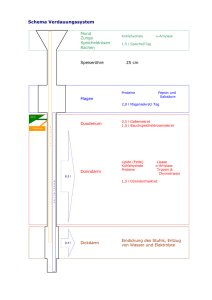
DIPLOMARBEIT
Werbung

FRIEDRICH-SCHILLLER-UNIVERSITÄT JENA BIOLOGISCH-PHARMAZEUTISCHE FAKULTÄT DIPLOMARBEIT zur Erlangung des akademischen Grades Diplom-Biologe Coxsackievirus B3-induzierte Veränderungen des Proteoms der Wirtszelle: Untersuchungen am Zellkulturmodell mit Hilfe der Zweidimensionalen Gelelektrophorese vorgelegt von Marc Carlsohn geboren am 15. Dezember 1975 in Jena Jena, April 2004 Selbständigkeitserklärung Hiermit erkläre ich, die eingereichte Diplomarbeit selbständig verfaßt und keine anderen Hilfsmittel und Quellen als die angegebenen benutzt zu haben. Alle Zitate sind gekennzeichnet, und alle Abbildungen enthalten nur die originalen Daten und sind in keinem Fall inhaltsverändernder Bildbearbeitung unterzogen worden. Alle existierenden Exemplare der vorliegenden Diplomarbeit sind in Wort und Bild übereinstimmend. Jena, den 23.04.2004 Marc Carlsohn Betreuer der Arbeit und als Gutachter eingesetzt: PD Dr. Thomas Munder Als Zweitgutachter bestimmt: Prof. Dr. Susanne Grabley Inhaltsverzeichnis Abkürzungsverzeichnis.........................................................................V 1 2 Einleitung.........................................................................................1 1.1 Das humanpathogene Coxsackievirus B3 ................................................... 1 1.2 Das Proteom .............................................................................................. 5 1.3 Die Zweidimensionale Gelelektrophorese .................................................. 7 1.4 Zielstellung der Arbeit ............................................................................. 12 Material und Methoden ................................................................13 2.1 Material ................................................................................................... 13 2.1.1 Zellkulturen...................................................................................... 13 2.1.2 Verwendeter Virusstamm ................................................................. 13 2.1.3 Medien............................................................................................. 13 2.1.4 Puffer und Lösungen ........................................................................ 13 2.1.4.1 Puffer und Lösungen für die Zellkultur......................................... 13 2.1.4.2 Lösungen für die TCA/Aceton-Fällung......................................... 14 2.1.4.3 Lösungen für die Bradford-Proteinbestimmung ............................ 14 2.1.4.4 Äquilibrierlösungen...................................................................... 14 2.1.4.5 Puffer und Lösungen für die SDS-PAGE (als zweite Dimension der 2D-Gelelektrophorese).......................................... 14 2.1.4.6 Lösungen zum Anfärben der Proteine im PAA-Gel ...................... 15 2.1.4.7 Puffer und Lösungen für den tryptischen In-Gel-Verdau von Proteinen ............................................................................... 15 2.1.4.8 Puffer und Lösungen für RT-PCR ................................................ 16 2.1.4.9 Lösungen für Western Blot........................................................... 16 2.1.5 Synthetische Oligonukleotide für die PCR........................................ 16 2.1.6 Kits, Enzyme und Marker................................................................. 16 2.1.7 Chemikalien ..................................................................................... 17 2.1.8 Materialien....................................................................................... 18 2.1.9 Geräte .............................................................................................. 18 2.2 Methoden................................................................................................. 19 2.2.1 2.2.1.1 Kultivierung der Zellen .................................................................... 19 Kulturbedingungen....................................................................... 19 I 2.2.1.2 Passagieren der Zellen .................................................................. 19 2.2.1.3 Infektion der Zellen mit CVB3H3 ................................................ 20 2.2.1.4 Elektronische Zellzählung und Vitalitätsbestimmung ................... 20 2.2.1.5 Ernte der Zellen............................................................................ 20 2.2.2 Aufarbeitung des Zellysats ............................................................... 20 2.2.2.1 Ultraschallbehandlung .................................................................. 20 2.2.2.2 Ultrafiltration ............................................................................... 21 2.2.2.3 Aceton/TCA-Fällung, verändert nach Görg et al........................... 21 2.2.3 Bradford-Proteingehaltsbestimmung ................................................ 21 2.2.4 Rehydratisierung und Beladung der IPG-Streifen ............................. 22 2.2.5 Isoelektrische Fokussierung.............................................................. 22 2.2.6 Äquilibrierung der IPG-Streifen ....................................................... 23 2.2.7 SDS-PAGE (als zweite Dimension der 2D-Gelelektrophorese)......... 24 2.2.7.1 Herstellung der Gele..................................................................... 24 2.2.7.2 Aufsetzen der IPG-Streifen auf die SDS-Polyacrylamidgele ......... 24 2.2.7.3 Laufbedingungen der SDS-PAGE ................................................ 24 2.2.8 Färben der Polyacrylamidgele .......................................................... 25 2.2.8.1 Coomassie-Blue-Färbung ............................................................. 25 2.2.8.2 Kolloidale Coomassie-Blue-Färbung ............................................ 25 2.2.8.3 Reversible Silberfärbung nach Mann ............................................ 25 2.2.9 2.2.9.1 Auswertung der Gele und Identifizierung einzelner Proteine ............ 26 Bildverarbeitung mit der Proteinanalyse-Software Proteomweaver............................................................................. 26 2.2.9.2 Trypsin-Verdau ausgeschnittener Proteinspots.............................. 27 2.2.9.3 Peptidmassenanalyse mittels SELDI-Technologie ........................ 28 2.2.9.4 Identifizierung der Proteine mit der Online-Anwendung ProFound...................................................................................... 29 3 2.2.10 Reverse Transkription der Gesamt-mRNA (RT-PCR) ...................... 29 2.2.11 Polymerase-Ketten-Reaktion (PCR) ................................................. 30 2.2.12 DNA-Agarosegel-Elektrophorese ..................................................... 31 2.2.13 Western Blot im Semi dry-Verfahren ............................................... 31 Ergebnisse......................................................................................33 3.1 Etablierung der Zweidimensionalen Gelelektrophorese ............................ 33 II 3.1.1 Optimierung einzelner Arbeitsschritte .............................................. 33 3.1.1.1 Vermeidung nachträglicher Proteindegradation ............................ 33 3.1.1.2 Reinigung der Proben ................................................................... 34 3.1.1.3 Vergleich von TCA/Aceton-Fällung und Ultrafiltration ................ 34 3.1.1.4 Proteingehaltsbestimmung............................................................ 35 3.1.1.5 Rehydratisierung der IPG-Streifen................................................ 35 3.1.1.6 Bedingungen der isoelektrischen Fokussierung............................. 36 3.1.1.7 SDS-PAGE-Bedingungen............................................................. 36 3.1.1.8 Vergleich verschiedener Färbemethoden ...................................... 37 3.1.2 Vorgeschlagenes Protokoll für Proteomuntersuchungen des Gesamtzellysats von HepG2-Zellen mittels 2D-Gelelektrophorese........ 38 3.1.3 3.2 Systembedingte Variabilität des Spotmusters einer Probe ................. 39 Nachweis Coxsackievirus B3-induzierter Änderungen des Proteinmusters von HepG2-Zellen ...................................................................... 40 3.2.1 Veränderungen im Proteom von HepG2-Zellen 12 Stunden nach Infektion mit CVB3H3............................................................. 42 3.2.1.1 Abschätzung des experimentellen Fehlers..................................... 44 3.2.1.2 Analyse des Proteinmusters CVB3H3-infizierter HepG2-Zellen und Vergleich mit dem Proteom nichtinfizierter Zellen................. 46 3.2.2 Analyse des ersten Experiments ....................................................... 48 3.2.2.1 Peptidmassenfingerprinting .......................................................... 48 3.2.2.2 Nachweis der infektionsabhängigen posttranslationalen Modifikation eines Proteins .......................................................... 50 3.2.2.3 Bestätigung der infektionsabhängigen Regulation der Mitogen-aktivierten Proteinkinase p38 durch RT-PCR............... 51 3.2.3 3.2.3.1 3.2.4 4 Analyse des zweiten Experiments..................................................... 52 Peptidmassenfingerprinting .......................................................... 52 Übereinstimmend regulierte Proteine................................................ 54 Diskussion ...................................................................................... 55 4.1 Etablierung der Zweidimensionalen Gelelektrophorese ............................ 55 4.1.1 Reproduzierbarkeit der 2D-Gelelektrophorese .................................. 55 4.1.2 Verbesserung des Auflösungsvermögens der Zweidimensionalen Gelelektrophorese ..................................................... 57 III 4.1.2.1 Solubilisierung der Proteine.......................................................... 57 4.1.2.2 Ultrafiltration oder TCA/Aceton-Fällung? .................................... 58 4.1.2.3 Bedingungen der Isoelektrischen Fokussierung ............................ 59 4.2 Coxsackievirus B3-induzierte Änderungen der Proteinausstattung von HepG2-Zellen ....................................................................................... 60 4.2.1 Die Zellkultur als Modell ................................................................. 61 4.2.2 Durch 2D-Elektrophorese detektierte Unterschiede .......................... 61 4.2.2.1 Fehlerraten beider Experimente .................................................... 62 4.2.2.2 Die Mitogen-aktivierte Proteinkinase p38 (MAPK p38) .......... 63 4.2.3 Ausblick........................................................................................... 65 5 Zusammenfassung .........................................................................67 6 Literatur ........................................................................................69 IV Abkürzungsverzeichnis 2D-PAGE zweidimensionale Polyacrylamid-Gelelektrophorese Abb. Abbildung A. bidest. bidestilliertes Wasser ACN Acetonitril A. dest. destilliertes Wasser AP Alkalische Phosphatase APS Ammoniumpersulfat AS Aminosäure(n) BCIP 5-Bromo-4-chloro-3-indolyl-phosphat-4-toluidin bp basepair(s) (Basenpaare) BSA bovine serum albumin (Rinderserumalbumin) CAR Coxsackie- und Adenovirus Rezeptor cDNA complementary DNA (komplementäre DNA) CHAPS 3-(3-Cholamidopropyl)dimethylammonio-1-propansulfat CVB3 Coxsackievirus B3 d day(s) (Tage) DCM dilatative Kardiomyopathie DEPC Diethylpyrocarbonat DMSO Dimethylsulfoxid DNA Deoxyribonucleic acid (Desoxyribonukleinsäure) dNTP Desoxynucleosid-5’-triphosphat dpi dots per inch (Bildpunkte pro Zoll) DTT Dithiothreitol EDTA Ethyldiamintetraessigsäure Ethidiumbromid 3,8-Diamino-5-ethyl-6-phenylphenanthridiumbromid IEF isoelectric focussing (Isoelektrische Fokussierung) IPG immobilized pH-gradient (immobilisierter pH-Gradient) kb Kilobasen kDa Kilo-Dalton LCM Laser Capture Microdissection (Mikrodissektion mit optischer Pinzette) V M Molar MALDI-TOF MS matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (matrixgestützte Laser-Desorption/Ionisations-Massenspektrometrie mit Flugzeitanalysator)) MAPK Mitogen-aktivierte Proteinkinase MDa Mega-Dalton min Minute(n) mind. mindestens MKK MAPK Kinase MKKK MKK Kinase MKS Maul- und Klauenseuche mRNA messengerRNA (Boten-RNA) N Normal NL nichtlinearer pH-Gradient (bei Herstellung der IPG-Streifen wurden bestimmte Bereiche des Gradienten aufgeweitet) OD optische Dichte PAA Polyacrylamid PAGE Polyacrylamid-Gelelektrophorese PBS mit Phosphat gepufferte NaCl-Lösung PCR Polymerase chain reaction (Polymerase-Kettenreaktion) pfu plaque forming units (plaquebildende Einheiten pro Flächeneinheit) pH pH-Wert p.i. post infection (nach Infektion) pI isoelektrischer Punkt RNA Ribonucleic acid (Ribonukleinsäure) rpm rounds per minute (Umdrehungen pro Minute) RT Raumtemperatur RT-PCR Reverse transcriptase-PCR (PCR mit reverser Transkriptase) SDS sodium dodecyl sulfate (Natriumdodecylsulfat) SDS-PAGE SDS-Polyacrylamid-Gelelektrophorese sec Sekunde(n) VI SELDI Surface enhanced laser desorption ionisation (Oberflächenbegünstigte Laser-Desorption/Ionisation) Tab. Tabelle TAE Tris-Acetat-EDTA TBS mit Tris gepufferte NaCl-Lösung TCA Trichloressigsäure TEMED Tetramethylendiamin TNFR Tumor necrosis factor receptor (Tumornekrosefaktorrezeptor) TOF time of flight (Flugzeitanalysator) Tris 2-Amino-2-(hydroxymethyl)-1,3-propandiol U unit (Einheit) v/v volume/volume (Volumen/Volumen) w/v weight/volume (Gewicht/Volumen) VII Einleitung 1 Einleitung 1.1 Das humanpathogene Coxsackievirus B3 Viren sind einfach gebaute Lebensformen mit in der Regel symmetrischer Struktur (helikal oder ikosaedrisch), die ohne ihre Wirtszellen nicht vermehrungsfähig sind. Sie verfügen über zwei Grundbestandteile: ein Nukleinsäuremolekül, die Erbinformation enthaltend, und eine die Nukleinsäure schützende Proteinhülle (Capsid). Das Capsid besteht aus einer definierten Zahl gleichartiger Capsidbausteine, den Capsomeren. Einige komplexere Viren sind zusätzlich von einer äußeren Lipidhülle (envelope) umgeben. Coxsackieviren gehören zur Familie der Picornaviridae, die derzeit ca. 9 Gattungen mit etwa 230 Serotypen umfaßt (Nestler, 2002). Zu den prominenten Vertretern der Picornaviren zählen neben den Coxsackieviren beispielsweise die Maul- und Klauenseuche hervorrufenden MKS-Viren, Rhinoviren, das Hepatitis A-Virus und Polioviren. „Picorna“ setzt sich aus dem griechischen Wort für klein („pikos“) und „RNA“ zusammen. Wie der Name schon sagt, besitzen die Picornaviren ein infektiöses RNAGenom in Plus-Orientierung, was bedeutet, daß die Virusproteine ohne Zwischenschritt von der RNA translatiert werden. In der Vergangenheit wurden Coxsackieviren anhand der unterschiedlichen bei einer Infektion hervorgerufenen Krankheitssymptome in die Subklasssen A mit 23 Serotypen und B mit 6 Serotypen unterschieden (Gauntt et al., 1979). Vor drei Jahren erfolgte eine Neuklassifizierung der Picornaviren auf der Grundlage molekularbiologischer Daten. Nach dieser Systematik werden Coxsackie A-Viren den Spezies Humane Enteroviren Typ A und C zugeordnet, die Coxsackie B-Viren gehören zu den Humanen Enteroviren vom Typ B (King et al., 2000). Das Virusgenom der Coxsackieviren kodiert für ein einzelnes Polyprotein von etwa 2100-2400 Aminosäuren Länge. Darin sind alle Proteine und Informationen enthalten, die das Virus zu seiner Vermehrung benötigt, unter anderem die vier Capsidproteine VP1 bis VP4 sowie einige Nichtstrukturproteine, wie zum Beispiel die RNA-abhängige Polymerase und die Proteasen 2A und 3C. Das Polyprotein wird noch während der Translation proteolytisch in seine einzelnen Komponenten gespalten. 1 Einleitung Zum Replikationszyklus ist folgendes bekannt: Nach der Bindung an einen Zelloberflächenrezeptor („Coxsackie und Adenovirus Rezeptor“, CAR) – ein Protein von 46kDa (LonbergHolm et al., 1976) – werden die Viruspartikel durch Endozytose in das Innere der Zellen aufgenommen. Vermutlich der Verstärkung der AdAbbildung 1-1 Röntgenstrukturanalyse der Virushülle. In den Furchen (cañons) werden die CAR/DAF-Rezeptorbindestellen vermutet. sorption der Viruspartikel an die Zielzellen (Selinka et al., 2002) dient die Bindung eines weiteren Oberflächenproteins, des als Korezeptor funktio- nierenden Decay Acceleration Faktors (DAF oder CD55). In einem „uncoating“ genannten Prozeß wird die Virus-RNA von ihrer Capsid-Proteinhülle befreit und gelangt anschließend durch Poren in der Vesikel-Membran in das Zytoplasma. Aufgrund der Plusstrangorientierung der RNA wird diese dort durch zelleigene Enzyme direkt translatiert. Es entsteht zunächst ein großes Vorläuferprotein, bestehend aus über 1200 AS-Resten, von welchem durch die viruseigene Protease 2A zunächst die vier Strukturproteine als Einheit abgespalten werden. Eine weitere Protease (Protease 2C) zerlegt diese Einheit dann in ihre Bestandteile, die Capsidproteine VP0, VP1 und VP3, wobei sich VP0 anschließend autokatalytisch zu VP2 und VP4 spaltet. Die Protease 2C ist auch für alle weiteren proteolytischen Spaltvorgänge am Polyprotein verantwortlich. Die RNA-abhängige Polymerase, ebenso ein wichtiger Bestandteil des PicornavirusPolyproteins, katalysiert die Synthese des Minus-Stranges der Virus-RNA, welcher wiederum als Matrize zur Replikation des Virusgenoms dient (Lindberg et al., 1987). Sind in einer infizierten Zelle genügend Bausteine für neue Viren vorhanden, werden diese durch „Self-assembly“ zu infektiösen Viruspartikeln zusammengebaut und diese nach außen abgegeben. Coxsackieviren sind säurestabil bis zu pH-Werten unter 3. Dies ermöglicht eine fäkalorale Übertragung, z.B. infolge mangelhafter Hygiene oder verunreinigten Trinkwassers, weil sie die Passage des Verdauungstraktes schadlos überstehen. Nach einer 2 Einleitung primären Vermehrung in den Dünndarmtonsillen gelangen die Viren in das Blut und von dort über infizierte Lymphozyten zu ihren Zielorganen. Coxsackieviren der Gruppe B verursachen neben leichten Erkrankungen des Respirationstraktes mit fiebrigen, grippeähnlichen Symptomen auch schwerwiegende akute und teilweise chronische Krankheitsbilder, die letalen Ausgang haben können. So sind Coxsackieviren der Gruppe B eine der Hauptursachen aseptischer Meningitis und wichtige Auslöser akuter und chronischer Myokarditis sowie akuter Pankreatitis, die zur vollkommenen Insuffizienz führen kann (Ramsingh, 1997). Serologische Untersuchungen zeigen, daß ca. 50% aller Myokarditis-Patienten eine Enterovirus-Infektion hatten bzw. haben (Martino et al., 1994). Die akute Phase einer Myokarditis ist gekennzeichnet durch diffus im Herzmuskel verteilte herdförmige Läsionen mit inflammatorischen Zellinfiltraten. Im Anfangsstadium der Myokarditis werden im Rahmen der unspezifischen Immunantwort Makrophagen und natürliche Killerzellen aktiviert. Später wandern T-Helferzellen und zytotoxische T-Lymphozyten ein (Abb. 1-2) und bilden die spezifische Immunantwort (Maisch et al., 2000). Die akute Erkrankung kann folgenlos ausheilen. Gelingt es allerdings dem Immunsystem nicht, das Virus vollständig zu eliminieren, kann das Virus bei verringerter Virulenz persistieren (Hufnagel et al., 1998). Durch immunologische Kreuzreaktionen sowohl mit viralen als auch mit myokardialen Antigenen kann es zur Chronifizierung der Erkrankung kommen (Schultheiss et al., 1998). In diesem Zusammenhang gibt es Hinweise auf Apoptoseprozesse: Durch Induktion des geordneten zellulären Selbstmordprogrammes wäre das Virus in der Lage, sich, geschützt vor dem Zugriff des Immunsystems, in den apoptoseinduzierten Zellfragmenten – den sogenannten „membrane bound bodies“ – weiterzuverbreiten. So konnte beispielsweise unter in-vitro-Bedingungen die Aktivierung des apoptotischen Proteins Caspase 3 als Folge der Infektion mit CVB3 nachgewiesen werden (Carthy et al., 1998). Zudem wurde in einem Hefe-Zweihybrid-Screening von CVB3-Proteinen mit einer cDNA-Bank aus HeLa-Zellen eine Interaktion zwischen dem Capsidprotein VP2 und dem humanen proapoptotischen Protein Siva nachgewiesen (Henke et al., 2000). An der Ausprägung der viralen Myokarditis – ursprünglich verstanden als überwiegend Immunsystem-vermittelt (Mason et al., 1995) – ist das Virus direkt wohl stärker beteiligt als angenommen. In infizierten Zellkulturen sind starke zytopathische Effekte nachweisbar (Carthy et al., 1998). Der Herzmuskel immunsupprimierter Mäuse 3 Einleitung zeigt ausgedehnte nekrotische Bereiche infolge der Infektion mit CVB3 (Chow et al., 1992). Als wichtige Langzeit-Folge-Erkrankung der akuten/chronischen Myokarditis wird das Krankheitsbild der Dilatativen Kardiomyopathie (DCM) angesehen. Infolge der durch die Infektion verursachten Schäden am Myokard treten fortschreitende Fibrosierungsprozesse auf (Abb. 1-2). Fibroblasten wandern in die nekrotischen Läsionen ein und bilden funktionsunfähige Narben. Die fibrotischen Ventrikelwände unterliegen einer zunehmenden Dilatation, und die Kontraktilität und die Pumpleistung des Herzens verringern sich deutlich. a) gesund b) akut infiziert (7d p.i.) c) gesund d) chronischer Verlauf Abbildung 1-2 Histologischer Befund einer CVB3-Infektion am Herzmuskel der Maus. Obere Reihe: Hämatoxylin-Eosin-Färbung. Gewebezerstörung und Infiltration inflammatorischer Zellen (dunkel angefärbt) in die Gewebsläsionen kennzeichnen die akute Phase. Untere Reihe: Chronifizierung der Erkrankung, fortschreitende Fibrosierung des geschädigten Gewebes (rot angefärbt). Bilder: A. Henke, FSU Jena Die Dilatative Kardiomyopathie ist durch eine hohe Letalität gekennzeichnet. Die Schäden sind oft so gravierend, daß sie zum plötzlichen Herztod des Patienten führen bzw. eine Herztransplantation notwendig machen. Auf die Ausprägung und den Schweregrad der Erkrankung hat neben Alter, Geschlecht und Immunstatus des Wirts auch die Virusvariante im Zusammenspiel mit dem jeweiligen Wirt einen erheblichen 4 Einleitung Einfluß (Leipner et al., 2000). Allein in den USA werden jährlich etwa 100.000 neue DCM-Fälle diagnostiziert (Figulla et al., 1995), in Deutschland sind es etwa 20.000. Obwohl Struktur und Genomorganisation der Coxsackieviren und auch eine Reihe Wirtszellproteine als Interaktionspartner im Verlauf der Infektion gut bekannt sind, sind doch die Pathogeneseprozesse wenig verstanden. Der virale Einfluß auf verschiedene Signalmoleküle wurde in mehreren Fällen schlaglichtartig beleuchtet (Carthy et al., 2003; Henke et al., 2000; Huber et al., 1999). Es ist jedoch wenig bekannt darüber, wie die einzelnen Moleküle im Zusammenspiel am komplexen Krankheitsgeschehen beteiligt sind. Für ein besseres Verständnis des viralen Replikationsverlaufs und des pathologischen Potentials der Coxsackieviren wird intensiv nach weiteren, bisher unbekannten Wirtszellproteinen gefahndet, die als Bindeglieder zwischen einzelnen Signalwegen fungieren. Die Kenntnis solcher Signalmoleküle ist auch von großem therapeutischem Interesse, weil sie in der Wirkstoffentwicklung potentielle Zielobjekte für neuartige Medikamente darstellen. Einen in diesem Zusammenhang vielversprechenden Ansatz beider Suche solcher Bindeglieder stellt die in der Molekularbiologie immer stärker in den Blickpunkt rükkende Proteomforschung dar, deren zentrales Werkzeug die Zweidimensionale Gelelektrophorese ist. 1.2 Das Proteom Der noch relativ junge Begriff proteome (dt. Proteom) wurde auf einer 2DElektrophorese-Tagung 1994 in Siena von dem Australier Marc Wilkins geprägt und ist ein Kunstwort, welches für „PROTEins expressed by the genOME“ steht (Wilkins et al., 1997). Damit gemeint ist die Gesamt-Proteinausstattung einer Zelle, eines Gewebes oder eines Organismus zu einem bestimmten Zeitpunkt. Die Analogie zu den Begriffen Genom und Transkriptom soll dies verdeutlichen. Anders als das Genom einer Zelle ist deren Proteom aber keine feststehende und unveränderliche Größe, sondern von Zelltyp zu Zelltyp verschieden. Zudem wird es von der Zelle in Reaktion auf Signale von außen, wie z.B. sich ändernde Mileubedingungen, ständig angepaßt, indem entbehrliche Proteine abgebaut und benötigte Proteine durch diverse Modifikationen aktiviert oder neu synthetisiert werden. 5 Einleitung Dabei gibt es eine ganze Reihe von Proteinen, kodiert von den sogenannten Haushaltsgenen, die ständig in der Zelle vorliegen. Andere sind (im Falle vielzelliger Organismen) auf bestimmte Gewebe- oder Zelltypen beschränkt, werden als Antwort und Anpassung an äußere Umstände de novo gebildet oder liegen als inaktive Vorstufen in der Zelle vor und werden aktiviert. Wenn man das Proteom einer Zelle letztlich als Momentaufnahme begreift, existieren in der Konsequenz endlos viele Proteome: nämlich für jeden zellulären Zustand genau eines. Bis zum Abschluß des humanen Genomprojektes wurden beim Menschen etwa 100.000-120.000 Gene vermutet. Diese Zahl mußte deutlich nach unten revidiert werden. Tatsächlich umfaßt das menschliche Genom nur ca. 30.000-35.000 Gene. In der Zahl der Gene unterscheiden wir uns damit nur unwesentlich von der Maus, deren Genom auf ebenfalls etwa 35.000 Gene geschätzt wird. Auch die in den Genen von Mensch und Maus verschlüsselte Information ist in erstaunlich hohem Maße übereinstimmend. So wurde von 731 vorhergesagten Genen des Maus-Chromosoms 16 nur für 14 Gene kein Homolog im humanen Genom gefunden, was einer Übereinstimmung von über 98 % auf der Ebene der Gene im untersuchten Umfang gleichkommt (Mural et al., 2002). Inwieweit sich die offensichtlichen phänotypischen Unterschiede zwischen beiden Säuger-Arten aus diesen Zahlen erklären lassen, ist umstritten. Zunehmend rückt deshalb die Ebene der Proteine in den Fokus der Aufmerksamkeit. Denn aus einem Gen können durch alternatives Spleißen und verschiedene posttranslationale Modifikationen (wie z.B. Glycosylierung, Acetylierung, Phosphorylierung) zahlreiche Proteinspezies entstehen. Die Gesamtheit der Proteinspezies des Menschen, sein „theoretisches Proteom“, wird auf etwa 300.000-1.000.000 geschätzt und ist damit weitaus höher als die Zahl der menschlichen Gene (Lottspeich and Zorbas, 1998). Diese Zahl, sowie die raumzeitlichen Expressions-, Modifikations- und Interaktionsmuster der Proteinspezies eines Organismus stellen, so wird vermutet, eine eigene Komplexitätsstufe oberhalb des Genoms dar (Lottspeich and Zorbas, 1998). Der Ansatz der globalen Erforschung des Proteoms in seiner Komplexität und Dynamik, d.h. die Charakterisierung möglichst vieler Proteine, die unter bestimmten Bedingungen in der Zelle vorliegen, hat sich sehr schnell zu einer eigenen Fachrichtung, der Proteomforschung bzw. Proteomik, entwickelt. Das raumzeitliche Expressionsmuster aller Proteine einer Zelle, eines Gewebes oder eines Organismus zu untersuchen und deren Erscheinen oder Verschwinden bestimmten Ereignissen, Zuständen oder Einflüssen zuzuordnen, ist also das Ziel der Proteomforschung. 6 Einleitung 1.3 Die Zweidimensionale Gelelektrophorese Um das komplexe Proteinmuster einer Zelle oder eines Organismus befriedigend darzustellen und reproduzierbar auszuwerten, ist eine sensitive und verläßliche Methodik unumgänglich. Zentraler Bestandteil des Methodenarsenals der Proteomik ist die zweidimensionale Gelelektrophorese. Sie ist derzeit die empfindlichste Methode zur Auftrennung komplexer Proteingemische (Görg et al., 2000). Das erste Mal wurden Proteine in zwei Dimensionen 1956 von Smithies und Poulik getrennt (Smithies and Poulik, 1956). Sie separierten Serum-Proteine mittels einer Kombination aus Papier- und Stärkegel-Elektrophorese. Das der modernen Form der 2D-Elekrophorese zugrundeliegende Prinzip wurde bereits 1975 durch Farrell, Klose und Scheele eingeführt (Klose, 1975; O´Farell, 1975; Scheele, 1975). Es besteht aus der Trennung eines Proteingemisches in zwei voneinander unabhängigen Schritten. Zunächst erfolgt eine Isoelektrische Fokussierung (IEF), bei der die Proteine in einem pH-Gradienten entsprechend ihrer isoelektrischen Punkte separiert werden. Als Isoelektrischer Punkt (pI) ist derjenige pH-Wert definiert, an dem alle positiven und negativen Teilladungen eines Proteins sich aufheben, d.h. seine Nettoladung gleich Null ist. In einem zweiten Schritt erfolgt die Trennung der derart separierten Proteine nach Molekulargewicht durch eine SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE). Dies geschieht senkrecht zur IEF (Abb. 1-3). Das entstehende, durch geeignete Detektionsmethoden (z.B. durch Anfärben der Proteine im Polyacrylamidgel) sichtbar zu machende, zweidimensionale Proteinmuster kann mehrere Tausend Proteine pro Gel umfassen. O´Farell separierte bereits über 1.000 Escherichia coli-Proteine und diskutierte die Nutzung der 2D-Gelelektrophorese zur Darstellung posttranslationaler Modifikationen (O´Farell, 1975). Ein optimales hochauflösendes und reproduzierbares Ergebnis hängt allerdings von sehr vielen Parametern ab, und über die Jahre sind zahlreiche Publikationen erschienen, die sich mit methodischen und gerätetechnischen Verbesserungen zur Erhöhung der Auflösung und der Reproduzierbarkeit beschäftigten. Erst in den letzten Jahren, vor allem bewirkt durch die Verwendung sogenannter Immobiline für die IEF, hat die 2D-Gelelektrophorese stark an Bedeutung gewonnen und ihre derzeitige überragende Stellung in der Proteomforschung eingenommen. Einige wichtige Innovationen auf diesem Wege sollen im Folgenden kurz dargestellt werden. 7 Einleitung Abbildung 1-3 Schematische Darstellung der 2D-Gelelektrophorese. Die Trennung eines Proteingemisches erfolgt zuerst entsprechend der individuellen isoelektrischen Punkte in einem pH-Gradient (erste Dimension – Fokussierung). Die fokussierten Proteine werden anschließend mit SDS beladen, auf ein SDS-Gel übertragen und senkrecht zur Fokussierung nach Molekülmasse getrennt (zweite Dimension). 1970 kombinierten Kenrick und Margolis eine native Isoelektrische Fokussierung mit einer Gradienten-SDS-PAGE, um Serum-Proteine zu separieren (Kenrick and Margolis, 1970). Fünf Jahre später erschienen die schon erwähnten Arbeiten von Klose, O´Farell und Scheele, in denen die 2D-Gelelektrophorese zu ihrer prinzipiell noch heute gültigen Form weiterentwickelt wurde, bei der die IEF unter denaturierenden Bedingungen, d.h. einer Harnstoff-IEF, erfolgt. Zur Erzeugung des für die IEF notwendigen pH-Gradienten wurden Trägerampholyte benutzt. Hierbei erfolgt zunächst die Synthese eines heterogenen Gemisches von Isomeren aliphatischer OligoaminoOligocarbonsäuren. Diese Puffer sind ein Spektrum kleinmolekularer Ampholyte mit eng benachbarten isoelektrischen Punkten. Natürlich vorkommende Ampholyte, wie Aminosäuren und Peptide, eignen sich aufgrund ihrer geringen Pufferkapazität an ihrem pI nicht (Westermeier, 1990). Den pH-Gradienten erzeugt man im elektrischen Feld: Die negativ geladenen Trägerampholyte wandern zur Anode, die positiv gelade- 8 Einleitung nen zur Kathode, wobei die Wanderungsgeschwindigkeit von der Höhe der jeweiligen Nettoladung abhängt. Sie arrangieren sich in der Reihenfolge ihrer isoelektrischen Punkte und geben den entsprechende pH-Wert an ihre Umgebung ab. Auf diese Weise erhält man einen stabilen, monoton steigenden pH-Gradienten. Dieser ist jedoch nicht vollkommen statisch. Vor allem verursacht durch längere Fokussierzeiten, wie sie bei engen Gradienten und in Anwesenheit hochviskoser Additiva wie Harnstoff oder nichtionischer Detergenzien notwendig sind, beginnt der Gradient in beide Richtungen, vor allem in die kathodische („Kathodendrift“), zu driften. In der Mitte entsteht ein Plateau mit Leitfähigkeitslücken. Ein Teil der Proteine, vor allem der basischen, geht verloren. Anfänglich wurde die IEF in Rundgelen durchgeführt. Diese wurden bald ersetzt durch rehydratisierte Flachgele. Hierbei werden zunächst die Gele hergestellt, anschließend getrocknet und erst bei Gebrauch rehydratisiert. Mehrere Vorteile sind damit verbunden: Die leeren Gele können gewaschen werden, APS, TEMED und verbliebene Acrylamid- und Bis-Monomere, welche die Fokussierung stören, können auf diese Weise aus dem Gel entfernt werden. Gele können leicht in größeren Mengen hergestellt und in trockenem Zustand bei -20 °C aufbewahrt werden. In der Rehydratisierungslösung sind dann die zur Erzeugung des pH-Gradienten notwendigen Ampholyte enthalten. Die Gele werden für 30 min vorfokussiert, um die Ampholyte zum pH-Gradienten auszurichten. Anschließend wird die Probenlösung mittels kleiner Silikontrichter („sample cups“) appliziert. Wie bereits erwähnt, war die Einführung der immobilisierten pH-Gradienten (kurz IPG) maßgeblich für die heutige gute Reproduzierbarkeit der 2D-Gelelektrophorese. Bei dieser Technik wird der pH-Gradient aus Acrylamid-Derivaten mit puffernden Gruppen, den Immobilinen, durch Kopolymerisation mit den Acrylamid-Monomeren in ein Polyacrylamidgel eingebaut. Ein Immobiline (Abb. 1-4) ist eine schwache Säure oder eine schwache Base. Man braucht mindestens zwei verschiedene Immobiline: eine Säure und eine Base. Ein pH-Gradient im Gel wird durch kontinuierliches Verändern des Immobiline-Mischungsverhältnisses erzielt, nach dem Prinzip einer SäureBase-Titration (Görg et al., 1987; Westermeier, 1990). In der Praxis werden immobilisierte pH-Gradienten durch lineares Mischen von zwei verschiedenen Polymerisationslösungen mit einem Gradientenmischer hergestellt. Der pH-Gradient bleibt absolut linear während der IEF und kann nicht durch die Proteine der Probe beeinflußt werden. Die Beladungskapazität der Gele ist dadurch deut9 Einleitung lich erhöht (Bjellqvist et al., 1993). Der Gradient ist vollkommen zeitstabil und kann nicht driften. Im Gegensatz zu mehreren 100 Trägerampholyt-Homologen wird der immobilisierte pH-Gradient mit wenigen, chemisch exakt definierten Substanzen erzeugt. H2C = CH–CO–NH–CH2–COOH pK = 3,6 H2C = CH–CO–NH–(CH2)2–N(C2H5)2 pK > 12 Abbildung 1-4 Beispiel für zwei Immobiline. Die Acrylamidgrundstruktur ist rot dargestellt. Die Technik der rehydratisierten Flachgele wird auch hier angewendet. Man stellt zwei Lösungen her: eine saure, durch Glycerin beschwerte, und eine basische, leichte. Die saure Lösung ist der saure, die basische der basische Endpunkt des pHGradienten. Auf eine Glasplatte der Gießkassette wird eine Trägerfolie aufgewalzt, beide Lösungen werden in einen Gradientenmischer mit Mischkammer und Reservoir gegeben, und die Gießkassette wird befüllt. Beim Gießen entsteht der lineare Dichtegradient durch kontinuierliche Überschichtung der immer leichter werdenden Gellösung. Nach seiner Polymerisation wird das Gel gewaschen, anschließend getrocknet und kann nun zu schmalen Gelstreifen zugeschnitten werden. Durch die Trägerfolie ist die Stabilität der Gelstreifen gewährleistet. Entscheidend für den Erfolg der 2D-Gelelektrophorese ist die Probenbehandlung. Neben einem schnellen und gründlichen Zellaufschluß, der z.B. mechanisch durch Mörsern, French pressing sowie Ultraschallbehandlung oder chemisch durch verschiedene Lysispuffer erfolgen kann, ist die Inhibierung zelleigener Proteinasen wichtig, um nachträgliche Proteindegradation oder -veränderung zu verhindern. Für ein hochauflösendes Ergebnis wie in Abb. 1-5 und die umfassende Darstellung möglichst vieler Elemente des Proteoms ist es notwendig, Proteine mit stark unterschiedlichen Eigenschaften gemeinsam in Lösung zu bringen. Weil schlecht gelöste Proteine die Gelporen verstopfen und zu horizontalen Streifen im 2D-Gel führen (Görg et al., 2000), werden nur vollständig gelöste Proteine ordnungsgemäß fokussiert. Dieses Problem ist Gegenstand vieler Arbeiten (Chevallet et al., 1998; Luche et al., 2003; Rabilloud, 1996; Rabilloud, 1999; Rabilloud et al., 1997; Rabilloud et al., 1990; Santoni et al., 10 Einleitung 2000; Vuillard et al., 1995). Besonders Proteine mit extremen isoelektrischen Punkten wie z.B. Histone, bzw. stark hydrophobe (Membran-)Proteine sind schwer löslich. Abbildung 1-5 Hochauflösende 2D-Gelelektrophorese unter Verwendung von IPG-Gelstreifen (pH-Bereich 4-7). Generell basieren die meisten Solubilisierungslösungen nach wie vor auf dem von O´Farell empfohlenen Lysispuffer, der eine hochmolare Harnstofflösung mit einem Detergenz (Triton X 100) und einem Reduktionsmittel (2-Mercaptoethanol) kombiniert. Abwandlungen ergeben sich aus der Einführung neuartiger wirksamerer nichtionischer bzw. zwitterionischer Detergenzien (z.B. aus der Gruppe der Sulfobetaine), chaotroper Substanzen wie Thioharnstoff und besser geeigneter Reduktionsmittel wie DTT. Die Aufrechterhaltung eines reduzierten Zustandes der Proteine während der Fokussierung ist deshalb wichtig, weil die Reoxidation von SH-Gruppen zu SHBrückenbildung und begleitend zu Löslichkeitsverlusten führen kann. Um Proteine, die nur in wenigen Kopien in der Zelle vorliegen, neben stark exprimierten Proteinen überhaupt darstellen zu können oder wenn nur ein Teil der Proteine einer Zelle interessiert (z.B. ribosomale Proteine), ist eventuell eine Vorfraktionierung der Probe sinnvoll. Eine Vorfraktionierung kann beispielsweise erreicht werden durch Ultrazentrifugation, präparative Zellelektrophorese oder Affinitätschromatographie. 11 Einleitung 1.4 Zielstellung der Arbeit Coxsackievirus B3, zur Familie der Picornaviridae gehörig, gilt als einer der häufigsten Erreger viraler Herzerkrankungen. Die Pathogenesemechanismen und VirusWirtszell-Interaktionen sind bisher allerdings nur ansatzweise aufgeklärt. Bis heute konnte noch keine geeignete Therapie oder Prävention bis zur klinischen Anwendung entwickelt werden. Im Rahmen dieser Arbeit sollten deshalb mit Hilfe der Zweidimensionalen Gelelektrophorese am Zellkulturmodell durch Proteinmustervergleich Wirtszellproteine identifiziert und nach Möglichkeit durch Peptidmassen-Fingerprinting am Massenspektrometer charakterisiert werden, die im Verlauf der Infektion differentiell exprimiert werden. Proteine, deren Abundanzen während des Infektionsgeschehens reguliert werden, sind einerseits potentiell (mittelbar bzw. unmittelbar) an Vorgängen bei der Virusreplikation beteiligt, andererseits können sie diese unter Umständen stören und werden deshalb inaktiviert. Der Überblick über solche Proteine wird helfen, einzelne bereits bekannte Details des Infektionsgeschehens in Zusammenhang zu stellen und bedeutet somit eine gute Ausgangsbasis zur Entflechtung der komplizierten Signalfolgen während der Virusreplikation. Für dieses Ziel sollte zunächst die Methode der 2D-Gelelektrophorese etabliert und für die Arbeit am Zellkulturmodell optimiert werden. Die Zweidimensionale Gelelektrophorese, die als die empfindlichste Methode zur Auftrennung komplexer Proteingemische gilt, ist in der Lage, die wichtigsten Komponenten eines beliebigen Proteoms befriedigend abzubilden. Aufgrund der Heterogenität, die solch ein Proteom aufweist (verschiedenste Abundanzen, Größen, Hydrophobizitäten und isoelektrische Punkte der enthaltenen Proteine), ist es jedoch nahezu unmöglich, diese Proteine alle gleichermaßen gut darzustellen. Die verläßliche Reproduzierbarkeit der Methode ist zudem von vielen Parametern (unter anderem der Probenbehandlung, den IEF- und SDS-PAGE-Bedingungen und der Färbemethode) abhängig. Für die geplanten Proteomuntersuchungen am Zellkulturmodell sollte daher ein Protokoll entwickelt werden, das bei einfacher Handhabung möglichst hohe Auflösung und gute Reproduzierbarkeit gewährleistet. 12 Material und Methoden 2 Material und Methoden 2.1 Material 2.1.1 Zellkulturen HepG2: humane hepatozelluläre Karzinomzellinie, ATCC-Nr. HB-8065, 1975 etabliert (Aden et al., 1979), epithelartige Zellen, adhärent als Monolayer wachsend, Verdopplungszeit 48-60 h, Ausbeute der konfluenten Kultur ca. 4 x 105 Zellen/cm2 Wachstumsfläche HeLa: humane Cervix-Karzinom-Zellinie, ATCC-Nr. CCL 2, 1951 etabliert (Gey, 1952), epithelartige Zellen, adhärent als Monolayer wachsend, Verdopplungszeit 48-60 h, Ausbeute der konfluenten Kultur ca. 5 x 104 Zellen/cm2 Wachstumsfläche 2.1.2 Verwendeter Virusstamm CVB3 Stamm „Nancy“, Variante Woodruff (CVB3H3), Titer: 1,6 x 108 pfu/ml (Knowlton et al., 1996) 2.1.3 Medien HepG2: 90 % RPMI 1640 + 10 % FCS, 4 mM L-Glutamin HeLa: 90 % RPMI 1640 + 10 % FCS, 2 mM L-Glutamin 2.1.4 Puffer und Lösungen 2.1.4.1 Puffer und Lösungen für die Zellkultur 10x Trypsin/EDTA-Lösung: 0,5 % (w/v) Trypsin, 0,2 % (w/v) EDTA PBS-Puffer: 10 mM PBS in Tablettenform, A. bidest. Lyse- und Solubilisierungs- 8 M Harnstoff, 2 M Thioharnstoff, 4 % CHAPS, puffer: 40 mM DTT Rehydratisierungslösung: 8 M Harnstoff, 2 M Thioharnstoff, 4 % CHAPS, 12 l/ml DeStreak™ 13 Material und Methoden 2.1.4.2 Lösungen für die TCA/Aceton-Fällung TCA-Lösung: 20 % (w/v) TCA in 50 % Aceton lösen, 20 mM DTT Aceton/DTT: 80 % Aceton, 20 mM DTT Rehydratisierungslösung: 8 M Harnstoff, 2 M Thioharnstoff, 4 % CHAPS, 12 l/ml DeStreak™ 2.1.4.3 Lösungen für die Bradford-Proteinbestimmung Bradford-Stammlösung: 100 ml Ethanol 96 %, 200 ml Phosphorsäure 88 %, 350 mg Serva Blue G, bei RT abgedunkelt lagern Bradford-Arbeitslösung: 425 ml A. bidest., 15 ml Ethanol 96 %, 30 ml Phosphorsäure 88 %, 30 ml Bradford-Stammlösung; bei RT und abgedunkelt 6 Wochen haltbar BSA-Lösung: 1 mg/ml BSA, A. bidest. 2.1.4.4 Äquilibrierlösungen Stammlösung: 6 M Harnstoff, 4 % SDS, 30 % Glycerin, 3,3 % Trenngelpuffer pH 8,8 Lösung I: Stammlösung mit 1 % DTT versetzen Lösung II: Stammlösung mit 4 % Jodacetamid und einigen mg Bromphenolblau versetzen 2.1.4.5 Puffer und Lösungen für die SDS-PAGE (als zweite Dimension der 2DGelelektrophorese) 10x Elektrodenpuffer 144 g Glycin, 30 g Tris, 10 g SDS ad 1.000 ml A. bidest. Trenngelpuffer: 181,66 g TRIS, 4 g SDS in 900 ml A. bidest. lösen, mit konz. HCl auf pH 8,8 titrieren, ad 1.000 ml mit A. bidest. Acrylamidlösung (10 %): 42 % (v/v) Acrylamid-Bis (37,5:1, 30 %), 25 % (v/v) Trenngelpuffer, 28 % (v/v) A. bidest., 0,56 M Glycerol 0,5 % (w/v) APS, 0,005 % (v/v) TEMED Glycerol-Lösung zum Un- 50 % (v/v) Glycerol, 25 % (v/v) Trenngelpuffer, 25 % terschichten: A. bidest., eine Spatelspitze Bromphenolblau Agaroselösung 0,5 %: 0,5 g Agarose in 100 ml 1x Elektrodenpuffer lösen 14 Material und Methoden 2.1.4.6 Lösungen zum Anfärben der Proteine im PAA-Gel Coomassie-Blue-Färbung Fixierlösung: 20 % Methanol, 1 % o-Phosphorsäure (85 %) A. bidest. Färbelösung: 0,1 % Coomassie, 10 % Essigsäure, 40 % Methanol, A. bidest. Entfärber: 10 % Essigsäure, A. bidest. Kolloidale Coomassie-Blue-Färbung Fixierlösung: 20 % Methanol, 1 % o-Phosphorsäure (85 %) A. bidest. Färbelösung: 20 % Methanol, 20 % Roti®-Blue (5x-Konz.), gut geschüttelt, A. bidest. Waschlösung: 20 % Methanol, A bidest. Lagerungslösung: 20 % Methanol, 5 % Glycerin (99 %), A. bidest. Silberfärbung nach Mann Fixierer: 40 % Ethanol, 10 % Essigsäure, A. bidest. Waschlösung: 30 % Ethanol, A. bidest. Sensitivierer: 0,02 % Natriumthiosulfatpentahydrat (NA2S2O3 x 5H2O), A. bidest. Färbelösung: 0,2 % Silbernitrat (AgNO3), 0,025 % Formaldehyd (HCHO), A. bidest. Entwickler: 3 % Natriumcarbonat, 0,05 % Formaldehyd, A. bidest. Stopplösung: 5 % Essigsäure, A. bidest. 2.1.4.7 Puffer und Lösungen für den tryptischen In-Gel-Verdau von Proteinen Puffer A: 50 % Methanol, 10 % Essigsäure, A. bidest. Puffer B: 100 mM Ammoniumbicarbonat, pH 8, A. bidest. Puffer C: 50 % Acetonitril, 100 mM Ammoniumbicarbonat, A. bidest. Puffer D: 100 % Acetonitril 10x Trypsin-Stammlösung: Trypsin in einer Konzentration von 0,1 mg/ml in 1 mM HCl lösen, Aliquots herstellen, bei -80 °C lagern Trypsin-Arbeitslösung Stammlösung mit 25 mM Ammoniumbicarbonat- (1x–5x): Puffer, pH 8 (Puffer B 1:4) verdünnen, sofort verbrauchen 15 Material und Methoden 2.1.4.8 Puffer und Lösungen für RT-PCR DEPC-H2O 0,1 % DEPC, A. bidest., 24 h rühren, 5 x autoklavieren 2.1.4.9 Lösungen für Western Blot Towbin-Blotpuffer 3 g Tris, 14,4 g Glycin, 0,05 % (w/v) SDS, ad 1 l A. bidest. TBS-T 20 mM Tris (pH 7,5), 150 mM NaCl, 0,1 % (v/v) Tween 20 Blockierlösung TBS-T mit 5 % (w/v) Magermilchpulver Ponceau-Rot-Färbelösung 0,1 % Ponceau-Rot in 5 % Essigsäure (v/v) AP-Puffer 100 mM Tris-HCl (pH 9,5), 100 mM NaCl, 5 mM MgCl2 AP-Färbelösung 66 l NBT-Stocklösung (5 % in DMFA), 33 l BCIPStocklösung (5 % in 70 % DMFA), 9,9 ml AP-Puffer 2.1.5 Synthetische Oligonukleotide für die PCR Name Sequenz Herkunft SivaF 5'-ATGCCCAAGCGGAGCTGCCCCTTCG-3' JenaBioscience SivaR 5'-TCAGGTCTCGAACATGGCACAGCTG-3' JenaBioscience ProF 5'-ATGGCTGCCAAAGTGTTTGAGTCCA-3' JenaBioscience ProR 5'-TCACTGGGGCAGCTGGAGGAGCACG-3' JenaBioscience DynF 5'-ATGCCCAGTGAAACTCTCTGGGAAA-3' JenaBioscience DynR 5'-AATCCCAAACAGCAACTCACAGGATGATCC-3' JenaBioscience p38F 5'-ATGTCTCAGGAGAGGCCCACGTTCT-3' JenaBioscience p38R 5'-TCAGGACTCCATCTCTTCTTGGTCA-3' JenaBioscience 2.1.6 Kits, Enzyme und Marker RNeasy Micro Kit (Quiagen, Hilden) Omniscript RT Kit (Quiagen, Hilden) Taq DNA Polymerase (Quiagen, Hilden) Protein- und DNA-Marker (Roth, Karlsruhe und Biorad, München) 16 Material und Methoden 2.1.7 Chemikalien Acetonitril Merck, Darmstadt Acrylamid-Bis 37,5:1 Serva, Heidelberg Agarose Invitrogen, Karlsruhe Ammoniumpersulfat (APS) Serva, Heidelberg Bromphenolblau Sigma, Deisenhofen CHAPS (3-(3-Cholamidopropyl)dimethylammonio-1-propansulfat) Sigma, Deisenhofen Coomassie Brilliantblau G250 Sigma, Deisenhofen Destreak™ Amersham Biotech, Freiburg Dimethylsulfoxid (DMSO) Roth, Karlsruhe dNTP´s Amersham Biotech, Freiburg DTT Sigma, Deisenhofen Ethidiumbromid (1%) Merck, Darmstadt Ethylendiamintetrasessigsäure (EDTA) Roth, Karlsruhe Fetales Kälber-Serum (FCS) Biochrom AG, Berlin Formaldehyd Roth, Karlsruhe Glycerin (plant, 99,5 %) Serva, Heidelberg Glycin Roth, Karlsruhe Guanidinhydrochlorid Sigma, Deisenhofen Harnstoff (EP-MB grade) Roche Diagnostics, Mannheim Jodacetamid Sigma, Deisenhofen L-Glutamin Sigma, Deisenhofen Natriumcarbonat Roth, Karlsruhe Natriumthiosulfatpentahydrat Merck, Darmstadt ortho-Phosphorsäure (85%) Merck, Darmstadt Pharmalyte™ (3-10, 4-6,5) Amersham Biotech, Freiburg Phosphate buffered saline (PBS; tablets) Sigma, Deisenhofen Roti®-Blue Roth, Karlsruhe RPMI 1640, Zellkulturmedium Gibco, USA SDS Roth, Karlsruhe Silbernitrat Sigma, Deisenhofen Silikonöl DC 200 Serva, Heidelberg 17 Material und Methoden TAE Buffer (50x) Invitrogen, Karlsruhe TEMED Merck, Darmstadt Thioharnstoff Sigma, Deisenhofen Trichloressigsäure (TCA) Merck, Darmstadt Triflouressigsäure (TFA) Merck, Darmstadt Tris Roth, Karlsruhe Trypsin (sequencing grade) Roche Diagnostics, Mannheim 2.1.8 Materialien Einmal-Küvetten 1,5ml (Roth, Karlsruhe) IPG-Streifen 24 cm; pH 4-7, 3-10, 3-10NL (Amersham Biosciences, Schweden) Kulturflaschen, div. Größen (Iwaki, Japan) Ultrafiltrationssystem Microcon-YM 10 (Millipore, USA) SELDI-Proteinchips: H4 und H50, hydrophobe Oberfläche (Ciphergen, USA) Immobilon™-P Transfer Membran (Millipore, USA) 2.1.9 Geräte Begasungsbrutschrank BB 6220 (Heraeus Instruments, Hanau) Concentrator 5301 (Eppendorf, Hamburg) Densitometer GS 800 (Bio-Rad, München) Elektrophorese- und Blotapparatur Multiphor™ II (Amersham Biosciences, Schweden) Geldokumentationsanlage Gene Genius, SynGene; Software: GeneSnap (Merck Eurolab, Darmstadt) Horizontale Gelelektrophorese-Apparatur Power Pac 200 (Bio-Rad, München) Laborzentrifuge 4K15C (Sigma, Taufkirchen) Mastercycler® gradient (Eppendorf, Hamburg) Mikroskop Axiovert 25 inkl. CCD-Kamera AxioCam HRC (Carl Zeiss GmbH, Jena) PCR-Gerät GeneAmp PCR-System 9700 (Perkin Elmer, Langen) pH-Meter CG825 (Schütt Labortechnik, Göttingen) Powersupply Powerpac 200 (Bio-Rad, München) Powersupply EPS 3501 XL (Amersham Biosciences, Schweden) 18 Material und Methoden Proteinspot-Excisionsgerät GelPal (Genetix, Großbritannien) Proteinanalysesoftware Proteomweaver (Definiens, München) Rehydratisierungstray (Amersham Biosciences, Schweden) Schüttelgerät Promax 1020 (Heidolph Instruments, Schwabach) SELDI-MS Protein-Chip-System (Ciphergen, USA) Steril-Sicherheitswerkbank HB 2448 (Holten LaminAir) Thermomixer comfort (Eppendorf, Hamburg) Tischzentrifuge 5415R (Eppendorf, Hamburg) Ultraschall-Gerät sonoplus HD 2070 mit Mikrospitze MS 73 (Bandelin electronic, Berlin) Ultrazentrifuge Optima MAX E (Beckman Coulter, USA) UV/VIS-Spektrometer Spectronic® 20 Genesys™ (Spectronic Instruments, USA) Vertikal-Gelelektrophoreseapparatur EttanDALTtwelfe™ inkl. Gelgießstand für 12 Gele (Amersham Biosciences, Schweden) Zellzahl- und Vitalitätsmeßgerät Vicell™ (Beckman Coulter, USA) 2.2 Methoden 2.2.1 Kultivierung der Zellen 2.2.1.1 Kulturbedingungen Die adhärent wachsenden Kulturen wurden in den entsprechenden Medien bei 37 °C, 5 % CO2 und gesättigt feuchter Atmosphäre im Begasungsbrutschrank kultiviert. Sämtliche Arbeiten wurden unter sterilen Bedingungen an einer Sterilwerkbank durchgeführt. 2.2.1.2 Passagieren der Zellen Subkonfluente Kulturen (ca. 95 % Konfluenz) wurden mit kalter PBS/EDTA-Lösung gewaschen, um tote Zellen und Reste des Kulturmediums zu entfernen. Anschließend wurde die Zellschicht mit vorgewärmter Trypsin/EDTA-Lösung benetzt (1 ml für 25 cm2, 3 ml für 75 cm2 und 5 ml für 150 cm2 Wachstumsfläche). Nach 3-minütiger Inkubation im Brutschrank wurde die Trypsinlösung abgegossen und die Zellen für maximal 10 weitere Minuten inkubiert. Lösten die Zellen sich vom Flaschenboden, wurde die Reaktion durch Zugabe von 2/5/8 ml auf 37 °C vorgewärmten Nährmediums abgestoppt. Durch mehrmaliges Pipettieren wurden die Zellen homogenisiert, 19 Material und Methoden vereinzelt, aufgenommen und im Verhältnis 1:4 in neue Flaschen verteilt (gesplittet). Abschließend wurden die Flaschen mit auf 37 °C vorgewärmtem Nährmedium aufgefüllt (25 cm2 auf 20 ml, 75 cm2 auf 40 ml und 150 cm2 auf 60 ml) und kultiviert wie beschrieben. 2.2.1.3 Infektion der Zellen mit CVB3H3 Diese Arbeiten wurden im Institut für Virologie der FSU Jena von Dr. A. Henke und Mitarbeitern durchgeführt. Von konfluenten Zellkulturen wurde zunächst das Medium entfernt. Anschließend erfolgte die Infektion mit 1-2 ml Virus-Suspension, die auf dem Zellrasen verteilt wurden. Um eine Adhäsion der Virionen an die Zellen zu ermöglichen, erfolgte eine 45-minütige Inkubation bei 37 °C. Regelmäßiges Schwenken verhinderte, daß der Zellrasen austrocknete. Schließlich wurde frisches Medium zugefügt und bei 37 °C weiterinkubiert. 2.2.1.4 Elektronische Zellzählung und Vitalitätsbestimmung Für die elektronische Zellzahl- und Vitalitätsbestimmung wurden die Zellen mit Trypsin/EDTA-Lösung vom Flaschenboden abgelöst (10 min bei 37 °C), die Zellsuspension bei 500 x g und 4°C für 5 min zentrifugiert und das Zellpellet in einem definierten Volumen PBS-Puffer resuspendiert. Mit Hilfe von Trypanblau wurde die Vitalität der Zellen sowie die Zellzahl im ViCell-Zellzähler bestimmt. 2.2.1.5 Ernte der Zellen Die Kulturflaschen wurden dem Brutschrank entnommen und der Zellrasen sofort viermal mit 4 °C kaltem PBS-Puffer gewaschen. Reste des Puffers wurden möglichst vollständig abgesaugt. Um die Zellen vom Flaschenboden zu lösen und zu lysieren, wurden diese mit 2 ml Solubilisierungslösung (bei 75 bzw. 150 cm2 Wachstumsfläche, 1 ml bei 25 cm2) versetzt. Der Puffer wurde zügig auf dem Flaschenboden verteilt, so lange, bis keine Zellen mehr zu erkennen waren. Da beide Puffer stark denaturierend wirken, werden in einem hohen Maße in der Probe enthaltene Proteinasen inhibiert. Dies ist wichtig, weil man nachträgliche Veränderungen am Proteom vermeiden möchte. 2.2.2 Aufarbeitung des Zellysats 2.2.2.1 Ultraschallbehandlung Die im Puffer aufgenommenen Zellen wurden in ein 15 ml Falcon-Reaktionsgefäß überführt und mit Ultraschall behandelt (10 x 5 sec bei Puls 2 und 50 %). Dies ge20 Material und Methoden schah vorrangig, um Membranreste und Nukleinsäuren mechanisch zu destruieren und daran assoziierte Proteine abzulösen. 2.2.2.2 Ultrafiltration Zunächst wurde das ultraschallbehandelte Lysat für 30 min bei 75.000 x g und 16 °C ultrazentrifugiert, um unlösliche Zellfragmente zu pelletieren. Der Überstand wurde abgenommen und jeweils 500l davon in einen Mikrofilter (regenerierte Zellulose, MW 10.000) gegeben, durch Zentrifugation (12.000 x g 16 °C) auf die Hälfte eingeengt und mit Rehydratisierungslösung auf das Ausgangsvolumen aufgefüllt. Dieser Waschvorgang wurde zweimal wiederholt. 2.2.2.3 Aceton/TCA-Fällung, verändert nach Görg et al. (Görg et al., 1997) Das ultraschallbehandelte Lysat wurde im Volumenverhältnis 2:1 mit TCA-Lösung vermischt und anschließend für 30 min bei -20 °C gefällt, gut geschüttelt und bei 4 °C für weitere 2 h gefällt. Der Überstand wurde anschließend für 30 min bei 16.000 x g und 4 °C abzentrifugiert, abgenommen und verworfen. Das Sediment wurde zweimal mit 1 ml -20 °C kaltem 80%igem Aceton/DTT gewaschen, indem das Pellet im Aceton/DTT durch Pipettieren homogenisiert und anschließend für 15 min bei 16.000 x g und 4 °C abzentrifugiert wurde. Das Aceton/DTT wurde abgenommen, das Pellet im Vakuum 10 min getrocknet, wieder in Rehydratisierungslösung aufgenommen, eine Stunde bei 25 °C und 500 rpm auf einem Thermomixer und über Nacht bei 4 °C inkubiert, um die Proteine möglichst quantitativ in Lösung zu bekommen. Unlösliche Bestandteile wurden durch 15-minütige Ultrazentrifugation bei 75.000 x g und 16 °C entfernt. 2.2.3 Bradford-Proteingehaltsbestimmung (Bradford, 1979) Für den quantitativen Vergleich des Proteinspotmusters ist die Ermittlung der Proteinkonzentration der zu vergleichenden Proben notwendig, um sicherzustellen, daß gleiche Proteinmengen auf die Gele geladen werden. Die Ermittlung des Proteingehaltes erfolgte mit dem Bradfordtest (Bradford, 1979). Aus der BSA-Lösung wurde eine Verdünnungsreihe mit Lösungen der Endkonzentrationen 1-6 g BSA/ml hergestellt: dazu wurden je 100 l A. bidest. auf die jeweiligen BSA-Mengen eingestellt und 900 l Bradford-Arbeitslösung hinzupipettiert. Die Bestimmung der Proteinkonzentration einer Probe erfolgte durch Zugabe von 1 l, 2 l und 4 l Probenlösung zu A. bidest., so daß auch hier Lösungen zu je 100 l 21 Material und Methoden Volumen vorlagen, die mit 900 l Bradford-Arbeitslösung zu 1 ml aufgefüllt wurden. Die Inkubationszeit betrug 15 min bei RT, danach wurde die Extinktion bei 595 nm bestimmt. Aus der Verdünnungsreihe wurde eine Eichkurve erstellt, aus der sich die Proteinkonzentrationen der vermessenen Proben errechnen bzw. ablesen ließen. Für jede unabhängige Messung wurde separat eine Eichkurve erstellt. 2.2.4 Rehydratisierung und Beladung der IPG-Streifen Bei der sogenannten aktiven Rehydratisierung wird die Probe im Zuge der Rehydratisierung auf die Streifen geladen. Aus den Proteingehalten der Proben ließen sich definierte Proteinmengen berechnen, mit denen die IPG-Streifen beladen wurden. Zur Rehydratisierung der 24 cm langen Streifen wurde ein Volumen von 500 l benötigt. Das heißt, die gewünschte Proteinmenge wurde mit Rehydratisierungslösung auf ein Gesamtvolumen von 500 l verdünnt. Die Probenlösungen wurden in ein Rehydratisierungstray pipettiert, die Gele von der Schutzfolie befreit und, mit der Gelseite nach unten zur Probenlösung zeigend, blasenfrei auf diese aufgelegt. Um Verdunstungserscheinungen zu minimieren wurden die Gele mit Silikonöl überschichtet. Die getrockneten und bei -20 °C gelagerten IPG-Streifen wurden mindestens über Nacht, in der Regel aber für 24 h, rehydratisiert. 2.2.5 Isoelektrische Fokussierung Die isoelektrische Fokussierung der Proteine erfolgte mit dem Multiphor™ II-System. Dieses besteht aus einer Kühlplatte, einer darauf liegenden Fokussierkammer, welche die Elektroden trägt und einer darin liegenden Schablone, deren Vertiefungen der korrekten Ausrichtung der IPG-Streifen dienen. Um eine optimale Wärmeableitung zu gewährleisten, wurden zwischen Fokussierkammer und Kühlplatte sowie zwischen Schablone und Fokussierkammer einige ml Silikonöl verteilt und die Fokussierkammer bzw. die Schablone möglichst luftblasenfrei aufgelegt. In die Vertiefungen der Schablone wurden die IPG-Streifen mittig, mit dem sauren Ende zur Anode zeigend, angeordnet und gegeneinander ausgerichtet. Auf die Enden der IPG-Streifen wurden restfeuchte Elektrodenpapierstreifen quer aufgelegt, so daß sie die äußeren IPG-Streifen ein kurzes Stück überragten. Auf die Filterpapierstreifen wurden dann die Elektrodenbrücken mittig aufgebracht. Um 22 Material und Methoden zu verhindern, daß die Elektroden- und Gelstreifen austrockneten, und um die Streifen vor CO2-Einflüssen zu schützen, wurde die gesamte Anordnung in der Fokussierkammer großzügig mit Silikonöl überschichtet. Die Fokussierung erfolgte bei 20 °C nach einem mehrstufigen Programm (Tabelle 2-1). Tabelle 2-1 Parameter für die isoelektrische Fokussierung (1. Dimension): IPG pH 4-7 Voltzahl in V Dauer in h 150 1,5 300 1,5 600 1 1.200 1 2.400 1 3.500 19 Gesamt: 71,9 kVh in 25 h IPG pH 3-10 Voltzahl in V Dauer in h 150 2 300 2 600 1,5 1.200 1 2.400 1 3.500 16 Gesamt: 61,4 kVh in 23,5 h IPG pH 3-10NL Voltzahl in V Dauer in h 150 2 300 2 600 1,5 1.200 1 2.400 1 3.500 16 Gesamt: 61,4 kVh in 23,5 h 2.2.6 Äquilibrierung der IPG-Streifen Um die Proteine erfolgreich aus dem IPG-Gelstreifen in das SDS-Gel der zweiten Dimension zu transferieren, ist eine Umpufferung, Äquilibrierung genannt, notwendig. Die Proteine müssen mit SDS beladen werden. Die Eigenladungen der Proteine werden effektiv überdeckt, es entstehen anionische Mizellen mit einer konstanten Nettoladung pro Masseneinheit. Die gestreckten, mit SDS beladenen Polypeptidketten bilden Ellipsoide und es ergibt sich eine lineare Beziehung zwischen dem Logarithmus der jeweiligen Molekulargewichte und den relativen Wanderungsstrecken dieser SDS-Polypeptid-Mizellen (Westermeier, 1990). Die Äquilibrierung verlief in zwei Schritten: im ersten Schritt wurden die Gelstreifen für 15 min in der Äquilibrierungslösung I inkubiert. Durch die Zugabe von DTT erfolgt eine nochmalige Reduzierung der Proteine. Im zweiten Schritt wurden die Gelstreifen für 15 min in der Äquilibrierungslösung II inkubiert. Die SH-Gruppen werden durch das Iodacetamid alkyliert und damit geschützt. Gleichzeitig wird das DTT, welches für vertikales „pointstreaking“ im silbergefärbten Gel verantwortlich gemacht wird (Görg et al., 1987), wieder entfernt. 23 Material und Methoden 2.2.7 SDS-PAGE (als zweite Dimension der 2D-Gelelektrophorese) 2.2.7.1 Herstellung der Gele Die für die vertikale SDS-PAGE benötigten Gele wurden unter Benutzung des zum Ettan DALTtwelfe™ -Systems gehörenden Gelgießstandes im Labor hergestellt. Die mit Abstandshaltern von 1 mm Stärke und einem Silikonscharnier versehenen Gelkassetten aus Glas wurden vor der Benutzung mit 70%igem Ethanol und einem fusselfreien Tuch poliert und in der benötigten Anzahl in den Gelgießstand eingesetzt, welcher anschließend mit Kunststoff-Attrappen in den Abmessungen der Gelkassetten aufgefüllt wurde. Die Acrylamidlösung wurde frisch hergestellt. Vor der Zugabe von TEMED und APS wurde die Lösung in eine Saugflasche überführt und für ca. 20 min in 3–4 Zyklen entgast. Nach der Zugabe von TEMED und APS und kurzem vorsichtigem Vermischen wurde die Lösung in den Gelgießstand eingebracht, unterschichtet mit Glycerol-Lösung und überschichtet mit 2 ml wassergesättigtem Butanol je Gelkassette. Die Polymerisationszeit betrug bei Raumtemperatur mindestens 4 h. 2.2.7.2 Aufsetzen der IPG-Streifen auf die SDS-Polyacrylamidgele Nach der Polymerisation wurden die Gelkassetten gründlich unter fließendem A. dest. gewaschen, um das Butanol sowie eventuell verbliebene Acrylamid-Monomere und TEMED-Reste zu entfernen. Die äquilibrierten IPG-Streifen wurden, mit der Gelseite zum Betrachter zeigend und mit dem sauren Ende nach links orientiert, vorsichtig zwischen die Glasplatten der Gelkassetten geschoben und auf die Geloberfläche aufgesetzt. Die Lösung eines Protein-Größenstandards wurde auf 4 x 4 mm messende Filterpapierecken getropft. Sobald die Ecken die Lösung aufgesaugt hatten, wurden diese neben den sauren Enden der IPG-Streifen plaziert. Die IPG-Streifen und Filterpapiere wurden blasenfrei mit 0,5%iger Agarose überschichtet und die Gelkassetten dabei randvoll befüllt. 2.2.7.3 Laufbedingungen der SDS-PAGE Nach dem Gelieren der Agarose wurden die Gelkassetten in die Elektrophoresekammer, deren untere Kammer bereits mit 1 x Laufpuffer befüllt war, eingesetzt, die gegen die untere Kammer abgedichtete obere Kammer mit 2 x Laufpuffer befüllt und der Trennvorgang durch Anlegen einer Spannung gestartet. Die Trennung der Proteine erfolgte bei 20 °C zu den in Tab. 2-2 angegebenen Bedingungen. 24 Material und Methoden Tabelle 2-2 Laufbedingungen 2. Dimension (SDS-PAGE, 20 °C): Zeit [h] Spannung [V] Einlaufen 4 30 Trennung 16 90 2.2.8 Färben der Polyacrylamidgele Die Gele wurden aus den Gelkassetten entnommen und wie unten beschrieben weiterbehandelt. Um Kontaminationen zu vermeiden, wurden die Gele nur mit Handschuhen berührt. 2.2.8.1 Coomassie-Blue-Färbung Die Gele wurden zunächst 60 min bei RT in der Fixierlösung und anschließend für 60 min in der Färbelösung geschwenkt. Nach dem Färbevorgang wurde wieder entfärbt, bis die Proteinspots sichtbar waren. Der Entfärber wurde dabei unter Umständen mehrfach gewechselt. 2.2.8.2 Kolloidale Coomassie-Blue-Färbung Zunächst wurden die Gele für 60 min bei RT in Fixierlösung geschwenkt, anschließend in frisch angesetzte Färbelösung überführt und darin über Nacht bei RT geschwenkt. Um Farbstoffreste zu entfernen, wurden die Gele am nächsten Morgen in die Waschlösung überführt und darin bis zur weiteren Verwendung aufbewahrt. 2.2.8.3 Reversible Silberfärbung nach Mann (Shevchenko et al., 1996) Die Silberfärbung für Proteine in Polyacrylamidgelen nach Mann ist eine sensitive und reversible Färbemethode. Die Proteine können mit Natriumthiosulfat wieder entfärbt werden. Nach Abdunklung des Labors und der Färbeschalen wurden die Gele für 60 min in den Fixierer gegeben und bei RT geschwenkt. Die Lösung wurde abgesaugt und die Gele anschießend für 2 x 20 min mit der Waschlösung und nochmals für 20 min mit A. dest. gewaschen, wobei die Lösungen durch Absaugen gewechselt wurden. Nun wurden die Gele für genau 1 min in der Natriumthiosulfat-Lösung inkubiert und daraufhin 2 x 20 sec mit A. dest. gewaschen. Nachfolgend wurde 4 °C kalte Färbelösung zugegeben und die Gele für 20 min in der Dunkelheit gefärbt, anschließend genau 20 sec mit A. dest. gewaschen und danach im Dunkeln mit Entwicklerlösung benetzt. Bei Schlierenbildung wurde die Lösung sofort entfernt und neu zugegeben. Die Gele 25 Material und Methoden wurden 1-5 min entwickelt (nach visuellem Eindruck), kurz in A. dest. gewaschen, 15 min in der Stopplösung inkubiert und abschließend 2 x 10 min mit A. dest. gewaschen. 2.2.9 Auswertung der Gele und Identifizierung einzelner Proteine 2.2.9.1 Bildverarbeitung mit der Proteinanalyse-Software Proteomweaver Um die Proteinspot-Muster verschiedener Proben vergleichen zu können, wurden von den fertig entwickelten 2D-Gelen digitale Bilder erzeugt. Dies geschah durch Scannen der Gele mit einem Densitometer im Transmissionsmodus bei einer Auflösung von 63,5 m (entsprechend 400 dpi). Die erzeugten Bilder wurden mit Proteomweaver 2.1, einer Proteinanalyse-Software, ausgewertet. Nach einer initialen Spoterkennung, deren Parameter einstellbar sind, und einem Matching genanntem Vorgang, bei dem die in den zu vergleichenden Bildern detektierten Spotmuster aufeinander abgestimmt werden, erfolgt der eigentliche Mustervergleich. Im Prinzip wird dabei das Proteinspot-Muster der Probe I, bestehend aus x angefärbten Proteinspots, mit dem Proteinspot-Muster der Probe II, bestehend aus y angefärbten Proteinspots, verglichen. Dabei werden Spots mit unterschiedlichen Intensitäten und Spots, die in einem der zu vergleichenden Muster fehlen, herausgefiltert. Das Programm rechnet dazu die Bilddaten in ein dreidimensionales Gebilde um: Die Proteinspots bilden, entsprechend ihrer Größe und Farbsättigung im 2D-Bild, die Berge in einer Ebene aus Hintergrundfärbung (siehe Abbildung 2-1). Diese „Landschaftsarchitektur“ liefert dem Programm die Eckdaten, die es braucht, um die zu vergleichenden Gelbilder zu „matchen“, d.h. virtuell übereinanderzulegen. Das Programm ist in der Lage, Positionsunterschiede einzelner Spots innerhalb einstellbarer Grenzen zu erkennen. Durch die Berechnung von Vektoren werden die zu vergleichenden 2D-Muster dann aufeinander abgestimmt. Erst im nächsten Schritt werden Proteinsspots untereinander in ihrer Intensität verglichen. 26 Material und Methoden Abbildung 2-1 Ausschnitt eines 2D-Gelbildes in der 3D-Ansicht. Die Berge im Bild stellen einzelne Proteinspots dar, wobei Höhe und Umfang der Berge mit Farbdichte und Größe der Spots korrespondieren. Die Empfindlichkeit dieses Detektionsschrittes war durch drei Parameter beeinflußbar: den Mindestdurchmesser, den ein Spot aufweisen muß, um erfaßt zu werden, seine aufsummierte Mindestintensität gegenüber dem Hintergrund und seinen Mindestkontrast gegen den Hintergrund. Um alle erkennbaren Spots zu erfassen, gleichzeitig nach Möglichkeit kleine Farbpartikel sowie Hintergrundrauschen von der Detektion auszuschließen, wurden für die vorliegenden Untersuchungen folgende Einstellungen zur Spotdetektion gewählt: als Mindest-Ø = 8 (2...10), als Mindestintensität = 5.000 (0...5.000) und als Mindestkontrast = 20 (2...50). Anschließend wurden die Spotmuster paarweise gematcht, in Experimenten mit mehr als zwei Gruppen zudem jedes Spotmuster mit jedem. Sowohl Proteinspots, die in einem der zu vergleichenden Gelbilder fehlten, als auch Proteinspots mit signifikanter (mehr als dreifacher) Abweichung in der Intensität wurden von der Analyse-Software herausgefiltert und in ein Analyse-Set überführt. Dieses wurde am Bildschirm manuell editiert. Spots, die nach dieser Sichtprüfung noch interessant erschienen, wurden aus dem entsprechenden Gel ausgeschnitten, entweder manuell mit dem Skalpell oder mit dem GelPal-Proteinspotexcisionsgerät. 2.2.9.2 Trypsin-Verdau ausgeschnittener Proteinspots Trypsin, als Trypsinogen im Pankreas gebildet und aktiv im Dünndarm, ist eine SerinProtease, die Proteine schneidet, indem sie Peptidbindungen C-terminal nach Lysin 27 Material und Methoden und Arginin hydrolytisch spaltet. Ein Protein wird also in eine charakteristische Zahl Peptide mit unterschiedlichen Massen geschnitten. Soweit dessen Sequenz vorher schon bekannt und in einer Proteindatenbank erfaßt war, läßt sich ein Protein durch das sich ergebende Peptidmassenmuster anschließend mit relativer Genauigkeit identifizieren. Die ausgeschnittenen Spots wurden in 0,5 ml-Eppendorf-Reaktionsgefäße überführt und auf dem Thermomixer 1 h mit 400 l Puffer A bei RT und 750 rpm entfärbt. Die Lösung wurde entfernt, durch frischen Puffer A ersetzt, und die Gelstückchen wurden bei gleichen Bedingungen für weitere 30 min inkubiert, wobei die Entfärbung möglichst vollständig erfolgen sollte. Der Puffer wurde entfernt, durch 400 l Puffer B ersetzt und die Spots wurden für zweimal 10 min auf dem Thermomixer bei RT und 750 rpm inkubiert. Puffer B wurde ersetzt durch 400 l Puffer C und in diesem wurden die Gelstückchen 1 h auf dem Thermomixer bei RT und 750 rpm inkubiert. Die Lösung wurde vollständig abgenommen und durch 50 l Puffer D ersetzt (Inkubationsdauer 15 min, auf dem Thermomixer bei RT und 750 rpm). Die Lösung wurde entfernt, die Gelstückchen sollten dehydratisiert weiß erscheinen. Die Gelstückchen wurden bis zur völligen Trocknung (ca. 10 min) im Vakuum abrotiert. Aus der 10x Trypsinlösung wurde die Arbeitslösung frisch hergestellt und von dieser 10 l pro Gelstück zugegeben. Die Inkubation erfolgte für 16 h bei 37 °C. 2.2.9.3 Peptidmassenanalyse mittels SELDI-Technologie SELDI (surface enhanced laser desorption ionisation) ist eine MALDI-TOF-basierte Proteinchip-Array-Technolgie. Proteinchips mit verschiedenen chromatographischen Oberflächen (Anionen-, Kationenaustauscher, hydrophobe Oberflächen, usw.) stehen zur Auswahl und können z.B. mit Zellysaten, Proben von Körperflüssigkeiten u.a. beladen werden. Durch nachfolgendes Waschen bleiben nur Proteine gebunden, die in hohem Maße mit der Chipoberfläche wechselwirken. Diese werden dann im Massenspektrometer analysiert. Typischerweise handelt es sich dabei um ein MALDI-MS (matrix-assisted laser desorption/ionisation mass spectrometry) mit angegliedertem Flugzeitanalysator (TOF – time of flight). Die Massenspektren verschiedener Proben (z.B. krank vs. gesund) werden anschließend verglichen. Unterschiede markieren krankheitsspezifische Proteine. Mit der Kenntnis solcher Marker läßt sich die SELDITechnologie beispielsweise zur klinischen Diagnose von Patientenproben einsetzen (protein profiling). 28 Material und Methoden Für die vorliegende Arbeit wurde SELDI zur Peptidmassenanalyse ausgewählter Proteine benutzt. Dies geschah in Kooperation mit Dr. Ch. Melle von der Core Unit Chip Applications, einer AG des Instituts für Humangenetik der Friedrich-SchillerUniversität Jena. Es kamen H4- und H50-Chips mit hydrophober Oberfläche zum Einsatz. Die H50-Chips wurden direkt vor Gebrauch durch 2 x 5-minütige Inkubation mit 5 l 50%igem ACN/Spot aktiviert. Anschließend wurde zweimal 1 l Peptidlösung/Spot aufgetragen und diese nach Eintrocknung mit zweimal 0,5 l 20%iger CHCA-Matrix überdeckt. In jedem Experiment wurde eine Leerprobe, d.h. ein gleichbehandeltes proteinfreies Gelstückchen mitgeführt, um Hintergrund- und Trypsin-Autolysis-Signale identifizieren zu können. Zur internen Kalibrierung jedes einzelnen Chips war das Mitführen eines Massenstandards, bestehend aus 6 Peptiden, notwendig. Die Kalibrierung erfolgte durch Abgleich der gemessenen Werte mit den exakt bekannten Massezahlen der Peptide des Standards. Die errechneten Massendifferenzen wurden auf alle mit dem Chip aufgenommenen Massenspektren übertragen und die Spektren entsprechend angepaßt. 2.2.9.4 Identifizierung der Proteine mit der Online-Anwendung ProFound Mit ProFound (http://129.85.19.192/profound_bin/WebProfound.exe) können verschiedene Proteindatenbanken (SwissProt, NCBI) durchsucht und die dort gelisteten Sequenzen „virtuell verdaut“ werden. In die Suchmaske eingegebene Peptidmassen werden mit den virtuell erzeugten Fragmentmustern verglichen. Die Proteine mit der höchsten Übereinstimmung zum eingegebenen Massenspektrum werden anschließend angezeigt. Die massenspektrometrisch ermittelten Peptidmassen wurden in die Suchmaske eingegeben. Um die Suche einzuengen, wurden zusätzliche, aus dem 2D-Muster ablesbare Daten wie ungefährer pI, ungefähres Molekulargewicht und Modifikationsstatus des zugehörigen Proteins, hinzugefügt und die Suche gestartet. 2.2.10 Reverse Transkription der Gesamt-mRNA (RT-PCR) Die Gewinnung der Gesamt-RNA aus dichtgewachsenem infiziertem bzw. nichtinfiziertem Zellrasen erfolgte mittels des kommerziell erhältlichen RNeasy-Mini-Kits von QIAGEN nach Angaben des Herstellers. 29 Material und Methoden Die Generierung von cDNA aus RNA erfolgte unter Verwendung der reversen Transkriptase des Molony Murine Leukemia Virus (M-MLV). Da nur die Umschreibung aller mRNA-Moleküle gewünscht war, wurde unter Ausnutzung der 3’-terminalen Poly-A-Sequenz, die alle zellulären mRNA-Spezies und auch das Coxsackievirus-RNA-Molekül besitzen, ein unspezifischer Oligo-d(T)-Primer verwendet. Es wurden 5 g RNA – das Volumen ergab sich aus photometrischer RNA-Konzentrationsmessung bei 260 nm – auf 10 l mit DEPC-H2O aufgefüllt und 1 l oligo-d(T)-Primer (0,5 g/l) zugegeben. Zur Denaturierung wurde zunächst 10 min bei 70 °C inkubiert. Nach 3 min auf Eis wurden folgende Lösungen hinzugegeben: 4 l 5x RT-Puffer 2 l dNTPs (10 mM) 2 l DTT (1 M) 1 l SUPERSCRIPT™ II-RT/M-MLV RT Die anschließende reverse Transkription erfolgte im Thermocycler mit folgenden Einstellungen: Primerannealing 10 min 25 °C Polymerisation 50 min 42 °C Denaturierung 5 min 90 °C Kühlung 1 Zyklus 4 °C Diese Arbeiten wurden bei Dr. A. Henke im Institut für Virologie der FriedrichSchiller-Universität durchgeführt. 2.2.11 Polymerase-Ketten-Reaktion (PCR) Zur Amplifikation der gewünschten cDNA-Moleküle (RT-Produkte) wurde die Polymerase-Ketten-Reaktion (PCR) benutzt. Die Arbeiten wurden im 50l-Maßstab durchgeführt. 30 Material und Methoden Folgender Ansatz wurde pipettiert: 2 l RT-Produkt (cDNA-Mischung, ca. 0,5g) 5 l PCR 10x-Puffer (inkl. 15 mM MgCl2) 5 l dNTP´s (0,2mM) 1,25 l 3'-Primer (2,5M) 1,25 l 5'-Primer (2,5M) 10 l Q-Solution 25 l A. bidest. 2,5 U Taq-Polymerase Als universales Programm wurde verwendet: Erst-Denaturierung 95 °C 3 min 1 Zyklus Denaturierung 95 °C 1 min 30 Zyklen Primer-Anlagerung 55 °C 1 min Verlängerung 72 °C 1 min Komplettierung 72 °C 10 min 4 °C Kühlung 2.2.12 DNA-Agarosegel-Elektrophorese Die elektrophoretische Auftrennung der PCR-Produkte erfolgte in 1%igen Agarosegelen mit 1x TAE-Elektrophoresepuffer bei 50–110 V. Den Agarosegelen wurde Ethidiumbromid (SL 10 mg/ml) in einer Endkonzentration von 0,3 g/ml zugegeben. Es wurden 5–10 l Probe pro Geltasche eingesetzt. Vor dem Auftragen auf das Agarosegel wurde die Probe im Verhältnis 1:5 mit Gelladepuffer gemischt. 2.2.13 Western Blot im Semi dry-Verfahren Für den spezifischen Nachweis eines Proteins wurden die Proteine aus dem PAA-Gel auf eine PVDF-Membran transferiert. Die Membran, das PAA-Gel und die Filterpapiere wurden in Towbin-Blotpuffer (Towbin et al., 1979) eingeweicht und dann in der Blot-Apparatur zu einem Sandwich geschichtet, so daß das Proteingel zur Anode und die Nitrocellulose zur Kathode zeigte. Durch Anlegen eines Stromes von 1 mA 31 Material und Methoden je cm2 Gelfläche wurden die Proteine für eine Stunde auf die Membran transferiert. Um die Proteine direkt nach dem Transfer auf der Membran sichtbar zu machen, wurde die Membran für 5 min mit Ponceau S-Lösung inkubiert und anschließend mit A. bidest. der überschüssige Farbstoff abgewaschen. Durch Zugabe der Blockierlösung und Inkubation für eine Stunde auf dem Kipptisch wurde die unspezifische Proteinbindung auf der Membran blockiert und die Ponceau S-Färbung wieder entfernt. Nach zweimaligem Waschen der Membran mit TBS-T wurde der primäre Antikörper, mit dem ein betreffendes Protein nachgewiesen werden sollte, 1:1.000 in Blockierlösung verdünnt und für eine Stunde auf die Membran gegeben. Anschließend wurde die Membran fünfmal für je 5min mit TBS-T gewaschen und danach der mit Alkalischer Phosphatase gekoppelte Spezies-spezifische Zweitantikörper zugefügt. Nach einer erneuten Inkubation für eine Stunde auf dem Kipptisch wurde wieder fünfmal mit TBS-T gewaschen. Zur Detektion der Proteine wurde die Membran in AP-Färbelösung inkubiert, bis Banden sichtbar wurden und die Färbelösung dann mit A. bidest. abgespült. 32 Ergebnisse 3 Ergebnisse 3.1 Etablierung der Zweidimensionalen Gelelektrophorese Die Ergebnisse und die Reproduzierbarkeit der 2D-Elelektrophorese sind stark von den Bedingungen, unter denen diese durchgeführt wird, abhängig. Dies bedeutet, daß eine Anpassung der Methode an den jeweiligen Einsatzzweck unumgänglich ist. Zunächst waren die notwendigen Einzelschritte nach den Hinweisen der Hersteller ausgeführt bzw. aus der vorhandenen Literatur übernommen worden. Zur Reinigung der Proben wurden zwei geeignete Methoden getestet und die Ergebnisse miteinander verglichen. Eigene Erfahrungen führten zur Anpassung der Methodik an die Untersuchungsobjekte: die humanen Zellinien HepG2 und HeLa. Die Ergebnisse dieses Optimierungsprozesses sowie die daraus erarbeitete „Anleitung für Proteomuntersuchungen an HepG2-Zellen“ werden nachfolgend präsentiert. 3.1.1 Optimierung einzelner Arbeitsschritte 3.1.1.1 Vermeidung nachträglicher Proteindegradation Um den nachträglichen Abbau von Proteinen zu verhindern, sollte die Ernte der Zellen möglichst schnell erfolgen. Der PBS-Puffer, mit dem Medium- und Serumreste vom Zellrasen gewaschen werden, sollte 4 °C kalt sein. Da die zellwandlosen Zellen mit Rehydratisierpuffer lysierbar sind, sollte dieser auch hierfür verwendet werden. Mehrere Vorteile sind damit verbunden: Die Probe liegt so bereits in der Rehydratisierungslösung vor und die Komponenten des Puffers sind mit der TCA/Aceton-Fällung verträglich. Da der Puffer stark denaturierend wirkt, kann – soweit die Proben rasch aufgearbeitet werden – auf die Zugabe von ProteinaseInhibitoren verzichtet werden. Nach der TCA/Aceton-Fällung, welche zuverlässig Proteinasen inaktiviert (Görg et al., 2000), war in den Proben auch nach längerer Lagerung (12 Wochen bei -80 °C) kaum Abbau von Proteinen feststellbar. 33 Ergebnisse 3.1.1.2 Reinigung der Proben Eine direkte Verwendung des Lysats für die isoelektrische Fokussierung war nicht möglich. DNA, Membranbestandteile und andere im Solubilisierungspuffer schlecht lösliche Verbindungen verstopfen die Poren der IPG-Streifen. Salze und kleinmolekulare geladene Verbindungen wie Oligonukleotide stören die Fokussierung durch zu hohe anfängliche Feldstärke. Dies hatte ein „Ausbrennen“ der Gele, bevorzugt am kathodischen Ende, bzw. Präzipitatbildung zur Folge. Es war also ein zusätzlicher Reinigungsschritt erforderlich. Zwei Methoden wurden in Betracht gezogen und verglichen: die Ultrafiltration der Proben und die TCA/AcetonProteinfällung nach Görg (Görg et al., 1997). 3.1.1.3 Vergleich von TCA/Aceton-Fällung und Ultrafiltration Die Abb. 3-1 zeigt Coomassie-gefärbte Gele, die mit je 250 l TCA/Aceton-gefälltem bzw. ultrafiltriertem Gesamtzellysat beladen wurden. Deutlich ist zu erkennen, daß nach TCA/Aceton-Fällung eine wesentlich höhere quantitative Ausbeute an Protein als nach Ultrafiltration der Proben erreicht wurde. pH 4 pH7 pH 4 pH7 Abbildung 3-1 Vergleich des Proteinmusters nach TCA/Aceton-Fällung und Ultrafiltration der Proben. Links: 250 l Gesamtzellysat HepG2, pH 4-7, TCA/Aceton-Fällung; rechts: 250 l Gesamtzellysat HepG2, pH 4-7, Ultrafiltration. Zudem war die Auflösung des 2D-Musters, die sich durch die Anzahl darstellbarer Proteine beschreiben läßt, bei TCA-Fällung um mehr als 25 % höher als bei Ultrafiltration des Zellysats (Abb. 3-2). Einzelne Proteinspots, die nach einer TCA-Fällung deutlich erschienen, waren nach paralleler Ultrafiltration der gleichen Probe kaum zu sehen oder nicht vorhanden. 34 Ergebnisse 2000 TCA-Fällung Ultrafiltration 1800 1600 Spots/Gel 1400 1200 TCA-Fällung 1000 800 600 400 200 0 Ultrafiltration Abbildung 3-2 Links: Vergleich der Anzahl der Spots bei TCA/Aceton-Fällung und Ultrafiltration der Proben in 3 Experimenten. Rechts: Auflösung und Abundanzen einzelner Proteine im Gel (je 250 l HepG2-Zellysat, gleiche Probe, paralleler Lauf, Ausschnitt) Im Vergleich zur TCA-Fällung waren die Proteine der äußeren pI-Bereiche nach Ultrafiltration der Proben undeutlich aufgelöst und horizontal streifig. Vor allem am kathodischen (basischen) Ende wurden über schmale Bereiche des IPG-Streifens die Proteine überhaupt nicht fokussiert (Abbildung 3-1) und teilweise waren Bereiche leer (d.h. proteinfrei). 3.1.1.4 Proteingehaltsbestimmung Die möglichst genaue Proteingehaltsbestimmung war entscheidend für die Vergleichbarkeit des 2D-Musters verschiedener Proben. Nur bei gleichen aufgetragenen Gesamtproteinmengen ließen sich reproduzierbare Aussagen über die Abundanzen einzelner Proteine treffen bzw. wurden 2D-Muster verschiedener Proben überhaupt erst vergleichbar. 3.1.1.5 Rehydratisierung der IPG-Streifen Wegen ihrer deutlich einfacheren Handhabbarkeit wurde in den vorliegenden Untersuchungen nur die sogenannte aktive Rehydratisierung (in-gel-rehydration; (Rabilloud et al., 1994) betrachtet. Hierbei wird die Probe bereits mit der Rehydratisierungslösung zu den Streifen gegeben. Wichtig war die Beachtung der maximalen Quellfähigkeit der IPG-Streifen: Die Aufnahmefähigkeit der 24 cm-Streifen liegt bei ca. 500 l Lösung/Streifen. Dieses Volumen sollte nicht überschritten werden, da vor allem große Proteine eher in Flüssigkeitsresten zurückbleiben, als sich in die Gelmatrix zu „zwängen“ (Görg et al., 2000). 35 Ergebnisse Die aktive Rehydratisierung sollte bei Raumtemperatur und mindestens über Nacht, besser noch für 24 h, erfolgen. Auch nach mehr als 16 h Inkubationsdauer wurden von den Streifen noch Flüssigkeitsreste aufgenommen. Aus bereits oben zitiertem Grund sollte die Rehydratisierung deshalb bis zur vollständigen Aufnahme der Probenflüssigkeit durch die Streifen erfolgen. 3.1.1.6 Bedingungen der isoelektrischen Fokussierung Je schmaler der pH-Gradient, desto länger die Fokussierdauer. IPG-Gele mit extrem engem pH-Gradient (sogenannte narrow interval IPGs) wurden nicht getestet. Wie aus Tab. 2-1 ersichtlich, wichen die optimalen Fokussierzeiten der verwendeten Streifen jedoch abhängig vom pH-Gradient deutlich voneinander ab. Für die 24 cm-Streifen mit einem pH-Bereich von 3–10 bzw. der nichtlinearen Variante 3–10NL waren 61 kVh Fokussierzeit ausreichend, die Streifen gleicher Größe mit einem pH-Bereich von 4–7 benötigten 71 kVh. Die Ursache liegt darin, daß die Proteine im flachen Gradient weite Strecken mit niedriger Eigenladung zurücklegen müssen. Da sich die Streifen durch die angelegten hohen Spannungen stark erwärmen, ist eine effektive Wärmeableitung notwendig. Als Kontaktflüssigkeit zwischen Kühlblock und Trägerfolie der Streifen wurde deshalb Silikonöl eingesetzt. Als Folge lokaler Überhitzungen durch den schlechten Kontakt zur Kühlplatte wurde über Luftblasen mehrmals dauerhafte oder vorübergehende Präzipitatbildung beobachtet. Darum sollten die Streifen möglichst luftblasenfrei in das Silikonöl gebettet werden. 3.1.1.7 SDS-PAGE-Bedingungen Für die gleichbleibende Qualität der Gele sind neben der Güte der für die Polymerisationslösung verwendeten Chemikalien und deren genauer Einwaage auch die Bedingungen während der Polymerisation entscheidend. Zu niedrige Temperaturen (z.B. bei Verlagerung des Gelgießstandes in den Kühlschrank) und zu kurze Polymerisationszeiten hatten nachteiligen Einfluß auf die Qualität der Gele. In der Gelmatrix findet eine langsame Nachpolymerisation statt (Westermeier, 1990). Deshalb sollten die SDS-PAA-Gele der zweiten Dimension immer einen Tag vor Verwendung hergestellt werden und bei Raumtemperatur auspolymerisieren. Für die Reproduzierbarkeit sehr wichtig waren die Bedingungen während der Elektrophorese. Starke angelegte Spannungen verkürzten zwar die zeitliche Dauer der Elektrophorese, bewirkten aber ein in die Länge gezogenes Spotmuster. Die einzelnen 36 Ergebnisse Punkte waren nicht klar rund, sondern zogen Spuren nach. Die Über-NachtElektrophorese bei 90 V stellte einen guten Kompromiß dar zwischen Spotqualität und möglichen Diffusionsverlusten aufgrund der verlängerten Trennungszeit. Außerdem hatte dies den Vorteil, daß bei unterschiedlicher Beladung der Laufkammer keine Anpassung des Programms an die Zahl der Gele notwendig war. Bei der anfänglich benutzten Laufkammertemperatur von 12 °C waren teilweise laufbedingte Unterschiede im 2D-Muster festzustellen. Die Kühlung der Kammer schaffte es nicht, die Temperatur in allen Punkten der Kammer über die gesamte Laufzeit konstant zu halten. Deshalb erfolgte die Trennung bei 20 °C, was auch Görg et al. empfehlen (Görg et al., 2000). 3.1.1.8 Vergleich verschiedener Färbemethoden In der vorliegenden Arbeit wurden einfache Coomassie-Blue-, kolloidale CoomassieBlue- und Silber-Färbung nach Mann (Shevchenko et al., 1996) getestet und die Färbeergebnisse verglichen. Die Färbung mit dem kolloidalen Comassie-Blue erreichte im visuellen Vergleich parallel gefärbter Gele nicht ganz die Sensitivität der Silbernitrat-Färbung. Allerdings war bei Färbung mit Silbernitrat eher die Sättigung erreicht. Coomassie wies dagegen einen weiteren linearen Bereich auf, was für die ausgeführten Proteomuntersuchungen, bei denen die relative Proteinmenge einzelner Spots über die Intensität der Färbung quantifiziert wurde, von Vorteil war. Es zeigte sich, daß die gewählte Färbemethode entscheidenden Einfluß auf die Struktur des 2D-Musters der untersuchten Proben und damit auf die Reproduzierbarkeit hatte, weil jedes Protein eine spezifische, von anderen Proteinen unterschiedliche Affinität zum jeweiligen Farbstoff besitzt (Westermeier, 1990). Einige Proteine, die nach kolloidaler Coomassie-Färbung deutliche Spots ergaben, waren im parallel silbergefärbten Gel kaum zu detektieren, wie Abb. 3-3 zeigt. Umgekehrt waren einige durch Silbernitrat angefärbte Spots im coomassiegefärbten Gel nicht aufzufinden. Verantwortlich sind die schon erwähnten unterschiedlichen Affinitäten der einzelnen Proteine zum jeweiligen Farbstoff. Nach unterschiedlichen Methoden gefärbte Gele waren deshalb nicht vergleichbar. Für die Coomassie-Färbung waren vergleichsweise wenige Arbeitsschritte notwendig. Die Anwendung war sehr einfach und wenig fehleranfällig. Die gefärbten Gele waren 37 Ergebnisse praktisch frei von störender Hintergrundfärbung, blieben stabil und längere Zeit lagerfähig. HepG2-Gesamtzellysat; 0,5 mg Protein/Gel, Ausschnitt, kolloidale Coomassie-Färbung HepG2-Gesamtzellysat; 0,5 mg Protein/Gel, Ausschnitt, Silberfärbung Abbildung 3-3 Vergleich von kolloidaler Coomassie- und Silbernitrat-Färbung. Sowohl im coomassiegefärbten als auch im silbergefärbten Gel sind Spots detektierbar, die im jeweils anderen fehlen oder deutlich unterschiedlich gefärbt erscheinen. Silbergefärbte Gele dagegen waren brüchig, was nachfolgende Schritte wie das Scannen stark erschwerte. Hintergrundfärbung war als teilweise deutlich wolkiger Schmier vorhanden, der die Auswertung behinderte. Unterschiedlich entwickelte Gele wiesen starke Intensitätsdifferenzen auf. Weil die optimale Entwicklungszeit für einzelne Gele beträchtlich schwankte, war eine feste Zeitvorgabe jedoch nicht möglich. Einzelne Spots waren in Richtung Kathode langgezogen, ein Phänomen, das mit verbliebenen DTT-Resten in Zusammenhang stehen könnte. Wegen der deutlich einfacheren Handhabung bei vergleichbarem Färbeergebnis wurde die Kolloidale Coomassie-Färbung in das Protokoll für Proteomuntersuchungen humaner HepG2-Zellen integriert. 3.1.2 Vorgeschlagenes Protokoll für Proteomuntersuchungen des Gesamtzellysats von HepG2-Zellen mittels 2D-Gelelektrophorese Nachfolgend wird das Protokoll, wie es in der vorliegenden Arbeit zur Anwendung kam, stichpunktartig vorgestellt. Die einzelnen Arbeitschritte wurden detailliert schon im Methodenteil der Arbeit unter 2.2 erläutert. 1. viermaliges Waschen des Zellrasens mit 4 °C kaltem PBS-Puffer 2. Lyse der HepG2-Zellen mit Solubilisierungslösung 38 Ergebnisse 3. Ultraschallbehandlung des Zellysats (Mikrospitze, 10 x 5 sec, Pulse 2, 55 %) 4. TCA/Aceton-Fällung 5. Resuspendierung des Proteinpellets in Rehydratisierungslösung 6. Sedimentation unlöslicher Fragmente durch Ultrazentrifugation der Probe (15 min, 75.000 g, 16 °C) 7. Bradford-Proteingehaltsbestimmung und Einstellung der Proben auf definierten Proteingehalt von 1 mg/ml mit Rehydratisierungslösung 8. aktive Rehydratisierung der 24 cm IPG-Streifen (500 l Probenvolumen – entsprechend 0,5 mg Protein, 24 h bei RT) 9. Isoelektrische Fokussierung (Multiphor II™-System, Streifen mit Silikonöl überschichtet, stufenweise Erhöhung der Spannung von 150 V auf 3.500 V, IPG 3-10, 3-10NL: ca. 72 kVh, IPG 4-7: ca. 61 kVh) 10. anschließend unverzüglich Äquilibrierung der IPG-Streifen zur Reduzierung/Alkylierung der Proteine und Beladung mit SDS (2 x 15 min) 11. anschließend SDS-PAGE als zweite Dimension (EttanDALTtwelfe™ -System, 3 h bei 30 V und ca. 16 h bei 90 V, Gele einen Tag vorher gießen) 12. Färbung der Proteine im Gel (kolloidale Coomassie-Färbung, 1 h fixieren, Färbung über Nacht, waschen in 20 % Methanol) Die gleiche Arbeitsanleitung war ohne Einschränkung auch zur Proteomanalyse von HeLa-Zellen nutzbar. Die Übertragung auch auf andere humane Zellinien ist daher wahrscheinlich unproblematisch. Die Methode in der vorliegenden Form ist außerdem auch für Proteomuntersuchungen an HepG2-Zellen im größeren Maßstab, wie z.B. zur Charakterisierung des Wirkmechanismus neuartiger Naturstoffe, geeignet. 3.1.3 Systembedingte Variabilität des Spotmusters einer Probe Um Unterschiede im 2D-Muster verschiedener Proben zu bewerten, ist es notwendig, den Fehler des Experiments zu kennen. Bei der 2D-Elektrophorese äußert sich dieser Fehler in nichtrealen signifikanten Intensitätsunterschieden einzelner Spots. Diese Artefakte können auf zwei Ebenen entstehen: bei der Probenaufarbeitung, -applikation und -trennung und später bei der computergestützten Auswertung der 2D-Muster. Sie sind unvermeidlich und der Grund, warum die Ergebnisse einer 2D-Elektrophorese durch mehrfache Wiederholung abgesichert werden müssen. 39 Ergebnisse Bei Versuchen zur Ermittlung dieses experimentellen Fehlers wurden identische Proben in gleicher Menge auf IPG-Streifen geladen, fokussiert und anschließend parallel im gleichen Lauf nach Molekülmasse getrennt. Die sich ergebenden 2D-Muster wiesen Unterschiede auf, die 5 bis 10 % aller detektierten Spots umfaßten. Das bedeutet, 5 bis 10 % aller detektierten Spots wurden im Vergleichsgel entweder nicht gefunden oder wurden dort mit signifikant unterschiedlicher Intensität erkannt. Die detektierten Unterschiede betrafen fast ausschließlich Spots nahe der Auflösungsgrenze und waren häufig die Folge abweichender Proteinmengen zwischen den Vergleichsgelen infolge Toleranzen bei Beladung der IPG-Streifen und Proteinverlusten während IEF, Äquilibrierung und SDS-PAGE (Görg et al., 2000). Zum Teil wurden Artefakte erst durch die Analyse-Software zu solchen gemacht, weil Proteinspots aufgrund von Verzerrungen im 2D-Muster falsch zugeordnet wurden, weil Hintergrundfärbung fälschlich zu einem Spot aufsummiert wurde oder weil ein Spot zwar in allen Vergleichsgelen vorhanden war, aber einmal nicht detektiert wurde. Beispiele hierfür werden in der Abb. 3-7 gezeigt. Da der Fehler bei diesen Versuchen deutlich variabel war, sollte jedes 2D-Elektrophorese-Experiment individuell auf seinen Fehler getestet werden. Dazu wird der Vergleich der parallelen Gele des Experiments (z.B. der Kontrollgele) empfohlen. 3.2 Nachweis Coxsackievirus B3-induzierter Änderungen des Proteinmusters von HepG2-Zellen Mit dem optimierten Protokoll sollten Einflüsse von Coxsackievirus B3 auf das Proteom von HepG2-Zellen untersucht werden. Dazu wurde der dichtgewachsene HepG2-Zellrasen mit einer CVB3H3-Suspension angeimpft und bis zu gerade sichtbaren zytopathischen Effekten weiterinkubiert. Nach 12 h Inkubationsdauer waren diese lichtmikroskopisch an abgerundeten Zellen und zerstörten Arealen eng begrenzten Umfanges im Zellrasen erkennbar. Die Proteine des Zellysats wurden in zwei 24 cm-IPG-Streifen fokussiert und anschließend nach Molekulargewicht im SDS-PAA-Gel aufgetrennt. In gleicher Weise erfolgte die Präparation einer nichtinfizierten Kontrolle. Wie Abb. 3-4 zeigt, unterschied sich das Proteom der HepG2-Zellen nach 12 h Infektionsdauer deutlich von dem der nichtinfizierten Kontrollzellen. 40 Ergebnisse Da also der Replikationszyklus der Coxsackieviren bis zur Freisetzung neuer Viruspartikel in HepG2-Zellen ca. 12 Stunden in Anspruch nahm, war die Frage interessant, ob bereits zu einem früheren Zeitpunkt der Replikation Veränderungen auf Proteomebene detektierbar sein würden. Deshalb wurde das Proteom 6 Stunden nach Infektion der HepG2-Zellen mit CVB3H3, vor sichtbaren mikroskopischen Veränderungen, analysiert. Bereits zu diesem Zeitpunkt waren Unterschiede in der Proteinausstattung zwischen infizierten und nichtinfizierten Zellen feststellbar (Abb. 3-4). Diese waren allerdings von deutlich geringerem Umfang als die nach 12 Stunden detektierten Veränderungen. Abbildung 3-4 Proteinmustervergleich CVB3H3-infizierter HepG2-Zellen. Links: nichtinfizierte Kontrolle, Mitte: 6 h nach Infektion, Rechts: 12 h nach Infektion. Das 2D-Muster infizierter Zellen (blau) wurde am Computer mit dem Spotmuster nichtinfizierter Kontrollzellen (orange) gematcht (in Übereinstimmung gebracht). Nichtregulierte Proteine erscheinen als schwarze Punkte, nach Virusinfektion neu bzw. verstärkt exprimierte Proteine sind blau und infolge der Infektion reprimierte bzw. degradierte Proteine erscheinen orange. Das blau-orange-Matching zweier Kontrollgele zeigt eine relativ hohe Übereinstimmung beider Spotmuster. Nach sechs Stunden sind bereits deutliche Einflüsse der Virusinfektion auf das Proteom der HepG2-Zellen an regulierten – blau bzw. orange gefärbten – Spots erkennbar. Zwölf Stunden nach Infektion der Zellen waren noch erheblich mehr Proteine in ihrer Abundanz verändert. Nach Auswertung der 2D-Muster konnte die vom Replikationszyklus des Virus abhängige Regulation mehrerer Wirtszellproteine gezeigt werden. Abbildung 3-5 zeigt die Regulation zweier potentiell zur Familie der Phosphodiesterasen gehörender Proteine. Die räumliche Nähe der beiden Spots zueinander legt nahe, daß es sich um zwei Isoformen eines Proteins handeln könnte. Für eine sichere diesbezügliche Aussage waren die SELDI-MS-Daten allerdings zu ungenau. 41 Ergebnisse Abbildung 3-5 Zeitabhängige Regulation mehrerer Proteine (bzw. verschiedener Isoformen eines Proteins) mit Homologie zur Familie der Phosphodiesterasen. Gegenüber der Kontrolle ist 6 h nach Infektion eine signifikante Zunahme der Intensitäten der beiden Spots rechts im Bild erkennbar; 12 Stunden nach Infektion sind die Spots fast vollständig verschwunden. Die in der Wirtszelle auf Proteomebene stattfindenden Veränderungen während der Infektion und Replikation der Viruspartikel in eine zeitliche Abfolge zu bringen, stellt also einen interessanten und vielversprechenden Ansatz dar, dessen detaillierte Untersuchung die Kenntnisse über den Replikationsvorgang und die beteiligten Komponenten der Wirtszelle entscheidend erweitern kann. 3.2.1 Veränderungen im Proteom von HepG2-Zellen 12 Stunden nach Infektion mit CVB3H3 Ausführlich wurde im Rahmen der vorliegenden Arbeit das Stadium 12 Stunden nach Infektion untersucht. Zur Infektion der HepG2-Zellen wurde Kulturflaschen mit dichtgewachsenem Zellrasen (> 95 % Konfluenz) das Medium entnommen, die Zellen mit 1-2 ml (je nach Flaschengröße) CVB3H3-Suspension überschichtet, nach 45minütiger Inkubation unter leichtem Schwenken erneut mit Medium versetzt und für 12 Stunden weiterkultiviert. Die Behandlung der Kontrollzellen erfolgte in gleicher Weise, statt Virus-Suspension erhielten diese ein äquivalentes Volumen virusfreien PBS-Puffers. Das Experiment wurde zweimal unabhängig ausgeführt. Die Proteine des jeweils gewonnenen Zellysats wurden in zwei unabhängigen Läufen pro Experiment je zweimal aufgetrennt, so daß sowohl von der nichtinfizierten Kontrolle als auch von den infizierten Zellen je 8 Gele zur Auswertung zur Verfügung standen. Nach dem Scannen der Gele wurden die Bilddateien mit der Proteinanalysesoftware Proteomweaver ausgewertet. Die Abbildung 3-5 zeigt am Computer orange und blau 42 Ergebnisse getönte und nach dem Matching virtuell übereinandergelegte 2D-Muster eines Kontrollgels und eines Gels mit dem Zellysat des 12 Stunden-Infektionsstadiums aus dem Experiment I. Zur detaillierten Auswertung war diese Darstellungsoption nicht geeignet. Abbildung 3-6 Das Proteom von nichtinfizierten und CVB3H3-infizierten, 12 Stunden postinfektional kultivierten HepG2-Zellen im orange-blau-Matching. Die Punkte des Kontrollgels wurden orange eingefärbt, die Punkte des Gels mit dem Proteom der infizierten Zellen blau. Proteine mit unveränderter Abundanz erscheinen übereinandergelegt schwarz, Spots mit größerer Intensität im 2D-Muster der Kontrolle erscheinen ± orange und Spots mit größerer Intensität im 2D-Muster des 12 Stunden-Stadiums ± blau. Ausgewählte Spots sind vergrößert dargestellt. Allerdings können auf diese Weise Abweichungen im Spotmuster unmittelbar erfaßt und anschaulich gemacht werden. In der Intensität gleichbleibende d.h. nichtregulierte Spots erscheinen schwarz, Spots mit einer Intensitätsabnahme zwischen Kontrolle und 12 Stunden-Stadium (z.B. infolge Degradation) erscheinen ± orange und Spots mit 43 Ergebnisse einer Intensitätszunahme (z.B. infolge verstärkter Expression) erscheinen ± blau. Sowohl negativ als auch positiv regulierte Spots sind zu erkennen, potentiell unmittelbar bzw. mittelbar als Folge der Infektion. Da jedoch auch durch die Probenaufarbeitung verursachte Intensitätsschwankungen einzelner Proteine mitunter ein signifikantes Niveau erreichten (siehe folgender Abschnitt), sind wahrscheinlich nicht alle in Abb. 3-6 sichtbaren Unterschiede tatsächlich die Folge der Infektion. 3.2.1.1 Abschätzung des experimentellen Fehlers Da Intensitätsunterschiede und Differenzen im Spotmuster selbst zwischen Gelen mit identischer Probe unvermeidlich waren, war es für die Bewertung und Einordnung der Ergebnisse der Proteinanalyse wichtig, diesen internen Fehler der 2D-Elektrophorese etwa abschätzen zu können. In beiden Experimenten wurden deshalb die Gele der nichtinfizierten Kontrollzellen untereinander verglichen. Diese waren mit identischer Probe gleicher Menge beladen worden, und sollten demzufolge paarweise untereinander identische 2D-Muster aufweisen. Tatsächlich vorhandene Abweichungen sind artifiziell und lassen erkennen, wie groß der interne Fehler des jeweiligen Experiments war. Tabelle 3-1 zeigt jeweils vergleichend die Daten des ersten Laufs beider Experimente. Tabelle 3-1 Festgestellte Abweichungen zwischen Gelen mit nichtinfiziertem Zellysat. Die Gele waren mit identischer Probe in gleicher Menge beladen worden. Exemplarisch sind die Ergebnisse des jeweils ersten Laufs beider Experimente dargestellt. Anzahl Abweichungen zum Intensitätsunter- im Vergleichsgel Spots/Gel Vergleichsgel (in %) schiede nicht vorhanden Kontrollgel 1 1861 237 (12,7 %) 7 230 Kontrollgel 2 1842 278 (15,1 %) 5 273 Kontrollgel 1 2422 136 (5,6 %) 3 133 Kontrollgel 2 2394 151 (6,3 %) 3 148 Experiment I: Experiment II: Der Fehler des ersten Experiments war mit durchschnittlich 13,9 % tendenziell größer als der des zweiten Experiments mit durchschnittlich 6,0 %. Das bedeutet, daß bei der anschließenden Analyse der 2D-Muster CVB3H3-infizierter und nichtinfizierter 44 Ergebnisse HepG2-Zellen rund 14 bzw. 6 % Abweichung infolge des internen Fehlers der 2DElektrophorese zu erwarten waren. Als signifikant unterschiedlich in der Intensität wurden 12 Spots im ersten und 6 im zweiten Experiment erkannt. Damit erreichten artifizielle Intensitätsunterschiede nur in wenigen Fällen ein signifikantes Niveau (Abb. 3-7d). Zum Teil war dies auf fehlerhaftes Matching, d.h. auf von der Software falsch zugeordnete Spots zurückzuführen (Abb. 3-7c). Die weitaus meisten Abweichungen ergaben sich daraus, daß Spots nur in einem der beiden Vergleichsgele detektiert wurden. In einigen Fällen wurde unzulässig Hintergrundrauschen bzw. Proteinschmier zu Spots aufsummiert (Abb 3-7a), die im Vergleichsgel folgerichtig nicht zu finden waren. Meist handelte es sich jedoch um Proteine an der Auflösungsgrenze, die zwar als Schatten in beiden Vergleichsgelen vorhanden waren, aber von der Software nur einmal detektiert wurden (Abb 3-7b). Abbildung 3-7 Verschiedene Arten des Zustandekommens artifizieller Unterschiede zwischen 2D-Mustern aus einer einzigen Probe. Bei a) und c) handelt es sich nicht um echte Unterschiede: beide sind Interpretationsfehler der Analyse-Software. Die Fälle b) und d) sind zurückzuführen auf artifizielle Abundanzunterschiede, die während der Aufarbeitung entstanden sind. a) Intensitätsunterschied infolge aufsummierten Hintergrundes; b) Spot vorhanden, aber nur einmal detektiert; c) Intensitätsunterschied infolge Falschmatching eines Spots; d) echter signifikanter Intensitätsunterschied eines Spots. Ursache für die mangelnde Spoterkennung war, daß die eingestellten Parameter zur Spoterkennung (Spotdurchmesser, Intensität, Kontrast) einen Schwellenwert definierten, der im Gel A erreicht, im Gel B jedoch unterschritten wurde. Die Erhöhung 45 Ergebnisse der Empfindlichkeit minderte zwar dieses Problem, ging aber einher mit vielen aus bloßem Hintergrundrauschen detektierten Pseudospots. 3.2.1.2 Analyse des Proteinmusters CVB3H3-infizierter HepG2-Zellen und Vergleich mit dem Proteom nichtinfizierter Zellen Die aus beiden Experimenten vorhandenen Gele wurden den beiden Gruppen „nichtinfizierte Kontrolle“ und „CVB3-infiziert“ zugeordnet und die zusammengehörenden Gele eines Laufs paarweise verglichen. Aus Spots, die nur in einem der beiden Gele aufzufinden waren, sowie aus Spots mit signifikanter Abweichung in der Intensität wurde ein Analyse-Set erstellt. Als signifikante Abweichung galt der mindestens dreifache Intensitätsunterschied eines Spots. Der paarweise Vergleich der 2D-Muster CVB3H3-infizierter und nichtinfizierter Zellen erbrachte im ersten Experiment 1367 und in der unabhängigen Wiederholung 691 signifikante Unterschiede (Tab. 3-2). Dies entsprach einer prozentualen Abweichung von 29 % für das erste und 15 % für das zweite Experiment. Das Proteom der infizierten Zellen war damit im ersten Experiment deutlich abweichender von der Kontrolle als im zweiten Experiment, was zum Teil durch den größeren Fehler des ersten Experiments erklärbar ist. Die Abb. 3-8 zeigt Beispiele der detektierten Unterschiede. Der Grund für die Zunahme bzw. Abnahme der Intensitäten einzelner Spots zwischen Kontrollgel und Gel der infizierten Zellen ist als unmittelbare bzw. mittelbare Folge der CVB3H3Infektion interpretierbar. Eine Zunahme ist erklärbar durch die alleinige Expression bzw. die verstärkte Expression des entsprechenden Proteins in den infizierten Zellen. Außerdem bewirken Modifikationen eines Vorläuferproteins, die Veränderungen der beiden Trennparameter (pI und Molekülmasse) zur Folge haben, daß der entsprechende modifizierte Zustand ausschließlich im Gel der infizierten Zellen detektierbar ist. Das Vorläuferprotein muß dann allerdings an anderer Stelle im Kontrollgel zu finden sein und im Gel der infizierten Zellen an Intensität abnehmen, weil es als Folge der Infektion in seinen modifizierten Zustand überführt wird. Eine Intensitätsabnahme kann außerdem durch den vollständigen oder teilweisen Abbau des entsprechenden Proteins hervorgerufen werden. Wurden Spots nur einmal, d.h. ausschließlich im Gel einer Kontrolle bzw. eines 12 Stunden-Stadiums detektiert (Abb 3-8a und c), bedeutet dies jedoch nicht zwangsläufig, daß die entsprechenden Proteine auf einen Zustand beschränkt vorkamen. 46 Ergebnisse a) nur in infiziert b) nur in Kontrolle c) signifikant stärker in infiziert d) signifikant stärker in Kontrolle Abbildung 3-8 Beispiele für die festgestellten Intensitätsunterschiede. Jeweils links im Bild der Ausschnitt eines Kontrollgels, rechts der gleiche Ausschnitt eines Gels des 12 StundenStadiums der CVB3H3-Infektion. Die Abundanz eines entsprechenden Proteins war immerhin soweit verändert, daß es in einem der Vergleichsgele nicht aufgelöst werden konnte. Allerdings werden die meisten der in der Zelle vorkommenden Proteine wegen ihres geringen Expressionsniveaus durch die 2D-Elektrophorese nicht erfaßt. Die zunächst automatisch generierten Analysedaten (Tab. 3-2) wurden manuell editiert, weil der begrenzte Durchsatz der SELDI-Technologie nicht die Bearbeitung aller vorhandenen Unterschiede erlaubte. Tabelle 3-2 Ermittelte Abweichungen im 2D-Muster zwischen Kontrollgelen und Gelen der infizierten Zellen beider Experimente und Anzahl weiterbearbeiteter Spots. Unterschiede (Software): nur in Kontrolle nur in infiziert Kontr. stärker infiziert stärker gesamt [%] Exp. I 709 549 62 47 1367 29 Exp. II 354 307 14 16 691 15 Subauswahl (weiterbearbeitete Spots): nur in Kontrolle nur in infiziert Kontr. stärker infiziert stärker gesamt [%] Exp. I 8 30 5 8 51 1 Exp. II 14 23 2 4 43 1 47 Ergebnisse Dabei wurden offensichtlich durch die Analysesoftware verursachte artifizielle Unterschiede (infolge fehlerhafter Detektion bzw. Falschmatching) aus dem Analyse-Set entfernt. Die im Set verbliebenen Proteinspots (Tab. 3-2) wurden aus den Gelen ausgeschnitten, mit Trypsin verdaut und mittels Peptidmassenfingerprinting am SELDIMassenspektrometer analysiert. 3.2.2 Analyse des ersten Experiments 3.2.2.1 Peptidmassenfingerprinting Zieht man die ermittelten 14 % falschunterschiedlichen Spots von den 29 % Gesamtunterschied ab, ergeben sich rein rechnerisch 15 % echte Unterschiede. In der Praxis wurde mit 51 Spots nur gut 1 % zur weiteren Bearbeitung ausgewählt. Die Massenspektren der mit Trypsin verdauten Spots wurden bearbeitet, indem Signale, die allen Spektren gemeinsam waren (wie Matrix- und Trypsin-Autolyse-Peaks), eliminiert wurden. Die verbliebenen Signale wurden als Peptidmassen interpretiert und ergaben jeweils charakteristische Fragmentmuster. Mit der Online-Anwendung ProFound wurden die in den Proteindatenbanken NCBI und SwissProt gelisteten Proteine nach der Ähnlichkeit ihrer Peptidmuster zu denen der regulierten Proteine durchsucht. Einem Teil der massenspektrometrisch analysierten Proteinspots konnte auf diese Weise ein bestimmtes Protein zugeordnet werden (Tab. 3-3). Bei allen identifizierten Proteinen handelte es sich um Proteine der Wirtszelle. Die Treffergenauigkeit war für MAPK p38, Prohibitin, Dynein light intermediate chain und Tubulin 1-chain besonders hoch. Die CVB3H3-abhängige Regulation dieser vier Proteine wurde deshalb zusätzlich auf Transkriptionsebene überprüft (Abschnitt 3.2.2.3). Sieben der identifizierten Proteine wurden ausschließlich bzw. mit signifikant höherer Intensität in den Gelen der infizierten Zellen gefunden. Diese Proteine wurden also vermutlich infektionsabhängig verstärkt exprimiert bzw. entstanden durch posttranslationale Modifikationen aus Vorläufern. Sechs der identifizierten Proteine wurden ausschließlich bzw. mit signifikant höherer Intensität in den Gelen der nichtinfizierten Kontrollzellen gefunden, was die vollständige bzw. teilweise Degradation oder Modifikation dieser Proteine im Infektionsverlauf wahrscheinlich macht. 48 Ergebnisse Tabelle 3-3 Im Experiment I regulierte und durch SELDI-Peptidmassenanalyse identifizierte Proteine. Während in der linken Spalte die Proteine abgebildet sind, die infektionsabhängig entstehen bzw. verstärkt in Erscheinung treten, werden in der rechten Spalte die Proteine aufgelistet, die infektionsabhängig vollständig bzw. teilweise verschwinden. Bei zusammengehörigen Bildpaaren wird jeweils links der Zustand in der nichtinfizierten Kontrolle dargestellt. Experiment I Intensitätszunahme infolge CVB3H3- Intensitätsabnahme infolge CVB3H3- Infektion Infektion MAPK p38 (SAPK 2a, MAPK 14) Solute carrier family 39 Hypothetical protein FLJ20651 Apolipoprotein A-IV Precursor Unnamed protein product BAB14332.1 Dynein light intermediate chain Ubiquitously-expressed transcript, Iso- Sulfated glycoprotein 2 form 2 49 Ergebnisse Tabelle 3-3 Fortsetzung Prohibitin 3’5’ Cyclic-nucleotide phosphodiesterase CTP synthetase 2 Hypothetical protein KIAA0546 Tubulin 1-chain 3.2.2.2 Nachweis der infektionsabhängigen posttranslationalen Modifikation eines Proteins Es gelang der Nachweis der infektionsabhängigen posttranslationalen Modifikation eines Proteins. Zwei Proteinspezies mit gleichem Molekulargewicht, die jeweils nur im Gel der nichtinfizierten Kontrollzellen bzw. im Gel der infizierten Zellen detektiert wurden, unterschieden sich in ihrem isoelektrischen Punkt um ca. 0,3 pH-Einheiten (Abb. 3-9). Wie die massenspektrometrische Untersuchung beider Proteinspots ergab, handelte es sich um ein und dasselbe Protein. Datenbankvergleiche erbrachten jedoch keine gute Übereinstimmung des Fragmentmusters mit denen bereits bekannter Proteine, so daß die Identifizierung und funktionelle Einordnung dieses Proteins im Rahmen der vorliegenden Arbeit nicht möglich war. Eine mögliche Erklärung für den pI-Shift bietet jedoch die einfache Phosphorylierung des Proteins, die eine Ansäuerung um ca. 0,3 pH-Einheiten bewirkt (F. Lottspeich, mündliche Mitteilung). 50 Ergebnisse pH 4,7 pH 5 a) nichtinfiziert pH 4,7 pH 5 b) 12 h CVB3H3-infiziert Abbildung 3-9 Nachweis der CVB3H3-abhängigen posttranslationalen Modifikation eines Wirtszellproteins mit der Technik der 2D-Gelelektrophorese. Bei dem rot umrandeten Proteinspot handelt es sich in beiden Bildern um das gleiche Protein. Der infektionsabhängige pI-Shift könnte die Folge einer Phosphorylierungsreaktion sein. 3.2.2.3 Bestätigung der infektionsabhängigen Regulation der Mitogenaktivierten Proteinkinase p38 (MAPK p38) durch RT-PCR Die gefundene infektionsabhängige Regulation von Prohibitin, Dynein light intermediate chain, Tubulin 1-chain und MAPK p38 wurde zusätzlich auf Transkriptionsebene überprüft. 1 2 3 4 5 6 7 8 9 10 11 12 M Dies geschah durch RT-PCR. Mit dieser Methode sind quantitative Untersuchungen des Transkriptoms möglich. Dazu wurde die GesamtRNA von CVB3H3- infizierten bzw. nichtinfizierten HepG2-Zellen isoliert und in cDNA umgeschrieben. Die Zahl identischer Abbildung 3-10 Agarosegel der RT-PCR-Produkte von Prohibitin, Dynein light intermediate chain, Tubu- mRNA-Transkripte wird bei diesem einfachen Kopiervor- lin 1-chain und MAPK p38. 1;2 = MAPK p38 , gang in die gleiche Zahl 3-6 = Kontrollen: 3;4 = -Aktin, 4;6 = Kapsidpro- cDNA-Moleküle übersetzt, tein VP1, 7;8 = SIVA, 9;10 = Dynein light interme- so daß die ursprünglichen diate chain, 11;12 = Tubulin 1-chain. mRNA-Abundanzen unver- 51 Ergebnisse ändert übernommen werden. Nach einer PCR-Reaktion und der anschließenden Trennung der PCR-Produkte im DNA-Agarosegel ist an der Bandenstärke das ursprünglich in vivo vorhandene Transkriptionsniveau der interessierenden Gene anhand der entsprechenden amplifizierten mRNA-Moleküle ablesbar. Für MAPK p38 konnte ein sehr deutlicher Anstieg des Expressionsniveaus infolge der Infektion mit CVB3H3 auch auf Transkriptionsebene gezeigt werden (Abb.3-10), was übereinstimmt mit der Detektion des als MAPK p38 identifizierten Proteinspots ausschließlich im Gel der infizierten Zellen.Für die Proteine Prohibitin, Dynein und Tubulin 1-chain war die im 2D-Gel auf Proteinebene erkennbare Regulation nicht in gleicher Weise durch RT-PCR auf mRNA-Ebene nachvollziehbar. 3.2.3 Analyse des zweiten Experiments 3.2.3.1 Peptidmassenfingerprinting Die ausgewählten und ausgeschnittenen 43 Proteinspots wurden analog zu den Spots des ersten Experiments mit Trypsin verdaut. Die Massen der resultierenden Peptidfragmente wurden per SELDI-MS bestimmt. Wegen eines Defekts am Detektor des SELDI-Gerätes waren diese Untersuchungen jedoch nicht erfolgreich. Deshalb wurden die Spots der entsprechenden Parallelgele ausgeschnitten und der Arbeitsgruppe Proteinanalytik von Prof. F. Lottspeich, Max-Planck-Institut für Biochemie, Martinsried bei München zur Untersuchung am Massenspektrometer (MALDI-TOF-TOF) zugesandt. Neben einigen Spots, die nicht identifizierbar waren, wurden verschiedene Isoformen von Aktin und Tubulin detektiert, die infektionsabhängig in höherer Konzentration vorlagen. Erneut wurde beispielsweise eine infektionsabhängige Intensitätssteigerung des Tubulin 1-Spots nachgewiesen. Diese gefundenen Veränderungen besitzen allerdings vermutlich geringe Aussagekraft, denn als Zellstrukturproteine sind Aktin und Tubulin einem ständigen Auf- und Abbau unterworfen, unabhängig von einem pathogenen Befall der Zelle. Der im ersten Experiment als MAPK p38 identifizierte und im zweiten Experiment in gleicher Weise regulierte Spot konnte im zweiten Experiment wegen unzureichender Signalstärke leider nicht erneut identifiziert werden. 52 Ergebnisse Tabelle 3-4 Im Experiment II regulierte und durch MALDI-Peptidmassenanalyse identifizierte Proteine. Während in der linken Spalte die Proteine abgebildet sind, die infektionsabhängig entstehen bzw. verstärkt in Erscheinung treten, werden in der rechten Spalte die Proteine aufgelistet, die infektionsabhängig vollständig bzw. teilweise verschwinden. Bei zusammengehörigen Bildpaaren wird jeweils links der Zustand in der nichtinfizierten Kontrolle dargestellt. Experiment II Intensitätszunahme infolge CVB3H3- Intensitätsabnahme infolge CVB3H3- Infektion Infektion nicht zu identifizieren (MAPK p38) ATP-dependent RNA helicase DDX19 (DEAD-box protein 19) Tubulin 1-chain Protein disulfide isomerase, Precursor (PDI) ATP-dependent DNA Helicase II, 80 kDa Plasma retinol-binding protein, Precursor -Aktin, -Aktin Eukaryotischer Translationsinitiationsfaktor 3, Untereinheit 6 (eIF-3 p48) 53 Ergebnisse Tabelle 3-4 Fortsetzung UTP-Glucose-1-phosphate uridylyltransferase 2 3.2.4 Übereinstimmend regulierte Proteine In Abhängigkeit der Infektion mit CVB3H3 entstandene Intensitätsunterschiede einzelner Proteinspots sollten reproduzierbar in allen Wiederholungen des Experiments in gleicher Weise wiederkehren. Mit hoher Übereinstimmung wurde die infektionsabhängige Intensitätssteigerung des als MAPK p38 identifizierten Spots detektiert. Insgesamt wurden sieben der 51 ausgewählten Spots des ersten Experiments unter den 43 ausgewählten Spots des zweiten Experiments gleichreguliert an gleicher Stelle im 2D-Muster wiedergefunden. Abgesehen von MAPK p38 konnten diese Proteine bisher nicht identifiziert werden, weil keine entsprechenden Fragmentmuster in den Datenbanken gefunden wurden. Es ist also davon auszugehen, daß die gefundenen Proteine bisher unbekannt sind bzw. bisher nicht sequenziert wurden. 54 Diskussion 4 Diskussion 4.1 Etablierung der Zweidimensionalen Gelelektrophorese Im Rahmen dieser Arbeit wurde zunächst die Zweidimensionale Gelelektrophorese etabliert und für die geplanten Proteomuntersuchungen an humanen Leberkarzinomzellen optimiert. Die Aufgabe dabei war, ein Protokoll zu entwickeln, das einfach anzuwenden ist und gute Reproduzierbarkeit mit einer möglichst hohen Auflösung kombiniert. 4.1.1 Reproduzierbarkeit der 2D-Gelelektrophorese Obwohl bereits nahezu 30 Jahre bekannt, hat die 2D-Gelelektrophorese erst in den letzten Jahren breite Anwendung erfahren, nicht zuletzt aufgrund zahlreicher Detailverbesserungen, welche die Reproduzierbarkeit der Methode deutlich erhöhten. Trotzdem ist die Handhabung relativ schwierig geblieben. In erster Linie existiert kein allgemeingültiges Rezept, das die Proteine aus Proben verschiedener Herkunft gleichermaßen gut auftrennt. Zudem ist nach wie vor eine gewisse Kunstfertigkeit im Umgang mit den Gerätschaften erforderlich. Die Reproduzierbarkeit der Ergebnisse eines Experiments ist an die Vergleichbarkeit der Spotmuster des Experiments und seiner unabhängigen Wiederholungen gebunden. Wie die unter 3.1 vorgestellten Ergebnisse deutlich machen, können auch kleine Abweichungen in den Versuchsbedingungen großen Einfluß auf die Qualität des 2DMusters haben. Eine gute Reproduzierbarkeit ist deshalb erst mit einem auf die jeweilige Fragestellung abgestimmten Protokoll gegeben und hängt in starkem Maße von der einwandfreien Gleichhaltung der äußeren Bedingungen ab. Mit dem unter 3.1.2 vorgestellten Protokoll wurde für die anvisierten Untersuchungsobjekte – humane Zellinien wie HepG2 oder HeLa – bei relativ einfacher Handhabung eine zuverlässige Reproduzierbarkeit erreicht. Die Vergleichbarkeit der Spotmuster der bis zu 12 Gele, die mit der vorhandenen Ausrüstung in einem 2DElektrophorese-Lauf möglich sind, war sehr gut. Trotzdem waren Abundanzschwankungen einzelner Proteine bei parallel aufgearbeiteten und vollkommen gleichbehandelten Proben nicht zu vermeiden. Die Ursachen sind systembedingt und können sehr vielgestaltig sein. Zum einen sind der Genauigkeit gerätetechnisch Grenzen gesetzt: 55 Diskussion Alle verwendeten Geräte einschließlich Densitometer und Analysesoftware arbeiten mit einem inneren Fehler. Beispielsweise sind in der Vertikalkammer bauartbedingt kleine Temperaturdifferenzen zwischen verschiedenen Gelen unvermeidlich, was zu Abweichungen im Spotmuster führen kann. Auch Pipetten arbeiten mit einem inneren Fehler. Infolge des Pipettenfehlers gering voneinander abweichende Proteinmengen verschiedener Proben können verursachen, daß ein Protein an der Auflösungsgrenze im Gel der Probe I gerade noch erkennbar, aber im Gel der Probe II nicht mehr vom Hintergrund zu unterscheiden ist. Zum anderen ist die manuelle Ausführung der notwendigen Arbeitsschritte als Fehlerursache in Betracht zu ziehen. Als besonders fehleranfällige Arbeitsschritte stellten sich die Proteingehaltsbestimmung, die isoelektrische Fokussierung und die Übertragung der IPG-Streifen auf die SDS-PAA-Gele der zweiten Dimension heraus. Die Beachtung der zu den angeführten Punkten im Ergebnisteil unter 3.1.1 erarbeiteten Hinweise und die möglichst vollständige Gleichhaltung aller beeinflußbaren experimentellen Bedingungen verringerte die Fehlergröße beträchtlich. Bei der vergleichenden Analyse verschiedener Spotmuster im experimentellen Teil der Arbeit war dieser absolute Fehler zu berücksichtigen. So war die Abgrenzung tatsächlicher Abundanzunterschiede von experimentellen Artefakten teilweise schwierig. Durch Wiederholung der Experimente konnten Artefakte allerdings erfaßt und von tatsächlichen regulierten Proteinen (mit differentem Expressionsniveau) unterschieden werden. Die 2D-Muster der Gele verschiedener Läufe waren direkt nur eingeschränkt vergleichbar, in erster Linie deshalb, weil die Proteinanalysesoftware als Folge von laufbedingten Unterschieden des Spotmusters teilweise Probleme hatte, einzelne Proteinspots einander zuzuordnen. Proben, deren 2D-Muster verglichen werden sollten, wie z.B. CVB3-infizierte und nichtinfizierte Zellen eines Experiments, wurden deshalb immer parallel behandelt und parallel, d.h. in einem Lauf, in der zweiten Dimension aufgetrennt. Die Reproduzierung eines Experiments erfolgte mehrmals unabhängig, jeweils mit unabhängiger Auswertung. Die herausgefilterten Unterschiede im Spotmuster wurden verglichen und annotiert. Erscheint ein Unterschied zuverlässig in allen Wiederholungen (mind. fünfmalig; F. Lottspeich, mündliche Mitteilung) des Experiments, gilt dieser als real, d.h. ein experimentelles Artefakt kann ausgeschlossen werden. 56 Diskussion Es war nicht möglich, die Ergebnisse der in der vorliegenden Arbeit ausgeführten Experimente auf diese Weise statistisch abzusichern. Einige der unter 3.2 gezeigten Daten tragen deshalb vorläufigen Charakter und bedürfen weiterer Verifizierung. 4.1.2 Verbesserung des Auflösungsvermögens der Zweidimensionalen Gelelektrophorese 4.1.2.1 Solubilisierung der Proteine Für Proteomuntersuchungen sollte die Solubilisierung der Proteine einer Probe so umfassend wie möglich erfolgen, denn nur vollständig gelöste Proteine werden ordnungsgemäß fokussiert. Der Probenpuffer/Solubilisierungspuffer stellt immer einen Kompromiß dar zwischen der möglichst umfassenden Solubilisierung aller Proteine einer Probe und der Verträglichkeit der verwendeten Chemikalien mit den Gegebenheiten der IEF. Beispielsweise bleibt die Verwendung des sehr effektiven anionischen Detergenz SDS für die 2D-Gelelektrophorese die Ausnahme, weil die gebildeten negativ geladenen Protein-SDS-Mizellen die Eigenladungen und damit die isoelektrischen Punkte der Proteine vollständig überdecken. Die Fokussierung unter Anwesenheit von SDS kann überhaupt nur bei kathodischem Probenauftrag, wobei die Probe während der IEF über kleine Trichter (sample cups) appliziert wird, funktionieren. Die negativ geladenen Protein-SDS-Mizellen müssen dann während des Laufs in Gegenwart von Harnstoff aufgelöst werden. Allerdings gilt der kathodische Probenauftrag als problematisch, weil in hohem Maße mit Proteinaggregation an der Auftragsstelle verbunden (Westermeier, 1990). Eine beispielsweise zur Solubilisierung von Membranproteinen (Santoni et al., 2000) verwendete Variante besteht aus der initialen Verwendung von SDS im Solubilisierungsgemisch. Vor der Fokussierung wird das SDS durch Verdünnung der Probe mit einem nicht- bzw. zwitterionische Detergenzien enthaltenden Puffer von den Proteinen verdrängt (Harder et al., 1999). Trotzdem kann SDS auch in geringen Konzentrationen unscharf fokussierte Proteine und damit horizontale Streifen im 2D-Muster verursachen. Zur Verbesserung der Löslichkeit bei gleichzeitiger Verträglichkeit mit der IEF sind zahlreiche Arbeiten veröffentlicht worden. Häufig wird die stark chaotrope Substanz Thioharnstoff als Bestandteil des Solubilisierungspuffers empfohlen (Rabilloud, 1998; Rabilloud et al., 1997). 57 Diskussion Als Detergenzien wurden traditionell häufig Triton X-100 und Nonidet P-40 benutzt. Wegen deren Unverträglichkeit mit der Silberfärbung (starke Hintergrundfärbung) kommt heute vielfach das zwitterionische Sulfobetain CHAPS zur Anwendung. Aber auch neuartige zwitterionische Detergenzien, z.B. aus der Gruppe der Amidosulfobetaine (Chevallet et al., 1998) wurden getestet. Als Reduktionsmittel wird häufig DTT eingesetzt. Allerdings liegt es im alkalischen Milieu geladen vor und wandert deshalb während der Fokussierung Richtung Anode. Es kommt zur Unterversorgung des basischen Bereichs des Gels mit DTT, was zur Reoxidation von SH-Gruppen und damit einhergehend zu reduzierter Löslichkeit verschiedener Proteine führen kann (Görg et al., 2000). Vor allem für extrem basische Gradienten (bis > pH 12) wird deshalb das ungeladene Tributylphosphin (TBP) empfohlen (Santoni et al., 2000). Für die vorliegende Arbeit wurde als Lyse-, Solubilisierungs- und Rehydratisierungslösung zugleich der Thioharnstoff-Puffer von Rabilloud (Rabilloud, 1998) in leicht modifizierter Form verwendet. Als chaotrope Substanz enthält dieser Thioharnstoff, als Detergenz das zwitterionische CHAPS und als Reduktionsmittel DTT. Die unzureichende Auflösung des basischen Bereichs bei Verwendung von DTT konnte bestätigt werden. Statt DTT wurde deshalb das seit kurzer Zeit kommerziell erhältliche Reagenz DeStreak (Amersham, Freiburg) nach Herstellerangabe der Rehydratisierungslösung zugesetzt, was eine deutlich verbesserte Auflösung des basischen Bereichs zur Folge hatte. Die Ultraschallbehandlung des Lysats war nützlich, weil Membranen und hochmolekulare Nukleinsäuren wirksam zerkleinert wurden und die Probe dadurch weniger viskos erschien. An Membransystemen oder Nukleinsäuren assoziierte Proteine werden mechanisch abgetrennt und gelangen so besser in Lösung (Görg et al., 2000). 4.1.2.2 Ultrafiltration oder TCA/Aceton-Fällung? Beide Methoden sind in der Handhabung einfach und sicher. Die TCA/AcetonFällung ist im Vergleich zur Ultrafiltration mit geringerem Material- und Kostenaufwand verbunden. Vom Zeitbedarf her ähneln sich beide Methoden: jeweils 4 h sind zu veranschlagen. Im folgenden werden die Auswirkungen beider Methoden auf das 2D-Muster des HepG2-Proteoms diskutiert. Die Abbildung 3-2 macht deutlich, daß die Auflösung des Spotmusters, die sich durch die Anzahl darstellbarer Proteine beschreiben läßt, bei 58 Diskussion TCA-Fällung um mehr als 25 % höher ist als bei Ultrafiltration des Zellysats. Einzelne Proteinspots, die nach der TCA-Fällung deutlich erschienen, waren nach paralleler Ultrafiltration der gleichen Probe kaum zu sehen oder nicht vorhanden (Abb. 3-2). Möglicherweise verantwortlich dafür ist der vor der Ultrafiltration notwendige Ultrazentrifugationsschritt, der unlösliche Bestandteile der Probe abtrennt. Dies sind hauptsächlich Teile der Zellmembran bzw. der zellinternen Membransysteme einschließlich integrierter bzw. assoziierter Proteine. Diese Proteine werden durch die TCA/AcetonFällung potentiell zugänglich für die 2D-Analyse, weil hier in Gegenwart von Aceton die Lipidschichten von den Membranproteinen entfernt werden (Rabilloud, 1996). Damit ist die Methode geeignet, Membranproteine anzureichern. Da die Spots jedoch im einzelnen nicht durch massenspektrometrische Analyse identifiziert wurden, ist nicht sicher zu entscheiden, ob die nach Ultrafiltration der Probe fehlenden Proteine als experimentelle Artefakte während der Fällung (z.B. durch pI-Shift infolge Carbamylierung oder Degradation) erzeugt wurden. In der Literatur finden sich allerdings keine Hinweise auf derart destruktive Eigenschaften der TCA/Aceton-Fällung. Der Grund für die bessere Auflösung der äußeren pH-Bereiche bei der TCA/AcetonFällung liegt in der im Vergleich zur Ultrafiltration besseren Reinigungsleistung begründet. Denn Lipide und Salze behindern die IEF erheblich und werden bei der Fällung durch Waschen des Proteinpellets mit Aceton effektiv entfernt (Görg et al., 1997), was durch Ultrafiltration der Proben vermutlich nicht in diesem Maße gegeben ist. Bei der TCA-Aceton-Fällung sind allerdings qualitative Proteinverluste durch inkomplette Fällung bzw. durch unvollständige Resolubilisierung des getrockneten Pellets im Anschluß an die Fällung nicht auszuschließen (Görg et al., 1997). Theoretisch ebenso möglich sind artifizielle Modifikationen einzelner Proteine, woraus sich ein verändertes Laufverhalten dieser Proteine ergeben kann. Wegen der insgesamt deutlichen Vorteile gegenüber der Ultrafiltration wurde die TCA/Aceton-Fällung in das Protokoll für Proteomuntersuchungen an HepG2-Zellen integriert. 4.1.2.3 Bedingungen der Isoelektrischen Fokussierung Für ein hochauflösendes Ergebnis der 2D-Gelelektrophorese war vor allem die bestmögliche Isoelektrische Fokussierung der Proteine entscheidend. Neben den bereits 59 Diskussion diskutierten Maßnahmen, die letztlich alle der verbesserten Fokussierung dienten, waren die Bedingungen während der Fokussierung selbst ebenso wichtig. Fokussierzeiten: Fokussiert wurde so lange, bis bei maximaler angelegter Spannung kein weiterer Stromstärkeabfall mehr stattfand, bis also eine Art Fließgleichgewichtszustand (steady state) erreicht war. Die anfänglich höhere Leitfähigkeit der Streifen wird mit der Zeit, wenn in der Probe verbliebene Ionen ausgewandert sind und die ersten niedermolekularen Proteine ihren pI erreicht haben, immer geringer. Deshalb wurde, um die Stromstärke über die Dauer der Fokussierung etwa konstant zu halten, die Spannung stufenweise angehoben (siehe Tabelle 2-1). Zu hohe anfängliche Stromstärken resultieren in Präzipitatbildung und verstärkter Elektroendoosmose, was zu Leitfähigkeitslücken bzw. zerstörten Gelen führen kann (Westermeier, 1990). Proben mit relativ hoher verbliebener Salzkonzentration können zunächst eine Stunde bei 50 V „entsalzt“ werden (Görg et al., 2000). Temperatur: Die pH-Gradienten der IPG-Streifen variieren in Abhängigkeit der Temperatur. Görg und Mitarbeiter konnten zudem temperaturabhängige ungerichtete Positionsverschiebungen einzelner Proteine nachweisen (Görg et al., 1991). Daher ist die Temperaturkontrolle bei der Isoelektrischen Fokussierung für die Reproduzierbarkeit der 2D-Gelelektrophorese wichtig. Erhöhte Temperaturen bei der IEF wirken sich positiv auf die Auflösung des 2D-Musters aus (Görg et al., 1991). Entgegen der früher empfohlenen 10 °C wird heute meist bei 20 °C fokussiert. Die meisten kommerziell erhältlichen Streifen sind dementsprechend für diese Temperatur konfektioniert. 4.2 Coxsackievirus B3-induzierte Änderungen der Proteinausstattung von HepG2-Zellen Nachdem die 2D-Elektrophorese etabliert und die Parameter soweit optimiert waren, daß das Proteom von HepG2-Zellen reproduzierbar in hoher Auflösung dargestellt werden konnte, wurde die 2D-Elektrophorese benutzt, um die Mechanismen der Coxsackievirus-Infektion am Zellkulturmodell auf Proteomebene zu studieren. Dabei war zunächst die Frage interessant, inwieweit die 2D-Elektrophorese in der Lage ist, die bei einer Infektion auftretenden Änderungen des Proteoms der Wirtszelle zu visuali- 60 Diskussion sieren. Die Frage, ob potentiell für die Replikation und Pathogenese des Virus wichtige Änderungen lokalisierbar wären, war der nächste Schritt. 4.2.1 Die Zellkultur als Modell Um den Verlauf der Virusinfektion im Herzmuskel detaillierter untersuchen zu können, wird häufig die Maus als Tiermodell genutzt. Zahlreiche Tiere mit unterschiedlichem genetischen Hintergrund stehen zur Verfügung und eine Reihe humanpathogener Virusstämme sind an das murine System adaptiert worden. Das murine Modell erweist sich aber für Proteomuntersuchungen als nur bedingt geeignet, da externe, proteomverändernde Einflüsse (z.B. Streß) schwer auszuschließen sind. Weil die befallenen Gewebsareale diffus im Herzmuskel verteilt vorliegen, wären Gewebsproben befallener Herzen daher inhomogen und würden sich aus infizierten und nichtinfizierten Myozyten, verschiedenen Immunzellen und anderen angrenzenden Zelltypen zusammensetzen. Moderne Mikromanipulationsmethoden wie die Laser Capture Microdissection (LCM) liefern zwar weitgehend homogenes Material, die erhaltenen Mengen wären für eine präparative Analyse größerer 2D-Gele aber vollkommen unzureichend. Die Zellkultur stellt dagegen ein vergleichsweise einfach zu überwachendes Modellsystem dar. Größere Mengen homogenen Materials können problemlos bereitgestellt werden. Allerdings werden die Verhältnisse im Organismus sehr stark vereinfacht nachgestellt. Soll aber der Verlauf der CVB3-Infektion per se untersucht werden, ist diese Vereinfachung zulässig. Die verwendeten HepG2-Zellen entstammen einem hepatozellulären Karzinom und werden hinsichtlich ihrer erhaltenen physiologischen Eigenschaften als relativ natürlich angesehen (Aden et al., 1979; Knowles and Aden, 1983; Knowles et al., 1980). Sie sind durch CVB3H3 infizierbar, es kommt zur Virusreplikation und das Virus verursacht zytopathische Effekte im Zellrasen. Damit sind HepG2-Zellen als zelluläres Modell zum Studium der CVB3-Infektion geeignet. 4.2.2 Durch 2D-Elektrophorese detektierte Unterschiede Wie die vorliegenden Proteomuntersuchungen der CVB3H3-infizierter HepG2-Zellen zeigen, kann die 2D-Elekrophorese einen wichtigen Beitrag zur Aufklärung an Virusreplikation und -ausbreitung beteiligter Wirtszellproteine leisten. 61 Diskussion Es konnten deutliche Unterschiede im Proteom nichtinfizierter und CVB3H3infizierter HepG2-Zellen nachgewiesen werden. Auch die infektionsabhängige Modifikation eines Wirtszellproteins aus der Familie der Phosphodiesterasen, möglicherweise durch Phosphorylierung eines Aminosäurerestes, wurde belegt. Zudem wurde die infektionsabhängige Induktion der Mitogen-aktivierten Proteinkinase p38 (MAPK p38) durch gesteigerte Expression dokumentiert. 4.2.2.1 Fehlerraten beider Experimente Wie der Vergleich der Kontrollgele ergab, lag der Fehler des ersten Experiments bei etwa 14 % und der des zweiten Experiments (einer unabhängigen Wiederholung) bei etwa 6 %. Das heißt, 14 bzw. 6 % aller detektierten Spots wiesen signifikante Intensitätsunterschiede auf, obwohl in den Vergleichsgelen dieselbe Probe in gleicher Menge und unter identischen Bedingungen aufgetrennt wurde. Für den Vergleich zwischen Kontrollgel und Gel der infizierten Zellen bedeutete dies, daß etwa 14 % bzw. 6 % der detektierten Intensitätsunterschiede durch experimentelle Artefakte hervorgerufen wurden. Von den sich bei 29 % (Exp. I) bzw. 15 % (Exp. II) Gesamtunterschied rechnerisch ergebenden 15 % bzw. 9 % echten Unterschieden wurde jedoch mit 51 (Exp. I) bzw. 43 Spots (Exp. II) nur ein Bruchteil weiterbearbeitet, weil die Auswahl durch visuelle Auslese stark eingeschränkt werden mußte. Das bedeutet, neben offensichtlichen Artefakten wurden wahrscheinlich zahlreiche echte Unterschiede aussortiert. Die geringe Übereinstimmung beider Experimente hinsichtlich in gleicher Weise regulierter Spots liegt zumindest teilweise hierin begründet. Denn auf Übereinstimmung überprüfbar waren nur die endgültigen, manuell editierten Analyse-Sets. Es besteht die Möglichkeit, daß ausgewählte Spots des ersten Experiments im zweiten Experiment in gleicher Weise reguliert waren, aber zugunsten anderer aus dem Analyse-Set aussortiert wurden. Die Bearbeitung deutlich größerer Mengen verbot sich jedoch aus wirtschaftlichen Gesichtspunkten, denn die SELDI-Technik arbeitet sehr kostenintensiv und ist für einen vergleichsweise geringen Probendurchsatz konstruiert. Moderne MALDI-MS-Geräte bearbeiten Chips im 96 well- Format (oder größer) mit hoher Geschwindigkeit und Genauigkeit und bieten damit die Möglichkeit, auch mehrere Hundert Spots eines Gels zu analysieren. Sofern die experimentellen Möglichkeiten vorhanden sind, sollten deshalb alle Spots mit detektierten Unterschieden massenspektrometrisch analysiert und tatsächlich re- 62 Diskussion gulierte Proteine von Artefakten durch mehrfache Wiederholung des Experiments unterschieden werden. 4.2.2.2 Die Mitogen-aktivierte Proteinkinase p38 (MAPK p38) Sowohl im Experiment I als auch in der unabhängigen Wiederholung (Exp. II) wurde im 2D-Gel CVB3H3-abhängig eine starke Intensitätszunahme des MAPKinase p38Proteinspots detektiert. Zunächst konnte nicht geklärt werden, ob diese Intensitätszunahme auf eine Aktivierung infolge Phosphorylierung oder auf die gesteigerte Transkription des MAPK p38-Gens zurückzuführen war. Durch RT-PCR konnte die infektionsabhängige Induktion der MAPK p38-Gens auf Transkriptionsebene nachgewiesen werden, weshalb eine infektionsabhängige Zunahme der Proteinmenge wahrscheinlich ist. Mitogen-aktivierte Proteinkinasen sind evolutionär konservierte Enzyme, die Zelloberflächenrezeptoren mit regulatorischen Proteinen verbinden. Die p38-Signalwege werden im allgemeinen durch inflammatorische bzw. streßauslösende Stimuli aktiviert und sind in diskreten parallelen Signalkaskaden organisiert, in denen eine MAPK Kinase Kinase (MKKK) eine dualspezifische MAPK Kinase (MKK) durch Phosphorylierung aktiviert, und diese wiederum aktiviert die MAPK durch doppelte Phosphorylierung an einem Threonin- und einem Tyrosin-Rest in einem Thr-Gly-TyrMotiv (Clark et al., 2003). Von p38 MAPKinasen weiß man, daß sie vielfältige regulatorische nachgeordnete Proteine durch Phosphorylierung aktivieren. Als direkt bzw. indirekt durch p38 aktivierte Zielobjekte sind verschiedene Transkriptionsfaktoren (unter anderem MEF 2C/2A, p53, CREB, SRF, STAT1, SAP1), Proteinkinasen (wie MK2, MK3/3pK, MNK1, MSK1/2) und andere Proteine wie cdc25B, die Hitzeschockproteine Hsp25/27 und der Translationsinitiationsfaktor eIF-4E bekannt (Shi and Gaestel, 2002). Das bedeutet, die p38 MAPKinase-Kaskade ist die zentrale Schaltstelle eines weit verzweigten Signalnetzwerkes. Die durch MAPK p38 aktivierten Transkriptionsfaktoren MEF 2C und 2A (myocyte enhancer factors) binden die Promotorregionen zahlreicher muskelspezifischer Proteine. Dem MAPK p38-Weg wird deshalb eine Rolle bei der Kardiogenese, einschließlich Kardiomyozyten-Differenzierung, -Apoptose und -Hypertrophie zugesprochen (Davidson and Morange, 2000). In Monozyten resultiert die Aktivierung von MEF 2C in vermehrter Transkription des unmittelbar frühen Gens c-jun (Han et al., 1997), weshalb vermutet wird, daß MAPK p38 über die Regulation der Produkti- 63 Diskussion on von c-Jun an Abwehrprozessen und Entzündungsreaktionen beteiligt ist. Die dauerhafte Aktivierung von MAPK p38 in transgenen Mäusen mit herzspezifisch konstitutiv aktivierten MAPKinase Kinasen (MKK3 und MKK6) führte zum frühzeitigen Tod der Tiere. Das pathologische Bild äußerte sich in stark vergrößerten Vorkammern, deutlicher interstitieller Fibrosierung, verminderter Myozytenkontraktilität und einem für Herzschwäche charakteristischem Expressionsprofil verschiedener fetaler Gene (Liao et al., 2001). In Primärkulturen von Myozyten neonataler Ratten verursachte die Aktivierung der MAPK p38 durch MKK3 eine Induktion des programmierten Zelltodes, während die Aktivierung von p38 der Apoptose-Induktion antagonistisch entgegenwirkte (Ravingerova et al., 2003; Wang et al., 1998). Eine CVB3-abhängige Aktivierung der MAP Kinase p38 wurde bisher nicht beschrieben. Allerdings ist bekannt, daß die Transduktion von Adenovirus-Vektoren in humane Nierenepithel-Zellen (REC) über die Aktivierung von MAPK p38 zur Expression des pro-inflammatorischen Chemokins IP 10 (interferon-inducible protein 10) führt. Die Aktivierung der MAPK p38 erfolgt jedoch unabhängig vom CAR (Coxsackie- und Adenovirus-Rezeptor) (Tibbles et al., 2002). Der Kerntransport der Adenovirus-RNA über den mikrotubuliabhängigen Dynein/Dynactin-Motorkomplex scheint ebenfalls durch die p38 MAPK-Kaskade (einschließlich der nachgeordneten Proteinkinase MK2) vermittelt zu sein (Suomalainen et al., 2001). Die Nutzung des Dynein/Dynactin-Multiprotein-Motorkomplexes als Transportband Richtung Zellkern ist in vielen Virusfamilien verbreitet. Von Coxsackieviren ist eine Interaktion zwischen Virushülle und einer 8 kDa-Untereinheit des Komplexes, Dynein light chain 8 (LC8), bekannt (Martinez-Moreno et al., 2003). Somit könnte die in HepG2-Zellen ermittelte Coxsackievirus-induzierte Aktivierung von MAPK p38 in Herzmuskelzellen einerseits direkt in das Infektionsgeschehen involviert sein. Denkbar wäre hier zum Beispiel eine Rolle bei der Vermittlung des Kerntransports der Viruspartikel bzw. der CVB3-RNA, wie bei Adenoviren nachgewiesen. Andererseits ist die Aktivierung im Zuge einer Abwehrreaktion beispielsweise unter Vermittlung pro-inflammatorischer Signale denkbar. Bei Chronifizierung der Erkrankung könnte MAPK p38 am komplexen Geschehen der Ausprägung einer dilatativen Kardiomyopathie beteiligt sein. Da im Zusammenhang einer CVB3Infektion auch über apoptotische Vorgänge berichtet wurde (Carthy et al., 1998; Hen- 64 Diskussion ke et al., 2000), ist auch in diesem Zusammenhang eine Beteiligung des p38 MAPKSignalweges nicht auszuschließen. Zur genaueren Charakterisierung der Rolle von MAPK p38 bei CVB3-Infektionen sollte zunächst durch Einsatz eines klassischen MAPK-Inhibitors wie SB203580, einem Pyridinyl-Imidazol, im Zellkulturmodell überprüft werden, ob bei Inhibierung von MAPK p38 die Infektionsfähigkeit bzw. die Replikationsrate von CVB3 herabgesetzt sind. Sollte dies der Fall sein, wäre eine direkte Beteiligung des MAPK p38-Signalweges am Infektionsgeschehen wahrscheinlich. Sollten keine Einflüsse auf die Vermehrungsrate feststellbar sein, ist es wahrscheinlich, daß MAPK p38 indirekt durch Streß- bzw. pro-inflammatorische Stimuli aktiviert wird. In der vorliegenden Arbeit wurde eine Induktion der MAPK p38 infolge gesteigerter Transkription nachgewiesen. Um die phosphorylierte, physiologisch aktive Form der MAPK p38 im infizierten Gewebe direkt nachzuweisen, sollte der MAPK p38-Phosphorylierungsgrad vor und während der Infektion z. B. über Western Blotting mit gegen PP-MAPK p38 gerichteten Antikörpern untersucht werden. Von großem Interesse wären auch Kenntnisse über den genauen Aktivierungsmechanismus sowie über stromab aktivierte Effektormoleküle. Sollte sich eine Beteiligung des MAPK p38-Weges an der CVB3-induzierten Ausprägung einer chronischen Myokarditis bzw. Dilatativen Kardiomyopathie bestätigen, wäre dies von großem therapeutischem Interesse, denn es würden sich neue Ziele für eine medikamentöse Behandlung der Erkrankung ergeben. 4.2.3 Ausblick Die vorliegende Arbeit bietet mehrere Ansatzpunkte für weiterführende Untersuchungen. Bereits in der vorangegangenen Diskussion wurden einige ungelöste Fragen aufgegriffen. An dieser Stelle sollen diese Fragen und mögliche Strategien zu ihrer Beantwortung kurz zusammengefaßt werden. Zunächst bedürfen die vorliegenden Ergebnisse weiterer Verifizierung. Das beschriebene Experiment der zweidimensionalen elektrophoretischen Untersuchung des Proteoms CVB3H3-infizierter HepG2-Zellen sollte zu diesem Zweck und zum ergänzenden Erkenntnisgewinn noch mehrmals wiederholt werden. Zusätzlich sollten diese Untersuchungen auch auf andere infizierbare humane Zellkulturen ausgeweitet werden. Gleichen sich die Ergebnisse in verschiedenen Zelltypen, wird die Übertragung 65 Diskussion auf das natürliche System plausibler. Von großem Wert wäre in diesem Zusammenhang die Gewinnung einer Myozyten-Primärkultur aus der Maus. Die MAPK p38, die, wie in der vorliegenden Arbeit gezeigt werden konnte, im Zellkulturmodell CVB3H3-abhängig induziert wird, könnte eine wichtige Rolle bei Pathogenese spielen. Wie bereits angedeutet, sollte zunächst geklärt werden, ob die Inhibierung des Enzyms die Infektionsfähigkeit des Virus herabsetzt. Ebenso eine Untersuchung des Phosphorylierungsgrad der MAPK p38 vor bzw. nach Infektion mit CVB3 erfolgen, denn die Aktivierung der Proteinkinase erfolgt durch doppelte Phosphorylierung. Um zu erfahren, auf welchem Weg die Induktion erfolgt, wäre eine Analyse des Promotors zu erwägen. Dies könnte Bindestellen für Proteine offenbaren, deren Beteiligung am Infektionsgeschehen bereits bekannt ist bzw. wichtige Hinweise über mögliche Aktivatoren liefern. 66 Zusammenfassung 5 Zusammenfassung Die vorliegende Arbeit beinhaltete zunächst die Etablierung der Zweidimensionalen Gelelektrophorese und die Anpassung und Optimierung der Parameter an die effiziente, reproduzierbare und hochauflösende Untersuchung des Proteoms von humanen Zellinien, insbesondere der Leberkarzinomzellinie HepG2. Da die Sauberkeit der Probe für ein hochauflösendes Ergebnis von besonderer Bedeutung ist, wurden zwei verschiedene Reinigungsmethoden, die Ultrafiltration und die TCA/Aceton-Fällung nach Görg, verglichen. Zur Anfärbung der Protein im Polyacrylamid-Gel wurden die kolloidale Coomassie- und die reversible Silberfärbung nach Mann ausführlich getestet. Die Ergebnisse dieses Optimierungsprozesses ermöglichten die Anfertigung einer Anleitung zur hochauflösenden und reproduzierbaren zweidimensionalen Trennung des Proteoms von HepG2-Zellen. Ausführliche Beachtung fanden die Grenzen des Systems, die zu einem Systemfehler führen, welcher zwischen 5 % und 10 % liegt. Die 2D-Elektrophorese wurde anschließend benutzt, um das Proteom Coxsackievirus B3H3-infizierter HepG2-Zellen zu untersuchen. Coxsackieviren der B-Gruppe gelten als die häufigsten Erreger der humanen Myokarditis. Die virusinduzierte Myokarditits ist charakterisiert durch das Auftreten von Herzmuskelnekrosen und die Infiltration inflammatorischer Zellen in die nekrotischen Bereiche. Die Chronifizierung der Erkrankung führt zum kardialen Remodelling und äußert sich im pathologischen Bild einer Dilatativen Kardiomyopathie mit terminaler Herzinsuffizienz. Mit Hilfe der 2D-Elektrophorese konnten zahlreiche virusinduzierte Änderungen der Proteinausstattung der HepG2-Wirtszellen nachgewiesen werden. Eine wichtige Erkenntnis stellt die CVB3H3-infektionsabhängige Induktion der Mitogen-aktivierten Proteinkinase p38 dar. Dieses Signalmolekül ist in wichtige zelluläre Regelprozesse in einer Vielzahl von Geweben eingebunden. In kardialen Myozyten ist es unter anderem an Differenzierungsprozessen beteiligt, und es spielt auch bei pathologischen Gewebsveränderungen wie Fibrosierungsprozessen oder Myozyten-Hypertrophien in Reaktion auf Bluthochdruck eine wesentliche Rolle. Die Vermutung einer Beteiligung der MAPK p38 am akuten virusinduzierten Entzündungsprozeß bzw. der Konversi- 67 Zusammenfassung on der Entzündung in das klinische Bild der Dilatativen Kardiomyopathie ist insoweit begründet. 68 Literatur 6 Literatur Aden, D.P., Fogel, A., Plotkin, S., Damjanov, I. and Knowles, B.B. (1979) Controlled synthesis of HBsAg in a differentiated human liver carcinoma-derived cell line. Nature, 282, 615-616. Bjellqvist, B., Sanchez, J.C., Pasquali, C., Ravier, F., Paquet, N., Frutiger, S., Hughes, G.J. and Hochstrasser, D. (1993) Micropreparative two-dimensional electrophoresis allowing the separation of samples containing milligram amounts of proteins. Electrophoresis, 14, 1375-1378. Bradford, M. (1979) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 72, 248-254. Carthy, C.M., Granville, D.J., Watson, K.A., Anderson, D.R., Wilson, J.E., Yang, D., Hunt, D.W. and McManus, B.M. (1998) Caspase activation and specific cleavage of substrates after coxsackievirus B3-induced cytopathic effect in HeLa cells. J Virol, 72, 7669-7675. Carthy, C.M., Yanagawa, B., Luo, H., Granville, D.J., Yang, D., Cheung, P., Cheung, C., Esfandiarei, M., Rudin, C.M., Thompson, C.B., Hunt, D.W. and McManus, B.M. (2003) Bcl-2 and Bcl-xL overexpression inhibits cytochrome c release, activation of multiple caspases, and virus release following coxsackievirus B3 infection. Virology, 313, 147-157. Chevallet, M., Santoni, V., Poinas, A., Rouquie, D., Fuchs, A., Kieffer, S., Rossignol, M., Lunardi, J., Garin, J. and Rabilloud, T. (1998) New zwitterionic detergents improve the analysis of membrane proteins by two-dimensional electrophoresis. Electrophoresis, 19, 1901-1909. Chow, L.H., Beisel, K.W. and McManus, B.M. (1992) Enteroviral infection of mice with severe combined immunodeficiency. Evidence for direct viral pathogenesis of myocardial injury. Lab Invest, 66, 24-31. 69 Literatur Clark, A.R., Dean, J.L. and Saklatvala, J. (2003) Post-transcriptional regulation of gene expression by mitogen-activated protein kinase p38. FEBS Lett, 546, 37-44. Davidson, S.M. and Morange, M. (2000) Hsp25 and the p38 MAPK pathway are involved in differentiation of cardiomyocytes. Dev Biol, 218, 146-160. Figulla, H.R., Stille-Siegener, M., Mall, G., Heim, A. and Kreuzer, H. (1995) Myocardial enterovirus infection with left ventricular dysfunction: a benign disease compared with idiopathic dilated cardiomyopathy. J Am Coll Cardiol, 25, 1170-1175. Gauntt, C.J., Trousdale, M.D., LaBadie, D.R., Paque, R.E. and Nealon, T. (1979) Properties of coxsackievirus B3 variants which are amyocarditic or myocarditic for mice. J Med Virol, 3, 207-220. Gey, G.O.e.a. (1952) Tissue culture studies of the proliferative capacity of cervical carcinoma and normal epithelium. Cancer Res, 12, 264-265. Görg, A., Obermaier, C., Boguth, G., Csordas, A., Diaz, J.J. and Madjar, J.J. (1997) Very alkaline immobilized pH gradients for two-dimensional electrophoresis of ribosomal and nuclear proteins. Electrophoresis, 18, 328-337. Görg, A., Obermaier, C., Boguth, G., Harder, A., Scheibe, B., Wildgruber, R. and Weiss, W. (2000) The current state of two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis, 21, 1037-1053. Görg, A., Postel, W., Friedrich, C., Kuick, R., Strahler, J.R. and Hanash, S.M. (1991) Temperature-dependent spot positional variability in two-dimensional polypeptide patterns. Electrophoresis, 12, 653-658. Görg, A., Postel, W., Weser, J., Günther, S., Strahler, J.R., Hanash, S.M. and Somerlot, L. (1987) Electrophoresis, 8, 122-124. Han, J., Jiang, Y., Li, Z., Kravchenko, V.V. and Ulevitch, R.J. (1997) Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature, 386, 296-299. 70 Literatur Harder, A., Wildgruber, R., Nawrocki, A., Fey, S.J., Larsen, P.M. and Gorg, A. (1999) Comparison of yeast cell protein solubilization procedures for twodimensional electrophoresis. Electrophoresis, 20, 826-829. Henke, A., Launhardt, H., Klement, K., Stelzner, A., Zell, R. and Munder, T. (2000) Apoptosis in Coxsackie B3-caused diseases: interaction between the capsid protein VP2 and the proapoptotic protein Siva. J Virol, 74, 4284-4290. Huber, M., Watson, K.A., Selinka, H.C., Carthy, C.M., Klingel, K., McManus, B.M. and Kandolf, R. (1999) Cleavage of RasGAP and phosphorylation of mitogenactivated protein kinase in the course of coxsackievirus B3 replication. J Virol, 73, 3587-3594. Hufnagel, G., Pankuweit, S. and Maisch, B. (1998) Therapy of dilated cardiomyopathies with and without inflammation. Med Klin (Munich), 93, 240-251. Kenrick, K.G. and Margolis, J. (1970) Isoelectric focusing and gradient gel electrophoresis: a two-dimensional technique. Anal Biochem, 33, 204-207. King, A.M.Q., Brown, F., Christian, P., Hovi, T., Hyypiä, T., Knowles, N.J., Lemon, P.D., Minor, P.D., Palmenberg, A.C., Skern, T. and Stanway, G. (2000) Picornaviridae. In: van Regenmortel, M.H.D.et al. (eds.), Virustaxonomy. Seventh report of the international committee for the taxonomy of viruses. Academic Press, New York, San Diego, pp. 657-673. Klose, J. (1975) Protein mapping by combined isoelectric focusing and electrophoresis in mouse tissues. A novel approach to testing for induced point mutations in mammals. Humangenetik, 26, 231-243. Knowles, B.B. and Aden, D.P. (1983) Human hepatoma derived cell line, process for preparation thereof, and uses therefor. US Patent 4,393.133, U.S.A. Knowles, B.B., Howe, C.C. and Aden, D.P. (1980) Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science, 209, 497-499. 71 Literatur Knowlton, K.U., Jeon, E.S., Berkley, N., Wessely, R. and Huber, S. (1996) A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J Virol, 70, 78117818. Leipner, C., Grun, K., Borchers, M. and Stelzner, A. (2000) The outcome of coxsakkievirus B3-(CVB3-) induced myocarditis is influenced by the cellular immune status. Herz, 25, 245-248. Liao, P., Georgakopoulos, D., Kovacs, A., Zheng, M., Lerner, D., Pu, H., Saffitz, J., Chien, K., Xiao, R.P., Kass, D.A. and Wang, Y. (2001) The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A, 98, 12283-12288. Lindberg, A.M., Stalhandske, P.O. and Pettersson, U. (1987) Genome of coxsackievirus B3. Virology, 156, 50-63. Lonberg-Holm, K., Crowell, R.L. and Philipson, L. (1976) Unrelated animal viruses share receptors. Nature, 259, 679-681. Lottspeich, F. and Zorbas, H. (1998) Bioanalytik. Spektrum Akademischer Verlag, Heidelberg, Berlin. Luche, S., Santoni, V. and Rabilloud, T. (2003) Evaluation of nonionic and zwitterionic detergents as membrane protein solubilizers in two-dimensional electrophoresis. Proteomics, 3, 249-253. Maisch, B., Portig, I., Ristic, A., Hufnagel, G. and Pankuweit, S. (2000) Definition of inflammatory cardiomyopathy (myocarditis): on the way to consensus. A status report. Herz, 25, 200-209. Martinez-Moreno, M., Navarro-Lerida, I., Roncal, F., Albar, J.P., Alonso, C., Gavilanes, F. and Rodriguez-Crespo, I. (2003) Recognition of novel viral sequences that associate with the dynein light chain LC8 identified through a pepscan technique. FEBS Lett, 544, 262-267. 72 Literatur Martino, T.A., Liu, P. and Sole, M.J. (1994) Viral infection and the pathogenesis of dilated cardiomyopathy. Circ Res, 74, 182-188. Mason, J.W., O'Connell, J.B., Herskowitz, A., Rose, N.R., McManus, B.M., Billingham, M.E. and Moon, T.E. (1995) A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N Engl J Med, 333, 269-275. Mural, R.J., Adams, M.D., Myers, E.W., Smith, H.O., Miklos, G.L., Wides, R., Halpern, A., Li, P.W., Sutton, G.G., Nadeau, J., Salzberg, S.L., Holt, R.A. et al. (2002) A comparison of whole-genome shotgun-derived mouse chromosome 16 and the human genome. Science, 296, 1661-1671. Nestler, M. (2002) Apoptoseprozesse in der Pathogenese viraler Infektionen: Interaktion des Kapsidproteins VP2 von Coxsackievirus B3 mit dem proapoptotischen Protein SIVA. Dissertation. Friedrich-Schiller-Universität, Jena. O´Farell, P.H. (1975) High resolution separation two-dimensional electrophoresis of proteins. J Bio Chem, 250, 4007-4021. Rabilloud, T. (1996) Solubilization of proteins for electrophoretic analyses. Electrophoresis, 17, 813-829. Rabilloud, T. (1998) Use of thiourea to increase the solubility of membrane proteins in two-dimensional electrophoresis. Electrophoresis, 19, 758-760. Rabilloud, T. (1999) Solubilization of proteins in 2-D electrophoresis. An outline. Methods Mol Biol, 112, 9-19. Rabilloud, T., Adessi, C., Giraudel, A. and Lunardi, J. (1997) Improvement of the solubilization of proteins in two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis, 18, 307-316. Rabilloud, T., Gianazza, E., Catto, N. and Righetti, P.G. (1990) Amidosulfobetaines, a family of detergents with improved solubilization properties: application for isoelectric focusing under denaturing conditions. Anal Biochem, 185, 94-102. 73 Literatur Rabilloud, T., Valette, C. and Lawrence, J.J. (1994) Sample application by in-gel rehydration improves the resolution of two-dimensional electrophoresis with immobilized pH gradients in the first dimension. Electrophoresis, 15, 15521558. Ramsingh, A.I. (1997) Coxsackieviruses and pancreatitis. Front Biosci, 2, 53-62. Ravingerova, T., Barancik, M. and Strniskova, M. (2003) Mitogen-activated protein kinases: a new therapeutic target in cardiac pathology. Mol Cell Biochem, 247, 127-138. Santoni, V., Molloy, M. and Rabilloud, T. (2000) Membrane proteins and proteomics: un amour impossible? Electrophoresis, 21, 1054-1070. Scheele, G.A. (1975) Two-dimensional gel analysis of soluble proteins Characterisation of guinea pig exocrine pancreatic proteins. J Bio Chem, 250, 5375-5385. Schultheiss, H.P., Pauschinger, M. and Kuhl, U. (1998) Pathogenesis of inflammatory cardiomyopathies. Med Klin (Munich), 93, 229-235. Selinka, H.C., Wolde, A., Pasch, A., Klingel, K., Schnorr, J.J., Kupper, J.H., Lindberg, A.M. and Kandolf, R. (2002) Comparative analysis of two coxsackievirus B3 strains: putative influence of virus-receptor interactions on pathogenesis. J Med Virol, 67, 224-233. Shevchenko, A., Wilm, M., Vorm, O. and Mann, M. (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem, 68, 850858. Shi, Y. and Gaestel, M. (2002) In the cellular garden of forking paths: how p38 MAPKs signal for downstream assistance. Biol Chem, 383, 1519-1536. Smithies, O. and Poulik, M.D. (1956) Two-dimensional electrophoresis of serum proteins. Nature, 176, 1256-1266. Suomalainen, M., Nakano, M.Y., Boucke, K., Keller, S. and Greber, U.F. (2001) Adenovirus-activated PKA and p38/MAPK pathways boost microtubulemediated nuclear targeting of virus. Embo J, 20, 1310-1319. 74 Literatur Tibbles, L.A., Spurrell, J.C., Bowen, G.P., Liu, Q., Lam, M., Zaiss, A.K., Robbins, S.M., Hollenberg, M.D., Wickham, T.J. and Muruve, D.A. (2002) Activation of p38 and ERK signaling during adenovirus vector cell entry lead to expression of the C-X-C chemokine IP-10. J Virol, 76, 1559-1568. Towbin, H., Staehelin, T. and Gordon, J. (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A, 76, 4350-4354. Vuillard, L., Marret, N. and Rabilloud, T. (1995) Enhancing protein solubilization with nondetergent sulfobetaines. Electrophoresis, 16, 295-297. Wang, Y., Huang, S., Sah, V.P., Ross, J., Jr., Brown, J.H., Han, J. and Chien, K.R. (1998) Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Biol Chem, 273, 2161-2168. Westermeier, R. (1990) Elektrophorese-Praktikum. VCH, Weinheim. Wilkins, M.R., Williams, K.L., Appel, R.D. and Hochstrasser, D.F. (eds.). (1997) Proteome Research: New Frontiers in Functional Genomics. Springer-Verlag, Heidelberg. 75 Danksagung Diese Arbeit wurde in der Zeit von Mai 2003 bis April 2004 am Hans-Knöll-Institut für Naturstoff-Forschung e.V. Jena in der Abteilung Molekulare Naturstoff-Forschung durchgeführt. Zuallererst möchte ich mich herzlich bei Frau Prof. Dr. Susanne Grabley für die Möglichkeit, in ihrer Abteilung die vorliegende Diplomarbeit anzufertigen, bedanken. Zudem wurde durch Frau Prof. Grabley die Aufgabe übernommen, das Zweitgutachten zu dieser Arbeit zu erstellen. Auch dafür danke ich herzlich. Mein besonderer Dank gilt Herrn PD. Dr. Thomas Munder, der mir stets als kompetenter Betreuer zur Seite stand und in seiner lockeren Art ein optimistisches und ergebnisorientiertes Arbeitsklima schuf. Die Zusammenarbeit mit dem Institut für Virologie der Friedrich-Schiller-Universität war eine wichtige Grundlage für die Bearbeitung des vorgestellten Themas. Vor allem danke ich hier Herrn PD. Dr. Andreas Henke für seine Unterstützung bei virologischen Arbeiten. Ebenso unverzichtbar war die Zusammenarbeit mit der Core Unit Chip Applications des Instituts für Humangenetik der FSU. Die Benutzung des dortigen SELDI-Gerätes ermöglichte die massenspektrometrische Vermessung relevanter Proteine, wofür ich vor allem Herrn Dr. Christian Melle dankbar bin. Alle Mitarbeiter und Nachwuchswissenschaftler der Abteilung Molekulare Naturstoff-Forschung haben mich in allen Phasen der Arbeit unterstützt, besonders durch die angenehme Arbeitsatmosphäre. Dafür danke ich Alexander Raßmann, Janka Teutschbein, Frau Dr. Katleen Gura, Katharina Mihatsch, Sebastian Günther, Dörthe Peter und Andreas Busch. Durch die kritische Revision des Manuskripts trug Janka Teutschbein in besonderem Maße zum Gelingen dieser Arbeit bei. Hierfür vielen Dank. Ganz besonders möchte ich mich bei meiner Familie bedanken, deren finanzielle und ideelle Unterstützung das Studium und dessen erfolgreichen Abschluß erst ermöglichten. Dafür danke ich vor allem meiner Frau Christiane, meinen Eltern sowie meinem Schwager Alexander, der mir am Computer eine große Hilfe war.