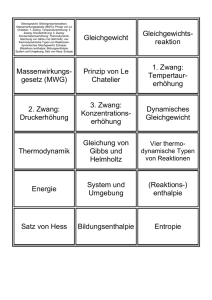
Physikalische und chemische Thermodynamik Das Leerscript
Werbung

Physikalische und chemische Thermodynamik Das Leerscript Hoeppe - PC Leerscript -1- Inhalt: 1 1.1 1.2 1.2.1 1.2.2 1.2.3 1.2.4 1.2.5 1.3 1.4 1.4.1 1.4.2 1.5 1.5.1 1.5.2 1.5.3 1.6 1.7 Grundlagen Einleitung Temperatur und Wärme Vorläufige Definition Temperaturmessung Spezifische Wärme und Wärmemessungen Wärmeleitung Der 0. Hauptsatz der Thermodynamik Zustandsfunktionen Das ideales Gas Phänomenologische Theorie Kinetische Gastheorie Weitere Ergebnisse der kinetischen Gastheorie Freiheitsgrade, spezifische Wärme, Gleichverteilungssatz, Maxwellverteilung Freie Weglänge, Stoßhäufigkeit, Wärmeleitung von Gasen Zustandsgleichungen realer Stoffe Luft 2 2.1 2.2 2.3 2.4 Energieumsatz Erster Hauptsatz Enthalpie Idealisierte Zustandsänderungen Reaktionswärme 3 3.1 3.2 3.3 3.4 3.5 Entropieumsatz Vorüberlegungen Zweiter Hauptsatz der Thermodynamik Dritter Hauptsatz, Absolutberechnung der Entropie Reaktionsentropie Partialdruckabhängigkeit der Entropie 4 4.1 4.2 Der Ablauf physikalisch-chemischer Prozesse Freie Energie und Freie Enthalpie Chemische Reaktionen 5 5.1 5.2 5.2.1 5.3 5.3.1 5.3.2 5.3.3 5.3.4 Freie Enthalpie und Gleichgewichte Reaktionsgleichgewichte Phasengleichgewichte Phasengleichgewichte reiner Stoffe Elektrolytgleichgewichte, Normalspannungen Löslichkeitsprodukt Säure-Base Gleichgewichte Normalspannungen Nernstsche Gleichung Hoeppe - PC Leerscript 4 5 11 13 17 21 26 29 34 37 43 47 52 61 62 63 65 68 72 79 81 -2- PT4-PC 6 6.1 6.1.1 6.1.2 6.1.3 6.1.3.1 6.1.3.2 6.1.3.3 6.1.4 Gemische Ideale Mischungen 86 Ideale Gasgemische Ideale Flüssigkeitsgemische Gleichgewicht eines Stoffes bei zusätzlicher Komponente Dampfdruckerniedrigung/Siedepunktserhöhung/Gefrierpunktserniedrigung. Druckabhängigkeit des Dampfdrucks Osmotischer Druck Gibbssche Phasenregel 6.2 6.2.1 6.2.1.1 6.2.1.2 6.2.1.3 Nichtideale, reale Mischungen Partielle molare Größen Volumen Mischungs- und Lösungsenthalpie Partielle molare Freie Enthalpie = Chemisches Potential 98 6.3 6.3.1 6.3.1.1 6.3.1.2 6.3.1.3 6.3.1.4* 6.3.2 6.3.2.1 6.3.2.2 6.3.2.3* Destillation Destillation idealer Mischungen Siedevorgang ohne Entfernung des Dampfes Siedevorgang mit Entfernung des Dampfes Einfache Destillation Fraktionierte Destillation - Rektifikation Rektifikation nach McCabe-Thiele Destillation realer Mischungen Minimumazeotrop Maximumazeotrop Azeotroprektifikation 106 6.4 6.4.1 6.4.2 6.4.3 6.4.4 6.5 Mischungen von FK und Schmelzen Lückenlose Mischkristallbildung Reale Systeme mit Mischungslücke Systeme mit großer partieller Mischungslücke Systeme ohne Mischkristallbildung Phasendiagramme ternärer Mischungen 115 7 7.1 7.2 7.2.1 7.2.2 7.2.3 7.2.4 7.2.5 7.3 Grenzflächengleichgewichte Allgemeine Betrachtungen Oberflächenspannung Oberflächenenergie und Oberflächenspannung Benetzung Laplace Gleichung Kapillarwirkung Keimbildung Adsorption 8 Ergänzungen Hoeppe - PC Leerscript 118 119 120 125 -3- 1 1.1 Grundlagen Einleitung Makroskopische Größen sind makrosk. Observable in einem makroskopischen System, aber „mikroskopische Größen“ müssen nicht klein sein sondern beschreiben wenige Teilchen eindeutig. (Eigentlich gibt es gar keine „mikroskopischen Größen“, sondern allenfalls mikroskopische Betrachtungen, bei welcher der Zustand eines jeden Teilchens wie auch in der klassischen Mechanik eindeutig definiert ist. Laplacescher Dämon) Ein System ist zwangsläufig nur noch makroskopisch beschreibbar, wenn es aus sehr vielen Teilchen besteht, hilfreich ist hier die Einheit mol: 1 Mol = Stoffmenge, welche die gleiche Zahl von Teichen enthält wie 12,0000 g des Kohlenstoff-Nuklids 12C Diese Zahl ist die Avogadrozahl / Loschmidtsche Zahl: Intensive Größe: Extensive Größe: mengenunabhängig mengenabhängig NA = NL = 6,022141023 mol-1 Bsp.: p, T, M, Vm, h, ... Bsp.: m, Q, V, n, H, ... Molzahl n Molmasse M Molvolumen Vm Dichte Masse m Übergang zu intensiven Größen molare Größen: Nicht nur die Teilchenzahl sondern auch die Anzahl von möglichen Einflüssen (Variablen) bestimmt die Komplexität eines Problems. Diese lässt sich reduzieren, indem man sich auf die Beschreibung eines tatsächlich oder gedachten räumlich begrenzten Bereich unserer Umwelt, dem System, beschränkt. Man unterscheidet: Offenes System: Geschlossenes System: E Abgeschlossenes System: E E n n Hoeppe - PC Leerscript n -4- 1.2 1.2.1 Temperatur und Wärme Vorläufige Definition A) Wärme: - ist eine Eigenschaft von Materie, die bei Berührung den Sinneseindruck „warm“ oder „kalt“ vermittelt. - kann auf andere Stoffe (Körper) durch ‚Wärmeleitung’ übertragen werden - ist eine extensive Größe - kann durch Arbeit erzeugt werden ist ... B) Temperatur: - ist ein Maß für die Stärke des warm/kalt Sinneseindrucks - geht mit einer „thermischen Ausdehnung“ der meisten Stoffe und Körper einher - bestimmt Stärke und Richtung einer ‚Wärmeleitung’ - ist eine intensive Größe ist ... Der Zusammenhang von Wärme und Temperatur wird mit Einführung der (spezifischen) Wärmekapazität in 1.2.3 deutlich werden. 1.2.2 Temperaturmessung Das Messprinzip basiert meist auf dem Effekt der thermischen Ausdehnung, d.h. der mittlere Abstand der Atome /Moleküle wird mit zunehmender Bewegung, also mit der Temperatur, größer. a) Bimetallstreifen Entscheidend: Die thermische Ausdehnung ist materialspezifisch! Linearer thermischer Ausdehnungskoeffizient : L= Hoeppe - PC Leerscript -5- Beispiele: Aluminium Kupfer Stahl Keramik Diamant ‚Invar‘ = 24 ·10-6 ·K-1 = 24 ppm ·K-1 = 16,8 ppm ·K-1 = 8 ~ 16 ppm ·K-1 = ~ 6 ppm ·K-1 = 1,3 ppm ·K-1 = ~ 0 ppm ·K-1 b) Flüssigkeitsthermometer: hier: Volumenausdehnungskoeffizient : V = Beispiele: Quecksilber = 181 ppm ·K-1 Wasser = 207 ppm ·K-1 Ethanol = 1100 ppm ·K-1 Für kleine und gilt der Zusammenhang: = 3· Das folgt direkt aus V ~ L3 unter Vernachlässigung höherer Potenzen von . c) Widerstandsthermometer Der elektrische Widerstand eines Leiters ist T-abhängig. Widerstandsmessung geeigneter Bauteile (NTC, PTC) entspricht T- Messung. d) Thermoelemente Infolge unterschiedlicher (T-abhängiger) Elektronendiffusion in zwei verschiedenen Metallen, bildet sich eine messbare Thermospannung aus. e) Pyrometer Analyse der Intensität (und evtl. spektralen Verteilung) der von einem heißen Körper (>1000°C) emittierten elektromagnetischen Strahlung. (vgl. Stefan-Boltzmann ; Plancksches Strahlungsgesetz, Farbtemperatur’ ) (Besonderheit: kein direkter thermischer Kontakt, d.h. Berührung nötig!) Hoeppe - PC Leerscript -6- f) Einheiten Die Celsiusskala ist definiert über Schmelzpunkt (0°C) und Siedepunkt (100°C) von Wasser bei Normdruck. Kelvinskala (absolute Temperatur): 1 K ist definiert als der 1/273,16 Teil der Temperatur des Tripelpunktes von Wasser; damit gilt: T [K] = 1.2.3 Spezifische Wärme und Wärmemessungen Die Wärmekapazität C gibt an welche Wärmemenge Q einer Stoffmenge zugeführt werden muß, um bei dieser eine Temperaturänderung T zu bewirken: Die Temperatur T wird damit bis auf die Proportionalitätskonstante C ein Maß für die Wärmemenge einer Stoffmenge bzw. eines Körpers. Als spezifische Wärmekapazität c (kurz: spezifische Wärme) bezeichnet man die auf die Stoffmenge bezogenen Wärmekapazität eines Körpers (oder einer Stoffmenge): Auf die Masse bezogen: Auf die Molzahl bezogen: Betrachtet man zwei nach außen isolierte Stoffmengen 1 und 2 mit anfänglich unterschiedlichen Temperaturen T1 und T2 so beobachtet man (infolge von Wärmeleitung) einen Temperaturausgleich: Aus der Energieerhaltung folgt: Hoeppe - PC Leerscript Q = Q1 + Q2 = 0 -7- Q1 = m1 c1 T1 = m1 c1 ( TM -T1 ) Q2 = m2 c2 T2 = m2 c2 ( TM -T2 ) m1 c1 ( TM -T1 ) + m2 c2 ( TM -T2 ) = 0 ( m1 c1 + m2 c2 ) TM = m1 c1 T1 + m2 c2 T2 und schließlich für die Mischtemperatur TM: Messung von Wärmekapazitäten: Werden die Massen und Temperaturen gemessen, so ist z.B. die unbekannte Wärmekapazität c2 experimentell bestimmbar wenn c1 (z.B. Wasser) bekannt ist. Auch durch chemische Reaktionen entstehende Wärmemengen werden durch die Temperaturerhöhung einer definierten Wassermenge in einem Kaloriemeter bestimmt. 1.2.4 Wärmeleitung Beobachtung: Sind zwei Körper in thermischem Kontakt, so gleichen sich ihre Temperaturen durch einen Wärmestrom an. Die Geschwindigkeit des Temperaturausgleichs ist abhängig von - Temperaturdifferenz und - Güte des thermischen Kontakts ( Fläche und Material) Spezifische Wärmeleitfähigkeit ist über folgenden Zusammenhang definiert: Wärmestromdichte q Hoeppe - PC Leerscript -8- Für kleine x und T gilt bzw. vektoriell In der Praxis ist oft der Wärmestrom pro Zeit, d.h. z.B. Heizleistung, Kühlleistung, Verlustleistung etc. am interessantesten: Beispiele für spezifische Wärmeleitfähigkeiten: Kupfer V2A-Stahl Styropor Ruhende Luft = 384 = 15 = 0,036 = 0,026 W· W· W· W· m-1 ·K-1 m-1 ·K-1 m-1 ·K-1 m-1 ·K-1 Messung einer Wärmeleitfähigkeit: - (kleine) konstante Heizleistung P bei T1 - (starke) Kühlung bei T2 auf T2 bei definiertem A und x ist aus P und T bestimmbar Eindimensionale Betrachtung z.B. in der Bauphysik: Wärmewiderstand Rth : Rth : x A T P Q Rth - Schichtaufbau einer Wand entspricht Serienschaltung der Rth,i. - Summe verschiedene Flächen (Wände, Fenster, etc.) entspricht Parallelschaltung. Wärmedurchgangskoeffizient k : k : 1 Rth A P k A T - k erlaubt eine ‚faire’ flächenunabhängige Beurteilung von z.B. Wandaufbauten, Isolationen oder Fenstern. Wärmeübergangskoeffizient : Beim Wärmeübergang von einem Festkörpers zu einem fluiden Medium (Wasser, Luft) tritt an Stelle von k der Wärmeübergangskoeffizient P A T wobei z.B. stark von der Strömungsgeschwindigkeit abhängt. Hoeppe - PC Leerscript -9- Wärmetransport durch Materialtransport: In fluiden Medien welche durch eine Erwärmung eine Dichteänderung erfahren, wird der Wärmetransport durch Konvektion dominiert. (Stichworte: Heizung, Wetter, Heatpipe) Wärmetransport durch Elektromagnetische Strahlung: Materie gibt je nach ihrer Temperatur Energie in Form von Wärmestrahlung ab. Empirisch gilt das Stefan Boltzmann Gesetz: Prad A T 4 ~ A T 4 Umgekehrt nimmt Materie aber nach den gleichen Gesetzmäßigkeiten auch Wärme auf, entscheidend ist also letztlich die Bilanz. In Festkörpern ist der Strahlungstransport bei Raumtemperatur eher vernachlässigbar. (Stichworte: Stefan-Boltzmann-Konstante, IR, Thermometer, Thermoskanne) Wärmeleitung und Diffusion: Will man den raum-zeitlichen Verlauf der Wärmeleitung (nichtstationäre Zustände) beschreiben, muss man die sog. Wärmeleitungsgleichung lösen: T T 0 t c Diese gleicht nicht nur formal dem 2. Fickschen Diffusionsgesetz, da sich Wärmeleitung (in einem Festkörper) auch als Diffusion von Bewegungsenergie auffassen bzw. beschreiben lässt. Anmerkung: Als Ursache für die Wärmeleitung und Diffusion wird gerne die Zunahme der Entropie entsprechend dem 2. Hauptsatz der Thermodynamik genannt. Verstehen lässt sich das aber erst mit der statistischen Interpretation der Entropie, welche im 3. Kapitel behandelt wird. 1.2.5 Der 0. Hauptsatz der Thermodynamik Wenn sich die Temperaturen zweier Systeme infolge von Wärmeleitung und- strahlung angeglichen haben, befinden sich diese miteinander im thermischen Gleichgewicht. Makroskopisch findet zwischen diesen Systemen kein Energieaustausch mehr statt. Ist ein System 1 jeweils mit zwei Systemen 2 und 3 im thermischen Gleichgewicht so folgt zwangsläufig, dass auch die Systeme 2 und 3 miteinander im Gleichgewicht sind. Diese Aussage des 0. Hauptsatzes schreibt sich kurz: Hoeppe - PC Leerscript - 10 - 1.3 Zustandsgleichungen Ein System mit n unabhängigen Größen bzw. Variablen wird mit einem Punkt im n-dimensionalen Zustandsraum n eindeutig beschrieben. Bsp.: Nur zwei unabhängige Variable (z.B. x1 und x2) Zustandsraum = Fläche. Die Variable x3 ist mit x1 und x2 über die Zustandsgleichung eindeutig festgelegt! (Alle anderen Größen, welche das System betreffen, sind auf x1 und x2 zurückführbar.) Die Zustandsgleichung definiert eine Zustandsgröße F eindeutig als Funktion der n unabhängigen Variablen x1, x2, ... xn eindeutig. F F ( x1 , x2 ,..., xn ) eindeutig Soweit die Zustandsgleichung auflösbar ist, ist die Wahl der unabhängigen Variablen beliebig. Im Gleichgewicht sind die Zustandsgrößen zeitlich konstant. Eine Zustandsänderung (=Prozess) ist immer Folge einer äußeren Störung. Die Änderung einer beliebigen Größe bei einem Prozess ist durch Summation bzw. Integration über die Änderungen d bei beliebig kleinen Schritten ds im Zustandsraum gegeben. Bei einem Prozess von Zustand 1 nach 2 ist also das Linienintegral (=Wegintegral) 2 1 d zu berechnen. WEG Die Änderung einer Zustandsgröße F ist unabhängig von Art bzw. Weg des Prozesses im Zustandsraum, sondern nur von Anfangszustand 1 und Endzustand 2 abhängig. Dies entspricht der Aussage, dass ein Zustandsgröße eindeutig mit einem Punkt im Zustandsraum definiert ist. Hoeppe - PC Leerscript - 11 - Da eine Zustandsgröße F eindeutig ist, ist das Linienintegral F F2 F1 dF WEG unabhängig von gewählten Weg / Prozess im Zustandsraum. Dies ist in der Anwendung sehr wichtig, da bei der Berechnung eines Prozesses ein beliebiger (einfacher!) Weg gewählt werden kann, soweit man sich auf Zustandsgrößen beschränkt. Für einen geschlossenen Weg (Kreisprozess) folgt damit zwangsläufig dF 0 . Beispiel: p(V,T) ist Zustandsgröße Linienintegral ist wegunabhängig, d.h. 2 p p 2 p1 dp 1 dp dp dp WEG I WEG II WEG III bzw. dp 0 Die Wegunabhängigkeit des Linienintegrals entspricht der Stetigkeit der Zustandsfunktion F(x1, x2, ..,xn) bzgl. aller unabh. Variablen. Damit existiert für F(xi) ein totales Differential mit Eine Veränderung einer Zustandsgröße dF lässt sich also immer als totales Differential schreiben, was insbesondere bei Herleitungen sehr nützlich ist. Nicht jede Größe ist eine Zustandsgröße. Zum Beispiel sind die Volumenarbeit W und die Wärmemenge Q keine Zustandsgrößen. Diese Größen können sich daher bei einem Kreisprozess auch ändern. Hoeppe - PC Leerscript - 12 - 1.4 1.4.1 a) Ideales Gas Phänomenologische Theorie Gesetz von Boyle Mariotte für T = const: b) Gesetz von Charles für p = const: Absolute Temperatur T: Volumenausdehnungskoeffizient von Luft : c) Gesetz von Avogadro: Unabhängig von der Teilchenart skaliert das Volumen eines (idealen) Gases bei konstantem Druck und Temperatur linear mit der Teilchenzahl: Bei Standardbedingungen für Gase, d.h. p = 1 bar und T = 0 °C haben alle (id.) Gase das gleiche Molvolumen von ~ 22,4 Liter. Hoeppe - PC Leerscript - 13 - Zusammenfassend: Gesetz von Boyle Mariotte: + Gesetz von Charles: + Gesetz von Avogadro: Zustandsgleichung des idealen Gases: molare Gaskonstante: R = Rm = 8,31451 J/(molK) = 0,0831451 barl /(molK) Ideales Gas gute Näherung falls: Zustandsdiagramme als Schnitt durch Zustandsfläche: T = const. Isothermen : Hoeppe - PC Leerscript V = const. p = const. Isochoren: Isobaren: - 14 - 1.4.2 Kinetische Gastheorie Vereinfachtes Modell eines Gases: - Gas bestehe aus lauter gleichen sehr kleinen harten Kugeln - Teilchenabstand >> Teilchengröße (d.h. das Eigenvolumen ist vernachlässigbar) - Bis auf elastische Stöße gebe es keine Wechselwirkungen - Die Bewegung sei vollkommen ungeordnet (d.h. „kein Wind“) - Alle Teilchen sind in erster Näherung gleich schnell Betrachte quaderförmigen Ausschnitt eines Gasvolumes mit N Teilchen pro Volumen V, wobei innerhalb der Zeit t aufgrund ihrer endlichen Geschwindigkeit nur Teilchen aus dem Volumen V* die Wand der Fläche A erreichen können: In der Zeit t erreicht jedes 2. Teilchen mit |Vx| > 0 aus V* = l* ·A die Wand A. Die Zahl der Stöße N* ist dann N 1N 1N V vx t A 2V 2V Die Impulsübertragung p pro Stoß ist wegen p’x = - p x : p = 2 p x = 2 m vx Die zeitliche Änderung des Impulses entspricht einer Kraft Fi = p x/t auf die Wand: F N 2 m vx N p x 1 N v x t A A m v x2 2V t t V Die Teilchen erzeugen daher zusammen auf die Wand den Druck p = F/A : p Eigentlich bewegen sich die Teilchen mit der Geschwindigkeit v in beliebiger Richtung. Im Mittel gilt sicher v x2 v y2 v z2 N m v x2 V v x2 v y2 v z2 und damit v 2 3 v x2 bzw. v x2 Damit ergibt sich aus dem Ausdruck für den Druck von oben: Hoeppe - PC Leerscript - 15 - 1 2 v 3 1 2 1 2 p V N m v 2 N m v 2 N E kin 3 3 2 3 Nach Boltzmann entspricht die Temperatur bis auf einen Faktor der mittleren kinetischen Energie der Teilchen: E kin 3 k BT 2 *) Damit folgt unmittelbar das ideale Gasgesetz, p V 2 3 N k B T N k B T n N A k B T n Rm T 3 2 wenn man die (Boltzmann-) Konstante kB über N A k B Rm definiert. *) Anmerkung: Für mehratomige Gase wird dieser Ausdruck im nächsten Kapitel noch geringfügig abgeändert. _______________________________________________________________ Nach diesem Ergebnis können wir eine endgültige Definition der Begriffe Wärme und Temperatur wagen: A) Wärme Q Wärme ist die (gespeicherte) kinetische Energie der ungeordneten Bewegung der Atome und Moleküle eines makroskopischen Systems. B) Temperatur T Die Temperatur T ist ein Maß für die mittlere kinetische Energie der ungeordneten Bewegung der Atome und Moleküle eines makroskopischen Systems. Hoeppe - PC Leerscript - 16 - 1.5 1.5.1 Weitere Ergebnisse der kinetischen Gastheorie Freiheitsgrade, spezifische Wärme, Gleichverteilungssatz Gleichverteilungssatz nach Boltzmann: Die kinetische Energie verteilt sich (im Mittel) gleichmäßig auf die Freiheitsgrade f aller Moleküle. (also nicht zwangsläufig auf die Moleküle!) Unter ‚Freiheitsgrad’ versteht man eine (mikroskopische) Energiespeichermöglichkeit: Translation in drei Raumrichtungen: Rotation o 2 atomiges (lineares) Molekül (in zwei Raumrichtungen): o 3 oder mehratomiges (nicht-lineares) Molekül (in drei Raumrichtungen): Schwingungen o 2 oder mehratomiges Molekül ftrans = frot = frot = fschw = Da Schwingung und Rotation der Quantisierung unterliegen (QM), können diese Freiheitsgrade „einfrieren“, wenn die mittlere Energie eines Teilchens bzw. Freiheitsgrades nicht ausreicht um z.B. eine Schwingung anzuregen. Dies ist bei Schwingungen für Moleküle i.d.R. bereits bei Raumtemperatur der Fall! Die Zahl der Freiheitsgrade zeigt sich unmittelbar bei der spezifischen molaren Wärmekapazität. Im Vorgriff auf Kapitel 2 gilt zusammenfassend: Stoff Freiheitsgrade cV cp 1-atomiges Gas: 3 3 5 2-atomiges Gas (tiefe Temperaturen): 5 5 7 2-atomiges Gas (hohe Temperaturen): 7 7 9 Ergänzung: Dulong-Petit’ Regel für FK: 6 6 Hoeppe - PC Leerscript /2 R /2 R /2 R /2 R /2 R /2 R /2 R 6 /2 R - 17 - Ideale Gase sind per Definition frei von Wechselwirkungen. Die innere Energie eines idealen Gases ist daher auch ausschließlich in der Bewegung der Teilchen enthalten, und damit nach Boltzmann durch den Ausdruck 1 f f U u N A E kin N A k B T Rm T n 2 2 gegeben und somit makroskopisch betrachtet nur von der Temperatur abhängig! Entsprechend der Definition der spez. Wärmen bzgl. der makroskopischen Inneren Energie U und Enthalpie ergibt sich hiermit in hervorragender Übereinstimmung mit dem Experiment: Spezifische Wärme bei const. Volumen: cV 1 U u n T V T V Spezifische Wärme bei const. Druck: cp 1 H h n T p T p id id f R 2 f 2 R 2 Berechnung Wärmemenge dQ n c p ,V dT Q n c p ,V dT c p ,V const n c p ,V T ( vgl. Kap. 2 ) Zusammenhang cV und cp: Für Gase gilt: c p cV R Hoeppe - PC Leerscript Für FK u. Flüssigkeiten gilt: c p cV - 18 - 1.5.2 Maxwellverteilung Eine ausführliche und vollständige Statistik für die Teilchen eines Gases von Boltzmann und Maxwell liefert eine Energie- und die Geschwindigkeitsverteilung: 3 2 mv 2 2000 2500 m 2 2 k BT dN (v) v e f (v) dv dv 4 2 N k T B Maxwellverteilung: Beispiel für Stickstoff ( N2 ) bei 300 K und 2000 K: 0.002 0.0015 f( v , 300. K ) 0.001 f( v , 2000. K ) 0.0005 0 0 500 1000 1500 3000 v Wahrscheinlichste Geschwindigkeit: (Maximum der Kurve) ! d f (v ) 0 dv vw v 2 RT M Mittlere Geschwindigkeit: v v f (v)dv 0 2k B T m 8 RT M Mittlere quadratische Geschwindigkeit: v2 v 2 f (v)dv v2 0 Es gilt damit der Zusammenhang: Hoeppe - PC Leerscript v 2 : v : vw 3RT M 1,73 : 1,60 : 1,41 - 19 - 1.5.3 Freie Weglänge, Stoßhäufigkeit und Wärmeleitung von Gasen Auch wenn die Teilchen in einem Gas sehr klein im Vergleich zum mittleren Teilchenabstand sind, bewegen sie sich doch sehr schnell. Daher kommt es häufig zu Stößen. Beschreibt man die Teilchenorte durch Punkte im Raum, wird die Teilchengröße durch den effektiven Teilchendurchmesser d im Stoßquerschnitt berücksichtigt: Stoßquerschnitt: d2 Als Stoßzylinder VZ bezeichnet man das von mit der mittleren Teilchengeschwindigkeit in der Zeit t überstrichene Volumen: VZ v t 2 v t (Der Faktor Wurzel 2 ergibt sich aus einer aufwendigeren Rechnung, welche auch die Bewegung der ‚getroffenen’ Teilchen berücksichtigt.) Die Zahl von Stößen in der Zeit t ergibt sich dann aus der Teilchenzahl in VZ : VZ N n p p VZ N A VZ N A ... 2 v t V V R T k BT Die Stoßhäufigkeit Z = / t ist: Z 2 v p k BT Die Zeit zwischen 2 Stößen entspricht dem Kehrwert der Stoßhäufigkeit womit für die k BT 1 mittlere freie Weglänge gilt. l v v Z 2 p Diese ist besonders wichtig im Zusammenhang mit elektrischen Überschlägen in Gasen, in der Vakuumtechnik und der Wärmeleitfähigkeit von Gasen. Wärmeleitkoeffizient für Gase: Hoeppe - PC Leerscript 1 N cV v l 3 V ~ T cV M - 20 - 1.6 Zustandsdiagramme realer Stoffe Allgemeines Zustandsdiagramm: TRP : Tripelpunkt kr. : Kritischer Punkt pD(T) : Dampfdruckkurve pm(T) : Schmelzdruckkurve psub(T): Sublimationsdruckkurve Anomalie des Wassers: 1) Verflüssigung bei Druckerhöhung, weil: - 2) Schnellkochtopf funktioniert, weil : - „Trockeneis“: Sublimation von CO2 bei Normaldruck, weil: - Hoeppe - PC Leerscript - 21 - Bestimmung von Phasengrenzlinien Phasengrenzlinien werden bestimmt in dem man Druck und Temperatur verändert, bis man einen Phasenübergang beobachtet. Aufgrund der dabei umgesetzten latenten Wärme (vgl. Kap. 2.2.2) geschieht das i.d.R. nicht schlagartig, so dass eine Messung von p und T möglich ist. Ein gutes Beispiel ist hier ‚Eiswasser’: Befinden sich ausreichend viele Eiswürfel in flüssigem Wasser (und ist der Wärmeübergang nach außen gering, z.B. durch ein Dewargefäß), dann hat das Wasser-Eis-Gemisch (nach wenigen Minuten) eine Temperatur von sehr genau 0°C. Durch geeignete äußere Zwangsbedingungen kann auch ein Gleichgewicht zwischen zwei Phasen erreicht werden, wodurch eine Messung sehr genau durchgeführt werden kann. Variiert man z.B. das Volumen bei gegebener Temperatur, erreicht man im pVDiagramm ein Gebiet, in dem flüssige und gasförmige Phase koexistieren, soweit die Temperatur unterhalb der kritischen Temperatur Tkrit ist. (vgl. auch Kap 5.2.1) T: Isothermen Tkrit: kritische Isotherme g: gasförmig l + g : 2 Phasenmischgebiet Beispiel Dampfdruckkurve: Messablauf: 1) Wasser in geschlossenen Behälter geben 2) Luft mit Vakuumpumpe abpumpen 3) Behälter verschließen 4) Der sich nach kurzer Zeit einstellende Druck entspricht dem Dampfdruck der Flüssigkeit (z.B. Wasser), soweit eine flüssige Phase noch vorhanden ist(!). 5) Behälter temperieren und Druck = Dampfdruck als Funktion der Temperatur messen. Hoeppe - PC Leerscript - 22 - 1.6.0 Zustandsgleichungen realer Gase Reale Stoffe bestehen aus Teilchen/Molekülen endlicher Größe, so dass diese sich bei Abkühlung und/oder Druckerhöhung so nahe kommen, dass Wechselwirkungen nicht mehr vernachlässigt werden können. Dies äußert sich z.B. in Abweichungen vom idealen Gasgesetz und der Beobachtung, dass sich Gase bei hinreichend tiefen Temperaturen verflüssigen lassen. Ziel ist es zunächst, eine verbesserte Gasgleichung für reale Gase zu erhalten, welche z.B. auch Wasserdampf bei Temperaturen knapp oberhalb der Siedetemperatur gut beschreibt. Größe der Teilchen, Art und Stärke der Wechselwirkung sind stoffspezifisch, weshalb jede Art von Korrektur(faktor) auch stoffspezifisch bestimmt werden muss. 1.6.1 Virialansatz Die pragmatischste Methode zur Beschreibung eines realen Gases ist die Einführung eines rein empirischen Korrekturfaktors Z über , welcher stoffspezifisch die Abweichung eines realen Gases vom idealen Gasgesetz beschreibt. Der Realfaktor Z = Z(p, T) ist dabei nicht nur stoffspezifisch sondern zudem eine Funktion von Druck und Temperatur wie die experimentell bestimmten Faktoren im Beispiel unten zeigen. Anstatt für jeden Stoff Unmengen von Zahlen zu tabellieren, wird Z als Potenzreihe in p als Funktion von T angegeben: Z (T ) 1 B (T ) 10 3 p C (T ) 10 6 p 2 D (T ) 10 9 p 3 Die temperaturabhängigen Parameter B, C und D (A1) sind die Virialkoeffizienten und in Tab. B für einige Stoffe aufgelistet. Hoeppe - PC Leerscript - 23 - 1.6.2 Van der Waals Gleichung Van der Waals (1837 – 1923) kommt aufgrund mikroskopischer Überlegungen zu einer physikalisch begründeten Korrektur der idealen Gasgleichung: 1. Die Berücksichtigung des Eigenvolumens der Gasteilchen mit einem Term b (Das für die Bewegung verfügbare Volumen wird um b verringert, bzw. das Gesamtvolumen um b vergrößert.) 2. Eine (offenbar immer) anziehende Wechselwirkung zwischen den Teilchen führt zu einer Verringerung des (gemessenen) Drucks. Der Wechselwirkungsparameter a wird in der Korrektur durch das Molvolumen zum Quadrat geteilt, da die WW nur bei kleinen Molvolumina (Teilchenabständen) relevant ist. Den Term a/Vm2 nennt man auch Binnendruck. Van der Waals Gleichung: Die Korrekturterme a und b sind hierbei Stoffkonstanten, und nicht von Druck oder Temperatur abhängig ( vgl. Tab. A ). Zudem lassen sie sich im pV-Diagramm aus der waagerechten Isothermen durch den kritischen Punkt bestimmen. Sie sind damit vollständig (und einfach!) aus dem kritischen Druck und Temperatur bestimmbar. Nach einer relativ kurzen Rechnung folgt: b R Tkrit 8 pkrit a 27 b 2 p krit Die mit der v.d.W. Gleichung berechneten Isothermen beschreiben sogar recht gut den 2-Phasen Koexistenzbereich (l+g), wenn man den unphysikalischen ‚Schlenker’ mit Hilfe der sog. Maxwellkonstruktion ( eingeschlossene Flächen ober- und unterhalb der Geraden sollen gleich groß sein) durch ein Waagerechte ersetzt. Im Gegensatz zu den Virialkoeffizienten ist die v.d.W. Gleichung nicht nur physikalisch begründet, sondern ohne weitere Messungen viele Daten bekannter Gase verfügbar. Zudem sind auch qualitative Rechnungen bei realen Gasen möglich (vgl. Kap. 2.3.5). Hoeppe - PC Leerscript - 24 - 1.6.3 Gleichung von Redlich und Kwong Die Gleichung von Redlich und Kwong ist eine Verfeinerung der v.d.W. Gleichung und führt quantitativ zu einer etwas besseren Beschreibung realer Gase. p a' (V m b ' ) R T T Vm (V m b ' ) Die Parameter a’ und b’ ( Tab. C) lassen sich analog zur v.d.W. Gleichung aus den Daten des kritischen Punktes bestimmen. Hier gilt: b' 0,08664 Hoeppe - PC Leerscript R Tkrit pkrit a ' 56,95 b'2 pkrit Tkrit - 25 - 1.7 Luft Die Luft unserer Atmosphäre besteht aus einem komplexen Gemisch von Gasen, in Vol% oder Mol% gilt für trockene Luft ca: N2 ~78%; O2 ~ 21%; Ar ~ 0,9%; CO2 ~ 0,038%; sonstige Gase jeweils < 20 ppm. Zusätzlich, je nach Temperatur, Bodennähe und Vorgeschichte, enthält Luft noch Wasserdampf in der Größenordnung von 1 % und natürlich auch Schwebstoffe (Aerosole, Staub) wodurch die physikalischen Eigenschaften der Luft wesentlich beeinflusst werden. Insbesondere die Variation des Wasserdampfgehaltes durch Verdunstung und Kondensation ist mit den damit verbundenen Übergangswärmen wetterbestimmend. 1.7.1 Die Barometrische Höhenformel beschreibt den Luftdruck als Funktion der Höhe, welcher durch den Schweredruck der Luftsäule auf dem betrachteten Volumenelement entsteht. Im Gegensatz zu Flüssigkeiten ist das Gasgemisch Luft gut kompressibel, weshalb der Luftdruck ‚nach oben’ abnimmt. Man betrachtet daher den Druckbeitrag dp, der durch eine Luftschicht dh durch ihren Schweredruck entsteht, berücksichtigt dabei die auch von der Höhe abhängige Dichte (h) und erhält schließlich nach partieller Integration den Luftdruck als Funktion der Höhe h: dp (h) g dh nRT n M R R T T V V M M p für T = const gilt also: dp 0 p0 p const p ( h ) p0 ( h) 0 p (h) g dh , partielle Integration Die barometrische Höhenformel kann nur für (vergleichsweise grobe) Abschätzungen verwendet werden, da weder die Temperatur noch die Zusammensetzung der Luft konstant sind. Wesentlich ist hierbei auch der Wasserdampfgehalt, der aufgrund des Wettergeschehens stark variiert. Hoeppe - PC Leerscript - 26 - 1.7.2 Luftfeuchtigkeit Warum verdampft (verdunstet) Luft, bei Temperaturen unterhalb des Siedepunktes? Dies lässt sich nicht nur verstehen, wenn man das Phänomen innerhalb der kinetischen Gastheorie mikroskopisch betrachtet. Auch makroskopisch kommt man zu einer richtigen Beschreibung, wenn man berücksichtigt, dass der entstehende Wasserdampf nicht als Reinstoff sondern als Gemisch vorliegt: Entsprechend dem Gesetz von Dalton p pi mit i pi ni RT V müssen wir daher statt des Gesamtdruckes von ca. 1 bar den Partialdruck des Wasserdampfes ansetzen, womit die Siedetemperatur entsprechend der Dampfdruckkurve für H2O sinkt. Wasser verdunstet an Luft also so lange, bis der (temperaturabhängige) Sättigungsdampfdruck psat(T) erreicht ist, welcher bis auf Korrekturen zweiter Ordnung dem Wasserdampfpartialdruck pD(T) entspricht. Aufgrund des tages- und jahreszeitenbedingten ständigen Temperaturwechsels in der Atmosphäre, Konvektion von Luftmassen oder z.B. Lüftung beheizter Räume kommt es daher zu einem ständigen Wechsel von Verdunstung und Kondensation. Maße für den Wasserdampfgehalt von Luft, d.h. die Luftfeuchte: Absolute Luftfeuchte [g/m³]: : mH 2O V Die maximal mögliche Luftfeuchte max = sat [g/m³] bzw. der Sättigungsdampfdruck psat [mbar] sind (vgl. Dampfdruckkurve) temperaturabhängig (!): Hoeppe - PC Leerscript - 27 - Relative Luftfeuchte rel [%]: rel : p HO max psat 2 Der Taupunkt Td [K, °C] , entspricht der Temperatur, bei der bei Abkühlung Kondensation einsetzt bzw. einsetzen würde. Mit der Temperaturabhängigkeit von max (vgl. Grafik oben) ist damit auch die absolute Feuchte definiert. In der Bauphysik versteht man unter dem Taupunkt den Ort (z.B. in einem Wandaufbau), an welchem Kondensation auftritt. Dies ist sinnvoll, da sowohl Temperatur als auch die absolute Feuchte hier stärker mit dem Ort als mit der Zeit variieren. Hoeppe - PC Leerscript - 28 - 2 2.1 Energieumsatz bei Stoffumwandlungen Erster Hauptsatz Die Innere Energie U eines Systems ist ... Eine Änderung der inneren Energie U erfolgt nur über einen - Wärmetransport Q und/oder über - Arbeit W im mechanischen Sinne (,d.h. Prozesse die sich auf das bloße Heben und Senken von Gewichten abbilden lassen). (Unter Wärme versteht man die Summe der stochastisch verteilten kinetischen Energie, unter Wärmetransport den Energietransport aufgrund mikroskopisch stochastischer Prozesse wie z. B. Wärmeleitung oder Wärmestrahlung; also Energietransport, welcher sich nicht auf das bloße Heben und Senken von Gewichten abbilden lässt .) Für ein geschlossenes System gilt daher: Diese Aussage wird oft schon als 1.HS bezeichnet, da sie die Energieerhaltung aus Gründen der Eindeutigkeit impliziert. Deutlicher ist jedoch: Für ein abgeschlossenes System gilt: Es existiert kein Perpetuum Mobile 1. Art, d.h. ... Folge des 1. HS: U ist Zustandsgröße, d.h. dU 0 . „Beweis“ durch Annahme des Gegenteils: Wäre U keine Zustandsgröße, wäre auch ein Weg/Kreisprozess im ZR denkbar für den das Wegintegral nicht null wäre. Somit wäre auch ein Perpetuum Mobile 1. Art möglich. Es existiert ein totales Differential für U. Wird der Zustand eines geschlossenen Systems eindeutig mit V und T definiert so lautet das totale Differential für U (V,T) : andererseits im Folgenden Hoeppe - PC Leerscript - 29 - 2.1.1 Volumenarbeit Ändert sich das Volumen eines Systems in Folge irgendeiner Störung so wird stets mechanische Arbeit im Sinne von „Kraft x Weg“ geleistet. Eine beliebige Volumenänderung lässt sich immer aus vielen kleinen Kolbenartigen Volumenänderungen an der Oberfläche des Systems zusammengesetzt denken. Betrachte daher die geleistete Arbeit eines idealen Gases welches sich in einem Zylinder ausdehnt und einen Kolben gegen eine äußere Kraft F nach außen schiebt: Volumenarbeit W: Da sich , wie in diesem Beispiel, der Druck des Gases während des Prozesses verändert muss die Volumenarbeit differentiell definiert werden. (Vgl. Beispiele unten) 1) Berechnung Volumenarbeit bei isothermer Expansion eines idealen Gases: Verfolge Prozess im pV-Diagramm, dem sog. Arbeitsdiagramm: Das Integral pdV ist hier direkt als Fläche unter der Isothermen interpretierbar *. „Isotherm“ bedeuted im Diagramm längs einer Isothermen, z.B. aufgrund fehlender thermischen Isolation oder beliebig langsam, so dass die Temperatur stets der (konstanten) Umgebungstemperatur entspricht. Das heißt nicht, dass kein Wärmefluss stattfinden würde, im Gegenteil: Ein Wärmefluss muss stattfinden, damit sich die Temperatur nicht ändert! (vgl. auch „innere Energie ideales Gas“ nächstes Kapitel.) Hoeppe - PC Leerscript - 30 - Hier wird auch die Vorzeichenkonvention noch einmal deutlich: Bei der Expansion ist V positiv und damit W negativ: Das System verrichtet Arbeit, gibt also Energie nach außen ab womit die innere Energie des Systems kleiner wird. *) Verläuft der Prozess nicht isotherm, also im pV-Diagramm entlang eines anderen Weges von Zustand 1 nach Zustand 2, ergibt sich für die Volumenarbeit offensichtlich ein anderes Ergebnis. Das (Weg-) Integral über pdV ist offensichtlich wegabhängig und die Volumenarbeit daher auch offensichtlich keine Zustandsgröße! 2) Berechnung Volumenarbeit bei isobarer Expansion eines idealen Gases: Hierbei handelt es sich eher um einen Spezialfall, da sich bei einem idealen Gas der Druck bei Expansion normalerweise immer ändert. Betrachtet man aber z.B. den Dampf einer verdampfenden Flüssigkeit näherungsweise als ideales Gas, dann wird z.B. gegen den konstanten Druck der Atmosphäre Volumenarbeit geleistet: Der Prozess verläuft hier im pV-Diagramm entlang einer Isobaren. Da p während des Prozesses konstant bleibt, reduziert sich das Integral auf eine einfache Multiplikation mit der Volumenänderung V. Neben der Volumenarbeit kann an/von einem System auch andere Art von Arbeit geleistet werden, z.B. elektrische oder magnetische Arbeit bei der Polarisation von dielektrischen oder magnetischen Materialien oder durch elektrischen Strom, was insbesondere für die Elektrochemie sehr wichtig ist. Soweit nicht explizit auf solche andere Arten von Arbeit verwiesen wird, ist im Folgenden mit Arbeit die Volumenarbeit gemeint. Hoeppe - PC Leerscript - 31 - 2.1.2 Spezifische Wärmekapazität Unter Wärmekapazität versteht man die Wärmespeicherfähigkeit eines Körpers bzw. einer Stoffmenge. Sie bestimmt welche Wärme einem Körper zugefügt werden muss, um eine Temperaturänderung von 1 K zu bewirken. Da wir mittlerweile von Volumenausdehnung und der damit verbundenen Volumenarbeit wissen, definieren wir jetzt etwas strenger als in Kap. 1.2.3 die Wärmekapazität bei konstantem Volumen: bzw. die spezifische (molare) Wärmekapazität Da mit der Einschränkung „bei konstantem Volumen“ auch jegliche Volumenarbeit ausgeschlossen ist, entspricht der Wärmeumsatz hier der Änderung der inneren Energie des Systems. Die allgemeine Definition von cV unter der Verwendung der Zustandsgrößen U und T lautet daher: Spezifische Wärme bei konstantem Volumen: ( vgl. totales Differential von U = U(V,T) in Kap.2.1 ! ) Für isochore Prozesse (das Volumen bleibt konstant) gilt damit dU = dQ = ncVdT *, im Allgemeinen gilt immer und für ein T-unabhängiges cV folgt: *) dU = dQ gilt streng nur, wenn neben der Volumenarbeit auch keine anderer Art von Arbeit bei dem Prozess verrichtet wird. 2.1.3 Die Innere Energie des idealen Gases Ideale Gase sind per Definition frei von Wechselwirkungen. Die innere Energie eines idealen Gases ist daher auch ausschließlich in der Bewegung der Teilchen enthalten, und nach Boltzmann (vgl. Kap.1.5.1) durch den Ausdruck 1 f f U u N A E kin N A k B T Rm T n 2 2 gegeben und somit makroskopisch betrachtet nur von der Temperatur abhängig! Hoeppe - PC Leerscript - 32 - Daher ist bei einem idealen Gas eine Änderung der inneren Energie U mit gegeben. Für ein beliebiges einatomiges Gas (f=3) ergibt sich daher für die molare Wärmekapazität . Für mehratomige Gase mit f > 3 berechnet sich cV entsprechend und ist nur von der Zahl der Freiheitsgrade und nicht z.B. von der Atomart abhängig. Auch „ideale“ Festkörper haben alle die gleiche molare Wärmekapazität von 3R, da man 3 x 2 Schwingungsfreiheitsgrade, also f=6, ansetzt. ( „Dulong-Petit“) Das für ein ideales Gas U=U(T) gilt zeigt sich prinzipiell auch im Experiment: Betrachte hierbei die Expansion eines idealen Gases auf zwei verschiedenen Arten: 1.) Adiabatische Expansion durch Kolbenverschiebung: Es wird Volumenarbeit geleistet, wegen thermischer Isolation kann aber keine Wärme ausgetauscht werden. U wird verringert T fällt 2. Adiabatische Expansion durch Überströmen: Es wird keine Volumenarbeit geleistet, und wegen thermischer Isolation auch keine Wärme ausgetauscht. U bleibt konstant T = const Das (schwierige) Experiment zeigt, dass die Temperatur bei nahezu idealen Gasen nahezu unverändert bleibt, bei realen Gasen aber nicht! ( vgl. Joule Thomson Effekt, Effusion). ( Warum sollte bei einem idealen Gas U auch vom Volumen abhängig sein ? ) Hoeppe - PC Leerscript - 33 - 2.2 Enthalpie Bei jedem Wärmeumsatz wird nach 2.1.1 aufgrund thermischer Ausdehnung auch Volumenarbeit geleistet, wenn das Volumen nicht durch geeignete Maßnahmen konstant gehalten wird. Dies ist jedoch allenfalls bei Gasen realisierbar, bei Festkörpern und Flüssigkeiten aufgrund der kleinen Kompressibilität kaum möglich. Zudem laufen viele Prozesse, insbesondere die meisten chemischen Reaktionen, bei Normaldruck p = po ab. Eine Änderung der inneren Energie U entspricht somit i. A. nicht dem Wärmeumsatz sondern entsprechend dem 1. HS gilt dU = dQ - pdV bzw. dQ = dU + pdV . Man definiert daher die neue Zustandsgröße Enthalpie H : Für den meistens gegebenen Fall von p=const ist damit dQ = dU + pdV + Vdp = dH und die umgesetzte Wärme durch eine Zustandsgröße beschrieben. Als unabhängige Variable wählt man für die Enthalpie praktischer Weise p und T: Totales Differential von H(p,T): Für konstanten Druck entspricht eine Änderung der Enthalpie dem Wärmeumsatz, daher definiert man die Spezifische Wärme bei konstantem Druck: Für const. Druck folgt damit für die bei einem Prozess umgesetzte differentielle Wärmemenge dQ: Für ein T-unabhängiges cp folgt: i. A. ist die spezifische Wärme jedoch deutlich temperaturabhängig (vgl. 2.2.1) und damit gilt für Q: Hoeppe - PC Leerscript - 34 - 2.2.1 Temperaturabhängigkeit der spezifischen Wärme Aufgrund von z.B. struktureller Veränderungen von Festkörpern ist dort eine Temperaturabhängigkeit der spezifischen Wärme zu erwarten, aber auch bei Gasen ändert sich aufgrund zunächst „seltsamer“ quantenmechanischer Effekte die spez. Wärme mit der Temperatur: Da die Energie von Schwingungs- und Rotationszuständen quantisiert ist, „frieren“ diese bei tiefen Temperaturen ein. Da die Energie entsprechend Boltzmann statistisch auf die Freiheitsgrade verteilt ist, ist der Temperaturverlauf für die (makroskopische) molare Wärmekapazität aber stetig. Die Translationsenergie ist dagegen nicht quantisiert. Hoeppe - PC Leerscript - 35 - Massen, Massenträgheitsmomente, Bindungskräfte und –längen etc. sind molekül- bzw. stoffspezifische Größen. Die T - Abhängigkeit von cp(T) wird meist mit einer Potenzreihe dargestellt: c p (T ) A B 10 3T C 10 5T 2 D10 6 T 2 Für einige Stoffe sind die Reihenentwicklungsparameter in Tab D dargestellt. Für einen Wärmeumsatz bei konstantem Druck gilt damit für Q : T 2 1 Q c p (T )dT n T1 T2 3 5 1 6 2 A B 10 T C 10 D 10 T dT 2 T T 1 Für die Reihenentwicklung von oben ist die allgemeine Lösung des Integrals: 1 1 1 2 2 3 3 3 6 5 1 c ( T ) dT A T T B 10 ( T T ) C 10 D 10 ( T T ) p 2 1 2 1 2 1 T T T 3 2 1 2 1 T2 Liegt zwischen den Temperaturen T1 und T2 eine Phasenumwandlung (ändert sich z.B. der Aggregatzustand) muss das obige Integral ‚stückweise’ mit den entsprechenden Parametern berechnet werden. Zudem muss der mit der Umwandlung verbundene Wärmeumsatz berücksichtigt werden (latente Wärme). 2.2.2 Latente Wärme Als latente (= versteckte) Wärme bezeichnet man die bei konstanter Temperatur umgesetzte Wärme, was i. A. mit einem Phasenübergang verbunden ist. (Damit sind nicht nur Änderungen des Aggregatzustandes gemeint sondern auch z.B. eine Umkristallisation eines Festkörpers oder der Übergang von der paramagnetischen in die ferromagnetische Phase.) Bsp: Schmelzen + Verdampfen Diese latente Wärme bzw. Umwandlungsenthalpie hu = ist die Energie bzw. Wärme, welche für die Phasenumwandlung ‚benötigt’ wird. Damit gilt für den gesamten Wärmeumsatz bei Temperaturänderung einer Stoffmenge von T1 nach T2: Hoeppe - PC Leerscript - 36 - 2.2.3 Zusammenhang der spezifischen Wärmen cv und cp Bei Flüssigkeiten und Festkörpern ist aufgrund ihrer vergleichsweise geringen Wärmeausdehnung der Unterschied von cp und cv meist vernachlässigbar. Für ideale Gase gilt nach Definition: Mit folgt Für ein einatomiges ideales Gas ergibt sich somit cV = 3/2 R und cp = 5/2 R , usw. (vgl. Kap. 1.5.1). 2.3 Idealisierte Zustandsänderungen Für die Berechnung der Änderung einer Zustandsgröße für einen beliebigen Prozess 12 ist es per Definition der Zustandsgrößen ( Wegunabhängigkeit des Linienintegrals! ) unerheblich, wie der Prozess tatsächlich oder im Detail stattfindet. Es ist ausreichend einen beliebigen einfach zu berechnenden Weg zu wählen, der vom Ausgangszustand 1 zum Endzustand 2 führt. Daher werden im Folgenden idealisierte Prozesse betrachtet, die sich bei Bedarf beliebig aneinanderreihen lassen. 2.3.1 Isochore Zustandsänderung Hier gilt V = const. bzw. dV = 0 dU = dH = dQ = dW = Hoeppe - PC Leerscript - 37 - 2.3.2 Isobare Zustandsänderung Hier gilt p = const. bzw. dp = 0 dU = dH = dQ = dW = 2.3.3 Isotherme Zustandsänderung Hier gilt T = const. bzw. dT = 0. Für ein ideales Gas gilt wegen U = U(T): dU = dH = dQ = dW = Bsp.: Isotherme Expansion eines id. Gases : Näherungsweise isotherme Prozesse sind i.d.R. sehr langsame Prozesse, so dass durch Wärmeleitung von oder zu einem sehr großen Wärmereservoir (d.h. die Umgebung) die Temperatur des Systems konstant gehalten wird. Hoeppe - PC Leerscript - 38 - 2.3.4 Adiabatische Zustandsänderung Für reversible Prozesse ist auch der Bergriff „isentrop“ gebräuchlich. Hier gilt Q = const. bzw. dQ = 0. dU = dH = dQ = dW = Noch gesucht ist p = p(V), d.h. die zugehörige Kurve im p-V-Diagramm (Adiabate), z. B. um die adiabatische Kompressibilität berechnen zu können, mit der sich wiederum z. B. die Schallgeschwindigkeit bestimmt. Für ein ideales Gas gilt wegen U = U(T): dW = dU = ncVdT. Leistet das System infolge Expansion Arbeit, so muss damit die innere Energie abnehmen. Da keine Wärme „nachfließen“ kann (und andere Arbeiten ausgeschlossen sein sollen) sinkt die Temperatur des Gases entsprechend. dU ncV dT pdV nRT dV V cV 1 1 dT R dV T V Integration für einen bel. adiabatischen Prozess 1 2 liefert: cV ln T2 V R ln 1 T1 V2 T ln 2 T1 cV V ln 1 V2 Mit cp = cv + R und dem sog. Adiabatenexponent R cp cV T2 V1 T1 V2 R cV folgt bzw. c p 7 / 2 R 7 14 Für Luft (N2, O2) gilt f = 5 und damit: = c 5 / 2 R 5 10 1,4 V Hoeppe - PC Leerscript - 39 - Näherungsweise adiabatische Prozesse sind i.d.R. sehr schnelle Prozesse, so dass durch (die langsame) Wärmeleitung kein Temperaturausgleich stattfinden kann. Da reale Prozesse nie ganz adiabatisch (Luft: = 1,4) oder ganz isotherm ( 1) ablaufen können, wird der tatsächliche Prozess am besten mit einem Polytropenexponenten beschrieben, wobei hier immer > > 1 gilt. Für die Polytrope gilt dann 2.3.5 p V const Isenthalpische Zustandsänderung - Joule Thomson Effekt Hier gilt H = const bzw. dH = 0. Diese Zustandsänderung betrifft vor allem technische Strömungsprozesse, also offene Systeme. Beispielhaft wird hier der sog. Joule-Thomson-Effekt behandelt: Betrachte ein Teilvolumen V eines strömenden Gases durch Drosselstelle, wobei ein Wärmeaustausch durch eine thermische Isolation verhindert wird, d.h. es gilt dQ=0: Die Drosselstelle erzeugt bei einem strömenden Gas eine Druckdifferenz p, d.h. es ist p2 < p1 und in Folge dessen auch V2 > V1. Bei der Energie und Arbeitsbilanz für diesen Strömungsprozess wird eine potentiell verrichtete Volumenarbeit und die innere Energie des Gases berücksichtigt: U 1 p1 V1 U 2 p 2 V2 d.h. H1 H 2 Offenbar ist H = 0 bzw. dH = 0 „isenthalpisch“ . Experimentelle Beobachtung: T ändert sich hinter Drosselstelle; oft Abkühlung! Hoeppe - PC Leerscript - 40 - A) Ideales Gas: Betrachtet man das strömende Gas als ideales Gas gilt für eine Enthalpieänderung dH d (U p V ) dU d ( p V ) n cV dT d (nRT ) ! dH (n cV nR) dT 0 dT 0 T const Mit einem idealen Gas kann also keine Änderung der Temperatur beobachtet bzw. erklärt werden. Bei der Expansion hinter der Drosselstelle wird ja auch keine Volumenarbeit verrichtet (vgl. „Effusion“ Kap. 2.1) und da U = U(T) und dQ = 0 aufgrund der Isolation gilt, darf sich T auch nicht ändern. B) Reales Gas (hier: Van der Waals): Bei einem realen Gas ist U nicht nur von T abhängig, sondern auch von den WW zwischen den Gasmolekülen und damit auch vom Volumen V ! Vereinfacht gilt dU n cV dT n dU WW n cV dT * dH (n cV nR) dT a dV V2 ! a dV 0 dT 0 ! V2 Aufgrund der Volumenzunahme wird die (attraktive) WW geringer, die für die Trennung der Moleküle notwendige Energie entstammt der kinetische Energie womit die Temperatur fällt! *) Dieser Ansatz ist (stark) vereinfacht, es fehlt hier noch die Korrektur für p im p·V Term der Enthalpie sowie die Korrektur des Volumens um das Eigenvolumen der Moleküle mit dem Parameter b. Eine etwas genauere Rechnung (vgl. z.B. Gerthsen, Kneser, Vogel; Physik) zeigt: Eine Abkühlung des Gases erhält man nur für T < Ti wobei die sog. Inversionstemperatur von den Konstanten a und b abhängt: Ti 2 a 27 TKrit R b 4 - Für Luft (N2 und O2 ) ist Ti 700 K Abkühlung ‚kein Problem’ - Für H2 ist Ti 200K Drosselung führt bei Raumtemperatur zu einer Erwärmung! Bei hohen Temperaturen verschwindet offensichtlich der Einfluss der WW (~1/V²) während die Korrektur des Molvolumens mit b nur linear mit V abnimmt. Hoeppe - PC Leerscript - 41 - Anwendung des Joule Thomson Effekts: Luftverflüssigung nach Linde Durch wiederholte Anwendung des Drosseleffekts wird Luft bis zur Verflüssigung bei ca. 83 K (bei Normaldruck) abgekühlt. Durch den Wärmetauscher wird verhindert, dass mit der gasförmigen Luft zuviel Wärme in den „Kaltbereich“ eingetragen wird. Kolben Ventile Wärmetauscher p 200 bar Drosselstelle p 20 bar Flüssige Luft Hoeppe - PC Leerscript - 42 - 2.4 Reaktionswärme Nicht nur physikalische Prozesse sondern insbesondere auch chemische Prozesse sind mit einem Wärmeumsatz verbunden, da sich die Bindungsenergie von den beteiligten Edukten und Produkten verändert. In der (chemischen) Thermodynamik wird nach den Details der Reaktion nicht gefragt, sondern („nur“) der Anfangszustand und der (potentielle) Endzustand miteinander verglichen. 2.4.1 Reaktionsgleichung Eine chemische Reaktion wird eindeutig durch die Reaktionsgleichung beschrieben, wobei die stöchiometrischen Faktoren i den Umsatz von jeweils 1 mol des entsprechenden Produkts bzw. Edukts beschreiben. Falls zweckmäßig kann die Reaktionsgleichung auf beliebige anderer Reaktionsmengen durch Multiplikation mit einem Reaktionsparameter umgestellt werden. Infolge der chemischen Reaktion bleibt zwar die Anzahl der Atome bzw. der chemischen Elemente erhalten, die Molzahl der Stoffe – d.h. der chemischen Verbindungen- kann sich durchaus ändern. Entsprechend Definition ändert sich die Molzahl von jedem Edukt/Produkt um -/+ ni = i . Die Änderung der Gesamtmolzahl ist insbesondere bei Gasreaktionen wichtig, da sich dadurch bei konstantem Volumen der Druck ändert (vgl. 2.4.2). 2.4.2 Bestimmung der Reaktionswärme Während ‚normalerweise’ eher der Druck als konstant angenommen werden kann, (Standard Laborbedingung) wird bei der Messung einer Reaktionswärme das Volumen konstant gehalten, da Edukte und Produkte ‚eingesperrt’ werden müssen. So kann die Reaktion möglichst vollständig ablaufen und der Wärmeumsatz kann in einem Kaloriemeter gemessen werden: Hoeppe - PC Leerscript - 43 - Wird in einem sog. Bombenkaloriemeter der Wärmeumsatz gemessen, so entspricht wegen der Randbedingung V = const Q = U. Für die Anwendung bei p = const wird aber die Änderung der Enthalpie H benötigt. Es gilt daher zu unterscheiden: a) Festkörper und Flüssigkeiten Ändern sich bei einer Reaktion nur die Molzahlen von Flüssigkeiten oder Festkörpern wird aufgrund der relativ kleinen und ähnlichen Molvolumina nur verschwindend wenig Volumenarbeit geleistet. Es gilt daher in guter Näherung: (vgl. A 2.12) b) Gase Ändert sich bei einer Reaktion die Gesamtmolzahl der Gase würde bei p=const Volumenarbeit geleistet werden; bei V=const ändert sich aber der Druck näherungsweise mit dem id. Gasgesetz entsprechend: p n RT V Für die Änderung der Enthalpie H =Q + Vp gilt daher (vgl. A 2.13) 2.4.3 Standardbildungsenthalpie Assoziiert man den Wärmeumsatz bei chemischen Reaktionen mit dem Verschwinden oder Erscheinen von chemischen Verbindungen (= Stoffen) und den damit verbundenen Bindungsenergien, so ist es sinnvoll jedem Stoff eine Standardbildungsenthalpie h0298 zuzuordnen. Die molare Standardbildungsenthalpie h0298 eines Stoffes entspricht der Wärmemenge die frei wird, wenn sich der jeweilige Stoff bei Standardzustand ( p = 1 bar und T = 298K ) aus den Elementen bildet. Für Elemente in ihrer natürlichen Form gilt dabei: ( Tab. F ) Hoeppe - PC Leerscript - 44 - Da mit cp(T) die Änderung der Enthalpie bei einer Temperaturänderung eindeutig definiert ist, ist auch die molare Enthalpie eines Stoffes für bel. Temperaturen eindeutig festgelegt: (vgl. A 2.14, A 2.15) Sind nun mit Hilfe jeweils eines Experiments die molaren Standardenthalpien der Stoffe A, B, C und D bekannt, so lässt sich für eine beliebige Reaktion zwischen diesen Stoffen der Wärmeumsatz durch einfache Differenzbildung bestimmen! 2.4.4 Reaktionsenthalpie Mit den oben eingeführten molaren Standardenthalpien definiert sich die (molare) Reaktionsenthalpie Rh einfach durch die Differenz der vorhandenen Standardenthalpien (also Bindungsenergien) vor und nach der Reaktion: Reaktionsenthalpie Rh bei Standardbedingungen p = po = 1 bar; T = 298 K : Reaktionsenthalpie Rh bei Standarddruck p = po = 1 bar und beliebiger TemperaturT: Die h0i,T sind dabei die Enthalpien der i-ten Komponente bei der Temperatur T. Hoeppe - PC Leerscript - 45 - Achtung: Die Reaktionsenthalpie Rh0T ist nicht gleich der Standardreaktionsenthalpie Rh0298 , obwohl dies auf den ersten Blick zu vermuten wäre, schließlich werden z.B. die Edukte von 298K auf die Reaktionstemperatur T aufgeheizt und die aus den gleichen Elementen bestehenden Produkte (also auch die gleiche Masse) anschließend wieder auf 298 K abgekühlt. Durch die chemische Reaktion verändert sich aber i. A. die spezifische Wärmekapazität, weshalb sich die umgesetzten Wärmen bei Aufheizen und Abkühlen unterscheiden! Zudem verschiebt sich durch etwaige Phasenumwandlungen bei Produkten und Edukten die Wärmebilanz. Für den speziellen Fall, dass bei keiner beteiligten Komponente zwischen 298 K und T eine Phasenumwandlung auftritt, lässt sich die Reaktionswärme auch mit der untenstehenden „verkürzten Formel“ berechnen: T h h 0 R T 0 R 298 c p (T ) dT 298 mit c p (T ) A B 10 3 T C 10 5 1 D 10 6 T 2 2 T A i Ai Pr i Ai Ed B i Bi Pr i Bi Ed usw. (vgl. A 2.21) 2.4.5 Reaktionsenthalpie bei Reaktionen in wässriger Lösung Löst sich ein Stoff in Wasser (oder einem anderen Lösungsmittel) liegt dieser in einer anderen Form (~ Phase) vor. Der Lösungsvorgang lässt sich in folgende mit einem Wärmeumsatz verbundene Teilschritte beschreiben: - (Verdampfen und) Ionisierung des zu lösenden Stoffes, Gitterenergie endotherm - Entassoziation der H2O Moleküle, endotherm - Einlagerung der „nackten“ Ionen in Lösungsmittel, Solvationsenergie exotherm Die Energiebilanz dieses Lösungsvorgangs wird durch die Lösungswärme Lh beschrieben. Da jeder Stoff in Form von Ionenpaaren in Lösung geht, ist eine Darstellung von Reaktionsenthalpien für Reaktionen der Ionen untereinander zunächst nicht eindeutig. Mit der Definition wird die Eindeutigkeit wieder hergestellt. ( vgl. Bsp. Lösen von HCl in Wasser; Tabelle G ) Hoeppe - PC Leerscript - 46 - 3 3.1 3.1.1 Entropieumsatz bei Stoffumwandlungen Vorüberlegungen Reversible und irreversible Prozesse Reversibel und irreversibel sind Begriffe einer makroskopischen Betrachtung von Prozessen. Dabei bedeutet: reversibel: irreversibel: Def.: Man bezeichnet einen Prozess 12, der ein System vom Zustand 1 nach Zustand 2 überführt, als reversibel, wenn ein Prozess 21 existiert, der das System in den Ausgangszustand 1 überführt, ohne das im System oder in der Umgebung irgendeine Änderung verbleibt. Viele (streng genommen eigentlich alle) spontan ablaufende Prozesse sind entsprechend der Beobachtung irreversibel, und damit leider auch beliebig schwer vollständig zu beschreiben. Zum Glück ist das auch meistens gar nicht nötig: Es genügt, wenn der Prozess innerhalb des Systems eindeutig beschrieben wird. Mit der Vernachlässigung der Umgebung beschränken wir uns auf die Beschreibung von geschlossenen oder offenen Systemen, bei abgeschlossenen Systemen hat die Umgebung ja per Definition sowieso keinen Einfluss. Irreversible Prozesse in nicht abgeschlossenen Systemen: Da die Zustandsgrößen eines Systems eindeutig mit der Zustandsgleichung gegeben sind, spielt es für diese folglich auch keine Rolle auf welchem Weg (Prozess) das System in einen bestimmten Zustand gelangt ist. Ist ein Prozess in einem nicht abgeschlossenen System tatsächlich irreversibel, kann man die Änderung der Zustandsgrößen dennoch (einfach) beschreiben, in dem man einen äquivalenten reversiblen Prozess findet, der zum gleichen Endzustand führt. (Dies ist prinzipiell immer möglich, da infinitesimale (d.h. einzelne mikroskopische) Prozesse stets reversibel sind.) Mit Hilfe der Zustandsgröße Entropie, welche später eingeführt wird, lässt sich dann sogar rechnerisch entscheiden, ob ein Prozess in einem abgeschlossenen System prinzipiell reversibel oder irreversibel ist, und damit ob dieser Prozess spontan (d.h. „ohne Hinzutun“) ablaufen wird oder nicht. Im Folgenden werden daher Beispiele für solche äquivalenten Prozesse betrachtet: Hoeppe - PC Leerscript - 47 - abgeschlossenes System geschlossenes System Gewicht fallen lassen irreversibel Gewicht senken reversibel Effusion irreversibel Kolbenexpansion reversibel Diffusion irreversibel Semipermeabler Kolben reversibel Hoeppe - PC Leerscript - 48 - 3.1.2 Carnotprozess: Der Carnotprozess dient der idealisierten theoretischen Beschreibung von Wärmekraftmaschinen, also Maschinen die Wärme in Arbeit umwandeln (oder umgekehrt). Eine solche Maschine soll - zyklisch arbeiten ( Kreisprozess im Zustandsraum und pV-Diagramm) - ideal, d.h. reibungs- und damit verlustfrei arbeiten ( reversible Prozesse) - mit einem (bei Wärmezufuhr expandierenden) idealen Gas betrieben werden. Damit für einen Zyklus netto Arbeit geleistet wird, müssen mindestens zwei verschiedene Prozesse durchgeführt werden: Erfolgt die Expansion bei Kolbenvorschub isotherm, so muss für die folgende Kompression zunächst die Temperatur des Gases gesenkt werden, damit hierfür nicht wieder die gleiche Menge Arbeit nötig ist, die bei der Expansion geleistet wurde. Dies wird z. B. durch eine adiabatische Expansion gewährleistet. N. L. S. Carnot (1796-1832) betrachtete eine (leicht zu berechnende) Abfolge von isothermen und adiabatischen Prozessen 1 2 3 4 1 ... wie folgt: Ein in umgekehrter Reihenfolge ablaufender Kreisprozess entspricht einer Kraftwärmemaschine (z.B. Wärmepumpe oder Kühlschrank). Im Folgenden wird der Wärme- und Arbeitsumsatz jedes einzelnen Teilschrittes betrachtet und anschließend das Verhältnis von zugeführter Wärme zu erhaltener Arbeit, also der Wirkungsgrad, bestimmt: Hoeppe - PC Leerscript - 49 - 1 2: isotherme Expansion bei T = T0 Q12 W12 nRT0 ln 2 3: adiabatische Expansion V2 V1 W23 U 23 ncV T23 ncV (Tu T0 ) 3 4: isotherme Kompression bei T = Tu Q34 W34 nRTu ln 4 1: adiabatische Kompression V4 V3 W41 U 41 ncV T41 ncV (T0 Tu ) Gesamtenergiebilanz für Kreisprozess: Wges W12 W23 W34 W41 V1 V ncV (Tu T0 ) nRTu ln 4 ncV (T0 Tu ) V2 V3 V V Q12 Q34 nRT0 ln 1 nRTu ln 4 V2 V3 Wges nRT0 ln Qges U ges Wges Qges 0 entsprechend dem 1. HS. Interessanter ist der Wirkungsgrad , allgemein Nutzen/Aufwand, hier also: das Verhältnis zugeführter Wärme Q zu erhaltener Arbeit W: : Hoeppe - PC Leerscript Wges Q12 V1 V nRTu ln 4 V2 V3 V nRT0 ln 1 V2 nRT0 ln - 50 - Die ln-Terme in obigem Ausdruck lassen sich eliminieren, da für die Adiabaten TV -1 = const gilt: T0V2 1 1 T0V1 TuV3 TuV4 1 1 T0 V3 Tu V2 T0 V4 Tu V1 1 1 V3 V4 V2 V1 bzw. und damit V4 V1 V3 V2 V ln 4 V3 V ln 1 V2 Damit ergibt sich als Wirkungsgrad für den Carnot’schen Kreisprozess: Carnot W ges Q12 T0 Tu T0 Da Tu = 0 grundsätzlich unmöglich ist, gilt also immer < 1 ! Damit eine Maschine zyklisch arbeiten kann, muss ein Teil der zugeführten Wärme bei der Temperatur T0 wieder bei Tu abgegeben werden. Eine vollständige Umwandlung der zugeführten Wärme in Arbeit ist also prinzipiell unmöglich. Jeder andere technische Kreisprozess kann durch eine Abfolge von infinitesimal kleinen isothermen und adiabatischen Prozessen approximiert werden, daher ist bei einem beliebigen anderen Kreisprozess auch kein größerer Wirkungsgrad möglich. Im Gegenteil: Bei realen Maschinen ist aufgrund von Reibung etc. der Wirkungsgrad noch geringer! Aus obiger Erkenntnis folgt bereits eine mögliche Formulierung des 2. HS der Thermodynamik: Es existiert kein Perpetuum Mobile 2. Art, d.h. keine periodisch arbeitende Maschine, die nichts weiter tut, als Wärme in Arbeit umzuwandeln. Hoeppe - PC Leerscript - 51 - 3.2 3.2.1 Zweiter Hauptsatz der Thermodynamik - Entropie Reduzierte Wärme und Entropie Um zu einer quantitativen mathematischen Formulierung des 2. HS zu gelangen, betrachten wir nochmals genauer die umgesetzten Wärmemengen Qi einer Carnotmaschine, wobei sich diese samt der Wärmereservoirs bei T0 und Tu in einem abgeschlossenen System 2 befinden soll: Die Gesamtenergiebilanz für den Kreisprozess bzgl. des (kleineren) geschlossenen Systems 1 in welchem sich nur die Carnotmaschine befindet ergibt U = 0. Für die umgesetzten Wärmemengen Q0 bei T0 und Qu bei Tu gilt (vgl. 3.1.2): Offensichtlich gilt: bzw. Für jeden anderen (reversiblen) Umsatz von Wärmemengen (weitere Carnotmaschinen oder andere reversible Prozesse) gilt das Gleiche, und daher allgemein: Die Summe der reduzierten Wärmen Qi/Ti ist in einem abgeschlossenen System null, d.h. eine Erhaltungsgröße. Hoeppe - PC Leerscript - 52 - Wenn sich in einem abgeschlossenen System eine Größe nicht ändert, so existiert offensichtlich eine entsprechende Zustandsgröße, die mit dem Zustand des Systems eindeutig bestimmt ist. R. Clausius (1822-1888) definierte diese neue Zustandsgröße Entropie S mit . Eine Entropieänderung S ergibt sich also aus der Summe der reversibel umgesetzten reduzierten Wärmen. 3.2.2 Quantitative Formulierung des 2.HS In einem abgeschlossen System gilt falls darin nur reversible Prozesse ablaufen. Aus Erfahrung gilt für beliebige Prozesse in einem abgeschlossenen System: Das bedeutet, dass bestimmte (irreversible) Prozesse nur in einer Richtung ablaufen können, da dS < 0 und damit S < 0 in einem abgeschlossenen System unmöglich ist! Damit wird, im Gegensatz zu allen anderen physikalischen Gesetzen, die prinzipiell umkehrbar (zeitumkehrinvariant) sind, die Richtung des Zeitablaufs festgelegt. Da sich für irreversible Prozesse in einem abgeschlossenen System stets auch äquivalente reversible Prozesse in einem geschlossenem System finden lassen, (vgl. Kap 3.1.1) ist auch für diese eine Entropieänderung S berechenbar. Allgemein gilt dann für einen beliebigen (potentiellen) Prozess in einem abgeschlossenen System: Hoeppe - PC Leerscript S = 0 S > 0 S < 0 - 53 - 3.2.3. Berechnung von Entropieänderungen reversibler Prozesse a) adiabatische Prozesse: b) isotherme Prozesse: Für die Berechnung von S ist ein expliziter Ausdruck für dQ des jeweiligen Prozesses erforderlich. Betrachte daher als Beispiel die isotherme Expansion eines idealen Gases: Wegen dU = 0 (isotherm) gilt hier mit dem 1. HS dQ = -dW = pdV (vgl. Kap. 2.3.3). Beachte: Für die Expansion ist V2 > V1 und damit S > 0. Läuft diese Expansion in einem abgeschlossenen System ab (z.B. Effusion), so ist dieser Prozess irreversibel! Wie schon in Kap. 3.1.1 dargestellt, ist ein Diffusionsprozess auf die isotherme Expansion eines id. Gases abbildbar. Somit ist auch ein Diffusionsprozess (in einem abgeschlossenen System) irreversibel. Effusion und Diffusion laufen also spontan, d.h. ohne hinzutun ab. c) isochore Prozesse: Betrachte hier die Erwärmung einer Stoffmenge mit konstanter Wärmekapazität cV in infinitesimal kleinen Schritten, d.h. in diesem Grenzfall reversibel. (vgl. e) Wärmeleitung!) Hoeppe - PC Leerscript - 54 - d) isobare Prozesse: Betrachte hier wie zuvor die Erwärmung einer Stoffmenge, jetzt mit konstanter Wärmekapazität cp in infinitesimal kleinen Schritten. (vgl. e) Wärmeleitung!) e) Wärmeleitung: Während eine Erwärmung wie unter c) und d) diskutiert zwangsläufig nur in einem nicht abgeschlossenen System stattfinden kann, betrachten wir jetzt die Wärmeleitung in einem abgeschlossenen System: Anfangs seien die Temperaturen TA und TB der Teilmengen A und B unterschiedlich, infolge Wärmeleitung findet ein Temperaturausgleich auf die Mischungstemperatur T statt (vgl. Kap. 1.2.3). Da die Entropie eine extensive Größe ist, können wir die Gesamtentropieänderung S aus den Entropieänderungen der Teilsysteme A und B bestimmen. Für diese ergibt sich SA und SB aus den Temperaturänderungen wie unter d (oder c) beschrieben: Da T zwischen TA und TB liegt, ist ein Entropiebeitrag negativ und der andere positiv. Für gleiche Wärmekapazitäten, also nAcp,A = nBcp,B = C, ergibt sich (vgl. Kap. 1.2.3) als Mischungstemperatur einfach das arithmetische Mittel und damit: Hoeppe - PC Leerscript - 55 - Da das arithmetische Mittel (im Zähler des ln-Terms) immer größer ist als das geometrische Mittel (im Nenner), ist das Argument des ln-Terms immer größer 1 und damit S > 0. Wärmeleitung ist also prinzipiell ein irreversibler Prozess! f) Mischungsentropie: Die Vermischung zweier unterschiedlicher Stoffe ( Diffusion) lässt sich auf die isotherme Expansion beider Stoffe auf das Gesamtvolumen abbilden: Berechnen wir die Entropieänderungen der Teilsysteme Gas A und Gas B wie unter b) beschrieben, ergibt sich für die Gesamtentropieänderung Die Entropie nimmt für beide Teilsysteme und damit für das Gesamtsystem zu. Die Vermischung von Stoffen ist in einem abgeschlossenen System somit irreversibel, und läuft daher spontan ab. g) Phasenübergänge: Da Phasenübergänge bei konstantem Druck und Temperatur ablaufen, berechnet sich hier die Entropieänderung ohne Integration einfach aus der Umwandlungswärme: Hoeppe - PC Leerscript - 56 - 3.2.4 Statistische Interpretation der Entropie Boltzmann (1844-1906) führte alternativ zu Formulierung von Clausius eine statistische Definition der Entropie ein (vgl. 3.2.5). Dabei ist entscheidend, mit welcher Wahrscheinlichkeit P ein makroskopischer Zustand eingenommen wird. Diese ergibt sich wiederum aus der Zahl mikroskopischer Zustände , welche zu dem gleichen makroskopischen Zustand führen. Der makroskopische Zustand mit den meisten Realisierungsmöglichkeiten (d.h. mit der größten Unordnung) ist der wahrscheinlichste und derjenige welcher letztlich beobachtet wird. Daher streben (abgeschlossene) Systeme (z.B. bei Variation durch rein zufällige Teilchenbewegung) grundsätzlich einen Zustand maximaler Unordnung an. Die Entropie eines Zustandes ist nach Boltzmann mit S k B ln definiert. Die Zahl mikroskopischer Realisierungsmöglichkeiten bezeichnet man auch als ‚statistisches Gewicht’ und entspricht einer (nicht normierten) Wahrscheinlichkeit des Zustandes. Ein „Streben nach Unordnung“ entspricht damit einer Entropieerhöhung und somit dem 2. HS der Thermodynamik! Betrachten wir das mikroskopische Modell eines id. Gases aus N Teilchen, welche sich rein zufällig in einem Kasten bewegen. Der makroskopische Zustand sei durch die Anzahl der Teilchen links ( bzw. rechts) beschrieben: Wie wahrscheinlich ist es, dass sich die Teilchen gleichmäßig auf beide Seiten verteilen? Wie wahrscheinlich ist es, dass sich alle Teilchen links befinden? 1 Teilchen: Hoeppe - PC Leerscript - 57 - 2 Teilchen: 3 Teilchen: 4 Teilchen: Hoeppe - PC Leerscript - 58 - 100 Teilchen Offensichtlich ist es sehr wahrscheinlich, dass sich die Teilchen gleichmäßig auf beide Seiten verteilen. Offensichtlich ist es äußerst unwahrscheinlich, dass sich alle Teilchen links befinden, bzw. derart unwahrscheinlich, dass wir „unmöglich“ sagen. 3.2.5 Äquivalenz der Definitionen nach Clausius und der nach Boltzmann Betrachte nochmals die isotherme Expansion des id. Gases makroskopisch nach Clausius a), und mikroskopisch nach Boltzmann b): a) Clausius Berechnung wie unter 3.2.3.b: 2 2 2 V dW dQ pdV nR S dV n R ln 2 T T T V V1 1 1 1 1 2 b) Boltzmann Hoeppe - PC Leerscript - 59 - Wahrscheinlichkeit P, dass sich ein Teilchen am Ende im Volumen V1 befindet: V P1 1 V2 N Teichen: V P1 1 V2 N V 1 1 V2 N (Wobei wir hier ausnutzen, dass das statistische Gewicht bis auf eine Normierungskonstante gleich der Wahrscheinlichkeit ist.) Wahrscheinlichkeit P, dass sich ein Teilchen am Ende im Volumen V2 befindet: V P2 2 V2 N Teichen: V P2 2 V2 N N V 2 2 V2 (Wobei das statistische Gewicht proportional der jeweiligen Wahrscheinlichkeit P ist.) Entropieänderung: S S 2 S1 k B ln 2 k B ln 1 k B ln 2 k B ln N 1 V1 V 2 N V2 k B ln V1 V V V N k B ln 2 n N A k B ln 2 n R ln 2 V1 V1 V1 Offensichtlich liefern beide Definitionen das gleiche Ergebnis für die Entropieänderung! Allerdings fällt auf, dass die Definition nach Boltzmann anscheinend auch eine Absolutberechnung der Entropie erlaubt, während mit der Definition nach Clausius nur eine Entropieänderung definiert ist. Dieser „Mangel“ wird mit dem dritten Hauptsatz behoben. Hoeppe - PC Leerscript - 60 - 3.3 Dritter Hauptsatz und Absolutberechnung der Entropie Man beobachtet experimentell ein Verschwinden der spezifischen Wärmekapazitäten bei Annäherung an den absoluten Nullpunkt. Damit werden auch Entropiedifferenzen immer kleiner. (Erklären lässt sich dies nur mit dem quantenmechanischen ‚Einfrieren’ von Freiheitsgraden.) Nernst (1864-1941) formulierte aufgrund dieser Beobachtung das heute sog. Nernstsche Wärmetheorem: Aufgrund von Ergebnissen der statistischen Physik formulierte Planck (1858-1947) später deutlich schärfer den 3. H.S. der Thermodynamik nachdem die Entropie eines idealen kristallinen Festkörpers für T 0 verschwindet. Diese Festlegung ermöglicht die Absolutberechnung der Entropie so(T) mit für beliebige Temperaturen T. Zweckmäßigerweise wird die Standardentropie so298 = so(T = 298K, p = po) definiert: ( Tab. F) Damit vereinfacht sich die so(T) Berechnungen bei „handelsüblichen“ Temperaturen: ( so: Tab. F, hu: Tab. E) Wird zur Beschreibung von cp(T) wieder die gleiche Reihenentwicklung Hoeppe - PC Leerscript - 61 - c p (T ) A B 10 3T C 10 5T 2 D10 6T 2 verwendet, berechnet sich das Integral in obiger Gleichung entsprechend T2 T 2 cP A 3 5 1 6 dT B 10 C 10 D 10 T dT 3 T T T T T 1 1 mit der allgemeinen Lösung 1 cP T2 1 1 1 2 2 5 3 6 dT A ln B 10 ( T T ) C 10 D 10 T T 2 1 2 1 T T 2 T 2 2 T1 2 T1 1 2 T2 3.4 Reaktionsentropie Rs Mit den in 3.3 eingeführten Standardentropien berechnet sich eine Änderung der molaren Entropie in Folge einer chemischen Reaktion Rs völlig analog zu den Reaktionsenthalpien: bei Standardbedingungen: bei Standarddruck: Falls bei keinem Stoff Umwandlungspunkte zwischen 298 K und T liegen, lassen sich die Summen bzw. Integrale zu einer „verkürzten Formel“ zusammenfassen: T s s o R T o R 298 298 mit c p (T ) T dT c p (T ) A B 10 3 T C 10 5 1 D 10 6 T 2 2 T A i Ai Pr i Ai Ed B i Bi Pr i Bi Ed usw. Hoeppe - PC Leerscript - 62 - 3.5 (Partial-) Druckabhängigkeit der Entropie Obwohl chemische Reaktionen üblicherweise bei Standarddruck betrachtet werden, muss i. A. beachtet werden, dass die Entropie eine Funktion von T und p ist. Dies wird insbesondere bei der Betrachtung von Gasgemischen deutlich: Nach dem Gesetz von Dalton ergibt sich der Gesamtdruck p des Gemischs aus der Summer der Partialdrücke pi der Komponenten. Der Partialdruck einer Komponente ist der Druck, der sich bei Abwesenheit der anderen Komponenten einstellen würde. Erzeugen wir ein Gasgemisch mit dem Gesamtdruck p = po , so verringert sich zwangsläufig der Druck jeder Komponente ausgehend von po auf pi , wodurch sich die molare Entropie jeder Komponente (vgl. 3.2.3b) ändert. Beschreiben wir diese Vermischung (bei T=const, aber beliebig) durch isotherme Expansionen idealer Gase auf das gemeinsame Volumen, dann gilt für jede Komponente: 2 2 V 1 1 1 1 s S dS nR dV R ln 2 n n1 n1 V V1 p p R ln 1 R ln 2 p2 p1 Mit dem Ausgangsdruck p1 = po und dem Partialdruck einer Komponente p2 = pi folgt p si si ( p 2 pi ) si ( p1 p o ) si ( pi ) si0 R ln oi p Da i. A. die Entropie noch von T abhängt, ergibt sich als molare Entropie der i-ten Komponente einer Mischung: Für ein Gemisch ist die Gesamtentropie S ni si also nicht gleich der Summe der Entropien der reinen Komponenten, sondern muss um den „Mischungsanteil“ mit den lnTermen ergänzt werden! Eine etwas allgemeinere Darstellung erhält man, wenn man das Mischungsverhältnis nicht über die Partialdrücke sondern über die Molenbrüche xi angibt: Hoeppe - PC Leerscript - 63 - Aus pi ni RT V und p0 nRT V folgt pi ni xi po n und damit Planck konnte über die statistische Definition der Entropie zeigen, dass der obige Ausdruck nicht nur für die Vermischung von idealen Gasen gilt, sondern genauso für Mischungen von Flüssigkeiten und Festkörpern. Für die Gesamtentropie eines Systems gilt damit allgemein Achtung: Mit obiger Verallgemeinerung finden wir jetzt eine Abhängigkeit der Entropie von den Molenbrüchen, auch wenn der (Gesamt-) Druck p = po = 1 bar ist. Der Index ’’ o ’’ bei den soi bekommt jetzt eine andere Bedeutung: Er steht jetzt nicht nur für den Standarddruck sondern in der Verallgemeinerung für „reine Komponente“. Die soi sind also die molaren Entropien der reinen (d.h. unvermischten) Komponenten bei p = po = 1 bar. Anmerkung: Der durch die ln-Terme beschriebene „Mischungsanteil“ der Entropien hat eine sehr wichtige Konsequenz für den Ablauf von chemischen Reaktionen, da im Verlauf einer Reaktionen zwangsläufig ‚auf halbem Wege’ Edukte und Produkte parallel und damit vermischt vorliegen. Dies führt zur Ausbildung von Gleichgewichten, die in Kap. 5 behandelt werden. Hoeppe - PC Leerscript - 64 - 4 4.1 Der Ablauf physikalischer und chemischer Prozesse Freie Energie und Freie Enthalpie Josiah Willard Gibbs (1839-1903) gelang es die zwei Prinzipien, nach dem ein System einem Zustand 1) minimaler Energie und 2) maximaler Entropie entgegenstrebt zusammenzufassen, in dem er eine neue Zustandsgröße Freie Energie (bzw. die Freie Enthalpie) einführte. Beide o. g. Prinzipien lassen sich damit zu einem allgemeinen Prinzip der Minimierung der freien Energie (freien Enthalpie) zusammenfassen. 4.1.1 Freie Energie F Insbesondere in der Festkörperphysik kann das Volumen von Stoffen in guter Näherung als konstant betrachtet werden, d.h. die thermische Ausdehnung und damit verbundene Volumenarbeit kann vernachlässigt werden. Für spontan ablaufende Prozesse gilt nach Gibbs Freie Energie F Für eine Änderung von F gilt entsprechend Definition dF = d.h. für T = const entspricht eine Änderung der freien Energie der (reversibel) umgesetzten Arbeit. Andererseits ist F = F(V,T) (ohne Beweis) eine Zustandsgröße, und damit gilt für dF und nach Koeffizientenvergleich folgen die sog. Fundamentalgleichungen: Hoeppe - PC Leerscript - 65 - 4.1.2 Freie Enthalpie G Insbesondere in der Chemie kann das Volumen von Stoffen nicht als konstant betrachtet werden, stattdessen gilt meist p = const. Für diesen Fall wurde bereits in Kap. 2.2 anstelle der inneren Energie die Enthalpie eingeführt. Dies führt jetzt an Stelle der freien Energie zur sog. freien Enthalpie ( oder Gibbs Potential). Für spontan ablaufende Prozesse gilt nach Gibbs Freie Enthalpie G Für eine Änderung von G gilt entsprechend Definition dG = Auch G = G(p,T) ist (ohne Beweis) eine Zustandsgröße, und damit gilt für dG und nach Koeffizientenvergleich folgen die sog. Fundamentalgleichungen für G: Aus der Vereinigung der beiden Prinzipien (Energie Min) + (Entropie Max) zu G Min folgt für einen Prozess oder eine Reaktion von 1 nach 2: G = G2 - G1 < 0 G = G2 - G1 > 0 G = G2 - G1 = 0 Hoeppe - PC Leerscript - 66 - 4.1.3 (Partial-) Druckabhängigkeit der Freien Enthalpie g(p,T) Die Druckabhängigkeit der Freien Enthalpie folgt direkt aus der Druckabhängigkeit der Entropie, wobei wir hier auch letztlich wieder den Partialdruck pi einer Komponente bei dem konstantem Gesamtdruck p = po betrachten werden. Bsp.: Isotherme Expansion eines id. Gases: Für T = const aber beliebig folgt aus dG = Vdp - SdT unmittelbar dG Vdp nRT dp p Mit p1 = po und p2 = p folgt bzw. molar Im Allgemeinen ist g = g(p,T) wobei statt p im Fall eines Gemischs der Partialdruck pi der jeweiligen Komponente i anzusetzen ist: Auch hier gilt nach Planck verallgemeinert mit den Molenbrüchen xi der Komponenten: mit Die gesamte Freie Enthalpie eines beliebigen Gemischs bei dem Gesamtdruck p=po berechnet sich dann entsprechend G ni g i ni g i0 (T ) RT ln xi i i und ist also nicht einfach die Summe der freien Enthalpien der reinen Komponenten. Hoeppe - PC Leerscript - 67 - 4.2 Chemische Reaktionen Die Definition einer molaren freien Enthalpie erlaubt auf einfache Weise die Anwendung des Minimumprinzips für die freie Enthalpie (G MIN) auf beliebige chemische Reaktionen. Nach Einführung der Freien Reaktionsentropie analog zur Reaktionsenthalpie und -entropie wird sich schließlich zeigen, dass die von der Entropie stammenden ‚ln-Terme’ einen besonderen Einfluss auf den Ablauf von chemischen Reaktionen haben. 4.2.1 Freie Reaktionsenthalpie Rg Analog zur Reaktionsenthalpie und –entropie wird die molare freie Reaktionsenthalpie definiert, Wobei die gi der Komponenten i.A. p und T abhängig sind und im Besonderen auch von ihren jeweiligen Partialdrücken pi bzw. Molenbrüchen xi abhängen. Im Folgenden betrachten wir nur Reaktionen bei einem Gesamtdruck p = po, d.h. die Beiträge der einzelnen Komponenten berechnen sich nach g i ( xi , T ) g i0 (T ) RT ln xi . Eine grobe (aber auch später wichtige) Vereinfachung für Rg erhält man, wenn man zunächst nur die Beiträge der reine Komponenten berücksichtigt, d.h. sämtliche „lnTerme“ vernachlässigt: Standard Gibbs-Reaktionsenthalpie Rgo (Achtung: Der Index “ o “ steht hier also nicht für Standarddruck, sondern dafür, dass nur die Beiträge der reinen Komponenten berücksichtigt werden.) Entsprechend G = H - T·S bzw. g = h - T·s berechnet sich Rgo (T) mit und falls bei keinem Stoff Umwandlungspunkte zwischen 298 K und T liegen, gilt mit cp(T): T c p (T ) 0 c p (T ) dT T R s 298 dT T 298 298 T R g Rh 0 0 298 Die freie Reaktionsenthalpie Rgo liefert also ein gute Abschätzung über die Hoeppe - PC Leerscript - 68 - Änderung der molaren freien Enthalpie in Folge einer Reaktion und erlaubt somit eine grobe Aussage (vgl. 4.1.2) darüber, ob eine Reaktion spontan ablaufen wird oder nicht. Für Rgo < 0 wird g (bzw. G) kleiner bzw. minimal, wenn die Reaktion von links nach rechts abläuft. Ist für ein potentielle (gedachte) Reaktion Rgo > 0, so wird diese nicht ablaufen. Das Vorzeichen von Rgo zeigt also an, ob eine Reaktion von „links nach rechts“ oder (eher) umgekehrt abläuft. 4.2.2 1. Ulich’sche Näherung: Ganz offensichtlich ist Rgo eine Funktion der Temperatur, womit auch die Richtung einer Reaktion evtl. von der Temperatur abhängig ist. Eine relativ gute Abschätzung bzgl. der Temperaturabhängigkeit von Rgo erhält man mit der Annahme, dass die spezifischen Wärmen von Edukten und Produkten sich kaum unterscheiden, d.h. cp(T) 0. Damit vereinfacht sich die Berechnung von Rgo(T) erheblich: Obige Gleichung ist nicht zu verwechseln mit dem Fall einer Reaktion unter Standardbedingungen: 0 0 0 R g 298 R h298 298 K R s 298 Hoeppe - PC Leerscript - 69 - 4.2.3 Richardson Diagramme Bisher haben wir nur das Vorzeichen von Rgo einer Reaktion betrachtet und daraus auf die Reaktionsrichtung geschlossen. Welche Information steckt denn im Betrag von Rgo ? „Je negativer desto eher (stärker) von links nach rechts“? Ja, so ähnlich kann man sich das vorstellen und am deutlichsten wird diese Aussage, wenn man verschiedene alternative Reaktionen miteinander vergleicht. In einem Richardson Diagramm werden die Rgo(T) unter Verwendung der 1. Ulichschen Näherung für verschieden Reaktionen über der Temperatur aufgetragen. Genauer: Für verschiedene alternative Reaktionen, in dem Beispiel unten jeweils mit O2: Richardson Diagramm 0 Standardreaktionsenthalpie -100 C + O2 -> CO2 2 Ni + O2 -> 2 NiO 2 C + O2 -> 2 CO 4/3 Cr + O2 -> 2/3 Cr2O3 4/3 Fe + O2 -> 2/3 Fe2O3 2 Fe + O2 -> 2 FeO 3/2 Fe + O2 -> 1/2 Fe3O4 -200 -300 -400 -500 -600 -700 200 400 600 800 1000 1200 1400 1600 1800 2000 T/K Verschiedene Reaktionen zeigen verschiedene Rgo Verschiedene Reaktionen zeigen verschiedene Temperaturabhängigkeit von Rgo Die Reaktion mit kleinerem Rgo läuft bevorzugt ab. Die Reaktion mit kleinerem Rgo läuft auch ab, falls nur das Produkt einer Reaktion mit größerem Rgo vorliegt, d.h. z.B.: Bei hohen Temperaturen wird NiO durch CO reduziert, bei Raumtemperatur eher umgekehrt! ( Heßscher Satz!) Eine Erweiterung des obigen Diagramms ist das Jeffes-Richardson Diagramm, welches zudem das Ablesen erlaubt, bei welcher Temperatur und welchem Sauerstoffpartialdruck ein Stoff oxidiert wird. ( vgl. heterogene Gasgleichgewichte) Hoeppe - PC Leerscript - 70 - 4.2.4 Unvollständig ablaufende Reaktionen Zerlegt man eine Reaktion A B in (sehr viele) Teilschritte und berechnet jeweils neu das jeweilige Rg für den nächsten Teilschritt unter Berücksichtigung der „ln-Mischungsterme“ (d.h. der Mischungsentropie Ms ) so ändert sich irgendwann das Vorzeichen von Rg. Damit ändert sich auch die Reaktionsrichtung, d.h. die Reaktion kommt letztlich zum Stillstand bevor sie vollständig abgelaufen ist. Im dem Minimum der freien Enthalpie G (oder g) ist für einen sehr kleinen Reaktionsschritt immer Rg = 0 und die Reaktion kommt deshalb zum Stillstand. Die Gleichgewichtsbedingung lautet also Die zuvor vernachlässigten ln-Terme, die durch die Vermischung aller Komponenten einen zusätzlichen Beitrag zur freien Enthalpie leisten, sind also entscheidend wichtig! Erst durch sie kommt es zu einer Ausbildung eines Gleichgewichts. Da diese ‚Mischungsterme’ letztlich von dem Entropieanteil der freien Enthalpie stammen, kann man auch vereinfacht sagen: Die Entropie bzw. das Streben nach Unordnung sorgt dafür, dass Reaktionen nie vollständig ablaufen. Die Frage wo das Gleichgewicht liegt, bzw. welche Konzentrationen die jeweiligen Komponenten im Gleichgewicht haben, wird in Kapitel 5 behandelt. Hoeppe - PC Leerscript - 71 - 5 Freie Enthalpie und Gleichgewichte In diesem Kapitel wird die Gleichgewichtsbedingung Rg = 0 auf verschiedene Probleme angewendet. Während die Standard Gibbs Enthalpie Rgo vollständig mit den thermodynamischen Standardwerten der Stoffe definiert ist, sind die Molenbrüche xi der Komponenten und damit die ln-Terme zunächst unbekannt. Ziel ist es, genau diese für den Endzustand beliebiger Prozesse, also dem jeweiligen Gleichgewichtszustand, zu berechnen. 5.1 5.1.1 Reaktionsgleichgewichte Homogene Gasgleichgewichte Hier sind alle Reaktionspartner/Komponenten gasförmig. (Bsp: HJ oder NH3 Synthese) Betrachte allgemein die Reaktion aA + bB dD + eE Die Freie Reaktionsenthalpie berechnet sich nach: Fordert man mit Rg = 0 das Gleichgewicht, beschreibt die ’Abkürzung’ K für das Argument des ln-Terms die Zusammensetzung des Systems im Gleichgewicht. Man nennt K daher die Gleichgewichtskonstante für diese Reaktion. Hoeppe - PC Leerscript - 72 - Auflösen obiger Gleichung nach K ergibt: RT ln K Rg0 bzw. mit Die Zusammensetzung des Systems im Gleichgewicht ist also mit K gegeben, welches sich wiederum eindeutig aus der Standard Gibbs Enthalpie Rgo berechnet! Da K i. A. durch mehrere Variablen bestimmt ist, muss für die Berechnung der Zusammensetzung mit den einzelnen Molenbrüche xi noch deren Verknüpfung durch die Reaktionsgleichung berücksichtigt werden. (vgl. A5.1) Bei Gasreaktionen werden anstelle der Molenbrüche xi auch oft die Partialdrücke pi verwendet. Für die freie Enthalpie gilt dann p Dd p Ee p 0 R g ( p i , T ) d g D e g E a g A b g B R g (T ) RT ln p Aa p Bb 0 mit d e a b . Das Verhältnis der Partialdrücke bezeichnet man üblicher Weise als Gleichgewichtskonstante Kp , wobei diese jetzt nicht mehr einheitenlos ist. Die Beschreibung der Mengenverhältnisse über die Konzentrationen führt analog zur Gleichgewichtskonstanten Kc . Zusammenfassend gilt mit xi p i ci p c0 und den Bezugsgrößen p o 1 bar und c0 1 mol l bzgl. der Partialdrücke: p Dd p Ee K p a b K p0 p A pB bzgl. der Konzentrationen: p0 c0 c d c e [ D] d [ E ]e K K C Da Eb p c A c B [ A]a [ B]b bzgl. der Molenbrüche: p xd xe K x Da Eb K 0 x A xB p Hoeppe - PC Leerscript - 73 - Für Reaktionen ohne Molzahländerungen, d.h. = 0 gilt: Kp = Kc = Kx = K (dimensionslos). Für 0 gilt: Kp K p 0 K bar und für id. Gase p Kc K 0 RT 1 K p RT (bzgl. Einheiten vgl. hierzu Kap. 5.1.3 !) Allgemein gilt: kleines Rgo großes Rgo 5.1.2 Heterogene Gasgleichgewichte Hier sind nicht alle Reaktionspartner/Komponenten gasförmig, wodurch die Komponenten auch nicht ‚automatisch’ alle vermischt vorliegen. Für die Molenbrüche der unvermischten, d.h. reinen Komponenten, ist dann in guter Näherung jeweils “1“ anzusetzen, wodurch die jeweiligen ln-Terme verschwinden. Die Stoffmengen bzw. Molenbrüche von z.B. (reinen) Festkörpern finden daher auch keinen Eingang in die Gleichgewichtskonstante, wohl aber deren Standardwerte in die Standard Gibbs Enthalpie Rgo . Beispiel: Brennen von Kalk CaCO3 CaO + CO2 Freie Reaktionsenthalpie mit xi 1 für Festkörper und xi = pi/po für Gase: R g ( pi , T ) .... R g ( pi , T ) K reduziert sich also auf den Partialdruck von CO2 gemessen in bar. Dieser berechnet sich entsprechend R go K p K p0 e RT p0 Wobei sich hier nur auf die Komponenten in der Gasphase bezieht. Hoeppe - PC Leerscript - 74 - Die Reaktion läuft also ab, bis p(CO2) = Kp erreicht ist, unabhängig von den Stoffmengen CaO und CaCO3 ! Da die Standard Gibbs Enthalpie von der Temperatur abhängig ist, gilt dies auch für K und damit für die Lage des Gleichgewichts. Für obiges Beispiel ergibt sich (vgl. A5.2) a) T = 700 K: Rgo700 = + 64,3 kJ/mol Kp, 700 = 1,59 · 10-5 bar Der CO2-Partialdruck von Luft (~0,04% ) ist größer als Kp , es findet keine Reaktion statt. b) T = 900 K: Rgo900 = + 33,1 kJ/mol K p, 900 = 0,012 bar Kp ist deutlich größer als 4·10-4 bar, die Reaktion läuft ab, bis Kp erreicht wird, vorausgesetzt es ist genügend CaCO3 vorhanden, und der Ofen wird nicht belüftet. c) T = 1200 K: Rgo1200 = - 12,33 kJ/mol K p, 1200 = 3,44 bar Der CO2-Partialdruck kann kaum größer als 1 bar werden, solange der Ofen nicht druckdicht ist. Kp ist also stets größer als der aktuelle CO2-Partialdruck, die Reaktion läuft vollständig ab. Die Temperaturabhängigkeit des Gleichgewichts wird nochmals ausführlicher in Kap. 5.1.3 behandelt. Beispiel: Boudouard Gleichgewicht CGrafit + CO2 2 CO Besonders wichtig ist dieses Gleichgewicht im Zusammenhang mit der Aufkohlung von Eisen bzw. der Härtung von Stahl. Hierbei liegt der Kohlenstoff aber nicht als Reinstoff vor; die Konzentration (genauer „Aktivität a“ ) des C in Eisen geht hier mit ins Gleichgewicht ein: CGrafit, in Eisen + CO2 2 CO Die Kohlenstoffkonzentration im Eisen ist also neben der Temperatur wesentlich vom CO (und CO2) Partialdruck in der Atmosphäre abhängig! Hoeppe - PC Leerscript - 75 - Beispiel: Jeffes Richardson Diagramm / Ellingham Diagramm Beschreiben z.B. die Stabilität von Oxiden in Abhängigkeit des Sauerstoffpartialdrucks und der Temperatur ( Hochtemperaturkeramiken). Falls keine anderen sauerstoffhaltigen Gase vorhanden sind gilt ja Rgo(T) = RT · ln (pO2) ... Grafik aus: Salmang, Scholze; Keramik, Springer 2007 Hoeppe - PC Leerscript - 76 - 5.1.3 Verschiebung des Gleichgewichts Wie bereits aus den bisherigen Beispielen ersichtlich war, ist die Lage des Gleichgewichts einer spezifischen Reaktion nicht nur von der Reaktion selber, d.h. der Reaktionsgleichung, sondern auch von den äußeren Bedingungen, d.h. p und T, abhängig. Damit lässt sich z.B. bei der Stoffsynthese die Gleichgewichtslage gezielt verschieben. A) Der Einfluss der Temperatur erfolgt direkt über die Temperaturabhängigkeit von K = K(T). Da der ln eine monoton und stetig steigende Funktion ist, genügt es für eine qualitative Betrachtung die Temperaturabhängigkeit von ln(K) zu betrachten: K (T ) ? T Aus der part. diff. von ln(K) nach T (und Gibbs-Helmholtz-Gl.) folgt die van’t Hoffsche Reaktionsisobare: d.h. das Vorzeichen von Rho entscheidet, ob K mit T steigt oder fällt: - für exotherme Reaktionen (Rho < 0) wird K mit der Temperatur kleiner! - das Gleichgewicht verschiebt sich mit steig. T auf die Seite der Edukte - umgekehrtes gilt für endotherme Reaktion Allgemein lässt sich das Prinzip von Le Chatelier (1888) und Braun (1887) formulieren: Ein System ... (vgl. A5.2 Brennen von Kalk, A5.3 Ammoniaksynthese) Hoeppe - PC Leerscript - 77 - B) Der Einfluss des Drucks erfolgt nicht über die Gleichgewichtskonstante K, da Rgo per Definition nur von den Standardwerten go, welche bei p = po definiert sind, abhängt. Die Lage des Gleichgewichts ist aber über die Bestimmungsgleichung für die Konzentrationen bzw. Molenbrüche vom Gesamtdruck p des Systems abhängig. Dazu betrachten wir jetzt die Druckabhängigkeit der Gleichgewichtskonstanten Kx. Man betrachtet auch hier statt Kx wieder lnKx: K x ( p) ? p ln K x ( p) ... p Nach part. Diff. von ln(Kx) nach p folgt wobei RVo die Volumenänderung (bei Standardbedingungen) in Folge der Reaktion beschreibt. Offensichtlich ist das insbesondere bei Gasreaktionen mit Molzahländerung der Fall ( po · RVo = Rn · RTo )! Hier entscheidet das Vorzeichen von RVo , ob Kx mit p steigt oder fällt: - für Reaktionen mit RVo < 0 wird Kx mit dem Druck größer! - das Gleichgewicht verschiebt sich mit steig. p auf die Seite der Produkte - umgekehrtes gilt für Reaktion mit RVo > 0 Auch hier gilt offensichtlich das Prinzip von Le Chatelier und Braun! Da die Standardwerte bei Standarddruck p = po = 1 bar definiert sind, ist die Gleichgewichtskonstante Kp bzgl. der Einheit po zahlenmäßig gleich K. Für Gleichgewichte bei einem anderen Gesamtdruck (=Ausgangsdruck), muss Kc bzw. Kx entsprechend umgerechnet werden! Damit wird die Druckabhängigkeit des Gleichgewichts ‚automatisch’ richtig dargestellt. (vgl. Kap. 5.1.1 und A5.3: Ammoniaksynthese) C) Der Einfluss der Stöchiometrie erfolgt ebenfalls nicht über K: Bei einer nichtstöchiometrischen Ausgangszusammensetzung, verändert sich K nicht, da die Beiträge der „überzähligen“ Stoffe bei der Berechnung von Rgo keinen Beitrag liefern. Bei der Berechnung der Zusammensetzung des Systems im Gleichgewicht geht die Ausgangszusammensetzung jedoch in die Bestimmungsgleichungen der Konzentrationen im Gleichgewicht ein! Dadurch kann die Lage des Gleichgewichts und damit z.B. die Ausbeute einer Synthese gezielt verändert werden. (vgl. A5.4 , Jodwasserstoffsynthese) 5.2 Phasengleichgewichte Hoeppe - PC Leerscript - 78 - Betrachtet man einen Phasenübergang I II , z.B. das Schmelzen oder Verdampfen eines Stoffes, so gilt für diesen Prozess allgemein (analog zu einer chemischen Reaktion), dass die freie Enthalpie im Gleichgewicht ein Minimum annimmt, also: Prozess: Stoff (I) Gleichgewicht: G = G(II) – G(I) = 0 Währendes Gleichgewicht: dG = dG(II) – dG(I) = 0 Stoff (II) Bei der Anwendung der Gleichgewichtsbedingung ist darauf zu achten, welche Variablen in beiden Phasen den gleichen Wert annehmen und - insbesondere bei Gemischen - bzgl. welcher Komponente/Phasen sich ein Gleichgewicht ausbilden kann. 5.2.1 Phasengleichgewichte reiner Stoffe Bei einem Reinstoff ist mit p und T eindeutig eine Phase festgelegt, die Koexistenz zweier Phasen, d.h. ein Gleichgewicht, ist daher i. A. praktisch nur für kurze Zeit bzw. näherungsweise möglich. Ein stabiles Gleichgewicht kann sich jedoch ausbilden, wenn mit dem Phasenübergang eine Rückkopplung auf p oder T verbunden ist, wodurch das System direkt auf einer Phasengrenze verbleibt. Ein Beispiel ist ein geschlossener Behälter, in dem sich ein stabiles l-g-Gleichgewicht ausbildet. Abhängig von der Temperatur verbleibt das System auf einem Punkt der Dampfdruckkurve, womit auch eine Vermessung des Dampfdrucks einfach möglich wird. Ursache ist letztlich das i. A. deutlich unterschiedliche Molvolumen beider Phasen. Hoeppe - PC Leerscript - 79 - 5.2.1.1 Lokale Beschreibung der Dampfdruckkurve Für einen Reinstoff liege wie oben allgemein beschrieben ein l-g Gleichgewicht vor, d.h. das System befinde sich auf einem Punkt der Dampfdruckkurve. Kontaktvariable: p(g) = p(l) T(g) = T(l) Gleichgewicht: G = 0 G(g) = G(l) Währendes Gleichgewicht: dG = 0 dG(g) = dG(l) und Clausius Clapeyrone: dp hu dT T (Vm , g Vm , l ) Die Gleichung von Clausius Clapeyrone liefert also die Tangentensteigung der Dampfdruckkurve als Funktion der Umwandlungsenthalpie und Molvolumina. (vgl. A5.5) 5.2.1.2 Dampfdruckkurven Eine vollständige Beschreibung der Dampfdruckkurve erhält man, solange man hinreichend weit weg vom kritischen Punkt bleibt, mit den Näherungen Molvolumen der flüssigen Phase vernachlässigbar: Vml << Vmg , Umwandlungswärme (= Verdampfungswärme): hu = const Dampf verhalte sich wie ideales Gas: p Vmg = RT Ausgehend von der Gleichung von Clausius Clapeyrone: Hoeppe - PC Leerscript - 80 - p h h B ln D u u : A RT RT0 T p0 Augustsche Dampfdruckformel Trägt man also den ln(pD) über 1/T auf, lassen sich die Dampfdruckkurven mit den Fitparametern A und B beschreiben (vgl. A5.6). Damit gilt für pD(T) explizit: p D (T ) p 0 e 5.3 hu 1 1 R T T0 Elektrolytgleichgewichte, Normalspannungen Elektrolyte sind Stoffe die als Ionen vorliegen oder zu Ionen dissoziieren können. Bei Lösungsvorgängen wird das Gleichgewicht einer gesättigten Lösung mit dem Löslichkeitsprodukt, bei der Dissoziation von Säuren und Basen in Lösung mit der Säure- bzw. Basenkonstante beschrieben. Elektrochemische Spannungen treten eigentlich in Folge eines Nicht gleichgewichtzustandes auf. Wird jedoch ein Stromfluss verhindert, lässt sich unter Berücksichtigung der anliegenden Spannung der Zustand wie ein Gleichgewicht beschreiben und auch die auftretenden Normalspannungen aus den thermodynamischen Standarddaten der beteiligten Stoffe berechnen. Hoeppe - PC Leerscript - 81 - 5.3.1 Löslichkeitsprodukt Betrachte den Lösungsvorgang eines Stoffes A, A(s) b B+ aq + c C-aq wobei dem Lösungsmittel soviel Stoff A zugegeben wird, bis eine gesättigte Lösung entsteht. Da der ungelöste Stoff A in guter Näherung als Reinstoff vorliegt, handelt es sich hier um ein heterogenes Gleichgewicht und Rg berechnet sich entsprechend: R g b g B c g C g A ... R g 0 RT ln a B a C b c R g 0 RT ln K Bei Vernachlässigung der Aktivitätskoeffizienten mit ai = ci · ci/co ci/co gilt für die Gleichgewichtskonstante: K c Bb cCc co( bc ) [ B ]b [C ]c ( mol ) b c l Das Produkt der Konzentrationen der gelösten Stoffe bezeichnet man als Löslichkeitsprodukt, K L : [ B ]b [C ]c K ( mol ) bc l womit sich also die (maximale) Ionenkonzentration der jeweiligen gesättigten Lösung berechnen lässt. (vgl. A5.8) Hoeppe - PC Leerscript - 82 - 5.3.2 Säure-Base Gleichgewichte Durch die Abgabe eines Protons wird der Säurerest im selben Augenblick zu einem potentiellen Protonenakzeptor und damit zu einer Base. Der Dissoziationsvorgang einer gelösten Säure oder Base führt daher immer zu einem Säure-Base Gleichgewicht. Die Dissoziation einer n-molaren Säure HA führt zu einer H+ Ionenkonzentration die immer kleiner ist als die Molarität n der ‚Einwaage’. Die Stärke einer Säure (,d.h. die H+ Ionenkonzentration bzw. der pH-Wert,) wird also neben der Molarität n wesentlich durch den Dissoziationsgrad bestimmt, welcher aber wiederum von der Molarität abhängt. Eindeutig bestimmt wird dieses Gleichgewicht mit Molarität und Säurekonstante Ka , die bis auf Einheiten der Gleichgewichtskonstanten K des Dissoziationsprozesses entspricht: HAaq H+ aq + A-aq a ( H ) a ( A ) c ( H ) c ( A ) 1 [ H ] [ A ] mol 1 co K ( l ) a HA c HA HA ( ) ( ) [ ] [ H ] [ A ] Ka [ HA] Achtung: Die Konzentration von HA im Nenner entspricht nicht der Molarität, sondern der verbliebenen Konzentration von HA nach Einstellung des Gleichgewichts! Die Säurekonstanten Ka bzw. ihre logarithmische Darstellung pKa sind für die meisten Säuren als Tabellenwerte verfügbar (vgl. Tab. M). Dabei gilt nach Definition: pK a : lg K a K a 10 pK a pH : lg [ H ] Dissoziationsgrad: Hoeppe - PC Leerscript [ A ] : [ HA]0 - 83 - 5.3.3 Normalspannungen Die Elektrochemische Spannung zwischen zwei galv. Halbzellen beschreibt die relative Neigung der beteiligten Stoffe in Lösung zu gehen (vgl. Daniellelement): A|A+||B|B+ Halbzellenpaar: Befinden sich alle Komponenten im Standardzustand, misst man eine Spannung U0 = Eo E 0 E 0 ( B | B ) E 0 ( A | A ) Die zugehörige Reaktionsgleichung für die komplette galvanische Zelle lautet A + B+ A+ + B Beobachtung: Die Reaktion läuft für positives E0 nach rechts ab. Mit Beachtung der elektrisch geleisteten Arbeit, lässt sich zeigen dass Rg0 = - z F E0 gilt. (z = Ladungszahl; Faradaykonstante F = NA e = 96485 Cmol-1) Mit der Wasserstoffnormalelektrode (mit E0 0) als Standard-Bezugs-Potential sind die Standard- bzw. die Normpotentiale E0 von Stoffen definiert. A|A+||½ H2|H+ entspricht A + H+ A+ + ½ H2 Elektrochemische Spannungsreihe Rg0 E zF 0 Auch die Elektrochemische Spannungsreihe lässt sich also mit Betrachtung eines (erzwungenen) Gleichgewichts auf die thermodynamischen Standardgrößen der beteiligten Stoffe zurückführen. (vgl. A5.9, A5.10) Hoeppe - PC Leerscript - 84 - 5.3.4 Nernstsche Gleichung Entspricht die Elektrolytkonzentration einer Halbzelle nicht dem Standardzustand von 1 mol/l, sind wie bei allen Reaktionsgleichgewichten die entsprechenden Mischungsterme, d.h. die ln-Terme, zu berücksichtigen. Befinden sich beide Halbzellen nicht im Standardzustand, wird die gemessene Spannung U = E mit der Nernstschen Gleichung beschrieben: a( A z ) a( B) z RT E E ln z zF a ( A) a ( B ) 0 c( A z ) x( B) z 0,059 V 0 E log z z x( A) c( B ) Die gemessenen Spannungen sind also von den Aktivitäten/Konzentrationen aller beteiligten Komponenten abhängig, was z.B. die sinkende Spannung einer sich leerenden Batterie erklärt. Bei Messung einer Me-Me+ Halbzelle mit bel. Elektrolytkonzentration und reiner Me-Elektrode (links) gegen die H-Normalelektrode (rechts) ergibt sich für das Halbzellenpotential 0 E Me E Me 0,059 V 0,059 V 0 log( a ( Me z )) E Me log(c( Me z ) z z l mol ) Die Nernstsche Gleichung beschreibt also die konzentrationsabhängigen elektrochemischen Spannungen. Das Argument des ln-Terms enthält dabei die Konzentrationen und entspricht der Gleichgewichtskonstanten des jeweiligen meist heterogenen Reaktionsgleichgewichts. _______________________________________________________________ Hoeppe - PC Leerscript - 85 - 6 Gemische Gemische bestehen per Definition aus vermischten verschiedenen Stoffen, zwischen welchen keine chemische Reaktion stattgefunden hat. Dennoch können WW (attraktiv oder repulsiv) existieren, die entscheidenden Einfluss auf z.B. die Phasengrenzlinien haben. Man unterscheidet daher zwischen ideale Gemischen (keine WW) und realen Gemischen (mit WW). Viele grundlegenden Effekte lassen sich bereits mit der Näherung idealer Gemische beschreiben, wie im Folgenden gezeigt werden soll. 6.1 Ideale Mischungen Eine Mischung von verschiedenen Stoffen heißt ideal wenn gilt: V Vi ni Vm ,i i i H H i ni hi i 6.1.1 i Ideale Gasgemische Liegen mehrere Gase getrennt nebeneinander vor (a), so haben diese gleiche Temperatur und Druck, befinden sich aber jeweils in verschiedenen Volumina. Liegen sie vermischt vor (b), so haben sie die gleiche Temperatur, das gleiche Volumen aber verschiedene Partialdrücke. Für den Molenbruch des Gesamtsystems gilt im unvermischten Zustand (a) : Hoeppe - PC Leerscript xi ni Vi n V - 86 - xi und im vermischten Zustand (b) : Nach Dalton gilt: p pi ni i i ni p i n p RT RT n V V Bei der Vermischung der Gase ändert sich die Enthalpie entsprechend Definition eines idealen Gemischs nicht, wohl aber die Entropie und damit die freie Enthalpie (vgl. hierzu auch Kap. 3.5, Druckabhängigkeit der Entropie): Aus den Definitionen M S n S i i Mischung M G n G i i Mischung n S i unvermisch t i n G i unvermisch t i M H T M S folgt die molare Mischungsentropie und die M s id M s xi R ln xi i molare Freie Mischungsenthalpie: M g id xi RT ln xi i 6.1.2 Ideale Flüssigkeitsgemische Jetzt soll ein Gemisch von zwei Substanzen betrachtet werden, welches gleichzeitig in der flüssigen Phase und der gasförmigen Phase besteht. Die Komponenten der flüssigen Phase stehen dabei jeweils im Gleichgewicht mit ihrer Dampfphase: Hoeppe - PC Leerscript - 87 - Die Zusammensetzung der Gasphase ist abhängig von der flüssigen Phase. Es gilt in sehr guter Näherung das Raoultsche Gesetz id pi xli poi id x gi xli poi p womit die Partialdrücke und damit die Molenbrüche der Gasphase xgi mit den Molenbrüchen der flüssigen Phase xli verknüpft sind. Nach Dalton ergibt sich der Gesamtdruck als Summe der Partialdrücke, womit der Gesamtdruck p letztlich eine eindeutige Funktion der Molenbrüche in der flüssigen Phase (und der Dampfdrücke poi der reinen Komponenten) ist. Für zwei Komponenten A und B gilt damit: p p A p B xlA poA xlB poB (1 xlB ) poA xlB poB Leichter messbar sind sicher die Drücke, Auflösen nach den Molenbrüchen xli ergibt: xlA p poB poA poB xlB p poA poB poA Für die Zusammensetzung der Gasphase des Gemischs folgt mit Dalton und Raoult: x gA p pA xlA 0 A p p x gB p pB xlB 0 B p p Sind die Dampfdrücke poi der reinen Komponenten unterschiedlich, ergeben sich damit unterschiedliche Molenbrüche in der flüssigen und gasförmigen Phase, was auch die Grundlage für die Stofftrennung durch Destillation ist! Hoeppe - PC Leerscript - 88 - Da bei einem Verdampfungsprozess offensichtlich von der leichterflüchtigen Komponente (die mit dem größeren Dampfdruck) mehr als von der anderen in die Gasphase übergeht, wird dadurch ihre Konzentration in der flüssigen Phase geringer. Entsprechend obiger Gleichung für den Gesamtdruck fällt aber der Gesamtdampfdruck des Flüssigkeitsgemischs damit wieder. Aufgrund dieses Rückkopplungsmechanismus bzgl. der Molenbrüche kann sich bei Gemischen (ohne äußere Zwangsbedingung!) für einen Gesamtdruckbereich zwischen den Dampfdrücken der reinen Flüssigkeiten ein stabiles Gleichgewicht ausbilden! Da sich die Molenbrüche der flüssigen und der Gasphase bei Ausbildung des Gleichgewichts je nach Gesamtdruck ändern, und evtl. wegen dem Fehlen einer Phase gar nicht definiert sind, beschreibt man das Gemisch sinnvoller Weise mit einem p-x-Diagramm, wobei mit x hier der Gesamtmolenbruch X beschrieben ist. Die Grenzlinien dieses Bereichs, d.h. des Zweiphasenmischgebiets, bestimmen sich aus den folgenden Forderungen: 1. Grenzlinie l : l + g ( Siedelinie ) - (Fast) Alles ist noch flüssig, d.h. XB = xlB - p ist der Gesamtdampfdruck des „ersten verdampften Tröpchens“ p p ( X B xlB ) p0 A ( p0 B p0 A ) X B 2. Grenzlinie l + g : g ( Taulinie ) - (Fast) Alles ist schon gasförmig, d.h. XB = xgB - p ist der Gesamtdampfdruck des „letzten verdampften Tröpchens“ p p( X B x gB ) Hoeppe - PC Leerscript p0 A 1 ( p0 B p0 A ) XB p0 B - 89 - Entsprechend der Definition der Phasengrenzlinien, lässt sich für einen bestimmten Gesamtdruck p die Zusammensetzung der flüssigen Phase xlB an der Siedelinie und die Zusammensetzung der Gasphase xgB an der Taulinie direkt ablesen! Die Verbindungslinie zwischen beiden Schnittpunkten für einen gegebenen Druck p heißt Konode. Üblicherweise wird das dadurch entstehende Zwei-Phasen-Mischgebiet nicht bei T = const in Abhängigkeit des Drucks, sondern bei p = const in Abhängigkeit der Temperatur betrachtet; die Darstellung über den Gesamt molenbruch X einer Komponente in einem T-X-Diagramm nennt man Siedediagramm . Dieses folgt direkt aus Umrechnung der Phasengrenzlinien unter Berücksichtigung der Temperaturabhängigkeit der Dampfdruckkurven poi = poi(T): Bei gegebener Temperatur T (und konstantem Druck p) bildet sich längs der Konode ein Gleichgewicht aus, die Schnittpunkte mit Siedelinie und Taulinie beschreiben wieder definitionsgemäß die Molenbrüchen der beiden Phasen. Mit Druck und Temperatur (sowie den Dampfdrücken/Siedetemperaturen der reinen Komponenten) ist die Zusammensetzung eines Zweikomponentengemischs also eindeutig festgelegt, soweit beide Phasen koexistieren. Die Aufteilung der Stoffmengen auf die zwei Phasen folgt aus dem Verhältnis der Molenbrüche xli und xgi zum Gesamtmolenbruch X unter Aufstellung einer entsprechenden Mengenbilanz: Hoeppe - PC Leerscript - 90 - Wie auch schon bei einem reinen Stoff, wird bei einer großen Gesamtstoffmenge in einem Behälter weniger verdampfen, als bei einer kleinen, da der Gesamtdruck schneller erreicht ist. Bei einer großen Gesamtstoffmenge wird z.B. der Molenbruch der flüssigen Phase daher kaum verändert und ungefähr dem Gesamtmolenbruch entsprechen. Aus n n g nl und n B X B n X B (n g nl ) folgt das sog. Hebelgesetz, welches die Aufteilung der Stoffmengen auf die beiden Phasen beschreibt: nl X xl n g x g X wobei hier der Übersichtlichkeit wegen der Index B bei den Molenbrüchen meist weggelassen wird. (Üblicherweise wird zur Beschreibung eines Zweikomponentengemischs der Molenbruch der leichterflüchtigen Komponente (LFK) verwendet.) Wird also einem Gemisch wie im Diagramm dargestellt bei konstantem p und T eine Komponente zusätzlich zugegeben, ändert sich entsprechend der Gesamtmolenbruch nicht aber die Molenbrüchen in den Phasen. Dies wird durch die unterschiedliche Aufteilung der Stoffmengen (Hebel) auf die l und g Phase ermöglicht. Bei realen Mischungen müssen noch die WW zwischen den Mischungskomponenten (Mischungswärmen) berücksichtigt werden, wobei diese i. A. nicht nur von p und T sondern auch vom Mischungsverhältnis in den einzelnen Phasen abhängig sind. Dadurch werden Siede- und Taulinie zum Teil stark verändert, die hier beschriebene Systematik und insbesondere das Hebelgesetz sind aber unverändert gültig! Hoeppe - PC Leerscript - 91 - 6.1.3 Gleichgewicht eines Stoffes bei zusätzlicher Komponente Ist die zusätzliche Komponente nur in einer Phase eines betrachteten Gleichgewichts vorhanden, so wird dieses entsprechend beeinflusst: Wird z.B. ein Salz in Wasser gelöst, so wird die Dampfphase darüber praktisch kein Salz enthalten; auch wird bei Abkühlung das Salz kaum in der festen Phase eingebaut, sondern verbleibt hauptsächlich in der flüssigen Phase (heterogenes Gleichgewicht). Aus diesen „Extremfällen von nur teilweise ausgebildeten Mischungen“ folgen: 6.1.3.1 Dampfdruck- und Gefrierpunktserniedrigung / Siedepunktserhöhung Betrachte eine Lösung als Extremfall einer Mischung mit xA Molenbruch der gelösten Komponenten (z.B. Salz) xB Molenbruch des Lösungsmittels (z.B. Wasser) dann gilt sicherlich a) xA << xB und p0A << p0B Dampfdruckerniedrigung (des Lösungsmittels) Nach Dalton und Raoult gilt: p p A p B xlA p0 A xlB p0 B xlB p0 B (1 xlA ) p0 B d.h. für die Dampfdruckerniedrigung pD gilt p D p 0 B p p 0 B (1 xlA ) p 0 B xlA p 0 B X A p 0 B p D X gel p 0 Lösungsmit tel Sie ist also nur vom Dampdruck des Lösungsmittels und der Menge (nicht der Art) des gelösten Stoffes abhängig. Hoeppe - PC Leerscript - 92 - b) Siedepunktserhöhung (des Lösungsmittels) Entsprechend untenstehender Grafik führt eine Dampfdruckerniedrigung je nach Steigung der Dampfdruckkurve zu einer Siedepunktserhöhung. Für eine kleine Menge des gelösten Stoffes lässt sich die Steigung der Dampfdruckkurve mit der Gleichung von Clausius-Clapeyrone (vgl. Kap. 5.2.1.1) beschreiben. Die Dampfphase soll zudem näherungsweise als ideales Gas beschrieben werden: Mit p dp hu p0 B T dT R TS2 und p X A p0 B folgt R TS2 TS XA hu Und mit Verwendung der Molalität mA des gelösten Stoffes an Stelle des Molenbruchs: R TS2 M B TS m A : ES m A hu Die sog. Ebullioskopische Konstante ES fasst nur Eigenschaften des Lösungsmittels zusammen, und mA beschreibt (lediglich) die Molalität des gelösten Stoffes. ( vgl. Tab. I ) Hoeppe - PC Leerscript - 93 - c) Gefrierpunktserniedrigung (des Lösungsmittels) Bisher wurde mit p0A << p0B angenommen, dass der gelöste Stoff nicht in der gasförmigen Phase vorkommt. Jetzt soll zusätzlich angenommen werden, dass das Lösungsmittel als reine Phase ausfriert, d.h. der gelöste Stoff soll immer in flüssigen Phase verbleiben. Damit bleibt die Sublimationsdruckkurve im Phasendiagramm zwangsläufig unverändert! Mit Absenkung der Dampfdruckkurve wird aber der Tripelpunkt des Gemischs nach unten links und damit auch die Schmelzdruckkurve nach links zu tieferen Temperaturen verschoben (vgl. Grafik oben). Da die Schmelzdruckkurve praktisch unverändert steil, d.h. fast senkrecht verbleibt entspricht die Verschiebung des Tripelpunktes auf der Temperaturskala der gesuchten Gefrierpunktserniedrigung TM. Nach etwas Geometrie folgt: R TTr R TTr M B Tm XA m A : EG m A hm hm 2 2 TTr ist die Tripeltemperatur, hm die Schmelzwärme, MB das Molgewicht des Lösungsmittels, XA der Molenbruch und mA die Molalität des gelösten Stoffes. EG bezeichnet man als Kryoskopische Konstante. ( vgl. Tab. K ) Hoeppe - PC Leerscript - 94 - 6.1.3.2 Druckabhängigkeit des Dampfdrucks Besteht andererseits die Gasphase aus zwei oder mehr eindeutig gasförmigen Komponenten (Luft), so werden diese sich kaum in der flüssigen Phase (Wasser) finden. Der Gesamtdruck ist daher deutlich größer als z.B. der Dampfdruck von H2O bei 300 K. Das führt zu einer Druckabhängigkeit des Dampfdrucks bzw. zu einer Gesamtdruckabhängigkeit des Partialdruckes: Betrachte also den Dampfdruck von Wasser bei anwesendem „Inertgas“ Luft, welches wegen sich wegen p0,N2 p0,O2 >> p0,H2O nur in der Gasphase befinden soll. Die Gleichgewichtsbedingung g = 0 für den Phasenübergang (vgl. Kap. 5.2) ist daher auch nur für die Komponente H2O und nicht für die anderen anzuwenden: „links“ reine Komponente H2O, flüssig „rechts“ H2O in einem Gasgemisch ! p g ( H 2O , l ) g 0 ( H 2O , l ) g 0 ( H 2O , g ) RT ln D g ( H 2O , g ) p Mit der Bedingung für währendes Gleichgewicht, dgi = dgii, folgt mit T, V,..= const: ! g 0 ( H O , g ) p g 0 ( H 2 O , l ) 2 dp dp dRT ln D p p p und wegen dG = VdP-SdT hier: ! Vm ,l dp Vm , g Mit id Vm , g Vm , l RT p dp RT d ln D p V m ,l V m , g RT ! d pD ln dp p RT geeigneter Substitution und partieller Integration folgt schließlich p p p 0 D p ln D0 pD und damit pD p e 0 D Vm , l RT p pD ist der resultierende Dampfdruck, wenn „Inertgase“ mit einem Druck von insgesamt p vorliegen und pD0 den Dampfdruck der reinen Komponente beschreibt. (Gleiches gilt bei einem zusätzlichen Druck z.B. durch Oberflächenspannungen, vgl. Kap. 7 !) Hoeppe - PC Leerscript - 95 - 6.1.3.3 Osmotischer Druck Eine ungleichmäßige Verteilung einer zweiten Komponente kann auch durch spezielle Strukturen erzeugt werden. Liegt z.B. eine gelöste Substanz nur auf einer Seite einer semipermeablen Membran vor, kann sich nur bzgl. des Lösungsmittels ein Gleichgewicht ausbilden. Die Folge ist der sog. osmotische Druck. Auf der linken Seite (1) befinde sich das Lösungsmittel B und ein darin gelöster Stoff A, auf der rechten Seite nur das Lösungsmittel B. Die Gleichgewichtsbedingung g = 0 für das Lösungsmittel lautet daher g 1 g 01 RT ln x B g 02 g 02 RT ln 1 g 2 ! und damit für währendes Gleichgewicht dg 1 dg 01 d RT ln x B d g 02 dg 2 ! Mit dg01,2 = Vm1,2dp1,2 – s dT ; T = const also dT = 0 und Vm1 Vm2 folgt nach Integration der osmotische Druck als Druckdifferenz: p1 p 2 RT RT ln( x B ) ln(1 x A ) : Vm Vm Wegen xA << 1 gilt ln(1-xA) - xA O(xAn2) und damit in guter Näherung die Van’t Hoffsche Gleichung RT RT xA nA Vm V für den osmotischen Druck, welcher auch wieder nur von der Konzentration, nicht aber der Art des gelösten Stoffes auf der linken Seite abhängt. Hoeppe - PC Leerscript - 96 - 6.1.4 Gibbssche Phasenregel Nach der Gibbs’schen Phasenregel können bei einem Gemisch aus K Komponenten P Phasen mit F Freiheitsgraden koexistieren. F K P2 Das führt u. a. dazu, dass auch ohne äußere Zwangsbedingung zwei oder mehrer Phasen im Gleichgewicht koexistieren können. Bei der Anwendung der Gleichgewichtsbedingung ist darauf zu achten, bzgl. welcher Komponente/Phasen sich ein Gleichgewicht ausbilden kann (vgl. z.B. Osmose). Hoeppe - PC Leerscript - 97 - 6.2 Nichtideale, reale Mischungen Bei realen Mischungen sind die i.A. immer vorhandenen WW zwischen den Molekülen zu beachten. Dabei sind nicht nur die WW zwischen den verschiedenen Komponenten sondern auch die WW zwischen Teilchen einer Komponente zu berücksichtigen: Grundsätzlich lassen sich folgende drei Fälle unterscheiden: i) 2EAB < EAA + EBB: exotherm: Wärme wird frei Volumen wird kleiner Mischung begünstigt (Bsp: Schwefelsäure-Wasser) ii) 2EAB > EAA + EBB: endotherm: Mischung kühlt ab Volumen wird größer Mischung erschwert (Bsp: Aceton-Schwefelkohlenstoff) iii) 2EAB >> EAA + EBB: stark endotherm: Mischung unter Abkühlung und Volumenvergrößerung nur durch z.B. starkes Umrühren realisierbar; i.A. aber Mischungslücke (Bsp: Phenol-Wasser) Zu beachten ist, dass die Größe der WW auch vom Mischungsverhältnis und der Einfluss der Entropie auf die Mischbarkeit von der Temperatur abhängen! Hoeppe - PC Leerscript - 98 - Eine Mischung von verschiedenen Stoffen heißt ideal wenn id V Vi n i V m , i i und i id H H i ni hi i gilt. i Aufgrund von i. A. immer vorhandenen WW zwischen den Molekülen, müssen wir für reale Mischungen für H und V allgemeiner V V E Vi V E niVm ,i H H E H i H E ni hi i i i i formulieren, wobei VE ein aufgrund der WW auftretendes Zusatz- oder Excessvolumen und HE eine Zusatz- oder Excessenthalpie beschreiben. 6.2.1 Partielle molare Größen Um zu einer Darstellung ähnlich der bei idealen Mischungen zu gelangen, werden die vom Mischungsverhältnis abhängigen Zusatzterme auf sog. partielle molare Größen abgebildet. Grundsätzlich ist dies für jede Zustandsgröße möglich, das Vorgehen sei hier am Beispiel der Enthalpie H erläutert: Wird die bei einer Mischung auftretende Excessenthalpie den einzelnen Komponenten zugeordnet gilt H H E H i ni hiE ni hi ni hiE hi ni hi i i i i i Erzeugt man eine Mischung „stückweise“ durch Zugabe sehr vieler sehr kleiner Anteile der Mischungskomponenten und hält man dabei das Mischungsverhältnis immer konstant so gilt demnach auch dH hi dni i Aus dem Vergleich mit dem totalen Differential für H = H(p=const, T=const, ni) folgt H dH i ni dni folgt H hi n i p ,T , j i was die Bezeichnung partielle molare Größe erklärt und letztlich hi* definiert. Betrachtet man nun hi* als zusätzliche Variable und schreibt das totale Differential für H = H(hi*, ni) so folgt daraus nach Koeffizientenvergleich unmittelbar die Hoeppe - PC Leerscript - 99 - Gleichung von Gibbs-Duhem, n dh i 0 i welche für alle partiellen molaren Größe in analoger Weise gilt. 6.2.1.1 Partielles Molvolumen Analog zum Beispiel Enthalpie gilt hier V V E Vi niVmE,i niVm ,i ni VmE,i Vm ,i niVm,i i i i i i Erzeugt man wieder eine Mischung „stückweise“ durch Zugabe sehr vieler sehr kleiner Anteile der Mischungskomponenten und hält man dabei das Mischungsverhältnis immer konstant so gilt demnach auch dV Vm,i dni i Aus dem Vergleich mit dem totalen Differential für V = V(p=const, T=const, ni) folgt V dni dV i ni mit V Vm,i n i p ,T ,n j i womit die partielle molare Größe Vmi* definiert ist. Mischt man z.B. Alkohol und Wasser erhält man ein Gesamtvolumen was nicht der Summe der Einzelvolumina entspricht, sondern aufgrund der ‚unterm Strich’ attraktiven WW kleiner ist (vgl. Grafik unten). Die partiellen molaren Volumina von Alkohol und Wasser für das jeweilige Mischungsverhältnis (und natürlich nur für diese Mischung) sind dementsprechend kleiner als die Molvolumina der reinen Komponenten, und offensichtlich deutlich vom Mischungsverhältnis abhängig. Hoeppe - PC Leerscript - 100 - 6.2.1.2 Mischungs- und Lösungsenthalpie Hier ist meist nicht die Gesamtenthalpie, sondern der Wärmeumsatz in Folge einer Mischung, also die Excessenthalpie interessant, welche auch Mischungswärme MH oder Lösungswärme LH = HL genannt wird. Entsprechend der Einführung in 6.2.1 gilt M H H E H H id ni hi ni hi ni hi hi ni hiE i i i i und als mittlere molare Mischungsenthalpie definiert man: HE 1 h ni hiE xi hiE n n i i E Hoeppe - PC Leerscript - 101 - Mischt man einen Festkörper durch Auflösen in einem Lösungsmittel erhält man mit der Lösungswärme einen (neben der Entropie) weiteren konzentrationsabhängigen Beitrag zur freien Enthalpie und damit z.B. zur Löslichkeit eines Salzes. Bei einem solchen Lösungsvorgang ist es üblich, mit der differentiellen Lösungswärme hdL und der differentiellen Verdünnungswärme hdV zwischen den wesentlichen Beiträgen des gelösten Stoffes und des Lösungsmittels zu unterscheiden: Betrachte Zweikomponentensystem mit Festkörper A und Lösungsmittel B: H E n A hAE nB hBE bzw. h E x A hAE xB hBE Integrale Lösungswärme hiL= Lösungswärme auf Molzahl des gelösten FK bezogen: HE n hiL hAE B hBE : hAE n hBE hdL n hdV nA nA Für gewöhnlich nimmt die integrale Lösungswärme mit der Konzentration ab; die Abnahme wird mit der Steigung der Kurve hiL(n) beschrieben. Mit der Gleichung von Gibbs-Duhem lässt sich zeigen, dass die Steigung der Kurve hiL(n) identisch gleich der differentiellen Verdünnungswärme hdV ist. Als erste Lösungswärme hdL0 bezeichnet man den Grenzwert von hiL bei unendlicher Verdünnung. Zur experimentellen Bestimmung von hdL und hdV aus der Messung von hiL(n) Hoeppe - PC Leerscript - 102 - 6.2.1.3 Partielle molare Freie Enthalpie = Chemisches Potential Entsprechend der Definition G = H-TS der Freien Enthalpie werden WW in einer Mischung die zur einer Excessenthalpie führen auch den Wert von G verändern. Da die Entropie letztlich eine rein statistische Größe ist, haben WW hier keinen Einfluss. Der Wert der Mischungsentropie unterscheidet sich bei realen Mischungen nicht vom Wert bei idealen Mischungen. Daher gilt letztlich giE = hiE mit G E H E M H ni hi hi ni g i g i ni g iE i i i Die Größe gi* beschreibt hierbei die molare freie Enthalpie der Komponente i , welche auch alle WW in einer spezifischen Mischung enthält. Damit ist der Beitrag einer Komponente gi* zur gesamten freien Enthalpie G nicht nur eine Funktion des Mischungsverhältnisses, also Molenbruchs, sondern auch von der jeweiligen Mischung! Die Definition von gi* erfolgt daher praktischer Weise als partielle molare Größe. Mit G = G(p, T, ni) lautet das totale Differential von G G G G dp dG dni dT p T n i T ,ni p ,ni i p ,T ,n j i Wird also der Anteil der i-ten Komponente um ein dni verändert, ändert sich die gesamte freie Enthalpie mit dem Faktor G g i n i p ,T ,n j i der sog. partiellen molaren Enthalpie, oder auch oft mit µi gi* als chemisches Potential bezeichnet. Der Begriff „chemisches Potential“ erklärt sich durch das Prinzip G MIN bzw. die Gleichgewichtsbedingung RG = 0 oder Rg = 0 : Bei einer chemischen Reaktion, oder auch einem Phasenübergang o. Ä., ändern sich zwangsläufig die ni von mindestens zwei Komponenten. Bei einer Reaktion A B gilt dnA = - dnB und nach der Gleichgewichtsbedingung außerdem µA·dnA + µB·dnB = 0. Im Gleichgewicht gilt daher immer µA = µB. Ungleiche chemische Potentiale von Stoffen innerhalb eines Systems führen somit immer zu einer chemischen Reaktion, bzw. i. A. zu einem Prozess welcher das System ins Gleichgewicht bringt. Hoeppe - PC Leerscript - 103 - Abbilden der WW auf den „Konzentrationsterm“ der Entropie durch Einführung einer Aktivität im Sinne einer „scheinbaren Konzentration“: Für ideale Mischungen gilt für die molare freie Enthalpie: g i ( xi , T ) g i0 (T ) RT ln xi hi0 (T ) T s i0 (T ) RT ln xi Werden jetzt zusätzlich (komponentenweise) die WW mit molaren (konzentrationsabhängigen) Exzessenthalpien hiE = giE berücksichtigt gilt: g i g i ( xi , T ) g iE g i0 (T ) RT ln xi g iE Schreibt man die Korrekturterme giE formal mit g iE RT ln( xi ) um , so lassen sich alle konzentrationsabhängigen Terme mit einer Aktivität a a ( x i ) xi xi zusammenfassen. Für die partielle molare freie Enthalpie gilt dann g i g i0 (T ) RT ln x i RT ln xi g i0 (T ) RT ln a i womit diese formal die gleiche Gestalt wie im Fall ohne WW hat. An die Stelle des Molenbruchs tritt jetzt die sog. Aktivität, welche somit also eine Art „effektive Konzentration“ beschreibt. Die Aktivitätskoeffizienten xi sind (auf beliebig komplexe Weise) konzentrationsabhängig und müssen experimentell bestimmt werden (vgl. z.B. Tab. H). Sie werden bei Lösungen auch oft als Funktion der Molalität mi und bei Gasen als Funktion der Partialdrücke angegeben. Bei Gasen spricht man von einer Fugazität fi und Fugazitätskoeffizienten i : p g i g i0 (T ) RT ln f i g i0 (T ) RT ln i oi p Für die gesamte freie Enthalpie des Systems gilt mit den oben eingeführten Aktivitäten jetzt G n i i g i n g i 0 i (T ) RT ln a i i d.h. bis auf die ai an Stelle der xi der gleiche Ausdruck wie bisher. Hoeppe - PC Leerscript - 104 - Besonderheit bei Lösungen bzw. Elektrolyten: Die Angabe der stoffspezifischen Standardwerte h i0, s i0 und damit auch von gi0 = h i0 - T s i0 ist bei gelösten Stoffen für Reinstoffe zwangsläufig nicht möglich. An Stelle eines Reinstoffs wird daher hier eine Konzentration bzw. Molalität von m = 1 mol/kg als Standardzustand definiert. Wenn nun die Konzentration/Molalität oder ganz allgemein die Aktivität in Einheiten von 1 mol/kg angegeben wird, verschwindet der konzentrationsabhängige ln-Term im Standardzustand: g i ( m i 1 mol kg ) g i0 (T ) RT ln 1 g i0 (T ) Zudem ist nicht jeder Stoff so gut löslich, dass sich eine Standardkonzentration mit einer Molalität m= 1 mol/kg einstellen ließe. Daher wird der Standardzustand letztlich durch eine hypothetische Lösung der Molalität 1 mol/kg definiert, in welcher sich die gelösten Stoffe ideal, d.h. mit Aktivitätskoeffizienten = 1, verhalten. Dieses ideale Verhalten gilt aber andererseits nur für unendliche Verdünnung also mi 0. Daher muss zur Bestimmung der Standardwerte bei unendlicher Verdünnung gemessen und anschließend auf die Standardkonzentration umgerechnet werden. Auf diese Weise ist die Angabe der Standardwerte für beliebige Elektrolyte in gleicher Weise möglich. Hoeppe - PC Leerscript - 105 - 6.3 Destillation In diesem Kapitel soll gezeigt werden, wie unter Ausnutzung der Anreicherung einer Komponente eines Gemischs in einer Phase bei einem Siedevorgang, wie in Kap. 6.1.2 bereits grundsätzlich gezeigt, eine praktikable Stofftrennung realisiert werden kann. Betrachtet werden sollen hier nur Zweistoffsysteme, zunächst als ideale Mischung und abschließend für den Fall realer Mischungen. Zur Vereinfachung sollen im Folgenden die Molenbrüche der Dampfphase mit y und der flüssigen Phase mit x geschrieben werden; der Gesamtmolenbruch mit einem großen X: x gi xli poi p y i xi poi p Das Hebelgesetz bzgl. der Komponente B lautet damit nl ( X B x B ) n g ( y B X B ) Die Trennbarkeit eines Gemischs durch Destillation wird durch die unterschiedliche Flüchtigkeit, also dem Verhältnis der Molenbrüche x und y der beiden Komponenten bestimmt. Man definiert den sog. Trennfaktor mit yB y y (1 x B ) A B xB x B (1 y B ) xA der im Falle eines idealen Gemisch identisch mit dem Verhältnis der Partialdrücke der reinen Komponenten ist: id x B poB / p x p /p p oB A oA xB poA xA Hoeppe - PC Leerscript - 106 - 6.3.1 6.3.1.1 Destillation idealer Mischungen Siedevorgang ohne Entfernung des Dampfes Wird der Dampf des Gemischs bei einem Siedevorgang nicht entfernt, bleibt zwangsläufig der Gesamtmolenbruch XB konstant. Wird also ein Gemisch ausgehend von der Temperatur T0 langsam erhitzt, tritt bei Erreichen der Siedelinie bei T1 eine Stofftrennung ein. Die zugehörige Zusammensetzung des Dampfes y1 wird durch den Schnittpunkt der Konode mit der Taulinien gegeben. Bei weiterer Erwärmung verarmt die Flüssigkeit an der leichterflüchtigen Komponente (LFK), womit auch der Molenbruch der LFK im der Dampfphase immer weiter abnimmt. Bei T3 verdampft schließlich das letzte Tröpfchen und der Molenbruch der Dampfphase ist dann zwangsläufig mit dem Gesamtmolenbruch identisch. Hoeppe - PC Leerscript - 107 - 6.3.1.2 Siedevorgang mit Entfernung des Dampfes Einfache Destillation Wird jetzt im Gegensatz zum vorherigen Kapitel das gleiche Gemisch ausgehend von T0 langsam erhitzt, und die Dampfphase kontinuierlich entfernt, verarmt das Gesamtsystem zwangsläufig an der LFK. Damit ändert sich auch kontinuierlich der Gesamtmolenbruch XB des Systems. Das System „rutscht“ daher entlang der Siedelinie zu immer höheren Temperaturen. Erst bei T = TSA ist das letzte Tröpfchen verdampft. Als Destillat bezeichnet man die mit einem Kühler wieder kondensierte Dampfphase, welche mit der LFK (lediglich) angereichert ist. Besonders wichtig sind die folgenden Punkte: - Bei allen T < TSA ist die LFK noch in der flüssigen Phase enthalten, da XB > 0. -Eine Reindarstellung einer Komponente ist offensichtlich nicht möglich, weder in der verbleibenden flüssigen noch in der rekondensierten Dampfphase. -Wird zwecks Anreicherung der LFK destilliert, muss der Prozess bei einer sinnvoll zu wählenden Temperatur beendet werden, da der Dampf immer weniger der LFK enthält. Bei der Temperatur T3 (vgl. Grafik unten) ist die Zusammensetzung des Dampfes y3 nur noch so groß, wie die ursprüngliche Konzentration XB0 des flüssigen Ausgangsmaterials. - Die höchst erreichbare Konzentration der LFK ist mit y1 gegeben. Diese wird aber nur erreicht, wenn man sich mit einem „winzigen Tröpfchen Destillat“ begnügt. Hoeppe - PC Leerscript - 108 - 6.3.1.3 Fraktionierte Destillation - Rektifikation Ist das Ergebnis einer einfachen Destillation bzgl. der Anreicherung der LFK nicht ausreichend, so kann man das Destillat natürlich einfach nochmals destillieren. Da der Gesamtmolenbruch der LFK im Destillat jetzt größer ist, wird auch das hiermit erzeugte nächste Destillat eine noch größere Konzentration der LFK haben. Im Siedediagramm bewegt man sich dann (bestenfalls) entlang einer ‚Treppenkurve’ (vgl. Grafik unten) zu höheren Konzentrationen der LFK. Die Anzahl der Treppenstufen entspricht den (minimal) nötigen Destillationsschritten, um ausgehend von der Startkonzentration XB,S eine gewünschte Produktkonzentration XB,P zu erreichen. Siedediagramm Gleichgewichtsdiagramm Eine alternative Darstellung dieser Mehrfachdestillation (Fraktionierte Destillation/ Rektifikation) erhält man mit dem sog. Gleichgewichtsdiagramm (vgl. Grafik oben). Hier werden nur die letztlich interessierenden Molenbrüche yB und xB gegeneinander aufgetragen; die „Dicke“ der Siedelinse wird auf die (gekrümmte) Gleichgewichtskurve y(x) abgebildet, und das Verwenden des Destillats für den nächsten Destillationsschritt (z.B. x2 = y1) wird mit der Hauptdiagonalen beschrieben. Auch hier lassen sich die (minimal) benötigten Destillationsschritte an den Treppenstufen abzählen. Das Gleichgewichtsdiagramm ist besonders wichtig im Zusammenhang mit der quantitativen Beschreibung einer Destillationsanlage, vgl. Kap. 6.3.1.4. Hoeppe - PC Leerscript - 109 - Die oben beschriebene Mehrfachdestillation lässt sich in einer Apparatur zusammenfassen. Man spricht dann von einer Rektifikationskolonne. Eine besonders anschauliche Bauart ist eine sog. Glockenbodenkolonne (vgl. Grafik unten), wobei mit jedem Boden ein weiterer Destillationsschritt realisiert wird. Glockenbodenkolonne nach Wedler Unter der theoretische Bodenzahl versteht man die Anzahl von Destillationsschritten, d.h. Treppenstufen im Gleichgewichtsdiagramm, die unter ‚idealen Verhältnissen’ die LFK ausgehend von XB,S bis zu XB,P anreichern. Diese ist also ein Maß für die Leistungsfähigkeit einer Apparatur. Bei einer einfacher zu bauenden Füllkörperkolonne sind die Destillationsschritte „homogen über die Säule verteilt“ und die Bodenzahl ist nicht direkt abzählbar. Durch eine experimentelle Bestimmung der Leistungsfähigkeit einer solchen Apparatur ist aber die (äquivalente) theoretische Bodenzahl bestimmbar. Mit Hilfe des Gleichgewichtsdiagramms eines Gemischs und der theoretischen Bodenzahl einer Apparatur, lässt sich dann abhängig von der Startkonzentration XB,S das Resultat XB,P vorhersagen, oder umgekehrt bei gegebener Rohstoff- und Produktspezifikation durch ‚schlichtes Treppenstufenzeichen’ die Dimensionierung der Anlage prinzipiell bestimmen bzw. abschätzen. Hoeppe - PC Leerscript - 110 - 6.3.1.4* Rektifikation nach McCabe-Thiele Die theoretische Beschreibung von Destillationsprozessen nach McCabe und Thiele erlaubt u. a. die Berechnung der Mindestbodenzahl, welche im vorherigen Kapitel grafisch bestimmt wurde. Wesentlicher ist aber die Beschreibung der Auswirkung einer Produktentnahme auf den Destillationsvorgang: Die Entnahme von Destillat führt letztlich zu einer Störung des Gleichgewichts längs der Konoden zwischen ‚benachbarten’ Böden und damit zu einer schlechteren Trennwirkung der Anlage. (vgl. Verschiebung des Gesamtmolenbruchs, wenn mehr als ein winziges Tröpfchen Destillat erzeugt wird!) Durch Aufstellung einer Mengenbilanz bzgl. der Stoffmengenströme innerhalb einer Kolonne, kann die Auswirkung einer Produktentnahme quantitativ beschrieben, und damit die Auslegung von Anlagen optimiert werden. Zudem wird durch die Unterscheidung und Beschreibung von der sog. ‚Auftriebs-’ und ‚Abtriebskolonne’ auch die Modellierung von kontinuierlich arbeitenden Anlagen ermöglicht. Mehr hierzu in der Veranstaltung PCMU. Hoeppe - PC Leerscript - 111 - 6.3.2 Destillation realer Mischungen Die Destillation realer Mischungen funktioniert genau so wie die idealer Mischungen, da insbesondere das Hebelgesetz auch für reale Mischung gültig ist. Durch die WW werden aber die Siedediagramme zum Teil stark verändert, wodurch sich in der Praxis deutliche Abweichungen und/oder neue Effekte ergeben. 6.3.2.1 Minimumazeotrop Wird durch eine WW z.B. die Bildung einer Mischung A-B erschwert, da sie energetisch ungünstig ist, wird die Phase mit dem größeren Teilchenabstand – also die Gasphase - begünstigt. Taulinie und Siedelinie werden daher im Siedediagramm nach unten geschoben und können sich bei hinreichend starker WW sogar berühren, und es kommt zur Ausbildung eines azeotropen Punkts, hier Minimumazeotrop: Ein Destillationsprozess lässt sich in einem solchen Siedediagramm analog zum Fall idealer Mischungen durch Einzeichnen von Konoden bzw. ‚Treppenstufen’ beschreiben: Die linke Seite des Zweiphasenmischgebietes ähnelt einer gewöhnlichen Siedelinse, allerdings mit dem entscheidenden Unterschied, dass „die Spitze unten rechts“ nicht bei XB = 1 sondern bei XB = Xaz liegt. Ein Destillationsprozess kann als Ergebnis daher maximal die Konzentration Xaz liefern! Liegt die Startkonzentration rechts vom azeotropen Punkt Xaz läuft der Destillationsprozess spiegelverkehrt auf der rechten Seite ab, so dass die Konzentration der (eigentlich) LFK B zum Kolonnenkopf abnimmt und den selben Wert Xaz annimmt, wie bei einem Start von der linken Seite. [vgl. Übung!] Azeotrope Punkte sind bei Destillationsprozessen daher unerwünscht und nur durch relativ aufwendige „Tricks“ zu umgehen, die in Kap. 6.3.2.3 kurz angesprochen werden sollen. Hoeppe - PC Leerscript - 112 - 6.3.2.2 Maximumazeotrop Wird durch eine WW andererseits die Bildung einer Mischung A-B erleichtert, wird die Phase mit dem kleineren Teilchenabstand – also die Flüssigphase - begünstigt. Taulinie und Siedelinie werden daher im Siedediagramm nach oben geschoben und können sich bei hinreichend starker WW sogar berühren, und es kommt auch hier zur Ausbildung eines azeotropen Punkts, hier Maximumazeotrop: Auch hier lässt sich durch Einzeichnen von Konoden bzw. ‚Treppenstufen’ der Siedeprozess beschreiben: Eine Anreicherung der LFK B wird offensichtlich nur auf der rechten Seite, d.h. für XBS > Xaz, erreicht. Man hat hier (zumindest auf der rechten Seite) also zunächst prinzipiell kein Problem mit der Qualität des Produktes, jedoch hat man sicher ein Ausbeuteproblem, da das im Sumpf verbleibende Gemisch immer einen hohen Gehalt beider Komponenten enthält. Beginnt man auf der linken Seite, wird zunächst die Komponente A im Kolonnenkopf angereichert, bis auch hier das im Sumpf verbleibende Gemisch die azeotrope Zusammensetzung Xaz erreicht. In beiden Fällen erreicht das Gemisch in der flüssigen Phase letztlich die azeotrope Konzentration und siedet somit unverändert, d.h. unter Abgabe einer Dampfphase mit der konstanten Konzentration Xaz und der konstanten (maximalen) Siedetemperatur. Daher auch der Name Azeotrop : der aus dem griechischem stammende Begriff bedeutet so viel wie „unverändert siedend“ (, vgl. z.B. Atkins). Hoeppe - PC Leerscript - 113 - 6.3.2.3* Azeotroprektifikation Unter Azeotroprektifikation versteht man die Destillation von azeotropen Gemischen unter Anwendung spezieller Verfahren, mit welchen sich die azeotropen Punkte bzw. deren negative Auswirkungen auf den Destillationsprozess umgehen lassen. Dabei werden hauptsächlich zwei Strategien angewendet: - Destillation bei verschiedenen Drücken: Da die Siedetemperaturen jeder Komponente (individuell) druckabhängig sind, wird das Siedediagramm mit dem Druck deutlich verändert. Zudem sind die Wechselwirkungen, welche einen azeotropen Punkt erzeugen, i. A. temperaturabhängig. Da mit dem Druck die Siedetemperatur des Gemischs verändert wird, wird auch der azeotrope Punkt zumindest verschoben. So kann z.B. Ethanol mit einer deutlich höheren Konzentration als 96 Gew% (azeotroper Punkt bei 1 bar) durch die sog. Zweidruckrektifikation hergestellt werden, bei der zwei Rektifikationskolonnen unter verschiedenem Druck arbeitend kombiniert werden. - Destillation mit einem Schleppmittel: Durch Zugabe eines sog. Schleppmittels, z.B. Benzol bei einem Ethanol-Wasser-Gemisch, entsteht eigentlich eine ternäre Mischung mit komplexer Wechselwirkung zwischen den Komponenten. Da das Schleppmittel i.d.R. nur in kleiner Konzentration zugegeben wird, betrachtet man aber oft nur das veränderte Siedediagramm des jeweiligen binären Gemischs in welchem der azeotrope Punkt stark verschoben oder ganz verschwunden ist. Im obigen Beispiel bildet das Benzol mit Ethanol und Wasser ein leicht siedendes ternäres Azeotrop, wodurch das Wasser aus einem ethanolreichen Gemisch „herausgeschleppt“ wird, und vergleichsweise sehr reines Ethanol im Sumpf der Kolonne verbleibt. Überreste des Schleppmittels müssen dann ggf. durch weitere Destillationsschritte entfernt werden. Man spricht bei diesen Verfahren auch von Extraktivrektifikation. Hoeppe - PC Leerscript - 114 - 6.4 Mischungen von FK und Schmelzen Was einführend zu den Phasengleichgewichten von l-g-Gemischen gesagt wurde, gilt analog auch s-l Gemische, also Schmelzvorgänge. Zu beachten ist allerdings, dass die Ausbildung eines Gleichgewichts oft dadurch erschwert oder gar nicht möglich ist, da eine Vermischung der betrachteten Komponenten in der s Phase nur vergleichsweise langsam erfolgen kann. Schmelz- bzw. Erstarrungsvorgänge sind daher eher komplizierter, da zusätzlich die Abkühlgeschwindigkeit zu berücksichtigen ist (Kristallseigerung). Andererseits ergeben sich hierdurch eine Vielzahl von Methoden um Werkstoffeigenschaften gezielt einzustellen, wie z. B. Weichglühen, Kaltverfestigen, oder Härten bis hin zu anisotropen oder Gradientenwerkstoffen. Im Folgenden sollen zunehmend schlechter mischbare binäre Mischungen betrachtet werden. 6.4.1 Lückenlose Mischkristallbildung Das Phasendiagramm von sehr gut mischbaren Komponenten, z.B. das System Germanium-Silizium, gleicht der Siedelinse einer idealen Mischung. Aufgrund lückenloser Mischkristallbildung existiert nur eine feste (s) Phase. Schmelzdiagramm eines binären Gemischs (analog zu Siedelinse) eines gut mischbaren Systems, d.h. mit lückenloser Mischkristallbildung. Bsp.: Tm,Si = 1683 K + Tm,Ge = 1210 K Bei unendlich langsamer Abkühlung entspricht der Weg im Phasendiagramm dem Fall „Sieden ohne Entfernung des Dampfes. Bei einer realen Abkühlgeschwindigkeit werden die zuerst kristallisierten festen Phasen bei weiterer Abkühlung nicht mehr vollständig ins Gleichgewicht mit den folgenden festen Phasen gelangen. In einem Schmelztigel wird nach kompletter Erstarrung zwangsläufig mehr oder weniger ein „Gradientenkristall“ entstehen (Zonenmischkristall, inhomogener Mischkristall). Die Tatsache, dass sich die niedriger schmelzende Komponente B in der l-Phase anreichert, wird bei dem Zonenschmelzverfahren ausgenutzt, bei welchem z.B. hochreine Si-Kristalle für die Wafer-Herstellung erzeugt werden. Hoeppe - PC Leerscript - 115 - Schematische Darstellung des Zonenschmelzverfahrens 6.4.2 Reale Systeme mit Mischungslücke Die bei Festkörpern i. A. noch stärkeren WW führen oft zu in der l-Phase schlecht und in der s-Phase gar nicht mischbaren Systemen (Mischungslücke). Ähnlich der Ausbildung eines azeotropen Punktes bei l-g-Gemischen kommt es hier zur Ausbildung eines eutektischen Punktes bzw. eines Eutektikums. Schmelzdiagramm eines binären Gemischs mit Mischungslücke in der festen Phase Bsp.: Gold-Nickel-Legierung mit Tm,Au = 1337 K + Tm,Ni = 1726 K Bei Abkühlung einer Schmelze beliebiger Zusammensetzung wird bei Erreichen der Liquiduskurve eine feste Phase entsprechender Zusammensetzung (Konode) ausgeschieden. Die Zusammensetzung der Schmelze läuft bei unvollständiger Einstellung des Gleichgewichts („rasche“ Abkühlung) damit zwangsläufig in Richtung des Wertes des Eutektikums xE. Wird xE erreicht, erstarrt bei weiterer Abkühlung die Restschmelze zu einem Mischkristall der Zusammensetzung xE. Wird bei noch tieferen Temperaturen die Mischungslücke erreicht, ist der Mischkristall thermodynamisch instabil und es kommt prinzipiell zur Ausbildung verschiedener Phasen deren Zusammensetzung durch Einzeichnen einer Konode in das 2-Phasen-Mischgebiet bestimmt ist (Segregatbildung). Der Begriff Eutektikum stammt aus dem griechischem und bedeutet gut schmelzend . Hoeppe - PC Leerscript - 116 - 6.4.3 Systeme mit großer partieller Mischungslücke Bei sehr schlechter Mischbarkeit in der festen Phase, reicht die Mischungslücke bis an die Soliduskurve heran. Das bedeutet, dass die Schmelze nach Erreichen des Eutektischen Punktes entsprechend dem Hebelgesetz gleichzeitig in zwei verschiedene feste Phasen erstarrt. Die waagerechte Konode durch den eutektischen Punkt nennt man auch Eutektikale, eine Mischung der Zusammensetzung xE eutektisches Gemisch. Läuft die Schmelze hier in xE, so kommt es zu einer „schlagartigen“ Verfestigung unter Ausbildung eines sehr feinkörnigen (auch lammellenartigen) Gemenges von Mischkristallen und , dem sog. Eutektikum. Schmelzdiagramm mit Eutektikum, und großer Mischungslücke. Bsp.: Silber-Kupfer-Legierung mit Tm,Ag = 1235 K + Tm,Cu = 1357 K Weitere eutektische Gemische sind z.B. Lötzinn ( Pb(33)-Sn(67), TE = 183°C ) oder auch Streusalz ( NaCl(23)-H2O(77), TE = –21,1 °C ). 6.4.4 Systeme ohne Mischkristallbildung Bei extrem schlechter Mischbarkeit der festen Phasen existieren keine Mischkristalle und die Soliduskurve besteht nur aus der Eutektikalen und den Senkrechten zu den Tm. Schmelzdiagramm mit Eutektikum, und vollständiger Mischungslücke, d.h. ohne Mischkristallbildung. Hoeppe - PC Leerscript - 117 - In einem solchen System läuft (auch bei extrem langsamer Abkühlung) der Rest der Schmelze immer in den eutektischen Punkt. Je nach Ausgangszusammensetzung der Schmelze kommt es daher bei Abkühlung zu einem mehr oder weniger großen Anteil der Phase, welche Eutektikum genannt wird. Hier besteht das Eutektikum aus einem feinkörnigen oder lammellenartigen Gemenge von Kristalliten der reinen Phasen A und B . 6.5 Phasendiagramme ternärer Gemische Ternäre Gemische bestehen aus 3 Komponenten. Wegen xA + xB + xC = 1 können damit Mischungsverhältnisse eindeutig in einer zweidimensionalen Ebene dargestellt werden. Meist wird dafür eine ebene symmetrische Dreiecksdarstellung gewählt: Im Dreiecksdiagramm wird ein Mischungsverhältnis eindeutig durch einen Punkt beschrieben. Bsp: A(40%); B(20%); C(40%) Bestimmte Eigenschaften der Mischung in bestimmten Bereichen werden dann z.B. in Form von Farben oder „Höhenlinien“ dargestellt, oder wie unten als Projektion eines dreidimensionalen Diagramms. Liquidusfläche eines ternären Systems: Das ternäre Eutektikum E liegt meist bei der tiefsten Temperatur; als Grenzfälle sind auch die Eutektika der binären Mischungen AB, BC und AC enthalten. Hoeppe - PC Leerscript - 118 - 7 Grenzflächengleichgewichte Grenzflächen sind auch Oberflächen, an welchen viele Prozesse stattfinden wie z.B.: - Verdampfen - Adsorption, Chemiesorption ( Chromatografie) - chem. Reaktionen - Korrosion ( Beschichtungen, Oberflächentechnik) Je kleiner einer Phase bzw. ein Teilchen ist, desto größer wird der Einfluss von Oberflächeneffekten (Reaktivität). Stichworte sind hier - Kolloide, Kolloidchemie (molekular dispers/kolloid dispers ; Sol-Gel-Technik) - Biochemie (molekulare Doppelschichten, Membranen) - Nanotechnolgie - Klebetechnologie 7.1 Allgemeine Betrachtungen Sind Komponenten einer Mischung aufgrund einer WW nicht mischbar, bilden sich zwangsläufig zwischen den auftretenden Phasen Grenzflächen aus. Die gleichen WW die zur Phasenausbildung führen, bedingen zwangsläufig auch eine Minimierung der Grenzfläche, weil dadurch wieder die energetisch ungünstigen WW zwischen den verschiedenen Komponenten an der Grenzfläche minimiert wird. Für die Freie Enthalpie G = G(p, T, A) folgt G G G dp dG dT dA V dp S dT dA A p T p ,T T , A p, A bzw. die freien Energie F = F(V, T, A) F F F dF dT dA p dV S dT dA dV V T , A T V , A A V ,T wobei die Oberflächenenergie ( oft auch „Oberflächenspannung“) beschreibt. Mit der Minimierung von F (anschaulich auch mit der „Oberflächenspannung“) lässt sich z.B. relativ leicht die Benetzung (Kontaktwinkel) von festen Oberflächen durch Flüssigkeiten erklären. Für gekrümmte Oberflächen (Blasen, Tropfen etc.) folgt aus der Minimierung von F die Laplacegleichung, mit welcher sich z.B. die Kapillarwirkung oder der Siedeverzug einfach erklären lassen. Hoeppe - PC Leerscript - 119 - 7.2 7.2.1 Oberflächenspannung Oberflächenenergie und Oberflächenspannung Entsprechend Definition entspricht die Oberflächenenergie der Arbeit, welche nötig ist, um eine Oberfläche zu erzeugen. Im Falle von Flüssigkeiten lässt sich direkt mit einer Oberflächenfilmwaage ( Langmuirsche Waage) bestimmen: F m g FOF dW ( m g 2 l ) ds dW m g dh dA Betrachtet man, z.B. bei einem Tropfen einen Schnitt durch eine Oberfläche kann man formal Arbeit Kraft Weg Kraft Fläche Weg Weg Weg Schreiben, und lässt sich im Schnitt als Oberflächenspannung verstehen und bei Auftreten verschiedener Oberflächen bequem wie ein Kräftegleichgewicht vektoriell behandeln (vgl. Benetzung). 7.2.2 Benetzung Bei Vorhandensein bzw. Kontakt dreier Phasen, z. B. fest, flüssig und gasförmig, ergeben sich zwangsläufig auch drei verschiedene Oberflächen. Die Konkurrenz der jeweiligen Oberflächenenergien führt z.B. zu einer Vergrößerung der energetisch günstigeren Oberfläche und damit bei (festen Volumina) zu einem bestimmten Kontaktwinkel K am Tripelpunkt zwischen den Phasen, welcher als Maß für die Benetzbarkeit eines Festkörpers mit einer Flüssigkeit dient: Man spricht von nicht benetzend für K > 90°, für benetzend für K < 90° und von spreitend für K 0°. Hoeppe - PC Leerscript - 120 - Die drei zu unterscheidenden relevanten ’Oberflächenspannungen’ (z.B. bei einem Wassertropfen auf einer Glasplatte) sind: lg = wesentlich abhängig von WW innerhalb l, da Gas dünn (Kohäsion) sg wesentlich abhängig von WW innerhalb s, da Gas dünn ls abhängig von WW zwischen l - und s-Phase (Adhäsion) Die vektorielle Behandlung der Oberflächenspannungen als Kräftegleichgewicht im Schnitt bestimmt den Kontaktwinkel K , welcher neben der Oberflächenspannung der Flüssigkeit aufgrund der geringen Teilchendichte in der Gasphase wesentlich von ls , also der Adhäsion abhängt: sg ls cos k cos k sg ls Wird der Wert einer Oberflächenspannungen z.B. in Tabellen angegeben, wird als angrenzende Gasphase üblicherweise der gleiche Stoff mit dem zur angegebenen Temperatur gehörigen Dampfdruck angenommen. Gleiches gilt bei Oberflächenspannungen für Festkörper, die für die Ausbildung der Gefügestruktur eines polykristallinen Stoffes (z.B. Keramiken) entscheidend wichtig ist. 7.2.3 Laplace Gleichung Die Minimierung von G bzw. F führt zur Ausbildung von sphärischen Tropfen (Flüssigkeit in Gas) oder Blasen / intergranulare Poren (Gas in Flüssigkeit oder FK) und damit zu einer gekrümmten Ober- bzw. Grenzfläche. Abhängig vom Krümmungsradius der Oberfläche kommt es, anschaulich durch die Oberflächenspannung, auf der konkaven Seite zu einer Erhöhung des Drucks relativ zum Druck auf der konvexen Seite. Dies führt z.B. über die Druckabhängigkeit des Dampfdrucks (vgl. Kap. 6.1.3) zum Siede- oder Kondensationsverzug (vgl. Keimbildung) oder bei Vorhandensein einer dritten Phase über die mehr oder weniger gute Benetzung zur Kapillarwirkung. Im Gleichgewicht gilt mit T = const dF = 0 und damit: Hoeppe - PC Leerscript - 121 - ! dF pdV dA 0 pdV dA Betrachten wir als Bsp. einen Tropfen mit dem Radius r, dessen Oberfläche und Volumen sich bei Vergrößerung des Radius um dr entsprechend mit vergrößern: dA 4 ( r dr ) 2 4 r 2 ... 8 r dr dV A dr 4 r 2 dr Laplacegleichung: p 4 r 2 dr 8 r dr p 2 r pin pex Die Laplacegleichung beschreibt für beliebige gekrümmte Oberflächen die (lokale) Druckdifferenz in Folge der Kohäsion in Abhängigkeit von Oberflächenspannung und Krümmungsradius r. 7.2.4 Kapillarwirkung Die Kapillarwirkung entsteht als Folge der Kombination von Adhäsion (Benetzung Festkörper/Wand durch Flüssigkeit) und Kohäsion (Druckdifferenz an gekrümmter Oberfläche entsprechend Laplacegleichung). Nach Einstellung des Gleichgewichts zwischen Laplacedruck und Schweredruck der Flüssigkeit beobachtet man in Abhängigkeit von den Materialien (Benetzung, Oberflächenspannung) und Durchmesser der Kapillare eine mehr oder weniger stark ausgeprägte Kapillarascension (Grafik: links) oder Kapillardepression (Grafik rechts). Hoeppe - PC Leerscript - 122 - Betrachten wir die Ausbildung einer Kapillarascension und die daraus folgende Steighöhe der Flüssigkeit in der Kapillare in drei Schritten: 1) 2) 3) Tauche Kapillare in Flüssigkeit. Adhäsion/Benetzung stellt Kontaktwinkel und damit Krümmungsradius innerhalb der Kapillare ein. Druck p* auf konvexer Seite (unten) kleiner. Flüssigkeit steigt in Kapillare auf wodurch p* um den Schweredruck gh erhöht wird, bis sich ein Gleichgewicht bei p’’ = p (Umgebungsdruck) einstellt: ! 2 gh p p ' ' p ' gh p r 2 gh r Nimmt man mit k 0 vereinfacht eine sehr gute Benetzung an, ist die Oberfläche in der Kapillare mit Innendurchmesser d eine Halbkugel und es gilt r = d/2. Bei guter Benetzung kann also auf einfache Weise mit Messung der Steighöhe in einer Kapillare die Oberflächenspannung einer Flüssigkeit bestimmt werden: 1 gh d 4 Da die Kapillarwirkung also nichts mit einer „Saugwirkung“ zu tun hat, können z.B. so auch Bäume, die deutlich größer als 10 m sind, bis in die Spitzen mit Wasser versorgt werden! Hoeppe - PC Leerscript - 123 - 7.2.5 Keimbildung Der (stoffspezifische) Dampfdruck einer Flüssigkeit ist nicht nur von der Temperatur abhängig, sondern auch vom Gesamtdruck, der auf der flüssigen Phase lastet z.B. in Folge Anwesenheit anderer Gase. Der Dampfdruck über der Flüssigkeit steigt dann mit Vm ,l pD p e RT 0 D p (vgl. Kap. 6.1.3), wobei p = pges – pD0 dann positiv ist. Gleiches gilt wenn der Gesamtdruck auf der Seite der Gasphase vom Laplacedruck, der an einer gekrümmten Oberfläche entsteht erhöht wird. Bei einer auf Seite der Gasphase konkaven Oberfläche kann aber durch den Laplacedruck p auch negativ werden womit der Dampfdruck pD verringert wird. Für z.B. hinreichend kleine Tröpfchen mit dem Radius r ist p durch den Laplacedruck pL = 2/r gegeben, der an einer gekrümmten Oberfläche mit dem Krümmungsradius r entsteht und einige bar betragen kann. ( vgl. Übung) Wegen pL ~ r-1 wird der Dampfdruck über dem Tröpfchen also umso größer je kleiner es ist. Wird z.B. feuchte Luft abgekühlt und es müsste eigentlich zur Kondensation kommen wird daher die Bildung der ‚ersten winzigen Tröpfchen’ deutlich erschwert oder ganz verhindert. Das erklärt die Existenz von übersättigtem Dampf, wie er z.B. in einer Nebelkammer zum Nachweis von Elementarteilchen verwendet wird. In der Natur (Nebel, Wolkenbildung, Tau, Reif ) ist dieser Effekt meist nicht so deutlich zu beobachten, da kleinste Schwebeteilchen als Kondensationskeime dienen, da eine bestimmte Menge von H2O Molekülen an einem einigermaßen gut benetzbaren Teilchen mit einem größeren Krümmungsradius und damit einem geringeren Laplaceund Dampfdruck kondensieren kann (vgl. Grafik). Auch sehr kleine Dampfblasen in einer siedenden Flüssigkeit ‚haben es schwer’: Hier ist das Wasser auf der konvexen Seite und der durch pL stark verringerte Dampfdruck ( p < 0 pD << pD0 ) auf der konkaven Seite lässt kleine Blasen „zusammenschnurren“, wodurch überhitztes Wasser entstehen kann. Nur größere Dampfblasen können entstehen und wachsen dann rasch an, was zum „blubbernden Kochen“ führt. Dieser Siedeverzug lässt sich durch z.B. durch Siedesteinchen vermeiden, welche als „Verdampfungskeime“ wirken. Eine ähnliche Wirkung haben auch Oberflächenrauhigkeit oder Schmutzpartikel in einem Sektglas. Hoeppe - PC Leerscript - 124 - 7.3 Adsorption Von Adsorption spricht man, wenn es in Folge einer schwachen (meist physikalischen) Wechselwirkung zu einer vorübergehenden Anlagerung von Molekülen (Adsorbtiv) an einer Oberfläche (Adsorbens) kommt. Das Adsorbtiv befindet sich dabei in einer flüssigen oder gasförmigen Phase meist in geringerer Konzentration und liegt mit dieser als Gemisch vor (Bsp.: Luftfeuchte). Man unterscheidet bzgl. der (attraktiven) Wechselwirkung zwischen Physikalischer Adsorption = Physisorption Van-der-Waals-Kräfte. weniger selektiv, mehrschichtige Belegung möglich Adsortionsenthalpie ~ 20 kJ/mol ( immer exotherm ) Chemischer Adsorption = Chemiesorption: Bindungsähnliche Zustände selektiv, i.d.R. monomolekular Adsortionsenthalpie ~ 200 kJ/mol Die Stoffmengenverhältnisse, d.h. die Menge von adsorbierten Molekülen (Adsorbat) im Verhältnis zu den nicht adsorbierten Molekülen (Adsorbtiv), der Adsorptionsgrad, wird neben den chemisch-physikalischen Eigenschaften aller beteiligten Phasen wesentlich durch die Temperatur bestimmt und daher durch sog. Adsorptionsisothermen beschrieben: a) Langmuirsche Adsorptionsisotherme Zahl adsorbierter Moleküle: N Zahl max. möglicher ad. Moleküle: Nmax Partialdruck (Konz.) des Adsorptivs: p Adsorptionsrate: ra Desorptionsrate: rd N Bedeckungsgrad N max ra ~ p (1 ) ra : a p (1 ) rd ~ rd : a ' Gleichgewicht: rd = ra N N max Hoeppe - PC Leerscript p b p a p (1 ) a ' N a p p : N max a ' a p b p wobei der systemspez. Parameter b = b(T) temperaturabhängig ist. - 125 - 120 Rel. Adsoprtion N 100 80 60 40 T1 20 T2 > T1 0 0 0,2 0,4 0,6 0,8 1 Konzentration bzw . Druck p Langmuirsche Adsorptionsisothermen eines Systems für zwei versch. Temperaturen: Da nur eine einfache Bedeckung der Oberfläche angenommen wird, existiert für p ein Grenzwert, bei dem die Oberfläche zu 100% bedeckt ist. b) Adsorptionsisotherme nach Freundlich Insbesondere im Fall der Physisorption kann die Oberfläche auch durch mehrere Schichten Adsorbat bedeckt sein, was z. B. mit der Adsorptionsisotherme nach Freundlich beschrieben werden kann: Die systemspez. Parameter a und m sind auch hier i. A. temperaturabhängig. N a p1 / m 160 Rel. Adsoprtion N 140 120 100 80 60 Langmuir 40 Freundlich 20 0 0 0,2 0,4 0,6 0,8 1 Konzentration bzw . Druck p Adsorptionsisotherme nach Freundlich im Vergleich zur Langmuirschen Adsorptionsisothermen: Aufgrund der Mehrschichtbelegung existiert hier kein Grenzwert für p . Hoeppe - PC Leerscript - 126 - c) Adsorptionsisotherme nach Brunauer, Emmet und Teller Auch bei der Adsorptionsisotherme nach Brunauer, Emmet und Teller (BET) wird eine Mehrschichtbelegung angenommen. Sie wurde speziell für den Fall der Stickstoffadsorption an Pulvern oder porösen Festkörpern entwickelt und dient im sog. BET Verfahren der Bestimmung der spezifischen Oberfläche (m²/g), womit z.B. auch die mittlere Teilchengröße von sehr feinen Pulvern ermittelt werden kann. BET Verfahren (vereinfacht): Wird eine bestimmte Menge Gas (Stickstoff) in ein definiertes Volumen gegeben stellt sich entsprechend dem id. Gasgesetz der Druck p = n·RT/V ein. Befindet sich neben dem Gas z.B. auch ein Pulver im gleichen Behälter, beobachtet man insbesondere bei tieferen Temperaturen eine stärkere Druckabnahme als nach dem id. Gasgesetz erwartet, da durch Adsorption ein beträchtlicher Teil der Stickstoffmoleküle der Gasphase entzogen werden und daher nicht zum gemessenen Druck beitragen. Der verringerte Druck wird daher grob mit p = (n-nads)·RT/V beschrieben. Die Zahl adsorbierter Moleküle nads ist direkt proportional der Oberfläche der Probe und mit bekannter Adsorptionsisotherme kann die spezifische Oberfläche berechnet werden. Schematisch zum BET Verfahren: Zwei Proben unterschiedlicher Teilchengröße bei gleicher Temperatur und gleicher Menge Stickstoff. Die Kenntnis der Adsorptionsisothermen sind z.B. im Zusammenhang mit der Luftfeuchtigkeit sehr wichtig bei Trocknungsprozessen oder der Bauphysik. In der chemischen Analytik werden die stoffspezifischen Adsorptionsisothermen in einer Vielzahl von Verfahren der Chromatographie zur Analytik komplexer Stoffgemische ausgenutzt. ________________________________________________ Hoeppe - PC Leerscript - 127 -