Fortbildung-2009-05-Fremdstoffmetabolismus-Teil-1
Werbung

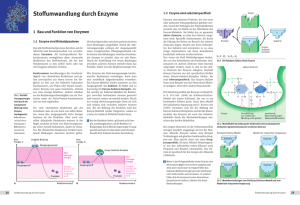
PHARMAZEUTISCHE WISSENSCHAFT Wim Wätjen, Ellen Fritsche Die Rolle des Fremdstoffmetabolismus in Pharmakologie und Toxikologie Teil 1: Phase-I-Reaktionen lisiert und dann als inaktive Transformationsprodukte ausgeschieden werden müssen. Viele Arzneimittel, wie z.B. Acetylsalizylsäure oder Taxol sind Abkömmlinge solcher Naturstoffe. Xenobiotika, die eine geringe Wasserlöslichkeit aufweisen, können vom Organismus nicht direkt eliminiert werden, sondern müssen in einer ersten Phase (Phase I) durch den Einbau bzw. die Demaskierung funktioneller Gruppen funktionalisiert werden. Diese Modifikationen sind nötig, um lipophile Fremdstoffe effektiv ausscheiden zu können und ihre Akkumulation im Organismus zu verhindern (Abb. 1). Um seinem Zweck als universelle Entgiftungsmaschinerie nachzukommen, muss das FME die besondere Eigenschaft besitzen, eine sehr große Anzahl von Substraten unterschiedlichster chemischer Struktur zu metabolisieren. Zudem muss es in der Lage sein, neue, unbekannte Substanzen zu verstoffwechseln. Diese Anforderungen schließen eine „Ein Enzym - ein Substrat“- Situation, wie sie von klassischen biochemischen Stoffwechselwegen bekannt ist, aus. Die Evolution hat diese Aufgabe gelöst, indem sie das FME aus „promiskuitiven“ Enzymen zusammengesetzt hat, die jedoch jeweils eine geringe Substratspezifität aufweisen. Abbildung 1: Ausscheidung von Fremdstoffen (schematische Darstellung) Während hydrophile Stoffe direkt eliminiert werden, müssen lipophile Substanzen vor ihrer Ausscheidung in hydrophilere Metabolite überführt werden. Diese Substanzen werden zunächst in der Phase I oxidiert, die polaren Intermediate in der Phase II konjugiert und als hydrophile Metabolite ausgeschieden. Fremdstoffe mit funktionellen Gruppen, wie z.B. das Phenol, können direkt durch Phase II Enzyme in hydrophile Metabolite überführt und eliminiert werden. Einige funktionalisierte Phase I-Metabolite können auch hydrophil genug sein, um renal ausgeschieden zu werden. Bestimmte lipophile Substanzen, wie z.B. das „Sevesogift“ Tetrachlorodibenzo(p)dioxin werden kaum metabolisiert und akkumulieren im Fettgewebe. 8 1. Chemische Reaktionen der Phase I Die chemischen Reaktionen des FME der Phase I beinhalten hauptsächlich oxidative Prozesse, wobei die Einführung eines Sauerstoffatoms in den Fremdstoff die Schlüsselreaktion darstellt (Abb. 3 & 4). Ziel der Phase I Reaktionen ist es, den lipophilen Fremdstoff durch Einführung bestimmter Gruppen zu funktionalisieren und damit eine Interaktion mit den Enzymen der Phase II zu ermöglichen. Eine allgemeine Übersicht der Enzyme der Phase I findet sich in Tab. 1. Obwohl Phase I Reaktionen die erste Stufe des körpereigenen Entgiftungssystems darstellen, kann es durch Phase I Reaktionen zur ‚Aktivierung’ von an sich inerten Fremdstoffen kommen. Einige Beispiele für Substanzen, die erst durch metabolische Aktivierung in toxische Verbindungen umgewandelt werden, sind in Abb. 2 gezeigt. Abbildungen: Wätjen/Fritsche Das Fremdstoff-metabolisierende Enzymsystem (FME) ist ubiquitär über die meisten Spezies verbreitet. Es wird vermutet, dass es sich vor ca. 2,5 Milliarden Jahren zur Entgiftung von Sauerstoff entwickelte. Die anschließende Weiterentwicklung der Enzymsysteme wird als Folge der evolutionären Trennung von Tier- und Pflanzenreich vor ca. 1,8 Milliarden Jahren erklärt. Tiere mussten in der Lage sein, sekundäre Pflanzeninhaltstoffe wie z. B. Blütenpigmente, chemo-taktische Schreck- und Lockstoffe sowie Phytoalexine zu metabolisieren. Diese Theorie wird von der Beobachtung unterstützt, dass es vor etwa 400 Millionen Jahren eine „explosionsartige“ Vermehrung der tierischen Cytochrom P450 (CYP)2 Familie, gab, als die ersten Tiere begannen, das Land zu besiedeln und anfingen sich von Landflora zu ernähren. Bei dem Konsum von Pflanzen wird der Organismus mit zehntausenden von Pflanzeninhaltstoffen konfrontiert, die metabo- Zertifizierte Fortbildung Cytochrom P450 Die große Gruppe der CYP Enzyme setzt sich beim Menschen aus derzeit 18 bekannten Familien zusammen. Zu einer Familie, die mit der ersten Zahl klassifiziert wird (z.B. CYP1), gehören CYP Enzyme mit einer Sequenzhomologie von mehr als 40%. Innerhalb dieser Familien werden CYPs mit Homologien von mehr als 55% durch einen Buchstaben in (beim Menschen 43) Unterfamilien gruppiert (z.B. CYP1A). In diesen Unterfamilien werden einzelne Isoenzyme zusammengefasst und durch eine weitere Zahl (z.B. CYP1A1) definiert. Bisher sind beim Menschen 57 Isoenzyme bekannt, die Anzahl variiert jedoch erheblich zwischen einzelnen Spezies. Da es hohe genetische Variabilitäten innerhalb der CYP Enzyme einer Spezies gibt, werden die verschiedenen Allele durch ein Sternchen und folgende Nummerierung identifiziert (z.B. CYP1A1*1). Dabei ist das *1 Allel in der Regel der Wildtyp und alle weiteren Allele (*2, *3, etc.) werden in der Reihenfolge ihrer Entdeckung durchnummeriert. tor Nuclear Translocator) und bindet dann an XREs der DNA, was zur Induktion bestimmter Proteine, wie z.B. CYP 1A1 führt (Abb. 5). Der Metabolismus von Benzo(a)pyren durch CYP1A1 ist in Abb. 6 dargestellt und zeigt, dass es sowohl zu einer Entgiftung als auch zur Giftung von B(a)P kommen kann. Die generelle CYP1-Aktivität kann über die Ethoxyresorufin O-Deethylierung gemessen werden. Während es für CYP1A1/1B1 keine spe- Die CYP Enzymaktivitäten des menschlichen Körpers sind nicht allein der Leber vorbehalten. Fast alle Zellen sind in der Lage, in gewissem Umfang Xenobiotika zu metabolisieren. Da sich die Expression der meisten CYP Enzyme - und damit ihre Aktivität Organ- und Substrat-spezifisch induzieren lässt, unterscheidet man zwischen basaler und induzierbarer Aktivität. Die Induktion durch ein Substrat erfolgt dabei auf der Ebene des Zellkerns: Ein Substrat bindet einen zytosolisch lokalisierten Rezeptor (z.B. AhR, CAR, PXR), welcher dann als Transkriptionsfaktor über spezifische Sequenzen der DNA (XRE: „Xenobiotic responsive element“) zur Geninduktion des CYPs führt. Die Leber besitzt zwar die höchsten basalen CYP Aktivitäten, jedoch sind auch in vielen extrahepatischen Organen CYP Enzyme basal exprimiert oder lassen sich induzieren. CYP Enzyme des FM gehören zu den Familien 1-3, auf welche dieses Kapitel näher eingehen wird. Diese Enzyme stellen quantitativ den größten Anteil des Phase-I-Stoffwechsels von Fremdstoffen dar. Abbildung 2: Beispiele für Fremdstoffe, die erst durch Metabolisierung (Cytochrom P450) in reaktive, toxische Substanzen umgewandelt werden (Benzo(a)pyren, n-Hexan, Vinylchlorid und Tetrachlorodibenzo(p)dioxin (TCDD)) Die CYP1 Familie Zu dieser Familie gehören CYP1A1, 1A2 sowie 1B1. Während das CYP1A2 das Leber-spezifische Enzym der CYP1 Familie darstellt, sind CYP1A1 und CYP1B1 auch extrahepatisch exprimiert. Die Expression dieser Familienmitglieder wird durch den AhR (Arylhydrocarbon Rezeptor) kontrolliert. Der AhR bindet hauptsächlich polyzyklische aromatische Kohlenwasserstoffe welche z.B. im Tabakrauch enthalten sind oder das als ‚Seveso-Gift’ bekante Dioxin (TCDD). Der durch den Fremdstoff aktivierte AhR transloziert in den Zellkern, dimerisiert dort mit einem weiteren Protein (ARNT: Arylhydrocarbon Recep- Oxidoreduktasen Cytochrom-P450-abhängige Monooxygenasen (CYP) Flavin-abhängige Monooxygenasen (FMO) Monoaminoxidasen (MAO) Cyclooxygenasen (COX) Dihydrodioldehydrogenasen DT-Diaphorase (NQOR) Alkohol- und Aldehyd-dehydrogenasen (ADH, ALDH) Hydrolasen Esterasen Amidasen Glucuronidasen Epoxidhydrolasen (EH) Tabelle 1: Phase-1-Enzym Abbildung 3: CYP-vermittelte Reaktionen Zu den Cytochrom P450-vermittelte Reaktionen zählen die aromatische und aliphatische Hydroxylierung, N-, O- S-Desalkylierung, Epoxidierung, Dehydrogenierung, N-Hydroxylierung, N-Oxidation, oxidative Desaminierung, oxidative Desulfurierung und Sulfoxidation. In dieser Abbildung sind beispielhaft einige Reaktionen dargestellt. 9 PHARMAZEUTISCHE WISSENSCHAFT CYP 1A1 A2 Theophyllin, Clozapin, Coffein, Benzo(a)pyren Amitriptylin, Theophyllin, Clozapin, Coffein, Haloperidol CYP 2A6 B6 Nicotin Clopidogrel, Cyclophosphamid, Methadon, Bupropion, Ifosfamid Tolbutamid, Verapamil, Paclitaxel Warfarin, Diclofenac, Tamoxifen, Amitriptylin, Tamoxifen, Losartan, Naproxen, Rosiglitazon Verapamil Omeprazol, Diazepam, Propanolol, Warfarin, Imipramin, Phenytoin Codein, Imipramin, Tamoxifen, Desipramin, Fluoxetin, Risperidon, Timolol, Haloperidol, Chlorpromazin, Amitriptylin Ethanol, Paracetamol, Tetrachlorkohlenstoff, Halothan, Enfluran C8 C9 C18 C19 D6 E1 CYP 3 A4 A5 Clarithromycin, Ciclosporin, Coffein, Docetaxel, Lovastatin, Sildenafil, Simvastatin, Verapamil, Nifedipin, Midazolam, Cyclophosphamid, Vincristin, Saquinavir, Lidocain Verapamil, Nifedipin, Ciclosporin, Midazolam, Cyclophosphamid Tabelle 2: Pharmakologisch/toxikologisch relevante CYP-Enzyme und deren Substrate (Auswahl) Abbildung 4: Der katalytische Zyklus des Cytochrom P450. Die Bindung des Substrats (X-H) unterstützt den Übergang des Hämin-Eisens in die high-spin-Konfiguration (1). Die folgende Reduktion des Hämins zu Häm wird durch die Cytochrom P450 Reduktase katalysiert (2). Nach Bindung von molekularem Sauerstoff an das Häm (3) wird der Komplex durch einen zweiten Ein-Elektron-Reduktionsschritt aktiviert. Das Elektron wird dazu entweder durch die CYP Reduktase oder durch Cytochrom b5 geliefert (4). Die heterolytische Spaltung der O-O Bindung geschieht unter Wasserabspaltung (5). Der angenommene Zwischenkomplex hat hohes Sauerstoffübertragungspotential und führt zur Oxygenierung des Substrats (6). Das Reaktionsprodukt wird vom Enzym freigesetzt (7). Die Intermediate (gestrichelte Kästchen) sind hypothetisch, da sie analytisch schwer nachweisbar sind. Abbildung 5: Ah-Rezeptor-Aktivierung durch TCDD (schematische, vereinfachte Darstellung) Der durch den Fremdstoff aktivierte AhR transloziert in den Zellkern, dimerisiert dort mit einem weiteren Protein (ARNT: Arylhydrocarbon Receptor Nuclear Translocator) und bindet dann an XREs der DNA, was zur Induktion bestimmter Proteine, wie z.B. CYP 1A1 führt. 10 zifischen Markersubstrate gibt, lässt sich für die Bestimmung der CYP1A2-Aktivität das Koffein nutzen. Das Besondere an CYP1A1/1B1 ist, dass diese Enzyme bisher in fast allen extrahepatischen Organen nachgewiesen werden konnten. Die CYP2 Familie Die CYP2 Familie ist die größte CYP Familie der Säugetiere. Zu ihr gehören die Unterfamilien CYP2A, CYP2B, CYP2C, CYP2D und CYP2E. Die wichtigsten Vertreter der CYP2A Familie des Menschen sind die leberständigen CYP2A6 und 2A7 Enzyme (beide machen <5% des Leber-CYPs aus) sowie das extrahepatisch exprimierte CYP2A13. CYP2A6 ist das Hauptenzym der 7-Hydroxylierung von Cumarin, welches daher als Modellsubstrat zur Messung der CYP2A6 Enzymaktivität herangezogen wird. Ebenso setzt es Nikotin zu Cotinin um und ist somit das einzige bisher bekannte Nikotin metabolisierende Enzym der Leber. Des Weiteren ist CYP2A6 an der Aktivierung einiger Prokarzinogene wie Aflatoxin B1, 6-Aminochrysen sowie im Tabakrauch enthaltener Nitrosamine beteiligt. Das unter anderem im Respirationstrakt identifizierte CYP2A13 scheint das effizienteste CYP Enzym für die Aktivierung des Tabak-spezifischen Karzinogens, 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanon (NNK) zu sein. Die Induktion von CYP2A6 durch Rifampizin und Phenobarbital (PB) wird durch den PXR (Pregnan X Rezeptor) vermittelt. Beim Menschen existieren verschiedene CYP2A6 Allele, von denen einige (CYP2A6*2, *4, *5) keine Enzymaktivitäten haben. Eine mögliche Schutzwirkung solcher CYP2A6 Allele vor Xenobiotika-induzierten Erkrankungen, wie z.B. Tabakrauch-induzierten Tumorerkrankungen, ist bisher noch nicht geklärt. Das CYP2B6 des Menschen macht 2 - 10% des menschlichen Leber CYPs aus und metabolisiert Antitumormittel wie Cyclophosphamid, Abbildungen: Wätjen/Fritsche Alle drei Gene der CYP1 Familie sind polymorph exprimiert. Im Gegensatz zum fehlenden Phänotyp der CYP1A1 Polymorphismen, variiert die CYP1A2 Aktivität der Leber zwischen verschiedenen Individuen um den Faktor 60. Zertifizierte Fortbildung Abbildung 6: Der Metabolismus von Benzo(a)pyren (schematische Darstellung) Erläuterung im Text (Abk.: GST: Glutathion-S-Transferase, UGT: UDP-Glucuronosyltransferase). Pestizide wie Methoxychlor, PCBs sowie einige Pro-Karzinogene wie das Aflatoxin B1 und Tabakrauch-spezifische Nitrosamine. PB ist der bekannteste Induktor des CYP2B und involviert den Rezeptor CAR (konstitutiver Androstan Rezeptor). Die Gruppe der strukturell sehr unterschiedlichen PB-ähnlichen CYP2B Induktoren ist sehr groß und umfasst u.a. Medikamente, Pestizide, Lösemittel und bestimmte PCBs. Zudem variiert die Enzymexpression interindividuell um den Faktor 20 bis 250. Die CYP2C8, CYP2C9 und CYP2C19 Enzyme machen ca. 20% des menschlichen CYPs aus. Sie metabolisieren eine große Zahl von Pharmazeutika, wie z.B. nicht steroidale Antiphlogistika (NSAIDs), Antidiabetika, orale Antikoagulantien, Protonenpumpenblocker und Antidepressiva. CYP2C8, CYP2C9 und CYP2C19 sind nicht nur in der Leber, sondern auch extrahepatisch exprimiert und können durch PB oder Rifampizin induziert werden. In der CYP2C Familie gibt es klinisch relevante Polymorphismen, so die CYP2C9*2 und CYP2C9*3 Allele, welche mit reduzierter enzymatischer Aktivität assoziiert sind. Auch die CYP2C19*2 und CYP2C19*3 Allele gehen mit eingeschränkter Enzymaktivität einher, während das CYP2C19*17 Allel ultraschnelle Metabolisierer hervorbringt. Die klinische Relevanz für die langsamen CYP2C19 Metabolisierer wird an der verminderten Ausscheidung und damit höheren Plasmakonzentration des Protonenpumpenblockers Omeprazol deutlich, welcher in Patienten mit diesem Phänotyp höhere Wirksamkeiten entfaltet. Abbildung 7: Die durch MAO katalysierte Desaminierung von Dopamin Gezeigt ist hier die MAO-B-vermittelte Umwandlung von Dopamin in Dihydroxyphenylalaninaldehyd (DOPAL), Ammoniak und Wasserstoffperoxid. 11 PHARMAZEUTISCHE WISSENSCHAFT irrreversible Inhibition Azol-Fungizide, Grapefruitsaft, Bergamottin, Dihydroxybergamottin Induktion Barbiturate, Carbamazepin, Dexamethason, Phenytoin, Rifampicin, Johanniskraut-Extrakte Tabelle 3: Klinisch relevante Modulatoren des CYP3A4 Ein für den Metabolismus von Arzneistoffen ebenfalls sehr bedeutsames CYP Enzym ist das CYP2D6. Es sind mittlerweile mehr als 200 Stoffe bekannt, die zum Teil ausschließlich durch dieses Enzym verstoffwechselt werden. Zu diesen gehören u.a. neben Herz-Kreislauf Medikamenten viele im zentralen Nervensystem wirksame Pharmaka wie Analgetika, Antitussiva, trizyklische Antidepressiva und Antipsychotika. In den 1970iger Jahren fielen in klinischen Studien Patienten auf, welche adverse Reaktionen gegenüber dem Sympatholytikum Debrisoquin sowie dem Antiarrythmikum Spartein zeigten. Die adversen Reaktionen waren auf eine Unfähigkeit der Patienten zurückzuführen, diese Substanzen oxidativ zu verstoffwechseln. Spätere genetische Analysen identifizierten eine große Anzahl von CYP2D6 Polymorphismen (44 Allele), welche für interindividuelle Unterschiede im Arzneimittelstoffwechsel verantwortlich gemacht werden können. Durch das zusätzliche Vorhandensein von Genduplikationen ergab sich für das CYP2D6 die Einteilung in langsame, normale, schnelle sowie ultraschnelle Metabolisierer. Im Gegensatz z.B. zur CYP1 Familie metabolisiert das CYP2E1 kleine Moleküle wie Ethanol, Paracetamol oder Benzol. Bei der Oxidation entstehen häufig toxische Intermediate, was besonders am Beispiel des Paracetamols gut demonstriert ist. Paracetamol ist das Medikament, das bei Überdosierung Leberschäden verursacht. Eine Leberschädigung setzt normalerweise erst bei einer massiven Überdosierung ein (ca. 10 g Paracetamol), jedoch können bei chronischem Alkoholkonsum in seltenen Fällen auch geringere Dosen schon zum akuten Leberversagen führen. Diese Sensibilisierung kann man u.a. auf eine Induktion von CYP2E1 zurückführen, welche zu einer ver- stärkten Bildung des hepatotoxischen Metaboliten NAPQI (N-Acetylp-Benzochinonimin) führt. Die CYP3 Familie Der Hauptanteil des hepatischen CYP des Menschen setzt sich aus Mitgliedern der CYP3A Familie (3A4 und 3A5) zusammen, wobei das CYP3A4 mit 30% der gesamten Leber CYPs dominiert. Neben der hepatischen Expression ist auch das extrahepatische Vorkommen der CYP3A Familie gut dokumentiert; die CYP3A4/3A5 Isoenzyme sind in Teilen des Respirations- sowie des Gastrointestinaltrakts exprimiert und können in diesen Organen induziert werden. Die strukturelle Bandbreite der CYP3A4 Substrate ist sehr groß. So nimmt das CYP3A4 am Metabolismus verschiedenster Pharmaka wie Makrolidantibiotika, Steroidhormone, Calciumantagonisten vom Dihydropyridin Typ, Fungizide sowie Antikonvulsiva teil. Auch toxikologisch ist das CYP3A4 von Bedeutung, da es Prokarzinogene wie das Benzo(a)pyren-7,8-Dihydrodiol oder das Aflatoxin B1 zu aktivieren vermag. Obwohl das CYP3A4 basal bereits einen großen Teil des Leber CYPs ausmacht, lässt es sich durch pharmakologische Induktoren wie PB, Dexamethason, Pregnenoloncarbonitril, Rifampizin sowie Johanniskraut bis auf 60% des gesamten Leber CYPs induzieren. CYP3A Enzymaktivitäten lassen sich jedoch nicht nur steigern, sondern durch Pharmaka oder Naturstoffe auch effektiv hemmen. Beispiele solcher extrem potenten Inhibitoren sind Azol-Fungizide und Grapefruitsaft. Sowohl die Steigerung als auch die Hemmung von CYP3A4 Enzymaktivitäten führen bei dieser großen Substratbreite zu unerwünschten pharmakologischen Wechselwirkungen. So kann die gleichzeitige Einnahme von CYP3A4 Induktoren (z.B. Antikonvulsiva oder Tuberkulostatika) mit Kontrazeptiva (z.B. Ethinylestradiol) zum Versagen der empfängnisverhütenden Wirkung der ‚Pille’ führen, da das induzierte CYP3A4 das Kontrazeptivum rasch zu unwirksamen Plasmakonzentrationen metabolisiert. Umgekehrt werden nach Trinken eines Glases Grapefruitsaft durch Hemmung des CYP3A4 Metabolismus verschiedene pharmakokinetische Parameter von Pharmaka, wie z.B. Nifedipin, bis zum Fünffachen gesteigert. Das Trinken von Grapefruitsaft ist daher im Beipackzettel vieler Arzneimittel als Warnhinweis aufgeführt. Wegen dieser großen Substratbreite sowie seiner ausgeprägten Expression ist das CYP3A4 das wichtigste Enzym des Arzneimittelstoffwechsels. 1.2 Flavin-haltige Monooxygenasen (FMO) FMO metabolisieren ein breites Spektrum an Substraten. Zu diesen gehören Nikotin, Phenothiazin-Derivate oder Cimetidin. Die Produkte der FMO-Reaktionen sind somit meist N- oder S-Oxide, die in der Regel ein geringeres pharmakologisches und toxikologisches Potential als ihre Ausgangssubstanzen haben. + Abbildung 8: Die Reaktion der Alkohol- und Aldehyddehydrogenase Unter Reduktion von NAD wird Ethanol zum Acetaldehyd metabolisiert, welcher durch die Aldehyddehydrogenase zur Essigsäure oxidiert wird. 12 Abbildungen: Wätjen/Fritsche reversible Inhibition Clarithromycin, Ciclosporin, Danazol, Fluoxetin, Indinavir, Ketokonazol, Ritonavir,Verapamil Zertifizierte Fortbildung nase (ADH) zum Acetaldehyd, dem eigentlich toxischen Metaboliten, verstoffwechselt (Abb. 8). Das CYP2E1 trägt nur in geringem Maße (2-8%) zum Ethanolstoffwechsel bei. Der entstehende Acetaldehyd wird dann weiter durch die Aldehyddehydrogenase (ALDH) zu Essigsäure metabolisiert. Abbildung 9: Reaktionen von Hydrolasen Hydrolyse der Esterbindung (Procain) bzw. eines Epoxides (Carbamazepinepoxid) 1.3 Monoaminoxidasen (MAO) Die zwei Monoaminoxidasen (MAO-A und -B) sind bis auf wenige Ausnahmen in fast allen Geweben des Menschen exprimiert. MAO metabolisieren biogene Amine wie Serotonin, Dopamin und Noradrenalin. Auch Fremdstoffe mit ähnlichen strukturellen Charakteristika werden von MAO umgesetzt. Die von MAO katalysierte chemische Reaktion ist die oxidative Desaminierung (Abb. 7). Die Menge des bei dieser Reaktion entstehenden Wasserstoffperoxids kann toxikologisch z.B. bei der Progredienz des Morbus Parkinson nach jahrelanger L-DOPA Therapie relevant sein. Cyclooxygenasen (COX) Die Arachidonsäure metabolisierenden Cyclooxygenasen (COX-1 und COX-2) sind die Schlüsselenzyme der Prostaglandinsynthese. Der Metabolismus zum Prostaglandin (PG)H2 verläuft in zwei Reduktionsreaktionen über die Bildung von PGG2. Während dieser können Xenobiotika (z.B. phenolische Verbindungen) co-oxidiert und somit zu DNA-reaktiven Verbindungen aktiviert werden. Pharmakologisch sind die COX Enzyme jedoch eher als Zielmoleküle für NSAIDs von Interesse. Dehydrogenasen und Reduktasen Die toxikologisch bedeutsamsten Dehydrogenasen sind die Alkoholund Aldehyddehydrogenasen, da sie Ethanol verstoffwechseln. Ethanol wird hauptsächlich von der leberständigen Alkoholdehydroge- Hydrolasen Zu der Gruppe der Hydrolasen gehören die Esterasen, wie z.B. die Cholinesterase und Epoxidhydrolasen (Abb. 9). Hydrolasen addieren H2O an ein Xenobiotikum und sorgen so für eine starke Veränderung der pharmakologischen Eigenschaften (Inaktivierung der Acetylcholinwirkung, Spaltung von Glucuroniden). Epoxid Hydrolasen (EH) hydrolysieren Epoxide zu Dihydrodiolen. Da Epoxide meist sehr reaktiv sind und daher eine Gefährdung für zelluläre Makromoleküle darstellen, sind die EH als Entgiftungsenzyme bedeutsam. Die Biotransformation von Substanzen durch Enzyme des Fremdstoffmetabolismus hat eine große Bedeutung für die Elimination von lipophilen Fremdstoffen, die ansonsten im Organismus akkumulieren würden. Diese Fremdstoffe werden zunächst durch Enzyme der Phase I funktionalisiert, d.h. es werden durch Oxidation funktionelle Gruppen in das Molekül eingeführt. Die Fremdstoffe können nun in der Phase II des Fremdstoffmetabolismus mit hydrophilen Substanzen konjugiert und danach ausgeschieden werden. Enzyme der Phase II sind z.B. UDP-Glucuronosyltransferasen, N-Acetyltransferasen, Sulfotransferasen und Glutathion-S-Transferasen, über die in einem zweiten Fortbildungsbeitrag im Apothekenmagazin näher eingegangen wird. Zusammenfassung Die Biotransformation von Substanzen durch Enzyme des Fremdstoffmetabolismus (FSM) hat eine große Bedeutung für die Elimination von lipophilen Fremdstoffen, die ansonsten im Organismus akkumulieren würden. Diese Fremdstoffe werden zunächst durch Enzyme der Phase I funktionalisiert. Wichtige Enzyme der Phase I sind: Cytochrom-P450-abhängige Monooxygenasen, Flavin-abhängige Monooxygenasen, Monoaminoxidasen, Cyclooxygenasen, Alkoholdehydrogenasen, aber auch Esterasen und Epoxidhydrolasen. Die Fremdstoffe können danach in der Phase II des FSM mit hydrophilen Substanzen konjugiert und ausgeschieden werden. Aktivitäten der Enzyme des FSM unterliegen interindividuellen Variationen, welche durch genetische Varianzen bedingt sind (Polymorphismen). Neben einer Entgiftung kann der FSM auch zu einer ‚Giftung’ von Fremdstoffen führen. Die Autoren PD Dr. rer. nat. Wim Wätjen wurde in Bremen geboren und studierte Chemie an der Universität Bremen, seine Promotion erfolgte im Jahr 2000. Seit 2001 arbeitet er im Institut für Toxikologie der Heinrich-Heine-Universität Düsseldorf, wo er sich im Jahre 2006 habilitierte. Forschungsgebiet von Herrn Wätjen sind Untersuchungen zu toxischen Effekten und zellulärem Metabolismus von Naturstoffen. Er ist Fachtoxikologe der Deutschen Gesellschaft für Pharmakologie und Toxikologie, Gutachter für verschiedene internationale Fachzeitschriften und in nationalen Gremien (BfRKommission für Lebensmittelzusatzstoffe) tätig. PD Dr. med. Ellen Fritsche wurde in Düsseldorf geboren und studierte an den Universitäten Regensburg und Düsseldorf Humanmedizin. Nach ihrer Promotion 1998 am damals Medizinischen Institut für Umwelthygiene in Düsseldorf verbrachte sie 3 Jahre am National Institute of Environmental Health Sciences in den USA. Seit 2002 arbeitet sie in der molekularen Toxikologie am Institut für Umweltmedizinische Forschung in Düsseldorf, wo sie sich im Jahre 2008 habilitierte. Ihre Forschungsschwerpunkte liegen in der Signaltransduktion des Arylhydrokarbon Rezeptors, der Untersuchung von Toxizitäten auf das sich entwickelnde Nervensystem sowie der Etablierung von in vitro Verfahren als Alternativen zu Tierversuchen. Sie ist Gutachterin für verschiedene internationale Fachzeitschriften und in internationalen Gremien zur Entwicklung von Tierversuchsersatzmethoden zur Erfassung von Entwicklungsneurotoxizität vertreten. 13 Fortbildungs-Fragebogen 5/2009 Faxnummer: 02 08 / 6 20 57 41 Hier finden Sie die Fortbildungsfragen zum Hauptartikel. Bei Beantwortung und Faxantwort erhalten Sie einen Fortbildungspunkt auf dem Postweg. Sie erhalten den Fortbildungspunkt für die Kategorie „Bearbeiten von Lektionen“ (rezertifiziert durch die Bundesapothekerkammer, Veranstaltungs-Nr.: BAK 2009/081). Es ist pro Aufgabe nur eine Antwort richtig. Die Lösungen werden Ihnen zusammen mit dem Fortbildungspunkt mitgeteilt. Bitte tragen Sie unbedingt Ihre Postanschrift und Ihre Telefon-Nummer (für evtl. Rückfragen) in das Faxformblatt ein! Faxnummer: 02 08 / 6 20 57 41 1. Welche Aussage über eine Vergiftung mit dem Analgetikum Paracetamol ist richtig? A) 앮 Personen mit chronischem Alkoholkonsum reagieren weniger sensitiv auf Paracetamol-Überdosierungen. B) 앮 Das Analgetikum Paracetamol verursacht eine Leberschädigung über COX-1-vermittelte Synthese von reaktiven Sauerstoffspezies. C) 앮 Der Leberschaden kommt durch einen oxidierten Metaboliten zustande (N-Acetyl-p-Benzochinonimin). D) 앮 Paracetamol wird hauptsächlich durch CYP 2D6 metabolisiert. E) 앮 Paracetamol führt zu Zellnekrosen durch Inhibition des Ah-Rezeptors. 2. Welches der folgenden fremdstoffmetabolisierenden Enzyme ist kein Phase-I-Enzym ? A) 앮 Cyclooxigenase B) 앮 Epoxid-Hydrolase C) 앮 N-Acetyltransferase D) 앮 Alkohol-Dehydrogenase E) 앮 Flavin-abhängige Monooxigenase 3. Welche der folgenden Zuordnungen (Cytochrom-Familie und entsprechendes Substrat) ist falsch ? A) 앮 CYP 2E1 ↔ Ethanol B) 앮 CYP 2B6 ↔ Cyclophosphamid C) 앮 CYP 2D6 ↔ Amitryptilin D) 앮 CYP 3A4 ↔ Paracetamol E) 앮 CYP 2A6 ↔ Nikotin 4. Welche Aussage über den Fremdstoffmetabolismus ist falsch? A) 앮 Cytochrom P450-Enzyme werden nur in der Leber exprimiert. B) 앮 Durch Enzyminduktion kann eine starke Erhöhung bestimmter CYPs verursacht werden. C) 앮 durch genetische Polymorphismen existiert eine breite Variabilität in der Enzymaktivität von CYP2D6. D) 앮 Tetrachlordibenzodioxin (TCCD) wird kaum verstoffwechselt und akkumuliert im Fettgewebe des Menschen. E) 앮 Die Aktivierung des Ah-Rezeptors durch TCDD kann eine CYP1A1-Induktion bewirken. 5. Welches des folgenden Enzyme wird durch Ethanol induziert? A) 앮 CYP 2E1 B) 앮 CYP 2B6 C) 앮 CYP 2D6 D) 앮 CYP 3A4 E) 앮 CYP 2C19 Berufsbezeichnung: 앮 Apotheker/in 6. Bei der metabolischen Aktivierung von Benzo(a)pyren durch Cytochrom P450 entsteht als ultimater krebserzeugender Metabolit ein: A) 앮 Carbokation B) 앮 Diolepoxid C) 앮 Chinon D) 앮 O-Glucuronid E) 앮 Nitrenium-Ion 7. Welche der folgenden Aussagen zur Enzyminduktion ist richtig? A) 앮 Eine Enzyminduktion kommt durch Phosphorylierung eines Apoproteins der Cytochrome P450 zustande. B) 앮 Phenobarbital induziert Cytochrom P450 2E1. C) 앮 Die Induktion von Cytochrom P450 1A1 kann zu verstärkter metabolischer Aktivierung von polyzyklischen aromatischen Kohlenwasserstoffen (z.B. Benzo(a)pyren) führen. D) 앮 Der Enzyminduktor Phenobarbital erhöht die Aktivität von Cytochrom P450-Enzymen über einen allosterischen Mechanismus (Bindung nicht an der Substratbindetasche des Enzyms) E) 앮 Die Enzyminduktion kommt über G-Protein-vermittelte Signalwege nach Aktivierung des Ah-Rezeptors zustande. 8. Welche der folgenden Substanzen ist kein CYP3A4-Inhibitor? A) 앮 Grapefruitsaft B) 앮 Ciclosporin C) 앮 Ketokonazol D) 앮 Ritonavir E) 앮 Rifampicin 9. Welche Aussage zum Metabolismus des Antiarrhythmikums Spartein ist falsch? A) 앮 Es existieren gentische Polymorphismen des Sparteinabbauenden Enzyms CYP2D6. B) 앮 Spartein inhibiert CYP2D6 und verursacht so individuell schwere adverse Reaktionen. C) 앮 Eine schnelle Metabolisierung kann durch Genduplikation von CYP2D6 hervorgerufen werden. D) 앮 In der Bevölkerung existieren langsame, normale, schnelle und ultraschnelle Metabolisierer von Spartein. E) 앮 Durch Polymorphismen von CYP2D6 können adverse Reaktionen von Spartein hervorgerufen werden. 10. Welche Aussage über Cytochrom P450 Monooxigenasen ist nicht richtig? A) 앮 Cytochrom P450 Monooxigenasen sind Enzyme der Phase I des FSM. B) 앮 Cytochrom P450 Enzyme besitzen eine Hämgruppe. C) 앮 Die höchsten basalen CYP-Aktivitäten sind in der Leber zu finden. D) 앮 Einige Cytochrom P450-Enzyme können durch Fremdstoffe induziert werden. E) 앮 Cytochrom P450 Monooxigenasen führen zu einer Erhöhung der Lipohilie von Xenobiotika. 앮 PTA BITTE UNBEDINGT IHRE POSTANSCHRIFT HIER EINTRAGEN! Ja, ich möchte das ApothekenMagazin regelmäßig erhalten! Bitte ankreuzen Lösen Sie – exklusiv für Abonnenten – den ABO-Fragebogen in dieser Ausgabe und Sie erhalten einen zusätzlichen Fortbildungspunkt! 14 Ich abonniere das Apotheken-Magazin zum Jahresvorzugspreis von 25,– EUR (10 Ausgaben inkl. MwSt. und Versand, Inland). Das Abonnement gilt für ein Jahr und kann danach jederzeit gekündigt werden. Wichtig: Dieses Angebot gilt nur in der Bundesrepublik Deutschland. Gebr. Storck GmbH & Co. Verlags-oHG · Duisburger Straße 375 (C-Gebäude) 46049 Oberhausen · Telefon 02 08-8 48 02 24 · Fax 02 08-8 48 02 42 Apothekenstempel