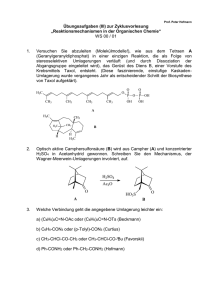
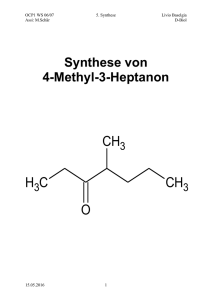
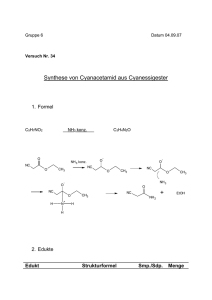
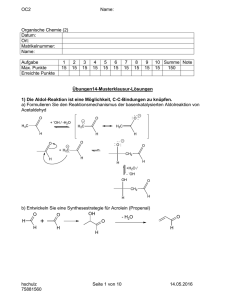
Skript
Werbung

Organische Chemie (II) für die Studiengänge BC und T Hartmut Schulz Sommersemester 2005 Skript: Biotechnologie-Server „Public Folders“ S:\Schulz Public\OC2\oc2bct-05-03-15 Oder angel-Plattform Organische Chemie (II) 1. Einführung 1.1 Voraussetzungen zum Verständnis der Vorlesung 1.2 Anmerkungen zum sicheren Arbeiten im Labor 1.3 Arbeiten im Chemielabor 2. Reaktionsmechanismen in der organischen Chemie 2.1 Radikalische Substitution (SR) (S.18) 2.2 Nucleophile Substitution (SN) (S.39) 2.3 Eliminierung (E) (S.71) 2.4 Elektrophile Addition an die C-C-Doppelbindung (AE) (S.100) 2.5 Elektrophile Addition an konjugierte p-Systeme (S.118) 2.6 Reaktivität des Allyl-Systems (S.125) hschulz, oc2bct 2 Organische Chemie (II) 2.7 Nucleophile Addition an die C-C-Doppelb. (AN) (S.131) 2.8 Radikalische Addition an die C-C-Doppelb. (AR) (S.135) 2.9 Cycloadditionen (S.138) 2.10 Elektrophile Addition an die C-C-Dreifachb. (AE) (S.157) 2.11 Nucleophile Addition an die C-C-Dreifachb. (AN) (S.163) 2.12 Hydroborierung von Doppel- und Dreifachb. (S.164) 2.13 Elektrophile Substitution am Aromaten (SE) (S.170) 2.14 Nucleophile Substitution am Aromaten (SN) (S.227) 3. Darstellung und Eigenschaften von metallorganischen Verbindungen (S.239) 3.1 Grignard-Reagenzien 3.2 Metallalkyle 3.3 Metallocene hschulz, oc2bct 3 Organische Chemie (II) 4. Reaktivität der Carbonylverbindungen (S.247) 4.1 Reaktivität der Carbonylgruppe (S.247) 4.1.1 Katalytische Hydrierung der C-O-Doppelb. (S.253) 4.1.2 Ionische Addition an die C-O-Doppelb. (AN) (S.254) 4.1.3 Wittig-Reaktion zur Synthese von Alkenen (S.287) 4.1.4 Additions-Eliminierungs-Mechanismus bei Carbonsäurederivaten (S.291) 4.2 Reaktivität von Carbonylverbindungen am a-C-Atom (S.298) 4.2.1 Keto-Enol-Tautomerie (S.305) 4.2.2 Aldol-Reaktion (S.311) 4.2.3 Halogenierung von Carbonylverbindungen (S.320) 4.2.4 Alkylierung von Carbonylverbindungen (S.326) 4.3 a,b-ungesättigte Carbonylverbindungen (S.327) hschulz, oc2bct 4 Organische Chemie (II) 5. Kohlensäure und ihre Derivate (S.330) 6. Reaktivität von Aminen (S.337) 6.1 Selektive Darstellung von prim. Aminen (S.338) 6.2 Alkene durch Elimierung von Aminen (S.341) 6.2.1 Hofmann-Eliminierung 6.2.2 Cope-Eliminierung 6.3 Nitrosierung von Aminen (S.353) 7. Farbstoffe (S.362) 8. Stereochemie ohne asymmetrische Zentren, asymmetrische Synthesen und Trennung von Enantiomeren (S.373) hschulz, oc2bct 5 Organische Chemie (II) 9. Polymere Werkstoffe (S.396) 9.1 Die Struktur der Polymere (S.401) 9.2 Mechanismus der Polymerisation (S.418) 9.2.1 Radikalische Polymerisation (S.419) 9.2.2 Kationische Polymerisation (S.424) 9.2.3 Anionische Polymerisation (S.429) 9.2.4 Polymerisation mittels Übergangsmetall komplexen (S.435) 9.2.5 Polykondensation (S.446) 9.2.6 Polyaddition (S.460) hschulz, oc2bct 6 Organische Chemie (II) 10. Naturstoffe (S.458) 10.1 10.2 10.3 10.4 10.5 10.6 10.7 Terpene Steroide Alkaloide Fette, Öle und Wachse Kohlenhydrate Aminosäuren und Proteine Nucleinsäuren hschulz, oc2bct 7 1. Einführung 1.1 Voraussetzung zum Verständnis der Vorlesung OC (II) Grundlagen der allgemeinen Chemie Chemische Bindungen MO-Theorie (Grundlagen) Aufbauprinzipien des Periodensystems Säure/Base-Reaktionen Redox-Reaktionen Reaktionskinetik (Grundlagen) Reaktionsgleichungen hschulz, oc2bct 8 Grundlagen der Organischen Chemie Stoffklassen und funktionelle Gruppen Physikalische und chemische Eigenschaften der Stoffklassen Nomenklatur und gängige Trivialnamen Struktur- und Strichformeln Stereochemie hschulz, oc2bct 9 1.2 Anmerkungen zum sicheren Arbeiten im Labor Literaturempfehlung Laborordnung Sicheres Arbeiten in Chemischen Laboratorien, Einführung für Studenten Sicherheit - Handbuch für das Labor, D. Bernabei, Hrsg. E. Merck The organic chem lab survival manual, J.W. Zubrick hschulz, oc2bct 10 Persönliche Schutzausrüstung Feuerlöscher Baumwoll-Labormantel Löschdecken Notduschen und Augenduschen Erste-Hilfe-Schränke Fluchtwege Hauptschalter für Gas und Elektrizität Feuermelder und Notruftelefon Schuhe ggf. Handschuhe Sicherheitseinrichtungen im Labor Schutzbrille geschlossene Verbot von Essen, Trinken, Rauchen im Labor gründliches Händewaschen hschulz, oc2bct und 11 Verhalten bei Unfall oder Brand Erste Hilfe Absichern des Unfallorts Versorgung der Verletzten Weisungen beachten Verletzte nicht alleine lassen In Sicherheit bringen Gefährdete Personen warnen Hilflose Personen mitnehmen Türen schliessen zum Sammelplatz gehen Keine Aufzüge benutzen hschulz, oc2bct Notruf: 0-112 Wer meldet? Was ist passiert? Wo ist es passiert? Wie viele Verletzte? Weitere Menschen in Gefahr? 12 Umgang mit Chemikalien Informationen vor der Handhabung Brennbare Stoffe Brandfördernde Stoffe Selbstentzündliche Stoffe Explosionsgefährliche Stoffe Betriebsanweisungen Sicherheitsdatenblätter R-, S-, E-Sätze Gefahrensymbole MAK-Werte TRK-Werte WGK Flammpunkt Zündtemperatur hschulz, oc2bct Handhabung nicht berühren oder einatmen Kontaminationen vermeiden kleine Ansatzmengen Sichere Aufbewahrung Umweltverträgliche Reaktionsplanung 13 1.3 Arbeiten im Chemielabor Literaturempfehlung Organikum, Wiley-VCH, Kapitel A und B Aufbau einer Glasapparatur Schliffe fetten und auf Dichtheit prüfen, Schliffklammern verwenden Jedes Glasteil mit mindestens einer Stativklammer sichern, spannungsfrei! Grösse der Apparatur dem Ansatz anpassen: Gesamtfüllmenge maximal 2/3 des Kolbenvolumens! Art und Grösse des Rührers der Apparatur und der Reaktion anpassen hschulz, oc2bct 14 Unter der Apparatur muss genügend Platz sein, um Heiz- oder Kühlbad bequem entfernen oder installieren zu können Druckaufbau vermeiden Apparatur muss zu jedem Zeitpunkt mindestens eine Öffnung zur Aussenatmosphäre haben: kein Stopfen auf Rückflusskühler! Hitzestau vermeiden Bei exothermen Reaktionen Kühlbad bereithalten Effektives Rühren gewährleisten: kein Magnetrührer bei Fällungsreaktionen! Sicherstellen, dass Reaktion schneller erfolgt als Eduktzugabe hschulz, oc2bct 15 Heizen oder Kühlen erst beginnen, wenn die Apparatur geprüft, alle Chemikalien vorgelegt und der Rührer eingeschaltet ist nie Heizen ohne Rührer oder Siedesteinchen Wasserzufuhr zum Rückflusskühler oder zur Destillationsbrücke vor dem Heizen einschalten und erst nach dem Abkühlen auf Raumtemperatur abschalten hschulz, oc2bct 16 Methoden Erhitzen am Rückfluss Filtrieren und Waschen von Feststoffen Trocknen von Flüssigkeiten und Feststoffen Umkristallisieren Extrahieren Destillieren Schmelzpunktbestimmung Brechungsindexbestimmung hschulz, oc2bct 17 2. Reaktionsmechanismen in der organischen Chemie 2.1 Radikalische Substitution (SR) R-H + X2 R-X + HX X = F, Cl, Br Die Iodierung von Alkanen ist endotherm und findet deshalb nicht statt! Radikalkettenmechanismus hschulz, oc2bct 18 Chlorierung von Methan Experimentelle Beobachtungen Bei Raumtemperatur findet in der Dunkelheit keine Reaktion statt Bei Temperaturen >300°C kommt es zu einer Reaktion auch bei Dunkelheit Bestrahlung mit UV-Licht führt zu einer Reaktion bereits bei Raumtemperatur Die Aktivierungsenergie für eine radikalische Substitution kann durch Licht oder Wärme zugeführt werden! Dissoziationsenergien der beteiligten Bindungen H H H C H + Cl-Cl H DH0 440 (kj/mol) H C H + H-Cl Cl 240 355 430 DH0= - 105 kj/mol Exotherme Reaktion Cl-Cl-Bindung ist die schwächste Bindung Reaktionsmechanismus der Radikalischen Substitution 1. Durch Licht oder Wärme werden Bindungen homolytisch gespalten Die schwächste Bindung bricht bevorzugt Spaltung der Cl2-Bindung, DH0= +240 kj/mol Cl-Cl D oder hn Cl + Cl Chlor-Radikale endotherm 2. Das reaktive Chlor-Radikal greift Methan an Bildung von Chlorwasserstoff Bildung eines Methyl-Radikals DH0= +10 kj/mol H Cl +H C H H Cl-H + C H H H Methyl-Radikal schwach endotherm 3. Das reaktive Methyl-Radikal greift ein Chlormolekül an stark exotherm Bildung von Chlormethan Bildung eines Chlor-Radikals DH0= -115 kj/mol H H Cl-Cl + C H H H C H + Cl Cl 4. Das verbleibende Chlor-Radikal greift ein weiteres Methan-Molekül an = Schritt 2 Kettenreaktion Kettenreaktion der radikalischen Substitution Kettenstart: Bildung von Radikalen (Schritt 1) X2 Kettenfortpflanzung (Schritt 2 und 3) CH4 + X CH3 + X2 2X HX + CH3 X + XCH3 Kettenabbruch CH3 + X CH3 + CH3 X+ X CH3X CH3-CH3 X-X Energiebilanz der Halogenierung von Methan DH0 H3C-H + X-X (kj/mol) H3C-X +H-X DH 0 (kj/mol) X=F 440 155 455 570 -430 X=Cl 440 240 355 430 -105 X=Br 440 190 290 365 -25 X= I 440 150 235 300 +55 I2 reagiert nicht mit Alkanen, da die Reaktion endotherm ist hschulz, oc2bct 25 Halogenierung von Alkanen Die Reaktivität der Halogene gegenüber Alkanen nimmt innerhalb der Hauptgruppe von oben nach unten ab Mehrfachhalogenierungen sind möglich F2>Cl2>Br2>I2 I2 reagiert nicht mit Alkanen, da die Reaktion endotherm ist Die Produkt-Zusammensetzung wird durch das Mengenverhältnis der Edukte beeinflusst Sind verschiedene Wasserstoffe im Molekül vorhanden erhält man immer eine Mischung verschiedener Produkte! Alkane mit equivalenten Wasserstoffen CH3 CH3 CH3-C-CH3 Cl2 Licht oder Wärme CH3 Neopentan (Überschuss) CH3-C-CH2 Cl CH3 Neopentylchlorid +HCl Halogenierung von Propan CH3CH2CH3 Propan (Überschuss) CH3CH2CH2 Cl + 1-Chlorpropan 45% Cl2 hn, 25°C CH3CHCH3 Cl 2-Chlorpropan 55% Als statistische Verteilung erwartet man ein Verhältnis von 6:2 Sekundäre C-H-Bindungen sind reaktiver als primäre Halogenierung von Isobutan H Cl2 CH3 C CH3 H CH3 CH3 C CH2 Cl CH3 Isobutylchlorid, 63% hn, 25°C Cl + CH3 C CH3 CH3 tert-Butylchlorid, 37% Als statistische Verteilung erwartet man ein Verhältnis von 9:1 Tertiäre C-H-Bindungen sind reaktiver als primäre Die C-H-Bindungsstärke und die Stabilität der Radikale 410 Ethyl primär H7C3-H 410 n-Propyl primär (CH3)2HC-H 395 Isopropyl sekundär (CH3)3C-H 390 t-Butyl tertiär hschulz, oc2bct Methyl Zunehmende Stabilität der Radikale H5C2-H Kategorie Abnehmende C-H-Bindungsstärke Radikal H3C-H DH0 (kj/mol) 440 30 Stabilität von Alkylgruppen Radikale CH3 < primär < sekundär < tertiär Carbenium-Ionen CH3+ hschulz, oc2bct < primär < sekundär < tertiär 31 Die Struktur der Alkylradikale Die Geometrie der Alkylradikale liegt zwischen einer Tetraeder- und einer planaren Anordnung Hybridisierung zwischen sp3 und sp2 R R R R R Carbanion sp3 R R Radikal sp3/sp2 R R Carbeniumion sp2 Umhybridisierung von Methan bei der Bildung des Methylradikals H H H H sp3 hschulz, oc2bct - H H H H sp3/sp2 33 Stabilisierung der Radikale durch Hyperkonjugation H H Isopropylradikal H H Ethylradikal H H H H H H H H t-Butylradikal H H H2C CH2 H2C H Wechselwirkung der gefüllten sp3-Hybridorbitale mit dem teilweise gefüllten p-Orbital: Delokalisation von Elektronendichte = Stabilisierung des Radikals hschulz, oc2bct 34 Die radikalische Substitution an Alkanen Kettenreaktion Im allgemeinen wenig selektiv (Bromierungen sind selektiver als Chlorierungen) C-H-Bindungen haben unterschiedliche Energie und folglich unterschiedliche Reaktivität die Stabilität der Radikale nimmt deshalb in folgender Reihe zu: Primäre C-H > sekundäre C-H > tertiäre C-H primär < sekundär < tertiär Alkylradikale sind annähernd planar und werden durch sp2/sp3-Hybridisierung beschrieben Praktische Bedeutung der radikalischen Halogenierung von Alkanen Fluor ist sehr reaktiv: gefährlich, wenig selektiv Chlor ist billig, aber als aggressives Gas schwierig zu handhaben. Einsatz in der Industrie Brom ist recht selektiv, als Flüssigkeit gut zu handhaben, aber teuer. Einsatz im Labor hschulz, oc2bct 36 Halogenierungsmittel für den Laborgebrauch N-Halogen-Succinimid (X=Cl, Br) O O R-H + N CCl4 Radikalinitiator X R-X + N O H O Radikalinitiatoren CN CN H3C N CH3 O N D CH3 O hschulz, oc2bct + 2 H3C N N CH3 CH3 O O O CN D 2 O 2 + 2 CO2 37 Autoxidation von Ether zu hochexplosiven Peroxiden H H2 C H2 C H3C O hn CH3 H2 C C H3C O + H CH3 + O2 H H H2 C C H3C O O + H5C2OC2H5 CH3 - H5C2OC2H4 H2 C C H3C O CH3 O OH O - C2H5OH CH3 CH3 CH HOO O O O CH CH3 CH O OC2H5 n Schwerflüchtiges polymeres Peroxid hschulz, oc2bct 38 2.2 Nucleophile Substitution (SN) Die C-X-Bindung in substituierten Alkanen besitzt ein Dipolmoment aufgrund einer ENDifferenz zwischen C und X Wenn das Kohlenstoffatom eine positive Partialladung trägt, kann es von elektronenreichen Reaktanden (Nucleophilen) angegriffen werden R hschulz, oc2bct d+ C R R d- X 39 Nucleophile Substitution Angriff eines anionischen Nucleophils auf ein Halogenalkan Nu - + R-X Nukleophil „kernliebend“ Nu-R + X Elektrophil „elektronenliebend“ - Abgangsgruppe Angriff eines neutralen Nucleophils auf ein Halogenalkan Nu + R-X + [Nu-R] + X - Begriffsbestimmungen Basizität thermodynamische - A + H2 O K Grösse AH + OH- K=Gleichgewichtskonstante Nucleophilie kinetische Grösse k Nu- + R-X hschulz, oc2bct Nu-R + X- k=Geschwindigkeitskonstante 41 Polarisierbarkeit ein Mass für die Deformierbarkeit der Elektronenhülle eines Atoms oder eines Moleküls grosse Atome und Moleküle haben grosse, diffuse Elektronenhüllen. Die Elektronen sind leichter verschiebbar leicht polarisierbare Gruppen stabilisieren den Übergangszustand und sind deshalb sowohl gute Eintritts- als auch Abgangsgruppen hschulz, oc2bct 42 Nucleophile und elektrophile Gruppen Nucleophile haben Elektronenüberschuss Freie Elektronenpaare Neutral oder negativ geladen Stärke der Nucleophilie nimmt i. d. R. mit der Basenstärke zu, aber: F-<< Cl- < Br- < I- entgegen der Basenstärke! Nucleophilie nimmt mit der Polarisierbarkeit zu (Im Periodensystem von oben nach unten: NH3 < PH3) Im Periodensystem nimmt die Nucleophilie von links nach rechts ab (5.-7. Hauptgruppe): NH3 > H2O >> HF Negative Ladung hat einen Verstärkungseffekt: NH2 > NH3 Solvatation beeinträchtigt die Nucleophilie: in polaren und vor allem in protischen Lösungsmitteln werden Anionen stärker abgeschirmt als neutrale Teilchen Nucleophile und elektrophile Gruppen Elektrophile haben Elektronenmangel Positiv polarisiert in einer polaren Bindung Umgeben von -I-Substituenten Neutral oder positiv geladen Eine nucleophile Substitution kann unterschiedlich beschrieben werden Nucleophiler Angriff auf ein elektrophiles Zentrum Elektrophiler Angriff auf ein nucleophiles Zentrum Beispiele nucleophiler Substitutionen Substrat Nucleophil CH3Cl + OH CH3CH2I + H3CO CH3CHCH2CH3 Br +I - - Produkt Abgangsgruppe - CH3OH + Cl CH3CH2OCH3 +I CH3CCH2CH3 I + Br CH3CHCH2CN CH3 +I - - CH3CHCH2I CH3 + CN CH3CH2Br + H3CS CH3CH2SCH3 + Br CH3CH2I + NH3 CH3CH2NH3+ +I CH3Br + P(CH3)3 H3CP(CH3)3+ + Br - - - Mechanismus der nucleophilen Substitution: SN2 CH3Cl + NaOH D, H2O CH3OH + NaCl Reaktionskinetik Reaktionsgeschwindigkeit ist abhängig von CH3Cl- und OH--Konzentration Geschwindigkeitsgesetz 2. Ordnung v = k[CH3Cl][OH-] N Bimolekulare Reaktion Im Übergangszustand (geschwindigkeitsbestimmender Schritt) sind beide Edukte beteiligt Konzertierter, einstufiger Prozess S 2-Reaktion SN2- Reaktionsmechanismus HO Rückseite OH H H Cl H H dHO H H H # dCl H H HO Kein Angriff auf der Vorderseite + Cl Inversion Walden-Umkehr Veränderung der potentiellen Energie während einer SN2-Reaktion d- H H Nu d- # X H E - H Ea H Nu + H X H H - Nu DG H +X Reaktionskoordinate hschulz, oc2bct 48 SN2-Reaktionsmechanismus Bindungsbruch und Bindungsbildung erfolgen gleichzeitig Angriff des Nucleophiles auf das elektrophile Zentrum erfolgt von der Rückseite Es erfolgt eine Inversion der Konfiguration am elektrophilen Zentrum Die SN2-Reaktion ist stereospezifisch! Inversion der Konfiguration bei SN2- Reaktionen H3C Cl H H HOcis-1-Chlor-3-methylcyclopentan SN 2 H3C H H OH trans-3-Methylcyclopentanol Inversion der Konfiguration bei SN2-Reaktionen H C6H13 Br SN2 C6H13 HO H + HO- CH3 (R)-(-)-2-Bromoctan [a]D25 = -34.25° CH3 (S)-(+)-2-Octanol [a]D25 = +9.90° Einflüsse auf die Reaktionsgeschwindigkeit bei SN2 Lösungsmittel Solvatation setzt die Reaktionsgeschwindigkeit herab Kleine und geladene Nucleophile sind in polaren Lösungsmitteln stark solvatisiert protische Lösungsmittel solvatisieren stärker als aprotische CH3I + Cl LM krel CH3OH 1 hschulz, oc2bct - LM krel HCONH2 12 CH3Cl + I HCONHCH3 45 - HCON(CH3)2 1 200 000 52 Lösungsmitteleffekte Unpolare Lösungsmittel Kohlenwasserstoffe, CCl4 Keine Wechselwirkung mit Ionen oder polaren Gruppen Reaktivität der Ionen oder polaren Gruppen wird nicht vermindert, aber Löslichkeit der Reaktanden ist gering SN2 wird unterstützt, SN1 behindert Polare, protische Lösungsmittel H2O, ROH, RNH2 Sehr gute Solvatation (Stabilisierung) von Ionen Herabsetzung der Reaktivität von Ionen, aber gute Löslichkeit der Reaktanden SN1 wird unterstützt, SN2 behindert hschulz, oc2bct 53 Lösungsmitteleffekte Polare aprotische Lösungsmittel THF, Acetonitril, Aceton Mässige Solvatation, mässige Stabilisierung von Ionen und mässige Löslichkeit Oft werden Kationen und Anionen unterschiedlich stark solvatisiert O + THF KI O O K + O + O I O O O K+ stark solvatisiert = gute Löslichkeit hschulz, oc2bct I- schwach solvatisiert = gute Nucleophilie 54 Einflüsse auf die Reaktionsgeschwindigkeit bei SN2 Die Struktur des Substrats Sekundär tertiär H3C-X Relative Geschwindigkeit 30 H3CH2C-X 1 (H3C)2HC-X 0.02 (H3C)3C-X 0 Die Struktur des Nucleophils sterische Hinderung verringert die Reaktionsgeschwindigkeit SN2-Reaktionsgeschw. nimmt ab Primär Halogenalkan Stabilität des Carbenium-Ions und die sterische Hinderung nimmt zu Kategorie Mechanismus der nucleophilen Substitution: SN1 CH3 Aceton/H2O CH3 CH3C-Cl + H-OH CH3C-OH + HCl CH3 CH3 Reaktionskinetik Reaktionsgeschwindigkeit ist nur abhängig von der (CH3)3CCl-Konzentration Geschwindigkeitsgesetz 1. Ordnung v = k[(CH3)3CCl] Monomolekulare Reaktion N Am Übergangszustand (geschwindigkeitsbestimmender Schritt) ist H2O nicht beteiligt zweistufiger Prozess S 1-Reaktion SN1- Mechanismus Schritt 1: Geschwindigkeitsbestimmend! H3C CH3 Cl H2O langsam CH3 -Cl H3C + CH3 CH3 Carbenium-Ion sp2 Hybridisierung trigonal planar Schritt 2: H3C + CH3 CH3 schnell O H H CH3 H3C + O H CH3 H Schritt 3: CH3 H3C O H + O H H schnell CH3 H + + H3O CH3 H3C O H CH3 tert-Butanol Veränderung der potentiellen Energie während einer SN1-Reaktion R R # R R X # Nu R R E R - Nu + R Ea X R3C+ X Nu R R Nu R - +X R Reaktionskoordinate hschulz, oc2bct 60 SN1-Reaktionsmechanismus Bindungsbruch erfolgt als erster Schritt unabhängig von der Bindungsbildung Es bildet sich ein (planares) Carbenium-Ion Angriff des Nucleophiles auf das elektrophile Zentrum kann von beiden Seiten erfolgen Es bildet sich eine racemische Mischung Die SN1-Reaktion ist nicht stereospezifisch! Racemisierung R1 R2 + R3 Nu - SN1 SN2 Bindungsbruch erfolgt als erster Schritt unabhängig von der Bindungsbildung Bildung eines planaren Carbenium-Ions Bindungsbruch und Bindungsbildung erfolgen gleichzeitig Bildung eines Übergangszustandes mit 5 Substituenten um den zentralen Kohlenstoff Angriff des Nucleophils Angriff des Nucleophils kann von beiden Seiten erfolgt immer von der des planaren Carbenium- Rückseite Ions erfolgen Keine Stereospezifität: Stereospezifität: Umkehr der Enantiomeren-reine absoluten Konfiguration: Edukte gehen in Inversion (Walden-Umkehr) Racemate über hschulz, oc2bct 62 Reaktivität von R-X gegenüber nucleophiler Substitution R SN1 SN2 CH3 nicht beobachtet (das MethylKation ist zu energiereich) Häufig Schnelle Reaktion primär nicht beobachtet (primäre Carbenium-Ionen sind zu energiereich) sekundär Relativ langsam Polare protische Lösungsmittel Gute Abgangsgruppen Sperrige Substituenten Häufig Polare protische Lösungsmittel Sterisch gehinderte Reste Gute Abgangsgruppen Häufig Schnelle Reaktion Langsam wenn Verzweigung an C2 Relativ langsam Aprotische, unpolare Lösungsmittel Gute Nucleophile tertiär Extrem langsam Einflüsse auf den SN1- und SN2Reaktionsmechanismus Das elektrophile Zentrum Grosse Substituenten führen zu einer sterischen Hinderung Besonders relevant des SN2-Angriffes bei sekundären Kohlenstoffen Zunahme der Stabilität von Carbenium-Ionen: primär < sekundär < tertiär Das Nucleophil Schwache Nucleophile begünstigen die SN1-Reaktion sterisch gehinderte Nucleophile begünstigen die SN1Reaktion Einflüsse auf SN1 und SN2 Gute Abgangsgruppen begünstigen SN1 Stark polarisierbare Gruppen sind gute Abgangsgruppen, da sie die negative Ladung gut aufnehmen können F- < Cl- < Br- < I- Austrittsvermögen nimmt zu! Alkylsulfat- und Alkylsulfonat-Ionen schwache Basen Kleine neutrale Moleküle sind i.d.R. gute Abgangsgruppen (NH3 < H2O) O H3C O S O O - O H3C S O O - O F3C S O - O Methylsulfat-Ion Methansulfonat-Ion Trifluormethan(Mesylat-Ion) sulfonat-Ion (Triflat-Ion) O H3C S O p-Toluolsulfonat-Ion (Tosylat-Ion) O - Einflüsse auf den SN1- und SN2Reaktionsmechanismus Das Lösungsmittel Polare Lösungsmittel stabilisieren das Carbenium-Ion und die (oft negativ geladene) Abgangsgruppe und begünstigen dadurch die SN1-Reaktion Polare, protische Lösungsmittel haben einen deutlich grösseren stabilisierenden Effekt Unpolare Lösungsmittel begünstigen die SN2-Reaktion In unpolaren Lösungsmitteln ist die Reaktivität des Nucleophils grösser SN-Reaktionen mit Retention Bestimmte Edukte reagieren mit einem Nucleophil unter Erhalt der Konfiguration am Chiralitätszentrum Retention Es findet keine Inversion statt Es findet keine Racemisierung statt Die Kinetik folgt einem Geschwindigkeitsgesetz erster Ordnung Am geschwindigkeitsbestimmenden Schritt ist das Nucleophil nicht beteiligt Nachbargruppeneffekte hschulz, oc2bct 67 Retention als Folge der doppelten Inversion Br H C O C H3C OH OH- (R)-a-Brompropionsäure Br H C O C H3C Br H H3C C C O O 1. Inversion - Br OH OH H C O H 2. Inversion C H3C O H3C C C O O H+ O OH H C O C H3C (R)-Milchsäure hschulz, oc2bct OH OH- H H3C C O C O a-Lacton 68 Ambidente Nucleophile Zwei Zentren eines Nucleophiles können mit einem Elektrophil reagieren Die beiden Zentren stehen in Konjugation miteinander Ambidente Nucleophile sind i.d.R. polar Die Zentren (nucleophile Positionen) unterscheiden sich bzgl. EN, Grösse und Polarisierbarkeit Beispiele ambidenter Nucleophile O C C N Cyanid hschulz, oc2bct N O N N O O Nitrit 69 Umsetzung von Alkylhalogeniden mit Cyanid Unter SN2-Bedingungen reagiert bevorzugt das stärker polarisierbare Zentrum R-CH2-Br + NaCN Kolbe-Nitrilsynthese R-CH2-CN + NaBr Nitril Unter SN1-Bedingungen reagiert bevorzugt das Zentrum mit höherer Elektronendichte (höhere EN) R3C-Br + NaCN R3C-NC + NaBr Isonitril hschulz, oc2bct 70 2.3 Eliminierung Eliminierungen können stattfinden Ohne Teilnahme anderer Reaktionspartner, in der Regel thermisch Unter dem Einfluss von Basen oder LösemittelMoleküle Mit Reduktionsmittel aus vicinalen disubstituierten Verbindungen – z.B. 1,2-Dihalogenverbindungen mit Metallen hschulz, oc2bct 71 2.3 Eliminierung 1,2-Eliminierung (b-Eliminierung) Eliminierung zweier Atomgruppen an vicinalen C-Atomen 1,1-Eliminierung (a-Eliminierung) Eliminierung zweier Atomgruppen am gleichen C-Atom hschulz, oc2bct 72 b – Eliminierung (1,2-Eliminierung) Bildung von Alkenen durch Abspaltung kleiner Moleküle aus substituierten Alkanen H Base B- + H-X X Die Substrate für Eliminierungen sind die gleichen wie die Substrate für nucleophile Substitutionen Eliminierung und Substitution sind Konkurrenzreaktionen hschulz, oc2bct 73 Unimolekulare Eliminierung (E1): Konkurrenz zur Substitution CH3 H3C Br H3C CH3 E1 + C CH3 H3C + BrCarbeniumIon SN1 +CH3OH CH3 H2C + H++ Br- CH3 20% 2-Methylpropen hschulz, oc2bct H3C H3C O CH3 + H++ Br- CH3 80% 2-Methoxy-2-methylpropan 74 Stabilisierungsmöglichkeiten eines Carbenium-Ions Bildung eines Carbenium-Ions unter den gleichen Bedingungen, wie bei der SN1 beschrieben Das Carbenium-Ion kann auf drei Reaktionswegen stabilisiert werden Addition des Nucleophiles = SN1 Abstraktion eines Protons durch das Nucleophil (Base) = E1 Umlagerung zu einem stabileren Carbenium-Ion und anschliessende Reaktion unter SN1 oder E1 hschulz, oc2bct 75 Abstraktion eines Protons aus einem Carbenium-Ion Kohlenstoffatom mit positiver Ladung übt –I-Effekt aus Benachbarte C-H-Bindungen werden polarisiert Basen können ein Proton abstrahieren, wobei sich ein Alken bildet R1 R2 R4 + C + R3 - H-B d H B hschulz, oc2bct R2 R4 R1 R3 76 Umlagerung von Carbenium-Ionen Weniger stabile Carbenium-Ionen können durch 1,2-Hydrid-Verschiebung in stabilere CarbeniumIonen umlagern Die neu entstanden stabileren Carbenium-Ionen können sich dann durch Addition eines Elektrophiles oder durch Eliminierung eines Protons stabilisieren H H Br H3C CH3 -Br- CH3 H SN1 hschulz, oc2bct H3C H CH3 CH3 H Sekundäres Carbenium-Ion H3C CH3 CH3 H Tertiäres Carbenium-Ion 77 Umlagerung von Carbenium-Ionen unter Veränderung des Kohlenstoffgerüsts Sind keine Wasserstoffatome in a-Stellung vorhanden, können auch Alkylgruppen wandern Wagner-Meerwein-Umlagerungen Neopentyl-Umlagerung: CH3 CH3 SN2 / H 2O Br H3C H3C C H2 H3C H3C Sterische OH Hinderung des C SN2-Angriffs H2 CH3 SN1 -Br- hschulz, oc2bct H3C H3C H3C CH2 CH3 Primäres Carb.-Ion -H+ H2 O OH H3C H3C C H2 CH3 Tertiäres Carb.-Ion C H2 CH3 H 78 Einfluss der Base (des Nucleophils) auf das Produktverhältnis H3C C CH3 H3C (CH3)3CX E1 + OHschnell SN1:E1 95 : 5 95 : 5 95 : 5 hschulz, oc2bct + X- SN1 + CH3CH2OH / H2O CH3 X- + HOH + H2C X Cl Br I + CH3 (CH3)3COCH2CH3 + (CH3)3COH + H+ + X- Zugabe von ca. 1 Äquivalent einer starken Base: fast 100% E1 79 Konkurrenz zwischen E1 und SN1 E1 ist bevorzugt bei schwachen Nucleophilen mit geringem Zusatz einer starken Base (z.B. H2O + OH-) voluminösen Basen (sterische Hinderung des Angriffs am C+) voluminösen Substituenten am Carbenium-Ion (sterische Hinderung des Angriffs am C+) hohen Temperaturen (>100°C) SN1 ist bevorzugt bei starken Nucleophilen mit schwächerem Basencharakter (hohe Polarisierbarkeit) niedrigeren Temperaturen (ca. 0°C) hschulz, oc2bct 80 Bimolekulare Eliminierung (E2) R1 R2 X R2 R4 + H-B + X- B- H R3 R4 R1 R3 Folgende Vorgänge laufen gleichzeitig ab: Bildung der B-H-Bindung Lösen der H-C-Bindung Bildung der Doppelbindung (sp3 wird zu sp2) Lösen der C-X-Bindung Austritt des Protons bevorzugt in anti-Stellung Konzertierte anti-Eliminierung hschulz, oc2bct 81 Konkurrenz zwischen E2 und SN E2 ist bevorzugt bei starken Base voluminösen Basen (Angriff an der Peripherie des Substrats) voluminösen Substituenten des Substrats (z.B. sekundäre und tertiäre Halogenalkane) SN ist bevorzugt bei starken Nucleophilen mit schwächerem Basencharakter (hohe Polarisierbarkeit) sterisch anspruchslosen Nucleophilen und Substraten (z.B. primäre und sekundäre Halogenalkane) hschulz, oc2bct 82 Günstige Bedingungen für eine Eliminierung Das Substrat ist sterisch anspruchsvoll (sterische Hinderung) CH3 Die Base (das Nucleophil) H3C O ist sterisch anspruchsvoll CH3 t-Butanolat Das Nucleophil ist eine starke Base (RO-, R2N-) CH3 H3C hohe Temperatur N H3C hschulz, oc2bct Diisopropylamid CH3 83 Eliminierung versus Substitution R-X Schwaches Nukleophil (H2O) Nukleophil, schwach basisch (I-) CH3CH2Br (primär) keine Reaktion SN2 SN2 E2 (CH3)2CHBr (sekundär) SN1 SN2 E1/E2 E2 (CH3)3CBr (tertiär) SN1/E1 SN1/E1 E1/E2 E1/E2 hschulz, oc2bct Stark basisch, Stark basisch und keine sterische ster. gehindert Hinderung (CH3)3CO(HO-) 84 Basenstärke des Nucleophils Sterische Hinderung am C-Atom Sterische Hinderung am stark basischen Nucleophil Substitution wahrscheinlicher Eliminierung wahrscheinlicher Schwache Basen H2O, ROH, PR3, X , RS , N3 , NC , RCOO Sterisch ungehindert primäre Halogenalkane Starke Basen HO , RO , H2N , R2N Sterisch ungehindert* HO , CH3O , C2H5O , H2N * Substitution möglich hschulz, oc2bct Sterisch gehindert verzweigte primäre, sekundäre, tertiäre Halogenalkane Sterisch gehindert** (CH3)3CO , [CH(CH3)2]2N ** Eliminierung stark bevorzugt 85 Die Orientierung der Doppelbindung bei einer Eliminierung B H H3C H CH2 CH3-CH-C Br 69% H CH3 CH3 Saytzev-Produkt: thermodynamisch bevorzugt - H-Br CH3 2-Brom-2-methylbutan H3C CH2 H H 31% CH3 Hofmann-Produkt: kinetisch bevorzugt Saytzev und Hofmann Orientierung Saytzev-Produkt: Doppelbindung hat Maximale Anzahl Alkyl-Gruppen Br -HBr Hofmann-Produkt: Doppelbindung hat maximale Anzahl Wasserstoffe Hofmann-Eliminierung 95% Hofmann Produkt CH2 H3C H CH2 CH2 CH3 + CH3-CH2-C N CH3 CH3 CH3 CH3 -N(CH3)3, -H+ H3C CH CH3 CH3 5% Saytzev-Produkt Die Stereochemie der E2-Reaktion: alle 5 Atome liegen in einer Ebene Antiperiplanarer Übergangszustand ist bevorzugt Synperiplanarer Übergangszustand kann bei cyclischen Verbindungen erzwungen werden B: H L B: H L Bevorzugung des antiperiplanaren Übergangszustands H Br Der antiperiplanare ÜZ entsteht aus der thermodynamisch stabileren gestaffelten Konformation BrH während der synperiplanare ÜZ aus der weniger stabilen ekliptischen Konformation entsteht Konzertierte anti-Eliminierung CH3 H cis bezüglich Abgangsgruppe CH3 H ROtrans bezüglich OTs Abgangsgruppe trans-2-Methylcyclohexyltosylat (a,a) 3-Methylcyclohexen Keine Reaktion zu 1-Methylcyclohexen Konzertierte anti-Eliminierung: Bevorzugung des Saytzev-Produkts CH3 H CH3 H OTs cis-2-Methylcyclohexyltosylat (a,e) RO- 64% Saytzev Produkt 1-Methylcyclohexen CH3 36% Hofmann Produkt 3-Methylcyclohexen Dehydratisierung von Alkoholen: Wasser-Eliminierung nach E1 oder E2 H HH HH OH CH3CH2OH H H H H H+, D -H2O konz.H2SO4, 170°C -H2O CH2=CH2 Unimolekulare Dehydratisierung: E1 HO H R C C H + H+ - H+ O + H R C C - H2O + H2O R + H+ - H+ H H + R C C Unimolekulare Dehydratisierung: eine reversible Reaktion Leichtigkeit der Dehydratisierung von Alkoholen: R= tertiär > sekundär > primär Umlagerung von Carbenium-Ionen führt zu den thermodynamisch stabilsten Alkenen CH3 OH H3C C CH2 C H H sekundär + H3C C CH2 C H H CH3 CH3 Umlagerung Saytzev, 54% CH3 H H3C C CH C H CH3 + H+, - H2O tertiär CH3 H+ + - H+ CH3 + H H3C C CH2 C H CH3 Bimolekulare Dehydratisierung von primären Alkoholen: E2 R CH2 CH2 OH H + + H+ (H2SO4) - - H2O, -H+ R CH CH2 O H H RCH2CH2OH oder HSO4 H+ H + R CH2 CH2 O H R CH CH2 + RCH2CH2OH2+ oder H2SO4 Bei tieferen Temperaturen (<100°C) überwiegt SN2-Reaktion zu Ethern Bei höheren Temperaturen (180°C) reagieren auch Ether unter Eliminierung zu Alkenen Monomolekulare Eliminierung E1cB (E1 conjugierte Base) Bei E2 erfolgt der Abgang des Protons durch den Angriff der Base und der Abgang der zweiten Atomgruppe gleichzeitig Bei E1 erfolgt der Abgang der zweiten Atomgruppe im ersten Schritt unter Bildung eines Carbenium-Ions Bei E1cB erfolgt der Abgang des Protons durch den Angriff einer Base im ersten Schritt unter Bildung eines Carbanions (=conjugierte Base) X X -HB C -X- H B hschulz, oc2bct 97 Beispiel für E1cB Starke Base BIHohe CH-Acidität des Substrats Br Br Br F Br F -HB F F F Br F -F C H Br - F F B Die Bildung des Carbanions erfolgt schnell Die Eliminierung der Abgangsgruppe erfolgt langsam Die Reaktionsgeschwindigkeit ist nur von der Konzentration des Carbanions abhängig hschulz, oc2bct 98 a- Eliminierung (1,1-Eliminierung) Beide Abgangsgruppen werden vom gleichen Zentralatom abgespalten Cl Cl C Cl - OH Cl H C schnell Cl Cl CO + HCO2+ Cl- H2O/OH - Cl schnell C Cl - Cl - langsam Dichlorcarben hschulz, oc2bct 99 2.4 Elektrophile Addition an die C-C-Doppelbindung H-X H X Halogenalkane H-OH H OH Alkohole X-X X X Alkene Dihalogenalkane Exotherme Reaktionen hschulz, oc2bct 100 Die C-C-Doppelbindung C-C-Doppelbindungen, die +I-Substituenten tragen, sind elektronenreich und können elektrophil angegriffen werden Doppelbindungen, die -I-Substituenten tragen, sind elektronenarm und können nucleophil angegriffen werden R R NC R R NC Elektronenreiche Doppelbindung hschulz, oc2bct CN CN Elektronenarme Doppelbindung 101 Reaktivität der C-C-Doppelbindung gegenüber Elektrophilen X Zunehmende Reaktivität durch +ISubstituenten Abnehmende Reaktivität durch -I- und -M-Substituenten Abnehmende Reaktivität durch sterisch anspruchsvolle Substituenten H HOOC H < H H < R H < < H H H H H H H H R H R R R R R R R R < R H hschulz, oc2bct < H H < R H 102 Elektrophile Addition von HX an Alkene 1. Schritt: elektrophiler Angriff von H+ H+ H C CarbeniumIon + 2. Schritt: nucleophiler Angriff von X- H C + +X- H X Reaktionsbedingungen: tiefe Temperatur (ca. 0°C) Bei hoher Temperatur erfolgt Rückreaktion (Eliminierung) hschulz, oc2bct 103 Regioselektiver Angriff an der Doppelbindung H H+ H3C C H CH2 H3C C + CH2 + Br- H sekundäres Br H H3C C CH2 H Carbenium-Ion H H3C C C H H + H + Br- H H3C C H Br CH2 primäres Carbenium-Ion Der elektrophile Angriff eines Protons führt zum stabileren Carbenium-Ion Es werden deshalb keine primären Alkylderivate erhalten hschulz, oc2bct 104 Regel von Markovnikov (1870) Wird HX an ein Alken addiert, lagert sich das Wasserstoffatom an die Stelle der Doppelbindung an, die bereits die meisten Wasserstoffatome hat Es bildet sich bevorzugt der Übergangszustand, der zum stabileren Carbenium-Ion führt CH2=CHCH3 höhere Anzahl H H-Br hschulz, oc2bct CH3CHBrCH3 MarkovnikovAdditionsprodukt 105 Elektrophile Addition von Wasser an die C-C-Doppelbindung Industrielle Methode zur Herstellung von Alkoholen Katalyse durch starke Mineralsäuren Addition folgt der Regel von Markovnikov CH3 H3C C CH2 + HOH 2-Methylpropen hschulz, oc2bct H+ CH3 H3C C CH2 OH H tert-Butylalkohol 106 Mechanismus der AE von Wasser an Alkene Schritt 1: CH3 Addition eines Protons CH3 langsam H3C C CH2 H H + O H + H3C C CH2 +H2O H Geschwindigkeitsbestimmender Schritt: Bildung des stabilsten Carbenium-Ions hschulz, oc2bct 107 Schritt 2: Addition eines Wassermoleküls CH3 + CH3 schnell H3C C CH2 H3C C CH3 + O H O H H H Protonierter Alkohol H Schritt 3: Proton-Transfer auf Wassermolekül CH3 H3C C + schnell CH3 O H O H H H hschulz, oc2bct CH3 H3C C O H CH3 +H3O+ Alkohol 108 Die Hydratisierung von Alkenen und die Dehydratisierung von Alkoholen sind Gleichgewichtsprozesse! Eliminierung konz. H2SO4, hohe Temperatur Alkohol Alken + H2O H2SO4, Überschuss H2O niedrige Temperatur Elektrophile Addition hschulz, oc2bct 109 Elektrophile Addition von Halogenen an die Doppelbindung Halogene reagieren mit Alkenen und Alkinen bereits bei Raumtemperatur – auch im Dunkeln! Entfärbung von Brom dient als Nachweis der Mehrfachbindung C C + Br2 Raumtemperatur im Dunkeln, CCl4 C C Br Br Dibromid hschulz, oc2bct 110 Schritt 1: Induzierte Polarisierung des Halogenmoleküls durch die Doppelbindung Heterolytische Spaltung des Halogenmoleküls Bildung eines intern stabilisierten CarbeniumIons (=Halonium-Ion) und eines Halogenid-Ions C C d+ Br d- Br hschulz, oc2bct C C + Br + Br Bromid-Ion Bromonium-Ion 111 Schritt 2: Angriff des Bromid-Ions an einem der beiden Kohlenstoffatome Angriff erfolgt von der Rückseite SN2-Reaktion Br - Br C C + Br Bromonium-Ion hschulz, oc2bct C C Br Dibromid 112 Halogen-Addition an Alkene Stereospezifische Reaktion anti-Addition an die Doppelbindung Zweiter Schritt ist eine SN2-Reaktion Fluorierungen sind sehr stark exotherm! Iodierungen sind thermodynamisch nicht begünstigt Reaktionsführung: RT oder Kühlung Inerte Lösungsmittel (Halogenalkane) hschulz, oc2bct 113 Stereospezifische Addition von Brom an trans-Buten H3C C C H H Raumtemperatur + Br2 CH3 trans-Buten CCl4 CH3 H Br C (2R, 3S) 2,3-Dibrombutan (eine Mesoverbindung) C Br hschulz, oc2bct H CH3 114 Stereospezifische Addition von Brom an trans-Buten CH3 H C C H H3C Br CH3 H C C H3C H Br d+ Br d- Br (a) Br (a) (b) CH3 (b) H C C H3C + H Br Bromoniumion hschulz, oc2bct (chiral) (2R, 3S) 2,3-Dibrombutan (meso) Br CH3 H C C H3C H Br (2R, 3S) 2,3-Dibrombutan (meso) 115 Stereospezifische Addition von Brom an cis-Buten H3C C C H3C H H cis-Buten + Br2 Raumtemperatur CCl4 CH3 H Br C CH3 Br H C C C H Br CH3 (2R, 3R) 2,3-Dibrombutan hschulz, oc2bct Br H CH3 (2S, 3S) 2,3-Dibrombutan 116 Stereospezifische Addition von Brom an cis-Buten H H C C H3C CH3 Br H H C C H3C CH3 Br (2R, 3R) d+ Br d- Br (a) Br (a) (b) H H (b) C C H3C + CH3 Br Bromonium-Ion hschulz, oc2bct (meso=achiral) 2,3-Dibrombutan (chiral) Br H H C C H3C CH3 (2S, 3S) Br 2,3-Dibrombutan (chiral) 117 2.5 Elektrophile Addition an konjugierte Doppelbindungssysteme Alternierende Folge von Doppel- und Einfachbindungen Konjugierte Systeme sind stabilisiert =Resonanzenergie! Hydrierungswärme DH°= -255 kj/mol H2C CH CH2 CH CH2 1,3-Butadien DH°= -240 kj/mol H2C CH CH CH2 Butadien ist durch Konjugation der Doppelbindungen um ca. 15 kj/mol stabilisiert (=Resonanzenergie) 1,4-Pentadien hschulz, oc2bct 118 MO-Betrachtung von Butadien p-Bindung 134 pm schwache p-Überlappung 147 pm p-Bindung 134 pm H H H H H H H H H hschulz, oc2bct H H H 119 Linearkombination der pz-Orbitale im Butadien E + - + - - + - + + - - + - + + - + + - - - + + + + + + - - - - p3 antibindend p2 bindend - hschulz, oc2bct p4 antibindend p1 bindend 120 Molekülstruktur des 1,3-Butadiens H H H H H H s-trans (transoid) H H H H H H s-cis (cisoid) Aufgrund sterischer Hinderung ist die cisoide Form um ca. 12 kj/mol energiereicher, d.h. instabiler hschulz, oc2bct 121 Elektrophile Addition von HCl an Butadien 1 H2C 2 3 4 CH CH CH2 Angriff an C1 +H+ H + H2C HC CH CH2 H +H+ + H2C CH CH CH2 Primäres nicht delokalisiertes Kation: wird nicht gebildet! + CH CH CH2 H3C +Cl- +Cl- Cl H3C HC CH CH2 1,2-Addition: kinetisch kontrolliert! hschulz, oc2bct H3C CH CH CH2 Cl 1,4-Addition: thermodyn. kontrolliert! 122 Elektrophile Addition von Brom an 1,3,5-Hexatrien + Br2 -Br- + (A) + Br Br + Br (B) 1,2-Addition kin. kontrolliert hschulz, oc2bct + Br + Br Br Br + Br - (C) + Br - Br Br Br Br 1,6-Addition 1,4-Addition kin. kontrolliert thermodyn. kontrolliert 123 Reaktivität konjugierter p-Systeme Konjugierte p-Systeme sind thermodynamisch stabiler als nicht konjugierte (Resonanzstabilisierung) Sie sind trotzdem kinetisch reaktiver. Die Aktivierungsenergie für elektrophile Additionen ist bei konjugierten Systemen geringer aufgrund der stark delokalisierten kationischen Zwischenstufen Ea1 E nicht konjugiert konjugiert hschulz, oc2bct Ea2 Ea1 > Ea2 124 2.6 Reaktivität des Allyl-Systems C-H-Bindung an der Methylgruppe des Propens ist schwach H H2C C C H H H DH° (kj/mol) 364 CH3 H3C C H CH3 389 CH3 H3C C H H 397 H H3C C H H 410 3-Chlorpropen bildet unter SN1-Bedingungen leicht ein kationisches Intermediat – im Unterschied zu gesättigten primären Halogenalkanen! H H H +CH OH + 3 CH3OH, D H2C C C Cl -Cl H2C C C H2C C C O CH3 + -H H H H H H H Allyl-Kation hschulz, oc2bct 125 Reaktivität des Allyl-Systems Der pKa-Wert der Methyl-C-H-Bindungen im Propen ist deutlich niedriger als in Propan (pKa=40 vs. 50) H H2C C C H H H H H2C C C H H + H+ Allyl-Anion Die Bildung von Allyl-Radikalen, Allyl-Kationen und Allyl-Anionen ist begünstigt! hschulz, oc2bct 126 Resonanzstabilisierung des Allyl-Systems H . H2C CH CH2 H2C CH CH2 Allyl-Radikal H2C . C CH2 H + H2C CH CH2 + H2C CH CH2 Allyl-Kation C H2C + CH2 H H2C CH CH2 H2C CH CH2 Allyl-Anion hschulz, oc2bct C H2C - CH2 127 MO-Beschreibung des Allyl-Systems p-Bindung pz-Orbitale H H H C C C H s-Gerüst H Die drei p-Orbitale der Allyl-Gruppe überlappen und ergeben eine symmetrische Struktur mit delokalisierten Elektronen! hschulz, oc2bct 128 Linearkombination der pz-Orbitale im Allylsystem Knotenebene E + - + - + - + - - + + + + - - - hschulz, oc2bct p3 antibindend p2 nichtbindend p1 bindend + . - 129 Hydrolyse isomerer Allylchloride: SN1 H H H3C C C Cl H3C C C CH2 Cl -Cl H3C C C + thermo- +H2O dynamisch -H+ kontrolliert H3C C C + CH2 CH2 H H Unsymmetrisch! H H H3C C C CH2 OH Stabilers Endprodukt hschulz, oc2bct H H H H - CH2 - -Cl +H2O kinetisch -H+ kontrolliert OH H3C C C H H CH2 130 2.7 Nucleophile Addition an die C-C-Doppelbindung Elektronenziehende Substituenten erniedrigen die Elektronendichte der C-C-Doppelbindung Substituenten mit -I und -M-Effekt sind besonders effektiv: CHO > COR > COOR > CN > NO2 Der Mechanismus der AN ist identisch mit dem der AE. Lediglich die Vorzeichen der Ladungen sind vertauscht. hschulz, oc2bct 131 Addition von Alkohol an elektronenarme Alkene (Basenkatalyse) 1. Schritt: nucleophiler Angriff von RO RO - - CN CN RO C Carbanion 2. Schritt: Protonierung des Carbanions CN RO C hschulz, oc2bct + H+ CN RO H 132 Die Cyanethylierung Die Doppelbindung in Acrylnitril wird leicht durch nucleophile Reagenzien (z.B. Anionen) angegriffen. Die Addition wird durch Protonierung des Carbanions abgeschlossen Formal wurde eine Cyanethyl-Gruppe an das Nucleophil gebunden. In weiteren Reaktionsschritten kann die Cyanogruppe in andere funktionelle Gruppen umgewandelt werden H2C=CH-CN +ROH +PhOH +H2S +RNH2 hschulz, oc2bct RO-CH2-CH2-CN PhO-CH2-CH2-CN HS-CH2-CH2-CN RNH-CH2-CH2-CN 133 Michael-Addition Nucleophile Addition an eine Doppelbindung, wobei das angreifende Nucleophil ein Carbanion ist. COOR Malonsäureester CH2 COOR + RO- ROH COOR COOR COOR HC CH2 CH2 C HC N + H2C + ROH / - RO- CH C N Acrylnitril COOR 2-Cyanoethylmalonsäureester (3-Propannitrilmalonsäureester) hschulz, oc2bct 134 2.8 Radikalische Addition an C-CDoppelbindungen (AR) Radikalische Hydrobromierung Kettenstart R-O-O-R 2 R-O R-O + H-Br R-O-H + Br Das H-Br Molekül wird durch „Radikal-Initiatoren“ homolytisch gespalten, und es entsteht ein freies Brom-Radikal Die Doppelbindung wird von dem entstandenen Br-Radikal angegriffen und nicht von einem möglichen H-Radikal! hschulz, oc2bct 135 Kettenwachstum H C CH2 Br H3C CH2 .C Br CH2 H CH3CH2 Stabileres Radikal H3C CH2 .C H H-Br CH2 Br H H3C CH2 C H + Br Br CH2 „Anti-Markovnikov“ Kettenabbruch hschulz, oc2bct durch Radikal-Rekombinationen 136 Radikalische Addition (AR) Zugang zu „anti-Markovnikov“-Produkten Die radikalische Addition von HCl und HI ist endotherm und deshalb nicht begünstigt Synthetische Bedeutung haben die radikalische Additionen von HBr, RSH und einige Halogenalkane hschulz, oc2bct 137 2.9 Cycloadditionen Bei der Cycloaddition reagieren zwei Moleküle miteinander, wobei das HOMO des einen mit dem LUMO des anderen in Wechselwirkung tritt Das Reaktionsprodukt ist eine cyclische Verbindung Entsprechend der Anzahl der Atome, die von jedem Edukt in den Ring eingebracht wird, unterscheidet man [2+1]-, [3+2]-,[4+2]-Cycloadditionen hschulz, oc2bct 138 Diels-Alder-Cycloaddition: [4+2] D Dien Dienophil Konjugierte Diene reagieren mit Alkenen zu Cycloalkenen Voraussetzung ist eine cisoide Konformation Konzertierte Reaktion: sie läuft in einem Schritt ab [4+2]-Cycloaddition hschulz, oc2bct 139 Grenzorbitalbetrachtung der [4+2]-Cycloaddition D LUMO HOMO und LUMO haben passende Symmetrie HOMO hschulz, oc2bct Reaktion erfolgt unter thermischen Bedingungen 140 Reaktivität von Dienen und Dienophilen Unsubstituierte Alkene liefern nur geringe Ausbeuten an cyclischen Reaktionsprodukten Die Reaktivität wird drastisch erhöht, wenn Dien und Dieneophil unterschiedliche Elektronendichte haben Elektronenreiches Dien Elektronenarmes Dienophil hschulz, oc2bct 141 Typische Diene und Dienophile Diene Dienophile NC NC CN CN O H H CN CN CO2CH3 H H CO2CH3 CO2CH3 O H3CO2C CO2CH3 H O H O H OMe CO2CH3 O hschulz, oc2bct 142 Stereochemie der Diels-AlderReaktion Die Konfiguration des Diens und des Dienophils wird beibehalten, d.h. die DielsAlder-Reaktion ist stereospezifisch H CO2CH3 + H H CO2CH3 CO2CH3 + H3CO2C hschulz, oc2bct 150°C/20h H 200°C/3h CO2CH3 CO2CH3 H H CO2CH3 H H CO2CH3 cisProdukt transProdukt 143 Exo- und Endo-Produkte O O + O O O O O O H O H O O H O endo-Produkt O kinetisch kontrolliert hschulz, oc2bct O H exo-Produkt thermodyn. kontrolliert 144 Beispiele von Diels-AlderReaktionen Alkine als Dienophile CO2CH3 100°C/6h + CO2CH3 H H Dimerisierung von Cyclopentadien + hschulz, oc2bct RT/langsam 145 [2+1]-Cycloadditionen Addition von Carbenen an Doppelbindungen R3 R3 R1 + R4 hschulz, oc2bct R1 C C R3 R2 + R2 C R1 R2 R4 R4 146 [2+1]-Cycloadditionen Elektronische Eigenschaften von Carbenen Elektronensextett an einem zweibindigen Kohlenstoff H H 103° H Singulett-Carben 136° H Triplett-Carben Grundzustand von Methylen (CH2) ist ein TriplettCarben. Einige Halogencarbene liegen aber im Singulett-Grundzustand vor. hschulz, oc2bct 147 [2+1]-Cycloadditionen Singulett-Carbene gehen [2+1]-Cycloadditionen ein Konzertierte Reaktion Stereoselektive syn-Addition Triplett-Carbene reagieren radikalisch in einem Stufen-Mechanismus der radikalischen Zwischenstufe um sBindungen möglich Keine Stereoselektivität Rotationen hschulz, oc2bct 148 Dichlorcarben Darstellung aus Chloroform durch aEliminierung von HCl Dichlorcarben kann nicht isoliert werden, sondern reagiert in Lösung mit Alkenen unter Bildung eines Cyclopropan-Derivats CHCl3 R3 Cl + R4 hschulz, oc2bct + KOH - HCl CCl2 R3 C Cl C Cl Cl R4 149 [2+2]-Cycloaddition Thermische Aktivierung führt nicht zur Dimerisierung von Alkenen Bindende Wechselwirkung wird durch antibindende Wechselwirkung aufgehoben Thermische Energie reicht nicht aus, um Elektronenübergänge zu ermöglichen LUMO HOMO hschulz, oc2bct 150 [2+2]-Cycloaddition Photochemische Aktivierung führt zur Dimerisierung unter Bildung von Cyclobutanderivaten Übergang eines Elektrons vom HOMO ins LUMO durch photochemische Anregung (p g p*-Übergang) Zwei einfach besetzte p*-Orbitale können konstruktiv überlappen, da sie die gleiche Symmetrie haben LUMO (p*) hn hn HOMO (p) hschulz, oc2bct 151 [2+2]-Cycloaddition C6H5 C6H5 C6H5 H H C6H5 hn H COOH Truxinsäure H HOOC COOH COOH Zimtsäure C6H5 C6H5 H HOOC COOH H hn H COOH Truxillsäure H C6H5 hschulz, oc2bct HOOC C6H5 152 [3+2]-Cycloaddition 1,3-Dipolare Cycloadditionen Stereospezifische B C A R2 R4 hschulz, oc2bct R1 R3 syn-Addition B A C R1 R2 R4 R3 153 [3+2]-Cycloaddition Ozonolyse: Bestimmung der Lage der Doppelbindung O O O O R2 - 78°C R1 H R1 H R3 Primärozonid R2 R3 O H R1 H O O R2 R3 R2 O R3 O O Sekundärozonid O R2CHO + R1-CO-R3 Aldehyd + Keton O R1 Reduktion Oxidation R2COOH + R1-CO-R3 Carbonsäure + Keton hschulz, oc2bct 154 [3+2]-Cycloaddition Addition von Diazoverbindungen N H2C H N N H2C H H H H CN N H - N2 CN 3-Cyano1-pyrazolin CN hschulz, oc2bct 155 Diazomethan Darstellung H C O S O N NO + KOH - H 2O N N H + H C N SO3-K+ N H Eigenschaften von Diazomethan Giftig, carcinogen In reiner Form explosiv: nur in Lösung handhaben In Lösung (Ether) ist Diazomethan bei Temperaturen um 0°C einige Tage stabil hschulz, oc2bct 156 2.10 Elektrophile Addition an die C-C-Dreifachbindung Die C-C-Dreifachbindung wird elektrophil angegriffen, da sie ein Zentrum mit hoher Elektronendichte ist. Die Elektronen sind aber stärker gebunden als in der C-C-Doppelbindung, d.h. sie haben eine niedrigere Energie. Alkine reagieren deshalb bereitwilliger unter nucleophiler Addition hschulz, oc2bct 157 Hydrierung der Dreifachbindung Stereospezifische Hydrierung zu cis-Alkenen: heterogene Katalyse H H H7C3 C C C3H7 LindlarKatalysator H H7C3 H H7C3 100% cis4-Octen Lindlar-Kat: 5%Pd/CaCO3, Bleiacetat, Chinolin hschulz, oc2bct 158 Stereospezifische Hydrierung zu trans- Alkenen: Ein-Elektronen-Reduktionen H7C3 C C C3H7 Na, NH3(l) H7C3 H H C3H7 86% trans4-Octen Hydrierung durch heterogene Katalyse mit Pd/Aktivkohle liefert Alkane hschulz, oc2bct 159 Mechanismus der Hydrohalogenierung von Alkinen 1. Schritt: Protonierung des Alkins unter Bildung eines Alkenyl-Kations 2. Schritt: Reaktion des Halogenids mit dem Kation unter Bildung des (Z)- oder (E)-Alkens H + R C C R +H R H R + H R R X R +X + C C R - X Die HX-Addition folgt der Regel von Markovnikov Das Alken kann mit weiterem HX zu Dihalogenalkanen weiter reagieren hschulz, oc2bct 160 Halogenierung von Alkinen Der Mechanismus ist identisch zur Halogenaddition an C-C-Doppelbindungen Bildung eines Halonium-Ions Angriff von X von der Rückseite liefert das trans-Alken R C C R X2, CCl4 R X X X2, CCl4 R X X R R X X Vicinales Dihalogenalken Die vicinalen Dihalogenalkene können isoliert werden hschulz, oc2bct 161 Die Hydratisierung von Alkinen: AE R H R H HOH, HOH, H+, H+, HgSO4 HgSO4 H OH H H O R R R R H OH H H O H H H H Enol Keton Acetaldehyd HO R H O H H HOH, H+, HgSO4 Keto-Enol-Tautomerie hschulz, oc2bct H R H R H Methylketon 162 2.11 Nucleophile Addition an die C-C-Dreifachbindung H H OR + R-O - OR H H +ROH C C H H H -R-O - Carbanion Stark basische Anionen reagieren mit Vinylierung Alkinen unter Addition zu Carbanionen RO - - , RS , NR2 - Die ebenfalls stark basischen Carbanionen werden durch ein Proton abgesättigt Ist kein Proton vorhanden, erfolgt eine anionische Polymerisation hschulz, oc2bct 163 2.12 Hydroborierung von Doppelund Dreifachbindungen BH3 liegt in Substanz als hochreaktives, selbstentzündliches Dimer B2H6 vor. Es kann aber in Form eines Boran-EtherKomplexes leicht gehandhabt werden H B H hschulz, oc2bct H H B H THF H B 2 3 O + H 164 Mechanismus der Hydroborierung # + H2B-H H2B H H2B H VierzentrenÜbergangszustand Stereoselektive Addition: syn Regioselektive Addition: das Boratom wird an das sterisch weniger gehinderte C-Atom addiert hschulz, oc2bct 165 Hydroborierung von Alkenen H H +BH3 H R R H H H H BH2 +2 H H R H R H H 3 H B H Bildung von Trialkylboran Bor addiert an das C-Atom mit den meisten Wasserstoffen (geringste sterische Hinderung) hschulz, oc2bct 166 Umwandlung der Alkylborane Oxidation und anschliessende Hydrolyse liefert Alkohole nach antiMarkovnikov R H H B 3 H H2O2, NaOH, H2O H R H H H H O H Reaktion mit Halogenen liefert Halogenalkane nach anti-Markovnikov R H H 3 H H hschulz, oc2bct B X2 R H H H H X 167 Hydroborierung von Alkinen R C C R +H-BR2 H BR2 1.H2O2 H 2.NaOH/H2O OH R R R R Hydroborierung mit sterisch H gehindertem Dialkylboran liefert H Alkenyl-dialkylboran R Oxidation und Hydrolyse liefert das entsprechende Enol, das zum Keton bzw. Aldehyd umlagert hschulz, oc2bct O R 168 Anti-Markovnikov-Addition von Wasser an 1-Alkine durch Hydroborierung B H R B 2 O R H OH H H Aldehyd hschulz, oc2bct 2 R H H2O2, HO- H R Enol 169 2.13 Elektrophile Substitution am Aromaten Typische Reaktion für Aromaten Alkene gehen unter vergleichbaren Bedingungen Additionen ein (AE) oder polymerisieren! H E + - +E X hschulz, oc2bct + - +H X 170 Beispiele für die elektrophile Substitution am Aromaten X2, FeX3 X Halogenierung X = Cl, Br HNO3/H2SO4 NO2 SO3 H2SO4 SO3H hschulz, oc2bct Nitrierung Sulfonierung 171 Beispiele für die elektrophile Substitution am Aromaten RCl, AlCl3 R Friedel-Crafts Alkylierung O RCOCl, AlCl3 hschulz, oc2bct C-R Friedel-Crafts Acylierung 172 Mechanismus der SE am Aromaten Schritt 1: Bildung eines nicht-aromatischen Carbenium-Ions (s-Komplex) E+ H + langsam, geschwindigkeitsbestimmend E E E H H H + E = + H + Arenium-Ion (s-Komplex) hschulz, oc2bct 173 Schritt 2: Abspaltung eines Protons unter Rückbildung des aromatischen Systems + E H Areniumion (s Komplex) hschulz, oc2bct E schnell -H+ Substituiertes Benzol 174 Energiediagramm für eine elektrophile Substitution am Aromaten (Ea) DG° hschulz, oc2bct 175 Halogenierung von Benzol Benzol reagiert mit Halogenen nur in der Gegenwart von Katalysatoren Als Katalysatoren werden Lewis-Säuren eingesetzt (Elektronenmangelverbindungen) Die Lewis-Säuren können „in situ“ gebildet werden, z.B. 2 Fe + 3 Br2 2 FeBr3 hschulz, oc2bct Cl2, FeCl3 Cl Br2, FeBr3 Br Chlorbenzol + HCl Brombenzol + HBr 176 Mechanismus der Halogenierung von Benzol Schritt 1: Bildung des Elektrophils, Aktivierung von Brom durch eine Lewis-Säure Br Br Br + FeBr3 + Br hschulz, oc2bct + Br FeBr3 - Br FeBr3 177 Schritt 2: Elektrophiler Angriff auf Benzol durch das aktivierte Brom Br + Br FeBr3 langsam, geschwindigkeitsbestimmend H + FeBr4- + Br H Br Br H H + + Arenium-Ion (s Komplex) hschulz, oc2bct 178 Schritt 3: Deprotonierung des Arenium-Ions unter Rückbildung des aromatischen Systems und des Katalysators + Br H + Br - FeBr3 Energiebilanz: Br-Br +193 kj/mol CPhenyl-H +465 kj/mol CPhenyl-Br -339 kj/mol H-Br -366 kj/mol -47 kj/mol hschulz, oc2bct Br + HBr + FeBr3 Reaktivität der Halogene gegenüber Benzol: F Cl Br I Explosion! exotherm wenig exotherm endotherm 179 Aromatische Nitrierung Schritt 1: Erzeugung des NO2+-Kations durch Protonierung von Salpetersäure durch die stärkere Schwefelsäure O O + - N O H + H OSO3H O O + + - N O H + HSO4- O H + - H N O H O hschulz, oc2bct O + N O + HOH Nitronium-Ion 180 Schritt 2: elektrophiler Angriff des NitroniumIons unter Bildung des s-Komplexes + O N H O langsam, geschwindigkeitsbestimmend + NO2 H s-Komplex Schritt 3: Abspaltung eines Protons NO2 + HSO4- H2SO4 + hschulz, oc2bct 181 Aromatische Sulfonierung Das elektrophile Agens ist das SO3 Es kann in konz. H2SO4 gelöst werden = rauchende Schwefelsäure SO3 reagiert mit Wasser zu H2SO4 H2SO4 reagiert bei RT nicht mit Benzol Sulfonierungen sind reversibel! Dies wird präparativ ausgenutzt hschulz, oc2bct 182 Mechanismus der aromatischen Sulfonierung O H S O O O + O - O S S H O H O O Umkehrung der Sulfonierung: SO3H H2O, 100°C, H2SO4 als Katalysator hschulz, oc2bct H + H2SO4 183 Friedel-Crafts-Alkylierung + R-X AlX3 R + HX Knüpfung von C-C-Bindungen Darstellung von Alkylbenzolderivaten Als alkylierende Agenzien wirken Halogenalkane, Alkohole, Alkene u.a. Reaktivität der Halogenalkane nehmen von R-F nach R-I ab Typische Lewis-Säuren: AlBr3 > AlCl3 > FeCl3 > SbCl5 > BF3 hschulz, oc2bct 184 Mechanismus der Friedel-CraftsAlkylierung Schritt 1 Primäre Halogenalkane (Alkohole) werden aktiviert (siehe elektrophile Halogenierung) = Lewis-Säure-Base-Reaktion Aus sekundären und tertiären Halogenalkanen entstehen meistens freie Carbenium-Ionen R CH2 X + AlX3 R R C X + AlX3 R hschulz, oc2bct d+ + R CH2 X AlX3 R + R C + X-AlX3R 185 Schritt 2 Elektrophiler Angriff des positivierten C-Atoms oder des Carbenium-Ions am Benzol R + d + H2C X AlX3 CH2R + AlX4+ Schritt 3 Abspaltung CH2R + H H hschulz, oc2bct des Protons + X-AlX3- + HX + AlX3 CH2R 186 Friedel-Crafts-Alkylierungen mittels Alkoholen oder Alkenen H H2 C H3CH2C + CH3 BF3, 60°C, 9h C H - HOH OH + HF, 0°C H3C hschulz, oc2bct C H CH3 187 Präperative Grenzen der Friedel-CraftsAlkylierung Mehrfach-Alkylierungen am Aromaten sind begünstigt Ein alkylierter Benzolring ist elektronenreicher als ein unsubstituierter Ring (+I-Effekt) Das Alkylierungsprodukt ist deshalb reaktiver als das Edukt! Durch Umlagerungen von instabileren zu stabileren Carbenium-Ionen sind viele Alkylbenzolderivate nicht zugänglich Produktgemische! hschulz, oc2bct 188 Präperative Grenzen der Friedel-CraftsAlkylierung Desaktivierte Aromaten (mit elektronenziehenden Substituenten) werden nur sehr langsam und in schlechten Ausbeuten alkyliert NO2 hschulz, oc2bct + N(CH3)3 COOH COR CF3 189 Friedel-Crafts-Acylierung + H3C C O AlCl3, 80°C Cl Acetylchlorid C O CH3 Reaktion eines Aromaten mit einem Acylhalogenid oder Anhydrid Lewis-Säure-Katalyse Vorteile gegenüber der Fried.-Crafts-Alkylierung Reaktionsprodukt ist desaktiviert, so dass keine Mehrfach-Acylierung stattfindet Selektiver Angriff des Carbonyl-C-Atoms (keine Produktgemische aufgrund von Umlagerungen) hschulz, oc2bct 190 Mechanismus der Friedel-CraftsAcylierung Schritt 1 Bildung eines positiven Acylium-Ions durch Lewis-Säure-Base-Reaktion + O R C X + AlCl3 + R C O O AlCl3 RC X + RC O O + R C X AlCl3 + XAlCl3- Acylium-Ion hschulz, oc2bct 191 Schritt 2 Elektrophiler Angriff des Acylium-Ions am Benzol (Bildung des s-Komplexes) H Abspaltung eines Protons H + R C O + hschulz, oc2bct Schritt 3 O O C C R R - H+ 192 Komplexbildung des Katalysators mit dem Reaktionsprodukt O C R + AlCl3 + AlCl3 O C O C R + 3H2O R + Al(OH)3 + 3 HCl Lewis-Säuren bilden mit dem basischen CarbonylSauerstoff einen Säure-Base-Komplex Friedel-Crafts-Acylierungen erfordern deshalb mindestens ein Äquivalent Katalysator Wässerige Aufarbeitung liefert das freie Keton und das Hydroxid des Katalysators hschulz, oc2bct 193 Friedel-Crafts-Acylierung durch ein Anhydrid O O O + -AlCl O + AlCl3 O + + 3 O + O O O O O -AlCl 3 -AlCl 3 O O -AlCl -H+ 3 O O H2O O O OH hschulz, oc2bct 194 Reaktivität substituierter Benzole bei der SE Die bereits vorhandenen Substituenten aktivieren oder desaktivieren das aromatische System Die bereits vorhandenen Substituenten kontrollieren die Geschwindigkeit einer weiteren Substitution Die bereits vorhandenen Substituenten kontrollieren die Regioselektivität (Orientierung) einer weiteren Substitution hschulz, oc2bct 195 Aktivierende und desaktivierende Erstsubstituenten Donor ortho ortho para Elektronendonor Erhöht Akzeptor die Elektronendichte im aromatischen System Erhöht die Reaktionsgeschwindigkeit der ZweitSubstitution Dirigiert in ortho- und paraStellung hschulz, oc2bct meta meta Elektronenakzeptor Erniedrigt die Elektronendichte im aromat. System Erniedrigt die Reaktionsgeschwindigkeit der ZweitSubstitution Dirigiert in meta-Stellung 196 Der induktive Effekt des Erstsubstituenten Elektronegativität des Substituenten Wirkt über das s-Gerüst Nimmt mit der Entfernung vom aromat. System schnell ab Alkylgruppen wirken durch Induktion und Hyperkonjugation elektronenschiebend (aktivierend) CH3 hschulz, oc2bct CF3 197 Der Resonanzeffekt des Erstsubstituenten (Mesomerie-Effekt) Substituenten mit p-Bindungen und/oder freien Elektronenpaaren Wirkt über das p-Gerüst Vergrösserung des delokalisierten pSystems Resonanzeffekte können aktivieren (+M-Effekt) oder desaktivieren (-M-Effekt) Resonanzeffekte können induktive Effekte übertreffen und umgekehrt hschulz, oc2bct 198 Aktivierung durch +I-Effekt CH3 CH3 E CH3 E E+ H orthoAngriff + H + CH3 CH3 CH3 + metaAngriff CH3 + E E+ CH3 paraAngriff E+ hschulz, oc2bct E H H CH3 + H CH3 E + H E H + CH3 CH3 + + E H E H E 199 Aktivierung durch +I-Effekt para und ortho-Angriff des Zweitsubstituenten s-Komplex hat positive Ladungskonzentration am ipsoKohlenstoffatom der Bindung zum Erstsubstituenten Charakter eines tertiären Carbenium-Ions Es resultiert ein stabilisiertes Kation meta-Angriff Es entsteht ein weniger stabilisiertes Kation Es können nur Resonanzformeln mit sekundärem Carbenium-Ion-Charakter gezeichnet werden Reine +I-Erstsubstituenten: - dirigieren in ortho/para-Position - erhöhen die Reaktionsgeschwindigkeit hschulz, oc2bct 200 Nitrierung von Toluol CH3 CH3 HNO3/ H2SO4 NO2 CH3 + CH3 + NO2 NO2 o-Nitrotoluol p-Nitrotoluol 59% 37% m-Nitrotoluol 4% Methylgruppe ist aktivierend statistischer Effekt (2 x ortho, 1 x para) ortho-Angriff kann sterisch gehindert sein hschulz, oc2bct 201 Desaktivierung durch -I-Effekt CF3 E+ CF3 CF3 E E H orthoAngriff + H + CF3 CF3 CF3 + metaAngriff CF3 + E E+ CF3 paraAngriff E+ hschulz, oc2bct E H H CF3 + H CF3 E + H E H + CF3 + CF3 + E H E H E 202 Desaktivierung durch -I-Effekt para und ortho-Angriff des Zweitsubstituenten s-Komplex hat positive Ladungskonzentration am ipsoKohlenstoffatom der bereits polarisierten Bindung zum Erstsubstituenten Es resultiert ein stark destabilisiertes Kation meta-Angriff Es entsteht ein weniger stark destabilisiertes Kation, da die positive Ladung bevorzugt ortho und para zum Erstsubstituenten delokalisiert werden kann Reine –I-Erstsubstituenten: - dirigieren in meta-Position - reduzieren die Reaktionsgeschwindigkeit hschulz, oc2bct 203 Induktive und Resonanzeffekte im Benzolamin (Anilin) + NH2 NH2 + NH2 + NH2 - - -I NH2 hschulz, oc2bct Resonanzeffekt überwiegt Negative Ladung in ortho- und para-Stellung konzentriert: ortho/para-dirigierend! -I-Effekt wenig ausgeprägt 204 Induktive und Resonanzeffekte im Hydroxybenzol (Phenol) OH OH + OH - + OH + - - -I OH hschulz, oc2bct NH2- und OH- wirken aktivierend Reaktionsgeschw. der ZweitSubstitution höher als bei Benzol ortho/para-dirigierend 205 Aktivierung durch +M-Effekt NH2 NH2 E H orthoAngriff + + E H +NH2 E H + NH2 + NH2 + E E E H H NH2 H + +NH2 NH2 NH2 + paraAngriff + hschulz, H oc2bct E E H NH2 metaAngriff NH2 + H E H E H E 206 Aktivierung durch +M-Effekt para und ortho-Angriff des Zweitsubstituenten s-Komplex hat positive Ladungskonzentration am ipsoKohlenstoffatom der Bindung zum Erstsubstituenten Positive Ladung kann auf Erstsubstituent weiter delokalisiert werden Es resultiert ein stabilisiertes Kation meta-Angriff Es entsteht ein weniger stabilisiertes Kation Es können nur Resonanzformeln mit sekundärem Carbenium-Ion-Charakter gezeichnet werden Erstsubstituenten mit +M-Effekt - dirigieren in ortho/para-Position - erhöhen die Reaktionsgeschwindigkeit hschulz, oc2bct 207 Bromierung von Phenol und Anilin OH OH Br Br +Br2/H2O 2,4,6-Tribromphenol 100% Br NH2 NH2 Br Br +Br2/H2O hschulz, oc2bct 2,4,6-Tribromanilin 100% Br 208 Desaktivierung durch -M-Effekt O C OH O C OH - O C OH O C OH + + + Desaktivierung durch -I- und durch Resonanzeffekt Reaktionsgeschwindigkeit der ZweitSubstitution niedriger als bei Benzol meta-dirigierend hschulz, oc2bct 209 Desaktivierung durch -M-Effekt O C OH E C OH O C + OH C H OH + E H hschulz, oc2bct C OH O + OH C E E H H OH C + O C OH + E O H + O + metaAngriff O E H orthoAngriff paraAngriff O E E H H + OH O O + OH OH O C C C + + H E H E H E 210 Desaktivierung durch -M-Effekt para und ortho-Angriff des Zweitsubstituenten s-Komplex hat positive Ladungskonzentration am ipsoKohlenstoffatom der Bindung zum Erstsubstituenten Positive Ladung kann kaum auf Erstsubstituent weiter delokalisiert werden, da C=O-Bindung bereits polarisiert ist Es resultiert ein stark destabilisiertes Kation meta-Angriff Es entsteht ein weniger stark destabilisiertes Kation Erstsubstituenten mit -M-Effekt - dirigieren in meta-Position - erniedrigen die Reaktionsgeschwindigkeit hschulz, oc2bct 211 Die Nitro-Gruppe: desaktivierend und meta-dirigierend NO2 NO2 HNO3/ H2SO4 NO2 NO2 NO2 + + NO2 o-Dinitro benzol (6%) NO2 p-Dinitro benzol (1%) m-Dinitro benzol (93%) -M-Effekt: meta-dirigierend! niedrige Reaktionsgeschwindigkeit! hschulz, oc2bct 212 Induktive und Resonanzeffekte in Halogenbenzol + X X +X - +X - - -I X hschulz, oc2bct Halogene wirken desaktivierend, da der -I-Effekt überwiegt Reaktionsgeschwindigkeit der ZweitSubstitution niedriger als bei Benzol ortho/para-dirigierend 213 Halogen-Substituenten -I-Effekt bei Fluor ist so stark, dass der aromatische Ring Elektronendichte abgibt und folglich desaktiviert wird Vom Fluor zum Iod nimmt die EN und damit der -I-Effekt ab Die freien Elektronenpaare am Halogen können in das delokalisierte p-Elektronensystem einbezogen werden. Der zunehmende Atomdurchmesser vom Fluor zum Iod macht aber eine effektive Überlappung mit dem aromatischen System immer ungünstiger Halogene sind schwach desaktivierend, dirigieren aber in ortho/para-Stellung hschulz, oc2bct 214 Einfluss des Erstsubstituenten auf die Regioselektivität des Zweitsubstituenten Stark aktivierend -NH2, -NHR, -NR2, -OH, -O-, -OR Schwach aktivierend Alkyl, Phenyl Schwach desaktivierend -F, Stark desaktivierend NO2 CF3 O C OH O C OR SO3H + NR3 O C R C N -Cl, -Br, -I ortho/paradirigierend hschulz, oc2bct metadirigierend 215 Einfluss des Erstsubstituenten auf die Reaktionsgeschwindigkeit Geschwindigkeitsbestimmender Schritt ist die Bildung des Arenium-Kations (s-Komplex) Die Aktivierungsenergie (Ea) ist um so niedriger, je stärker das Arenium-Ion durch den Erstsubstituenten stabilisiert werden kann Niedrige Ea bedeutet schnelle Reaktion! hschulz, oc2bct 216 Energiediagramme für die Bildung von Arenium-Ionen bei der SE desaktivierender Erst-Substituent hschulz, oc2bct aktivierender Erst-Substituent 217 SE an disubstituierten Benzolen Substituenteneffekte sind additiv Doppelt aktivierte Positionen sind besonders reaktiv Im Konfliktfall setzt sich der stärker aktivierende Substituent durch Resonanz-Aktivierung übertrifft gewöhnlich die Aktivierung durch induktive Effekte Bei Dritt-Substitutionen gewinnen sterische Effekte deutlich an Bedeutung! hschulz, oc2bct 218 Konkurrenz zwischen aktivierenden und desaktivierenden Substituenten ortho/para-dirigierende Substituenten sind stärker aktivierend als meta-Substituenten desaktivierend Die Dritt-Substitution wird deshalb durch den aktivierenden Substituenten bestimmt + NR3 CF3 m desaktivierend E+ Br E+ hschulz, oc2bct CH3 o,p aktivierend 219 Sterische Effekte bei der Drittsubstitution Die Position zwischen Substituenten in meta-Stellung ist oft sterisch gehindert Wenn andere Positionen verfügbar sind, werden diese bevorzugt substituiert E+ Cl o,p Br o,p E+ hschulz, oc2bct 220 Nitrierung von 1-Brom-3-chlorbenzol Cl Cl Cl Cl HNO3/ H2SO4 O2N + Br Br NO2 + Br Br NO2 2-Brom4-Brom1-Brom4-chlor2-chlor3-chlor1-nitrobenzol 1-nitrobenzol 2-nitrobenzol (62%) (37%) (1%) hschulz, oc2bct 221 Synthesestrategien für substituierte Benzole Dirigierende Substituenteneinflüsse müssen berücksichtigt werden Reihenfolge der Substitutionsschritte ist entscheidend Ggf. müssen Substituenten mit Schutzgruppen maskiert werden Reversible Substitutionen können durchgeführt werden, um den dirigierenden Effekt zu nutzen hschulz, oc2bct 222 Synthese von 3-Bromanilin m-dirigierend NO2 o,p-dirigierend NH2 NO2 Br2, FeBr3 Fe, HCl Br hschulz, oc2bct O CF3 COOH Br 223 Synthese von 1,2-Dinitrobenzol mit Hilfe einer Schutzgruppe o,p-dirigierend O HN C CH3 NH2 O H3C C Cl HNO3 Pyridin O HN C CH3 O HN C CH3 NO2 + NO2 Nach Trennung der Isomere + +H , H2O NO2 m-dirigierend NH2 NO2 hschulz, oc2bct O CF3 COOH NO2 224 Ortho-disubstituierte Benzole durch reversible Sulfonierung o,pdirigierend C(CH3)3 C(CH3)3 SO3, konz. H2SO4 C(CH3)3 NO2 HNO3, H2SO4 mSO3H dirigierend C(CH3)3 NO2 +H+, H2O, D SO3H hschulz, oc2bct 225 Substitutionen an Alkylbenzolen Alkylbenzole können mit Halogenen an der Seitenkette (SR) oder am aromatischen Ring (am „Kern“, SE(Ar)) reagieren Bedingungen für Substitution an der Seitenkette: Hohe Temperatur SSS-Regel: Lichtenergie Siedehitze, Sonnenlicht, Seitenkettensubstitution Bedingungen für Substitution am Kern: Niedrige Temperatur Katalysator hschulz, oc2bct KKK-Regel: Kälte, Katalysator, Kernsubstitution 226 2.14 Nucleophile Substitution am Aromaten (SN,Ar) Der unsubstituierte aromatische Ring ist ein Zentrum hoher Elektronendichte und wird nur elektrophil angegriffen Durch mehrere elektronenziehende Substituenten (-I, -M) wird aber die Elektronendichte so weit reduziert, dass ein nucleophiler Angriff möglich wird Zwei verschiedene SN2-Mechanismen: Additions-Eliminierungs-Mechanismus Eliminierungs-Additions-Mechanismus hschulz, oc2bct 227 Nucleophile aromatische ipso-Substitution Die Substitution einer anderen Gruppe als H wird als ipso-Substitution bezeichnet Cl Beispiele: NO OH NO2 Na2CO3, HOH, 100°C HNH2, D NO2 NaOCH3, CH3OH, D OMe NH2 NO2 NO2 NO2 NO2 hschulz, oc2bct 2 NO2 228 Additions-Eliminierungs-Mechanismus der S 2 N ,Ar Schritt 1: Addition Nu Nu O O N O O + O N O Cl O N Cl O O Nu + N N O N + O + O O + O - O + O hschulz, oc2bct Nu + - N O N Nu N Cl O + + N Nu + - Cl O Cl - -O Cl + N O O + O N O 229 Schritt 2: Eliminierung (nur eine Resonanzformel gezeichnet) Nu Cl Nu O N + O N O + O + Cl O N + O O N - + O Für maximale Delokalisierung müssen die elektronenziehende Substituenten in ortho- oder para-Stellung sein hschulz, oc2bct 230 Eliminierungs-Additions-Mechanismus der SN2,Ar, über Arin-Zwischenprodukt Halogenarene ohne weitere elektronenziehende Substituenten reagieren nach einem EliminierungsAdditions-Mechanismus (Arin-Mechanismus) Schritt 1: Eliminierung X H Schritt - + NH2 - HNH2 X + Cl Arin (Benz-in) 2: Addition + NH2 - - NH2 NH2 + H-NH2 - NH2- H Anilin hschulz, oc2bct 231 Die nucleophile Substitution von Fluor in elektronenarmen Aromaten Sanger-Reagenz Das Fluor-Atom wird leicht durch eine Aminogruppe einer Aminosäure substituiert Identifizierung des N-terminalen Endes eines Peptids NO2 O2N R F hschulz, oc2bct O + NO2 R O - HF H2N NHR´ O2N NH NHR´ 232 Darstellung von Phenol und Anilin OH Phenol Cl 1. NaOH,H2O,340°C,15 MPa 2. H+,H2O NH2 1. KNH2, NH3 (l) 2. H+,H2O hschulz, oc2bct Anilin 233 Das Cumol-Phenol-Verfahren nach Hock H3C CH2 + CH CH3 CH H3PO4 CH3 Cumol Friedel-CraftsAlkylierung O2 (Luft) OOH H3C CH3 C OH O H3C CH3 Aceton hschulz, oc2bct H2SO4 + Phenol Cumolhydroperoxid 234 Mechanismus des Cumol-PhenolVerfahrens 1.Schritt: Radikalische Oxidation des Cumols mit Luftsauerstoff O H3C CH CH3 H3C C O CH3 H3C + O2 - HOO. C CH3 + O2 HO O H3C C H3C CH3 H3C C CH CH3 CH3 Cumolhydroperoxid hschulz, oc2bct 235 2. Schritt: sauere Spaltung zum Phenol H3C HO O O H3C C H3C CH3 + H+ - H2 O C + H3C C O Umlagerung H3C OH O CH3 CH3 - H+ OH2 C CH3 + H2O O CH3 hschulz, oc2bct 236 Kohlendioxid als Elektrophil: die Kolbe-Schmitt-Reaktion Darstellung von Phenolcarbonsäuren O OH O H + CO2 COO- COO- 125°C/4-7bar + H+ OH COOH Salicylsäure hschulz, oc2bct 237 Darstellung von Phenolaldehyden: Reimer-Tiemann-Synthese O O O H + ICCl2 CCl2 CHCl2 OH CHO + H+/H2O - 2 HCl Salicylaldehyd hschulz, oc2bct 238 3. Darstellung und Eigenschaften von metallorganischen Verbindungen Halogenalkane, Halogenalkene und Halogenarene reagieren mit bestimmten Metallen unter Bildung metallorganischer Verbindungen Die elektronischen Verhältnisse am Kohlenstoff werden dabei umgekehrt Halogenalkan Metallorganische d- d+ R3C-M hschulz, oc2bct d+ dR3C-X Kohlenstoff ist elektrophil Verbindung R3C - M+ Carbanion Kohlenstoff ist nucleophil 239 3.1 Grignard-Reagenzien Synthese von AlkylmagnesiumVerbindungen elementares Magnesium wird in Ether mit dem entsprechenden Halogenalkan umgesetzt O R-X + Mg0 (C2H5)2O R-Mg-X O Vorsicht: die Reaktion muss anspringen bevor zuviel Halogenalkan zudosiert wird hschulz, oc2bct 240 Reaktionen der Grignard-Reagenzien Hydrolyse Alkylrest reagiert als Base MgBr + D2O D + BrMgOD Transmetallierung Alkylrest reagiert als Nucleophil 3 CH3MgCl + SiCl4 (CH3)3SiCl + 3 MgCl2 Chlortrimethylsilan hschulz, oc2bct 241 Reaktionen der Grignard-Reagenzien mit Carbonylverbindungen d- O d+ C - + d MX + O M X R C d- O H +H+, H2O C R R O H H O R OH 1.R´-Mg-X, 2.H2O H R O R OH 1.R-Mg-X, 2.H2O R 1.R´´-Mg-X, 2.H2O R´ hschulz, oc2bct H R´ H H primärer Alkohol sekundärer Alkohol OH R tertiärer R´´ Alkohol R´ 242 3.2 Metallalkyle Alkalimetalle reagieren mit Halogenalkanen zu Metallalkylen, z.B. Alkyllithium-Verbindungen CH3Br + 2 Li C4H9Br + 2 Li Ether, 0°C THF, 0°C CH3Li + LiBr Methyllithium (MeLi) C4H9Li + LiBr Butyllithium (BuLi) höhere Alkalimetalle reagieren analog. Die entsprechenden Metallalkyle sind hoch reaktiv, haben aber nur geringe synthetische Bedeutung. Der ionische Charakter nimmt von oben nach unten zu (Li Cs) hschulz, oc2bct 243 Weitere Metallalkyle D Pb(C2H5)4 Pb + 4 C2H5 Bleitetraethyl zerfällt bei hohen Temperaturen in Radikale. Es wurde bis vor kurzem Benzin zugesetzt als Antiklopfmittel Si(CH3)4 Standard für die NMR-Spektroskopie RxSiCly (x+y=4) Herstellung im Direktverfahren nach Rochow/Müller aus RCl und Si/Cu. Durch Hydrolyse entstehen Silicone hschulz, oc2bct 244 3.3 Metallocene H 2 H + 2 Na 2 - Na+ + H2 CyclopentadienylNatrium (Cp-Natrium) 2 - Na+ + MCl2 M = Fe M = Co M = Ni hschulz, oc2bct Ferrocen Cobaltocen Nikelocen M + 2 NaCl Metallocen Komplexverbindung Sandwich-Verbindung 245 Cyclopentadienyl-Metall-Halogenide Verbindungen dieses Types sind neuartige Katalysatoren für die AlkenPolymerisation Zr Cl Cl hschulz, oc2bct 2 CH3Li Zr CH3 + 2 LiCl CH3 246 4. Reaktivität der Carbonylverbindungen 4.1 Reaktivität der Carbonylgruppe 4.1.1 Katalytische Hydrierung der Carbonylgruppe 4.1.2 Ionische Additionen an die Carbonylgruppe 4.1.2.1 Reaktivität von Aldehyden und Ketonen gegenüber Wasser 4.1.2.2 Reaktivität von Aldehyden und Ketonen gegenüber Alkoholen 4.1.2.3 Reaktivität von Aldehyden und Ketonen gegenüber Aminen 4.1.2.4 Addition von Cyanwasserstoff an Aldehyde und Ketone 4.1.2.5 Oxidative Nachweise für Aldehyde hschulz, oc2bct 247 4.1.3 Wittig-Reaktion zur Synthese von Alkenen 4.1.4 Additions-Eliminierungs-Mechanismus bei Carbonsäurederivaten 4.2 Reaktivität von Carbonylverbindungen am a-C-Atom 4.2.1 Keto-Enol-Tautomerie 4.2.2 Aldol-Reaktion 4.2.3 Halogenierung von C-H-aciden Carbonylverbindungen 4.2.4 Alkylierung von C-H-aciden Carbonylverbindungen 4.3 a,b-ungesättigte Carbonylverbindungen hschulz, oc2bct 248 Carbonylverbindungen O O R C R´ Aldehyd Keton R C Carbonsäure O R C R C X CarbonsäureHalogenid O C R Anhydrid hschulz, oc2bct OH O O O R C R C H O R C O OR´ CarbonsäureEster O R C R´ NR´2 CarbonsäureAmid a,b-ungesättigte Carbonyle 249 Die Struktur der Carbonylgruppe Kohlenstoff- und Sauerstoffatom sind sp2-hybridisiert Bindungswinkel am C-Atom ca. 120° C=O-Gruppe und die beiden an Kohlenstoff gebundenen Atome liegen in einer Ebene Die p-Bindung hat Elektronendichte oberhalb und unterhalb dieser Ebene R C R hschulz, oc2bct s-Bindung p-Bindung O 250 Die Struktur der Carbonylgruppe H Bindungswinkel und Bindungslängen im Acetaldehyd 112 pm 121° 120 pm C 114° O 154 pm H3C 125° Polarität der Carbonylgruppe C O nucleophiler Angriff hschulz, oc2bct + C O - d+ = d- C O elektrophiler Angriff 251 Reaktive Zentren in Carbonylverbindungen sauer: CH-acide Verbindungen Angriff von oben H nucleophiler Angriff C d+ Od elektrophiler Angriff Angriff von unten hschulz, oc2bct 252 4.1.1 Katalytische Hydrierung der Carbonylgruppe O H O H2, Ru, 160°C, 30 bar H H H O H O H2, Raney-Ni, 80°C, 5bar H3C H3C H syn-Addition Aldehyde und Ketone sind schwerer katalytisch zu hydrieren als Alkene Deshalb kann man aus ungesättigten Carbonylen selektiv gesättigte Carbonyle machen hschulz, oc2bct 253 4.1.2 Ionische Addition an die Carbonylgruppe R d+ dC O H d+ d- + H- Nu R O H C H Nu Zwei mögliche Reaktionswege Im ersten Schritt erfolgt ein nucleophiler Angriff von Nu- am CarbonylKohlenstoffatom Im ersten Schritt erfolgt ein elektrophiler Angriff von H+ am CarbonylSauerstoffatom hschulz, oc2bct 254 Mechanismus der ionischen Addition Reaktion des Carbonyls mit einem starken Nucleophil unter Abwesenheit von Säure Schritt 1: Angriff des Nucleophiles - R Nu R´ Nu C O d+ d- trigonal planar hschulz, oc2bct R - C O R´ Tetraedrales Zwischenprodukt (Alkoxid-Ion) 255 Schritt 2: das stark basische Alkoxid-Ion abstrahiert ein Proton von H-Nu Nu R C O R´ H-Nu Nu R C O H + NuR´ Alkoxidion hschulz, oc2bct 256 Reaktion des Carbonyls mit einem schwach basischen Nucleophil unter Anwesenheit von Säure (Säure-Katalyse) Schritt 1: Elektrophile Protonierung R d+ dC O - R +HA/-A + + C O H C O H R´ R´ R R´ Carbenium-/Oxonium-Ion mit positiviertem C-Atom hschulz, oc2bct 257 Schritt 2: Angriff des Nucleophiles am positivierten C-Atom und Abstraktion eines Protons H Nu + R +Nu-H + C O H R C O H R´ R´ Nu R -H+ C O H R´ hschulz, oc2bct 258 Ionische Hydrierung der Carbonylgruppe Li+ H3Al-H + C O H C O +3 H - + AlH3 Li C O + +HOH Li H C O 4 Al C O H -Al(OH)3 -LiOH Stark exotherm Irreversibel C=C-Doppelbindungen werden nicht angegriffen: Herstellung von ungesättigten Alkoholen! hschulz, oc2bct 259 Irreversible Addition von stark basischen Nucleophilen an die Carbonylgruppe O 1.LiAlH4, 2.H2O H R O R OH 1.LiAlH4, 2.H2O R´ R O R 1.R-Mg-X, 2.H2O O H R OH O R 1.R´´-Mg-X, 2.H2O R´ hschulz, oc2bct H OH R 1.R´-Mg-X, 2.H2O H primärer Alkohol sekundärer Alkohol H R´ H H R OH H R´ H primärer Alkohol H sekundärer Alkohol OH R´´ R R´ tertiärer Alkohol 260 Reversible Addition von schwach basischen Nucleophilen OH + H-OH C C O + H-OR + H-SR + H-NHR OH OH C C C H Hydrat geminales Diol +HSR -H2O OR Halbacetal/ C Acetal OR SR Thioacetal C SR NHR -H2O Schiffsche C NR Base (Imin) OR OH SR OH +HOR -H2O H C O + H-NR2 hschulz, oc2bct OH C NR2 -H2O NR2 C C Enamin 261 Beeinflussung der Reaktivität der Carbonylgruppe durch die Substituenten Zunehmende Reaktivität... mit zunehmender Elektrophilie des CarbonylKohlenstoffatoms Mit abnehmender sterischer Hinderung O R C < O R C < OH O < R C O O O R C < NR2 O < H hschulz, oc2bct H C R C O < OR´ R H Cl3C C R O O < C < H R C Cl 262 Reaktivität von Aldehyden und Ketonen gegenüber Wasser OH +H2O C O C -H2O OH geminales Diol nicht isolierbar Gleichgewichtsreaktion: Reaktion ist reversibel! Ketone: Gleichgewicht liegt links Formaldehyd und Aldehyde mit -I-Substituenten: Gleichgewicht liegt rechts Andere Aldehyde: Gleichgewichtskonstante ca. 1 Isolierung von geminalen Diolen nur möglich, wenn starke -I-Substituenten vorhanden sind (z.B. Chloralhydrat)! hschulz, oc2bct 263 Einfluss stark elektronenziehender Gruppen (starker -I-Effekt) Cl Cl O C Cl H Trichlorethanal hschulz, oc2bct + H2O Cl Cl C Cl OH OH H Chloralhydrat (2,2,2-Trichlorethan-1,1-diol) isolierbar, stabil 264 Mechanismus für die basenkatalysierte Addition von Wasser an die C=O-Gruppe Schritt 1: Nucleophiler Angriff von OH- am Carbonyl-C-Atom R C O HO OH- R - C O R´ R´ Schritt 2: Übertragung eines Protons von Wasser auf das Alkoxid-Ion unter Rückbildung des Katalysators HO HO H-OH R C O H + OHR C O R´ hschulz, oc2bct R´ 265 Mechanismus für die säurekatalysierte Addition von Wasser an die C=O-Gruppe Schritt 1: R C O R´ hschulz, oc2bct Elektrophiler Angriff von H+ am Carbonyl-O-Atom H-OH2+ R R´ R R´ + C O H + C O H 266 Schritt 2: Nucleophiler Angriff von Wasser am Carbenium-C-Atom + H H O H-O-H + R R C O H C O H R´ R´ Schritt 3: Deprotonierung unter Rückbildung des Katalysators + H H O HO H-O-H R C O H + H3O+ R C O H R´ R´ hschulz, oc2bct 267 Reaktivität von Aldehyden und Ketonen gegenüber Alkoholen OH +HOR C O OH C OR -HOR +HOR/-HOH +HOH/-HOR C OR Halbacetal OR C OR Gleichgewichtsreaktion: alle Reaktionsschritte sind reversibel! Offenkettige Halbacetale haben gleiche Stabilität wie geminale Diole Ringförmige Halbacetale sind stabiler Acetale sind i.d.R. isolierbar hschulz, oc2bct Acetal 268 Mechanismus für die basenkatalysierte Halbacetal-Bildung Schritt 1: Nucleophiler Angriff von OR- am Carbonyl-C-Atom R C O OR- RO R - C O R´ R´ Schritt 2: Übertragung eines Protons vom Alkohol auf das Alkoxid-Ion unter Rückbildung des Katalysators RO RO H-OR R C O H + ORR C O R´ R´ oc2bct hschulz, 269 Säurekatalysierte Halbacetal-Bildung und Weiterreaktion zum Acetal Schritt 1: R C O R´ hschulz, oc2bct Elektrophiler Angriff von H+ am Carbonyl-O-Atom H-OH2+ R R´ R R´ + C O H + C O H 270 Schritt 2: Nucleophiler Angriff von Alkohol am Carbenium-C-Atom + H R O H-O-R + R R C O H C O H R´ R´ Schritt 3: Deprotonierung unter Bildung des Halbacetals + H R O RO H-O-H R C O H + H3O+ R C O H R´ R´ hschulz, oc2bct 271 Schritt 4: Elektrophiler Angriff von H+ an der OHGruppe des Halbacetals und Abstraktion von H2O RO R RO H-OH2+ C O H R´ SN1 R R´ hschulz, oc2bct + C O H R R´ H -HOH + C O R R R´ + C O R 272 Schritt 5: Nucleophiler Angriff von Alkohol am Carbenium-C-Atom + R R´ + R O R C O R R´ H-O-R C O R Schritt 6: + H Deprotonierung unter Bildung des Acetals H R O R C O R R´ hschulz, oc2bct RO H-O-H R C O R + H3O+ R´ 273 Bildung cyclischer Halbacetale durch intramolekularen Ringschluss Spannungsarme 5- und 6-Ringe sind stabil Bei der Bildung von intramolekularen Halbacetalen nimmt die Entropie nur wenig ab, im Gegensatz zu der starken Abnahme bei intermolekularer Halbacetalbildung! CH2OH H O H H H OH H C O OH H OH Aldehyd-Form hschulz, oc2bct H+ oder OH- Glucose CH2OH O H H H OH H C *O H OH H OH Cyclische Halbacetal-Form (99%) 274 Bildung von Halbacetalen und Acetalen Gleichgewichtsreaktionen: alle Schritte sind reversibel! Basenkatalyse ergibt Halbacetale Säurekatalyse ergibt Halbacetale und Acetale Alkoholüberschuss begünstigt Acetalbildung Offenkettige Halbacetale sind i.d.R. instabil Cyclische Halbacetale (5-, 6-Ringe) sind stabil Acetale sind relativ stabil vor allem im basischen Milieu (Verwendung als Schutzgruppe für C=O) Acetale aus Aldehyden sind stabiler als Acetale aus Ketonen Acetal-Hydrolyse durch Wasserüberschuss und Säure-Katalyse hschulz, oc2bct 275 Bildung cyclischer Acetale aus Diolen Aldehyde und Ketone bilden mit Ethylenglycol spannungsarme 5-Ringe Die entstehenden cyclischen Ether sind weniger reaktiv als Carbonyle und im basischen Milieu stabil Mit Wasserüberschuss findet die Rückreaktion zur Carbonyl-Verbindung statt (Säurekatalyse) Cyclische Acetale sind deshalb Schutzgruppen für Carbonylgruppen R C O R hschulz, oc2bct + HO CH2 HO CH2 H + R C R O CH2 + H2 O O CH2 276 Thioacetale R C O + 2 HS CH2 CH3 H + R S CH2 CH3 C R R R C O R + HS CH2 H + HS CH2 R C R S CH2 CH3 S CH2 S CH2 + H2O + H2O H2/Raney-Nickel Thioacetale sind in wässerigen Säuren stabil Reduktion zur Methylengruppe durchH2/Ni hschulz, oc2bct R R H H 277 Reaktivität von Aldehyden und Ketonen gegenüber Aminen Addition von Ammoniak oder primären Aminen führt zur Bildung von Iminen C O + H H N R C N R + H 2O Imin Kondensation: Zwei Moleküle verbinden sich unter Abspaltung von Wasser hschulz, oc2bct 278 Kondensation mit sekundären Aminen führt zur Bildung von Enaminen H R C C O+ H N R R R R R C C N R R R R + H 2O Enamin Tertiäre Amine reagieren nicht mit Aldehyden und Ketonen hschulz, oc2bct 279 Mechanismus für die Bildung von Iminen C O + H H H+ R N H C O N R C -H+ C + H N R N Imin R + C N H H N R C OH Halbaminal +H+ H -HOH N R C + OH H R Iminium-Ion Imine kommen als Z/E-Isomere vor! hschulz, oc2bct 280 Mechanismus der Enamin-Bildung H N H R C C O + R R R H R R C R H R C R C C N R R R + -H R H R Enamin hschulz, oc2bct R C R R+ R N H C R O + R C N R R H Iminium-Ion N R R C C OH R R Halbaminal +H+ -HOH H R C + R C N R R R R R N R C + OH R H 281 Reaktion mit speziellen Amin-Derivaten Hydroxylamin H N OH + H C N Oxim OH C O + C H H N N Hydrazin N N C H H C N N H Hydrazon H + C O Azin hschulz, oc2bct 282 Addition von Cyanwasserstoff an Aldehyde und Ketone R C O + R HCN H O H C H R C O R + R HCN CN OH C CN R Cyanhydrin hschulz, oc2bct 283 Mechanismus der Cyanhydrinbildung C O + C N - CN C O - CN +H+ C O H Gleichgewichtsreaktion, die durch Basen leicht umgekehrt wird (Entzug von H+) CN C O H - C O + HCN +OH C O - + HOH + CN hschulz, oc2bct 284 Reaktivität der Cyanhydrine OH H 1. LiAlH4 2. H+,H2O OH R C C R N R C C NH2 R H 1-Amino-2-alkanol H+,H2O OH R C R OH C O 2-HydroxyDurch die Cyanhydrin-Bildung carbonsäure wird die ursprüngliche Kohlenstoffkette um ein C verlängert Durch Reduktion oder Hydrolyse sind weitere funktionelle Gruppen zugänglich hschulz, oc2bct 285 Oxidative Nachweise für Aldehyde Fehling-Nachweis von Aldehyden O O R C + Cu2+ NaOH, Tartrat, H2O Cu2O + R C ziegelroter H OH Niederschlag Tollens-Nachweis von Aldehyden O R C + Ag+ H hschulz, oc2bct O NH3, H2O Ag + R C Silberspiegel OH 286 4.1.3 Wittig-Reaktion zur Synthese von Alkenen Carbonylverbindungen reagieren mit Phosphor-Ylid (Phosphoran) in einer konzertierten Reaktion zu Alkenen C-C-Doppelbindungen können so gezielt aufgebaut werden Produktgemische wie bei der Eliminierung üblich sind hier nicht zu erwarten Die Reaktion ist nicht stereoselektiv. Es entstehen deshalb Mischungen aus Z- und EAlkenen hschulz, oc2bct 287 Bildung von Phosphor-Ylide (C6H5)3P + R Triphenylphosphan CH2 X SN Halogenalkan R CH2 + P(C6H5)3 + X - Alkyltriphenylphosphoniumhalogenid - + R C P(C6H5)3 + R CH2P(C6H5)3 X- + BuLi THF H R C P(C6H5)3 Ylid + BuH + LiX Ylen H hschulz, oc2bct 288 Mechanismus der Wittig-Reaktion - + R C P(C6H5)3 + H R C P(C6H5)3 H H R1 R C C R2 (H5C6)3 P O R1 C O R2 H R1 R R2 + (C6H5)3P=O Oxaphosphacyclobutan (Oxaphosphetan) hschulz, oc2bct 289 Beispiel einer Wittig-Reaktion O + CH2=P(C6H5)3 hschulz, oc2bct D, THF + (C6H5)3P=O 290 4.1.4 Additions-EliminierungsMechanismus bei Carbonsäurederivaten O R C + H-Nu L L=X, -OR, -NR2,... O H O L C R Nu R + H-L Nu Tetraedrisches Zwischenprodukt Der Carboxy-Kohlenstoff ist elektrophil und wird folglich durch Nucleophile angegriffen Im Unterschied zu Aldehyden und Ketonen besitzen Carbonsäurederivate gute Abgangsgruppen Carbonsäurederivate reagieren deshalb mit Nucleophilen unter Substitution und nicht unter Addition hschulz, oc2bct 291 Reaktivität von Carbonsäure-Derivaten bei nucleophilen Additions-Eliminierungs-Reaktionen O O R C < R C hschulz, oc2bct R C OR´ O O R < NR´2 OH < O C O O < C R´ R C X 292 Mechanismus für die säurekatalysierte Veresterung und Esterhydrolyse Schritt 1: Protonierung der Carboxygruppe O R C OH +H O H+ R C OH H H O O R C OH R C + + OH Schritt 2: Nucleophiler Angriff durch Alkohol +H O O H + H-O-R R C OH hschulz, oc2bct R C OH +O H R - H+ O H R C OH O R Tetraedrisches Zwischenprodukt 293 Schritt 3: Wasserabspaltung O H O H R C OH O R +H O H R C O+ H O R +H+ -H+ H O -HOH +HOH O -H+ +H+ R C O R R C O R + Additions-Eliminierungs-Reaktion: Addition von Alkohol gefolgt von Eliminierung von Wasser R C O R hschulz, oc2bct 294 Estersynthese Carbonsäure + Alkohol ergibt Ester + Wasser Gleichgewichtsreaktion Steuerung durch Entzug bzw. Überschuss von Wasser Der Sauerstoff im Ester stammt vom Alkohol O R C O H + H-O-R hschulz, oc2bct O R C O R + HOH 295 Intramolekulare Veresterung bei Hydroxysäuren: Lacton-Bildung Bildung von cyclischen Estern besonders bevorzugt, wenn sich spannungsarme 5- und 6-Ringe bilden können O O H H2SO4,H2O O OH O O 10% O H2SO4,H2O 90% O OH H O 5% hschulz, oc2bct 95% + H 2O CH3 + H2 O 296 „Veresterung“ von zwei Säuren: Anhydrid-Bildung Kondensation: O O R C O H + H O C R´ R O O C C O R´ + H2O Technische Synthese: O O - + R C O Na + Cl C R´ Acylchlorid hschulz, oc2bct -NaCl R O O C C O R´ Anhydrid 297 Cyclische Anhydride O COOH 230°C O + H2 O COOH Maleinsäure COOH O Maleinsäureanhydrid Anhydrid O HOOC Fumarsäure O O Phthalsäureanhydrid hschulz, oc2bct 298 Carbonsäurehalogenide (Alkanoylhalogenide, Acylhalogenide) Herstellung durch Reaktion von Carbonsäure mit anorganischen Säurechloriden Ausgangssubstanz für viele nucleophile Substitutionen O O O + Cl R C OH R=H hschulz, oc2bct S -SO2, -HCl R Cl Cl +PCl5, -OPCl3, -HCl +PBr3, -H3PO3 R O O Cl R Br 299 Reaktivität der Säurehalogenide O R´OH OR´ R O NH3 O NH2 R R Ester Amid Cl O NR2H O R´COONa R hschulz, oc2bct Dialkylamid NR2 R O O R´ Anhydrid 300 Imide und Lactame Imide sind die Stickstoffanaloga der Anhydride COOH + NH3 COOH COO-NH4+ COO-NH4+ O 290°C -2 H2O,-NH3 NH O Lactame sind die Stickstoffanaloga der Lactone O O D, -HOH H2N OH NH 86% hschulz, oc2bct 301 4.2 Reaktivität von Carbonylverbindungen am a-C-Atom Wasserstoffatome am a-Kohlenstoffatom von Carbonylverbindungen sind sauer a-Wasserstoffe: pKa ca. 20 b-Wasserstoffe: pKa ca. 40-50 H b bH hschulz, oc2bct Ha bH a H O H O b H H b H H b a a Ha Ha H 302 C-H-Acidität von Carbonylverbindungen a-Wasserstoffe sind acide wegen dem – I-Effekt des Carbonyl-Sauerstoffatoms Abstraktion eines Protons führt zu einem resonanzstabilisierten Anion B- H O C C -HB O C C Carbanion hschulz, oc2bct O C C Enolat-Ion 303 Protonierung des Enolat-Ions O - C C C C Carbanion +H+ C C Keto-Form hschulz, oc2bct = d- C C Enolat-Ion +H+ H O O O d- Keto-EnolTautomerie HO C C Enol-Form 304 4.2.1 Keto-Enol-Tautomerie Tautomere sind Konstitutionsisomere, die sich nur durch die Konnektivität eines Wasserstoffatoms unterscheiden Tautomere liegen miteinander im Gleichgewicht Bei Monocarbonylverbindungen liegt das Gleichgewicht i.d.R. weit auf Seiten der Keto-Form Keto-Form ist begünstigt, da die C-O-p-Bindung stärker ist als die C-C-p-Bindung H O HO C C C C Keto-Form Enol-Form hschulz, oc2bct 305 Keto-Enol-Tautomerie bei Monocarbonylverbindungen Ketoform O Acetaldehyd HO C H C H3C H H2C (sehr gering) (~ 100%) O Aceton Enolform HO C CH3 H3C C CH3 H2C (1,2*10-4%) (> 99%) O OH Cyclohexanon hschulz, oc2bct ( 98,8%) ( 1,2%) 306 Keto-Enol-Tautomerie bei Dicarbonylverbindungen b-Dicarbonylverbindungen Enol-Form ist durch Resonanz und durch interne Wasserstoffbrückenbildung begünstigt Dicarbonylverbindungen ohne resonanzstabilisierte Enol-Form verhalten sich wie Monocarbonylverbindungen O O H3C C CH2 C CH3 2,4-Pentadion (24%) hschulz, oc2bct OH O H3C C CH C CH3 Enolform (76%) 307 Stabilisierung von b-Carbonylverbindungen H H3C O O C C C H Wasserstoffbrücken CH3 H3C H O O C C C CH3 H Stabiler, spannungsarmer 6-Ring hschulz, oc2bct 308 Mechanismus für die basenkatalysierte Enolisierung Schritt 1: Abspaltung eines Protons in a-Stellung durch eine Base O OH C H O -H2O C Enolation Keton Schritt 2: Protonierung des Enolat-Ions O C Enolation hschulz, oc2bct H2O -OH- OH C Enol 309 Mechanismus für die säurekatalysierte Enolisierung Schritt 1: Protonierung des CarbonylSauerstoffatoms + ( c h i r a l ) + O-H H O H O C H -H2O C H H Keton Schritt 2: Deprotonierung in a-Stellung O + C H OH H C + H + O H H H2O hschulz, oc2bct Enol 310 4.2.2 Aldol-Reaktion O 2 H3C C 10% NaOH, H2O H Acetaldehyd 5°C H3C OH O CH C CH2 H 3-Hydroxybutanal “Aldol” Addition eines Aldehyds an ein zweites Aldehydmoleküls Das Produkt hat zwei funktionelle Gruppen: Aldehyd und Alkohol = Aldol Aldole können leicht Wasser abspalten unter Bildung von ungesättigten Aldehyden hschulz, oc2bct 311 Mechanismus der basenkatalysierten Aldol-Reaktion Schritt 1: Deprotonierung des Aldehyds in a-Stellung O H O + H CH2 C H O O CH2 C H H2C C H + H2O Enolation hschulz, oc2bct 312 Schritt 2: Nucleophiler Angriff des EnolatIons am Carbonyl-C-Atom eines zweiten Aldehydmoleküls CH3 C H CH2 C O O O O H H3C CH CH2 C H Alkoxidion Schritt 3: Protonierung des Alkoxid-Ions unter Rückbildung des basischen Katalysators O H3C CH CH2 hschulz, oc2bct O C H +H2O/-OH- O H O H3C CH CH2 C H Aldol 313 Dehydratisierung von Aldolen: Aldolkondensation Aldole werden leicht dehydratisiert (Säure- oder Basenkatalyse) Dehydratisierung ist bei hoher Temperatur begünstigt Bildung von a,b-ungesättigten Aldehyden mit konjugierten Doppelbindungen O OH Ph CH CH C H H hschulz, oc2bct -H2O +H2O O Ph CH CH C H 3-Phenylpropenal Zimtaldehyd 314 Gekreuzte Aldol-Reaktion H3C O OH O C C C H OH - + CH3-CH2 H3C H CH2 H O OH O C C H C CH3-CH2 H H CH3 OH - H3C hschulz, oc2bct CH OH O C C H CH CH3 H CH3-CH2 OH O C H C CH2 H 315 Die Aldolreaktion in der organischen Synthese Knüpfung einer C-C-Bindung Erzeugung eines Produkts mit zwei funktionellen Gruppen Gekreuzte Aldolreaktionen (mit zwei unterschiedlichen Aldehyden) ergeben Produktgemische Synthetisch interessant sind gekreuzte Aldolreaktionen wenn ein Aldehyd nicht enolisierbar ist Aldolreaktionen mit Ketonen sind leicht endotherm und laufen deshalb nur durch äusseren Zwang ab hschulz, oc2bct 316 Claisen-Kondensation: Herstellung von 1,3-Diketonen Eng verwandt mit der Aldol-Reaktion Mindestens ein Edukt ist ein Carbonsäureester Reaktion mit CH-acider Verbindung unter Abspaltung von Alkohol Acetylaceton-Synthese: O O + H3C OC2H5 H3C O C2H5O-Na+ CH3 H3C O C H2 CH3 + C2H5OH hschulz, oc2bct 317 Claisen-Kondensation Acetessigester-Synthese O O + C2H5O-Na+ H3C O H2C OC2H5 OC2H5 O + H2C OC2H5 OC2H5 O O H3C Na+ H2C H3C OC2H5 O CH2 OC2H5 OC2H5 - C 2 H 5 O- O H3C + C 2 H 5 O- O C H hschulz, oc2bct OC2H5 - C2H5OH O H3C O C H2 OC2H5 318 Cannizzaro-Reaktion Aldehyde ohne a-ständige H-Atome können kein Aldol bilden In Gegenwart von starken Basen unterliegen sie einer Disproportionierung zum entsprechenden Alkohol und Carbonsäure O 2 + NaOH OH CH2 H Benzaldehyd hschulz, oc2bct Benzylalkohol O + O Na+ Natrium-Benzoat 319 4.2.3 Halogenierung von Carbonylverbindungen Halogenierungen erfolgen am a-C-Atom Ausmaß der Halogenierung hängt von der Art der Katalyse (sauer oder basisch) ab H O H R C C C H H X O H R + X2 - HX R C C C R H H Elektrophile Substitution hschulz, oc2bct 320 Mechanismus für die säurekatalysierte Halogenierung Schritt 1: Bildung des Enols O H3C C CH3 OH H+ H2C CH3 Schritt 2: Elektrophiler Angriff von Brom an der Doppelbindung OH H2C CH3 Br hschulz,Br oc2bct OH - Br- + H2C C Br CH3 +O H H2C C Br CH3 321 Schritt 3: Deprotonierung +O H H2C C Br CH3 O -H+ BrH2C C CH3 Unter saurer Katalyse ist die Zweithalogenierung deutlich langsamer, da die Ausbildung der Enol-Form erschwert ist. Der –I-Effekt des Halogens reduziert die Basizität des Carbonyl-Sauerstoffatoms! hschulz, oc2bct 322 Mechanismus für die basenkatalysierte Halogenierung Schritt 1: Bildung des Enolat-Ions O O R C CH2 H + OH- CH2 + HOH R Schritt 2: Elektrophiler Angriff von Brom an der Doppelbindung - Br O CH2 R hschulz, oc2bct Br O R C CH2 Br + Br323 Schritt 3: Vollständige Substitution aller C-H-acider Wasserstoffe O R C H CHBr - + OH- - HOH O CH Br R +Br2 /- Br- O R C CBr3 +Br2 + OH- - Br- - HOH O R C H CBr2 Unter basischer Katalyse ist die Zweit- und Dritthalogenierung schneller, da die Acidität der a-Wasserstoffe durch den –I-Effekt der Halogene erhöht wird. hschulz, oc2bct 324 Haloform-Reaktion Methylketone werden im basischen Milieu zunächst zu Trihalogenmethylketonen halogeniert Basische Hydrolyse führt zur Bildung von Trihalogenmethan (Haloform) und Carbonsäure Iodierung führt so zur Bildung von gelbem Iodoform als Niederschlag: Nachweis für Methylketone O R C CJ3 + OH- O R C OH hschulz, oc2bct O +H2O CJ3 -OH- R C OH + HCJ3 325 4.2.4 Alkylierung von Carbonylverbindungen Alkylierungen am a-C-Atom sind bei Ketonen erfolgreich Ketone mit mehreren aciden Wasserstoffen ergeben Produktgemische Aldehyde reagieren unter diesen Bedingungen bevorzugt unter Aldoladdition O O 1. NaH, Toluol, D H CH2CH=C(CH3)2 2. (CH3)2C=CHCH2Br H C H5C6 5 6 -H-H -NaBr 2-Methyl-1-phenylPhenyl-2-(2,5-dimethylhex-4-en) 1-propanon -keton hschulz, oc2bct 326 4.3 a,b -ungesättigte Carbonylverbindungen Darstellung durch Aldol-Kondensation oder durch Wittig-Reaktion das konjugierte Enon-System ist resonanzstabilisiert b,-ungesättigte Carbonylverbindungen isomerisieren deshalb leicht zu den konjugierten a,b-ungesättigten Systemen O CH3CH CH C H hschulz, oc2bct O CH3CH CH C + - - H + CH3CH O CH C H 327 1,4-Addition von Nucleophilen an a,bungesättigte Carbonylverbindungen Wasser, Alkohole und Amine können 1,4-Additionen eingehen (Addition von Wasser = Rückreaktion der Aldolkondensation) Saure oder basische Katalyse Das zunächst gebildete Enol tautomerisiert zur Carbonylverbindung hschulz, oc2bct 328 Mechanismus der basenkatalysierten Hydratisierung a,b -ungesättigter Carbonylverbindungen - O O O H H-OH - HO HO HO - +OH hschulz, oc2bct 329 5. Kohlensäure und ihre Derivate Kohlensäure: Hydrat des Kohlendioxids O CO2 + H2O HO OH Kohlensäurederivate Cl O O O Cl Phosgen Kohlensäuredichlorid hschulz, oc2bct C2H5O OC2H5 Diethylcarbonat Kohlensäurediethylester H2N OC2H5 Urethan Carbamidsäureethylester 330 Kohlensäurederivate O H2N NH S NH2 Harnstoff Kohlensäurediamid H2N NH2 Thioharnstoff Thiokohlensäurediamid N C H2N NH2 Guanidin N Diphenylcarbodiimid = Derivat des Kohlendioxids hschulz, oc2bct 331 Instabile Kohlensäurederivate Kohlensäurederivate, die eine OHGruppe enthalten sind instabil O H2N OH NH3 + CO2 Carbamidsäure O RO OH ROH + CO2 Kohlensäuremonoester hschulz, oc2bct 332 Herstellung von Kohlensäurederivaten O 200°C Aktivkohle CO + Cl2 Cl + RCOOH O R C Cl HO C +ROH -HCl - CO2, HCl O + H 2O -HCl O Cl RO + R2OH -HCl Cl C Cl +H2NR´ -HCl O O CO2 + HCl hschulz, oc2bct RO C OR´ RO C NHR´ 333 Harnstoff-Synthesen O NH4+ O Cl Cl D O C 150°C -H2O O Wöhler, 1828 NH2 +H2O + O NH4 H2N NH2 Ammoniumcarbamat OH +NH 3 C N Cyanamid O NH2 Carbamidsäure hschulz, oc2bct N Ammoniumcyanat +2NH3 -2HCl H2N C CO2 + NH3 334 Reaktionen des Harnstoffs Herstellung von Barbitursäure O NH2 O RO C CH2 + NH2 RO HN RO-Na+/110°C O -2ROH HN OH C H2N OH OH hschulz, oc2bct C HN O + C N H OH O O C O N C Herstellung von Harnsäure HN HO CH2 C O N C O OH O H2N C H N N N OH O C O N H N H HO N N H 335 Biuret-Reaktion Nachweis-Reaktion der –CO-NH-Gruppe O H2N NH2 D -NH3 H2N HN C O Isocyansäure H2N N NH2 Biuret O NH HN HN NH2 H 2O O O O Cu + Cu2+/KOH NH + 2K NH HN O O Blauvioletter Kupferkomplex hschulz, oc2bct 336 6. Reaktivität von Aminen 6.1 Selektive Darstellung von primären Aminen 6.2 Alkene durch Elimierung von Aminen 6.2.1 Hofmann-Eliminierung 6.2.2 Cope-Eliminierung 6.3 Nitrosierung von Aminen hschulz, oc2bct 337 6. 1 Selektive Darstellung von primären Aminen Reaktion von Ammoniak mit Halogenalkanen (SN) führt immer zu Gemischen aus Primären, sekundären und tertiären Aminen Reduktion von Nitrilen oder Carbonsäureamiden O R C N LiAlH4 R CH2 NH2 LiAlH4 R NH2 hschulz, oc2bct 338 Hofmann-Abbau zur selektiven Darstellung von primären Aminen, die ein C-Atom weniger enthalten, als die ursprünglichen Carbonsäure-Verbindungen O + NaOH + Br2 H3C NH2 Acetamid hschulz, oc2bct H3C NH2 Methylamin + NaBr + CO2 339 Gabriel-Synthese zur selektiven Darstellung von primären Aminen O NH O + KOH - H2O N O O SN K+ + R-Cl - KCl O N R O N-Alkyl-pthalimid Phthalimid O NH NH + H2N-R + NH2-NH2 (Hydrazin) Primäres Amin O hschulz, oc2bct 340 6.2 Alkene durch Eliminierung von Aminen Basische Bedingungen Keine Eliminierung, da die nicht protonierte Aminogruppe eine schlechte Abgangsgruppe ist hschulz, oc2bct 341 Alkene durch Eliminierung von Aminen Sauere Reaktionsbedingungen Keine Eliminierung, da die Basizität des Säure-Anions oder des Wassers nicht ausreicht, ein Proton am bKohlenstoffatom zu abstrahieren - im Unterschied zu Alkoholen! HSO4- H C C H2SO4 H C OH + H2O OH2 HSO4- H C H H2SO4 C C NH2 hschulz, oc2bct C C + NH3 NH3 342 Alkene durch Eliminierung von Aminen Hofmann-Eliminierung Erschöpfende Alkylierung des Amins zum Tetraalkylammonium-Salz – Methylierung bevorzugt, da aus der Methylgruppe keine Eliminierung stattfinden kann und folglich einheitlichere Eliminierungsprodukte entstehen Umwandlung des Ammonium-Salzes in das Hydroxid Eliminierung von Trialkylamin und H+ durch Erhitzen (ca. 150°C) hschulz, oc2bct 343 6.2.1 Die Hofmann-Eliminierung von primären Aminen 1.Schritt: Erschöpfende Methylierung NH2 - + 3 CH3-I N(CH3)3 I + 2 HI 2. Schritt: Umwandlung in ein Hydroxid 2 - N(CH3)3 I hschulz, oc2bct + Ag2O, + H2O - 2 AgI 2 N(CH3)3 OH 344 Hofmann-Eliminierung 3. Schritt: Hofmann-Eliminierung (E2) HO- H H D, ca. 150°C C2H5 C H hschulz, oc2bct C H N(CH3)3 + H2O + N(CH3)3 345 Stereochemie der HofmannEliminierung Überwiegende Bildung des kinetisch kontrollierten Alkens mit maximaler Anzahl von H-Substituenten an der Doppelbindung (Hofmann-Produkt) Die Bildung des thermodynamisch kontrollierten Saytzeff-Produkts erfolgt nur in geringen Mengen, da der entsprechende anti-periplanare Übergangszustand sterisch sehr ungünstig ist Der Unterschied in der Aktivierungsenergie ist so gross, dass fast nur das kinetisch bevorzugte Produkt gebildet wird! hschulz, oc2bct 346 Eliminierung von (2-Butyl)trimethylammoniumhydroxid Produktverteilung bei der Hofmann-Eliminierung H3C CH2 CH CH3 150°C H3C Saytzeff (5%) N(CH3)3 OH CH CH CH3 + H3C CH2 CH CH2 Hofmann (95%) Produktverteilung bei einer entsprechenden Saytzeff-Eliminierung H3C CH2 CH CH3 Cl hschulz, oc2bct NaOCH3 H3C CH CH CH3 + H3C Saytzeff (67%) CH2 CH CH2 Hofmann (33%) 347 Sterische Betrachtung der verschiedenen anti-periplanaren Konformationen C3-C2-Bindung CH3 H H H N(CH3)3 H3C C1-C2-Bindung H H3C H H CH3 N(CH3)3 instabil, aber benötigt für E2 HOCH2CH3 H CH3 H3CH2C H N(CH3)3 hschulz, oc2bct H H H H N(CH3)3 Stabiler, aber keine E2 möglich H H H H H3C H N(CH3)3 Alle 3 gestaffelten Konformationen ermöglichen E2 348 Oxidation von Aminen Primäre Amine werden von Oxidationsmitteln leicht angegriffen unter Bildung komplexer Produktgemische Sekundäre Amine ergeben Hydroxylamin und weitere Oxidationsprodukte R1 R1 N H + H2O2 R2 N OH + H2O R2 Tertiäre Amine reagieren einheitlich zu Aminoxiden (CH3)3N + H2O2 hschulz, oc2bct (H3C)3N O + H2O 349 6.2.2 Cope-Eliminierung in Aminoxiden Aminoxide gehen eine Eliminierungsreaktion ein unter Bildung eines Alkens Es bildet sich ein Hofmann-Produkt Der Reaktionsmechanismus ist mit der Hofmann-Eliminierung vergleichbar Cope-Eliminierungen laufen aber bereits bei milderen Reaktionsbedingungen ab Es bildet sich ein cyclischer Übergangszustand, der zu einer syn-Eliminierung führt hschulz, oc2bct 350 Reaktionsmechanismus der CopeEliminierung - O d+ d H N(CH3)2 O H N(CH3)2 R1 C C H H R2 Aminoxid R1 C C H H Übergangszustand (syn) R1 R2 + H hschulz, oc2bct R2 HO N(CH3)2 H 351 Stereoselektivität der Cope-Eliminierung - HO-NR2 syn-Eliminierung hschulz, oc2bct 352 6.3 Nitrosierung von Aminen NO+ (Nitrosonium-Ion) reagiert mit Aminen zu unterschiedlichen Produkten Primäre Amine reagieren zu Diazonium-Salzen – Aliphatische Diazonium-Salze sind sehr instabil und neigen zur Explosion – Aromatische Diazonium-Salze sind in Lösung stabil. Reine Produkte neigen ebenfalls zur Explosion Sekundäre Amine reagieren zu N-Nitrosaminen Viele Vorsicht: Diazonium-Salze nicht isolieren! Nitrosamine sind stark cancerogen Tertiäre Amine reagieren zum N-NitrosoammoniumIon (typische Gleichgewichtsreaktion) hschulz, oc2bct 353 Darstellung von Diazonium-Salzen R NH2 + NaNO2 + 2 HCl R N N Cl Alkyldiazonium-Chlorid + 2 H2O + NaCl Ar NH2 + NaNO2 + 2 HCl Ar N N Cl Aryldiazonium-Chlorid + 2 H2O + NaCl Reaktives Agenz: Nitrosonium-Ion NO+ NaNO2 + HCl H+ H O N O HNO2 + NaCl H H O N O N O N O + H2O hschulz, oc2bct 354 Mechanismus der Darstellung von Diazonium-Salzes H R N H O N R N H N O + H 2O - H 3 O+ R N N O H H N-Nitrosamin R N N O H R H N N OH N OH +H+ H -H+ hschulz, oc2bct R N 355 Mechanismus der Darstellung von Diazonium-Salzes H +H+ R N N R OH N N O H -H2O R hschulz, oc2bct N N 356 Reaktivität des Diazonium-Ions R N R+ N Reaktion als Elektrophil + N N Umlagerung Eliminierung unter H+-Abstraktion Alkyldiazonium-Ionen sind hochreaktiv und ergeben komplexe Produktgemische Aryldiazonium-Ionen reagieren selektiv hschulz, oc2bct 357 Reaktivität des Aryldiazonium-Ions C N OH Hydrolyse D, H2SO4/H2O X SandmeyerReaktion CuCN N Reduktion mit H3PO2 Sandmeyer-Reaktion CuX (X=Cl, Br) N Fluorierung +HBF4/-N2,-BF3,-H+ Iodierung KI F I hschulz, oc2bct 358 Die Diazokupplung Elektrophile aromatische Substitution Ar1 N Cl + H N Ar2 Aktivierter Aromat Ar1 N N Ar2 + HCl Darstellung von Methylorange O HO S N N + H N(CH3)2 O O -H+ HO S N N N(CH3)2 O hschulz, oc2bct 359 Nitrosamine aus sekundären Aminen H R1 N O R1 N R2 H N R2 N O -H+ R1 N N O R2 N-Nitrosamin Nitrosamine sind stabil, da sie kein weiteres H-Atom am Stickstoff haben zur Weiterreaktion Nitrosamine sind cancerogen NaNO2 zur Fleischkonservierung („Nitritpöckelsalz“) kann im Verdauungstrackt Amine nitrosieren hschulz, oc2bct 360 Nitrosierung von tertiären Aminen R3 R3 R1 + N R2 N O R1 N N O R2 N-Nitrosammonium-Ion Keine Stabilisierungsmöglichkeit Gleichgewichtsreaktion hschulz, oc2bct 361 7. Farbstoffe Farbigkeit von Substanzen Eine Substanz erscheint farbig, wenn ein bestimmter Teil des sichtbaren Lichts absorbiert wird. Die sichtbare Farbe ist dann die entsprechende Komplementärfarbe Wird alles sichtbare Licht absorbiert, erscheint die Substanz schwarz Wird alles sichtbare Licht reflektiert, erscheint die Substanz weiss Wird alles sichtbare Licht durchgelassen (keine Absorbtion und keine Reflektion) erscheint die Substanz farblos hschulz, oc2bct 362 Farben des Sonnenlichts Wellenlängenbereich (nm) < 400 Absorbierte Spektralfarbe ultraviolett Komplementärfarbe (unsichtbar) 400 – 435 435 – 480 480 – 490 violett blau (indigo) blau oder türkis gelbgrün gelb orange 490 – 500 500 – 560 560 – 580 580 – 595 595 – 605 605 – 750 > 750 blaugrün grün gelbgrün gelb orange rot ultrarot rot purpur violett blau blau oder türkis blaugrün (unsichtbar) hschulz, oc2bct 363 Wellenlänge der absorbierten Strahlung Bei der Absorption von Strahlung werden Elektronen aus besetzten MOs in unbesetzte MOs übertragen Übergang vom Grundzustand in einen angeregten Zustand Die Wellenlänge der absorbierten Strahlung korreliert mit dem Energieunterschied von besetztem MO und unbesetztem MO den Übergang HOMO g LUMO ist die geringste Energie erforderlich (= grösste Wellenlänge) Je geringer die Energiedifferenz zwischen HOMO und LUMO, desto langwelliger das absorbierte Licht Für E = hn = hc/l hschulz, oc2bct 364 Wellenlänge der absorbierten Strahlung Isolierte Doppelbindungen haben ein DE zwischen HOMO und LUMO, das einer Absorption im UV-Bereich entspricht In konjugierten Doppelbindungssystemen ist DE kleiner als in isolierten Doppelbindungen Je grösser ein kunjugiertes System, desto kleiner DE zwischen HOMO und LUMO und je langwelliger das Absorbtionsmaximum Verschiebung des Absorbtionsmaximums zu längeren Wellen nennt man batochromer Effekt (Farbvertiefung) hschulz, oc2bct 365 Chromophore Gruppen Chromophore (=farbgebende) Gruppen sind dadurch gekennzeichnet, dass ihre Elektronen relativ leicht zum Übergang in ein höheres Energieniveau angeregt werden können p g p*-Übergange n g p*-Übergänge (n=freie Elektronenpaare) Bekannte Chromophor -N=N- (Azo) -N=O (Nitroso) -NO2 (Nitro) hschulz, oc2bct >C=O (Carbonyl) >C=NH (Carbimino) >C=C< (en) 366 Teilstrukturen von Farbstoffen Mehrere Chromophore Gruppen Oft zusätzliche aromatische Systeme zur Vergrösserung der Delokalisation und damit Verkleinerung von DE zwischen HOMO und LUMO Zusätzliche auxochrome Gruppen (Hilfsgruppen) -NH2, -OH, -NR2 haben batochromen Effekt Durch Salzbildung geht dieser batochrtome Effekt verloren (-NH3+, -O-) Die neurale Struktur und die ionische Struktur ist deshalb oft unterschiedlich gefärbt (pH-Indikator) hschulz, oc2bct 367 Einteilung der Farbstoffe nach dem Chromophor Azofarbstoffe (ca. die Hälfte der verwendeten Farbstoffe) O H2N HO N N NH2 S N N N(CH3)2 O Methylorange (rot) Na-Salz ist gelb 2,4-Diaminoazobenzol Chrysoidin (orange) NH2 NH2 N N N N SO3H SO3H Kongorot (blau) Di-Na-Salz ist rot hschulz, oc2bct 368 Antrachinonfarbstoffe O chinoides System O OH OH O O Anthrachinon O Alizarin (rot) O KOH, NaNO3 200°C 2 NH NH2 O O O HN 2-Amino-anthrachinon Indanthren (blau) O hschulz, oc2bct 369 Weitere Farbstoffe mit einem chinoiden System H N -2 H N Cl (H3C)2N S N(CH3)2 H Methylenblau (farblose Leukoverbindung) hschulz, oc2bct Cl +2H (H3C)2N S N(CH3)2 Methylenblau (eine mesomere Grenzstruktur) 370 Triarylmethanfarbstoffe O - O R R OOC R R +2NaOH -2H2O HO O R R=H R=Br OH R Fluorescein (rot) Eosin (rot) hschulz, oc2bct - O O R O R gelbgrüne Fluoreszenz in verd. Lsg rote Lösung (gut wasserlöslich) 371 Triarylmethanfarbstoffe O O H2SO4 -H2O +2 O O C OH O HO +2 OH- - Phenolphthalein (farblose Form) - OOC OH OOC +2 H+ - O O O O- Phenolphthalein (rote, chinoide Form) hschulz, oc2bct 372 8. Stereochemie ohne asymmetrische Zentren, asymmetrische Synthesen und Trennung von Enantiomeren Isomere gleiche Summenformel unterschiedliche Struktur Konstitutionsisomere Stereoisomere (Strukturisomere) unterschiedliche Konnektivität gleiche Konnektivität aber unterschiedliche Anordnung im Raum hschulz, oc2bct Enantiomere Diastereomere Bild und Spiegelbild sind nicht deckungsgleich stehen nicht im Sinne von Bild und Spiegelbild zueinander in Beziehung 373 Voraussetzung für das Auftreten von Chiralität Asymmetrisches Zentrum Atom mit vier verschiedenen Substituenten, die in der Richtung der Ecken eines Tetraeders ausgerichtet sind Keine intramolekulare Spiegelebene COOH COOH H C* OH HO C* H (S,R)-Weinsäure (R,R)-Weinsäure Optisch aktiv HO C* H COOH hschulz, oc2bct HO C* H Optisch inaktiv (meso-Form) COOH 374 Atropisomerie (Behinderungsisomerie) die Rotation um s-Bindungen durch sterische Hinderung eingefroren, kann es Enantiomere geben, auch wenn kein asymmetrisches Atom im Molekül vorhanden ist Ist Br Br Br Br I Br Br I I I I I 2,2´-Dibrom-6,6´-diiodbiphenyl Racemisierung hängt von der grösse der Substituenten in 2,2´- bzw. 6,6´-Stellung und von der Temperatur ab hschulz, oc2bct 375 Br Br I I Br Br I hschulz, oc2bct I 376 Enantiomere des trans-Cyclooctens H H H H s Bild und Spiegelbild sind nicht deckungsgleich = Enantiomere Stark gespanntes Ringsystem, da um die Doppelbindung keine freie Drehbarkeit herrscht hschulz, oc2bct 377 Stereoisomerie bei Kumulenen R1 R3 R4 Allen sp2 R3 R1 C R4 sp C sp2 R2 R1 C C R2 R2 C C R3 R4 s In Allen-Derivaten mit vier verschiedenen Substituenten (R1, R2, R3, R4) sind Bild und Spiegelbild nicht deckungsgleich = Enantiomere hschulz, oc2bct 378 Stereoisomerie bei Kumulenen R3 R1 C R4 R3 R4 C C (Z/E)-Butatrien C R2 R1 R2 sp2 sp sp sp2 Verbindungen mit gerader Anzahl kumulierter Doppelbindungen können chiral sein Verbindungen mit ungerader Anzahl kumulierter Doppelbindungen können Z/EIsomerie zeigen (Diastereomere) hschulz, oc2bct 379 Voraussetzung für das Auftreten von Chiralität Keine Symmetrie-Elemente im Molekül Keine Spiegelebenen Keine Symmetriezentren hschulz, oc2bct 380 Nomenklatur in der Stereochemie R/S-Nomenklatur nach CIP International gültig Definition basiert auf der Ordnungszahl der Substituenten am chiralen Zentrum D/L-Nomenklatur Von Pionieren der Stereochemie entwickelt (E. Fischer, Wohl) Wird heute noch bei Zuckern und Aminosäuren parallel zur R/S-Nomenklatur verwendet hschulz, oc2bct 381 D/L-Nomenklatur Bezugssystem ist der Glycerinaldehyd H H O OH CH2OH H HO O H CH2OH D(+)-Glycerinaldehyd L(-)-Glycerinaldehyd (R)(+)-Glycerinaldehyd (S)(-)-Glycerinaldehyd Jede Verbindung deren experimentell ermittelte Konfiguration am Chiralitätszentrum der des D(+)-Glycerinaldehyds entspricht, gehört zur D-Reihe Der Drehungssinn des pol. Lichts ist davon unabhängig Bei Zuckern erfolgt die Zuordnung zur D- oder L-Reihe nach dem am weitesten von der C=O-Gruppe entfernten asymmetrischen C-Atom hschulz, oc2bct 382 Stereoselektivität in der Entwicklung von pharmazeutischen Wirkstoffen Enzyme sind Biokatalysatoren Enzyme sind Proteine, die aus 20 verschiedenen Aminosäuren der L-Reihe aufgebaut sein können Enzyme sind chirale Moleküle, mit stereoselektiven aktiven Zentren COOH H C* NH2 CH3 COOH H2N C* H CH3 D-Alanin L-Alanin R-Form S-Form Enantiomere wechselwirken mit dem aktiven Zentrum eines Enzyms unterschiedlich hschulz, oc2bct 383 Wechselwirkung des aktiven Zentrums eines Enzyms mit enantiomeren Wirkstoffen H H H2N HOOC HOOC H2N L-Leucin [(S)-Leucin] basisch sauer Aktives Zentrum eines Enzyms hschulz, oc2bct D-Leucin [(R)-Leucin] H HOOC H2N Nur das L-Enantiomere passt in das aktive Zentrum des Enzyms 384 Enantiomere mit unterschiedlicher pharmakologischer Wirkung Das „falsche“ Enantiomer kann folgende Auswirkungen haben: Das Enantiomer ist inert und reagiert überhaupt nicht mit dem Enzym Das Enantiomer reagiert in der gleichen Weise wie das „richtige“ Enantiomer, zeigt aber i.d.R. eine geringere Aktivität Das Enantiomer zeigt eine ganz andere Reaktivität (es reagiert z.B. mit einem anderen aktiven Zentrum). Es ist evtl. ein Wirkstoff für eine andere Fehlfunktion oder es ist toxisch. O HN H O O O HN O N N O H O S-Thalidomid Mildes Beruhigungsmittel O R-Thalidomid verursacht Fehlbildungen des Fötus In pharmazeutischen Wirkstoffen müssen auch die Effekte aller möglichen Enantiomere getestet werden. Idealerweise sollte im Medikament nur das Enantiomere mit der besten Wirkung enthalten sein. hschulz, oc2bct 385 Herstellung enantiomeren-reiner Wirkstoffe „Standard-Synthesen“ unter Herstellung eines Racemats, das anschliessend in Enantiomere getrennt wird Kristallisation und manuelle Auslese der Kristalle, falls beide Enantiomere getrennt auskristallisieren (das geschieht aber nur selten!) Reaktion des Racemats mit einem enantiomeren-reinen Reagenz ergibt Diastereomere, die getrennt werden können Chromatographische Aufarbeitung an stereoselektiven stationären Phasen Reaktion mit Mikroorganismen oder isolierten Enzymen (biotechnologische Methoden) – Ein Enantiomer wird verbraucht (Stoffwechsel des Mikroorganismus) – Ein Enantiomer wird in ein Derivat umgewandelt hschulz, oc2bct 386 Herstellung enantiomeren-reiner Wirkstoffe „Stereoselektive Synthesen“ unter ausschliesslicher Herstellung des gewünschten Enantiomers Asymmetrische Synthesen mittels stereospezifischer synthetischer Katalysatoren (Übergangsmetall-Komplexe) Asymmetrische Synthesen mittels Biokatalysatoren (Enzyme, Mikroorganismen) = Biotechnologie hschulz, oc2bct 387 Enantiomeren-reine Kristalle durch spontane Kristallisation Racemische Lösungen kristallisieren i.d.R. auch als Racemate aus (die Elementarzelle enthält gleich viele R- und S-Enantiomere) In Ausnahmefällen kann bei einer bestimmten Temperatur ein Gemenge aus enantiomeren-reinen Kristallen ausfallen (Konglomerat) Pasteur (1844) Isolierung der Enantiomere des Natriumammoniumsalzes der Traubensäure (= racemische Weinsäure) Handauslese der spiegelbildlichen Kristalle und anschliessendes Ansäuern führte zu reiner (+)- und (-)Weinsäure hschulz, oc2bct 388 Chemische Spaltung des Racemats durch einen enantiomeren-reinen Reaktand Racemische Säuren reagieren mit einer enantiomeren-reinen Base zu diastereomeren Salzen (Pasteur 1858) Racemische Basen reagieren mit einer enantiomeren-reinen Säure zu diastereomeren Salzen (Pasteur 1858) Hydrolyse (+)-Säure (+)-Base (-)-Säure (++)-Salz (-+)-Salz Trennung Hydrolyse hschulz, oc2bct (+)-Säure (-)-Säure 389 Biochemische Spaltung (Pasteur 1858) Enzyme (Mikroorganismen) reagieren bevorzugt mit einem bestimmten Enantiomer, das andere wird oft nicht angegriffen Das reagierende Enantiomer kann vollständig abgebaut werden (Stoffwechsel des Mikroorganismus, Verlust von 50% Reaktionsausbeute!) oder als Derivat isoliert und zurückgewonnen werden Der Pilz Penicillium glaucum reagiert nur mit der (+)-Weinsäure und nicht m it der (-)Weinsäure. Aus Traubensäure kann dadurch reine (-)-Weinsäure isoliert werden hschulz, oc2bct 390 Enzymatische Trennung der Enantiomere bei der Ibuprofen-Herstellung H3C H H OCH3 H3C CH3O CH3 O O H3C CH3 (S)-Ibuprofen-Methylester CH3 (R)-Ibuprofen-Methylester Pferdeleber-Esterase H2O, pH=7,2, RT H3C OCH3 H3C H3C H H O (S)-Ibuprofen-Methylester CH3 HO CH3 O CH3 (R)(-)-Ibuprofen Trennung von Ester und Säure Hydrolyse des Esters zu (S)(+)-Ibuprofen hschulz, oc2bct 391 Biokatalytische Trennung der D,L-Methionin-Enantiomere H2N H H COOH CH3S D-Methionin R-Form NH2 HOOC SCH3 L-Methionin S-Form N-Acetylierung O Racemisierung HN CH3S O CH3 H3C H COOH D-(N-Acetyl)methionin H NH HOOC SCH3 L-(N-Acetyl)methionine Biokatalysator O HN CH3S - Asperagillus Oryzae: Microorganismus, Pilz - Hydrolase: Enzym, isoliert aus den Pilzzellen und auf einem Ionenaustauscherharz fixiert CH3 H COOH D-(N-Acetyl)methionin hschulz, oc2bct Biokatalysator H HOOC NH2 SCH3 L-Methionin 392 Stereospezifische Synthese von Ephidrin D-Glucose O Hefe H3C COOH O HO O + HC H COOH H Hefe HO CH3 3 O Erster biokatalytischer Schritt führt ausschliesslich zum R-Enantiomer Hefe: Saccharomyces cerevisiae + H CH3 H3C NH2 H2/Pt (R)-2-Keto-1hydroxypropylbenzol hschulz, oc2bct Brenztraubensäure Pyruvinsäure NHCH3 H L-Ephidrin (1R, 2S) Zweiter chemischer Schritt führt vorwiegend zum 1R,2S -Enantiomer durch Induktion des benachbarten Zentrums 393 Stereoisomere der Ascorbinsäure OH HO H * HO * HO *H O O O H OH O H CH2OH CH2OH L-Ascorbinsäure (R,S) D-Ascorbinsäure (S,R) Kommt in einigen Bakterien vor Verhindert Skorbut nicht Häufig in der Natur (Vitamin C) Anti-Skorbut HO O O HO OH HO *H H *H CH2OH L-Isoascorbinsäure (S,S) Kommt in der Natur nicht vor Verhindert Skorbut nicht hschulz, oc2bct * OH H s OH * * O OH O CH2OH D-Isoascorbinsäure (R,R) Kommt in der Natur nicht vor Geringe Wirkung gegen Skorbut 394 Technische Vitamin C-Synthese H O CH2OH H HO OH H H H +2H OH Pt-Kat. H OH HO OH H H OH H OH CH2OH CH2OH D-Glucose D-Sorbit CH2OH CH2OH acetobacter suboxydans -2H H HO H OH H O HO OH H O HO Pt-Kat. H HO H OH H CH2OH 2-Keto-L-gulonsäure hschulz, oc2bct H L-Sorbose O HO O HO OH CH2OH CH2OH COOH + O 2 / - H 2O H - H 2O O + H2O H HO HO H CH2OH L-Ascorbinsäure 395 9. Polymere Werkstoffe Ungesättigte Verbindungen können nicht nur mit „kleinen“ Molekülen unter Addition reagieren, sondern auch mit ihresgleichen H n CH2 CH H2C C R R n Aus Monomeren werden dadurch Oligomere und schliesslich Polymere monos, griechisch, allein meros, griechisch, Teil oligos, griechisch, wenig, klein poly, griechisch, viel hschulz, oc2bct Makromoleküle 396 Die Polymerisation Aktivierung der Doppelbindung durch Initiatoren (Aktivatoren, Katalysatoren) Radikalische Polymerisation Kationische Polymerisation Anionische Polymerisation Polymerisation an Übergangsmetallkomplexen Additionsreaktionen multifunktioneller Edukte Kondensationen: Verknüpfung von Monomeren unter Austritt kleiner Moleküle Bildung von linearen oder verzweigten Ketten hschulz, oc2bct 397 Klassifizierung der Polymere Homopolymere (Unipolymere) bestehen aus einer Sorte von Monomeren: AAAAA Copolymere bestehen aus mehreren Sorten von Monomeren: Bipolymeren, Terpolymere,...: ABABABABAB Im Copolymer können die ursprüngl. Monomere rein statistisch verteilt sein: ABABBABAABAAABBA Block-Copolymere: Monomere sind blockweise angeordnet: AAAAABBBAAAABBBBBAAAAA hschulz, oc2bct 398 Klassifizierung der Polymere Biopolymere Polyisoprene, Polysaccharide, Polypeptide (Proteine), Polynukleotide Synthetische Polymere Organische Polymere in der Kette (im Polymer-Rücken) sind C-Atome enthalten Anorganische Polymere in der Kette sind keine C-Atome enthalten: Polymerer Schwefel, Siliciumfluorid, Silicone (z.B. Polydiphenylsiloxan), Polyphosphate hschulz, oc2bct 399 Nomenklatur der Polymere Trivialnamen sind weit verbreitet Benennung nach dem entsprechenden Monomer: Polystyrol, Polyacrylnitril, Polyvinylchlorid (PVC) Benennung nach dem Strukturelement, aus dem das Polymer besteht: Polyethylenterephtalat, Polyphenylenoxid IUPAC-Nomenklatur Strukturelement -CH2- Trivialname Polyethylen -CH=CH-CH2-CH2- 1,4-Polybutadien Polyformaldehyd -O-CH2-CClH-CH2hschulz, oc2bct Polyvinylchlorid IUPAC-Name Poly(methylen) Poly(1-butenylen) Poly(oxymethylen) Poly(1-chlorethylen) 400 9.1 Die Struktur der Polymere Die Polymerisationsreaktion führt immer zu einem Produktgemisch Ketten unterschiedlicher Länge Kettenverzweigungen: unterschiedlicher Ort und Ausmass Konstitutionsisomerie bei unsymmetrischen Monomeren und bei Copolymeren Konfigurationsisomere hschulz, oc2bct 401 Der Polymerisationsgrad Anzahl der im Makromolekül enthaltenen Grundbausteine P = M / M0 M =Molmasse des Makromoleküls M0=Molmasse des Grundbausteins die einzelnen Makromoleküle einer Substanz haben i.d.R. unterschiedliche Polymerisationsgrade, da ihre Molmassen herstellungsbedingt unterschiedlich sind (unterschiedliche Kettenlänge!) hschulz, oc2bct 402 Molmassenverteilung in einem Polymer Ein Mass für die Häufigkeit, mit der eine bestimmte Molmasse in einer Substanz auftritt charakterisiert durch bestimmte Parameter wie z.B. Mittelwert und Streuung Verschiedene Mittelwerte zur Charakterisierung der Molmassenverteilung Zahlenmittel Mn = S Ni Mi / S Ni – Ni = Anzahl der Makromoleküle mit Molmasse Mi – bestimmbar durch Osmose-Experimente Massenmittel Mw = S mi Mi / S mi – mi = Masse der Makromoleküle mit Molmasse Mi – bestimmbar durch statische Lichtstreuung hschulz, oc2bct 403 Molmassenverteilung in einem Polymer Weiter Mittelwerte: Zentrifugenmittel Mz Viskositätsmittel Mh Die Uneinheitlichkeit U ein Mass für die Breite der Molmassenverteilung U = (Mw / Mn) - 1 U hängt mathematisch mit der Standardabweichung s zusammen U = 0: alle Makromoleküle besitzen die gleiche Molmasse: monodisperse Substanz U>0: polydisperse Substanz hschulz, oc2bct 404 Molmassenverteilung in einem Polymer Technisch interessante Polymere haben Uneinheitlichkeiten von U=0,01-4, in einigen Fällen kann U>30 sein kleine U-Werte erhält man z.B. bei der anionischen Polymerisation grosse U-Werte erhält man bei der radikalischen Polymerisation oder der Polykondensation hschulz, oc2bct 405 Die Konstitution eines Polymers Art und Anordnung der Grundbausteine Struktur bzgl. Linearität und Verzweigung statistische oder regelmässige Anordnung der verschiedenen Grundbausteine bei Copolymeren statistische oder regelmässige Anordnung eines unsymmetrischen Grundbausteins bei Homo- oder Copolymeren hschulz, oc2bct 406 Konstitutionsisomere Homopolymere mit unsymmetrischen Grundbausteinen 1 2 H H2C C Schwanz R Kopf 1 1 2 2 R R Kopf-Schwanz 1 R 2 R 1 2 1 R Kopf-Kopf 2 2 1 R SchwanzSchwanz Copolymere mit gleicher Anzahl der jeweiligen Grundbausteine, aber unterschiedlicher Sequenz -ABABABABAB-AABBAABBAB- hschulz, oc2bct 407 Konstitution von Copolymeren Statistische Bipolymere -ABABBABAABBBABABAAA periodische Bipolymere -AABAABAABAABAABAAB alternierende Bipolymere -ABABABABABABAB- Gradientbipolymere -AAABAAABAABABBABBBABBBBBB Blockbipolymere -AAAAAAAAAAABBBBBBBBBBBAAAAAAAAhschulz, oc2bct 408 Pfropf- oder Graftpolymere -ABAAB- -ABAABABBBBAAB-BBBBBB- -BBBB- -BBBBBB- -AAAAAAA- Hauptkette ist meist ein Homopolymer oder ein statistisches Copolymer Die Seitenketten werden in einer zweiten Reaktionsstufe an die Hauptkette polymerisiert hschulz, oc2bct 409 Molekularstruktur von Polymeren Unverzweigte Polymere die Monomere enthalten nur zwei reaktionsfähige funktionelle Gruppen, bzw. nur eine Doppelbindung, die unter Addition angegriffen werden kann Durch Reaktion der Enden einer Kette können Ringpolymere entstehen Verzweigte Polymere einige oder alle Monomere enthalten mehr als zwei reaktionsfähige funktionelle Gruppen hschulz, oc2bct 410 Verzweigt Polymere Kammpolymer Baumpolymer hschulz, oc2bct Sternpolymer Dendrimer 411 Netzwerke Durch intramolekulare Bindungen wird aus Makromolekülen ein einziges Riesenmolekül = Netzwerk chemische Bindungen, zB. Schwefelvulkanisation von Kautschukpolymeren physikalische Bindungen: Wasserstoffbrücken, coulombsche oder van-der-Waals-Kräfte, Verhackung von Polymerketten hschulz, oc2bct 412 Vulkanisation: ein wichtige Vernetzungsreaktion Sx+ + S8 Sx Sx+ + C CH SySx + CH2 C CH CH2 C CH2 CH2 + C CH CH + H C CH S8 CH C CH C CH CH CH C CH CH S8 S8+ + C C CH CH2 S8 CH CH2 + + C CH CH C CH2 CH2 H C CH hschulz, oc2bct CH 413 Konfiguration von Polymeren Ein Makromolekül enthält i.d.R. sehr viele asymmetrische C-Atome die räumliche Anordnung der Substituenten an den asymmetrischen C-Atomen kann statistisch verteilt sein = ataktische Polymere sich nach einer gewissen Regelmässigkeit ändern = taktische Polymere – alle gleich – alternierend hschulz, oc2bct = isotaktische Polymere = syndiotaktische Polymere 414 Taktizität der Polymere Isotaktische Anordnung H CH3 H H CH3 n CH3 CH3 CH3 CH3 Polypropylen Kopf-Schwanz-Verknüpfung Syndiotaktische Anordnung CH3 H CH3 H3C CH3 CH3 CH3 CH3 hschulz, oc2bct CH3 CH3 H H R R H R H H HH Ataktische Anordnung CH3 R HH H H CH3 CH3 CH3 415 Taktizität im Polybutadien cis-1,4-Polybutadien (Z)-1,4-Polybutadien cis-taktisch trans-1,4-Polybutadien (E)-1,4-Polybutadien trans-taktisch 1,2-Polybutadien isotaktisch 1,2-Polybutadien syndiotaktisch hschulz, oc2bct 416 Die Konformation eines Polymers Kettenförmige Polymere, die nur van-der Waals-Kräfte aufeinander ausüben liegen als statistisches Knäuel vor Proteine bilden durch Wasserstoffbrückenbindungen eine Faltblatt oder Helixstruktur Polynucleotide bilden ebenfalls eine Helixstruktur hschulz, oc2bct 417 9.2 Mechanismus der Polymerisation Thermodynamische Voraussetzungen DGP = DHP - TDSP DGP muss negativ sein. Die Entropie nimmt aber bei der Polymerisation immer ab. DHP muss deshalb so stark negativ sein, dass es den positiven TDSP-Term überkompensiert Kinetische Voraussetzungen ausreichende Reaktionsgeschwindigkeit Abwesenheit von Konkurrenzreaktionen genügende Löslichkeit hschulz, oc2bct 418 9.2.1 Die radikalische Polymerisation Radikale können an die Doppelbindungen addieren Die daraus entstehenden neuen Radikale können an weitere Doppelbindungen addieren Reaktionsbedingungen Hohe Temperatur (>100°C) ggf. hoher Druck (ca. 100 bar) Molekulargewicht und Verzweigungsgrad können durch Zusätze kontrolliert werden (Radikalbildner) hschulz, oc2bct 419 Mechanismus der radikalischen Polymerisation Initiierung (Kettenstart) RO OR R O + H2C CH2 2 R O ROCH2 CH2 Kettenwachstum ROCH2 CH2 + H2C CH2 ROCH2CH2CH2 CH2 +(n-1) H2C CH2 RO CH2CH2 n CH2CH2 hschulz, oc2bct 420 Kettenabbruch RO CH2CH2 mOR 2 RO CH2CH2 nCH2 CH2 RO CH2CH2 n CH2 CH3 + RO CH2CH2 n CH Kettenverzweigung +R -R-H CH2 CH CH2 H H CH2 C CH2 CH2 CH2 H CH2 C CH2 + H2C CH2 H2C hschulz, oc2bct 421 Initiatoren für die radikalische Polymerisation Peroxide O O O O O O Dibenzoylperoxid Di-t-butylperoxid O O H t-Butylhydroperoxid aliphatische Azoverbindungen CH3 N N H3C CN hschulz, oc2bct CH3 CH3 Azobisisobutyronitril AIBN CN 422 Polymere durch radikalische Polymerisation Monomer Ethen Chlorethen (Vinylchlorid) Tetrafluorethen Struktur H2C CH2 Methylmethacrylat F2C CF2 H C CH2 H H2C C hschulz, oc2bct C CH3 H2C C 2-Methylpropen (Isobutylen) Polyvinylchlorid (PVC) Teflon H2C CHCl Vinylbenzol (Styrol) Propennitril (Acrylnitril) Polymer Polyethylen N O Polystyrol (Styropor) Polyacrylnitril Orlon Plexiglas C OMe H2C C CH3 CH3 Elastol 423 9.2.2 Die kationische Polymerisation Elektrophile Addition eines InitiatorKations an die Doppelbindung das daraus entstehende Carbenium-Ion darf nicht durch das Initiator-Anion oder durch das Lösungsmittel abgefangen werden, sondern muss schneller mit weiterem Monomer reagieren keine nucleophilen Lösungsmittel Reaktionsbedingungen tiefe hschulz, oc2bct Temperatur (< 0°C) 424 Mechanismus der kationischen Polymerisation Startreaktion (Initiierung) HClO4 + H2C=CHR + H3C-CHR + ClO4 - Kettenwachstum + H3C-CHR + n H2C=CHR + H (CH2-CHR )nCH2-CHR hschulz, oc2bct 425 Umlagerungen durch Hydrid- oder Alkylgruppenwanderung können tertiäre Carbenium-Ionen entstehen bei sterisch anspruchsvollen Monomeren kann dann die Kettenfortpflanzung so langsam werden, dass Kettenabbruch eintritt Kettenabbruch i.d.R. durch Reaktion mit dem InitiatorGegenion Gezielter Abbruch durch Addition von Wasser hschulz, oc2bct 426 Initiatoren für die kationische Polymerisation Protonsäuren HClO4, H2SO4, HI, Trifluormethylsulfonsäure Lewis-Säuren mit Spuren von protischen Lösungsmitteln (z.B. Wasser) - BF3 + H2O H+BF3OH weitere Lewissäuren: AlCl3, AlBr3, TiCl4 grössere Mengen an Wasser führt zum Kettenabbruch hschulz, oc2bct 427 Industriell hergestellte KationPolymerisate Polymer Hauptanwendungsgebiete Isobutylen-IsoprenCopolymer (Butylkautschuk) Polyisobutylen (oligomer) Elastomer: Schläuche, Luftschläuche, Isolierungen Polyvinylether Kleber Polytetrahydrofuran Blöcke für Polyurethan Polyformaldehyd Konstruktionswerkstoff hschulz, oc2bct Kleber, Öle, Additive 428 9.2.3 Die anionische Polymerisation Nucleophile Addition eines Initiator-Anions an die Doppelbindung das daraus entstehende Carbanion darf nicht durch das Initiator-Kation oder durch das Lösungsmittel abgefangen werden, sondern muss schneller mit weiterem Monomer reagieren keine protischen Lösungsmittel, kein Sauerstoff Reaktionsbedingungen tiefe Temperatur (< 0°C) hschulz, oc2bct 429 Mechanismus der anionischen Polymerisation Startreaktion (Initiierung) Li+ - - Bu + H2C=CHR Bu-H2C-CHR + Li+ Kettenwachstum - Bu-H2C-CHR + n H2C=CHR Bu (CH2-CHR )nCH2-CHR hschulz, oc2bct 430 Umlagerungen sind bei Carbanionen nicht so häufig zu beobachten wie bei Carbenium-Ionen Kettenabbruch wenn keine Verunreinigungen wie Sauerstoff, Wasser oder CO2 vorhanden sind findet kein Kettenabbruch statt die Reaktion ist beendet wenn kein Monomer mehr vorhanden ist wird weiteres Monomer zugegeben fängt die Reaktion wieder an = Lebendes Polymer Gezielter Abbruch durch Addition von Wasser oder Alkoholen hschulz, oc2bct 431 Initiatoren für die anionische Polymerisation Organometallverbindungen der 1. Hauptgruppe Butyllithium, Phenylnatrium, Octylkalium in polaren Lösungsmitteln liegt ein echtes + Ionen-Paar vor: Li Bu in unpolaren Lösungsmitteln liegt eine stark polarisierte kovalente Bindung vor Alkoholate (NaOCH3), Metallamide (NaNH2) Alkalimetalle (Elektronenübertragung) hschulz, oc2bct 432 Monomere für die anionische Polymerisation Monomere mit elektronenarmen Doppelbindungen (elektronenziehenden Substituenten) Styrol, Vinylketone, Acrylverbindungen, Diene Ringsysteme Alkenoxide, RO - Lactame, Lactone O + H C CH 2 2 O O RO O O + N hschulz, oc2bct NH N NH O 433 Industriell hergestellte AnionPolymerisate Polymer Hauptanwendungsgebiete cis-1,4-Polybutadien Elastomer: Reifen Blockcopolymere (Styrol-Diene) Polycyanoacrylat thermoplastische Elastomere, Schuhsohlen Kleber Polyalkylenglycole Blöcke für Polyurethan Polyformaldehyd Konstruktionswerkstoff hschulz, oc2bct 434 9.2.4 Polymerisation mittels Übergangsmetallkomplexen Das Monomer koordiniert an den Metallkomplex das koordinierte Monomer wird in eine Metall-Alkyl-Bindung eingeschoben (Insertion) stereospezifische Polymerisation, da das Monomer in einer bestimmten Orientierung koordiniert und eingeschoben wird hschulz, oc2bct 435 Ziegler-Natta-Katalysatoren Katalysatorsysteme bestehend aus Übergangsmetallverbindung der 3. - 8. Nebengruppe metallorganische Verbindung oder Hydrid der 1. - 4. Hauptgruppe typisches Beispiel: TiCl4 + AlEt3 EtTiCl3 + AlEt2Cl dieses System ist teilweise unlöslich = heterogene Katalyse hschulz, oc2bct 436 Mechanismus der Polymerisation an Übergangsmetallkomplexen H3C Kettenfortpflanzung H CH2 H3C H2C CH3 Cl H3C CH3 H H3C Cl H Cl H Cl H H H H Cl H Cl Cl Cl Cl CH2 Cl Cl C H2 C CH 3 H Cl Kettenabbruch H2C R C CH2 CH 3 Cl RCH2 H Cl Cl Cl hschulz, oc2bct H Cl + CH3 CH2 Cl Cl Cl 437 Polyisopren-Synthese: Versuch der Kopie des Naturkautschuks AlEt3/TiCl4 2 -CH4 Isopren + n Katalysator n Z-1,4-Polyisopren E-1,4- hschulz, oc2bct 1,2 3,4 438 Struktur von Polyisopren Produkt ist abhängig von der Reaktionsführung radikalische Polymerisation Anionische Polymerisation mit versch. Katalysatoren metallkomplex-katalysierte Polymerisation Z-1,4 (%) E-1,4 (%) 1,2- (%) 3,4- (%) Radikalisch 22 65 6 7 Lithiumalkyle 92 1 0 7 Natriumalkyle 0 45 7 48 Kaliumalkyle 0 52 8 40 AlR3/TiCl4 98 0 0 2 Naturkautschuk 98 0 0 2 Im Vergleich zur AlR3/TiCl4-Type hat NR ein etwas höheres mittleres Molekulargewicht und hat geringe Mengen von organischen Nebenbestandteilen, die die Verarbeitbarkeit erleichtern. hschulz, oc2bct 439 Polyethylen Kationische Polymerisation von Ethylen Lewis-Katalysatoren (AlCl3, BF3) in Lösung bei 30-50 bar niedermolekulare Polymere (M ca. 400 g/mol) Radikalische Polymerisation Hochdruckverfahren: 1000-2000 bar/200°C Initiator: Sauerstoff lineare und verzweigte Ketten (M >10 000) rel. niedere Dichte: LDPE (low density polyethylen) hschulz, oc2bct 440 Polyethylen Übergangsmetall-Komplex katalysierte Polymerisation Normaldruck und niedrige Temperatur (70°C) fast unverzweigte Ketten bis zu 80% kristallin Molmasse zwischen 1x104 und 6x106 höhere Dichte: HDPE (high denisty) hschulz, oc2bct 441 Polypropylen Polymerisation durch Ziegler-NattaKatalysatoren Stereospezifische Polymerisation ausschliesslich Kopf-Schwanz-Verknüpfung 95% isotaktische Anordnung der Methylgruppe Schmelzpunkt 176°C H CH3 H H n hschulz, oc2bct CH3 CH3 CH3 CH3 CH3 442 Polyacetylen trans-Polyacetylen cis-Polyacetylen Radikalische, kationische und anionische Katalyse führt vorwiegend zu cyclischen Produkten, vor allem zu Benzol Polymerisation gelingt mit bestimmten Metallkomplex-Katalysatoren Bildung von vorwiegend cis- oder transPolyacetylen hängt vom Katalysator und den Reaktionsbedingungen ab hschulz, oc2bct 443 Synthese von Polyacetylen H H3C H H3C H2C CH2 H H C Cl C H Cl Cl Cl Cl Cl Cl Cl Cl Cl H Cl Cl H2 C CH 3 H H H H H C Cl Cl Cl hschulz, oc2bct C Cl H H2 C H C C CH3 H 444 Eigenschaften von Polyacetylen Konjugiertes Polyen cis-Polyacetylen (PA) ist ein Isolator trans-PA ist ein Halbleiter durch Dotierung von cis-PA mit Iod oder AsF5 erhält man PA-Filme mit guter elektrischer Leitfähigkeit PA ist oxidationsempfindlich hschulz, oc2bct Polymer s (-1cm-1) Polyethylen 10-18 (E)-PA 10-9 (E)-PA (dot. I2) 102 PA (dot. AsF5) 103 Graphit 104 Kupfer 6,5 x 106 s = spez. Leitfähigkeit 445 9.2.5 Polykondensation Multifunktionelle Monomere oder Oligomere können in einer Additionsreaktion zu Polymeren reagieren Bei der Polykondensation wird dabei ein niedermolekulares Produkt eliminiert hschulz, oc2bct 446 Die Polyamid-Synthese O O n C R HO C + n OH HO H N R´ N H O - (n-1) H2O H C R H O H H C N R´ N H n Nylon 66 Polykondensat aus Adipinsäure und Hexamethylendiamin O HO C hschulz, oc2bct O C H N H N H n 447 Polyamide aus Aminocarbonsäuren O n NH + n H2O n H2N - n H2O e-Aminohexansäure OH e-Caprolactam H2N H - (n-1) H2O N OH O n-1 6-Polyamid, Perlon O O Durch intermolekulare Kondensation von Aminocarbonsäuren entstehen Polymere Durch intramolekulare Kondensation entstehen Lactame Lactame können auch direkt kationisch oder anionisch polymerisiert werden hschulz, oc2bct 448 p-Aramid: Kevlar oder Twaron O O n C Cl - (n-1) HCl C Cl + n Cl H N H H N H O O C C N H N H n H Fasern übertreffen die Festigkeit von Stahl bei 1/5 der Dichte! Verwendung im Automobilbau (Bremsbeläge) und Raumfahrt hschulz, oc2bct 449 Polypeptide a-Aminosäuren bilden unter Polykondensation Polypeptide (Proteine) H R1 + N C C H H R2 H O OH N C C H H H O O N C C + OH R3 H H OH a-Aminocarbonsäuren H - 3 H2O R1 O N C H H C R2 O N C H H C R3 O N C C OH H H Tripeptid hschulz, oc2bct 450 Resonanz induzierte Planarität der Peptidbindung H O N C R C H hschulz, oc2bct H R C N C H O H O N C R C H H R + C N C H O 451 Die Polyester-Synthese O O n C R HO C + n H OH O O HO - (n-1) H2O R´ OH O C R C O R´ O H n PET: Polyethylenterephthalat Polykondensat aus Terephthalsäure und Ethylenglycol HO O O C C O O H n hschulz, oc2bct 452 Polyester aus Hydroxycarbonsäuren O n O + n H2O O n HO - n H2O e-Hydroxyhexansäure e-Lacton OH - (n-1) H2O HO O OH O n-1 O Durch intermolekulare Kondensation von Hydroxycarbonsäuren entstehen Polymere Durch intramolekulare Kondensation entstehen Lactone Lactone können auch direkt kationisch oder anionisch polymerisiert werden hschulz, oc2bct 453 Alkydharze Dreidimensionale Polyester-Netzwerke durch Polykondensation von mehrwertigen Alkoholen mit Dicarbonsäuren oder deren Anhydriden O n OH HO OH +m O - H2O O O O OO O O OO O O OH O OH O O O O OH hschulz, oc2bct OO O O O OO O O OH 454 Polyether n + n H2O O SnCl4 + wenig Wasser n HO - n H2O Ethylenoxid HO OH Glycol OH O n-2 - (n-1) H2O O Polyethylenglycol (PEG) Durch intermolekulare Kondensation von Polyolen entstehen Polymere Durch intramolekulare Kondensation entstehen Epoxide Epoxide werden i.d.R. direkt kationisch oder anionisch polymerisiert werden hschulz, oc2bct 455 Kronenether: cyclische Polyether Cl OH O O KOH, THF -2 HCl + O O OH Cl O O O O O O O KMnO4, C6H6 O O + K O O + MnO4 - O [18]Krone-6 hschulz, oc2bct 456 Kronenether: cyclische Polyether Struktur erinnert an eine Krone sehr starke Solvatation von Kationen passender Grösse Wirt (Kronenether)-Gast (Kation)Beziehung Ionische Verbindungen können damit in unpolaren Lösungsmitteln gelöst werden hschulz, oc2bct 457 Phenoplaste (Bakelite) Polykondensation von Phenol und Formaldehyd benannt nach ihrem Entdecker Bakeland (1909) erster technisch eingesetzter Kunststoff HCl H2C O OH H2C O H OH OH CH2 O H + + H2C O H + (A) - H+ CH2 O H OH H -H2O hschulz, oc2bct + +H+ H CH2 O CH2 O H + H2C O H -H+ OH + OH + CH2 +(A) -H+ OH CH2 CH2 O H 458 Polycarbonate O CH3 HO CH3 C CH3 OH Cl Cl HO Phosgen C CH3 OH Bisphenol A O CH3 HO C CH3 CH3 O O n C CH3 OH Polycarbonat (Makrolon) Verwendung zur Herstellung von CDs, Schutzhelme, Essbesteck usw. hschulz, oc2bct 459 9.2.6 Polyaddition Addition von Alkoholen an Isocyanate R N C O H O R´ R N H C O O R´ O R N C O R´ H Urethan (Carbamidsäureester) hschulz, oc2bct O R N C R´ H Zum Vergleich: Amid 460 Polyurethane (PU) n OH HO +n O C N N C O H O HO C O N O N C H O O H n Katalysator: tertiäre Amine, zinn-organische Verbindungen (Dibutylzinndiacetat) Polymerisationsgrad meist gering (ca. 50 Einheiten) Trifunktionelle Edukte ergeben Netzwerke hschulz, oc2bct 461 Polyurethane Thermoplastische Polyurethane wichtigstes System ist das Additionsprodukt von Hexamethylendiisocyanat und 1,4-Butandiol Fasern, Filme Schmelztemperatur bis ca. 200°C, danach erfolgt Zersetzung Polyurethanelastomere Polyurethanschäume durch kontrollierten Zusatz von Wasser wird ein Teil der Isocyanogruppen hydrolysiert und es bildet sich während der Polyaddition CO2: R-NCO + H2O R-NH2 + CO2 hschulz, oc2bct 462 10. Naturstoffe Organische Verbindungen, die von lebenden Organismen gebildet werden Einteilung der Naturstoffe nach verschiedenen Gesichtspunkten Chemische Struktur Physiologische Wirkung Organismus- oder Pflanzenspezifität Biochemische Herkunft Die chemische Struktur reicht von kleinen Molekülen bis hin zu hochkomplizierten Polymeren H H H H H H hschulz, oc2bct H H 463 Wichtige Naturstoffgruppen Naturstoffe Terpene Steroide Alkaloide Fette, Öle und Wachse Kohlenhydrate Aminosäuren und Proteine Vitamin A Cholesterin Atropin Margarine Saccharose Insulin ß-Carotin Vitamin D Cocain Seifen Vitamin C Glucagon Campher Cortison Morphin nichtionische Tenside Stärke Thyroxin Naturkautschuk Sexualhormone Strychnin Stearinsäure Cellulose Penicillin hschulz, oc2bct 464 10.1 Terpene Diels-AlderReaktion zweier Isopren-Einheiten * H Dipenten (Racemat) (+)-Limonen (-)-Limonen Das Grundgerüst der Terpene ist formal aus Isopren-Einheiten aufgebaut (Isoprenoide) Einteilung in Acyclische oder aliphatische Terpene Monocyclische Terpene Bi- und oligocyclische Terpene hschulz, oc2bct 465 Terpene O * * * * * * OH O * * Dipenten Menthol L-Campher D-Campher ß-Carotin CH2OH CHO Vitamin A (Retinol) hschulz, oc2bct 11-cis-Retinal 466 Acyclische Terpene Geraniol des Geranium- und Rosenöls OH Hauptbestandteil OH Nerol Citronellol * (-)-Citronellol im Rosenöl (+)-Citronellol im Zitronenöl Racemat-Form im Geraniumöl OH Ipsenol (-)-Ipsenol ist ein Pheromon der Borkenkäfer hschulz, oc2bct HO H 467 Monocyclische Terpene Menthol 8 Stereoisomere sind synthetisch zugänglich In der Natur kommt nur (-)-Menthol vor (Hauptbestandteil des Pefferminzöls) 7 1 6 5 9 2 3 * * OH 4 HO * 8 hschulz, oc2bct 10 H CH(CH3)2 H3C H H (1R,3R,4S)-(-)-Menthol 468 Bicyclische Terpene Pinangruppe a- und b-Pinen sind die Hauptbestandteile des Terpentinöls Terpentinöl wird durch Wasserdampfdestillation aus dem Harz von Pinien (Kiefern) gewonnen Terpentinöl ist ein hervorragendes Lösungs-mittel für Öle und Harze (Einsatz in der Lackindustrie) hschulz, oc2bct a-Pinen b-Pinen 469 Bornangruppe Wichtigster Vertreter ist Campher Von den 4 möglichen Stereoisomeren existiert nur ein Enantiomerenpaar. Die Ringspannung für das andere Paar ist zu gross (+)-Campher kommt im Holz des Campherbaums vor (-)-Campher ist Bestandteil des Kamillenöls Verwendung als Weichmacher für Celluliod und als Antiseptikum Industrielle Darstellung aus a-Pinen O * O * * O * * * L-(-)-Campher D-(+)-Campher (1S,4S)-Campher (1R,4R)-Campher hschulz, oc2bct 470 Carotinoide Polyenfarbstoffe Carotin Vorkommen in grünen Blättern, in vielen Blüten und Früchten Gewinnung aus Mohrrüben (3 Isomere) Dunkelrote kristalline Substanz, Smp. ca. 180°C b-Carotin (=Provitamin A) wird im tierischen Organismus in zwei Moleküle Vitamin A gespalten hschulz, oc2bct 471 Carotin a-Carotin H b-Carotin -Carotin hschulz, oc2bct 472 Vitamin A (Retinol) Die oxidative Spaltung von b-Carotin im lebenden Organismus führt zu Vitamin A Vitamin A (Retinol) kann in vitro oder in vivo zu trans-Retinal oxidiert werden Oxidation CH2OH 2 in vitro: KMnO4 in vivo: Retinol-Dehydrogenase O 2 hschulz, oc2bct H trans-Retinal 473 Retinal und der Sehvorgang Trans-Retinal wird in den Lichtrezeptorzellen zu cis-Retinal isomerisiert Cis-Retinal passt in das aktive Zentrum des Proteins Opsin und reagiert mit einem Amin-Substituenten zu einem Imin (Rhodopsin) Trifft ein Photon auf Rhodopsin isomerisiert der cis-Retinal-Teil sehr schnell zum trans-Isomer Das trans-Retinal passt schlecht in das aktive Zentrum des Opsins. Es kommt zu Konformationsänderungen und schliesslich zur Hydrolyse der Imin-Bindung Diese Reaktionsfolge induziert ein Nervensignal, das als Licht wahrgenommen wird Das freie trans-Retinal wird wieder zu cis Retinal isomerisiert und steht wieder für den gleichen Prozess zur Verfügung hschulz, oc2bct 474 Retinal und der Sehvorgang CHO Retinalisomerase trans-Retinal O R + cis-Retinal H2N CHO H -H2O R N H cis-Retinal hschulz, oc2bct Opsin Rhodopsin 475 Naturkautschuk Gewinnung aus „hevea brasiliensis“ (6 mio jato) Biosynthese: O >98% Z-1,4-Polyisopren <2% Z-3,4-Polyisopren 2 NADP+ HO C ~CoA 3 2 NADPH H3C CH3 OH O OH Mevalonsäure „aktives Isopren“ CO2+H2O H3P2O7 H3P2O7 3 ATP Dimethylallyldiphosphat Isopentenyldiphosphat Enzymatische Polymerisation n Z-1,4-Polyisopren hschulz, oc2bct 476 10.2 Steroide Wie die Terpene sind auch die Steroide formal aus Isopren-Einheiten aufgebaut Einteilung in Sterole (Sterine, z.B. Cholesterin) Gallensäuren Sexualhormone Corticoide (z.B. Cortison) Grundgerüst der Steroide: Gonan (Steran) hschulz, oc2bct 477 Einige wichtige Steroide * * * * HO * * HO COOH * * HO OH Cholesterin (Cholesterol) OH OH Cholsäure HO Östradiol O Testosteron hschulz, oc2bct HO Vitamin D2 478 10.3 Alkaloide Stickstoffhaltige Naturstoffe mit vorwiegend basischen Eigenschaften (Alkali = Alkaloide!) Liegen in den Pflanzen meist in Form ihrer Carbonsäure-Salze vor (Salze der Oxal-, Essig-, Milch-, Wein-, Citronensäure usw.) Alkaloide haben i.d.R. ein heterocyclisches Ringsystem Sie sind i.d.R. optisch aktiv und in der Natur fast immer linksdrehend Alkaloide zeigen oft bereits in geringsten Konzentrationen physiologische Wirkung und werden als Heilmittel, Rausch- oder Genussmittel eingesetzt Einige Alkaloide sind sehr starke Gifte hschulz, oc2bct 479 Eine kleine Auswahl an Alkaloiden Nicotin Hauptalkaloid in den Blättern und Wurzeln der Tabakpflanze Wirkt in kleinen Mengen anregend auf das Nervensystem, blutdrucksteigernd, wirkt als Neurotransmitter In höherer Dosierung erfolgt Lähmung des Atemzentrums (LD ca. 100 mg) hschulz, oc2bct N H N CH3 (-)-Nicotin (S)-3-[N-Methyl2-pyrrolidyl]pyridin 480 Coniin Äusserst giftiges Alkaloid des Schierlings („Schierlingsbecher“) Piperin des schwarzen Pfeffers, das den scharfen Pfeffergeschmack verursacht Kristalline Substanz (Smp. 129°C), schwer wasserlöslich N H (+)-Coniin (S)-2Propylpiperidin Hauptalkaloid hschulz, oc2bct O O N O Piperin 481 Atropin H3C Alkaloid der Tollkirsche (Racemat) Kristalline Substanz (Smp. 115°C) Antidot gegen Insektizide und Kampfstoffe, die sich von der Phosphorsäure ableiten, im Auge wirkt es pupillenerweiternd Kokain Hauptalkaloid der Blätter des N Kokastrauches Kristalline Substanz (Smp. 98°C) Wirkt lähmend auf die peripheren Nerven (Lokalanesthetikum) Rauschmittel hschulz, oc2bct N OH H O O Atropin CH3 COOCH3 H O O H (2R.3S)-(-)-Kokain 482 Morphin Ein Alkaloid des Opiums Wasserlösliche kristalline H C 3 Substanz (Smp. 254°C) N Das Hydrochlorid wird als sehr starkes Schmerzmittel eingesetzt Längere Einname führt zur Sucht Heroin Synthethisch hergestelltes Diacetylmorphin hschulz, oc2bct H OH O OH Morphin 483 Ergotamin (Prolin) Vertreter der (a-HydroxyAlanin) HO H Mutterkorn-Alkaloide N (Mutterkorn ist ein auf O (PhenylGetreide lebender parasitärer H3C O N alanin) Pilz) HN O Diverse physiologische CH3 Wirkungen (z.B. a-Blocker, N O H Uteruskontraktion) Wichtigster Lysergsäurediethylamid (LSD) Synthetisch herstellbar Ruft im menschen bereits in geringsten Konzentrationen (30 µg) Halluzinationen hervor hschulz, oc2bct (Lysergsäure) N H Ergotamin 484 10.4 Fette, Öle und Wachse Natürliche Fette und Öle sind Glycerinester der höheren geradzahligen Fettsäuren (Glyceride) Tierische Fette enthalten hauptsächlich gemischte Glyceride von Palmitin-, Stearin- und Ölsäure O H2C O C (Palmitinsäure: C16) O HC O C O H2C O C hschulz, oc2bct (Stearinsäure: C18) 9 (Ölsäure: C18´) Triglycerid 485 Pflanzliche Öle enthalten vor allem Glycerinester mit mehrfach ungesättigten Fettsäuren Sonnenblumen-, Soja-, Raps-, Oliven-, Kokos-, Palm-, Lein-, Rizinusöl... Ungesättigte Fettsäuren sind vor allem Öl-, Linol- und Linolensäure (C18´, C18´´, C18´´´) Je höher der Gehalt an ungesättigten Fettsäuren, desto höher ist der Schmelzpunkt von Fetten und Ölen Rindertalg: bei RT fest Schweine-Gänseschmalz: bei RT pastös Walöl, Sojaöl: bei RT niedrig visköse Öle hschulz, oc2bct 486 Gewinnung der Fette und Öle Tierische Fette werden aus dem Gewebe ausgeschmolzen Pflanzliche Öle werden aus den aus den Samenkapseln oder Früchten ausgepresst, oder die gemahlenen Samen werden mit einem organischen Lösungsmittel extrahiert Fetthärtung Die meisten Fette liegen bei RT als Öle vor Durch katalytische Hydrierung der C-C-Doppelbindungen (H2/Ni, 180°C, 5 bar) entstehen Glyceride mit vorwiegend gesättigten Fettsäuren, die bei RT fest sind Margarine: Wasser in Öl-Emulsion von gehärteten Fetten hschulz, oc2bct 487 Fettspaltung (Verseifung) O H2C O C (CH2)16CH3 O HC O C (CH2)16CH3 H2C OH O + 3 NaOH HC OH + + Na O C (CH2)16CH3 O H2C O C (CH2)16CH3 H2C OH Natriumstearat = Kernseife Kaliumstearat = Schmierseife Hydrolytische Spaltung der Fette in Fettsäuren und Glycerin durch überhitzten Wasserdampf im Autoklaven (170°C, basischer Katalysator, z.B: CaO) hschulz, oc2bct 488 Wachse Ester von Fettsäuren mit höheren, primären, einwertigen Alkoholen Myricylpalmitat Bestandteil des Bienenwachses O H3C hschulz, oc2bct (CH2)29 O C (CH2)14 CH3 489 10.5 Kohlenhydrate (Zucker) „Hydrate des Kohlenstoffs“ Cn(H2O)n Es gibt auch natürlich vorkommende Zucker mit einer abweichenden Summenformel, einschliesslich Heteroatome wie z.B: N oder S O O H H * H * HO * H * H OH HO H hschulz, oc2bct OH OH H H C H C OH HO C H H C OH FischerProjektion H C OH CH2OH D(+)-Glucose 490 Einteilung der Kohlenhydrate Zucker können als Oxidationsprodukte von mehrwertigen Alkoholen aufgefasst werden Polyhydroxyaldehyde = Aldosen – Oxidation einer primären OH-Gruppe Polyhydroxyketone = Ketosen – Oxidation einer sekundären OH-Gruppe O H C Aldose H C OH D(+)-Glycerinaldehyd CH2OH hschulz, oc2bct CH2OH C O CH2OH Ketose Dihydroxyaceton 491 Einteilung der Kohlenhydrate Einteilung nach der Anzahl der C-Atome C2-Zucker = Biose (Aldobiose) C3-Zucker = Triose (Aldotriose, Ketotriose) C4-Zucker = Tetrose (Aldotetrose, Ketotetrose) C5-Zucker = Pentose (Aldopentose, Ketopentose) C6-Zucker = Hexose (Aldohexose, Ketohexose) O H C CH2OH Biose O H C H C OH CH2OH Triose hschulz, oc2bct O H C H C OH HO C H CH2OH Tetrose 492 Einteilung der Kohlenhydrate Monosaccharide(Einfachzucker, saccharon = Zucker) Pentosen (C5H10O5) Hexosen – Ribose O H C H C OH H C OH – Glucose (Traubenzucker) – Fructose (Fruchtzucker) O H C H C OH HO C H H C OH H C OH CH2OH H C OH D(-)-Ribose hschulz, oc2bct (C6H12O6) CH2OH D(+)-Glucose CH2OH C O HO C H H C OH H C OH CH2OH D(-)-Fructose 493 Einteilung der Kohlenhydrate Oligosaccharide (Mehrfachzucker) Acetalartige Verknüpfung von 2-6 Monosacchariden Disaccharide – Saccharose (Rohrzucker) – Maltose (Malzzucker) – Lactose (Milchzucker) Tri-, Tetra-, Penta-, Hexasaccharide Polysaccharide Stärke und Glycogen Cellulose Chitin hschulz, oc2bct 494 Die absolute Konfiguration der Kohlenhydrate Zucker haben viele asymmetrische C-Atomen Historisch unterscheidet man zwischen der D- und L-Reihe (R/S-Nomenklatur ist auch möglich) Zuordnung anhand der Lage der von der C=OGruppe am weitesten entfernten OH-Gruppe in der Fischer-Projektion – OH rechts = D-Reihe – OH links = L-Reihe Bezugssystem ist der Glycerinaldehyd hschulz, oc2bct O H C H C OH CH2OH O H C HO C H CH2OH D(+)-Glycerin- L(-)-Glycerin- aldehyd aldehyd 495 O L-Glucose H C H C OH HO C H C OH CH2OH H C HO C H H C OH HO C H HO C H CH2OH O H C C H C H C OH H C OH hschulz, oc2bct O Diastereomere CH2OH D-Manose Enantiomerenpaar H C OH Aldohexosen haben H 4 asymmetrische C-Atome und kommen deshalb in 24=16 Stereoisomeren (8 Enantiomerenpaare) vor O D-Glucose = (2R,3S,4R,5R)-Aldohexose HO L-Glucose = HO (2S,3R,4S,5S)-Aldohexose H Enantiomerenpaar Die Stereoisomere der Aldohexosen D-Glucose H C H C OH H C OH HO C H HO C H CH2OH L-Manose 496 Ringstruktur der Monosaccharide Bei Pentosen und Hexosen ist die intramolekulare Halbacetalbildung begünstigt, da sich spannungsarme 5- oder 6-Ringe bilden können Es entsteht ein zusätzliches asymmetrisches C-Atom: a- und b-Form CH2OH O OH H H OH H C * OH H H OH b-D-Glucose hschulz, oc2bct CH2OH H O H H H OH H C O OH H OH Aldehyd-Form CH2OH O H H H OH H C * OH OH H OH a-D-Glucose 497 Disaccharide Die cyclische Halbacetalform der Monosaccharide kann mit Alkohol zum Acetal weiterreagieren (Glycosid) Als Alkohol kann ein weiterer Monosaccharid fungieren CH2OH CH2OH O H O H H H + OH H H HO CH2OH HO OH OH H OH OH H a-D-Glucose hschulz, oc2bct b-D-Fructose CH2OH O H H a H OH H OH H CH2OH O b O OH H H OH HO CH2OH H Saccharose glycosidische Bindung 498 Polysaccharide CH2OH CH2OH O H H H OH H + OH HO H O H H H OH H 4 1 H OH Maltose a(1,4)-glycosidische Bindung CH2OH CH2OH O H O H H H OH H O H OH OH + n GlucoseEinheiten CH2OH HO O H Glucose H H OH H HO OH O H H H OH H a H OH O H H OH HO CH2OH CH2OH OH hschulz, oc2bct O H H H OH H H OH H O H OH OH n H Stärke (Amylum) OH 499 Polysaccharide Stärke (Amylum, wird von Pflanzen erzeugt) Amylose (20% der Stärke) – Unverzweigte Ketten von a(1,4)-glycosidisch verknüpften D-Glucose-Einheiten (M= 17000-225000 g/mol = 100-1400 Glucose-Einheiten) – Amylose liegt in einer spiralförmigen Helix vor Amylopektin (80% der Stärke) – Verzweigte Ketten von D-Glucose-Einheiten, a(1,4)- und a(1,6)-glycosidisch verknüpft (M = 2x105-106 g/mol) Glycogen (tierische „Reservekohlenhydrate“) Ähnelt dem Amylopektin, die Ketten sind aber noch stärker verzweigt (M = 105-107 g/mol) hschulz, oc2bct 500 Cellulose Ketten von b(1,4)-glycosidisch verknüpften D-Glucose-Einheiten Unverzweigte CH2OH H O H H Chitin OH O H H OH H H H O H CH2OH OH O H OH H OH H H H O H HO H O H H O H OH H CH2OH OH OH n OH CH2OH Unverzweigte Ketten von N-Acetyl-D-glucosaminEinheiten, die b(1,4)-glycosidisch verknüpft sind O O CH2OH O H H HO H NH H3C O hschulz, oc2bct OH H H H O CH2OH OH H H H O H H NH H3C O H O H OH H O CH3 H HN O H H O H OH H CH2OH CH3 H HN n OH CH2OH 501 Vitamin C = L-Ascorbinsäure Natürliches Vorkommen Physiologische Wirkung Hagebutte (250-1400mg/100g) Paprika (ca. 100mg/100g) Citrusfrüchte (ca. 50mg/100g) Verwendung als Antioxidans in der Nahrungsmittelindustrie CH2OH H OH O CH2OH CH2OH H H OH O O H OH +2H H O O OH O -2H O H HO Redox-Reaktionen Cofaktor in vielen Hydroxylierungsreaktionen, z.B. Prolin zu Hydroxyprolin (Verantwortlich für die Stabilität von Kollagen, Mangel: Skorbut!) Tagesbedarf: ca. 75 mg O H O O H Ascorbinsäure hschulz, oc2bct Dehydroascorbinsäure 502 Technische Synthese von Vitamin C H O CH2OH H HO OH H H H +2H OH Pt-Kat. H OH HO OH H H OH H OH CH2OH CH2OH D-Glucose D-Sorbit CH2OH CH2OH Acetobacter suboxydans -2H H HO H OH H O HO OH H O HO Pt-Kat. H HO H OH H CH2OH 2-Keto-L-gulonsäure hschulz, oc2bct H L-Sorbose O HO O HO OH CH2OH CH2OH COOH + O2 / - H 2O H - H2O O + H2O H HO HO H CH2OH L-Ascorbinsäure 503 Grundprinzip der biotechnologischen Herstellung von L-Sorbose Bakterien des Types Acetobacter suboxydans gewinnen Energie durch partielle Oxidation von Alkoholen zu Säuren bzw. Ketone Voraussetzungen Ausreichende Sauerstoffversorgung Geeigneter Temperaturbereich Geeignete Nährlösung Industrielle Durchführung in Bioreaktoren Oberflächenverfahren (diffusionskontrolliert, langsam) Tankverfahren (Submersverfahren) hschulz, oc2bct 504 10.6 Aminosäuren und Proteine Proteine sind hochmolekulare kolloide Naturstoffe, die sich aus a-Aminosäuren zusammensetzen Die Verknüpfung der Aminosäuren erfolgt über die Peptidbindung –CO-NHDer tierische Organismus kann nicht alle zum Aufbau der Proteine benötigten Aminosäuren selbst herstellen. Sie müssen deshalb durch die Nahrung zugeführt werden (=essentielle Aminosäuren) hschulz, oc2bct 505 Proteinogene Aminosäuren Proteine sind aus 20 verschiedenen Aminosäuren aufgebaut Es sind ausschliesslich a-Aminosäuren der L-Reihe COOH COOH H2N H H2N H CH3 H COOH COOH H2N H H H2N CH2 CH3 CH3 H H CH3 CH3 Glycin L(+)-Alanin L(+)-Valin L(-)-Leucin COOH H2N H H3C H CH2 CH3 L(-)-Iso- leucin hschulz, oc2bct 506 Proteinogene Aminosäuren COOH COOH H2N H2N H H H2C OH H OH CH3 L(-)-Threonin L(-)-Serin COOH COOH COOH H2N H2N H2N H CH2 CH2 H H2C SH H H2C S CH3 CH2 L(-)-Methionin CH2 L(+)-Cystein HN L(+)-Arginin COOH H2N H CH2 CH2 COOH CH2 H2C NH2 L(+)-Lysin hschulz, oc2bct H2N H CH2 NH COOH COOH H2N NH2 COOH H2N H L(+)-AsparaginCH2 säure CONH2 L(-)-Asparagin H CH2 CH2 COOH H2N CH2 COOH L(+)-Glutamin- säure H CH2 CONH2 L(+)-Glutamin 507 Proteinogene Aminosäuren COOH H2N COOH COOH H2N H H HN OH H2C H2C L(-)-Phenylalanin L(-)-Tyrosin L(-)-Prolin COOH COOH H2N H H H2N H CH2 CH2 N NH L(-)-Tryptophan hschulz, oc2bct NH L(-)-Histidin 508 Polypeptide a-Aminosäuren bilden unter Polykondensation Polypeptide (Proteine) H R1 + N C C H H R2 H O OH N C C H H H O O N C C + OH R3 H H OH a-Aminocarbonsäuren H - 3 H2O R1 O N C H H C R2 O N C H H C R3 O N C C OH H H Tripeptid hschulz, oc2bct 509 Polypeptide Peptidbindung: planare Struktur H O N C R C H R C H N C H O O N C C R H R + C N C H O Peptidkette H R1 N H H H N H O H N R2 H R3 O H N H O H N R4 H R5 O H N H O Proteine Dreidimensionale Strukturen, bestehend aus meist mehreren Peptidketten (>100 Aminosäure-Einheiten) hschulz, oc2bct 510 Insulin Peptidhormon, das in der Bauchspeicheldrüse gebildet wird Blutzuckersenkende Wirkung Eines der kleinsten Proteinmoleküle mit 51 Aminosäuren und 2 Polypeptidketten S A-Kette S Gly Ile Val Glu Gln Cys Cys Thr Ser Ile Cys Ser Leu Tyr Gln Leu Glu Asn Tyr Cys Asn 1 21 S S S S 1 Phe Val Asn Gln His Leu Cys Gly Ser His Leu Val Glu Ala Leu Tyr Leu Val Cys Gly B-Kette 30 Thr Glu Lys Pro Thr Tyr Phe Phe Gly Arg Aminosäuresequenz von menschlichem Insulin (Schwein: in Position 30 der B-Kette Ala statt Thr) hschulz, oc2bct 511 10.7 Nucleinsäuren Bestandteil aller Zellkerne Der Bauplan der Zellproteine ist in der Sequenz von Nucleinsäuren codiert Nucleinsäuren sind aus Nucleotiden aufgebaute Makromoleküle Das Gerüst ist ein Polyester bestehend aus einer alternierenden Folge von Zuckerresten (Ribose oder Desoxyribose) und Phosphorsäureresten Die einzelnen Nucleotide unterscheiden sich durch die am Zucker sitzende Base hschulz, oc2bct 512 Nucleotide N N OH N N HO P O CH2 N HN H2N N N H H Guanin H O O CH3 HN N O O H NH2 O NH2 N H Cytosin O N H Thymin H OH H Desoxyadenosinmonophosphat Die vier DNA-Basen Purin-Basen: Adenin, Guanidin Pyrimidin-Basen: Cytosin, Thymin hschulz, oc2bct 513 Polynucleotide (Nucleinsäuren) NH2 N N OH N N 5´ P O CH2 O 1´ O H H NH2 H H 3´ H O 5´ HO P O CH2 N N O O 1´ O H H H H 3´ OH H Desoxycytidinmonophosphat Verknüpfung der Nucleotide über die Phosphorsäure HO Ein Zuckerrest ist dabei über die 5´-OHGruppe, der andere Zuckerrest über die 3´OH-Gruppe mit der Phosphorsäure verknüpft Desoxyadenosinmonophosphat Dinucleotid hschulz, oc2bct 514 Struktur der Nucleinsäuren Durch Wasserstoffbrückenbildung zwischen den Basen der NucleotidEinheiten bilden sich Doppelstränge Diese Doppelstränge sind spiralförmig gedreht (a-Helix) hschulz, oc2bct 515
![6.3.1 1-Oxa-spiro[2.5]octan - Institut für Organische Chemie](http://s1.studylibde.com/store/data/001356875_1-96e669e5c88ad586db9f9f199d424d05-300x300.png)