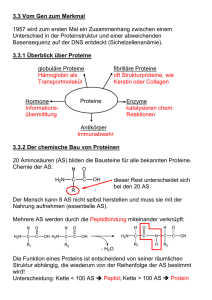
10 Proteine
Werbung

10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 1 (2008/2009) November 3, 2008 __________________________________________________________________________________________ CHEMOKINE 1. Chemokine und organspezifische Entzündungen Abschnitt 10.1.3. Seite 116 Nature Medicine 8: 117-118, 2002 Spalte rechts unten Die Hinweise, dass das System der spezifischen Abwehr kompartimentiert sein könnte, mehren sich. An einer solchen K o m p a r t i m e n t i e r u n g sind gewebespezifische Chemokinrezeptoren und Adhäsionsmoleküle beteiligt, welche die Wanderung («Homing») der Tund B-Lymphozyten steuern. Man spricht neuerdings von einem «Tropismus» (1) der Gedächtnis-T-Lymphozyten für ein Gewebe («Affinität» zu einem bestimmten Gewebe) und (2) von Medikamenten zur Blockade von Chemokinen. Ein Pathogen, welches das erste Mal die Haut schädigt, wird von den dendritischen Zellen der Haut abgefangen und in die regionären Lymphknoten transportiert. Dort werden Peptide des Pathogens den naiven T-Lymphozyten präsentiert. Dadurch differenzieren sich die naiven Lymphozyten in Effektor- oder Gedächtnis-T-Lymphozyten. Zu dieser Differenzierung gehört auch, dass die stimulierten Gedächtnis-T-Lymphozyten Adhäsionsmoleküle und Chemokinrezeptoren ausbilden, um damit im Rahmen der Abwehr den Weg in das geschädigte Hautareal zurückfinden zu können. Die Signale, welche die Bildung dieser Adhäsionsmoleküle und Chemokinrezeptoren induzieren, sind noch nicht bekannt. Die zusätzliche Ausbildung von Adhäsionsmolekülen und Chemokinrezeptoren bei der Differenzierung der T-Lymphozyten scheint aber einem allgemeinen Prinzip zu entsprechen: In den regionären Lymphknoten der Haut bilden die T-Lymphozyten bei ihrer Stimulierung das Adhäsionsmolekül Cutaneous lymphocyteassociated antigen (CLA), in den regionären Lymphknoten des Magendarm-Traktes das Integrin α4β7. So werden beispielsweise Antikörper gegen das Integrin α4β7 zur Behandlung des Morbus Crohn (chronische idiopathische Kolonschleimhautentzündung) eingesetzt. Wenn in einer Körperregion auf den Endothelzellen der «Code» E-Selectin, Chemokin CCL27 und CCL17 vorhanden ist, werden die CLA-, CCR10- und CCR4-positiven und stimulierten Gedächtnis-T-Lymphozyten in dieser Region «hängenbleiben». In der Haut werden die benötigten Chemokine, welche zu den Chemokinrezeptoren passen, von den Keratinozyten, Fibroblasten und Endothelzellen gebildet, wenn das Pathogen die Haut das zweite Mal geschädigt hat. Gleichzeitig exprimieren die Endothelzellen in diesem Moment vermehrt die Adhäsionsmoleküle E-Selektin und VCAM (vascular cell adhesion molecule, ein Adhäsionsmolekül der Immunglobulin-ähnlichen Superfamilie). Die Synthese der Chemokine in den Endothelzellen 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 2 (2008/2009) November 3, 2008 __________________________________________________________________________________________ wird durch das Auftreten der proinflammatorischen Zytokine Tumornekrosefaktor α (TNF-α), Interferon γ (IFN-γ) und Interleukin 1 (IL-1) induziert. Die «Hautselektivität» der T-Lymphozyten wird wesentlich durch die Kombination «CLA-CCR4» bestimmt. Tab.1 Chemokine der Haut und entsprechende Chemokinrezeptoren auf T-Lymphozyten sorgen für eine organspezifische Abwehrreaktion in der Haut. (siehe Text). __________________________________________________________________________________________ Organ Adhäsionsmolekül Ligand Chemokine Chemokinrezeptoren (auf den T-Lymphozyten) (auf den Endothelzellen) (in der Haut) (auf den T-Lymphozyten) __________________________________________________________________________________________ Haut CLA E-Selektin CCL271 CCL172 CCR103 CCR4 Magendarm Integrin α4β7 MadCAM-1 __________________________________________________________________________________________ 1 2 3 2. Das Chemokin CCL27 diffundiert in die extrazelluläre Matrix (ECM) der Haut und bindet dort an Moleküle der ECM, an Fibroblasten und auch an Endothelzellen. Das Chemokin CCL17 wird in den Epithelzellen der Haut (Keratinozyten) und der Lungen gebildet, nicht aber in den Epithelzellen von Schleimhäuten. Die Tumorzellen des malignen Melanoms exprimieren vermehrt CCR10 (siehe Abb.1). Diese Beobachtung erklärt, warum die malignen Melanome gehäuft in die Haut metastasieren. Chemokine und Metastasierung maligner Tumoren Abschnitt 10.1.3. Seite 116 N Engl J Med 345: 833-834, 2001 Spalte rechts unten Bei der Metastasierung dringen die Tumorzellen wie die Leukozyten durch die Gefässwände hindurch. Dies hat zur Idee geführt, dass maligne Tumoren bei ihrer Ausbreitung im Organismus über Chemokine und Chemokinrezeptoren gesteuert werden könnten. Bisher sind zwei Tumormodelle untersucht worden. Menschlichen Mammakarzinomzellen zeigen eine erhöhte Expression der beiden Chemokinrezeptoren CXCR4 und CCR7. CXCR4 hat CXCL12 [auch stromal cell derived factor 1 (SDF-1), siehe S.117-119 im Buch] zum Liganden, CCR7 das Chemokin CCL21. Dieses Chemokin CCL21 wurde bisher ausschliesslich in Lymphknoten gefunden. Das Mammakarzinom metastasiert vor allem in die regionären Lymphknoten in die Lungen, in die Leber und ins Knochenmark. In Extrakten aus der Lunge, der Leber und dem Knochenmark konnte bei Mammakarzinomen eine vermehrte Konzentration des CXCL12 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 3 (2008/2009) November 3, 2008 __________________________________________________________________________________________ gefunden werden. Diese Beobachtungen sind ein starker Hinweis darauf, dass an der Metastasierung maligner Tumoren auch Chemokine und Chemokinrezeptoren beteiligt sind und den Tumorzellen den «Weg weisen» (Abb.1). Da auf dysplastischen Vorläuferzellen des Mammakarzinoms keine vermehrte Expression des CXCR4 beobachtet wird, besteht die Vermutung, dass der CXCR4-Phänotyp zu einem wichtigen Faktor der Früherkennung der Mammakarzinome werden könnte. Eine analoge Beobachtung einer Beteiligung von Chemokinen und ihrer Rezeptoren an der Metastasierung maligner Tumoren konnte beim malignen Melanom gemacht werden: Die malignen Melanome exprimieren verstärkt die Chemokinrezeptoren CCR7 (wie die Mammakarzinome) und CCR10. Der Ligand für CCR10 kommt vor allem in der Haut vor. Klinisch sind in der ersten Phase der Metastasierung der malignen Melanome die Metastasen tatsächlich vor allem in der Haut und in den Lymphknoten anzutreffen. 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 4 (2008/2009) November 3, 2008 __________________________________________________________________________________________ Abb.1 Regulation der Metastasierung durch Chemotaxine und ihre Rezeptoren CXCL12 ist identisch mit dem Stromal cell derived factor 1 (SDF-1). In den Zellen der Mammakarzinome werden die Chemokinrezeptoren CXCR4 und CCR7 in vermehrtem Ausmass gebildet. Der Ligand von CCR7, das Chemokin CCL21, wurde ausschliesslich in Lymphknoten gefunden. Die gehäufte Metastasierung der Mammakarzinome in die Lymphknoten kann über den Rekrutierungsmechanismus mit einer Bindung zwischen dem CCR7 (auf den Karzinomzellen) und dem CCL21 (im lymphatischen Gewebe) erklärt werden. Mammakarzinom CXCR4 CXCL12 Knochenmark CXCL12 Lungen CXCL12 Leber CCL21 Lymphknoten CCL27 Haut CCR7 Malignes Melanom CCR7 CCR10 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 5 (2008/2009) November 3, 2008 __________________________________________________________________________________________ TRANSKRIPTIONSFAKTOREN Abschnitt 10.1.6 Tabelle 10.6 Seite 124 oben Tab.2 Die wichtigsten Transkriptionsfaktoren. __________________________________________________________________________________________ TranskriptionsTranskription Transkription Transkription Faktoren assoziiert mit ... aktiviert durch gehemmt durch __________________________________________________________________________________________ Activator Protein-1 (AP-1, Heterodimer aus Fos- und JunProteinen) Nuclear factor κB (NF−κB) STATs (Signal Transduction Activated Transscription factors) STAT 1 STAT 2 STAT 3 STAT 4 STAT 5 STAT 6 Zellproliferation Blockade aktivierter GR Phorbol-Ester TNF-α IL-1β Stickstoff-Monoxid-Synthese Cyclooxygenase Chemokinen (IL-8, RANTES) TNF-α Wasserstoff-Peroxid Ozon Virusinfekte Adhäsionsmolekülen (ICAM-1, VCAM-1) Blockade aktivierter GR malignen Tumoren Glukokortikoide Zytokine (via Janus-Kinasen) INF-α, IFN-γ INF-α Zytokine der «IL-6-Familie» NF-κB AP-1 INF-α, IL-12 IL-4, IL-5 cAMP-Response Hemmung des AP-1 und der Element Binding GR Protein (CREB) __________________________________________________________________________________________ GR TNF IL RANTES ICAM VCAM Glukokortikoid-Rezeptoren Tumornekrosefaktor Interleukin Regulated upon Activation, Normal T-Expression presumably Secreted Intercellular Adhesion Molecule Vascular Cellular Adhesion Molecule 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 6 (2008/2009) November 3, 2008 __________________________________________________________________________________________ ZYTOKINE 1. Zytokine vom Typ der IL-6-Zytokine Abschnitt 10.1.3 Seite 115 Biochem J 1334: 297-314, 1998 Spalte rechts Mitte Z y t o k i n e sind Polypeptide mit niedrigem Molekulargewicht. Sie sind wichtige Kommunikatoren zwischen Zellen. Sie werden nicht - wie die Hormone - intrazellulär in Granula gespeichert, sondern nach einer Stimulation der Zellen sofort synthetisiert und freigesetzt. Weil sie gleichzeitig auf verschiedene Zielzellen wirken können, werden sie manchmal als «pleiotrop» bezeichnet. Eine Gruppe von Zytokinen benützt für ihre Bindung an Rezeptoren die Rezeptoruntereinheit gp130 (siehe Zusatzscriptum «Signaltransduktion»). Diese Gruppe von Zytokinen wird als «Zytokine des IL-6-Typs» bezeichnet. Ihr werden neben dem IL-6 die folgenden weiteren Proteine zugeordnet: IL-11, der Leukemia Inhibitory Factor (LIF), das Oncostatin M (OSM), der Ciliary Neurotrophic Factor (CNTF) und das Cardiotrophin 1 (CT-1; S.206). Im Blut zirkulieren löslich Moleküle des IL-6-Rezeptors (IL-6R) und der Untereinheit gp130. Die Zytokine vom Typ der IL-6-Zytokine übertragen ihr Signal über eine Aktivierung der intrazellulären Januskinasen (JAK) und eine Aktivierung von Transkriptionsfaktoren der STATFamilie (Signal Transducers and Activators of Transcription; siehe Scriptum STATs). Die JAK gehören in die Gruppe der «Nicht-Rezeptor-Tyrosinkinasen» «Januskinasen»). (siehe Zusatzscriptum 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 7 (2008/2009) November 3, 2008 __________________________________________________________________________________________ 2. Tumornekrosefaktor-α Abb.2 Die Wirkungen des Tumornekrosefaktors α (TNF-α) Wenn die Makrophagen stimuliert werden (z.B. durch Endotoxine gramnegativer Bakterien), sezernieren sie in vermehrtem Ausmass die Zytokine TNF-a und das Interleukin 6 (IL-6) sowie Stickstoffmonoxid (NO). Das IL-6 ist einer der potentesten Stimulatoren der Sekretion von Proteinen der Phase der akuten Antwort durch die Hepatozyten. Gramnegative Bakterien LPS-bindendes Protein Lipopolysaccharide (LPS) Adhäsion NGR CD14 Proteine der Phase der akuten Antwort IL-6 NO Monozyten/ Makrophagen Vasodilatation Aktivierung der Plättchen Proliferation der glatten Muskelzellen Wachstum der Endothelzellen Tumornekrosefaktor α Endothelzellen Adhäsion von neutrophilen Granulozyten Sekretion von IL-1 Direkte Schädigung Synthese prothrombotischer Faktoren 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 8 (2008/2009) November 3, 2008 __________________________________________________________________________________________ PRIONEN-KRANKHEIT Abschnitt 10.1.3 Seite 122 Nature Medicine 7: 289-290, 2001 Nature Medicine 7: 410-411, 2001 Spalte links unten Die pathogenen Prion-Proteine werden im Magendarmtrakt aufgenommen, gelangen über die Payer-Plaques des Dünndarmes in die lymphatischen Organe (Lymphknoten, Tonsillen, Milz). Damit sich in einer Zelle pathogene Prionen vermehren können, ist es absolut notwendig, dass die Zelle normale Prionen besitzt. So sind denn auch Mäuse, denen das Gen für das normale Prion-Protein fehlt, oder Mäuse, denen die B-Lymphozyten fehlen, vollständig resistent gegen die Krankheit. Ein möglicher Therapieansatz könnte demzufolge auf die Verminderung der Produktion normaler Prion-Proteine durch die Nervenzellen oder durch andere Zellen (z.B. dendritische Zellen) ausgerichtet sein. Im April 2001 konnte festgestellt werden, dass Mäuse, denen die Komponenten C1q, C2 und C3 des Komplements fehlen, die Symptome der Prionen-Krankheit stark verzögert entwickeln. Das pathogene Prion-Protein wird nach seinem Eintritt in den Organismus sehr wahrscheinlich an Komplementkomponenten gebunden. Dieser Komplex wird dann über die Rezeptoren für Komplement (CR-1 und CR-2) von den dendritischen Zellen in den lymphatischen Organen aufgenommen. Die dendritischen Zellen bilden physiologischerweise das nichtpathogene Prion-Protein, sodass in den dendritischen Zellen eine Vermehrung des pathogenen Prion-Proteins stattfinden kann. In den Amyloidplaques des Gehirns können bei der PrionenKrankheit neben dem pathogenen Prion-Protein tatsächlich auch Komponenten des Komplementsystems gefunden werden. Sehr überraschend war die Entdeckung, dass der Erythroid differentiation-related factor (EDRF), ein Protein, welches in den Vorläuferzellen der Erythrozyten vorkommt, im lymphatischen Gewebe von Mäusen mit der Prionen-Krankheit reduziert ist. Der EDRF kann aber nicht nur im lymphatischen Gewebe, sondern auch im Knochenmark und in Blutproben nachgewiesen werden. Die Störung des EDRF wurde entdeckt, weil systematisch nach Störungen von Proteintranskriptionen bei der Prionen-Krankheit in lymphatischen Organen gesucht worden war. Diese Methode der «blinden» Suche nach veränderten Transkriptionen wurde bereits mit dem Begriff «Transcriptomics» versehen. Die Entdeckung der Reduktion des EDRF bei der PrionenKrankheit weckt die Hoffnungen auf einen zuverlässigen Frühtest für die Krankheit. 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 9 (2008/2009) November 3, 2008 __________________________________________________________________________________________ NEURODEGENERATIVE ERKRANKUNGEN Abschnitt 10.2.1. Seite 135 Spalte rechts unten New England Journal of Medicine 344: 1516-1524, 2001 Prionen Nature Medicine 7: 1280-1282, 2001 Alpha-synuclein Nature 413: 691-694, 2001 Morbus Huntington Nature 414: 159-160, 2001 Alzheimer und Entzündung Nature 415: 377-379, 2002 Mobrus Huntington Tab.3 Proteine und Gene, welche an den verschiedenen neurodegenerativen Krankheiten beteiligt sind. __________________________________________________________________________________________ Krankheit Protein Pathologische Ablagerungen Gen Mutation __________________________________________________________________________________________ Prion-Krankheiten Alzheimer-Krankheit PrPRES Aβ Prionprotein-Amyloid Aβ Ablagerungen Parkinson-Krankheit Tau α-Synuclein Neurofibrilläre Tangles Lewy-Körper prnp app ps1 ps2 Punktmutationen Punktmutationen Frontotemporale Demenz Tau Filamente Punktmutationen Deletionen Pick-Krankheit Tau Pickkörperchen tau Punktmutationen Amyotrophe Lateralsklerose Neurofilament Neuronale Aggregate sod1 Punktmutationen Huntington-Krankheit Huntingtin Kerneinschlüsse hd PolyglutaminExpansionen Spinocerebelläre Ataxie Ataxin Kern- und Zytoplasmaeinschlüsse sca PolyglutaminExpansionen __________________________________________________________________________________________ Aβ prnp app ps1 ps2 parkin sod1 hd sca Amyloid β-Protein Gen für das Prionprotein Gen für das Amyloidprecursor-Protein Gen für Presenilin 1 Gen für Presenilin 2 Gen für Parkin Gen für die Superoxid Dismutase Typ 1 (Die SOD1 liegt im Zytoplasma.) Gen für das Huntingtin Gen für das spinocerebelläre Ataxin snca parkin tau Punktmutationen Punktmutationen Punktmutationen Punktmutationen 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 10 (2008/2009) November 3, 2008 __________________________________________________________________________________________ 1. Alzheimer-Krankheit An der Entstehung der deterministischen (familiären oder autosomal-dominant vererbten) Alzheimer-Krankheit sind die Preseniline 1 und 2 sowie die γ-Sekretase und β-Sekretase (Abb.10.12, S.134) beteiligt. Die Preseniline 1 und 2 bilden mit der γ-Sekretase Komplexe. Die γSekretase spaltet das Amyloidprecursor-Protein (APP) in seiner transmembranösen Domäne und führt so zur Bildung von Amyloid β (Aβ ) (Abb.10.12, S.134). Weil die γ-Sekretase die transmembranöse Domäne des APP an verschiedenen Stellen spalten kann, entstehen verschieden lange Aβ-Peptide. Es konnte gezeigt werden, dass (1) bei der Alzheimer-Krankheit vor allem das Aβ-Peptid mit 42 Aminosäuren (Aβ42) gebildet und dieses Peptid bedeutend schneller abgelagert wird als kürzere Peptide wie das Aβ40. Gleichzeitig konnte beobachtet werden, dass die beiden nichtsteroidalen antiinflammatorischen Medikamente (NSAID) Ibuprofen und Indomethacin die Bildung von Aβ 42 einzudämmen vermögen, sodass der Quotient Aβ42/Aβ40 substanziell abnimmt. In retrospektiven klinischen Studien konnte nachgewiesen werden, dass es nach Gabe von Indomethacin bei Patienten mit der Alzheimer-Krankheit tatsächlich zu einer Verlangsamung des Verlustes der kognitiven Funktionen kommt. Mit Aspirin konnte dieser Effekt nicht erzeugt werden. Vorläufig kann noch nicht erklärt werden, warum einige NSAIDs eine reduzierte Bildung von Aβ42 bewirken. 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 11 (2008/2009) November 3, 2008 __________________________________________________________________________________________ Alzheimer-Krankheit und Apolipoprotein E Abschnitt 10.2.1. Seite 135 Spalte links unten Abb.3 Funktion des Apolipoprotein E in den Neuronen. Das Apolipoprotein E3 bindet in den Neuronen vorübergehend das Tau-Protein und beugt dadurch einer Selbstaggregation des Tau-Proteins und somit einer Störung seiner Funktion als Mikrotubulus-Stabilisator vor. Anders das Apolipoprotein E4: Es besitzt nur eine schwache Affinität für das Tau-Protein. Dies erlaubt dem Tau-Protein eine Selbstaggregation. Folge davon ist die Ausbildung von neurofibrillären Tangles und eine vermehrte Phosphorylierung des Tau-Proteins. Tau-Protein ApoE4 Synthese ApoE4 ApoE4 ApoE4 Homodimerisierung Mikrotubuli Neurofibrilläre Tangles NEURON 2. Parkinson-Krankheit α-Synuclein ist ein präsynaptisches Protein in den Nervenzellen. Es ist nicht gefaltet und bindet reversibel an die vesikulären Membranen. α-Synuclein liegt normalerweise in gelöster Form im Zytoplasma vor. Es kann zusammen mit Dopamin-Quinon Oligomere und Protofibrillen bilden (Abb.4), wenn es im Übermass vorhanden oder defekt ist. Diese Protofibrillen sind toxisch. Der Organismus versucht, die Protofibrillen in den Lewy-Körpern zu kompartimentieren und unschädlich zu machen (siehe Kapitel 1: Zellschädigung). Das α -Synuclein wird 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 12 (2008/2009) November 3, 2008 __________________________________________________________________________________________ physiologischerweise von Parkin, einer Ubiquitin-Ligase, proteolysiert und abgebaut. Da beim Morbus Parkinson diese Proteolyse nicht richtig funktioniert, wird in den Neuronen vermehrt αSynuclein abgelagert. Der Morbus Parkinson wird deshalb den Synucleinopathien zugerechnet. Abb.4 Die Wirkung von α-Synuclein (siehe Text). Wenn zu viel oder defektes α-Synuclein vorhanden ist, wird es unter Mithilfe von Parkin ubiquiniert. αSynuclein dient dazu, Dopamin zu neutralisieren. Dies erfolgt über die Bildung von Dopamin-Quinon und eine anschliessende Autophagozytose dieses Dopamin-Quinon unter Mithilfe des α-Synuclein. Aus DopaminQuinon können sich durch eine Oligomerisierung Protofibrillen bilden. Diese sind toxisch. Sie werden in LewyKörper kompartimentiert. Die Bildung von Lewy-Körpern kann durch mutiertes α-Synuclein gehemmt werden. Dann bleiben die toxischen Protofibrillen im Zytoplasma liegen. Defektes α-Synuclein Proteolyse über Ubiquitin Exzess von α-Synuclein Parkin Mutiertes α-Synuclein Exzess von Dopamin DopaminQuinon kann hemmen AUTOPHAGIE DopaminQuinon LEWY-KÖRPER PROTOFIBRILLEN (toxisch) Die Parkinson-Krankheit kann zu einer Demenz führen. Eine Parkinson-Krankheit steht hinter ungefähr 20%-30% der verschiedenen Demenzformen. 3. Huntington-Krankheit Bei dieser Form der neurodegenerativen Erkrankung kommt es zu einem Untergang von Neuronen im Striatum. Diese Neuonen hemmen Neuronen, welche an den koordinierten Bewegungsabläufen direkt beteiligt sind. Wenn diese Kontrolle ausfällt, treten in vermehrtem Ausmass unkoordinierte Bewegungen auf, welche die Patienten erheblich einschränken. 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 13 (2008/2009) November 3, 2008 __________________________________________________________________________________________ Die Ursache des Morbus Huntington liegt in einer pathologischen Wiederholung der Sequenz der drei Nukleotide CAG im Gen, welches das Huntingtin (Huntington-Protein) kodiert. Folge davon ist, dass das Huntingtin nicht nur eine Glutaminsäure aufweist, sondern eine ganze Kette von Glutaminsäuren. Dadurch wird das Huntingtin in seiner Funktion blockiert; Patienten mit mehr als 40 Glutaminen entwickeln eine Huntington-Krankheit. Das Huntingtin kontrolliert in den Synapsen der Nervenzellen die Endozytose von Neurotransmittern und deren Sekretion, somit also die Kommunikation zwischen den Neuronen. Eine Mutation des Proteins hat zwei Folgen: (1) Diese Kommunikation zwischen den Neuronen kommt zum Erliegen und (2) die Neuronen gehen durch eine Apoptose zugrunde. An der Endozytose der Neuronen im Striatum sind namhaft die beiden Proteine «Clathrin» und «Adaptorprotein 2» beteiligt; Clathrin interagiert direkt mit dem Zytoskelett. Die Interaktion dieser beiden Proteine mit dem Huntingtin wird durch ein drittes Protein, das Hip1, vermittelt: Alle drei Proteine bilden physiologischerweise mit dem Huntingtin einen Komplex (Abb.5a). Wenn das Huntingtin mutiert ist, kann der erwähnte Komplex nicht mehr gebildet werden und es kommt dabei auch zu einer Auflösung der Bindung zwischen dem Huntingtin und dem Hip1 (Abb.5b). Das freie Hip1 aktiviert über das Protein Hippi die Caspase 8, welche die Kaskade der Caspasen in Bewegung setzt, sodass es zu einer Apoptose des Neurons kommt. Weil die an diesem Prozess beteiligte Caspase 3 auch das pathologischen Huntingtin zu spalten vermag, kommt es gleichzeitig zu Ablagerungen von Fragmenten des Huntingtin in den Neuronen. 10 Proteine Allgemeine Pathologie für Studierende der Zahnmedizin 14 (2008/2009) November 3, 2008 __________________________________________________________________________________________ Abb.5 Die Wirkung von Huntingtin. Die Störung, welche der Huntigton-Krankheit zugrunde liegt, ist eine pathologische Wiederholung der Sequenz der drei Nucleotide CAG im Gen, welches das Huntingtin kodiert. Folge davon ist die Ausbildung eines Strangs von Glutaminsäuren (G) am Huntingtin und demzufolge eine Fehlfunktion des Huntingtins. a b Actinfilament Sekretion von Neurotransmittern Endozytose Clathrin Adaptorprotein2 Hip1 Hippi Huntingtin G Actinfilament G G G Caspase 8 Caspase 3 Clathrin Adaptorprotein2 Huntingtin Hip1 Hippi APOPTOSE Caspase 8 Selbstaggregate Caspase 3