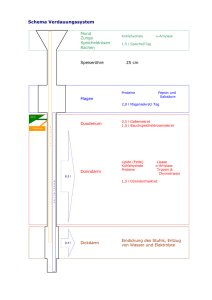
Kapitel 10_2005
Werbung

KAPITEL 10 PROTEINE ___________________________________________________________________________ 10.1. 10.1.1. 10.1.2. 10.1.3. 10.1.4. 10.1.5. 10.1.6. 10.2. 10.2.1. 10.2.1.1. 10.2.1.2. 10.2.1.3. 10.2.1.4. 10.2.2. 10.2.3. ZUSAMMENFASSUNG 1 VIELE PROTEINE SIND AN SICH STÄNDIG WIEDERHOHLENDEN SCHLÜSSELPROZESSEN BETEILIGT. 2 Die Histokompatibilitätsantigene sind die Identifikationskarten der individuellen Zelle. Es gibt zwei Klassen von Transportproteinen: die Transportproteine, welche Energie benötigen, und die Kanalproteine, welche keine Energie benötigen. Zytokine sind lösliche Proteine oder Glykoproteine, welche hauptsächlich als chemische Kommunikatoren auf kleinem Raum wirken. Prionen sind nicht-mikrobielle Proteine, welche Ursache ansteckender Krankheiten sein können. Die Immunglobuline sind die humoralen Mediatoren der Immunantworten. Transkriptionsfaktoren sind intrazelluläre Proteine, welche an regulatorische Sequenzen der DNA binden. 2 5 6 12 15 17 DEFEKTE DER SYNTHESE, DES ABBAUS ODER DER AUSSCHEIDUNG VON PROTEINEN KÖNNEN SICH IN FORM VERSCHIEDENER KRANKHEITEN MANIFESTIEREN. 20 Intrazelluläre Proteinablagerungen können Hinweis auf eine veränderte Proteinsynthese oder eine abnorme Struktur der Proteine sein. 21 Viruskrankheiten. Störungen des Zyto- oder Membranskeletts. l-Antitrypsin-Mangel Alzheimer Krankheit. 22 26 28 29 Unter der Amyloidose werden extrazelluläre Ablagerungen nicht-löslicher, fibrillärer Proteine in verschiedenen Organen bei verschiedenen Krankheiten verstanden. «Hyalin» und «Fibrinoid» sind teils extrazelluläre, teils intrazelluläre Ablagerungen von Proteingemischen. 35 38 10.3. 10.4 BEI MALIGNEN TUMOREN KÖNNEN PROTEINE AN UNGEWÖHNLICHEN ORTEN ODER IN UNGEWÖHNLICHER MENGE ERSCHEINEN. 39 PROTEINE KÖNNEN MITTELS IMMUNHISTOCHE-MISCHER METHODEN IM HISTOLOGISCHEN SCHNITT ODER ZYTOLOGISCHEN PRÄPARAT SICHTBAR GEMACHT WERDEN. 39 ZUSAMMENFASSUNG Störungen von Proteinen können in erheblichem Ausmass Körperfunktionen beeinträchtigen. Sie können sich auch morphologisch manifestieren; Beispiele dafür sind die Mallory Bodies in den Hepatozyten, die Speicherung von Vorstufen des 1-Antitrypsins (AT) beim 1-AT-Mangel in den Hepatozyten, die «Viruseinschlüsse» in verschiedenen Zellen bei Viruserkrankungen und Amyloidablagerungen. Zu den wichtigsten Proteinen, welche die Funktion der Zellen in ihrem Umfeld definieren, sichern und steuern, gehören: die Histokompatibilitäts-Antigene, die Proteine im Dienste der Signaltransduktion, die Transport- und Kanalproteine, die Proteine der «Phase der akuten Antwort», Zytokine, Chemokine (eine Untergruppe der Zytokine), Immunglobuline, Transkriptions- und Wachstumsfaktoren. Die Histokompatibilitätsantigene sind die Identifikationskarten der individuellen Zelle. Man unterscheidet zwei Sets von Histokompatibilitätsantigenen: die Proteine der Klasse 1 des Major Histocompatibility Complex (MHCP-1) und jene der Klasse 2 (MHCP-2). Die Transportproteine spielen grundsätzlich die Rolle membrangebundener Enzyme. Ein Teil der Transportproteine benötigt für ihre Funktion Energie. Ein Vertreter dieser Gruppe ist das P-Glycoprotein, auch Multidrug Resistance Protein (MDR) genannt. Zytokine sind lösliche Proteine oder Glykoproteine, welche hauptsächlich als chemische Kommunikatoren auf kleinem Raum wirken. Sie moderieren schwergewichtig entzündliche Prozesse (z.B. der Tumornekrosefaktor-a), die Entstehung von Thromben und der disseminierten intravasalen Gerinnung und sie kontrollieren Intensität und Dauer der verschiedenen Immunantworten (Interleukine). Die Chemokine (eine Familie der Zytokine) stehen in erster Linie im Dienst der Chemotaxis, der Rekrutierung von Leukozyten und der Migration von Lymphozyten und dendritischen Retikulumzellen. Prionen sind nicht-mikrobielle Proteine, welche ansteckende, degenerative Erkrankungen des zentralen Nervensystems hervorrufen. Transkriptionsfaktoren sind intrazelluläre Proteine, welche an regulatorische Sequenzen der DNA binden. Sie bilden als tertiäre Messengers das letzte Glied in der Signaltransduktion. Defekte in der Proteinsynthese können ein Zuviel oder ein Zuwenig an Proteinen bewirken. Bei einer Abbaustörung oder verminderten Ausscheidung kommt es zu einer intrazellulären oder extrazellulären Ablagerung (z.B. bei Virusinfekten, bei Krankheiten, die mit einer Störung des Zyto- oder Membranskeletts einhergehen, beim 1-AT-Mangel und bei der Alzheimer Krankheit). Bei der Alzheimer-Krankheit sind pathologische Proteinablagerungen extrazellulär in den Plaques des Hirngewebes (Amyloid Protein ) und intraneuronal (fibrilläre Tangles, phosphoryliertes Tau-Protein) vorhanden. Die Amyloidose steht für extrazelluläre Ablagerungen nicht-löslicher, fibrillärer Proteine in verschiedenen Organen bei verschiedenen Krankheiten. Ursache der AL-Amyloidose (Amyloid light chain) ist meistens eine monoklonalen Überproduktion der oder Leichtketten, einer AA-Amyloidose (Amyloid associated) eine chronische Entzündung. «Hyalin» und «Fibrinoid» sind teils extrazelluläre, teils intrazelluläre Ablagerungen von Proteingemischen. Bei malignen Tumoren können Syndrome auftreten, die nicht direkt auf eine Destruktion des umgebenden Gewebes oder die Fernwirkung des Tumors via Metastasen zurückzuführen sind, sondern auf Proteine, welche vom Tumorgewebe pathologischerweise synthetisiert und als Hormone oder Antigene wirksam werden können. Die Syndrome werden als paraneoplastische Syndrome bezeichnet. Proteine können mittels immunhistochemischer Methoden im histologischen Schnitt oder zytologischen Ausstrichpräparat sichtbar gemacht werden. Proteine haben vielfältige Aufgaben, vor allem als intra- und extrazelluläre Bausteine, Rezeptoren, Oberflächenproteine, Transmitter oder Sekretionsprodukte. Defekte der Proteine führen zu Beeinträchtigungen der Körperfunktionen und manifestieren sich morphologisch, z.B. als Mallory Bodies in den Hepatozyten, Ablagerungen von Vorstufen des 1-Antitrypsins beim 1-Antitrypsin-Mangel in den Hepatozyten oder als «Viruseinschlüsse» in verschiedenen Zellen bei Viruserkrankungen. 10.1. VIELE PROTEINE SIND AN SICH STÄNDIG WIEDERHOLENDEN SCHLÜSSELPROZESSEN BETEILIGT. Zu den Proteinen, welche die Funktion der Zellen in ihrem Umfeld definieren, sichern und steuern, gehören: die Histokompatibilitäts-Antigene, die Proteine im Dienste der Signaltransduktion, darunter die Transkriptionsfaktoren, die Transport- und Kanalproteine, die Proteine der Phase der akuten Antwort, Zytokine, Chemokine, Immunglobuline und Wachstumsfaktoren. Die Proteine im Dienste der Signaltransduktion sind im Kapitel 5 (Signaltransduktion), die Proteine der Phase der akuten Antwort im Kapitel 1 (Zellschädigung) und die Wachstumsfaktoren im Kapitel 7 (Wundheilung) im Detail besprochen. 10.1.1. Die Histokompatibilitätsantigene sind die Identifikationskarten der individuellen Zelle. Zellen können sich gegenseitig durch «Schnüffeln» oder durch Berühren erkennen. Die Informationen, die sie durch «Schnüffeln» wahrnehmen, werden ihnen durch extrazelluläre Signalmoleküle (first messengers) vermittelt. Die Kontakte durch Berühren setzen voraus, dass die Zellen miteinander in Kontakt treten können. Alle Zellen haben auf ihrer Oberfläche ein Set von Markern in Form von Glykoproteinen zur Verfügung, welche ihnen erlauben, sich gegenseitig zu identifizieren. Diese Marker werden Histokompatibilitäts-Antigene oder HLA (Human Leucocyte Antigen) genannt. Die Gene, welche die HLA kodieren, sitzen in Form eines Clusters auf dem kurzen Arm des Chromosoms 6. Der Cluster wird als Major Histocompatibility Complex (MHC) oder als HLA-Komplex bezeichnet (Tab.10-1). Auf dem gleichen Chromosom liegen auch drei Gene für Komponenten des Komplementsystems und das Gen für den Tumornekrosefaktor (TNF). Der MHC-Komplex kodiert zwei Klassen von Proteinen: die MHC-Protein Klasse 1 (MHCP-1) und Klasse 2 (MHCP2). Die MHC-Gene, welche die MHCP-2 codieren, werden auch als «Immunantwortgene» bezeichnet. Die Expression dieser Gene ist Voraussetzung (1) für die Aktivierung von TLymphozyten durch die Antigen-präsentierenden Zellen und (2) die Selektion der TLymphozyten bei der Reifung im Thymus. Es sind Defekte der MHCP-2 bekannt. Ursache dieser Defekte sind Störungen der Gene der Regulationsproteine, welche die Expression der MHC-Gene für die Proteine der Klasse 2 steuern. Tab.10-1 Man unterscheidet drei Sets von Proteinen des MHC-Komplexes: die Proteine der Klasse 1 (MHCP-1) und die Proteine der Klasse 2 (MHCP-2). Zu den Proteinen der Klasse 3 gehören unter anderem Proteine der Komplementkaskade, die Hitzeschock-Proteine und die Tunornekrosefaktoren. ___________________________________________________________________________ MHCP-1 MHCP-2 ___________________________________________________________________________ Genlocus Struktur des Proteins A, B, C Glykoprotein ß2-Mikroglobulin DR, DQ, DP -Kette -Kette Vorkommen auf den meisten kernhaltigen somatischen Zellen auf Blutplättchen auf den meisten immunkompetenten Zellen Monozyten Makrophagen B-Lymphozyten aktivierte T-Lymphozyten auf Spermatozoen auf Endothelzellen und Fibroblasten nach Induktion durch -Interferon auf den antigen-präsentierenden Zellen Präsentation des Antigens gegenüber ... Zytotoxischen T-Lymphozyten (Killer-Zellen) Helfer T-Lymphozyten Vielfalt Herkunft des präsentierten Peptids Gross In der Zelle hergestellt (virale, Tumor- oder eigene Proteine) Klein Aus phagozytierten Plasmamembranen und extrazellulären Proteinen PräsentationsMechanismsu via Golgi-Apparat (nach intrazellulärer Synthese) via Endosomen und Lysosomen (nach Phagozytose) Primäre Funktion Vermittlung der Zytolyse Antigen-Präsentation Mikrobielles Agens Virus Bakterium ___________________________________________________________________________ Die MHC-Proteine zeigen drei wichtige Eigenschaften: (1) Sie regulieren die Interaktion zwischen den Effektorzellen der Immunantwort. Sie stellen den «Personalausweis» oder «Pass» der Effektorzellen dar. Sensibilisierte zytotoxische T-Lymphozyten zerstören z.B. Zellen, welche mit einem Virus infiziert sind, nur dann, wenn die infizierten Zellen das gleiche MHCP-1 besitzen wie die zytotoxischen T-Lymphozyten. Diese Bedingung wird als HLA-Restriktion bezeichnet. (2) Die Expression verschiedener MHCP-1 und MHCP-2 ist mit einem erhöhten Risiko für verschiedene Krankheiten verbunden (Abb.10-1). Die Ursache dieses Phänomens ist nicht definitiv geklärt. (3) Bei Organtransplantationen wird angestrebt, dass die MHCP-1 und MHCP-2 des Empfängers und Spenders eine möglichst weitgehende Übereinstimmung zeigen. Die MHCP der Zellen eines transplantierten Organs können beim Empfänger als Antigene wirken und eine Abstossungsreaktion (Host versus graft- oder Graft versus host-Reaktion) hervorrufen. Abb.10-1 Verschiedene Krankheiten manifestieren sich häufiger bei Patienten, welche verschiedene Allele für verschiedene MHCP besitzen: Das relative Risiko (Häufigkeit der Manifestation einer Krankheit bei exponierten Personen verglichen mit der Häufigkeit der Krankheitsmanifestation bei nicht exponierten Personen) für diese Krankheiten ist erhöht. IDDM: Diabetes mellitus Typ I. Reiter Syndrom: Krankheit mit Konjunktivitis, Uretheritis, Polyarthritis und Hautexanthem. 10.1.2. Es gibt zwei Klassen von Transportproteinen: die Transportproteine, welche Energie benötigen, und die Kanalproteine, welche keine Energie benötigen. Die Transportproteine (auch Carrierproteine genannt) beziehen die Energie über eine Hydrolysierung von vorher gebundenen ATP-Molekülen. Die Carrierproteine werden deshalb der Familie der ATP-bindenden «Kassetten-Proteine» (ABC) zugeordnet. Den Namen «KassettenProteine» erhielten diese Proteine wegen ihrer Funktionsweise: Wenn ATP in das Protein «hineingeschoben» wird (wie eine Kassette in ein Wiedergabe-Gerät), tritt das Protein in Aktion. Die Carrierproteine spielen grundsätzlich die Rolle von membrangebundenen Enzymen. Man kann die Carrierproteine - abhängig von ihrem Energiebedarf - wiederum in zwei Gruppen einteilen: (1) in die Gruppe, bei der die benötigte Energie von der Menge der transportierten Moleküle abhängt und (2) in die Gruppe, bei der Energie nur für den Start der Funktion der Moleküle (Öffnung des Ionenkanals) benötigt wird. Zwei wichtige Vertreter der ersten Gruppe sind die Ionen-ATPasen und das P-Glykoprotein. Die Na+- und K+-Ionen-ATPase z.B. pumpt aktiv Na+-Ionen aus der Zelle heraus und K+-Ionen in die Zelle hinein. Auf der dem Zytoplasma zugewandten Oberfläche besitzt das membrangebundene Protein Bindungsstellen für Na+ und ATP, auf der Richtung Extrazellularraum gelegenen Oberfläche die Bindungsstelle für K +. Einige Ca2+-Ionen-Pumpen sind ebenfalls membrangebundene ATPasen. Das P-Glycoprotein, auch Multidrug Resistance Protein (MDR) genannt, wurde 1986 entdeckt. Die Bezeichnung «P» erhielt es, weil vorerst vermutet wurde, dass das Protein die Permeabilität der Zellmembran steuere (P: Permeability). Das P-Glycoprotein gehört jedoch heute - wie der «Cystic Fibrose Transmembrane Conductance Regulator» (CFTR, siehe Kapitel 9: Kohlenhydrate) - eindeutig in die Familie der «Kassettenproteine». Das Protein ist physiologischerweise in verschiedenen Zellen anzutreffen: in Epithelzellen (Hepatozyten, Zellen der Nebennierenrinde, der Gallecanaliculi, des Gastrointestinaltraktes, der Nierentubuli und Lungenalveolen), in Zellen der Plazenta, in Endothelzellen der Blut-Hirn-Schranke und des Hodens sowie in Stammzellen des Knochenmarks. Die Hauptaufgabe des P-Glykoproteins ist die Elimination von fremden Substanzen aus der Zelle. So ist das Protein z.B. für die Resistenz des Malaria-Erregers Plasmodium falciparum gegenüber Chloroquin verantwortlich. Zellen maligner Tumoren können das P-Glykoprotein vermehrt exprimieren. Zwischen der Konzentration der P-Glykoproteine in einem Tumor und der Antwort auf eine Chemotherapie gegen den Tumor ist eine Korrelation mit negativem Vorzeichen zu beobachten: Die Chemotherapie hat nur einen geringen Effekt, wenn viel P-Glykoprotein auf den Tumorzellen vorhanden ist. Denn das P-Glykoprotein transportiert das Chemotherapeuticum wieder aus der Zelle heraus, bevor es seine Wirkung entfalten kann. Cyclosporin, Verapramil und Kortikosteroide können diese Pumpfunktion des P-Glykoproteins hemmen. Tumoren, welche viel P-Glykoprotein exprimieren, zeigen auch mehr Gefässinvasionen und Lymphknotenmetastasen als Tumoren mit weniger «P-Glykoprotein-Pumpen». Die Kanalproteine benötigen für ihre Funktion keine Bioenergie. Sie bilden hydrophile Poren, welche durch die Membran hindurchreichen. Alle Kanalproteine gestatten den gelösten Molekülen einen passiven Membrandurchtritt (auch als «passiver Transport» oder «erleichterte Diffusion» bezeichnet). Ist das zu transportierende Molekül ungeladen, treibt der Konzentrationsgradient den passiven Transport an; ist das Molekül geladen, beeinflussen sowohl der Konzentrationsgradient als auch die elektrostatische Potentialdifferenz seinen Transport. 10.1.3. Zytokine sind lösliche Proteine oder Glykoproteine, welche hauptsächlich als chemische Kommunikatoren auf kleinem Raum wirken. Die Zytokine moderieren verschiedene wichtige Prozesse: die akute und chronische Entzündung, die Entstehung der Thromben und die Immunreaktionen. Die Zytokine werden hauptsächlich in Leukozyten gebildet, können aber auch von anderen Zellen synthetisiert werden. Die meisten Zytokine werden auf eine Stimulation hin direkt synthetisiert und sezerniert; einige werden jedoch in intrazellulären Granula oder in der extrazellulären Matrix (ECM) gespeichert und stehen deshalb zum Zeitpunkt einer Stimulation sofort zur Verfügung. Die Zytokine wirken autokrin, parakrin oder endokrin; sie sind grundsätzlich in nur geringen Konzentrationen vorhanden. Die Zytokine können nach zwei Kriterien eingeteilt werden: (1) nach ihrer chemischen Struktur (Tab.10-2) und (2) nach ihrer Wirkung oder Herkunft. Von ihrer Struktur her werden sechs Familien von Zytokinen unterschieden, von ihrer Wirkung oder Herkunft her sieben Gruppen (Interferone, inflammatorische Zytokine, Lymphozyten-abhängige Zytokine, Makrophagen-abhängige Zytokine, Chemokine, hämatopoietische Wachstumsfaktoren und transformierende Wachstumsfaktoren). Tab.10-2 Die Zytokine werden in sechs Familien eingeteilt. ___________________________________________________________________________ Zytokin-Familie Vertreter ___________________________________________________________________________ Hämatopoietine IL-2 bis IL-7, IL-9, IL-10, IL-12, IL-13, IL-15 G-CSF, GM-CSF, M-CSF Erythropoietin CNTF INF-, INF-, INF- Epidermale Wachstumsfaktoren EGF, TGF-, Zytokine mit «-Kleeblatt-Konfiguration» IL-1 aFGF, bFGF Tumornekrosefaktoren TNF-, TNF- Zytokine mit «Cystein-Knoten» NGF, TGF-, PDGF, VEGF Chemokine ___________________________________________________________________________ CSF CNTF aFGF bFGF G-CSF GM-CSF IFN IL M-CSF PDGF TNF VEGF Kolonie-stimulierende Faktoren Ciliarer neurotrophischer Faktor Sauerer Fibroblasen-Wachstumsfaktor Basischer Fibroblasten-Wachstumsfaktor Granulozyten-Kolonie-stimulierender Faktor Granulozyten-Makrophagen-Kolonie-stimulierender Faktor Interferon Interleukin Makrophagen-Kolonie-stimulierender Faktor Plättchen-Wachstumfaktor Tumornekrosefaktor Vaskulärer endothelialer Wachstumfaktor Chemokine (Tab.10-3) sind spezielle Zytokine, welche primär im Dienst der selektiven Rekrutierung von Leukozyten für die spezifische und unspezifische Abwehr stehen und so die Migration von Zellen aus den Blutgefässen in den extravaskulären Raum kontrollieren. Sie lösen diese Aufgabe dadurch, dass sie eine gerichtete Bewegung der von ihnen beeinflussten Zellen bewirken (Abb.10-2). Man ordnet die Chemokine - entsprechend ihrer Struktur - drei Gruppen zu: (1) Die C- (-) Chemokine sind ausschliesslich chemotaktisch für Lymphozyten. (2) Die CC(-) Chemokine sind vorwiegend chemotaktisch für Lymphozyten, Monozyten, basophile und eosinophile Granulozyten; und (3) die CXC- (-) Chemokine wirken besonders auf neutrophile Granulozyten. Die für die Chemokine verwendeten Abkürzungen leiten sich von der chemischen Struktur der Moleküle ab: CC bedeutet, dass die ersten beiden Cystein-Moleküle des Chemokins unmittelbar hintereinander liegen; bei den CXC-Zytokinen liegt zwischen ihnen eine andere Aminosäure. Die meisten Chemokine werden unter pathologischen Bedingungen von Zellen verschiedener ortständiger Gewebe und von infiltrierenden Leukozyten gebildet. Neuerdings sind zwei weitere Funktionen der Chemokine entdeckt worden: die Hemmung und Stimulation der Angiogenese und der Myelopoiese. Tab.10-3 Die verschiedenen Chemokine stehen vor allem Dienste der Chemotaxis (siehe Kapitel 23: Entzündungsreaktionen) und der Migration und Rekrutierung von Leukozyten, darunter auch der immunkompetenten Zellen. Die Tabelle ist nicht vollständig. ___________________________________________________________________________ Biologische Aktivität ChemokinRezeptoren ___________________________________________________________________________ Migration naiver T-Zellen zu den Lymphknoten und Peyer Plaques naiver T-Zellen innerhalb der lymphatischen Gewebe der Gedächtnis T-Zellen 2 in die lymphatischen Gewebe der Gedächtnis T-Zellen in die Haut der Gedächtnis T-Zellen in die Darmmukosa der Gedächtnis T-Zellen in Entzündungsherde Chemokine TCA-4, MIP 1 CCR7 SDF-1 CXCR4 TCA-4, MIP CCR7 MDC CCR4 TECK CCR9 MCP CCR2 RANTES, MIP CCR5 der TH1-Zellen (Effektorzellen) MCP CCR2 RANTES, MIP CCR5 IP-10 CXCR3 der TH2-Zellen (Effektorzellen) Eotaxin CCR3 MDC CCR4 SDF-1 CXCR4 B-Zellen TCA-4, MIP CCR7 SDF-1 CXCR4 der dendritischen Zellen in die lymphatischen Gewebe TCA-4, MIP CCR7 der dendritischen Zellen in die Haut MIP CCR6 der dendritischen Zellen in Entzündungsherde MCP CCR2 RANTES, MIP CCR5 IL-8 CXCR1 von hämatopoietischen Stammzellen SDF-1 CXCR4 von prä-B-Zellen SDF-1 CXCR4 Rekrutierung von Monozyten PBF-4 RANTES, MIP CCR5 MCP CCR2 IL-8 CXCR1 neutrophilen Granulozyten IL-8 CXCR1 MIP CCR6 PBP, PBF-4 eosinophilen Granulozyten Eotaxin CCR3 RANTES, MIP CCR5 basophilen Granulozyten IL-8 CXCR1 Fibroblasten PBP ___________________________________________________________________________ Tab.10-3 (Fortsetzung) ___________________________________________________________________________ Biologische Aktivität ChemokinRezeptoren ___________________________________________________________________________ Aktivierung von Monozyten Lymphozyten neutrophilen Granulozyten eosinophlen Granulozyten basophilen Granulozyten (Histaminfreisetzung) Proliferation von hämatopoietischen Stammzellen prä-B-Zellen Endothelzellen Chemokine MCP TCA-4, MIP IL-8 MIP RANTES, MIP RANTES CCR2 CCR7 CXCR1 CCR6 CCR5 CCR5 MIP CCR6 MIP CCR6 IL-8 CXCR1 PBF-4 Blutplättchen PBF-4 glatten Muskelzellen PBF-4 ___________________________________________________________________________ 1 Bei den MIP und MCP wird nicht zwischen den einzelnen Typen unterschieden. 2 Die Gedächtnis-T-Zellen werden in zwei Subgruppen unterteilt: in die CCR7-Rezeptor positiven (CCR7+) und CCR7-Rezeptor negativen (CCR7-). Die CCR7+ Gedächtniszellen exprimieren das L-Selektin, was ein Hinweis dafür ist, dass sie vor allem in die Lymphknoten wandern; die CCR7- besitzen das L-Selektin nicht und finden sich vor allem an den Wirkungsorten in peripheren, nicht lymphatischen Geweben. Die CCR7 + werden deshalb auch als «zentrale Gedächtniszellen» bezeichnet, die CCR7- als «Effektor-Gedächtniszellen». IP-10 MIP MCP PBP PBF-4 RANTES SDF-1 TCA-4 TECK Inducible Protein-10 (10 Kilodaltons) Makrophagen inflammatorisches Protein (MIP-1, MIP-2, MIP-3) Monozyten chemotaktisches Protein (MCP-1, MCP-2, MCP-3) Plättchen basisches Protein Plättchenfaktor-4 (Oncostatin) Regulated upon activation, normal T-expression presumably secreted Stroma cell Derived Factor-1 Thymus derived chemotactic agent Thymus expressed chemokine Molekularpathologisch ebenso wichtig wie die Chemokine selber sind deren Rezeptoren (Heparin-ähnliche Glykosaminoglykane) auf der Oberfläche vieler Zellen und in der ECM, an welche sie binden. Die Verteilung dieser Rezeptoren entspricht einer «Fahrrinne», in der die immunkompetenten Zellen (Lymphozyten, Antigen-präsentierende Zellen) an ihren Einsatzort gelangen können. Wenn verschiedene Chemokine und Chemokinrezeptoren benötigt werden, um die Zellen richtig zu lotsen, spricht man von «Multistep navigation». Abb.10-2 Die Chemokine lösen eine gerichtete Zellbewegung aus, entweder (1) über eine Anheftung der Zelle an ein Substrat oder (2) eine Abkoppelung der Zelle von einem Substrat. Die Zelle bewegt sich dann entweder über die Ausbildung von Pseudopodien oder über Kontraktionen des Zytoskeletts. Die Chemokine binden an spezifische Rezeptoren. Diese Rezeptoren werden auch von infektiösen Agentien [z.B. Herpesviren und Human Immunodeficiency Viren (HIV)] benützt. Beim Eintritt der HIV in die Zellen spielen zwei Faktoren eine Schlüsselrolle: (1) Kofaktoren zum Glykoprotein 120 (gp120), welches auf der Oberfläche der Viren liegt, und (2) Suppressorfaktoren, welche durch aktivierte CD8-Lymphozyten gebildet werden und die Virusreplikation in aktivierten CD4-Lymphozyten hemmen (Abb.10-3). Die Kofaktoren des gp120 auf der Oberfläche der vom HIV angegriffenen Zellen sind die beiden Chemokinrezeptoren CXCR4 und CCR5. CXCR4 (auch «Fusin» genannt) wird von den T-tropen Viren (Viren, welche sich nur in transformierten T-Lymphozyten vermehren) benützt, CCR5 von den HIV, welche nur Monozyten und Makrophagen zu infizieren vermögen (M-trope Viren). Es ist inzwischen bekannt, dass eine homozygote Mutation des CCR5 vor einem Infekt mit dem Mtropen HIV schützt und eine heterozygote Mutation den Ausbruch des AIDS um zirka zehn Jahre zu verzögern vermag. Ein relativer Schutz vor einem Infekt mit einem T-tropen HIV wird auch durch eine Mutation des Liganden des CCR5 (SDF-1: Stroma cell Derived Factor-1) gewährleistet. Der mutierte Ligand löst eine verstärkte Bildung von normalem SDF-1 aus. Die so erhöhte Konzentration des SDF-1 reicht aus, um den CXCR4 kompetitiv zu blockieren und die Bindung des HIV an die T-Lymphozyten zu verunmöglichen. Auf eine Ansteckung mit dem HIV hin folgt gewöhnlich eine lange Latenzzeit. Dahinter verbirgt sich wahrscheinlich eine durch CD8+-Lymphozyten vermittelte Suppression der Virusinfizierten CD4+Zellen (Abb.10-3). Diese Suppression wird durch einen «Cell Antiviral Factor» (CAF), welcher von den die CD8+-Zellen abgegeben wird, vermittelt. Diese antivirale Funktion der CD8+-Zellen wird durch das IL-2 der nicht-infizierten TH1-Lymphozyten verstärkt, weil IL2 die CD8+-Zellen stimuliert. Das von den TH1-Zellen ebenfalls sezernierte INF- blockiert die TH2-Zellen und reduziert so die durch IL-10 vermittelte Hemmung der Sekretion des CAF. Unter diesen Bedingungen können die CD8+-Zellen genügend CAF bilden. Beim Fortschreiten der Krankheit stellt sich aber allmählich ein Überwiegen der Expression der Zytokine der TH2Zellen ein («Dominanz der TH2-Zellen»). Dies führt gleichzeitig zu einer Hemmung der TH1Zellen und zu einer verminderten Sekretion des CAF durch die CD8+-TH-Zellen. Abb.10-3 Die infizierten CD4+Zellen verharren in einem stabilen Status, weil sie durch CD8 +-Lymphozyten supprimiert werden. Dies ist durch eine «Dominanz der TH1-Zellen» gewährleistet. Weicht diese «Dominanz der TH1-Zellen» einer «Dominanz der TH2-Zellen» kommt es zu einer Reduktion der antiviralen Aktivität der CD8+-Lymphozyten (siehe Text). Es konnte neuerdings nachgewiesen werden, dass verschiedene von Viren gebildete Proteine mit Interleukinen interagieren können. Diese viralen Proteine werden als Virokine bezeichnet. So kann das Vacciniavirus ein Protein sezernieren, welche IL-1 bindet, oder das Myxomavirus ein Protein, welches an den Rezeptor für das IFN- andockt, oder das Cytomegalievirus ein Protein, welches dem Rezeptor des Makrophagen inhibitierenden Proteins 1 (MIP-1) sehr ähnlich ist. Der Tumornekrosefaktor (TNF) ist der Hauptmediator für die Entzündungs- und Schockreaktionen des Organismus. Es werden zwei Typen des TNF unterschieden: (1) Der TNF (Kachektin) wird von Makrophagen gebildet. Er wird als Antwort auf unspezifische Reize verschiedener Bakterien sezerniert. (2) Der TNF- (Lymphotoxin) wird von den T- und BLymphozyten sowie den Fibroblasten als Antwort auf einen Stimulus durch ein Antigen oder einen mitogenen Reiz gebildet. Die potentesten Stimulatoren für den TNF- sind bakterielle Toxine [vor allem Lipopolysaccharide gramnegativer Bakterien (LPS)] und INF-. Die LPS werden im Blut an das LPS-bindende Protein (LPSBP) gekoppelt transportiert). Der TNF kommt sowohl in membrangebundener als auch in löslicher Form vor. Der TNF- kann an zwei verschiedene Rezeptoren binden: an den Tumornekrosefaktor-Rezeptor-1 (TNFR1) und an den TNFR2. Diese beiden Rezeptoren gehören in die Familie der Rezeptoren, zu welcher auch der Rezeptor für den Nerven-Wachstumsfaktor und für das Fas-Protein (Fas-Ligand genannt) gezählt werden. Der Name «Tumornekrosefaktor» stammt von der ursprünglichen Beobachtung her, dass Tumoren bei Patienten mit schweren bakteriellen Infekten nekrotisch werden können. Dieses Phänomen ist gut erklärbar: Bakterielle Endotoxine können - an das LPSBP gekoppelt - über eine Bindung an das CD14-Oberflächenprotein der Makrophagen die Makrophagen zu einer verstärkten Abgabe des TNF-, des IL-6 und von Stickoxid (NO) stimulieren. Der TNF- schädigt unter anderem die Endothelzellen der Gefässe in der Umgebung seines Wirkungskreises (Tab.10-4), sodass sich Mikrothromben oder eine disseminierte intravasale Gerinnung begleitet von einer Hypoxie entwickeln können. Da der TNF- aber auch neutrophile Granulozyten zur Bildung von freien Radikalen anregt, wird die durch die Hypoxie bereits eingeleitete Gewebedestruktion noch verstärkt. Der TNF- vermag ebenfalls einen direkten Effekt auf den Hypothalamus auszuüben mit dem Resultat eines verminderten Appetites. Andererseits scheint der TNF- die Bildung des Transkriptionsfaktors MyoD, welcher für die Differenzierung und Ausreifung der Myozyten von Bedeutung ist, zu hemmen, und zwar über eine Aktivierung des NF-B (Nuclear Factor-B): Der aktivierte NF-B supprimiert die mRNA des MyoD. Dies Fakten erklären teilweise den Gewichtsverlust (Kachexie) bei Patienten mit einem malignen Tumor. Das NO induziert eine Vasodilatation, welche zu einem «warmen», nicht durch einen Blutverlust bedingten Schock (Endotoxin-Schock) führen kann. Tab.10-4 Der TNF-a hat sehr verschiedene und vielfältige Funktionen (siehe Text). Im Vordergrund steht die Funktion als Hauptmediatoren der Entzündung. ___________________________________________________________________________ Ort der Wirkung Art der Wirkung ___________________________________________________________________________ Zellen (generell) Endothelzellen Direkte zytotoxische Wirkung (Mechanismus nicht defnitiv klar) Reduktion der antithrombotischen Wirkung des Oberflächenproteins Thrombomodulin Gesteigerte Synthese prothrombotischer Faktoren (Thromboplastin und Gerinnungsfaktor IX): Folge ist eine intravasale Gerinnung Expression von Adhäsionsmolekülen für neutrophile Granulozyten (ELAM-1, ICAM-1, VCAM-1): Folge ist ein ermehrtes Rolling der neutrophilen Granulozyten Reduktion der Synthese der Lipoprotein-Lipase: Folgen sind (1) eine eduktion der Triglyzeride und freien Fettsäuren in den Lipozyten mit der Möglichkeit, dass sich eine Kachexie entwickelt und (2) eine gesteigerte Synthese von Interleukin-1 (IL-1) Neutrophile Granulozyten Chemotaxis Degranulierung Stimulierung der Bildung freier Radikale ___> Gewebenekrosen Verstärkung der Adhärenz an die Endothelzellen und an andere neutrophile Granulozyten Fettstoffwechsel Hemmung der Fettsäuren-Synthetase Hemmung der Acetyl-Coenzym A-Carboxylase Quergestreifte Muskulatur Hemmung des Transkriptionsfaktors MyoD (siehe Text) Thymozyten Proliferation Fibroblasten Aktivierung Zytotoxische T-Lymphozyten Reifung Gesteigerte Expression der MHCP Gesteigerte Expression von Interleukin-6 (IL-6) Epithelzellen der intrahepatischen Cholestase Gallengänge ___________________________________________________________________________ 10.1.4. Prionen (protein related infectious agent) sind nicht-mikrobielle Proteine, welche Ursache ansteckender Krankheiten sein können. Die Prionen-Erkrankungen gehören in die grosse Gruppe der degenerativen Erkrankungen des zentralen Nervensystems. Die Krankheiten weisen die folgenden Merkmale auf: (1) Es sind keine mikrobiellen Partikel nachweisbar. (2) Es fehlt primär eine Entzündungsoder immunologische Reaktion. (3) Die Krankheit ist mit Plaques im Hirngewebe assoziiert. Um diese Plaques herum ist das Hirngewebe vakuolisiert. Dieses Phänomen hat der Krankheit auch den Namen «spongioforme Encephalopathie» eingebracht (Abb.10-4). (4) In den Plaques um die vakuolisierten Areale herum oder diffus im Hirngewebe wird ein abnormales Protein abgelagert. (5) Eine Schädigung von Nucleinsäuren (z.B. durch eine Bestrahlung des Hirngewebes) hemmt das Auftreten der Krankheit nicht. (6) Das Gen, welches das nicht-infektiöse Protein kodiert, ist bekannt. Es ist physiologischerweise vor allem in Lymphozyten und Neuronen aktiv. Abb.10-4 Das Hirngewebe von Patienten, welche an einer Prionenkrankheit leiden, zeigt schwammförmige Vakuolen. Davon stammt die für die Krankheit auch verwendete Bezeichnung «spongioforme Enzephalopathie» ab [spongia (lateinisch) = der Schwamm]. Man unterscheidet zwei Formen von Prionen: die normalen (harmlosen, PrPC-Form) und die pathogenen (PrPRES-Form). Die pathogenen Prionen können beim Menschen vier Krankheitsbilder hervorrufen: (1) die Creutzfeldt-Jakob-Krankheit (CJD), (2) die Fatale Familiäre Schlaflosigkeit (FFI), das (3) Gerstmann-Sträussler-Scheinker-Syndrom (GSS) und (4) Kuru, eine CJD-ähnliche Erkrankung der Eingeborenen Neuguineas, die angeblich durch rituellen Kannibalismus weitergegeben wird. Die CJD tritt mit einer Häufigkeit von zirka 1 zu 1'000'000 Personen pro Jahr auf. Die Latenzzeit zwischen dem Auftreten der pathogenen Prionen und dem Ausbruch der Krankheit wird auf 5-7 Jahre geschätzt. Das normale PrP (PrPC) des Menschen kann in zwei Ausprägungen vorhanden sein: Entweder ist an der Position 129 die Aminosäure Methionin oder die Aminosäure Valin vorhanden. Neuere Untersuchungen zeigten, dass das PrPC zwei verschieden strukturierte Zonen aufweist: (1) wendeltreppenartige Abschnitte (-Helices) und (2) zwei als -Faltblätter bezeichnete, leiterähnliche Abschnitte. Eine der «Wandeltreppen» verbindet in Form eines Zwischenbogens die beiden «Leiterstangen» und ist deshalb vom übrigen Teil des Proteins etwas abgewandt. Dadurch erhält das ganze Molekül eine spezielle Beweglichkeit. Es sind Hinweise darauf vorhanden, dass es in pathogenen Prionen am «Zwischenbogen» zur Bildung eines zusätzlichen -Faltblattes («Leiterstange») gekommen ist. Eine solche Umfaltung könnte durch eine «falsche» Aminosäure im «Zwischenbogen» nach einer Mutation verursacht sein. Bereits vor 30 Jahren wurde zur Erklärung der Pathogenese der Prionen-Erkrankungen die «Protein-only»-Hypothese aufgestellt. Diese Hypothese postuliert, dass eine Konformationsänderung des PrPC in den Neuronen durch gleichzeitig vorhandene PrPRES hervorgerufen wird und sich schneeballartig fortsetzen kann. Ausgangspunkt dieses Prozesses ist ein Kontakt zwischen einem PrPRES und einem PrPC. Dieser Kontakt bewirkt, dass sich das normale PrPC «umfaltet» und zu einem PrPRES wird. Das PrPC ist also mit einem Substrat in einem chemischen Prozess zu vergleichen, welcher vom PrPRES katalysiert wird (Abb.10-5). Der genaue Mechanismus dieser katalytischen Konversion (auch als «Matrizenmodell» bezeichnet) ist noch nicht bekannt. Das neu entstandene PrPRES tritt wiederum mit einem normalen PrPC in Kontakt. Der Prozess setzt sich fort, bis die Menge an PrPRES in den Neuronen ein kritisches Ausmass erreicht hat und eine Vakuolisierung der Neuronen eintritt. Diese Vakuolisierung ist wahrscheinlich Ausdruck einer Schädigung der intraneuronalen Lysosomen. Abb.10-5 Die «Protein-only-Hypothese» postuliert, dass pathologische Prion-Proteine (PrPRES) aus normalen PrionProteinen (PrPC) durch Umfaltung einer a-Helix in den PrPC)unter Einwirkung der PrPRES entstehen können. In Tiermodellen konnte gezeigt werden, dass die Prionenkrankheit ausbleibt, wenn im Hirngewebe kein normales Prion vorhanden ist. Für die «Protein-only»-Hypothese spricht, dass meistens eine gute Übereinstimmung zwischen der Menge an PrPRES und dem sogenanntem «infektiösen Agens» festgestellt werden konnte. Zusätzlich war nachweisbar, dass die Abwesenheit des normalen PrPC zu einem vollständigen Schutz gegenüber einer Infektion mit Prionen führt. Andererseits steht der Beweis noch aus, dass PrPRES auch in vitro PrPC in einen infektiösen Erreger umzuwandeln vermag. Alternativ zum vorgestellten Modell der Konversion wird ein «Kristallisationsmodell» diskutiert. Dieses Modell unterscheidet sich vom Modell der Konversion durch die Annahme, dass die physiologische Bildung von PrPRES pathologisch gesteigert sei. 10.1.5. Die Immunglobuline Immunantworten. sind die humoralen Mediatoren der Das Immunsystem ist einer der wichtigsten Verteidigungsmechanismen des Organismus' gegen Bakterien, Viren, Pilze und andere Fremdmoleküle, die Infektionen oder eine Zellschädigung anderer Art verursachen können. Die Reaktion des Immunsystems läuft grundsätzlich in zwei Phasen ab: (1) Erkennung und Identifikation der gefährlichen oder eingedrungenen Moleküle und (2) Auslösung einer spezifischen Abwehr. Die Abwehr kann wiederum über zwei Achsen erfolgen: (1) über Antikörper (AK), die an das Antigen (AG) binden und dadurch das AG neutralisieren oder (2) über die Mobilisierung von Zellen (Effektorzellen), welche das AG und seinen Träger aufspüren und den Träger dann zerstören (siehe Kapitel 24: Immunpathologie). Die AK an sich sind für die Bekämpfung eines Krankheitserregers nicht sehr wirksam. Mit den Signalen aber, die sie auslösen können, werden spezialisierte Zellen und weitere Proteine rekrutiert, um die Zellen, welche das AG enthalten, zu zerstören. Die AK sind Immglobuline mit einer spezifischen Affinität zu den AG. Entsprechend ihrer Struktur und Wirkungen werden verschiedene Typen von Immunglobulinen unterschieden (Tab.10-5). Das Immunglobulin M (IgM) wird fast ohne Zeitverzug unmittelbar nach der Konfrontation des Organismus mit einem AG gebildet. Allerdings ist IgM ein «minderwertiger» AK, der schnell wieder abgebaut wird. Seine Schutzwirkung ist begrenzt, doch sorgt er für eine Art «erste Hilfe», solange noch keine wirkungsvolleren AK verfügbar sind. Bedeutend hochwertiger sind die IgG-AK. Deren Produktion kommt allerdings erst zirka zehn bis vierzehn Tage nach dem ersten Kontakt mit dem AG in Gang. Tab.10-5 Überblick über die verschiedenen Antikörper. ___________________________________________________________________________ Eigenschaften Antikörperklasse _________________________________________________________ IgM IgD IgG IgA IgE ___________________________________________________________________________ Schwere Ketten Leichte Ketten oder oder oder oder oder Zahl der Grundeinheiten 5 1 1 1 oder 2 1 % des gesamten 10% < 1% 75% 15% <1% Immunglobulins Komplementaktivierung ++++ ++ «Erste_Hilfe» ++++ Sekundäre ++++ Immunantwort Bindung an Rezeptoren + ++++ von ACP Durchquerung der + Plazenta Bindung an + Makrophagen und neutrophile Granulozyten Bindung an + Mastzellen, basophile und eosinophile Granulozyten Fixation der Komple+ + ment C1-Komponente Transport durch + + Epithelzellen In Körpersekreten* + ___________________________________________________________________________ APC * Antigen-präsentierende Zellen: Makrophagen, neutrophile Granulozyten Speichel, Bronchial- und Urogenitalsekret, Milch Die Form der Immunglobulin-Moleküle ist für ihre Wirkung von grosser Bedeutung. IgM besteht aus fünf einzelnen Untereinheiten, verfügt also über zehn potentielle Bindungsstellen mit AG. Sein Wirkungsspektrum ist darum sehr breit. Weil das Molekül aber relativ gross ist, bleibt seine Wirkung weitgehend auf den Blutkreislauf beschränkt; es kann nur sehr beschränkt in die Gewebe hineindiffundieren. IgG ist der AK der ausgereiften Immunantwort. Er besteht (im Gegensatz zu IgM und IgA) wie IgD und IgE aus nur einer Einheit. IgG ist ein kleines, «zähes» Molekül mit einer Lebensdauer von zirka einem Monat. Diese Eigenschaften kommen z.B. dem Fötus zugute, wenn gegen Ende der Schwangerschaft die Mutter ihre IgG auf den Fötus überträgt. Auf diese Weise ist das Neugeborene während seiner ersten Lebenswochen vor Infektionskrankheiten geschützt. IgA besteht aus zwei Untereinheiten und ist vor allem auf Schleimhäuten, die mit der Umwelt direkt in Kontakt kommen, vorhanden. Es kann Krankheitserreger abfangen, bevor sie in den Körper eindringen. Bei modernen Immunisierungskonzepten spielt die Anregung der Produktion von IgA eine wichtige Rolle, weil man auf diese Weise eine Infektion im frühestmöglichen Stadium verhindern will. Das Molekül ist gegen proteolytische Enzyme, die in fast allen Körpersekreten vorhanden sind, durch eine ins Molekül integrierte J-Kette und ein Polypeptid, Sekretions-Komponente genannt, geschützt. 10.1.6. Transkriptionsfaktoren sind intrazelluläre Proteine, welche an regulatorische Sequenzen der DNA binden. Die Transkriptionsfaktoren (TF) (Tab.10-6) bilden als tertiäre Messengers das letzte Glied in der Signaltransduktion: Sie übertragen das Signal aus dem Zytoplasma in den Zellkern. Dort lagern sie sich üblicherweise an die Promotor-Region des entsprechenden Gens und gleichzeitig an die Polymerase an. Die Bindungsstellen an der DNA werden Transscription Factor Response Elements (TFREs) genannt. Tab.10-6 Die Transkriptionsfaktoren stellen das Endglied der Signaltransduktion dar. ___________________________________________________________________________ TranskriptionsTranskription Transkription Transkription Faktoren assoziiert mit ... aktiviert durch gehemmt durch ___________________________________________________________________________ Activator Protein-1 Zellproliferation Phorbol-Ester (AP-1, Heterodimer Blockade aktivierter GR TNF- aus Fos- und JunIL-1 Proteinen) Nuclear factor B Stickstoff-Monoxid-Synthese TNF- (NF-B) Cyclooxygenase Wasserstoff-Peroxid Chemokinen (IL-8, RANTES) Ozon Virusinfekte Adhäsionsmolekülen (ICAM-1, VCAM-1) Blockade aktivierter GR STATs (Signed Zytokine (IL-2, IL-6, IL-5) Transduction Activated via Janus-Kinasen Transscription factors) STAT-1 STAT-2 STAT-3 Glukocortikoide ___________________________________________________________________________ Tab.10-6 (Fortsetzung) ___________________________________________________________________________ TranskriptionsTranskription Transkription Transkription Faktoren assoziiert mit ... aktiviert durch gehemmt durch ___________________________________________________________________________ cAMP-Response Hemmung des AP-1 und der Element Binding GR Protein (CREB) ___________________________________________________________________________ GR TNF IL RANTES ICAM VCAM Glucocorticoid-Rezeptoren Tumornekrosefaktor Interleukin Regulated upon Activation, Normal T-Expression persumably Secreted Intercellular Adhesion Molecule Vascular Cellular Adhesion Molecule TF können auf verschiedene Arten aktiviert werden: (1) via Rezeptoren auf der Zelloberfläche oder (2) durch eine Bindung mit intrazytoplasmatischen Liganden (z.B. Glucocorticoide, Schilddrüsenhormone, Vitamin D). Die Hauptaufgabe der aktivierten TF besteht in einer Weitergabe eines kurzdauernden externen Signals an den Zellkern, um länger dauernde Mechanismen des Zellwachstums, der Zelldifferenzierung und der Zellfunktion in Gang zu setzen. Manche der TF kommen in vielen Zellen vor, andere wiederum sind zellspezifisch. Ein wichtiger, in die Entzündung involvierter Transkriptionsfaktor ist der Nuclear Factor-B (NF-B). Er wurde ursprünglich als Regulator der Expression des Gens für die Leichtketten in B-Lymphozyten der Maus entdeckt. Der NF-B liegt in einer aktivierten und einer inaktivierten Form vor. In der aktivierten Form entspricht er einem heterodimeren Molekül bestehend aus zwei Proteinen, dem P65-Protein (auch relA genannt) und dem P50-Protein (Abb.10-6a). In der inaktiven Form ist NF-B an die Proteine IB und IB gebunden und befindet sich im Zytoplasma der Zellen. Nach einer Zellstimulation wird das Molekül IB durch eine spezifische Kinase phosphoryliert. Dadurch wird es vom NF-B abgekoppelt. Der frei gewordene NF-B diffundiert in den Zellkern und übt dort seine Wirkung aus. Abb.10-6a Der Nuclear factor-kB (NF-kB) ist ein heterodimeres Molekül, welches aus den beiden Proteinen P50 und P65 besteht. Er wird durch das Molekül IkBa gehemmt. Im Zellkern wirkt NF-kB als Transkriptionsfaktor für verschiedene Proteine. Er wird durch eine Phosphorylierung von IkBa aus dem Komplex mit IkBa freigesetzt. Cyclosporin hemmt die Aktivierung des NF-kB. Der Transkriptionsfaktor NF-B reguliert die Expression verschiedener Proteine (proinflammatorische Zytokine, Chemokine, inflammatorische Enzyme, Adhäsionsmoleküle und Rezeptoren) und kann via verschiedene Stimuli aktiviert werden [Zytokine, Proteinkinase CAktivatoren, Oxidantien (z.B. Ozon), Viren, immunogene Stimuli, Lipopolysaccharide und Ultraviolettstrahlen]. Eine Aktivierung von NF-B kann eine koordinierte, gleichzeitige Expression verschiedener Gene wichtiger Entzündungsmediatoren bewirken. Dies ist beispielsweise bei einer akuten Entzündung für die Rekrutierung und Aktivierung der neutrophilen Granulozyten der Fall. Dazu werden das Adhäsionsmolekül E-Selectin, das Chemokin Interleukin-8 und der Tumornekrosefaktor benötigt. NF-B kann aber auch die Stickoxid-Synthase exprimieren oder die Cyclooxygenase-2, ein Enzym, welches für die Synthese von Prostaglandinen und Thromboxan A2 zuständig ist. Der NF-B steuert seine Wirkung selber: Das Auftreten des NF-B induziert die Synthese von IB, weil das IB-Gen eine Erkennungssequenz für B besitzt. Das IB tritt in den Zellkern ein, bindet dort an den NF-B, transportiert den NF-B aus dem Zellkern ins Zytoplasma und beendet so die Arbeit des NF-B im Zellkern. Glukokortikoide können die Wirkung der NF-B bei chronischen Entzündungen hemmen. Dies geschieht wahrscheinlich auf zwei Arten (Abb.10-6b). (1) Die Glukokortikoide werden im Zytoplasma an den Glukokortikoid-Rezeptor gebunden. Dann wird der Komplex in den Zellkern transportiert. Dort wirkt er direkt als Transkriptionsfaktor, in dem er an das Glukokortikoid-Response Element des IB-Gens bindet und die Expression von IB induziert. IB blockiert den NF-B.(2) Der Komplex bestehend aus dem Glukokortikoid und Glukokortikoid-Rezeptor bindet im Zellkern direkt an den NF-B (P50 + P65). Dadurch wird die Bindung des NF-B an die DNA verunmöglicht . 10.2. DEFEKTE DER SYNTHESE, DES ABBAUS ODER DER AUSSCHEIDUNG VON PROTEINEN KÖNNEN SICH IN FORM VERSCHIEDENER KRANKHEITEN MANIFESTIEREN. Defekte in der Proteinsynthese können ein Zuviel oder ein Zuwenig an Proteinen bewirken; bei einer Abbaustörung oder verminderten Ausscheidung kommt es zu einer intrazellulären oder extrazellulären Ablagerung und dadurch zur Beeinträchtigung von Organfunktionen. Auch die Enstehung maligner Tumoren ist schlussendlich auf eine Störung von Proteinen zurückzuführen: Die bei malignen Tumoren nicht korrekt funktionierenden Proteine sind oft an der Steuerung und Kontrolle des Zellzyklus oder an der Zellproliferation beteiligt (siehe Kapitel 21: Tumoren). Abb.10-6b Die Glukokortikoide können den Nuclear Factor-kB (NF-kB, P50 + P65) auf zwei Arten hemmen: (1) direkt durch eine Bindung des Komplexes Glukokortikoid/Glukokortikoid-Rezeptor an den NF-kB und (2) indirekt durch eine Induktion der Expression des NF-kB-Inhibitors (IkBa). 10.2.1. Intrazelluläre Proteinablagerungen können Hinweis auf eine veränderte Proteinsynthese oder eine abnorme Struktur der Proteine sein. Pathologische intrazelluläre Proteinablagerungen sind morphologisches Schlüsselmerkmal bei verschiedenen Krankheitsbildern (Tab.10-7), vor allem bei Virusinfekten, bei Krankheiten, welche mit einer Störung des Zyto- oder Membranskeletts einhergehen, beim 1-Antitrypsinmangel (AT) und bei der Alzheimer Krankheit. Tab.10-7 Pathologische intrazelluläre Proteinablagerungen kommen in verschiedenen Organen vor und können morphologisches Begleitsymptom verschiedener Krankheiten sein. ___________________________________________________________________________ Typ der Typ des Organ Ursache Beispiele von Krankheiten Ablagerung Proteins ___________________________________________________________________________ Intrazellulär Hyalin Haut Virusinfekt Hyalin Leber Strukturanomalien des Zytoskeletts Hirn Erythrocyten Extrazellulär Hyalin Hyalin Leber Nierentubuli Plasmazellen Amyloid multipel Amyloid Hirn Hyalin multipel Hyalin Nierenrinde Fibrinoid Strukturanomalien des Membranskeletts Gestörte Proteinsynthese Proteinurie Gesteigerte Funktion der Plasmazellen Tumoren des lymphopoietischen Systems Chronische Entzündungen Gestörte Synthese und gestörter Katabolismus von Proteinen Fusion von Kollagenfasern Störungen der mesangialen Matrix der Glomerula Störungen der Blutgefässwand Nekrose in Bindegewebe Verruca vulgaris (Papillomavirus) Condyloma accuminatum (Papillomavirus) Zytomegalie (Herpesvirus) Herpes genitalis Molluscum contagiosum (Variolavirus) Aethylabusus Alzheimer Krankheit Hereditäre Sphärozytose 1-Antitrypsinmangel Nephrotisches Syndrom Chronische Entzündungen Plasmocytom kanAmyloidose Amyloidose Alzheimer Krankheit Narben, Ulcera Diabetes mellitus, Glomerulonephritis Hypertonie Ulcera Rheumatismus nodosus Sklerodermie Lupus erythematodes Dermatomyositis Nekrose in Blutgefäss- Polyarteriitis nodosa wänden Nekrotisierende Arteriitis ___________________________________________________________________________ 10.2.1.1. Viruskrankheiten. Viren sind bewegliche, in Proteine eingehüllte Gene. Sie enthalten entweder RNA oder DNA. Viren synthetisieren ihre eigenen Proteine selber oder lassen sie von den infizierten Zellen synthetisieren und lagern sie in den infizierten Zellen ab. Die meisten Viren besitzen zwei verschiedene Hüllen: die Hülle unmittelbar um die Nukleinsäure herum (Capsid) und die Virushülle (äussere Hülle) (Abb.10-7). Zur Herstellung der äusseren Hülle fügen die Viren die neu synthetisierten Proteine ihrer Membran in die Zellmembran der Wirtszelle ein. Anschliessend knospen die virale Nukleinsäure, das Capsid, die Lipid-Doppelschicht der Zellmembran und die in die Zellmembran eingelagerten viralen Membranproteine als ein neu generiertes Virus aus der Zelle aus (Abb.10-8). Abb.10-7 Die Viren besitzen eine Proteinhülle unmittelbar um ihre DNA oder RNA herum (Capsid). Daneben verfügen sie über eine zweite äussere Hülle, in die zusätzlich Glykoproteine eingelagert sind. Das Human Immunodeficiency Virus (Erreger von AIDS, ein RNA-Virus) dockt mit diesen Proteinen seiner äusseren Hülle an die Rezeptoren der von ihm infizierten Zellen an. Viren gelangen über spezifische Lipoprotein-Rezeptoren der Wirtszelle in die Wirtszelle hinein. Der Eintritt in die Zellen kann über die apikale Zellmembran (z.B. Influenzavirus) oder über die basolaterale Zellmembran (z.B. Stomatitisvirus) erfolgen. Die Transkription der DNA-Viren erfolgt in zwei Schritten: (1) Im ersten Schritt werden die «frühen» Proteine synthetisiert. Diese werden für die Synthese der neuen viralen DNA benötigt. (2) Im zweiten Schritt werden die Gene der «späten» Proteine transkribiert. Diese «späten» Proteine sind Bestandteile der beiden Hüllen (Capsid und äussere Virushülle). Die Polymerasen, welche für die Transkription der DNA gebraucht werden, stammen meistens von der infizierten Wirtszelle. Die Replikation der RNA-Viren erfolgt über die viruseigene reverse Transkriptase (eine RNAabhängige DNA-Polymerase). Abb.10-8 Für den Austritt der Viren aus einer Zelle bedienen sich die Viren der Zellmembran, indem sie die Proteine der Virushülle vorübergehend in der Zellmembran der Wirtszelle deponieren. Ein Virus kann eine Wirtszelle auf fünf verschiedene Arten beeinflussen. (1) Der Virusinfekt limitiert sich selbst, strukturelle Veränderungen in der Zelle bleiben aus («latenter» Infekt) (Abb.10-9a). (2) Das Virus induziert den Untergang der Wirtszelle (Abb.10-9b). Der Zelluntergang kann verschiedene Ursachen haben: (i) eine starke, intrazelluläre Vermehrung von Viruspartikeln, (ii) eine Unterbrechung normaler synthetischer Aktivitäten der Wirtszelle durch Proteine, welche durch das Virusgenom exprimiert worden sind, oder (iii) eine immunologische zytotoxische Abwehrreaktion, welche durch virale, in die Zellmembran eingelagerte Proteine ausgelöst wird. (3) Das Virus induziert Änderungen an den Oberflächenproteinen der Zellmembran. Dies ist vor allem typisch für Viren der Gruppe der Paramyxoviren. Folge solcher Veränderungen sind Fusionen zwischen infizierten und nichtinfizierten Zellen, die sich in Form von Riesenzellen manifestieren (Abb.10-9c). Ein Beispiel dafür sind die Warthin-Finkeldy Riesenzellen in Lymphknoten von Patienten mit Masern. (4) Im Zellkern oder im Zytoplasma der Wirtszellen bilden sich Einschlusskörper. Diese können aus viralen Proteinen bestehen, aber auch aus vermehrt synthetisierten zellulären Proteinen (Abb.10-9d und Tab.10-8). (5) Einige Virusinfekte können eine Proliferation der Wirtszelle induzieren (Abb.10-9e). Beispiele dafür sind die verschiedenen Tumoren, welche durch einen Virusinfekt hervorgerufen werden oder die infektiöse Mononukleose, eine Lymphadenitis vor allem junger Erwachsener. Abb.10-9 Es können fünf verschiedene Effekte von Viren auf die Wirtszelle unterschieden werden: (a) kein Effekt, (b) Zelluntergang (häufig), (c) Bildung von Riesenzellen, (d) entweder intrazytoplasmatische oder intranukleäre Viruseinschlüsse und (5) eine Proliferation der Wirtszellen. Tab.10-8 Virusinfekte können sich morphologisch durch intrazytoplasmatische oder intranukleäre Einschlüsse in den Wirtszellen manifestieren. ___________________________________________________________________________ Virus Einschlüsse Organbefall Krankheit ___________________________________________________________________________ DNA Adenoviren Hep-a-DNA Virus Hepatitis B Virus Herpesviren Herpes simplex Virus Varizellavirus Zytomegalievirus Papovaviren Papillomavirus Poxviren Orthopox-Virus Variolavirus Intranukleär Intrazytoplasmatisch Leber Hepatitis Intranukleär Genitaltrakt Mundhöhle Gingiva-Stomatitis Genitaltrakt Intrauterine Infekte Intrazytoplasmatisch Intrazytoplasmatisch Intrazytoplasmatisch Haut Haut Verruca vulgaris Condyloma accuminatum Intrazytoplasmatisch Haut Molluscum contagiosum Intranukleär Intranukleär RNA Paramyxoviren Intrazytoplasmatisch Respirationstrakt Mumpsvirus Intrazytoplasmatisch Glandula parotis Reoviren (respiratory, enteric, orphan) Intrazytoplasmatisch Darmtrakt Retroviren Ocornaviren HIV Intrazytoplasmatisch Acute Immunodeficiency Rhabdoviren Intrazytoplasmatisch Syndrome (AIDS) ___________________________________________________________________________ HIV Human Immunodeficiency Virus 10.2.1.2. Störungen des Zyto- oder Membranskeletts. Der Raum einer Zelle wird durch die Proteine des Zytoskleletts strukturiert. Diese Proteine verbinden Organellen an verschiedenen Orten in der Zelle und dienen als «Schienen» für den Transport von Molekülen zwischen den Organellen. Zusätzlich stellen sie ein stützendes Zellgerüst dar. Defekte des Zytoskeletts manifestieren sich hauptsächlich in einer Störung der Lokomotion der Zellen (z.B. bei der Chemotaxis) und der Phagozytose. Das Zytoskelett wird von den Actinfilamenten (Mikrofilamente), den Intermediärfilamenten und den Mikrotubuli gebildet. Die Actinfilamente sind zweisträngige, helikale Polymere des Proteins Actin. Actin macht zirka 5% der gesamten Proteinmenge einer Zelle aus. Die Actinfilamente sind dünn und biegsam. Sie kommen vor allem in der «Zellrinde», unmittelbar unter der Zellmembran vor. Sie verleihen der Zelle durch ihr Zusammenwirken mit den Mikrotubuli eine Polarität. Die Actinfilamente haben auch Kontakt mit den Adhäsionsplaques der Zellen und beeinflussen auf diese Art und Weise die Lage der Zelle bezüglich ihrer Umgebung. Die Intermediärfilamente sind Polymere aus Faserproteinen. Sie verleihen der Zelle die mechanische Stabilität. Sie können in drei Gruppen unterteilt werden (Tab.10-9). Tab.10-9 Zu den Intermediärfilamenten gehören die Keratine, die vimentinartigen Proteine und die neuronalen Intermediärfilamente. Der immunhistochemische Nachweis der einzelnen Intermediärfilamente stellt ein wichtiges Hilfsmittel zur Klassifizierung von Tumoren in der pathologisch-anatomischen Diagnostik dar. ___________________________________________________________________________ Gruppe Polypeptid Vorkommen ___________________________________________________________________________ Keratine Typ I (sauer) Typ II (neutral/basisch) Epithelzellen Epithelzellen Vimentinartige Proteine Vimentin Zellen mesynchemaler Herkunft Desmin Muskelzellen Saures fibrilläres Gliaprotein Astrozyten, Schwannzellen Peripherin Neuronen Neuronale Intermediärfilamente Neurofilament-Proteine Neuronen ___________________________________________________________________________ Die Mikrotubuli sind gerichtete und dynamische Strukturen, deren eines Ende relativ schnell wachsen kann, und deren anderes Ende in das Zentrosom eingebettet ist. Es sind Polymere aus Tubulinmolekülen. Pathogenetische Veränderungen des Zytoskeletts werden oft durch eine nicht geregelte Bindung von Molekülen an die Proteine des Zytoskeletts vermittelt (Tab.10-10). So führen Phalloidine und Cytochalasin B zu strukturellen Veränderungen der Epithelzellen in den Gallekanaliculi und können auf diesem Weg die Funktion der Gallekanaliculi stören und eine Hyperbilirubinämie bewirken. Tab.10-10 Die pathologischen Veränderungen am Zytoskelett können verschiedene Ursachen haben. ___________________________________________________________________________ ZytoskelettKrankheit/ Stoff, der an die ZytoWirkung Komponente Störung skelettkomponente bindet ___________________________________________________________________________ Mikrotubuli Alzheimer Krankhheit Protein MAP-1 Protein MAP-2 Tau-Protein Phosphoryliertes Tau-Protein Chediak-Higashi-Syndrom: gestörte Phagozytose Stabilisierung Destabilisierung Defekte Polymerisierung Tubulin Diarrhoe Hemmung der Zellteilung Hemmung der Zellteilung Hemmung der Zellteilung Colchizin 1 Vinblastin Vincristin Griseofulvin Hemmung der Polymerisierung Actin Gestörte Phagozytose Cytochalasin B Biliäre Störung Phalloidine 2 Verminderung der Polymerisierung Stabilisierung der Polymerisierung Neurofilamente Neuropathien Antabus Aluminium ___________________________________________________________________________ MAP 1 2 Mikrotubuli assoziierte Proteine Colchizin wird als Medikament gegen die Gicht eingesetzt. Gegenmittel sind grosse Mengen von rohem Fleisch, dessen Actinfilamente Phalloidin binden können. In der Leber kann es bei Alkoholabusus zu einer Aggregation von Intermediärfilamenten kommen. Diese Aggregate manifestieren sich lichtmikroskopisch als die sogenannten «Mallory bodies». Frank B.Mallory hat diese «hyalinen Massen» 1911 erstmals beschrieben. Ähnliche Aggregate werden in Hepatozyten bei Adipositas, nach Dünndarmresektionen und bei biliären Störungen, in Alveolarwandepithelien bei Asbestose und in einigen Leber- und Lungenkarzinomen beobachtet. Beim Crooke-Hyalin in den Zellen der Adenohypophyse handelt es sich ebenfalls um Aggregate von Intermediärfilamenten. Sie entstehen bei hohen Plasmakonzentrationen von ACTH (adrenocorticotropes Hormon) und sind auf eine übermässige Bildung von Intermediärfilamenten in den von ACTH gehemmten Zellen zurückzuführen. Die Zilien der Flimmerepithelzellen und Spermien stehen in engem Kontakt zu den Mikrotubuli. Störungen der Funktion der Zilien können durch Infekte, Hypersensitivitätsreaktionen oder Nikotin hervorgerufen werden, aber auch genetisch bedingt sein. Die wichtigste genetische Störung ist das «Syndrom der immobilen Zilien». Es besteht aus den drei Leitsymptomen: Bronchiektasen infolge schwerer Pneumonien, chronische Sinusitis und Situs inversus verschiedener Organe. Oft ist auch eine Infertilität vorhanden. Vom Zytoskelett ist das Membranskelett zu unterscheiden. Es besteht aus einem filamentösen Netzwerk von Proteinen, die an der inneren Oberfläche der Zellmembran verschiedener Zellen, vor allem der Erythrozyten, angeordnet sind. Die wichtigsten Proteine des Membranskeletts sind: Spectrin, Actin, Protein 4.1 und Ankyrin. Die Interaktion zwischen diesen Proteinen definiert z.B. die Stabilität der Erythrozyten. Erythrozyten sind während ihrer Zirkulation ständig Tubulenzen und Scherkräften ausgesetzt. Sie müssen deshalb flexibel und dauerhaft «konstruiert» sein. Dies gewähren die Membranskelett-Proteine. Sind sie defekt, können Krankheiten entstehen. So kommt es zu einer (hereditären) Sphärozytose, wenn die Bindung von Spectrin an ein anderes Membranprotein gestört ist. Folge davon ist eine Einschränkung der Membranelastizität: Es resultieren «unflexible» Erythrozyten (Sphärozyten), welche schneller als normale Erythrozyten zerstört werden. Symptome der Krankheit sind: Hämolyse, prähepatischer Ikterus, Erythrophagie in der Milz, Splenomegalie, Hämosiderose und Gallensteine (bei 50-80% der Patienten). 10.2.1.3. l-Antitrypsin-Mangel l-Antitrypsin (AT) ist ein in der Leber synthetisiertes Glykoprotein, welches die bei einer Zellschädigung oder einer Entzündung auftretenden Proteasen unter Kontrolle halten sollte. Die wichtigsten der durch AT überwachten Enzyme sind: Trypsin, Chymotrypsin, PankreasElastase, Haut-Kollagenase, Renin, Urokinase, Hageman-Faktor sowie neutrale Proteasen der neutrophilen Granulozyten (Elastase, Kollagenase, Proteasen gegen Basalmembranen). AT kommt in Lymphe, Speichel, Stuhl, Muttermilch, Duodenalsekret, (Synovialflüssigkeit), Zervixschleim und Samenflüssigkeit vor. Galle, Synovia Ein l-AT-Mangel ist morphologisch an PAS-positiven, tropfenförmigen Ablagerungen im Zytoplasma der Hepatozyten zu erkennen. Die Ablagerungen liegen im glatten und rauhen endoplasmatischen Retikulum (ER) und sind Diastase-resistent (im Gegensatz zu Glykogen). Die PAS positiv angefärbten Moleküle sind Vorstufen des definitiven AT. Bei diesen defekten Molekülen ist die Glutaminsäure in einem Peptidfragment durch Lysin ersetzt. Dadurch kommt es im ER zu einer Störung der Ankoppelung der Moleküle an die Sialinsäure, welche für die Abgabe der Moleküle an den Golgi-Apparat wichtig ist. Als einzige wirksame Therapie des AT-Mangels bleibt nur die Lebertransplantation übrig. Das l-AT-Molekül weist viele molekulare Varianten auf, welche von mindestens 25 Allelen auf dem Pi (Proteinase Inhibitor) Locus des Chromosoms 14 bestimmt werden. Der absolute l-AT-Mangel wird autosomal-co-dominant vererbt. Er führt zu einem panlobulären Lungenemphysen und bei zirka 10% der Patienten zusätzlich zu einer Leberzirrhose; rund 50% der kindlichen Leberzirrhosen beruhren auf einem AT-Mangel. Die genaue Pathogenese der Leberzirrhose beim AT-Mangel ist nicht klar. Ein relativer AT-Mangel kann bei Nikotinabusus auftreten. Grund dafür ist eine Inaktivierung des AT durch Thiolproteasen im Kondensat des Zigarettenrauches. Ein zusätzlicher Grund sind Infiltrate von neutrophilen Granulozyten in der Bronchusschleimhaut, welche bei den mit dem Abusus einhergehenden rezidivierenden Bronchitiden auftreten. Diese sezernieren vermehrt Proteasen und führen so zur Störung des Gleichgewichtes zwischen den Proteasen und Antiproteasen. Ein relativer l-AT-Mangel ist an der Entstehung des Lungenemphysems bei Rauchern namhaft mitbeteiligt . 10.2.1.4. Alzheimer Krankheit. Eine immer grösser werdende Zahl älterer Menschen sind von einer progressiv verlaufenden Demenz, der Alheimer Krankheit, betroffen. Sie führt zu einem kognitiven Defizit, einer zunehmenden Vergesslichkeit, einer sinkenden Aufmerksamkeit und launenhaften Stimmungsschwankungen, bei denen sich Aggressionen und Frustrationsgefühle abwechseln. Das Morbiditätsrisiko für 75-85-Jährige beträgt im Mittel 10%. Das Krankheitsbild wurde 1907 erstmals von Alzheimer mit folgenden Worten beschrieben: «Über die ganze Hirnrinde zerstreut, besonders zahlreich in den oberen Schichten, findet man miliare Herdchen, welche durch Einlagerung eines eigenartigen Stoffes in die Hirnrinde bedingt sind. Der Stoff lässt sich ohne Färbung erkennen». Der erwähnte Stoff ist heute als Amyloid Protein- (A) bekannt. A kann auf zwei Arten in den extrazellulären Raum gelangen: entweder durch eine Freisetzung aus zugrunde gegangenen Neuronen oder eine aktive Sekretion aus den Neuronen. Als Risikofaktoren für eine Alzheimer Krankheit gelten heute: Alter, Trisomie 21, das Vorhandensein des 4-Allels des Apolipoproteins E, Mutationen im Gen, welches das Amyloid Precursor Protein (APP) kodiert, ein Schädel-Hirntrauma in der Anamnese oder eine Herpes simplex-Encephalopathie. Morphologische Charakteristika der Alzheimer Demenz sind: eine Hirnatrophie, vorwiegend der Frontal- und Temporallappen, extrazelluläre Plaques im Hirngewebe und intraneuronale Proteinablagerungen (fibrilläre Tangles) (Abb.10-10 und Tab.10-11). Die Hirnatrophie ist Folge eines Verlustes von Neuronen. Abb.10-10 Die senilen Plaques sind extrazelluläre Proteinablagerungen. Im Zentrum bestehen sie aus dem Ab. Am Rand der Plaques sind Mikrogliazellen und Astrozyten vorhanden. Histologie-Bild Tab.10-11 Die wichtigsten morphologischen Merkmale einer Alzheimer Demenz sind Hirnatrophie, Plaques und neuronale Tangles. __________________________________________________________________________ Morphologische Veränderungen PLAQUES TANGLES ___________________________________________________________________________ Lokalisation Extrazellulär Grosshirnrinde Hippocampus Intrazellulär Neuronen Zusammensetzung Monomere Aggregate aus A (A) Amyloid Precursor Protein (APP) Proteine der Phase der akuten Antwort Amyloid P C-reaktives Protein 1-Antitrypsin 2-Makroglobulin Immunglobulin G Komplementproteine 1 Clusterin (Antikomplement-Protein) Glycosaminoglykane Apolipoprotein E Protein Tau (im Übermass phosphoryliert) 1-Chymotrypsin Ubiquitin Proteoglykane Fibroblasten-Wachstumsfaktor Apolipoprotein E an der Peripherie Geschwollene und fragementierte Axone Mikrogliazellen Astrozyten (Makrogliazellen) Ausdehnung 100-150 m Korrelation mit dem Mässig stark Stark Schweregrad der Demenz ___________________________________________________________________________ A 1 Amyloid Protein Das Vorhandensein von Komponenten des Komplementsystems ist ein Hinweis auf eine verstärkte Lyse von Neuronen. Bei der Pathogenese der Alzheimer-Krankheit kommt drei Proteinen eine Schlüsselrolle zu: dem APP, A und dem Tau-Protein. Das APP ist ein Membran-assoziiertes Protein. Es wird im Golgi-Apparat und glatten endoplasmatischen Reticulum der Neuronen und Astrozyten synthetisiert, dann an die Zelloberfläche transportiert und via Endozytose wieder in die Neuronen aufgenommen. An einzelnen Stellen steht das APP in enger Nachbarschaft zum Protein Presenilin-1 (PS-1). Die Funktion des PS-1 ist nicht klar: Entweder ist es am intrazellulären Transport des APP beteiligt oder es beeinflusst Enzyme, welche das APP metabolisieren. Das APP scheint als Membranprotein über Interaktionen mit Second Messengers in die Regulation des Zellwachstums und in die Steuerung der Funktion der Synapsen (Gedächtnisbildung) involviert zu sein. Zusätzlich ist es ein potenter Proteasen-Hemmer. Das Gen, welches das APP kodiert, liegt auf dem Chromosom 21. Schon seit langem ist bekannt, dass Patienten mit einer Trisomie 21 («Mongoloismus») bereits im Alter von 40-50 Jahren Symptome einer Alzheimer Krankheit entwickeln können. Das APP kann an drei Stellen durch Enzyme aufgespalten werden: an der -, - und Stelle. Die entsprechenden Enzyme sind: die -, - und -Sekretase. Physiologischerweise wird das APP an der -Stelle proteolysiert. Dabei entstehen harmlose Fragmente. Bei der Alzheimer Krankheit dagegen wird das APP vor allem an der - und -Stelle gespalten (Abb.10-11). Dabei entsteht das A. Die -Sekretase wurde erst vor kurzem identifiziert und wird nun BACE genannt (-site APP Cleaving Enzyme). Bereits wird nach Medikamenten gesucht, welche das Enzym zu hemmen vermögen. Abb.10-11 Das Amyloid Precursor Protein (APP) wird im Golgi-Apparat und im endoplasmatischen Retikulum (ER) synthetisiert. Es wird vorerst aus der Zelle abgegeben und wieder über eine Endozytose in die Zelle aufgenommen. Das APP kann an drei Stellen proteolysiert werden (siehe Text). Das A liegt im Zentrum der Plaques. Es zeigt drei Hauptwirkungen: (1) Es kann die Neuronen stimulieren. Diese Funktion übt das Molekül mit einem Segment aus, welches grosse Ähnlichkeiten mit der Substance P zeigt. (2) Es kann neurotoxisch wirken. Diese Eigenschaft hat das Molekül dadurch, dass es in der Membran der Neuronen Ca2+-Ionen-Kanäle bilden kann. (3) Es aktiviert den klassischen Weg des Komplementsystems. Das Tau-Protein gehört in die Gruppe der Mikrotubuli-assoziierten Proteine (MAPs). Die MAPs haben zwei Aufgaben: (1) Sie dienen dazu, die Mikrotubuli zu stabilisieren und so gegen einen Zerfall zu schützen. (2) Sie wahren im neuralen Gewebe die Form der Zellen und die Integrität der Synapsen; im Zytoplasma bilden sie aus Tubulin aufgebaute Röhren. Die Funktion des Tau-Proteins wird wahrscheinlich durch das Apolipoprotein E überwacht und gesteuert. Das Apolipoprotein E wird vor allem in den Astrozyten, aber auch in Neuronen gebildet. Es hat zwei Bindungsstellen, eine für Proteine (z.B. auch für Proteine von Rezeptoren), die andere für Lipide. Es bestehen starke Hinweise darauf, dass im Zytoplasma der Neuronen das Apolipoprotein E3 vorübergehend mit dem Tau-Protein einen Komplex bildet. Dieser Komplex dient dazu, das Tau-Protein an einer Homodimerisierung zu hintern, bevor es mit den Mikrotubuli in Kontakt tritt. Wenn der Tau-Apolipoprotein E-Komplex mit einem Mikrotubulus in Berührung gekommen ist, wird das Tau-Protein aus dem Komplex entlassen, damit es - seiner Aufgabe entsprechend - mit dem -Tubulin interagieren kann. Im Gegensatz zum Apolipoprotein E3 bindet das Apolipoprotein E4 das Tau-Protein nur schwach. Überwiegt in den Neuronen das Apolipoprotein E4, so wird das Tau-Protein praktisch nicht mehr daran gehindert, mit sich selber zu aggregieren. Folge davon ist eine intrazelluläre Ablagerung und eine vermehrte Phosphorylierung des Tau-Proteins. Das Tau-Protein steht unter diesen Bedingungen nicht mehr als Stabilisator der Mikrotubuli zur Verfügung. Die Ablagerungen des Tau-Proteins ist morphologisch nach Jahren in Form von intraneuronalen Tangles sichtbar. Ablagerungen von hyperphosphoryliertem Tau-Protein werden als Tauopathien bezeichnet (Tab.10-12). Tab.10-12 Proteinablagerungen sind oft Ursache neurodegenerativer Erkrankungen des zentralen Nervensysems. Dazu werden auch die Tauopathien (Ablagerungen von hyperphosphoryliertem Tau-Protein) gerechnet. ___________________________________________________________________________ Ort der Proteinablagerung Neurodegenerative Krankheit ___________________________________________________________________________ Intraneuronal Intranukleär Zytoplasmatisch (neuritisch) Extrazellulär Huntington Krankheit Amyotrophe Lateralsklerose Parkinson Krankheit Tauopathien Alzheimer Krankheit Trisomie 21 Pick Krankheit Prion Krankheit Alzheimer Krankheit Trisomie 21 ___________________________________________________________________________ Das Apolipoprotein E bindet nicht nur an das Tau-Protein, sondern auch an das A. Dazu verwendet das Molekül seine zweite Bindungsstelle für Lipide. Diese Bindung ist bei der E4Isoform des Apolipoproteins stärker als bei der E3-Isoform. Es wird angenommen, dass der Komplex A/Apolipoprotein dazu dient, in den Astrozyten synthetisiertes A in die Neuronen abzutransportieren. Wird von den Astrozyten zu viel A gebildet oder ist zu viel Apolipoprotein E4 vorhanden, kann nicht alles A in die Neuronen gelangen, weil (1) die Kapazität der Rezeptoren für das Apolipoprotein auf der Oberfläche der Neuronen limitiert ist und (2) der Komplex A/Apolipoprotein E4 instabil ist, sodass sich das A extrazellulär wieder vom Komplex ablösen kann und liegen bleibt. Folge dieser Störungen ist eine verstärkte extrazelluläre Ablagerung von A und Apolipoprotein E in den Plaques. Die Alzheimer Krankheit kann familiär gehäuft oder sporadisch auftreten. Den familiär gehäuften Formen der Erkrankung liegen deterministische Genveränderungen zugrunde. Deterministisch bedeutet, dass die Krankheit autosomal dominant vererbt wird und sich bereits früh (im Alter von 40-60 Jahren) manifestieren kann. Die genetische Dysfunktion führt zu einer pathologisch verstärkten Ablagerung von Proteinen (z.B. A, Presenilin) im Hirngewebe (Tab.10-13). Tab.10-13 Die Alzheimer Krankheit kann familiär gehäuft oder sporadisch auftreten. Bislang sind drei Genveränderungen bekannt, welche bei der familiären Form der Krankheit vorkommen. Für das Auftreten der sporadischen Form prädisponiert ein homozygotes Vorhandensein des e4 Allels des Apolipoproteins E in den Astrozyten. ___________________________________________________________________________ Genveränderung Gen Lokalisation auf Expression in Protein in Chromosom ___________________________________________________________________________ Deterministisch APP-Gen Presenilin 1 Presenilin 2 21 14 1 Neuronen Neuronen Neuronen Zellmembran Zellmembran Zellmembran Astrozyten 1 Zytoplasma Mikrogliazellen Zytoplasma Neuronen Zytoplasma ___________________________________________________________________________ Sporadisch 1 Apolipoprotein E 19 Das Apolipoprotein E wird hauptsächlich in Astrozyten exprimiert. Bei der sporadischen Form der Alzheimer Krankheit steht das Gen für das Apolipoprotein E im Zentrum. Das Gen weist drei Allele auf: 2, 3 und 4, sodass sechs verschiedene Genotypen definiert werden können (Tab.10-14). Diese Genotypen kommen unterschiedlich häufig vor. Der Genotyp mit dem höchsten Risiko für die Entstehung einer Alzheimer Krankheit ist der 4/4-Genotyp. Das Risiko, an einer Alzheimer Demenz zu erkranken, beträgt für 4-Homozygote 3.58. In den USA weisen 64% der Patienten mit einer Alzheimer Krankheit diesen Genotypen auf. Die Genotypen 2/2 und 2/3 scheinen dagegen für die Alzheimer Krankheit sogar protektiv zu sein. In den Plaques von Patienten mit einem 4Allel ist eine stärkere Akkumulation des A zu beobachten und kommen mehr neurofibrilläre Tangles vor als in einem Kollektiv ohne 4-Allel. Tab.10-14 Die drei verschiedenen Allele des Apolipoproteins E erlauben die Definition von sechs verschiedenen Genotypen. Diese Genotypen sind in der Bevölkerung unterschiedlich häufig ausgeprägt. ___________________________________________________________________________ Genotyp Häufigkeit Beginn der sporadischen Bindung des Apolipoproteins E an in den USA (%) Alzheimer Krankheit ... das Tau-Protein ... das A ___________________________________________________________________________ 2/2 <1 + 2/3 10 im Alter von > 90 Jahren 2/4 8 3/3 60 + + 3/4 20 4/4 2 im Alter von < 70 Jahren +++ ___________________________________________________________________________ 10.2.2. Unter der «Amyloidose» werden extrazelluläre Ablagerungen nichtlöslicher, fibrillärer Proteine in verschiedenen Organen bei verschiedenen Krankheiten verstanden. Lichtmikroskopisch erscheint Amyloid als homogene, eosinophile interzellulär gelegene Substanz. Sie besteht zu 90% aus Proteinen, zu 10% aus Glykoproteinen. Makroskopisch erscheinen Organe mit Amyloideinlagerungen glasig-wachsartig. Mikroskopisch erscheinen Amyloidablagerungen in der Kongorot-Färbung orange und brechen (wegen ihrer -fibrillären Struktur) doppelt. Elektronenmikroskopisch manifestiert sich Amyloid als lockeres Maschenwerk von Fibrillen mit einer -Faltblattstruktur. Die Amyloidose wird deshalb gelegentlich auch als -Fibrillose bezeichnet. Im Säugetier-Organismus kommen Amyloidablagerungen physiologischerweise nicht vor. Die moderne Klassifikation der Amyloidose basiert auf der Natur der VorläuferProteine, aus denen das Amyloid gebildet wird (Tab.10-15). Die Vorläufer-Proteine sind Proteine, welche im Blutplasma zirkulieren. Tab.10-15 Die moderne Klassifikation der Amyloidosen richtet sich nach den Vorläufer-Proteinen der fibrillären, extrazellulär abgelagerten Amyloid-Proteine. ___________________________________________________________________________ AmyloidoseVorläufer-Protein Fibrilläres Protein Klinische Symptome Typen (Amyloid-Protein) ___________________________________________________________________________ oder Leichtketten Kardiomyopathie Hepatomegalie Proteinurie (Bence Jones Proteine) Makroglossie Orthostasestörungen Neuropathie des autonomen und peripheren Nervensystems Ekchymosen Carpaltunnelsyndrom Abnormales Transthyretin Transthyretin Neuropathie des autonomen und peripheren Nervensystems (gastrointestinale Symptome) Kardiomyopathie Glaskörpertrübung Serum Amyloid A Protein Amyloid A Protein Infektionskrankheiten (als Ursache dieses Typs der Amyloidose) Hepatosplenomegalie Proteinurie Niereninsuffizienz Orthostasestörungen AL Monoklonale Immunglobulin(primär) Leichtketten ATTR (familiär) AA (sekundär) Andere (familiär) AApo A-1 Apolipoprotein A-1 Polyneuropathie Nephropathie Agel Gelsolin Gelsolin Dystrophie der Kornea Neuropathie AFib Fibrinogen A Fibrinogen A Nephropathie Arterielle Hypertonie ALys Lysozyme Lysozyme Nephropathie Hepatomegalie AE Prohormone Polypeptide Medulläres Schilddrüsenkarzinom Ablagerungen im endokrinen Pankreas ___________________________________________________________________________ AL ATTR AA Bence Jones Proteine AE Apolipoprotein A-1 Amyloid Light Chains: Immunoglobulin-Leichtketten-Amyloid Amyloid Trans-Thy-Retin: Familiäres, Transthyretin-assoziiertes Amyloid Amyloid Associated: Amyloid A-Protein Freie monoklonale Leichtketten im Urin Endokrines Amyloid Ursache der AL-Amyloidose ist meistens eine monoklonalen Überproduktion der oder Leichtketten durch die Plasmazellen (z.B. bei multiplen Myelomen oder anderen lymphopoietischen malignen Tumoren). Die AL-Amyloidose ist der am häufigsten auftretende Amyloidose-Typ. Die familiären Amyloidosen werden autosomal-dominant vererbt. Im Vordergrund der familiären Amyloidosen steht die Amyloid trans-thy-retin (ATTR)-Amyloidose. Transthyretin ist ein Transportprotein für Thyroxin. Es wird vor allem in der Leber synthetisiert, jedoch auch im Plexus chorioideus. In der Familienanamnese sind oft unklare neurologische Krankheiten (z.B. Störungen der Motoneurone) anzutreffen. Das Serum Amyloid A-Protein (SAA) gehört zu den Proteinen der Phase der akuten Antwort. Die häufigsten chronischen Krankheiten, bei denen eine AA-Amyloidose beobachtet werden kann, sind: Tuberkulose, chronische Osteomyelitis und Bronchiektasen. Es bestehen verschiedene Modelle einer möglichen Pathogenese der Amyloidablagerungen. Fest steht, dass die Pathogenese multifaktoriell und für die verschiedenen Amyloidtypen unterschiedlich ist. Diskutiert werden folgende vier Modelle: (1) Die VorläuferMoleküle (z.B. leichte Immunglobulin-Ketten) werden durch einen Ersatz von Aminosäuren destabilisiert und dann abgelagert. (2) Lokal stimulieren chemische Einflüsse wie pHVeränderungen die Amyloidbildung. (3) Die Makrophagen nehmen die Vorläuferproteine auf, bilden intrazellulär daraus Fibrillen, erschöpfen sich und gehen zugrunde; die Fibrillen werden dabei freigesetzt und extrazellulär deponiert. (4) Die Makrophagen und Endothelzellen bauen die Voräufer-Proteine partiell ab. Fragmente der Proteine werden in die Umgebung abgegeben und aggregieren dort in Kombination mit Glykosaminoglykanen der extrazellulären Matrix (Abb.1012). Am häufigsten kommt es bei einer Amyloidose zu Amyloid-Ablagerungen in den Nieren. Das Amyloid erscheint hauptsächlich in den Glomerula, jedoch auch in den Gefässwänden. Bei einem Gefässbefall sind Zeichen einer Ischämie mit einer konsekutiven Tubulusatrophie und interstitiellen Fibrose zu beobachten. Eine Nierenamyloidose kann zu einem nephrotischen Syndrom führen. In der Leber treten die Amyloidablagerungen zuerst im Disse'schem Raum auf, später im Interstitium des Parenchyms und in der Wand der Sinus. Eine Leberfunktionsstörung ist erst bei sehr ausgedehnten Ablagerungen zu beobachten. Im Herzen wird das Amyloid vor allem subendokardial, im Myocard des Septum interventriculare, in der Wand der Vorhöfe und der Gefässe abgelagert (Abb.10-13). Folge der Herzamyloidose können Veränderungen des Elektrokardiogramms (Low voltage) und Herzrhythmusstörungen sein. Die senile ATTRAmyloidose (senile Herzamyloidose) bleibt klinisch oft unerkannt. Abb.10-12 Das AL-Amyloid wird möglicherweise dadurch gebildet, dass die Makrophagen und Endothelzellen Immunglobulin-Leichtketten aufnehmen und inkomplet verdauen. Nicht abgebaute Protein-Fragmente werden an die Umgebung abgegeben und aggregieren dort. ein ähnlicher Prozess läuft mit dem Serum Amyloid A-Protein ab. Fragmente dieses Proteins werden als AA-Amyloid extrazellulär abgelagert. Abb.10-13 Bei älteren Patienten kann es - aus unerklärten Gründen - zu einer Ablagerung von Amyloid zwischen die Herzmuskelfastern und in die Wand der interstitiellen Gefässe kommen. Die wolkigen Ablagerungen sind extrazellulär gelegen und sehen sehr homogen aus. Sie können zu Rhythmusstörungen führen. Eine Amyloidose wird meistens über eine Biopsie aus der Zunge, dem Rektum, der Sehnenscheiden oder Nieren diagnostiziert. 10.2.3. «Hyalin» und «Fibrinoid» sind teils extrazelluläre, teils intrazelluläre Ablagerungen von Proteingemischen. «Hyalin» ist ein unspezifischer deskriptiver morphologischer Begriff. Er kann für jede morphologische Veränderung, welche sich in der Hämatoxilin-Eosin-Färbung als homogene, rosafarbige, glasige, intra- oder extrazelluläre Ablagerung darstellt, verwendet werden. Als intrazelluläres Hylin können die tröpfchenförmigen resorptiven Proteinablagerungen bezeichnet werden, welche z.B. bei einem nephrotischen Syndrom in den Nierentubuluszellen beobachtet werden. Amyloidablagerungen erfüllen zwar die Kriterien des Hyalins, werden aber vom Hyalin unterschieden, weil das Amyloid färberisch, biochemisch und elektronenoptisch identifizierbar ist. «Fibrinoid» wird eine homogene extrazelluläre Proteinablagerung genannt, welche sich einerseits mit Eosin (einem sauren Farbstoff) intensiv anfärbt, andererseits Färbeeigenschaften des Fibrins aufweist und eine zelluläre Reaktion des umgebenden Gewebes auslöst. Fibrinoide Proteinablagerungen werden in Demarkationszonen von Haut- oder Schleimhautnekrosen (z.B. Magenulcus, Hautulcus), im Bindegewebe bei verschiedenen Autoimmunkrankheiten und in Blutgefässwänden bei Vasculitiden beobachtet. Die fibrinoiden Ablagerungen bestehen hauptsächlich aus Zelltrümmern, Fragmenten der extrazellulären Matrix und Bestandteilen des Blutplasmas. 10.3. BEI MALIGNEN TUMOREN KÖNNEN PROTEINE AN UNGEWÖHNLICHEN ORTEN ODER IN UNGEWÖHNLICHER MENGE ERSCHEINEN. Maligne Tumoren können Syndrome bewirken, die nicht direkt auf eine Destruktion des umgebenden Gewebes oder die Fernwirkung des Tumors via Metastasen zurückzuführen sind, sondern auf Proteine, welche vom Tumorgewebe pathologischerweise synthetisiert und als Hormone oder Antigene wirksam werden. Die Syndrome werden als paraneoplastische Syndrome bezeichnet. Werden Hormone nur im Tumorgewebe, nicht aber im Ursprungsgewebe, aus dem der Tumor entstanden ist, gebildet, spricht man von einer ektopischen Hormonproduktion. Vor allem bekannt ist die ektopische Sekretion von ACTH bei Bronchuskarzinomen. Folge davon ist ein Cushing Syndrom. 10.4 PROTEINE KÖNNEN MITTELS IMMUNHISTOCHE-MISCHER METHODEN IM HISTOLOGISCHEN SCHNITT ODER ZYTOLOGISCHEN PRÄPARAT SICHTBAR GEMACHT WERDEN. Die zellulären und extrazellulären Proteine, welche immunhistochemisch dargestellt werden können, werden Epitope genannt. Eine immunhistochemische Darstellung von Epitopen erfordert zwei Schritte: (1) Das Epitop wird monoklonalen Antikörpern, welche gegen das Epitop gerichtet sind, ausgesetzt. (2) An den Antikörper, der mit dem Epitop reagiert, wird ein gegen ihn gerichteter zweiter Antikörper gebracht. An diesen Antikörper ist ein Farbstoff gekoppelt. Dadurch kann das Epitop im histologischen Schnitt oder zytologischen Präparat sichtbar gemacht werden. Der Nachweis spezifischer Epitope im Gewebe eines malignen Tumors kann von wegweisender Bedeutung für eine korrekte Tumorklassifizierung sein (Tab.10-16, Abb.10-14). Wird die Methode des immunologischen Nachweises von Epitopen an zytologischen Präparaten eingesetzt, spricht man von Immunzytochemie. Tab.10-16 Zelluläre Epitope können von wegweisender diagnostischer Bedeutung als pathologisch-anatomische «Tumormarker» sein. ___________________________________________________________________________ Struktur Element Protein/Amin Zellen ___________________________________________________________________________ Zytoskelett Intermediärfilamente Keratine CK 7 CK 20 Vimentin Desmin Neurofilamente Epithelzellen Kolon Ovar Mesenchymale Zellen Quergestreifte und Herzmuskulatur Neuronen Gliales fibrilläres saures Protein Neuronen Glatte Muskulatur Actin Vesikel Membran-Proteine Synaptophysin Neuroendokrine Zellen Sekretgranula Matrix-Proteine Chromogranine A,B,C Leu 7 (CD57) Neuroendokrine Zellen Neuroendokrine Zellen Zytoplasma Enzyme S 100-Protein Neurale und neurogene Zellen -Fetoprotein Epithelzellen Carcino-embryonales Antigen Epithelzellen (CEA) Prostata-spezifisches Antigen Prostata-Epithelzellen MelanA Melanozyten HMB45 Melanozyten Neuron-spezifische Enolase Neuroendokrine Zellen Prostatische alkalische Prostata-Epithelzellen Phosphatase Plazentare alkalische Phosphatase (PLAP) Hormone Amine 5-Hyroxytryptamin Neuroendokrine Zellen ___________________________________________________________________________ Tab.10-16 (Fortsetzung) ___________________________________________________________________________ Struktur Element Protein/Amin Zellen ___________________________________________________________________________ Zellmembran Adhäsionsmoleküle Glykoproteine Leucocytic common antigen (LCA, CD 45) CD3 CD5 CD10 CD20 CD34 CD68 CD79 CD2 Faktor VIII assoziiertes Antigen Epitheliales Membran-Antigen (EMA), B72.3 Ber-EP4 Granulozyten, Lymphozyten, Plasmazellen T-Lymphozyten T-Lymphozyten B-Lymphozyten B-Lymphozyten Endothelzellen Monozyten, Makrophagen B-Lymphozyten T-Lymphozyten Endothel Epithelzellen Epithelzellen Zellkern Rezeptoren Östrogen-Rezeptor Progesteron-Rezeptor Androgen-Rezeptor Proteine der Proliferation Ki 67-Antigen, Cyclin D1 Transkriptionsfaktoren Myc Suppressorproteine P53, P27 Extrazelluläre Matrix Fibronectin Laminin Kollagene ___________________________________________________________________________ CK CD Zytokeratin Cluster of Differentiation (Cluster Design protein) Abb.10-14 In Lymphknoten, welche im Rahmen einer Neck dissection wegen eines Plattenepithelkarzinoms der Mundhöhle untersucht wurden, fanden sich im Randsinus kleine, relativ monomorphe, teilweise tubulär angeordete Zellen. Die Monomorphie liess differentialdiagnostisch an ein neuroendokrines Karzinom denken. Die immunhistochemischen Untersuchungen ergaben, dass in den Halslymphknoten tatsächlich zusätzlich zu den Metastasen des Plattenepithelkarzinoms auch Metastasen eines medullären Schilddrüsenkrazinos vorhanden waren.