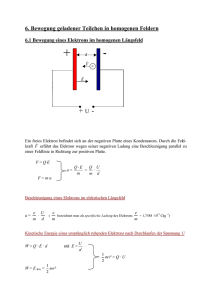
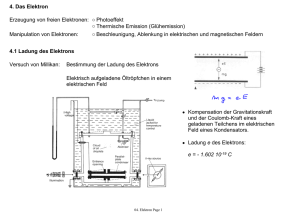
QC Skript - Direkt zur Suche Alt+5
Werbung

Quantenchemie Eine Einführung Altmodisches Drama, das sich jede Nacht im [Kr]4d10 abspielt. Es kann mit Hilfe des englischen PSE gelesen werden. (aus Dickerson/Geis: Chemie – eine lebendige und anschauliche Einführung) Matthias Küng und Thomas Hari © 1998 bis 2002 Seite 2 Quantenchemie – eine Einführung Einige Zitate zu Beginn ... Ich mag sie nicht und es tut mir leid, dass ich jemals etwas damit zu tun hatte. Erwin Schrödinger, 1887-1961 Nothing is real. John Lennon, 1940-1980 Wer über die Quantentheorie nicht entsetzt ist, der hat sie nicht verstanden. Niels Bohr, 1885-1962 Lieber oder vielmehr geliebter geliebter Bohr! ... Besonders reizend finde ich Ihre Angst, Sie könnten den Preis vor mir bekommen – das ist ächt bohrisch. Ihre neuen Untersuchungen [zum Aufbau des Periodensystems] haben meine Liebe zu Ihrem Geist noch vergrössert Albert Einstein, 1879 – 1955, über Niels Bohr Es war immer schwierig, die Sicht der Dinge zu verstehen, die sich in der Quantentheorie zeigt. Wenigstens für mich, denn ich bin gerade so alt, dass ich den Punkt noch nicht erreicht habe, an dem für mich alles offensichtlich ist. Ich werde immer noch nervös dabei. Ihr wisst doch, wie das ist, jede neue Idee braucht eine Generation oder zwei, bevor es offenbar wird, dass eigentlich gar kein Problem vorliegt. Ich kann das eigentliche Problem nicht definieren, also vermute ich, dass es so ein Problem nicht gibt. Doch ich bin nicht sicher, dass es kein wirkliches Problem gibt. Richard Feynman, 1918 – 1988 Die naturwissenschaftlichen Kenntnisse werden zwar in der Schule gelehrt; sie tragen auch einiges zum Verständnis der Natur, aber wenig zum Verständnis der Kultur bei. [...] [Und] so bedauerlich es manchem erscheinen mag: Naturwissenschaftliche Kenntnisse müssen zwar nicht versteckt werden, aber zur Bildung gehören sie nicht. Dietrich Schwanitz, Literaturprofessor, 1940 – 2004 Doch im Laufe seiner Arbeit am Text muss Schwanitz aufgefallen sein, dass sein Wissen mindestens eine riesige Lücke aufweist und er nicht wirklich alles, was man wissen muss parat hat. Da war er wieder, der Hochmut eines literarisch und philosophisch Gebildeten gegenüber den Leistungen der Naturwissenschaften. Wie viel Stoff zum Nachdenken wird hier geliefert. Warum schaut der gebildete Mensch, der offenbar souverän die abendländische Literatur und Kunst im Griff hat und goutiert, beharrlich in die falsche Richtung, wenn es um Naturwissenschaft geht? Ernst Peter Fischer, Wissenschftshistoriker, *1947 Ich denke, ich kann davon ausgehen, dass niemand die Quantenmechanik versteht. Ein Philosoph hat einmal behauptet: „Naturwissenschaft setzt notwendig voraus, dass gleiche Umstände immer auch gleiche Auswirkungen haben.“ Nun, dem ist nicht so! Atome sind völlig unmöglich – vom klassischen Standpunkt aus betrachtet. Richard Feynman, 1918 – 1988 Das Universum ist nicht seltsamer, als wir uns vorstellen, es ist auch seltsamer, als wir uns überhaupt vorstellen können. J.B.S. Haldane „To be or not to be“, das ist nicht die Frage; es ist die Antwort. Fred Alan Wolf Ohne Quantenmechanik ist ein Mensch nicht in der Lage, seine Umwelt und die Bedingungen seiner Existenz zu übersehen. (Er ist es wahrscheinlich auch mit der Quantenmechanik nicht vollständig!) Er mag antike Schriftsteller in der Originalsprache oder Heidegger in deutscher Sprache verstehen, er ist trotzdem ungebildet: Er hat die wichtigste geistige Revolution unseres Jahrhunderts und einen wesentlichen Inhalt seiner Kultur verpasst. Erstaunlicherweise wird dieses Bildungsgut unseren Jungen in den Mittelschulen immer noch vorenthalten. E. Schumacher Das wellenmechanische Atommodell kommt mir als etwas Interessantes vor; im allgemeinen würde ich die Quantenchemie als eines der interessantesten, in der Schule behandelten Gebiete der Chemie und der Naturwissenschaften überhaupt bezeichnen. SchülerIn einer Prima des Literargymnasiums Neufeld (Bern) Ich schätze es, dass wir uns mit der Quantenchemie beschäftigten bzw. auseinandersetzten. Wir erhielten so einen Einblick in die moderne naturwissenschaftliche Denkweise. Dies ist nötig, um die Auswirkungen dieser Modellvorstellung auf unsere Zeit zu verstehen. SchülerIn einer Prima des Literargymnasiums Neufeld (Bern) Quantenchemie – eine Einführung Seite 3 Inhaltsverzeichnis 1 WORUM GEHT ES ? ........................................................................................................ 5 2 PROLOG – NICHTS IST REAL ODER AUF DER SUCHE NACH SCHRÖDINGERS KATZE .............................................................................................................................. 5 3 WELLEN ........................................................................................................................... 7 4 DER WELLE-TEILCHEN-DUALISMUS.......................................................................... 11 4.1 Das Doppelspaltexperiment mit Gewehrkugeln .................................................................................................... 12 4.2 Doppelspaltexperiment mit Wasserwellen ............................................................................................................. 14 4.3 Doppelspaltexperiment mit Elektronen ................................................................................................................. 15 5 HEISENBERGS UNSCHÄRFERELATION UND SCHRÖDINGERS GLEICHUNG ....... 17 6 DAS TEILCHEN (E-) IM KASTEN................................................................................... 19 7 WASSERSTOFFATOM .................................................................................................. 22 7.1 Orbitale ..................................................................................................................................................................... 24 7.2 Der erste Energiezustand (Grundzustand) des Elektrons im Wasserstoffatom ................................................. 25 7.2.1 Das Quadrat der Wellenfunktion: Wahrscheinlichkeitsdichte und radiale Wahrscheinlichkeits-dichte ............ 26 7.2.2 Aufenthaltsraum (Ladungswolke) des Elektrons im Grundzustand ................................................................... 28 7.2.3 Zusammenfassung des Zustandes 1s.................................................................................................................. 29 7.3 Der zweite Energiezustand des Elektrons im Wasserstoffatom ........................................................................... 30 7.3.1 Der 2s-Zustand ................................................................................................................................................... 30 7.3.2 Der 2p-Zustand................................................................................................................................................... 33 7.3.3 Die höheren Energiezustände des Wasserstoffatoms ......................................................................................... 35 8 ATOME MIT MEHR ALS EINEM ELEKTRON: DER AUFBAU DES PSE ..................... 38 9 MOLEKÜLBINDUNG...................................................................................................... 40 9.1 Bindender Zustand................................................................................................................................................... 40 9.2 Antibindender Zustand ........................................................................................................................................... 42 9.3 Die Bindung in Molekülen....................................................................................................................................... 43 9.4 Systematische Darstellung der Bildung einer Einfachbindung............................................................................ 46 9.4.1 9.5 Homonukleare Moleküle der 2. Periode............................................................................................................. 46 Wechselwirkung zwischen den Orbitalen – das Modell der Hybridisierung ....................................................... 49 9.5.1 Die Hybridisierung von s- und p-Orbitalen........................................................................................................ 49 9.5.2 Weitere Hybridisierungsarten - Mehrfachbindungen ......................................................................................... 51 9.6 Bindungsverhältnisse weiterer Moleküle ............................................................................................................... 54 Quantenchemie – eine Einführung Seite 4 10 FARBEN ......................................................................................................................... 57 10.1 Organische Farbstoffmoleküle................................................................................................................................ 59 10.1.1 Fluoreszierende Farbstoffe................................................................................................................................. 63 10.2 Anorganische Farbstoffe ......................................................................................................................................... 65 10.3 Beispiele .................................................................................................................................................................... 68 11 LITERATUR .................................................................................................................... 71 12 ANHANG ........................................................................................................................ 74 7.1.1 7.2 Polarkoordinaten ................................................................................................................................................ 74 Der erste Energiezustand (Grundzustand) des Elektrons im Wasserstoffatom ................................................. 74 7.2.1 Vereinfachte Wellenfunktion 1s und räumliche Darstellung ............................................................................. 75 7.2.2 Die Energie des Wasserstoffatoms im Grundzustand 1s.................................................................................... 76 7.2.3 Das Quadrat der Wellenfunktion: Wahrscheinlichkeitsdichte und radiale Wahrscheinlichkeits-dichte ............ 76 7.2.4 Aufenthaltsraum (Ladungswolke) des Elektrons im Grundzustand ................................................................... 79 7.2.5 Zusammenfassung des Zustandes 1s.................................................................................................................. 80 7.3 Der zweite Energiezustand des Elektrons im Wasserstoffatom........................................................................... 80 7.3 Der zweite Energiezustand des Elektrons im Wasserstoffatom........................................................................... 81 7.3.1 Der 2s-Zustand ................................................................................................................................................... 81 7.3.2 Der 2p-Zustand .................................................................................................................................................. 84 Quantenchemie – eine Einführung Seite 5 1 Worum geht es ? Die Quantenchemie ist eine vergleichsweise junge Wissenschaft. Ihre Anfänge begannen vor etwa 80 Jahren mit den Arbeiten von Heitler und London. 1927 konnten sie zum ersten mal quantenmechanisch das Rätsel der kovalenten Bindung des Wasserstoffmoleküls (H2) lösen. Die Quantenchemie hat sich seither ungeheuer weiterentwickelt. Ohne Quantenchemie und Quantenphysik gäbe es weder Atomphysik noch Molekularbiologie, blieben chemische Bindungen ohne Erklärung, wären weder Laser noch Computer denkbar. Es spielt keine Rolle, wenn Sie die physikalischen Grundlagen der gezeigten Phänomene noch nicht kennen. Der Zweck dieses Einschubes besteht darin, Ihnen den Ursprung der Modellvorstellung der Elektronenwolken als Aufenthaltsräume der Elektronen in der Atomhülle anzudeuten, bevor wir mit diesem Modell arbeiten werden. 2 Prolog – Nichts ist real oder auf der Suche nach Schrödingers Katze Die Katze aus unserem Titel ist ein fiktives Tier, doch Schrödinger war ein realer Mensch. Erwin Schrödinger war ein österreichischer Wissenschaftler, der Mitte der 20-er Jahre des 20. Jahrhunderts dazu beigetragen hat, die Gleichungen eines Wissenschaftsgebietes zu entwickeln, das wir heute als Quantenmechanik bezeichnen. Wissenschaftsgebiet ist eigentlich nicht der richtige Ausdruck, denn die Quantenmechanik ist die Grundlage aller modernen Naturwissenschaften. Die Gleichungen beschreiben das Verhalten sehr kleiner Objekte - die, allgemein gesagt, so gross wie ein Atom oder kleiner sind -, und sie allein machen die Welt des sehr Kleinen verständlich. Ohne diese Gleichungen könnten die Physiker keine Atomkraftwerke (oder Atombomben) planen, keine Laser bauen, nicht erklären, warum die Sonne nicht erkaltet. Ohne die Quantenmechanik wäre die Chemie noch im dunklen Mittelalter, und von der Molekularbiologie, vom Verstehen der DNS und von Gentechnik könnte gar keine Rede sein. Die Quantentheorie ist die grösste wissenschaftliche Errungenschaft; sie ist weitaus bedeutsamer und von sehr viel direkterem praktischem Nutzen als die Relativitätstheorie. Dabei macht sie einige ganz merkwürdige Vorhersagen. Die Welt der Quantenmechanik ist in der Tat so merkwürdig, dass sogar Albert Einstein sie unverständlich fand und sich weigerte, sämtliche Implikationen der von Schrödinger und seinen Kollegen entwickelten Theorie anzuerkennen. Einstein, und mit ihm viele Wissenschaftler, fühlten sich wohler in der Annahme, die Gleichungen der Quantenmechanik seien so etwas wie ein mathematischer Kunstgriff, der für das Verhalten atomarer und subatomarer Teilchen zufällig einen leidlich brauchbaren Anhaltspunkt liefert, der jedoch eine tiefere Wahrheit verbirgt, die eher der Realität in unserem üblichen Sinn entspricht. Der Quantenmechanik zufolge ist nämlich nichts real, und wir können nichts über das Verhalten von Dingen aussagen, die wir nicht beobachten. Schrödingers sagenumwobene Katze zitiert man, um die Unterschiede zwischen Quantenwelt und der gewöhnlichen Welt zu verdeutlichen. In der Welt der Quantenmechanik gelten die physikalischen Gesetze, die wir aus der uns vertrauten Welt kennen, nicht mehr. Die Vorgänge werden vielmehr durch Wahrscheinlichkeiten bestimmt. Nehmen wir zum Beispiel ein radioaktives Atom; vielleicht zerfällt es und emittiert dabei, sagen wir, ein Elektron, vielleicht aber auch nicht. Mit einer bestimmten Versuchsanordnung kann man eine Wahrscheinlichkeit von genau fünfzig Prozent dafür erreichen, dass eines der Atome einer radioaktiven Substanz innerhalb einer bestimmten Frist zerfällt und dass der Zerfall, wenn es tatsächlich zu ihm kommt, von einem Detektor registriert wird. Schrödinger, der über die Folgerung der Quantenmechanik genauso beunruhigt war wie Einstein, wollte ihre Absurdität aufzeigen und ersann ein Gedankenexperiment, bei dem sich in einem abgeschlossenen Raum oder Quantenchemie – eine Einführung Seite 6 Behälter eine lebende Katze sowie eine Phiole mit Gift befindet. Falls der radioaktive Zerfall tatsächlich stattfindet, zerbricht die Phiole und die Katze stirbt. In der gewöhnlichen Welt besteht eine Wahrscheinlichkeit von fünfzig Prozent, dass die Katze getötet wird, und man kann, ohne in den Behälter hinein zu schauen, ganz getrost sagen, dass die Katze darin entweder tot oder lebendig sein wird. Aber hier stossen wir auf die Merkwürdigkeit der Quantenwelt. Nach der Theorie ist keine der beiden Möglichkeiten, die für die radioaktive Substanz und damit für die Katze bestehen, in irgendeiner Weise real, sofern sie nicht beobachtet wird. Der Atomzerfall hat weder stattgefunden, noch hat er nicht stattgefunden, und die Katze ist weder getötet worden, noch ist sie nicht getötet worden, sofern wir nicht in den Behälter hineinschauen, um zu sehen, was passiert ist. Ein Theoretiker, der die unverfälschte Quantenmechanik vertritt, würde sagen, die Katze befinde sich in einem unbestimmten Zustand, sie sei weder tot noch lebendig, solange nicht ein Beobachter in dem Behälter nachschaut, wie sich die Dinge entwickeln. Nichts ist real, falls es nicht beobachtet wird. Für Einstein und andere war diese Vorstellung ein Greuel. „Der Herrgott würfelt nicht“, sagte er im Hinblick auf die Theorie, nach der die Welt eine Ansammlung der Resultate von im Grunde willkürlichen „Entscheidungen“ auf der Quantenebene ist. Davon, dass Schrödingers Katze sich in einem unwirklichen Zustand befindet, wollte er nichts wissen; er meinte, den Dingen müsse ein „Uhrwerk“ zugrunde liegen, das dafür sorgt, dass sie in einem ganz fundamentalen Sinne Realität besitzen. Er hat jahrelang über Versuche nachgegrübelt, durch die sich das Wirken dieser fundamentalen Realität zeigen lassen sollte, aber erst nach seinem Tode wurde es möglich, einen entsprechenden Versuch durchzuführen. Vielleicht ist es gut so, dass er nicht mehr erlebt hat, wohin eine der von ihm angeregten Überlegungen führt. Im Sommer 1982 schloss ein Forscherteam unter der Leitung von Alain Aspect an der Universität Paris-Sud eine Reihe von Experimenten ab, welche die fundamentale Realität unter der unwirklichen Welt der Quanten aufdecken sollten. Der Realität, die allem zugrunde liegt - dem fundamentalen Uhrwerk - hatte man den Namen „verborgene Variablen“ gegeben. Bei dem Experiment ging es um das Verhalten von zwei Photonen, also „Lichtteilchen“, die von einer Quelle in entgegengesetzter Richtung davonfliegen. Die beiden Photonen aus einer einzigen Quelle können mit Hilfe von zwei Detektoren beobachtet werden, die eine Polarisation genannte Eigenschaft messen. Der Quantentheorie zufolge existiert diese Eigenschaft nicht, solange sie nicht gemessen wird. Nach der Vorstellung von „verborgenen Variablen“ weist aber jedes Photon vom Augenblick seiner Erzeugung an eine „wirkliche“ Polarisation auf. Da die beiden Photonen gleichzeitig emittiert werden, besteht zudem ein Zusammenhang zwischen ihrer jeweiligen Polarisation. Die Art des Zusammenhangs, die tatsächlich gemessen wird, hängt jedoch davon ab, welche der beiden erwähnten Realitätsvorstellungen man vertritt. Die Ergebnisse dieses entscheidenden Experiments waren eindeutig. Man fand nicht jenen Zusammenhang, der nach der Theorie von den verborgenen Variablen zu erwarten war, sondern im Gegenteil den Zusammenhang, den die Quantenmechanik vorhersagte. Ausserdem stellte man fest, dass die Messung, die an einem Photon vorgenommen wird, sich - wie ebenfalls von der Quantentheorie vorhergesagt - direkt auf die Eigenschaften des anderen Photon auswirkt. Beide sind unentwirrbar durch eine Wechselwirkung miteinander verbunden, obwohl sie sich mit doppelter Lichtgeschwindigkeit voneinander entfernen, und wir aus der Relativitätstheorie wissen, dass kein Signal sich schneller als Licht fortpflanzen kann. Die Experimente beweisen, dass es eine der Welt zugrunde liegende Realität an sich nicht gibt. „Realität“ im üblichen Sinne ist keine angemessene Vorstellung über das Verhalten der fundamentalen Teilchen, aus denen das Universum sich zusammensetzt. Andererseits scheinen diese Teilchen gleichzeitig unzertrennlich in einem unteilbaren Ganzen verbunden zu sein, so dass jedes weiss, was mit den übrigen geschieht. Die Suche nach einer Erklärung für das Verhalten von Schrödingers Katze war die Suche nach der Quanten-Realität. Nach diesem kurzen Abriss könnte man meinen, Quantenchemie – eine Einführung Seite 7 die Suche sei fruchtlos gewesen, da es eine Realität im üblichen Sinne des Wortes nicht gibt. Aber damit ist die Geschichte noch nicht ganz zu Ende, und es könnte sein, dass wir auf jener Suche zu einem neuen Verständnis jener Realität gelangen, welche die herkömmliche Interpretation der Quantenmechanik übersteigt und dennoch einschliesst. Es ist allerdings ein langer Weg. Er begann mit einem Wissenschaftler, den es wohl noch stärker gegraust hätte als Einstein, hätte er die Antworten sehen können, die wir inzwischen auf jene Fragen gefunden haben, über die er sich bereits den Kopf zerbrach. Isaac Newton hat, als er vor dreihundert Jahren die Natur des Lichts studierte, nicht ahnen können, dass er sich schon auf dem Wege befand, der zu Schrödingers Katzenparadoxien führt. 3 Wellen Zu Beginn möchten wir einen Überblick über die für die Quantenchemie wichtigsten Teile der Wellenlehre 1 geben . Als Welle bezeichnet man eine von einem Erreger aus gehende Störung. Dabei wird nicht Materie weitergeleitet, sondern Energie. Ist die Störung zeitlich und räumlich gleichmässig (Sinuswelle), spricht man von einer harmonischen Welle. Als Schwingung bezeichnet man eine Bewegung, die in periodischer Folge um die Ruhelage erfolgt. Ein solches Teilchen, das eine Schwingung ausführt, bezeichnet man seinerseits als Oszillator. Bei einer Welle unterscheidet man zwischen zwei Geschwindigkeiten. Die Geschwindigkeit der einzelnen Oszillatoren ist die Teilchengeschwindigkeit, bei der Ausbreitungsgeschwindigkeit versteht man die Geschwindigkeit, mit der sich eine Welle ausbreitet. Abbildung 1: Momentanbild einer harmonischen Welle 1 Ausführlicher finden Sie die Wellentheorie in Ihrem Physikbuch Seite 8 Quantenchemie – eine Einführung Bei einer harmonischen Welle gelten folgende physikalischen Beziehungen: c= λ T c=λf und dabei gilt: f= 1 T λ (Lambda) ist die Wellenlänge (kürzester Abstand zweier phasengleich schwingender Oszillatoren), c die Ausbreitungsgeschwindigkeit, T (Schwingungsdauer) die Zeit während der eine Welle die Strecke λ zurücklegt -1 und die Frequenz f gibt an, wie viele Schwingungen pro Zeiteinheit erfolgen. Ihre Einheit ist Hertz (sec ). Die Energie einer Welle berechnet sich folgendermassen: E = ½ m f2 s2max Sie ist also proportional zum Quadrat der maximalen Auslenkung. Die Ausbreitungsgeschwindigkeit hängt von den physikalischen Eigenschaften des Mediums ab, in dem sich die 2 Welle bewegt. So breiten sich Schallwellen in Helium schneller aus, da Helium leichter ist als Luft. Eine menschliche Stimme ertönt daher höher (grössere Frequenz) wenn mit Helium gesprochen wird (λ kann nicht verändert werden, da sie durch die Stimmbänder gegeben ist). Als zweites Beispiel sei die Messung der Meerestemperatur erwähnt. Schallwellen breiten sich in warmem Wasser schneller aus als in kälterem Wasser. So kann die Erhöhung der Wassertemperatur infolge des Treibhauseffektes genau bestimmt werden. Zwei Wellen, die sich aufeinander zu bewegen, überlagern sich. Sind es zwei gleich grosse Wellenberge, die aufeinander treffen, ergibt dies eine Verdoppelung der Amplitude (maximale Höhe einer Welle). Dies bezeichnet man als konstruktive Interferenz. Trifft hingegen ein Wellenberg auf ein gleich grosses Wellental, löschen sich die Wellen aus, was man als destruktive Interferenz bezeichnet: Abbildung 2: Konstruktive Interferenz 2 Schallwellen sind zwar Longitudinalwellen, es gelten aber die gleichen Gesetze wie für Transversalwellen Seite 9 Quantenchemie – eine Einführung Abbildung 3: Destruktive Interferenz Bringt man ein Seil zum Schwingen, werden die Wellen vom anderen Ende zurückgeworfen und überlagern sich mit den neu ausgesandten Wellen. Laufen nun zwei Wellen, die gleiche Frequenz, Wellenlänge und Amplitude aufweisen, aufeinander zu, so bildet sich eine (scheinbar) stehende Welle als Ergebnis der Interferenz aus. Die Orte, an denen die Oszillatoren zu keinem Zeitpunkt ausgelenkt werden, nennt man Schwingungsknoten. Der Abstand zwischen zwei Schwingungsknoten entspricht der halben Wellenlänge. Die Bereiche zwischen den Knoten nennt man Schwingungsbäuche. L n=1 n=2 n=3 n=4 Abbildung 4: Stehende Welle eines schwingenden Seils Nur bestimmte Frequenzen des Erregers führen zu stehenden Wellen auf dem Seil. Da das Seil an zwei Punkten fix ist, nämlich an der Wand und bei der Person, müssen dort immer Knoten vorhanden sein. Auf der Seite 10 Quantenchemie – eine Einführung übrigen Länge des Wellenträgers variiert die Anzahl der Knoten je nach Frequenz des Erregers. Man spricht in diesem Zusammenhang von Eigenschwingung bzw. Eigenfrequenz des Wellenträgers. Dadurch ergibt sich zwangsläufig folgende Wellenlänge bei einer stehenden Welle: 2L λ= n L ist der Abstand zwischen den beiden fixen Enden, n ist der Schwingungszustand das Seils. Eine stehende Welle kann sich aber nicht nur in einer Dimension, wie bei einem Seil oder einer Gitarre ausbilden, sondern auch in zwei Dimensionen, wie dies bei einer Trommel geschieht. In diesem Fall 3 entsprechen die Bäuche einer Fläche, die Knoten einer Linie . Abbildung 5: Eine mit Pulver bestreute Kesselpauke bringt sechs von ihren vielen möglichen Schwingungsmuster zum Vorschein 3 Vgl. dazu http://www.fhbb.ch/pages/abteilungen/chemie/Stmue/Wellen.htm oder die entsprechenden Dateien auf dem O: Seite 11 Quantenchemie – eine Einführung Abbildung 6: Schwingungsformen einer quadratischen „Pauke“ 4 Der Welle-Teilchen-Dualismus Die ersten Anläufe der Physiker zu einer Quantentheorie waren meist Versuche, die Natur des Lichts zu verstehen. Bereits im 17. Jahrhundert hatte Isaac Newton vorgeschlagen, Licht als einen Strom von Teilchen anzusehen - vergleichbar dem Kugelhagel eines Maschinengewehrs. Zwar gab es vereinzelt auch andere Ansichten, doch war Newtons Autorität derart gross, dass seine Theorie bis ins 19. Jahrhundert hinein der Kritik standhielt. Damals konnten Thomas Young und andere schliesslich überzeugend darlegen, dass das Teilchenbild des Lichts falsch sein musste. Sie favorisierten statt dessen die Idee, Licht als eine Art Wellenbewegung aufzufassen. Eine typische Eigenschaft von Wellen ist das Auftreten von „Interferenz“, wie man die Erscheinungen bei der Überlagerung zweier Wellen bezeichnet. Young verschaffte sich mit seiner berühmten „Doppelspalt“-Vorrichtung zwei Lichtquellen und konnte damit Interferenzmuster bei Licht beobachten. Bei einem Laserstrahl, der nichts anderes als Licht einer einzigen Wellenlänge ist, ist dieses Interferenzmuster besonders deutlich sichtbar. Lange sollten sich die Physiker über dieses Ergebnis nicht freuen. Gegen Ende des 19. Jahrhunderts zeigten sich in einigen Experimenten Phänomene, die mit der Wellentheorie des Lichts nicht erklärt werden konnten. Das berühmteste dieser Experimente betraf den sogenannten „photoelektrischen Effekt“. Ultraviolettes Licht, das man auf eine negativ geladene Metallplatte richtete, entlud die Platte - sichtbares Licht jedoch zeigte keinerlei Wirkung, auch wenn seine Intensität erhöht wurde. Anschaulich können wir diese Tatsache mit einem Tennisball und einer Bleikugel vergleichen, die wir gegen eine Wand werfen. Der Tennisball hinterlässt absolut keine Spuren an der Wand, auch wenn wir die Wurfintensität erhöhen. Die Bleikugel hingegen hinterlässt schon nach einem Wurf ein Loch in der Wand. Es war Albert Einstein, der dieses Rätsel als erster auflöste, im selben Jahr übrigens, in dem er seine Relativitätstheorie veröffentlichte, durch die er später berühmt wurde. Seine Erklärung des photoelektrischen Effekts liess das Teilchenbild des Lichts wieder aufleben. Die Metallplatte entlud sich demnach, weil deren Elektronen durch einzelne kleine Energiepakete herausgeschlagen wurden, in denen die Lichtenergie konzentriert ist - sie werden heute „Photone“ genannt. Einstein zufolge sind die Photonen des sichtbaren Lichts energieärmer als die des ultravioletten Lichts, so dass man soviel sichtbares Licht auf das Metall richten kann wie man will - keines der sichtbaren Photonen besitzt genügend Energie, um ein Elektron herauszuschlagen. Die Verwirrung unter den Physikern war gross, und es dauerte einige Jahrzehnte, bis mit dem Aufkommen der Quantenmechanik in den zwanziger Jahren des 20. Jahrhunderts durch die bahnbrechenden Arbeiten von Werner Heisenberg, Ernst Schrödinger, Paul A. M. Dirac und anderen ein Weg aus dieser Sackgasse gefunden wurde. Diese Theorie konnte die so widersprüchlich anmutenden Eigenschaften des Lichts, den Aufbau der Atome, aber auch vieles andere mehr, erfolgreich erklären. Der Preis für diesen Erfolg ist jedoch hoch. Wir müssen jegliche Hoffnung aufgeben, die Vorgänge auf atomarer Ebene mit unseren gängigen Begriffen und Vorstellungen wie Teilchen und Wellen anschaulich beschreiben zu können. Ein Photon verhält sich anders als alles, was uns bisher je begegnet ist. Das heisst aber nicht, dass die Quantenchemie – eine Einführung Seite 12 Quantenmechanik nicht mit klaren, genau definierten Begriffen arbeiten würde oder keine aussagekräftigen Prognosen stellen könnte - im Gegenteil: Die Quantenmechanik ist die einzige Theorie, die wir kennen, die eindeutige und erfolgreiche Voraussagen für Systeme atomarer und subatomarer Grössenordnung machen kann, ganz ähnlich wie dies die Klassische Mechanik für das Verhalten von Billardkugeln, Raketen oder Planeten tut. Die Schwierigkeit mit Quantenobjekten wie Photonen liegt darin, dass wir uns kein präzises anschauliches Bild von ihrer Bewegung machen können. Wir müssen uns sozusagen mit der Feststellung begnügen, dass das Verhalten der Photonen die typisch quantenmechanischen Züge trägt. In einer Hinsicht allerdings hat es die Natur gut mit uns gemeint. Vom klassischen Standpunkt aus gesehen sind Photon und Elektron grundlegend verschiedene Objekte; in der Quantenwelt verhalten sich Elektron und Photon jedoch in bemerkenswerter Weise ähnlich: Das seltsame, typisch quantenmechanische Verhalten ist ihnen, wie allen anderen Quantenobjekten, gemeinsam – wenigstens ein kleiner Trost dafür, dass wir die Quantenwelt nicht bildhaft darstellen können. Die Geschichte der verschiedenen Versuche, die Beschaffenheit des Elektrons zu verstehen, entbehrt nicht einer gewissen Komik. 1897 hatte J. J. Thomson das Verhältnis von elektrischer Ladung zur Masse des Elektrons experimentell bestimmen können und dadurch das Elektron als fundamentales Teilchen der Natur in die Physik eingeführt; 30 Jahre später gelang es seinem Sohn G. P. Thomson etwa gleichzeitig mit Davisson und Germer in den USA, den Wellencharakter des Elektrons in einer Reihe sehr schöner Experimente schlüssig nachzuweisen. Den Wissenschaftshistoriker Max Jammer veranlasste dies zu der Bemerkung, man sei geneigt zu sagen, dass Thomson senior den Nobelpreis bekam, weil er gezeigt hatte, dass das Elektron ein Teilchen ist, und Thomson junior dafür, dass es eine Welle ist. Auch William Bragg, der zusammen mit seinem Sohn im Jahr 1915 für Kristalluntersuchungen mit Röntgenstrahlen den Nobelpreis bekam, brachte das Dilemma der Physiker auf den Punkt, als er verzweifelt ausrief, dass er montags, mittwochs und freitags die Korpuskulartheorie des Lichts lehre, dienstags, donnerstags und samstags jedoch die Wellentheorie. Im folgenden wird anhand des Doppelspaltexperimentes gezeigt, wie Elektronen einerseits als Teilchen, andererseits als Wellen aufgefasst werden können. Dazu zuerst zwei Experimente mit ähnlichem Versuchsaufbau. 4.1 Das Doppelspaltexperiment mit Gewehrkugeln In unserem ersten Experiment benutzen wir als „Quelle“ ein hin und her schwingendes Maschinengewehr, das die Kugeln mit gleicher Geschwindigkeit, aber zufällig verteilt in einen Raumkegel feuert. Eine Panzerplatte mit zwei parallelen Spalten dient als „Doppelspaltblende“, und eine Reihe kleiner Sandbüchsen als „Detektor“, um die Kugeln aufzufangen. Das Gewehr feuert seine Kugeln mit einer gleichbleibenden Ausstossrate ab, und wir können die Kugeln zählen, die während eines vorgegebenen Zeitraums in einer beliebig ausgewählten Detektorbüchse ankommen. Die Kugeln können entweder geradewegs durch die Spalte hindurch fliegen oder auch an ihrer Kante abprallen, auf alle Fälle aber werden wir sie in einer der Büchsen wiederfinden. Die Kugeln sollen so hart sein, dass sie beim Versuch nicht auseinanderbrechen und wir keine halben Kugeln in den Büchsen vorfinden. Zudem werden nie zwei Kugeln gleichzeitig ankommen - wir benutzen ja nur ein Gewehr, und jede Kugel ist ein einzelnes, für sich identifizierbares Massestückchen. Seite 13 Quantenchemie – eine Einführung Abbildung 7: Eine schematische Darstellung eines Doppelspaltexperiments mit Gewehrkugeln In Abb. 7 ist links der experimentelle Aufbau skizziert, rechts sind die Ergebnisse für die drei verschiedenen Experimente nebeneinander aufgetragen. Kugeln, die Spalt 1 passiert haben, sind als weisse Kreise gezeichnet, während die durch Spalt 2 hindurchgegangenen Kugeln durch schwarze Kreise dargestellt sind. Die erste - mit P1 überschriebene - Kolonne zeigt die Verteilung der im Detektor ankommenden Kugeln für den Fall, dass Spalt 1 geöffnet und Spalt 2 geschlossen ist. Die zweite Kolonne, mit P2 bezeichnet, zeigt die ganz ähnliche Verteilung bei geschlossenem Spalt 1 und geöffnetem Spalt 2. Die maximale Anzahl an Kugeln erhält man beide Male in den Auffangbüchsen, die um die direkte Verlängerung der Flugbahn durch den jeweils offenen Spalt liegen. Die letzte Verteilung (P12) zeigt das Ergebnis mit zwei geöffneten Spalten. Ob eine Kugel durch den ersten oder zweiten Spalt hindurchgegangen war, ist eine Sache des Zufalls, und dementsprechend sind die weissen und schwarzen Kugeln in den Auffangbüchsen völlig durcheinandergeworfen. Wichtig ist hier die Beobachtung, dass - wenn beide Spalte geöffnet sind - in jeder der Büchsen die Zahl der Kugeln gleich ist der Summe der beiden entsprechenden Anzahlen, wenn jeweils nur der eine oder der andere Spalt geöffnet ist. Das muss so sein, denn es ist klar, dass die Kugeln nur die Wahl haben, durch einen der beiden Spalte zu fliegen, um zum Detektor zu gelangen. Es gilt also: P12 = P1 + P2 Seite 14 Quantenchemie – eine Einführung 4.2 Doppelspaltexperiment mit Wasserwellen Ein Stein, den wir in ein grosses Wasserbecken werfen, dient uns als „Quelle“ für die Wasserwellen; als „Doppelspaltblende“ legen wir einen Damm, der an zwei Stellen durchbrochen ist, quer über die Wasseroberfläche, und eine Reihe kleiner, auf dem Wasser treibender Bojen, deren Auf- und Abbewegung uns ein Mass für den Energiegehalt der Welle an der betreffenden Stelle gibt, bildet unseren „Wellendetektor“. Die Wasserwellen breiten sich von ihrem Ausgangspunkt aus, bis sie den Damm erreichen. Auf der anderen Seite des Damms bilden sich an den beiden Dammlücken zwei neue, kreisförmige Wellenzüge, die sich von dort weiter ausbreiten; die Bewegung der Wasseroberfläche am Detektor resultiert also aus der Überlagerung dieser beiden Wellenbewegungen. Schauen wir uns die Reihe der Bojen genauer an, so sehen wir, dass an manchen Stellen ein Wellenkamm, der vom Dammspalt 1 ausging, auf einen Wellenkamm von Spalt 2 trifft, wodurch sich eine sehr heftige Auf- und Abbewegung der entsprechenden Boje ergibt. An anderen Stellen wiederum wird ein Wellenkamm von einem der beiden Spalte mit einem Wellental des anderen zusammentreffen, so dass sich die Bojen dort überhaupt nicht bewegen; irgendwo zwischen diesen beiden Extremen liegen die Verhältnisse für die Bojen an den anderen Positionen. Die Auf- und Abbewegung der Bojen als Ganzes entspricht dem Bild einer stehenden Welle: Zwischen den periodisch auftretenden festen Knoten dieser Welle, an denen die Bojen in Ruhe sind, bilden sich die Wellenbäuche, in denen die Bojen zwischen ihren jeweiligen maximalen Auslenkungen hin- und herschwingen. Wenn wir die pro Zeiteinheit, also pro Sekunde an einem Ort ankommende Energie als Intensität (I) bezeichnen, ergibt sich zwischen der Intensität und der maximalen Wellenhöhe (h) folgenden Zusammenhang (abgesehen von Proportionalitätsfaktoren): I = h2 (vgl. Energie-Amplituden-Beziehung) Abbildung 8: Wasserwellen an einem Damm mit zwei Spalten Seite 15 Quantenchemie – eine Einführung Abbildung 9: Das Doppelspaltexperiment mit Wasserwellen Analysiert man das Experiment, sieht das Resultat wie in Abb. 9 dargestellt aus. Die von den beiden Dammspalten ausgehenden Wellenkämme sind schematisch in die Abbildung eingezeichnet (vergleiche auch Abb. 8). Wenn lediglich Spalt 1 geöffnet ist, erhält man die nur wenig schwankende Intensitätsverteilung der ersten Kolonne (I1); sie ist der Verteilung P1 ganz ähnlich, die wir für die Gewehrkugeln in Abb. 7 erhielten. Das Maximum liegt wieder in der Verlängerung der Linie, die Quelle und Spalt 1 miteinander verbindet. Die Verteilung I2 in der zweiten Kolonne, bei der Spalt 1 geschlossen und Spalt 2 geöffnet war, erscheint gegenüber der ersten lediglich etwas verschoben. Die letzte Kolonne I12 jedoch zeigt die völlig anders geartete Intensitätsverteilung für den Fall, dass beide Spalte geöffnet sind: Im Gegensatz zur entsprechenden Verteilung P12 für Gewehrkugeln ergibt sie sich gerade nicht aus der Summe der beiden Verteilungen I1 und I2, die man mit jeweils nur einem geöffneten Spalt erhielt. Die resultierende Intensität (I12) berechnet sich vielmehr nach: I12 = (h1 + h2)2 = h12 + 2 h1h2 + h22 Diese stark schwankende Intensitätskurve ist eine typische Interferenzerscheinung. 4.3 Doppelspaltexperiment mit Elektronen Wir benutzen als „Quelle“ eine sogenannte Elektronen-„Kanone“, einen glühenden Metalldraht, aus dem Elektronen „verdampfen“ und an den eine elektrische Spannung angelegt wird, um die Elektronen zu beschleunigen. Eine dünne Metallplatte mit zwei schmalen Spalten dient uns als „Blende“. Die auf der anderen Quantenchemie – eine Einführung Seite 16 Seite ankommenden Elektronen registrieren wir mit einem „Detektorschirm“, der mit einer phosphoreszierenden Substanz beschichtet ist; diese gibt bei jedem Aufprall eines Elektrons einen Lichtblitz ab. Abbildung 10: Das Doppelspaltexperiment mit Elektronen Wie beim Experiment mit Gewehrkugeln finden wir auch hier, dass die Elektronen als einzelne Materieklümpchen gleicher Grösse ankommen, die - jeweils durch einen Lichtblitz im Detektor angezeigt - zu einem bestimmten Zeitpunkt an einer bestimmten Stelle lokalisiert werden. Fahren wir die Intensität der Elektronenkanone herunter, verdampfen also weniger Elektronen pro Minute, sehen wir immer noch Lichtblitze derselben Helligkeit, nur dass weniger Elektronen während einer Minute ankommen. Ganz analog zum Gewehrkugelexperiment können wir die Lichtblitze, die wir an einer bestimmten Stelle im Detektor in einem festen Zeitintervall beobachten, zählen und somit ein Mass für die Ankunftswahrscheinlichkeit der Elektronen in Abhängigkeit vom Auftreffort gewinnen. Falls nur Spalt 1 geöffnet ist, erhalten wir die Verteilung P 1. Entsprechend die Verteilung P2, wenn Spalt 2 geöffnet ist. Die ganze Eigentümlichkeit der Quantenmechanik offenbart sich uns in dieser einen Verteilung! Die Kurve P12 ist nichts anderes als das charakteristische Interferenzmuster von Wellen, obgleich doch - wie wir gesehen haben - die Elektronen wie einzelne Kugeln ankommen. P12 ist also nicht die Summe von P1 und P2; deshalb kann man nicht sagen, durch welchen Spalt ein bestimmtes Elektron gekommen ist. Um diese Unsicherheit anzudeuten, sind die Elektronen, die ja immer noch wie einzelne Kügelchen ankommen, halb weiss, halb schwarz aufgezeichnet. Das ist seltsam genug; schauen wir uns das Ergebnis jedoch genauer an, dann wird die Sache noch mysteriöser. Betrachten wir dazu eine Stelle auf dem Detektorschirm, an der das Interferenzmuster ein Minimum aufweist, wenn beide Spalte Seite 17 Quantenchemie – eine Einführung geöffnet sind. An dieser Stelle finden wir tatsächlich weniger Elektronen vor, als wir erhielten, wenn wir das Experiment mit nur einem geöffneten Spalt wiederholten! Wir würden dann nämlich die ebenfalls in Abb. 10 eingezeichneten „Einzelspalt“-Verteilungen bekommen, die mit denen der Wasserwellen identisch sind. Wie verträgt sich das aber mit der Tatsache, dass sich die Elektronen bei ihrer Ankunft wie feste Materiekügelchen verhielten? Sollte sich das Elektron etwa in zwei Hälften aufgespalten haben, die jeweils durch einen der beiden Spalte hindurchgegangen waren? Nein - Elektronen werden immer als Ganzes beobachtet, wie die Gewehrkugeln: sie sind an einer bestimmten Stelle entweder ganz da oder nicht da. Seit es die Quantenmechanik gibt, haben sich immer wieder Menschen den Kopf darüber zerbrochen, ob man nicht einen Ausweg aus diesem Dilemma finden könne. Soweit wir wissen, gibt es ihn nicht. Es scheint so, als würden die Elektronen als Teilchen in der Elektronenkanone starten und als Teilchen im Detektor ankommen, doch ist ihre Verteilung dort so, als würden sie sich unterwegs als Wellen fortpflanzen. Wie wir gesehen haben, können wir Interferenz mathematisch sehr einfach beschreiben; im Falle der Wasserwellen ergab sich die Interferenzkurve einfach aus der Addition der jeweiligen Auslenkungen der beiden Wellen, die von Spalt 1 beziehungsweise Spalt 2 ausgingen und zum Detektor gelangten. Die gemessene Intensität oder Energie der Welle war dabei mit dem Quadrat der maximalen Wellenauslenkung (Amplitude) verknüpft. Ganz analog können wir nun die Interferenz von Elektronen mathematisch beschreiben. In diesem Fall messen wir allerdings nicht die Intensität einer realen Wellenbewegung, sondern die Ankunftswahrscheinlichkeit für ein Elektron; übertragen wir unsere einfache mathematische Überlegung auf die Elektroneninterferenz, so müssen wir entsprechend nach einer Art Wellenamplitude für Elektronen suchen. Was aber bedeutet die Amplitude einer solchen „Elektronenwelle“? Das Quadrat dieser Wellenamplitude entspricht der Aufenthaltswahrscheinlichkeit des Elektrons an der betreffenden Stelle, und man nennt diese Grösse daher „quantenmechanische Wahrscheinlichkeitsamplitude“; wir bezeichnen sie im folgenden mit dem Symbol a. Die Gleichungen für die Ankunftswahrscheinlichkeit der Elektronen sehen dann genauso aus wie die Gleichungen für die Intensitäten der Wasserwellen, mit Wahrscheinlichkeiten P (von englisch probability) und Quantenamplituden a anstatt Intensitäten I und Wellenauslenkungen h. Wir erhalten dann jeweils für den Fall zweier geöffneter Spalte beziehungsweise eines geöffneten Spalts die Gleichungen P1 = a 1 2 P2 = a 2 2 P12 = (a1 + a2)2 Wie zuvor addieren sich auch hier nur die zugrundeliegenden Wellenauslenkungen, nicht jedoch die gemessenen Wahrscheinlichkeiten. So bleibt uns also nur die Schlussfolgerung, dass Elektronen im Hinblick auf ihre räumliche Verteilung im Detektor wie Wellen miteinander interferieren, andererseits aber dort wie Gewehrkugeln als diskrete Materieteilchen einzeln registriert werden. Das ist eigentlich gemeint, wenn man davon spricht, dass sich Quantenobjekte manchmal wie eine Welle und manchmal wie ein Teilchen verhalten. Sie mögen das geheimnisvoll finden - es ist geheimnisvoll. Wir können das Rätsel der Quantenmechanik nicht weiter auflösen; alles was wir tun können, ist zu beschreiben, wie sich uns das Verhalten der Quantenobjekte darstellt. Genau das leistet die Quantenmechanik. 5 Heisenbergs Unschärferelation und Schrödingers Gleichung Kann man denn nicht durch eine geeignete Anordnung der Messapparatur den Weg eines Elektrons bestimmen? Was würde dies bedeuten? Damit ein Elektron „sichtbar“ gemacht werden kann, d.h. sein Ort Seite 18 Quantenchemie – eine Einführung bestimmt werden kann, muss Licht, also Photonen, hinter die Spalten geschickt werden. Eine Gewehrkugel würde dies nicht gross stören, hingegen ein Elektron kann durch das Photon aus „seiner Bahn“ geworfen werden. Tatsächlich beobachtet man bei einer derartigen Versuchsanordnung, dass das typische Interferenzmuster verschwunden ist. Die Verteilung präsentiert sich vielmehr gleich wie bei den Gewehrkugeln! Für mikroskopische Objekte ist also dieser Stoss – Wechselwirkung zwischen Messapparatur und Messobjekt – nicht mehr vernachlässigbar. Dies ist, in einfachen Worten ausgedrückt, der Inhalt der Heisenbergschen 4 Unschärferelation. Ort und Geschwindigkeit eines Quantenteilchens kann nicht gleichzeitig festgestellt werden . Die Heisenbergsche Unschärferelation drückt die Beziehung zwischen den jeweiligen Unschärfen der Orts- und Impulsmessung in folgender Weise aus: ∆x ∆p = h (Plank’sches Wirkungsquantum: 6.67 10-34 Js) Die Vorstellung einer Materiewelle (Wellen- und Teilcheneigenschaften, Louis de Broglie) ist nötig, um die Eigenschaften von atomaren Teilchen zu verstehen. 1926 gelang es schliesslich Erwin Schrödinger eine Gleichung zu finden, mit deren Hilfe die Wellenfunktionen (Ψ) für Elektronen ermittelt werden können. Ihr Zweck war es, das Verhalten eines subatomaren Teilchens in derselben Weise zu beschreiben, wie dies bei makroskopischen Teilchen durch die klassische Mechanik geschieht. Die Schrödinger-Gleichung sieht auf den ersten Blick sehr kompliziert aus. Sie sagt aber nur aus, dass die Summe der kinetischen Energie (Ekin) und der potentiellen Energie (Epot) die gesamte Energie (Eges) des Elektronensystems ergibt: Eges = E (Ψ) = EKin + EPot ∂2Ψ ∂2Ψ ∂2Ψ + V (Ψ) ⋅ + + 8 ⋅ π 2 ∂x 2 ∂y 2 ∂z 2 h2 ∇ 2 (Laplace-Operator) oder: ∇2Ψ + 8π 2 m (E − V ) = 0 h2 Ψ: Wellenfunktion des betrachteten Elektrons. Damit lässt sich die maximale Amplitude der Materiewelle an jedem Ort (x, y, z) berechnen. V: Potentielle Energie m: Masse des Teilchens h: Planksches Wirkungsquantum Die Lösung der Wellengleichung ist die Wellenfunktion Ψ. Für reale Wellen entspricht Ψ der Amplitude der Welle und besitzt demgemäss bei der vorliegenden Anwendung keine physikalische Realität. Jedoch so, wie beispielsweise die Intensität einer Lichtwelle durch das Quadrat ihrer Amplitude dargestellt wird, ist die Aufenthaltswahrscheinlichkeit eines Teilchens proportional dem Quadrat seiner Wellenfunktion ( Ψ ). 2 4 vgl. dazu Fussnote 14 im Chemiebuch S. 44 Seite 19 Quantenchemie – eine Einführung Der herausragende theoretische Physiker Richard Feynman meinte einmal zur Schrödinger-Gleichung: „Woher haben wir diese Gleichung? Nirgendwoher. Es ist unmöglich, sie aus irgend etwas Bekanntem herzuleiten. Sie ist Schrödingers Kopf entsprungen“. Als Wellenmechanik oder Quantenmechanik bezeichnet man nun das Teilgebiet der Quantenphysik, dessen Aufgabe es ist, Werte von Ψ zu ermitteln. Grundlage dazu ist die Schrödinger-Gleichung. Mit Hilfe dieser Gleichung lassen sich die verschiedenen Grössen (Aufenthaltswahrscheinlichkeit, Impuls, Energie, Ionisierungsenergien etc.) berechnen. Ausgehend von der Wellenmechanik gelang es, das wellenmechanische Atommodell zu entwickeln, das zu einem grundlegenden Verständnis der chemischen Bindung führt. Die Beschreibung eines Elektrons einmal durch ein Teilchen und ein anderes Mal durch ein Wellenmodell führte zu einer neuen Beschreibung einfacher Systeme, wie eindimensionaler Kasten und Quaderhohlraum. Diese einfachen Analogien können dann auf die Atome und schliesslich auf Atomverbände übertragen werden, wobei die Komplexität der Betrachtung zunimmt, so dass mathematische Näherungsverfahren verwendet werden müssen. 6 Das Teilchen (e-) im Kasten Allgemein kann die Lösung der Wellengleichung für ein Atom sehr schwierig oder sogar unmöglich sein. Das nun folgende Gedankenmodell dient, wie oft bei solchen Modellen, ein komplexes System auf das wesentlichste zu reduzieren. Ein Teilchen, das in einem Kasten eingeschlossen ist, verhält sich in gewisser Weise ähnlich wie ein Elektron, das in einem dreidimensionalen Atom gebunden ist. Eine noch grössere Analogie zum Kasten besteht etwa zu einem linearen Molekül mit konjugierten Doppelbindungen, in denen sich ein Elektron über die gesamte Länge des Moleküls bewegen kann. Betrachten wir ein Elektron in einen 1-dimensionalen Kasten wie er in Abbildung 11 dargestellt ist. Man stellt sich dabei vor, dass das Elektron zwischen zwei parallelen (unendlich hohen) Wänden im Abstand L reflektiert wird. In den Bereichen I und III (ausserhalb des Kastens) soll die potentielle Energie unendlich gross sein (das Teilchen kann den Kasten nicht verlassen), und innerhalb soll sie null sein. Die klassische, Newton‘sche Mechanik sagt für ein solches Teilchen voraus, dass die Aufenthaltswahrscheinlichkeit überall gleich gross ist und dass die kinetische Energie des Teilchens jeden beliebigen Wert annehmen kann. Die Wellenmechanik führt zu ganz anderen Ergebnissen: ∞ ∞ I II III V=∞ V=0 V=∞ 0 Abbildung 11: Ein Elektron eingeschlossen in einem Kastenpotential x L Seite 20 Quantenchemie – eine Einführung 5 Nach de Broglie , der allen bewegten Teilchen einen Wellencharakter zuordnete („alles hat seine Wellenlänge“), können wir dem Elektron eine stehende Materiewelle zuordnen. Die undurchdringlichen Kastenwände reflektieren diese Elektronenwelle. Damit sich die Welle nicht durch Interferenz auslöscht (das Elektron würde damit nicht mehr existieren), sind nur ganz bestimmte Schwingungszustände möglich, wie dies auch bei einem Seil der Fall ist (vgl. Abb. 4). Wie findet man nun die Wellenfunktion für die angenommene Materiewelle? 1. Die Materiewelle bildet eine stehende Welle mit Knoten an den Wänden im eindimensionalen Kasten (Randbedingungen) à Man sucht nun eine Wellenfunktion, die diese Randbedingungen und die Schrödinger-Gleichung erfüllt: Ψn(x) 2. Durch Einsetzen der Wellenfunktion in die Schrödinger-Gleichung erhält man einen Ausdruck für die möglichen Energiezustände der Materiewelle des Elektrons 3. Die Wahrscheinlichkeit, das Elektron an einer Stelle des Kastens anzutreffen, ist proportional zu Ψ2n (x) (Quadrat der Amplitude) An den Kastenwänden müssen sich Knoten befinden, die Wellenfunktion muss die Schrödinger-Gleichung 2 erfüllen und das Quadrat der Amplitude (Ψ (x)) muss über den ganzen Kasten integriert 1 ergeben, was nichts anderes bedeutet, als dass das Elektron mit 100 %-iger Sicherheit irgendwo im Kasten ist. Lösungen, die diese Bedingungen erfüllen sind in der folgenden Abbildung dargestellt: 5 De Broglie verknüpfte dabei in seiner Dissertation die Einsteinsche Masse-Energie-Beziehung E = m c2 mit der Planckschen Gleichung für die Energie von Lichtquanten E = h · f. De Broglie nahm an, dass dem Licht einer bestimmten Frequenz f Photonen einer bestimmten Masse m zugeordnet werden müssen, die sich mit Lichtgeschwindigkeit c fortbewegen. c c⋅h h Mit m · c2= h · f und λ = ergibt sich λ = ; es folgt: λ = f m⋅c m ⋅ c2 Diese Beziehung, die zunächst nur für die Photonen galt, übertrug de Broglie auf bewegte Teilchen, deren Geschwindigkeit kleiner als c ist (v < c) und deren Teilchen den Impuls p = m·v haben. h h Die Wellenlänge berechnet sich dann zu λ = = m⋅v p Seite 21 Quantenchemie – eine Einführung Abbildung 12: Das Teilchen im Kasten: (a) Wellenfunktionen und (b) Wahrscheinlichkeitsfunktionen; bzw. Aufenthaltswahrscheinlichkeiten (nach F.L.Pilar „Elementary Quantum Chemistry“, Mc Graw-Hill, New York, 1968) Wie lassen sich nun die aufgezeichneten Graphen der Wellenfunktion interpretieren? Zum einen sind nur ganz bestimmte Schwingungszustände für die stehende Materiewelle möglich. Zum anderen lassen sich Aussagen über die Amplitude dieser Elektronenwelle machen 1. Entgegen der klassischen Voraussage ist die Aufenthaltswahrscheinlichkeit für das Teilchen nicht konstant, sondern eine Funktion von x. Ausserdem hängt die Wahrscheinlichkeit, das Teilchen in einem bestimmten Bereich des Kastens zu finden, von der Energie des Teilchens ab. 2. Ein weiterer Unterschied zu den klassischen Voraussagen ist die Tatsache, dass nur bestimmte Energie-Werte (Quantisierung) erlaubt sind, die mit der Quantenzahl n verknüpft sind. Die Energie null (n = 0) ist nicht erlaubt, andernfalls wäre Ψ = 0, die Lösung also trivial – die Wahrscheinlichkeit, das 2 Teilchen zu finden wäre null (Ψ = 0); es gäbe also gar kein Teilchen im Kasten. 3. Ausserdem wächst die Energie mit n à En ≈ n . 2 2 Um sich über quantisierte Energieniveaus und deren Wellenfunktionen verständigen zu können, ist es zweckmässig, sie in irgendeiner Weise zu kennzeichnen. Wir führen deswegen eine „Quantenzahl“ n ein und ordnen dem Grundzustand die Zahl n = 1 zu, dem ersten angeregten Zustand die Zahl n = 2, und so weiter. In unserem einfachen Beispiel scheint dies trivial; für die meisten anwendungsorientierten Probleme, die natürlich nicht mehr so einfach zu lösen sind wie unser Beispiel des Elektrons im Kasten, ist diese Kennzeichnung der Wellenfunktionen und Energien durch Quantenzahlen jedoch von tieferer Bedeutung. Nichtsdestoweniger bleiben unsere prinzipiellen Überlegungen weiter gültig: Die Schrödinger-Gleichung für realistischere Situationen zu lösen, läuft also darauf hinaus, die Schwingungsmuster für kompliziertere Systeme zu berechnen. Die Abb. 6 zeigt solche „Klangfiguren“ für ein uns vertrauteres Schwingungsobjekt. Hier kommen wir nun noch zu einem charakteristischen weiteren Merkmal quantenmechanischer Systeme, das in unserem Kastenbeispiel nicht auftaucht. Schauen wir uns dazu in Abb. 5 und 13 die Schwingungsmodi quadratischen „Pauke“ einer an: Das quantenmechanische Analogon - ein Elektron in einem „zweidimensionalen Kasten“ - hat exakt dieselben Lösungen. Wenn wir jetzt die Wellenfunktionen anhand ihrer Energien durchnumerieren, stossen wir auf eine Schwierigkeit. Für den niedrigsten Energiezustand gibt es Abbildung 13: 2-dimensionale Wellenfunktionen mit dazugehörigen Wahrscheinlichkeitsdichten; oben und Mitte links: Grundzustand; oben und Mitte rechts: in einer Schwingungsrichtung angeregt; unten links und rechts: in beiden Schwingungsrichtungen angeregt. Seite 22 Quantenchemie – eine Einführung genau eine mögliche Wellenfunktion, die wir durch eine einzige Quantenzahl n = 1 kennzeichnen. Für den ersten angeregten Zustand n = 2 haben wir jedoch die Wahl zwischen zwei unabhängigen Schwingungsrichtungen x und y: Wir können entweder in x-Richtung den ersten Oberton anregen und in yRichtung den Grundton oder umgekehrt. Beide Möglichkeiten führen zu exakt derselben Energie. Wir benötigen daher eine weitere Quantenzahl, um die beiden zu diesem Energiewert gehörenden Wellenfunktionen voneinander zu unterscheiden. Zum Beispiel können wir durch den Zusatz x oder y spezifizieren, welche Schwingungsrichtung „angeregt“ ist; wir unterscheiden also die Wellenfunktionen 2x und 2y. In einer solchen Situation, in der es mehr als einen möglichen Quantenzustand mit derselben Energie gibt, sprechen wir von „Entartung“ (oder „entarteten Zuständen“). Ähnliche Entartungen werden wir vorfinden, wenn wir uns mit den Wellenfunktionen des Wasserstoffatoms beschäftigen. Es sollte uns dann nicht mehr überraschen, dass wir bei diesem - dreidimensionalen - Problem (hier befindet sich das Elektron in einem Quaderhohlraum) mindestens drei Quantenzahlen benötigen werden, um alle möglichen Quantenzustände eindeutig zu kennzeichnen. 7 Wasserstoffatom Um in der Quantenmechanik der chemischen Elemente etwas tiefer schürfen zu können, müssen wir noch mehr über die Wellenfunktionen und Quantenzahlen wissen, die man für die Beschreibung des Wasserstoffatoms braucht. Wir werfen einen Blick auf die Wasserstoff-Wellenfunktionen, das heisst auf die Wahrscheinlichkeitswellen für die Quantenzustände des Elektrons, die den Energieniveaus des Wasserstoffs entsprechen. Wie schon erwähnt, müssen wir noch weitere Quantenzahlen einführen. Die Überlegungen zum ein- (und analog zum dreidimensionalen Kasten) lassen sich auf das Wasserstoffatom übertragen, wenn man die Wände des Quaderhohlraumes durch elektrostatische Anziehungskräfte ersetzt (Randbedingung). Im Zentrum des Atoms befindet sich die positive Kernladung, in der Elektronenhülle die negative Elektronenladung. Das Elektron im Wasserstoffatom lässt sich, gemäss der Wellenmechanik, mit einer dreidimensionalen stehenden Elektronenwelle beschreiben. Entsprechend der Modellvorstellung einer stehenden Materiewelle, kann das Elektron im Wasserstoffatom nur ganz bestimmte (diskrete) Energiezustände annehmen. Das rechnerische Vorgehen entspricht demjenigen des eindimensionalen Kastens: 1. „Suchen“ der Funktion der Materiewelle; Interpretation der Wellenfunktion 2. Einsetzen der Wellenfunktion in die Schrödinger-Gleichung, um die Energiezustände des Systems Elektron-Proton zu erhalten 3. Quadrieren der Wellenfunktion; Interpretation der neuen Funktion; Aussage über die Elektronendichte (Aufenthaltswahrscheinlichkeit) des Elektrons im entsprechenden Energiezustand. Man stellt fest, dass die Lösung für das Wasserstoffatom drei Quantenzahlen n, l und ml erfordert, wie es für ein dreidimensionales System zu erwarten ist. Jede Lösung, die sich für eine bestimmte Kombination von n, l und ml ergibt, wird Eigenfunktion genannt und stellt ein Orbital des Wasserstoffatoms dar. Diese zusätzlichen Quantenzahlen ergeben sich aus der Quantisierung einer anderen klassischen Grösse, des Drehimpulses. In den folgenden Absätzen wollen wir versuchen, diese neuen Quantenzahlen etwas eingehender zu charakterisieren. Wie sein Name schon sagt, hängt der Drehimpuls mit dem gewöhnlichen Impuls zusammen; er spielt eine wichtige Rolle bei der Beschreibung von Systemen, in denen Teilchen um ein Zentrum kreisen, wie zum Beispiel die Planeten um die Sonne. Stellen wir uns vor, wir würden ein Stück Seil an Seite 23 Quantenchemie – eine Einführung einem Ball befestigen, das andere Seilende in die Hand nehmen und nun den Ball immerzu um uns herumschleudern. Der Drehimpuls des Balls ist dann einfach das Produkt aus dem gewöhnlichen Impuls des Balls (Masse mal Geschwindigkeit) und der Seillänge, also: L=rxp (Drehimpuls = Seillänge mal gewöhnlicher Impuls.) Für konstante Seillänge gilt: Je schneller der Ball rotiert, desto grösser ist sein Drehimpuls. Den Drehimpuls eines Objekts zu kennen, ist deshalb wichtig, weil er - wie die Energie - sowohl in klassischen als auch in Quantensystemen insgesamt erhalten bleibt. Was passiert nun in unserem Gedankenexperiment, wenn wir, während der Ball weiter kreist, das Seil verkürzen? Da sein Drehimpuls erhalten bleibt, muss, wenn die Seillänge r abnimmt, der gewöhnliche Impuls p des Balls grösser werden - der Ball rotiert schneller. Auch in der quantenmechanischen Behandlung des Wasserstoffatoms ist der Drehimpuls eine Erhaltungsgrösse. Die Quantentheorie lässt jedoch nicht jeden beliebigen Wert für den Drehimpuls zu: Der quantenmechanische Drehimpuls ist - ähnlich wie die Energie - quantisiert. Genau davon war Bohr ausgegangen, als er seine stabilen Elektronenbahnen postulierte: Der Drehimpuls wird durch zwei neue Quantenzahlen l und m beschrieben: Der Index l legt den jeweiligen Betrag des Drehimpulses fest, während die Quantenzahl m - grob gesprochen - die räumliche Orientierung der Drehung ausdrückt. In zwei Dimensionen kann man sich die Quantisierung des Drehimpulses in etwa so vorstellen, wie es in Abb. 14 gezeigt wird. Dreidimensional kann das Elektron - ähnlich einem Kreisel – in den drei verschiedenen Raumrichtungen rotieren. Stellen wir uns noch einmal den Ball am Seil vor und lassen ihn um die vertikale z-Achse rotieren: Der Ball umkreist diese Achse also in der dazu senkrechten Ebene, die von der x- und der y-Achse aufgespannt wird. Quantenmechanisch entspricht diese Situation dem Zustand m = +1, dessen Wahrscheinlichkeitsverteilung ringförmige rotations- symmetrisch zur z-Achse ist und sich hauptsächlich um die x-y-Ebene konzentriert. Der Zustand m = -1 entspricht einer Rotation in entgegengesetzter Richtung - um die negative z-Achse -, so dass die Bereiche hoher Aufenthaltswahrscheinlichkeit Abbildung 14: Quantisierung des Drehimpuls Seite 24 Quantenchemie – eine Einführung wiederum um die x-y-Ebene herum zu liegen kommen. Beim m = 0-Zustand schliesslich sind sie in z-Richtung orientiert und haben die Form einer Hantel, was in etwa einer Rotationsachse irgendwo in der x-y-Ebene entspricht. (Eine exakte Rotatationsrichtung lässt sich nicht angeben; hier kommt wieder die quantenmechanische Unbestimmtheit ins Spiel!). 7.1 Orbitale6 Eine Wellenfunktion, die ein Elektron in einem Elektronensystem beschreibt, nennt man Orbital. Je nachdem, ob es sich um ein Elektron eines Atoms oder eines Moleküls handelt, spricht man von Atom- oder Molekülorbital. Die aus der Schrödinger-Gleichung berechneten Werte von ψ lassen sich nicht messen. Die Orbitale sind mathematische Funktionen ohne anschauliche Bedeutung, die nicht beobachtbar (nicht observabel) sind. Mittels solcher Wellenfunktionen können aber z.B. Energien von Atomen und Molekülen, lonisierungsenergien, Bindungsenergien u.a. berechnet werden. 7 2 Hingegen erkannte Born , dass das Quadrat der Wellenfunktion (Ψ ) eine anschauliche Bedeutung hat: es ist 8 ein Mass für die Wahrscheinlichkeit, das Elektron an einer bestimmten Stelle zu finden , was sich auch experimentell bestätigen liess. Das Quadrieren der Wellenfunktion liefert deshalb Aussagen darüber, wo und mit welcher Wahrscheinlichkeit das Elektron zu finden ist. Da sich diese Wahrscheinlichkeit auf ein Volumenelement dV bezieht, gibt ψ die Wahrscheinlichkeitsdichte an (auch Elektronendichte, Ladungsdichte 2 9 oder Aufenthaltswahrscheinlichkeit pro Volumeneinheit genannt). Es ist also möglich, Räume zu ermitteln, in denen sich Elektronen mit einer bestimmten Wahrscheinlichkeit aufhalten (z.B. 90%). Ein solcher Raum wird als Wahrscheinlichkeitsraum eines Elektrons oder als „Elektronenwolke“ bezeichnet, denn man kann die Wahrscheinlichkeitsverteilung als eine in bestimmter Weise über das Atom verteilte „Wolke“ negativer Ladung veranschaulichen. Diese Ladungswolken besitzen an den Stellen grösster Aufenthaltswahrscheinlichkeit (d.h. dort, wo sich ein Elektron am häufigsten aufhält) ihre grösste Dichte. Die Darstellungen solcher Wolken kann man sich als Summierung zahlreicher „Momentaufnahmen“ des Elektrons vorstellen; ein Punkt entspricht dabei dem Ort des Elektrons in einem bestimmten Augenblick. Dort, wo die Punkte dichter liegen, hält sich das Elektron häufiger auf. Oft zeichnet man die Ladungswolken mit Umgrenzungslinien, die dann eine bestimmte prozentuale Aufenthaltswahrscheinlichkeit (meist 90%) einschliessen. Es muss betont werden, dass die Beschreibung des Verhaltens eines Elektrons durch eine Wellenfunktion nicht zur Vorstellung verleiten darf, „das Elektron sei eine Welle“ oder „das Elektron bewege sich wellenförmig“ oder „das Elektron schwinge“ bzw. es „nehme bestimmte Schwingungszustände ein“; es handelt sich vielmehr um eine Möglichkeit seine Aufenthaltswahrscheinlichkeit zu berechnen. Haben wir vorher von Quantenzahlen gesprochen, werden diese Zahlen in der Chemie zum Teil mit Buchstaben abgekürzt. Die Hauptenergieniveaus n entsprechen den Schalen (K, L, M, usw.) der Elektronenhülle. Die Quantenzahl l entspricht den Orbitalen s, p, d und f, wobei diese Orbitale entartet sein können. l = 0 entspricht dem s-Orbital, l = 1 dem p-Orbital usw. Die Schalen enthalten jeweils alle n – 1 Orbitale. Beispielsweise hat die M-Schale (n = 3) alle Orbitale, die einem l von 3 – 1 = 2 entsprechen, also l = 0 (s-Orbital), l = 1 (p-Orbitale) und l = 2 (d-Orbitale). Die Quantenzahl m entspricht der Entartung der Orbitale, wobei m = –l, ....., 0,....., +l möglich sind. So haben die d-Orbitale (l = 2) für m die Möglichkeiten 6 7 8 Bearbeiten Sie auch Kapitel 28 in Ihrem Chemiebuch M. Born. deutscher Physiker(1882-1970). Nobelpreis 1954 Vgl. dazu das Elektron am Doppelspalt Seite 25 Quantenchemie – eine Einführung m = -2, -1, 0, 1, 2. Es gibt also für die d-Orbitale 5 entartete Zustände. Diese Zustände werden in der Chemie 10 mit den Raumindizes x, y und z angegeben . 7.2 Der erste Energiezustand (Grundzustand) des Elektrons im Wasserstoffatom Wie sehen aber nun diese Orbitale aus? Nach Max Born ist das Quadrat der Amplitude der angenommenen Materiewelle proportional zur Aufenthaltswahrscheinlichkeit des Elektrons in einem bestimmten Raumvolumen. Auf Grund des Coulomb-Potentials kann man annehmen, dass die Elektronendichte nahe des Kerns gross ist und mit zunehmendem Abstand im Unendlichen gegen Null strebt. Somit bietet sich für die Wellenfunktion eine abnehmende Exponentialfunktion an. Löst man also die SGL für das 1s-Orbital, erhält man folgendes Resultat: Ψ = e − a⋅r Wobei Ψ: Amplitude der stehenden Welle r: Abstand vom Atomkern a: Zahlenfaktor; Konstante, in der u.a. die Planksche Wirkungskonstante und die elektrische Elementarladung enthalten sind e: Eulersche Zahl, 2.718 Grafisch dargestellt: Ψ = e − a ⋅r Abbildung 15: Graphische Darstellung der Wellenfunktion Ψ für den Grundzustand des H-Atoms Die Amplitude der angenommenen Materiewelle hat in der Nähe des Atomkerns einen grossen Wert; die Funktionswerte werden mit zunehmendem Abstand kleiner und streben im Unendlichen gegen Null. Auch hier sei nochmals erwähnt, dass diesen Amplituden keine physikalische Realität zukommt! Da die Wellenfunktion 1s nur von der Variablen r abhängig ist, ist der Verlauf von Ψ1s nach allen Raumrichtungen gleich und die 1s-Funktion lässt sich auf einfache Art räumlich darstellen. Man zeichnet alle Punkte für r, die den gleichen Funktionswert Ψ1s besitzen. Der entsprechende geometrische Ort ist ein Kreis in der Zeichenebene, bzw. eine Kugeloberfläche bei Berücksichtigung aller Raumrichtungen. 9 In Analogie zur Dichte: Masse pro Volumeneinheit vgl. Merkblatt „Die Quantenzahlen“ 10 Seite 26 Quantenchemie – eine Einführung Ψ = e − a ⋅r Abbildung 16: Geometrischer Ort aller Punkte P mit gleichem Funktionswert. Links: in der Zeichenebene; rechts: räumlich 7.2.1 Wird Das Quadrat der Wellenfunktion: Wahrscheinlichkeitsdichte und radiale Wahrscheinlichkeitsdichte die Wellenfunktion des Grundzustandes quadriert, so ergibt sich daraus die sogenannte Wahrscheinlichkeitsdichte (Elektronendichte, Ladungsdichte) mit der sich die Aufenthaltswahrscheinlichkeit in einem bestimmten Volumenelement dV berechnen lässt. Je nach Betrachtungsweise unterscheidet man neben der Wahrscheinlichkeitsdichte die radialen Wahrscheinlichkeitsdichte. Die Wahrscheinlichkeitsdichte Die Wahrscheinlichkeitsdichte (Elektronendichte, Ladungsdichte) vergleicht die Aufenthaltswahrscheinlichkeit des Elektrons in gleichen Volumenteilen dV, wenn diese verschiedene Abstände vom Kern aufweisen. Das Quadrat der Wellenfunktion 1s: Ψ 2 = e −2 a ⋅ r Die Aufenthaltswahrscheinlichkeit in einem bestimmten Volumenteil dV mit einem bestimmten Abstand vom Atomkern berechnet sich nach dW = Ψ dV = e 2 -2a·r dV. Um Ψ graphisch darstellen zu können, muss man die 2 Wahrscheinlichkeit dW für verschiedene Abstände r berechnen und so die erhaltenen Punkte miteinander verbinden. Untenstehende Abbildung zeigt, dass die Wahrscheinlichkeit, das Elektron in einem Raumelement dV in der Nähe des Kerns anzutreffen sehr gross ist. Seite 27 Quantenchemie – eine Einführung Abbildung 17: Links Graphische Darstellung der Elektronendichte im Grundzustand des H-Atoms Rechts: Elektronendichte für Ψ2 im Grundzustand des H-Atoms Die Wahrscheinlichkeitsdichte des Wasserstoffelektrons im Grundzustand nimmt mit zunehmendem Kernabstand stark ab (im Abstand von 0.182 pm ist die Wahrscheinlichkeitsdichte bereits auf 1/1000 des maximalen Wertes abgesunken); sie wird jedoch erst im Unendlichen Null. Dies bedeutet, dass auch in grösserer Entfernung vom Kern eine zwar extrem kleine, aber doch endliche Wahrscheinlichkeit dafür besteht, das Elektron dort anzutreffen. Mit anderen Worten, das Wasserstoffatom hat (wie natürlich auch die anderen Atome) nach aussen theoretisch keine scharfe Umgrenzung. Die radiale Wahrscheinlichkeitsdichte (radiale Aufenthaltswahrscheinlichkeit, radiale Elektronendichte) Das Kraftfeld des Atomkerns ist dreidimensional und kugelsymmetrisch. Deshalb ist es sinnvoll, auch die sogenannte radiale Wahrscheinlichkeitsdichte zu betrachten: Die radiale Wahrscheinlichkeitsdichte (radiale Elektronendichte) vergleicht die Aufenthaltswahrscheinlichkeit des Elektrons in Kugelschalen gleicher Dicke dr, wenn diese verschiedene Abstände vom Kern aufweisen. Anschaulich in zwei Dimensionen ist dies in der folgenden Abbildung dargestellt: r dr r1 r r2 r dr r1 r2 r Abbildung 18: oben: schematische Darstellung der radialen Wahrscheinlichkeitsdichte; unten: In welchem Abstand vom Atomkern des Wasserstoffatoms befindet sich die Kugelschale mit der grössten Aufenthaltswahrscheinlickeit des Elektrons? Quantenchemie – eine Einführung Seite 28 Nimmt man nun diese Zählung vor, erhält man folgende radiale Verteilung des Elektrons im 1s-Zustand: Abbildung 19: Graphische Darstellung der radialen Wahrscheinlichkeitsdichte des Elektrons im Wasserstoffatom (Grundzustand) Stellt man diese Graphik räumlich dar, erhält man die in Abb. 20 dargestellte kugelsymmetrische Verteilung der Elektronen im 1s-Zustand. Abbildung 20: Räumliche Darstellung das Aufenthaltsraumes (Ladungswolke) des Elektrons im Wasserstoff; 1s-Zustand 7.2.2 Aufenthaltsraum (Ladungswolke) des Elektrons im Grundzustand Es ist oft zweckmässig, den Umriss des Raumes darzustellen, in dem sich das Elektron gemäss der Wahrscheinlichkeitsdichte mit einer bestimmten Wahrscheinlichkeit aufhält (z.B. 90 %). Diesen Raum bezeichnet man als Aufenthaltsraum des Elektrons. Das Teilchenmodell steht dabei „Pate“: Dieser Aufenthaltsraum ist identisch mit dem Raum, der einen bestimmten Betrag der Elektronendichte umschliesst. Da das Wellenmodell in dieser Betrachtung überwiegt spricht man von einer Ladungswolke. Für den 1s-Zustand entspricht dies einer Kugel (vgl. Baars/Christen „Chemie – Theorie und Praxis, Sauerländer, 1996/98, Kapitel 4 und Kapitel 28, und Duden Chemie Kapitel 3.1). Seite 29 Quantenchemie – eine Einführung 7.2.3 Zusammenfassung des Zustandes 1s Zusammenfassung des Grundzustandes 1s Oben links: Wellenfunktion 1s, Zeichenbene Oben rechts: Wellenfunktion 1s, räumlich Mitte links: Quadrat der Wellenfunktion 1s; bezogen auf das konstante Volumen dV à Wahrscheinlichkeitsdichte Mitte rechts: Quadrat der Wellenfunktion 1s; bezogen auf das Volumen von Kugelschalen 4πr2 mit konstanter Dicke dr à radiale Wahrscheinlichkeitsdichte unten: Ladungswolke bzw. Aufenthalstraum (Raum in dem sich das Elektron zu z.B. 90 % aufhält Seite 30 Quantenchemie – eine Einführung 7.3 Der zweite Energiezustand des Elektrons im Wasserstoffatom Als Lösung der SGL findet man für den zweiten Energiezustand (n = 2) des Wasserstoffatoms vier Funktionen, welche die angenommene Materiewelle des Elektrons beschreiben. Eine 2s-Wellenfunktion (l = 0; m = 0) und drei 2p-Wellenfunktionen (l = 1; m = -1, 0, +1). 2 bedeutet zweiter Energiezustand; p und s symbolisieren die räumliche Form der entsprechenden Wellenfunktion 7.3.1 Der 2s-Zustand 7.3.1.1 Die Wellenfunktion des 2s-Zustandes Die Wellenfunktion Ψ des 2s Zustandes lautet in ihrer vereinfachten Form wie folgt: Ψ2 s = (2 − r ) ⋅ e − r Im Vergleich zur Wellenfunktion Ψ1s besitzt sie also eine Nullstelle für r = 2, wie in Abbildung 21 ersichtlich ist. Abbildung 21: links: Graphische Darstellung der Wellenfunktion 2s entlang einer Raumrichtung und rechts: Geometrischer Ort aller Punkte mit gleichem Funktionswert Ψ2s (räumlich) Die Wellenfunktion Ψ2s ist nur von der Variablen r abhängig, ihr Verlauf ist nach allen Raumrichtungen hin also gleich. Die räumliche Darstellung ist deshalb auch hier sehr einfach und führt mit ähnlichen Überlegungen wie wir bei der Wellenfunktion 1s angestellt haben (Suche nach Punkten für r mit selbem Funktionswert Ψ2s) zu der Darstellung in Abbildung 21 rechts. Wir erkennen darin die Knotenfläche (Kugelfläche) bei r = 2 in der räumlichen Darstellung. 7.3.1.2 Das Quadrat der Wellenfunktion 2s; Wahrscheinlichkeitsdichte und radiale Wahrscheinlichkeitsdichte Wie im Grundzustand unterscheidet man auch hier die beiden Wahrscheinlichkeitsdichten. Die Aufenthaltswahrscheinlichkeit in einem bestimmten Volumenteil dV mit einem bestimmten Anstand r vom Atomkern (Wahrscheinlichkeitsdichte) sieht grafisch dargestellt folgendermassen aus: Seite 31 Quantenchemie – eine Einführung Abbildung 22: Geometrischer Ort aller Punkte mit der gleichen Elektronendichte, links in der Zeichenebene und rechts räumliche Darstellung. Vergleicht man die Aufenthaltswahrscheinlichkeit des Elektrons in Kugelschalen gleicher Dicke dr mit unterschiedlichen Abständen vom Atomkern, so erhält man die radiale Wahrscheinlichkeitsdichte mit folgendem Funktionsbild: Abbildung 23: Graphische Darstellung der radialen Wahrscheinlichkeitsdichte. Zusammenfassend lässt sich also sagen, dass im ersten angeregten Zustand 2s sich das Elektron an beliebigen Stellen im Wasserstoffatom (mit unterschiedlichen Wahrscheinlichkeiten) aufhält. Eine Ausnahme bildet die Kugelschale mit dem Abstand r0, in der die Aufenthaltswahrscheinlichkeit Null ist (Knotenfläche). Mit grösster Wahrscheinlichkeit hält sich das Elektron in der Kugelschale beim Maximum auf. Seite 32 Quantenchemie – eine Einführung 7.3.1.3 Zusammenfassung des Zustandes 2s Zusammenfassung des Grundzustandes 2s Oben links: Wellenfunktion 2s, Zeichenbene Oben rechts: Wellenfunktion 2s, räumlich Mitte links: Quadrat der Wellenfunktion 2s; bezogen auf das konstante Volumen dV à Wahrscheinlichkeitsdichte Mitte rechts: Quadrat der Wellenfunktion 2s; bezogen auf das Volumen von Kugelschalen 4πr2 mit konstanter Dicke dr à radiale Wahrscheinlichkeitsdichte unten: Ladungswolke bzw. Aufenthalstraum (Raum in dem sich das Elektron zu z.B. 90 % aufhält Quantenchemie – eine Einführung 7.3.2 Seite 33 Der 2p-Zustand Die drei 2p-Wellenfunktionen, die sich ebenfalls als Lösungen für den zweiten Energiezustand ergeben, sind im Gegensatz zu den 2s-Wellenfunktionen nicht kugel- sondern achsensymmetrisch. Analoge Überlegungen, wie wir sie bei den Zuständen 1s und 2s gemacht haben ergeben untenstehende zusammenfassende Betrachtung 11 des Zustandes 2p , wobei zu beachten ist, dass die charakteristische „Hantel“ der p-Wolke zwei Bereiche hat, die man mit + und – bezeichnet (nicht mit den elektrischen Ladungen zu verwechseln!): Legende zur folgenden Seite: Zusammenfassung des ersten angeregten Zustandes 2p A: Graphische Darstellung der Wellenfunktionen 2p entlang der Koordinatenachsen, von links nach rechts: 2px, 2py, 2pz. B: Geometrischer Ort aller Punkte (räumlich) mit dem gleichen Funktionswert Ψ C: Wolkendarstellung der Wahrscheinlichkeitsdichten entlang der drei Raumachsen D: Graphische Darstellung der radialen Wahrscheinlichkeitsdichte für das Elektron im Wasserstoffatom im Zustand 2p E: Aufenthaltsräume (Ladungswolken) des Elektrons im Wasserstoffatom für die Zustände 2px. 2py und 2pz. 11 Für interessiert sei auf das Leitprogramm „Quantenchemie“ von G. Baars et al. unter http://dcbwww.unibe.ch/fachdidaktik/lpqc.htm verwiesen (Verlag ETHZ, Abteilung für das höhere Lehramt). Seite 34 Quantenchemie – eine Einführung A B C D E Seite 35 Quantenchemie – eine Einführung 7.3.3 Die höheren Energiezustände des Wasserstoffatoms Anhand der Linienspektren haben wir schon einmal besprochen, dass Elektronen in einen höheren Energiezustand angehoben werden können. Das Spektrum des „Wasserstofflichtes“ hat gezeigt, dass beim Übergang von einem angeregten Zustand in den Grundzustand nur ganz bestimmte Energien abgegeben werden können. Dies hat uns zu der Annahme geführt, dass das Wasserstoffatom nur ganz bestimmte (diskrete) Energiezustände einnehmen kann. Wie wir gesehen haben, existieren für diese Zustände bestimmte Wellenfunktionen, aus denen sich die Energien des Systems Proton – Elektron und die Aufenthaltswahrscheinlichkeiten des Elektrons berechnen lassen. Die Energiezustände bezeichnet man mit kleinen ganzen Zahlen, den sogenannten Hauptquantenzahlen; Symbol: n. Sie entsprechen den Hauptenergieniveaus (Elektronenschalen). Im Wasserstoffatom liefern die 2 Berechnungen für die Quantenzahlen n jeweils n Wellenfunktionen, welche der SGL genügen. Für den Grundzustand also 1 Lösung, die 1s-Wellenfunktion; der erste angeregte Zustand besitzt jedoch bereits 4 2 Lösungen (2 =4), eine 2s und drei 2p-Wellenfunktionen. Der nächst höhere Zustand hat also 9 verschiedene Lösungen, eine 3s-, drei 3p-, und fünf 3d-Wellenfuktionen etc. Zusammenfassend ergeben sich für das Wasserstoffatom verschiedene Energieniveaux, welche mit der jeweiligen Hauptquantenzahl n (entsprechend der Schalennummer) bezeichnet werden. Innerhalb eines jeden 2 Energieniveaus lassen sich n verschiedene Funktionen unterscheiden mit n-1 Knotenflächen. Die Energiewerte innerhalb eines bestimmten Energieniveaus sind identisch; man spricht von entarteten Energiezuständen. E [eV] -13.6 s p d 4 3 16 9 2 4 1 1 3 2 3 2 1 1 0 0 f Anzahl Funktionen Ψ (n2) Hauptquantenzahl n Anzahl Knotenflächen pro Funktion Abbildung 24: Energiezustände des Wasserstoffatoms für n = 1 bis n = 4. Auf den beiden folgenden Seiten sind für n = 1 bis 3 die Wellenfunktionen und deren graphische Darstellung abgebildet: Quantenchemie – eine Einführung Abbildung 25: links: rechts: Seite 36 Wellenfunktionen des Elektrons im Wasserstoffatom für die ersten drei Energiezustände (Schalen) Die entsprechenden Wahrscheinlichkeitsdichten Quantenchemie – eine Einführung Abbildung 26: Räumliche Darstellung der radialen Wahrscheinlichkeitsdichten; Orbitale Seite 37 Seite 38 Quantenchemie – eine Einführung 8 Atome mit mehr als einem Elektron: Der Aufbau des PSE In Vielelektronensystemen kommt es zu komplizierten Wechselwirkungen zwischen den Elektronen einerseits und den Elektronen mit dem Atomkern andererseits. Das hat wichtige Konsequenzen für die quantenmechanische Behandlung: - Die einzelnen Elektronen sind prinzipiell nicht unterscheidbar und lassen sich hinsichtlich ihrer Energie und Elektronendichte nicht mehr einzeln erfassen. - Man verwendet wasserstoffähnliche Wellenfunktionen - Die Wellenfunktionen der stehenden Wellen für irgendein Elektron in einem Elektronenkollektiv werden als Orbitale bezeichnet. Die dazu passenden Energiewerte und Elektronendichten sind die Orbitalenergien; bzw. die Orbitalelektronendichten. Beide Grössen sind fiktiv; nicht observabel. - Mit Hilfe dieser Orbitale lassen sich die Gesamtenergie und die Gesamtelektronendichte eines Vielelektronensystems berechnen. Diese beiden Grössen sind observabel, d.h. messbar. - Innerhalb einer Elektronenschale ist die beim Wasserstoffatom beobachtete Entartung aufgehoben. Die Verwendung wasserstoffähnlicher Wellenfunktionen für die Beschreibung der Elektronen in einem Mehrelektronensystem ergibt die charakteristische Gliederung der Elektronenschalen höherer Atome. Da nach dem Pauli-Prinzip in einem Mehrelektronensystem zwei Elektronen nicht in allen Eigenschaften (Quantenzahlen) übereinstimmen können, kann man mit einer Wellenfunktion maximal zwei Elektronen beschreiben. Jedes Elektron ist charakterisiert durch den Energiezustand (Hauptquantenzahl n), die Art der Wellenfunktion (Quantenzahl l à s,p,d,...), durch die sogenannte magnetische Quantenzahl (ml à px, py, pz, ...) und durch den Spin s. Der Spin kann, ähnlich der Eigenrotation der Erdkugel, als eine Art Kreiselbewegung angesehen werden. Ihm werden willkürlich die Werte ±½ (dargestellt durch /) zugeordnet. Zwei Elektronen, die sich durch dieselbe Wellenfunktion beschrieben werden, unterscheiden sich nur noch im Drehimpuls der Eigenrotation. Stehen in einem Atom energetisch gleichwertige Wellenfunktionen zur Verfügung, so werden diese mit den neu hinzukommenden Elektronen (bei zunehmender Ordnungszahl) zuerst einzeln besetzt (Hundsche Regel). Diese Regel findet auch im alltäglichen Leben ihre Anwendung. Denken Sie nur an einen Bus oder einen Vorortszug am Morgen in der Schweiz. Auch hier werden die Abteile zuerst einzeln gefüllt. Scheinbar braucht es mehr Energie, sich in ein bereits (halb-) besetztes Abteil zu setzen ... Seite 39 Quantenchemie – eine Einführung Mit den Orbitalen hat man also ein Ordnungsprinzip zur Verfügung, das es erlaubt, die Elektronenhüllen höherer Atome zu gliedern. Die Elektronen lassen sich auf die verschiedenen Orbitale verteilen. Um den Aufbau des Periodensystems verstehen zu können, sind, wie oben erwähnt, drei einfache Regeln zu beachten: 1. Energieärmster Zustand Es werden immer zuerst die energieärmsten Orbitale mit Elektronen aufgefüllt. 2. Pauli Prinzip Ein Orbital kann maximal 2 Elektronen aufnehmen. 3. Hundsche Regel Stehen in einem Atom energetisch gleichwertige Orbitale zur Verfügung, so werden diese mit den neu hinzukommenden Elektronen zuerst einzeln besetzt. Mit diesen einfachen Regeln können alle bekannten Elemente in einer Tabelle dargestellt werden. Diese Tabelle ist das Periodensystem. Betrachten wir nun zwei Beispiele für die Elektronenkonfiguration eines Atoms im Grundzustand. Unsere 2 2 2 Beispiele sind Kohlenstoff mit der Elektronenkonfiguration 1s 2s 2p , Natrium mit der Elektronenkonfiguration 2 2 6 1 2 2 6 2 6 1 1s 2s 2p 3s und Kalium mit der Elektronenkonfiguration 1s 2s 2p 3s 3p 4s . E 3d 4s 3p 3s 2p 2s C: 1s22s22p2 1s Na: 1s22s22p63s1 K: 1s22s22p63s23p64s1 Abbildung 27: Elektronenkonfigurationen von C (links), Na (Mitte) und Kalium (rechts) Die Energie der Orbitale wächst allgemein in der Reihenfolge s < p < d < f. Aufwendige Rechnungen haben ergeben, dass bei höheren Hauptquantenzahlen eine Änderung der Orbitalfolge eintreten kann. Die Energie eines gegebenen Orbitals hängt von der Kernladung (Ordnungszahl) ab, durch welche die verschiedenen Orbitale in unterschiedlichem Masse beeinflusst werden. Daher gibt es eigentlich keine eindeutige Reihenfolge der Orbitalenergien, die für alle Elemente gültig ist. Dennoch hat sich folgende Ordnung als ausserordentlich brauchbar erwiesen: 1s < 2s < 2p < 3s < 3p < 4s < 3d < 4p < 5s < 4d < 5p < 6s < 5d ≅ 4f < 6p < 7s < 6d ≅ 5f 2 2 6 2 6 1 So hat das Element Kalium (Z = 19, Abb. 27, rechts) die Konfiguration 1s 2s 2p 3s 3p 4s obwohl die 3d-Orbitale noch verfügbar sind: das 4s-Orbital liegt energetisch tiefer als die 3d-Orbitale. Seite 40 Quantenchemie – eine Einführung 9 Molekülbindung Der Wasserstoff existiert normalerweise in Form von H2-Molekülen. Warum verbinden sich nun eigentlich Wasserstoffatome zu Wasserstoffmolekülen? Wir betrachten zwei Wasserstoffatome, die sich einander nähern und verwenden dazu die Vorstellung der Materiewelle. Im Bereich zwischen den beiden Atomen kommt es zu einer Überlagerung der beiden Materiewellen. Konstruktive und destruktive Interferenz ist die Folge dieser Überlagerung (vgl. Kapitel 4 im Duden Chemie). 9.1 Bindender Zustand Überlagern sich die beiden Materiewellen im Bereich zwischen den Atomkernen, so kann es dort zu einer konstruktiven Interferenz kommen, was den bindenden Zustand im Wasserstoffmolekül ergibt. Es entsteht eine neue Materiewelle, die durch eine neue Wellenfunktion beschrieben wird. Damit lassen sich die zwei Elektronen im so entstandenen Wasserstoffmolekül beschreiben. Aus einer Atomwellenfunktion, oder Atomorbital (AO) wurde ein Molekülwellenfunktion, oder Molekülorbital (MO). Ψ1+2 = Ψ1 + Ψ2 Abbildung 28: Wellenfunktion des Systems H2 im Grundzustand zwischen den Kernen links: Wellenfunktion der einzelnen H-Atome rechts: Verlauf der Wellenfunktion im Molekül H2 Aus der neuen Wellenfunktion resultiert schliesslich die Wahrscheinlichkeitsdichte Ψ 2 1+2 (bezogen auf gleiche Volumenteile in unterschiedlichen Abständen von den Protonen) zwischen den beiden Kernen. Ψ21+2 = (Ψ1 + Ψ2)2 = Ψ12 + Ψ22 + 2Ψ1Ψ2 Abbildung 29: Wahrscheinlichkeitsdichte im Wasserstoffmolekül (links) und dazugehörige Wolkendarstellung (rechts) Seite 41 Quantenchemie – eine Einführung Die Darstellungen der Wahrscheinlichkeitsdichte des Grundzustandes im Wasserstoffmolekül macht deutlich, weshalb dieses System existieren kann. Die Aufenthaltswahrscheinlichkeit in den Volumenelementen V1 und V2 entspricht jeweils Ψ , während sie im Volumenelement V3 Ψ1 + Ψ2 + 2Ψ1Ψ2 beträgt, also viermal so gross ist. 2 2 2 Diese erhöhte Aufenthaltswahrscheinlichkeit führt somit zu einem bindenden Effekt zwischen Proton 1 und Proton 2. Natürlich bewirken die elektrostatischen Kräfte der Elektronen in den Volumina V1 und V2 mit den Protonen eine Vergrösserung des Abstandes der beiden Wasserstoffkerne. Nachdem die Elektronen aber im Volumenelement V3 viermal so häufig anzutreffen sind, überwiegt die bindende Kraft. Die Protonen werden solange zur Mitte hingezogen, bis die abstossende Kraft zwischen den positiven Teilchen gleich gross ist wie die anziehende Kraft der Elektronen auf die Protonen. Ein Gleichgewichtszustand stellt sich ein, der charakterisiert ist durch ein Energieminimum (Vergleichen Sie dazu auch Abb. 56.2 im Buch von G. Baars und H.U. Christen „Chemie – Theorie und Praxis). Beim H2-Molekül findet man einen Protonenabstand von ca. -10 0.7·10 -1 m und eine Bindungsenergie von –436 kJ·mol . Die Wellenmechanik liefert somit eine Erklärung für die Existenz von H2-Molekülen, die deutlich weniger reaktionsfähig sind als die beiden isolierten Wasserstoffatome. Deren Bildung aus dem Molekül erfordert einen Energieaufwand. Nähern sich zwei Wasserstoffatome einander, so können sich die beiden Elektronen jeweils im Bereich beider Protonen aufhalten. Es kommt (modellhaft) zu einer Addition der Amplituden der beiden angenommenen Elektronenwellen (konstruktive Interferenz). Damit ist der Grund für die Bestandfähigkeit einer Bindung zwischen zwei Nichtmetallatomen gegeben: Die Wechselwirkung der Atomkerne mit den Elektronen erzeugt eine Reduktion der Elektronendichte auf den der Bindung abgewandten Seiten zugunsten einer Akkumulation von negativer Ladung zwischen den beiden zu bindenden Kernen. Diese Zunahme ist gerade so gross, dass die Kern-Kern Abstossungskraft kompensiert wird beim Gleichgewichtsabstand der gebundenen Atome. Die Atombindung kommt demnach durch Coulomb’sche elektrostatische Anziehung zustande. Für den Grundzustand des Wasserstoffmoleküls gilt zusammenfassend also: Elektron 1: Elektron 2: Ψ1+2 = Ψ1 + Ψ2 Ψ1+2 = Ψ1 + Ψ2 Die Wahrscheinlichkeitsdichte zwischen den Atomkernen ist gegeben durch Elektron 1: Elektron 2: Ψ21+2 = (Ψ1 + Ψ2)2 = Ψ12 + Ψ22 + 2Ψ1Ψ2 Ψ21+2 = (Ψ1 + Ψ2)2 = Ψ12 + Ψ22 + 2Ψ1Ψ2 Die chemische Bindung wird also erzeugt durch die erhöhte Aufenthaltswahrscheinlichkeit der beiden Elektronen zwischen den Protonen. In diesem Fall spricht man von einem gemeinsamen Elektronenpaar bzw. von einer Atombindung (Elektronenpaarbindung, kovalente Bindung, gemeinsame, doppelt besetzte Elektronenwolke; symbolisiert durch Striche in Lewisformeln). Die Kräfte, welche die Atome innerhalb der Moleküle zusammenhalten, werden auf diese Weise erklärt und lassen sich berechnen. Experimente bestätigen diese Werte. Seite 42 Quantenchemie – eine Einführung 9.2 Antibindender Zustand Statt einer konstruktiven ist auch eine destruktive Interferenz von zwei Wellenfunktionen möglich: Ψ1-2 = Ψ1 - Ψ2 Ψ1-2 = Ψ1 - Ψ2 Elektron 1: Elektron 2: Abbildung 30: Wellenfunktion des Systems H2 im Grundzustand zwischen den Kernen. links: Wellenfunktion der einzelnen H-Atome; rechts: Verlauf der Wellenfunktion im Molekül H2 Aus der neuen Wellenfunktion resultiert schliesslich die Wahrscheinlichkeitsdichte Ψ 2 1-2 zwischen den beiden Kernen. Ψ21-2 = (Ψ1 - Ψ2)2 = Ψ12 + Ψ22 - 2Ψ1Ψ2 Ψ21-2 = (Ψ1 - Ψ2)2 = Ψ12 + Ψ22 - 2Ψ1Ψ2 Elektron 1: Elektron 2: Abbildung 31: Wahrscheinlichkeitsdichte der Elektronen im H2-Molekül (antibindender Zustand) Im Bereich zwischen den beiden Protonen kommt es zu einer Verminderung der Aufenthaltswahrscheinlichkeit der Elektronen. Man spricht in diesem Zusammenhang von einem antibindenden Zustand, der energiereicher ist als der bindende Zustand. Die Interferenz der beiden 1s-Wellenfunktionen ergeben somit im Wasserstoffmolekül zwei neue Wellenfunktionen, von denen eine energetisch tiefer liegt (konstruktive Interferenz) als die andere (destruktive Interferenz). Diese beiden neuen Energieniveaus liegen relativ eng beieinander. Energiezufuhr kann ein Elektron des Wasserstoffmoleküls leicht in den antibindenden Zustand anheben. Die antibindenden Zustände sind in Farbstoffmolekülen von grosser Bedeutung. Die Energie des sichtbaren Lichtes reicht bei ihnen aus, um ein Elektron in einen antibindenden Zustand anzuheben. Dies bedeutet wellenmechanisch die Überführung der betreffenden Elektronenwelle in den nächsthöheren Seite 43 Quantenchemie – eine Einführung Schwingungszustand. Damit kommt es zu einer selektiven Absorption bestimmter Wellenlängen des sichtbaren Lichtes und deshalb zum Farbeindruck. 9.3 Die Bindung in Molekülen Bilden Atome höherer Elemente Moleküle, so ist eine Ueberlagerung von s- und p-Wellenfunktionen nötig. Auf der äussersten Schale eines Atoms existieren maximal 8 Elektronen. Diese werden mit einer s- und drei p-Wellenfunktionen beschrieben. Es gibt nun einfache Regeln, die Möglichkeit der Bildung von Bindungen zwischen Atomen abzuschätzen. 1. Die Wellenfunktionen (AO) müssen sich räumlich soweit nähern können, dass eine nennenswerte Überlagerung ("Überlappung") möglich ist. Wellenfunktionen, die Elektronen innerer Schalen beschreiben, überlagern sich praktisch nicht. 2. Hinsichtlich der Bindungsachse im Molekül müssen die Wellenfunktionen gleiche Symmetrie besitzen. 3. Die Wellenfunktionen können sich nur dann überlagern, wenn sie ähnliche Energien aufweisen. 4. Das Pauli Prinzip und die Hundsche Regel sind zu beachten. 12 Es gibt folgende Kombinationsmöglichkeiten von Atomorbitalen zu Molekülorbitalen : _ + s + s + s s Schnitt durch die räumliche Darstellung von s-Atomorbitalen (s-AO) 12 _ + ψσ*s antibindendes MO + ψσs bindendes MO Schnitt durch die räumliche Darstellung der Molekülorbitale (σ-MO) Bei der hier verwendeten Darstellungsweise benützt man einen Schnitt durch den geometrischen Ort aller Punkte mit einem bestimmten Funktionswert ψ. Seite 44 Quantenchemie – eine Einführung _ + px _ _ + px _ + _ Schnitt durch die räumliche Darstellung von px-Atomorbitalen + Schnitt durch die räumliche Darstellung der Molekülorbitale + _ + + _ + _ pz pz + + _ _ pz pz Schnitt durch die räumliche Darstellung von pz- (bzw. py-) Atomorbitalen _ Ψσpx bindendes MO px _ _ + Ψσ*px antibindendes MO px + _ + Ψπ*pz antibindendes MO + _ Ψπpz bindendes MO Schnitt durch die räumliche Darstellung der Molekülorbitale Seite 45 Quantenchemie – eine Einführung + _ s + _ + s _ + Ψσ*spx antibindendes MO px + + px + _ Ψσspx bindendes MO Schnitt durch die räumliche Darstellung von s- und p-Atomorbitalen Schnitt durch die räumliche Darstellung der Molekülorbitale + + _ s pz oder py Schnitt durch die räumliche Darstellung von s- und pz- (bzw. py-) Atomorbitalen keine Überlagerung möglich Definition der σ- und π-Molekülorbitale: σ-Orbitale: Symmetrisch bzgl. Kernverbindungsachse. σ-Molekülorbitale entstehen aus s-Orbitalen oder aus px-Orbitalen. π-Orbitale: Nicht symmetrisch bzgl. Kernverbindungsachse. π-Molekülorbitale entstehen aus py- und pz-Orbitalen. Seite 46 Quantenchemie – eine Einführung 9.4 Systematische Darstellung der Bildung einer Einfachbindung 9.4.1 Homonukleare Moleküle der 2. Periode Bei homonuklearen, zweiatomigen Molekülen der 2. Periode muss man folgende Regeln beachten: Es gibt jeweils von Atom 1 und Atom 2 folgende Orbitale: für die inneren Elektronen: (1s)1, (1s)2 à keine nennenswerte Überlappung. für die Valenzelektronen: à Es lassen sich Molekülorbitale bilden (2s, 2px, 2py, 2pz)1, (2s, 2px, 2py, 2pz)2 In einem neutralen homonuklearen Molekül, das aus Atomen mit der Ordnungszahl Z besteht, muss man 2 Z Elektronen unterbringen (bei geladenen Teilchen entsprechend mehr oder weniger). Die Energieniveaus der p-Molekülorbitale hängen u. a. von der Kernladung der betreffenden Atome ab. Für die Elemente Bor, Kohlenstoff und Stickstoff liegt die Energie der beiden πp - Molekülorbitale tiefer als die des σp - Molekülorbitals, wie Rechnungen gezeigt haben. Bevor wir einige Beispiele von Elementen der zweiten Periode betrachten, müssen wir die Bindungsordnung definieren: Sie entspricht der Anzahl Elektronen in bindenden Zuständen minus Anzahl Elektronen in antibindenden Zuständen dividiert durch zwei. Die Zahl der Elektronenpaare, die effektiv zu einem Zusammenhalt der Atome beitragen, berechnet sich somit folgendermassen: Elektronen in bindenden Molekülorbitalen – El. in antibindenden Molekülorbitalen Bindungsordnung (BO) = 2 Bindende und antibindende Molekülorbitale heben sich in ihrer Wirkung gegenseitig auf. 9.4.1.1 Li2 Dieses Molekül existiert in geringen Konzentrationen im dampfförmigen Lithium. antibindendes MO E Ψσ∗s Ψ(1) 2s Ψ(2) 2s Abbildung 32: Ψσs bindendes MO 9.4.1.2 Elektronenverteilung im Li2-Molekül (Orbitalschema). Die inneren 4 Elektronen sind an der Bindung nicht beteiligt. Be2 Die beiden 2s Atomorbitale repräsentieren jeweils zwei Elektronen, die damit das bindende σ2s und das antibindende σ*2s MO besetzen. Da das antibindende Orbital stärker destabilisierend wirkt als das bindende Seite 47 Quantenchemie – eine Einführung 13 stabilisierend , kann das Be2 Molekül nicht existieren. Experimentelle Untersuchungen haben diese Theorie bestätigt. antibindendes MO Ψσ∗s E Ψ(1) 2s Ψ(2) 2s Abbildung 33: Ψσs Elektronenverteilung im angenommenen Be2-Molekül (Orbitalschema). bindendes MO 9.4.1.3 B2; C2; N2 Sind p-Orbitale vorhanden, so stehen in den gebildeten Molekülen zu den beiden σ- noch prinzipiell sechs weitere Molekülorbitale zur Verfügung: σpx, σ*px, πpy, π*py, πpz und π*pz. Die Energieniveaus der p-Orbitale hängen u. a. von der Kernladung der betreffenden Atome ab. Für die Elemente Bor, Kohlenstoff und Stickstoff liegt die Energie der beiden πp-Molekülorbitale tiefer als die des σp-Molekülorbitals. σ* px 2p π* pz π* py 2p σ px π py 2s π pz σ* s 2s σs AO (1) MO AO (2) Abbildung 34: Aufspaltung der MO’s ausgehend von den AO’s im B2, C2 und N2 Sie können nun (mit verschiedenen Farben) die Aussenelektronen (der 2. Schale) der Bor- bzw. Kohlenstoffbzw. Stickstoffatome in die Atom- bzw. Molekülorbitale einzeichnen. 13 Die höhere Entropie der beiden einzelnen Atome im Vergleich zum hypothetischen Molekül kann nicht durch Bindungsenergie überwunden werden. Seite 48 Quantenchemie – eine Einführung Betrachten wir als Beispiel das Stickstoffmolekül. Pro Stickstoffatom stehen 7 Elektronen zur Verfügung, die nach dem Pauli-Prinzip und der Hund’schen Regel in die Atomorbitale eingefüllt werden. In die MO’s werden nun alle 14 Elektronen wiederum nach den bekannten Kriterien eingefüllt. Die Bindungsverhältnisse erhält man, indem man alle antibindenden Orbitale von den bindenden Orbitalen abzieht. Dabei erhalten wir die Bindungsordnung 3, was der Dreifachbindung in der Lewis-Struktur entspricht. 9.4.1.4 O2, F2, Ne2 In angenommenen den Molekülen der Elemente Sauerstoff, Fluor und Neon sind die beiden π2p-Molekülorbitale mit dem σ2p-Molekülorbital im Vergleich zu den vorhergehenden Molekülen energetisch vertauscht. σ* px 2p π* py π* pz π py π pz 2p σ px 2s σ* s 2s σs AO (1) MO AO (2) Abbildung 35: Aufspaltung der MO’s ausgehend von den AO’s im O2, F2 und Ne2 Auch in dieses Schema können Sie mit verschiedenen Farben die Aussenelektronen in die Atom- bzw. Molekülorbitale einzeichnen. Füllen wir die 8 Elektronen für das Sauerstoffatom ein, erhalten wir zu unserer Überraschung bei den MO zwei einfach besetzte Elektronenwolken. Zwar erhalten wir auch hier die Bindungsordnung zwei, was der Doppelbindung in der Lewis-Struktur entspricht, aber Sauerstoff ist quantenchemisch betrachtet ein Diradikal. Tatsächlich beobachtet man in Experimenten, dass Sauerstoff paramagnetisch 14 ist. Dieser Effekt wurde zwar schon vor der Quantentheorie experimentell gefunden, er konnte jedoch nicht erklärt werden, da nach der einfachen Theorie, dass zwei einfach besetzte Wolken eine doppelt besetzte Wolke mit unterschiedlichen Spins der Elektronen ergeben, keinen Paramagnetismus vorhersagt. Der diradikale Charakter des Sauerstoffs erklärt auch seine ausgesprochen hohe Reaktivität. Der menschliche Körper muss sich zum Beispiel vor den schädlichen Wirkungen des Sauerstoffs auf Zellmembranen mit Vitamin E (α-Tocopherol), einem sogenannten 14 Eine paramagnetische Substanz hat die Tendenz, in ein Magnetfeld hineinzuwandern. Den Paramagnetismus beobachtet man bei Substanzen mit ungepaarten Elektronenspins. Seite 49 Quantenchemie – eine Einführung Radikalfänger schützen. Ein Mangel an Vitamin E kann zu einer erhöhten Zerstörung der Erythrocyten führen. Im Tierversuch führt Vitamin E-Mangel zu schuppiger Haut, Muskelschwäche und Sterilität. Zurück zu den Elektronenkonfigurationen und somit zum letzten zweiatomigen Molekül, dem Fluor. Wir können wiederum bei den AO je ein Elektron hinzufügen, und erhalten bei den MO die Bindungsordnung eins, was der Einfachbindung entspricht. Hier haben wir es also wieder mit einem Molekül zu tun, das die bekannten Theorien nicht widerlegt 9.5 Wechselwirkung zwischen den Orbitalen – das Modell der Hybridisierung Befassen wir uns mit mehratomigen Molekülen, so müssen wir unsere bisher erarbeiteten Konzepte erweitern. Dies wird bei der genaueren Betrachtung der Bindungsverhältnisse im Methan-Molekül (CH4) offensichtlich: Wendet man das oben beschriebene Verfahren auf den Kohlenstoff mit der Elektronenkonfiguration 2 2 1 1 1s 2s 2px 2py (s. Abb. 27 und 34) an, so würde man aus der Reaktion von Kohlenstoff mit Wasserstoff offensichtlich das Molekül CH2 erwarten, da am Kohlenstoff nur zwei einfach besetzten 2p-Orbitale vorhanden sind. Nun entsteht aber effektiv das Molekül mit der Zusammensetzung CH4 (Methan) mit vier äquivalenten σ-C-H-Bindungen die im Winkel von 109.5 (regelmässiges Tetraeder) zueinander stehen. Die Lösung dieses Problems ist das Modell der „Hybridisierung“, welches in den folgenden Abschnitten kurz erklärt werden soll. Es ist an dieser Stelle deutlich darauf hingewiesen, dass das Phänomen der Hybridisierung nicht experimentell nachweisbar ist. Es handelt sich dabei lediglich um eine Modellvorstellung, welche in eleganter Weise die Bindungsverhältnisse mehratomiger, vielfach organischer Moleküle, zu beschreiben vermag. Dieses Modell findet denn auch in der organischen Chemie seine grösste Akzeptanz und Verbreitung. Es ist jedoch unter Fachleuten nicht unbestritten 9.5.1 Die Hybridisierung von s- und p-Orbitalen Der Schlüssel zum oben dargestelltem Problem liegt darin, dass sich die Wellenfunktionen der s- und p-Orbitale „vermischen“. Man verzichtet auf die Unterscheidung von s- und p-Wellenfunktionen und geht von vier gleichwertigen Wellenfunktionen aus, die man durch eine Linearkombination aus den ursprünglichen 15 Wellenfunktionen erhalten hat . Mathematisch betrachtet ist eine Linearkombination nichts anderes als eine Addition von Vektoren, die ihrerseits noch mit einer reellen Zahl multipliziert werden können. In der Chemie entstehen durch diese Linearkombination vier Atomorbitale (AO) mit genau der gleichen Form. Ihre Gestalt ist vergleichbar mit den p-Orbitalen, mit dem Unterschied, dass ein Teil der Hantel auf Kosten des anderen Teils stark anwächst. Der grössere Hantelteil weist dabei in eine Ecke eines Tetraeders (Abb. 36). Dieses sogenannte sp3-hybridisierte (entstanden aus einem s- und drei p-Orbitalen) C-Atom enthält nun neben einem 3 1s-Orbital in der zweiten Schale vier gleichwertige sp -(Hybrid)orbitale. Die resultierenden vier Wellenfunktionen sind nun also alle auf dem gleichem Energieniveau und werden je einzeln, gemäss der Hund’schen Regel, mit einem Elektron gefüllt (Abb. 37). Nun haben wir wieder vier gleichwertige einfach besetzte AO, die tetraedrisch angeordnet sind, und je eine σ-Bindung mit einem Wasserstoff eingehen können, und so das Molekül CH4 bilden können. 15 http://www.bcpl.net/~kdrews/hybrids.html Seite 50 Quantenchemie – eine Einführung 2p HYBRIDISIERUNG 3 2sp 2s Abbildung 36: Bildung von vier sp3-Hybridorbitale aus einem s-Orbital und drei p-Orbitalen beim C-Atom . Die vier sp3-Orbitale sind entartet; ihr Energiegehalt liegt zwischen der Energie des 2s- und der drei 2p-Orbitale aus denen sie gebildet wurden. Siehe auch die folgende Abbildung E E Hybridisierung 2p 2s 1s Abbildung 37: Energieschema der Hybridisierung im C-Atom 2sp3 1s Seite 51 Quantenchemie – eine Einführung Auf der Grundlage dieser Konzepte sind die Bindungen im Methan einfach zu erklären. Die vier σ-Bindungen sind äquivalent, in gleicher Weise ausgerichtet und bilden einen regelmässigen Tetraeder. Mit ähnlichen Vorgehensweisen lassen sich auch die Bindungen in anderen Molekülen erklären. 9.5.2 Weitere Hybridisierungsarten - Mehrfachbindungen Andere Typen von Hybridorbitalen entstehen durch das Verschmelzen von AO in unterschiedlichen Anteilen. 3 Die für die Chemie wichtigsten drei Typen der Hybridisierung sind die tetraedrische (sp , Bindungswinkel 2 109.5 °), die trigonal-planare (sp , Bindungswinkel 120 °) und die lineare (sp, Bindungswinkel 180 °). Sie führen zu den Konzepten der Mehrfachbindungen. Betrachten wir die Struktur des Ethens. Aus experimentellen Untersuchungen weiss man, dass alle sechs Atome des Ethens in einer Ebene liegen und dass die HCH- und die CCH-Bindungswinkel 120 ° betragen. 2 Diese Geometrie deutet darauf hin, dass die C-Atome sp -hybridisiert sind, wobei sich jeweils ein Elektron in jedem der drei Hybridorbitale „befindet“. Das vierte Elektron muss daher ein unhybridisiertes 2p-Orbital beschreiben: 2p 2pz HYBRIDISIERUNG 2 2sp 2s Abbildung 38: Hybridisierung eines s- mit zwei p-Orbitalen Dieses unhybridisierte p-Orbital steht senkrecht zu der Ebene, die durch die Hybridorbitale aufgespannt wird (Abb. 39). Die beiden Kohlenstoffatome sind durch eine σ-Bindung von jeweils einem sp -Hybridorbital der 2 C-Atome miteinander verbunden. Die H-Atome bilden σ-Bindungen mit den zwei verbleibenden 2 sp -Hybridorbitalen. Die Elektronen in den zwei verbleibenden 2pz-Orbitalen bilden das Elektronenpaar einer π-Bindung. Daraus folgt, dass die C=C-Doppelbindung aus einer σ-Bindung der Csp -Hybridorbitale und einer 2 π-Bindung der zwei C2p-Orbitale besteht. Die beiden Orbitallappen der π-Bindung befinden sich ober- und unterhalb der Molekülebene. Diese Vorstellung macht keine Mühe, wenn wir uns vergegenwärtigen, dass es sich bei den Orbitalen um Ein-Elektronen-Wellenfunktionen handelt. Seite 52 Quantenchemie – eine Einführung σ-Bindung π-Bindung Abbildung 39: Bindungsverhältnisse im C2H4-Molekül Durch die grössere Distanz der p-Orbitale und somit der schlechteren Überlagerung im Vergleich der σ-Bindung, ist die Bindungsenergie der π-Bindung kleiner als diejenige der σ-Bindung (vgl. Tabelle der Bindungsenergien, Baars/Christen, Seite 61). Dieser Umstand ist auch im Energieschema des Ethen sichtbar: die Aufspaltung der Orbitale bei einer π-Bindung ist weniger gross als bei einer σ-Bindung (Abb. 40). E σ∗ p π∗ p σ∗ sp2 sp2 π σ σ∗ 1s 1s σ σ C C 1s H Abbildung 40: Aufspaltung der MO’s bei der Bildung einer Doppelbindung im Ethen. Die Aufspaltung von zwei sp2-Orbitalen des linken C und einem sp2-Orbital des rechten C ist nicht gezeigt, kann aber gleich dargestellt werden wie die Wechselwirkung mit dem gezeigten H. Die Energien sind qualitativ, nicht quantitativ zu verstehen. Seite 53 Quantenchemie – eine Einführung Die Struktur von Ethin kann in ähnlicher Weise beschrieben werden. Um eine lineare Geometrie zu erreichen, müssen die C-Atome sp-hybridisiert sein. Es befindet sich jeweils ein Elektron in jedem der zwei sp- Hybridorbitale und je eines in jedem der beiden senkrecht dazu stehenden unhybridisierten 2p-Orbitalen: 2p 2p HYBRIDISIERUNG 2sp 2s Abbildung 41: Hybridisierung eines s- mit einem p-Orbital Zwei σ- und eine π-Bindung oder eine σ- und zwei π-Bindungen? Grundsätzlich gilt, dass zwischen zwei Atomen nur eine σ-Bindung möglich ist. Also muss das Ethin zwei π-Bindungen haben. Da die zwei p-Orbitale eines C-Atoms rechtwinklig aufeinander stehen, sind die Elektronen der σ-Bindung oben, unten, links und rechts von π-Elektronen umgeben. Dies resultiert in einer Art Röhre um die σ-Bindung herum (Abb. 43). Die Aufspaltung im Energieschema verhält sich ähnlich derjenigen im Ethen (Abb. 42). MO‘s E AO‘s σ∗ AO‘s p π∗ p sp π sp σ σ∗ 1s 1s σ C C Abbildung 42: Aufspaltung der MO’s bei der Bildung einer Dreifachbindung im Ethin. Die Wechselwirkung je eines sp-AO’s mit dem 1s-Orbital des H ist nicht gezeigt. Die Energien sind qualitativ, nicht quantitativ zu verstehen. Seite 54 Quantenchemie – eine Einführung py py spz spz px px Abbildung 43: Hybridisierung und Elektronenwolken im Ethin 9.6 Bindungsverhältnisse weiterer Moleküle Betrachten wir zum Schluss noch den ursprünglichen Grund dieses kleinen Einschubs, nämlich das Benzol 16 2 mit seinen delokalisierten Elektronen. Im Benzol ist jedes der sechs C-Atome, wie beim Ethen, sp -hybridisiert. Die verbleibenden p-Orbitale, eines pro C-Atom, stehen nun rechtwinklig auf der Ringebene. Die Elektronen der sp -Hybridorbitale bilden Paare und gehen sechs σ(C-C)-Bindungen ein. Jeweils ein H-1s-Elektron bildet mit 2 dem dritten sp -Hybridorbital der Kohlenstoffe ein Elektronenpaar und es entstehen 6 σ(C-H)-Bindungen. Der 2 sich so bildende Ring bringt die unhybridisierten 2p-Elektronen der Kohlenstoffatome so nahe zusammen, dass eine Wechselwirkung stattfindet und sich die Orbitale seitlich mit einem ihrer Nachbarn zu einer π-Bindung überlappen können. Anders als bei einer „normalen“ Doppelbindung können nun in einem solchen System die π-Elektronen über alle C-Atome delokalisiert sein. Daraus resultieren zwei Ringe von insgesamt 6 delokalisierten π-Elektronen, die dem Molekül eine besondere Stabilität verleihen (Abb. 44). 16 Bearbeiten Sie die Aufgaben und die Links unter folgender Adresse: http://www.merian.fr.bw.schule.de/Bobeth/chemie/organik/benzol1.htm Quantenchemie – eine Einführung Abbildung 44: Seite 55 Oben: σ-Bindungen im Benzolmolekül; links: p-Orbitale senkrecht zu den σ-Bindungen; rechts: delokalisierte π-Bindungen Quantenchemie – eine Einführung Seite 56 Die dazugehörigen bindenden und antibindenden Molekülorbitale haben dabei folgende Formen: Abbildung 45: 17 MO von Benzol, zu finden unter: http://mc.net/~buckeroo/ARSY.html17; Das unterste Niveau hat keine, das erste eine, das zweite zwei und das dritte drei Knotenlinien. Die unteren zwei Niveaus sind bindend, die oberen antibindend. Molekülorbitale (können bewegt werden) sind auch unter http://www.chem.arizona.edu/~salzmanr/orbitals.html zu finden (Erfordert Plug-in „Chime“; kann unter http://www.mdli.com/support/chime/default.html heruntergeladen werden) Quantenchemie – eine Einführung Seite 57 10 Farben Was ist Farbe, und was macht einen Stoff farbig? Die Antworten sind unterschiedlich, in den meisten Fällen handelt es sich aber um eine Absorption gewisser Wellenlängen. Verbindungen, die elektromagnetische Wellen im sichtbaren Bereich absorbieren, vermitteln in unserem Gehirn den Farbeindruck. Obschon jedes Molekül in einem ganz bestimmten Bereich des elektromagnetischen Spektrums die Wellen absorbiert, gibt es doch ganz bestimmte Kriterien, damit wir eine Farbe wahrnehmen: die Absorption muss zwischen 380 nm und 720 nm liegen. Unsere Augen „sehen“ jedoch nicht die absorbierten Wellen, sondern die übrig bleibenden Anteile des Spektrums. Abbildung 46: Die roten und blauen Anteile des weissen Lichts werden absorbiert und für chemische Reaktionen verwendet. Übrig bleibt die Farbe grün. Seite 58 Quantenchemie – eine Einführung Die absorbierten Wellen bewirken, dass ein Elektron von einem Orbital in ein höheres gehoben wird. Da die Energien zwischen den Orbitalen quantisiert sind, sind nur ganz bestimmte Übergänge möglich. Nämlich diejenigen, die der Energie der entsprechenden Photonen entsprechen. Ein Überblick zwischen den absorbierten und den „gesehenen“ Wellen ist in der nebenstehenden Abbildung dargestellt. Doch was macht ein Teilchen zu einem Farbstoff oder einem Pigment? 18 Abbildung 47: 18 Beziehung zwischen absorbierter Wellenlänge, sichtbare Farbe und Energie In der Farbstoffchemie unterscheidet man zwischen den wasserlöslichen Farbstoffen, die unter anderem für Textilien Verwendung finden und den unlöslichen Pigmenten, die als Lackfarbstoffe auf den Markt kommen. Seite 59 Quantenchemie – eine Einführung 10.1 Organische Farbstoffmoleküle19 Vergleicht man die folgenden Stoffe, wird schnell einmal klar, dass die Delokalisation von Elektronen in einem Molekül von entscheidender Bedeutung ist. Je grösser die Delokalisation in einem Molekül, desto höher ist die Wellenlänge des absorbierten Lichts, oder desto kleiner ist die Energie, um die Elektronen auf das nächst höhere Niveau zu heben. Molekül Lewis-Formel Absorptionspeak absorbierte beobachtete “Farbe” Farbe Benzol 255 nm UV farblos Naphthalin 315 nm UV farblos Anthracen 380 nm UV farblos Naphtacen 480 nm blau orange Pentacen 580 nm gelb indigo Wechseln sich in einem Molekül Doppel- und Einfachbindungen ab, handelt es sich dabei um ein delokalisiertes System, bei dem alle am System beteiligtem Atome sp -hybridisiert sind. Neben dem σ-Gerüst bleibt eine p2 Elektronenwolke übrig, die senkrecht zum Grundgerüst steht, wie das Beispiel 1,3-Butadien zeigt: Abbildung 48: σ-Gerüst und p-Wolken des 1,3-Butadien Daraus resultieren die entsprechenden π-Orbitale und eine Delokalisation über das ganze Molekül: C Abbildung 49: 19 C Delokalisierte Elektronen im 1.3-Butadien vgl. dazu Kapitel 10.2 im Duden Chemie und Kapitel 29 im Baars / Christen C C Seite 60 Quantenchemie – eine Einführung Da eine Delokalisation mit der Lewis-Formel nicht genau wiederzugeben ist, muss man sich oft mit den Grenzstrukturen behelfen, wie das Beispiel Indigo zeigt: O H O N H N N + N H O O H H O N N H Abbildung 50: O Grenzstrukturen des Indigo. Da das Molekül symmetrisch ist, können die Grenzstrukturen zwei Mal gezeichnet werden. Tritt ein Atom bei irgend einer Grenzstruktur mit einer Doppelbindung auf, kann man davon ausgehen, dass es sicher zum delokalisierten System beiträgt. Für Indigo 20 würde sich dementsprechend folgende Delokalisation ergeben: O H N N H Abbildung 51: O Delokalisation im Indigomolekül Betrachtet man das Absorptionsspektrum dieses Moleküls, bemerkt man, dass lediglich der blaue Anteil des Lichts nicht absorbiert wird (siehe Abb. 54). Will man mit Indigo färben, muss das Molekül unter basischen Bedingungen reduziert werden, damit es wasserlöslich wird. Während die Erhöhung des pH-Wertes früher mit Urin21 gemacht wurde, geschieht dies heute mit Ammoniak oder NaOH. Zur Reduktion verwendet man heute Dithionit. Bei der Reduktion verkleinert sich jedoch das delokalisierte System im Molekül. Die Absorption verschiebt zu kürzeren Wellenlängen: wir sehen die Farbe gelb: 20 21 Bilder zu dieser Pflanze sind zu sehen unter: http://hep.itp.tuwien.ac.at/~kreuzer/strings.html http://m.aei-potsdam.mpg.de/startrek/sld019.htm http://www.feininger.de/ Die verwendeten Ausdrücke „blau machen“ und „blau“ für alkoholisiert, sind wahrscheinlich auf die Färbemethode mit Indigo zurückzuführen. Damit möglichst viel Urin produziert werden konnte, wurde ausgiebig Alkohol, der diuretisch wirkt (Vasopressin- (=Antidiuretin-)ausschüttung wird gehemmt), konsumiert. Dies war natürlich angenehmer als die übliche Arbeit zu verrichten! Seite 61 Quantenchemie – eine Einführung O H N N O H Abbildung 52: Delokalisation der Elektronen in der wasserlöslichen Form des Indigo Aber nicht nur das delokalisierte System, sondern auch die daran angehängten Gruppen spielen eine wichtige Rolle bei der Absorption von Licht. Das 6,6’-Dibromindigo ist eine purpurne Farbe, die im Altertum sehr kostbar war. Es war nur dem jeweiligen Cäsar erlaubt, Kleider mit dieser Farbe zu tragen. Das 6,6’-Dibromindigo wurde 22 aus einer Schnecke gewonnen . Dazu benötigte man 6000 bis 8000 Tiere um ein Gramm Farbstoff zu gewinnen. Die beiden angehängten Bromatome bewirken aufgrund ihrer relativ hohen EN eine Verschiebung zu tieferen Wellenlängen (vgl. Abbildung 54), da Elektronen aus dem delokalisierten System gezogen werden: O H N N Br H Abbildung 53: Br O Lewis-Formel des Purpur aus der Purpurschnecke OD Indigo 6,6’-Dibromindigo λ (nm) Abbildung 54: Absorptionsspektrum von Indigo und Dibromindigo Will man aber die zwischen den Orbitalen liegenden Energie berechnen, stösst man beim Indigo auf einige Schwierigkeiten. Man muss deshalb davon ausgehen, dass dieses Molekül keine vollständige Delokalisation aufweist. Grund dafür können die in sich abgeschlossenen Benzolringe sein. Nicht so bei idealen Farbstoffmolekülen, d.h. Molekülen mit einer vollständigen Delokalisation. Bei ideal konjugierten Doppelbindungen geht man vom linearen Kasten aus, um die Energien zwischen den Orbitalen zu berechnen. Dabei ist die Länge des Moleküls der Kasten, in dem die Elektronenwellen Platz haben. Die Anregungsenergie berechnet sich nach: 22 zu sehen unter www.seilnacht.tuttlingen.com/ Lexikon/Purpur.htm Seite 62 Quantenchemie – eine Einführung ( z + 1) 8mc λ= •d2 • (N + 1) h 2 λ: absorbierte Wellenlänge m: Masse des Elektrons (9.11 • 10 c: Lichtgeschwindigkeit (2.998 • 10 m/s) h: Plank’sches Wirkungsquantum (6.626 • 10 d: Mittlere Bindungslänge (1.39 • 10 z: Anzahl Atomkerne N: Anzahl delokalisierte Elektronen -31 kg) 8 -34 -10 Js) m) _________________________________________________________________________________________________________________________ Einschub Nach de Broglie (alles hat seine Wellenlänge) gilt für bewegte Teilchen: m • v = h λ Nach Schrödinger gilt für die Energie aller Systeme Eges = Ekin + Epot; da die potentielle Energie gleich bleibt, interessiert uns nur die kinetische Energie, die sich ganz normal nach E kin = hier für v de Broglie ein, ergibt dies E = 1 mv 2 berechnen lässt. Setzt man 2 h2 2mλ2 Die Wellenlänge ist in einem Kasten quantisiert, da nur stehende Wellen mit der Beziehung λ = kommen. Setzt man dies bei der kinetischen Energie ein, ergibt sich Für das HOMO gilt n = E = h2 8mL 2 2L in Frage n •n2 N N + 1 ist (N: Anzahl π-Elektronen). , während das LUMO auf n = 2 2 Die Differenz berechnet sich nach 2 2 h 2 N h2 h 2 N − = ∆E = + 1 8mL 2 2 8mL 2 2 8mL 2 2 2 N h2 N + 1 − = (N + 1) 2 2 8mL 2 è Anregungsenergie des Moleküls h ist das Plank’sche Wirkungsquantum (6.626 · 10 -34 Js) und m die Masse eines Elektrons (9.11 · 10 -31 kg). Für die Länge des Kastens wird das konjugierte π-System betrachtet. Die mittlere Bindungslänge beträgt 1.39 · 10 -10 m. Bei beiden Enden wird noch je eine Bindungslänge dazugezählt (oder man zählt die Kohlenstoffkerne (z), zählt auf beiden Seiten noch eine Bindungslänge (d) dazu und multipliziert mit der mittleren Bindungslänge: L = (z + 1) · d; Das Resultat ist das gleiche). Mit ∆E = hc hc oder λ = ergibt sich die ausserhalb des Einschubs erwähnte Formel. ∆E λ Mit anderen Worten: je länger der Kasten, desto kleiner wird die Anregungsenergie. Die Wellenlänge verschiebt sich in den sichtbaren Bereich. _________________________________________________________________________________________________________________________ Seite 63 Quantenchemie – eine Einführung Ein Beispiel dafür sind die Cyaninfarbstoffe: R N CH CH CH + n N R Aber auch die Polyene werden gut verstanden, obschon bei ihnen ein Korrekturfaktor eingeführt werden muss, der aber sehr wohl erklärbar ist. Beispiele für Polyene sind die Carotinoide. OH HO Abbildung 55: Carotinoide werden in die Carotine (oben: β -Carotin) und in die sauerstoffhaltigen Xanthophylle (unten: Lutein) eingeteilt. Während α- und β-Carotin eine Vitaminfunktion für den Menschen einnehmen (Sehprozess: vgl. Retinal) kommen Xanthophylle neben den Carotinen in Blättern vor und helfen Lichtenergie „einzufangen“. Die gelben und roten Farben der Blätter im Herbst sind auf die Carotinoide zurückzuführen. Im Sommer wird ihre Farbe vom Chlorophyll überdeckt. Dieses wird aufgrund seines Magnesiumions von den Pflanzen in den Stamm zurück transportiert, damit es im nächsten Frühling wieder zur Verfügung steht (vergleichen Sie dazu Ihr Biologiebuch). 10.1.1 Fluoreszierende Farbstoffe Diese Farbstoffe absorbieren Wellenlängen häufig im UV- oder Blaubereich. Fluorescein sollte uns deshalb rot erscheinen. Zwischen dem Grundzustand und dem durch die Absorption dieses Photons erreichte angeregte Zustand gibt es noch ein weiteres Niveau. Das Elektron fällt bei seinem Weg auf den Grundzustand zuerst auf dieses Niveau. Die Energie gibt es dabei in Form von Wärme ab. Erst beim zweiten Schritt fällt nun das Elektron in den Grundzustand zurück. Dabei gibt es die Energie in Form von grünem Licht ab, ist also ein sogenannter aktiver Farbstoff: Seite 64 Quantenchemie – eine Einführung O O HO O OH Wärme angeregte Elektronenzustände 485 nm Licht: 570 nm Grundzustand Abbildung 56: Lewis-Formel von Fluorescein (oben) und Asorptions- und Emissionsverhältnisse im Fluorescein (unten) Seite 65 Quantenchemie – eine Einführung 10.2 Anorganische Farbstoffe Während bei den organischen Farbstoffen die delokalisierten Elektronen eine Rolle spielen, kommen bei den anorganischen Farbstoffen die d-Wolken ins Spiel. Das Prinzip ist folgendes: In der Mitte befindet sich ein sogenanntes Zentralion, häufig ein Übergangsmetall. Um das Zentralion befinden sich die Liganden. Je nach Substanz, d.h. Zusammensetzung des Zentralions und der Liganden, wird das Metallion von unterschiedlichen Seiten und unterschiedlicher Stärke koordiniert. Abbildung 57: 23 Dabei sind folgende ausgewählte Situationen möglich: Alle d-Wolken sind um das Zentralion gezeichnet. Dunkel die dz2- und die dx2-y2-Wolken, hell die dxy-, dxz- und dyz-Wolken. Das Zentralion kann oktaedrisch (links) oder tetraedrisch (rechts) oder quadratisch planar (rechts ohne Liganden aufderz-Achse) von den Liganden koordiniert werden. Je nach Anordnung der negativen oder negativ polarisierten Liganden gegen die Elektronen enthaltenden, also auch negativen Wolken, befinden sich die d-Orbitale auf einem höheren oder tieferen Energieniveau. Die Aufspaltung der Orbitale, abhängig vom Ligandenfeld, ist auf der folgenden Seite zusammengefasst: 23 Lesen Sie Kapitel 8 im Duden Chemie Seite 66 Quantenchemie – eine Einführung Energie d z2 d x2 – y2 dxy dyz dxz Energie dxy dyz dxz d z2 d x2 – y2 Energie d x2 – y2 d z2 d x2 – y2 dxy d z2 dxy dyz dxz dyz Abbildung 58: dxz Aufspaltung der d-Orbitale im Oktaederfeld (oben), Tetraederfeld (Mitte) und quadratisch planaren Feld (unten). Das quadratisch planare Feld ergibt sich aus dem Oktaederfeld, dem die zwei Liganden in der z-Achse weggenommen werden. Seite 67 Quantenchemie – eine Einführung Beim Oktaederfeld muss zusätzlich zwischen high spin und low spin Komplexen unterschieden werden. Während bei high spin Komplexen die Differenz zwischen den d-Orbitalen relativ klein ist und die Elektronen über die fünf Orbitale nach Pauli aufgeteilt werden, ist bei low spin Komplexen die Energiedifferenz so gross, dass die Paarungsenergie (zweites Elektron in das gleiche Orbital) kleiner ist. Die Elektronen werden in die unteren Orbitale verteilt (vgl. folgende Abb.). Eine Unterscheidung zwischen high und low spin macht nur Sinn, wenn die d-Orbitale 4, 5, 6 oder 7 Elektronen enthalten. high spin low spin d z2 d x2 – y2 Energie d z2 d x2 – y2 dxy dyz dxz dxy schwaches Feld dxz starkes Feld 3+ 3- Bsp.: Fe[(H2O)6] Abbildung 59: dyz Bsp.: Fe[(CN)6] Je nach Ligand und Zentralion werden die höheren und tieferen d-Orbitale energetisch weiter auseinandergezogen. Sowohl die Zentralionen als auch die Liganden können nach zunehmender Stärke eingeordnet werden. - - 2- - - - - - 2- - - - I < Br < S < SCN < Cl < N3 , F < Harnstoff, OH < O < H2O < NCS < NH3 < NO2 < CN < CO Energie wird grösser; absorbierte Wellenlänge wird kleiner und 2+ Mn 2+ < Ni < Co 2+ < Fe 2+ 2+ <V 3+ < Fe 3+ < Co 3+ < Mn 3+ < Mo 3+ < Rh < Ru 3+ 4+ < Pd < Ir 3+ < Pt 4+ Es kommt also sowohl auf das Zentralion als auch auf den Liganden an, welche Farbe von dem 2+ entsprechenden Komplex absorbiert wird. Während Mn 3+ Co vor allem high spin Komplexe bildet, trifft man beim vorwiegend low spin Komplexe an. Je nach Stärke des Liganden werden andere Farben absorbiert, da die Anregungsenergie jeweils unterschiedlich hoch ist. Das folgende Beispiel soll diesen Zusammenhang illustrieren. Es handelt sich dabei um einen low spin Komplex: Seite 68 Quantenchemie – eine Einführung Komplex Absorbierte Wellenlänge (nm) Energiedifferenz 700 klein 3- [CoF6] 3- 640 3+ 600 [Co(CO3)3] [Co(H2O)6] 2+ [Co(NH3)5Cl] 535 3+ [Co(NH2)6] [Co(CN)5Br] 475 3- 415 3- [Co(CN)6] 310 gross Jedes dieser Zentralionen und alle Liganden können aber auch eine tetraedrische oder quadratisch planare Anordnung einnehmen. Dabei gibt es Tendenzen, die aber nicht überall stimmen müssen. So kann man sagen, dass Halogene eine tetraedrische Anordnung bevorzugen, neutrale Liganden eine oktaedrische Anordnung, 2+ 2+ und die Zentralionen Ni , Pt 2+ und Pd häufig eine quadratisch planare Umgebung haben. 10.3 Beispiele Beispiele von organischen und anorganischen Farbstoffen und Pigmenten finden Sie auf den folgenden Seiten: http://www.cis.tugraz.at/orgc/hoegroup/naturst/skript/farbstoffe_low.html http://www.educeth.ch/chemie/diverses/pigmente/pigmente/ http://www.uni-bayreuth.de/departments/ddchemie/umat/komplexfarbe/komplexfarbe.htm (mit Anordnung im Komplex) http://www.seilnacht.tuttlingen.com/farbe.htm http://www.seilnacht.tuttlingen.com/Lexikon/FLexikon.htm http://neon.chem.ox.ac.uk/vrchemistry/complex/allbottlesmsiedefault.html Seite 69 Quantenchemie – eine Einführung Ein Beispiel für Systeme, die sowohl einen organischen Teil als auch ein Zentralion besitzen, sind der organische delokalisierte Porphyrinring mit unterschiedlichen Zentralionen. Befindet sich ein Fe 2+ in der Mitte, handelt es sich um das Sauerstofftransport- und -speichersystem im Blut bzw. Muskel. Der Porphyrinring befindet sich im Innern des Hämoglobins oder Myoglobins. Das Fe 2+ ist an vier Stellen durch den Porphyrinring koordiniert, an einer Stelle vom Stickstoff eines Histidins und an der sechsten Stelle, falls vorhanden, vom Sauerstoffmolekül. R N N 2+ Mg Fe N N COOH COOH N N 2+ N N O O O O O O O R = CH3: Chlorophyll a R = CHO: Chlorophyll b 2+ Porphyrinring Fe N His Abbildung 60: N Porphyrinringe des Häm (links) und des Chlorophylls (rechts). Das Histidin ist ein Teil des Hämoglobins. Enthält das Hämoglobin ein Sauerstoff (Oxy-Form) bildet das Häm einen low spin Komplex. Die sechs Elektronen sind alle gepaart, was dazu führt, dass das Häm diamagnetisch 24 ist. In der Desoxy-Form, also ohne 25 Sauerstoff, bildet das Häm einen high spin Komplex. Der Komplex ist paramagnetisch . Diese unterschiedlichen Formen haben auch einen Einfluss auf die Farbe des Blutes. Vollständig mit Sauerstoff 24 richtet sich in einem magnetischen Feld nicht aus Seite 70 Quantenchemie – eine Einführung - gesättigtes arterielles Blut ist hellrot, während venöses Blut dunkelrot bis violett erscheint. Cyanidionen (CN ; Blausäure: HCN; Cyankali: KCN) und CO können im Hämoglobin den Platz des Sauerstoffs einnehmen. Obwohl es sich dabei um ein chemisches Gleichgewicht handelt, liegt das Gleichgewicht stark auf der Seite der beiden Gifte. Ein weiteres Beispiel eines Gleichgewichts, das stark auf einer Seite liegt, ist die im Praktikum 2+ durchgeführte Konkurrenzreaktion von Ammoniak mit Wasser beim Cu -Ion. 2+ Cu H + 6 O [ Cu(H2O)6]2+ H hellblau +4NH3 -2H O -4 H22O [ Cu(NH3)4(H2O)2]2+ dunkelblau Tetraamminkupfer(II)-Komplex b) Hexaaquakupfer(II)-Komplex 2+ OH2 OH2 H2O NH3 H3N Cu H2O 2+ OH2 Cu OH2 OH2 H3N NH3 OH2 2+ Chlorophyll hat genau das gleiche delokalisierte System wie Häm, jedoch in der Mitte ein Mg . Dieser Unterschied bewirkt, dass Chlorophyll grün ist. Im Herbst wird das Chlorophyll aus den Blättern zurück gezogen, damit das Magnesiumion im nächsten Frühling wieder zur Verfügung steht. Übrig bleiben die Carotinoide. Das Blatt erscheint gelb oder rot. 25 richtet sich in einem magnetischen Feld aus Quantenchemie – eine Einführung Seite 71 11 Literatur - Baars, G. et al. Quantenchemie – ein Leitprogramm in Chemie, Herausgeber: Günter Baars und Walter Caprez, ETH Zürich, Institut für Verhaltenswissenschaft & Universität Bern, Abteilung für das höhere Lehramt - Baars, G. et al. Das Denken in Modellen – Skriptum zum Unterricht - Hey, T. & Walters P. (1990/98) Das Quantenuniversum; Die Welt der Wellen und Teilchen, Heidelberg, Spektrum-Verlag - Atkins, Peter W. (1988) Physikalische Chemie, 1. vollst. Durchges. U. berichtigter Nachdr. D. 1 Aufl., Weinheim, Basel, Cambridge, New York (NY), VCH-Verlag - Atkins, Peter W. (1998) Chemie – einfach alles, 2. korr. Auflage, Weinheim, New York, Chichester, Brisbane, Singapore, Toronto, Wiley-VCH-Verlag - Huheey, James E. (1988) Anorganische Chemie, Prinzipien von Struktur und Reaktivität, Berlin, New York, de Gruyter - Christen, H.R. & Baars, G. (1997) Chemie, Verlag Sauerländer, Aarau und Diesterweg Verlag, Frankfurt am Main - Mortimer, Charles E. (1996) Chemie; das Basiswissen der Chemie, 6. völlig neu bearb. Aufl., Stuttgart, New York, Thieme - Dickerson, Richard E. (1986) Chemie – eine lebendige und anschauliche Einführung, Weinheim, Deerfield Beach, Florida, Basel, VCH-Verlag Seite 72 Quantenchemie – eine Einführung Wichtige Begriffe – kurz erklärt Photon komprimierte, diskrete Energieeinheit (“Teilchen”) der elektromagnetischen Strahlung Photonenenergie E=h Wellenmechanik Die Wellenmechanik baut auf dem Gedanken der Materiewellen von de Broglie auf, die dieser aufgrund des Einsteinschen Konzepts des Welle-Teilchen-Dualismus für Elektronen in Atomhüllen postulierte. Als Wellenmechanik bzw. Quantenmechanik bezeichnet man das Teilgebiet der Quantenphysik, dessen Aufgabe es ist, Werte für ψ (Amplitude der Materiewelle) zu ermitteln. Ausgehend von der Wellenmechanik wird das wellenmechanische Atommodell entwickelt. Wahrscheinlichkeitswelle Die Bahn eines atomaren, sich bewegenden Teilchens, folgt Gesetzen, die die mit dem Teilchen verbundene Welle vorschreibt. Diese Welle wird als Wahrscheinlichkeitswelle verstanden. Beziehung nach de Broglie h: Planck’sches Wirkungsquantum λ= h mv λ: m: Masse v: Geschwindigkeit Materiewelle Elektromagnetische Strahlung, sowie materielle Teilchen kann man sowohl Wellen- als auch Teilcheneigenschaften zuschreiben. In diesem Zusammenhang spricht man von Materiewellen. Wahrscheinlichkeitsdichte Die Wahrscheinlichkeitsdichte vergleicht die Aufenthaltswahrscheinlichkeit des Elektrons in gleichen Volumenteilen dV, wenn diese verschiedene Entfernungen vom Kern aufweisen. Mit Hilfe der Wahrscheinlichkeitsdichten (Elektronendiche, Ladungsdichte) lässt sich die Aufenthaltswahrscheinlichkeit eines Elektrons in bestimmten Volumenelementen berechnen. Radiale Wahrscheinlichkeitsdichte Die radiale Wahrscheinlichkeitsdichte vergleicht die Aufenthaltswahscheinlichkeit des Elektrons in Kugelschalen gleich Dicke dr, wenn diese verschiedene Abstände vom Kern haben. Mit Hilfe der radialen Wahrscheinlichkeitsdichte lässt sich die Aufenthaltswahrscheinlichkeit in einer bestimmten Kugelschale berechnen. Aufenthaltsraum/Ladungswolke Raum, in dem sich das Elektron mit z.B. 90%iger Wahrscheinlichkeit aufhält. Raum, der 90% der Elektronenladung umschliesst. Hauptquantenzahlen Zahlen, die den Energiezustand von Elektronen in einem Atom beschreiben; sie entsprechen den Elektronenschalen. Entartete Energiezustände Identische Energiewerte innerhalb eines Energieniveau Quantenchemie – eine Einführung Seite 73 Pauli Prinzip Keine zwei Elektronen dürfen in allen Quantenzuständen übereinstimmen. Jede Elektronenwellenfunktion kann so maximal nur zwei Elektronen mit unterschiedlichem Spin beschreiben Hund’sche Regel Die im Atombau mit zunehmender Ordnungszahl hinzukommenden Elektronen werden zunächst die zur Verfügung stehenden energetisch gleichwertigen Plätze in einer Elektronenschale einzeln besetzen (Schweizerbusprinzip). Hybridisierung (Lat.: Hybrida, Mischling) Ein von Linus Pauling eingeführter Begriff. Man versteht darunter eine Linearkombination verschiedener AO am gleichen Atom zu sogenannten Hybridorbitalen. Bei der Hybridisierung handelt es sich um keinen physikalisch observablen Effekt, sondern lediglich um ein Hilfsmittel (Modell) zur Erklärung der Elektronenstruktur eines Moleküls. Orbital Ein-Elektronen-Wellenfunktion für Elektronen. Atombindung Erhöhte Aufenthaltswahrscheinlichkeit von mindestens zwei Elektronen zwischen zwei Atomen. Als Folge von konstruktiver Interferenz zwischen zwei Elektronenwellenfunktionen (Addition der Amplituden) wird die Elektronendichte erhöht. Komplex Ein aus Zentralteilchen und Liganden aufgebautes Teilchen, das durch die anziehenden Kräfte zwischen nichtbindenden Elektronenpaaren von Liganden und positiver Ladung des Zentralteilchens zusammengehalten wird. Ligandenfeld Das elektrische Feld, das die nichtbindenden Elektronenpaare der Liganden erzeugen. Ligandenfeldaufspaltung Energiedifferenz zwischen den energiereicheren und den energieärmeren d-Orbitalen im Komplex. Seite 74 Quantenchemie – eine Einführung 12 Anhang 7.1.1 Polarkoordinaten Die Wellenfunktionen werden wegen der Kugelsymmetrie des Coulomb-Potentials im Polarkoordinaten ausgedrückt. Die Beziehungen sind unten dargestellt: z · Q P(x,y,z) P(r,θ,φ) θ r θ y φ · · x Wobei die Polarkoordinaten in Abhängigkeit der Raumachsen x, y und z lauten: cos(θ ) = 7.2 z r sin(θ ) = Q r sin(φ ) = y r ⋅ sin(θ ) cos(φ ) = x r ⋅ sin(θ ) Der erste Energiezustand (Grundzustand) des Elektrons im Wasserstoffatom Wie sehen aber nun diese Orbitale aus? Nach Max Born ist das Quadrat der Amplitude der angenommenen Materiewelle proportional zur Aufenthaltswahrscheinlichkeit des Elektrons in einem bestimmten Raumvolumen. Auf Grund des Coulomb-Potentials kann man annehmen, dass die Elektronendichte nahe des Kerns gross ist und mit zunehmendem Abstand im Unendlichen gegen Null strebt. Somit bietet sich für die Wellenfunktion eine abnehmende Exponentialfunktion des Typs Ψ = k·e -r/C an. Mit der Bedingung ∞ ∫ Ψ dτ = 1 2 0 2 (dτ = 4πr dr; Volumen der Kugelschale der Dicke dr), was nichts anderes heisst, als dass sich das Elektron irgendwo im Raumvolumen von 0 bis ∞ aufhalten muss. Dadurch kann die Konstante k durch die Konstante C ausgedrückt werden und es folgt folgende Wellenfunktion: Ψ= 1 π ⋅C3 ⋅e − r C Seite 75 Quantenchemie – eine Einführung Diese abgewandelte Wellenfunktion wird nun in die SGL (Schrödinger-Gleichung) eingesetzt, um die Gesamtenergie zu berechnen. Diese soll minimal sein. Das bedeutet, dass man die erste Ableitung von EGES Null setzt und auf diese Weise die Konstante C bestimmt: dE h2 = 0 → C = 4πε 0 ⋅ 2 2 ≡ a0 = 0.53 ⋅ 10 −10 m dC 4π me0 1 Für den Grundzustand (Symbol 1s ) findet man eine Wellenfunktion, die mit den oben gemachten Angaben wie folgt aussieht: Ψ1s = Wobei: Ψ: r: 1 π ⋅ a 03 ⋅e − r a0 Amplitude der angenommenen, stehenden Materiewelle Abstand des Elektrons vom Atomkern a0: Bohrscher Radius; 0.53·10 -10 m e0: Elementarladung h: Planksches Wirkungsquantum m: Elektronenmasse 7.2.1 ε0: Influenzkonstante e: Eulersche Zahl, 2.718 Vereinfachte Wellenfunktion 1s und räumliche Darstellung Lässt man von der Wellenfunktion die konstanten Faktoren weg, so ergibt sich eine vereinfachte Funktion, die vollständig zur qualitativen Diskussion der wesentlichen Erscheinungen genügt: Ψ = e − a ⋅r Abbildung 61: Graphische Darstellung der Wellenfunktion Ψ für den Grundzustand des H-Atoms Die Amplitude der angenommenen Materiewelle hat in der Nähe des Atomkerns einen grossen Wert; die Funktionswerte werden mit zunehmendem Abstand kleiner und streben im Unendlichen gegen Null. Auch hier sei nochmals erwähnt, dass diesen Amplituden keine physikalische Realität zukommt! Seite 76 Quantenchemie – eine Einführung Da die Wellenfunktion 1s nur von der Variablen r abhängig ist, ist der Verlauf von Ψ1s nach allen Raumrichtungen gleich und die 1s-Funktion lässt sich auf einfache Art räumlich darstellen. Man zeichnet alle Punkte für r, die den gleichen Funktionswert Ψ1s besitzen. Der entsprechende geometrische Ort ist ein Kreis in der Zeichenebene, bzw. eine Kugeloberfläche bei Berücksichtigung aller Raumrichtungen. Ψ = e − a ⋅r Abbildung 62: Geometrischer Ort aller Punkte P mit gleichem Funktionswert. Links: in der Zeichenebene; rechts: räumlich 7.2.2 Die Energie des Wasserstoffatoms im Grundzustand 1s Die Energie des Grundzustandes 1s berechnet sich durch das Einsetzen der Wellenfunktion Ψ1s in die SGL. Als Ergebnis erhält man: 1 e02 E1s = − ⋅ 4πε 0 2 a0 Das negative Vorzeichen bedeutet, dass bei der Bildung des Wasserstoffatoms aus den beiden unabhängigen Teilchen Proton und Elektron Energie frei wird à exothermer Vorgang. 7.2.3 Wird Das Quadrat der Wellenfunktion: Wahrscheinlichkeitsdichte und radiale Wahrscheinlichkeitsdichte die Wellenfunktion des Grundzustandes quadriert, so ergibt sich daraus die sogenannte Wahrscheinlichkeitsdichte (Elektronendichte, Ladungsdichte) mit der sich die Aufenthaltswahrscheinlichkeit in einem bestimmten Volumenelement dV berechnen lässt. Je nach Betrachtungsweise unterscheidet man neben der Wahrscheinlichkeitsdichte die radialen Wahrscheinlichkeitsdichte. Die Wahrscheinlichkeitsdichte Die Wahrscheinlichkeitsdichte (Elektronendichte, Ladungsdichte) vergleicht die Aufenthaltswahrscheinlichkeit des Elektrons in gleichen Volumenteilen dV, wenn diese verschiedene Abstände vom Kern aufweisen. Das Quadrat der Wellenfunktion 1s: Ψ 2 = e −2 a ⋅ r Seite 77 Quantenchemie – eine Einführung Die Aufenthaltswahrscheinlichkeit in einem bestimmten Volumenteil dV mit einem bestimmten Abstand vom Atomkern berechnet sich nach dW = Ψ dV = e 2 -2a·r dV. Um Ψ graphisch darstellen zu können, muss man die 2 Wahrscheinlichkeit dW für verschiedene Abstände r berechnen und so die erhaltenen Punkte miteinander verbinden. Untenstehende Abbildung zeigt, dass die Wahrscheinlichkeit, das Elektron in einem Raumelement dV in der Nähe des Kerns anzutreffen sehr gross ist. Abbildung 63: Links Graphische Darstellung der Elektronendichte im Grundzustand des H-Atoms Rechts: Elektronendichte für Ψ2 im Grundzustand des H-Atoms Die Wahrscheinlichkeitsdichte des Wasserstoffelektrons im Grundzustand nimmt mit zunehmendem Kernabstand stark ab (im Abstand von 0.182 pm ist die Wahrscheinlichkeitsdichte bereits auf 1/1000 des maximalen Wertes abgesunken); sie wird jedoch erst im Unendlichen Null. Dies bedeutet, dass auch in grösserer Entfernung vom Kern eine zwar extrem kleine, aber doch endliche Wahrscheinlichkeit dafür besteht, das Elektron dort anzutreffen. Mit anderen Worten, das Wasserstoffatom hat (wie natürlich auch die anderen Atome) nach aussen theoretisch keine scharfe Umgrenzung. Die radiale Wahrscheinlichkeitsdichte (radiale Aufenthaltswahrscheinlichkeit, radiale Elektronendichte) Das Kraftfeld des Atomkerns ist dreidimensional und kugelsymmetrisch. Deshalb ist es sinnvoll, auch die sogenannte radiale Wahrscheinlichkeitsdichte zu betrachten: Die radiale Wahrscheinlichkeitsdichte (radiale Elektronendichte) vergleicht die Aufenthaltswahrscheinlichkeit des Elektrons in Kugelschalen gleicher Dicke dr, wenn diese verschiedene Abstände vom Kern aufweisen. Für die radiale Wahrscheinlichkeitsdichte gilt: dW = Ψ ·4π r dr. Die Aufenthaltswahrscheinlichkeit innerhalb 2 2 2 2ar 2 2 einer derartigen Kugelschale berechnet sich also folgendermassen: dW = e ·4π r dr, wobei dr die Dicke der 2 2 2 2 Kugelschale, 4π r die Oberfläche einer Kugel und 4π r dr das Volumen einer Kugelschale darstellen: r dr r1 r2 r Quantenchemie – eine Einführung Seite 78 Abbildung 64: oben: schematische Darstellung der radialen Wahrscheinlichkeitsdichte; unten: In welchem Abstand vom Atomkern des Wasserstoffatoms befindet sich die Kugelschale mit der grössten Aufenthaltswahrscheinlickeit des Elektrons? Nimmt man nun diese Zählung vor, erhält man folgende radiale Verteilung des Elektrons im 1s-Zustand: Abbildung 65: Graphische Darstellung der radialen Wahrscheinlichkeitsdichte des Elektrons im Wasserstoffatom (Grundzustand) Stellt man diese Graphik räumlich dar, erhält man die in Abb. 20 dargestellte kugelsymmetrische Verteilung der Elektronen im 1s-Zustand. Abbildung 66: Räumliche Darstellung das Aufenthaltsraumes (Ladungswolke) des Elektrons im Wasserstoff; 1s-Zustand Quantenchemie – eine Einführung 7.2.4 Seite 79 Aufenthaltsraum (Ladungswolke) des Elektrons im Grundzustand Es ist oft zweckmässig, den Umriss des Raumes darzustellen, in dem sich das Elektron gemäss der Wahrscheinlichkeitsdichte mit einer bestimmten Wahrscheinlichkeit aufhält (z.B. 90 %). Diesen Raum bezeichnet man als Aufenthaltsraum des Elektrons. Das Teilchenmodell steht dabei „Pate“: Dieser Aufenthaltsraum ist identisch mit dem Raum, der einen bestimmten Betrag der Elektronendichte umschliesst. Da das Wellenmodell in dieser Betrachtung überwiegt spricht man von einer Ladungswolke. Für den 1s-Zustand entspricht dies einer Kugel (vgl. Baars/Christen „Chemie – Theorie und Praxis, Sauerländer, 1996/98, Kapitel 4 und Duden Chemie Kapitel 3.1). Seite 80 Quantenchemie – eine Einführung 7.2.5 Zusammenfassung des Zustandes 1s Zusammenfassung des Grundzustandes 1s Oben links: Wellenfunktion 1s, Zeichenbene Oben rechts: Wellenfunktion 1s, räumlich Mitte links: Quadrat der Wellenfunktion 1s; bezogen auf das konstante Volumen dV à Wahrscheinlichkeitsdichte Mitte rechts: Quadrat der Wellenfunktion 1s; bezogen auf das Volumen von Kugelschalen 4πr2 mit konstanter Dicke dr à radiale Wahrscheinlichkeitsdichte unten: Ladungswolke bzw. Aufenthalstraum (Raum in dem sich das Elektron zu z.B. 90 % aufhält Seite 81 Quantenchemie – eine Einführung 7.3 Der zweite Energiezustand des Elektrons im Wasserstoffatom Als Lösung der SGL findet man für den zweiten Energiezustand (n = 2) des Wasserstoffatoms vier Funktionen, welche die angenommene Materiewelle des Elektrons beschreiben. Eine 2s-Wellenfunktion (l = 0; m = 0) und drei 2p-Wellenfunktionen (l = 1; m = -1, 0, +1). 2 bedeutet zweiter Energiezustand; p und s symbolisieren die räumliche Form der entsprechenden Wellenfunktion 7.3.1 Der 2s-Zustand 7.3.1.1 Die Wellenfunktion des 2s-Zustandes Die Wellenfunktion Ψ des 2s Zustandes lautet in ihrer vereinfachten Form wie folgt: Ψ2 s = (2 − r ) ⋅ e − r Im Vergleich zur Wellenfunktion Ψ1s besitzt sie also eine Nullstelle für r = 2, wie in Abbildung 21 ersichtlich ist. Abbildung 67: links: Graphische Darstellung der Wellenfunktion 2s entlang einer Raumrichtung und rechts: Geometrischer Ort aller Punkte mit gleichem Funktionswert Ψ2s (räumlich) Die Wellenfunktion Ψ2s ist nur von der Variablen r abhängig, ihr Verlauf ist nach allen Raumrichtungen hin also gleich. Die räumliche Darstellung ist deshalb auch hier sehr einfach und führt mit ähnlichen Überlegungen wie wir bei der Wellenfunktion 1s angestellt haben (Suche nach Punkten für r mit selbem Funktionswert Ψ2s) zu der Darstellung in Abbildung 21 rechts. Wir erkennen darin die Knotenfläche (Kugelfläche) bei r = 2 in der räumlichen Darstellung. 7.3.1.2 Die Energie des Wasserstoffatoms im 2s-Zustand Durch Einsetzen der Wellenfunktion 2s in die SGL lässt sich der Energiezustand des Wasserstoffatoms für den ersten angeregten Zustand 2s berechnen: 1 e02 1 E2 s = − ⋅ ⋅ 4πε 0 2 a0 4 Sie ist damit um den Faktor ¼ grösser als die Energie im Grundzustand 1s. Seite 82 Quantenchemie – eine Einführung 7.3.1.3 Das Quadrat der Wellenfunktion 2s; Wahrscheinlichkeitsdichte und radiale Wahrscheinlichkeitsdichte Wie im Grundzustand unterscheidet man auch hier die beiden Wahrscheinlichkeitsdichten. Die Aufenthaltswahrscheinlichkeit in einem bestimmten Volumenteil dV mit einem bestimmten Anstand r vom dW = Ψ22s ⋅ dV = ( 2 − r ) 2 ⋅ e −2 r ⋅ dV Atomkern (Wahrscheinlichkeitsdichte) berechnet sich nach: und dargestellt als Graph: Abbildung 68: Geometrischer Ort aller Punkte mit der gleichen Elektronendichte, links in der Zeichenebene und rechts räumliche Darstellung. Vergleicht man die Aufenthaltswahrscheinlichkeit des Elektrons in Kugelschalen gleicher Dicke dr mit unterschiedlichen Abständen vom Atomkern, so erhält man die radiale Wahrscheinlichkeitsdichte: dW = Ψ22s ⋅ 4πr 2 dr = ( 2 − r ) 2 ⋅ e −2 r ⋅ 4πr 2 dr Was zu folgendem Funktionsbild führt: Abbildung 69: Graphische Darstellung der radialen Wahrscheinlichkeitsdichte. Zusammenfassend lässt sich also sagen, dass im ersten angeregten Zustand 2s sich das Elektron an beliebigen Stellen im Wasserstoffatom (mit unterschiedlichen Wahrscheinlichkeiten) aufhält. Eine Ausnahme bildet die Kugelschale mit dem Abstand r0, in der die Aufenthaltswahrscheinlichkeit Null ist (Knotenfläche). Mit grösster Wahrscheinlichkeit hält sich das Elektron in der Kugelschale beim Maximum auf. Seite 83 Quantenchemie – eine Einführung 7.3.1.4 Zusammenfassung des Zustandes 2s Zusammenfassung des Grundzustandes 2s Oben links: Wellenfunktion 2s, Zeichenbene Oben rechts: Wellenfunktion 2s, räumlich Mitte links: Quadrat der Wellenfunktion 2s; bezogen auf das konstante Volumen dV à Wahrscheinlichkeitsdichte Mitte rechts: Quadrat der Wellenfunktion 2s; bezogen auf das Volumen von Kugelschalen 4πr2 mit konstanter Dicke dr à radiale Wahrscheinlichkeitsdichte unten: Ladungswolke bzw. Aufenthalstraum (Raum in dem sich das Elektron zu z.B. 90 % aufhält Seite 84 Quantenchemie – eine Einführung 7.3.2 Der 2p-Zustand Die drei 2p-Wellenfunktionen, die sich ebenfalls als Lösungen für den zweiten Energiezustand ergeben, sind im Gegensatz zu den 2s-Wellenfunktionen nicht kugel- sondern achsensymmetrisch. Das bedeutet, dass sie winkelabhängig sind. Die vereinfachten Wellenfunktionen lauten folgendermassen: Ψ2 p = r ⋅ e − r ⋅ sin θ ⋅ cos φ x Ψ2 p = r ⋅ e − r ⋅ sin θ ⋅ sin φ y Ψ2 p = r ⋅ e − r ⋅ cos θ z Die Wellenfunktionen lassen erkennen, dass nun gleiche Funktionswerte Ψ in unterschiedlichen Abständen vom Atomkern auftreten (im Gegensatz zu den s-Funktionen), je nach dem welche Raumrichtung (d.h. welcher Winkel) gewählt wird. Analoge Überlegungen, wie wir sie bei den Zuständen 1s und 2s gemacht haben ergeben 26 untenstehende zusammenfassende Betrachtung des Zustandes 2p , wobei zu beachten ist, dass die charakteristische „Hantel“ der p-Wolke zwei Bereiche hat, die man mit + und – bezeichnet (nicht mit den elektrischen Ladungen zu verwechseln!): Legende zur folgenden Seite: Zusammenfassung des ersten angeregten Zustandes 2p A: Graphische Darstellung der Wellenfunktionen 2p entlang der Koordinatenachsen, von links nach rechts: 2px, 2py, 2pz. B: Geometrischer Ort aller Punkte (räumlich) mit dem gleichen Funktionswert Ψ C: Wolkendarstellung der Wahrscheinlichkeitsdichten entlang der drei Raumachsen D: Graphische Darstellung der radialen Wahrscheinlichkeitsdichte für das Elektron im Wasserstoffatom im Zustand 2p E: Aufenthaltsräume (Ladungswolken) des Elektrons im Wasserstoffatom für die Zustände 2px. 2py und 2pz. 26 Für interessiert sei auf das Leitprogramm „Quantenchemie“ von G. Baars et al. unter http://dcbwww.unibe.ch/fachdidaktik/lpqc.htm verwiesen (Verlag ETHZ, Abteilung für das höhere Lehramt). Seite 85 Quantenchemie – eine Einführung A B C D E