Kristallstruktur und Schwingungsspektrum des Tetrammingold(III
Werbung

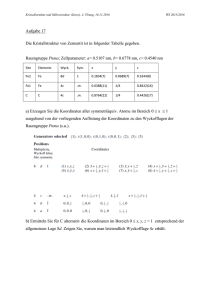
Kristallstruktur und Schwingungsspektrum des Tetrammingold(III)-nitrats Crj'stal Structure and Vibrational Spectrum of Tetraamminegold(III)-Nitrate MARTIN WEISHAUPT u n d JOACHIM STRÄHLE Institut für Anorganische Chemie der Universität Tübingen (Z. Naturforsch. 31b, 554-557 [1976]; eingegangen am 27. Januar 1976) Tetraamminegold(III) Nitrate, X-ray, Crystal Structure, IR, Raman [Au(NH3) 4 ](NO3crystallizes in the orthorhombic space group Cmmm, a = 7.243, b= 13:805, c = 5.302 Ä, Z = 2. The crystal structure is built up by NC>3~ anions and square planar [Au(NH3)4]3+ cations with Au-N bond lengths of 2.02 Ä. The vibrational spectra of the compound are discussed. Im Rahmen von Arbeiten über Gold-StickstoffVerbindungen haben wir uns auch für das Tetrammingold(III)-nitrat interessiert und an Einkristallen die Kristallstruktur der Verbindung bestimmt. [Au(NH 3 ) 4 ](N0 3 )3 wurde erstmals 1915 von E. W E I T Z 1 dargestellt und untersucht. Neuere Untersuchungen befassen sich insbesondere mit dem Elektronenspektrum 2 und der Stabilität 3 - 4 des Tetrammingold (III)-ions. Darstellung der Einkristalle und Strukturbestimmung Die Darstellung des Tetrammingold(III)-nitrats erfolgte in Anlehnung an das von SKIBSTEDT und B J E R R U M 3 - 4 beschriebene Verfahren aus einer mit NH 4 N03 gesättigten HAuCl4-Lösung, in die wir unter genauer pH-Wert-Kontrolle langsam NH 3 Gas einleiteten. Zentimeterlange Einkristalle erhielten wir durch Umkristallisieren der Verbindung in Wasser. Aufgrund der auf Einkristallaufnahmen beobachteten orthorhombischen Symmetrie und der integralen Auslöschung, hkl nur für h-\-k = 2n vorhanden, kommen folgende Raumgruppen in Frage: C222, Cmm2, C 2 mm und Cmmm, von denen im weiteren Verlauf der Strukturbestimmung die Raumgruppe Cmmm als die wahrscheinlichste angenommen wurde. Sonderdruckanforderungen an Prof. Dr. J. STRÄHLE, Institut für Anorganische Chemie der Universität Tübingen, Auf der Morgenstelle 18, D-7400 Tübingen. Eine Verfeinerung der Gitterkonstanten an Hand von 33 koinzidenzfreien Linien einer Guinieraufnahme nach der Methode der kleinsten Fehlerquadrate ergab folgende Werte: a = 7,243 ± 0,002 Ä, b = 13,805 ± 0,006 Ä, c = 5,302 ± 0,002 Ä. Die Abschätzung des Volumenbedarfs einer Formeleinheit [Au(NH 3 ) 4 ](N0 3 )3 mit Hilfe der BiLTz'schen Volumeninkremente5 führte zur Annahme von zwei Formeleinheiten pro Elementarzelle und der berechneten Dichte von ^ = 2,81 g/ cm 3 . Für die Strukturbestimmung wurden mit monochromatischer MoK a -Strahlung bis zu einem Beugungswinkel von 0 = 30° 703 symmetrieunabhängige Reflexe gemessen. Die Größe des hierzu verwendeten Kristalls betrug 0,06 x 0,04 x 0,02 mm. Die Lage der Goldatome und der Ammin-Stickstoffatome entnahmen wir einer dreidimensionalen Patterson-Synthese. Die Verfeinerung dieser Teilstruktur, die die Nitratgruppen noch nicht enthielt, führte unter Benutzung anisotroper Temperaturfaktoren für die Goldatome in der Raumgruppe Cmmm auf einen R-Wert von 10,7%. Zur Bestimmung der Lage der Nitratgruppen berechneten wir eine Differenz-Fouriersynthese, aus der vier der sechs in der Elementarzelle vorhandenen Nitratgruppen eindeutig entnommen werden konnten, und zwar mit N(2) und 0(1) in der Punktlage 4j und Unauthenticated Download Date | 8/20/17 8:47 AM M. WEISHAUPT-J. STRÄHLE • KRISTALLSTRUKTUR VON Au(NH3)4(N03)3 555 Tab. I Atomkoordinatsn von. [Au (NH3)4](N03)3 in der Raumgruppe Cmmm sowie individuelle isotrope und anisotrope Temperaturparameter der Atome. Die in Klammern angegebenen Standardabweichungen beziehen sich aul die letzten Stellen der Zahlenwerte. Die Komponenten ßij des Schwingungstensors sind als 104-fache Werte aufgeführt. Die Parameter B beruhen auf dem Temperaturfaktor der Form exp(—B • sin2 0/A2), die Parameter ß\j auf dem Ansatz exp[—(ßn • h 2 + ß^ • Je2 + ß33 • Z2)]. Für das Gold ist der Parameter B nach H A M I L T O N 6 aus den Werten ßij berechnet worden. Atom Punkt läge Au N(l) N(2) 0(1) 0(2) N(3) 0(3) 0(4) 2a 8p 4j 4j 8 n 4 h* 4 h* 8 0* X y 2 B 0 0,195(2) 0 0 0 0,456(7) 0,298(8) 0,531(6) 0 0,105(1) 0,233(1) 0,140(2) 0,274(1) 0 0 0 0 0 0,5 0,5 0,713(4) 0,5 0,5 0,729(7) 1,4 2,5(2) 2,3(3) 3,7(4) 4,2(3) 3,3(8) 4,5(6) 4,1(8) ßn [Ä2] ß%2 69(2) 23(1) /S33 97(3) * Statistisch halbe Besetzung. 0(2) in der Punktlage 8n. Die noch fehlenden zwei Nitratgruppen waren jeweils mit halbem Gewicht doppelt vorhanden [N(3) und 0(3): Punktlage 4h, 0(4): Punktlage 80]. Diese Tatsache bedeutet, daß entweder für diese zwei Nitratgruppen eine statistisch halbe Besetzung in der bisher angenommenen Raumgruppe Cmmm vorliegt oder ein Symmetrieabbau zur Raumgruppe C2mm vorgenommen werden muß. Diese Möglichkeiten wurden mit Hilfe einer Parameterverfeinerung überprüft. Die Rechnung in der zentrosymmetrischen Raumgruppe Cmmm ergab dabei einen R-Wert von 6,3%, während im Fall der nichtzentrosymmetrischen Raumgruppe C2mm der i?-Wert 5,7% betrug. Dennoch nehmen wir an, daß die Struktur in der Raumgruppe Cmmm beschrieben werden muß, da die Verfeinerung nur unter dieser Voraussetzung zu einer sinnvollen Geometrie der Nitratgruppen führt und außerdem die Standardabweichungen der Abstände und Winkel hierbei um etwa den Faktor 10 besser sind. Entsprechend beziehen sich auch die in Tab. I angegebenen Ortsparameter und Temperaturfaktoren der Atome auf die Raumgruppe Cmmm. Diskussion der Struktur Wie aus Abb. 1 hervorgeht, ist die Kristallstruktur von [Au(NH3)4](N03)3 aus [Au(NH 3 ) 4 ] 3+ -Kationen und N03~-Anionen aufgebaut. Die Tetrammingold- + öco 0(1) N(2) 0(2) QpC CXO—Ö OX>X OXD Abb. 1. Darstellung der Kristallstruktur des [Au(NH3)4](N03)3 in Projektion parallel [001]. Die Kreise stellen in der Reihenfolge zunehmender Größe die Atome Gold, Stickstoff und Sauerstoff dar. Um die [Au(NH3)4]3+Gruppen besser hervorzuheben, sind die dazugehörigen Atome in der Höhe z = 1,0 eingezeichnet worden. Unauthenticated Download Date | 8/20/17 8:47 AM M. WEISHAUPT-J. STRÄHLE • KRISTALLSTRUKTUR VON Au(NH 3 ) 4 (N0 3 )3 556 556 (III)-Gruppen sind parallel (001) angeordnet und längs der c-Achse translatorisch identisch übereinander gestapelt. Die Nitrationen hingegen liegen entweder parallel (100) [Nitratgruppe I : N(2) 0(1) 0(2) 0(2')] oder parallel (010) [Nitratgruppe I I : N(3) 0(3) 0(4) 0(4')]Während sich alle [Au(NH3)4]3+-Gruppen in der Höhe z — 0 befinden, liegen die Mittelpunkte der Nitrationen in der Höhe z — 0,5. Eine günstige Anordnung wird offensichtlich dadurch erreicht, daß die Nitratgruppen mit einem Sauerstoffatom auf die Mitte eines aus vier NH 3 Gruppen gebildeten Rechtecks weisen. Die Zahl der in der Struktur vorhandenen Nitrationen reicht aber nicht aus, um diese Anordnung vollständig zu verwirklichen. Ein Ausgleich dafür wird durch die statistische Anordnung der Nitratgruppe II erreicht. Die Tetrammingold-Gruppen besitzen die Punkt Symmetrie mmm (D2h). Sie sind daher exakt planar und weisen vier gleichlange Au-N-Abstände von 2,02 Ä auf (Tab. II). Die Stickstoff-Gold-StickstoffWinkel liegen nahe bei 90° (Winkel N(1 )-Au-N(l') = 91,4°), so daß praktisch eine quadratische Anordnung vorliegt. Der gemessene Au-N-Abstand entspricht recht gut dem Erwartungswert für eine kovalente Gold-Stickstoff-Einfachbindung7. Außerdem liegen gleichgroße Met all-Stickstoff-Abstände Tab. II. Interatomare Abstände und Winkel in [Au(NH 3 )4](N0 3 ) 3 . Die in Klammern angegebenen Standardabweichungen beziehen sich auf die letzten Stellen der Zahlenwerte. Zur Zuordnung der Werte siehe Abb. 1. Abstände [Ä] 2,02(1) ) 2,90(2) ') 2,83(2) 1,29(3) 1,26(2) 2,17(3) ) 2,26(3) 1,14(7) 1,33(4) 2,08(6) ) 2,43(5) 3,04(1) 3,13(2) 3,16(2) 3,11(2) 3,17(4) 2,85(4) Winkel [Grad] N(l) - Au N(l) - Au -N(l') - N(l") 91,4(6) 88,6(6) 0 ( 1 ) - N ( 2 ) - 0(2) 0 ( 2 ) - N ( 2 ) - 0(2') 117(1) 127(2) 0 ( 3 ) - N ( 3 ) - 0(4) 0 ( 4 ) - N ( 3 ) - 0(4') 114(3) 132(5) auch in anderen quadratisch-planaren Tetramminmetall-Ionen vor. So findet man zum Beispiel im [Cu(NH 3 ) 4 ](PtCl 4 ) 8 einen Cu-N-Abstand von 2,04 Ä und im [Pt(NH 3 ) 4 ](PtCl 4 ) 9 einen Pt-N-Abstand von 2,06 Ä. Die Nitrationen haben die Punktsymmetrie mm 2 (C2V), wobei die zweizählige Achse durch die Atome N(2) und 0(1) bzw. durch N(3) und 0(3) verläuft. Die Abstände und Winkel innerhalb der zwei kristallographisch verschiedenen Nitratgruppen unterscheiden sich nur wenig voneinander. Sie stimmen weitgehend mit entsprechenden Daten in anderen Nitraten überein 10 ' 11 . In diesem Zusammenhang muß jedoch berücksichtigt werden, daß die Abstände und Winkel in der Nitratgruppe II aufgrund der statistischen Orientierung sicher weniger genau sind. Die kürzesten N-0-Abstände zwischen Kation und Anion liegen mit einer Ausnahme [N(l I V )-0(3) = 2,85 Ä] zwischen 3,04 und 3,17 Ä und damit oberhalb des van der Waals'schen Berührungsabstands, so daß eine Wasserstoffbrückenbindung zwischen Ammin-Stickstoff und Nitrat-Sauerstoff in diesen Fällen wohl ausgeschlossen werden kann. Für diese Annahme sprechen auch die im IR- und Ramanspektrum beobachteten N-H-Valenzschwingungsfrequenzen (Tab. III). Die Tatsache, daß der Abstand N ( l I V ) - 0 ( 3 ) kürzer ist als die vergleichbaren anderen N-0-Abstände, beruht sicherlich auf der geringeren Genauigkeit, mit der die statistisch verteilte Nitratgruppe II lokalisiert werden konnte. IR- und Ramanspektrum von [Au(NH3NO3 In Tab. III sind die im IR- und Ramanspektrum des [Au(NH 3 )4](N0 3 ) 3 beobachteten Schwingungsfrequenzen und ihre Zuordnung zusammen mit Literaturdaten für das N 0 3 - - I o n und das NH 3 Molekül angegeben. Zur Aufnahme des IR-Spektrums wurde ein KBr-Preßling der Substanz verwendet. Zur Registrierung des Ramanspektrums konnte nur eine konzentrierte wäßrige Lösung benutzt werden, da sich das feste [Au(NH3)4](N03)3 sehr rasch im Laserstrahl zersetzte. In Übereinstimmung mit der Tatsache, daß in der Struktur des [Au(NH 3 ) 4 ](N0 3 ) 3 isolierte N0 3 -Ionen vorliegen, beobachtet man auch im Festkörperinfrarotspektrum genau die für das N0 3 - -Ion zu erwartenden Frequenzlagen. Das Auftreten der IR-verbotenen symmetrischen N0 3 -Valenzschwingungsfrequenz als sehr schwache Absorption bei 1050 cm - 1 kann dadurch erklärt werden, daß die Unauthenticated Download Date | 8/20/17 8:47 AM M. WEISHAUPT-J. STRÄHLE • KRISTALLSTRUKTUR VON Au(NH 3 )4(N0 3 )3 557 Tab. I I I . I R - und Ramanspektrum des [Au(NH3)4](N03)3 und Vergleich mit Literaturdaten 13 . IR Frequenz Raman [cm- 1 ] [Au(NH 3 ) 4 ](N0 3 )3 Intensität m s st, pol s 545 556 570 720 SS 720 822 920 935 1050 m m, Sch m-st SS 1050 1365 sst sst, br SS 1360 1575 3100 3240 m-s sst sst NH 3 Frequenz [ c m - 1 ] Typ NO3Frequenz [ c m - 1 ] B I E vs AUN4 E U VAS A U N 4 A I G vs AUN4 Q Ö N03 720 N0 830 3 ÖS N H 3 933 966 1050 vs NO 3 vas N 0 3 <5as NH 3 FS N H 3 Vas NH 3 1390 1628 3337 3415 Abkürzungen: ss = sehr schwach, s = schwach, m = mittel, st — stark, sst = sehr stark, br = breit, Sch = Schulter, pol = polarisiert. Lagesymmetrie für die Nitratgruppe in der Kristall struktur (C2v) gegenüber der Symmetrie des freien Nitrations (Dah) erniedrigt ist. Die N-H-Valenzschwingungsfrequenzen sind gegenüber den Werten für das freie NH3-Molekül um ca. 200 Wellenzahlen erniedrigt. Der gleiche Effekt wurde auch bei anderen Metallamminen mit quadratisch-planarer Konfiguration beobachtet. So liegen die asymmetrische und symmetrische N-HValenzschwingungsfrequenz im [Pt(NH3)4](PtCLi) bei 3277 bzw. 3182 cm- 1 9. In Übereinstimmung mit anderen Autoren12 kann man diese Frequenzerniedrigung damit begründen, daß der Ammin-Stickstoff eine partielle positive Ladung trägt. Das Vorliegen einer Wasserstoff-Brückenbindung, das ebenfalls zu einer Frequenzerniedrigung führen würde, halten wir für weniger wahrscheinlich. Für das quadratisch-planare AuNi-Gerüst sind in dem registrierten Bereich oberhalb 400 cm - 1 unter Annahme der Punktsymmetrie D411 nur drei Au-N1 2 E . W E I T Z , Liebigs W . R . M A S O N und 90, 5721 [1968]. 3 L. H. SKIBSTEDT Ann. Chem. 410, 117 [1915]. H . B . G R A Y , J. Amer. Chem. Soc. und Scand. A 28, 740 [1974]. 4 5 6 7 8 J. BJERRUM, Acta Chem. L . H . SKIBSTEDT u n d J . B J E R R U M , Acta Chem. Scand. A 28, 764 [1974]. W . B I L T Z , Raumchemie der festen Stoffe, Leopold Voss, Leipzig 1934. W . C. H A M I L T O N , Acta Crystallogr. 12, 609 [1959]. L. P A U L I N G , The Nature of the Chemical Bond, Ithaca, New York 1960. M . B U K O V S K A und M . A . P O K A I - K O S H I T S , Kristallographija 0, 137 [I960]. Valenzschwingungsfrequenzen zu erwarten. Dabei sind wegen des Alternativverbots die Schwingungen in Aig und B2g nur im Ramanspektrum und die Schwingung in E u nur im IR-Spektrum zu beobachten. Die in der Tabelle angegebene Zuordnung der Frequenzen zu Aig und B2g ist aufgrund einer Polarisationsmessung eindeutig möglich. Die gemessenen Werte von 570,556 und 545 cm - 1 für die Schwingungen in Aig, E u und B 2g entsprechen den im [Pt(NH3)4]2+-Ion beobachteten Frequenzlagen von 538, 510 und 526 cm -113 > 14 . Die kristallographischen Berechnungen wurden mit dem Programm system X-RAY 67 von J. M. S T E W T A B T et al. an der Rechenanlage CDC 3300 des Zentrums für Datenverarbeitung der Universität Tübingen ausgeführt. Wir danken der Deutschen Forschungsgemeinschaft und dem Fonds der Chemischen Industrie für die großzügige Unterstützung dieser Arbeit und Herrn Priv.-Doz. Dr. J. W E I D L E I N , Stuttgart, für die Anfertigung des Ramanspektrums und wertvolle Diskussionen. 9 M . ATOJI, J . W . RICHARDSON u n d R. E. RUNDLE, K . NAKAMOTO, M . MARGOSHES u n d R . E . RUNDLE, J. Amer. Chem. Soc. 79, 3017 [1957]. 10 N . E L L I O T T , J. Amer. Chem. Soc. 59, 1380 [1937]. 1 1 J . R . C. D U K E und F . J . L L E W E L L Y N , Acta Crystallogr. 3, 305 [1950]. 12 13 14 J. Amer. Chem. Soc. 77, 6480 [1955]. H . S I E B E R T , Anwendungen der Schwingungsspektroskopie in der Anorganischen Chemie, Springer, Berlin 1966. K . H. S C H M I D T U. A. M Ü L L E R , Inorg. Chem. 14, 2183 [1975]. Unauthenticated Download Date | 8/20/17 8:47 AM