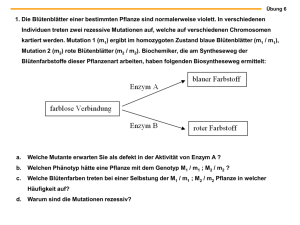
III.1 Versuchsteil A
Werbung

Block II: „Biochemische und genetische Analysen des mikrobiellen Stoffwechsels“ Physiologie, Biochemie, Organisation und Regulation des β Ketoadipatstoffwechselweges in Acinetobacter baylyi BD413 Yvonne Voges und Melanie Thompson Inhalt I. Einleitung ............................................................................................................. 1 II. Material und Methoden ........................................................................................ 4 II.1…..Versuchsteil A ........................................................................................ 4 II.2…..Versuchsteil B ........................................................................................ 5 II.3…..Versuchsteil C ........................................................................................ 8 III. Ergebnisse ........................................................................................................ 9 III.1…..Versuchsteil A ....................................................................................... 9 III.2…..Versuchsteil B ..................................................................................... 13 III.3…..Versuchsteil C ..................................................................................... 20 IV. Diskussion ...................................................................................................... 27 IV.1…..Versuchsteil A ..................................................................................... 27 IV.2…..Versuchsteil B ..................................................................................... 29 IV.3…..Versuchsteil C ..................................................................................... 33 V. Literatur .............................................................................................................. 35 VI. Anhang ........................................................................................................... 36 I. Einleitung Natürlich vorkommende Aromaten finden sich vor allem in pflanzlichen Organismen, so macht Lignin, ein komplexes aromatisches Polymer, ca. 25% der terrestrischen Biomasse der Erde aus (1). Die Umsetzung dieser und weiterer aromatischen Komponenten ist ein wichtiger Bestandteil des Kohlenstoffkreislaufes und wird in erster Linie durch Mikroben vollbracht. Aromaten, bzw. genauer Benzol und seine Derivate, sind chemisch gesehen cyclische Systeme mit (4+2) -Elektronen. Der flache Benzolring wird durch -Bindungen gebildet. Über und unter dieser Ebene befindet sich das Aufenthaltsgebiet der -Elektronen. Der aromatische Ring ist energetisch eine sehr günstige Form der Bindung, zurückzuführen auf die hohe Resonanzenergie, die die Kohlenstoff-Kohlenstoff-Bindungen stabilisiert. Dieses Phänomen, auch als Resonanzstabilität bezeichnet, sorgt im Umkehrschluss dafür, dass die Spaltung des aromatischen Ringes hohe Energieinvestitionen verlangt und damit Mikroorganismen vor eine große Herausforderung stellt. Der mikrobielle Abbau von Aromaten erfolgt grob in zwei Schritten: Aktivierung des aromatischen Ringes und anschließende Spaltung. Anaerob wird der Aromat durch Reduktion am Ringsystem aktiviert und anschließend durch weitere Reduktion oder auf hydrolytischem Weg gespalten. Aerob erfolgt die Aktivierung des Aromaten durch Oxidation, wobei mittels Mono- oder Dioxygenasen entweder ein, oder beide Sauerstoffatome des O2-Moleküls in den Aromaten eingebaut werden. Die Ringspaltung geschieht unter enzymatischer Hilfe mittels Oxygenasen, die den Aromaten entweder in ortho1- , meta2- oder para3- Position spalten. Charakteristisch für beide Aromaten-Stoffwechselwege ist jedoch zum einen die sog. Konvergenz und zum anderen die substratabhängige Induzierbarkeit. Die Umsetzung verschiedenster Aromaten über denselben Stoffwechselweg unter Zuhilfenahme vorher erzeugter, zentraler Intermediate wird als Konvergenz bezeichnet. So resultiert der erste Schritt des Aromatenstoffwechsels, die Aktivierung des Aromaten, auch bezeichnet als 1 Bei der Spaltung in ortho-Position setzt das Enzym zwischen den beiden Hydroxylgruppen des aktivierten Aromaten an (auch intradiol-Spaltung). 2 Die Spaltung in meta-Position vollzieht sich neben einer der Hydroxylgruppen des aktivierten Aromaten (auch extradiol-Spaltung). 3 Findet die Spaltung des Aromaten zwischen einem C-Atom mit Carboxylrest und einem benachbarten C-Atom mit Hydroxylgruppe statt, spricht man von Spaltung in para-Position. 1|Seite peripherer Stoffwechselweg oder upper pathway, aerob in den zentralen Intermediaten Brenzcatechin (engl. Catechol), Protocatechuat oder Gentisinsäure, und anaerob im zentralen Intermediat Benzoyl-CoA. Im zweiten Schritt, der Spaltung des Aromaten, werden die erzeugten Intermediate im zentralen Stoffwechselweg, auch bezeichnet als lower pathway, weiter abgebaut. Produkte sind nicht-zyklische Verbindungen, die im letzten Schritt zu zentralen Stoffwechselprodukten abgebaut und in den Tricarbonsäure-Cyclus (TCC) eingeschleust werden. Im weiteren Verlauf wird das Augenmerk auf den aeroben ortho-Spaltungsweg gelegt, auch bekannt als β-Ketoadipat-Stoffwechselweg, benannt nach einem der Schlüsselintermediate: β-Ketoadipat. Exemplarisch wird dieser Stoffwechselweg Analysiert, wobei dies auf der Ebene der DNA bzw. der Gene, der Ebene der RNA und auf Protein-Ebene geschieht. Im ersten Teil der Analyse (Versuchsteil A) wird das Wachstum des Modelorganismus Acinetobacter baylyi Stamm BD413 auf unterschiedlichen aromatischen Substraten des β-Ketoadipatstoffwechsels überprüft und eine vergleichende Wachstumskinetik bestimmt. Hierzu wird die Zunahme der Zelldichte einer Kultur über den Zeitraum einiger Stunden beobachtet und gezielt zu bestimmten Zeitpunkten des Wachstums die Lebendzellzahl anhand ausplatierter Proben bestimmt. Diese Zeitpunkte umfassen die lag-Phase4, die exponentielle Phase5 und die stationäre Phase6 und können mit Hilfe einer Wachstumskurve (Auftragung der optischen Dichte im Bezug auf die Zeit) abgeschätzt werden. Mit Hilfe der Wachstumskurve können dann auch die Wachstumsrate (µ) und die Verdopplungszeit (td) der Kultur bestimmt werden. Parallel hierzu werden aus den einzelnen Phasen weitere Proben entnommen um die Transformationsfrequenz in Abhängigkeit der Wachstumsphase und der Kohlenstoff-Quelle zu bestimmen. Transformiert wird mit chromosomaler DNA einer A. baylyi Mutante, die eine Kanamycin-Resistenz in einem essentiellen Gen des natürlichen 4 In der lag-Phase geht das Wachstum zunächst nur langsam vonstatten, da die Mikroorganismen einen Moment brauchen, bis sie sich an die neuen Kulturbedingungen gewöhnt haben. Die Zelldichte nimmt hier nur langsam zu, erkennbar in einer Wachstumskurve anhand der sehr flachen Steigung. 5 Die exponentielle Phase hat ihren Namen aufgrund der exponentiellen Zunahme der Zelldichte, maximale Teilungsaktivität ist erreicht. Erkennbar ist diese Phase in einer Wachstumskurve daran, dass die Steigung der Kurve linear erfolgt. 6 Wenn das Substrat knapp, oder aber die Anreicherung toxischer Stoffwechselintermediate zu groß wird, bleibt die Zelldichte aufgrund gleicher Anzahl teilender und sterbender Zellen konstant. Erkennbar ist diese Phase in einer Wachstumskurve daran, dass die Steigung der Kurve stark abflacht und konstant bleibt. 2|Seite Transformationssystems besitzt. Die Fähigkeit freie DNA aufzunehmen und ins Genom einzubauen wird als natürliche Transformation, ein Mikroorganismus der diese Fähigkeit besitzt, als genetisch kompetent bezeichnet. Acinetobacter spp. kommen in der Natur im Wasser, in Böden und lebenden Organismen vor. Es sind Gram-negative, Oxidase-negative, nicht-bewegliche strikt aerobe Bakterien, die sich zumeist paarweise in Form von Kokken zusammenlagern (2). Die Eignung des Organismus Acinetobacter spp. für diesen Versuch rührt daher, dass dieser Genus ein hohes Maß an genetischer Kompetenz aufweist, und damit für Transformationsversuche eingesetzt werden kann. Die Kanamycin-Resistenz im Gen des Transformationssystems der Donor-Mutante hat zwei Vorteile: Erstens ist die Mutante selbst nicht mehr in der Lage DNA aus der Umgebung ins Genom einzubauen, und zweitens kann auf die eingebaute Resistenz selektiert werden 7, und so die Anzahl transformierter Klone (auch: Transformanden) genau bestimmt werden. Aus dem Quotienten aus der Anzahl der Transformanden und der Lebendzellzahl kann im letzten Schritt die Transformationsfrequenz bestimmt werden. Der zweite Teil der Analyse (Versuchsteil B) findet auf genetischer Ebene statt. Hierbei wird die Organisation der Gene des β-Ketoadipatstoffwechselweges unter Zuhilfenahme verschiedener Mutanten analysiert. Die einzelnen Organismen werden auf die Fähigkeit untersucht, auf unterschiedlichen Intermediaten des βKetoadipatstoffwechsels zu wachsen, um so vorhandene Mutationen einzugrenzen oder gar zu identifizieren. Anschließend wird zum einen mit den bereits identifizierten Mutanten, zum anderen mit Hilfe unterschiedlicher rekombinanter Plasmide ein Transformationsschnelltest durchgeführt. Im ersten Fall kommt es zu einem positiven Transformationserfolg, wenn sich die Mutationsorte der beiden Mutanten unterscheiden, die Fähigkeit ein bestimmtes Intermediat umzusetzen konnte dann zurückerlangt werden. Zu einem negativen Transformationserfolg kommt es, wenn beide Mutanten einen Defekt im selben Gen aufweisen, es findet keine Reparatur dieses Genes statt, die Fähigkeit zur Umsetzung eines bestimmten Intermediates kann nicht wiedererlangt werden. Die Transformation mit Hilfe der verschiedenen rekombinanten Plasmide erlaubt eine auf wenige hundert Basenpaare genaue 7 Auf die Kanamycin-Resistenz wird selektiert, indem die vermeintlich transformierten Organismen auf einen LB/Kanamycin-Agar ausplatiert werden. Die Klone, die die Resistenz eingebaut haben, zeigen Wachstum, solche Klone, die die angebotene DNA nicht in ihr Genom eingebaut haben, werden ausselektiert. 3|Seite Eingrenzung des Mutationsortes. Mutanten mit weiterhin unbekanntem Mutationsort werden im letzten Schritt durch Amplifikation des defekten Gens, anschließende gelelektrophoretische Auftrennung und Vergleich der Fragmentgröße mit dem intakten Gen des Wildtyps, identifiziert. Der dritte und letzte Teil der Analyse (Versuchsteil C) untersucht die Regulation des β-Ketoadipatstoffwechsels und findet auf Protein-Ebene statt. Hierbei soll ermittelt werden, ob die Induktion bzw. Regulation des β-Ketoadipatstoffwechselweges auf Ebene der Enzymaktivität oder auf Ebene der Transkription stattfindet. Untersucht wird eines der Schlüsselenzyme des β-Ketoadipatstoffwechselweges, die Protocatechuat-3,4-Dioxygenase, die Protocatechuat zu β-Carboxy-cis-cis-Muconat umsetzt. Die Enzymaktivität kann dabei auf zwei unterschiedlichen Ebenen betrachtetet werden: Zum einen im Bezug auf die Menge an umgesetztem Substrat, zum anderen, auf die Menge an entstandenem Produkt in einer bestimmten Zeitspanne. Der β-Ketoadipatstoffwechsel wird hierbei exemplarisch analysiert, die beschriebenen Methoden können jedoch auf jeden beliebigen Stoffwechselweg angewendet werden. II. Material und Methoden Die Versuche wurden vorwiegend wie im Praktikumsskript angegeben durchgeführt, bis auf einige Änderungen, welche im Folgenden nochmals aufgeführt werden: II.1 Versuchsteil A Hier wird bei der Ermittlung der Transformandenzahl nach dem Inkubieren der Probe mit der chromosomalen DNA einer kanamycinresistenten Mutante, folgende Verdünnungsstufen auf LB/Kanamycin-Agar ausplattiert: lag-Phase: 100, 10-1 und 10-2 4|Seite exp. und stat. Phase: 10-2, 10-3 und 10-4 Eine weitere Abwandlung zum Skript, ist dass wir bezüglich der Probenentnahme, für die Ermittlung der Teilungsrate und Generationszeit g, während der exponentiellen Wachstumsphase statt zwei, drei Proben entnommen, und diese jeweils auf den verschiedenen Kohlenstoffquellen ausplattiert haben. Hier wurden die Proben mit einer Verdünnung von 10-7, 10-8 und 10-9 auf den Platten ausgestrichen. II.2 Versuchsteil B Für die „Mutantenstudien“ wurden nur sechs Mutanten (Stamm 25, 28, 29, 31, 32 und 275) untersucht, welche verschiedene Mutationen im oberen Teil des pHydroxybenzoatabbauweges vorweisen. Hier wurde Acinetobacter baylyi -Stamm 30 nicht in Bezug auf bevorzugte Kohlenstoffquellen, sowie Komplementationsanalysen, als auch bezüglich der Komplementation mit rekombinanten Plasmide untersucht. 2. Komplentationsanalysen der Mutanten Bei der Komplementationsanalyse der Mutanten wurde bei der Zusammenstellung des TE-Puffers, 0,6g Tris und 0,37g EDTA abgewogen und auf 100ml mit Wasser aufgefüllt Isolierung der chromosomalen DNA Die Konzentrationsbestimmung der isolierten chromosomalen DNA wird an einem Gerät namens Nano-Drop durchgeführt. Hier ermittelt ein Computerprogramm die Reinheit der isolierten DNA, was für die Weiterverarbeitung von Bedeutung ist. Dabei ist das Verhältnis zwischen der Extinktion bei 260nm (Spektrum der DNA) und der Extinktion bei 280nm (Spektrum Proteine) entscheidend. Entspricht die Division dieser beiden Werte ein Ergebnis von 1,5 bis 1,8, so ist die isolierte Probe relativ 5|Seite gereinigt von den Proteinen, wobei ein vollständiges Herauslösen der Proteine nahezu ausgeschlossen ist. Der Komplementationstest (Transformationsschnelltest) der Mutanten 28 und 275 wurde auf drei Kohlenstoffquellen durchgeführt: Protocatechuat, Quinat und auf pHydroxybenzoat. Isolierung der Plasmid-DNA Für die Isolierung der Plasmid-DNA wurde der Peq-Lab Plasmid Kit verwendet. Die Vorgehensweise ist wie folgt: aus der 6ml Übernachtkultur eines rekombinanten E.coli-Stammes, werden zweimal 2ml in Eppendorfgefäße überführt und für zehn Minuten bei 8000Upm zentrifugiert. Daraufhin wird der Überstand verworfen und das Pellet in 125µl von Puffer A1 (RNase und EDTA) resuspendiert. Die gelösten Pellets werden in ein Eppendorfgefäß überführt und es wird 250µl von Puffer A2 (SDS, NaOH alkalische Zelllyse) hinzu gegeben und acht Mal invertiert. Die Proben werden für zwei Minuten bei Raumtemperatur inkubiert und daraufhin mit 350µl von Puffer A3 (Kaliumacetat (sauer) Neutralisation und Renaturierung der Plasmide (Proteine, RNA und chromosomale DNA renaturieren nicht und fallen aus) Überstand enthält hauptsächlich Plasmide) ein weiteres Mal wird acht Mal invertiert. Es wird nochmals für zehn Minuten bei 11000Upm zentrifugiert. In dieser Zeit werden die Säulen auf 2ml Eppendorfgefäße gesetzt und der Überstand (enthält Plasmid-DNA) wird auf diese Säule gegeben. Das Pellet mit enthaltenen Zelltrümmern und Proteinen wird verworfen. Die Säulen werden mit den Eppendorfgefäßen für zwei Minuten bei 11000Upm in die Zentrifuge gegeben, wobei nun die DNA an die Säule bindet. Danach wird der Durchfluss verworfen und es wird 500µl von Puffer A4 (EtOH zum Waschen der DNA) hinzu gegeben. Die Proben werden ein weiteres Mal für zwei Minuten zentrifugiert (11000Upm), der Durchfluss wird verworfen und es wird 750µl von Puffer A5 (EtOH zum Waschen der DNA) auf die Säule gegeben. Daraufhin wird wieder für zwei Minuten bei 11000Upm zentrifugiert, und der Durchfluss wird wiederum verworfen, die Säule wird zum Trocknen nochmals für zwei Minuten in die Zentrifuge gegeben. Die Säule wird danach auf ein steriles 2ml-Eppendorfgefäß gesetzt und mit 50µl autoklaviertem Wasser durchspült. Es wird für eine weitere Minute zentrifugiert. 6|Seite Die Plasmid-DNA befindet sich nun im Durchfluss und wird am Nano-Drop hinsichtlich der DNA-Konzentration untersucht. Die nun isolierte Plasmid-DNA wird elektrophoretisch in einem Agarosegel aufgetrennt. Dies dient somit als Kontrolle, dass es sich bei den Plasmiden pZR2, pZR210, pZR211, pZR2105, pZR2106, pZR2111 und pZR2113 auch wirklich um unterschiedliche lange DNA-Fragmente handelt. Der Transformationsschnelltest mit der Plasmid-DNA wurde nur noch auf den Protocatechuat-Platten durchgeführt. 3. Isolierung und Analyse eines Mutationsortes mittels PCR In diesem Versuchsabschnitt wurde das PCR-Programm geändert: 95°C für 2 min - Denaturierung der Matrizen-DNA 95°C für 1 min - Denaturierung der Matrizen-DNA 45°C für 1 min - Anlagerung der Starter-Oligonukleotide 35 Wiederholungen 72°C für 2 min - Elongation__________________ 72°C für 10 min - Renaturierung der DNA 7|Seite - Denaturierung - „Primer Annealing“ der MatrizenDNA Die Starteroligonukleotide lagern sich an DNA an 95°C 95°C 45°C 2 min - Elongation 72°C 72°C 1 min für 1 min 2 min 10 min Dieser Vorgang wird 35mal wiederholt II.3 Versuchsteil C 1. Induktion der Protocatechuat-3,4-Dioxygenase-Aktivität Proteinbestimmung nach Bradford Hier wurde bei der Messung der Absorption bei 595nm das Photometer auf Luft geeicht. Ermittlung der spez. Aktivität der Protocatechuat-3,4-Dioxygenase Bei diesem Versuchsschritt wurden die Messungen am Schreiber mit je einmal 5µl, 10µl, 20µl und 30µl durchgeführt. Zusätzlich kamen 1ml Tris-Puffer, als auch jeweils 25µl der 10mM Protocatechuat-Lösung hinzu. β-Galactosidase-Test Hier wurde die Aktivität nach Miller mit folgender Formel berechnet: 8|Seite Aktivität nach Miller = (1000 ∙ E420) / (OD600 ∙ 0,2 ∙ t) E420 = Extinktion nach Stoppen der Reaktion OD600 = Optische Dichte nach Inkubation (60min bei 30°C) t = Reaktionsdauer: Start => Stopp der Reaktion III. Ergebnisse III.1 Versuchsteil A Das Wachstum von Acinetobacter baylyi BD413 auf unterschiedlichen KohlenstoffQuellen (hier: Quinat) wurde über den Zeitraum mehrerer Stunden verfolgt um mit Hilfe der ermittelten Wachstumskurve (Abb. 1) eine vergleichende Wachstumskinetik zu erstellen. [OD600] 1 0,1 50 100 150 200 250 300 350 400 450 Zeit [min] Abbildung 1: Wachstumskurve für Acinetobacter baylyi BD413 für die Kohlenstoff-Quelle Quinat 9|Seite Mit Hilfe der Wachstumskurve (Abb. 1) können nun die verschiedenen Wachstumsparameter (Wachstumsrate µ und Verdopplungszeit td) berechnet werden. Unter Zuhilfenahme einer Ausgleichsgeraden, werden zwei Punkte innerhalb des exponentiellen Wachstums, x0 und xt, markiert. Der Punkt x0 liegt am Beginn der exponentiellen Phase und gibt die optische Dichte zu einem bestimmten Zeitpunkt t 0 an. Xt, auf der anderen Seite, liegt am Ende der exponentiellen Phase, und beschreibt die optische Dichte zu einem bestimmten (späteren) Zeitpunkt t. Die Werte x0, xt, t0 und t sind im Graphen ersichtlich, mit ihnen kann die Wachstumsrate µ berechnet werden: (A) Für Acinetobacter baylyi BD413 mit der Kohlenstoff-Quelle Quinat ergab sich folgende Wachstumsrate : Dies entspricht einer Zunahme der optischen Dichte um 0,0113 pro Minute. Die Verdopplungzeit td wird wie folgt berechnet: (B) Für Acinetobacter baylyi BD413 mit der Kohlenstoff-Quelle Quinat ergab sich folgende Verdopplungszeit : 10 | S e i t e Die Zelldichte hat sich damit innerhalb etwa einer Stunde verdoppelt. Für die Berechnung der Teilungsrate und der Generationszeit g wurde jeweils eine Proben zum Zeitpunkt t0 und t entnommen, in einer Verdünnungsreihe verdünnt und zur Bestimmung der Lebendzellzahl auf LB-Medium ausplattiert. Die Zellzahl zum Zeitpunkt t0 entspricht dabei N0, die zum Zeitpunkt t entspricht N. Da die Zellzahl für Quinat nicht bestimmbar war, wurden die folgenden Parameter anhand der Ergebnisse für das LB-Medium exemplarisch berechnet. Die Teilungsrate wird mit Gleichung (C) ermittelt: (C) Die Generationszeit g ergibt sich aus dem reziproken Wert der Teilungsrate: (D) 11 | S e i t e Die Wachstumsparameter für Acinetobacter baylyi BD413 hinsichtlich der verschiedenen Kohlenstoff-Quellen werden zusammengetragen und miteinander verglichen (Tab. 1). Tabelle 1: Vergleich der Wachstumsraten, Verdopplungszeiten, Generationszeiten und Teilungsraten hinsichtlich unterschiedlicher Kohlenstoff-Quellen C-Quelle Protocatechuat Quinat p-Hb Succinat LB Wachstumsrate μ 1,8 ∙ 10-2 7,9 ∙ 10-3 9,2 ∙ 10-3 1,8 ∙ 10-2 1,47 ∙ 10-2 75,3 39 47,1 --- 27,6 48,5 --- 0,036 0,0206 1,1 ∙ 10-2 [min-1] Verdopplungszeit td 38,5 [min] Generationszeit g 61,5 --- [min] Teilungsrate [min-1] 87 ----- --- ----- Parallel zur Ermittlung der Wachstumskinetik und der Wachstumsparameter, wurde die Transformationsfrequenz in Abhängigkeit der Kohlenstoff-Quellen, als auch der Wachstumsphase bestimmt. Hierzu wurden während der lag-Phase, zum Zeitpunkt t0 in der exponentiellen Phase und in der stationären Phase Proben entnommen und mit chromosomaler DNA einer Kanamycin-resistenten Mutante transformiert. Durch anschließende Selektion auf LB/Kanamycin-Platten kann die Anzahl der Transformanden und damit die Transformationsfrequenz8 genau berechnet werden. Die nachfolgende Tabelle (Tab. 2) zeigt die erhaltenen Ergebnisse, GZZ ist beschreibt die Gesamt- oder auch Lebendzellzahl, TZZ die Transformandenzellzahl und T.Frequenz die Transformationsfrequenz bezogen auf die jeweilige KohlenstoffQuelle: 8 Die Transformationsfrequenz berechnet sich als Quotient aus Transformanden und Lebendzellzahl. 12 | S e i t e Tabelle 2: Transformationsfrequenz in Abhängigkeit von der Kohlenstoff-Quelle und der Wachstumsphase C-Quelle Proto- Quinat Quinat p-Hb Succinat LB catechaut GZZ [ml-1] 4,2 ∙ 107 4,13 ∙ 109 4,17 ∙ 109 8,4 ∙ 109 3,18∙ 109 1,21 ∙ 1010 TZZ [ml-1] --- 1 ∙ 104 --- --- 3,33∙ 102 7,66 ∙ 109 T.Frequenz --- 2,42 ∙ 10-6 --- --- 1,05∙10-7 6,33 ∙ 10-7 GZZ [ml-1] 5,7 ∙ 109 1,37 ∙ 109 3,8 ∙ 108 4,16∙ 106 3,89∙ 108 5,38 ∙ 108 TZZ [ml-1] 6 1 ∙ 102 --- 26 10 6,93 ∙ 102 T.Frequenz 1,17 ∙ 10-9 7,3 ∙ 10-8 --- 6,25 ∙10-6 2,5∙10-8 1,29 ∙ 10-6 GZZ [ml-1] 2 ∙ 107 4,15 ∙ 109 3 ∙ 108 1,33∙ 1010 5,56∙ 109 4,58 ∙ 109 TZZ [ml-1] 2 ∙ 102 --- 2 ∙ 103 2,12 ∙ 103 1,23∙ 104 6,88 ∙ 104 T.Frequenz 1 ∙ 10-5 --- 6,67 ∙10-6 1,59 ∙ 10-7 2,21∙10-6 1,5 ∙ 10-5 stat. Phase lag-Phase exp. Phase Die Gesamtzellzahl setzt sich aus den transformierten und untransformierten Zellen einer Probe zusammen. Grundsätzlich ist hierbei zu erkennen, dass die Gesamtzellzahl größer ist als die Transformandenzellzahl. Im Allgemeinen ist zu erkennen, dass die Transformationsfrequenz in der exponentiellen Phase am höchsten ist (Tab. 2). Hierbei liegen die Werte im Mittel bei stationären, in der lag- und in der in der exponentiellen Phase. Dies bedeutet für die stationäre und die lag-Phase, dass ungefähr eine von 1 Million Zellen und in der exponentiellen Phase 6 von 1 Million Zellen transformiert wurden. Im Bezug auf die unterschiedlichen Kohlenstoff-Quellen liegen die Transformationsfrequenzen im Mittel bei Quinat, für p-Hydroxybenzoat (pHb), für Protocatechuat, für für Succinat und für LB-Medium. Hierbei sind keine drastischen Diskrepanzen zu erkennen. III.2 Versuchsteil B Der Test bezüglich des Wachstums auf unterschiedlichen Kohlenstoff-Quellen der einzelnen Mutanten zeigte folgendes Ergebnis: 13 | S e i t e Tabelle 3: Wachstum der einzelnen Mutanten auf verschiedenen C-Quellen C-Quellen Quinat Protocatechuat p-Hydroxy- Mutante Succinat benzoat WT + + + + 25 + + + + 28 - + + + 29 - - - + 31 - - - + 32 - - - + 275 + + - + Tabelle 3 fasst die Fähigkeit der einzelnen Mutanten zusammen auf unterschiedlichen Kohlenstoff-Quellen zu wachsen. Der Wildtyp sowie Mutante 25 wachsen auf allen angebotenen Kohlenstoffquellen. Mutante 28 zeigte kein Wachstum auf Quinat, war jedoch in der Lage auf Protocatechuat, p-Hydroxybenzoat als auch auf Succinat zu wachsen. Die Mutanten 29, 31, und 32 waren lediglich imstande auf Succinat zu wachsen. Mutante 275 zeigte die Fähigkeit auf Quinat, Protocatechuat und Succinat zu wachsen, allerdings fehlte ihm diese im Bezug auf pHydroxybenzoat. Da von einer Mutation eines der Gene ausgegangen wird, können sowohl Mutante 28 als auch Mutante 275 keinen Defekt im pca-Operon aufweisen und infolge dessen für weitere Komplementationsanalysen eingesetzt werden. Die Ergebnisse dieser Komplementationsanalysen sehen wie folgt aus: Tabelle 4: Komplementationsanalyse mittels chromosomaler DNA von Mutante 28 bzw. 275 Donor Rezi- Mutante 28 Mutante 275 P Q H P Q H 25 + + + + + + 29 + + + + + + pient 14 | S e i t e 31 + + + + + + 32 + + + + + + Tabelle 4 zeigt, dass nach erfolgter Komplementation die Mutanten 25, 29, 31 und 32 in der Lage waren auf Protocatechuat (P), Quinat (Q) sowie p-Hydroxybenzoat (H) zu wachsen. Damit hat eine Transformation der DNA eines vorher defekten Genes mit DNA desselben, jedoch intakten Genes stattgefunden. Zur genauen Identifizierung des Mutationsortes wurden die Mutanten 29, 31 und 32 mit verschiedenen rekombinanten Plasmiden inkubiert. Diese Plasmide tragen unterschiedliche Fragmente des pca-Operons, sodass eine genaue Zuordnung des Mutationsortes möglich ist. Die Plasmide trugen folgende DNA-Fragmente (Abb. 2): Abbildung 2: DNA- bzw. Gen-Fragmente der rekombinanten Plasmide (3) Trennt man die Plasmide gelelektrophoretisch auf, werden die Längenunterschiede verdeutlicht (Abb. 3): 15 | S e i t e 1. 2. 3. 4. 5. 6. 7. 8. Abbildung 3: Gelelektrophoretische Auftrennung der Plasmide. Die Reihenfolge der Auftrennung lautet wie folgt: 1. Marker, 2. pZR2, 3. pZR210, 4. pZR211, 5. pZR2105, 6. pZR2106, 7. pZR2111 und 8. pZR2113 (Abb. 3). Im Anschluss an die gelelektrophoretische Auftrennung wurde die Konzentration der Plasmid-DNA und ihre Reinheit mit Hilfe eines Gerätes namens Nano-Drop überpüft (Tab. 5). Tabelle 5: DNA-Konzentration und Reinheit der Plasmide Plasmid Konzentration der DNA Reinheit [OD260/OD280] [ng/µl] pZR2 47,4 2,06 pZR210 57,7 1,9 pZR211 83,8 -- pZR2105 29,9 1,9 pZR2106 138,5 pZR2111 70,3 1,8 pZR2113 27,3 1,9 Die Beziehung OD260/OD280 gibt das Verhältnis von DNA (Absorption bei 260nm) zum noch vorhandenen Proteinanteil (Absorption bei 280nm) an. Liegt dieses 16 | S e i t e zwischen 1,5-1,8 ergibt sich daraus eine hohe Reinheit, da wenig unerwünschtes Protein in der extrahierten DNA-Probe vorhanden ist. Die Konzentration der DNA wird benötigt, um etwa immer die gleiche Menge in den Komplementationsversuchen einzusetzen. Wie in Abbildung 2 erkennbar, trägt das Plasmid pZR2 den größten Ausschnitt des pca-Operons der hier vorhandenen Plasmide; die Gene pcaC, pcaH und pcaG sind intakt. Alle weiteren Plasmide tragen nur Fragmente dieser Gene, wobei die Plasmide pRZ211, pZR2102, pZR2103, pZR2105, pZR2106, pZR2107, pZR2108, pZR2109, pZR2111 und pZR2113 unterschiedlich lang sind. Anhand dieser Abstufungen kann die Mutation auf wenige hundert Basenpaare genau eingegrenzt werden. In diesem Versuch wurden nur die Plasmide pZR2, pZR210, pZR211, pZR2105, pZR2106, pZR2111 und pZR2113 verwendet. Tabelle 6: Eingrenzung der Mutation mit Hilfe unterschiedlich langer rekombinanter Plasmide Mutante Plasmid 29 31 32 pZR2 + + - pZR210 - - - pZR211 + - - pZR2105 + - - pZR2106 + - + pZR2111 + - - pZR2113 - - - Mit Hilfe der Ergebnisse in Tabelle 6 kann nun der Ort der jeweiligen Mutation genau ermittelt werden. Der Vergleich dieser Ergebnisse mit den in Abbildung 2 dargestellten rekombinanten Plasmiden bzw. den darauf liegenden Genen/GenFragmenten, erlaubt die gezielte Zuordnung der Mutation zu einem bestimmten Gen. Mutante 29 wurde komplementiert durch das Plasmid pZR2, sowie die Plasmide pZR211, pZR2105, pZR2106 und pZR2111. Die Plasmide pZR210 und pZR2113 konnten den Defekt nicht ausgleichen. Mutante 31 wurde ausschließlich durch das Plasmid pZR2 komplementiert, keines der weiteren verwendeten Plasmide brachte einen Ausgleich im defekten Gen. Mutante 32 zeigte bereits vor der aufgetragenen 17 | S e i t e Plasmid DNA, also vor erfolgter Komplementation, Wachstum. Dieses Ergebnis kann nicht als Komplementation bezeichnet werden, kein vorhandener Defekt wurde ausgeglichen, da die Mutante scheinbar intakte Gene aufwies. Eindeutig war die Komplementation nur durch das Plasmid pZR2106 zu beurteilen. (Tab. 6) Die Art des Gendefekts von Mutante 25 konnte nur durch gelelektrophoretische Auftrennung und Vergleich mit dem Wildtyp ermittelt werden (Abb. 4). Legende Abbildung 4 1. Bande Kontrolle (forward Primer) 2. Bande Marker 3. Bande Wildtyp (4) 4. Bande Wildtyp (B) 5. Bande Wildtyp (4) 6. Bande Mutante 25 (5) 7. Bande Mutante 25 (B) 8. Bande Kontrolle (reverse Primer) 9. Bande Mutante 25 (2) Abbildung 4: Gelelektrophoretische Auftrennung nach PCR: Vergleich von Mutante 25 mit dem Wildtyp Aufgrund vorangegangener Tests konnte das Regulatorgen pcaU als defekt ermittelt werden. Um welche Art von Defekt es sich handelt gibt die gelelektrophoretische Auftrennung Auskunft (Abb. 4). Der Vergleich des Regulatorgens pcaU von Mutante 25 mit dem Wiltyp zeigt, dass das DNA-Fragment der Mutante kürzer9 ist, als das des Wildtyps. Durch Ausmessen der Wanderungsstrecke des Markers [cm] (Abb. 4) und graphische Auftragung im Verhältnis zur Größe der einzelnen DNA-Fragmente [bp] kann die genaue Größe des DNA-Fragmentes von Mutante 25 bzw. des DNAFragementes des Wildtyps bestimmt werden. 9 Bei der Auftrennung durch Gelelektrophorese laufen die DNA-Fragmente vom negativen Pol (oben) zum positiven Pol (unten) und bilden durch anschließende Färbung mit Hilfe von Ethidiumbromid sichtbare Banden. Lange Fragmente wandern dabei weniger weit als kurze Fragmente, da erste eher in den Poren des Agarosegels stecken bleiben als letztere. 18 | S e i t e Abbildung 5: 1kb DNA-Ladder, Gene Ruler, Fermentas Länge des Fragments [bp] 10 1 0 1 2 3 4 5 6 7 8 Wanderungsstrecke [cm] Abbildung 6: Eichkurve des Markers Vergleich der DNA-Fragmente des pcaU-Gens des Wildtyps mit ca. 750bp und von Mutante 25 mit ca. 280bp zeigen auch hier, dass das defekte Gen von Mutante 25 ca. 470bp kürzer ist, als das Wiltyp-Gen (Abb. 6 und Anhang). 19 | S e i t e III.3 Versuchsteil C In diesem Versuchsteil soll die Regulation des β-Ketoadipatstoffwechsels analysiert werden. Geklärt werden soll die Fragestellung, ob die Regulation auf Transkriptionsoder Protein-Ebene stattfindet. Der erste Teil der Analyse umfasst die Bestimmung der Enzymaktivität anhand verschiedener Substratkonzentrationen. Mit Hilfe einer Eichgerade konnte durch die Proteinbestimmung nach Bradford die Proteinkonzentration des Rohextraktes bestimmt werden. 10 Absorption 8 6 4 2 0 0,0 0,1 0,2 0,3 0,4 0,5 0,6 Menge Protein [µg] Abbildung 7: Eichgerade für die Proteinbestimmung nach Bradford Mittels dieser Eichgerade (siehe Abb. 7 und Anhang) konnte nun die Proteinmenge des Rohextraktes anhand der Absorption bei 595nm, abgelesen werden. Die entsprechenden Werte sind in Tabelle 7 vorzufinden. 20 | S e i t e Tabelle 7: Absorption, Proteinmenge und Proteinkonzentration verschiedener Rohextraktmengen Rohextraktmenge 2µl 5µl 10µl Absorption (595nm) 0,739 0,836 0,945 0,718 0,835 0,919 Mittelwert (Absorption) 0,729 0,836 0,932 Proteinmenge [µg] 1,2 2,95 5,1 Proteinkonzentration [µg/µl] 6 5,9 5,1 Mittelwert (Proteinkonz.) 5,67 [µgl/µl] In Tabelle 7 ist zu erkennen, dass mit steigender Rohextraktmenge, die Absorption ebenfalls zunimmt (jeweils um etwa den Wert 0,1). Ein ähnliches Phänomen ist im Bezug auf die Proteinmenge zu beobachten, diese verdoppelt sich bei steigender Rohextraktmenge. Die Proteinkonzentration bleibt nahezu konstant. Zur Berechnung der spezifischen Aktivität der Protocatechuat-3,4-Dioxigenase wird der Extinktionskoeffizient von Protocatechuat benötigt. Dieser wurde anhand der Absorption verschiedener Protocatechuat-Konzentrationen bei 290nm ermittelt. Hierbei wird die gemessene Absorption (E) in folgende Gleichung eingesetzt: c entspricht der jeweiligen Protocatechuat-Konzentration und d ist die Schichtdicke der Küvette (in diesem Fall 1cm). Dabei ergibt sich nun für die Berechnung des Extinktionkoeffizienten, bei einer Protocatechuat-Konzentration von 0,1mM: Berechnet für die einzelnen Protocatechuat-Konzentrationen lautet das Ergebnis wie folgt: Tabelle 8: Bestimmung des Extinktionskoeffizienten von Protocatechuat Konz. von Protocatechuat Absorption (Extinktionskoeffiezient) 21 | S e i t e [290nm] 0,1mM 0,414 4,14 mM-1cm-1 0,08mM 0,275 3,45 mM-1cm-1 0,06mM 0,233 3,88 mM-1cm-1 0,04mM 0,157 3,93 mM-1cm-1 0,02mM 0,085 4,25 mM-1cm-1 Die Absorption der Proben nimmt mit geringerer Protocatechuat-Konzentration stetig ab (Tab. 8). Der Mittelwert des Extinktionskoeffiezienten von Protocatechuat, ergibt 3,93 mM-1cm-1 und wird für die spätere Berechnung der spezifischen Enzymaktivität verwendet. Im weiteren Verlauf wurde die Extrakproportionalität überprüft. Dieses Phänomen beschreibt das Verhältnis zwischen Proteinmenge und Enzymaktivität. Es besagt, dass bei Verdopplung der Proteinmenge eine doppelt so hohe Enzymaktivität resultiert. Nur wenn diese vorherrscht, ist ein Vergleich der spezifischen Aktivitäten in Abhängigkeit des Substrates, möglich. 0,22 0,20 Enzymaktivität [dE/min] 0,18 0,16 0,14 0,12 0,10 0,08 0,06 0,04 5 10 15 20 25 30 Rohextraktmenge [µl] Abbildung 8: Extraktproportionalität zwischen Extinktionsänderung pro Zeiteinheit und Rohextraktmenge 22 | S e i t e Wie in Abb. 8 erkennbar ergibt sich ein nahezu linearer Zusammenhang zwischen der Änderung der Extinktion pro Zeiteinheit und der Proteinmenge. Da anhand des Graphen (Abb. 8) die Extraktproportionalität bestätigt wurde, kann im nächsten Schritt die spezifische Aktivität der Protocatechuat-3,4.Dioxygenase ermittelt werden. Die spezifische Enzymaktivität wird im Folgenden an einem Beispiel berechnet: Im Anhang ist das Schreiber-Ergebnis vorzufinden. Anhand dieser Geraden kann nun die Extinktionsänderung pro Zeiteinheit (E) ermittelt werden. Bei 5µl Rohextrakt und einem Papiervorschub von 0,1mm/sec, ergibt sich eine Extinktionsänderung von E= 0,716 – 0,475 = 0,241. Diese Extinktionsänderung wird nun in Bezug zur Zeit gesetzt. Der zeitliche Verlauf kann anhand des Papiervorschubes ermittelt werden, da wie bereits schon oben erwähnt, der Papiervorschub 0,1mm/sec beträgt. Die Extinktionsänderung findet in einem Zeitraum von 4,67 Minuten statt. Wird nun die Extinktionsänderung (0,241) durch den Zeitraum (4,67min) dividiert, erhält man die Extinktionsänderung pro Zeiteinheit (E = 0,052min-1). Dieser Wert kann nun in die folgende Berechnung der spezifischen Enzymaktivität eingehen: V = Volumen des gesamten Messansatzes mit variierender Rohextraktmenge (1030µl) = Extinktionskoeffizient (aus Tab. 8: 3,93 mM-1cm-1) d = Schichtdicke der Küvette (1cm) v = Volumen des jeweiligen Rohextraktes (hier 5µl) P = Proteinkonzentration (siehe Tab. 7 5,67 µg/µl) 23 | S e i t e Eingesetzt ergeben diese Werte für die spezifische Aktivität: Um die Frage der Einheit der spezifischen Aktivität zu klären, werden folglich nur die Einheiten der einzelnen Komponenten aufgezeigt: Da dasselbe ist wie ist folgender Schritt möglich Unter Berücksichtigung aller Rohextraktmengen, ergibt sich ein Mittelwert für die spezifische Enzymaktivität von . 24 | S e i t e Zusammenfassend sehen die Enzymaktivitäten unter bestimmten KohlenstoffQuellen wie folgt aus: Abbildung 9: Enzymaktivitäten bei verschiedenen C-Quellen Die Enzymaktivität hinsichtlich der verschiedenen Kohlenstoff-Quellen zeigt, dass die höchste Aktivität bei Protocatechuat (Mittelwert: 0,37µmol/min∙mg Protein), die zweit höchste bei p-Hydroxybenzoat (0,33 µmol/min∙mg Protein) und die niedrigste bei Quinat (Mittelwert: 0,31 µmol/min∙mg Protein) zu beobachten ist (Abb. 9). Der zweite Teil der Analyse beschäftigt sich mit der Enzymaktivität hinsichtlich der resultierenden Produktmenge. Gemessen wurde die optische Dichte vor und nach Zugabe des Substrates, wobei letzteres die optische Dichte des Produktes beschreibt. Anhand dieser Werte, wurde die Aktivität nach Miller mit folgender Formel bestimmt: 25 | S e i t e E420 = Messung nach Stoppen der Reaktion bei 420nm OD600 = Messung nach Inkubation von 60min bei 30°C vor Zugabe des Substrates t = Zeit [min] vom Starten der Reaktion bis zum Stoppen Exemplarisch sieht diese Berechnung für eine Substratkonzentration von 10µM wie folgt aus: OD600 = 0,845 E420 = 0,039 Zeit vom Starten der Reaktion bis zum Stoppen = 3 Minuten 10 Sekunden Zusammengetragen ergeben sich folgende Werte: Tabelle 9: Aktivität nach Miller Protocatechuat- OD600 E420 Konzentration Aktivität nach Miller [U/OD600] 0 0,821 0,041 69,36 0,01µM 0,929 0,05 74,75 0,1µM 0,886 0,046 72,11 1µM 0,927 0,041 61,43 10µM 0,845 0,039 61,1 100µM 0,879 0,151 238,59 1mM 0,866 0,485 777,84 10mM 0,870 0,730 1165,39 26 | S e i t e Tabelle 9 zeigt, dass die Aktivität nach Miller bei einer Konzentration von 100µM sprunghaft ansteigt. Dies lässt sich anhand des folgenden Diagramms (Abb. 10) verdeutlichen: Abbildung 10: Aktivität nach Miller IV. Diskussion IV.1 Versuchsteil A Die Wachstumskurve von Acinetobacter baylyi BD413 (Abb. 1) zeigt deutlich die einzelnen Wachstumsphasen der Kultur. Die ersten 45 Minuten stellen die lag-Phase dar, der Anstieg der optischen Dichte vollzieht sich zunächst nur langsam. Von Minute 45 an, in einem Zeitraum von ca. drei Stunden, befand sich die Kultur in der exponentiellen Phase. Hierbei herrscht maximale Teilungsaktivität, erkennbar an einer nahezu linearen Beziehung der Zeit im Bezug auf die Zunahme der Zelldichte. 27 | S e i t e Nach insgesamt vier Stunden stieg die optische Dichte (und damit die Zelldichte der Probe) nur noch unwesentlich an, die Kultur befand sich in der stationären Phase. Hier hält sich die Anzahl teilender und sterbender Zellen die Waage. In der letzten Phase werden sowohl Nährstoffe als auch der zur Verfügung stehende Raum knapp, die Zellen beginnen stärker abzusterben, als dass sie in der Lage wären sich zu teilen. Diese Phase, auch Absterbephase genannt, ist in den letzten Messungen am Absinken der optischen Dichte erkennbar. Die mit Hilfe der Wachstumskurve berechneten Wachstumsparameter Wachstumsrate Literatur angegeben mit und Verdopplungszeit und damit ist sind in der . Diese Unterschiede in der Wachstumsrate bzw. der Verdopplungszeit können auf Unzulänglichkeiten bei der Inkubation der Zellen zurückzuführen sein. So kann die verminderte Intensität beim Schütteln der Kolben zu einer geringen Sauerstoffversorgung der Kulturen führen. Da Sauerstoff jedoch essentiell für die Aktivierung und Spaltung der Aromaten im aeroben Aromatenabbau ist, resultiert der Mangel an Sauerstoff in geringem Wachstum. Aufgrund massiver Kontamination der verwendeten LB-Agarplatten konnten Lebendzellzahl und Transformandenzellzahl nur unzureichend bestimmt werden (Tab. 1). Diese Unvollständigkeiten nehmen ebenfalls Einfluss auf die Berechnung der Generationszeit g sowie der Teilungsrate ν. Exemplarisch wurden diese Berechnung anhand der ermittelten Werte für das LB-Medium bestimmt. Die für das LB-Medium bestimmte Verdopplungszeit korreliert mit der exemplarisch berechneten Generationszeit von 48,54min (Tab. 1). Dies ist logisch, da eine neue Generation aus der Teilung vorhandener Zellen hervorgeht. Im Vergleich der Lebendzellzahl mit der Transformandenzellzahl ist ersichtlich, dass erstere grundsätzlich höher ist als letztere (Tab. 2). Dies rührt daher, dass nicht alle Zellen einer Probe transformiert werden. Da zur Bestimmung der Transformandenzellzahl jedoch auf Kanamycin-Resistenz selektiert wird, zeigen die LB/Kanamycin-Platten ausschließlich die transformierten Zellen an. Die Gegenüberstellung der Transformationsfrequenzen der verschiedenen Wachstumsphasen zeigt, dass die Transformationsfrequenz während der exponentiellen Phase am höchsten ist (Tab. 2). Literaturwerte verneinen dieses 28 | S e i t e Ergebnis jedoch, da die Transformierbarkeit von Acinetobacter spp. während der lagPhase am stärksten ist. Die für die lag-Phase ermittelten Werte weichen jedoch stark von den Referenzwerten ab (Tab. 2) und setzen sie in einen ähnlichen Rahmen wie die Werte für die stationäre Phase. Die für die exponentielle Phase ermittelten hohen Transformationfrequenzen könnten durch eine verfrühte Probeentnahme, nämlich am Ende der lag-Phase, zu erklären sein. Damit wären die für die exponentielle Phase ermittelten Transformationsfrequenzen eher der lag-Phase zuzuordnen, und stünden im Einklang mit den Literaturwerten. Die geringen Transformationsfrequenzen für die lag-Phase könnten auf eine zu geringe Zelldichte aufgrund der Sedimentation der Zellen zurückzuführen sein. IV.2 Versuchsteil B Im Wachstumsversuch wurde die Fähigkeit der Mutanten auf unterschiedlichen Kohlenstoff-Quellen zu wachsen untersucht (Tab. 3). Dies lässt Rückschlüsse auf den Mutationsort innerhalb der im β-Ketoadipat-Stoffwechselweg beteiligten Gene zu. Der Wildtyp, als erste Kontrolle, zeigt Wachstum auf allen untersuchten Intermediaten des β-Ketoadipat-Stoffwechselweges, weist somit keine Mutation in einem der beteiligten Gene (qui-, pca- bzw. pob-Gene) auf und ist demzufolge in der Lage alle notwendigen Enzyme zu synthetisieren. Als zweite Gegenprobe fungierte die Kohlenstoff-Quelle Succinat, ein Intermediat des Tricarbonsäure-Zyklus‘ (TCC), als positiv Kontrolle, ob die einzelnen Mutanten grundsätzlich lebensfähig sind. So lässt sich die Mutation bei den Mutanten 28 und 275 bereits hier genau eingrenzen, bei den Mutanten 25, 29, 31 und 32 waren weitere Tests zur Identifizierung des Mutationsortes erforderlich. Mutante 28 zeigte kein Wachstum auf Quinat, jedoch auf Protocatechuat, pHydroxybenzoat sowie Succinat. Dies lässt auf eine Mutation im QuinatStoffwechselweg schließen, wobei hier die Gene quiA, quiB und/oder quiC betroffen sind. Eine Mutation an dieser Stelle verhindert die entsprechende Umsetzung von Quinat zu Hydroquinat (quiA), von Hydroquinat zu Hydroshikimat (quiE/quiB) bzw. im letzten Schritt Hydroshikimat zu Protocatechuat (quiC) (Tab. 3). 29 | S e i t e Mutante 275 war imstande auf Quinat, Protocatechuat sowie Succinat, jedoch nicht auf p-Hydroxybenzoat zu wachsen. Bei dieser Form der Mutation handelt es sich um einen Defekt in den pob-Genen. Hierbei können pobS, pobR oder pobA betroffen sein. PobS codiert für einen Transkriptionsregulator, der die pobA-Expression reduziert. PobR fungiert als Transkriptionsaktivator und induziert in Anwesenheit von p-Hydroxybenzoat die Transkription von pobA. PobA wiederum codiert für die pHydroxybenzoat-Hydroxylase, die p-Hydroxybenzoat zu Protocatechuat umsetzt (Tab. 3). Nach erfolgter Komplementation mit Hilfe chromosomaler DNA der Mutanten 28 und 275 konnte die Mutation der verbleibenden Mutanten 25, 29, 31 und 32 auf das pcaOperon eingegrenzt werden. Das Rückerlangen der Fähigkeit auf Protocatechuat, Quinat und p-Hydroxybenzoat zu wachsen beruht auf der Komplementation der DNA im Bereich des pca-Operons. Auch das Wachstum auf Quinat und p-Hydroxybenzoat ist im Falle einer Mutation im pca-Operon eingeschränkt, da die vorhandenen Aromaten zwar bis zum Protocatechuat, jedoch nicht über dieses hinaus, umgesetzt werden können (Tab 4). Komplementation mit Hilfe der Plasmide zeigt, welches Plasmid in der Lage ist den Gendefekt zu komplementieren und grenzt damit den Mutationsort ein (Tab. 6). Mutante 29 wurde komplementiert durch das Plasmid pZR2, sowie die Plasmide pZR211, pZR2105, pZR2106 und pZR2111. Die Plasmide pZR210 und pZR2113 konnten den Defekt nicht ausgleichen (Tab. 6). Vergleicht man diese Ergebnisse mit der Plasmidkarte (Abb. 2) so weist Mutante 29 einen Defekt im pcaG-Gen auf. Entscheidend ist hierbei das Plasmid pZR2113, da dieses das letzte nicht mehr komplementierende Plasmid ist, wird der Gendefekt von Mutante 29 irgendwo vor dem Beginn dieses Plasmid liegen. Die Mutation kann auf den Bereich 1885 - 2003 Basenpaare eingegrenzt werden. Mutante 31 wurde ausschließlich durch das Plasmid pZR2 komplementiert, keines der weiteren verwendeten Plasmide brachte einen Ausgleich im defekten Gen (Tab. 6). Dieses Ergebnis lässt leider keine Kartierung der Mutation zu. Da das Plasmid pZR2 die Mutante komplementiert, liegt der Defekt in jedem Fall in einem der Gene pcaK, pcaC, pcaH oder pcaG und mindestens eines der weiteren Plasmide hätte Komplementation, und damit die Rückerlangung der Fähigkeit Protocatechuat oder 30 | S e i t e p-Hydroxybenzoat umsetzen zu können, zeigen müssen. Diese Unvollständigkeit könnte aufgrund einer zu geringen Menge an Plasmid-DNA, bzw. einer zu geringen Konzentration an Plasmid-DNA in der aufgebrachten Menge, oder aber durch zu starke Verunreinigung der Plasmid-DNA herrühren (Tab. 5). In allen Fällen wäre nicht genug Plasmid-DNA vorhanden gewesen um die aufgetragenen Mutanten, von denen über dies hinaus nicht alle Aufnahme und Einbau der DNA zeigen, zu komplementieren. Mutante 32 zeigte bereits vor der aufgetragenen Plasmid-DNA, also vor erfolgter Komplementation, Wachstum. Dieses Ergebnis kann nicht als Komplementation bezeichnet werden, kein vorhandener Defekt wurde ausgeglichen, da die Mutante scheinbar intakte Gene aufwies (Tab. 6). Eindeutig war die Komplementation nur durch das Plasmid pZR2106 zu beurteilen. Dieses Ergebnis ist auf Unzulänglichkeiten in der Präparation der Mutante 32 zurückzuführen. So könnte die Kultur der Mutante 32 mit einer weiteren Mutante verunreinigt worden sein. Da Acinetobacter spp. eine hohe genetische Kompetenz aufweist, wird diese Verunreinigung schon ausgereicht haben, um die Mutante zu Komplementieren, sodass eine Komplementation unter Einsatz der Plasmide dann überflüssig war. Die Ergebnisse, die dieser Komplementationstest eigentlich bringen sollte, sehen wie folgt aus (Tab. 10): Tabelle 10: Komplementation der Mutanten 31 und 32 mit Hilfe rekombinanter Plasmide Mutante Plasmid 31 32 pZR2 + + pZR210 - - pZR211 + + pZR2105 - + pZR2106 - + pZR2111 - - pZR2113 - 31 | S e i t e Mutante 31 wird also, zusätzlich zum Plasmid pZR2, durch das Plasmid pZR211 komplementiert (Tab. 10). Keines der weiteren Plasmide zeigt Komplementation. Damit liegt die Mutation in einem Bereich am Anfang des Plasmides pZR211. Das Plasmid pZR210 komplementiert nicht, damit liegt der Mutationsort nicht auf diesem Plasmid, sondern dahinter. Das Plasmid pZR2105 wäre die nächste Abstufung zum Plasmid pZR211, dieses komplementiert jedoch auch nicht, damit liegt die Mutation vor dem Beginn dieses Plasmides. Die Mutation kann damit auf einen Bereich von 1020 - 1334 Basenpaare eingegrenzt werden und liegt im pcaH-Gen. Mutante 32 sollte durch die Plasmide pZR2, pZR211, pZR2105 sowie pZR2106 komplementiert werden (Tab. 10). Dies grenzt die Mutation auf den Bereich Anfang des Plasmids pZR211 und den Beginn des Plasmides pZR2111 ein. Da letzteres nicht mehr in der Lage ist den Defekt auszugleichen, liegt der Mutationsort nicht im Bereich des Plasmides pZR2111. Da jedoch vorangegangene Plasmide in der Lage sind den Defekt auszugleichen, nachfolgende Plasmide nicht für einen Ausgleich der Mutation sorgen, ist ersichtlich, dass die Mutation vor dem Bereich des Plasmides pZR2111 liegen muss. Eingegrenzt werden kann die Mutation auf einen Bereich zwischen 1020 - 1884 Basenpaaren und könnte somit im pcaH- aber auch im pcaGGen liegen. Werden die Literaturwerte der Mutationsorte der einzelnen Mutanten herangezogen (3) können Unterschiede zu den hier erhaltenen Ergebnissen beobachtet werden. So handelt es sich bei Mutante 29 um einen Basenaustausch beim Basenpaar 1681. Hier findet sich anstelle des Tripletts AGT das Triplett GGT. Der Vergleich mit dem im Versuch ermittelten Mutationsort im Bereich von 1885 - 2003 Basenpaare bringt keine Übereinstimmung. Damit hätte die Komplementation mit Plasmid pZR2111 nicht mehr erfolgen sollen. Zu einem solchen Ergebnis kann es durch Vertauschen der Plasmide bzw. Verunreinigung des Plasmides pZR2111 mit einem noch komplementierenden Plasmid (bspw. pZR211, pZR2105, pZR2106) kommen. Mutante 31 weist eine Deletion zwischen Basenpaar 1333 und 1460 auf. Das im Versuch ermittelte Ergebnis (Tab. 6) machte eine Kartierung der Mutation unmöglich. Die im Vergleich in Tabelle 10 gezeigten erwarteten Ergebnisse stehen im Konsens zu dem in der Literatur (3) angegebenen Mutationsort. 32 | S e i t e Auch bei Mutante 32 kann aufgrund der im Versuch gesammelten fehlerhaften Ergebnisse (Tab. 6) nur auf die zu erwarteten Ergebnisse (Tab. 10) verwiesen werden. Laut Literatur (3) weist diese Mutante eine, der Erwartung nach bestätigten, Deletion zwischen den Basenpaaren 1588 und 1889 auf. Die Mutationsart der Mutante 25 konnte nur durch Amplifikation des defekten pcaUGens und anschließende gelelektrophoretische Auftrennung im Vergleich zum Wildtyp-Gen bestimmt werden. Die gelelektrophoretische Auftrennung der Mutante 25 im Vergleich zum Wildtyp zeigt schon hier (Abb. 4) einen deutlichen Unterschied in der Länge der Fragmente. Mit Hilfe einer auf den Marker geeichten Gerade (Abb. 6 und Anhang) lassen sich die Längen in Basenpaaren der DNA-Fragmente des Wildtyps und von Mutante 25 ablesen. So misst das pcaU-Gen-Fragment der Mutante ca. 280bp und ist damit um ca. 470bp kürzer als das 750bp messende Fragment des Wildtyps. Damit ist die Mutation im pcaU-Gen der Mutante 25 auf eine Deletion von ca. 470bp zurückzuführen. Laut Literatur (3) beträgt die Länge des Wiltyp-pcaU-Gens 802bp. Das pcaU-Gen der Mutante wurde um 522bp deletiert und misst somit 280bp. Die Differenzen zu den im Versuch ermittelten Werten sind minimal und wahrscheinlich auf Messfehler der Laufstrecken zurückzuführen. IV.3 Versuchsteil C Im ersten Teil dieses Versuches wurde die Enzymaktivität anhand der zeitlichen Umsetzung des Substrates Protocatechuat untersucht. Mit Hilfe einer Eichgeraden (Abb. 7 und Anhang) konnte durch die Proteinbestimmung nach Bradford die Proteinmenge des Rohextraktes bestimmt werden. Der Quotient aus Proteinmenge und Rohextraktmenge ergab die Proteinkonzentration (Tab. 7). Bei Zunahme der Rohextraktmenge nimmt ebenfalls die enthaltene Proteinmenge zu, weshalb sich die Absorption proportional erhöht. Die Proteinkonzentration bleibt bei allen Rohextraktansätzen nahezu konstant. Die Bestimmung des Extinktionskoeffizienten von Protocatechuat wird für die Berechnung der spezifischen Aktivität benötigt. Der in diesem Versuch ermittelte Wert liegt bei 3,93mM-1cm-1 und weicht nur unwesentlich vom Literaturwert mit 3,89mM-1cm-1 ab. Anhand unterschiedlicher Substratkonzentrationen wurde zunächst die Extraktproportionalität bewiesen (Abb. 33 | S e i t e 8). Diese besagt, dass bei Verdopplung der Proteinmenge sich die Enzymaktivität verdoppelt. Darum entsteht ein nahezu linearer Zusammenhang aus diesen beiden Größen. Die Bestimmung der spezifischen Aktivität zeigt, dass diese bei Protocatechuat am höchsten, gefolgt von p-Hydroxybenzoat und am niedrigsten bei Quinat ist (Abb. 9). Dieses Ergebnis ist nachvollziehbar, da Protocatechuat das direkte Substrat für die Protocatechuat-3,4-Dioxygenase ist, und somit eine sofortige Umsetzung erfolgen kann. P-Hydroxybenzoat ist nur einen Zwischenschritt vom Protocatechuat entfernt, dies könnte der Grund sein, weshalb die Aktivität der Protocatechuat-3,4-Dioxygenase im Verhältnis zum Substrat Protocatechuat etwas vermindert ist. Quinat ist bezogen auf den Stoffwechselweg, am weitesten vom Protocatechuat entfernt. Dies könnte eine mögliche Erklärung für die geringe Enzymaktivität der Protocatechuat-3,4-Dioxygenase sein. Es muss erst eine Umsetzung des Quinates über Hydroquinat und Hydroshikimat zu Protocatechuat erfolgen, bevor die Protocatechuat-3,4-Dioxygenase Aktivität zeigen kann. Im zweiten Versuchsteil wurde die Enzymaktivität anhand des erzeugten Produktes ermittelt. Es wurde eine Reportergenfusion des promotorlosen lacZ-Gens mit dem pcaI-Gen vorgenommen, welches direkt stromabwärts des Promotorbereichs des pca-Operons liegt. Die durch das lacZ-Gen codierte β-Galactosidase hat die Fähigkeit das angebotene Substrat ortho-Nitro-Phenol-Galactose (oNPG) zu spalten. Das resultierende Monomer ortho-Nitro-Phenol weist eine gelbe Färbung auf, die anhand der Messung der optischen Dichte quantifiziert werden kann. Dies lässt Rückschlüsse auf die Promotoraktivität und damit auf die Gesamt-Enzymaktivität zu. Die in diesem Versuchsteil ermittelten Aktivitäten nach Miller zeigen, einen sprunghaften Anstieg der Enzymaktivität ab einer Konzentration von 100µM Substrat (Tab. 9 bzw. Abb. 10). Eine weitere Erhöhung der Konzentration hat eine massive Zunahme der Enzymaktivität zur Folge. Die Transkription der Protocatechuat-3,4Dioxygenase ist Substrat-Induziert. Eine stärkere Induktion durch eine hohe Substratkonzentration hat eine Erhöhung der Enzymmenge zur Folge. Damit findet die Regulation der Protocatechuat-3,4-Dioxygenase bereits auf transkriptioneller Ebene statt. Die Konzentrationen unter 100µM zeigen nur eine geringe Enzymaktivität. Eine mögliche Erklärung wäre, dass ein bestimmter Schwellwert an Substrat (hier: Protocatechuat) vorhanden sein muss um die Expression der Enzyme 34 | S e i t e des Aromatenstoffwechsels (hier: Protocatechuat-3,4-Dioxygenase) zu induzieren. Energetisch gesehen ist dies nachvollziehbar, da Acinetobacter (und weitere Aromaten-umsetzende Mikroorganismen) eine ausreichende Konzentration an aromatischem Substrat braucht, damit sich die Umstellung auf den Aromatenstoffwechsel rentiert. Bei Abwesenheit von Protocatechuat wird darum auch das pca-Operon nicht transkribiert. In diesem Fall dürfte keine Enzymaktivität vorhanden sein. Die dennoch gemessene Aktivität (Tab. 9) könnte auf Pipetierfehler bzw. Verunreinigungen bei der Messung der optischen Dichte, zurückzuführen sein. V. Literatur Anleitung „Mikrobiologisches Praktikum, Block Physiologie, Biochemie, Organisation und Regulation des β -Ketoadipatstoffwechselweges in Acinetobacter baylyi BD413“ der Universität Frankfurt am Main (Hauptstudium, WS 2007/2008) Michael T. Madigan, John M. Martinko, Brock – Biology of Microorganisms, 11. Auflage, Pearson Prentice Hall, United States of America, 2006 Michael T. Madigan, John M. Martinko, Jack Parker, Brock - Mikrobiologie, 9.Auflage, Spektrum Akademischer Verlag, Heidelberg - Berlin, 2003 Hans G. Schlegel, Georg Fuchs, Allgemeine Mikrobiologie, 8.Auflage, Thieme Verlag, Stuttgart, 2006 (1) CS Harwood, RE Parales, 1996, The β-ketoadipate pathway and the biology of self-identity, Annu. Rev. Microbiol. 50: 553 - 590 (2) V Barbe et al, 2004, Unique features revealed by the genome sequence of Acinetobacter sp. ADP1, a versatile and naturally transformation competent bacterium, Nucleic Acids Res. 32: 5766 - 5779 (3) U Gerischer, LN Ornston, 1995, Spontaneous mutations in pcaH and -G, Structural Genes for Protocatechuate 3,4-dioxygenase in Acinetobacter calcoaceticus, J. Bacteriol. 177: 1336 - 1347 35 | S e i t e VI. Anhang Wachstumskurve von Acinetobacter baylyi BD413 auf Quinat (Vergl. Abb. 1) Eichkurve für die Bestimmung der DNA-Fragmentlängen von Wiltyp und Mutante 25 (Vergl. Abb. 6) Eichgerade für die Proteinbestimmung nach Bradford (Vergl. Abb. 7) Schreiber-Ergebnis (Versuchsteil C) 36 | S e i t e