3.4 Essigsäure-(–)-menthylester aus (–)
Werbung
-menthylester aus (–)")
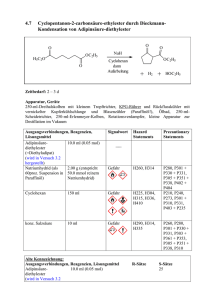
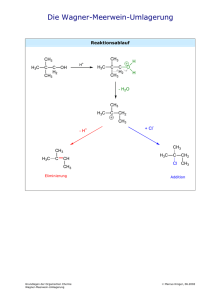
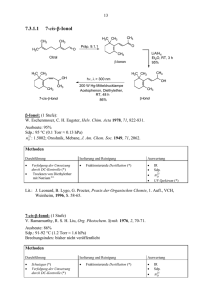
3.4 Essigsäure-(–)-menthylester aus (–)-Menthol und Essigsäureanhydrid CH3 CH3 O OH H3C + H3C O O CH3 O CH3 O O H2SO4 H3C + CH3 H3C OH CH3 Zeitbedarf: 2 – 3 d Apparatur, Geräte 100-ml-Dreihalskolben mit KPG-Rührer, Rückflusskühler und Trockenrohr, Ölbad, 10-mlSpritze, 250-ml-Scheidetrichter, Rotationsverdampfer, kleine Apparatur zur Destillation im Vakuum Ausgangsverbindungen, Reagenzien, Lösungsmittel (–)-Menthol*) 15.6 g (0.10 mol) Signalwort Achtung Hazard Statements H315 Precautionary Statements P262, P302 + P352 Essigsäureanhydrid (= Acetanhydrid) 10.5 ml (0.11 mol) Gefahr H226, H332, H302, H314, H335 P280, P301 + P330 + P331, P305 + P351 + P338 Schwefelsäure (96proz. = 18 M) 2 Tropfen Gefahr H290, H314 Cyclohexan 90 ml Gefahr H225, H304, H315, H336, H410 P280, P301 + P330 + P331, P309, P310, P305 + P351 + P338 P210, P240, P273, P301 + P310, P331, P403 + P235 Alte Kennzeichnung: Ausgangsverbindungen, Reagenzien, Lösungsmittel (–)-Menthol 15.6 g (0.10 mol) Xi Essigsäureanhydrid 10.5 ml (0.11 mol) C R-Sätze 36 10-20/22-34 konz. Schwefelsäure 35 2 Tropfen C S-Sätze 26-36/37/ 39-45 26-30-45 *) Statt (-)-Menthol können Sie auch rac-Menthol verwenden. Überlegen Sie, welche Konsequenzen dies für Ihre Produktmischung hätte! Cyclohexan 90 ml F, Xn, N 11-38-50/ 53-65-67 9-16-3360-61-62 Arbeitsvorschrift In dem Dreihalskolben suspendiert man unter Rühren (–)-Menthol in Essigsäureanhydrid (mit 10-ml-Spritze abmessen). Nach Zugabe von konz. Schwefelsäure erhitzt man 2.5 h mit einem ca. 90 – 100 °C heißen Ölbad. Nach Abkühlen des Gemischs gießt man es in den Scheidetrichter mit Eiswasser (50 ml), schüttelt und trennt die organische Phase ab. Man extrahiert die wässrige Phase mit Cyclohexan (2 x 30 ml). Die vereinigten organischen Phasen werden mit wässriger NatriumcarbonatLösung (1 M, 3 x 30 ml) und Wasser (2 x 30 ml) gewaschen. Danach trocknet man die organische Phase mit Natriumsulfat (ca. 5 g, ca. 0.5 h) (Vorgehensweise siehe Trocknen der Extrakte Teil A, Kapitel 8). Man dekantiert die Lösung durch einen Trichter mit Filter in einen 100-ml-Kolben und destilliert das Lösungsmittel am Rotationsverdampfer (45 °C, 220 hPa). In der Apparatur zur Destillation im Vakuum destilliert man das Rohprodukt (Ölbadtemperatur ca. 120 – 130 °C, 16 hPa). Man fängt als erste Fraktion einen Vorlauf auf, bis die Siedetemperatur konstant ist, und erhält dann eine farblose Flüssigkeit (85 – 95 %, Sdp. 102 – 103 °C / 21 hPa, nD20 = 1.4461) als Hauptfraktion. Steigt die Temperatur gegen Ende der Destillation, so nimmt man eine dritte Fraktion. Notieren Sie Siedepunkt und Menge der einzelnen Fraktionen.
![6.3.1 1-Oxa-spiro[2.5]octan - Institut für Organische Chemie](http://s1.studylibde.com/store/data/001356875_1-96e669e5c88ad586db9f9f199d424d05-300x300.png)