Molekulare Diagnostik
Werbung

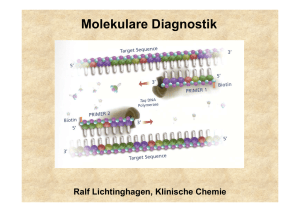
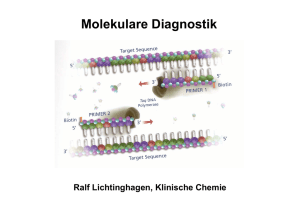
Molekulare Diagnostik Ralf Lichtinghagen, Klinische Chemie Gliederung •Methodische Grundlagen der DNA-Analytik •Anwendungen der molekularen Diagnostik: Assoziation von DNA-Polymorphismen mit Krankheiten oder Krankheitsrisiken Pharmakogenetik Tumordiagnostik auf Grundlage von molekularbiologischen Methoden Untersuchung von DNA •Southern-Blot (Gensondentechik) •Polymerase-Kettenreaktion (PCR) Restriktionsfragmentlängenpolymorphismus-PCR Allel-spezifische Oligonukleotid-Hybridisierung •DNA-Sequenzierung Southern Blot Southern Blot: Anwendungen Bsp: Sichelzellanämie Restriktionsenzym Mst II ..CCTNAGG.. β-Globingen ..CCTGAGG.. Mutation ..CCTGTGG.. Polymerase Chain Reaction (PCR) cycle 1 94°C ∼1min cycle 2 94°C ∼1min 72°C ∼1min 72°C ∼1min 55°C ∼1min 55°C ∼1min 1 1 1 1 1 2 2 1 2 denaturation annealing 2 2 1 2 2 1 2 elongation DNA-Template, Primer (sense, antisense) Nukleotide, Puffer, Mg2+, thermostabile DNA-Polymerase Temperaturen und Zeiten beispielhaft Bedeutung der PCR-Effizienz N = N0 x En, E [%] Ausbeute [%] *) 100 100 95 21 90 4 85 0,8 80 0,1 75 0,02 70 0,002 (E von 0-2) *)nach Effizienz (E) der Reaktion wichtig (oft 70-80%): 30 Zyklen Protein-Polymorphismus in einer Population Ursache: Akkumulation verschiedener Mutationen (Genpolymorphismen) im Genpool einer Population Die Phänotyp-Verteilung in einer Population hängt von der Art der zugrunde liegenden Mutation ab SNP: „single nucleotide polymorphisms“ Mutationstypen Ermittlung eines Genotyps durch die Analyse einer bekannten Mutation •RFLP (Restriktionsfragmentlängenpolymorphismus)-PCR (Häufigkeit des Einsatzes etwa 25%) •Allel-spezifische Oligonukleotid-Hybridisierung (Häufigkeit des Einsatzes etwa 50%) Genchip-Analyse zur gleichzeitigen Erfassung vieler unterschiedlicher Mutationen kommerziell bedingt erhältlich (z.B. CYP450-Array mit 33 versch. Mutationen) Ausstattung für RFLP-PCR RFLP-PCR Ermittlung der Punktmutation (C/T) bei Nukleotid 677 im MTHFR-Gen (Methylentetrahydrofolsäure-Reduktase) Bedeutung für den VitB12/Folsäure-Stoffwechsel (Artheroskleroserisiko über Einfluss auf Homocystein-Konzentration im Serum) HE N N N N N HO HE N M bp 300 200 100 •Amplifikation des DNA Bereiches •DNA-Fragment von 198 bp •Verdauung der DNA mit Hinf I •Wildtyp: 198 bp •Homozyg. Mutation: 175 bp •Heterozyg. Mutation: 175+198 bp Real-Time-PCR Bsp.: Glaskapillar-Thermocycler Real-Time-PCR Fluoreszenz-Resonanz-Energietransfer (FRET) zwischen Hybridisierungssonden •Methode geeignet zur Quantifizierung von PCR-Produkten Messung eines zunehmenden Fluoreszenzsignals während der laufenden PCR. Beginn des Signalanstiegs ist das Maß für die Ausgangskonzentration des betreffenden DNA-Bereiches. •Methode geeignet zur Mutationsanalyse Nach der abgeschlossenen PCR wird das Schmelzverhalten der FRETSonden geprüft. Mutation und Wildtyp haben aufgrund von BasenFehlpaarungen verschiedene Schmelzpunkte. Genotypisierung mittels Schmelzkurvenanalyse Schmelzkurve 45 –75 °C WT HE HO WT HO HE Im Beispiel: FRET-Sonden haben eine 100%ige Übereinstimmung mit der Wildtyp-Sequenz Ableitung -d[Fluoreszenz]/dT Genotypisierung mittels Sequenzierung der betroffenen Genregion Erforderlich bei: •Aufspüren unbekannter Mutationen •zahlreiche Polymorphismen/Gen Nukleotid 677 im MTHFR-Gen klassische Autoradiographie Enzymatische Reaktion (DNA-Polymerase): Einbau von Didesoxynukleotiden (ddNTP) in 4 getrennten Reaktionen, Statistisch verteilte Strangabbrüche, Fragmente elektroph. getrennt, Einbau markierter dNTP (dATP) Autoradiographie (32P), Fluoreszenzlabel Increasing role of environment and stochastic factors Assoziation von DNA-Polymorphismen mit Krankheiten oder Krankheitsrisiken •G6PD deficiency •Hemochromatosis •Acute intermittent porphyria • α1-Antitrypsin ZZ •Type I diabetes mellitus •Apo E-2/E-2 hyperlipoproteinämie •Phenylketonuria •Sickle-cell anemia •Cystic fibrosis •Huntington disease •Duchenne dystrophy •Tay Sachs Increasing role of genotype at other loci Häufig untersuchte Gene (Beispiele) • Alpha-1-Antitrypsin • Angiotensin converting enzyme (ACE) • Apolipoprotein B100 • Apolipoprotein E • Faktor II-Leiden (Prothrombin) • Faktor V-Leiden • HFE (HLA-H) • HLA (Klasse I und Klasse II, zur Typisierung) • Laktase (Laktosetoleranzstörung) • MTHFR • Mukoviszidose • UDP-Glucuronosyltransferase (Gilbert-Meulengracht) Angiotensin-converting enzyme (ACE) Insertions (I) / Deletions (D) –Polymorphismus im ACE-Gen 12 Häufigkeit 10 ID 8 6 4 II 2 0 0 20 DD 40 Referenzintervalle (Genotyp-abhängig) ACE Genotyp DD (ca. 25%) ACE Genotyp DI (ca. 50%) ACE Genotyp II (ca. 25%) 24-89 U/l 13-66 U/l 7-33 U/l 60 80 100 U/l Klinische Bedeutung: Labordiagnostik der Sarkoidose: S-ACE als Surrogatmarker erhöht (Polymorphismus beeinflusst Serumkonz.) Gendefekt: Risikofaktor kardiovaskulärer Erkrankungen?? α1-Antitrypsinmangel α1-Antitrypsin: Akute-Phase-Protein aus Hepatozyten, Aktivitätshemmung von Serinproteasen (PMN-Elastase) >40 Allele Wildtyp (PiM), Mangelallele: 7% Prävalenz Wichtige klinisch relevante Mangelallele: PiZ, PiS, PiNull Risiko korreliert mit vermind. Antitryp.-Konz. high risk: PiZZ, PiZNull, PiNullNull increased risk : PiSZ Erkrankungen der Leber (Kindesalter): Sekretionsstörung in Hepatozyten bei PiZZ Lungenemphysem (Erwachsenenalter): Folge einer nicht inhibierten PMN-ElastaseAktivität Hereditäre Hämochromatose (HH) Prävalenz in Mitteleuropa: 1:400, häufigste autos. rez. Erbkrankheit Manifestationsalter: 40-60 Jahre, Hepatomegalie, Leberzirrhose, D. mellitus, dunkle Hautpigmentierung Mutationen im HFE-Gen (Typ 1-Hämochromatose) Cys 282Tyr (C282Y) homozygot bei ca. 85-95% der HHPatienten, Compound-Heterozygotie mit His63Asp (H63D) bei 3-8% der HH-Patienten (Prävalenz dieses H63D-Polymorphismus: 13%) Typ 2-Hämochromatose (Hepcidin, Hämojuvelin): seltene juvenile Form, vor 30. Lebensjahr manifest, geht mit schwerer Kardiomyopathie und Hypogonadismus einher. Typ 3: (Transferrinrezeptor 2) Typ 4: (basolateraler Eisencarrier Ferroportin 1) Diagnoseweg bei V.a. hereditäre Hämochromatose Klinische Hinweise und Transferrinsättigung >60% und S-Ferritin >250-300 ng/l Gen-Test: Cys282Tyr-Mutation, gegebenenfalls His63Glu _ + Gentest negativ, aber klin. Verdacht, bzw. zur Bestimmung des Ausmaßes des Leberschadens Hämochromatose gesichert Leberbiopsie mit quantitativer Messung des Lebereisengehaltes Aderlasstherapie + Fettstoffwechsel Lipoproteinstoffwechsel maßgeblich durch Zelloberflächenrezeptoren mitbestimmt LDL-Rezeptor bindet neben ApoB100 auch Apo E Funktion: Regulation der Cholesterinkonzentration Cholesterinlieferung für zell. Bedarf, Hormonsynthese Familiäre Hypercholesterinämie: LDL-Rez.defekte: >100 bislang bekannte Mutationen (DNA-Sequenzierung) ApoB100: häufigste Mutation: G10699A (Arg3500Gln) Frequenz: 1 auf 700 Kaukasier bei Heterozygotie erhöhtes S-LDL-Cholesterin, gestörte Bindung an LDL-Rezeptor, Risiko kardiovaskulärer Erkrankungen Fettstoffwechsel Hyperlipoproteinämie Typ III: ApoE: ApoE2-Homozygotie ist Voraussetzung zur Ausprägung dieser Fettstoffwechselstörung Aber: Nur ein kleiner Teil der Homozygoten erkrankt!!! Ansonsten korreliert das Allel E2 mit niedrigeren Cholesterinspiegeln. ApoE-Allele: Isoformen: E2, E3 und E4 durch Mutat. C112R und R158C E2/2 (1%), E3/3(55%), E4/4, E2/3, E2/4, E3/4 (26%) Rezeptorbindung E2 bindet nicht , Aktivierung des hepat. LDL-Rezeptors E4-Partikel bewirken das Gegenteil, E4 ist somit potentiell artherogen, E2 protektiv Assoziation mit M. Alzheimer für E4 Thrombophiliediagnostik Studien gerade bei jungen Thrombophiliepatienten ergaben eine Korrelation von verschiedenen genetischen Polymorphismen vor allem in zwei Genen von Faktoren des Gerinnungssystems : Faktor V (G1691A) Prothrombin (Faktor II, G20210A) Außerdem konnte eine Korrelation mit einem Polymorphismus (thermolabiles Enzym) der Methylentetrahydrofolsäurereduktase (MTHFR; C611T) gezeigt werden. Thrombophiliediagnostik Faktor V: G1691A (Arg506Gln), autosom. dominant (nach Entdeckungsort Faktor V/Leiden genannt) Effekt : verlangsamte Spaltung durch aktiviertes Protein C (APC-Resistenz) Prävalenz: 1:20 für heterozygoten Defekt Weitverbreitester Risikofaktor für venöse Thrombosen Heterozygotie: ca. fünf- bis zehnfache Erhöhung des Thromboserisikos gegenüber der Normalbevölkerung Homozygotie: Risiko ca. 100fach erhöht Weitere Risikofaktoren (z.B. Kombinationsdefekte, Nikotinkonsum, Einnahme oraler Kontrazeptiva) können das Thromboserisiko zusätzlich erheblich erhöhen. (zusätzlich Ausschluss von Prothrombin-Genmutation, Antithrombin III-, Protein C-, Protein S-Mangel). Pharmakogenetik Beschäftigt sich mit den hereditären Grundlagen der Unterschiede in einer Population bezüglich dem Response auf ein Arzneimittel. DNA-Sequenzen variieren leicht von Individuum zu Individuum zahlreiche Polymorphismen Genetische und damit häufig auch Unterschiede in Proteinzusammensetzungen verursachen daher bei gleicher Medikation verschiedene systemische Konzentrationsniveaus. Insgesamt abhängig von Aufnahme, Absorption, Metabolismus (Phase I und Phase II Biotransformationsreaktionen), Clearance und Exkretion Pharmakogenetik Enzym Häufigkeit Cytochrom P450 CYP2D6 CYP2C19 CYP1A2 3-10% viele (Antidepressiva 2-5% Siehe MHH-Homepage/MHH-Internes/ Neuroleptika, Beta-Blocker.....) 12% CYP P450 Tabelle sowie N-Acteyltransferase NAT2 50% Thiopurinmethyltransf. TPMT 0,3% Medikamente Einrichtungen/Klinische Chemie/ Analysenspektrum/Pharmakogenetik/ Isoniazid, Sulfapyridin Dapson, Koffein, Hydralazin Probleme Meist verstärkte Wirkung Überempfindlichkeitsreaktionen Azathioprin, Mercaptopurin Leukopenie Dihydropyrimidin-Dehydr. 5% (Het.) (DPD, DPYD) 5-Fluoruracil verstärkte Wirkung Glukose-6-Phos.-Dehydr. 1% Sulfonamide, Primaquin Hämolyse Serum-Cholinesterase 0,04% Succinylcholin Apnoe Ca-Transport im ER 0,005% Inhalationsanästhetika Maligne Hyperthermie Pharmakogenetik: z.B. CYP 2D6 Sinn dieser Analysen abh. von klin. Bedeutung + Durchführbarkeit. Bsp: Die Cytochrom P450-Genfamilie (CYP) umfasst eine Vielzahl von Isoenzymen, die für die Verstoffwechslung diverser Substanzen verantwortlich sind. CYP 2D6: Von diesem Isoenzym (Debrisoquin-Hydroxylase) sind eine Reihe von Allelen bekannt, die zu einem Verlust der Enzymaktivität führen [CYP2D6*1 (Wildtyp)] („extensive (rapid) Metabolizer“). CYP2D6*3, *4, *5, *6 („poor (slow) Metabolizer (PM)“) sind in 98% aller Fälle an einem Verlust der CYP2D6-Aktivität beteiligt. Zwei Mangelallele müssen allerdings nachgewiesen werden. CYP2D6*2 („intermediate Metabolizer“) in Verbindung mit einem PoorMetabolizer-Allel. Genduplikationen führen zu „Ultrafast Metabolizer“. Betroffene Pharmaka: Antidepressiva (z.B. Amitriptylin), Neuroleptika (z.B. Haloperidol), Antiarrhythmika (z.B. Propafenon) Pharmakogenetik: z.B. CYP 2D6 UM: ultrafast metabolizer EM: extensive metabolizer IM: intermediate metabolizer PM: poor metabolizer Pharmakogenetik: z.B. CYP 2D6 200 40 inpatient treatment days 45 150 5 100 50 0 N= 25 30 non-polymorphism polymorphism EM PM/IM CYP-abhängige Schwierigkeiten bei der medikamentösen Therapie psychiatrischer Patienten spiegeln sich sogar in der stationären Behandlungsdauer wider. Höhere Behandlungskosten Mutationsnachweise in der molekulargenetischen Tumordiagnostik •Keimbahnmutationen •somatische Mutationen Tumordiagnostik Nachweis von Keimbahn-Mutationen Familiäre Disposition bei Krebserkrankungen: Hereditäre Tumoren: ca. 2% aller Krebserkrankungen Hinweise auf erbliche Disposition für Tumoren: Familie Zwei oder mehr Verwandte ersten Grades mit demselben Tumor Zwei oder mehr Verwandte mit seltenen Tumoren Evtl. drei oder mehr Tumoren in typischer Assoziation (z.B. Mamma und Ovar) Patient Multiple Tumoren in einem Organ oder in typischer Assoziation Assoziation von Tumoren mit anderen genetischen Auffälligkeiten oder kongenitalen Defekten Tumoren an ungewöhnlichem Ort oder in frühen Lebensjahren Tumordiagnostik Nachweis von Keimbahn-Mutationen Erkrankung Gen Fam. Retinoblastom Fam adenomatöse Polyposis (FAP) Heredit. kolorektale Karzinome o. Polyposis Fam. Brust-/Ovarialkrebs Fam. Brustkrebs Li Fraumeni Syndrom Multiple endokrine Neoplasie (MEN) Typ1 Fam. Medulläres Schilddrüsenkarzinom MEN Typ 2A und 2B Fam. Melanom Neurofibromatose Typ 1 Neurofibromatose Typ 2 Gorlin Sydrom, Basalzellkarzinom Von Hippel-Lindau Syndrom Fam. Wilms-Tumor rb apc mlh 1 brca 1 brca 2 p53 men 1 (menin) ret p16 nfl 1 nfl 2 ptch vhl wt 1 Tumordiagnostik Nachweis von Keimbahn-Mutationen Diagnostische und präventive Maßnahmen, die den Nachweis einer genetischen Tumordisposition rechtfertigen: Maßnahme Beispiele Präventive Diagnostik Koloskopie bei HNPCC und FAP Vermeidung invasiver diagnost. Maßnahmen bei Ausschluss einer disponierenden Mutation Ausschluss eines Risikos bei Mitgliedern von HNPCC- und FAPFamilien Präventive therapeutische Maßnahmen Thyreodetektomie bei Patienten aus MEN Typ II-Familien HNPCC: Hereditäres nicht-polypöses kolorektales Karzinom FAP: Familiäre adenomatöse Polyposis Coli MEN: Multiple endokrine Neoplasie Tumordiagnostik II Nachweis von somatischen Mutationen Genetische Instabilität in malignen Tumoren: Entweder kausal für Tumorgenese verantwortlich, und/oder für Tumorprogression verantwortlich! Wichtige Gene: p53, k-ras Mutationsanalyse zur Früherkennung von Tumoren. Tumorzellnachweise in Körperflüssigkeiten und Stuhl. Problem: Vielzahl möglicher Mutationen, Nachweis eines mutierten Allels unter einem Überschuss von Wildtyp-Allelen. Tumoren: z.B. Kolon-Karzinom, Pankreas-Karzinom Tumordiagnostik II Nachweis von somatischen Mutationen p53 Am häufigsten mutiertes Tumorgen (Tumorsuppressorgen kodiert für Transkriptionsfaktor). Mutationen (meist Punktmutationen/missense) fast ausschließlich im Bereich der DNA-bindenden Domäne (Exone 4-8). Mutiertes P53-Protein häufig mit verlängerter Halbwertszeit, immunhistochemisch im Tumorgewebe nachweisbar. Mutationen bzw. zelluläre Akkumulationen korrelieren mit geringerer Überlebenszeit des Patienten. (z.B. Mamma-Ca: unabhängiger prognostischer Parameter) Tumordiagnostik III Nachweis von somatischen Translokationen (Tumorgene an Bruchstellen von Chromosomen) Aus technischen Gründen Nachweis von Chromosomenanomalien in Tumoren hämatopoetischer Zellen leichter. Große Mehrzahl bisher gesicherter Chromosomenanomalien bei Leukämien und Lymphomen. Gen Rearrangement Erkrankung Rearrangements, kodierende Sequenz exprimiert und nicht verändert BCL2 t(14;18)(q32;q21) follikuläres Lymphom Rearrangements, Hybridgene BCR-ABL t(9;22) (q34;q11) CML, B-ALL Philadelphia-Chromosom zytogenetisch bei bis zu 90% der CML nachweisbar!!! Nachweis mittels PCR /RT-PCR mit Primern von beiden Seiten der chromosomalen Bruchstelle. Tumordiagnostik IV Nachweis/Quantifizierung zirkulierender Tumorzellen Nachweis residualer Tumorzellen im peripheren Blut, Knochenmark oder Blutprodukten über Marker (Transkripte, Methylierungsgrad der DNA), welche nicht in hämatopoetischen Zellen vorhanden sind. Target: meist Gene klassischer Tumormarker: z.B. AFP PSA CK19 alpha-Fetoprotein Prostataspezifisches Antigen Zytokeratin 19 Nachweis mittels RT-PCR o. quantitativer RT-PCR Nachweisgrenzen: ca. 1 Tumorzelle unter 106 Zellen Methylierungsgrad der DNA: (neuerer Trend). DNA von Tumoren ist häufig 5mC). (spez. in Promorbereichen) hypermethyliert (C Nachweis des Methylierungsgrades von ausgewählten Indikatorgenen zum sensitiven Nachweis einer Metastasenbildung. Experimentelle Doktorarbeit Gemeinschaftsprojekt der Abt. Gastroenterologie und der Klinischen Chemie Matrixdegradation bei chronischen Lebererkrankungen Funktionelle Rolle des MMP/TIMPSystems für die hepatische Regeneration und die Entwicklung chronischer Organschäden an der Leber. Adenovirale hepatische Überexpression von MMPs im Mausmodel. Techniken: Immunoassays, Western-Blot in situ-Techniken, (quantitative) RT-PCR, PCR, Genexpression, Genotypanalyse, rekombinante Proteinherstellung in prokaryonten und viralen Expressionssystemen, molecular cloning Kontakt: Prof. Dr. Ralf Lichtinghagen Tel: 3940; Pieper: 74-2272 Voraussetzungen: Februar 2006 Molekularbiologische Vorkenntnisse nicht hinderlich, Teamfähigkeit, Interesse am wissenschaftl. Arbeiten Dauer der Labortätigkeit: ca. 1 Jahr