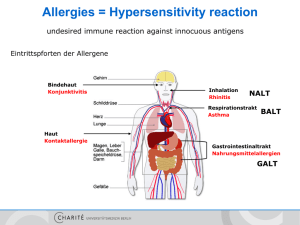
Die Rolle der autoreaktiven B-Zellen und Autoantikörper in der
Werbung

Die Rolle der autoreaktiven B-Zellen und Autoantikörper in der Pathophysiologie der Multiplen Sklerose The role of autoreactive B cells and autoantibodies in the pathophysiology of multiple sclerosis Dissertation zur Erlangung des naturwissenschaftlichen Doktorgrades der Graduate School of Life Sciences, Julius-Maximilians-Universität Würzburg, Neurowissenschaften Vorgelegt von Damiano Mario Rovituso aus Alimena (PA), Italien Würzburg, Mai 2016 Eingereicht am: ………………………………………………………… Mitglieder des Promotionskomitees: Vorsitzende/r: …………………………………………………………… 1. Betreuer: Prof. Dr. med. Stefanie Kürten 2. Betreuer: Prof. Dr. rer. nat. Thomas Hünig 3. Betreuer: Prof. Dr. Paul V. Lehmann Tag des Promotionskolloquiums: ……………………………………… Doktorurkunden ausgehändigt am: ……………………………………. Inhaltsverzeichnis Inhaltsverzeichnis Inhaltsverzeichnis ........................................................................................................................ I Zusammenfassung .................................................................................................................... III Summary.................................................................................................................................... V Abkürzungsverzeichnis ...........................................................................................................VII 1. Einleitung ............................................................................................................................ 1 1.1. Das Immunsystem ........................................................................................................ 1 1.1.1. Die Immunantwort ................................................................................................ 1 1.1.2. Toleranzinduktion und Immunerschöpfung ......................................................... 4 1.1.3. Autoimmunität ...................................................................................................... 9 1.2. Multiple Sklerose ....................................................................................................... 12 1.2.1. Geschichte der MS ............................................................................................. 12 1.2.2. Klinische MS-Symptomatologie ........................................................................ 13 1.2.3. Verlaufsformen und Therapieansätze ................................................................. 14 1.2.4. Epidemiologie und volkswirtschaftliche Relevanz ............................................ 16 1.2.5. Ätiologie und Pathogenese ................................................................................. 17 1.3. B-Zellen ..................................................................................................................... 20 1.3.1. B-Zell-Untergruppen .......................................................................................... 20 1.3.2. Die Rolle der B-Zellen in MS ............................................................................ 23 1.4. Biomarker .................................................................................................................. 27 2. Ziele der Arbeit ................................................................................................................. 29 3. Manuskripte....................................................................................................................... 30 3.1. B1 cells are unaffected by immune modulatory treatment in remitting-relapsing multiple sclerosis patients ..................................................................................................... 30 3.2. The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsing-remitting multiple sclerosis patients ....................................... 36 3.3. CEACAM1 mediates B cell aggregation and tolerance induction in central nervous system autoimmunity ............................................................................................................. 45 I Inhaltsverzeichnis 4. Diskussion ......................................................................................................................... 99 4.1. B1-Zellen sind in Patienten mit schubförmiger MS unabhängig von der MS-Therapie signifikant erniedrigt ........................................................................................................... 100 4.2. Die Anwesenheit von gehirnantigenspezifischen B-Zellen bei RRMS-Patienten korreliert mit dem Behandlungserfolg durch GA ............................................................... 102 5. 6. 4.3. CEACAM1-vermittelte Toleranzinduktion in RRMS-Patienten ............................. 105 4.4. Die Rolle autoreaktiver B-Zellen und B-Zell-Untergruppen in der MS .................. 110 Ausblick .......................................................................................................................... 114 5.1. Blutanalysen............................................................................................................. 114 5.2. Liquoranalysen......................................................................................................... 114 5.3. Längsschnittstudie ................................................................................................... 114 5.4. Zytozentrifugation ................................................................................................... 115 Literaturverzeichnis ......................................................................................................... 116 Lebenslauf ................................................................................................................................... i Publikationsliste ......................................................................................................................... v Eidesstattliche Erklärung ........................................................................................................... vi Affidavit ................................................................................................................................... vii Erklärung zu Eigenanteilen an Publikationen und Zweitpublikationsrechten......................... viii Statement of individual author contributions and of legal second publication rights ................ x Statement of individual author contributions to figures/tables/chapters included in the manuscripts .............................................................................................................................. xiii Danksagung .............................................................................................................................. xv II Zusammenfassung Zusammenfassung Multiple Sklerose (MS) ist die häufigste neurologische Erkrankung, die bei jungen Erwachsenen zu dauerhaften körperlichen Einschränkungen führt. Ein Kennzeichen der MS sind zeitlich und örtlich disseminierte entzündliche Läsionen im zentralen Nervensystem (ZNS). Die Läsionsart, die am häufigsten auftritt, ist u. a. durch Antikörperablagerungen charakterisiert. Die häufigste Verlaufsform der MS tritt in Schüben auf. Im Laufe der Erkrankung bilden sich die Symptome in der Mehrzahl der Patienten unvollständig zurück und es entwickelt sich ein chronischer Verlauf. Trotz intensiver Forschung ist die Ätiologie der MS bisher unbekannt Bis heute gibt es keine Biomarker, um den Therapieerfolg oder das Therapieversagen der MS-Basistherapeutika (Glatirameracetat und β-Interferon) zu bestimmen. Aktuelle Studien, bei denen B-Zellen depletiert wurden, zeigten eine signifikante Reduktion MS-typischer Läsionen und der Schubrate bei der schubförmigen MS. Man vermutet, dass autoreaktive B-Zellen vielfältige Aufgaben in der Pathogenese der MS übernehmen: sie produzieren Autoantikörper, präsentieren autoreaktiven T-Zellen Autoantigene und sezernieren Mediatoren, die zur Aktivierung anderer Immunzellen führen. Es ist noch unklar, welche B-Zell-Untergruppe bei der MS besondere Relevanz hat. Vor kurzem wurden B1-Zellen beim Menschen beschrieben. Eine Studie zeigte, dass die Anzahl der B1Zellen in unbehandelten MS-Patienten signifikant erniedrigt war. Des Weiteren wurden im ZNS von chronisch erkrankten MS-Patienten B-Zell-Aggregate nachgewiesen. Diese B-ZellAggregate ähneln sekundären lymphatischen Organen und könnten zur Progredienz der Erkrankung beitragen. Eine ex vivo-Studie zeigte, dass die B-Zell-Aggregat-Bildung durch das Adhäsionsmolekül CEACAM1-(carcinoembryogenic antigen-related cell adhesion molecule-1) vermittelt wird. Überdies ist die Koexpression von CEACAM1 und TIM-3 (T-cell immunoglobulin- and mucin-domain containing-3) für immunerschöpfte und tolerante T-Zellen charakteristisch. Schließlich konnte unsere Arbeitsgruppe zeigen, dass ZNS-reaktive B-Zellen nur im Blut von Patienten mit einem klinisch isolierten Syndrom und MS-Patienten nachweisbar waren. In meiner Studie habe ich den Einfluss von MS-Basistherapeutika und einer MSEskalationstherapie auf die B-Zell-Untergruppen untersucht. Dabei habe ich die naive B-Zell-, B-Gedächtniszell-, B1-Zell- und Plasmablasten-Zahl von gesunden Probanden sowie unbehandelten und behandelten MS-Patienten miteinander verglichen. Die B-ZellUntergruppen wurden durchflusszytometrisch untersucht. Die B1-Zell-Zahl war bei behandelten und unbehandelten MS-Patienten signifikant erniedrigt. In einer weiteren Studie konnte ich zeigen, dass die Anwesenheit von ZNS-reaktiven B-Zellen im Blut von III Zusammenfassung glatirameracetat-behandelten MS-Patienten mit dem Therapieerfolg assoziiert war. Die ZNSreaktiven B-Zellen wurden durch einen ZNS-Lysat-ELISPOT detektiert. Schließlich habe ich in einer dritten Studie die Expression von CEACAM1 und TIM-3 auf B-Zellen bei natalizumabbehandelten MS-Patienten durchflusszytometrisch untersucht. Im Vergleich zu gesunden Probanden zeigte sich, dass im Blut der MS-Patienten die CEACAM1+- und die CEACAM1+TIM-3+-B-Zell-Zahl signifikant erhöht war. Im Gegensatz dazu waren CEACAM1+TIM-3+-T-Helferzellen signifikant erniedrigt in behandelten MS-Patienten. Meine Arbeit belegt, dass die B1-Zell-Population unabhängig von der MS-Therapie in MSPatienten erniedrigt ist. Ungeklärt bleibt, ob diese Erniedrigung eine Folge oder eine Ursache der Erkrankung ist. B1-Zellen sind die Quelle von natürlichen Antikörpern in Mensch und Tier. Sie haben protektive Eigenschaften und sind bei der B-Zell-Toleranzinduktion beteiligt. Die protektiven Funktionen der natürlichen Antikörper könnten durch die Erniedrigung der B1Zell-Zahl ausbleiben. Zusätzlich waren B-Zellen mit einem immunerschöpften Phänotyp im Blut von MS-Patienten erhöht. Trotz Stimulation konnte kein Phänotyp bei T-Helferzellen induziert werden, der für tolerante und immunerschöpfte T-Zellen beschrieben worden ist. In zukünftigen Studien sollte man die B1-Zell-Zahl und die CEACAM1+TIM-3+-B- und -T-ZellZahl bei Patienten mit einem klinisch isolierten Syndrom im Liquor und im Blut untersuchen. Damit könnte man feststellen, ob B1-Zellen aus der Peripherie bei MS-Patienten in das ZNS migrieren. Die Anwesenheit ZNS-reaktiver B-Zellen im Blut von behandelten MS-Patienten zeigte sich in meiner Arbeit als ein Marker, um den Therapieerfolg zu dokumentieren. Eine weiterführende Querschnittstudie (COPSELECT) wird ZNS-reaktive B-Zellen mittels ZNSLysat-ELISPOT als zukünftige Therapie-Biomarker ausführlicher untersuchen. MS-Biomarker wären für den einzelnen Betroffenen von großer Bedeutung und hätten ebenfalls gesundheitsökonomisch eine hohe Relevanz. IV Summary Summary Multiple sclerosis (MS) is the most common neurological disorder that leads to permanent disability in young adults. MS is characterized by temporally and spatially disseminated inflammatory lesions in the central nervous system (CNS). The most frequently observed pattern is associated with immunoglobulin deposition. The most common form of MS displays a relapsing-remitting course. Over time remissions are incomplete and most patients develop a chronic course of the disease. Currently, there are no biomarkers to predict therapeutic success or treatment failure of MS first-line therapies (glatiramer acetate and β-interferon). Despite intensive research, the etiology of MS is still unknown. Recent studies in which B cells were depleted, showed a significant reduction of MS-typical lesions and relapse rate in the relapsingremitting MS. Presumably, autoreactive B cells have different roles in the pathogenesis of MS: they produce autoantibodies, are antigen presenting cells for autoreactive T cells and secrete mediators that activate other immune cells. It is unclear, which B cell subset is particularly relevant in MS. Recently B1 cells have been described in humans. Additionally, it was observed that the number of B1 cells was significantly reduced in untreated MS patients. Furthermore, B cell aggregates were found in the CNS of patients with a progressive form of MS. These B cell aggregates resembled secondary lymphoid organs and might contribute to the progression of the disease. An ex vivo study showed that B cell aggregate formation was mediated by the cell adhesion molecule CEACAM1 (carcinoembryogenic antigen-related cell adhesion molecule-1). Moreover, it was shown that immune exhausted and tolerant T cells co-express CEACAM1 and TIM-3 (T-cell immunoglobulin- and mucin-domain containing-3). Finally, our group has demonstrated that CNS-reactive B cells were detectable only in the blood of patients with a clinically isolated syndrom (CIS) or MS. In my thesis I have investigated the influence of different MS drugs on B cell subsets in the blood. I compared the numbers of naive B cells, memory B cells, B1 cells and plasmablasts of healthy subjects as well as untreated and treated MS patients. The B cell subsets were examined by flow cytometry. B1 cell numbers was significantly decreased in treated and untreated patients with MS. Second, I was able to show that the presence of CNS-reactive B cells in the blood of glatiramer acetate-treated MS patients was associated with a positive treatment response. CNS-reactive B cells were detected by CNS-lysate-ELISPOT. Finally, I investigated the expression of CEACAM1 and TIM-3 on B cells in natalizumab-treated MS patients by flow cytometry. CEACAM1+- and CEACAM1+TIM-3+-B cell numbers were significantly increased in the blood of MS patients. In contrast, CEACAM1+TIM-3+-T helper cells were significantly decreased in treated MS patients. V Summary My research demonstrates that the B1 cell population is decreased in the blood of MS patients regardless of MS therapy. It remains unclear whether this reduction is a consequence or a cause of the disease. B1 cells are the source of natural antibodies in humans and animals. They have protective properties and are involved in B cell tolerance induction. The protection by natural antibodies might be missing because of the low B1 cell counts in MS. In addition, B cell counts with an exhausted phenotype were significantly increased in the blood of MS patients. However, despite stimulation T helper cells from MS patients expressed neither an exhausted nor a tolerant phenotype. Future studies should examine the B1 cell-, CEACAM1+TIM-3+-B cell and CEACAM1+TIM-3+-T cell numbers in CIS patients in the blood and cerebrospinal fluid. It could help to determine whether B1 cells from the periphery migrate in the CNS in MS patients. The presence of CNS-reactive B cells in the blood of treated MS patients proved to be a marker in order to document the success of therapies. A study of a large cohort of patients (COPSELECT) will investigate the potential of CNS-reactive B cells by ELISPOT as MS biomarker in more detail. MS biomarkers are urgently needed to detect MS treatment responders early on and are of high socio-economic importance. VI Abkürzungsverzeichnis Abkürzungsverzeichnis A ACTH AICD AIRE APECEP APZ Adrenocorticotropes Hormon activation-induced cell death autoimmune regulator Autoimmun-Polyendokrinopathie-Candidiasis-ektodermaleDystrophie-Syndrom antigenpräsentierende Zellen B BAFF BAFF-R BTLA BZR B cell activation factor of the TNF family B cell activation factor of the TNF family receptor B- and T-lymphocyte attenuator B-Zell-Rezeptor C CCL21 CCR7 CD CD40L CEACAM1 CIS CNTF CTL CTLA-4 CVID CXCL13 C-C motif ligand 21 C-C motif receptor 7 cluster of differentiation CD40 Ligand, CD154 carcinoembryogenic antigen-related cell adhesion molecule-1 clinically isolated syndrom ciliary neurotrophic factor cytotoxic T lymphocytes cytotoxic T-lymphocyte-associated protein 4, CD152 common variable immunodeficiency chemokine (c-x-c motif) ligand 13 D DNS DZ Desoxyribonukleinsäure dendritische Zellen E EAE EBV ECTRIMS EDSS ELISA ELISPOT experimentelle allergische bzw. autoimmune Enzephalomyelitis Epstein-Barr-Virus European Committee for Treatment and Research in Multiple Sclerosis Expanded Disability Status Scale enzyme-linked immunosorbent assay enzyme-linked immunospot assay F Fab Fas FasL Fc FcγIIB FDZ antigen-binding fragment first apoptosis signal, CD95 Fas-Ligand auch CD95-Ligand crystallisable fragment Fc fragment of IgG follikulär dendritische Zellen VII Abkürzungsverzeichnis FoxP3 forkhead box P3 G GA GFAP Glatirameracetat glial fibrillary acidic protein H HIV human immunodeficiency virus, humanes Immundefizienz-Virus HLA human leucocyte antigen I i.m. intramuskulär IDO IFN-γ Ig Indoleamine-2,3-Dioxygenase Interferon-γ Immunglobuline IHC IL IPEX Immunhistochemie Interleukin Immunderegulation, Polyendokrinopahtie, Enteropathie, X-gekoppeltes Syndrom intravenöse Immunglobuline IVIG J JC John Cunningham K Kir4.1 einwärts rektifizierender Kaliumkanal 4.1 L LAG-3 LT lymphocyte-activation gene 3, CD223 lymphotoxin M MBP Myelin-Basisches Protein mCC1 MCR MHC murine Anti-CEACAM1-Antikörper Melanocortin-Rezeptoren major histocompatibility complex MP4 MRT MS mTEC MBP-PLP-Fusionsprotein Magnetresonanztomographie Multiple Sklerose medulläre Thymusepithelzellen N NAT NCAM NCR Nfl Natalizumab neural cell adhesion molecule negative checkpoint regulators Neurofilament O OKB OVA oligoklonale Banden Ovalbumin VIII Abkürzungsverzeichnis P PAMP PBMC PD-1 PI3K PLP pathogen-associated molecular patterns peripheral blood mononuclear cells programmed death-ligand 1 oder CD279 Phosphoinositid-3-Kinase, auch phosphatidylinositol-4,5bisphosphat 3-kinase Proteolipidprotein PML PNAd PPMS PRR progressive multifokale Leukenzephalopathie peripheral node addressin primär progrediente MS pathogen recognition receptors R RRMS RT-PCR relapsing-remitting MS reverse transcription polymerase chain reaction S s.c. SEB subkutan Staphylococcus aureus enterotoxin B SHP-1 SJL SLE Src homology region 2 domain-containing phosphatase-1 Swiss Jim Lambert systemischen Lupus erythematodes SPMS SSC sekundär progrediente MS side scatter T Tc TFH TGF-β TH TIL cytotoxic T lymphocytes follikuläre T-Helferzellen tansforming growth factor-β T-Helferzellen tumor-infiltrating lymphocytes TIM-3 TLO TLR TNF-α T-cell immunoglobulin- and mucin-domain containing-3 tertiary lymphoid organ toll-like receptor tumor necrosis factor-α Treg TSLR regulatorische T-Zellen time since last relapse TZR T-Zell-Rezeptoren V V(D)J VISTA V-(variable), D-(diversity) und J-(joining) Gensegmente V-domain Ig suppressor of T cell activation Z ZNS zentrales Nervensystem IX Einleitung 1. Einleitung 1.1. Das Immunsystem Das Abwehrsystem eines Lebewesens, das Immunsystem, das aus speziellen Organen, zellulären und humoralen Bestandteilen besteht, hat die Aufgabe Krankheitserreger und krankhaft veränderte körpereigene Zellen (z. B. Tumorzellen) zu erkennen und zu beseitigen. Auch nicht-infektiöse körperfremde und körpereigene Antigene können eine Immunreaktion auslösen. Antigene sind Substanzen, die von Immunzellen über ihre membrangebundenen antigenspezifischen Rezeptoren oder löslichen Immunglobuline (Ig) gebunden werden können und eine Immunantwort auslösen. Eine Immunantwort gegen körpereigene Antigene führt zur Autoimmunität. Das Immunsystem wird in einen angeborenen und einen adaptiven Teil eingeteilt. Das angeborene Immunsystem hat sich phylogenetisch früh entwickelt und wurde bei allen Vertretern der Deuterostomier gefunden 1. Die Bestandteile des angeborenen Immunsystems erkennen konservierte Strukturen von Eindringlingen und lösen eine sofortige Immunreaktion aus. Zur frühen angeborenen Immunantwort kommt eine verzögerte, adaptive Immunantwort hinzu, die sehr spezifisch ausgerichtet ist. Eine adaptive Immunität war bis vor einem Jahrzehnt nur in Vertebraten bekannt, wurde aber mittlerweile auch in Bakterien und Archaeen beschrieben 2. 1.1.1. Die Immunantwort Neben den physikalischen und chemischen Barrieren der Haut und der Schleimhäute, treffen Pathogene i. d. R. auf geweberesidente Makrophagen. Das angeborene Immunsystem erkennt pathogenassoziierte molekulare Muster (pathogen-associated molecular patterns, PAMP), die an den Mustererkennungsrezeptoren (pathogen recognition receptors, PRR) von Immunzellen des angeborenen Immunsystems binden. PAMP werden von pathogenen Mikroorganismen oder anderen Krankheitserregern exprimiert. PRR finden sich auch auf Zellen des adaptiven Immunsystems, z. B. den B-Zellen. Werden durch PRR der Makrophagen bakterielle, konservierte Strukturen erkannt, folgt eine Freisetzung von bestimmten Proteinen, den Zytokinen. Zytokine haben pleiotrope Funktionen, u. a. rekrutieren sie weitere Immunzellen. Zytokine wirken autokrin oder parakrin und induzieren Proliferations- oder Spezialisierungsschritte. Anschließend können neutrophile Granulozyten, die mittels bestimmter Zytokinen, den Chemokinen, ins Gewebe migrieren, gemeinsam mit den Makrophagen die Pathogene phagozytieren und zerstören. Die Phagozytose kann unter anderem durch das Vorhandensein von Plasmaproteinen induziert und verstärkt werden, die 1 Einleitung Teil des sogenannten Komplementsystems sind. Das Komplementsystem kann, als Ergebnis einer proteolytischen Kaskade, die Zielzelle zerstören. Auch unreife dendritische Zellen (DZ) finden sich in peripheren Geweben. Über Makropinozytose nehmen DZ Pathogene oder deren Bestandteile auf, wandern über afferente Lymphgefäße zu regionalen Lymphknoten und präsentieren diese Antigene als Peptidfragmente naiven T-Lymphozyten. Sie stellen damit ein wichtiges Bindeglied zwischen angeborener und adaptiver Immunantwort dar. Nachdem T-Zellen den Thymus verlassen haben, zirkulieren sie zwischen Blutgefäßen und sekundären lymphatischen Organen. Nach Antigenkontakt in der T-Zell-Zone der lymphatischen Organe bekommen naive T-Zellen das erste Signal zur Proliferation und Differenzierung. T-Zellen erkennen Antigene mit ihren antigenspezifischen T-Zell-Rezeptoren (TZR) im Kontext mit dem Haupthistokompatibilitätskomplex-Molekül (major histocompatibility complex, MHC, beim Menschen: human leucocyte antigen system, HLA-System) auf der Oberfläche von antigenpräsentierenden Zellen (APZ). Naive T-Zellen kann man anhand des TransmembranGlykoproteins CD3 (cluster of differentiation 3) charakterisieren und durch CD8 und CD4 in zwei große Gruppen unterteilen. Dabei beobachtet man das Phänomen der MHC-Restriktion, d. h. CD8+-T-Zellen, auch zytotoxische T-Lymphozyten (cytotoxic T lymphocytes, CTL auch Tc) genannt, erkennen intrazelluläre Antigene, die durch den MHC-Klasse-I-Komplex präsentiert werden. Andererseits erkennen CD4+-T-Zellen (T-Helferzellen, TH-Zellen) Antigene, die vom MHC-Klasse-II-Komplex präsentiert werden. MHC-Klasse-II-Komplexe werden von APZ und in geringerem Maße von Epithel- und Endothelzellen exprimiert. Zu den APZ gehören DZ, B-Zellen und Makrophagen. Sie können aktiv extrazelluläre Antigene aufnehmen und präsentieren. Im Gegensatz dazu können alle kernhaltigen Zellen intrazellulär lokalisierte Antigene präsentieren. Damit T-Zellen vollständig aktiviert werden, benötigen sie ein zweites, kostimulatorisches Signal. Kostimulatorische Signale erhalten T-Zellen, wenn akzessorische Rezeptoren, wie z. B. CD28-Moleküle die B7-Familie (u. a. CD80, CD86) auf APZ binden. Eine Antigenerkennung ohne zusätzliches kostimulatorisches Signal führt zu einer antigenspezifischen Unreaktivität (Anergie). Aktivierte antigenspezifische TH-Zellen können ihrerseits antigenspezifische B-Zellen in lymphatischen Geweben aktivieren, welche die gleichen Antigene erkennen (kognate T-Zell-Stimulierung). Reife B-Zellen migrieren aus dem Knochenmark und zirkulieren als naive B-Zellen zwischen Blut und Lymphe. Im Gegensatz zum TZR der T-Zellen erkennen die B-Zell-Rezeptoren (BZR) antigenspezifische dreidimensionale Strukturen. Nach Antigenbindung kommt es zur rezeptorvermittelten Endozytose des Antigens. Die B-Zelle kann nun über ihre MHC-Klasse-II-Komplexe TH-Zellen Antigene präsentieren. Die B-Zelle benötigt ebenfalls zwei Signale, um vollständig aktiviert zu werden. Das erste Signal (Antigenbindung) bekommt die B-Zelle über die Bindung des MHC2 Einleitung Peptid-Komplexes an den TZR. Das zweite Signal (Kostimulation) erhält die B-Zelle, wenn sie CD40L (CD40 Ligand, CD154) auf aktivierten TH-Zellen bindet. Die Bindung an CD40L durch CD40 induziert die Expression bestimmter Interleukin-(IL) Rezeptoren und verstärkt die B7Expression auf B-Zellen. B-Zellen können auch durch Komplementbestandteile (C3bSpaltprodukte) TH-Zell-unabhängig aktiviert werden. T-Zellen sezernieren Zytokine, die u. a. für die Expansion und Antikörperproduktion von B-Zellen nötig sind (drittes Signal). Nachdem T- und B-Zellen ihr drittes Signal erhalten haben, treten sie in eine Phase der klonalen Expansion ein, d. h. es folgt eine Proliferationsphase mit dem Ergebnis, dass eine antigenspezifische, klonale Population entsteht. Frank Macfarlane Burnet postulierte in seiner Klon-Selektionstheorie, wie der Organismus gegen körperfremde und unbekannte Stoffe eine Immunantwort generieren kann 3,4. Die molekularen Prozesse, die zu einer großen Rezeptordiversität führen, sind mittlerweile weitestgehend verstanden. Die Rezeptordiversität erfolgt aufgrund einer somatischen DNS-(Desoxyribonukleinsäure) Rekombination, die zufällig abläuft und als kombinatorische Diversität bezeichnet wird. Am Ende der klonalen Expansion finden sich T- und B-Gedächtniszellen, T- und B-Effektorzellen und antikörpersezernierende Plasmazellen. Gedächtniszellen werden durch den erneuten Kontakt mit ihren spezifischen Antigenen restimuliert, was zu einer schnellen adaptiven Immunantwort führt. Während CTL infizierte Zellen eliminieren, differenzieren sich TH-Zellen zu verschiedenen TH-Zell-Untergruppen. proinflammatorische oder Entsprechend antiinflammatorische ihrer Untergruppe zytokinvermittelte können sie Effektorfunktionen ausführen. TH-Zellen sind nötig, damit B-Zellen einen Ig-Klassenwechsel durchführen. Der Klassenwechsel eines B-Zell-Klons betrifft nicht nur den oberflächengebundenen BZR, sondern auch die sezernierte Form des BZR, den antigenspezifischen Antikörper. Antikörper sind Hauptbestandteil der adaptiven humoralen Immunantwort. Jeder Antikörper besteht aus zwei identischen schweren und leichten Ketten, die durch kovalente Disulfidbrücken verbunden sind. Antikörper werden auch in ein antigenbindendes Fragment (antigen-binding fragment, Fab) und ein kristallisierbares Fragment (crystallisable fragment, Fc) eingeteilt. Während die Antigenerkennung durch den Fab-Teil stattfindet, vermittelt der Fc-Teil die Effektorfunktionen. Zu den Effektorfunktionen gehören die Aktivierung der Komplementkaskade (klassischer Aktvierungsweg) und Opsonierung, d. h. die erleichterte Phagozytose von Pathogenen durch z. B. Makrophagen. Antikörper können auch Pathogene oder deren Bestandteile binden und verhindern damit, dass sie an Zellen binden können (Neutralisation). 3 Einleitung 1.1.2. Toleranzinduktion und Immunerschöpfung Ein Hauptmerkmal der Immunantwort ist die Fähigkeit zwischen Fremdstoffen und körpereigenen Stoffen zu unterscheiden. Körpereigene Stoffe werden toleriert, d. h. eine Immunantwort gegen Selbstantigene wird i. d. R. nicht induziert. Nach Burnets KlonSelektionstheorie kann es aber zu Spezifitäten kommen, die gegen Selbstantigene gerichtet sind 4. Immunreaktionen gegen Selbstantigene bezeichnet man als Autoimmunität. Erkrankungen die daraus resultieren, werden Autoimmunerkrankungen genannt. Die Toleranzinduktion ist aber kein passiver, sondern ein mehrstufiger, aktiver Prozess. Er wird eingeteilt in zentrale und periphere Toleranz. Die zentrale Toleranzinduktion findet in den primären lymphatischen Organen (Knochenmark und Thymus) während der Ontogenese der Lymphozyten statt. Obwohl die zentrale Toleranz während der Reifung der Lymphozyten induziert wird, gewährt sie keinen absoluten Schutz und wird deshalb durch die periphere Toleranz erweitert. Ein Grund kann darin liegen, dass nicht alle immundominanten Epitope im Thymus exprimiert werden 5. Die periphere Toleranz wird außerhalb der primären lymphatischen Organe in der Peripherie induziert. Zu den peripheren Toleranzmechanismen gehören die Induktion von Anergie, die Ignoranz gegenüber kryptischen und sequestrierten Antigenen, die klonale Deletion und die Immunregulation durch regulatorische T-Zellen (Treg). Anergie kann durch fehlende kostimulatorische Signale induziert werden. Die Ignoranz von Antigenen ist von der Konzentration des Antigens abhängig. Durch eine kurzfristig wiederholte Aktivierung durch das Antigen oder durch hohe Konzentrationen des Antigens werden antigenspezifische Zellen mittels Apoptose entfernt. Treg induzieren periphere Toleranz mittels Zytokin-Sekretion und interzellulären Kontakt. Neben diesen Mechanismen können körpereigene Stoffe durch physikalische Barrieren gegenüber dem lymphatischen System segregiert sein. Negative Immun-Checkpoints (negative checkpoint regulators, NCR), also Proteine, welche die Immunantwort negativ modulieren, sind wesentlicher Bestandteil der ZellZell-Interaktion und relevant, um die periphere Toleranz aufrecht zu erhalten 6. Die bekanntesten NCR, die auf T-Zellen exprimiert werden, sind CTLA-4 (cytotoxic T-lymphocyteassociated protein 4 oder CD152) und PD-1 (programmed death-ligand 1 oder CD279). Mittlerweile gehören TIM-3 (T-cell immunoglobulin- and mucin-domain containing-3 oder Hepatitis A virus cellular receptor 2 (HAVCR2)), LAG-3 (lymphocyte-activation gene 3 oder CD223), BTLA (B- and T-lymphocyte attenuator oder CD272) und VISTA (V-domain Ig suppressor of T cell activation) ebenfalls zu den NCR 7. TIM-3 ist auf aktivierten TH1-Zellen und Tc1-Zellen exprimiert 8. TH1- und Tc1-Zellen sind CD4+- bzw. CD8+-T-Zellen, die große Mengen des proinflammatorischen Zytokins IFN-γ (Interferon-γ) sezernieren. Im Gegensatz dazu sezernieren TH2- und Tc2-Zellen antiinflammatorische Zytokine, z. B. IL-4, IL-5, IL-10 4 Einleitung und IL-13 9,10. NCR wie TIM-3, markieren nicht nur tolerante, sondern auch immunerschöpfte Zellen 8. Das antiinflammatorische Zytokin IL-10 inhibiert die IL-2-Produktion und wirkt auf die T-Zell-Proliferation hemmend 11. Immunerschöpfung beschreibt das Phänomen, das bei einer chronischen Virusinfektion beobachtet wird und mit einer graduellen Abnahme der TZell-Effektorfunktionen einhergeht 12. Im Laufe der schrittweise ablaufenden Immunerschöpfung beobachtet man, dass die Sezernierung von proinflammatorischen Zytokinen abnimmt, aber die Expression von NCR wie PD-1 gesteigert wird 12. Eines der ersten Zeichen einer Immunerschöpfung bei Zellen ist die verringerte Produktion von IL-2 12. Obwohl das Phänomen der Immunerschöpfung bei CD8+-T-Zellen als erstes beschrieben wurde, ist es mittlerweile ebenfalls bei CD4+-T-Zellen beobachtet worden 12,13. Immunerschöpfung wird nicht nur durch chronische virale Infektionen induziert, sondern auch bei tumorinfiltrierenden Lymphozyten (tumor-infiltrating lymphocytes, TIL) beobachtet 14. Huang et al. zeigten, dass murine CD8+-TIL PD1+TIM-3+CEACAM1+ (carcinoembryogenic antigen-related cell adhesion molecule-1) Merkmale einer Immunerschöpfung aufweisen: eine verringerte intrazelluläre IL-2- und TNF-α-Expression 15. Nach einer subkutanen Implantation von Kolontumorzellen in Mäuse, führte die Blockade beider Moleküle (CEACAM1, TIM-3) zu einem verlangsamten Tumorwachstum. Außerdem zeigten Huang et al., dass HIV-(human immunodeficiency virus, humanes Immundefizienz-Virus) Patienten im Vergleich zu gesunden Probanden signifikant erhöhte CEACAM1+TIM-3+CD4+-T-Zell-Zahlen hatten 15. Sowohl CEACAM1+TIM-3+CD4+- als auch CEACAM1+TIM-3+CD8+-T-Zellen aus HIV-Patienten sezernierten signifikant weniger IFN-γ 15. Im Vergleich zu anergen Zellen verlieren immunerschöpfte Zellen nicht die Fähigkeit ihre Effektorfunktionen auszuführen 14. Immunerschöpfung ist also kein aktiver Toleranzmechanismus, führt aber zu anergieähnlichen Phänotypen. Humane CD4+-T-Zellen koexprimieren CEACAM1 und TIM-3 nachdem sie in vitro durch Anti-CD3- und Anti-CD28-Antikörper aktiviert wurden 15. Zusätzlich zeigten die Autoren, dass die CEACAM1 Expression ebenfalls vermindert war, wenn das Gen für TIM-3 inhibiert war. Im OVA-(Ovalbumin) spezifischen Toleranz-Modell zeigten Huang et al., dass OT-II Rag2-/--CEACAM-/--Tiere keine T-Zell-Toleranz gegenüber OVA aufwiesen 15 . Nach Stimulation durch Staphylococcus aureus enterotoxin B (SEB) exprimierten T-Zellen aus CEACAM-/- Tieren kein TIM-3. Huang et al. schlossen aus diesen Ergebnissen, dass die doppelte Expression von CEACAM1 und TIM-3 tolerante und immunerschöpfte T-Zellen markiert. Mittlerweile modifizierte kristallographische Modelle bestätigen eine theoretische CEACAM1-TIM-3-Bindung, es bleibt aber unklar, ob es weitere Faktoren gibt, die diese Bindung unterstützen 16 . Außerdem kann nach diesem neuen Modell nicht mehr eindeutig 5 Einleitung zwischen parallel (cis) und antiparallel (trans) angeordneten Interaktionen unterschieden werden 16. 1.1.2.1. T-Zell-Toleranz T-Zellen wandern aus dem Knochenmark aus und bevölkern den Thymus. Dort reifen sie heran und verlassen diesen als reife naive T-Zellen. Die zentrale T-Zell-Toleranz findet im Thymus statt, weil dort autoreaktive T-Zellen deletiert werden 17. Im doppel-positiven Stadium (CD4+CD8+) präsentieren medulläre Thymusepithelzellen (mTEC) und DZ an MHCKomplexen gebundene körpereigene Peptide 18. Durch den Transkriptionsfaktor AIRE (autoimmune regulator), der von mTEC und thymusständigen B- Zellen exprimiert wird, können auch gewebespezifische Peptide präsentiert werden 18,19. Nicht alle immundominanten Epitope werden im Thymus exprimiert 55. Z. B. kann man bei C57BL/6-Mäusen durch Proteolipidprotein-(PLP) Injektionen keine Multiple Sklerose-(MS) ähnliche Erkrankung induzieren, weil sie im Gegensatz zu SJL-(Swiss Jim Lambert) Mäusen PLP-Isoformen, die im zentralen Nervensystem (ZNS) vorkommen, im Thymus exprimieren 5. Die Bindungsstärke zwischen dem MHC-Peptid-Komplex und TZR entscheidet über das Überleben und die Differenzierung der T-Zellen. Ist die Bindungsstärke schwach, kommt es zum „Tod durch Vernachlässigung“ (death by neglect). Bei intermediärer Bindungsstärke erhalten die T-Zellen Überlebenssignale und differenzieren sich in der Rinde zu einfach-positiven CD4+- oder CD8+T-Zellen (positive Selektion, MHC-Restriktion) 18,20. Eine starke Bindung zwischen TZR und dem MHC-Peptid-Komplex führt zur Apoptose der T-Zelle und zur Deletion autoreaktiver TZellen aus dem T-Zell-Repertoire (negative Selektion) 21. Es bleibt umstritten, ob natürliche Treg, die eine Bindungsstärke zwischen der positiven und negativen Selektion aufweisen, aus dem Repertoire der autoreaktiven T-Zellen entstehen oder, ob sie bereits in einem früheren Stadium (CD4-CD8-, doppel-negativ Stadium) durch TZR-unabhängige Antigenbindung Überlebenssignale erhalten 22. Letzteres wäre unabhängig von der Bindungsstärke zwischen TZR und dem MHC-Peptid-Komplex. Nicht alle positiv selektierten T-Zellen, die den Thymus verlassen, sind selbsttolerant wird kontrovers diskutiert 25 23,24 . Wie viele selbstreaktive T-Zellen den Thymus verlassen, . Treg (CD4+CD25+-T-Zellen) spielen eine wichtige Rolle in der Aufrechterhaltung der peripheren Toleranz, weil sie Immunantworten modulieren, autoreaktive Zellen inhibieren oder Apoptose induzieren 26. Außerdem haben sie Einfluss auf die angeborene und die adaptive Immunantwort. Die Funktionen der Treg umfassen vier Mechanismen: die Sekretion antiinflammatorischer Zytokine (IL-10, TGF-β (tansforming growth factor- β), die granzym-perforin-vermittelte Zytotoxizität, die zytokinmangel-vermittelte Apoptose und die 6 Einleitung Inhibierung von DZ 27. Treg hemmen T-Zellen in ihrer Proliferation, indem sie durch ihren hochaffinen IL-2-Rezeptor (CD25), den sie konstitutiv exprimieren, kompetitiv IL-2 binden 28. Der dadurch entstehende Zytokinmangel kann in benachbarten T-Zellen Apoptose induzieren 28. Treg exprimieren NCR wie CTLA-4, die eine vielfach höhere Affinität zu den Oberflächenmolekülen CD80/86 aufweisen als zum kostimulatorischen Oberflächenmolekül CD28 29. CTLA-4 auf Treg supprimiert T-Zellen, indem es die Produktion von IDO (Indoleamine-2,3-Dioxygenase) in DZ induziert und über Trans-Endozytose CD80/CD86 von der Oberfläche von APZ entfernt 30,31. IDO baut die essentielle Aminosäure Tryptophan zu N-Formylkynurenin ab und hemmt damit die Proliferation der umliegenden T-Zellen 30. Eine Immunantwort wird dadurch beendet, dass reife antigenspezifische T-Zellen nach wiederholter Aktivierung Fas (first apoptosis signal, auch CD95) und FasL (Fas-Ligand auch CD95-Ligand) koexprimieren, wodurch es zur klonalen Kontraktion kommt 32. Zusätzlich kann die aktivierungsinduzierte Apoptose (activation-induced cell death, AICD), die durch autokrine und parakrine Fas/FasL-Bindung vermittelt wird, ebenfalls aktivierte autoreaktive T-Zellen entfernen 32,33. Ein weiterer wichtiger peripherer T-Zell-Toleranzmechanismus ist die fehlende Kostimulation durch APZ und die anschließende Anergie-Induktion 34. 1.1.2.2. B-Zell-Toleranz B-Zellen sind ebenfalls hämatopoetischen Ursprungs, verbleiben aber im Knochenmark und verlassen dieses als reife naive B-Zellen. Die erste Station der B-Zell-Toleranz findet im Knochenmark statt (zentrale Toleranz). Die B- und T-Zell-Rezeptorvielfalt basiert auf einer zufälligen Anordnung der V-(variable), D-(diversity) und J-(joining) Gensegmente (B-Zellen: schwere Kette, T-Zellen: β-Kette) bzw. V- und J-Gensegmente (leichte Kette, α-Kette) und wird als V(D)J- oder somatische Rekombination bezeichnet 35. Die Mechanismen der zentralen Toleranz während der B-Zell-Reifung im Knochenmark gleichen denen der negativen und positiven Selektion der T-Zellen im Thymus. Werden Selbstantigene von unreifen B-Zellen im Knochenmark gebunden, wird der B-Zell-Rezeptor (BZR) einem „receptor editing“ unterzogen, d. h. es folgt eine sekundäre Umlagerung der V- und J-Gensegmente der leichten Kette 36. Es gibt Hinweise, dass zwischen 50 % bis 70 % aller B-Zellen vor dem receptor editing im Knochenmark autoreaktiv sind 37. Es entsteht eine neue leichte Kette, die mit der schweren Kette eine neue Spezifität ausbilden kann. Ist diese Spezifität immer noch gegen körpereigene Antigene gerichtet, führt die Bindung des BZR an körpereigene Antigene zum Zelltod, bevor die autoreaktiven B-Zellen das Knochenmark verlassen können 36. Ein Drittel aller autoreaktiven B-Zellen wird durch das sekundäre Umlagern der leichten Kette entfernt 37. Die 7 Einleitung erneute Gen-Umlagerung wird mittlerweile als Hauptmechanismus der zentralen B-ZellToleranz betrachtet. Neben diesem Mechanismus können autoreaktive B-Zellen in den Zustand der Anergie oder der immunologischen Ignoranz versetzt werden. Ignorante B-Zellen erkennen ihr Antigen nicht, weil es sich z. B. im Zellinneren befindet und es deshalb nicht zugänglich ist. Im Gegensatz zur Anergie, sind immunologisch ignorante B-Zellen leichter aktivierbar und werden durch CD22-Signale inhibiert 38. Hingegen dazu regulieren anerge B-Zellen im Knochenmark das Oberflächenmolekül CD19 und ihren BZR herunter 36. In der Peripherie sind B-Zellen, die aus dem Knochenmark emigrieren, von Überlebenssignalen abhängig. Diese Signale können BZR-abhängig oder BZR-unabhängig durch Zytokine vermittelt werden, z. B. durch Signale des BAFF-Rezeptors (B cell activation factor of the TNF family receptor) 39 . Anerge und ignorante autoreaktive B-Zellen, mit einer geringen Avidität zu körpereigenen Antigenen, haben im Gegensatz zu naiven B-Zellen eine reduzierte Lebensdauer 40,41 . Die eigeschränkte Lebensdauer kann verlängert und die Anergie aufgehoben werden, wenn autoreaktive B-Zellen starke Überlebenssignale erhalten 42 . Autoreaktive B-Zellen sind im Vergleich zu nicht autoreaktiven B-Zellen stärker vom Überlebensfaktor BAFF abhängig 43 . Dieser Umstand ist ein weiterer Mechanismus, um die Toleranz aufrecht zu halten. Obwohl selbstreaktive Zellen mehrere Checkpoints durchlaufen und einen erhöhten Schwellenwert haben, um auf Überlebenssignale zu reagieren, finden sich in gesunden Menschen autoreaktive T-Zellen und autoreaktive IgM+- und IgG+-B-Gedächtniszellen (IgM+-memory B cells, classswitched memory B cell) 44,45. Das Vorhandensein von autoreaktiven B-Gedächtniszellen wird durch die somatische Hypermutation erklärt, die während der follikulären Keimzentrumsreaktion in sekundären lymphatischen Organen stattfindet 46. Bei der somatischen Hypermutation werden Zellen selektiert, bei denen DNS-Mutationen die Spezifität und Affinität zum Antigen verbessern. Dieser Prozess wird Affinitätsreifung genannt und kann zu neuen, u. a. auch autoreaktiven Spezifitäten führen. Ignorante autoreaktive B-Zellen oder autoreaktive B-Zellen, die nach der Affinitätsreifung entstanden sind, können von follikulären T-Helferzellen (TFH) körpereigene oder körperfremde Antigene präsentiert bekommen. Dies kann zum Toleranzbruch führen. Um zufällige Toleranzbrüche zu verhindern, wird vermutet, dass Signale durch den inhibitorischen B-Zell-Korezeptor FcγIIB (Fc fragment of IgG) Aktivierungssignale hemmen 47. Aus FcγIIB-Knockout-Experimenten weiß man, dass in defizienten Tieren die Anzahl der autoreaktiven B-Zellen nach der Affinitätsreifung im Lymphknoten stark erhöht ist 47. Die Anzahl der autoreaktiven B-Zellen in der Milz und der Plasmazellen im Knochenmark war davon nicht betroffen 47. Es wurde vermutet, dass weitere unbekannte FcγIIB-Rezeptor-unabhängige Toleranz-Mechanismen existieren 47. Die Interaktion zwischen B- und T-Zellen ist wichtig für die Erhaltung der peripheren B-Zell8 Einleitung Toleranz, denn reife B-Zellen aus CD40L-defizienten Patienten und Patienten mit genetischen Defekten der HLA-Expression (bare lymphocyte syndrom) produzierten große Mengen an Autoantikörpern 48. Die Autoren vermuten, dass Treg über CD40L-CD40-Bindung und MHCKlasse-II-Peptid-BZR-Bindung die periphere B-Zell-Toleranz kontrollieren 48. Ein weiterer unbekannter Toleranzmechanismus wird zwischen dem Stadium der frühen reifen naiven BZelle und dem späteren IgM+-Gedächtniszell-(auch Marginalzonen-B-Zellen genannt) Stadium vermutet 49. Ungefähr 20 % aller naiven B-Zellen, die frisch aus dem Knochenmark emigrieren, sind autoreaktiv 49. Durch Ausfall der zentralen oder peripheren Toleranzmechanismen können Autoimmunerkrankungen entstehen. Das Versagen der Toleranzmechanismen kann man auf genetische oder äußerliche Einflüsse zurückführen. 1.1.3. Autoimmunität Lange galt eine Immunantwort auf körpereigene Stoffe als unvorstellbar 50. Das Konzept der Autoimmunität wurde zu Beginn des 19. Jahrhunderts kontrovers diskutiert. Paul Ehrlich postulierte 1901, dass Individuen Antikörper gegen Fremdantigene besitzen, aber dass der „horror autotoxicus“, also die Furcht vor der Selbstzerstörung, die Existenz von autoreaktiven Antikörpern unmöglich macht 3. Die Präsenz von Antikörpern gegen Selbstantigene war nach Paul Ehrlichs Auffassung dysteleologisch und somit wäre ihr Vorhandensein unmöglich 50 . Aber ausgerechnet Paul Ehrlichs Seitenkettentheorie wurde zu einer der Grundlagen der KlonSelektionstheorie, die bekanntlich Spezifitäten gegen Selbstantigene postulierte und somit das Vorhandsein von autoreaktiven Antikörpern erlaubte 3. In seiner Seitenkettentheorie oder Rezeptortheorie postuliert Ehrlich, dass alle Zellen des Körpers Seitenketten bzw. Rezeptoren besitzen, die Bakterientoxine binden können. Durch deren schädliche Wirkung auf die Zellen kommt es zur vermehrten Produktion weiterer Seitenketten. Durch erneuten Kontakt mit dem für die Seitenkette spezifischen „Gift“ wird die Zelle trainiert, d. h. dass sie immer mehr spezifische Seitenketten bzw. Rezeptoren produziert und schließlich die Seitenketten abstößt. Paul Ehrlich unterschied spezifische Immunkörper und unspezifische Faktoren. Immunkörper waren die freien Seitenketten und die unspezifischen Faktoren nannte er Komplement. Nach der Seitenkettentheorie wird das Komplement mit Hilfe der Immunkörper an das Bakterium gebunden und löst es auf 51. Diese theoretischen Annahmen wurden teilweise bestätigt. Nach Frank Macfarlane Burnet wird die Selbsttoleranz durch den Kontakt zwischen dem Immunsystem mit körpereigenen Antigenen während eines sehr frühen Stadiums der individuellen Entwicklung erworben 4. Autoimmunerkrankungen sind eine Folge aus dem Zusammenbruch der Selbsttoleranz. Es gibt viele Hypothesen zur Ätiologie der 9 Einleitung Autoimmunerkrankungen. Zum Bruch der Selbsttoleranz kann es kommen, wenn Antigene durch Traumata oder durch entzündliche Prozesse zugänglich werden. Die Immunantwort kann sich gegen neue Antigene richten, die zuvor unzugänglich (kryptische, sequestrierte Antigene) waren, oder die Immunantwort kann sich auf köpereigene Antigene, die durch entzündliche Prozesse zugänglich werden, ausweiten (epitope spreading) 52,53. Homologien von Aminosäuresequenzen oder strukturelle Ähnlichkeiten zwischen körperfremden und körpereigenen Antigenen (molekulare Mimikry) können ebenfalls Autoimmunerkrankungen auslösen 53–55. Zusätzlich können tolerante Immunzellen durch proinflammatorische Mediatoren aktiviert werden (bystander activation). Molekulare Mimikry kann bei Patienten, die mit A-Streptokokken (β-hämolysierenden Streptokokken) infiziert sind, zu rheumatischem Fieber führen. Rheumatisches Fieber ist dadurch gekennzeichnet, dass es zu vielfältigen Immunreaktionen gegen verschiedene Organe kommt. Zum Beispiel finden sich in Patienten Antikörper gegen A-Streptokokken, die eine Kreuzreaktivität mit dem Gewebe des Herzmuskels (Myosin) aufweisen 55. Virusinfizierte Zellen können APZ stimulieren und diese wiederum können autoreaktive T-Zellen, die vorab Antigenkontakt hatten (pre-primed), vollständig aktivieren 55. Außerdem können virusspezifische T-Zellen in das betroffene Gewebe migrieren und dort durch die Sezernierung von zytotoxischen Granula (von CTL) oder durch Zytokin-Sezernierung (von TH-Zellen) Zellen eliminieren 55. In solch einem inflammatorischen Milieu beginnen T-Zellen und Makrophagen Mediatoren zu sezernieren, die zur bystander activation von benachbarten Zellen führen kann 55 . Autoimmunerkrankungen können durch Fehler bei der zentralen und peripheren Toleranzinduktion entstehen. Die Gründe für den teilweisen Ausfall der zentralen Toleranz können in genetischen Defekten liegen. So ist die Ursache für das seltene rezessiv vererbte Autoimmun-Polyendokrinopathie-Candidiasisektodermale-Dystrophie-Syndrom (APECEP) ein Defekt auf dem AIRE-Gen 56. Das Gen AIRE codiert den AIRE-Transkriptionsfaktor und dieser wird in Stromazellen des Thymusmarks und von B-Zellen im Thymus exprimiert 19. Thymus B-Zellen unterscheiden sich von B-Zellen aus der Peripherie durch ihre AIRE-Expression 19. AIRE ist notwendig, um gewebespezifische Proteine, die nicht im Thymus vorkommen, wie z. B. Insulin, zu exprimieren. Es gibt körpereigene immundominate Epitope, die nicht im Thymus exprimiert werden 5. Wenn sequestrierte oder kryptische Antigene zugänglich werden, die im Thymus nicht präsentiert werden, kann es zu Autoimmunreaktionen kommen, weil die verantwortlichen autoreaktiven Klone durch die negative Selektion nicht aus dem Repertoire entfernt wurden. Einige Individuen haben eine genetische Prädisposition an Autoimmunerkrankungen zu erkranken. Es konnte gezeigt werden, dass einige HLA-Genotypen mit dem Auftreten von Autoimmunerkrankungen assoziiert sind. So ist bekannt, dass das relative Risiko an MS zu 10 Einleitung erkranken erhöht ist, wenn man Träger des HLA-DR2-Haplotyps ist. HLA-DR*1501 ist als das Allel identifiziert worden, das den stärksten Zusammenhang zeigt 57. Andererseits kann die periphere Toleranzinduktion ausfallen, weil negative Immun-Checkpoints, also Proteine, welche die Immunantwort negativ modulieren, defekt sind 6. Ungenügende oder fehlende TIM-3 Expression kann in Autoimmunerkrankungen wie der MS beobachtet werden 58,59. Kommen Defekte in Treg vor, führt dies ebenfalls zum Ausfall der peripheren Toleranz. Ist das Gen, das den Transkriptionsfaktor FoxP3 (forkhead box P3) kodiert defekt, kommt es zum XChromosom-gekoppelten, Polyendokrinopahtie, rezessiven Enteropathie, Autoimmunsyndrom X-gekoppeltes IPEX (Immunderegulation, Syndrom) 60. Man kann Autoimmunerkrankungen in organspezifische und systemische Erkrankungen unterteilen 53. Bei organspezifischen Krankheiten werden einzelne Organe angegriffen, z. B. ist die Ursache des Diabetes mellitus Typ 1 die Immunreaktion gegen insulinproduzierenden β-Zellen des Pankreas 61. Eine weitere organspezifische Erkrankung ist die MS, bei der sich die Immunreaktion auf das ZNS beschränkt 62. Das primäre Antigen, das eine Reaktion gegen die Myelinscheide initiiert, ist unbekannt 63. Ebenfalls unbekannt sind die Antigene in der Synovialmembran der Gelenkkapsel, die zur rheumatoiden Arthritis (RA) führen 64 . Die RA wird zu den systemischen Autoimmunerkrankungen gezählt, weil die Reaktionen in diesem Fall gegen unbekannte und ubiquitär vorkommende Antigene gerichtet sind. Im Gegensatz zur RA sind beim systemischen Lupus erythematodes (SLE) die Antigene bekannt, die zum Ausbruch der Erkrankung führen. Bei Patienten mit SLE wurden Antikörper gegen körpereigene Zellkernbestandteile nachgewiesen 65. Die immunpathogenen Mechanismen bei Autoimmunerkrankungen können nach Hypersensitivitätsreaktionen des Typ-II (Antikörper gegen Zelloberflächen- oder Matrixantigene), des Typ-III (Immunkomplexerkrankungen) oder des Typs-IV (T-Zell-vermittelt) eingeteilt werden 66. Traditionell werden die MS und der Diabetes mellitus Typ 1 als Typ-IV-vermittelte Erkrankungen eingestuft, anders als die RA, die sowohl als Typ III- als auch als Typ-IV-vermittelte Erkrankung eingeteilt wird 66 . Neben genetischen Prädispositionen finden sich Umweltfaktoren, die in Zusammenhang mit Autoimmunerkrankungen stehen. Umweltfaktoren können durch Lebensgewohnheiten, Infektionen oder Vitaminmangel zur Autoimmunität führen bestimmte 66 . Es gibt Hinweise darauf, dass Nikotinabusus, hohe Antikörpertiter gegen Epstein-Barr-Virus-(EBV)Antigene und Vitamin D-Mangel MS-Risikofaktoren sind 67. MS gehört zu den häufigsten chronischen ZNS Erkrankungen bei jungen Erwachsenen. Außerdem gilt sie als die häufigste neurologische Ursache bei jungen Erwachsenen, die zu bleibenden Behinderungen führt 63. Der nächste Abschnitt wird sich näher mit der MS befassen. 11 Einleitung 1.2. Multiple Sklerose 1.2.1. Geschichte der MS In den 1830er Jahren lieferten Robert Carswell und Jean Cruveilhier die ersten pathologischen Studien, in denen sie disseminierte Plaques im Hirngewebe, die sie durch Autopsien von Patienten mit Paresen gewonnen hatten, beschrieben 68. Zu diesem Zeitpunkt war die MS noch nicht als eigenständige Erkrankung erkannt worden. Erst 1849 gelang es Friedrich von Frerichs als einer der Ersten, die klinischen und pathologischen Befunde zusammenzufassen. Er erkannte, dass es sich um eine eigenständige Erkrankung handelte, die er „Hirnsklerose“ bezeichnet 68. Seine Arbeit blieb weitestgehend unbeachtet. Kurze Zeit später erkannte Eduard Rindfleisch, dass es sich bei den Plaques um entzündliche Schädigungen der Nerven handelte 68. Allgemein setzte sich aber Jean-Martin Charcots Erstbeschreibung durch, die er 1868 in einer Serie von Vorlesungen am Hôpital de la Salpêtrière einem breiten Publikum vorstellte. In seinen Vorlesungen beschrieb er „sclerose en plaques“ im ZNS und assoziierte diese mit klinischen Symptomen, die er als Krankheitsbild zusammenfasste 68. Wie viele seiner Zeitgenossen, die durch die Entdeckungen von Louis Pasteur beeindruckt waren, glaubte auch Charcot, dass es sich bei der MS um eine Infektion durch Mikroorganismen handelte 68. Nystagmus, Intentionstremor und skandierende Sprache galten als charakteristisch für die Erkrankung und bis heute nennt man diese Symptomgruppe „Charcot-Trias“ 66 . Erst in den 1930er Jahren stellte man nach Tierexperimenten von Thomas Rivers und seinen Kollegen fest, dass es sich bei der MS um eine Autoimmunerkrankung handelte 69. Dabei war die Erkenntnis auch darauf zurückzuführen, dass keine Krankheitserreger im Gehirn der Versuchstiere nachgewiesen werden konnten und man die Hypothese, dass Infektionen eine mögliche Ursache waren, verwarf 69. Ab der zweiten Hälfte des 20. Jahrhunderts wurden akute MS-Schübe mit Adrenocorticotropin (auch Adrenocorticotropes Hormon, ACTH) behandelt, bis man in den 1980er Jahren die Hochdosistherapie mit synthetischen Glukokortikosteroiden einführte. ACTH ist ein funktionelles Peptid, das ein hochaffiner universeller Agonist für alle fünf Melanocortin-Rezeptoren (MCR1-MCR5) ist 70. Das aus dem Hypophysenvorderlappen stammende Hormon wirkt u. a. auf die Nebennierenrinde während Stressphasen stimulierend, die daraufhin Glukokortikosteroide, z. B. Kortisol, freisetzt 71. Die Behandlung führt zu einer Verbesserung der Symptome und zu einer Verkürzung der Schubdauer innerhalb von wenigen Wochen 71. Die Tatsache, dass es intra- und interindividuelle Schwankungen in der KortisolFreisetzung gibt, führte Anfang der 1980er Jahre dazu, dass Methylprednisolon- und Prednisolonpräparate, beides synthetische Glukokortikosteroide, vermehrt in der Schubtherapie eingesetzt wurden 71. Diese Behandlungsform hat sich bis heute bewährt. Hochdosistherapien 12 Einleitung mit synthetischen Glukokortikosteroiden (Glukokortikosteroid-Pulstherapie) verkürzen nur die Schubdauer, haben aber keinen positiven Einfluss auf den weiteren Verlauf der Erkrankung 72. Glukokortikosteroide hemmen die Aktivität von APZ und T-Zellen, die Migration von Lymphozyten ins ZNS, die intrathekale IgG-Synthese, proinflammatorische Zytokine und sie dichten die Bluthirnschranke während eines MS-Schubs ab 73,74. Außerdem wirkt die Glukokortikosteroid-Pulstherapie während eines akuten MS-Schub inhibierend auf die Arachidonsäuremetaboliten, hemmt die Degranulierung lysosomaler Enzyme und stimuliert antiinflammatorische Zytokine 75. Erst in den 1990er Jahren wurden die ersten Medikamente zugelassen, die zur Schubprophylaxe eingesetzt wurden. Es folgt eine kurze Übersicht der zugelassenen MS-Therapien in Deutschland. 1.2.2. Klinische MS-Symptomatologie Die Diagnose MS wird als gesichert angesehen, wenn ein Patient an fokalen neurologischen Symptomen leidet, die mindestens 24 Stunden anhalten und die weiße Hirnsubstanz betreffen 66,75. Diese neurologischen Symptome müssen in Bezug auf Zeit und Ort disseminiert auftreten. MS-Schübe können bis zu einer Woche dauern und unterschiedliche klinische Symptome zeigen, weil verschiedene Orte des ZNS von der Entzündung betroffen sein können. Die McDonald-Kriterien wurden 2001 vorgestellt und haben sich mittlerweile im klinischen Alltag und in der Forschung etabliert 76. Diese Kriterien wurden 2005 und 2010 modifiziert 77,78. Neben der klinischen Manifestation (klinische Zeichen von Läsionen und anamnestisch gesicherten Schüben) und nach Ausschluss von Differenzialdiagnosen werden Ergebnisse von Zusatzuntersuchungen benutzt, um die Diagnose zu stützen. Die Magnetresonanztomographie (MRT) kann das Kriterium der zeitlichen und örtlichen Dissemination untersuchen, die Lumbalpunktion kann eine Erhöhung des Gesamteiweißes nachweisen, eine anschließende isoelektrische Fokussierung kann oligoklonale Banden (OKB) identifizieren 66. OKB sind Banden monoklonaler IgG-Antikörper im Liquor und liefern einen Hinweis auf entzündliche Prozesse im ZNS. Zusätzlich erlauben Untersuchungen durch visuell und/oder akustisch evozierte Potenziale den Nachweis einer möglichen verzögerten Latenz der kortikalen bzw. pontomedullären evozierten Potentiale 66. Die häufigsten Befunde bei MS-Patienten sind Visusstörungen, Pyramidenbahnzeichen, abnorme Reflexe, Muskelschwäche, Spastiken und Sensibilitätsstörungen (Parästhesien) 66. Bei fast 64 % der MS-Patienten finden sich zerebelläre Symptome 66 . Die häufigsten MS-Symptome sind zu Beginn der Erkrankung Schwäche und Sensibilitätsstörungen 66. Mit abnehmender Häufigkeit fanden sich Miktions- oder Defäkationsstörungen, Hirnstammsymptome, psychische Probleme, kognitive Störungen und 13 Einleitung anfallsartige Phänomene 66. Die Hälfte der unbehandelten MS-Patienten ist 10 Jahre nach der Erstdiagnose auf eine Gehhilfe angewiesen, 15 % benötigen einen Rollstuhl 79 . Um den Schweregrad der Behinderung anzugeben, wurde eine Leistungs-/Behinderungsstärke-Skala eingeführt (Expanded Disability Status Scale, EDSS), die physiologische neurologische Befunde (0) bis hin zum Tod (10) als Folge der MS einschließt. Die MS wird als zweistufiger Prozess betrachtet: eine frühe Phase, die durch Inflammationsprozesse bestimmt wird und eine neurodegenerative Phase 80. Während die erste Phase individuell unterschiedlich lange Verläufe aufweist, ist die spätere Phase unabhängig davon und zeigt einen zeitlich gleichförmigen Verlauf 81. Durch die Erfolge der B-Zell-Depletionstherapien, nicht nur bei der schubförmigen MS (relapsing-remitting MS, RRMS) sondern auch bei der primär progredienten MS (PPMS), wird aktuell die zweiphasige Natur der Erkrankung kontrovers diskutiert 82 . Es scheint, dass entzündliche Prozesse auch in der chronischen Phase eine wichtige Rolle spielen. 1.2.3. Verlaufsformen und Therapieansätze Die Therapieansätze umfassen heute neben der Immunsuppression und Immunmodulation auch die gezielte Depletion bestimmter Immunzellen. Nach den Leitlinien für Diagnostik und Therapie in der Neurologie sind 2016 zehn Arzneimittel zur Langzeitbehandlung der RRMS zugelassen: Alemtuzumab, Azathioprin, Dimethylfumarat, Fingolimod, Glatirameracetat (GA), Interferon β-1a (intramuskulär, i.m. und subkutan, s.c.), Interferon β-1b (s.c.), Mitoxantron, Natalizumab (NAT) und Teriflunomid 75. β-Interferone und GA werden als Basistherapeutika eingesetzt 83. β-Interferone und GA beeinflussen die Zunahme der Behinderung negativ und reduzieren die Anzahl neuer Läsionen 84–89. Beide Medikamente sind ebenfalls zur Behandlung von Patienten mit einem klinisch isolierten Syndrom (clinically isolated syndrom, CIS) zugelassen 75 . Beide Präparate-Klassen zeigen keinen Unterschied in ihrer Wirksamkeit 90,91 . Es fehlen bis heute Prädiktoren, die den Therapieerfolg oder das Therapieversagen zuverlässig vorhersagen. Die Leitlinien der Deutschen Gesellschaft für Neurologie empfehlen bei leichten Schüben einen Wechsel des Basistherapeutikums 75. CIS-Patienten haben ein erhöhtes Risiko an MS zu erkranken 92. Innerhalb von zehn Jahre erkranken 67 % der CIS Patienten mit MRTBefunden an MS, dagegen konvertieren nur 11 % der CIS Patienten ohne MRT-Läsionen 93. In späteren Phasen der RRMS kommt es zu Überlagerungen von Behinderungen und unvollständigen Remissionen nach Krankheitsschüben 66. In dieser Phase der Erkrankung wird von einer sekundär progredienten MS (SPMS) gesprochen 66. β-Interferon ist z. Zt. das einzige immunmodulierende Basistherapeutikum in Deutschland, das zur Behandlung der SPMS zugelassen ist 75. Neben der RRMS und der SPMS gibt es eine weitere Verlaufsform, die mit 14 Einleitung oder ohne Krankheitsschüben primär fortschreitend ist; die PPMS 66. Bei hochaktiver RRMS und beim Versagen der Basistherapeutika wird NAT, ein α4-Integrin-Antagonist, eingesetzt. NAT ist ein monoklonaler Antikörper, der die T-Zell-Aktivierung und Lymphozytenmigration durch das Adhäsionsmolekül VLA-4 (very late antigen-4, auch α4β1-Integrin) verhindert 94. NAT alleine oder mit β-Interferon zusammen verabreicht, reduziert effektiv die Schubrate (nur NAT: 50 %) bzw. die MRT-Aktivitäten (NAT und β-Interferon: 90 %) im Vergleich zu Patienten, die mit einem Placebo behandelt wurden 95,96. NAT kann bei gleichzeitiger JC-(John Cunningham) Virusinfektion eine letale progressive multifokale Leukenzephalopathie (PML) auslösen 97. Besonders nach längerer Therapie mit NAT erhöht sich das Risiko einer PML 98. Ein weiterer monoklonaler Antikörper ist Alemtuzumab bzw. Campath-1H. Alemtuzumab depletiert alle CD52+-Zellen 99. Die Immunzell-Verhältnisse werden durch die Depletion verändert und möglicherweise werden Toleranzmechanismen aktiviert oder wiederhergestellt 99. Bei 30 % bis 40 % der Patienten, die mit Alemtuzumab behandelt wurden, kommt es zu sekundären Autoimmunerkrankungen, die größtenteils die Schilddrüsenfunktion betreffen 99. In einer Studie mit Patienten, bei denen es trotz β-Interferon- oder GA-Behandlung zu Schüben gekommen war, zeigten 20 % der Patienten ohne Alemtuzumab-Behandlung im Vergleich zu 13 % der Patienten aus der Alemtuzumab-Gruppe eine Verschlechterung des Schweregrades der Behinderung 100 . Fingolimod, ein Sphingosin-1-phosphat-Analogon, wird ebenfalls als Basistherapie bei RRMS-Patienten eingesetzt 75. Es hemmt die Emigration aus den lymphatischen Organen 101 . Die Behandlung zeigt unerwünschte Effekte auf das Herz (Bradykardie) und steht in Verdacht Hauttumore zu fördern 102. Mittlerweile sind auch unter Fingolimod-Therapie Fälle von PML beschrieben worden 102. β-Interferone, GA, NAT und Fingolimod wirken immunmodulierend. Neben diesen Medikamenten werden auch Immunsuppressiva eingesetzt. Mitoxantron ist als Eskalationstherapie für Patienten mit RRMS und SPMS, bei Versagen oder bei Unverträglichkeit einer immunmodulatorischen Vortherapie zugelassen 75. Es behindert die Aktivierung von T-Zellen und verhindert die Proliferation von B- und T-Zellen 66 . Weitere Immunsuppressiva, die bei RRMS eingesetzt werden, sind die Reservepräparate Azathioprin, Cyclophosphamid und Methotrexat 66,75 fehlendem Behandlungserfolg mit β-Interferone eingesetzt werden . Azathioprin kann bei 75 . Außerdem werden intravenös Immunglobuline (IVIG) verabreicht oder Plasmapharesen durchgeführt 103. IVIG werden zur Schubprophylaxe eingesetzt, wenn andere MS-Therapien versagt haben 66. Plasmapharesen werden als Schubtherapie beim Versagen der Methylprednisolon-Behandlung durchgeführt und zeigten bei einer bestimmten Gruppe von MS-Patienten eine Verbesserung der neurologischen Einschränkungen 75,103. Teriflunomid ist ein Basistherapeutikum, das oral eingenommen werden kann und im Verdacht steht, teratogen zu sei 75. Teriflunomid wirkt 15 Einleitung proliferationshemmend auf aktivierte B- und T-Zellen 75. Dimethylfumarat ist ein weiteres immunmodulierendes orales Basistherapeutikum, das erst 2014 zugelassen wurde und bei Langzeitbehandlung gastrointestinale Unverträglichkeit als Nebenwirkung zeigt 75. Die erfolgreichsten Studien wurden aber mit therapeutischen Antikörpern durchgeführt, die BZellen depletieren. In einigen MS-Zentren werden diese Therapieoptionen off-label eingesetzt. Rituximab ist ein Antikörper, der alle CD20+-B-Zellen depletiert 104 . Plasmazellen werden durch Rituximab nicht depletiert 105. In kleinen Studien zeigte Rituximab als Add-on-Therapie zu β-Interferon und GA einen stabilisierenden Effekt auf die Behinderungsstärke 106 . Bar-Or et al. zeigten in einer Phase-I-Studie, dass 80 % der RRMS-Patienten nach 72 Wochen schubfrei waren und nach Rituximab-Behandlung keine MRT-Aktivität nachweisbar war 104. Außerdem wurde in einer zweiten, größeren placebokontrollierten Studie gezeigt, dass Rituximab signifikant die Schubrate reduziert und eine sistierende Krankheitsaktivität im MRT induzierte 105. Die Behandlung mit Rituximab wird als Eskalationstherapie empfohlen 75. Neben leichten Infusionsreaktionen kommt es bei der Rituximab-Behandlung zu milden bis schweren Infektionen 75. Interessanterweise ändert sich nach Rituximab-Behandlung der relative Anteil autoreaktiver B-Zellen nicht 107. Chamberlain et al. untersuchten für diesen Zweck die Antikörperspezifität gegen körpereigene Antigene durch indirekte Immunfluoreszenz und mittels Enzymimmunoassay (enzyme-linked immunosorbent assay, ELISA) 107 . Ein anderer anti-CD20-Antikörper, Ocrelizumab, ist humanisiert und hat in drei Phase-III-Studien, die beim weltweit größten MS-Kongress ECTRIMS (European Committee for Treatment and Research in Multiple Sclerosis) 2015 in Barcelona vorgestellt wurden, als erstes Medikament erstmals auch positive Effekte bei PPMS-Patienten gezeigt 108. Im Vergleich zu β-Interferonbehandelten RRMS-Patienten zeigten ocrelizumab-behandelte RRMS-Patienten eine Reduzierung aktiver MS-Läsionen um > 90 % und der jährlichen Schubrate um > 45 % 108 . Auch schon frühere Ocrelizumab-Studien zeigten ähnliche Erfolge im Vergleich zur β-Interferon-Behandlung 109 . Ein Anti-CD20-Antiköper, der ein anderes Epitop des CD20- Antigens auf B-Zellen bindet als Rituximab oder Ocrelizumab, ist Ofatumumab. Die Ergebnisse einer kleinen Studie zeigten, dass durch Ofatumumab eine Reduktion aktiver MSLäsionen um 99,8 % erreicht wurde 110. Die Ergebnisse der B-Zell-Depletionsstudien belegen eindrucksvoll, dass B-Zellen eine große Rolle in der Pathogenese der MS einnehmen. 1.2.4. Epidemiologie und volkswirtschaftliche Relevanz In Deutschland gab es 2010 199.505 Menschen mit MS. Diese Informationen stammen aus einer der ersten gesamtdeutschen epidemiologischen MS-Studie 111. Peters et al. zeigten, dass 16 Einleitung das epidemiologische Ausmaß der Erkrankung drastisch unterschätzt wurde. Die Grundlage für diese Studie lieferte der morbiditätsorientierte Risikostrukturausgleich in der gesetzlichen Krankenversicherung, der Diagnosen und ambulante Arzneimittelverordnungen von über 90 % der Bevölkerung umfasst. Die tatsächliche Prävalenz von 289/100.000 übertraf deutlich die früheren Angaben, die aus kleineren Stichproben extrapoliert worden waren. Die Studie beschrieb auch bekannte Phänomene, z. B. dass in allen Altersklassen Frauen häufiger als Männer an MS erkrankt waren. Die mittlere Prävalenz bei Frauen betrug 382/100.000 111 . Im Gegensatz dazu lag die Prävalenz im gleichen Zeitraum bei Männern bei 167/100.000. Das Geschlechterverhältnis lag bei 2,3:1. Des Weiteren waren die Patienten im Mittel 49,4 Jahre alt. Bei einem Drittel der Erkrankten wurde 2010 RRMS (33 %) diagnostiziert, bei 7 % der MSErkrankten mit einer spezifischen Diagnose wurde SPMS und PPMS diagnostiziert. Daneben gab es im Untersuchungszeitraum Patienten, die zu mehreren Kategorien gezählt wurden. Insgesamt hatten 63 % der Erkrankten eine gesicherte Diagnose und die Leistungsausgaben in dieser Gruppe lagen 2010 im Durchschnitt bei 13.060 Euro. Keine eindeutige Diagnose hatten 37 % der Erkrankten. In dieser Gruppe befanden sich Patienten mit einer MS-Erstdiagnose und Patienten, deren MS-Verlaufsform nicht näher definiert werden konnte. Die Leistungsausgaben bei dieser Gruppe lagen im Durschnitt bei 7.267 Euro. Insgesamt erhielten 49 % der MSErkrankten eine MS-relevante Pharmakotherapie 111. Im Barmer GEK Arzneimittelreport von 2014 wurden MS-relevante Immunmodulatoren als einer der großen Kostenfaktoren im Arzneimittelbereich ausgemacht 112 . Bei Jahrestherapiekosten zwischen 16.000 und 20.000 Euro (β-Interferone, GA) hat die MS-Erkrankung eine gesundheitsökonomisch hohe Relevanz. Nach aktuellem Kenntnisstand ist die Immunopathogenese der MS multifaktoriell und nicht vollständig verstanden. Die Suche nach Biomarkern, die bestimmte MS-Patientengruppen identifizieren, ist bis heute fruchtlos geblieben. Bei der Hälfte der Patienten, die mit β-Interferonen oder GA behandelt werden, wird keine signifikante Schubprophylaxe erzielt 83. Obwohl MS-Patienten heute viele MS-Therapieoptionen offen stehen, gibt es keine Biomarker, die den Therapieerfolg prognostizieren können. 1.2.5. Ätiologie und Pathogenese Die ersten Belege, dass es sich bei MS um eine Autoimmunerkrankung handelt, wurden 1933 von Thomas Rivers geliefert 69. Das heute bekannteste und am weitesten verbreitete Tiermodell der MS, die experimentelle allergische bzw. autoimmune Enzephalomyelitis (EAE), basiert auf seinen ersten Arbeiten. Es war bekannt, dass Injektionen mit Suspensionen von FremdKaninchengehirn in Kaninchen zu Paralysen führten 69. Rivers überprüfte, ob Injektionen von 17 Einleitung gesundem Kaninchengewebe in Rhesusaffen ähnliche Effekte zeigten. Obwohl keine Infektion nachweisbar war, konnten bei einigen Tieren Immunzellinfiltrate und Demyelinisierungen nachgewiesen werden 69. Ein erster Zusammenhang zwischen der Fähigkeit eine ZNSPathologie zu induzieren und dem Myelingehalt in den Injektionen wurde schon früh vermutet. Mehrere Injektionen waren auf ein Jahr verteilt nötig, um eine ZNS-Erkrankung herbeizuführen. Elvin Kabat versetzte die Injektionen mit Adjuvanzien, die zuvor von Jules Freund entwickelt worden waren 69. Nun konnte die Erkrankung mit einer einzigen Injektion induziert werden. Der endgültige Beleg, dass es sich um eine autoimmune Reaktion handelte, lieferten Versuche von Elvin Kabat und Isabel Morgan, die durch Injektionen mit homologem Hirngewebe ebenfalls eine ZNS-Entzündung herbeiführten 69. Die Immunpathogenese der MS ist bis heute nicht vollständig verstanden. Es wird vermutet, dass autoreaktive T-Zellen in den peripheren Lymphknoten durch unbekannte Auslöser aktiviert werden 80. Zu den Faktoren, die autoreaktive T-Zellen aktivieren könnten, zählt die molekulare Mimikry 54,55. Es wurden in MSPatienten sowohl Myelin-Basisches Protein-(MBP) spezifische CD4+- und CD8+-T-Zellen als auch MBP-spezifische Antikörper nachgewiesen, die mit EBV-Antigenen kreuzreagierten 113,114. Nach der Aktivierung erfolgt die Extravasion durch die Blut-HirnSchranke mit Hilfe von Adhäsionsmolekülen und Metalloproteasen auf T-Zellen und Endothelzellen 115. Dieser Umstand wird therapeutisch genutzt, z. B. verhindert NAT die VLA-4-vermittelte Zelladhäsion von Immunzellen an Endothelzellen der Blut-HirnSchranke 94. DZ und Mikroglia können den transmigrierten T-Zellen ZNS-Antigene präsentieren 116,117. Der Grund für die Reaktivierung der T-Zellen im ZNS ist nicht vollständig verstanden. Die Reaktivierung hat aber zur Folge, dass es zur klonalen Proliferation kommt und zur Sekretion von proinflammatorischen Zytokinen. Einerseits werden weitere Immunzellen wie Makrophagen und B-Zellen rekrutiert, andererseits kommt es zur Amplifikation der Immunantwort gegen Myelinproteine und folglich zur Zerstörung der Myelinschicht 118 . Seit Kurzem ist bekannt, dass das murine ZNS über ein Lymphsystem verfügt, das mit den zervikalen Lymphknoten verbunden ist 119. Makrophagen, die Myelinbestandteile phagozytiert haben, können in zervikale Lymphknoten migrieren, naive T-Zellen aktivieren und infolgedessen Entzündungsprozesse aufrechterhalten 120 . Erste Hinweise, dass es sich bei der MS nicht um eine ausschließlich T-Zell-vermittelte Autoimmunerkrankung handelt, lieferten Ergebnisse einer randomisierten Placebo-kontrollierten Doppelblindstudie mit einem chimären monoklonalen Anti-CD4-Antikörper (cM-T412), die zu keiner signifikanten Reduzierung der Krankheitsaktivität beim MS-Patienten führten 121. In einer kürzlich veröffentlichten Studie, die genetische krankheitsassoziierte Risikovarianten zur MS-Erkrankung ermittelte, waren Gene, die die Aktivierung und Differenzierung von TH-Zellen regulieren, am häufigsten vertreten 122. 18 Einleitung Schon früh erkannte man, dass die Zytokine IL-12 und IFN-γ, die für eine TH1-Immunantwort charakteristisch sind, eine wichtige Rolle in der EAE und MS einnahmen 123. In Versuchen aus den 1980er und 1990er Jahren zeigte sich, dass durch adoptiven Zelltransfer ZNS-spezifischer TH1-Zellen eine EAE induziert werden konnte 124. Im Gegensatz dazu zeigten Studien mit IFN-γ-defizienten Tieren, dass diese Tiere einen schwereren Krankheitsverlauf als Wildtyptiere zeigten 125. Außerdem konnte man zeigen, dass in IL12p35-/-- aber nicht in IL12p40-/--Knockout-Mäusen EAE induziert werden konnte 126. Das Zytokin IL-23, das von APZ sezerniert wird und ähnliche Effekte auf TH-Zellen hat wie IL-12, teilt sich die Untereinheit p40 mit dem Zytokin IL-12 127. Eine erst kürzlich beschriebene Untergruppe von TH-Zellen, die TH17-Zellen, die durch die Expression und Sezernierung von IL-17 charakterisiert werden, benötigen IL-23 zur Differenzierung 128,129. In einer Studie, in welcher der IL-17-Rezeptor geblockt oder IL-17 neutralisiert wurde, waren die EAE- Krankheitssymptome während der akuten und der chronischen Phase abgeschwächt 130. TH17Zellen könnten auch bei MS eine wichtige Rolle spielen. TH17-Zellen sind in erhöhten Konzentrationen im Liquor von RRMS-Patienten, besonders während eines Schubereignisses, nachgewiesen worden 131,132. Trotz dieser Beobachtungen führte eine Phase-II-Studie mit AntiIL12/23p40-Antikörpern (Ustekinumab) bei RRMS-Patienten zu keiner signifikanten Reduzierung Kontrastmittel-aufnehmender Läsionen 133 . Die enttäuschenden Ergebnisse der vorgestellten Phase-II-Studie und die Erfolge durch die B-Zell-Depletion haben B-Zellen weiter in den Fokus der aktuellen MS-Forschung gerückt. 19 Einleitung 1.3. B-Zellen 1.3.1. B-Zell-Untergruppen B-Zellen können in verschiedenen Untergruppen gleichzeitig in gesunden Individuen nachgewiesen werden. Die unreifen B-Zellen, die aus dem Knochenmark emigrieren, sind kurzlebig und noch funktionell unreif. Erst nachdem sie sekundäre lymphatische Organe kolonisiert haben, werden sie zu reifen naiven B-Zellen. Nachdem naive B-Zellen Antigenkontakt hatten, können sie zu kurzlebigen IgM-produzierenden Plasmablasten werden oder durchlaufen in der Keimzentrumsreaktion unterschiedliche Phasen. Das Ergebnis der Keimzentrumsreaktion sind hochaffine Antikörper, B-Gedächtniszellen und langlebige Plasmazellen, die man nach Infektionen und Impfungen nachweisen kann 134. Während BGedächtniszellen im Körper patrouillieren und bei einer erneuten Infektion mit einem bekannten Pathogenantigen T-Zell-unabhängig eine immediate Immunreaktion auslösen, wandern Plasmablasten zum Knochenmark und sezernieren spezifische Antikörper 135. Somit sorgen sie für einen dauerhaften humoralen Schutz während und noch einige Zeit nach Infektionen und Impfungen 135. Mit Hilfe von Oberflächenmarkern können B-ZellUntergruppen charakterisiert werden. CD19 und CD20 finden sich auf allen B-ZellDifferenzierungsstadien, auf Pro-(CD19) bzw. Prä-(CD20) B-Zellen im Knochenmark bis hin zu B-Gedächtniszellen in den sekundären lymphatischen Geweben 136. Charakteristisch für B-Gedächtniszellen sind der IgD-Verlust auf der Zelloberfläche und die Expression des Oberflächenmoleküls CD27 137. 20 Einleitung B-Zell-Untergruppe Typ Oberflächenmarker Bm1-Bm5-Klassifizierung 135,138 unreife naive B-Zellen (virgin naïve cells) IgD+CD38- Bm1 aktivierte naive B-Zellen (activated naïve cells) naive B-Zellen IgD+CD38+ Bm2 reife naive B-Zellen (pre-germinal centre cells) Follikuläre-B-Zellen (germinal centre (GC) cells) B-Gedächtniszellen IgD+CD38++ Zentroblasten Bm3 Zentrozyten Bm4 frühe B-Gedächtniszellen späte B-Gedächtniszellen IgD-CD38++ IgD-CD38+ Bm5 IgD-CD38- B-Gedächniszell-CD27-Klassifizierung 139,140 B-Gedächtniszellen doppel-negative B-Gedächtniszellen IgD-CD27- non-switched B-Gedächtniszellen IgD+CD27+ IgM-onlyB-Gedächtniszellen IgM+IgD-CD27+ switched B-Gedächtniszellen IgM-IgD-CD27+ CD24/CD38-Klassifizierung 141 naive B-Zellen Übergangs-(transitional) B-Zellen Übergangs-(transitional) B-Zellen/reife naive B-Zellen T1 IgD-CD27CD24highCD38high T2 IgD-CD27CD24highCD38high T3 IgD-CD27CD24+/lowCD38+/low Plasmablasten IgD-CD27highCD38high Plasmazellen IgD-CD27CD38highCD138high Tabelle 1 Die wichtigsten B-Zell-Untergruppen nach Oberflächenmarker charakterisiert. 21 Einleitung Man kann B-Zell-Untergruppen nach der Expression der Oberflächenmoleküle CD38 und IgD klassifizieren (Bm1-Bm5-Klassifizierung) 135,138. Eine Alternative bietet die Einteilung durch die Expression von IgD und CD27 139,140. Man kann die B-Zell-Untergruppen in verschiedene Übergangstyp-B-Zellen (transitional) (T1-3), Plasmablasten und Plasmazellen einteilen, wenn weitere Oberflächenmaker einbezogen werden 141 . Die wichtigsten B-Zell-Untergruppen und die dazugehörigen Oberflächenmarkerkombinationen sind in Tabelle 1 aufgelistet. Im Gegensatz zu den IgM-only-B-Gedächtniszellen, ist der Ig-Klassenwechsel von der Klasse M zur Klasse G T-Zell-abhängig. Der Ursprung der IgM-only-B-Gedächtniszellen ist unbekannt. Es wird vermutet, dass sie nach dem ersten Antigenkontakt im Follikel entstehen und danach diesen verlassen 134. Reife B-Zellen sind die Quelle für IgG+-B-Gedächtniszellen, die während einer follikulären Keimzentrumsreaktion nach einer zweiten Aktivierung entstehen 134. Plasmablasten sind unreife Vorstufen von Plasmazellen und können u. a. durch das Fehlen von CD20 und eine hohe CD27-Expressionsdichte charakterisiert werden 142. Plasmazellen entstehen ebenfalls durch die Keimzentrumsreaktion und exprimieren das Oberflächenmolekül CD138 143. Plasmazellen benötigen keinen Antigenkontakt, um große Mengen spezifischer Antikörper zu produzieren, während B-Gedächtniszellen BZR-abhängig Antikörper produzieren. Kürzlich wurde das Human Immunology Project gegründet, um einen Standard zur durchflusszytometrischen Phänotypisierung von peripheren mononuklearen Blutzellen (peripheral blood mononuclear cells, PBMC) vorzuschlagen 144. Danach werden die wichtigsten B-Zell-Untergruppen im Blut folgendermaßen eingeteilt: B-Zellen (CD3−CD19+), naive B-Zellen (CD3−CD19+CD27−), B-Gedächtniszellen (CD3−CD19+CD27+IgD+ oder IgD−), Plasmablasten (CD3−CD19+CD27+CD20−CD38+) und transitional-B-Zellen (CD3−CD19+CD24highCD38high) 144. Neben den konventionellen B-Zellen gibt es B1-Zellen, die ontogenetisch früher auftauchen und ihren Ursprung nicht im Knochenmark haben 145. Humane B1-Zellen sind durch die Expression folgender B1-Zell-Oberflächenmoleküle, die murine B1Zellen beschreiben, charakterisiert worden: CD20, CD27 und CD43 146,147. Neben den murinen B1-Zell-ähnlichen Merkmalen wie einer tonischen BZR-Aktivierung und der Fähigkeit allogene T-Zellen zu aktivieren, sind B1-Zellen in der Lage spontan kreuzreaktive IgMAntikörper zu produzieren 146. Man beobachtete bei humanen B1-Zell-Antikörpern eine geringere somatische Hypermutationsrate im Vergleich zu den Antikörpern, die von konventionellen B-Zellen produziert wurden 146. Murine B1-Zellen werden in der Splanchnopleura und in der fetalen Leber produziert 145 . Sie bevölkern hauptsächlich Pleura- und Peritonealhöhlen und zeigen wie B-Zellen, die sich in der Marginalzone der Milz befinden, Merkmale der unspezifischen und spezifischen Immunabwehr 145 . Die Antikörperproduktion muriner und humaner B1-Zellen erfolgt spontan und ist T-Zell-unabhängig 145,146. Der Ursprung 22 Einleitung humaner B1-Zellen ist unbekannt. Der Phänotyp ist kontrovers diskutiert worden. So ist vorgeschlagen worden, dass es sich beim B1-Phänotyp um Prä-Plasmablasten handelt 148 . B- Zell-Untergruppen spielen in den verschiedenen Phasen der MS eine jeweils spezielle, noch nicht völlig verstandene Rolle. Im Blut unbehandelter MS-Patienten waren die naiven CD27CD86+-B-Zell-Zahlen im Vergleich zu β-Interferon-behandelten MS-Patienten und gesunden Probanden signifikant erhöht 149. Während eines Schubereignisses waren die CD27-CD86+-BZell-Zahlen im Vergleich zur Remissionsphase erhöht 149. Es wurde vorgeschlagen, dass die Erhöhung auf die krankheitsbedingte Verschiebung des naiven B-Zell-/B-GedächtniszellVerhältnisses zurückzuführen sei 150 . Knippenberg et al. konnten keine Unterschiede in der Verteilung der B-Zell-Untergruppen (Bm1-Bm5-Klassifizierung) im Blut von β-Interferonbehandelten RRMS-Patienten zwischen Remissionsphase und Schubphase feststellen 150. Switched memory B-Zellen (CD19+CD27+IgD−) waren in RRMS-Patienten im Gegensatz zu gesunden Probanden in der Remissionsphase erniedrigt 150. Eine aktuelle Studie zeigt, das humane B1-Zellen bei unbehandelten MS-Patienten während der Remission signifikant erniedrigt sind 151. 1.3.2. Die Rolle der B-Zellen in MS 1.3.2.1. Autoantikörper Lange wurde die Rolle der B-Zellen in der Pathogenese der MS unterschätzt und u. a. auf die Produktion von Autoantikörpern reduziert. Elvin Kabat konnte 1942 durch Liquoranalysen als erster eine intrathekale Ig-Produktion nachweisen und 1959 beschrieb Armand Lowenthal OKB im Liquor 152. Bei 95 % aller MS-Erkrankten können OKB im Liquor nachgewiesen werden, die auf eine intrathekale Antikörperproduktion hinweisen 153. Die Anwesenheit von OKB bei MS-Patienten korrelierte in mehreren Studien mit einem schweren Verlauf der Erkrankung und war ein unabhängiger Prädiktor an MS zu erkranken 92,154,155 . Ob es sich um pathologische Antikörper handelt, ist immer noch umstritten, denn in frühen MS-Läsionen kommt es durchaus zum Untergang von Oligodendrozyten ohne Beteiligung von T-Zellen, Makrophagen oder Antikörpern 156. Eine weitere Studie zeigte, dass Antikörper von B-Zell-Klonen aus dem Liquor von MS-Patienten mit keinem der bekannten Myelinproteinen reagierten 157. Dennoch sind Autoantikörper an der Demyelinisierung beteiligt 158,159. Myelinspezifische Antikörper sind sehr umstritten als prognostische Marker. Berger et al. zeigten, dass myelinspezifische Antikörper bei CIS-Patienten ein prognostischer Marker für die Progression zur definitiven MS sein könnten 160. Das Vorhandensein von myelinspezifischen Antikörpern wurde ebenfalls im Zusammenhang mit einem verkürztem Zeitintervall zwischen ersten klinischen Symptomen 23 Einleitung und klinisch diagnostizierter MS gebracht 161. Andere Arbeitsgruppen konnten die Ergebnisse von Berger et al. und Tomassini et al. nicht reproduzieren und fanden keinen Zusammenhang zwischen dem myelinspezifischen Antikörperstatus und der Häufigkeit an MS zu erkranken 162,163. Die verschiedenen Ergebnisse sind eher auf verschiedene Studienkohorten als auf methodische Unterschiede zurückzuführen. Autoantikörper können viele Quellen haben, z. B. können sie aus dem Pool der natürlichen Antikörper stammen, die in der Maus von B1Zellen produziert werden 145. Natürliche Antiköper zeigen eine breite Spezifität mit konservierten pathogenen Strukturen und sie kreuzreagieren mit körpereigenen Antigenen 145,164. Im Gegensatz zu konventionellen Antikörpern ist die Halbwertszeit von natürlichen Antikörpern verringert 164. Natürliche Antikörper gehören größtenteils zur IgMAntikörperklasse, daneben gibt es aber auch natürliche IgA- und IgG-Antikörper 164,165. In einer kürzlich publizierten Studie konnte nachgewiesen werden, dass humane natürliche IgM-, IgGund IgA-Antikörper während der ersten sechs Lebensmonate nachgewiesen werden konnten 165. Beim Menschen vermutet man, dass die Quelle natürlicher Antikörper entweder IgM-Gedächtniszellen oder die erst kürzlich beschriebenen humanen B1-Zellen sind 134,146 . Einen weiteren Beleg, dass B-Zellen und Antikörper eine wichtige Rolle in der Pathogenese der MS einnehmen, lieferten immunhistochemische (IHC) Analysen MS-typischer Läsionen. Lucchinetti et al. haben vier MS-Läsionsformen beschrieben 159. Von den vier Läsionsmustern war die häufigste Läsionsform (Läsionsmuster Typ II) mit Antikörper- und KomplementAblagerungen assoziiert 159 . Läsionsmuster Typ I zeigte Demyelinsierung assoziiert mit T- Zellen und Makrophagen, Typ III und IV wiesen Oligodendrogliopathie auf, Typ IV zeigte zusätzlich degenerative Prozesse, die sich an den Rändern der Läsion befanden 159. Keegan et al. beobachteten in einer retrospektiven post mortem-Studie, dass nur Patienten mit histologischem Läsionsmuster vom Typ II von Plasmapheresen profitierten 103. IgM- und IgGAntikörper können die Komplementkaskade des klassischen Wegs aktivieren und führen u. a. zur Zelllyse 166. Die demyelinisierende Antikörperwirkung wird durch die IgG-Klasse und die darauffolgende Komplementaktivierung bestimmt 167. Antikörper der Klasse IgG1, -2 und -3, aber nicht der Klasse IgG4 können die klassische Komplementkaskade aktivieren 168. 1.3.2.2. Antigenpräsentation und Zytokinproduktion Die ersten B-Zell-Depletionstherapien zeigten, dass der Anteil von Autoantikörpern an der Pathogenese der MS nicht so groß war, wie vorerst angenommen. In einer Phase-II-Studie konnte gezeigt werden, dass die Ig-Titer nach Rituximab-Behandlung nicht verändert waren, obwohl eine signifikante Reduktion aktiver Läsionen und der Schubrate bei MS-Patienten 24 Einleitung beobachtet wurde 105. Es wurde vermutet, dass ein Teil des therapeutischen Effekts auf die kurzfristige Depletion einer sehr kleinen CD20+-T-Zell-Population zurückzuführen war 169 . Palanichamy et al. zeigten, dass diese seltene T-Zell-Untergruppe bei MS-Patienten erhöht war 169. B-Zellen sind nicht nur Vorläufer für Plasmazellen, sondern modulieren T-ZellFunktionen durch Antigenpräsentation und Kostimulation oder nehmen Einfluss auf ihre Umgebung durch Zytokinproduktion (bystander activation). Obwohl in MS-Läsionen alle drei Typen von APZ (Makrophagen, DZ, B-Zellen) nachgewiesen wurden, konnte man im Tiermodell der MS zeigen, dass die Antigenpräsentation durch DZ ausreichend war, um ZNSPathologien zu induzieren 116. Aktuellere Studien mit MHC-Klasse-II-Knockout-Tieren zeigten, dass die Antigenpräsentation durch MHC-Klasse-II-Komplexe auf B-Zellen hinreichend war, um EAE zu induzieren 170. Studien belegten, dass B-Zellen eine wichtige Rolle bei der Antigenpräsentation spielten, insbesondere wenn das Antigen nur in geringen Mengen verfügbar war 171 . Harp et al. beobachteten, dass die Bindung an PRR alleine nicht genügte, um humane myelinspezifische B-Zellen zu aktivieren, so dass sie ihrerseits kognate T-Zellen aktivierten 172. CD40L- und IL-4-Signale waren nötig, um myelinspezifische T-Zellen vollständig zu aktivieren 172 . Die Studie von Harp et al. zeigte, dass die Antigenpräsentation alleine nicht entscheidend war, um autoreaktive T-Zellen zu aktivieren, sondern dass die von B-Zellen produzierten Zytokine und kostimulatorische Signale ebenfalls eine große Rolle spielten. Bar-Or et al. zeigten in ex vivo-Experimenten, dass B-Zellen aus unbehandelten MSPatienten eine veränderte Zytokinproduktion aufwiesen 173. Zu diesem Zweck wurden alle BZellen mittels Quervernetzung (antigenähnliche Aktivierung) des und BZR mit anschließender Anti-humanen-IgG/IgM-Antikörpern CD40L-Inkubation (T-Zell-Hilfe) stimuliert 173. Es folgte eine dritte Stimulation (drittes Signal) entweder durch TLR9-(toll-like receptor) Agonisten (pathogenassoziierte Aktivierung) oder durch IFN-γ (TH1-Zell-vermittelte Aktivierung) 173 . Bar-Or et al. beobachteten, dass die Produktion der proinflammatorischen Zytokine LT (lymphotoxin) und TNF-α (tumor necrosis factor-α), durch B-Zellen aus RRMSPatienten nach TH1-assoziierter und pathogenassoziierter Aktvierung signifikant erhöht war im Vergleich zu Proben von gesunden Probanden 173. Die IL-10-Produktion aus B-Zellen bei RRMS-Patienten war nach pathogenassoziierter Aktvierung signifikant erniedrigt 173 . Andere Arbeitsgruppen zeigten, dass die LT-α- und TNF-α-Produktion von naiven B-Zellen oder BGedächtniszellen zwischen RRMS-Patienten und gesunden Probanden vergleichbar war, wenn die Zellen kein drittes Signal erhielten 174,175. Naive B-Zellen aus RRMS-Patienten hatten eine signifikant erhöhte IL-10-Produktion im Vergleich zu Zellen aus gesunden Probanden, wenn sie nur kostimulatorische Signale (CD40L) ohne BZR-Beteiligung erhielten 174. Interessanterweise waren die IL-10-Spiegel bei naiven B-Zellen aus RRMS-Patienten 25 Einleitung signifikant erniedrigt, wenn der BZR zusätzlich quervernetzt wurde 174 . Knippenberg et al. zeigten, dass IL-10-produzierende B-Zellen in RRMS-Patienten erniedrigt waren 150 . Duddy et. al. beobachteten, dass größtenteils CD27- B-Zellen aus gesunden Probanden IL-10 aber CD27+-B-Zellen vermehrt LT und TNF-α produzierten 175. Die B-Gedächtniszellen spielen eine besondere Rolle, denn wenn sie mit T-Zellen zusammen kultiviert wurden, dann fand man in einigen, aber nicht in allen Proben von RRMS-Patienten eine myelinspezifische CD4+-T-ZellProliferation 174. Im Gegensatz dazu führte nach der Ko-Kultivierung keine der Proben aus gesunden Probanden zu einer myelinspezfischen T-Zell-Proliferation 174. Es wird vermutet, dass B-Zellen aus MS-Patienten besonders effektiv T-Zellen stimulieren können. So findet man während eines Schubereignisses signifikant erhöhte CD80+- und CD86+-B-Zell-Zahlen im Blut von MS-Patienten 149,176. Wie wichtig die Anwesenheit der B-Zellen für die T-Zell-Pathogenität ist, zeigen B-Zell-Depletionsstudien. Nach Rituximab-Behandlung zeigten T-Zellen, die aus MS-Patienten gewonnen wurden, eine reduzierte proinflammatorische Immunantwort 173. Diese konnte wiederhergestellt werden, wenn die Zellen mit LT und TNF-α aus autologen BZellen inkubiert und kultiviert wurden 173. 1.3.2.3. Ektopische follikelähnliche Strukturen Meningeale ektopische follikelähnliche Strukturen, die in Autopsien von SPMS-Patienten in verschiedenen post mortem-Studien nachgewiesen wurden, haben in den letzten Jahren die Rolle der B-Zellen in der MS bekräftigt 177,178. Diese Strukturen enthielten neben proliferierenden B-Zellen auch Plasmazellen, T-Zellen und DZ 179. Man fand in MS-Läsionen Hinweise dafür, dass es im ZNS zur klonalen Expansion und zur somatischen Hypermutation von B-Zellen kommt 180 . Ektopische follikelähnliche Strukturen sind mit einem schwereren Verlauf der Erkrankung, einem jüngeren Alter bei Erstdiagnose, einer schnelleren Konversion zu einer progredienten Verlaufsform und einer verkürzten Lebenserwartung assoziiert 181. Diese Strukturen sind aus anderen Autoimmunerkrankungen bekannt, wie z. B. der RA 182. Sie bilden sich i. d. R. während der chronischen Phase der Erkrankung im betroffenem Organ aus und bestehen aus organisierten B- und T-Zell-Zonen, die in ihrem Aufbau sekundären lymphatischen Organen ähneln 183. Man vermutet, dass diese Strukturen eine wichtige Rolle in der chronischen Phase der MS spielen, da sie eine dauerhafte Immunantwort „vor Ort“ gewährleisten können. Bis heute ist nicht eindeutig geklärt, ob ektopische follikelähnliche Strukturen die Ursache der Progredienz oder die Folge entzündlicher Prozesse sind. 26 Einleitung 1.4. Biomarker Es ist immer noch unklar, welche Krankheitsmechanismen der MS zugrunde liegen. Es gibt viele Hinweise darauf, dass MS-Patienten wahrscheinlich in Subtypen eingeteilt werden können. Die aktuelle medizinische Forschung verfolgt einen personalisierten Ansatz, d. h. es wird untersucht, ob bestimmte Patientengruppen besser auf spezielle Medikamente ansprechen als andere. Bis heute sind keine Biomarker bekannt, die unterschiedliche MS-Patientengruppen klassifizieren. In der Vergangenheit gab es viele Bemühungen MS-spezifische Biomarker zu finden 184. Im klinischen Alltag werden drei Biomarker genutzt: OKB, Läsionen in der weißen Substanz des ZNS und der JC-Virustiter. OKB sind nicht mehr Teil der McDonald-Kriterien, um eine RRMS zu diagnostizieren, werden aber bei der PPMS-Diagnose eingesetzt 78. Trotzdem haben OKB prognostische Qualitäten, denn OKB-positive CIS-Patienten haben eine höhere Wahrscheinlichkeit an MS zu erkranken 92. Li et al. beobachteten, dass es eine positive Korrelation zwischen MRT-Läsionen und Schubrate, Alter bei Krankheitsbeginn und Behinderung gab 185. Interessanterweise verliert sich dieser Zusammenhang ab einer bestimmten Behinderungsstufe (EDSS 4,5), die nach den Autoren als Erkrankungs-„Plateau“ beschrieben wird 185. Weitere klinische MS-Marker sind JC-Virustiter, die bei NATbehandelten Patienten routinemäßig kontrolliert werden 75. Das Risiko, an einer PML zu erkranken, steigt mit der Behandlungsdauer (> 24 Monate) 75. Seropositiven Patienten wird eine alternative Eskalationstherapie empfohlen 75. Einer der bekanntesten Biomarker, die ZNS- und Neurodegeneration belegen, ist der Nachweis von Neurofilament (Nfl), das löslich im Liquor oder im Serum nachgewiesen werden kann. Nfl wird nach axonaler Schädigung freigesetzt 186. Im Serum von RRMS-Patienten sind die Nfl-Konzentrationen im Vergleich zu Proben aus gesunden Probanden erhöht und korrelieren mit dem Nfl-Spiegel im Liquor 186 . In einer Folgestudie zeigte sich, dass MS-Patienten während eines Schubs einen erhöhten Nfl-Spiegel im Serum im Vergleich zu Patienten in der Remissionsphase hatten 186. Außerdem korrelieren die Nfl-Spiegel mit der Läsionslast in der weißen Substanz 186. Folgende Proteine, die als Biomarker die neuronale und gliale Schädigung widerspiegeln und evtl. Marker für die Progression der Erkrankung sein könnten, sind z. Zt. Gegenstand aktueller Forschung: GFAP (glial fibrillary acidic protein), MBP, S100B (kalziumbindendes Protein, Astrozyten Proliferationsmarker), Tau-Protein (Zytoskelett-Protein), NCAM (neural cell adhesion molecule), NGF (nerve growth factor), CNTF (ciliary neurotrophic factor) und Ferritin im Liquor 184. Allerdings wird die Erhöhung dieser Faktoren im Liquor von MS-Patienten eher als Epiphänomen nach einem Schubereignis betrachtet und nicht als prognostischer Marker 184. Andere Marker könnten lösliche Oberflächenmoleküle sein, die nach Aktivierung von 27 Einleitung Makrophagen abgetrennt werden (sCD163) und im Plasma und Liquor von MS-Patienten erhöht sind 184. Weitere potentielle MS-Biomarker sind: YKL-40 (Chitinase-3-like 1) und CXCL13 (chemokine (c-x-c motif) ligand 13) 184. Antikörper gegen Myelin- oder Virusbestandteile im Plasma oder im Liquor von MS-Patienten waren immer wieder Gegenstand von Kontroversen. Myelinspezifische Antikörper sind bei MS-Patienten und gesunden Individuen nachgewiesen worden, konnten aber als Biomarker nicht bestätigt werden 160–162,187. Eine aktuelle Studie zeigte, dass Anti-Kir4.1-(einwärts rektifizierender Kaliumkanal 4.1) Antikörper bei einem Großteil der MS-Patienten, aber nicht bei gesunden Probanden, nachgewiesen werden konnten 188. Allerdings könnten Anti-Kir4.1-Antikörper bei MS-Patienten ein Epiphänomen sein, da Kir4.1 auch von Parietalzellen im Darm exprimiert wird 189 . Darmepithel-Läsionen, die durch Kortison-Behandlung verursacht werden, könnten dazu führen, dass es zur Produktion von Anti-Kir4.1-Antikörpern kommt 189. In zwei Folgestudien konnten die Ergebnisse von Srivastava et al. nicht reproduziert werden 190,191 . Anti-Kir4.1-Antikörper sind deshalb als Biomarker nicht geeignet. Eine Studie, die in unserer Arbeitsgruppe durchgeführt wurde, zeigte, dass ZNS-reaktive B-Zellen im Blut von CISPatienten (60 %) und RRMS-Patienten (79 %), aber nicht im Blut von gesunden Probanden nachgewiesen werden konnten 192 . In einer weiteren Studie aus unserer Arbeitsgruppe zeigte sich, dass die Anwesenheit von ZNS-reaktiven B-Zellen während eines Schubs mit einer signifikant kürzeren schubfreien Phase assoziiert war 193 . In meiner Studie, die ich in dieser Doktorarbeit vorstellen werde, konnte ich zeigen, dass die Anwesenheit bzw. Abwesenheit von ZNS-reaktiven B-Zellen im Blut von RRMS-Patienten mit dem Therapieerfolg bzw. Therapieversagen assoziiert war 194 . Es bleibt weiterhin ein wichtiges Ziel, verlässliche MS- Biomarker zu finden, die helfen, eine personalisierte Therapie zu empfehlen. 28 Ziele der Arbeit 2. Ziele der Arbeit Die vorliegende Arbeit untersucht folgende Fragen: I. Haben die RRMS-Basistherapeutika GA und β-Interferon Einfluss auf die signifikant erniedrigte B1-Zell-Zahl im Blut? Hat das RRMS- Eskalationstherapeutikum NAT Einfluss auf die signifikant erniedrigte B1-ZellZahl? Haben die gängigen MS-Therapien Einfluss auf die B-Zell-Untergruppen im Blut von MS-Patienten? II. Ist die Anwesenheit von ZNS-reaktiven B-Zellen ein Maß für den Therapieerfolg bzw. das Therapieversagen mit den RRMS-Basistherapeutika GA und β-Interferon? Gibt es Unterschiede bei GA-behandelten RRMS-Patienten, die ZNS-reaktive BZellen im Blut haben, wenn man die Patienten mit einen milden Krankheitsverlauf (EDSS < 3) mit Patienten, die einen schwereren Krankheitsverlauf aufweisen (EDSS ≥ 3), vergleicht? III. Ist die Expression von CEACAM1 auf B-Zell-Untergruppen bei RRMS-Patienten, die mit NAT behandelt wurden, verändert? Ist die Koexpression der NCR TIM-3 und CEACAM1 auf B- und T-Zellen in RRMS-Patienten, die mit NAT-behandelt wurden, verändert? Die Beantwortung dieser Fragen soll folgende Arbeitshypothesen überprüfen: Man kann durch die Anwesenheit von ZNS-reaktiven B-Zellen, einer erhöhten CEACAM1+-B-Zell-Zahl und einer erniedrigten B1-Zell-Zahl im Blut eine Untergruppe von MS-Patienten definieren, die besonders von einer MS-Therapie, die gezielt B-Zell-Funktionen moduliert (z. B. GA) oder B-Zellen depletiert (z. B. Rituximab), profitiert. 29 Manuskripte 3. Manuskripte 3.1. B1 cells are unaffected by immune modulatory treatment in remittingrelapsing multiple sclerosis patients Das Manuskript “B1 cells are unaffected by immune modulatory treatment in remittingrelapsing multiple sclerosis patients” wurde als short communication im Journal Journal of Neuroimmunology im Juli 2014 veröffentlicht. Die Planung der Untersuchungen und die Entwicklung des Studiendesigns sind hauptsächlich durch mich erfolgt. Außerdem war ich verantwortlich für die Sammlung, Erfassung, Analyse und Interpretation der Daten. Ein Teil der Daten der GA-behandelten und unbehandelten MS-Patienten, wurde durch die Doktorandin Stefanie Heller gesammelt und erfasst. Ich habe den ersten Entwurf des Manuskripts angefertigt. Alle Teile des Manuskripts (Einleitung, Material und Methoden, Ergebnisse und Diskussion) sind von mir verfasst worden. Alle Abbildungen und Tabellen sind ebenfalls von mir entworfen und angefertigt worden. Stefanie Kürten hat bei der Bearbeitung und bei der Formatierung des Manuskripts mitgewirkt. Reprinted from Journal of Neuroimmunology, 272, 1-2 (2014), Damiano Rovituso, Stefanie Heller, Michael Schroeter, Christoph Kleinschnitz, Stefanie Kuerten, B1 cells are unaffected by immune modulatory treatment in remitting–relapsing multiple sclerosis patients, pp. 86-90, Copyright 2014, with permission of Elsevier. 30 Manuskripte 31 Manuskripte 32 Manuskripte 33 Manuskripte 34 Manuskripte 35 Manuskripte 3.2. The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsing-remitting multiple sclerosis patients Das folgende Manuskript “The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsing-remitting multiple sclerosis patients” wurde im September 2015 im Open-Access-Journal Scientific Reports veröffentlicht. Die Planung der Untersuchungen wurden von Stefanie Kürten durchgeführt. Ich war hauptverantwortlich für die Sammlung, Erfassung, Analyse und Interpretation der Daten, bis auf die Daten der β-Interferonbehandelten MS-Patienten. Diese wurden hauptsächlich von der Doktorandin Cathrina E. Duffy erhoben und analysiert. Ich habe den ersten Entwurf des Manuskripts angefertigt. Alle Teile des Manuskripts (Einleitung, Material und Methoden, Ergebnisse und Diskussion) sind von mir verfasst worden. Alle Abbildungen und Tabellen sind ebenfalls von mir entworfen und angefertigt worden. Stefanie Kürten hat bei der Bearbeitung und bei der Formatierung des Manuskripts mitgewirkt. 36 Manuskripte 37 Manuskripte 38 Manuskripte 39 Manuskripte 40 Manuskripte 41 Manuskripte 42 Manuskripte 43 Manuskripte 44 Manuskripte 3.3. CEACAM1 mediates B cell aggregation and tolerance induction in central nervous system autoimmunity Das folgende Manuskript “CEACAM1 mediates B cell aggregation and tolerance induction in central nervous system autoimmunity” ist am 27. April 2016 an das Journal Scientific Reports zur Veröffentlichung eingereicht worden. Der Peer-Review-Prozess wurde am 24. April 2016 beendet. Die folgenden Kommentare werden von mir in den folgenden vier Wochen bearbeitet: Reviewer #1 (Remarks to the Author): This study investigates the role of CEACAM1 on the formation of B cell aggregates in the CNS, which are observed in patients with multiple sclerosis. The authors utilized a B cell-dependent EAE model of MS to demonstrate the presence of CEACAM1+ B cells in CNS B cell aggregates and subsequently showed that administration of a CEACAM1 antibody significantly reduced EAE clinical score and the overall percent of B cell aggregates without affecting antibody responses or lymph node architecture, implicating CEACAM1 in the formation of these aggregates. These results were mirrored in human MS patients, which had increased frequencies of CEACAM1+ naïve, memory, and B1 B cells as well as CEACAM1+ B cells within brain infiltrates. Antibody targeting of CEACAM1 was also successful at preventing in vitro B cell aggregation in both healthy control and RRMS samples. This paper also identified a novel population of TIM3+ CEACAM+ B cells, which were shown to be elevated in MS patients. 1. The observation of a previously unidentified TIM3+ B cell population could have important implications for understanding the role of B cells in autoimmunity and how it impacts their function. As this is the first study identifying TIM3+ B cells, more extensive validation beyond flow cytometry and PCR would be useful for supporting this discovery. This could include incorporation of TIM3 into immunohistochemistry, singlecell imaging flow cytometry, or RNA sequencing. 2. While this paper sufficiently demonstrated that CEACAM1 is involved in the formation of B cell aggregates in the CNS, the link between CEACAM1 and tolerance was not adequately demonstrated. The reduction of the clinical score in EAE after treatment with a CEACAM1 antibody suggests that the prevention of new aggregates could be responsible for the disease alleviation rather than a tolerance mechanism mediated by the immunomodulatory effects of CEACAM1 and/or TIM3. The reduced clinical score signifies CEACAM1 is contributing to the disease state but there is not enough evidence for or discussion of how this is mediated through improved tolerance. Thus, removal of the word tolerance from the title would seem appropriate. 45 Manuskripte 3. The main issue is that MS patients categorized as in remission were on anti-VLA-4 therapy, while active patients were not. The difficulty in obtaining untreated MS patients is understood, and these data are interesting. However, the graphs should be labelled with the therapy that the patient is on and the potential effects of depleting VLA-4 positive cells in the circulation should be discussed. Die Umsetzung der Kommentare und Hinweise werden im Kapitel Ausblick (5.4.) besprochen. Das Studiendesign und die Methoden sind von Stefanie Kürten entwickelt worden. Die Bachelorstudentin Laura Scheffler und ich waren zu gleichen Teilen an der Sammlung, Analyse und Interpretation der Daten beteiligt. Ich habe selbständig die Daten, die im Abschnitt „The percentage of CEACAM1+ B cells was increased in patients with RRMS“ vorgestellt werden, gesammelt, analysiert und interpretiert. Ich habe diesen Abschnitt selbständig verfasst. Außerdem habe ich den Teil der „Diskussion“ verfasst, der die Ergebnisse, die im zuvor genannten Abschnitt diskutiert. Die dazugehörigen Abbildungen (Figure 3 und Supplementary Figures 3, 4, 5 und 6) und Tabellen (Supplementary Tables 1, 2, 3 und 4) wurden ebenfalls von mir selbständig angefertigt. Die Doktorandin Sandra Lauer-Schmaltz war bei der Sammlung und der Analyse der Daten, die in den Tabellen 1, 2 und 3 präsentiert wurden, beteiligt. Sie und ich haben zu gleichen Teile die Experimente, die in den Abbildungen Figure 3 und Supplementary Figures 4 und 5 dargestellt sind, durchgeführt. 46 Manuskripte 47 Manuskripte 48 Manuskripte 49 Manuskripte 50 Manuskripte 51 Manuskripte 52 Manuskripte 53 Manuskripte 54 Manuskripte 55 Manuskripte 56 Manuskripte 57 Manuskripte 58 Manuskripte 59 Manuskripte 60 Manuskripte 61 Manuskripte 62 Manuskripte 63 Manuskripte 64 Manuskripte 65 Manuskripte 66 Manuskripte 67 Manuskripte 68 Manuskripte 69 Manuskripte 70 Manuskripte 71 Manuskripte 72 Manuskripte 73 Manuskripte 74 Manuskripte 75 Manuskripte 76 Manuskripte 77 Manuskripte 78 Manuskripte 79 Manuskripte 80 Manuskripte 81 Manuskripte 82 Manuskripte 83 Manuskripte 84 Manuskripte 85 Manuskripte 86 Manuskripte 87 Manuskripte 88 Manuskripte 89 Manuskripte 90 Manuskripte 91 Manuskripte 92 Manuskripte 93 Manuskripte 94 Manuskripte 95 Manuskripte 96 Manuskripte 97 Manuskripte 98 Diskussion 4. Diskussion Die Ätiologie der MS ist unbekannt, aber besonders Disanto et al. haben auf eine Kausalitätsbeziehung zwischen B-Zellen und MS anhand der von Bradford Hill vorgeschlagenen Kriterien hingewiesen 195 . Bradford Hill entwickelte neun Kriterien, welche die Ursache-Wirkungs-Beziehung in der Medizin und Epidemiologie überprüfen sollten 196. Für diesen Zweck untersuchte er das Kriterium der Stärke, der Folgerichtigkeit, der experimentellen Überprüfung, der Zeitlichkeit, der Plausibilität, der Stimmigkeit, der Analogie, der Spezifität und das Kriterium des biologischen Gradienten. Disanto und seine Kollegen sehen bis auf das Kriterium der Spezifität alle Kriterien erfüllt 195. Das Kriterium der Stärke wird für Disanto et al. dadurch erfüllt, dass in 95 % aller MS-Patienten OKB nachgewiesen wurden 197. Die BZell-Depletionsstudien mit Rituximab und Ocrelizumab sind zu vergleichbaren Ergebnissen gekommen und zeigten ähnliche Effekte bei klinischen Parametern, somit erfüllen sie die Kriterien der Folgerichtigkeit und der experimentellen Überprüfung 105,109. B-Zell-Anomalien sind in allen Phasen der Erkrankung nachgewiesen worden (Kriterium der Zeitlichkeit), z. B. findet sich schon zu Beginn der Erkrankung bei etwa 70 % der MS-Patienten eine IgGVermehrung im Liquor 66. Das Kriterium des biologischen Gradienten ist erfüllt, da u. a. OKBpositive CIS-Patienten eine höhere Wahrscheinlichkeit haben, an MS zu erkranken 92. Das Kriterium der Plausibilität ist dadurch erfüllt, dass B-Zellen in anderen Autoimmunerkrankungen eine Rolle spielen (z. B. in der RA) und MS-Risikofaktoren einen Einfluss auf die Proliferation und Aktivierung von B-Zellen haben können. So führt Hypovitaminosis D in ex vivo-Experimenten zu einer verstärkten B-Zell-Proliferation und ist mit einem schwererem Krankheitsverlauf assoziiert 198. Disanto et al. stellen fest, dass B-Zellen als mögliche Verursacher nicht im Widerspruch mit dem Verlauf und der Biologie der Erkrankung stehen (Kriterium der Stimmigkeit). Das Kriterium der Analogie wird dadurch erfüllt, dass andere Zellarten, wie T-Zellen, ähnlich zur Pathogenese beitragen können. Da BZellen auch bei anderen Erkrankungen eine Rolle spielen, wie B-Zell-Depletionsstudien z. B. bei RA- oder SLE-Patienten belegen, ist das Kriterium der Spezifität nicht erfüllt 199. In der hier vorgelegten Arbeit wurden die Kriterien der Stärke, der Folgerichtigkeit, der experimentellen Überprüfbarkeit und der Stimmigkeit analysiert. Die Assoziationen zwischen Wirksamkeit der Therapie und der Anwesenheit von Autoantikörpern bei RRMS-Patienten ist ein Beleg für die Kausalität zwischen B-Zellen und Erkrankung (Kriterium der Stärke). Die hier vorgestellten Daten zeigen eine signifikante Erniedrigung der B1-Zellen bei MS-Patienten und stimmen mit zuvor publizierten Daten überein. Somit sind die Kriterien der Folgerichtigkeit und der experimentellen Überprüfbarkeit erfüllt. Meine Studien zeigten, dass die Unterschiede in den 99 Diskussion B-Zell-Untergruppen, die Anwesenheit von ZNS-reaktiven B-Zellen und die veränderte Oberflächenmarker-Expression zumindest bei einem Teil der untersuchten Patienten mit den klinischen Daten korrelieren (Kriterium eines biologischen Gradienten). Die drei Studien werden in den folgenden Abschnitten im Einzelnen und zusammenhängend diskutiert. Im nächsten Kapitel folgt ein Ausblick auf u. a. aktuelle Projekte, die sich den hier vorgestellten Studien anschließen. 4.1. B1-Zellen sind in Patienten mit schubförmiger MS unabhängig von der MS-Therapie signifikant erniedrigt Die Studie mit dem Titel „B1 cells are unaffected by immune modulatory treatment in remittingrelapsing multiple sclerosis patients“ überprüfte, ob MS-Basistherapeutika und MSEskalationstherapien einen Einfluss auf die relative Verteilung der B-Zell-Untergruppen hatten. Die Untersuchung der B-Zell-Untergruppen mittels polychromatischer Durchflusszytometrie zeigte, das die B1-Zell-Zahlen signifikant erniedrigt waren, unabhängig davon, ob es behandelte oder unbehandelte RRMS-Patienten waren. Zuvor hatten Tørring et al. gezeigt, dass unbehandelte RRMS-Patienten erniedrigte B1-Zell-Zahlen hatten 151. Weil Covens et al. beobachten konnten, dass der B1-Phänotyp aus CD43--B-Zellen induziert werden konnte, wurden die Zellen in meiner Studie mit IL-2 und dem TLR7/8-Agonisten R-848 (auch Resiquimod oder 4-Amino-2-(ethoxymethyl)-a,a-dimethyl-1H-imidazol[4,5-c]quinoline-1ethanol) inkubiert 148. Die Inkubation mit IL-2 und R-848 führt dazu, dass sich ein Teil der BGedächtniszellen zu Plasmablasten ausdifferenziert 200 . Die Analyse der Größe und Struktur des Zellkerns und der Menge der Vesikel wird über das Seitwärtsstreulicht (side scatter, SSC) bestimmt (Granularität). Ich konnte eine erhöhte Granularität der B1-Zellen feststellen. Die Beobachtung stimmte mit den Beschreibungen aus den zuvor publizierten Studien überein 148,151,201. Die Plasmablasten-(CD3-CD20-CD27high vs. SSC) Zahlen aus unbehandelten RRMS-Patienten waren im Vergleich zu Proben aus gesunden Probanden, GA-, β-Interferonund NAT-behandelten RRMS-Patienten signifikant erhöht. Da trotzt unveränderten Plasmablasten-Zahlen bei behandelten Patienten eine signifikante Erniedrigung der B1-Zellen zu beobachtet war, konnte ich ausschließen, dass es sich bei den B1-Zellen um Vorstufen von Plasmablasten handelte. Es zeigte sich, dass die anderen B-Zell-Untergruppen (naive B-Zellen, B-Gedächtniszellen) durch die verschiedenen Behandlungen verändert wurden. Die Analyse der naiven B-Zellen und Gedächtniszellen in meiner Studie ergab, dass GA- und β-Interferone gegensätzliche Wirkungen auf diese Untergruppen zeigten. Im Vergleich zu Proben aus gesunden Probanden und unbehandelten RRMS-Patienten waren durch GA-Behandlung die 100 Diskussion CD19+-B-Zell-Zahlen erniedrigt und die CD27-IgD+-(non-switched) Zahlen erhöht 202 . Die naiven B-Zellen (CD20+CD27-CD43-CD3-) waren nach GA-Behandlung im Vergleich zu Proben aus unbehandelten und β-Interferon-behandelten RRMS-Patienten erniedrigt. Es wird vermutet, dass GA T-Zell-abhängig naive B-Zellen aktiviert und dadurch zur beobachteten Erniedrigung führt 203. Mehrere Studien belegten, dass durch GA-Behandlung GA-spezifische Antikörper gebildet wurden 204,205 . Der relative Anteil naiver B-Zellen könnte durch die GA- Behandlung verringert werden, weil sich GA-spezifische B-Gedächtniszellen ausbilden. Haas et al. zeigten, dass die naiven B-Zell-Zahlen nach β-Interferon-Behandlung, nicht aber nach GA-Behandlung erhöht waren 206. Es konnte gezeigt werden, dass β-Interferon-Behandlung die Aktivierungskapazität von B-Zellen erniedrigt, was die signifikante Erhöhung der naiven BZell-Zahlen im Vergleich zu GA-behandelten Proben im meiner Studien erklären könnte 207 . Ramgolam et al. beobachteten, dass die erniedrigte Aktivierungsschwelle über die Aktivierungsmarker CD40 und CD80 vermittelt wurde, während die Antigenpräsentation über die MHC-Klasse-I- und II-Komplexe davon nicht betroffen war. In einer aktuellen Studie führte die β-Interferon-Behandlung zur Erhöhung von B-Zellen, die einen Übergangsphänotyp (CD19+CD24highCD38high) aufwiesen 208. Es kann nicht ausgeschlossen werden, dass es sich bei meinen naiven B-Zellen ebenfalls um B-Zellen des Übergangsphänotyps handelt. In diesem Falle würden meine Beobachtungen mit denen von Schubert et al. übereinstimmen 208. Skarica et al. zeigten, dass die B-Zell-Zahl (CD20+-B-Zellen) und die IgD+-Zell-Zahl (unreife B-Zellen oder naive B-Zellen) nach NAT-Behandlung signifikant erhöht waren 209. Die BGedächtniszell-Zahlen in meiner Studie waren nach NAT-Behandlung signifikant erhöht, so wie in der Studie von Hass et al. für un- und switched-B-Gedächtniszellen beschrieben 206. Da die NAT-Behandlung die Transmigration u. a. der B-Zellen behindert, könnte dies die erhöhten B-Gedächtniszell-Zahlen im Blut der NAT-behandelten RRMS-Patienten in meiner Studie erklären 94,210. Eine weitere Erklärung für die erhöhten Werte könnte sein, dass NAT-behandelte B-Zellen eine reduzierte Verweildauer in der Milz haben 211,212 . In meiner Studie wurden die B-Zell-Untergruppen durch die unterschiedlichen Behandlungen wie in der Literatur beschrieben beeinflusst, mit Ausnahme der B1-Zell-Untergruppe. Es wird vermutet, dass B1Zellen aktiv an entzündlichen Prozessen im ZNS beteiligt sind 151. Es wurde die Hypothese aufgestellt, dass B1-Zellen aktiv in das ZNS migrieren und deshalb die B1-Zell-Zahlen im Blut verringert sind 151 . Es kann nicht ausgeschlossen werden, dass die B1-Zell-Zahlen schon vor Krankheitsbeginn erniedrigt sind. B1-Zellen können im Gegensatz zu naiven B-Zellen und Gedächtniszellen spontan das antiinflammatorische Zytokin IL-10 produzieren. Dadurch könnten B1-Zellen eine wichtige Rolle in der Immunregulation spielen 213. Es ist bekannt, dass stimulierte B-Zellen (Quervernetzung des BZR, TLR9-Agonist) aus MS-Patienten weniger 101 Diskussion IL-10 produzieren, dafür aber vermehrt das proinflammatorische Zytokin IL-6 202. In mehreren Studien konnte nachgewiesen werden, dass B-Zellen aus MS-Patienten signifikant erniedrigte IL10-Spiegel haben 150,175 . Eine ex vivo-Studie zeigte, dass die IL-10-Produktion nach Quervernetzung des BZR und nach Kostimulation durch CD40L naiver (CD27-) B-Zellen signifikant erniedrigt war 174 . Man könnte spekulieren, dass die niedrigen B1-Zell-Zahlen im Blut eine mögliche Ursache für den verringerten IL-10-Spiegel in MS-Patienten sein könnten. In der Gruppe der GA-behandelten Patienten konnte man eine inverse Korrelation zwischen B1-Zell-Zahl, der Zeitspanne bis zum letzten Schubereignis und dem Alter der Patienten feststellen. Diese Beobachtungen implizieren, dass humane B1-Zellen eine wichtige Rolle in der Pathogenese der MS spielen. Meine Studie zeigt eindrücklich, dass GA und β-Interferon unterschiedliche Wirkungen auf die relative Verteilung der B-Zell-Untergruppen haben. 4.2. Die Anwesenheit von gehirnantigenspezifischen B-Zellen bei RRMSPatienten korreliert mit dem Behandlungserfolg durch GA In unserer Arbeitsgruppe konnten wir zeigen, dass B-Zellen gegen ZNS-Antigene bei einem Großteil der CIS- und MS-Patienten nachweisbar waren 192 . Im Blut von gesunden Probaden konnten keine ZNS-reaktiven B-Zellen detektiert werden 192. Beide Basistherapeutika, GA und β-Interferon, sind vergleichbar in der Reduktion MS-typischer klinischer Parameter wie MRTLäsionslast und Schubrate 90,91. Gegenwärtig gib es keine prognostischen Marker, um GA- bzw. β-Interferon-Therapie-Versager zu identifizieren. Als bester Prädiktor, um Patienten zu identifizieren, die von einer β-Interferon-Therapie nicht profitieren, wurde der Nachweis von neuen hyperintensen-(„hellen“) Läsionen in der T2-Wichtung, die innerhalb eines Jahres auftreten, vorgeschlagen 214. β-Interferon-behandelte Patienten, die eine oder mehrere neue Läsionen hatten, erlebten in den darauffolgenden fünf Jahren eine signifikante Progredienz der Erkrankung 214. Es hat große Bemühungen gegeben, genetische Biomarker, die den Behandlungserfolg durch β-Interferon vorhersagen können, zu finden 83. Bis heute konnte kein Gen identifiziert werden, das als Biomarker für den Behandlungserfolg durch β-Interferon in Frage kommt. Einen ersten Ansatz, die Wirksamkeit der GA-Behandlung mittels ELISPOT (enzyme-linked immunospot assay) zu dokumentieren, wurde bereits in einer Studie aus dem Jahre 2007 untersucht 215 . Valenzuela et al. beobachteten, dass die Produktion TH2-typischer Zytokine durch B-Zellen im Verhältnis zu den TH1-typischen Zytokinen nach GA-Behandlung erhöht war 215 . Wie bereits erwähnt, führt die GA-Behandlung zur Produktion von GA- spezifischen-Antikörpern 204,205,216. Nach Langzeit-GA-Behandlung (> 2 Jahre) waren die GAspezifischen IgG4-Antikörper-Spiegel erhöht 216 . Außerdem korrelierten IgG4-Antikörper102 Diskussion Spiegel und IgG4/IgG1- bzw. IgG4/IgG2-Verhältnisse negativ mit der Anzahl der Schübe 216. IgG4-Antikörper werden nach wiederholten Immunisierungen mit Antigenen, besonders Antigene die eine IgE-Antwort auslösen, produziert 217. Außerdem aktivieren IgG4-Antikörper nicht die antikörperinduzierte Komplementkaskade 218. Man vermutet, dass die B-ZellFunktionen durch GA so verändert werden, dass GA-spezifische B-Zellen TH2-Zellen aktivieren und diese die IgG4-Antikörper-Produktion induzieren. Im Gegensatz zur β-Interferon-Behandlung werden durch GA-Behandlung keine neutralisierenden Antikörper gebildet 219. Höhere GA-spezifische Antikörper-Spiegel im Blut von Patienten sind mit einer niedrigen Schubfrequenz assoziiert worden 219. Karussis et al. berichten, dass die zu Beginn der Studie niedrigen MBP-spezifischen Antikörper-Titer durch die GA-Behandlung nicht verändert wurden 216. GA bindet kompetitiv an MBP-spezifische-MHC-Klasse-II-Komplexe auf THZellen 220. Die Daten von Karussis et al. könnten als Beleg betrachtet werden, dass die GABehandlung nicht zu erhöhten MBP-spezifischen Antikörper-Spiegeln durch Kreuzreaktivität führt. Im Gegensatz dazu ist der Effekt der β-Interferon-Behandlung nicht auf die Modulierung der Antikörperproduktion zurückzuführen, sondern eher auf die Reduzierung der Expression kostimulatorischer Oberflächenmoleküle (CD40 und CD80) und die daraus resultierende verringerte T-Zell-Aktivierung 176,207. Außerdem zeigte sich, dass die β-Interferon-Behandlung die B-Zell-Sekretion von Zytokinen inhibierte, die nötig sind, um TH17-Zellen zu bilden 207. Die Wirkungsweisen auf B-Zellen durch GA bzw. β-Interferone sind also sehr unterschiedlich. Die Ergebnisse der ZNS-Lysat-ELISPOT-Studie, die bei einem Großteil der MS- und CISPatienten ZNS-reaktive B-Zellen nachgewiesen hatte, belegte ebenfalls, dass ein Teil der MSPatienten keine ZNS-reaktiven B-Zellen im Blut hatte 192. Man könnte somit annehmen, dass einige MS-Patienten von B-Zell-Therapeutika mehr profitieren als andere. Es fehlen Biomarker, die den Therapieerfolg prognostizieren können. Myelinspezifische Antikörper sind als Biomarker nicht eindeutig und führten in der Vergangenheit zu kontroversen Beobachtungen. Einerseits war die Anwesenheit von myelinspezifischen Antikörpern im Blut von CIS-Patienten mit einer erhöhten Wahrscheinlichkeit assoziiert an MS zu erkranken 160,161. Andererseits konnten andere Arbeitsgruppen diese Ergebnisse nicht reproduzieren 162,187 . Das ZNS-Lysat, das in meiner Studie mit dem Titel „The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsing-remitting multiple sclerosis patients“ genutzt wurde, besteht aus einer Mischung aller im ZNS vorkommenden Antigene anstatt eines ausgewählten Myelinproteins. Ein weiterer Autoantikörper wurde vor kurzem als Biomarker vorgestellt: Anti-Kir4.1-Antikörper. Anti- Kir4.1-Antikörper sind im Blut von 47 % der MS-Patienten, in 0,9 % der Patienten mit anderen neurologischen Erkrankungen aber in keinem der Proben aus gesunden Probanden, nachgewiesen worden 188. Die Ergebnisse konnten 103 Diskussion aber von anderen Arbeitsgruppen nicht reproduziert werden 190,191. Man fand Anti-Kir4.1Antikörper in der gleichen Größenordnung bei MS-Erkrankten und gesunden Probanden oder sie waren nicht nachweisbar 190. Unsere Arbeitsgruppe hatte darauf hingewiesen, dass Kir4.1 auch von Parietalzellen exprimiert wird und die Anwesenheit von Anti-Kir4.1-Antikörpern eine Folge von epitope spreading sein könnte 189. Im Gegensatz zu anderen Tests (z. B. ELISA) wurden in meiner Studie gezielt B-Zell-Klone detektiert, die gegen eine Vielzahl von unbekannten ZNS-Antigenen gerichtet waren. Ein weiterer Vorteil gegenüber anderen Methoden ist, dass man für die Analyse nur eine Blutprobe benötigt und keinen Liquor entnehmen muss. Außerdem zeigte der ZNS-Lysat-ELISPOT eine hohe Spezifität, denn 79 % aller RRMS-Patienten aber keiner der getesteten gesunden Probanden oder der Patienten mit anderen neurologischen oder autoimmunen Erkrankungen hatte ZNS-spezifischen B-Zellen im Blut 192. In der hier zugrundeliegenden Studie wurde die Anwesenheit von ZNS-Antigenspezifischen B-Zellen als kategorische, nominale Variable betrachtet, d. h. wir haben die Patienten eingeteilt in ELISPOT responder (positiver Nachweis von ZNS-Antigen-spezifischen B-Zellen im ELISPOT) und ELISPOT non-responder (kein Nachweis). Die kategorische (metrische) Variable, die Anzahl der ELISPOT-Spots, wurde nicht beachtet. In der Gruppe der RRMS-Patienten, die mit GA-behandelt und ELISPOT responder waren, wurde eine positive Korrelation zwischen der Zeitspanne seit dem letzten Schubereignis (time since last relapse, TSLR) und der Behandlungsdauer beobachtet. Dagegen fand sich kein Zusammenhang zwischen TSLR und Behandlungsdauer in der GA-behandelten ELISPOT non-responder Gruppe. Ein anderes Bild zeigte sich bei der Untersuchung der TSLR und der Behandlungsdauer in der Gruppe der β-Interferon-behandelten RRMS-Patienten. Die RRMSPatienten, die ZNS-reaktive B-Zell-Antworten im ELISPOT aufwiesen, zeigten keine Korrelation zwischen TSLR und Behandlungsdauer, während die RRMS-Patienten ohne ZNSreaktive B-Zellen eine positive Korrelation aufwiesen. Man kann schlussfolgern, dass RRMSPatienten mit ZNS-reaktiven B-Zellen von einer GA-Behandlung profitieren. Unabhängig von der Anwesenheit ZNS-spezifischer B-Zellen im Blut von RRMS-Patienten, wurden die Behandlungsdauer und die TSLR auf ihren statistischen Zusammenhang überprüft. In beiden Gruppen (GA- und β-Interferon-behandelte RRMS-Patienten) wurde eine positive Korrelation zwischen beiden Parametern beschrieben. In der Gruppe der ELISPOT-responder wurden die RRMS-Patienten mit einem milden (EDSS < 3) und einem schweren (EDSS ≥ 3) Krankheitsverlauf auf ihre Beziehung zwischen Behandlungsdauer und TSLR überprüft. Es zeigte sich, dass es eine positive Korrelation zwischen der Behandlungsdauer und der TSLR in den Patienten gab, die einen milden Krankheitsverlauf hatten, nicht aber in der Gruppe mit schwerem Verlauf. Diese Beobachtung entspricht den Daten aus der Literatur, so z. B. ist die 104 Diskussion Schubrate in den ersten zwei Jahren ein prognostischer Marker für die Schwere der Erkrankung, aber dieser statistische Zusammenhang geht im Verlauf verloren 221. Außerdem wurde ein Zusammenhang zwischen einem fulminanten Verlauf zu Beginn der Erkrankung und einer verstärkten Progredienz festgestellt 222. Leray et al. beschreibt zwei Phasen in der MS, die unabhängig voneinander sind 223. Die dynamische erste Phase ist im interindividuellen Verlauf heterogen, die zweite Phase verläuft dagegen interindividuell uniform. Schübe während der frühen Phasen sind gemäß Leray et al. unabhängige Prädiktoren für eine spätere Progredienz 223. Meine Studie zeigt, dass der GA- bzw. β-Interferon-Behandlungserfolg mit einem einfachen ZNS-Lysat-ELISPOT nachgewiesen werden kann. Trotz unterschiedlicher Wirkungen auf B-Zellen, hat die GA- und β-Interferon-Behandlung vergleichbar modulierende Wirkungen auf Immunzellen. So wirken GA- und β-Interferon-Behandlung u. a. auch auf die Normalisierung der Expression von negativen Ko-Rezeptoren wie z. B. TIM-3 59. Koguchi et al. hatten T-Zell-Klone aus dem ZNS von MS-Patienten auf ihre TIM-3 Expression untersucht und festgestellt, dass die Expression im Vergleich zur Expression bei gesunden Probanden erniedrigt war 58. Yang et al. beobachteten bei ex vivo-Experimenten mit T-Zellen aus MS-Patienten, dass die Immunregulation durch TIM-3 reduziert war 59. Die im folgenden Abschnitt diskutierten Ergebnisse aus der Studie, in der ich die TIM-3-Expression auf B- und T-Zellen untersucht habe, stammen aus NAT-behandelten RRMS-Patienten. Damit konnte ich immunmodulierende Effekte durch GA- und β-Interferon weitestgehend ausschließen. 4.3. CEACAM1-vermittelte Toleranzinduktion in RRMS-Patienten Das vorliegende Manuskript mit dem Titel „CEACAM1 mediates B cell aggregation and tolerance induction in central nervous system autoimmunity“, das im folgenden Abschnitt diskutiert werden soll, präsentiert auch Ergebnisse, die von anderen Mitgliedern der Arbeitsgruppe produziert wurden. Im Manuskript wurden Effekte von Anti-CEACAM1Antikörpern auf den Krankheitsverlauf, die zerebrale Pathologie und auf die mögliche Inhibition der B-Zell-Aggregat-Bildung (ex vivo und in vivo) im EAE-Tiermodell untersucht. Anschließend wurden Lymphozyten aus NAT-behandelten RRMS-Patienten auf die Expression von Toleranzmarkern, die in Verbindung mit der Bildung von B-Zell-Aggregaten stehen, untersucht. Mittels RT-PCR (reverse transcription polymerase chain reaction) konnten Magliozzi et al. schon 2004 zeigen, dass die Expression von CXCL13, einem Chemokin, das B-Zellen zu lymphatischen Follikel rekrutiert und von BAFF, einem wichtigen Überlebensfaktor für B-Zellen, im ZNS-Gewebe von Mäusen mit schubförmiger und chronischer EAE erhöht waren 224. Durch zusätzliche IHC-Analysen wurde gezeigt, dass in den 105 Diskussion Meningen einiger Tiere mit einem chronischen Verlauf ektopische follikelähnliche Strukturen, bestehend aus einem Netzwerk aus B-Zellen, CXCL13+- und FDC-M1+-follikulär dendritischen Zellen (FDZ), nachgewiesen werden konnten 224. Serafini et al. zeigte durch IHCAnalysen von post mortem-Hirngewebeschnitten, dass einige SPMS-Patienten ebenfalls meningeale ektopische follikelähnliche Strukturen besaßen, bestehend aus einem Netzwerk aus CD20+-B-Zellen, CD3+-T-Zellen, CD21+CD35+-FDZ, PNAd+ (peripheral node addressin) hochendothelialen Venolen (high endothelial venules, HEV), CD138+-Plasmazellen und den Chemokinen CXCL13 und CCL21 (C-C motif ligand 21) 177 . CCL21 ist ein Ligand für den Rezeptor CCR7 (C-C motif receptor 7), der wiederum für die Migration von B- und T-Zellen in sekundäres lymphatisches Gewebe benötigt wird 182 . Die im Liquor von MS-Patienten gefundenen B-Gedächtniszellen und Zentroblasten unterstützen die Vermutung, dass es sich dabei um tertiäres lymphatisches Gewebe (tertiary lymphoid organ, TLO) handeln könnte 225. TLO können unabhängig von sekundären lymphatischen Organen Immunantworten induzieren, wenn eine chronische Infektion vorliegt 226. Dabei werden TLO bevorzugt im Gewebe, das eine dauerhafte Immunantwort generieren muss, induziert 226. Die Anwesenheit von BGedächtniszellen und Zentroblasten könnte durch intrathekal-ablaufende Immunantworten in TLO erklärbar sein. Die Aufteilung in B- und T-Zell-Zonen, die Anwesenheit von FDZ und lymphatisch relevanten Chemokinen sowie Plasmazellen ist charakteristisch für TLO 183,226,227. Aus anderen Autoimmunerkrankungen weiß man, dass ein chronisch entzündlicher Verlauf u. a. mit der Anwesenheit von TLO im Zielorgan assoziiert ist 182,183,226. Außerdem stehen ektopische follikelähnliche Strukturen in Verbindung mit einem früheren Beginn der Erkrankung, einem höheren Behinderungsgrad, einer Mikroglia-Aktivierung mit einer stärkeren kortikalen Pathologie und einer verstärkten Demyelinisierung 177,179,181 . In unserer Arbeitsgruppe wurden Versuche unternommen, die Natur dieser B-Zell-Aggregate durch ein B-Zell-abhängiges MS-Tiermodell zu untersuchen. Kuerten et al. konnten mittels eines MBPPLP-Fusionsproteins (MP4) EAE in C57BL/6-Mäusen induzieren und in mehreren Studien beobachten, dass die Erkrankung durch myelinspezifische Antikörper und Komplementaktivierung vermittelt wurde 228–230. Mit dem Auftreten von klinischen Symptomen wurden ebenfalls B-Zell-Aggregate im ZNS von MP4-immunisierten Mäuse nachgewiesen 231,232. Die B-Zell-Infiltrate wiesen Charakteristika auf, die typisch für ektopische follikelähnliche Strukturen waren CD138+-Zellen identifiziert 231 231,233 . Es wurden u. a. B- und T-Zell-Zonen, PNAd+- und . Über Gensequenzierungsanalysen fand man einen TH17-Zell- abhängigen Klassenwechsel der B-Zellen in diesen follikelähnlichen Strukturen 233. Außerdem konnte gezeigt werden, dass die ektopischen follikelähnlichen Strukturen im ZNS der Mäuse mit fortschreitendem Verlauf der Erkrankung phänotypisch tertiärem lymphatischem Gewebe 106 Diskussion entsprachen 231. Diese Beobachtungen entsprachen den Ergebnissen von post mortem-Studien mit MS-Hirngewebe, die den Nachweis von ektopischen follikulären Strukturen auch in frühen Phasen der MS belegten 120 . Eine mögliche Erklärung, wie diese B-Zell-Aggregate entstehen könnten, wurde durch eine ex vivo-Studie aufgezeigt: Lobo et al. zeigten, dass es zur CEACAM1-abhängigen und CD19-vermittelten B-Zell-Aggregation kam, nachdem humane BZellen (Daudi-Zellen, Plasmablasten) mittels löslichem IgM-Antikörper stimuliert wurden 234. Außerdem konnte gezeigt werden, dass CEACAM1 ein negativer Ko-Rezeptor für BZRvermittelte intrazelluläre Signale ist 234. CEACAM1 inhibiert die BZR-vermittelten intrazellulären Signale dadurch, dass SHP-1 (Src homology region 2 domain-containing phosphatase-1) rekrutiert und die PI3K-(Phosphoinositid-3-Kinase, auch phosphatidylinositol4,5-bisphosphat 3-kinase) induzierte Aktivierungskaskade inhibiert wird 234. CEACAM1 wird auf aktivierten T-Zellen, Endothelzellen und lymphatischen Gefäßen, sowie konstitutiv auf Epithelzellen, Monozyten, Granulozyten, DZ und Tumorzellen exprimiert 235 . In Studien mit murinen B-Zellen war CEACAM1 für eine optimale virusspezifische Antikörperbildung nötig 236. In CEACAM1-Knockout-Mäusen wurden signifikant erniedrigte virusspezifische neutralisierende Antikörper detektiert 236 . Nach adoptivem virusspezifischem B-Zell-Transfer aus Wildtyptieren wurden normale virusspezifische Antikörpertiter in CEACAM1-KnockoutMäusen gemessen 236 Überlebenssignale 236 . Außerdem induzierte die CEACAM1-Expression in murinen B-Zellen . Diese Ergebnisse widersprachen den Ergebnissen von Lobo et al. die zeigten, dass die CEACAM1-Expression eher negative Effekte auf Daudi-Zellen hatte 234. Lobo et al. zeigten, dass CEACAM1 auf Daudi-Zellen ein negativer Korezeptor für den BZR ist 234. CEACAM1 kann homophile oder heterophile (mit anderen CEACAM-Klassen) Bindungen eingehen, die entweder parallel (cis) oder antiparallel (trans) angeordnet sind 237. Durch adhäsionsvermittelte homophile trans-Bindung werden homophile cis- Konformationsänderungen induziert, die für die intrazelluläre Signalweitergabe verantwortlich sind 237. In der vorliegenden Studie haben wird den Einfluss von Anti-CEACAM1-Antikörpern auf die B-Zell-Aggregation mit Hilfe der B-Zell-vermittelten MP4-EAE analysiert. Zuerst prüfte die Masterstudentin Laura Scheffler die CEACAM1-Expression auf B-Zellen in ZNSInfiltraten von Tieren mit akut und chronisch verlaufender EAE. Nur in Proben von Tieren, die einen chronischen Verlauf zeigten, konnten B-Zell-Aggregate nachgewiesen werden und nahezu alle B-Zellen darin exprimierten CEACAM1. Diese B-Zell-Aggregate ähnelten durch ihre CEACAM1-Expression B-Zellen in den Keimzentren sekundärer lymphatischer Organe. Die zuvor von Lobo et al. beschriebene CD19-vermittelte B-Zell-Aggregation wurde durch murine Anti-CEACAM1-(mCC1) Antikörper, in ex vivo-Experimenten, signifikant inhibiert. Außerdem wurden MP4-immunisierte Mäuse nach Krankheitsbeginn mit Anti-mCC1107 Diskussion Antikörpern behandelt. Die so behandelten Tiere zeigten im Vergleich zur Kontrollgruppe bereits nach 13 Tagen eine signifikante Verbesserung ihrer klinischen Symptome. Anschließend wurden die Infiltrate durch IHC analysiert und charakterisiert. Es stellte sich heraus, dass nur die B-Zell-Aggregate, nicht aber die Summe der Infiltrate, durch die mCC1Behandlung abgenommen hatte. Die Untersuchung der B-Zell-Aggregatgröße zeigte keinen Unterschied zwischen behandelten und unbehandelten Tieren, folglich hatte die mCC1Behandlung keinen Einfluss auf bereits bestehende Aggregate. Man könnte vermuten, dass die Behandlung die Neuformation von B-Zell-Aggregaten verhindert. Die mCC1-Behandlung hatte keinen Einfluss auf den Anti-MP4-Antikörper-Titer und ebenfalls nicht auf die Struktur der sekundären lymphatischen Organe. Letztere zeigten in IHC-Analysen keine Veränderungen. Von mir zusätzlich durchgeführte durchflusszytometrische Studien zeigten, dass nach ex vivo mCC1-Antikörper-Inkubation weder kostimulatorische (CD80, CD86, CD40) noch negative Ko-Rezeptor-(CD22) Marker auf B-Zellen verändert waren. Das war auch unabhängig davon, ob die B-Zellen vor oder nach dem Aggregationsprotokoll mit Anti-mCC1-Antikörpern inkubiert wurden. Wir schlussfolgerten, dass die Wirkung des mCC1-Antikörpers spezifisch war und die Inhibition der Aggregate auf sterische Hinderungen beschränkt war. Die Doktorandin Marie Wunsch konnte im Rahmen dieser Studie mit Hilfe von humanen post mortem-ZNS-Gewebeproben von MS-Patienten IHC-Analysen anfertigen. Sie konnte zeigen, dass in zwei der Proben B-Zell-Aggregate vorhanden waren und sich diffuse B-Zell-Infiltrate in allen Proben nachweisen ließen. Die Hälfte der B-Zellen aus den Aggregaten und diffusen Infiltraten exprimierte CEACAM1. CEACAM1 ist auch auf humanen B-Zellen (naiven BZellen und B-Gedächtniszellen) im Blut von gesunden Probanden nachgewiesen worden 236 . Aktuelle Studien zeigten, dass CEACAM1 eine wichtige Rolle bei der Induktion der peripheren T-Zell-Toleranz spielt. CEACAM1 und TIM-3 kennzeichnen immunerschöpfte TH-Zellen nach einer Virusinfektion 15. Huang et al. konnten zeigen, dass CEACAM1 die TIM-3-vermittelte T-Zell-Toleranz induziert 15. Die Ergebnisse aus den post mortem-Studien und die CEACAM1Expression auf B-Zellen in der Peripherie legen nahe, dass die CEACAM1-Expression auf Immunzellen im Blut von MS-Patienten im Vergleich zu gesunden Probanden unterschiedlich sein könnte. Die Peripherie könnte der Ort sein, der für die Entstehung von CEACAM1+-BZell-Aggregaten eine wichtige Rolle spielt. Palanichamy et al. vermuteten, dass die follikelähnlichen Strukturen im ZNS die Orte der Affinitätsreifung sein könnten, wohingegen die primäre Antigenerkennung und B-Zell-Aktivierung in der Peripherie stattfinden 238. Neueste Daten aus ultrahochdurchsatz-basierten Sequenzierungsstudien (deep sequenzing) belegten eine klonale Verwandtschaft zwischen B-Zell-Untergruppen, wie z. B. B-Gedächtniszellen, die aus Liquor und Blut isoliert wurden 238 . Ich vermutete, dass während eines Schubereignisses 108 Diskussion die CEACAM1+-B-Zellen im Blut von RRMS-Patienten signifikant reduziert sein könnten. Im Sinne dieser Annahme habe ich B-Zell-Untergruppen aus dem Blut von RRMS-Patienten in Remission und während eines Schubereignisses untersucht. Die CEACAM1+-B-Zell-Zahlen (naive B-Zellen, B-Gedächtniszellen und B1-Zellen) in RRMS-Patienten während der Remission waren im Vergleich zu gesunden Probanden signifikant erhöht. Dagegen fand sich kein Unterschied in der CEACAM1+-B-Zell-Untergruppen-Zahl während eines Schubereignisses oder in der Remissionsphase. Nachdem die B-Zellen stimuliert wurden (durch den TLR7/8 Agonisten R-848), waren die CEACAM1+-B-Zell-Zahlen bei RRMS-Patienten im Vergleich zu gesunden Probanden ebenfalls erhöht. Eine Analyse der B-Zell-Untergruppen zeigte, dass nur die CEACAM1+-B1-Zellen nach der Stimulation signifikant erhöht waren. Die Analyse der CEACAM1+CD4+- und CD8+-T-Zellen zeigte keine Unterschiede zwischen Proben aus Patienten und gesunden Probanden. Meine durchflusszytometrischen Analysen ergaben, dass B-Zellen aus gesunden Probanden und MS-Patienten TIM-3 exprimierten. Die B-Zell-TIM-3 Expression konnte durch RT-PCR bestätigt werden. CEACAM1+-TIM-3+ BZell- und CEACAM1+TIM-3+-CTL-Zahlen waren bei RRMS-Patienten im Vergleich zu Proben aus gesunden Probanden signifikant erhöht. B-Zellen und CTL aus RRMS-Patienten wiesen einen Phänotyp aus, der nach chronischer Virusinfektion oder nach CEACAM1vermittelter TIM-3 Toleranzinduktion bei TH-Zellen beschrieben worden war 15. Es wurde kein Unterschied zwischen RRMS-Patienten und gesunden Probanden festgestellt, wenn die CEACAM1+TIM-3+-TH-Zell-Zahlen verglichen wurden. Nachdem TH-Zellen aus RRMSPatienten und gesunden Kontrollen durch Anti-CD3/CD28-Antikörper für 72 Stunden stimuliert worden waren, exprimierten TH-Zellen TIM-3 und CEACAM1. Diese Ergebnisse stimmten erwartungsgemäß mit Daten aus vorherigen Studien überein 15. Interessanterweise waren die CEACAM1+-TH-Zell-Zahlen nach Anti-CD3/CD28-Antikörper-Stimulation bei RRMS-Patienten im Vergleich zu gesunden Kontrollen reduziert. Ich konnte aber keinen Unterschied bei den CEACAM1+-TH-Zell-Zahlen nach polyklonaler Stimulation zwischen RRMS-Patienten und gesunden Probanden feststellen. Diese Ergebnisse implizieren eine Beteiligung aktivierter B-Zellen bei der CEACAM1-Induktion auf TH-Zellen. Es wurde eine signifikante Reduktion der CEACAM1+TIM-3+-TH-Zahl bei RRMS-Patienten im Vergleich zu gesunden Probanden beobachtet. Huang et al. beschrieben CEACAM1+TIM-3+-TH-Zellen als immuntolerant 15 . Meine Untersuchung zeigte, dass TH-Zellen aus RRMS-Patienten nicht CEACAM1 und TIM-3 koexprimieren können. Die einzelne Expression ist dagegen nicht per se gestört. Zusammenfassend kann man feststellen, dass B-Zellen und CTL aus RRMSPatienten einen immunerschöpften Zustand aufwiesen und dass man bei TH-Zellen aus RRMSPatienten trotz unspezifischer Stimulation keine Toleranz induzieren konnte. Die erhöhte 109 Diskussion CEACAM1+-B-Zell-Zahl könnten eine Folge der reduzierten CEACAM1+TIM-3+-TH-ZellZahl sein, weil CEACAM1 nicht nur auf gleichen Zellen (cis) TIM-3, sondern auch auf anderen Zellen (trans) rekrutieren kann 15. Eine TIM-3-Expression auf B-Zellen war bisher unbekannt. Diese neue Beobachtung eröffnet neue Möglichkeiten in der B/T-Zell-Interaktion. Die Erhöhung der CEACAM1+-B-Zellen könnte eine direkte Folge einer gestörten B- und T-ZellInteraktion sein. Nach den vorliegenden Daten könnten B-Zellen aktiv TH-Zell-Toleranz durch CEACAM1 und TIM3 induzieren. Dieser Mechanismus könnte bei MS-Patienten gestört sein. 4.4. Die Rolle autoreaktiver B-Zellen und B-Zell-Untergruppen in der MS Zusammenfassend kann ich feststellen, dass in meinen Studien bei RRMS-Patienten die B1Zell-Zahl signifikant erniedrigt und die CEACAM1+-B-Zell-Zahl signifikant erhöht war. Außerdem zeigten meine Untersuchungen, dass B-Zellen aus RRMS-Patienten, die mit NATbehandelt wurden, den Phänotyp einer immunologischen Erschöpfung aufwiesen. Überraschenderweise ist die Anwesenheit ZNS-reaktiver B-Zellen im Blut von behandelten Patienten ein Indiz für einen Therapieerfolg bzw. Versagen der Therapie mit GA bzw. β-Interferon. Dies wirft die Frage auf, welche Rolle autoreaktive B-Zellen und Autoantikörper in der Pathogenese der MS spielen? Warum ist die B1-Zell-Zahl im Blut von RRMS-Patienten erniedrigt? Warum sind immunologisch erschöpfte B-Zellen im Blut von RRMS-Patienten erhöht? In Hinblick auf diese Fragen möchte ich im folgenden Abschnitt meine Ergebnisse zusammenfassend diskutierten. Das Neue in meiner Arbeit zum Themengebiet „B1-Zellen in der MS“ ist der Umstand, dass die Behandlung keinen Einfluss auf die signifikant erniedrigte B1-Zell-Zahl hatte. Humane B1-Zellen sind im Nabelschnurblut nachgewiesen worden 146. Außerdem konnte nachgewiesen werden, dass humane B1-Zellen spontan IgM-Antikörper produzierten, die polyreaktiv bzw. kreuzreaktiv waren 146. Sie könnten also die Quelle natürlicher Antiköper sein, wie sie aus der Maus bekannt sind 239. Humane natürliche Antiköper gehören zur IgM-, IgG- oder IgA-Isotypklasse 165,240. Alle drei Isotypklassen können schon in den ersten Lebensmonaten nachgewiesen werden 165. Von murinen B1-Zellen, die ausführlich untersucht und beschrieben wurden, weiß man, dass sie früh in der Ontogenese des Individuums gebildet werden 145. Humane und murine B1-Zellen sekretieren spontan und konstitutiv natürliche Antikörper, welche die erste Antikörper-Abwehr gegen virale und bakterielle Infektionen bilden 146,241 . Natürliche IgM-Antikörper unterstützen DZ bei der Erkennung von opportunistischen Erregern oder unterstützen die Gedächtnisantwort des Immunsystems gegen Influenzaviren 239,242 . Damit schließen sie eine zeitliche Lücke, die zwischen Infektion und Keimzentrumsreaktion entsteht. Ihre zweite Aufgabe besteht darin, apoptotische 110 Diskussion Zellmembranen zu erkennen und Zelldebris mit Hilfe von DZ zu entsorgen 243,244 . Aus dem RA-Tiermodell ist bekannt, dass das Vorhandensein von natürlichen Antikörpern nötig ist, damit B- und T-Zellen nach Injektionen mit apoptotischen Zellen IL-10 produzieren 245. Damit nehmen sie einen wichtigen Platz in der Aufrechterhaltung der individuellen GewebeHomöostase ein. Eine ältere Studie mit Mäusen zeigte, dass fehlende IgM-Titer im Serum zu einer erniedrigten Fremdantigen-, aber einer verstärkten Selbstantigen-Antwort führten 246. Folglich könnten natürliche IgM-Antikörper vor Autoimmunität schützen 246. Eine weitere murine Studie zeigte, dass natürlich vorkommende IgM-Antikörper vor Autoimmunität schützen, indem sie die zentrale B-Zell-Toleranz unterstützen 247. Es wurde vermutet, dass natürliche Antikörper an B-Zellen im Knochenmark binden und so bei der Toleranzinduktion mitwirken 247. Außerdem vermutete man anhand von Daten aus einem virusvermittelten MSMausmodell, dass natürliche Antikörper möglicherweise remyelinisierende Eigenschaften besitzen 248. Die Daten zu humanen B1-Zellen und natürlichen Antiköpern sind leider dürftig. Trotzdem darf man annehmen, dass Patienten, die signifikant erniedrigte B1-Zell-Zahlen haben, ebenfalls erniedrigte natürliche Antikörper-Spiegel aufweisen. An dieser Stelle muss erwähnt werden, dass als Quelle für natürliche Antikörper auch andere B-Zell-Untergruppen in Frage kommen könnten, z. B. IgM-Gedächtniszellen 134. Myelinspezifische IgM-Antikörper sind in gesunden Individuen und MS-Patienten nachgewiesen worden 249 . Mittlerweile haben andere Arbeitsgruppen ebenfalls humane B1-Zellen und natürliche Antikörper untersucht. Tørring et al. zeigten, dass unbehandelte RRMS-Patienten signifikant erniedrigte B1-ZellZahlen aufwiesen 151 . Ist die B1-Zell-Erniedrigung MS-spezifisch? In anderen Erkrankungen wurden ebenfalls erniedrigte B1-Zell-Zahlen nachgewiesen und sind damit ein Hinweis darauf, dass die Reduktion nicht MS-spezifisch ist 250. Es wurde eine signifikant erniedrigte B1-ZellZahl bei Patienten mit variablem Immundefektsyndrom (common variable immunodeficiency, CVID) detektiert 250 . Interessanterweise wurde eine starke positive Korrelation zwischen B1- Zell-Zahlen und IgM-Titer beobachtet SLE-Patienten erhöht 250 . Im Gegenteil dazu waren die B1-Zell-Zahlen bei 251 . Welche Gründe könnten dazu führen, dass B1-Zellen im Blut von MS-Patienten erniedrigt sind? Eine mögliche Erklärung wurde von Tørring et al. vorgeschlagen: Die B1-Zellen sind durch die entzündlichen Prozesse ins ZNS migriert und deshalb in der Peripherie erniedrigt 151. Aus Studien weiß man, dass B-Zell-Chemokine im Liquor von CIS- und MS-Patienten erhöht waren 252. Kadhemi et al. berichteten von signifikant erhöhten CXCL13-Titern in CIS- bzw. MS-Patienten 252. Signifikant erhöhte CXCL13-Spiegel im Liquor von CIS-Patienten waren mit einer höheren Wahrscheinlichkeit an MS zu erkranken assoziiert und es wurden während eines Schubereignisses hohe CXCL13-Spiegel nachgewiesen 252,253. Wie zuvor schon von Griffin et al. beschrieben, habe auch ich einen 111 Diskussion negativen Zusammenhang zwischen Alter und B1-Zell-Zahl beobachtet, sowohl bei gesunden Probanden als auch bei MS-Patienten 146. Der altersbedingte Abfall der B1-Zell-Zahl und die vermehrte ZNS-Migration könnten zu der niedrigen B1-Zell-Zahl in MS-Patienten führen. Es wurde spekuliert, ob es sich bei humanen B1-Zellen um eine Vorstufe von Plasmablasten handelt 148. Interessanterweise wurden im Liquor von CIS- und MS-Patienten hohe Plasmablasten- und B-Gedächtniszell-Zahlen detektiert 225,238. B-Gedächtniszellen exprimieren wie B1-Zellen CD27 140,146. Könnte es sich bei den erhöhten B-Gedächtniszellen im Liquor von CIS- und MS-Patienten evtl. auch um B1-Zellen handeln? Sind die erhöhten immunerschöpften B-Zell- und CTL-Zahlen in MS-Patienten die Folge einer ungenügenden viralen und bakteriellen Abwehr durch fehlende natürliche Antikörper? Diese Frage kann anhand der vorliegenden Daten nicht geklärt werden. Eine weitere Beobachtung aus meiner Studie war, dass die TH-Zell-Zahlen aus MS-Patienten mit einem Phänotyp, der für tolerante TH-Zellen beschrieben worden ist, nach Stimulation signifikant erniedrigt waren. Im Gegenteil dazu waren CEACAM1+-B-Zellen im Blut von MS-Patienten signifikant erhöht. Wenn man bedenkt, dass CEACAM1 homophile Bindungen (cis und trans) eingeht und CEACAM1 TIM-3 auf THZellen rekrutiert, könnte man spekulieren, dass CEACAM1+TIM-3+-TH-Zellen bei der Induktion der B-Zell-Toleranz eine wichtige Rolle spielen. Aus bislang unbekannten Gründen ist diese T-Zell-Hilfe in der MS gestört und möglicherweise kommt es deshalb zur signifikanten Erhöhung der CEACAM1+-B-Zell-Zahl in der MS. Ist bei MS die negative Feedbackschleife zwischen TH-Zellen und B-Zellen nach symptomlosen viralen oder bakteriellen Infektionen gestört? Wird die Immunantwort der CEACAM1-CEACAM1-Interaktion zwischen B- und TZellen beendet? Ist diese Interaktion in der MS gestört? Diese Fragen können zu diesem Zeitpunkt nicht beantwortet werden. Auffällig ist nur, dass B-Zell-Aggregate im ZNS CEACAM1+-B-Zellen enthielten. Dieser Umstand und die erhöhten CEACAM1+-B-ZellZahlen im Blut verleiten zur Annahme, dass entzündliche Prozesse in der Peripherie die Bildung ektopischer follikelähnlicher Strukturen im ZNS induzieren könnten. Die hier präsentierten ZNS-Lysat-ELISPOT Studien mit GA- und β-Interferon-behandelten RRMS-Patienten zeigten, dass es eine Assoziation zwischen ZNS-reaktiven B-Zellen im Blut und dem Therapieerfolg, zumindest in einer Gruppe von RRMS-Patienten, gibt. Schon länger ist bekannt, dass nicht jeder Patient ähnliche Läsionsmuster aufweist, sondern dass man unterschiedliche Läsionsmuster findet 159 . Zu diesem Zweck wurde Hirngewebe von SPMS- Patienten untersucht. Das Läsionsmuster, das am häufigsten auftrat, war u. a. durch Komplement- und Antikörperablagerungen gekennzeichnet 159 . In keiner anderen Läsionsart wurden Antikörper nachgewiesen 159. Diese Ergebnisse implizieren, dass MS-Patienten zumindest in zwei Gruppen eingeteilt werden könnten, abhängig davon, ob die Läsionen 112 Diskussion Antikörperablagerungen aufweisen oder nicht. Die Ergebnisse von Lucchinetti et al. waren Gegenstand einiger Kontroversen, denn diverse Studien betrachteten die verschiedenen Läsionsarten als zeitliche Bestandsaufnahmen einer kohärenten Abfolge ein und derselben Läsionsart 156 . Andere Arbeitsgruppe zeigten, dass die Läsionen aller MS-Patienten in späten Krankheitsphasen (Median = 22,9 Jahre Krankheitsdauer) eine Antikörperbeteiligung aufwiesen 254,255 . Eine kürzlich publizierte Studie favorisierte eine Trennung in verschiedene Gruppen von Beginn an der Erkrankung 256. Anhand longitudinal gewonnener Biopsien konnte gezeigt werden, dass sich bei 21 von 22 MS-Patienten ein einheitliches Läsionsmuster im Verlauf der Erkrankung wiederfand 256 . Meine ELISPOT-Studie zeigte, dass es eine Gruppe von GA-behandelten Patienten gab, deren Therapieerfolg mit der Anwesenheit von ZNSreaktiven Antikörpern assoziiert war. Dieser Umstand scheint zunächst nicht sehr plausibel, weil Autoantikörpern eine pathologische Rolle zugeschrieben wird. Man fand im Blut von gesunden Individuen und MS-Patienten im gleichen Maße niedrige myelinspezifische IgM- und IgG-Antikörper-Zahlen 249. Außerdem konnten myelinspezifische Antikörper-Titer in CISPatienten als Biomarker nicht bestätigt werden 162,163,187. Auch Anti-Kir4.1-Antikörper sind als MS-Biomarker vorgeschlagen worden, die Ergebnisse sind aber ebenfalls nicht von anderen Arbeitsgruppen bestätigt worden und daher umstritten 188–191. Es ist bekannt, dass die LangzeitBehandlung mit GA bei MS-Patienten u. a. zur Produktion von GA-spezifischen IgG4Antikörper führt 216 . Man vermutet, dass ein Teil des Therapieerfolgs auf der Induktion von GA-spezifischen IgG4-Antikörpern beruht, da deren Anwesenheit mit einer TH2-Antwort assoziiert ist 168,217,218 . Man könnte die Anwesenheit ZNS-reaktiver B-Zellen als Biomarker verstehen. Im Gegensatz zur GA-Therapie werden in der β-Interferon-Therapie neutralisierende Antikörper gebildet, wodurch der Therapieeffekt reduziert wird 257. Außerdem modulieren βInterferone T-Zellen dadurch, dass sie die Zytokinsekretion von B-Zellen beeinflussen. Eine direkte B-Zell-Wirkung durch β-Interferone ist nicht beschrieben worden. Könnte in Zukunft ein einfacher ELISPOT-Test den Therapieerfolg oder Misserfolg von β-Interferon oder GA voraussagen? Um diese Frage zu klären, müssten weitere Studien mit einer höheren Anzahl an Probanden und Erkrankten durchgeführt werden. Es wäre im Sinne vieler Erkrankten, einen einfachen Test zu etablieren, der den Therapieerfolg prognostiziert. Somit ist es notwendig die B-Zell-Untergruppen und deren Rolle in der MS noch intensiver zu erforschen. Ich hoffe, dass meine Arbeit einen kleinen Beitrag auf dem Weg zu einer personalisierten MS-Therapie geliefert hat. 113 Ausblick 5. Ausblick 5.1. Blutanalysen Erfreulicherweise findet meine Studie mit dem Titel „The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsing-remitting multiple sclerosis patients“ eine Fortsetzung in einer von Teva Pharmaceutical Industries Ltd. geförderten Studie mit dem Namen COPSELECT. Diese Studie wird GA- und β-Interferon-behandelte RRMSPatienten untersuchen, aber auch RRMS-Patienten einschließen, welche die Therapie gewechselt haben (GA β-Interferon bzw. β-Interferon GA). Die Anzahl der analysierten Patienten wird erhöht (> 200) und die Studie wird Patienten aus diversen MS-Zentren einschließen. Die Studiendauer ist auf 12 Monate angesetzt. Die Rekrutierung der Patienten und die Verwaltung der Daten wird durch die NeuroTransData (NTD) GmbH bereitgestellt. Der Assay wird von mir durchgeführt und ausgewertet. 5.2. Liquoranalysen Um die Rolle der B1-Zellen in der Pathogenese der MS intensiver zu untersuchen, müsste man bei CIS- und RRMS-Patienten B1-Zellen aus dem Liquor mittels Durchflusszytometer untersuchen. Die Analyse müsste sowohl Patienten während der Remission als auch während eines Schubs erfassen. Außerdem könnte man B-Zellen, die aus dem Liquor isoliert wurden, mit Hilfe des ZNS-Lysat-ELISPOT untersuchen. Werden die B1-Zell- und ZNS-reaktiven BZell-Zahlen im Liquor durch die MS-Therapien beeinflusst? Sind niedrige B1-Zell-Zahlen im Blut dadurch zu erklären, weil sie während eines Schubs ins ZNS migrieren? 5.3. Längsschnittstudie Schließlich müsste CEACAM1+TIM-3+-B- man und die natürlichen -T-Zellen bei Antikörper-Titer, gesunden und B1-Zell-Zahlen, CIS-Patienten in einer Längsschnittstudie untersuchen. Dadurch könnte man erfahren, ob niedrige natürliche Antikörper-Titer, B1-Zell- oder CEACAM1+TIM-3+-B- und -T-Zell-Zahlen mit einem erhöhten Risiko an MS zu erkranken korrelieren. Man könnte den ZNS-Lysat-ELISPOT nutzen, um zu Beginn der Studie MS-Patienten-Untergruppen (ELISPOT-responder, nonresponder) zu definieren. 114 Ausblick 5.4. Zytozentrifugation Einer der Kommentare, die wir nach dem Peer-Review-Prozess zu unserem Manuskript „CEACAM1 mediates B cell aggregation and tolerance induction in central nervous system autoimmunity“ erhalten haben, beschäftigte sich mit der TIM-3-Expression auf B-Zellen. Um die Forderung nach weiterführenden Experimenten, welche die TIM-3-Expression auf B-Zellen belegen, nachzukommen, werde ich in den nächsten Wochen mittels Zytozentrifugation Einzelzellfärbungen anfertigen. Meine Daten zeigen, dass es sich bei TIM-3+-B-Zellen um eine seltene B-Zell-Population handelt. Die Zytozentrifugation erlaubt es, auch seltene Zellen zu analysieren, weil durch diese Methode viele Zellen auf eine kleine Fläche konzentriert werden. 115 Literaturverzeichnis 6. 1. Literaturverzeichnis Litman, G. W., Cannon, J. P. & Dishaw, L. J. Reconstructing immune phylogeny: new perspectives. Nat. Rev. Immunol. 5, 866–79 (2005). 2. Koonin, E. V & Wolf, Y. I. Evolution of the CRISPR-Cas adaptive immunity systems in prokaryotes: models and observations on virus-host coevolution. Mol. Biosyst. 11, 20–7 (2015). 3. Ribatti, D. Sir Frank Macfarlane Burnet and the clonal selection theory of antibody formation. Clin. Exp. Med. 9, 253–8 (2009). 4. Burnet, F. M. The clonal selection theory of acquired immunity. (Vanderbilt University Press, 1959). at <https://archive.org/details/clonalselectiont00burn> 5. Klein, L., Klugmann, M., Nave, K. A., Tuohy, V. K. & Kyewski, B. Shaping of the autoreactive T-cell repertoire by a splice variant of self protein expressed in thymic epithelial cells. Nat. Med. 6, 56–61 (2000). 6. Racke, M. K., Ratts, R. B., Arredondo, L., Perrin, P. J. & Lovett-Racke, A. The role of costimulation in autoimmune demyelination. J. Neuroimmunol. 107, 205–15 (2000). 7. Le Mercier, I., Lines, J. L. & Noelle, R. J. Beyond CTLA-4 and PD-1, the Generation Z of Negative Checkpoint Regulators. Front. Immunol. 6, 418 (2015). 8. Sakuishi, K., Jayaraman, P., Behar, S. M., Anderson, A. C. & Kuchroo, V. K. Emerging Tim-3 functions in antimicrobial and tumor immunity. Trends Immunol. 32, 345–9 (2011). 9. Delfs, M. W., Furukawa, Y., Mitchell, R. N. & Lichtman, A. H. CD8+ T cell subsets TC1 and TC2 cause different histopathologic forms of murine cardiac allograft rejection. Transplantation 71, 606–10 (2001). 10. Raphael, I., Nalawade, S., Eagar, T. N. & Forsthuber, T. G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 74, 5–17 (2015). 11. Taga, K. & Tosato, G. IL-10 inhibits human T cell proliferation and IL-2 production. J. Immunol. 148, 1143–8 (1992). 12. Kahan, S. M., Wherry, E. J. & Zajac, A. J. T cell exhaustion during persistent viral infections. Virology 479-480, 180–93 (2015). 116 Literaturverzeichnis 13. Crawford, A. et al. Molecular and transcriptional basis of CD4+ T cell dysfunction during chronic infection. Immunity 40, 289–302 (2014). 14. Delgoffe, G. M. & Powell, J. D. Feeding an army: The metabolism of T cells in activation, anergy, and exhaustion. Mol. Immunol. 68, 492–6 (2015). 15. Huang, Y.-H. et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 517, 386–90 (2015). 16. Huang, Y.-H. et al. Corrigendum: CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2016). doi:10.1038/nature17421 17. Anderton, S. M. & Wraith, D. C. Selection and fine-tuning of the autoimmune T-cell repertoire. Nat. Rev. Immunol. 2, 487–98 (2002). 18. Anderson, G. & Takahama, Y. Thymic epithelial cells: working class heroes for T cell development and repertoire selection. Trends Immunol. 33, 256–63 (2012). 19. Yamano, T. et al. Thymic B Cells Are Licensed to Present Self Antigens for Central T Cell Tolerance Induction. Immunity 42, 1048–61 (2015). 20. Klein, L., Hinterberger, M., Wirnsberger, G. & Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 9, 833– 44 (2009). 21. Palmer, E. Negative selection--clearing out the bad apples from the T-cell repertoire. Nat. Rev. Immunol. 3, 383–91 (2003). 22. Hsieh, C.-S., Lee, H.-M. & Lio, C.-W. J. Selection of regulatory T cells in the thymus. Nat. Rev. Immunol. 12, 157–67 (2012). 23. Bouneaud, C., Kourilsky, P. & Bousso, P. Impact of negative selection on the T cell repertoire reactive to a self-peptide: a large fraction of T cell clones escapes clonal deletion. Immunity 13, 829–40 (2000). 24. Danke, N. A., Koelle, D. M., Yee, C., Beheray, S. & Kwok, W. W. Autoreactive T cells in healthy individuals. J. Immunol. 172, 5967–72 (2004). 25. Thorlacius-Ussing, G., Sørensen, J. F., Wandall, H. H. & Pedersen, A. E. Auto-reactive T cells revised. Overestimation based on methodology? J. Immunol. Methods 420, 56–9 (2015). 117 Literaturverzeichnis 26. André, S., Tough, D. F., Lacroix-Desmazes, S., Kaveri, S. V & Bayry, J. Surveillance of antigen-presenting cells by CD4+ CD25+ regulatory T cells in autoimmunity: immunopathogenesis and therapeutic implications. Am. J. Pathol. 174, 1575–87 (2009). 27. Peterson, R. A. Regulatory T-cells: diverse phenotypes integral to immune homeostasis and suppression. Toxicol. Pathol. 40, 186–204 (2012). 28. Pandiyan, P., Zheng, L., Ishihara, S., Reed, J. & Lenardo, M. J. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat. Immunol. 8, 1353–62 (2007). 29. Dilek, N. et al. Targeting CD28, CTLA-4 and PD-L1 costimulation differentially controls immune synapses and function of human regulatory and conventional T-cells. PLoS One 8, e83139 (2013). 30. Mellor, A. L. & Munn, D. H. Ido expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol. 4, 762–774 (2004). 31. Qureshi, O. S. et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600–3 (2011). 32. Zhang, J., Xu, X. & Liu, Y. Activation-induced cell death in T cells and autoimmunity. Cell. Mol. Immunol. 1, 186–92 (2004). 33. Moreno, M. et al. Activation-induced cell death in T lymphocytes from multiple sclerosis patients. J. Neuroimmunol. 272, 51–5 (2014). 34. Mahnke, K., Schmitt, E., Bonifaz, L., Enk, A. H. & Jonuleit, H. Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol. Cell Biol. 80, 477–83 (2002). 35. Outters, P., Jaeger, S., Zaarour, N. & Ferrier, P. Long-Range Control of V(D)J Recombination & Allelic Exclusion: Modeling Views. Adv. Immunol. 128, 363–413 (2015). 36. Lang, J. et al. Receptor editing and genetic variability in human autoreactive B cells. J. Exp. Med. 213, 93–108 (2016). 37. Wardemann, H. et al. Predominant autoantibody production by early human B cell precursors. Science 301, 1374–7 (2003). 38. Pillai, S., Mattoo, H. & Cariappa, A. B cells and autoimmunity. Curr. Opin. Immunol. 23, 721–31 (2011). 118 Literaturverzeichnis 39. Schiemann, B. et al. An essential role for BAFF in the normal development of B cells through a BCMA-independent pathway. Science 293, 2111–4 (2001). 40. Hartley, S. B. et al. Elimination from peripheral lymphoid tissues of self-reactive B lymphocytes recognizing membrane-bound antigens. Nature 353, 765–9 (1991). 41. Fulcher, D. A. & Basten, A. Reduced life span of anergic self-reactive B cells in a doubletransgenic model. J. Exp. Med. 179, 125–34 (1994). 42. Thien, M. et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity 20, 785– 98 (2004). 43. Lesley, R. et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 20, 441–53 (2004). 44. Tiller, T. et al. Autoreactivity in human IgG+ memory B cells. Immunity 26, 205–13 (2007). 45. Cai, G. & Hafler, D. A. Multispecific responses by T cells expanded by endogenous selfpeptide/MHC complexes. Eur. J. Immunol. 37, 602–12 (2007). 46. McHeyzer-Williams, L. J. & McHeyzer-Williams, M. G. Antigen-specific memory B cell development. Annu. Rev. Immunol. 23, 487–513 (2005). 47. Tiller, T. et al. Development of self-reactive germinal center B cells and plasma cells in autoimmune Fc gammaRIIB-deficient mice. J. Exp. Med. 207, 2767–78 (2010). 48. Hervé, M. et al. CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J. Exp. Med. 204, 1583–93 (2007). 49. Tsuiji, M. et al. A checkpoint for autoreactivity in human IgM+ memory B cell development. J. Exp. Med. 203, 393–400 (2006). 50. Langman, R. E. & Cohn, M. A short history of time and space in immune discrimination. Scand. J. Immunol. 44, 544–8 (1996). 51. Maehle, A.-H. A binding question: the evolution of the receptor concept. Endeavour 33, 135–40 (2009). 52. Vanderlugt, C. L. & Miller, S. D. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2, 85–95 (2002). 53. Moll, I. et al. Autoimmunerkrankungen. Duale R. Dermatologie (Georg Thieme Verlag, 2010). doi:10.1055/b-002-11376 119 Literaturverzeichnis 54. Chastain, E. M. L. & Miller, S. D. Molecular mimicry as an inducing trigger for CNS autoimmune demyelinating disease. Immunol. Rev. 245, 227–38 (2012). 55. Fujinami, R. S., von Herrath, M. G., Christen, U. & Whitton, J. L. Molecular Mimicry, Bystander Activation, or Viral Persistence: Infections and Autoimmune Disease. Clin. Microbiol. Rev. 19, 80–94 (2006). 56. Nagamine, K. et al. Positional cloning of the APECED gene. Nat. Genet. 17, 393–8 (1997). 57. Hoppenbrouwers, I. A. & Hintzen, R. Q. Genetics of multiple sclerosis. Biochim. Biophys. Acta 1812, 194–201 (2011). 58. Koguchi, K. et al. Dysregulated T cell expression of TIM3 in multiple sclerosis. J. Exp. Med. 203, 1413–8 (2006). 59. Yang, L., Anderson, D. E., Kuchroo, J. & Hafler, D. A. Lack of TIM-3 immunoregulation in multiple sclerosis. J. Immunol. 180, 4409–14 (2008). 60. Wildin, R. S. & Freitas, A. IPEX and FOXP3: clinical and research perspectives. J. Autoimmun. 25 Suppl, 56–62 (2005). 61. Murphy, K., Tavers, P. & Walport, M. in Janeway´s Immunobiol. 599–654 (Garland Science, Taylor and Francis Group, 2008). 62. Nylander, A. & Hafler, D. a. Multiple sclerosis. J. Clin. Invest. 122, 1180–8 (2012). 63. Compston, A. & Coles, A. Multiple sclerosis. Lancet 372, 1502–17 (2008). 64. Verheul, M. K., Fearon, U., Trouw, L. A. & Veale, D. J. Biomarkers for rheumatoid and psoriatic arthritis. Clin. Immunol. 161, 2–10 (2015). 65. Sherer, Y., Gorstein, A., Fritzler, M. J. & Shoenfeld, Y. Autoantibody explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin. Arthritis Rheum. 34, 501–37 (2004). 66. Mattle, H. & Mumenthaler, M. in Neurologie 351–369 (Georg Thieme Verlag, 2013). 67. Mouhieddine, T. H. et al. Risk factors for multiple sclerosis and associations with antiEBV antibody titers. Clin. Immunol. 158, 59–66 (2015). 68. Murray, T. J. The history of multiple sclerosis: the changing frame of the disease over the centuries. J. Neurol. Sci. 277 Suppl , S3–8 (2009). 69. Van Epps, H. L. Thomas Rivers and the EAE model. J. Exp. Med. 202, 4–4 (2005). 120 Literaturverzeichnis 70. Berkovich, R. & Agius, M. A. Mechanisms of action of ACTH in the management of relapsing forms of multiple sclerosis. Ther. Adv. Neurol. Disord. 7, 83–96 (2014). 71. Stöhr, M. Kortison-Stoßtherapie bei multipler Sklerose. (Springer-Verlag, 2013). at <https://books.google.com/books?id=jwTNBgAAQBAJ&pgis=1> 72. Brusaferri, F. & Candelise, L. Steriods for multiple sclerosis and optic neuritis: a metaanalysis of randomized controlled clinical trials. J. Neurol. 247, 435–442 (2000). 73. Sloka, J. S. & Stefanelli, M. The mechanism of action of methylprednisolone in the treatment of multiple sclerosis. Mult. Scler. 11, 425–32 (2005). 74. Deckx, N., Lee, W.-P., Berneman, Z. N. & Cools, N. Neuroendocrine immunoregulation in multiple sclerosis. Clin. Dev. Immunol. 2013, 705232 (2013). 75. Diener, H.-C. & Weimar, C. in Leitlinien für Diagnostik und Thrapie der Neurol. 1–52 (Thieme Verlag, 2014). at <http://www.dgn.org/images/red_leitlinien/LL_2012/pdf/03 0-050l_S2e_Multiple_Sklerose_Diagnostik_Therapie_2014-08_verlaengert.pdf> 76. McDonald, W. I. et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Ann. Neurol. 50, 121–7 (2001). 77. Polman, C. H. et al. Diagnostic criteria for multiple sclerosis: 2005 revisions to the ‘McDonald Criteria’. Ann. Neurol. 58, 840–6 (2005). 78. Polman, C. H. et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann. Neurol. 69, 292–302 (2011). 79. Courtney, A. M., Treadaway, K., Remington, G. & Frohman, E. Multiple sclerosis. Med. Clin. North Am. 93, 451–76, ix–x (2009). 80. Steinman, L. Multiple sclerosis: a two-stage disease. Nat. Immunol. 2, 762–4 (2001). 81. Confavreux, C. & Vukusic, S. Age at disability milestones in multiple sclerosis. Brain 129, 595–605 (2006). 82. Steinman, L. & Zamvil, S. S. Beginning of the end of two-stage theory purporting that inflammation then degeneration explains pathogenesis of progressive multiple sclerosis. Curr. Opin. Neurol. 29, 340–44 (2016). 83. Mahurkar, S., Suppiah, V. & O’Doherty, C. Pharmacogenomics of interferon beta and glatiramer acetate response: a review of the literature. Autoimmun. Rev. 13, 178–86 (2014). 121 Literaturverzeichnis 84. Johnson, K. P. et al. Copolymer 1 reduces relapse rate and improves disability in relapsing-remitting multiple sclerosis: results of a phase III multicenter, double-blind placebo-controlled trial. The Copolymer 1 Multiple Sclerosis Study Group. Neurology 45, 1268–76 (1995). 85. Ford, C. C. et al. A prospective open-label study of glatiramer acetate: over a decade of continuous use in multiple sclerosis patients. Mult. Scler. 12, 309–20 (2006). 86. Ford, C. et al. Continuous long-term immunomodulatory therapy in relapsing multiple sclerosis: results from the 15-year analysis of the US prospective open-label study of glatiramer acetate. Mult. Scler. 16, 342–50 (2010). 87. Johnson, K. P. et al. Extended use of glatiramer acetate (Copaxone) is well tolerated and maintains its clinical effect on multiple sclerosis relapse rate and degree of disability. Copolymer 1 Multiple Sclerosis Study Group. Neurology 50, 701–8 (1998). 88. The IFNB MS Study Group. Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial. The IFNB Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. Neurology 45, 1277–85 (1995). 89. The IFNB MS Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebocontrolled trial. The IFNB Multiple Sclerosis Study Group. Neurology 43, 655–61 (1993). 90. Mikol, D. D. et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): a multicentre, randomised, parallel, openlabel trial. Lancet. Neurol. 7, 903–14 (2008). 91. O’Connor, P. et al. 250 microg or 500 microg interferon beta-1b versus 20 mg glatiramer acetate in relapsing-remitting multiple sclerosis: a prospective, randomised, multicentre study. Lancet. Neurol. 8, 889–97 (2009). 92. Kuhle, J. et al. Conversion from clinically isolated syndrome to multiple sclerosis: A large multicentre study. Mult. Scler. 21, 1013–24 (2015). 93. O’Riordan, J. I. et al. The prognostic value of brain MRI in clinically isolated syndromes of the CNS. A 10-year follow-up. Brain 121 , Pt 3, 495–503 (1998). 122 Literaturverzeichnis 94. Steinman, L. Blocking adhesion molecules as therapy for multiple sclerosis: natalizumab. Nat. Rev. Drug Discov. 4, 510–8 (2005). 95. Polman, C. H. et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 354, 899–910 (2006). 96. Rudick, R. A. et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N. Engl. J. Med. 354, 911–23 (2006). 97. Hoepner, R., Faissner, S., Salmen, A., Gold, R. & Chan, A. Efficacy and side effects of natalizumab therapy in patients with multiple sclerosis. J. Cent. Nerv. Syst. Dis. 6, 41–9 (2014). 98. Kappos, L. et al. Natalizumab treatment for multiple sclerosis: updated recommendations for patient selection and monitoring. Lancet. Neurol. 10, 745–58 (2011). 99. Ruck, T., Bittner, S., Wiendl, H. & Meuth, S. Alemtuzumab in Multiple Sclerosis: Mechanism of Action and Beyond. Int. J. Mol. Sci. 16, 16414–16439 (2015). 100. Coles, A. J. et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet 380, 1829–39 (2012). 101. Chun, J. & Hartung, H.-P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 33, 91–101 102. Khatri, B. O. Fingolimod in the treatment of relapsing-remitting multiple sclerosis: longterm experience and an update on the clinical evidence. Ther. Adv. Neurol. Disord. 9, 130–47 (2016). 103. Keegan, M. et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 366, 579–82 (2005). 104. Bar-Or, A. et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, openlabel, phase I trial. Ann. Neurol. 63, 395–400 (2008). 105. Hauser, S. L. et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 358, 676–88 (2008). 106. Cross, A. H., Klein, R. S. & Piccio, L. Rituximab combination therapy in relapsing multiple sclerosis. Ther. Adv. Neurol. Disord. 5, 311–9 (2012). 123 Literaturverzeichnis 107. Chamberlain, N. et al. Rituximab does not reset defective early B cell tolerance checkpoints. J. Clin. Invest. 126, 282–7 (2016). 108. Dunant, N., Engels-Lange, U., Kráčala, Š., Rüppel, N. & Schmitt, C. Roche - Roche’s ocrelizumab first investigational medicine to show positive pivotal study results in both relapsing and primary progressive forms of multiple sclerosis. 29.10.2015 (2015). at <http://www.roche.com/media/store/releases/med-cor-2015-10-08.htm> 109. Kappos, L. et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 378, 1779–87 (2011). 110. Sorensen, P. S. et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology 82, 573–81 (2014). 111. Petersen, G., Wittmann, R., Arndt, V. & Göpffarth, D. Epidemiologie der Multiplen Sklerose in Deutschland Regionale Unterschiede und Versorgungsstruktur in Abrechnungsdaten der gesetzlichen Krankenversicherung. Nervenarzt 85, 990–8 (2014). 112. BARMER GEK Krankenkasse - Arzneimittelreport 2014. Online (2014). at <https://presse.barmer- gek.de/barmer/web/Portale/Presseportal/Subportal/Infothek/S tudien-und-Reports/Arzneimittelreport/Arzneimittelreport-2014/Arzneimittelreport2014.html> 113. Cheng, W. et al. Cross-reactivity of autoreactive T cells with MBP and viral antigens in patients with MS. Front. Biosci. 17, 1648–58 (2012). 114. Gabibov, A. G. et al. Combinatorial antibody library from multiple sclerosis patients reveals antibodies that cross-react with myelin basic protein and EBV antigen. FASEB J. 25, 4211–21 (2011). 115. Engelhardt, B. & Ransohoff, R. M. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. 26, 485–95 (2005). 116. Greter, M. et al. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 11, 328–34 (2005). 117. Sosa, R. A. & Forsthuber, T. G. The critical role of antigen-presentation-induced cytokine crosstalk in the central nervous system in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Interferon Cytokine Res. 31, 753–68 (2011). 118. Becher, B., Bechmann, I. & Greter, M. Antigen presentation in autoimmunity and CNS inflammation: how T lymphocytes recognize the brain. J. Mol. Med. 84, 532–43 (2006). 124 Literaturverzeichnis 119. Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–41 (2015). 120. Lucchinetti, C. F. et al. Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 365, 2188–97 (2011). 121. Van Oosten, B. W. et al. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MRmonitored phase II trial. Neurology 49, 351–7 (1997). 122. Beecham, A. H. et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 45, 1353–60 (2013). 123. Luchtman, D. W., Ellwardt, E., Larochelle, C. & Zipp, F. IL-17 and related cytokines involved in the pathology and immunotherapy of multiple sclerosis: Current and future developments. Cytokine Growth Factor Rev. 25, 403–13 (2014). 124. Rangachari, M. & Kuchroo, V. K. Using EAE to better understand principles of immune function and autoimmune pathology. J. Autoimmun. 45, 31–9 (2013). 125. Chu, C.-Q., Wittmer, S. & Dalton, D. K. Failure to Suppress the Expansion of the Activated Cd4 T Cell Population in Interferon γ–Deficient Mice Leads to Exacerbation of Experimental Autoimmune Encephalomyelitis. J. Exp. Med. 192, 123–128 (2000). 126. Becher, B., Durell, B. G. & Noelle, R. J. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J. Clin. Invest. 110, 493–7 (2002). 127. Oppmann, B. et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–25 (2000). 128. Aggarwal, S., Ghilardi, N., Xie, M.-H., de Sauvage, F. J. & Gurney, A. L. Interleukin23 Promotes a Distinct CD4 T Cell Activation State Characterized by the Production of Interleukin-17. J. Biol. Chem. 278, 1910–1914 (2002). 129. Hofstetter, H., Gold, R. & Hartung, H.-P. Th17 Cells in MS and Experimental Autoimmune Encephalomyelitis. Int. MS J. 16, 12–8 (2009). 130. Hofstetter, H. H. et al. Therapeutic efficacy of IL-17 neutralization in murine experimental autoimmune encephalomyelitis. Cell. Immunol. 237, 123–30 (2005). 131. Brucklacher-Waldert, V., Stuerner, K., Kolster, M., Wolthausen, J. & Tolosa, E. Phenotypical and functional characterization of T helper 17 cells in multiple sclerosis. Brain 132, 3329–41 (2009). 125 Literaturverzeichnis 132. Durelli, L. et al. T-helper 17 cells expand in multiple sclerosis and are inhibited by interferon-beta. Ann. Neurol. 65, 499–509 (2009). 133. Segal, B. M. et al. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet. Neurol. 7, 796–804 (2008). 134. Capolunghi, F., Rosado, M. M., Sinibaldi, M., Aranburu, A. & Carsetti, R. Why do we need IgM memory B cells? Immunol. Lett. 152, 114–120 (2013). 135. Crespo, M., Heidt, S., Redondo, D. & Pascual, J. Monitoring B cell subsets and alloreactivity in kidney transplantation. Transplant. Rev. 29, 45–52 (2015). 136. Pescovitz, M. D. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am. J. Transplant 6, 859–66 (2006). 137. Sanz, I., Wei, C., Lee, F. E.-H. & Anolik, J. Phenotypic and functional heterogeneity of human memory B cells. Semin. Immunol. 20, 67–82 (2008). 138. Jackson, S. M., Wilson, P. C., James, J. A. & Capra, J. D. Human B cell subsets. Adv. Immunol. 98, 151–224 (2008). 139. Klein, U., Rajewsky, K. & Kuppers, R. Human Immunoglobulin (Ig)M+IgD+ Peripheral Blood B Cells Expressing the CD27 Cell Surface Antigen Carry Somatically Mutated Variable Region Genes: CD27 as a General Marker for Somatically Mutated (Memory) B Cells. J. Exp. Med. 188, 1679–1689 (1998). 140. Agematsu, K., Hokibara, S., Nagumo, H. & Komiyama, A. CD27: a memory B-cell marker. Immunol. Today 21, 204–206 (2000). 141. Kaminski, D. A., Wei, C., Qian, Y., Rosenberg, A. F. & Sanz, I. Advances in human B cell phenotypic profiling. Front. Immunol. 3, 302 (2012). 142. Avery, D. T. et al. Increased Expression of CD27 on Activated Human Memory B Cells Correlates with Their Commitment to the Plasma Cell Lineage. J. Immunol. 174, 4034– 4042 (2005). 143. O’Connell, F. P., Pinkus, J. L. & Pinkus, G. S. CD138 (syndecan-1), a plasma cell marker immunohistochemical profile in hematopoietic and nonhematopoietic neoplasms. Am. J. Clin. Pathol. 121, 254–63 (2004). 126 Literaturverzeichnis 144. Maecker, H. T., McCoy, J. P. & Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 12, 191–200 (2012). 145. Baumgarth, N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 11, 34–46 (2011). 146. Griffin, D. O., Holodick, N. E. & Rothstein, T. L. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70-. J. Exp. Med. 208, 67–80 (2011). 147. Griffin, D. O. & Rothstein, T. L. Human B1 cell frequency: isolation and analysis of human B1 cells. Front. Immunol. 3, 122 (2012). 148. Covens, K. et al. Characterization of proposed human B-1 cells reveals pre-plasmablast phenotype. Blood 121, 5176–83 (2013). 149. Niino, M., Hirotani, M., Miyazaki, Y. & Sasaki, H. Memory and naïve B-cell subsets in patients with multiple sclerosis. Neurosci. Lett. 464, 74–8 (2009). 150. Knippenberg, S. et al. Reduction in IL-10 producing B cells (Breg) in multiple sclerosis is accompanied by a reduced naïve/memory Breg ratio during a relapse but not in remission. J. Neuroimmunol. 239, 80–6 (2011). 151. Tørring, C. et al. The B1-cell subpopulation is diminished in patients with relapsingremitting multiple sclerosis. J. Neuroimmunol. 262, 92–9 (2013). 152. Holmøy, T. The discovery of oligoclonal bands: a 50-year anniversary. Eur. Neurol. 62, 311–5 (2009). 153. Freedman, M. S. et al. Recommended standard of cerebrospinal fluid analysis in the diagnosis of multiple sclerosis: a consensus statement. Arch. Neurol. 62, 865–70 (2005). 154. Villar, L. M. et al. Intrathecal IgM synthesis predicts the onset of new relapses and a worse disease course in MS. Neurology 59, 555–9 (2002). 155. Joseph, F. G. et al. CSF oligoclonal band status informs prognosis in multiple sclerosis: a case control study of 100 patients. J. Neurol. Neurosurg. Psychiatry 80, 292–6 (2009). 156. Henderson, A. P. D., Barnett, M. H., Parratt, J. D. E. & Prineas, J. W. Multiple sclerosis: distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 66, 739–53 (2009). 157. Owens, G. P. et al. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid. Ann. Neurol. 65, 639–49 (2009). 127 Literaturverzeichnis 158. Linington, C., Engelhardt, B., Kapocs, G. & Lassman, H. Induction of persistently demyelinated lesions in the rat following the repeated adoptive transfer of encephalitogenic T cells and demyelinating antibody. J. Neuroimmunol. 40, 219–24 (1992). 159. Lucchinetti, C. et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47, 707–17 (2000). 160. Berger, T. et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N. Engl. J. Med. 349, 139–45 (2003). 161. Tomassini, V. et al. Anti-myelin antibodies predict the clinical outcome after a first episode suggestive of MS. Mult. Scler. 13, 1086–94 (2007). 162. Pelayo, R. et al. Antimyelin antibodies with no progression to multiple sclerosis. N. Engl. J. Med. 356, 426–8 (2007). 163. Lim, E. T. et al. Anti-myelin antibodies do not allow earlier diagnosis of multiple sclerosis. Mult. Scler. 11, 492–4 (2005). 164. Wootla, B. et al. Naturally Occurring Monoclonal Antibodies and Their Therapeutic Potential for Neurologic Diseases. JAMA Neurol. 72, 1346–53 (2015). 165. Hamanova, M., Chmelikova, M., Nentwich, I., Thon, V. & Lokaj, J. Anti-Gal IgM, IgA and IgG natural antibodies in childhood. Immunol. Lett. 164, 40–3 (2015). 166. Sarma, J. V. & Ward, P. A. The complement system. Cell Tissue Res. 343, 227–35 (2011). 167. Piddlesden, S. J., Lassmann, H., Zimprich, F., Morgan, B. P. & Linington, C. The demyelinating potential of antibodies to myelin oligodendrocyte glycoprotein is related to their ability to fix complement. Am. J. Pathol. 143, 555–64 (1993). 168. Vidarsson, G., Dekkers, G. & Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 5, (2014). 169. Palanichamy, A. et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J. Immunol. 193, 580–6 (2014). 170. Molnarfi, N. et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 210, 2921–37 (2013). 128 Literaturverzeichnis 171. Rivera, A., Chen, C. C., Ron, N., Dougherty, J. P. & Ron, Y. Role of B cells as antigenpresenting cells in vivo revisited: antigen-specific B cells are essential for T cell expansion in lymph nodes and for systemic T cell responses to low antigen concentrations. Int. Immunol. 13, 1583–93 (2001). 172. Harp, C. T., Lovett-Racke, A. E., Racke, M. K., Frohman, E. M. & Monson, N. L. Impact of myelin-specific antigen presenting B cells on T cell activation in multiple sclerosis. Clin. Immunol. 128, 382–91 (2008). 173. Bar-Or, A. et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann. Neurol. 67, 452–61 (2010). 174. Harp, C. T. et al. Memory B cells from a subset of treatment-naïve relapsing-remitting multiple sclerosis patients elicit CD4(+) T-cell proliferation and IFN-γ production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur. J. Immunol. 40, 2942–56 (2010). 175. Duddy, M. et al. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J. Immunol. 178, 6092–9 (2007). 176. Genç, K., Dona, D. L. & Reder, A. T. Increased CD80(+) B cells in active multiple sclerosis and reversal by interferon beta-1b therapy. J. Clin. Invest. 99, 2664–71 (1997). 177. Serafini, B., Rosicarelli, B., Magliozzi, R., Stigliano, E. & Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 14, 164–74 (2004). 178. Magliozzi, R. et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann. Neurol. 68, 477–93 (2010). 179. Howell, O. W. et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 134, 2755–71 (2011). 180. Baranzini, S. E. et al. B cell repertoire diversity and clonal expansion in multiple sclerosis brain lesions. J. Immunol. 163, 5133–44 (1999). 181. Magliozzi, R. et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 130, 1089–104 (2007). 182. Weyand, C. M. & Goronzy, J. J. Ectopic germinal center formation in rheumatoid synovitis. Ann. N. Y. Acad. Sci. 987, 140–9 (2003). 129 Literaturverzeichnis 183. Aloisi, F. & Pujol-Borrell, R. Lymphoid neogenesis in chronic inflammatory diseases. Nat. Rev. Immunol. 6, 205–17 (2006). 184. Housley, W. J., Pitt, D. & Hafler, D. A. Biomarkers in multiple sclerosis. Clin. Immunol. 161, 51–8 (2015). 185. Li, D. K. B. et al. MRI T2 lesion burden in multiple sclerosis: a plateauing relationship with clinical disability. Neurology 66, 1384–9 (2006). 186. Kuhle, J. et al. Serum neurofilament light chain in early relapsing remitting MS is increased and correlates with CSF levels and with MRI measures of disease severity. Mult. Scler. (2016). doi:10.1177/1352458515623365 187. Kuhle, J. et al. Lack of association between antimyelin antibodies and progression to multiple sclerosis. N. Engl. J. Med. 356, 371–8 (2007). 188. Srivastava, R. et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N. Engl. J. Med. 367, 115–23 (2012). 189. Wunsch, M., Rovituso, D. M. & Kuerten, S. KIR4.1 Antibodies as Biomarkers in Multiple Sclerosis. Front. Neurol. 5, 62 (2014). 190. Nerrant, E. et al. Lack of confirmation of anti-inward rectifying potassium channel 4.1 antibodies as reliable markers of multiple sclerosis. Mult. Scler. 20, 1699–703 (2014). 191. Brickshawana, A. et al. Investigation of the KIR4.1 potassium channel as a putative antigen in patients with multiple sclerosis: a comparative study. Lancet. Neurol. 13, 795– 806 (2014). 192. Kuerten, S. et al. Identification of a B cell-dependent subpopulation of multiple sclerosis by measurements of brain-reactive B cells in the blood. Clin. Immunol. 152, 20–4 (2014). 193. Hohmann, C. et al. Categorization of multiple sclerosis relapse subtypes by B cell profiling in the blood. Acta Neuropathol. Commun. 2, 138 (2014). 194. Rovituso, D. M. et al. The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsing-remitting multiple sclerosis patients. Sci. Rep. 5, 14265 (2015). 195. Disanto, G., Morahan, J. M., Barnett, M. H., Giovannoni, G. & Ramagopalan, S. V. The evidence for a role of B cells in multiple sclerosis. Neurology 78, 823–32 (2012). 196. Höfler, M. The Bradford Hill considerations on causality: a counterfactual perspective. Emerg. Themes Epidemiol. 2, 11 (2005). 130 Literaturverzeichnis 197. Davenport, R. D. & Keren, D. F. Oligoclonal bands in cerebrospinal fluids: significance of corresponding bands in serum for diagnosis of multiple sclerosis. Clin. Chem. 34, 764–5 (1988). 198. Haas, J. et al. Hypovitaminosis D upscales B-cell immunoreactivity in multiple sclerosis. J. Neuroimmunol. 294, 18–26 (2016). 199. Edwards, J. C. W. & Cambridge, G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat. Rev. Immunol. 6, 394–403 (2006). 200. Pinna, D., Corti, D., Jarrossay, D., Sallusto, F. & Lanzavecchia, A. Clonal dissection of the human memory B-cell repertoire following infection and vaccination. Eur. J. Immunol. 39, 1260–70 (2009). 201. Suchanek, O., Sadler, R., Bateman, E. A., Patel, S. Y. & Ferry, B. L. Immunophenotyping of putative human B1 B cells in healthy controls and common variable immunodeficiency (CVID) patients. Clin. Exp. Immunol. 170, 333–41 (2012). 202. Ireland, S. J. et al. The effect of glatiramer acetate therapy on functional properties of B cells from patients with relapsing-remitting multiple sclerosis. JAMA Neurol. 71, 1421– 8 (2014). 203. Varkony, H. et al. The glatiramoid class of immunomodulator drugs. Expert Opin. Pharmacother. 10, 657–68 (2009). 204. Sellebjerg, F. et al. Glatiramer acetate antibodies, gene expression and disease activity in multiple sclerosis. Mult. Scler. 18, 305–13 (2012). 205. Farina, C. et al. Treatment with glatiramer acetate induces specific IgG4 antibodies in multiple sclerosis patients. J. Neuroimmunol. 123, 188–92 (2002). 206. Haas, J. et al. B cells undergo unique compartmentalized redistribution in multiple sclerosis. J. Autoimmun. 37, 289–99 (2011). 207. Ramgolam, V. S. et al. B cells as a therapeutic target for IFN-β in relapsing-remitting multiple sclerosis. J. Immunol. 186, 4518–26 (2011). 208. Schubert, R. D. et al. IFN-β treatment requires B cells for efficacy in neuroautoimmunity. J. Immunol. 194, 2110–6 (2015). 209. Skarica, M., Eckstein, C., Whartenby, K. A. & Calabresi, P. A. Novel mechanisms of immune modulation of natalizumab in multiple sclerosis patients. J. Neuroimmunol. 235, 70–6 (2011). 131 Literaturverzeichnis 210. Krumbholz, M., Meinl, I., Kümpfel, T., Hohlfeld, R. & Meinl, E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology 71, 1350–4 (2008). 211. Planas, R., Jelčić, I., Schippling, S., Martin, R. & Sospedra, M. Natalizumab treatment perturbs memory- and marginal zone-like B-cell homing in secondary lymphoid organs in multiple sclerosis. Eur. J. Immunol. 42, 790–8 (2012). 212. Zanotti, C. et al. Peripheral accumulation of newly produced T and B lymphocytes in natalizumab-treated multiple sclerosis patients. Clin. Immunol. 145, 19–26 (2012). 213. Griffin, D. O. & Rothstein, T. L. Human ‘orchestrator’ CD11b(+) B1 cells spontaneously secrete interleukin-10 and regulate T-cell activity. Mol. Med. 18, 1003–8 (2012). 214. Prosperini, L., Gallo, V., Petsas, N., Borriello, G. & Pozzilli, C. One-year MRI scan predicts clinical response to interferon beta in multiple sclerosis. Eur. J. Neurol. 16, 1202–9 (2009). 215. Valenzuela, R. M. et al. Clinical response to glatiramer acetate correlates with modulation of IFN-gamma and IL-4 expression in multiple sclerosis. Mult. Scler. 13, 754–62 (2007). 216. Karussis, D., Teitelbaum, D., Sicsic, C. & Brenner, T. Long-term treatment of multiple sclerosis with glatiramer acetate: natural history of the subtypes of anti-glatiramer acetate antibodies and their correlation with clinical efficacy. J. Neuroimmunol. 220, 125–130 (2010). 217. Aalberse, R. C., van der Gaag, R. & van Leeuwen, J. Serologic aspects of IgG4 antibodies. I. Prolonged immunization results in an IgG4-restricted response. J. Immunol. 130, 722–6 (1983). 218. Aalberse, R. C., Stapel, S. O., Schuurman, J. & Rispens, T. Immunoglobulin G4: an odd antibody. Clin. Exp. Allergy 39, 469–77 (2009). 219. Brenner, T. et al. Humoral and cellular immune responses to Copolymer 1 in multiple sclerosis patients treated with Copaxone. J. Neuroimmunol. 115, 152–60 (2001). 220. Lalive, P. H. et al. Glatiramer acetate in the treatment of multiple sclerosis: emerging concepts regarding its mechanism of action. CNS Drugs 25, 401–14 (2011). 221. Scalfari, A. et al. The natural history of multiple sclerosis: a geographically based study 10: relapses and long-term disability. Brain 133, 1914–29 (2010). 132 Literaturverzeichnis 222. Weinshenker, B. G. et al. The natural history of multiple sclerosis: a geographically based study. 2. Predictive value of the early clinical course. Brain 112 (Pt 6), 1419–28 (1989). 223. Leray, E. et al. Evidence for a two-stage disability progression in multiple sclerosis. Brain 133, 1900–13 (2010). 224. Magliozzi, R., Columba-Cabezas, S., Serafini, B. & Aloisi, F. Intracerebral expression of CXCL13 and BAFF is accompanied by formation of lymphoid follicle-like structures in the meninges of mice with relapsing experimental autoimmune encephalomyelitis. J. Neuroimmunol. 148, 11–23 (2004). 225. Corcione, A. et al. B-cell differentiation in the CNS of patients with multiple sclerosis. Autoimmun. Rev. 4, 549–54 (2005). 226. Neyt, K., Perros, F., GeurtsvanKessel, C. H., Hammad, H. & Lambrecht, B. N. Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol. 33, 297–305 (2012). 227. Ruddle, N. H. Lymphatic vessels and tertiary lymphoid organs. J. Clin. Invest. 124, 953– 9 (2014). 228. Kuerten, S. et al. MBP-PLP fusion protein-induced EAE in C57BL/6 mice. J. Neuroimmunol. 177, 99–111 (2006). 229. Kuerten, S. et al. Myelin-reactive antibodies mediate the pathology of MBP-PLP fusion protein MP4-induced EAE. Clin. Immunol. 140, 54–62 (2011). 230. Hundgeburth, L. C. et al. The complement system contributes to the pathology of experimental autoimmune encephalomyelitis by triggering demyelination and modifying the antigen-specific T and B cell response. Clin. Immunol. 146, 155–64 (2013). 231. Kuerten, S. et al. Tertiary lymphoid organ development coincides with determinant spreading of the myelin-specific T cell response. Acta Neuropathol. 124, 861–73 (2012). 232. Kuerten, S., Javeri, S., Tary-Lehmann, M., Lehmann, P. V & Angelov, D. N. Fundamental differences in the dynamics of CNS lesion development and composition in MP4- and MOG peptide 35-55-induced experimental autoimmune encephalomyelitis. Clin. Immunol. 129, 256–67 (2008). 233. Batoulis, H. et al. Central nervous system infiltrates are characterized by features of ongoing B cell-related immune activity in MP4-induced experimental autoimmune encephalomyelitis. Clin. Immunol. 158, 47–58 (2015). 133 Literaturverzeichnis 234. Lobo, E. O., Zhang, Z. & Shively, J. E. Pivotal advance: CEACAM1 is a negative coreceptor for the B cell receptor and promotes CD19-mediated adhesion of B cells in a PI3K-dependent manner. J. Leukoc. Biol. 86, 205–18 (2009). 235. Gray-Owen, S. D. & Blumberg, R. S. CEACAM1: contact-dependent control of immunity. Nat. Rev. Immunol. 6, 433–46 (2006). 236. Khairnar, V. et al. CEACAM1 induces B-cell survival and is essential for protective antiviral antibody production. Nat. Commun. 6, 6217 (2015). 237. Klaile, E. et al. The CEACAM1 N-terminal Ig domain mediates cis- and trans-binding and is essential for allosteric rearrangements of CEACAM1 microclusters. J. Cell Biol. 187, 553–67 (2009). 238. Palanichamy, A. et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci. Transl. Med. 6, 248ra106 (2014). 239. Gonzalez, S. F., Jayasekera, J. P. & Carroll, M. C. Complement and natural antibody are required in the long-term memory response to influenza virus. Vaccine 26, I86–I93 (2008). 240. Quách, T. D. et al. Distinctions among Circulating Antibody-Secreting Cell Populations, Including B-1 Cells, in Human Adult Peripheral Blood. J. Immunol. 196, 1060–9 (2016). 241. Ochsenbein, A. F. et al. Control of early viral and bacterial distribution and disease by natural antibodies. Science 286, 2156–9 (1999). 242. Rapaka, R. R. et al. Conserved natural IgM antibodies mediate innate and adaptive immunity against the opportunistic fungus Pneumocystis murina. J. Exp. Med. 207, 2907–19 (2010). 243. Kaveri, S. V, Silverman, G. J. & Bayry, J. Natural IgM in immune equilibrium and harnessing their therapeutic potential. J. Immunol. 188, 939–45 (2012). 244. Chen, Y., Park, Y.-B., Patel, E. & Silverman, G. J. IgM antibodies to apoptosisassociated determinants recruit C1q and enhance dendritic cell phagocytosis of apoptotic cells. J. Immunol. 182, 6031–43 (2009). 245. Notley, C. A., Brown, M. A., Wright, G. P. & Ehrenstein, M. R. Natural IgM is required for suppression of inflammatory arthritis by apoptotic cells. J. Immunol. 186, 4967–72 (2011). 134 Literaturverzeichnis 246. Ehrenstein, M. R., Cook, H. T. & Neuberger, M. S. Deficiency in Serum Immunoglobulin (Ig)m Predisposes to Development of Igg Autoantibodies. J. Exp. Med. 191, 1253–1258 (2000). 247. Nguyen, T. T. T., Elsner, R. A. & Baumgarth, N. Natural IgM prevents autoimmunity by enforcing B cell central tolerance induction. J. Immunol. 194, 1489–502 (2015). 248. Lang, W., Rodriguez, M., Lennon, V. A. & Lampert, P. W. Demyelination and remyelination in murine viral encephalomyelitis. Ann. N. Y. Acad. Sci. 436, 98–102 (1984). 249. Lampasona, V. et al. Similar low frequency of anti-MOG IgG and IgM in MS patients and healthy subjects. Neurology 62, 2092–2094 (2004). 250. Kraljevic, K., Wong, S. & Fulcher, D. A. Circulating phenotypic B-1 cells are decreased in common variable immunodeficiency and correlate with immunoglobulin M levels. Clin. Exp. Immunol. 171, 278–82 (2013). 251. Wu, Y.-Y. et al. Concordance of increased B1 cell subset and lupus phenotypes in mice and humans is dependent on BLK expression levels. J. Immunol. 194, 5692–702 (2015). 252. Khademi, M. et al. Cerebrospinal fluid CXCL13 in multiple sclerosis: a suggestive prognostic marker for the disease course. Mult. Scler. 17, 335–43 (2011). 253. Sellebjerg, F. et al. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology 73, 2003–10 (2009). 254. Breij, E. C. W. et al. Homogeneity of active demyelinating lesions in established multiple sclerosis. Ann. Neurol. 63, 16–25 (2008). 255. Barnett, M. H. & Prineas, J. W. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol. 55, 458–68 (2004). 256. Metz, I. et al. Pathologic heterogeneity persists in early active multiple sclerosis lesions. Ann. Neurol. (2014). doi:10.1002/ana.24163 257. Krumbholz, M. et al. Interferon-beta increases BAFF levels in multiple sclerosis: implications for B cell autoimmunity. Brain 131, 1455–63 (2008). 135 Lebenslauf Lebenslauf Geburtstag: 27. September, 1976 Geburtsort: Alimena (Italien) Nationalität: italienisch Beruflicher Werdegang seit 08/2013 Wissenschaftlicher Mitarbeiter am Institut I für Anatomie (Julius-Maximilians-Universität Würzburg) Assistent im Kursus der mikroskopischen Anatomie SS14, SS15 der makroskopischen Anatomie WS13/14 03/2012-07/2013 Wissenschaftlicher Mitarbeiter am Institut I für Anatomie (Universität zu Köln) Assistent im Kursus der mikroskopischen Anatomie SS12, SS13 der makroskopischen Anatomie WS12/13 i Lebenslauf Lebenslauf Ausbildung seit 08/2013 Doktorand in der Arbeitsgruppe von Prof. Dr. S. Kürten am Institut I für Anatomie (Julius-Maximilians-Universität Würzburg) Promotionsstudent an der Graduiertenschule Graduate School Life Science an der Julius-MaximiliansUniversität, Würzburg 10/2012-07/2013 Promotionsstudent im Interdisziplinären Promotionsstudiengang Molekulare Medizin der Universität zu Köln (IPMM) 03/2012-07/2013 Doktorand in der Arbeitsgruppe von PD Dr. S. Kürten am Institut I für Anatomie (Universität zu Köln) 11/2011 Diplom (1,6) 01/2011-10/2011 Diplomarbeit in der Arbeitsgruppe Neuroimmunologie (Universitätsklinikum Greifswald) “Mechanismen der Immunmodulation durch Cladribin: Phänotypische und Funktionelle Charakterisierung überlebender Lymphozytenpopulationen“ ii Lebenslauf Lebenslauf Ausbildung 10/2008-10/2011 Hauptstudium der Humanbiologie an der Ernst-Moritz-Arndt Universität, Greifswald Hauptfächer Immunologie, Biochemie des Menschen, Molekulare Mikrobiologie 10/2006-09/2008 Grundstudium der Humanbiologie an der Ernst-Moritz-Arndt Universität, Greifswald 06/2005 Abitur (2,1) 09/2001-07/2005 Berlin-Kolleg Extrakurrikulare Aktivitäten 09/2014-10/2014 Praktikum „ Optimization of a murine IgG B cell ELISPOT assay “ Cellular Technology Limited (CTL), Shaker Heights, Ohio, USA 11/2008-11/2010 Hilfswissenschaftliche Tätigkeit in der Arbeitsgruppe Neuroimmunologie (Universitätsklinikum Greifswald) “Schlaganfall in der DBH-/- Maus” iii Lebenslauf Lebenslauf Extrakurrikulare Aktivitäten 08/2009-10/2009 Praktikum in der Arbeitsgruppe von Prof. Dr. Radbruch Rheumaforschungszentrum Berlin “Optimierung der Transfektion und Transduktion für die Expression von miRNA in T-Helferzellen“ Deutsch Fließend Italienisch Muttersprache Englisch Fließend Französisch Grundkenntnisse Datum, Ort Damiano Mario Rovituso iv Publikationsliste Publikationsliste 1. Hundgeburth, L. C., Wunsch, M., Rovituso, D. M., Recks, M.S., Addicks, K., Lehmann, P. V., et al. The complement system contributes to the pathology of experimental autoimmune encephalomyelitis by triggering demyelination and modifying the antigenspecific T and B cell response. Clin. Immunol. 146, 155–64 (2013) 2. Kuerten, S., Pommerschein, G., Barth, S. K., Hohmann, C., Milles, B., Sammer, F.W., Duffy, C. E., Wunsch, M., Rovituso, D. M., Schroeter, M., Addicks, K., Kaiser, C. C., Lehmann, P. V. Identification of a B cell-dependent subpopulation of multiple sclerosis by measurements of brain-reactive B cells in the blood. Clin. Immunol. 152, 20–4 (2014) 3. Wunsch, M., Rovituso, D. M., Kuerten, S. KIR4.1 Antibodies as Biomarkers in Multiple Sclerosis. Front. Neurol. 5, 62 (2014) 4. Rovituso, D. M., Heller, S., Schroeter, M., Kleinschnitz, C., Kuerten, S. B1 cells are unaffected by immune modulatory treatment in remitting-relapsing multiple sclerosis patients. J. Neuroimmunol. 272, 86–90 (2014) 5. Rovituso, D. M., Duffy, C. E., Schroeter, M., Kaiser, C. C., Kleinschnitz, C., Bayas, A., et al. The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsing-remitting multiple sclerosis patients. Sci. Rep. 5, 14265 (2015). v Eidesstattliche Erklärung Eidesstattliche Erklärung Hiermit erkläre ich an Eides statt, die Dissertation „Die Rolle der autoreaktiven B-Zellen und Autoantikörper in der Pathophysiologie der Multiplen Sklerose“ eigenständig, d. h. insbesondere selbständig und ohne Hilfe eines kommerziellen Promotionsberaters, angefertigt und keine anderen als die von mir angegebenen Quellen und Hilfsmittel verwendet zu haben. Ich erkläre außerdem, dass die Dissertation weder in gleicher noch in ähnlicher Form bereits in einem anderen Prüfungsverfahren vorgelegen hat. Ort, Datum Unterschrift vi Affidavit Affidavit I hereby confirm that my thesis entitled “The role of autoreactive B cells and autoantibodies in the pathophysiology of multiple sclerosis” is the result of my own work. I did not receive any help or support from commercial consultants. All sources and/or materials applied are listed and specified in the thesis. Furthermore, I confirm that this thesis has not yet been submitted as part of another examination process neither in identical nor in similar from. Place, Date Signature vii Erklärung zu Eigenanteilen an Publikationen und Zweitpublikationsrechten „Dissertation unter Einschluss mehrerer publizierter Manuskripte“ in der GSLS – Erklärung zu Eigenanteilen an Publikationen und Zweitpublikationsrechten Publikation (Vollständiges Zitat): D. Rovituso, S. Heller, M. Schroeter, C. Kleinschnitz, and S. Kuerten, “B1 cells are unaffected by immune modulatory treatment in remitting-relapsing multiple sclerosis patients” J. Neuroimmunol., vol. 272, no. 1–2, pp. 86–90, Apr. 2014. Autoren-Initialen, Verantwortlichkeit abnehmend von links nach rechts Beteiligt an Planung der Untersuchungen DR SK SH MS CK Datenerhebung DR SK SH MS CK Daten-Analyse und Interpretation DR SK SH MS CK Schreiben des Manuskripts DR SK SH MS CK Publikation (Vollständiges Zitat): D. M. Rovituso, C. E. Duffy, M. Schroeter, C. C. Kaiser, C. Kleinschnitz, A. Bayas, R. Elsner, and S. Kuerten, “The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsingremitting multiple sclerosis patients.”, Sci. Rep., vol. 5, p. 14265, Jan. 2015. Autoren-Initialen, Verantwortlichkeit abnehmend von links nach rechts Beteiligt an Planung der Untersuchungen SK DR CED MS AB/CCK/RE Datenerhebung DR SK CED MS AB/CCK/RE Daten-Analyse und Interpretation DR SK CED MS AB/CCK/RE Schreiben des Manuskripts DR SK CED MS AB/CCK/RE Für alle in dieser „Dissertation unter Einschluss mehrerer publizierter Manuskripte“ verwendeten Manuskripte liegen die notwendigen Genehmigungen der Verlage und Co-Autoren für die Zweitpublikation vor. Mit meiner Unterschrift bestätige ich die Kenntnisnahme und das Einverständnis meines direkten Betreuers. Damiano M. Rovituso __________________________________________________________________________________________ Name Doktorand Datum, Ort Unterschrift viii Erklärung zu Eigenanteilen an Publikationen und Zweitpublikationsrechten „Dissertation unter Einschluss mehrerer publizierter Manuskripte“ in der GSLS – Erklärung zu Eigenanteilen an Publikationen und Zweitpublikationsrechten Publikation (Vollständiges Zitat): D. Rovituso, L. Scheffler, M. Wunsch, C. Kleinschnitz, S. Dörck, J. Ulzheimer, A. Bayas, L. Steinman, S. Ergün, and S. Kürten, “CEACAM1 mediates B cell aggregation and tolerance induction in central nervous system autoimmunity” Sci. Rep. (submitted April 2016) Autoren-Initialen, Verantwortlichkeit abnehmend von links nach rechts Beteiligt an Planung der Untersuchungen SK DR/LS MW Datenerhebung DR/LS SK MW JU AB/CCK/RE Daten-Analyse und Interpretation DR/LS SK MW AB SE Schreiben des Manuskripts SK/DR LS MW AB SE Für alle in dieser „Dissertation unter Einschluss mehrerer publizierter Manuskripte“ verwendeten Manuskripte liegen die notwendigen Genehmigungen der Verlage und Co-Autoren für die Zweitpublikation vor. Mit meiner Unterschrift bestätige ich die Kenntnisnahme und das Einverständnis meines direkten Betreuers. Damiano M. Rovituso __________________________________________________________________________________________ Name Doktorand Datum, Ort Unterschrift ix Statement of individual author contributions and of legal second publication rights “Dissertation Based on Several Published Manuscripts“ Statement of individual author contributions and of legal second publication rights Publication (complete reference): D. Rovituso, S. Heller, M. Schroeter, C. Kleinschnitz, and S. Kuerten, “B1 cells are unaffected by immune modulatory treatment in remitting-relapsing multiple sclerosis patients,” J. Neuroimmunol., vol. 272, no. 1–2, pp. 86–90, Apr. 2014. Participated in Author Initials, Responsibility decreasing from left to right Study Design Methods Development DR SK SH MS CK Data Collection DR SK SH MS CK Data Analysis and Interpretation DR SK SH MS CK Manuscript Writing Writing of Introduction Writing of Materials & Methods Writing of Discussion Writing of First Draft DR SK SH MS CK The doctoral researcher confirms that she/he has obtained permission from both the publishers and the co-authors for legal second publication. The doctoral researcher and the primary supervisor confirm the correctness of the above mentioned assessment. Damian M. Rovituso ___________________________________________________________________________ Doctoral Researcher’s Name Date, Place Signature Stefanie Kürten ___________________________________________________________________________ Primary Supervisor’s Name Date, Place Signature x Statement of individual author contributions and of legal second publication rights “Dissertation Based on Several Published Manuscripts“ Statement of individual author contributions and of legal second publication rights Publication (complete reference): D. M. Rovituso, C. E. Duffy, M. Schroeter, C. C. Kaiser, C. Kleinschnitz, A. Bayas, R. Elsner, and S. Kuerten, “The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsingremitting multiple sclerosis patients.”, Sci. Rep., vol. 5, p. 14265, Jan. 2015. Author Initials, Responsibility decreasing from left to right Participated in Study Design Methods Development SK DR CED MS AB/CCK/RE Data Collection DR SK CED MS AB/CCK/RE Data Analysis and Interpretation DR SK CED MS AB/CCK/RE Manuscript Writing Writing of Introduction Writing of Materials & Methods Writing of Discussion Writing of First Draft DR SK CED MS AB/CCK/RE The doctoral researcher confirms that she/he has obtained permission from both the publishers and the co-authors for legal second publication. The doctoral researcher and the primary supervisor confirm the correctness of the above mentioned assessment. Damian M. Rovituso ___________________________________________________________________________ Doctoral Researcher’s Name Date, Place Signature Stefanie Kürten ___________________________________________________________________________ Primary Supervisor’s Name Date, Place Signature xi Statement of individual author contributions and of legal second publication rights “Dissertation Based on Several Published Manuscripts“ Statement of individual author contributions and of legal second publication rights Publication (complete reference): D. Rovituso, L. Scheffler, M. Wunsch, C. Kleinschnitz, S. Dörck, J. Ulzheimer, A. Bayas, L. Steinman, S. Ergün, and S. Kürten, “CEACAM1 mediates B cell aggregation and tolerance induction in central nervous system autoimmunity”, Sci. Rep. (submitted April 2016) Participated in Study Design Methods Development Author Initials, Responsibility decreasing from left to right SK DR/LS MW Data Collection DR/LS SK MW JU AB/CK/SD Data Analysis and Interpretation DR/LS SK MW AB SE Manuscript Writing Writing of Introduction Writing of Materials & Methods Writing of Discussion Writing of First Draft SK/DR LS MW AB SE The doctoral researcher confirms that she/he has obtained permission from both the publishers and the co-authors for legal second publication. The doctoral researcher and the primary supervisor confirm the correctness of the above mentioned assessment. Damian M. Rovituso ___________________________________________________________________________ Doctoral Researcher’s Name Date, Place Signature Stefanie Kürten ___________________________________________________________________________ Primary Supervisor’s Name Date, Place Signature xii Statement of individual author contributions to figures/tables/chapters included in the manuscripts “Dissertation Based on Several Published Manuscripts“ Statement of individual author contributions to figures/tables/chapters included in the manuscripts Publication (complete reference): D. Rovituso, S. Heller, M. Schroeter, C. Kleinschnitz, and S. Kuerten, “B1 cells are unaffected by immune modulatory treatment in remitting-relapsing multiple sclerosis patients,” J. Neuroimmunol., vol. 272, no. 1–2, pp. 86–90, Apr. 2014. Figure Author Initials, Responsibility decreasing from left to right 1 DR SK 2 DR SK 3 DR SK Table 1 Author Initials, Responsibility decreasing from left to right DR SK Publication (complete reference): D. M. Rovituso, C. E. Duffy, M. Schroeter, C. C. Kaiser, C. Kleinschnitz, A. Bayas, R. Elsner, and S. Kuerten, “The brain antigen-specific B cell response correlates with glatiramer acetate responsiveness in relapsingremitting multiple sclerosis patients.,” Sci. Rep., vol. 5, p. 14265, Jan. 2015. Figure Author Initials, Responsibility decreasing from left to right 1 DR SK 2 DR SK 3 DR SK Table 1 Author Initials, Responsibility decreasing from left to right DR SK I also confirm my primary supervisor’s acceptance. Damiano M. Rovituso ___________________________________________________________________________ Doctoral Researcher’s Name Date, Place Signature xiii Statement of individual author contributions to figures/tables/chapters included in the manuscripts “Dissertation Based on Several Published Manuscripts“ Statement of individual author contributions to figures/tables/chapters included in the manuscripts Publication (complete reference): D. Rovituso, L. Scheffler, M. Wunsch, C. Kleinschnitz, S. Dörck, J. Ulzheimer, A. Bayas, L. Steinman, S. Ergün, and S. Kürten, “CEACAM1 mediates B cell aggregation and tolerance induction in central nervous system autoimmunity”, Sci. Rep. (submitted April 2016) Figure Author Initials, Responsibility decreasing from left to right 1 SK LS 2 LS SK 3 DR SK 4 MW SK 5 LS SK Suppl.Figure Author Initials, Responsibility decreasing from left to right 1 SK 2 LS 3 DR 4 DR 5 DR 6 DR Suppl. Table MW Author Initials, Responsibility decreasing from left to right 1-4, 6 DR 5 MW DR I also confirm my primary supervisor’s acceptance. Damiano M. Rovituso ___________________________________________________________________________ Doctoral Researcher’s Name Date, Place Signature xiv Danksagung Danksagung Ich möchte mich bei allen bedanken, die mich während meiner Promotion begleitet und unterstützt haben. Mein besonderer Dank gilt Frau Prof. Stefanie Kürten für die Möglichkeit in ihrer Arbeitsgruppe meine Arbeit anzufertigen. Ihre Begeisterung und ihr Fachwissen haben eine exzellente Arbeitsatmosphäre geschaffen. Ich bin dankbar, dass ich in einem Umfeld arbeiten durfte, in dem ich mich frei entfalten konnte. Ich möchte an dieser Stelle auch Prof. Thomas Hünig danken, der immer großes Interesse an meiner Arbeit gezeigt hat. Seine Seminarreihe „Immunmodulation“ hat mich immer wieder daran erinnert, was für ein Glück es ist, diesen beruflichen Weg zu gehen. Prof. Paul Lehmann möchte ich nicht nur für seine Betreuung in den letzten Jahren danken, sondern auch für die einmalige Gelegenheit bei ihm ein Praktikum absolvieren zu können. Seine Begeisterung für die Wissenschaft hat mich stets inspiriert. Für die Zusammenarbeit an den drei Manuskripten, die über die Jahre entstanden sind, möchte ich mich im Besonderen bei meinen Kollegen bedanken, die mit mir gearbeitet, gelacht und gelitten haben: Stefanie Heller, Cathrina E. Duffy, Laura Scheffler, Marie Wunsch und besonders Sandra Lauer-Schmalzt, die mich auch privat erträgt. Alla Ganscher und Eleonora Maier möchte ich danken, dass sie mir bei der Durchführung meiner Experimente geholfen haben, ohne die meine Arbeit nicht vollständig wäre. Meine Arbeit wäre nicht möglich gewesen ohne Patienten, die freiwillig bereit waren an meinen Studien teilzunehmen. Für den Patientenkontakt und die Hilfe bei der Rekrutierung ein großes Danke an Prof. Dr. Micheal Schroeter aus Köln, Prof. Dr. Mathias Mäurer aus Bad Mergentheim, Prof. Dr. Friedemann Paul aus Berlin, Prof. Dr. Christoph Kleinschnitz aus Würzburg und Prof. Dr. Markus Naumann aus Augsburg. Nur durch ihre Unterstützung konnte ich meine Arbeit beenden. Dr. med. Antonios Bayas möchte ich sehr danken, dass er immer Zeit und motivierende Worte für meine Arbeit fand. Ihm und seinem Team in Augsburg möchte ich danken, dass sie das Unmögliche möglich gemacht haben: Frau Stefanie Denkel und Frau Dorota Wojcieszek. Mein Dank geht aber auch an alle anderen Kollegen in meiner Arbeitsgruppe und den Kollegen aus der Arbeitsgruppe Edenhofer für die tollen Diskussionen, die Feste und für so viele lustige Momente. Danke für die unglaublich schöne Zeit mit euch! Für die liebe Hilfe bei den Korrekturen möchte ich mich herzlichst bei Sabine Katzschmann, Charlotte Samwer, Nanny Krause und Giovanna Pommerschein bedanken. Ich möchte Prof. Ergün und seiner Arbeitsgruppe danken, dass er mir die Möglichkeit gegeben hat, in seinem Haus meine Arbeiten durchzuführen. Viele haben mich unterstützt, besonders Herr Michael Christof. Ich möchte mich bedanken, dass er mir bei der Formatierung der Bilder in meinen Manuskripten und bei meiner Dissertation geholfen hat. Aber ohne mein privates Umfeld wäre diese Arbeit nicht vorstellbar: Benedikt Grütter und Isabell von Schorlemer sind für mich immer da gewesen und haben mich in den letzten vier Jahren immer moralisch unterstützt. Sie haben mir Essen, Wein und viel Liebe gegeben. Sina Meyer danke ich, weil sie an mich glaubte, als es noch keiner tat. Meiner Mutter, meiner Schwester und Maurizio danke ich, weil sie mich lieben und mein Zuhause sind. Christoph Kaiser danke ich für die schönste Zeit inmitten der härtesten und stressigsten Phase meines Lebens. Ich habe bestimmt einige Menschen vergessen. Sie mögen mir verzeihen. Sie sind nicht erwähnt worden, aber in meinem Herzen. Diese Arbeit ist Christa Neumann, meiner Grundschullehrerin und späteren Mentorin, und meinem Vater gewidmet. xv