Umweltfaktoren, die nicht immer 74 5 Diskussion phänotypische Unterschiede erklären können, umstritten, da sogar in isogenen Modellpopulationen trotz gleicher genetischer und äußerer Bedingungen eine phänotypische Variation persistieren kann (Cho et al. 2004). Außerdem ist unklar, warum sich das Krankheitsrisiko eines genetisch belasteten, adoptierten Kindes in seiner neuen, „gesunden“ Umgebung nicht verringert (Petronis 2004). Möglicherweise beruht auch die Diskordanz monozygoter Zwillinge, welche gleiches Erbmaterial aufweisen und ähnlichen Umgebungsfaktoren ausgesetzt sind, auf epigenetischen Mechanismen (Petronis 2003). Altersabhängige epigenetische Veränderungen wären eine Erklärung für das späte Ersterkrankungsalter und die epigenetische Dynamik könnte klinische Veränderungen wie Remission und Rückfälle begründen (Petronis 2003). Inzwischen gibt es Hinweise, dass epigenetische Mechanismen auf die Genexpression Einfluss nehmen und andauernde Veränderungen der Gehirnfunktion bewirken können (Tsankova et al. 2007). Besonders die Aktivität der Transkription ist von einer Interaktion der Transkriptionsfaktoren mit dem Chromatin abhängig und kann durch epigenetische Faktoren, wie Methylierung der Bindestellen oder Histonmodifikation, beeinflusst werden (Petronis 2003; Tsankova et al. 2007). Eine Hypothese zur Schizophrenie ist eine epigenetische Fehlfunktion, die zu Hypermethylierung und konsekutiver verminderter Genexpression in GABAergen Neuronen führt. Dies würde das Zusammenspiel mit anderen neuronalen Schaltkreisen behindern (Tsankova et al. 2007). Auch für einige andere Krankheiten, z. B. in der Krebsentstehung, sind epigenetische Prozesse von Bedeutung. Dementsprechend wird viel Hoffnung in die Entwicklung und Etablierung von Medikamenten gelegt, die epigenetische Faktoren beeinflussen. Durch Inhibitoren der DNA-Methylierung ist es möglich, Gene, die durch epigenetische Einflüsse ausgeschaltet sind, wieder zu induzieren. Auch HDACs (histone deacetylases), die Histone deacetylieren und auf diese Weise Gene ausschalten, können medikamentös inhibiert werden. So ist beispielsweise Valproat, welches bei bipolaraffektiv Erkrankten als Stimmungsstabilisator eingesetzt wird, ein HDAC-Inhibitor und kann die Transkription verschiedener Promotoren aktivieren (Göttlicher et al. 2001; Phiel et al. 2001). Selbstverständlich ist es zunächst unbedingt notwendig, zu wissen, welche Gene bei Schizophrenie und der bipolar-affektiven Erkrankung epigenetisch modifiziert werden, bevor man sichere Mittel findet, sie therapeutisch zu beeinflussen. 75 5 Diskussion 5.8 Sozioökonomische Folgen und Ausblick Schizophrenie und die bipolar affektive Erkrankung sind schwere mentale Erkrankungen, die bei Menschen zwischen 15 und 44 Jahren weltweit der acht- bzw. neunthäufigste Grund für behinderungsbedingt beeinträchtigte Lebensjahre sind. Sie führen zu einer Reduktion der Lebenserwartung und gehen mit einer erhöhten Suizidrate einher (World Health Organization 2001). Beide Erkrankungen führen zu einer Stigmatisierung und stellen neben dem persönlichen Schicksal des Betroffenen auch eine hohe sozioökonomische Bürde dar. In westlichen Ländern machen die direkten, also mit Diagnose, Therapie und Rehabilitation verbundenen Kosten für Schizophrenie mit 1,3-2,5% der gesamten Gesundheitsausgaben den größten Anteil an direkten Kosten für eine mentale Störung aus. Dazu kommen hohe indirekte Kosten durch Verlust an Produktivität des einzelnen Betroffenen bedingt durch Behinderung und vorzeitigen Tod (Rössler et al. 2005; Serretti et al. 2009). Die totalen Kosten für die bipolar-affektive Erkrankung werden in den USA auf 45 Mrd. $ pro Jahr (Zahlen für 1991) geschätzt (Wyatt und Henter 1995). Trotz der großen Belastung des Einzelnen und der gesamten Gesellschaft durch endogene Psychosen sind die Behandlungsmöglichkeiten teilweise eingeschränkt und bei einigen Patienten unzureichend. So liegt die allgemeine Rezidivrate SchizophrenieKranker im ersten Jahr nach Erstmanifestation trotz Behandlung bei ca. 40% (Müller 2004). Auch unter der Therapie mit Clozapin tritt eine Besserung der Symptome nur bei 30 - 60% der Betroffenen ein (Arranz et al. 2000). In Langzeitstudien zur bipolaraffektiven Erkrankung haben weniger als 50% der Patienten einen guten Behandlungserfolg gezeigt (Kleinman et al. 2003). Auch unter Dauertherapie mit Lithium liegt das Rezidivrisiko bei ca. 40% (Geddes et al. 2004). In einer jüngeren Studie über die Effektivität atypischer Neuroleptika und eines klassischen Neuroleptikums brachen ca. 70% der Probanden die Studie vorzeitig ab (Lieberman et al. 2005). Auch in einer älteren Metaanalyse lag die durchschnittliche Compliance-Rate bei antipsychotischer Medikation nur bei 58% (Wahlbeck et al. 2001). Dies betont die Notwendigkeit, das Ansprechen der Therapie und das Auftreten von Nebenwirkungen besser vorhersagen zu können, was das Ziel der Forschung nach spezifischen, auf genetischen Informationen basierenden Therapien ist (Nnadi und 76 5 Diskussion Malhotra 2007). Die Entdeckung, dass Patienten individuelle Reaktionen auf medikamentöse Behandlungen zeigen, ist die Grundlage der Pharmakogenetik. Diesbezüglich gibt es verschiedene Kandidatengene, die zum einen auf dem pharmakodynamischen und zum anderen auf dem pharmakokinetischen Ansatz basieren. Bei ersterem stehen hauptsächlich Medikamentenrezeptoren und deren nachgeschaltete Signalsysteme im Mittelpunkt, während letzterer vor allem metabolisierende Enzyme oder Proteine der Blut-Hirn-Schranke betrachtet (Nnadi und Malhotra 2007). Bisher wurden besonders Rezeptoren des dopaminergen und serotonergen Systems, zu denen Clozapin eine hohe Affinität hat, und das CytochromEnzym CYP2D6 untersucht (Malhotra et al. 2004; Reynolds 2007). Nicht nur das Ansprechen auf Medikamente ist individuell, auch das Auftreten von Nebenwirkungen wird wahrscheinlich von genetischen Mechanismen beeinflusst. Zu den Nebenwirkungen von Antipsychotika und Antidepressiva zählen Hyperprolaktinämie, extrapyramidalmotorische Symptome und anticholinerge Effekte bzw. Sedierung und Gewichtszunahme (Malhotra et al. 2004; Nnadi und Malhotra 2007; Reynolds 2007). Ziel der Forschung ist es, vorhersagen zu können, welcher Patient unter solchen, teilweise schweren Nebenwirkungen leiden wird und die Medikation dementsprechend anzupassen (Malhotra et al. 2004). So wird es zukünftig z. B. einen Test geben, der das Risiko von Clozapin-induzierter Agranulozytose durch den Nachweis des genetischen Markers HLA-DQB1 einschätzt (Nnadi und Malhotra 2007). Aufgrund des moderaten Effekts einzelner Gene auf die Medikamentenantwort sind genomweite Assoziationsstudien, in denen viele Gene bzw. SNPs untersucht werden, notwendig (Nnadi und Malhotra 2007). Die Replikation der Ergebnisse ist hier wiederum der entscheidende Faktor, um in Zukunft spezifischere Therapien entwickeln zu können (Malhotra et al. 2004). 77 6 Zusammenfassung 6 Zusammenfassung Schizophrenie und die bipolar-affektive Erkrankung sind mit einer Lebenszeitprävalenz von ca. 1% häufige psychiatrische Krankheitsbilder. Die genaue Ätiologie beider Krankheiten ist bisher noch nicht eindeutig geklärt, allerdings ist aus Familien-, Zwillings- und Adoptionsstudien bekannt, dass die genetische Komponente mit einer Heritabilität von jeweils ca. 80% die entscheidende Rolle spielt. Generell nimmt man für beide Krankheiten eine multifaktorielle Genese an, bei der eine genetische Anfälligkeit im Zusammenspiel mit Umweltfaktoren zur Krankheitsentstehung führt. Es bestehen für beide Krankheiten diverse pathophysiologische Modelle, besonders interessant ist dabei eine Dysregulation der Neurotransmitter. Neben Dopamin und GABA steht auch Glutamat, der häufigste exzitatorische Neurotransmitter im ZNS, im Verdacht, eine entscheidende Rolle bei der Entstehung der Schizophrenie zu spielen. Bei der bipolar-affektiven Erkrankung stehen besonders Veränderungen der monoaminergen Neurotransmission im Vordergrund. Eine Beteiligung des Glutamatsystems ist hier zwar weniger gut belegt, jedoch auch vorstellbar. Inzwischen sind sowohl für Schizophrenie als auch für die bipolar-affektive Erkrankung verschiedene Kandidatengene gefunden worden, die möglicherweise an der Krankheitsentstehung beteiligt sind. In dieser Arbeit wurde das Kandidatengen NOS1AP untersucht. NOS1AP interagiert mit der NOS-I und führt zu einer Translokation dieses Enzyms ins Zytosol, wodurch es dem Calciumeinstrom durch den NMDAR entzogen wird. Auf diese Weise ist es zu einem geringeren Grad aktiv und produziert weniger NO. NOS1AP ist also ein negativer Regulator der NOS-I. Diese funktionelle Verbindung mit dem NMDAR und der NOS-I, die beide im Verdacht stehen, an der Pathogenese der Schizophrenie beteiligt zu sein, machen NOS1AP zu einem interessanten Kandidatengen. Bestärkt wird dies durch positive Kopplungsbefunde für den Genlocus 1q22, auf dem das Gen für NOS1AP liegt. Auch in Assoziationsstudien konnte allelische und haplotypische Assoziation mit Schizophrenie innerhalb des NOS1AP-Gens nachgewiesen werden. Nicht zuletzt stammen auch aus funktionellen Studien Hinweise, die eine mögliche pathogenetische Bedeutung von NOS1AP untermauern. Bisher wurden keine Assoziationsstudien bezüglich NOS1AP bei Patienten mit bipolar-affektiver Erkrankung durchgeführt, allerdings wies eine 78 6 Zusammenfassung funktionelle Studie auf eine erhöhte Expression von NOS1AP bei betroffenen Patienten hin. In der vorliegenden Arbeit wurden 14 SNPs im Bereich des NOS1AP-Gens und daraus resultierende Haplotypen in einem unterfränkischen Fall-Kontroll-Sample untersucht. Dabei konnte für rs1538018, rs945713 und rs4306106 jeweils eine nominelle Assoziation mit Schizophrenie festgestellt werden. Auch nach Durchführung eines Permutationstestes blieb für rs1538018 und rs945713 mit p-Werten von 0,0924 bzw. 0,0788 ein statistischer Trend bestehen. Bei Betrachtung der Haplotypen ließ sich lediglich eine nominelle Assoziation des Haplotyps CTAGC (SNP 3–7) mit Schizophrenie nachweisen. Die geschlechtsspezifische Analyse ergab für die männlichen Patienten im Permutationstest eine grenzwertig signifikante Assoziation von rs1538018 und rs945713 mit p-Werten von 0,0706 bzw. 0,0588, während die Haplotypen TCGAT bzw. CTAGC aus Block 1 nur eine nominelle Assoziation zeigten. Bei den weiblichen Patienten ließ sich weder eine allelische noch eine haplotypische Assoziation nachweisen. Für die bipolar-affektive Erkrankung wurden keine Assoziationen, weder auf Einzelmarker- noch auf Haplotyp-Ebene festgestellt, eine Erklärung dafür ist die möglicherweise unzureichende Größe des Samples. Die grenzwertige Assoziation der SNPs mit Schizophrenie macht eine pathogenetische Beteiligung von NOS1AP an Schizophrenie denkbar. Beim Vergleich der vorliegenden Ergebnisse mit anderen Studien zeigen sich Überlappungen hinsichtlich der assoziierten SNPs. Manche Untersuchungen lieferten jedoch auch negative Befunde. Gründe dafür können die differierenden genetischen Hintergründe der Probanden sein, andererseits auch die phänotypische Heterogenität der Schizophrenie. Im Allgemeinen sind Assoziationsstudien mit Fall-Kontroll-Samples zwar geeignet, um Gene mit geringem Effekt aufzudecken, allerdings sind sie auch sehr anfällig für Populationsstratifikation, welche man auch bei der vorliegenden Studie nicht ausschließen kann. Dennoch sind Replikationsstudien von positiven Assoziationsbefunden notwendig, um besser einschätzen zu können, welchen Einfluss das einzelne untersuchte Gen tatsächlich für die Krankheitsentstehung hat. 79 7 Literaturverzeichnis 7 Literaturverzeichnis Aarnoudse AJ, Newton-Cheh C, de Bakker PI, Straus SM, Kors JA, Hofman A, Uitterlinden AG, Witteman JC, Stricker BH (2007): Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation 116(1), 10-16 Abi-Dargham A, Moore H (2003): Prefrontal DA transmission at D1 receptors and the pathology of schizophrenia. Neuroscientist 9(5), 404-416 Abou-Sleiman PM, Hanna MG, Wood NW (2006): Genetic association studies of complex neurological diseases. J Neurol Neurosurg Psychiatry 77(12), 13021304 Ackenheil M (2001): Neurotransmitters and signal transduction processes in bipolar affective disorders: a synopsis. J Affect Disord 62(1-2), 101-111 Adityanjee, Aderibigbe YA, Theodoridis D, Vieweg VR (1999): Dementia praecox to schizophrenia: the first 100 years. Psychiatry Clin Neurosci 53(4), 437-448 Alaerts M, Del-Favero J (2009): Searching genetic risk factors for schizophrenia and bipolar disorder: learn from the past and back to the future. Hum Mutat 30(8), 1139-1152 Albensi BC (2007): The NMDA receptor/ion channel complex: a drug target for modulating synaptic plasticity and excitotoxicity. Curr Pharm Des 13(31), 31853194 Alloy LB, Abramson LY, Urosevic S, Walshaw PD, Nusslock R, Neeren AM (2005): The psychosocial context of bipolar disorder: environmental, cognitive, and developmental risk factors. Clin Psychol Rev 25(8), 1043-1075 Andreasen NC (1999): A unitary model of schizophrenia: Bleuler's "fragmented phrene" as schizencephaly. Arch Gen Psychiatry 56(9), 781-787 Angst J (2007): The bipolar spectrum. Br J Psychiatry 190, 189-191 Angst J, Gamma A (2008): Diagnosis and course of affective psychoses: was Kraepelin right? Eur Arch Psychiatry Clin Neurosci 258 Suppl 2, 107-110 Arking DE, Pfeufer A, Post W, Kao WH, Newton-Cheh C, Ikeda M, West K, Kashuk C, Akyol M, Perz S et al. (2006): A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet 38(6), 644-651 Arranz MJ, Munro J, Birkett J, Bolonna A, Mancama D, Sodhi M, Lesch KP, Meyer JF, Sham P, Collier DA et al. (2000): Pharmacogenetic prediction of clozapine response. Lancet 355(9215), 1615-1616 Badner JA, Gershon ES (2002): Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry 7(4), 405-411 Barrett JC, Fry B, Maller J, Daly MJ (2005): Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21(2), 263-265 Bauer M, Pfennig A (2005): Epidemiology of bipolar disorders. Epilepsia 46 Suppl 4, 8-13 Baumann B, Normann C, Bielau H (2003): Neurobiologische Grundlagen bipolarer affektiver Erkrankungen. Nervenarzt 74(7), 607-623 Becker ML, Aarnoudse AJ, Newton-Cheh C, Hofman A, Witteman JC, Uitterlinden AG, Visser LE, Stricker BH (2008): Common variation in the NOS1AP gene is associated with reduced glucose-lowering effect and with increased mortality in users of sulfonylurea. Pharmacogenet Genomics 18(7), 591-597 80 7 Literaturverzeichnis Belmaker RH (2004): Bipolar disorder. N Engl J Med 351(5), 476-486 Belsham B (2001): Glutamate and its role in psychiatric illness. Hum Psychopharmacol 16(2), 139-146 Benes FM (2009): Neural circuitry models of schizophrenia: is it dopamine, GABA, glutamate, or something else? Biol Psychiatry 65(12), 1003-1005 Bernstein HG, Bogerts B, Keilhoff G (2005): The many faces of nitric oxide in schizophrenia. A review. Schizophr Res 78(1), 69-86 Berrettini W (2003): Evidence for shared susceptibility in bipolar disorder and schizophrenia. Am J Med Genet C Semin Med Genet 123C(1), 59-64 Berrettini WH (2000): Genetics of psychiatric disease. Annu Rev Med 51, 465-479 Berrettini WH (2001): The human genome: susceptibility loci. Am J Psychiatry 158(6), 865 Blum-Degen D, Heinemann T, Lan J, Pedersen V, Leblhuber F, Paulus W, Riederer P, Gerlach M (1999): Characterization and regional distribution of nitric oxide synthase in the human brain during normal ageing. Brain Res 834(1-2), 128-135 Boehning D, Snyder SH (2003): Novel neural modulators. Annu Rev Neurosci 26, 105131 Braff DL, Greenwood TA, Swerdlow NR, Light GA, Schork NJ (2008): Advances in endophenotyping schizophrenia. World Psychiatry 7(1), 11-18 Brieger P (2007): Bipolare affektive Störungen. Teil I: Diagnostik, Epidemiologie und Verlauf Fortschr Neurol Psychiatr 75(11), 673-682 Brugue E, Vieta E (2007): Atypical antipsychotics in bipolar depression: neurobiological basis and clinical implications. Prog Neuropsychopharmacol Biol Psychiatry 31(1), 275-282 Brunnhuber S, Frauenknecht S, Lieb K: Intensivkurs Psychiatrie und Psychotherapie. 5. Auflage; Elsevier, Urban und Fischer, München, Jena 2005 Brzustowicz LM (2008): NOS1AP in schizophrenia. Curr Psychiatry Rep 10(2), 158163 Brzustowicz LM, Hayter JE, Hodgkinson KA, Chow EW, Bassett AS (2002): Fine mapping of the schizophrenia susceptibility locus on chromosome 1q22. Hum Hered 54(4), 199-209 Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS (2000): Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21q22. Science 288(5466), 678-682 Brzustowicz LM, Simone J, Mohseni P, Hayter JE, Hodgkinson KA, Chow EW, Bassett AS (2004): Linkage disequilibrium mapping of schizophrenia susceptibility to the CAPON region of chromosome 1q22. Am J Hum Genet 74(5), 1057-1063 Burt DR, Creese I, Snyder SH (1976): Properties of [3H]haloperidol and [3H]dopamine binding associated with dopamine receptors in calf brain membranes. Mol Pharmacol 12(5), 800-812 Buselmaier W, Tariverdian G: Humangenetik. 4. Auflage; Springer, Heidelberg 2007 Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM (2007): Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8(10), 766-775 Cannon TD, Keller MC (2006): Endophenotypes in the genetic analyses of mental disorders. Annu Rev Clin Psychol 2, 267-290 Carrel D, Du Y, Komlos D, Hadzimichalis NM, Kwon M, Wang B, Brzustowicz LM, Firestein BL (2009): NOS1AP regulates dendrite patterning of hippocampal 81 7 Literaturverzeichnis neurons through a carboxypeptidase E-mediated pathway. J Neurosci 29(25), 8248-8258 Chang KC, Barth AS, Sasano T, Kizana E, Kashiwakura Y, Zhang Y, Foster DB, Marban E (2008): CAPON modulates cardiac repolarization via neuronal nitric oxide synthase signaling in the heart. Proc Natl Acad Sci U S A 105(11), 44774482 Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE et al. (2007): Replicating genotype-phenotype associations. Nature 447(7145), 655-660 Chiavegatto S, Dawson VL, Mamounas LA, Koliatsos VE, Dawson TM, Nelson RJ (2001): Brain serotonin dysfunction accounts for aggression in male mice lacking neuronal nitric oxide synthase. Proc Natl Acad Sci U S A 98(3), 12771281 Cho KS, Elizondo LI, Boerkoel CF (2004): Advances in chromatin remodeling and human disease. Curr Opin Genet Dev 14(3), 308-315 Chowdari KV, Mirnics K, Semwal P, Wood J, Lawrence E, Bhatia T, Deshpande SN, B KT, Ferrell RE, Middleton FA et al. (2002): Association and linkage analyses of RGS4 polymorphisms in schizophrenia. Hum Mol Genet 11(12), 1373-1380 Clinton SM, Meador-Woodruff JH (2004): Abnormalities of the NMDA Receptor and Associated Intracellular Molecules in the Thalamus in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology 29(7), 1353-1362 Cookson J (2005): Toward a clinical understanding of bipolar disorders: classification and presentation. Epilepsia 46 Suppl 4, 3-7 Coyle JT, Tsai G, Goff D (2003): Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann N Y Acad Sci 1003, 318-327 Craddock N, Forty L (2006): Genetics of affective (mood) disorders. Eur J Hum Genet 14(6), 660-668 Craddock N, Jones I (1999): Genetics of bipolar disorder. J Med Genet 36(8), 585-594 Craddock N, O'Donovan MC, Owen MJ (2005): The genetics of schizophrenia and bipolar disorder: dissecting psychosis. J Med Genet 42(3), 193-204 Crane GE (1961): The psychotropic effects of cycloserine: A new use for an antibiotic. Comprehensive Psychiatry 2(1), 51-59 Creese I, Burt DR, Snyder SH (1976): Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192(4238), 481483 de Laat W, Grosveld F (2003): Spatial organization of gene expression: the active chromatin hub. Chromosome Res 11(5), 447-459 Duan J, Sanders AR, Molen JE, Martinolich L, Mowry BJ, Levinson DF, Crowe RR, Silverman JM, Gejman PV (2003a): Polymorphisms in the 5'-untranslated region of the human serotonin receptor 1B (HTR1B) gene affect gene expression. Mol Psychiatry 8(11), 901-910 Duan J, Wainwright MS, Comeron JM, Saitou N, Sanders AR, Gelernter J, Gejman PV (2003b): Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum Mol Genet 12(3), 205216 Duman RS (2002): Synaptic plasticity and mood disorders. Mol Psychiatry 7 Suppl 1, S29-34 82 7 Literaturverzeichnis Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R et al. (2007): Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 447(7148), 1087-1093 Eastwood SL (2005): Does the CAPON gene confer susceptibility to schizophrenia? PLoS Med 2(10), e348 Egger G, Liang G, Aparicio A, Jones PA (2004): Epigenetics in human disease and prospects for epigenetic therapy. Nature 429(6990), 457-463 Erdely HA, Tamminga CA, Roberts RC, Vogel MW (2006): Regional alterations in RGS4 protein in schizophrenia. Synapse 59(8), 472-479 Erfurth A, Arolt V (2003): Das Spektrum bipolarer Erkrankungen. Nervenarzt 74(1), 55-70 Fang C, Tang W, Tang RQ, Wang L, Zhou GQ, Huang K, Li XW, Feng GY, He M, Du LZ et al. (2008): Family-based association studies of CAPON and schizophrenia in the Chinese Han population. Prog Neuropsychopharmacol Biol Psychiatry 32(5), 1210-1213 Fang M, Jaffrey SR, Sawa A, Ye K, Luo X, Snyder SH (2000): Dexras1: a G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron 28(1), 183-193 Faraone SV, Hwu HG, Liu CM, Chen WJ, Tsuang MM, Liu SK, Shieh MH, Hwang TJ, Ou-Yang WC, Chen CY et al. (2006): Genome scan of Han Chinese schizophrenia families from Taiwan: confirmation of linkage to 10q22.3. Am J Psychiatry 163(10), 1760-1766 Garcia JG, Ma SF (2005): Polymerase chain reaction: a landmark in the history of gene technology. Crit Care Med 33(12 Suppl), S429-432 Geddes JR, Burgess S, Hawton K, Jamison K, Goodwin GM (2004): Long-term lithium therapy for bipolar disorder: systematic review and meta-analysis of randomized controlled trials. Am J Psychiatry 161(2), 217-222 Gershon ES (2000): Bipolar illness and schizophrenia as oligogenic diseases: implications for the future. Biol Psychiatry 47(3), 240-244 Goff DC, Coyle JT (2001): The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 158(9), 1367-1377 Göttlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG et al. (2001): Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 20(24), 6969-6978 Greenwood TA, Kelsoe JR (2003): Promoter and intronic variants affect the transcriptional regulation of the human dopamine transporter gene. Genomics 82(5), 511-520 Griffin TJ, Smith LM (2000): Single-nucleotide polymorphism analysis by MALDITOF mass spectrometry. Trends Biotechnol 18(2), 77-84 Guix FX, Uribesalgo I, Coma M, Munoz FJ (2005): The physiology and pathophysiology of nitric oxide in the brain. Prog Neurobiol 76(2), 126-152 Gurling HM, Kalsi G, Brynjolfson J, Sigmundsson T, Sherrington R, Mankoo BS, Read T, Murphy P, Blaveri E, McQuillin A et al. (2001): Genomewide genetic linkage analysis confirms the presence of susceptibility loci for schizophrenia, on chromosomes 1q32.2, 5q33.2, and 8p21-22 and provides support for linkage to 83 7 Literaturverzeichnis schizophrenia, on chromosomes 11q23.3-24 and 20q12.1-11.23. Am J Hum Genet 68(3), 661-673 Guze SB, Robins E (1970): Suicide and primary affective disorders. Br J Psychiatry 117(539), 437-438 Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ et al. (2007): Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 357(9), 851-862 Harrison PJ (2002): The neuropathology of primary mood disorder. Brain 125(Pt 7), 1428-1449 Harrison PJ, Owen MJ (2003): Genes for schizophrenia? Recent findings and their pathophysiological implications. Lancet 361(9355), 417-419 Harrison PJ, Weinberger DR (2005): Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 10(1), 4068 Hata Y, Takai Y (1999): Roles of postsynaptic density-95/synapse-associated protein 90 and its interacting proteins in the organization of synapses. Cell Mol Life Sci 56(5-6), 461-472 Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P, BramonBosch E, Valmaggia L, Johns L, Broome M et al. (2009): Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry 66(1), 13-20 Hwu HG, Liu CM, Fann CS, Ou-Yang WC, Lee SF (2003): Linkage of schizophrenia with chromosome 1q loci in Taiwanese families. Mol Psychiatry 8(4), 445-452 Ioannidis JP, Ntzani EE, Trikalinos TA, Contopoulos-Ioannidis DG (2001): Replication validity of genetic association studies. Nat Genet 29(3), 306-309 Jaffrey SR, Benfenati F, Snowman AM, Czernik AJ, Snyder SH (2002): Neuronal nitric-oxide synthase localization mediated by a ternary complex with synapsin and CAPON. Proc Natl Acad Sci U S A 99(5), 3199-3204 Jaffrey SR, Snowman AM, Eliasson MJ, Cohen NA, Snyder SH (1998): CAPON: a protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron 20(1), 115-124 Jamison KR (1986): Suicide and bipolar disorders. Ann N Y Acad Sci 487, 301-315 Javitt DC (2004): Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry 9(11), 984-997, 979 Kato T (2001): Molecular genetics of bipolar disorder. Neurosci Res 40(2), 105-113 Kennedy JL, Farrer LA, Andreasen NC, Mayeux R, St George-Hyslop P (2003): The genetics of adult-onset neuropsychiatric disease: complexities and conundra? Science 302(5646), 822-826 Kim JS, Kornhuber HH, Schmid-Burgk W, Holzmuller B (1980): Low cerebrospinal fluid glutamate in schizophrenic patients and a new hypothesis on schizophrenia. Neurosci Lett 20(3), 379-382 Kiss JP, Vizi ES (2001): Nitric oxide: a novel link between synaptic and nonsynaptic transmission. Trends Neurosci 24(4), 211-215 Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST et al. (2005): Complement factor H polymorphism in age-related macular degeneration. Science 308(5720), 385-389 Kleinjan DA, van Heyningen V (2005): Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet 76(1), 8-32 84 7 Literaturverzeichnis Kleinman L, Lowin A, Flood E, Gandhi G, Edgell E, Revicki D (2003): Costs of bipolar disorder. Pharmacoeconomics 21(9), 601-622 Knable MB, Weinberger DR (1997): Dopamine, the prefrontal cortex and schizophrenia. J Psychopharmacol 11(2), 123-131 Kremeyer B, Garcia J, Kymalainen H, Wratten N, Restrepo G, Palacio C, Miranda AL, Lopez C, Restrepo M, Bedoya G et al. (2009): Evidence for a role of the NOS1AP (CAPON) gene in schizophrenia and its clinical dimensions: an association study in a South American population isolate. Hum Hered 67(3), 163-173 Kristiansen LV, Meador-Woodruff JH (2005): Abnormal striatal expression of transcripts encoding NMDA interacting PSD proteins in schizophrenia, bipolar disorder and major depression. Schizophr Res 78(1), 87-93 Kugaya A, Sanacora G (2005): Beyond monoamines: glutamatergic function in mood disorders. CNS Spectr 10(10), 808-819 Lander E, Kruglyak L (1995): Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11(3), 241-247 Laruelle M, Kegeles LS, Abi-Dargham A (2003): Glutamate, dopamine, and schizophrenia: from pathophysiology to treatment. Ann N Y Acad Sci 1003, 138-158 Lau CG, Zukin RS (2007): NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci 8(6), 413-426 Le Hellard S, Lee AJ, Underwood S, Thomson PA, Morris SW, Torrance HS, Anderson SM, Adams RR, Navarro P, Christoforou A et al. (2007): Haplotype analysis and a novel allele-sharing method refines a chromosome 4p locus linked to bipolar affective disorder. Biol Psychiatry 61(6), 797-805 Lehtinen AB, Newton-Cheh C, Ziegler JT, Langefeld CD, Freedman BI, Daniel KR, Herrington DM, Bowden DW (2008): Association of NOS1AP genetic variants with QT interval duration in families from the Diabetes Heart Study. Diabetes 57(4), 1108-1114 Levitt P, Ebert P, Mirnics K, Nimgaonkar VL, Lewis DA (2006): Making the case for a candidate vulnerability gene in schizophrenia: Convergent evidence for regulator of G-protein signaling 4 (RGS4). Biol Psychiatry 60(6), 534-537 Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, Williams NM, Schwab SG, Pulver AE, Faraone SV et al. (2003): Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: Schizophrenia. Am J Hum Genet 73(1), 34-48 Lewis DA, Levitt P (2002): Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci 25, 409-432 Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RS, Davis SM, Davis CE, Lebowitz BD et al. (2005): Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med 353(12), 1209-1223 Lunetta KL (2008): Genetic association studies. Circulation 118(1), 96-101 Maier W (2008): Common risk genes for affective and schizophrenic psychoses. Eur Arch Psychiatry Clin Neurosci 258 Suppl 2, 37-40 Malhotra AK, Murphy GM, Jr., Kennedy JL (2004): Pharmacogenetics of psychotropic drug response. Am J Psychiatry 161(5), 780-796 85 7 Literaturverzeichnis Manolio TA, Brooks LD, Collins FS (2008): A HapMap harvest of insights into the genetics of common disease. J Clin Invest 118(5), 1590-1605 McClellan JM, Susser E, King MC (2007): Schizophrenia: a common disease caused by multiple rare alleles. Br J Psychiatry 190, 194-199 McEwen BS (1999): Stress and hippocampal plasticity. Annu Rev Neurosci 22, 105122 McIntyre RS, Muzina DJ, Kemp DE, Blank D, Woldeyohannes HO, Lofchy J, Soczynska JK, Banik S, Konarski JZ (2008): Bipolar disorder and suicide: research synthesis and clinical translation. Curr Psychiatry Rep 10(1), 66-72 Michael N, Erfurth A, Ohrmann P, Gossling M, Arolt V, Heindel W, Pfleiderer B (2003): Acute mania is accompanied by elevated glutamate/glutamine levels within the left dorsolateral prefrontal cortex. Psychopharmacology (Berl) 168(3), 344-346 Miller SA, Dykes DD, Polesky HF (1988): A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16(3), 1215 Minton GO, Young AH, McQuade R, Fairchild G, Ingram CD, Gartside SE (2009): Profound changes in dopaminergic neurotransmission in the prefrontal cortex in response to flattening of the diurnal glucocorticoid rhythm: implications for bipolar disorder. Neuropsychopharmacology 34(10), 2265-2274 Miranda A, Garcia J, Lopez C, Gordon D, Palacio C, Restrepo G, Ortiz J, Montoya G, Cardeno C, Calle J et al. (2006): Putative association of the carboxy-terminal PDZ ligand of neuronal nitric oxide synthase gene (CAPON) with schizophrenia in a Colombian population. Schizophr Res 82(2-3), 283-285 Mirnics K, Middleton FA, Stanwood GD, Lewis DA, Levitt P (2001): Disease-specific changes in regulator of G-protein signaling 4 (RGS4) expression in schizophrenia. Mol Psychiatry 6(3), 293-301 Mueser KT, McGurk SR (2004): Schizophrenia. Lancet 363(9426), 2063-2072 Müller N (2004): Mechanisms of relapse prevention in schizophrenia. Pharmacopsychiatry 37 Suppl 2, S141-147 Murray RM, Lewis SW (1987): Is schizophrenia a neurodevelopmental disorder? Br Med J (Clin Res Ed) 295(6600), 681-682 Naqvi HA (2008): Schizophrenia: a concept. J Pak Med Assoc 58(3), 133-137 Nedvetsky PI, Sessa WC, Schmidt HH (2002): There's NO binding like NOS binding: protein-protein interactions in NO/cGMP signaling. Proc Natl Acad Sci U S A 99(26), 16510-16512 Nnadi CU, Malhotra AK (2007): Individualizing antipsychotic drug therapy in schizophrenia: the promise of pharmacogenetics. Curr Psychiatry Rep 9(4), 313318 Oswald P, Souery D, Kasper S, Lecrubier Y, Montgomery S, Wyckaert S, Zohar J, Mendlewicz J (2007): Current issues in bipolar disorder: a critical review. Eur Neuropsychopharmacol 17(11), 687-695 Petronis A (2003): Epigenetics and bipolar disorder: new opportunities and challenges. Am J Med Genet C Semin Med Genet 123C(1), 65-75 Petronis A (2004): The origin of schizophrenia: genetic thesis, epigenetic antithesis, and resolving synthesis. Biol Psychiatry 55(10), 965-970 Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS (2001): Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276(39), 36734-36741 86 7 Literaturverzeichnis Post W, Shen H, Damcott C, Arking DE, Kao WH, Sack PA, Ryan KA, Chakravarti A, Mitchell BD, Shuldiner AR (2007): Associations between genetic variants in the NOS1AP (CAPON) gene and cardiac repolarization in the old order Amish. Hum Hered 64(4), 214-219 Prasad KM, Chowdari KV, Nimgaonkar VL, Talkowski ME, Lewis DA, Keshavan MS (2005): Genetic polymorphisms of the RGS4 and dorsolateral prefrontal cortex morphometry among first episode schizophrenia patients. Mol Psychiatry 10(2), 213-219 Pulver AE (2000): Search for schizophrenia susceptibility genes. Biol Psychiatry 47(3), 221-230 Puri V, McQuillin A, Choudhury K, Datta S, Pimm J, Thirumalai S, Krasucki R, Lawrence J, Quested D, Bass N et al. (2007): Fine mapping by genetic association implicates the chromosome 1q23.3 gene UHMK1, encoding a serine/threonine protein kinase, as a novel schizophrenia susceptibility gene. Biol Psychiatry 61(7), 873-879 Puri V, McQuillin A, Datta S, Choudhury K, Pimm J, Thirumalai S, Krasucki R, Lawrence J, Quested D, Bass N et al. (2008): Confirmation of the genetic association between the U2AF homology motif (UHM) kinase 1 (UHMK1) gene and schizophrenia on chromosome 1q23.3. Eur J Hum Genet 16(10), 1275-1282 Puri V, McQuillin A, Thirumalai S, Lawrence J, Krasucki R, Choudhury K, Datta S, Kerwin S, Quested D, Bass N et al. (2006): Failure to confirm allelic association between markers at the CAPON gene locus and schizophrenia in a British sample. Biol Psychiatry 59(2), 195-197 Reif A, Herterich S, Strobel A, Ehlis AC, Saur D, Jacob CP, Wienker T, Topner T, Fritzen S, Walter U et al. (2006): A neuronal nitric oxide synthase (NOS-I) haplotype associated with schizophrenia modifies prefrontal cortex function. Mol Psychiatry 11(3), 286-300 Reynolds GP (2007): The impact of pharmacogenetics on the development and use of antipsychotic drugs. Drug Discov Today 12(21-22), 953-959 Rodriguez-Murillo L, Greenberg DA (2008): Genetic association analysis: a primer on how it works, its strengths and its weaknesses. Int J Androl 31(6), 546-556 Rosa A, Fananas L, Cuesta MJ, Peralta V, Sham P (2002): 1q21-q22 locus is associated with susceptibility to the reality-distortion syndrome of schizophrenia spectrum disorders. Am J Med Genet 114(5), 516-518 Rössler W, Salize HJ, van Os J, Riecher-Rössler A (2005): Size of burden of schizophrenia and psychotic disorders. Eur Neuropsychopharmacol 15(4), 399409 Rybakowski JK, Twardowska K (1999): The dexamethasone/corticotropin-releasing hormone test in depression in bipolar and unipolar affective illness. J Psychiatr Res 33(5), 363-370 Sanacora G, Zarate CA, Krystal JH, Manji HK (2008): Targeting the glutamatergic system to develop novel, improved therapeutics for mood disorders. Nat Rev Drug Discov 7(5), 426-437 Sapolsky RM (2000): Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 57(10), 925-935 Sauer S (2007): The essence of DNA sample preparation for MALDI mass spectrometry. J Biochem Biophys Methods 70(2), 311-318 87 7 Literaturverzeichnis Schmider J, Lammers CH, Gotthardt U, Dettling M, Holsboer F, Heuser IJ (1995): Combined dexamethasone/corticotropin-releasing hormone test in acute and remitted manic patients, in acute depression, and in normal controls: I. Biol Psychiatry 38(12), 797-802 Schosser A, Aschauer HN (2004): Auf der Suche nach Vulnerabilitätsgenen der Schizophrenie. Wien Klin Wochenschr 116(24), 827-833 Seeman P, Chau-Wong M, Tedesco J, Wong K (1975): Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Natl Acad Sci U S A 72(11), 4376-4380 Seeman P, Lee T (1975): Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science 188(4194), 12171219 Segurado R, Detera-Wadleigh SD, Levinson DF, Lewis CM, Gill M, Nurnberger JI, Jr., Craddock N, DePaulo JR, Baron M, Gershon ES et al. (2003): Genome scan meta-analysis of schizophrenia and bipolar disorder, part III: Bipolar disorder. Am J Hum Genet 73(1), 49-62 Serretti A, Mandelli L, Bajo E, Cevenini N, Papili P, Mori E, Bigelli M, Berardi D (2009): The socio-economical burden of schizophrenia: a simulation of costoffset of early intervention program in Italy. Eur Psychiatry 24(1), 11-16 Shastry BS (2005): Bipolar disorder: an update. Neurochem Int 46(4), 273-279 Shaw SH, Kelly M, Smith AB, Shields G, Hopkins PJ, Loftus J, Laval SH, Vita A, De Hert M, Cardon LR et al. (1998): A genome-wide search for schizophrenia susceptibility genes. Am J Med Genet 81(5), 364-376 Shinkai T, Ohmori O, Hori H, Nakamura J (2002): Allelic association of the neuronal nitric oxide synthase (NOS1) gene with schizophrenia. Mol Psychiatry 7(6), 560-563 Slatkin M (2008): Linkage disequilibrium--understanding the evolutionary past and mapping the medical future. Nat Rev Genet 9(6), 477-485 Stahl SM (2007): The genetics of schizophrenia converge upon the NMDA glutamate receptor. CNS Spectr 12(8), 583-588 Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, Fossdal R, Sigurdsson E, Sigmundsson T, Buizer-Voskamp JE et al. (2008): Large recurrent microdeletions associated with schizophrenia. Nature 455(7210), 232236 Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, Gunnarsdottir S, Ivarsson O, Chou TT et al. (2002): Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet 71(4), 877892 Stone JM, Morrison PD, Pilowsky LS (2007): Glutamate and dopamine dysregulation in schizophrenia--a synthesis and selective review. J Psychopharmacol 21(4), 440452 Strachan T, Read A: Molekulare Humangenetik. 3. Auflage; Elsevier, Spektrum, Akad. Verl., München, Heidelberg 2005 Suarez BK, Duan J, Sanders AR, Hinrichs AL, Jin CH, Hou C, Buccola NG, Hale N, Weilbaecher AN, Nertney DA et al. (2006): Genomewide linkage scan of 409 European-ancestry and African American families with schizophrenia: suggestive evidence of linkage at 8p23.3-p21.2 and 11p13.1-q14.1 in the combined sample. Am J Hum Genet 78(2), 315-333 88 7 Literaturverzeichnis Talkowski ME, Chowdari K, Lewis DA, Nimgaonkar VL (2006): Can RGS4 polymorphisms be viewed as credible risk factors for schizophrenia? A critical review of the evidence. Schizophr Bull 32(2), 203-208 Tandon R, Keshavan MS, Nasrallah HA (2008a): Schizophrenia, "just the facts" what we know in 2008. 2. Epidemiology and etiology. Schizophr Res 102(1-3), 1-18 Tandon R, Keshavan MS, Nasrallah HA (2008b): Schizophrenia, "just the facts": what we know in 2008 part 1: overview. Schizophr Res 100(1-3), 4-19 Tandon R, Nasrallah HA, Keshavan MS (2009): Schizophrenia, "just the facts" 4. Clinical features and conceptualization. Schizophr Res 110(1-3), 1-23 Thomas DC, Witte JS (2002): Point: population stratification: a problem for casecontrol studies of candidate-gene associations? Cancer Epidemiol Biomarkers Prev 11(6), 505-512 Tölle R, Windgassen K: Psychiatrie. 14. Auflage; Springer, Berlin, Heidelberg, New York 2006 Toro C, Deakin JF (2005): NMDA receptor subunit NRI and postsynaptic protein PSD95 in hippocampus and orbitofrontal cortex in schizophrenia and mood disorder. Schizophr Res 80(2-3), 323-330 Tosato S, Dazzan P, Collier D (2005): Association between the neuregulin 1 gene and schizophrenia: a systematic review. Schizophr Bull 31(3), 613-617 Tost J, Gut IG (2002): Genotyping single nucleotide polymorphisms by mass spectrometry. Mass Spectrom Rev 21(6), 388-418 Tost J, Gut IG (2005): Genotyping single nucleotide polymorphisms by MALDI mass spectrometry in clinical applications. Clin Biochem 38(4), 335-350 Tsankova N, Renthal W, Kumar A, Nestler EJ (2007): Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci 8(5), 355-367 Tsuang M (2000): Schizophrenia: genes and environment. Biol Psychiatry 47(3), 210220 Vawter MP, Freed WJ, Kleinman JE (2000): Neuropathology of bipolar disorder. Biol Psychiatry 48(6), 486-504 Wahlbeck K, Tuunainen A, Ahokas A, Leucht S (2001): Dropout rates in randomised antipsychotic drug trials. Psychopharmacology (Berl) 155(3), 230-233 Watson S, Gallagher P, Ritchie JC, Ferrier IN, Young AH (2004): Hypothalamicpituitary-adrenal axis function in patients with bipolar disorder. Br J Psychiatry 184, 496-502 White TJ, Arnheim N, Erlich HA (1989): The polymerase chain reaction. Trends Genet 5(6), 185-189 World Health Organization: The World health report 2001. Mental health - new understanding, new hope. World Health Organization, Genf 2001 Wratten NS, Memoli H, Huang Y, Dulencin AM, Matteson PG, Cornacchia MA, Azaro MA, Messenger J, Hayter JE, Bassett AS et al. (2009): Identification of a schizophrenia-associated functional noncoding variant in NOS1AP. Am J Psychiatry 166(4), 434-441 Wyatt RJ, Henter I (1995): An economic evaluation of manic-depressive illness--1991. Soc Psychiatry Psychiatr Epidemiol 30(5), 213-219 Xu B, Wratten N, Charych EI, Buyske S, Firestein BL, Brzustowicz LM (2005): Increased expression in dorsolateral prefrontal cortex of CAPON in schizophrenia and bipolar disorder. PLoS Med 2(10), e263 89 7 Literaturverzeichnis Zarate CA, Jr., Du J, Quiroz J, Gray NA, Denicoff KD, Singh J, Charney DS, Manji HK (2003): Regulation of cellular plasticity cascades in the pathophysiology and treatment of mood disorders: role of the glutamatergic system. Ann N Y Acad Sci 1003, 273-291 Zheng Y, Li H, Qin W, Chen W, Duan Y, Xiao Y, Li C, Zhang J, Li X, Feng G et al. (2005): Association of the carboxyl-terminal PDZ ligand of neuronal nitric oxide synthase gene with schizophrenia in the Chinese Han population. Biochem Biophys Res Commun 328(4), 809-815 90 8 Abbildungsverzeichnis 8 Abbildungsverzeichnis Abb. 1.1: Schematische Darstellung des NMDA-Rezeptors (Albensi 2007)............ 12 Abb. 1.2: Genomische Konfiguration der langen (oben) bzw. kurzen Isoform (unten) von NOS1AP. Darstellung der Exons durch nummerierte Balken (nach Xu et al. 2005) ................................................................................................. 26 Abb. 1.3: Interaktion der NOS-I mit NOS1AP und weiteren Adaptorproteinen (nach Nedvetsky et al. 2002) ............................................................................... 27 Abb. 3.1: Schematische Darstellung eines MALDI-ToF Massenspektrometers (Sauer 2007) .......................................................................................................... 44 Abb. 4.1: LD Struktur Schizophrenie Gesamtstichprobe (D’) .................................. 46 Abb. 4.2: LD Struktur bipolar-affektive Störung Gesamtstichprobe (D’) ................ 48 Abb. 5.1: Assoziation der untersuchten SNPs (rot) und SNPs anderer, ausgewählter Studien mit Schizophrenie. Umgekehrt logarithmische Auftragung (signifikant: -log(p;10) > 1,3) .................................................................... 64 Abb. 10.1: LD Struktur Schizophrenie Gesamtstichprobe (D’),vergrößerter Block 1 95 Abb. 10.2: LD Struktur Schizophrenie, Männer (D’).................................................. 95 Abb. 10.3: LD Struktur Schizophrenie, Frauen (D’) ................................................... 96 91 9 Tabellenverzeichnis 9 Tabellenverzeichnis Tab. 4.1: Schizophrenie Gesamtstichprobe - Einzelmarker...................................... 49 Tab. 4.2: Schizophrenie Gesamtstichprobe - Haplotypen ........................................ 50 Tab. 4.3: Schizophrenie Gesamtstichprobe – Haplotypen (vergrößerter Block 1) ... 51 Tab. 4.4: Schizophrenie Männer - Einzelmarker ...................................................... 52 Tab. 4.5: Schizophrenie Männer - Haplotypen ......................................................... 54 Tab. 4.6: Schizophrenie Frauen - Einzelmarker ....................................................... 55 Tab. 4.7: Schizophrenie Frauen - Haplotypen .......................................................... 56 Tab. 4.8: Bipolar-affektive Erkrankung Gesamtstichprobe - Einzelmarker ............. 57 Tab. 4.9: Bipolar-affektive Erkrankung Gesamtstichprobe - Haplotypen ................ 59 Tab. 10.1: HWE und MAF Schizophrenie Gesamtstichprobe – Einzelmarker .......... 93 Tab. 10.2: HWE und MAF Schizophrenie Männer - Einzelmarker ........................... 93 Tab. 10.3: HWE und MAF Schizophrenie Frauen - Einzelmarker ............................ 94 Tab. 10.4: HWE und MAF Bipolar-affektive Erkrankung Gesamtstichprobe Einzelmarker.............................................................................................. 94 92 10 Anhang 10 Anhang Hardy-Weinberg-Equilibrium und minore Allelfrequenz der untersuchten SNPs Tab. 10.1: HWE und MAF Schizophrenie Gesamtstichprobe – Einzelmarker SNP Nr. Marker 1 2 3 4 5 6 7 9 10 11 12 13 14 rs1572495 rs1538018 rs945713 rs1415263 rs4306106 rs3924139 rs4145621 rs1508263 rs3751284 rs7521206 rs905721 rs348624 rs1964052 Hardy-Weinberg-Equilibrium p-Wert 0,214 0,4001 0,7768 1,0 0,8917 0,9071 0,2199 0,8126 0,4479 0,76 0,5477 0,0065 1,0 Minore Allelfrequenz 0,096 0,268 0,41 0,394 0,214 0,359 0,454 0,452 0,356 0,343 0,344 0,102 0,124 Tab. 10.2: HWE und MAF Schizophrenie Männer - Einzelmarker SNP Nr. Marker 1 2 3 4 5 6 7 9 10 11 12 13 14 rs1572495 rs1538018 rs945713 rs1415263 rs4306106 rs3924139 rs4145621 rs1508263 rs3751284 rs7521206 rs905721 rs348624 rs1964052 Hardy-Weinberg-Equilibrium p-Wert 0,1417 0,3624 0,7458 0,2217 0,1811 0,4645 0,4324 0,7081 0,7398 0,8447 0,8042 0,1767 0,4621 93 Minore Allelfrequenz 0,092 0,277 0,418 0,405 0,217 0,377 0,453 0,469 0,377 0,339 0,344 0,092 0,12 10 Anhang Tab. 10.3: HWE und MAF Schizophrenie Frauen - Einzelmarker SNP Nr. Marker 1 2 3 4 5 6 7 9 10 11 12 13 14 rs1572495 rs1538018 rs945713 rs1415263 rs4306106 rs3924139 rs4145621 rs1508263 rs3751284 rs7521206 rs905721 rs348624 rs1964052 Hardy-Weinberg-Equilibrium p-Wert 0,9734 0,9151 0,3618 0,2303 0,2844 0,6042 0,4263 0,3719 0,555 0,8949 0,6115 0,0719 0,3371 Minore Allelfrequenz 0,1 0,258 0,401 0,381 0,21 0,339 0,456 0,433 0,332 0,347 0,345 0,113 0,128 Tab. 10.4: HWE und MAF Bipolar-affektive Erkrankung Gesamtstichprobe - Einzelmarker SNP Nr. Marker 1 2 3 4 5 6 7 9 10 11 12 13 14 rs1572495 rs1538018 rs945713 rs1415263 rs4306106 rs3924139 rs4145621 rs1508263 rs3751284 rs7521206 rs905721 rs348624 rs1964052 Hardy-Weinberg-Equilibrium p-Wert 0,0777 1,0 0,4448 0,6583 0,7126 0,5669 0,0522 0,5662 0,0972 0,8679 0,8893 0,0513 0,3907 94 Minore Allelfrequenz 0,094 0,247 0,381 0,375 0,203 0,337 0,442 0,451 0,354 0,349 0,35 0,097 0,128 10 Anhang Kopplungsungleichgewicht Schizophrenie Abb. 10.1: LD Struktur Schizophrenie Gesamtstichprobe (D’), vergrößerter Block 1 Abb. 10.2: LD Struktur Schizophrenie, Männer (D’) 95 10 Anhang Abb. 10.3: LD Struktur Schizophrenie, Frauen (D’) 96 Danksagung Ganz besonders möchte ich Herrn Prof. Dr. A. Reif für die Überlassung des Themas und die sehr gute Betreuung meiner Doktorarbeit danken. Er sorgte stets für ein nettes Arbeitsklima und hat mir mit seinen Anregungen bei der Verfassung dieser Arbeit sehr geholfen. Herrn Prof. Dr. J. Deckert danke ich für die Möglichkeit, diese Promotionsarbeit in seiner Klinik durchführen zu können. Herrn Priv.-Doz. Dr. C. Kleinschnitz danke ich für die Übernahme des Korreferates. Ich bedanke mich bei Frau Prof. Dr. C. Freitag für die Erstellung der Statistik. Mein herzlicher Dank gilt außerdem Frau T. Töpner, die den experimentellen Teil meiner Arbeit sehr gut betreute und mir im Labor jederzeit mit Rat und Tat zur Seite stand. Von ganzem Herzen danke ich meinen Eltern, die mich immer unterstützt und gefördert haben. Mein größter Dank gilt Matthias für seine unermüdlichen Aufmunterungen und seinen Rückhalt bei der Verfassung dieser Arbeit und weit darüber hinaus. Lebenslauf Persönliche Daten Name: Alexandra Kramer Geburtstag: 04.01.1983 Geburtsort: Göttingen Staatsangehörigkeit: deutsch Familienstand: ledig Schulbildung 08/1989 – 07/1993 Albani-Grundschule Göttingen 08/1993 – 07/1995 OS Lutherschule Göttingen 08/1995 – 06/2002 Theodor-Heuss-Gymnasium Göttingen Abschluss: Abitur Studium 10/2002 – 06/2009 Humanmedizin, Julius-Maximilians-Universität Würzburg 09/2004 1. Teil der ärztlichen Prüfung/Physikum 07/2009 2. Teil der ärztlichen Prüfung Beruf Seit 11/2009 Assistenzärztin Pädiatrie, Hannoversche Kinderheilanstalt: Kinderkrankenhaus auf der Bult, Hannover bzw. Klinikum Neustadt am Rübenberge