PC_Kurzscript
Werbung

Physikalische Chemie - „Kurzscript“
Inhalt:
1
1.1
1.2
1.3
1.4
1.5
1.6
Einstoffsysteme
Grundlagen
Zustandsfunktionen
Ideales Gas
Kinetische Gastheorie
Zustandsdiagramme
Zustandsgleichungen realer Stoffe
2
2.1
2.2
2.3
2.4
Energieumsatz bei Stoffumwandlungen
Erster Hauptsatz
Enthalpie
Das Verhalten von innerer Energie und Enthalpie bei Zustandsänderungen
Reaktionswärme
3
3.1
3.2
3.3
Entropieumsatz bei Stoffumwandlungen
Reversible und irreversible Prozesse
Zweiter Hauptsatz der Thermodynamik
Dritter Hauptsatz, Absolutberechnung der Entropie
4
4.1
4.2
Der Ablauf physikalisch-chemischer Prozesse
Freie Energie und Freie Enthalpie
Chemische Reaktionen
5
5.1
5.2
5.3
Freie Enthalpie und Gleichgewichte
Reaktionsgleichgewichte
Phasengleichgewichte
Normalspannungen
PC_Kurzscript
Hoeppe
-1-
1
1.1
Einstoffsysteme
Grundlagen
Makroskopische Größen = makrosk. Observable in makrosk. System
Intensive Größe:
Extensive Größe:
mengenunabhängig
mengenabhängig
Bsp.: p, T, M, Vm, h, ...
Bsp.: m, Q, V, n, H, ...
1 Mol = Stoffmenge, welche die gleiche Zahl von Teichen enthält
wie 12,0000 g des Kohlenstoff-Nuklids 12C
Avogadrozahl / Loschmidtsche Zahl: NA = NL = 6,02214⋅1023 mol-1
Molzahl n
n=
Molmasse M
Molvolumen Vm
m
M
Vm =
Dichte ρ
M =
V M
=
n
ρ
ρ=
m = n ⋅ M = ρ ⋅V
Masse m
m
n
m
m
M
= n =
V
V
Vm
n
Übergang zu intensiven Größen → molare Größen:
Molare Größe von Φ:
Begriffe:
Φm =
Φ
n
- reiner Stoff: Besteht aus (vielen) gleichen Atomen, Molekülen oder
zugeordneten Ionen. (technisch rein >96%, reinst > 99%)
- homogener Stoff: Hat in jedem Teilgebiet die gleiche chemische
Zusammensetzung und physikalischen Eigenschaften
- Phase: Teilgebiet eines Stoffes in welchem er homogen ist
- reine Phase: Phase, welche nur einen Stoff (und dessen Dissoziationsprodukte)
enthält
- Mischphase: Phase, welche aus vielen verschiedenen Stoffen besteht
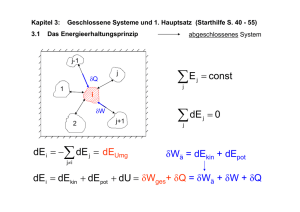
Offenes System:
Geschlossenes System:
E
Abgeschlossenes System:
E
E
n
n
PC_Kurzscript
Hoeppe
n
-2-
1.2
Zustandsgleichungen
System mit n unabhängigen Größen bzw. Variablen → Zustandsraum ℜn
Soweit Zustandsgleichung auflösbar, ist Wahl der unabhängigen Variablen beliebig.
Bsp.:
Hier: Nur zwei unabhängige Variable (z.B. x1 und x2) → Zustandsraum = Fläche,
die Variable x3 ist mit x1 und x2 eindeutig festgelegt!
p(V,T) ist Zustandsgröße → Linienintegral ist wegunabhängig, d.h.
2
∆p = p 2 − p1 = ∫ dp =
1
∫ dp = ∫ dp = ∫ dp
WEG I
WEG II
bzw.
WEG III
→ es existiert für p(V, T) ein totales Differential mit
∫ dp = 0
⎛ ∂p ⎞
⎛ ∂p ⎞
dp = ⎜
⎟ dV + ⎜
⎟ dT
∂
∂
V
T
⎝
⎠T
⎝
⎠V
Allgemein für Zustandsgröße Φ(x1, x2, ..,xn):
⎛ ∂Φ ⎞
⎛ ∂Φ ⎞
⎛ ∂Φ ⎞
⎟⎟ dxn
⎟⎟ dx2 + ... + ⎜⎜
⎟⎟ dx1 + ⎜⎜
dΦ = ⎜⎜
x
x
x
∂
∂
∂
⎝ 2 ⎠ xi ≠ 2
⎝ 1 ⎠ xi≠1
⎝ n ⎠ xi ≠ n
( Gegenbeispiel: Volumenarbeit W und Wärmemenge Q sind keine Zustandsgrößen.)
PC_Kurzscript
Hoeppe
-3-
1.3
Ideales Gas
p ⋅ V = const
Gesetz von Boyle Mariotte:
+
Gesetz von Charles:
+
Gesetz von Avogadro:
→
V = const ⋅ ( Θ + 273 ,15 ) = const ⋅ T
V = const ⋅ n
Zustandsgleichung des idealen Gases:
p ⋅V = n ⋅ R ⋅ T
Ideales Gas gute Näherung falls:
- p klein, d.h. keine WW
- Teilchenvolumen << V
- T >> Ts
Zustandsdiagramme als Schnitt durch Zustandsfläche:
T = const. →
V = const. →
p = const. →
Isothermen :
Isochoren:
Isobaren:
Molare Gaskonstante: Rm = 8,31451 J/molK = 0,0831451 bar l /molK
Absolute Temperatur T:
PC_Kurzscript
Hoeppe
T = ( Θ / °C + 273 ,15 ) ⋅ K
-4-
1.4
Kinetische Gastheorie
3
mv 2
⎛ m ⎞ 2 2 − 2 kBT
dN (v)
⎟⎟ v e
f (v) dv =
dv
= 4π ⎜⎜
N
⎝ 2π k BT ⎠
Maxwellverteilung:
Boltzmannkonstante kB
Bsp.: N2
kB =
Für ideale Gase:
Mittlere quadratische Geschwindigkeit:
Mittlere Geschwindigkeit:
3k BT
=
m
v2 =
v=
Wahrscheinlichste Geschwindigkeit:
Rm
J
= 1,3807 ⋅ 10 − 23
NA
K
3 N A k BT
=
N Am
8k BT
R T
≅ 1,60 m
πm
M
vW =
R T
2 k BT
≅ 1,41 m
m
M
v 2 : v : vw
≅ 1,73 : 1,60 : 1, 41
Stoßquerschnitt:
σ =π ⋅d2
mit d = effektiver Teilchendurchmesser
Stoßhäufigkeit:
Z = 2 ⋅σ ⋅ v ⋅
p
k BT
Mittlere freie Weglänge:
l =τ ⋅v =
1
⋅v =
Z
k BT
2 ⋅σ ⋅ p
r
Wärmeleitung / Wärmeleitkoeffizient: q = −λ ⋅ grad T
Wärmeleitkoeffizient für Gase: λ =
3 RmT
R T
≅ 1,73 m
M
M
RT
2 cV
⋅
⋅
~
3 N A ⋅ σ πM
Q
∆T
= −λ ⋅ A ⋅
∆t
∆x
T
⋅ cV v
M
( Ergänzung )
PC_Kurzscript
Hoeppe
-5-
Gleichverteilungssatz nach Boltzmann:
Die kinetische Energie verteilt sich (im Mittel)
gleichmäßig auf die Freiheitsgrade aller Moleküle
Stoff
E kin =
f
⋅ k BT
2
Freiheitsgrade
cp
cV
1-atomiges Gas:
3
3
5
2-atomiges Gas (tiefe T, d.h. T ≤ T0):
5
5
7
2-atomiges Gas (hohe T, d.h. T >> T0):
7
7
9
Ergänzung: Dulong-Petit’ Regel für FK:
6
6
/2 R
/2 R
/2 R
/2 R
/2 R
/2 R
Spezifische Wärme bei const. Volumen:
cV =
1 ⎛ ∂U ⎞
⎛ ∂u ⎞
⋅⎜
⎟ =⎜
⎟
n ⎝ ∂T ⎠V ⎝ ∂T ⎠V
Spezifische Wärme bei const. Druck:
cp =
1 ⎛ ∂H ⎞
⎛ ∂h ⎞
⋅⎜
⎟ =⎜
⎟
n ⎝ ∂T ⎠ p ⎝ ∂T ⎠ p
/2 R
≅
6
/2 R
→ Berechnung Wärmemenge
dQ = n ⋅ c p ,V ⋅ dT
→
∆Q = n ⋅ ∫ c p ,V ⋅ dT
c p ,V =const
=
n ⋅ c p ,V ⋅ ∆T
( vgl. Kap. 2 )
Zusammenhang cV und cp:
Für Gase gilt:
c p = cV + R
PC_Kurzscript
Hoeppe
Für FK u. Flüssigkeiten gilt:
c p ≅ cV
-6-
1.5
Zustandsdiagramme
Allgemeines Zustandsdiagramm:
TR P
: Tripelpunkt
kr.
: Kritischer Punkt
pD(T) : Dampfdruckkurve
pm(T) : Schmelzdruckkurve
psub(T): Sublimationsdruckkurve
Anomalie des Wassers:
1) Verflüssigung bei Druckerhöhung, weil:
- Schmelzdruckkurve hat negative Steigung,
d.h. für T = const ist Vm,l < Vm,s
2) Schnellkochtopf funktioniert, weil :
- für p > po sind T > 100°C möglich
„Trockeneis“:
→ Sublimation von CO2 bei Normaldruck, weil:
- Tripelpunkt bei 5,11 bar →
flüssiger Zustand wird nicht erreicht
PC_Kurzscript
Hoeppe
-7-
1.6 Zustandsgleichungen realer Stoffe
→ Korrekturen der Gleichung für ideale Gase
T: Isothermen
Tkrit: kritische Isotherme
g: gasförmig
l + g : 2 Phasenmischgebiet
rein empirisch:
Virialansatz:
p ⋅V = Z ⋅ n ⋅ R ⋅ T
( → Tab. B )
mit Z(T) = 1 + B(T)⋅10-3 ⋅p + C(T)⋅10-6 ⋅p2 + D(T)⋅10-9 ⋅p3
physikalisch motiviert:
van der Waals:
⎛
a
⎜⎜ p + 2
Vm
⎝
⎞
⎟⎟ ⋅ (Vm − b ) = R ⋅ T
⎠
( → Tab. A )
a/Vm2 ~ Binnendruck, a Maß für Wechselwirkung zwischen Atomen/Molekülen
b ~ Eigenvolumen
Aus der waagerechten Tangente im krit. Punkt folgt:
Verbesserung v. d. Waals:
Redlich-Kwong:
⎛
⎜p+
⎜
⎝
b=
R Tkrit
8 pkrit
a = 27 ⋅ b 2 ⋅ p krit
⎞
a'
⎟ ⋅ (V m − b ' ) = R ⋅ T
T ⋅ Vm (V m + b ' ) ⎟⎠
( → Tab. C )
PC_Kurzscript
Hoeppe
-8-
2
2.1
Energieumsatz bei Stoffumwandlungen
Erster Hauptsatz
Die Innere Energie U eines Systems ist die
Summe aller darin enthaltenen Energien.
Eine Änderung der inneren Energie U erfolgt nur über einen
- Wärmetransport ∆Q und/oder über
- Arbeit ∆W im mechanischen Sinne (,d.h. Prozesse die sich auf
das bloße Heben und Senken von Gewichten abbilden lassen).
(Unter Wärme versteht man die Summe der stochastisch verteilten kinetischen Energie)
Für ein geschlossenes System gilt daher:
∆U = ∆Q + ∆W
Diese Aussage wird oft schon als 1.HS bezeichnet, da sie die
Energieerhaltung aus Gründen der Eindeutigkeit impliziert. Deutlicher ist jedoch:
Für ein abgeschlossenes System gilt:
→
∆U = ∆Q + ∆W = 0
Es existiert kein Perpetuum Mobile 1. Art, d.h. keine
periodisch arbeitende Maschine, die mehr Energie abgibt
als ihr Betrieb benötigt.
Volumenarbeit W:
dW = − p ⋅ dV
Spezifische Wärme bei
konstantem Volumen:
cV =
→
∆W = − ∫ p ⋅ dV
1 ⎛ ∂U ⎞
⎛∂u ⎞
⎟ =⎜
⎜
⎟
n ⎝ ∂T ⎠ V ⎝ ∂ T ⎠ V
Für ein T-unabhängiges cV folgt:
dQ = ncV dT
→
∆Q = n ⋅ cV ⋅ ∆T
i.A. gilt jedoch für ∆Q:
dQ = ncV dT
→
∆Q = n ⋅ ∫ cV dT
Totales Differential der
inneren Energie U(V,T):
⎛ ∂U ⎞
⎛ ∂U ⎞
dU = ⎜
⎟ dT
⎟ dV + ⎜
⎝ ∂T ⎠V
⎝ ∂V ⎠T
id
Bei einem idealen Gas, ist U nur von der Temperatur abhängig: dU = ncV dT
PC_Kurzscript
Hoeppe
-9-
2.2
Enthalpie
Aus dem 1. HS folgt dQ = dU + pdV; d.h. für p=const ist dQ = dU + pdV + Vdp =: dH ...
Die meisten Prozesse laufen bei const. Druck
p = po ab, daher ist die Def. einer neuen
Zustandsgröße Enthalpie H sinnvoll:
⎛ ∂H
dH ( p, T ) = ⎜⎜
⎝ ∂p
Totales Differential von H(p,T):
Spezifische Wärme bei
konstantem Druck:
cp =
H := U + p ⋅ V
⎞
⎛ ∂H ⎞
⎟⎟ dp + ⎜
⎟ dT
T
∂
⎝
⎠p
⎠T
1 ⎛ ∂H ⎞
⎛∂h⎞
⋅⎜
⎟
⎟ =⎜
n ⎝ ∂T ⎠ p ⎝ ∂T ⎠ p
Für const. Druck folgt für die bei einem Prozess umgesetzte
differentielle Wärmemenge dQ:
dQ = n ⋅ c p dT
Für ein T-unabhängiges cpfolgt:
i.A. gilt jedoch für ∆Q:
∆Q = n ⋅ ∫ c p (T ) dT
T - Abhängigkeit von cp(T)
mit Potenzreihe dargestellt:
Damit gilt für ∆Q :
∆ Q = n ⋅ c p ⋅ ∆T
c p (T ) = A + B ⋅10 −3T + C ⋅10 5T −2 + D⋅10 −6T 2
T
T
2
2
1
⎡
⎤
∆Q = ∫ c p (T )dT = ∫ ⎢ A + B⋅10 − 3T + C ⋅10 5 2 + D ⋅10 − 6T 2 ⎥dT
T
⎦
T1
T1 ⎣
Allgemeine Lösung des Integrals:
T2
∫c
p
T1
⎧
⎛1 1⎞ 1
1
2
2
3
3 ⎫
(T )dT = ⎨ A ⋅ (T2 − T1 ) + B⋅10 −3 (T2 − T1 ) − C ⋅10 5 ⎜⎜ − ⎟⎟ + D ⋅10 −6 (T2 − T1 )⎬
2
⎝ T2 T1 ⎠ 3
⎭
⎩
Latente Wärme bzw. Umwandlungsenthalpie hu = Energie bzw. Wärme,
welche für Phasenumwandlung benötigt wird →
T2
∆Q( p = const ; T1 → T2 ) = ∫ c p (T )dT + ∑ hu
T1
PC_Kurzscript
Hoeppe
- 10 -
2.3
Zustandsänderungen
Isochore Zustandsänderungen:
V = const. ; d.h.
dV = 0 →
dU = dQ
dH = dU + Vdp
dQ = n cV dT
dW = 0
Isobare Zustandsänderungen:
p = const. ; d.h.
dp = 0 →
dU = dQ + dW
dH = dQ
dQ = n cp dT
dW = - pdV
Isotherme Zustandsänderung
T = const. ; d.h.
{ id. Gas }
dT = 0 →
dU = n cV dT { = 0 }
dH = dU + d(pV) { = 0 }
dQ = dU - dW { = pdV }
dW = - pdV
Adiabatische Zustandsänderung
Q = const. ; d.h.
p ⋅ V = const
→
p ⋅ V κ = const
{ id. Gas }
dQ = 0 →
dU = dW = -pdV
dH = Vdp
dQ = 0
dW = -pdV { = ncVdT }
Adiabatenexponent:
PC_Kurzscript
Hoeppe
→
κ=
cp
cV
- 11 -
2.4
Reaktionswärme
Reaktionsgleichung:
⎛
λ ⋅⎜
⎝
∑ν E
i
⎞
⎟
⎠
→ ∑ν i Pi
i
i
i
mit Reaktionsparameter λ folgt für Molzahländerung:
Bombenkaloriemeter (Gase ideal):
⎞
⎛
∆ n = λ ⋅ ∆ ν = λ ⋅ ⎜ ∑ ν i − ∑ν i ⎟
Ed
⎠
⎝ Pr
∆H = ∆Q + ∆n ⋅ R ⋅ T
Standardbildungenthalpie h0298:
( Tab. F )
Def.: Für Elemente in ihrer natürlichen Form gilt: h298 ≡ 0
0
T
→
h( p = p , T ) = h (T ) = h
o
0
298
o
+
∫c
p
(T ) ⋅ dT + ∑ hu
298
Reaktionsenthalpie ∆Rh
bei Standardbedingungen:
0
∆ R h298
= ∑ ν i ⋅ hi0, 298 − ∑ ν i ⋅ hi0, 298
Pr
Ed
∆ R hT0 = ∑ ν i ⋅ hi0,T − ∑ ν i ⋅ hi0,T
bei Standarddruck:
Ed
Pr
falls bei keinem Stoff Umwandlungspunkte zwischen 298 K und T liegen:
T
∆ h = ∆ Rh
0
R T
0
298
−
∫ ∆c
p
(T ) dT
298
mit
∆ c p (T ) = ∆A + ∆B ⋅10 −3 T + ∆C ⋅10 5
1
+ ∆D⋅ 10 −6 T 2
T2
Wässrige Lösungen ( → Ionenpaare ):
Def.:
PC_Kurzscript
Hoeppe
0
h298
( H aq+ ) ≡ 0
- 12 -
3
Entropieumsatz bei Stoffumwandlungen
3.1 Reversible und irreversible Prozesse
→ Begriffe einer makroskopischen Betrachtung!
reversibel: umkehrbar
irreversibel: nicht umkehrbar
Infinitesimale (mikroskopische) Prozesse sind stets reversibel, d.h.
Zustandsänderungen sind auf reversible Prozesse abbildbar.
3.2
Zweiter Hauptsatz der Thermodynamik
Carnotprozess:
- Kreisprozess für Wärmekraftmaschinen
→ idealer, d.h. verlustfreier Prozess
→ zyklisch arbeitende Maschine
→ Grenzfall realer Maschine
1 → 2:
isotherme Expansion bei T = T0
Q12 = −W12 = nRT0 ln
V2
V1
3 → 4:
isotherme Kompression bei T = Tu
Q34 = −W34 = nRTu ln
PC_Kurzscript
Hoeppe
V4
V3
2 → 3:
adiabatische Expansion
W23 = ∆U 23 = ncV ∆T23 = ncV (Tu − T0 )
4 → 1:
adiabatische Kompression
W41 = ∆U 41 = ncV ∆T41 = ncV (T0 − Tu )
- 13 -
Wirkungsgrad = Verhältnis zugeführter Wärme zu erhaltener Arbeit η :
η=
W ges
Q12
=
T0 − Tu
T0
2. HS:
Es existiert kein Perpetuum Mobile 2. Art, d.h. keine Maschine,
die nichts weiter tut, als Wärme in Arbeit umzuwandeln.
dS =
Definition der Entropie nach Clausius:
2.HS:
Für bel. Prozess gilt in einem
abgeschlossenem System:
dQ rev
T
dS ≥ 0
Berechnung von Entropieänderungen dS und ∆S für reversible Prozesse:
a) adiabatische Prozesse:
dQ = 0
→
dS = 0
→
∆S = 0
b) isotherme id. Prozesse: dS = dQ = − dW = pdV →
T
T
T
⎛V
∆ S = nR ln ⎜⎜ 2
⎝ V1
c) isochore id. Prozesse:
dS = n ⋅ cV
dT
T
→
⎛T ⎞
∆ S = n ⋅ cV ln ⎜⎜ 2 ⎟⎟
⎝ T1 ⎠
d) isobare id. Prozesse
dS = n ⋅ c p
dT
T
→
⎛T ⎞
∆ S = n ⋅ c p ln ⎜⎜ 2 ⎟⎟
⎝ T1 ⎠
e) Wärmeleitung:
∆Q A = − ∆Q B
f) Mischungsentropie:
g) Phasenübergänge:
PC_Kurzscript
Hoeppe
→
⎛T
∆ S = ∆ S A + ∆ S B = n A c p , A ln ⎜⎜
⎝ TA
⎛ V + VB
∆ S = ∆ S A + ∆ S B = n A R ln ⎜⎜ A
⎝ VA
∆S =
⎛ p ⎞
⎞
⎟⎟ = nR ln ⎜⎜ 1 ⎟⎟
⎝ p2 ⎠
⎠
⎛T ⎞
⎞
⎟⎟ + n B c p , B ln ⎜⎜ ⎟⎟
⎝ TB ⎠
⎠
⎞
⎛ V + VB
⎟⎟ + n B R ln ⎜⎜ A
⎠
⎝ VB
⎞
⎟⎟
⎠
∆ Q n ⋅ hu
=
T
Tu
- 14 -
Statistische Interpretation der Entropie:
„Wie groß ist die Wahrscheinlichkeit, dass eine zerbrochen Kaffeetasse
sich unter Wärmezufuhr zusammenfügt und auf den Tisch springt?“
„Wie groß ist die Wahrscheinlichkeit, dass eine gleiche Tasse vom Tisch fällt,
in exakt gleiche Bruchstücke zerspringt, welche an genau den gleichen Stellen liegen
bleiben wie die erste?“
Antwort: ’ 1023 ’ + ’ Fakultät ’
→ Boltzmann:
S = k B ⋅ ln Ω
(statistisches Gewicht Ω ≥ 1 entspricht statistischer Wahrscheinlichkeit eines makrosk. Zustandes)
3.3
Dritter Hauptsatz, Absolutberechnung der Entropie
cp (T→ 0) = 0
Beobachtung, bzw. Ergebnis QM:
→ Nernst:
lim ∆S = 0
T →0
lim S (id .FK ) = 0
→ Planck:
3. H.S. der Thermodynamik
T →0
3. H.S. ermöglicht Absolutberechnung der Entropie so(T) (bei Standardbedingungen):
T
s (T ) = ∫
0
c p (T )
T
0
dT + ∑
hu
Tu
Zweckmäßigerweise wir Standardentropie so298 = so(T = 298K) definiert:
298
s
0
298
=
∫
c p (T )
0
T
dT + ∑
hu
Tu
( → Tab. F)
Damit vereinfacht sich die so(T) Berechnungen bei „handelsüblichen“ Temperaturen:
T
s (T ) = s
0
0
298
+
∫
c p (T )
298
T
dT + ∑
hu
Tu
( → so: Tab. F, hu: Tab. E)
PC_Kurzscript
Hoeppe
- 15 -
Betrachte wieder die allg. Reaktion:
⎛
∑ν E
λ ⋅⎜
i
⎝
i
i
→ ∑ν i Pi
i
⎞
⎟
⎠
Reaktionsentropie ∆Rs
bei Standardbedingungen:
0
∆ R s 298
=
∑ν
0
i i , 298
s
Pr .
− ∑ ν i s i0, 298
Ed .
∆ R sT0 = ∑ ν i si0,T − ∑ ν i si0,T
bei Standarddruck:
Pr .
Ed .
falls bei keinem Stoff Umwandlungspunkte zwischen 298 K und T liegen:
T
∆ s =∆ s
0
R T
0
R 298
+
∫
∆ c p (T )
298
T
dT
∆c p (T ) = ∆A + ∆B ⋅10 −3 T + ∆C ⋅10 5
mit
1
+ ∆D⋅ 10 −6 T 2
2
T
(Partial-) Druckabhängigkeit der Entropie s(p,T):
⎛ p ⎞
si ( pi , T ) = si0 (T ) − R ⋅ ln ⎜⎜ oi ⎟⎟
⎝p ⎠
(Beachte: gilt auch Komponentenweise, für Gasgemisch → Index i, →Partialdrücke)
Gesetz von Dalton:
p = ∑ pi
i
Planck:
Für ein Gemisch setzt sich die Entropie nicht einfach als Summe der Entropien der
einzelnen reinen Komponenten zusammen. Mit Ersetzen der Partialdrücke von oben
mit den Molenbrüchen xi der Komponenten gilt verallgemeinert:
S = ∑ ni si = ∑ ni ( si0 − R ln xi )
i
mit
si ( xi , T ) = si0 (T ) − R ⋅ ln xi
i
(Dieser Zusammenhang bedingt, dass Reaktion unvollständig ablaufen → vgl. Mischungsentropie
Die obige Gleichung ist daher Vorraussetzung für die Berechnung von Gleichgewichten !)
PC_Kurzscript
Hoeppe
- 16 -
4
4.1
Der Ablauf physikalisch-chemischer Prozesse
Freie Energie und Freie Enthalpie
Josiah Willard Gibbs (1839-1903):
Freie Energie F
F = U - T·S
F = F(V,T) →
dF = d(U-T·S) = dU -SdT -TdS = dQ - pdV -SdT -TdS = -pdV -SdT
Freie Enthalpie G
G = H - T·S
G = G(p,T) →
dG = d(H - T·S) = dU + pdV + Vdp - TdS - SdT = ... = Vdp - SdT
Vereinigung der beiden Prinzipien (Energie → Min) + (Entropie → Max):
G → Min
∆G = G2 - G1 < 0 → spontane Reaktion /Prozess
∆G = G2 - G1 > 0 → keine Reaktion / Prozess*
∆G = G2 - G1 = 0 → Gleichgewicht
*) bzw. Umkehrreaktion
(Partial-) Druckabhängigkeit der freien Enthalpie g(p,T):
(folgt direkt aus (partial)druckabhängigkeit der Entropie)
⎛ p ⎞
g i ( pi , T ) = g i0 (T ) + RT ⋅ ln ⎜⎜ oi ⎟⎟
⎝p ⎠
Auch hier gilt nach Planck verallgemeinert mit den Molenbrüchen xi der Komponenten:
g i ( xi , T ) = g i0 (T ) + RT ⋅ ln xi
mit
PC_Kurzscript
Hoeppe
g i0 (T ) = hi0 (T ) − T ⋅ si0 (T )
- 17 -
4.2
Chemische Reaktionen
Freie Reaktionsenthalpie ∆Rg(p,T):
∆ R g = ∑ν i g i −∑ν i g i
Pr .
Ed .
Für Standarddruck p = po folgt die Standard Gibbs-Reaktionsenthalpie ∆Rgo (T):
∆ R g 0 = ∑ ν i g i0 − ∑ ν i g i0
Pr .
Ed .
Entsprechend g = h - T·s berechnet sich ∆Rgo (T)
∆ R g 0 = ∆ R g 0 (T ) = ∆ R h 0 (T ) − T ⋅ ∆ R s 0 (T )
Falls bei keinem Stoff Umwandlungspunkte zwischen 298 K und T liegen, gilt mit ∆cp(T):
T
∆ c p (T )
⎡
⎤
0
+ ∫ ∆ c p (T ) ⋅ dT − T ⎢ ∆ R s 298 + ∫
⋅ dT ⎥
T
298
298
⎣
⎦
T
∆ R g = ∆ Rh
0
0
298
Annahme: spezifische Wärme von Produkten ~ Edukten →
Ulich’sche Näherung:
∆cp(T) = 0
0
0
∆ R g 0 = ∆ R h298
− T ⋅ ∆ R s 298
Obige Gleichung ist nicht zu verwechseln mit dem Fall einer
Reaktion unter Standardbedingungen:
0
0
0
∆ R g 298
= ∆ R h298
− 298 K ⋅ ∆ R s 298
Das Vorzeichen von ∆Rg zeigt an, ob eine Reaktion von
„links nach rechts“ oder (eher) umgekehrt abläuft.
PC_Kurzscript
Hoeppe
- 18 -
Richardson Diagramme:
→ vgl. verschiedener Reaktionen mit gleichem Reaktionspartner (Bsp.: O2) bzgl. Vorzeichen
und Betrag von ∆Rgo(T) unter Verwendung der Ulich’schen Näherung.
→ Liefert Abschätzung für Reaktionen der einzelnen Produkte untereinander.
Bsp:
Richardson Diagramm
0
Standardreaktionsenthalpie
-100
C + O2 -> CO2
2 Ni + O2 -> 2 NiO
2 C + O2 -> 2 CO
4/3 Cr + O2 -> 2/3 Cr2O3
4/3 Fe + O2 -> 2/3 Fe2O3
2 Fe + O2 -> 2 FeO
3/2 Fe + O2 -> 1/2 Fe3O4
-200
-300
-400
-500
-600
-700
200
400
600
800
1000
1200
1400
1600
1800
2000
T/K
Achtung: Nur Abschätzung!
- ∆cp(T) i.A. ≠ 0
- Vermischung der Komponenten bzw. der Edukte und Produkte vernachlässigt!
Unvollständig ablaufende Reaktionen:
Zerlege Reaktion in (sehr viele) Teilschritte und berechne jeweils neu ∆Rg(T)
unter Berücksichtigung der „ln-Mischungsterme“ (d.h. der Mischungsentropie ∆Ms ):
→ Je unvollständiger die Reaktion (in beide Richtungen) desto größer der Einfluss dieser Terme!
→ 100% vollständige Reaktion praktisch unmöglich!
Reaktion:
A→B
goA(T), goB(T), ∆Rgo(T) , T⋅∆Ms
Reaktionsverlauf, bzw. -umsatz
PC_Kurzscript
Hoeppe
- 19 -
5
5.1
Freie Enthalpie und Gleichgewichte
Reaktionsgleichgewichte
∆Rg(p,T) = 0
Gleichgewichtsbedingung:
Homogene Gasgleichgewichte (→Bsp: HJ oder NH3 Synthese)
aA + bB → dD + eE
Betrachte Reaktion
→
⎛ p Dd ⋅ p Ee ⋅ p 0− ∆ν
∆ R g ( p, T ) = d ⋅ g D + e ⋅ g E − [a ⋅ g A + b ⋅ g B ] = ∆ R g (T ) + RT ⋅ ln⎜⎜
p Aa ⋅ p Bb
⎝
0
∆ν = d + e − a − b
(→ van’t Hoffsche Reaktionsisotherme)
’Abkürzung’ für Argument des ln-Terms: Gleichgewichtskonstante K
Auflösen nach K ergibt:
K (T ) = e
−
∆R g0
RT
Mit K sind Partialdrücke, Konzentration bzw.
Molenbrüche der Reaktionspartner definiert!
Üblich sind auch die Gleichgewichtskonstanten
bzgl. der Partialdrücke:
p Dd ⋅ p Ee
Kp = a b
p A ⋅ pB
bzgl. der Konzentrationen:
c Dd ⋅ c Ee [ D]d ⋅ [ E ]e
KC = a b =
c A ⋅ c B [ A]a ⋅ [ B]b
bzgl. der Molenbrüche:
x Dd ⋅ x Ee
Kx = a b = K
x A ⋅ xB
Für Reaktionen ohne Molzahländerungen ∆ν = 0 gilt: Kp = Kc = Kx = K (dimensionslos).
Für ∆ν ≠ 0 gilt:
Kp = K ⋅ p
PC_Kurzscript
Hoeppe
∆ν
0
= K ⋅ bar
∆ν
und
⎛ p ⎞
Kc = K ⋅ ⎜ 0 ⎟
⎝ RT ⎠
∆ν
⎛ 1 ⎞
= K p ⋅⎜
⎟
⎝ RT ⎠
∆ν
- 20 -
⎞
⎟⎟
⎠
Heterogene Gasgleichgewichte (→Bsp: Brennen von Kalk)
→ Im ln-Term erscheinen nur die gasförmigen Reaktionspartner
→ ∆ν bezieht sich nur auf die gasförmigen Reaktionspartner
Verschiebung des Gleichgewichts
→ mit der Temperatur: K = K(T) :
K (T ) = e
−
∆R g0
RT
(*)
Aus (*) folgt mit part. diff. nach T (und Gibbs-Helmholtz-Gl.) die
van’t Hoffsche Reaktionsisobare:
∂ ln K ∆ R h 0
=
∂T
RT 2
d.h. das Vorzeichen von ∆Rh0 entscheidet, ob K mit T steigt oder fällt:
- für exotherme Reaktionen (∆Rh0 < 0) wird K mit der Temperatur kleiner!
- Gleichgewicht verschiebt sich auf die Seite der Edukte
- umgekehrtes gilt für endotherme Reaktion
Allgemein lässt sich das Prinzip von Le Chatelier (1888) und Braun (1887)
formulieren:
Ein System verändert seinen Gleichgewichtszustand der Art,
dass es dem äußeren Zwang ausweicht, bzw. diesen mindert.
Ähnliches gilt bei Änderung des Drucks bzw. der Konzentrationen:
→ mit dem (Gesamt-) Druck:
betrifft Gasreaktionen mit Molzahländerung
van’t Hoffsche Reaktionsisochore:
∆V 0
∂ ln K
=−
RT
∂p
→ mit der Konzentration:
betrifft nichtstöchiometrische Ausgangszusammensetzung
Nur die Temperatur geht direkt in ∆Rgo und damit in K ein, Einfluss von Druck und
Konzentration über die Bestimmungsgleichung für gesuchtes Gleichgewicht.
PC_Kurzscript
Hoeppe
- 21 -
Elektrolytgleichgewichte (heterogenes Gleichgewicht)
A → b B+ aq + c C-aq
Betrachte ‚Reaktion’ Lösungsvorgang:
K berechet
sich mit
(
∆ R g = b ⋅ g B + + c ⋅ g C − − g A = ... = ∆ R g 0 + RT ⋅ ln a B + ⋅ a C −
Löslichkeitsprodukt:
b
c
)
= ∆ R g 0 + RT ⋅ ln (K )
K L := [ B + ]b ⋅ [C − ]c = K ⋅ ( mol
) b+c
l
(Ähnliches gilt für Säure-Basen-Gleichgewichte → pH-Wert, Ionenprodukt Wasser ...)
5.2
Phasengleichgewichte
Nach der Gibbs’schen Phasenregel können bei
einem Gemisch aus K Komponenten P Phasen
mit F Freiheitsgraden koexistieren.
Clausius Clapeyrone:
F = K −P+2
dp
hu
=
dT T (Vm , g − Vm , l )
speziell mit Vml << Vmg , hu = const und Dampf = id. Gas gilt für pD(T):
p D (T ) = p 0 ⋅ e
−
hu ⎛ 1 1
⎜ −
R ⎜⎝ T T0
⎞
⎟⎟
⎠
→ ‚Anfitten’ von Dampfdruckkurven mit Fitparametern A und B:
⎛p ⎞
h
h
B
ln ⎜⎜ D ⎟⎟ = − u + u =: A −
RT RT0
T
⎝ p0 ⎠
PC_Kurzscript
Hoeppe
- 22 -
5.3
Normalspannungen
Elektrochemische Spannung zwischen zwei galv. Halbzellen beschreibt Neigung
der beteiligten Stoffe in Lösung zu gehen (vgl. Daniellelement):
A|A+||B|B+
Halbzellenpaar:
∆E 0 = E 0 ( B | B + ) − E 0 ( A | A + )
Reaktion läuft für positives ∆E0 nach rechts ab
zugehörige
Reaktionsgleichung:
A + B+ → A+ + B
0
0
0
0
0
∆ R g 298
= g 298
( B ) + g 298
( A + ) − g 298
( A) − g 298
(B + )
Reaktion läuft für negatives ∆Rg0 nach rechts ab
Faradaykonstante: F = NA⋅ e = 96485 C⋅mol-1
Mit der Wasserstoffnormalelektrode als Standard-Bezugs-Potential sind die
Standardpotentiale E0 von Stoffen definiert.
A|A+||½ H2|H+
Rechte Seite H-Normalelektrode mit E0 = 0 :
→ Elektrochemische Spannungsreihe
∆Rg0
E =
z⋅F
0
entspricht Reaktionsgleichung: A + H
+
→ A+ + ½ H2
→ d.h. (unedle) Metalle mit E0 < 0 lösen sich in 1-mol HCl-Standardlösung auf,
(edle) Metalle mit E0 > 0 nicht.
PC_Kurzscript
Hoeppe
- 23 -