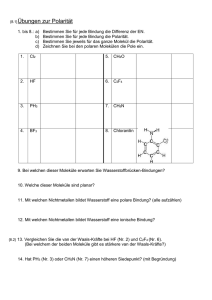
BPC I Praktikum
Werbung

BPC I Praktikum Theoretische Grundlagen zu den Experimenten (1) (2) (3) (4) (5) (6) (7) NMR ATR-FTIR ITC Enzymkinetik GC Fluoreszenz Docking NMR Kernspezifische Eigenschaften: Eigendrehimpuls, Spin und magnetisches Moment Elektronen haben ein intrinsisches magnetisches Moment. Auch Protonen haben ein magnetisches Moment, das jedoch ca. 2000mal schwächer ist als das des Elektrons. Daher spielt das kernmagnetische Moment für die Reaktivität eines Moleküls keine Rolle. Dennoch macht es ein wichtiges spektroskopisches Verfahren möglich. Das magnetische Moment wird von einem Spin (Quantenzahl des Drehimpulses) erzeugt. Während ein Elektron immer die Spinquantenzahl 1/2 aufweist, können Kernspins ein ganz-zahliges Vielfaches von 1/2 annehmen, da mehrere Protonen vorhanden sind, die jeweils eine Spinquantenzahl von 1/2 aufweisen. Der Eigendrehimpuls P = I ⋅ ( I + 1) der Atomkerne steht in direktem Zusammenhang mit der Spinquantenzahl I. Dabei gilt = h / 2π , wobei h = 6,626 ⋅ 10−34 Js das Plancksche Wirkungsquantum ist. I kann Werte von 0 bis 6 annehmen, wobei der Kernspin I=0 NMR Inaktivität bedeutet. Der Kerndrehimpuls P erzeugt immer ein magnetisches Moment µ = γ ⋅ P = gN ⋅ βN ⋅ P = gN ⋅ e⋅ ⋅P 2mPr oton Der Kerndrehimpuls P und das magnetische Moment µ sind über das gyromagnetische oder magnetogyrische Verhältnis γ proportional verknüpft. e = 1,6 ⋅ 10−19 C ist die Elementarladung und mPr oton = 1,672 · 10-27 kg = 1836,152 · m Elektron ist die Masse des Protons, die wesentlich größer ist als die des Elektrons, weshalb das kernmagnetische Moment wesentlich geringer ist. β N = 5,051 ⋅ 10−27 J / T ist das Kernmagneton und der Kern-g-Faktor g N eine für den jeweiligen Atomkern charakteristische dimensionslose Zahl. γ = g N ⋅ β N ist das gyromagnetische Verhältnis. Je größer der Wert für γ , desto nachweisempfindlicher ist die Substanz in der NMR Spektroskopie. Kerne mit einem großen γ Wert werden daher als NMR empfindlich, solche mit einem kleinen Wert für γ als NMR-unempfindlich bezeichnet. Anhand der Formel ist ersichtlich, dass Kerne ohne Spin kein magnetisches Moment aufweisen. Solche Kerne sind zum Beispiel 12C und 16O. Um sie im NMR Spektrometer zu detektieren, müssen diese Elemente Isotopenmarkiert werden, damit sie einen Spin aufweisen. Insgesamt beeinflusst sowohl die Form des Kerns als auch das Verhältnis zwischen Protonen und Neutronen im Kern die NMR Empfindlichkeit. Wechselwirkung zwischen Kerndrehimpuls und externem Magnetfeld Bringt man nun ein Teilchen mit Kernspin in ein externes, homogenes, statisches Magnetfeld B0, so wird sich sein Kerndrehimpulsvektor in diesem Feld aufgrund seines magnetischen Momentes ausrichten. Die Ausrichtung des zuvor zufallsverteilten Kerndrehimpulses entspricht einer Richtungsquantelung (siehe Abbildung 1). Der Drehimpulsvektor P nimmt einen bestimmten Winkel α zur Richtung des Magnetfelds B0 ein. Die Z-Komponente des Drehimpulsvektors pz= m ⋅ Abbildung 1 Übersicht über die Ausrichtung von P in B0 in Abhängigkeit von m steht in Verbindung mit der magnetischen Quantenzahl m. Der Drehimpuls orientiert sich also im Raum so, dass seine Komponente in Feldrichtung (PZ) ein m-Faches von ist. M kann Werte von I, I-1,… bis –I annehmen. Für die Z-Komponente des magnetischen Moments gilt somit µz = γ ⋅ m ⋅ Ein magnetisches Moment, das nicht parallel zum Magnetfeld ausgerichtet ist, erfährt eine Kraft. Das resultierende Drehmoment ist gegeben durch Γ = µ × B0 Und befindet sich somit senkrecht zur µ − B0 Ebene. So wird eine Bewegung von μ auf der Oberfläche eines Kegels um die Richtung des Magnetfeldes (Z-Komponente des gedachten Koordinatensystems) bewirkt (Präzession: siehe Abbildung 2). Abbildung 2 dicker Pfeil entspricht P. er rotiert entlang Die Frequenz ω0 = 2π ⋅ f = γ ⋅ B0 der Fläche eines Kegels um B0 Mit der das magnetische Moment um die Z-Achse präzidiert heißt Lamorfrequenz. Diese Frequenz ist kernspezifisch und nimmt linear mit der Magnetfeldstärke zu. Radiowellen dieser Frequenz bewirken die Resonanz des Spinsystems. Solche werden bei der Messung zusätzlich eingestrahlt um den Winkel α der Drehung so zu verändern, sodass der Vektor in der Nähe der xy-Ebene liegt, wo er messbar ist. Auf der Z-Achse ist der Kerndrehimpuls nicht zu messen, da das magnetische Moment sehr gering ist und vom viel größeren externen Magnetfeld überlagert wird. Die Anzahl der energetischen Niveaus Energetisch gesehen bedeutet eine Ausrichtung des Atomkerns eine Aufspaltung in Energieniveaus (Zeeman Effekt). Es gibt für einen Spin I , der zuvor zufällig im Raum ausgerichtet war, exakt 2 I + 1 verschiedene Energieniveaus in einem äußeren Magnetfeld B0. Für das Wasserstoffatom gilt I = 1 2 , wodurch 2 mögliche Energieniveaus möglich sind (siehe Abbildung 3). Der Abstand der Energieniveaus nimmt linear mit der Magnetfeldstärke zu. Für Wasserstoff gibt es den energetisch niedrigeren Zustand α bzw. + 1 2 , der Abbildung 3 Aufteilung der Energieniveaus. Energiedifferenz zwischen Niveaus steigt mit B0 parallel zum Magnetfeld ausgerichtet ist und den energiereicheren, antiparallelen Zustand β bzw. + 1 2 . Es herrscht ein thermisches Equilibrium der Absolutmagnetisierung, wenn alle Spins im α oder β Zustand vorliegen, diese Zustände aber untereinander wechseln können. Die Energie der Niveaus und ihre Verteilung Die Energie eines Teilchens E = − µ Z B0 = −γ ⋅ PZ ⋅ B0 = −γ ⋅ m ⋅ ⋅ B0 ist der Stärke des äußeren Magnetfeldes proportional. Da es auch stets 2I+1 verschiedene m Werte und damit Orientierungsmöglichkeiten für das magnetische Moment gibt, lassen sich die Energien der zwei möglichen Niveaus berechnen: 1 Eα / β = ± γ ⋅ ⋅ B0 2 Für das Wasserstoffatom ergibt sich so eine Energiedifferenz, das so genannte ZeemanSplitting von 1 1 ∆E = − γ ⋅ B0 ⋅ ⋅ + − − γ ⋅ B0 ⋅ ⋅ − = −γ ⋅ ⋅ B0 , 2 2 welche aufgebracht werden muss, um vom α in den β Zustand zu gelangen. Die Population der Spin ½ Teilchen teilt sich proportional zur Zeeman-Energie ΔE und zur thermischen Energie der Teilchen (führt zur Vermischung der Zustände) in beide Energieniveaus auf. Die Populationsverteilung entspricht einer Boltzmann-Verteilung ∆E − β = e kT α So kommt es zu einer Überschussmagnetisierung der Gesamtprobe. Es liegen mehr Spins im Zustand α als im Zustand β. Dies ist per NMR detektierbar, wobei zu beachten ist, dass das beobachtete Signal immer der Gesamtüberschuss-Magnetisierung aus allen Spins der Probe entspricht. Die Überschussmagnetisierung ist jedoch sehr gering und nimmt mit der Stärke des Magnetfelds zu. Einstellung der Überschussmagnetisierung Wird eine Probe in ein Magnetfeld eingebracht, stellt sich die Überschussmagnetisierung jedoch nicht sofort ein. Die räumliche Orientierung bleibt zunächst unverändert. Spins verhalten sich wie Magneten und beeinflussen sich daher über ihr Magnetfeld gegenseitig (dipolare Kopplung). Da sich die Teilchen in Lösung sehr schnell bewegen erfahren sie so ein leicht zeitlich fluktuierendes Magnetfeld. Dadurch fluktuiert auch die Drehachse der Spins. Das fluktuierende Magnetfeld setzt sich aus dem starken äußeren und den sie jeweils umgebenden Magnetfeldern zusammen. Wenn eine Komponente des Magnetfeldes mit der Lamorfrequenz um die Spins oszilliert, kann durch Resonanz-Effekt der Drehachsen-Fluktuation ein Übergang zwischen den Energieniveaus ausgelöst werden, indem die Drehachse immer stärker fluktuiert, bis sie letztlich nahezu parallel oder antiparallel zur Z-Achse steht. Dadurch, dass die Vektoren jedoch nicht kohärent um die Z-Achse präzidieren, ist die Nettomagnetisierung (siehe Abbildung 4) exakt auf der Z bzw. der –Z Achse Abbildung 4 dicker Pfeil entspricht der Nettomagnetisierung; sie ergibt sich aus den nicht kohärent rotierenden einzelnen Kerndrehimpulsvektoren. Im α Zustand liegen mehr Kerndrehimpulsvektoren vor. Hier liegt demnach die Überschussmagnetisierung vorzufinden. Wie bereits beschrieben ist hier jedoch aufgrund des externen Magnetfeldes keine Überschussmagnetisierung messbar. So wird mit der Zeit die energetisch günstigere Überschussmagnetisierung erzeugt. Dieser Prozess kann über einen exponentiellen Anstieg der Nettomagnetisierung t − T1 M Z (t ) = M eq 1 − e Abbildung 5 Einstellung der Überschussmagnetisierung, nach Einstellung des externen Magnetfeldes beschrieben werden (siehe Abbildung 5), wobei T1 „Spin Lattice Relaxation“ Konstante genannt wird. Sie muss mittels eines Experiments bestimmt werden. M eq ist die Magnetisierung im Equilibrium (Gleichgewicht). Messung der Überschussmagnetisierung, Radiofrequenz-Pulstechnik Wenn sich die Überschussmagnetisierung nun eingestellt hat, muss sie nun detektierbar gemacht werden, indem die Magnetisierung von der Z bzw. –Z Achse in die xy-Ebene geklappt wird. Dies geschieht durch einen 90° Puls. Ist der Kerndrehimpulsvektor in der xyEbene angelangt erzeugt die Magnetisierung ein zeitlich veränderliches Magnetfeld. Dieses erzeugt einen Strom in der fest installierten Detektorspule. Dieser erzeugt ein messbares FID Signal. Bei einem Puls handelt es sich um eine elektromagnetische Welle der Lamorfrequenz einer bestimmten Dauer und Intensität. Diese Welle entspricht einem mit der Lamorfrequenz rotierenden Vektor. Wechselwirkt dieser mit dem Kerndrehimpulsvektor, der ebenfalls mit der Lamorfrequenz rotiert, so kommt es zur Resonanz und der Winkel α des Kerndrehimpulsvektors zur Z-Achse verändert sich. longitudinale Relaxationszeit T1 und transversale Relaxationszeit T2 Diese Relaxationszeiten sind unterschiedlichen Ursprunges, bewirken jedoch beide eine Abnahme der Signalintensität mit der Zeit. Die T1 Relaxationszeit beschreibt die Zeit, die der Drehimpuls-Vektor benötigt seine xy-Komponenten zu verlieren, indem er zurück zur ZAchse relaxiert. Für das Verständnis um die T2 Relaxationszeit stellt man sich die in der NMR letztlich gemessene Nettomagnetisierung als Summe der Einzelmagnetisierungen vor. Alle Spins präzidieren mit ihrer Lamorfrequenz um die Z-Achse. Da die Lamorfrequenz jedoch Magnetfeldabhängig ist und sich die Spins in leicht unterschiedlichen magnetischen Umgebungen befinden, geht die Kohärenz der Einzelspins mit der Zeit verloren, weil die Lamorfrequenz fluktuiert. Diese Kohärenz macht jedoch die Überschussmagnetisierung und somit die Intensität des Signals aus. Liegt absolut keine Kohärenz vor, befindet sich die Nettomagnetisierung wieder auf der Z-Achse, weshalb sie nicht mehr zu detektieren ist. Beide Relaxationszeiten lassen sich durch Experimente bestimmen. Im Folgenden soll nur das „Inversion-Recovery“ Experiment zur Bestimmung von T1 erläutert werden. Inversion-Recovery Experiment zur Spin-Gitter Relaxation (T1) Zunächst wird durch einen 180° Puls die Nettoüberschussmagnetisierung um 180°, von der Z-Achse auf die -Z-Achse gedreht. Dazu wird eine Radiowelle mit der Resonanzfrequenz der zu drehenden Spins eingestrahlt. So wandert die Nettoüberschussmagnetisierung von oben entlang der Z-Achse nach unten, indem sich der Winkel α des Kerndrehimpulsvektors durch Resonanzeffekt vergrößert, bis die Rotation des Überschuss-Kerndrehimpulsvektors um die –Z Achse erfolgt (energetisch ungünstiger Zustand), anstatt um die Z-Achse (energetisch günstiger Zustand). Aus dieser energetisch invertierten Lage (die Boltzmann-Verteilung wurde umgekehrt) relaxiert das Spinsystem zurück in den Ausgangszustand, indem die Nettoüberschussmagnetisierung wieder entlang der Z-Achse nach oben wandert. Abbildung 6 Übersicht über das Inversion-Recovery Experiment. Zunächst schiebt ein 180° Puls die Überschussmagnetisierung auf –Z. Unterschiedliche Zeiten des Wartens vor dem 90° Puls bewirken ein unterschiedliches Signal nach diesem 90° Puls. Die Rückkehr in den energetischen Gleichgewichtszustand wird durch die Änderung der Schwingungszustände der Moleküle ermöglicht. Die Dauer der Relaxationszeit hängt somit auch von der Molekülbewegung ab. Stimmt die Molekülbewegung nicht mit der Resonanzfrequenz überein bewirkt ein fluktuierendes Magnetfeld zu einer höheren Wahrscheinlichkeit die Entstehung einer Rotations-Frequenz ungleich der Lamorfrequenz, wodurch die T1 Relaxationszeit groß wird. Wenn die Molekülbewegung und die Resonanzfrequenz ideal harmonieren, ist die Relaxationszeit minimal. Die Molekülbewegung hängt von der Temperatur (Molekülbewegung steigt) und der Viskosität (Molekülbewegung sinkt) des Lösungsmittels ab (Hierzu siehe auch Diskussionsteil). Nachdem eine gewisse Zeit nach dem 180° Puls vergangen ist, kann nun ein 90° Impuls eingestrahlt werden, der die Magnetisierung in die xy-Ebene bringt. Erst in dieser Position (transversal) kann die Magnetisierung beobachtet werden. Dieses Experiment muss nun für mehrere Zeiten τ zwischen dem 180° und dem 90° Impuls durchgeführt werden, bis diejenige Zeit gefunden ist, für die kein Signal mehr detektierbar ist. Diese Zeit wird benötigt, um die invertierte Anfangsnettomagnetisierung des Spins (-MZ) genau auf 0 zu bringen. Für die Inversion-Recovery (T1 Relaxation) lässt sich die Magnetisierung wie folgt beschreiben: t − T1 M Z (t ) = M eq 1 − 2e Der resultierende Graph ist in folgender Abbildung 7 zu sehen. Abbildung 7 Abhängigkeit der Signal-Intensität zwischen 180° Puls und 90° Puls a (τ ) von der Zeit τ Spin Echo Experiment zur Spin-Spin Relaxation (T2) Bei t = 0 werde die Magnetisierung mit einem π/2 bzw. 90° Puls in die x-Richtung gedreht und beginnt zu präzedieren. Die Inhomogenität des Feldes bewirkt, dass die Präzessionsfrequenz ebenfalls nicht einheitlich ist. Das bedeutet, dass die einzelnen Teilmagnetisierungen sich schon bald planar um B0 verteilt haben und sich gegenseitig kompensieren. In obigem Bild ist dies mit zwei Teilmagnetisierungen (rot und blau) dargestellt, die im rotierenden Koordinatensystem schneller (blau) bzw. langsamer (rot) als die mittlere Präzessionsfrequenz sind. Der mit der Verzögerung τ auf den ersten Impuls folgende π bzw. 180° Puls vertauscht die vorauseilenden Teilmagnetisierungen mit den zurückgebliebenen. Vorausgesetzt, die Präzessionsgeschwindigkeit bleibt konstant, treffen sich alle Teilmagnetisierungen wieder in der ursprünglichen Richtung, wenn die Verzögerung τ ein weiteres Mal abgelaufen ist. Das Messsignal sieht wie die rote Linie im oberen Diagramm in Abb. 1 aus. Das Zusammentreffen nach 2τ äußert sich als Wiederanstieg des Signals und wird als Echo bezeichnet. Die T2 läuft schneller ab als die T1: − Tt M Y (t ) = M eq e 2 FID Signal, Fouriertransformation, Frequenz-Spektrum Der Strom, welcher in der Spule erzeugt wird, ist auf dem Monitor als FID (free induction decay) zu sehen. Es handelt sich um eine Sinusförmige Kurve, welche die Intensität in Abhängigkeit der Zeit darstellt. Die Intensität entspricht den einer bestimmten Zeit zugeordneten Aufenthaltsorten (innerhalb der xy-Ebene) aller kohärenten Kerndrehimpulsvektoren, die um die Z-Achse rotieren. Wenn sich die Spule (siehe Abbildung 8) in Richtung der –x Achse befindet, erzeugt der Kerndrehimpulsvektor, wenn er sich auf der –x Achse befindet eine Maximalintensität, auf der x-Achse erzeugt er ebenso eine Maximalintensität, mit umgekehrtem Vorzeichen. Auf der y-Achse ist die Intensität jeweils 0. Die Sinuskurve schwächt aufgrund der Relaxation (Kerndrehimpulsvektor verlässt die xy- Abbildung 9 nach der FT resultierendes typisches NMR-Spektrum, deren Piks anhand der Farbmarkierung bestimmten Gruppen eines Moleküls zugeordnet werden konnten Ebene und verliert an Kohärenz) mit der Zeit ab. Dieses Zeitdomänensignal wird nun mittels Fouriertransformation in ein Frequenzspektrum umgewandelt. Dazu wird aus der erhaltenen FID-Funktion ein Produkt mit einer SinusFunktion und über dieses Produkt ein Integral gebildet. Sind beide Faktoren gleich so ist das Integral maximal, es resultiert ein Peak im Frequenzspektrum (siehe Abbildung 9). Sind sie nicht gleich, heben sie sich zu null auf. Jeder Peak bei einer bestimmten Frequenz kann im Frequenzspektrum einer bestimmten chemischen Gruppe zugeordnet werden Fourier Transformation Jedes Proton hat eine unterschiedliche Larmorfrequenz, je nachdem in welcher chemischen Umgebung sie sich befinden; Bei einer NMR Messung werden alle Frequenzen gleichzeitig eingestrahlt, weshalb auch alle H Atome angeregt werden, wodurch sich ihre Kerndrehimpulsvektoren mit ihrer Larmorfrequenz (nach einem 90° Puls) innerhalb der xy Ebene um die Z Achse drehen; jedes einzelne Proton induziert dadurch einen Wechselstrom in der Spule mit seiner Larmofrequenz, wodurch es im FID zu einer Überlagerung aller Larmorfrequenzen kommt; daher lassen sich aus dem zeitdomänen Signal des FID nicht einfach die Frequenzen ableiten; Die Fourier Transformation ist eine mathematische Operation, mit der die Zeitdomänen Information der F(t) Funktion (FID) in die Frequenzdomänen Information der F(omega) Funktion (Spektrum) umgewandelt wird; Das FID stellt eine periodische Funktion dar, die sich durch eine Reihe harmonischer Funktionen annähern lässt: Es wird das Produkt zwischen f(t) und dem sinusteil, sowie dem cosinusteil gebildet; Dabei werden alle Frequenzen einmal für den sinusteil (Punktsymmetrisch) und einmal für den cosinusteil (Achsensymmetrisch) durchprobiert; ist z.b. der sinusteil für eine bestimmte Frequenz gleich der Funktion f(t), so ergibt das Produkt der beiden Funktionen eine maximale Intensität; es resultiert somit ein Peak; genauso wird das mit dem cosinusteil gemacht; F(omega) ist eine komplexe Größe, weshalb man nach der FT zwei Spektren erhält; einen Realteil (durch den cosinusteil) und einen Imaginärteil (durch den sinusteil) Zeitfunktion (links) und Frequenzfunktion (rechts) für zwei um 90° phasenverschobene Signale. Die obere Frequenzfunktion ist das Absorptionsspektrum oder der Realteil, und die untere ist das Dispersionsspektrum oder der Imaginärteil. In der Praxis ergibt die Fouriertransformation zunächst eine Kombination der beiden Frequenzfunktionen (jedes Signal liefert 2 Peaks), aus denen man durch Phasenkorrekturen die reinen Absorptions- und Dispersionsspektren berechnet. Bei der Phasenkorrektur wird aus dem einen Teil das für die Praxis relevante Absorptionsspektrum berechnet (enthält 1 Peak je Signal), während der andere Teil, das Dispersionsspektrum, die um 90° phasenverschobenen Signale enthält. Nachteile und Vorteile der NMR Spektroskopie Nachteil: Der Energieabstand zwischen den Niveaus ist im Vergleich zu kT sehr gering. Dadurch wird der Nachweis der Energieabsorption erheblich erschwert, da die Niveaus annähernd gleich besetzt sind (siehe Boltzmann-Verteilung). Das NMR Signal wird so nur durch einen sehr kleinen Anteil der Kernspins erzeugt. Daher sind sehr starke Magneten notwendig, um eine Erhöhung der Energieabstände der Niveaus zu bewirken. Vorteil: Das Magnetfeld entspricht nicht dem angelegten Magnetfeld, sondern einem lokalen Magnetfeld. Dieses lokale Magnetfeld wird von der Elektronenverteilung an dem untersuchten Atom und an benachbarten Atomen beeinflusst. Die so genannte chemische Verschiebung bewirkt die Differenz zwischen dem externen und dem induzierten Magnetfeld. Daher weisen H-Atome in verschiedenen Molekülen auch verschiedene Lamorfrequenzen auf (siehe Abschnitt 12: chemische Verschiebung) Vorteil: Einzelne magnetische Dipole wechselwirken miteinander. Dies bewirkt die Aufspaltung der Energieniveaus von Systemen mit zwei Spins und die Entstehung von Multiplettspektren. Diese liefern unmittelbare Informationen über die Struktur eines Moleküls (Die Einführung in das Thema „Multiplettspektren“ in diesem Protokoll wurde übergangen, da es im Rahmen des Versuches keine Rolle spielt) Die chemische Verschiebung Die Ursache der chemischen Verschiebung ist die magnetische Suszeptibilität der Elektronen, die den jeweiligen Atomkern umgeben. Die magnetische Suszeptibilität gibt die Fähigkeit der Magnetisierung in einem externen Magnetfeld an. Das resultierende induzierte Magnetfeld führt zu einer teilweisen Abschirmung des externen Magnetfeldes. Die Abschirmwirkung wird von der Elektronendichte beeinflusst. Die chemische Verschiebung für ein isoliertes Atom: Die Richtung des induzierten Magnetfeldes ist der des externen entgegengesetzt und proportional. Man spricht von einer diamagnetischen Reaktion Binduziert = −σ ⋅ B0 Die Proportionalitätskonstante ist die Abschirmkonstante σ . Das Gesamt-Magnetfeld ist die Summe aus induziertem und externem Magnetfeld BGesamt = (1 − σ ) ⋅ B0 Somit ergibt sich eine neue Resonanzfrequenz in Abhängigkeit der Abschirmkonstante (welche durch die das betrachtende Atom umgebenen Elektronendichten bestimmt wird) ω0 = 2π ⋅ f = γ ⋅ B0 ⋅ (1 − σ ) Da der Wert für die Abschirmkonstante mit zunehmender Ordnungszahl und somit auch Elektronendichte ansteigt, verringert also eine ansteigende Elektronendichte die Resonanzfrequenz. Die chemische Verschiebung für ein Atom in einem Molekül σ ändert sich, sobald benachbarte Atome oder Gruppen die Elektronendichte des betrachtenden Atoms verringert oder erhöht. Ursache für diese chemische Verschiebung sind erstens die Elektronegativität benachbarter Gruppen und zweitens die Magnetfelder benachbarter Gruppen. Dies führt zu einer Frequenzverschiebung, die Rückschlüsse auf die Umgebung des Atoms und somit die Struktur des Moleküls zulässt. Hier wird die chemische Verschiebung δ definiert als dimensionslose Größe (ppm-1) zur Beschreibung der Frequenzverschiebung in Bezug auf eine festgelegte Referenzverbindung. Dabei gilt die Betrachtung immer einem bestimmten Atom, wie zum Beispiel 1H. δ (1H ) ppm = f − f ref f ref = (γ ⋅ B0 ⋅ (1 − σ ) ) − (γ ⋅ B0 ⋅ (1 − σ ref ) ) γ ⋅ B0 ⋅ (1 − σ ref ) = γ ⋅ B0 ⋅ (σ ref − σ ) ≈ σ ref − σ γ ⋅ B0 ⋅ (1 − σ ref ) Die Referenz ist im Falle der NMR Spektroskopie des 1H die Verbindung Tetramethylsilan (CH3)4Si. ATR-FTIR Spektroskopie Die spektroskopischen Techniken beruhen auf den Übergängen zwischen verschiedenen erlaubten Energie- bzw. Quantenzuständen von Molekülen oder Atomen aufgrund ihrer Wechselwirkung mit elektromagnetischer Strahlung. In den meisten Experimenten, wie auch hier, misst man eine Abschwächung oder Verstärkung der einfallenden Strahlung, die durch Anregung von Atomen oder Molekülen oder ihren Rückfall in Zustände niedrigerer Energie ausgelöst wurde, als Funktion der Wellenlänge oder Frequenz. Ein grundlegendes Ergebnis der Quantenmechanik ist die diskrete Natur der Energiespektren von Atomen und Molekülen. Solche können nur jene Energiebeträge aufnehmen und abgeben, welche der Differenz zwischen zwei Energieniveaus entsprechen. Das Energiespektrum ist somit für jede chemische Substanz einzigartig. Es muss nur in die Struktur der jeweiligen Substanz übersetzt werden. Da die Energiespektren quantenmechanischer Systeme diskret sind, setzen sich die Absorptions- und Emissionsspektren aus einzelnen Linien zusammen. Dabei ist jede Linie einem Übergang zwischen zwei erlaubten Energieniveaus eines Systems zugeordnet. Die Frequenz hängt mit den, am Übergang beteiligten, Energieniveaus über h ⋅ f = E2 − E1 zusammen. Bei der IR Spektroskopie regen Frequenzen im IR Bereich Schwingungen an (Übergang zwischen Schwingungsniveaus), wobei gleichzeitig auch Rotation angeregt wird (reine Rotationsanregung würde durch Strahlungen im Mikrowellenbereich geschehen) Die Spektroskopie erlaubt es mit 13 Größenordnungen der Photonenenergie zu arbeiten (von Mikrowellen bis Röntgenstrahlung): Spektroskopieart NMR Kernspinübergang Wellenlänge Radiowellen: λ = 10 nm Energieabstand E = 10 −8 kJ / mol 9 Rotationsspektroskopie Anregung der Translation Übergang im ? ? Mikrowellen: E = 10−2 kJ / mol Vibrationsspektroskopie Elektronenspektroskopie Rotationsniveau Übergang im Vibrationsniveau Elektronischer Übergang λ = 105 nm Infrarotwellen: λ = 103 nm Ultraviolett: λ = 450nm E = 103 kJ / mol E = 3 ⋅ 105 kJ / mol Spektroskopiker verwenden anstelle der Wellenlänge oder Frequenz die Wellenzahl, den Kehrwert der Wellenlänge in reziproken Zentimetern: ν~ = 1 / λ [cm −1 ] . Die Frequenz entspricht so: f = ν~ ⋅ c [ s −1 ] . Wechselwirkung von elektromagnetischer Strahlung mit Molekülen permanente und dynamische Dipolmomente Licht ist eine elektromagnetische Welle, die sich mit aufeinander senkrecht stehenden elektrischen und magnetischen Feldkomponenten fortpflanzt. Nehmen wir ein zweiatomiges, dipolares Molekül. Die beiden Atome, über die Bindung verbunden, schwingen um den Gleichgewichtsabstand x und erzeugen so ein periodisch in der Zeit veränderliches dynamisches Dipolmoment. Wenn nun das äußere elektrische Feld und die Schwingung des Dipolmoments (deren Frequenz sich ständig verändert) die gleiche Frequenz besitzen und sich in Phase befinden, kann das Molekül Energie aus dem Feld aufnehmen. Alle Schwingungen von Molekülen mit Symmetriezentrum, die symmetrisch zu diesem Zentrum erfolgen sind IR-inaktiv, da sie lediglich ein über die Zeit unveränderliches dynamisches Dipolmoment aufweisen. Die entsprechende Absorption ist zu schwach um im Spektrum sichtbar zu sein. Dafür lässt sich die Emission der entsprechend IR-inaktiven Bindung messen. Diese liefert ein Raman-Spektrum. Symmetrische Gruppen können demnach mittels Raman-Spektrum, asymmetrische mittels IR-Spektrum nachgewiesen werden. Die Größe des permanenten Dipolmoments ist abhängig von der Bindungslänge und dem Ausmaß der Ladungsverschiebung zwischen den zwei polarisierten Komponenten eines Dipols. Die Ladungsverschiebung hängt von den überlappenden Elektronendichten beider Atome ab und ist somit vom Kernabstand abhängig. Bei der Schwingung des Moleküls entsteht zusätzlich ein dynamisches Dipolmoment, wodurch sich das Gesamtdipolmoment verändert. Da die Amplitude der Schwingung nur einem kleinen Bruchteil der Bindungslänge entspricht, ist das dynamische Dipolmoment wesentlich kleiner als das permanente. Die Energieabsorption eines Moleküls im Infrarotbereich wird durch das dynamische Dipolmoment bestimmt (Die Energie wird von der Vibration aufgenommen). Die Energieabsorption eines Moleküls im Mikrowellenbereich wird durch das permanente Dipolmoment bestimmt (Die Energie wird von der Rotation aufgenommen). Da wesentlich weniger Energie für eine Anregung der Rotation ausreicht als für die Anregung der Vibration ist mit einem Energieübergang im Vibrationsbereich auch immer ein Übergang der Rotation verbunden. Genauer: wann ist eine Bindung IR aktiv und wann nicht? Damit ein Molekül IR-Strahlung absorbieren kann, muss sich das Dipolmoment durch die Schwingungs- bzw. Rotationsbewegungen ändern (also ein dynamischer Dipol vorliegen); dies ist für alle permanenten Dipole gewährleistet; aber auch solche Moleküle, die kein permanentes Dipol aufweisen, können ein dynamisches Dipolmoment haben; Anhand der Grundschwingungen eines linearen dreiatomigen Moleküls (z.B. wie CO2) wird die Änderung des Dipolmomentes dargestellt. Abbildung: symmetrische valenzschwingung (Dipolmoment ändert sich nicht) Tatsächlich ist die symmetrische Valenzschwingung des gestreckten Moleküls nicht infrarotaktiv, da diese Schwingung keine Änderung des Dipolmomentes bewirkt. Die beiden Sauerstoffatome bewegen sich simultan vom zentralen Kohlenstoffatom weg oder auf dieses zu. Es tritt keine periodische Gesamtänderung der Ladungsverteilung auf. Die Ladungsschwerpunkte der positiven und negativen Ladung fallen in jeder Phase der Schwingung zusammen. Das Dipolmoment ist gleich Null. Abbildung: antisymmetrscihe Valenzschwingung (Dipolmoment ändert sich) Während der antisymmetrischen Valenzschwingung hingegen ändert sich das Dipolmoment und es tritt Infrarotabsorption auf. Ein Sauerstoffatom entfernt sich vom Kohlenstoffatom, währenddessen sich das andere Sauerstoffatom dem zentralen Kohlenstoffatom nähert. Die Ladungsverteilung verändert sich damit periodisch. Abbildung: Deformationsschwingung (Dipolmoment ändert sich) Ähnlich ist es auch bei der Deformationsschwingung. Diese ist ebenfalls infarot-aktiv, da sich aufgrund der mit der Schwingung auftretenden gewinkelten Struktur die Dipolmomente während der Schwingung ändern. Betrachtet man zusätzlich die Raman-Aktivitäten der Schwingungen des oben gezeigten dreiatomigen linearen Moleküls, so erkennt man, dass keine Schwingung gleichzeitig Raman-aktiv und Infrarot-aktiv ist. Absorptions- und Emissionsraten Insgesamt gibt es drei unterschiedliche Wechselwirkungen zwischen Licht und Molekül. Bei der Absorption regt ein einfallendes Photon einen Übergang zu einem höheren Energieniveau an. Bei der stimulierten Emission fällt ein angeregter Zustand unter Aussendung eines Photons auf ein nieder energetisches Niveau zurück. Absorption und stimulierte Emission werden von einem Photon ausgelöst, das auf ein Molekül auftrifft. Bei der stimulierten Emission stimmen Phase und die Ausbreitungsrichtung mit dem einfallenden Photon überein (kohärente Photonenemission). Die spontane Emission ist ein zufälliges Ereignis, wobei die entsprechende Übergangsrate mit der Dauer des angeregten Zustands zusammenhängt. Die emittierten Photonen sind nicht kohärent. Ihre Phase und Ausbreitungsrichtung sind zufallsverteilt. Absorptionsrate und stimulierte Emission sind proportional zur Strahlungsdichte bei einer bestimmten Frequenz ρ ( f ) . Die spontane Emission ist von dieser unabhängig, da sie durch elektromagnetischen Feldern der Vakuumfluktuation ausgelöst wird und nicht von der im Versuch eingestrahlten Photonenmenge. Für ein genaueres Verständnis der spontanen Emission siehe Literatur der Quantenelektrodynamik (quantenfeldtheoretische Beschreibung des Elektromagnetismus). Jeder dieser drei Übergangsraten ist abhängig von der Anzahl der Moleküle im jeweiligen Zustand (Grundzustand oder angeregter Zustand: N1 oder N2). Es müssen immer so viele Übergänge vom Grundzustand in den angeregten Zustand geschehen, wie sie vom angeregten in den Grundzustand stattfinden: Stimuliert: B1→2 ⋅ ρ ( f ) N 1 = B2→1 ⋅ ρ ( f ) N 2 , Spontan: A1→2 ⋅ ρ ( f ) N 2 = A2→1 ⋅ ρ ( f ) N 2 wobei B und A Proportionalitätskonstanten darstellen. Die Proportionalitäts-konstanten stehen wie folgt im Zusammenhang B1→ 2 = B2 →1 , A1→2 = A2→1 A2 →1 8π ⋅ h ⋅ f 3 = und B2 →1 c3 Schwingungsmodi und Schwingungsübergänge, harmonische und anharmonische Schwingungen von Molekülen Die wahrscheinlichsten Übergänge in der Schwingungsspektroskopie Da Übergänge zwischen einem Zustand n=0 zu einem anderen Zustand n=1 nur dann stattfinden können, wenn Zustand n=0 auch in ausreichender Menge existiert, ist es wichtig zu wissen welche der unendlich vielen Schwingungsniveaus eine nennenswerte Besetzungswahrscheinlichkeit aufweist. Bei Raumtemperatur befinden sich fast alle Moleküle einer makroskopischen Probe im Schwingungsgrundzustand, da N1 << 1 N0 (bei höheren Temperaturen wird das Verhältnis größer; Die Berechnung kann mit der Boltzmann- Verteilung Ni = N0 ⋅ gi ⋅ e − E K B ⋅T durchgeführt werden). Daraus folgt, dass diese Moleküle Strahlung bestimmter Frequenzen von ihren Schwingungsgrundzuständen n=0 absorbieren. Nun ist die Frage, welche Endzustände möglich sind. Für einen harmonischen Oszillator gilt die Auswahlregel Δn=1. Da einzig der Schwingungsgrundzustand nennenswert besetzt ist, beobachtet man einen Übergang n=0 n=1. Oberschwingungen bzw. Oberton-Übergänge (Δn>1) sind wesentlich schwächer, unwahrscheinlicher, treten aber dennoch auf. Die verschiedenen Schwingungsmodi Im Allgemeinen lassen sich alle Schwingungen grob folgenden Typen zuordnen. Man unterscheidet erstens die Valenzschwingungen oder auch Streckschwingungen ν (Bindungslängen werden geändert, mehr Energie zur Anregung notwendig) von den Deformationsschwingungen oder auch Scherschwingungen (Bindungswinkel variieren, weniger Energie zur Anregung notwendig) Betrachtet man die Streckschwingungen, so ist eine Einteilung in symmetrische Schwingungen ν S (verlaufen unter Wahrung der Molekülsymmetrie) und antisymmetrische Schwingungen ν as (Symmetrieelemente gehen verloren) möglich. Betrachtet man hingegen die Scherschwingung, so unterscheidet man ebene (Schwingung innerhalb einer Ebene) von nichtebenen (die drei zueinander schwingenden Atome bilden keine Ebene) Schwingungen. Die rocking- (Pendel-) und wagging- (Kipp-) Schwingungen tragen das Kürzel γ , die bending- und twist- Schwingungen tragen das Kürzel δ . Spricht man von entarteten Schwingungen, so meint man unterschiedliche Schwingungen, die bei derselben Wellenlänge absorbieren und somit zu demselben Absorptionspeak beitragen. Im Versuch sollen vor allem antisymmetrische und symmetrische Streckschwingungen untersucht werden. Die Frequenzen für diese Schwingungen lassen sich mit folgenden Gleichungen berechnen f antisym = 1 2π k1 + 2k 2 µ f sym = 1 2π k1 µ Anhand der Formeln sieht man, dass der antisymmetrische Modus der Streckschwingung die höhere Frequenz, also die höhere Wellenzahl aufweist. Übersicht über Schwingungsmodi zur Analyse der Wasser- und Lipidspektren In der folgenden Tabelle sind alle möglichen Schwingungsmodi in Wasser aufgelistet. In der folgenden Tabelle sind alle möglichen Schwingungsmodi in Lipiden aufgelistet Der harmonische Oszillator in der klassischen Physik Es handelt sich um ein mechanisches System (z.B. zwei Massen über eine Feder verbunden) der klassischen Physik, das ein Potentialminimum besitzt und bei einer Auslenkung x aus diesem Minimum eine dieser Auslenkung proportionale Rückstellkraft F erfährt. Diese Tatsache wird im Hooke’schen Gesetz dargestellt: F ( x) = −kx , wobei k die Federkonstante des Oszillators darstellt. Ein harmonischer Oszillator besitzt ein quadratisches, also parabelförmiges Potential (ist die Funktion für die zur Streckung der Feder benötigte Energie): V ( x) = 1 2 kx 2 In Schwerpunktkoordinaten wandelt sich das Bild des Systems von zwei Massen, die mit einer Feder der Federkonstanten k verbunden sind, zu einem System einer reduzierten Masse, die mit einer Feder der gleichen Stärke mit einer unbeweglichen Wand verbunden ist. Diese Transformation wird durchgeführt, weil lediglich die relative Bewegung dieser beiden Massen in Bezug zueinander, nicht aber ihre individuellen Bewegungen interessieren. So reduziert sich die Beschreibung der Schwingung auf eine einzige Koordinate, dem Massenschwerpunkt xSP. Die Positionen x1/2 der Massen m1/2 sind nun nicht mehr nötig. Ein zweiatomiges Teilchen der reduzierten Masse µ= m x + m2 x 2 m1 ⋅ m2 , wobei x SP = 1 1 m1 + m2 m1 + m2 beschreibt im Oszillatorpotential eine Sinusschwingung der Frequenz: ω = 2πf = k µ Die Frequenzen die bei zweiatomigen Molekülen in der Schwingungsspektroskopie zu beobachten sind, hängen demnach von zwei Materialparametern ab: der Kraftkonstanten k und der reduzierten Masse µ (entsprechen den Materialparametern Rotationskonstante B und Bindungslänge r bei der Rotationsspektroskopie). Man kann das Potential daher auch schreiben als: V ( x) = 1 µω 2 x 2 2 In einem harmonischen Oszillator sind die Potentiale immer äquidistant. Außerdem gibt es keinen Grenzwert für die Potentiale. Eine unendlich große Energiemenge, welche von einem harmonischen Oszillator aufgenommen wird, führt zu einem unendlich großen Potential. Die im Potential n des klassischen harmonischen Oszillators gespeicherte Energie kann jeden beliebigen Wert annehmen (anders als im quantenmechanischen Fall). Es handelt sich um Potentiallinien, das heißt alle Abstände x um den Gleichgewichtsabstand herum, die sich im erlaubten Bereich (innerhalb der Potentialfunktion) befinden, haben eine gleiche Wahrscheinlichkeit. Der Zustand n=0 hat eine verschwindende Breite, weshalb keine Schwingung vorliegt (Die Energie ist 0). Der harmonische Oszillator in der Quantenphysik In der Quantenphysik können alle Teilchen auch als Wellen betrachtet werden. Daher lässt sich das um die Gleichgewichtslage schwingende Welle-Teilchen der Masse µ durch eine Menge von Wellenfunktionen Ψn (x) beschreiben. Um die einzelne Wellenfunktion für das Potential n zu erhalten und die entsprechenden erlaubten Energien zu ermitteln muss die Schrödinger Gleichung E n Ψn ( x) = − 2 d 2 Ψn ( x) kx 2 ⋅ + ⋅ Ψn ( x) 2µ 2 dx 2 gelöst werden. Die Lösung liefert die Eigenfunktionen der verschiedenen Energieniveaus, wie z.B. die Eigenfunktion des Niveaus n=0: α Ψ0 ( x) = π 1/ 4 ⋅ e −(1 / 2 )αx , wobei α = 2 kµ 2 Man erkennt, dass der niedrigste Zustand (absoluter Nullpunkt der Temperatur ist erreicht) eine von 0 verschiedene Energie besitzt, die so genannte Nullpunktenergie: E0 = 1 ω 2 Das Teilchen ist demnach bei dem absoluten Nullpunkt nicht exakt bei x=0 lokalisiert (im Gegensatz zum klassischen harmonischen Oszillator), sondern weist eine Nullpunktsschwingung entsprechend der nullten Eigenfunktion auf, wodurch es um x=0 herum verschmiert ist. Aus den Eigenfunktionen folgt direkt, dass im quantenmechanischen Fall nicht alle beliebige Energien möglich sind, sondern lediglich die für das Potential n nach folgender Beziehung entsprechende: 1 1 E (n) = hf n + = ω n + 2 2 In der Abbildung ist das Quadrat der Eigenfunktionen in einem Graphen, der das Potential (Energie) gegen die Verschiebung aus dem Gleichgewichtsabstand x=0 zeigt aufgetragen. Das Quadrat der Eigenfunktion entspricht der Wahrscheinlichkeitsdichte, also der Wahrscheinlichkeit zu der sich das Teilchen in einer bestimmten Auslenkung um den Gleichgewichtsabstand liegt (in der klassischen Physik handelt es sich um Linien, da alle Auslenkungen gleichwahrscheinlich sind). Die rote Parabel entspricht der Potentialfunktion. Außerhalb dieser liegen die verbotenen Zustände. Der anharmonische Oszillator Die Potentialfunktionen der Moleküle weichen vom einfachen harmonischen Potential ab. Eine analytische Annäherung an die Molekülpotentiale, die anharmonischen Charakters sind, stellt das Morse- Potential dar: V ( R) = De ⋅ [1 − e −α ⋅( R − Re ) ]2 . De ist die Dissoziationsenergie in Bezug auf das Potentialminimum. Re ist der entsprechende Kernabstand. Das heißt im anharmonischen Oszillator führt eine unendlich große Energiemenge zur Dissoziation des Moleküls. Die einzelnen Potentiale sind nicht mehr äquidistant. Deren Abstände nehmen mit zunehmender Energie ab (Energiedifferenz zwischen den einzelnen Schwingungszuständen wird kleiner). Der Steifeparamter α ist definiert durch: α = k / 2 De Die Bindungsenergie D0 wird in Bezug auf das niedrigste erlaubte Energieniveau definiert, das ungleich 0 ist. Die einzelnen Energieniveaus für dieses Potential sind mit 1 (h ⋅ f ) 2 1 ⋅n + E ( n) = h ⋅ f n + − 2 4 De 2 2 gegeben. Der zweite Term ist der Korrekturterm für den anharmonischen Oszillator. Die möglichen Energien werden in der Morse Potentialkurve als horizontale Linien im klassischen Fall oder als Quadrat der Eigenfunktionen im quantenmechanischen Fall dargestellt. Das Minimum der Funktion entspricht dem Gleichgewichtsabstand der Kerne, wie beim harmonischen Oszillator. Um das Potentialminimum herum stellt das Harmonische Potential (Parabel) deshalb eine gute Näherung an das Morse-Potential dar. Prinzip der FTIR Spektroskopie und Aufbau des Spektrometers Das FTIR Spektrometer ist eine Apparatur, bestehend aus einer Polychromatischen Lichtquelle, einem Michelson-Interferometer, einer Kammer, welche während der Messung die zu analysierende Probe enthält und einem Detektor. Das Michelson-Interferometer Bei Einem Interferometer handelt es sich um einen optischen Aufbau, der dazu dient Lichtbündel zu trennen, sie räumlich gegeneinander zu verschieben und wieder zu überlagern. Das einfachste Interferometer ist das Michelson-Interferometer. Der Strahlengang im Interferometer findet wie folgt statt. Das einfallende, parallele Lichtbündel (1) wird an der aktiven Schicht (3) eines Strahlenteilers (2) in zwei gleich intensive Teile aufgespaltet. Ein Teil reflektiert am Strahlenteiler zu einem festen Spiegel (4), der sich am Referenzarm des Interferometers befindet. Der andere Teil transmitiert zu einem sich ständig hin und her bewegenden Spiegel (5), der sich im Messarm befindet. Beide Teile werden am jeweiligen Spiegel zum Strahlenteiler zurück reflektiert und wiedervereinigt, wobei beide Teilbündel eine Laufzeitdifferenz bzw. Weglängendifferenz x = 2 ⋅ (d1 − d 2 ) aufweisen. Daher kommt es zur Interferenz. Durch den Einbau eines Interferometers ist es möglich aus einer Dauerlichtquelle aus polychromatischem Licht mittels dem beweglichen Spiegel einen Licht-Impuls pro Zeit los zusenden. bei gleichzeitiger Anregung der Probe mit allen Wellenlängen durch polychromatisches Licht, würde der Detektor die gemessenen Intensitäten nicht den einzelnen λ zuordnen können, da alle gleichzeitig ankommen. Die Spiegelposition im Interferrometer codiert für die einzelnen Wellenlängen. Dadurch kommen die Intensitäten in unterschiedlicher Reihenfolge am Detektor an und können den einzelnen Wellenlängen zugeordnet werden. Eine manuelle Einstrahlung aller einzelnen Wellenlängen nacheinander würde jedoch zu viel zeit in Anspruch nehmen! Monochromatische Interferenz Aus der Bewegung des Spiegels (5) folgernd, verschiebt sich die aus dem Messarm kommende Welle gegen die des Referenzarms. Beide Wellen interferieren zu einer resultierenden Welle. Im Fall der optischen Weglängendifferenz null beider Arme überlagern sich beide Wellen so konstruktiv, sie verstärken sich. Entspricht die optische Weglängendifferenz genau einer halben Lichtwellenlänge, so erfolgt die Überlagerung destruktiv (Auslöschung). Das Licht, welches das Interferometer letztlich verlässt, schwankt daher kosinusförmig über der Spiegelverschiebung. Wenn der eine der beiden Spiegel mit konstanter Geschwindigkeit kontinuierlich bewegt wird, dann variiert die Strahlungsintensität am Detektor nach der Kosinusfunktion 2π I ( x) = 0,5 I 0 ⋅ cos x = I (ν~ ) ⋅ cos(2πν~x) λ Hierin sind λ die Wellenlänge und x die Weglängen- bzw. Laufzeitdifferenz der elektromagnetischen Welle (auch Retardierung genannt). Polychromatische Interferenz Da Spektrometer Licht vieler Wellenlängen verarbeiten, entsteht die oben beschriebene Interferenz für jede Wellenlänge. Entsprechend überlagern sich die einzelnen Wellenlängen zusätzlich. Der Nullpunkt des Spiegelwegs (x=0) ist der Ort an dem beide Interferometerarme gleich lang sind. Dort besitzen alle Wellen die Phasendifferenz Null und überlagern sich deshalb konstruktiv. Die Intensität ist maximal. Bei größer werdender Weglängendifferenz interferieren die einzelnen Wellenlängen destruktiv. Es kommt zur Auslöschung. Überlagert man alle Wellenlängen (Kontinuum) des an der Messung beteiligten Lichts, ergibt sich die vom Detektor erfasste Intensität als Integral über die Bandbreite B. I ( x) = ∫ [I (ν~ ) cos(2πν~x)]dν~ B Die Bandbreite ist der nutzbare Wellenzahlbereich eines Spektrometers. Die Bandbreite umfasst alle von der Lichtquelle zum Detektor gelangenden und im spektralen Empfindlichkeitsbereich des Detektors liegenden Wellenzahlen. Die Bandbreite ist limitiert durch die Empfindlichkeit des Detektors, der verwendeten Fenster oder des verwendeten Strahlenteilers. ATR (abgeschwächte Totalreflexion) Bei dieser, im Praktikum verwendeten Methode wird die vom Interferometer kommende Strahlung durch einen Lichtwellenleiter geführt. Im Innenraum dieses Lichtwellenleiters findet die abgeschwächte Totalreflexion statt. Totalreflektion bedeutet, dass die gesamte Energie eines einfallenden Lichtstrahls an der Grenze zwischen zwei Medien (vom optisch dichteren zum optisch dünneren Medium bzw. vom Medium mit einem größeren Brechungsindex zum Medium mit einem kleineren Brechungsindex) reflektiert wird, wenn der für die Medien charakteristische Einfallswinkel überschritten wird. Bei der abgeschwächten Totalreflektion dringt der Lichtstrahl, abhängig vom Einfallswinkel, der Wellenlänge des Lichtstrahls und den Brechungsindizes der Medien, bis in eine bestimmte Tiefe in das optisch dünnere Medium, also der IR-Probe ein. λ dp = 2π ⋅ n1 n sin θ − 2 n1 2 2 Dort wird ein Teil der Strahlung absorbiert. Nur der nicht absorbierte Teil wird vom Detektor am Ende des Strahlengangs detektiert. Das Strahlenmodell hat an der Grenzfläche zwischen IR Probe und Lichtwellenteiler keine Gültigkeit. Es bildet sich an dieser Grenzfläche ein evaneszentes Feld, das in die Probe eindringt und exponentiell abfällt! Die Differenz zwischen eingestrahlter Intensität jeder bestimmten Wellenlänge und nicht absorbierter Intensität jeder bestimmten Wellenlänge bildet das Absorptionsspektrum, das erst mittels eines Rechners durch Fouriertransformation aus dem Interferogram errechnet werden muss. So ist das Absorptionsspektrum, dass mittels Reflektionstechnik erzeugt wird den gewöhnlichen Absorptionsspektren, die mittels Durchstrahltechnik erzeugt werden, nahezu identisch. Der Vorteil dieser Methode liegt darin, dass bei einem großen Proberaum (besseres Absorptionsvermögen) nur wenig Probe (nicht teuer) benötigt wird. Außerdem können Oberflächen, ohne zerstört zu werden und andere Proben, die für die Durchstrahltechnik zu stark absorbieren oder zu dick sind, analysiert werden Evaneszentes Feld Erfolgt zwischen zwei Medien (ATR-Kristall - optisch dichter und Probe - optisch dünner) aufgrund unterschiedlicher Brechungsindizes Totalreflexion, dann fällt die Strahlungsintensität hinter der Grenzfläche nicht abrupt ab, sondern dringt in die Probe ein und pflanzt sich parallel zur Grenzfläche als evaneszente Welle fort. Sie tritt an der Grenzfläche wieder aus und durchdringt den ATR-Kristall. Die Abschwächung des evaneszenten Feldes erfolgt exponentiell mit zunehmendem Abstand von der Grenzfläche. Absorbiert die Probe die Strahlung, beobachtet man eine Schwächung der Totalreflexion und es sind ATR-Spektren messbar. Aufgrund der vorhandenen Abhängigkeit der Eindringtiefe von der Wellenlänge (Absorptionsbanden werden im ATR-Spektrum mit größeren Wellenlängen intensiver) müssen ATR-Spektren korrigiert werden, um transmissionsähnliche Spektren zu erhalten. Die nicht propagierende Komponente des Nahfeldes wird als evaneszentes Feld bezeichnet. Es fällt exponentiell zur Oberflächennormalen des strahlenden Körpers ab. Jedes beleuchtete Objekt erzeugt also ein evaneszentes und ein propagierendes Feld [Cou 94]. Ein rein evaneszentes Feld kann man z.B. im Fall von Totalreflexion beobachten. Ein im Medium mit Brechungsindex n1 propagierender Lichtstrahl trifft auf die Grenzfläche zum , so daß der optisch dünneren Medium ( n2<n1) mit einem Einfallswinkel Strahl totalreflektiert wird. Aufgrund der Stetigkeitsbedingung kann auf der Seite des dünneren Mediums das Feld nicht abrupt Null sein, sondern es fällt exponentiell in den Halbraum n2 ab [Hec 98]. Sei z.B. die Grenzfläche in der (x,y)-Ebene bei z=0. Dann ist die zKomponente des Wellenvektors kz komplex und das evaneszente Feld ist senkrecht zur Oberfläche stark exponentiell gedämpft. Es ist i.a. bereits bei einem Abstand komplett verschwunden. Gerade dieses Feld enthält Informationen über Strukturen unterhalb der Auflösungsgrenze. Die Fouriertransformation Der Rechner muss nun die Fouriertransformation durchführen, um aus einem Interferogram ein Spektrum zu erstellen. Das Interferometer liefert eine Gleichung für die Absorptionsintensität in Abhängigkeit von der Retardierung x. Nun formen wir diese Gleichung so um, dass ein unbestimmtes Integral (Intervall ist unendlicher Wellenzahlbereich) entsteht, indem wir die Fensterfunktion D (ν~ ) multiplikativ im Integral ergänzen. Diese Funktion entspricht der Fourierkosinustransformation. Da es sich um eine gerade Funktion handelt, lässt sich diese invertieren: Diese Inverse Fourierkosinustransformation entspricht nun, da es sich um eine gerade Funktion handelt und somit die Sinusteile der Eulerschen Funktion entfallen, der komplexen Fouriertransformation Diese Formel liefert nun einen Graphen, der dem Einstrahlspektrum entspricht. Das Einstrahlspektrum einer Probe ist das Spektrum, das der Energieverteilung entspricht, die durch den Detektor aufgenommen wird. Im Einstrahlspektrum sind alle Gerätefunktionen wie die Energie-Verteilungsfunktionen der Lichtquelle, die Transmission des Spektrometers selbst und die Empfindlichkeit des Detektors enthalten. Um diese Gerätefunktionen zu filtern, bedarf es einer Aufnahme des Spektrums der leeren Probekammer. Dieses Spektrum wird vom Probenspektrum subtrahiert, um ein möglichst reines Spektrum zu erhalten. Das nun auf dem Bildschirm erscheinende Spektrum zeigt Peaks einer bestimmten Intensität an den Wellenzahlen, an denen die Probe zu dieser Intensität absorbiert. graphische /physikalische Erklärung der Fourriertransformation: Bei der Fouriertransformation in der IR-Spektroskopie wird wie folgt das Spektrum berechnet. I(x) ist das vom Detektor erzeugte FID. Es wird mit den cosinus-Funktionen einer jeden Frequenz multipliziert. Die Integrale des Produkts aus dem FID und jeder einzelnen Frequenz werden nun berechnet und gegenübergestellt. Wenn die Frequenz genau dem FID entspricht, so ist das Integral maximal. Je höher die Überlappung der entsprechenden Frequenz mit dem FID, desto größer wird das Integral. Findet keine Überlappung statt und ist der Unterschied zwischen FID und entsprechender Frequenz zu groß, so ist dass Integral des Produkts sehr klein oder null (negative Anteile und positive Anteile heben sich auf). Werden die Integrale nun in einem Graphen gegen die entsprechende Frequenz aufgetragen, so entsteht das Fouriertransformierte Signal, das Spektrum. In diesem Beispiel handelt es sich um einen einzigen Peak, da das maximale integral für eine einfache Kosinusfunktion erreicht wird und für alle niedrigeren und höheren Frequenzen dieser Kosinusfunktion eine geringere Überlappung erreicht wird. Für komplexere FIDs gibt es natürlich mehrere Maxima, da für keine einfache Frequenz eine perfekte Überlappung stattfinden wird. Das Lambert Beersche Gesetz Im Allgemeinen verwendet man in der Absorptionsspektroskopie elektromagnetische Strahlung einer geeigneten Wellenlänge, die auf ein Probevolumen geschossen wird. In den chemischen Substanzen im Probevolumen finden die Übergänge zwischen Rotations-, Schwingungs- und Elektronenzustände statt. Die Intensität I 0 (λ ) der einfallenden Strahlung wird beim Durchgang durch ein Längenelement der Länge d des Probevolumens abgeschwächt. Diese Abschwächung wird durch das Lambert-Beer’sche Gesetz beschrieben: I (λ ) = I 0 (λ ) ⋅ e −ε ( λ )⋅c⋅d E (λ ) = − log I (λ ) = ε (λ ) ⋅ c ⋅ d I 0 (λ ) Hier ist c die Konzentration des absorbierenden Mediums in mol/l. ε (λ ) ist der molare Absorptionskoeffizient. Er entspricht der Steigung der Geraden „spezifische Absorption bei der Wellenlänge λ vs. c“. Ermittelt werden kann er durch Photometrie einer Verdünnungsreihe des Stoffes. Das Absorptionsvermögen nimmt mit ε (λ ) , c und d zu und ist Proportional zu I (λ ) . I 0 (λ ) Durch die Methode der ATR ist die Länge d erheblich erhöht, weshalb die Absorption sehr hoch ist, obwohl nur sehr wenig Probe vorhanden ist Dichroismus Dichroismus ist ein optischer Effekt, der bewirkt, dass ein Mineral in Abhängigkeit von der Blickrichtung in zwei verschiedenen Farben erscheint. In der Infrarot Spektroskopie wird dieser Effekt zur Bestimmung von Orientierungen verwendet! Materialien haben eine oder mehrere ausgezeichnete optische Achsen. Verschiedene Eigenschaften unterscheiden sich hierbei, ob man sie parallel zur optischen Achse (außerordentlich) oder senkrecht dazu (ordentlich) betrachtet. Zeigt das Material zwei unterschiedliche Absorptionsverhalten bezüglich dieser Achsen, also wird z. B. je nach Polarisation des durchscheinenden Lichtes eine andere Farbe absorbiert, wird dieses Verhalten Dichroismus genannt. Das Wort Dichroismus leitet sich aus dem griechischen Wort Dichroos für „zweifarbig“ ab. Die Mehrfarbigkeit wird allgemeiner als Pleochroismus bezeichnet Der Dichroismus wurde 1939 von Peter Debye entdeckt. Hierbei wird unterschieden, welche Art von Polarisation das einfallende Licht hat. Es gibt den linearen Dichroismus, bei dem das Material von linear polarisiertem Licht durchschienen wird. Daneben wird mit Zirkulardichroismus die Differenz der Absorptionskoeffizienten für links bzw. rechts zirkular polarisiertes Licht beim Durchgang durch optische aktive Verbindungen beschrieben Zirkulardichroismus ist eine spezielle spektroskopische Messung, um die Struktur optisch aktiver Moleküle aufzuklären. Dazu verwendet man zirkular polarisiertes Licht. Ein optisch aktives Molekül absorbiert nun das zirkular rechts polarisierte Licht im Vergleich zu dem zirkular links polarisierten Licht unterschiedlich stark. Diese Differenz wird für jede Wellenlänge gemessen und man erhält das gewünschte Spektrum. Dieses sollte für die Enantiomere eines Moleküls spiegelbildlich ausfallen. Besonders gerne wird die Methode bei biologischen Substanzen wie Aminosäuren und Zuckern, aber auch bei Untersuchungen zu Helix-Strukturen von Peptiden benutzt. Hier sind allerdings die Röntgenstrukturanalyse und NMR-Spektroskopie weitaus besser bei der Strukturaufklärung. zirkular polarisiertes Licht Eine elektromagnetische Welle ist zirkular polarisiert, wenn der elektrische Feldvektor an jedem Punkt im Raum als Funktion der Zeit einen Kreis beschreibt. Eine zirkular polarisierte Welle lässt sich als eine Überlagerung zweier senkrecht aufeinander stehender, linear polarisierter Wellen mit gleichem Wellenvektor und gleicher Amplitude sowie einem Phasenversatz von 90 Grad darstellen. Erzeugen kann man eine zirkular polarisierte Welle durch Beleuchtung eines λ/4-Plättchens mit einer linear polarisierten Welle λ/n-Plättchen Eine Wellen- oder Verzögerungsplatte (auch: λ/n-Plättchen) ist ein optisches Gerät, das die Polarisation und Phase von passierendem Licht ändern kann. Ein λ/4-Plättchen ist eine spezielle Wellenplatte, die das Licht in einer Richtung um eine viertel Wellenlänge – bzw. π/2 – gegen die dazu senkrechte Richtung verzögert. Es kann aus linear polarisiertem Licht zirkular oder elliptisch polarisiertes Licht machen und aus zirkular polarisiertem Licht wieder linear polarisiertes. Ein λ/2-Plättchen ist ebenfalls eine spezielle Wellenplatte, die das Licht wie oben um eine halbe Wellenlänge – bzw. π – verzögert. Es kann die Polarisationsrichtung von linear polarisiertem Licht drehen. Die Verschiebung kommt dadurch zustande, dass das Licht in zwei senkrecht stehende Polarisationsrichtungen zerlegt werden kann, die die Wellenplatte mit unterschiedlicher Geschwindigkeit passieren, deren Phasen also gegeneinander verschoben werden. Ein solches Plättchen besteht typischerweise aus einem doppelbrechenden Kristall (z. B. Kalzit) mit passend gewählter Dicke und Ausrichtung Bei einer Wellenplatte handelt es sich um eine dünne Scheibe von optisch anisotropem Material, also Material, welches für unterschiedlich polarisiertes Licht verschiedene Ausbreitungsgeschwindigkeiten c/n (bzw. verschiedene Brechzahlen n) in verschiedenen Richtungen aufweist. Oft verwendete Materialien sind optisch einachsig, das heißt es gibt zwei zueinander senkrechte Achsen im Kristall, entlang derer sich die Brechzahlen unterscheiden. Man nennt diese ordentliche (das Licht ist senkrecht zur optischen Achse polarisiert) und außerordentliche Achse (das Licht ist parallel zur optischen Achse polarisiert). Die Schwingungsrichtung des Lichtes, bei der eine Welle die größere Ausbreitungsgeschwindigkeit hat, heißt „schnelle Achse“, die dazu senkrecht stehende Richtung entsprechend „langsame Achse“. Licht, welches parallel zur schnellen Achse polarisiert ist, benötigt weniger Zeit zum Durchlaufen des Mediums als Licht, welches senkrecht dazu polarisiert ist. Man kann sich das Licht in zwei Komponenten senkrecht (ordentlicher Strahl) und parallel (außerordentlicher Strahl) zur optischen Achse aufgeteilt vorstellen, die nach dem Durchlaufen des Plättchens eine Phasenverschiebung von aufweisen. Dabei ist d die Dicke des Plättchens und λ die Wellenlänge des eingestrahlten Lichtes. Die beiden Strahlen überlagern sich hinter dem Kristall (Interferenz) zum ausgehenden Licht. Durch die (kohärente) Überlagerung dieser beiden Strahlen ergibt sich dann eine neue Polarisation des Lichtes (Frequenz und Wellenlänge bleiben erhalten; siehe nächster Abschnitt). Wie man an der Formel sieht, ist eine solche Wellenplatte also immer nur für eine bestimmte Wellenlänge ausgelegt. Es sei noch bemerkt, dass die Aufspaltung in zwei Strahlen nur eine Art Rechentrick ist. In der Realität überlagern sich diese beiden Strahlen natürlich an jeder Stelle des Kristalls. Die Elektronen um die Kristallatome bilden lokale und momentane Dipole, die in einer Überlagerung der beiden Polarisationsrichtungen der Strahlen schwingen. Lipide und Membranen Die Glycerolipide, die aus einem Glycerinbaustein und daran veresterten Acylresten bestehen spielen bei dem Aufbau der Membranen und als Speicherstoff eine entscheidende Rolle. Speicherlipide sind Triacylglycerine, Membranlipide sind Diacylglyceride mit polarer Kopfgruppe als dritten Substituenten. Je nach Art des dritten Substituenten spricht man von Phospholipiden (Substituent: Phosphatguppe mit polarem Rest wie Cholin), Sulfolipiden (Substituent: Sulfatgruppe mit polarem Rest) oder Glykolipiden (Substituent: Saccharide). Ein weiterer Membranbaustein sind Sphingolipide, bei denen Sphingosin statt Glycerin das Grundgerüst darstellt. Hier befindet sich jeweils ein Acyl-Rest verestert an die Amidgruppe des Sphingosins. Auch hier unterscheidet man je nach polarer Kopfgruppe drei Typen voneinander: Ceramide (Substituent: Wasserstoff), Sphingomyelin (Substituent: Phosphatguppe mit polarem Rest wie Cholin) oder Glykosphingolipide (Substituent: Saccharide) Außerdem gibt es Plasmalogene Lipide. Solche unterscheiden sich von Phospholipiden lediglich durch den Rest in sn1 Position. Dieser enthält eine O-Alkyl oder eine O-Alkenyl Etherfunktion. Als dritte Hauptgruppe gehören zu den Lipiden die so genannten Isoprenoide bzw. Terpenoide. Jene lassen sich noch einmal in Carotinoide und Steroide einteilen. Steroide weisen als Grundgerüst vier miteinander verbundene Kohlenstoffringe auf. Das wichtigste Steroid für die Membranen ist Cholesterin. Carotinoide bestehen aus 8 Isopreneinheiten und kommen ausschließlich in Pflanzen vor. Man unterscheidet sie nochmals in Carotine und Xanthophylle, welche zusätzlich Sauerstofffunktionen tragen. Carotinoide sind Pigmente und Steroide werden häufig als Hormone verwendet. Die Fluidität der Membran hängt von der Membranzusammensetzung sowie von der Temperatur ab. Bei tiefen Temperaturen gibt es einen parakristallinen Zustand. Oberhalb einer Transition-Temperatur wird die Membran flüssig. Ungesättigte Fettsäuren erhöhen die Membranfluidität (behindern den parakristallinen Zustand, da die Doppelbindungen die gerade kette der Fettsäure abknicken und somit die restlichen geradkettigen Fettsäuren sterisch „wegschieben“), da sie den Schmelzpunkt der Membran herabsetzen. Langkettige Fettsäuren erniedrigen die Membranfluidität, da sie den Schmelzpunkt der Membran erhöhen. Der Cholesteringehalt der Membran beeinflusst ebenso die Membranfluidität. Unterhalb der Transition-Temperatur erhöht Cholesterin die Fluidität, während oberhalb der TransitionTemperatur die Membranfluidität erniedrigt wird. Cholesterin beeinflusst das Schmelzverhalten allosterisch, sodass der Phasenübergang nicht plötzlich stattfindet, sondern nach und nach (sigmoidal) über ein Temperaturintervall. Extremophile Organismen, welche bei extrem hohen Temperaturen leben, weisen eine ganz andere Membranzusammensetzung auf, als Organismen normaler Habitate. Dies ist lebensnotwendig. Damit die Membranen bei hohen Temperaturen noch eine ausreichende Integrität aufweisen, müssen sie einen großen Anteil an Cholesterin und gesättigte langkettige Fettsäuren aufweisen. Des Weiteren besitzen die Lipide der extremophilen Organismen Etherbrücken anstelle von Esterbrücken. Häufig kommen auch Monolayer anstelle von Bilayer vor. ITC Thermodynamik und Spontanität von Reaktionen Eine chemische Reaktion läuft immer dann spontan (exergonisch) ab, wenn ∆G , die Änderung der Gibbs Energie zwischen Edukten und Produkten negativ ist. ∆G = ∆H − T∆S für p=const. und T=const. Spontane Reaktionen werden demnach durch exotherme Enthalpien und Entropieerhöhungen angetrieben. Aufgrund der großen Dichte und Komplexität der Moleküle in einer Zelle, bedarf es einer hohen Interaktions-Selektivität. Der größte Teil dieser Selektivität liefert die Thermodynamik. Intramolekulare Interaktionen erlauben die Proteinfaltung, während intermolekulare Interaktionen die Substrat-Wahrnehmung der Enzyme ermöglicht. In diesem Praktikum wird der Frage nachgegangen, warum aus thermodynamischer Sicht die Bindung zwischen Ligand und Enzym zustande kommt. Es ist klar, dass ∆G für die Protein-Ligand Bindung negativ ist. Doch wird jener von der Enthalpie, der Entropie oder beidem angetrieben? Calmodulin (CaM) und RNAse A In der ITC kann z.B. die Faltung eines Proteins beobachtet werden, um zu ermitteln, ob der Faltungsprozess von der Enthalpie oder der Entropie getrieben wird. Dies lässt sich am Beispiel des Proteins Ca-Rinder-Calmodulin erforschen. Es handelt sich um ein 16kD Ca2+-bindindes Protein, das eine Schlüsselkomponente des Ca2+-second-messenger Systems darstellt. Es kontrolliert demnach viele biochemische Prozesse einer Zelle. In Abwesenheit von Calcium ist dieses Protein größtenteils ungefaltet. Ist Calcium vorhanden, so faltet sich das Protein schnell zu einer organisierten Struktur, je nachdem ob es 2 oder 4 Ionen gebunden hat. Diese Calcium-abhängige Faltung von Calmodulin wird in vielen biochemischen Reaktionsketten als regulatorischen An bzw. Ausschalter verwendet. So werden über Calmodulin regulierte Proteine (zelluläre Enzyme und transmembrane Ionentransporter) erst aktiviert, wenn ein aktiviertes Calmodulin an sie bindet und so eine Reaktionskaskade ein bzw. ausschaltet. Außerdem lässt sich mittels ITC die Frage beantworten, weshalb Liganden an Enzyme binden. Im Praktikum wird dieser Frage am Beispiel der Bindung von CMP an das Enzym RNAse nachgegangen. RNAse ist eine Endonuklease und gehört zu den Hydrolasen. Sie spaltet die bereits in die Proteinsequenz übersetzte single stranded RNA am Zuckergerüst von ungepaarten C und U Resten am 3’ Ende des Stranges in ihre einzelnen Monomere, damit diese neue RNA aufbauen können. CMP ist also ein Produkt der RNAse. Es handelt sich um ein relativ kleines Protein aus 124 Aminosäuren mit einem Molekulargewicht von 13,7kDa. Aufgrund der Tatsache, dass in unserem Experiment keine RNA in Lösung vorhanden ist, aber auch keine RNA aufgebaut werden kann, weil die dafür notwendige Energie fehlt, wird CMP von dem Enzym RNAse zwar gebunden, aber nicht umgesetzt. Die Bindung erfolgt mit einer bestimmten Affinität, sodass entweder Energie frei wird oder Energie benötigt wird, während die Protein-Ligand Bindung entsteht. Dieser Energiebetrag ist mittels ITC messbar. Funktion und Prinzip der ITC Es wird im Versuch RNAse mit CMP titriert und die Enthalpieänderung ∆H für die Ligandenbindung festgehalten. Die Daten, welche die Titration liefert, geben einen Aufschluss darüber, welche Kräfte die Ligandenbindung antreiben. Die Enthalpieänderung während der Interaktion von Makromolekülen kann über ITC am direktesten bestimmt werden. Bei einem typischen Experiment wird der Titrator L (bekannter Konzentration) in eine Lösung von M titriert und dabei die Änderung der Enthalpie des gesamten adiabatischen Systems gemessen. Die Assoziationskonstante Ka und die Stöchiometrie n des Komplexes kann ebenso durch ITC ermittelt werden. Wenn die ITC bei verschiedenen Temperaturen durchgeführt wird, erhält man zusätzlich die Änderung der Wärmekapazität bei konstantem Druck. Ein solches Kalorimeter besteht aus zwei identischen Zellen aus Wärmeleitendem Material, umgeben von einem adiabatischen Mantel (siehe Abbildung). Die Referenzzelle (enthält gepuffertes H2O) besitzt einen Heizer, der auf die gewünschte Temperatur für das Experiment eingestellt wird. Die Probenzelle (enthält die gepufferte Probe) besitzt zwei Heizer. Der erste Heizer ist identisch mit dem Heizer der Referenzzelle. Der zweite Heizer (Feedback Heizer) ist dafür verantwortlich auf thermische Fluktuationen, die während dem Experiment erzeugt werden, zu reagieren. Beide Zellen sind mit einem Thermoelement (meist: 2000-6000 Bimetall Kabeln; aus je mehr Bimetallkabeln dieses Thermoelement besteht, desto genauere Messungen von ΔT sind möglich) verbunden, das die Temperaturdifferenz zwischen den Zellen ermittelt. Die Probenzelle ist zugänglich mit einer langen Spritze. Das Experiment wird bei einer konstanten Temperatur durchgeführt, indem der Titrator L in der Spritze in die Lösung mit dem Makromolekül M der Probenzelle titriert wird. Der Inhalt der Probenzelle bewegt sich ständig, um den Mischvorgang aller Reaktanden zu beschleunigen. Nach jeder Zugabe einer kleinen Menge L, wird durch die Interaktion von L und M Hitze frei oder verbraucht. Diese Temperaturänderung wird über das Thermoelement gemessen. Die Temperaturdifferenz wird ausgedrückt in derjenigen elektrischen Kraft [J/s], die benötigt wird um eine konstante Temperatur zwischen der Proben- und der Referenzzelle mittels Feedback Heizer wieder herzustellen. Wenn die Temperatur in der Probenzelle mit der Zugabe des Titrators steigt, nimmt der Heizer die frei gewordene Energie des exothermen Mischprozesses auf und kühlt somit die Probenzelle. Wenn die Temperatur sinkt, muss der Heizer die Probenzelle auf die Anfangstemperatur erwärmen. Der Prozess ist hier endotherm. Es liegt nahe, dass die ersten Titrationen exothermer sein werden, als die letzten Titrationen, da je höher die Konzentration des Titrators in der Probenzelle ist, desto mehr L-M Komplexe liegen vor. Nach jeder Titration befinden sich weniger freie Proteine in Lösung, weshalb mit jeder Titration immer weniger der titrierten Liganden auch an ein Protein binden. Nach einer bestimmten Anzahl an Titrationen, wird kein zusätzlicher Ligand mehr an Proteine binden können. Das dynamische Gleichgewicht zwischen L-M und freiem L und M hat sich eingestellt. (siehe Abbildung). Warum sind nun zwei Zellen für die Messung notwendig? Nur mit einer Referenzzelle ist eine Messung bei geringen Temperaturunterschieden, also bei geringen Konzentrationen und Volumina der Probe möglich. Wenn man nur eine Zelle hätte, müsste man ΔT aus der Differenz der Temperaturen vor und nach der Titration messen. Die Referenz wäre hier jeweils die nicht adiabatische Umgebung. Nicht adiabatisch bedeutet, dass ein Wärmeaustausch mit an dem System angrenzenden Bereichen des Universums möglich ist. Es resultiert daher ein größerer absoluter Fehler bei der Messung, da die Wärmemenge der nicht adiabatischen Umgebung nicht konstant ist. Nur wenn die Messungen mit großen Konzentrationen und Volumina durchgeführt werden würden könnte dieser Absolutfehler relativ betrachtet gering ausfallen. Dies würde allerdings erheblich mehr kosten und aufwändiger sein. Die ITC Apparatur, welche wir im Praktikum nutzen, besteht aus einem „control block“, an dem drei wichtige Daten abzulesen sind: die momentane Temperatur am Thermoelement, die Zellen Feedback Temperatur und die Mantel Feedback Temperatur. Außerdem gibt es einen „measure block“, an dem die Main-Heater ein bzw. ausgeschaltet werden. Sie sorgen für eine konstante Anfangstemperatur im Mantel. Während der Messung wird der MainHeater immer ausgeschaltet. Protein-Ligand Wechselwirkung und ITC Im einfachsten Fall hat ein Protein nur eine Bindungsstelle, an die ein Ligand andocken kann. Die Stöchiometrie und die entsprechende Gleichgewichtskonstante lassen sich einfach beschreiben: P + L ⇔ PL K= [ PL] [ L] ⋅ [ P ] Die Gleichgewichtskonstante steht in direkter Verbindung zur freien Gibbs Energie K =e − ∆G RT Diese wiederum lässt sich in enthalpische und entropische Beiträge zerlegen ∆G = ∆H − T∆S = − RT ⋅ ln K Im Prinzip kann man die Reaktionsenthalpie mittels Van’t Hoff Gleichung ermitteln, da wir die Temperaturänderung und die Änderung der Gleichgewichtskonstante kennen. ∆H R0 ∂ ln K = 2 ∂T p RT RT 2 ∂ ln K ∆H R0 = ∂ T p In einer anderen Form enthält diese Beziehung sowohl die Reaktionsenthalpie, als auch die Entropie: ln K = − ∆H R0 1 ∆S R0 ⋅ + R T R In einem Graphen, der lnK gegen den Reziprokwert der Temperatur aufträgt, ist also − ∆H R0 ∆S R0 die Steigung und der Schnittpunkt mit der lnK Achse. R R Die Temperatur lässt sich im Experiment einfach einstellen und zuordnen. Nun muss der Wert für die Gleichgewichtskonstante zur entsprechenden Temperatur ermittelt werden. K kann nur ermittelt werden, wenn das Verhältnis zwischen Protein-Ligand Komplex und freiem Protein und freiem Liganden bekannt sind. K= [ PL] [ P][ L] Dieses Verhältnis lässt sich zum Beispiel über Absorptionsverschiebungen messen, aber auch zahlreiche andere Werte (z.B. Fluoreszenz, NMR, UV) lassen sich beobachten und liefern ein Verhältnis zwischen gebundenem und freiem Protein. So lässt sich die obige Gleichung wie folgt verallgemeinern. K= θD −θX θX = θX −θN 1−θX Dabei ist θ X der aktuell beobachtete Parameter (z.B. Absorption einer bestimmten Wellenlänge) und θ N und θ D jeweils die zu Beginn und am Ende der Interaktion beobachteten Parameter . Wir gehen im Experiment davon aus, dass jeder Temperatur Zuwachs von einem Wärmeeffekt Q bewirkt wird. Zu Beginn des Experiments ist die Temperatur konstant, weshalb Qinitial null zu setzen ist. Die Gesamtwärmemenge, die benötigt wird, damit alle Liganden binden ist die Bindungsenthalpie. K= Qtotal − qi ∆H bind − qi = qi − Qanfang qi Die Wärmemenge q, welche im Titrationsprozess i entsteht, steht in Zusammenhang mit dem Volumen der Zelle, der Konzentration des Protein-Ligand-Komplexes im Schritt i, der Anzahl der Bindungsstellen des Proteins n und natürlich der Bindungsenthalpie selbst. q i = n ⋅ [ PL]i ⋅ V Zelle ⋅ ∆H bind Die Wärmemenge des Titrationsprozesses i entspricht der Fläche unter dem Peak des entsprechenden Schrittes (das Integral über alle Piks demnach der Bindungsenthalpie). So ist auch die Gesamtwärmemenge bekannt. Nun wird die Gleichgewichtskonstante für die Ligandenbindung Kbind, die Bindungsenthalpie ∆H bind und der Wert für n im Computerprogramm so eingestellt, dass die gemessenen Werte am besten beschrieben werden können bzw. die erhaltene Kurve am besten gefittet wird. So sind wir auch in der Lage aus der Bindungsenthalpie die Bindungsentropie rechnerisch zu ermitteln. ∆S bind = (∆H bind − ∆Gbind ) T Nachdem alle Werte für mehrere Temperaturen bestimmt wurden, kann die Protein-Ligand Wechselwirkung aufgrund folgender Beziehungen thermodynamisch exakt beschrieben werden. ∆c p bind = ∂∆H bind ∂T∆Sbind = ∂T ∂T Die Wärmekapazität entspricht diejenige Wärmemenge, die nötig ist, um ein mol eines Stoffes um 1K zu erwärmen. Es handelt sich dabei um eine Materialkonstante. Ist ∆c p bind negativ bedeutet dies, dass mit zunehmender Temperatur immer weniger Wärmeenergie aufgebracht werden muss, um eine Erwärmung um 1K zu bewirken. 1. Bestimmung der einzelnen Parameter A.) Bestimmung von ΔH0 Die molare Reaktionsenthalpie ΔH0 kann direkt gemessen werden, wenn man sicher sein kann, dass der gesamte hinzu titrierte Ligand auch gebunden wird, also gilt: ∆[ L] gebunden = ∆[ L]titriert dies ist bei einem einfachen Bindungsmodell mit n unabhängigen gleichartigen Bindungsstellen am Beginn der Titration der Fall; Voraussetzung ist, dass n ⋅ K A ⋅ [ P] >> 1 (Protein muss hohe Affinität, und ausreichend vorhanden sein) Somit resultiert aus der Beziehung: q i = n ⋅ [ PL] i ⋅ V Zelle ⋅ ∆H bind ∆H bind = qi n ⋅ [ L]titriert ⋅ VZelle ∆H bind kann alternativ auch wie K und n über eine mathematische Näherung bestimmt werden; B.) Bestimmung von KA und n Zur Bestimmung der Bindungsparameter K und n wird die Auftragung qi vs. ∆[ L]titriert _ i (tatsächlich aufgetragen ist die Zeit t, welche ∆[ L]titriert repräsentiert) für einen Satz von Vorgaben für die Parameter ∆H bind , n und K rechnerisch simuliert (Annahme eines bestimmten Bindungsmodells); Die Parameter werden softwaregestützt solange variiert, bis eine gute Übereinstimmung der simulierten Kurve mit dem Experiment erzielt ist; für ∆H bind kann ein fester Wert eingesetzt werden, um die Anzahl an Variablen zu verringern (Berechnung siehe oben) C.) Bestimmung von ΔGbind und ΔSbind Über K können nun diese Werte anhand folgender Beziehungen errechnet werden: ΔSbind positiv: Ligand verdrängt Wassermoleküle aus Bindungstasche ΔSbind negativ: Flexibilität von Seitenketten wird eingeschränkt D.) Bestimmung von Δcp Wird das ITC Experiment für unterschiedliche Temperaturen durchgeführt, so kann Δcp aufgrund der T-Abhängigkeit von ΔH berechnet werden: Cp groß: apolare, hydrophobe Seitenketten in Bindungstasche Cp klein: polare, ionisierte Seitenketten in Bindungstasche Die resultierende Kurve in der vom Computer berechneten resultierenden Kurve ist ΔH die Differenz vom h öchsten Wert zum niedrigsten Wert der sigmoidalen Kurve; n ist der x Wert des Wendepunkts; K (Assoziation) lässt sich einfach für jeden Titrationsschritt berechnen: K = qtotal − qi ; je qi größer das K für den gesamtprozess, desto sigmoidaler ist die Kurve Molare Rate = [injizierter Ligand] / [Protein] Gegenüberstellung: Differential Scanning Calorimetry (DSC) In dieser Methode wird die Referanzzelle kontinuierlich aufgeheizt (0,5-1,5K/min). Die Temperatur der Probenzelle wird durch Heizen der Temperatur der Referenzzelle angepasst. Die Probenzelle enthält bereits eine bestimmte Menge Protein und Ligand (keine Titration). Es wird der Unterschied im Heizstrom zwischen Probenzelle und Referenzzelle in Abhängigkeit von der Temperatur gemessen; dadurch kann direkt die Änderung der Wärmekapazität bei konstantem Druck bestimmt werden; Enzymkinetik Geschwindigkeitsgesetz und Reaktionsordnung Im Gegensatz zur Thermodynamik, welche untersucht wann eine Reaktion stattfindet und wo das Gleichgewicht dieser Reaktion liegt, beschäftigt sich die Kinetik mit der Reaktionsgeschwindigkeit. Die Reaktionsgeschwindigkeit ist immer proportional zur Konzentration der Reaktanden: v = k ⋅ cn Die Geschwindigkeitskonstante k bildet dabei die Proportionalitätskonstante. Diese beinhaltet ebenso die Temperaturabhängigkeit der Geschwindigkeit. N ist die Reaktionsordnung, nach der die Reaktion klassifiziert wird. Die Reaktionsordnung ist im Geschwindigkeitsgesetz der Exponent der Produktkonzentration, der beschreibt von wie vielen Edukten die Produktbildung abhängt. Dabei spielen nur solche Edukte eine Rolle, deren Konzentration nicht konstant ist. Die Reaktion von Glucose und ATP zu Glucose-6-phosphat mittels Hexokinase ist eine Reaktion zweiter Ordnung, da die Produktbildung von den Konzentrationen von ATP und Glucose abhängt. Da in biologischen Systemen die ATP-Konzentration jedoch nahezu konstant gehalten wird, spricht man von einer Reaktion pseudo erster Ordnung. In der Kinetik wird der Verbrauch des Edukts A oder die Zunahme des Produkts P gemessen, weshalb eine Differentielle Schreibweise möglich ist (hier für eine Reaktion erster Ordnung): v=− d [ P] d [ A] = k ⋅ [ A] = k ⋅ [ A0 − P ] = + dt dt Enzymatisch katalysierte Reaktionen: Michaelis Menten Kinetik Im ersten Reaktionsschritt wird aus Substrat und Enzym ein Enzym-Substrat Komplex gebildet. Es handelt sich um eine vor gelagerte Gleichgewichtsreaktion. Im zweiten Reaktionsschritt werden aus dem Enzym-Substratkomplex das Enzym und das Produkt freigesetzt. Wendet man die Differentialschreibweise auf die Bildung des Enzymsubstratkomplexes an folgt: d [ ES ] = k1 ⋅ [ E ][ S ] + k − 2 [ E ][ P] − k −1 ⋅ [ ES ] − k 2 ⋅ [ ES ] dt Aufgrund der Tatsache, dass das Produkt in vivo schnell weiter reagiert und in vitro in kleinen Konzentrationen vorliegt, lässt sich der Term k-2 vernachlässigen: d [ ES ] = k1 ⋅ [ E ][ S ] − k−1 ⋅ [ ES ] − k2 ⋅ [ ES ] dt Durch die Annahme des quasi-stationären Zustands (die Konzentration des Enzym-SubstratKomplexes erreicht einen Sättigungswert und bleibt über einen gewissen Zeitraum konstant) resultiert: d [ ES ] = k1 ⋅ [ E ][ S ] − (k−1 + k2 ) ⋅ [ ES ] = 0 dt Nach einer äquivalenten Umformung erhalten wir die Michaelis-Menten Konstante k −1 + k 2 [ E ][ S ] = = KM k1 [ ES ] Nun betrachten wir die Geschwindigkeit der Produktbildung, also die Reaktionsgeschwindigkeit bezüglich des Produkts v= d [ P] = k2 [ ES ] − k− 2 [ E ][ P] dt Auch hier ist der Term k-2 aus genannten Gründen zu vernachlässigen v= d [ P] = k2 [ ES ] dt Der limitierende Faktor für den Reaktionsumsatz stellt die Enzymkonzentration dar, weil diese wesentlich geringer ist, als die Substratkonzentration. [ E ] frei = [ E ]total − [ ES ] Durch einsetzen und umformen der bisher erläuterten Gleichungen erhält man unter Berücksichtigung, dass die Maximalgeschwindigkeit v max = k 2 [ E ]total Ist, die Michaelis-Menten-Gleichung v = k 2 [ E ]total ⋅ [S ] K M + [S ] Diese Gleichung lässt sich auf alle Enzymreaktionen anwenden. Sie zeigt einen hyperbolischen Sättigungsverlauf, wobei ihre beiden Asymptoten dem Vmax (reale Asymptote bei positiver Reaktionsgeschwindigkeit) und dem –KM Wert (imaginäre Asymptote bei negativer Substratkonzentration) entsprechen. Wenn die Substratkonzentration dem KM Wert gleicht, liegt die Hälfte der Enzyme als Enzym-SubstratKomplex vor, was bedeutet, dass die Halbmaximale Reaktionsgeschwindigkeit erreicht ist. Man sieht, dass die Reaktionsgeschwindigkeit bei geringen Substratkonzentrationen proportional zu [S] verläuft. Bei hohen Substratkonzentrationen, ist sie jedoch [S]unabhängig. Es wird die Maximalgeschwindigkeit des Enzyms erreicht. Nun ist die Enzymkonzentration limitierend für die Gesamtreaktionsgeschwindigkeit. Messung der Daten und Auswertung Um die charakteristischen Werte KM und vmax eines Enzyms zu bestimmen, müssen einige Messungen durchgeführt werden. Um die Produktbildung zu verfolgen, bedient man sich der Photometrie. In manchen Reaktionen haben die Edukte andere Absorptionsmaxima als die Produkte. Dadurch lässt sich die Zunahme der relativen Absorption in dem Wellenlängenbereich, bei dem die Produkte absorbieren, direkt mit der Konzentrationszunahme der Produkte korrelieren. So erhält man einen Graphen, welcher die Extinktion gegen die Zeit darstellt. Die Anfangssteigung dieses Graphen entspricht der Reaktionsgeschwindigkeit. Die Steigungen später entsprechen zwar ebenso Reaktionsgeschwindigkeiten, sind aber nicht aussagekräftig, da die Geschwindigkeit erstens mit zunehmender Produktbildung abnimmt und zweitens durch den Substratverbrauch nicht mehr der Anfangssubstratkonzentration, zuzuordnen ist. Führt man diese photometrische Messung mit unterschiedlichen Substratkonzentrationen durch, lässt sich ein Diagram erstellen, dass die Substratkonzentration der Reaktionsgeschwindigkeit zuordnet. Die Anfangssteigung, also die Reaktionsgeschwindigkeit, ist dann am größten, wenn die Substratkonzentration sehr hoch ist. Insofern kann das Enzym die maximale Reaktionsgeschwindigkeit vmax erst bei einer unendlich hohen Substratkonzentration erreichen. Dieser Graph entspricht der Michaelis-Menten Kinetik. Allerdings lassen sich hier die charakteristischen Werte KM und vmax nur erahnen, da sie die Asymptoten des hyperbolen [E] = [ ES ] total wird. Ursache Graphen darstellen. Vmax wird in der Realität nie erreicht, da nie hierfür ist, dass die Affinität des Enzyms zum Substrat nie stark genug ist, damit alle Enzyme als Enzym-Substratkomplex vorliegen. Um dennoch die Werte exakt ermitteln zu können werden die Graphen linearisiert. Hier gibt es unterschiedliche Möglichkeiten, wobei jede seine Vor- und Nachteile hat. Diskutiert und angewendet werden im Praktikum die folgenden zwei Linearisierungen. Lineweaver-Burk Darstellung Diese Darstellung entspricht einer Linearisierung mittels doppelreziproker Darstellung. v = v max ⋅ [S ] K M + [S ] 1 1 K M + [S ] 1 1 = ⋅ = v v max [S ] v v max K ⋅ M + 1 [S ] 1 KM 1 1 = ⋅ + v v max [ S ] v max 1 1 v In dieser Darstellung entspricht der Schnittpunkt mit der v Achse genau max . Der KM 1 1 − K M . Die Steigung ist vmax . Problematisch bei dieser Schnittpunkt mit der [ S ] Achse ist Darstellung ist, dass die Geschwindigkeitswerte bei den niedrigen Substratkonzentrationen den größten Fehler aufweisen. Diese sind jedoch die aussagekräftigsten und daher wichtigsten Werte, da sie den Messdaten am Anfang des Michaelis-Menten-Plots entsprechen. Diese Daten liefern die genauesten Geschwindigkeits-Werte. Spätere Werte für die Geschwindigkeit sind verfälscht, da hier die Maximalgeschwindigkeit des Enzyms aufgrund bereits vorhandener Produkte nicht erreicht wird. sie sind deshalb wichtig, weil nur bei niedrigen Substratkonzentrationen die angenommene quasi-Stationarität auch tatsächlich vorhanden ist; größere Substratkonzentrationen haben einen höheren systematischen Fehler (Annahme falsch); die kleineren allerdings einen hohen statistischen Fehler! Eadie-Hofstee Darstellung Diese Darstellung basiert auf der Lineweaver-Burk Gleichung. Sie ergibt sich durch äquivalente Umformung v KM K vK M 1 1 v max = M + 1 v max = = + + v max + v v = −K M v v max ⋅ [ S ] v max [S ] v [S ] [S ] v Hier entspricht der Schnittpunkt mit der v Achse genau vmax. Der Schnittpunkt mit der [S ] vmax K Achse ist M . Die Steigung ist − K M . Problematisch bei dieser Darstellung ist, dass v in beide Koordinaten eingeht. Daher konvergieren alle Fehler im Ursprung. Die Fehler sind nicht mehr einzuschätzen. Theoretischer Hintergrund zur betrachteten Reaktion Die in diesem Praktikumsversuch betrachtete Reaktionsfolge wird von den Enzymen Hexokinase und Glucose-6-phosphat-Dehydrogenase katalysiert. Die Hexokinase-Reaktion beinhaltet die Umwandlung von Glucose unter ATP-Verbrauch zu Glucose-6-Phosphat (Eingangsreaktion in die Glykolyse). Die Glucose-6-phosphat-Dehydrogenase spielt im Pentosephosphatweg eine entscheidende Rolle. Sie wandelt das überschüssige Glucose-6Phosphat unter Erzeugung von NADPH+H+ zu 6-Phosphoglucono-δ-lacton um. Betrachtet man die Folge beider Reaktion, ist wichtig festzustellen, dass die Affinität der Hexokinase zu Glucose wesentlich geringer ist, als die Affinität der Glucose-6-phosphatDehydrogenase zu Glucose-6-Phosphat. Dies bedeutet erstens, dass die Hexokinase die Geschwindigkeit der Reaktionsfolge bestimmt und zweitens, dass somit die Kinetik der NADPH-Synthese im zweiten Schritt identisch ist mit der Glucose-Umsetzung im ersten Schritt. Somit lässt sich die NADPH-Synthese messen und mit der Glucose Umsetzung direkt korrelieren. Zweitens liegt in unserem Fall ATP und NADP+ im Überschuss vor. Somit ist sichergestellt, dass die gesamte Glucosemenge, welche im Reagenz vorhanden ist, auch zu Glucose-6-Phosphat und weiter zu 6-Phosphoglucono-δ-lacton unter NAPH+H+ Erzeugung reagiert. Die gebildete NAPH+H+ Menge entspricht also der Glucose-Menge die sich zu Beginn der Reaktion im Reagenz befand. Die NADPH-Synthese ist in einem Photometer direkt messbar über die Zunahme der spezifischen Extinktion bei 340nm (siehe folgende Abbildung) Gaschromatographie Das Prinzip der Chromatographie Die Chromatographie ist ein in der Chemie sehr häufig verwendetes Trennverfahren. Die Auftrennung eines Stoffgemisches erfolgt in allen chromatographischen Methoden durch die unterschiedliche Verteilung seiner Einzelbestandteile zwischen der stationären und der mobilen Phase. Dabei strömt die mobile Phase durch die stationäre Phase. Je nachdem, ob ein Einzelbestandteil mehr mit der mobilen oder mehr mit der stationären Phase wechselwirkt, desto weiter bzw. kürzer wandert er in der stationären Phase. Nachdem der Einzelbestandteil sichtbar gemacht wurde (Detektion am Ende der stationären Phase in der Gaschromatographie oder Anfärbung bei der Dünnschichtchromatographie), kann es aufgrund der Wanderungsstrecke innerhalb einer bestimmten Zeit bzw. der Wanderungsdauer auf der Strecke der stationären Phase einem bestimmten Stoff zugeordnet werden. Die Gaschromatographie selbst dient zur Auftrennung von unzersetzt verdampfbaren Stoffgemischen. Hier dient als mobile Phase ein Inertgas mit guten Strömungseigenschaften (z.B. Stickstoff, Helium oder Wasserstoff). Dieses Trägergas wird durch eine Säule mit definiertem Innendurchmesser geleitet. Die 25-50m lange und 0,5mm dünne Säule (Metall oder Quarzglas) ist mit einem bestimmten Material (Polysiloxane oder Polyethylen-Glykole), der stationären Phase, ausgekleidet und zu einer Spule aufgewickelt. Der Bau eines Gaschromatographen Ein GC besteht aus drei wesentlichen Bauteilen: Injektor, Trennsäule im GC Ofen und Detektor (siehe Abbildung 1) Abbildung 1 Aufbau eines Gaschromatographen. (1) Druckbehälter mit Trägergas; (2) Injektor; (3) Trennsäule zur Spule aufgewickelt; (4) Detektor; (5) Chromatogramm Der Injektor und die mobile Phase Die in einem niedrig siedenden Lösungsmittel gelöste Probe wird durch das Septum des in unserem Praktikum verwendeten Split-Injektors (siehe Abbildung 2) über eine Hamilton Pipette in die Vaporisations-Kammer eingespritzt. Diese wird auf ca. 170° erhitzt, damit die Probe schnellstmöglich vollständig verdampft. Das obere „Septum purge outlet“, dient dazu Überreste des durchstochenen Gummi-Septums abzusaugen, sodass es nicht in die Probe gerät. Das darauf folgende „Carrier Gas inlet“ ist die Verbindung zu einem externen TrägergasBehälter. Dieser enthält in unserem Fall Wasserstoff. Sobald die Probe in den Injektor Abbildung 2 Aufbau eines Split Injektors eingespritzt wurde, wird der Startbutton betätigt, und das Trägergas strömt ein. Es kommt zur Vermischung des Trägergases mit der Substanz. Der Split outlet dient dazu, überflüssiges Trägergas-Proben Gemisch zu entfernen. Nur ein sehr geringer Teil der Probe gelangt letztlich in die Trennsäule (column). Die Trennsäule und die stationäre Phase Die Substanzen werden durch das Trägergas (Säulenvordruck) in die Trennsäule transportiert, welche in den GC Ofen eingebaut ist. Der GC Ofen dient dazu, die Säule präzise zu temperieren. Das Proben-Trägergas Gemisch sublimiert zunächst in der Säule wieder, wenn die Temperatur des Ofens unterhalb der Siedetemperatur aller Bestandteile der Probe liegt. Nun wird ein bestimmtes Temperaturprogramm gefahren, das heißt der Ofen erwärmt sich langsam, sodass alle Bestandteile erst nach und nach verdampfen, sodass sie letztlich weiter entfernt voneinander wandern (dies verhindert ein Überschneiden der Peaks und erleichtert die Auswertung). Man unterscheidet zwei verschiedene Arten von Trennsäulen: beschichtete und gepackte. Beschichtete Trennsäulen haben im Inneren lediglich eine Schicht stationäre Phase aufgelagert. Bei gepackten Trennsäulen hingegen kleidet die stationäre Phase das gesamte Volumen der Trennsäule aus. Die Trennsäule selbst kann aus Metall oder Glas bestehen. In unserem Fall handelt es sich um eine beschichtete Säule aus Glas (FSOT: fused silica open tubular; siehe nebenstehende Abbildung 3). Sie besteht aus einer stabilisierenden Schicht aus fusioniertem Silikat und einem äußeren PolyimidMantel, der für die Flexibilität der Säule sorgt. Nur so kann sie zu einer Spule aufgewickelt werden. Die stationäre Phase ist auf die Silikat-Schicht aufgelagert. Gebräuchlich sind Polysiloxane und Polyethylen-Glykole (siehe Abbildung 4), wobei unsere Säule eine Polyethylen-Glykolschicht als stationäre Phase aufweist. Abbildung 3 Aufbau eines FSOTs Abbildung 4 links: Polysiloxane mit unterschiedlichen Resten; oben: Struktur der Polyethylen-Glykole Der Detektor und das Signal Am Ende der Säule befindet sich ein Detektor, der ein elektronisches Signal erzeugt, wenn eine Substanz das Trennsystem verlässt. So wird ein typisches Chromatogramm erzeugt. Es gibt zahlreiche Detektoren, welche jeweils nur selektiv verschiedene Stoffe detektieren können und auch nur mit bestimmten Trägergasen kompatibel sind. Im Gaschromatographen, der in unserem Versuch verwendet wurde, befindet sich ein Flammen-Ionisations-Detektor (FID; siehe nebenstehende Abbildung 5). Dieser kann lediglich C-C und C-H Abbildung 5 Aufbau eines FlammenBindungen detektieren. Dieser Detektor Ionisations-Detektors (FID) ionisiert die Moleküle der Probe, die aus der Trennsäule kommen und Sauerstoffmoleküle, die aus einem externen Behälter synthetischer Luft (80% Stickstoff, 20% Sauerstoff) mit der aus der Säule hervortretenden Probe vermischt werden. Außerdem wird Wasserstoff als Brenngas beigemischt. Durch die Ionisation der Sauerstoffmoleküle entstehen Sauerstoffradikale, die sofort mit den ionisierten Kohlenstoffatomen und Wasserstoffatomen reagieren. Die hierbei frei werdende spezifische Energie erzeugt einen spezifischen Strom, dessen Intensität gemessen und an den Computer zur Auswertung weitergegeben wird. Eine Messung erzeugt somit einen Punkt im Chromatogramm. Ein typisches Chromatogramm (siehe Abbildung 6) eines FI-Detektors ist im Folgenden abgebildet. Die Zuordnung der Peaks zu den Stoffen erfolgt aufgrund bekannter Retentionszeiten. Die Fläche unterhalb des Peaks entspricht der Menge des Stoffes innerhalb des Gemisches. Je breiter der Peak ist, desto verzerrter, also schlechter ist das Chromatographische Ergebnis (hierzu siehe Abschnitt 6: Die Güte der Trennung) Totzeit und Retentionszeit Die Zeit, die das Trägergas benötigt, um die ganze Säule zu durchströmen, nennt man die Totzeit t0 des Systems. Sie liegt bei ca. einer Minute. Im Idealfall wechselwirkt das Trägergas gar nicht mit der stationären Phase, so dass die Totzeit lediglich von Vordruck (mit dem das Trägergas durch die Säule Abbildung 6 typisches Chromatogramm mit einer Zuordnung der einzelnen Piks zu den zugehörigen gedrückt wird) und vom Strömungswiderstand der Säule abhängt. Verbindungen aufgrund der Retentionszeiten Die zu trennenden Stoffe müssen chemisch auf die stationäre Phase abgestimmt sein. Im Gegensatz zu den Trägergasen zeigen die meisten chemischen Stoffe daher eine Wechselwirkung mit der stationären Phase. Diese brauchen daher länger, um die Säule zu passieren. Die Totzeit beschreibt also auch diejenige Zeit, die sich ein Analyt in der mobilen Phase aufhält. Die Nettoretentionszeit beschreibt im Gegensatz dazu die Zeit, die sich ein Analyt in der stationären Phase aufhält. Die Retentionszeit t R ist die Zeit, die ein Analyt zum passieren der Säule benötigt, also die Summe aus Totzeit und Nettoretentionszeit. Die Retention einer Substanz durch die stationäre Phase wird durch drei Aspekte bestimmt: Die Stärke der Wechselwirkung (z.B. Van-der-Waals, H-Brücken oder Dipol-Moment Wechselwirkungen) der Substanz mit der stationären Phase beschreibt die Neigung in der stationären Phase zu bleiben. Der Siedepunkt der Substanz beschreibt die Neigung in der mobilen Phase zu bleiben. Die Diffusionseigenschaften der Substanz beschreiben die Beweglichkeit der Substanz in beiden Phasen. Von außen kann auf die Retention einer Substanz am einfachsten durch TemperaturVeränderungen am Ofen und Veränderungen der Flussrate der mobilen Phase eingewirkt werden. Eine Temperaturerhöhung bewirkt eine höhere kinetische Energie der Teilchen, weshalb alle Retentionszeiten verkürzt werden. Wird die Flussrate erhöht, so erhöht sich auch die Geschwindigkeit der mobilen Phase. Dementsprechend verkürzt sich die Retentionszeit. Mit der Verkürzung der Retentionszeit reduziert sich allerdings die Trenneffizienz, weshalb bei geringen Flussraten und bei geringst-möglichen Temperaturen (knapp oberhalb der Siedetemperatur) aufgetrennt wird. Biophysikalischer Hintergrund der Auftrennung Ist ein Stoff A dazu in der Lage sich zwischen zwei nicht mischbaren Phasen physikalisch zu verteilen, findet das Nernst’sche Verteilungsgesetz Anwendung. Es besagt, dass diese Verteilung zu einem Gleichgewicht mit dem Nernst’schen Verteilungskoeffizienten K= c A ( Phase2) c Aflüssig = gas c A ( Phase1) cA führt. K ist lediglich von der Temperatur abhängig. Dabei ist zu beachten, dass der Quotient aus der Menge X der Komponente A in der Phase 1 und der Menge X der Komponente A in der Phase 2 immer konstant ist X Aflüssig = const. X Agas Der Retentionsfaktor oder auch Kapazitätsfaktor k ' wird genutzt, um die Wanderungsrate eines Analyten anhand durch folgende Beziehung zu beschreiben. k' A = t R − t0 t0 Um die Trennung von zwei Substanzen auf der Säule zu beschreiben, wird ein Selektivitätsfaktor α= B k ' B t R − t0 = A k ' A t R − t0 definiert. Bei der Verwendung dieser Formel, soll Stoff A schneller diffundieren als Stoff B. Der Selektivitätsfaktor ist daher immer größer als 1. Die Güte der Trennung Die Güte der Trennung lässt sich anhand der resultierenden Peaks im Chromatogramm ersehen. Eine ideale Trennung zeigt Peaks mit verschwindender Breite und großer Intensität. Das heißt alle Moleküle eines Stoffes, der zuvor im Gemisch vorlag, kommen exakt zum selben Zeitpunkt am Ende der Trennkapillare an. Je breiter die Peaks, desto schlechter die Auftrennung. Je unsymmetrischer die Peaks sind, desto unsauberer (Probe wurde z.B. zu langsam injiziert) wurde gearbeitet. Um die Trenneffizienz anhand der Peakbreite theoretisch vorauszusagen wurden zwei Modelle entwickelt, die im Folgenden erläutert werden sollen. Neben der Peakbreite korreliert die Trenneffizienz außerdem mit der Retentionszeit. Je größer diese ist, desto besser ist die Auftrennung, da die Peaks der einzelnen Stoffe weiter voneinander entfernt sind und sich somit die Flächen weniger bis nicht überschneiden. Mit größerer Retentionszeit werden jedoch die Peaks breiter. Es muss also eine optimale Einstellung zwischen Retentionszeit durch u.a. Flussrate und Breite der Peaks gefunden werden, je nachdem wie weit die Peaks der Stoffe im Chromatogramm ohnehin auseinander liegen. Das Theoretische Plattenmodell Das theoretische Plattenmodell beschreibt die Vorstellung, dass eine Trennkapillare aus einer großen Anzahl aufeinander folgender theoretischer Platten besteht. In jeder Platte findet ein neuer Trennprozess statt, der eine neue Gleichgewichtseinstellung der zu trennenden Stoffe in der mobilen und stationären Phase bewirkt. Stellt man sich die Trennkapillare wie beschrieben vor, lässt sich die Trenneffizienz exakter beschreiben: Sie nimmt mit der Anzahl N der theoretischen Platten zu (je mehr, desto besser) und mit der Höhe (HETP = Höhenäquivalent) der theoretischen Platten ab (je kleiner, desto besser). Es resultiert folgende Beziehung zur Länge L der Trennkapillare HETP = L N Die Anzahl der theoretischen Platten kann aufgrund des Peaks im Chromatogramm berechnet werden. 5,55 ⋅ t R N= 2 b1 / 2 2 Hierzu ist die Halbwertsbreite b1/2 relevant. Sie lässt sich einfach anhand eines Peaks im Koordinatensystem ermitteln, wie die folgende Abbildung 7 zeigen soll. Die Halbwertsbreite ist der Abstand zwischen den zwei x Werten, deren zugehörige y Werte auf der halben Höhe der Amplitude liegen. Abbildung 7 Die Halbwertsbreite entspricht der Breite des Peaks auf der halben Intensität. Die Ratentheorie Das Plattenmodell geht davon aus, dass die Gleichgewichtseinstellung in jeder Platte sofort geschieht, also keine Zeit benötigt wird. Eine realistischere Beschreibung für die Prozesse innerhalb der Trennkapillare liefert die Ratentheorie. Sie konzentriert sich auf die Beschreibung der Zeit, die für das Lösen des zu trennendes Gemisches benötigt wird, bis sich ein Gleichgewicht zwischen stationärer und mobiler Phase eingestellt hat. Für die resultierende Peakbreite ist also die Elutionsrate (Elution ist der Transport einer Substanz durch eine chromatographische Säule durch kontinuierliche Zugabe von mobiler Phase) von großer Bedeutung. Außerdem spielen die Anzahl der unterschiedlichen Wege, die das Gas einschlagen kann eine wichtige Rolle. Die Raten-Theorie führte zu der Entwicklung der Van-Deemter Gleichung, welche das Höhenäquivalent mit 4 Parametern beschreibt: HETP = A + A: [m] B: [m*ml/min] B + Cv v C: [m*min/ml] v: [ml/min] v ist hier die durchschnittliche lineare Gasgeschwindigkeit der mobilen Phase (=Flussrate). Sie kann am Chromatographen eingestellt werden und ist somit bekannt. A, B und C sind spezielle Parameter. A ist die Wirbeldiffusionskonstante (Eddy Diffusion). Sie ist unabhängig von der Gasgeschwindigkeit und berücksichtigt, dass das Gas unterschiedliche Wege einschlagen kann. Diese Konstante spielt jedoch nur bei gepackten Säulen (Säulen, bei denen die stationäre Phase den Hohlraum vollständig auskleidet) eine bedeutende Rolle, da hier viel mehr Wege möglich sind. Bei beschichteten Säulen, wie wir sie im Praktikum verwendet haben, ist der Wert dieser Konstante sehr klein. B ist die Längsdiffusionskonstante (Longitudinale Diffusion). Um diese zu veranschaulichen, stellt man sich einen Stoff in der Kapillare als ovaler Tropfen vor, dessen Dichte in der Mitte am größten, vorne und hinten am geringsten ist. Wird nun die Flussrate erhöht, so driften die einzelnen Moleküle dieses Tropfen durch Ausgleichsdiffusion in Bereiche niedriger Dichte bzw. Konzentration weniger auseinander, da weniger Zeit dazu zur Verfügung steht. Der Tropfen vergrößert sich weniger. Die Breite des Peaks wird geringer. Die Flussrate verhält sich hier also antiproportional mit der Konstante zum Höhenäquivalent. Die Longitudinale Diffusion hängt ab von der Temperatur und dem Trägergas. C ist die Stoffübergangskonstante. Sie beschreibt de Interaktion des Stoffes in der stationären Phase und die Möglichkeit des Übergangs in die mobile Phase. Je schneller die Flussrate, desto größer ist der Widerstand für den Übergang von der stationären Phase in die mobile Phase. Der Übergang erfolgt daher statistisch gesehen später, was eine Verbreiterung der Peaks zur Folge hat. Mit der Flussrate nimmt auch der Widerstand, und somit die Konstante zu. Sie verhalten sich proportional zum Höhenäquivalent. Die Stoffübergangskonstante ist abhängig vom Trägergas und der Dicke der stationären Phase. Der Van-Deemter Plot Anhand dieser Beziehung lässt sich die ideale Flussrate, die einen minimalen Wert für HETP liefert, bestimmen. Es ist das Minimum des v -HETP Plots. (1) Bildung der Ableitungen: HETP (v) = A + B ∂HETP = − 2 +C v ∂v ∂HETP 2 B = 3 v ∂v∂v (2) Notwendige Bedingung: − und ∂HETP =0 ∂v B C B B + C = 0 C = 2 Cv 2 = B v = 2 v v (3) Hinreichende Bedingung: 2B B C (4) 3 > 0 Minimum bei v = ∂HETP ≠0 ∂v∂v v= B ∂HETP 2 B in = 3 C v ∂v∂v B C Berechnung der HETP Koordinate: v = HETP(v) = A + B + Cv v B B in HETP (v) = A + + Cv C v B1 / 2 B ⋅ C1 / 2 C ⋅ B1 / 2 B B B +C = A + 1/ 2 + C ⋅ 1/ 2 = A + + B C B1 / 2 C1 / 2 C B C1 / 2 C = A + B1 / 2 ⋅ C1 / 2 + C 1 / 2 ⋅ B1 / 2 = A + 2(C ⋅ B ) 1/ 2 = A + 2 CB In der folgenden Abbildung 8 ist der Graph gezeigt. Man kann erkennen wie sich das Höhenäquivalent aus den einzelnen Konstanten additiv zusammensetzt. Das Minimum der Ergebnis-Kurve (rot) entspricht exakt den Koordinaten [ B | A + 2 CB C ] Abbildung 8 Auftragung des Höhenäquivalents H gegen die Flussrate u. ein typischer Van-Deemter Plot ist hier rot gezeigt. Die drei Parameter, von denen der Van-Deemter Plot abhängt, sind ebenfalls eingezeichnet Es ist bei kleinen Flussraten gut die Abhängigkeit von der Diffusionskonstante und bei großen Flussraten gut die Abhängigkeit von der Stoffaustauschkonstanten zu erkennen. Anwendung der Gaschromatographie Die Gaschromatographie findet ihre Anwendung in Bereichen u.a. der Medizin (klinische Diagnostik: Auftrennung von Blut), Lebensmittelchemie (Typisierung von Lebensmitteln aufgrund ihrer Zusammensetzung) und Umweltanalytik (Untersuchung von AerosolTreibgasen). Dies sind nur einige Beispiele. In der Biochemie eignet sich die Gaschromatographie zur Analyse der Proteine. Es kann der prozentuale Gehalt der einzelnen Aminosäuren in einem Protein bestimmt werden. Da Gaschromatographen lediglich eine maximale Temperatur von 250°C unterstützen, Proteine jedoch eine erheblich höhere Siedetemperatur haben, ist eine Auftrennung der Proteine an sich nicht möglich. Auch Aminosäuren haben einen Siedepunkt von weit über 250°C. Meistens kommt es durch solch hohe Temperaturen auch zur Zersetzung, weshalb ebenso keine Gaschromatographische Auftrennung erfolgen kann. Mittels Totalhydrolyse eines Proteins und anschließender Derivatisierung der Carboxylgruppe mit einem Ester wird der Siedepunkt jedoch soweit herabgesetzt, dass eine Auftrennung möglich ist. Das Verhältnis der Integrale der einzelnen Peaks ergibt das Verhältnis zu dem die einzelnen Aminosäuren im Protein vorkommen. Fluoreszenz Fluoreszenz: Übergänge zwischen elektronischen und Schwingungs- Zuständen Bei der Fluoreszenz handelt es sich um eine Strahlung, ausgehend von einem fluoreszierenden Molekül. Sie wird ausgelöst, indem dieses Molekül zunächst ein Photon absorbiert und so vom elektronischen und Schwingungsgrundzustand in den ersten elektronischen Zustand und je nach Photonenenergie in einen höheren Schwingungszustand angeregt wird. Innerhalb von Femtosekunden relaxiert das System in den untersten Schwingungszustand des ersten angeregten elektronischen Zustandes zurück. Die freiwerdende Energie entspricht einer Wärmestrahlung (interne Konversion: internal conversion). Von hier aus wird ein Photon emittiert. Das System gelangt so in den elektronischen Grundzustand und durch weitere schnelle interne Konversion in den Schwingungsgrundzustand. Dadurch, dass durch die zweimalig auftretende interne Konversion Energie verloren geht, kann das emittierte Photon nicht dieselbe Energie aufweisen, wie das absorbierte. Es resultiert eine Rotverschiebung der Fluoreszenzstrahlung relativ zur absorbierten Strahlung. Dieses Phänomen wird als Stokes-Shift nach seinem Entdecker beschrieben Mittels des Franck Condon Prinzips kann der wahrscheinlichste Übergang zwischen verschiedenen Schwingungszuständen unterschiedlicher elektronischer Zustände berechnet werden. Die Berechnung geht davon aus, dass die Zeit für den Übergang der Elektronen zwischen den beiden Niveaus so schnell stattfindet (10-15s), dass sich der Kernabstand nicht ändern kann, da eine Kernschwingungsperiode länger dauert (10-13s). Die Elektronenbewegung ist aufgrund der geringeren Masse des Elektrons in einer solchen Geschwindigkeit möglich. Betrachtet man das Energieschema des quantenmechanischen Morse-Potentials (siehe Abbildung 1), erkennt man die direkte Konsequenz dieser Annahme. Die Elektronen aus dem Schwingungsgrundzustand landen nicht im Schwingungsgrundzustand des höheren elektronischen Niveaus, sondern in einem sich darüber befindenden Schwingungszustand. Der Pfeil, welcher für diesen Übergang steht, verläuft dabei senkrecht nach oben. Man spricht von einem vertikalen Übergang. Ob der Übergang nun hin zum Schwingungszustand n, n+1 oder n-1 stattfindet ist wahrscheinlichkeitsverteilt. Die Verteilung der Übergangsintensitäten (vom Schwingungsgrundzustand hin zu diesen oberen Schwingungszuständen des elektronisch höheren Niveaus) in einer makroskopischen Probe hängt davon ab, welcher Schwingungszustand bei einem vertikalen Übergang auch den wahrscheinlichsten (je größer die Amplitude der Eigenfunktion eines Schwingungsniveaus für einen bestimmten Abstand ist) Schwingungsabstand hat und in welchem Abstand sich das Elektron gerade im aktuellen Schwingungsgrundzustand befindet. Abbildung 1, Energieschema des quantenmechanischen Morse-Potentials; links: wahrscheinlichster Übergang nach dem Franck Condon Prinzip; rechts: Wanderung eines Elektrons zwischen den Zuständen bis zum Ausgangspunkt Auf der x Achse ist der Kernabstand aufgetragen; er ist abhängig von der Kernschwingung (kann vernachlässigt werden, da sie sehr langsam ist) und der Schwingung der Bindungselektronen; würde die Schwingung der Kerne zueinander schneller sein, als die Elektronen für den Übergang zwischen ihren elektronischen Niveaus benötigen, so würde kein vertikaler Übergang stattfinden; das Elektron würde also zufällig in einem beliebigen Schwingungszustand landen; angeregt werden mittels Fluoreszenz die Pi Elektronen in das Pi* Orbital; dadurch steigt der mittlere Abstand der Kerne voneinander an (Antibindendes Orbital hat geringere Überlappung); Die Lebensdauer des angeregten Zustands (Elektron im Pi*) beträgt ca. 10ns (d.h. nach Abstellen der Anregungsquelle erlischt die Fluoreszenz sofort); im Unterschied zur Phosphoreszenz bleibt der Spin des Elektrons bei allen Übergängen erhalten! Die Photonen-Emission findet immer aus dem S1 statt, weshalb das Emissionsspektrum unabhängig von der Anregungswelle ist; höhere elektronische zustände gehen immer durch interne Konversion (Wärmestrahlung) in den S1 Zustand über; Der Triplettzustand n liegt energetisch unterhalb des Singulettzustandes n. Ursache ist, dass für den Übergang in den unteren S Zustand vom Triplett-1 Zustand aus weniger Energie frei wird als vom Sigulett-2 Zustand aus. Es wird deshalb weniger Energie frei, da Energie dafür verloren geht, den Spin umzudrehen. Diesen Energiebetrag nennt man Spinpaarungsenergie. Auch das Jablonski-Diagramm veranschaulicht die Prozesse der Photonen -Absorption und –Emission, wobei Rotationsübergänge eine Ebene unter den Vibrationsübergängen sind und deshalb in diesem Diagram nicht abgebildet werden (siehe Abbildung 2). Im Folgenden soll auf den Prozess der Phosphoreszenz eingegangen werden. Abbildung 2, Jablonski Diagram der möglichen quantenmechanischen Übergänge Phosphoreszenz Phosphoreszenz ist ähnlich der Fluoreszenz ebenfalls eine Strahlung, ausgehend von phosphoreszierenden Molekülen. Während die Fluoreszenz-Strahlung direkt während der Einstrahlung von Licht messbar ist, und schon Mikrosekunden danach nicht mehr, so ermöglicht die Phosphoreszenz ein mitunter sehr langes Nachleuchten. Dieses entsteht dadurch, dass vom ersten elektronischen angeregten Zustand kein Photon emittiert wird, sondern zunächst ein Interkombinationsprozess (internal crossing), bei dem sich der Spin des Elektrons umkehrt, stattfindet (S1 Zustand geht in T1 Zustand über). Es handelt sich um ein höheres metastabiles Energieniveau (Triplett-Zustand), in dem das Elektron nun „gefangen“ ist. Interkombinationsprozesse sind eigentlich quantenmechanisch verboten und geschehen selten und zufällig, mit einer bestimmten Wahrscheinlichkeit. Die Rückkehr in den elektronischen Grundzustand ist nur durch einen weiteren Interkombinationsprozess möglich, der den Triplett-Zustand durch Spinumkehr und Photonen-Emission in den Singulett-Zustand überführt. Da die gesamte Phosphoreszenz-Strahlung aufgrund ihrer Abhängigkeit von einer Wahrscheinlichkeitsverteilung mitunter erst Sekunden oder länger nach der Einstrahlung des Lichts abgestrahlt wird, eignet sie sich nicht für unsere Messungen. Es würde eine zu lange Zeit beanspruchen. Typische Lebensdauern der Phosphoreszenz betragen millisekunden bis sekunden und werden daher eher durch mögliche Deaktivierungsprozesse begrenzt als durch die eigentliche Emission des Photons; Messung der Phosphoreszenz Lebensdauer ich sollte erklären, wie man die tripplett lebensdauer messen kann, wenn keine Phosphoreszenz stattfindet (das war so eine Knobelaufgabe, wo man keine Ahnung von haben muss ;)). Bin erst nach vielen Hilfestellungen draufgekommen. Man nutzt einfach zwei gepulste Laser, einen intensiven und einen schwächeren, der es nicht schafft in den Triplet zustand anzuregen. der schwächere Laser läuft im dauerfeuer, sodass man die ganze zeit ein Fluoreszenzsignal hat, dann kommt der starke Laserpuls. Dadurch werden Elektronen in den Tripletzustand angeregt. Dementsprechend schwächt sich die Fluoreszenz ab. wenn die Elektronen nun nach und nach wieder strahlungsfrei in den Grundzustand übergehen werden sie wieder von dem schwachen Laser angeregt wodurch das Fluoreszenzsignal wieder zunimmt. Als Konkurrenz zur Lumineszenz (Fluoreszenz + Phosphoreszenz) können weitere zwei Möglichkeiten der Relaxation in den Grundzustand stattfinden Fluoreszenzlöschung (Quenching) Fluoreszenzlöschung meint die strahlungslose Desaktivierung des angeregten Zustands des Fluorophors durch Quencher Moleküle, wodurch die Fluoreszenzintensität reduziert wird (geringere Quanten-Ausbeute). Fluoreszenzlöschung kann über verschiedene Prozesse erfolgen (Energietransfer, Elektronen- bzw. Protonentransfer, chemische Reaktionen im Anregungszustand); Die Löschung geschieht entweder durch Kollision des Fluorophors mit dem Quencher (dynamische Fluoreszenzlöschung) oder durch Komplexbildung zwischen beiden Molekülen (statische Fluoreszenzlöschung). Die Effektivität beider Prozesse ist von der Konzentration der Löschermoleküle abhängig. In beiden Prozessen ist der direkte molekulare Kontakt zwischen Fluorophor und Quencher erforderlich. Quenching-Messungen geben daher Aufschluss über die Zugänglichkeit eines Fluorophors (z.B. in einem Protein oder einer Membran) und über die Diffusionsgeschwindigkeiten des Quenchers. Fluorophore und Fluoreszenzintensität Man unterscheidet intrinsische von extrinsischen Fluorophoren. Erstere sind in vivo vorkommende fluoreszierende Moleküle, letztere sind synthetisch hergestellte. Alle Fluorophore weisen delokalisierte Pi Elektronensysteme auf, die in der Lage sind Photonen zu absorbieren; Wichtige intrinsische fluoreszierende Moleküle sind die aromatischen Aminosäuren Tryptophan, Tyrosin und Phenylalanin, sowohl die Kofaktoren FMN, FAD und NADH. In gefalteten Proteinen ist lediglich die Fluoreszenz der Tryptophane relevant (macht 90% der Fluoreszenzemission des Proteins aus), da die der Tyrosine gequencht wird (Energietransfer von Tyrosin auf Tryptophan oder durch Wasserstoffbrücken über die OH Gruppe auf die Lösungsmittelumgebung) und die der Phenylalanine sehr schwach ist. Als typische Quencher fungieren Sauerstoff, Acrylamid und Amine. Auch die Aminosäuren desselben Proteins können ein Quenchen der Tryptophanfluoreszenz bewirken, was zu einer Intensitätssteigerung der Fluoreszenz proportional zum Denaturierungsprozess führt. Fluoreszenzanisotropie Ein Fluorophor absorbiert bevorzugt Photonen, deren elektrische Feldvektoren parallel zum Übergangsmoment des Fluorophoren ausgerichtet sind. Die Übergangsmomente für Absorption und Emission eines Fluorophoren haben jeweils definierte Orientierungen. In einer isotropen Lösung nehmen die Moleküle alle Orientierungen mit gleicher Wahrscheinlichkeit ein. Strahlt man mit polarisiertem Licht ein, so werden von allen Molekülen nur diejenigen angeregt, deren Absorptionsübergangsmoment parallel zum ausgerichteten elektrischen Feldvektor des einfallenden Lichtes steht. Aus dieser Photoselektion, welche die Fluoreszenzintensität im Vergleich zur „normalen“ Fluoreszenz erheblich verringert, resultiert eine partiell polarisierte Fluoreszenzemission, die daraus resultiert, dass diese angeregten Moleküle ihrerseits Licht emittieren, deren elektrischer Feldvektor parallel zu ihrem Emissionsübergangsmoment steht. Zweck der Fluoreszenzspektroskopie Fluoreszenzspektroskopie ist eine sehr leistungsfähige Untersuchungsmethode, um dynamische Eigenschaften der Lösungen von Biomolekülen zu untersuchen. Dies liegt vor allem in der hierfür günstigen Lebensdauer der angeregten Zustände. Innerhalb dieser Zeitspanne können sich viele molekulare Prozesse ereignen, die das Erscheinungsbild des Fluoreszenzspektrums beeinflussen und damit Informationen über eben diese molekulare Prozesse, an denen man ja interessiert ist, geben. Die Parameter, die das Fluoreszenzspektrum beeinflussen sind von allen Prozessen, die innerhalb der Lebensdauer des angeregten Zustands stattfinden, abhängig. So z.B. Kollisionen mit Quenchern, Rotationsdiffusion, Bildung von Komplexen mit Lösungsmittelmolekülen oder anderen gelösten Stoffen sowie der Einfluss der Lösungsmittelrelaxation. Diese Prozesse können Moleküle mit einschließen, die zum Zeitpunkt der Anregung über 100 Å vom Fluorophoren entfernt sind. Rotationsdiffusion der Fluoreszenzanisotropie Wie oben erwähnt kann man durch Einstrahlung von polarisiertem Licht eine polarisierte Lichtemission erzeugen. Die Größe der theoretischen Polarisation, die durch den Winkel zwischen den Übergangsmomenten für Absorption und Emission bestimmt wird, kann im Experiment durch verschiedene Faktoren vermindert werden. Einer dieser Faktoren ist die Rotationsdiffusion. Rotieren die Moleküle in Lösung sehr viel langsamer als die Lebensdauer des angeregten Zustands beträgt, so tritt die Lichtemission schneller ein, als dass die Fluoreszenzanisotropie durch die Rotation merklich beeinflusst werden könnte. Rotieren die Moleküle sehr viel schneller, so mittelt sich der Effekt der Rotation auf das Fluoreszenzspektrum heraus. Ist die Rotationsgeschwindigkeit wie im Fall von Proteinen in wässriger Lösung in der Größenordnung der Fluoreszenzlebensdauern, so ändern sich die Orientierungen der Moleküle, während sie sich im angeregten Zustand befinden und damit auch die Orientierung des Emissionsübergangsmoments. Dadurch wird die gemessene Anisotropie bzw. Polarisation des emittierten Lichtes von der theoretischen (d.h. ohne Rotation) abweichen. Es folgt, dass Messungen von Fluoreszenzanisotropie empfindlich auf alle Faktoren sein werden, welche die Rotationsrate der Fluorophoren beeinflussen. Quenchen der Fluoreszenz durch molekularen Sauerstoff Stößt ein Fluorophor im angeregten Zustand mit einem Sauerstoffmolekül zusammen, so relaxiert das Fluorophor ohne die Emission eines Photons in den Grundzustand. Die durchschnittliche Weglänge, die ein Sauerstoffmolekül innerhalb von 10ns diffundieren kann beträgt nach Einstein Dx2 = 2Dt. Setzt man für den Diffusionskoeffizienten D den Wert für Sauerstoff ein (D = 2,5 10-5 cm2/s) so erhält man Dx = 70 Å. Dies ist mit der Dicke einer biologischen Membran oder dem Durchmesser eines Proteins vergleichbar. Mit Fluorophoren, die eine längere Lebensdauer aufweisen, kann die Sauerstoffdiffusion via gemessene Fluoreszenzintensität über (noch) längere Abschnitte verfolgt werden. Lösungsmittelrelaxation Die Lösungsmittelrelaxation bewirkt in polaren Lösungsmitteln eine Rotverschiebung der Fluoreszenz (unpolare Lösungsmittel verschieben das Fluoreszenzmaximum hin zu kleineren Wellenlängen). Dies geschieht, weil die angeregten elektronischen Zustände eine stärkere Polarisation (höheres Dipolmoment) aufweisen und daher mit den Molekülen des polaren Lösungsmittels wechselwirken (höherer Solvatationsgrad), was zu einer Energieerniedrigung des angeregten Zustandes führt. Diese Energieerniedrigung entspricht einer erheblichen Rotverschiebung (Stokes shift), welche abhängig ist von der Polarisierbarkeit und Viskosität des Lösungsmittels sowie von der Ladungsverteilung im Grund- und ersten angeregten Zustand des Fluorophors. Ist das Lösungsmittel sehr viskos, erfolgt die Lösungsmittelrelaxation nicht während der Lebensdauer des angeregten Zustands (Rotverschiebung wird daher reduziert); normalerweise erfolgt die Lösungsmittelrelaxation in einer Zeit von einer Pikosekunde; Wenn Tryptophane in der hydrophoben Tasche des Proteins liegen, bewirken sie also die Maximalintensität der Fluoreszenz bei einer anderen Wellenlänge als, wenn die Tryptophane an der Proteinoberfläche liegen, wie es im denaturierten Zustand der Fall ist. sind sie der polaren wässrigen Umgebung ausgesetzt, zeigen sie Fluoreszenzmaxima bei ca. 350-353 nm; ist Trp vom wässrigen Medium abgeschirmt zeigen sie eine Blauverschiebung im Fluoreszenzmaximum; So lässt sich der Denaturierungsprozess korrelieren mit der Zunahme der Rotverschiebung bzw. der Verschiebung des Fluoreszenzmaximums. Anders formuliert: die Fluoreszenzintensität bei einer höheren Wellenlänge nimmt zu, während die Fluoreszenzintensität bei einer niedrigeren Wellenlänge abnimmt. Damit lassen sich Aussagen über die Proteinstabilität machen; Fluoreszenz- Resonanz-Energietransfer (FRET) Es ist möglich, dass die Energie des angeregten Zustands eines Fluorophors (Donor-Fluorophor) direkt und strahlungslos (über Dipol-Dipol Wechselwirkung) auf ein anderes Molekül (Akzeptor-Fluorophor) übertragen wird. FRET kann über ein Quenchen (verringern) der Fluoreszenz des ersten Farbstoffes und die Zunahme der Fluoreszenzintensität des zweiten, nicht direkt angeregten Farbstoffes detektiert werden (siehe Abbildung 3: Prinzip der Wechselwirkung Abbildung 3). Damit ein FRET stattfinden kann, zwischen einem Fluorophorenpaar, müssen 3 Kriterien erfüllt werden. Auslösung des FRET 1.) Das Emissionsspektrum des Fluorophors muss dem Absorptionsspektrum des Moleküls entsprechen, das die Energie aufnimmt (Überlappung der Spektren) 2.) Der Abstand r zwischen den beiden Farbstoffen darf nicht größer als 6-8nm sein, da die Effizienz der Energieübertragung antiproportional zu r6 ist. 3.) Donor und Akzeptor müssen parallele elektronische Schwingungsebenen aufweisen (in Lösung aufgrund der Molekülbewegung meist erfüllt) Aufgrund dieser Bedingungen haben sich bestimmte FRET Fluorophorenpaare entwickelt, wie z.B. CFP (Zyan fluoreszierendes Protein) und YFP (gelb fluoreszierendes Protein), dessen überlappendes Spektrum in der folgenden Abbildung 4 gezeigt werden soll. Abbildung 4: grau hinterlegt die Überlappung des CFP Emissionsspektrums mit dem YFPAbsorptionsspektrum Aus der Größe der überlappenden Fläche lässt sich die maximale FRET-Effizienz herleiten. Außerdem hängt die FRET Effizienz vom Förster-Radius R0 und vom Abstand r des Fluorophorenpaars ab. R06 E= 6 R0 + r 6 Der Förster-Radius R0 beschreibt denjenigen Abstand zwischen beiden Fluorophoren, bei dem die Energieübertragung zu 50% erfolgt. Er entspricht für das Paar CFP-YFP 4,9nm und ist lediglich von den eingesetzten Fluorophoren abhängig (bewegt sich immer im nm Bereich): Der Förster Radius wird theoretisch berechnet und dann auf das FRET Experiment angewandt. Auch die Aminosäuren Tyrosin und Tryptophan lassen sich als FRET Fluorophorenpaare nutzen, weshalb Abstände zwischen solchen Aminosäuren innerhalb eines Proteins bestimmbar sind. FRET dient hauptsächlich zum Nachweis von Protein-Protein, ProteinNukleinsäure und Nukleinsäure-Nukleinsäure Wechselwirkungen. Die Interaktion zwischen zwei Proteinen kann beobachtet werden, indem an das erste Protein ein Donor-Fluorophor und an das zweite Protein ein Akzeptor-Fluorophor über Kopplungsreaktionen (meist über Thiobindungen an Cysteine) bindet. So entsteht ein FRET-Signal, dessen Effizienz je nach Abstand der modifizierten Proteine variiert (bewegen sie sich voneinander weg, wird die Effizienz geringer, bewegen sie sich aufeinander zu, wird die Effizienz höher). So kann der Abstand zwischen beiden Proteinen berechnet und über die Zeit Signaltransduktionswege oder Dimerisierungen aufgeklärt werden. Darüber hinaus ist die Vermessung von ProteinKonformationsänderungen möglich, wenn Donor- und Akzeptor-Fluorophor an dasselbe Protein gebunden sind. Die Konzentrationsbestimmung von Nukleinsäuren ist ebenso über FRET möglich. Dabei werden während der PCR (Polymerase Kettenreaktion) mit Donorbzw. Akzeptor-Fluorophoren markierte Oligonukleotide im Überschuss hinzu gegeben. Diese dienen als Hybridsonden, welche während eines PCR Zyklus spezifisch an die DNA binden und somit eine double-stranded Hybrid-DNA erzeugen, bei der sich Donor- und AkzeptorFluorophor sehr nahe kommen. So steigt mit Zunahme der double-stranded Hybrid-DNA Konzentration proportional auch die Fluoreszenz des Akzeptor-Fluorophors (siehe Abbildung 5). Abbildung 5: 1: vor einem PCR Zyklus liegen die Oligonukleotide frei vor; 2: danach sind die Hybridsonden in einer double-stranded DNA eingebaut und bewirken FRET Zur Untersuchung von Protein-Ligand WW dienen FRET, intrinsische Fluorophore und Anisotropie Messungen; Protein-Protein-WW können über Anisotropie Messungen nur schwer beobachtet werden, da Rotation zu langsam; Grün fluoreszierendes Protein (GFP) Das für unseren Versuch verwendete Protein trägt die Bezeichnung GFP. Es wurde aus der Leuchtqualle Aequoria victoria isoliert. Es besteht aus 238 Aminosäuren mit einem Molekulargewicht von 26,9 kDa und stellt somit ein eher kleines Protein dar. Strukturell besteht es aus einem mittels Enzym post-translational gebildetem Chromophor, das durch Zyklisierung und Oxidation dreier Aminosäure Reste entsteht (Ser65-Tyr66-Gly67) und einer β-Fass Struktur, welche sich um das Chromophor herum befindet (siehe Abbildung 6). Abbildung 6: Tertiärstruktur von GFP Das unmodifizierte GFP weist zwei Absorptionsmaxima auf. Eines befindet sich bei 395nm, das andere bei 475nm. Das Fluoreszenzmaximum liegt bei 509nm. Das besondere an GFP ist, dass es sich in Zellen überexprimieren lässt, weshalb es in der Fluoreszenzmikroskopie exakt lokalisiert werden kann. Nun lässt sich GFP mit beliebigen anderen Proteinen Genspezifisch fusionieren. Somit kann eine räumliche und zeitliche Verteilung eines jeden Proteins in einem Organismus über die empfindliche Laser-Fluoreszenzmikroskopie verfolgt werden. Es handelt sich heutzutage um eine Standardmethode der Zellbiologie, vor allem auch deshalb, weil GFP in nahezu allen eukaryotischen Zellen als nicht toxisch einzustufen ist. Auf eine Denaturierung des Proteins, folgt auch eine Zerstörung des Chromophors. Die Folge ist der Verlust der Fähigkeit Fluoreszenz zu zeigen. In der FRET wird GFP ebenso verwendet, wobei hier als Fluorophorenpaar Rhodamin 6G (siehe Abbildung 7) zum Einsatz kommt. Rhodamin 6G weist eine Quantenausbeute von 95% auf, was bedeutet, dass 95% aller absorbierten Photonen und Fluoreszenzphotonen umgewandelt werden. Außerdem ist die Wahrscheinlichkeit eines Interkombinationsprozess Abbildung 7: Struktur von Rhodamin 6G äußerst gering. Rhodamine liegen also selten im nicht fluoreszierenden, langlebigen Triplett-Zustand vor. Kinetik des Denaturierungsprozesses Die Denaturierung eines Proteins beschreibt den Verlust der Tertiärstruktur und der damit verbundenen biologischen Funktion. Sie kann durch Hitze, Säure, Base, Schwermetallionen, Detergentien (=Tenside) und anderen Denaturierungsmittel erfolgen. Die Denaturierung ist, solange nicht abgeschlossen, reversibel. Ist das Protein jedoch vollständig denaturiert, ist es auch evtl. irreversibel zerstört. Die Denaturierung zerstört neben der Tertiärstruktur auch die Sekundärstruktur und die Quartiärstruktur. Die Primärstruktur jedoch bleibt erhalten (keine Spaltung kovalenter Peptidbindungen). Der Denaturierungsprozess verläuft kooperativ (Effekt der Allosterie). Es resultiert somit ein sigmoidaler Zusammenhang zwischen der Intensität des Denaturierungsfaktors (zum Beispiel Konzentration des Detergentiums oder Temperatur) und dem Denaturierungsgrad des Proteins. In der Folgenden Abbildung 8 wird dies über die Veränderung der relativen Absorption bei 260nm, die wie später genauer erläutert wird mit dem Denaturierungsgrad direkt proportional korreliert, über ein Temperaturintervall verdeutlicht. Abbildung 8: sigmoidaler Zusammenhang zwischen der Intensität des Denaturierungsfaktors (hier Temperatur) und Denaturierungsgrad (hier indirekt über die relative Absorption bei 260nm dargestellt) Bekannte und häufig in der Biochemie verwendete Denaturierungsmittel sind Harnstoff und SDS (Natriumdodecylsulfat). In unserem Versuch findet Guanidiniumhydrochlorid (GuHCl) als Denaturierungsmittel seine Anwendung. Vorteile und Anwendung der Fluoreszenzspektroskopie (Zusammenfassung und Ergänzung) Fluoreszenzspektroskopie ist die wichtigste Spektroskopie Methode in der Biochemie. Sie erlaubt die Unersuchung von Einzelmolekülen und sehr kleinen Mengen Probe sowohl in vitro als auch in vivo. Es können thermodynamische (Bindungskonstanten, Faltungsgleichgewichte), kinetische (Faltungskinetik) und dynamische (Rotationskorrelationszeit) Parameter gemessen werden. Wie bereits beschrieben, können über FRET Abstände von Proteinen gemessen werden und somit Signaltransduktionswege und Dimerisierungen aufgeklärt werden. Darüber hinaus können Konformations-Veränderungen und Entfaltungsprozesse beobachtet werden. In der FCS (Fluoreszenz-Korrelations-Spektroskopie) können Diffusionskonstanten, Konzentrationen und Bindungen zwischen verschiedenen diffundierenden Spezies gemessen werden. Außerdem ist eine Einzelmolekülfluoreszenzspektroskopie möglich. Hier werden einzelne Moleküle sichtbar gemacht, indem mittels eines Konfokalmikroskopes die Größe des Detektionsvolumens auf unter einen Femtoliter begrenzt wird. Dadurch befindet sich bei geeigneter Verdünnung im Durchschnitt weniger als ein fluoreszenzaktives Teilchen im Detektionsvolumen. So können emittierte Photonen einem bestimmten Teilchen zugeordnet werden und charakterisieren somit die Eigenschaften des Teilchens, welche in der Analyse einer Probe verborgen bleiben würden. In einer Probe können evtl. nur wenige Einzelmoleküle den Hauptteil der gemessenen Fluoreszenzintensität ausmachen, während die anderen nur schwach fluoreszieren. Docking Computerchemie: Das strukturbasierte Design von Liganden Durch die genaue Kenntnis über die dreidimensionale Struktur von Proteinen, die durch die Aminosäuresequenz determiniert ist, und über ihre Wirkungsweise (geometrisch und chemisch komplementäre Anlagerung kleiner Moleküle, im folgenden als Liganden bezeichnet, an biologische makromolekulare Zielstrukturen, häufig Proteine, im folgenden als Rezeptoren bezeichnet, bewirkt einen Eingriff in die Abfolge von Stoffwechsel- und Signaltransduktionsprozessen und löst somit einen physiologischen Effekt aus) 1 ist es der modernen Wissenschaft möglich gezielter Wirkstoffe herzustellen. Früher wurden große Mengen an Substanzen willkürlich auf ihre Wirksamkeit getestet. Diesen Prozess, dessen Anwendung selbst heutzutage teilweise noch unverzichtbar ist, wird als Screening (häufig: HTS = Hochdurchsatz-Screening) bezeichnet. Heute ist bekannt, dass die Wirksamkeit bestimmter Stoffe auf ihre Inhibitions-Wirkung auf bestimmte Proteine zurückzuführen ist. Da außerdem bekannt ist, dass der Inhibitor exakt in die dreidimensionale Struktur des zu inhibitierenden Enzyms passen muss (Modell des Schlüssel-Schloss Prinzips bzw. des induced fits), lassen sich, wenn die Enzym-Struktur bereits aufgeklärt wurde, einige wenige ideale Inhibitoren am Computer designen, welche im Anschluss synthetisiert und im Experiment getestet werden. Der Materialverbrauch ist demnach erheblich geringer. Die Zukunft des Experimentierens wird also stark in silico liegen, wobei in vitro und in vivo Experimente den in silico Experimenten nur noch in reduzierter Form folgen. Diese in silico Experimente werden als strukturbasiertes Liganden-Design bezeichnet (im Gegensatz zu Wirkstoff-Design, da bei jenem außerdem die Optimierung von Faktoren wie Absorption, Verteilung, metabolische Stabilität, Ausscheidung [zusammen: ADME Eigenschaften] Lösungsmittelverhalten, toxikologisches Profil, pharmazeutische Eigenschaften und Synthesekosten u.a. eine Rolle spielen), da sie lediglich die Stärke eines Liganden in einem in vitro Experiment optimieren sollen. So können sich auch Liganden, die sich im strukturbasierten Design und im Folgenden in vitro Experiment als ideal erwiesen haben, aufgrund von geringer Bioverfügbarkeit und metabolischer Instabilität als nicht optimales Medikament herausstellen (wie der HIV-Protease Inhibitor DMP323). Der darauf folgend entwickelte Wirkstoff DMP450 zeigt eine hohe Plasmaproteinbindunng (hängt von Lipophilie und Ladung ab), welche die Wirkung der Liganden in vivo erheblich herabsetzt. Grob zusammengefasst ist der Weg zu einem idealen Wirkstoff durch mehrere Abschnitte charakterisiert, die im Folgenden noch einmal graphisch zusammengefasst werden sollen. Abbildung 1 Überblick über das Wirkstoff Design Das HTS kann zum Teil durch strukturbasiertes Liganden Design (in silico) ersetzt oder kombiniert werden, um den Arbeitsaufwand zu minimieren. Die Strukturaufklärung erfolgt mittels NMR oder Röntgenstrukturanalyse. Die Testung entspricht den in vitro Experimenten. Ist diese erfolgreich kommt die Voraussetzung für in silico Experimente: Strukturaufklärung Um in silico Experimente durchführen zu können, müssen die Strukturen der jeweiligen Moleküle und Enzyme exakt bekannt sein. Dabei spielt die Konformation der Atome im Molekül, sowie die Elektronendichte eine große Rolle. Solche Informationen werden durch röntgenographische Methoden (z.B. Protein-Ligand-Kristallographie), KryoElektronenmikroskopie und auch eingeschränkt (aufgrund der Größe von Proteinen ist mehrfache Isotopenmarkierung notwendig) durch NMR-spektroskopische Methoden ermittelt, welche Substanzbibliotheken durchmustern (Screening) und somit günstige Templates identifizieren, die anschließend in silico optimiert werden können, so dass die Bindungsstärke des Liganden maximal wird. Jene kann nun über kompetetive Inhibierungsoder Bindungsassays in vitro gemessen werden, was das in silico Experiment verifiziert und falsifiziert. Die Kristallographie spielt bei der Strukturaufklärung von Proteinen und Protein-Ligand Komplexen eine erhebliche Rolle, weshalb nun auf dieses Verfahren näher eingegangen werden soll. Die Züchtung des Kristalls eines Rezeptor-Substrat Komplexes ist leider nicht möglich, da das Substrat sehr schnell wieder frei gegeben wird. Daher sind Inhibitoren als Liganden dieser Rezeptoren nicht wegzudenken, um kristallographische Untersuchungen an solchen Komplexen durchführen zu können. Ist das zu untersuchende Protein (bzw. der Protein-Ligand Komplex) kristallisiert, folgt das eigentliche kristallographische Röntgenbeugungsexperiment, welches die Positionen und Intensitäten der im Beugungsmuster des Kristalls gemessenen Beugungen liefert. Nachdem die Strukturfaktoramplituden mittels Quadratwurzel der Intensitäten berechnet wurden und die Phasen der Strukturfaktoren bekannt sind, kann mittels Fouriertransformation eine dreidimensionale Matrix aus Zahlen, welche den lokalen Elektronendichten entsprechen, berechnet werden. Diese Matrix wird nun anhand eines bestimmten Atommodels mittels Kristallograph in eine Struktur übersetzt (große Elektronendichten bedeuten schwere Atome). Dieser Vorgang ist iterativ (ein Teil der Struktur wird aufgebaut und anschließend durch Veränderung der Atomkoordinaten mittels Verfeinerungsprogramm verbessert) und durch mehrere Randparameter eingeschränkt. Dieser iterative Prozess ist notwendig, da durch Röntgenstrahlung nur schwer zwischen Elementen ähnlicher Ordnungszahl unterschieden werden kann und daher manche Gruppen ein fast identisches Signal liefern. Die resultierende Struktur liefert nun ihrerseits eine Matrix-Karte aus Elektronendichten, in der weitere Einzelheiten erkannt werden können. Neben den Koordinaten weisen die Atome auch einen Temperaturfaktor auf, der die Fehlerordnung im Kristall charakterisiert. Er liefert demnach einen Hinweis auf die Verlässlichkeit des entsprechenden Modellteils. Ist er hoch, bedeutet dies, dass nur eine geringe Elektronendichte gemessen wurde und die Koordinaten daher ein hohes Fehlerintervall aufweisen. Die Auflösung von solchen Matrix-Karten liegt selten unterhalb von 1,5 Å, meist bei 2-3 Å. Erst ab einer Auflösung von 1,2 Å, kann die Position polarer H-Atome bestimmt werden. Der Fehler korreliert wie bereits erwähnt mit dem Temperaturfaktor, aber auch direkt mit der Auflösung. Bei einer Auflösung von 2,5 Å liegt der Fehler bei ca. 0,4 Å. Ein Temperaturfaktor von 20 Å2 bedeutet eine mittlere Verschiebung aus der Gleichgewichtsposition von 0,5 Å. Eine solche resultierende verbesserte kristallographische Matrix wird nun in so genannten PDB Dateien gespeichert, auf die ich später noch zu sprechen kommen werde (Einleitung Abschnitt 7). Qualität der Protein-Ligand Bindung Ob ein Ligand nun exakt in die Bindungstasche des Proteins passt, ist noch nicht ausschlaggebend für eine ideale Protein-Ligand Bindung. Die sterische Kompatibilität ist vielmehr Grundvorrausetzung für die Bindung. Hinzu kommt die energetische Erkennung zwischen den Partnern. Betrachtet wird im Folgenden die nicht-kovalente Assoziation eines Liganden an seinen Rezeptor. Raq + Laq ↔ RLaq Die Bindungsaffinität selbst ist durch die Bindungskonstante Ki zugänglich und setzt sich aus entropischen und enthalpischen Beträgen zusammen. Beide können die Ligandenbindung antreiben, wie Messungen an unterschiedlichen Protein-Ligand-Wechselwirkungen durch ITC bestätigt haben. Die Bindungskonstante entspricht der Dissoziationskonstante, welche gleich dem reziprokem Wert der Assoziationskonstante ist. KA = 1 1 [ RL] = = K D K I [ R ] ⋅ [ L] ∆G = − RT ln K A = ∆H 0 − T∆S Die Dissoziationskonstante K für die Ligandenbindung an ein Protein liegt in einem Bereich von 10-2 M (niederaffin: ΔG=-10kJ/mol) bis 10-12 M (hochaffin: ΔG=-70 kJ/mol) bei einer Temperatur von 298 K. Wodurch diese unterschiede in der Bindungsaffinität zustande kommen, soll im folgenden Abschnitt 4 behandelt werden. Wechselwirkungen zwischen Ligand und Protein enthalpische und entropische Beiträge Bei der nicht-kovalenten Bindung eines Liganden an seinen Rezeptor spielen elektrostatische (= Coulomb-) und Van-der-Waals Wechselwirkungen für die enthalpischen Beiträge zur Bindungsaffinität eine große Rolle. Entropische Beiträge liefern Solvatisierungseffekte des Lösungsmittels und Entstehung oder Verlust neuer Freiheitsgrade im Liganden oder Rezeptor. Außerdem können durch die Bindung induzierte konformelle Strukturveränderungen in Rezeptor und/oder Ligand ausschlaggebend für die Bindungsaffinität sein. kovalente Bindungen Wie so eben beschrieben, sollen nur nicht kovalente Wechselwirkungen zwischen Liganden und Rezeptoren betrachtet werden. Um einen Vergleich der Bindungsenergien und -längen zu ermöglichen, möchte ich hier dennoch auf die kovalenten Bindungen eingehen. Eine C-C Einfachbindung hat einen Energiegehalt von 340kJ/mol. Eine C=C Doppelbindung hingegen weist einen im Vergleich zum doppelten Energiegehalt einer C-C Einfachbindung geringeren Energiewert auf (ca. 610kJ/mol), da es zu einer Verzerrung der Elektronendichten kommt und sich deshalb die π-Orbitale nicht so gut wie die σ-Orbitale überlappen. Bei der CΞC Dreifachbindung resultiert somit ein noch geringerer Energiewert von ca. 800 kJ/mol. Die CC Einfachbindung ist 154 pm lang, die C=C Doppelbindung ist mit 133 pm wesentlich kürzer, da wegen der Orbital- Überlappung die Atome dichter liegen müssen. Eine C-H Bindung ist ca. 110 pm lang. Am stabilsten sind C-C Bindungen bei einem Winkel von 120°. Die Bedeutung der Wasserstoffbrücke Wasserstoffbrücken kommen durch die anziehende Wechselwirkung zwischen einem an ein elektronegativeres Atom gebundenes Wasserstoffatom und einem weiteren elektronegativem Atom (kann auch ein π-Elektronensystem sein) zustande. Die Länge der Wasserstoffbrücke liegt zwischen 1,7 (in reinem HF) und 3,2A (teilweise in Proteinen). Sie ist für einen Winkel zwischen den drei Punkten X~H-X (X ist elektronegatives Atom; - ist kovalente Bindung; ~ ist H-Brücke) von 180° am stabilsten, kann sich allerdings bis zu einem Winkel von 135° ausbilden. Das Coulomb’sche Gesetz findet bei der Beschreibung aller elektrostatischen Wechselwirkungen seine Anwendung und ist in folgender Gleichung dargestellt. V = 1 4π ⋅ ε 0 ⋅ ε M ⋅ q1 ⋅ q 2 r2 V ist das Potential mit dem sich die Teilchen anziehen, q sind die Ladungen der Teilchen und r ist der Abstand. ε 0 ist die Permittivität (= dielektrische Leitfähigkeit) des Vakuums, welche die Abschirmung elektrostatischer Wechselwirkungen beschreibt. In einem Medium ist die elektrostatische Bindungsstärke über ε 0 hinaus zusätzlich von der Dielektrizitätskonstanten ε M des umgebenden Mediums abhängig. Im Vakuum gilt ε M = 1 . Hohe Werte für ε M (etwa 80) sind an der Grenzfläche Protein-Lösungsmittel zu finden, weshalb hier die Wechselwirkung wesentlich geringer ist, als im Inneren des Proteins ( ε M Werte von 2-8 sind hier häufig). In polarer Umgebung sind Wasserstoffbrücken also schwächer, als in apolarer. Bei der Protein-Ligand Bindung müssen bereits bestehende H-Brücken des freien Proteins aufgebrochen werden, wobei neue H-Brücken zwischen Protein und Ligand entstehen. Ob der H-Brücken Austausch zur Bindungsaffinität beiträgt, hängt von den Enthalpien der HBrücken im freien Protein und im Protein-Ligand-Komplex ab. Eine Wechselwirkung zwischen einem Wasserstoffatom und einem elektronegativeren Partner in apolarer Umgebung liefert einen enthalpischen Beitrag von ca. ∆G = (−5 ± 2,5)kJ / mol . Die Energie, die aufgebracht werden müsste um Wasserstoffbrücken von H2O in reinem Wasser zu spalten, beträgt ∆G = (17,5 ± 2,5)kJ / mol . Im Allgemeinen sind die Energien der Wasserstoffbrückenbindungen von sehr vielen Faktoren abhängig (Länge der Bindung, Bindungswinkel, stärke der Polarisation des H-Atoms bzw. Differenz der Elektronegativitäten zwischen Wasserstoff und seinem elektronegativem Partner), weshalb jene zwischen 1 und 50 kJ/mol schwanken können. Salzbrücken und spezielle H-Brücken Die geladenen proteinogenen Aminosäuren weisen bei einem physiologischen pH-Wert von 7,4 die unterschiedlichsten Protonierungsgrade auf. Arginin und Lysin sind protoniert, während Asparagin- und Glutaminsäure deprotoniert vorliegen. Der Protonierungsgrad kann je nach lokaler elektrostatischer Veränderung in der Umgebung variieren. Sind Reste unterschiedlicher Ladung in direkter Reichweite, kommt es zur Ausbildung von Salzbrücken. Salzbrücken können auch spezielle Wasserstoffbrücken sein (siehe Abbildung). Die enthalpischen Beiträge von ladungsunterstützten Wasserstoffbrücken und Salzbrücken betragen ca. ∆G = (−15 ± 5)kJ / mol . Diese sind nicht zu verwechseln mit den Wasserstoffbrücken in polarer Umgebung, die wesentlich schwächer sind. Bei ladungsunterstützten H-Brücken liegen jeweils zwei unterschiedlich geladene Reste in einer sonst apolaren Umgebung vor, die miteinander wechselwirken. In polarer Umgebung würde diese Wechselwirkung erheblich abgeschwächt, da die Wassermoleküle ebenso mit den geladenen Resten wechselwirken (ständiger Umbau der H-Brücken). Beispiele für ladungsunterstützte H-Brücken sind in folgender Abbildung zu sehen. Abbildung 2 Ladungsunterstützte H-Brücken Es werden grob 2 Typen von H-Brücken unterschieden: solche in polarer Umgebung und solche in apolarer Umgebung. Letztere sind wesentlich stärker. Kommt bei H-Brücken in apolarer Umgebung hinzu, dass sie durch eine Ladung am Nachbaratom verstärkt werden, so In einem Salz-Kristall sind die Bindungsenergien wesentlich höher. Die NaCl Bindung im Kristall ist 250kJ/mol stark. In Lösung wird sie erheblich abgeschwächt, da die Wassermoleküle die Ladungen der Gegenionen voneinander abschirmen. weitere elektrostatische Wechselwirkungen So genannte π-π-Wechselwirkungen zwischen aromatischen Systemen in Ligand und den Seitenketten der aromatischen Aminosäuren des Proteins liefern ebenso einen bedeutenden Beitrag zur Affinität des Liganden an seinen Rezeptor. Außerdem sind die Wechselwirkungen von Kation und π-Elektronen-System nicht zu vergessen. Kation-πSysteme liefern einen um 10kJ/mol größeren Beitrag zur Bindungsaffinität als normale Salzbrücken. Außerdem können koordinative Bindungen funktioneller Gruppen von Liganden an Metallionen des Proteins die Affinität erheblich erhöhen. Van-der-Waals Wechselwirkung Die Van-der-Waals-Kräfte tauchen zwischen allen Molekülen auf und können durch das Lennard-Jones-Potential beschrieben werden. Es erklärt die intermolekularen Wechselwirkung zweier ungeladener nicht-kovalent verbundener Teilchen. Dabei spielen ein anziehender Faktor und ein abstoßender Faktor eine Rolle. Somit lässt sich das LennardJones-Potential in zwei Anteile gliedern. Der anziehende Anteil entspricht V =− C r6 In dieser Gleichung ist V das Potential der Wechselwirkung und r der Abstand zwischen den Teilchen. C entspricht einem externen Term, welcher stoffspezifische Konstanten (u.a. die Ionisierungsenergie) enthält. Der abstoßende Teil lässt sich durch V = C r 12 beschreiben. Es resultiert das so genannte Lennard-Jones-(12,6)-Potential, das nun wie folgt aus diesen beiden zusammengesetzten Gleichungen beschrieben werden kann σ 12 σ 6 V (r ) = 4ε ⋅ − r r Das Potentialminimum der resultierenden Kurve ist ε . Die Nullstelle (für V=0) der Kurve ist bei einem Abstand r = σ abzulesen. σ wird auch als Van-der-Waals Radius definiert. Es existiert auch die folgende Schreibweise für das Lennard-Jones-(12,6)-Potential: reqm V (r ) = ε ⋅ r 12 r − 2 ⋅ eqm r 6 Hier ist reqm = 21 / 6 σ der Abstand des Energieminimums vom Ursprung. Dies soll die folgende Abbildung noch einmal veranschaulichen. Abbildung 3 Das Lennard-Jones-Potential Die Abbildung zeigt die zwei Komponenten des Lennard-Jones-Potentials in einer unterbrochenen Linie und die resultierende Summe aus beiden Komponenten. Das Minimum ist bei V (r ) = ε abzulesen. Dieses Potentialminimum ist bei einem idealen Abstand reqm = 21 / 6 σ vorzufinden. Die Nullstelle ist σ und entspricht gleichzeitig den Van-der-Waals Radien. Die Van-der-Waals Wechselwirkung trägt mit einer Energie von ca. ∆G = (−2,5 ± 2)kJ / mol zur Bindungsaffinität bei. Aufgrund der hohen Anzahl mit der diese Wechselwirkung auftritt spielt diese eine bedeutende Rolle bei der Rezeptor-Ligand-Bindung. Der anziehende Term Die Ursache des anziehenden Terms des Lennard-Jones-Potentials sind die 3 verschiedenen Van-der-Waals Kräfte, und zwar die Keesom-Wechselwirkung zwischen zwei Dipolen, die Debye-Wechselwirkung zwischen einem Dipol und einem polarisierbaren Molekül und die Londonsche Dispersionswechselwirkung. Ursache ist jeweils das Auftreten eines fluktuierenden Dipolmomentes. Dazu kommt es, weil die Elektronen um einen Atomkern herum ständig in Bewegung sind, weshalb sich die Ladungsverteilung mit der Zeit ändert. Wird der Schwerpunkt der negativen Ladung vom Schwerpunkt der positiven Ladung, der sich immer im Kern befindet getrennt, so liegt ein Dipol vor. Wenn sich nun zwei Moleküle mit einem solchen temporären Dipol nahe genug kommen, können sie miteinander elektrostatisch wechselwirken. Dabei induziert die negativ polarisierte Seite des Teilchens 1 eine Abstoßung der Elektronen der zugewandten Seite des Teilchens 2. Der Minuspol eines temporären Dipols influiert (von Influenz = Ladungstrennung innerhalb eines ungeladenen Leiters durch ein elektrisches Feld) also beim Nachbarmolekül einen Pluspol. Es kommt zur Van-der-Waals Wechselwirkung zwischen dem temporärem Dipol des Teilchens 1 und dem influierten Dipol des Teilchens 2 durch Synchronisation der Elektronenbewegungen beider Teilchen. Der abstoßende Term Elektronen, die zu den Fermionen gehören, stoßen sich aufgrund der gleichen Ladung ab. Daher gilt je näher sich zwei Atome kommen, desto mehr überlappen sich ihre Orbitale und desto energetisch ungünstiger wird der Zustand. Es lässt sich berechnen, dass bei antisymmetrischen Wellenfunktionen die Wahrscheinlichkeit beide Elektronen am selben Ort vorzufinden null ist (im Gegensatz zu symmetrischen Wellenfunktionen). Außerdem ist der durchschnittliche Abstand beider Teilchen antisymmetrischer Wellenfunktionen größer als mit symmetrischen Wellenfunktionen. Ergo ist bei gleichsinnig geladenen Teilchen eine antisymmetrische Wellenfunktion erheblich günstiger. Das Pauli Prinzip fasst dies in einem Postulat zusammen: „Wellenfunktionen, die ein Mehrelektronensystem beschreiben, müssen beim Austausch von zwei beliebigen Elektronen das Vorzeichen wechseln. Solche Wellenfunktionen sind de facto antisymmetrisch.“ Wenn die Wellenfunktionen der überlappenden Elektronen-Orbitale antisymmetrisch sind, so liegt automatisch eine parallele Spinkonstellation vor. Fermionen (Teilchen mit halbzahligem Spin), die denselben Raum belegen, dürfen jedoch nicht in allen vier Quantenzahlen übereinstimmen (Pauli-Verbot, welches sich direkt aus dem Pauli Prinzip ableiten lässt). Daher ist die Überlappung der Orbitale mit Elektronen gleichen Spins quantenmechanisch verboten, weshalb es zur sehr starken Pauli Repulsion zwischen Atomen mit geringen Abständen zueinander kommt. Solvatisierungseffekte Bei der Betrachtung der entropischen Beiträge zur Qualität der Protein-Ligand Wechselwirkung spielen die Lösungsmittelmoleküle eine entscheidende Rolle. Diese bewirken Solvatisierungs- und Desolvatisierungsprozesse, im Falle von Wasser (De-)Hydratisierungsprozesse. Wassermoleküle bilden in reiner Phase des flüssigen Zustands ein Netzwerk aus H-Brücken-Donoren und -Akzeptoren. Dabei bildet ein Wassermolekül 3-4 H-Brücken aus. Befindet sich ein Stoff, z.B. ein Protein in wässriger Lösung, so bildet sich eine Hydrathülle. Die Wassermoleküle dieser Hülle wechselwirken mit dem Protein, anstatt untereinander H-Brücken auszubilden. Im Allgemeinen kommt es bei der Hydratisierung einer Substanz zu einer Verringerung der Anzahl an H-Brücken. Die resultierenden H-Brücken (z.B. der Solvatationssphäre von Argon: ∆G = 2,6kJ/mol ) sind jedoch stärker als diejenigen in der reinen Wasserphase ( ∆G = 2kJ/mol ). Daher kompensiert sich der enthalpische Beitrag nahezu zu null (wenige starke statt viele schwache H-Brücken). Da die Wassermoleküle der Hydrathülle in ihrer Bewegungsfreiheit erheblich eingeschränkt sind, erniedrigt sich jedoch die Entropie (höhere Ordnung). Wenn nun ein Ligand eine hydrophobe Oberfläche des Proteins bedeckt, werden Solvensmoleküle freigesetzt. Die Protein-Ligand-Bindung wird also entropisch getrieben. Dies wird auch als Hydrophober Effekt beschrieben, der u.a. auch ausschlaggebend für die Proteinfaltung und die Ausbildung von Biomembranen ist. Da der Beitrag des hydrophoben Effekts zur freien Enthalpie ΔG proportional zu der begrabenen hydrophoben Fläche ist, lässt sich dieser Wert auf ca. ∆G = (−0,175 ± 0,075)kJ/molA 2 quantifizieren. Nicht zu verwechseln mit diesem entropischen Beitrag ist der Verbrückungs-Effekt, den Wassermoleküle der Hydrathülle um das aktive Zentrum herum aufweisen können. Dieser ist enthalpischer Natur. Die Wassermoleküle im unbesetzten aktiven Zentrum verbrücken Protein und Ligand miteinander (solvensvermittelte Wechselwirkung), bevor es zu einer Bindung zum Protein-Ligand-Komplex kommt. Die Überführung eines Wassermoleküls aus der umgebenden Wasserphase in das Bindungsepitop (Rezeptor-Wasser-Ligand Komplex) setzt sich aus dem entropischen ungünstigen Betrag von - T∆S = 9kJ/mol bei 298K und dem enthalpisch günstigen Betrag von ∆H = −16kJ/mol zusammen. Die solvensvermittelte Wechselwirkung zwischen Protein-Wasser und Wasser-Ligand liefert also einen energetischen Beitrag von ∆G = −7 kJ/mol zur Bindungsaffinität. Die Wassermoleküle des Bindungsepitops dienen darüber hinaus als Mediatoren zur Anpassung an die unterschiedlichen Ligand-Strukturen, während die Proteinstruktur nahezu, aber nicht absolut (wie in Abschnitt 4.8 gezeigt) unverändert bleibt. Mobilität und Beweglichkeit durch Freiheitsgrade Bei der Bindung von Ligand und Rezeptor gehen immer auch 3 Freiheitsgrade der Translation und 3 der Rotation verloren, wodurch 6 neue Vibrationsfreiheitsgrade entstehen. Unter Anwendung der Trouton’schen Regeln resultiert, dass eine solche Einschränkung ein Entropieverlust von ∆S = −420J/molK bedeutet. Die Praxis zeigt jedoch, dass es sogar möglich ist, dass Liganden ihre Mobilität im gebundenen Zustand beibehalten und durch zusätzliche Proteinbewegung die Ligand-Bindung sogar entropisch gefördert wird (in diesem Fall sind keine elektrostatischen Wechselwirkungen möglich, da der Ligand innerhalb der Bindungstasche rotiert und ist eine Selektivität bei der katalysierten Reaktion nahezu auszuschließen). Torsionsfreiheitsgrade, welche nicht zu den 3n Freiheitsgraden eines n-atomigen Moleküls zählen, spielen eine große Rolle bei der Betrachtung der Bindungsaffinität. Jede frei drehbare Bindung weist einen solchen Freiheitsgrad auf. Bei der Bindung geht dieser verloren. Es muss also erreicht werden, dass diese Torsionsfreiheitsgrade von ideal bindenden Inhibitoren schon in Lösung eingeschränkt werden (Doppelbindungen, Ringsysteme, sterische Hinderung). intramolekulare Konformationsänderungen Um eine ideale Bindung eines Inhibitors zu ermöglichen, wird jener so designt, dass er bereits in Lösung in derjenigen Konformation vorliegt, in der er auch an den Rezeptor bindet. So wird durch die Bindung keine neue, energetisch ungünstigere Konformation erzwungen, die eine größere Spannung aufweist und somit die Bindungsaffinität reduzieren würde. Neben der Konformation des Inhibitors spielen auch Strukturveränderungen des Rezeptors eine Rolle. Obwohl früher geglaubt wurde, dass Proteine starr sind (Hypothese des starren Rezeptors), hat sich dies als großer Irrtum herausgestellt. Die Dynamik von Protein-Ligand Wechselwirkungen wurde bei der Analyse von Proteinen (z.B. HIV-Revertase) entdeckt, welche mehrere strukturell unterschiedliche Liganden (Nevirapin ist Inhibitor, der sich strukturell sehr von den Substraten α-APA und HEPT unterscheidet) aufweisen. Abbildung 4 Substrate der HIV-Revertase Die drei Strukturen, welche an das Enzym HIV-Revertase am selben aktiven Die Bindung aller dieser drei Liganden ans aktive Zentrum erfolgt durch unterschiedliche konformelle Veränderungen, sowohl des Liganden als auch des Proteins, sodass immer dieselben drei Aminosäurereste hydrophob mit den Liganden wechselwirken können. So werden die Cα Positionen in diesem Fall um bis zu 2,7 Å verschoben. Die elektrostatischen Wechselwirkungen variieren hingegen stark. Dies ist ein Hinweis darauf, dass hydrophobe Wechselwirkungen für die sterische Komplementarität, die elektrostatischen hauptsächlich für die Erkennung aus größerer Entfernung verantwortlich sind. Das Enzym DFPase Im Praktikumsversuch wird ein Inhibitor mittels strukturbasiertem Designverfahren für das Enzym DFPase entwickelt. Die DFPase kommt unter anderem in Kopfganglien und dem Riesenaxon (Axone der Kalmaren, die in der Regel 100-1000mal größer sind als bei Mammalia) vor und besteht aus 314 Aminosäuren mit einem Molekulargewicht von 35 kDa. Es wurde aus loligo vulgaris extrahiert, einem häufig vorkommenden Kalmar. Die Struktur der DFPase Die DFPase ist ein globuläres Protein mit einem in einer mittels Oberflächenstrukturmodellierten Abbildung gut sichtbarem aktivem Zentrum (siehe Abbildung). In der nativen Form enthält die DFPase zwei Calciumionen, wobei eines für die strukturelle Stabilität des Enzyms, das andere für die katalytische Aktivität benötigt wird. Die Oberflächenstruktur des aktiven Zentrums zeigt, dass die Struktur des Enzyms sehr gut mit der Struktur des Substrats wechselwirken kann, weshalb es zu einer relativ hohen Affinität zum Substrat kommt. Abbildung 5 Oberflächenstruktur des aktiven Zentrums der DFPase mit gedocktem DFP Die Oberflächenstruktur zeigt die äußersten Grenzen der Orbitale der einzelnen Atome des Enzyms. Die verschiedenen Farben kennzeichnen die Atomart, zu dem die Orbitale gehören (rot: Sauerstoff, cyan: Wasserstoff, blau: Stickstoff, grau: Kohlenstoff, gelb: Schwefel, grün: Kalzium) Die Sekundärstruktur des Enzyms entspricht sechs antiparallelen β Faltblättern, die zusammen einen sechsblättrigen Β-Propeller bilden. Abbildung 6 Sekundärelemente der DFPase, Aufsicht Es sind die zwei Calcium Ionen im Zentrum des Enzyms und die i ll l β F l bl k Nervenkampfstoffe: Die Substrate der DFPase Die Funktion und der Reaktionsmechanismus des Enzyms sind noch nicht vollständig aufgeklärt, wohl aber ist bekannt, welche Substrate umgesetzt werden können. Die DFPase spaltet Organophosphat-Verbindungen wie unter anderem DFP (Diisopropylfluorophosphat), aber auch Tabun, Sarin, Soman und Cyclohexylsarin (siehe Abbildung 7). Viele dieser Organophosphat-verbindungen gehören zur Gruppe der Ultragifte, die als Nervenkampfstoffe verwendet werden. Zur Anwendung kommen diese als Aerosol, es handelt sich nicht um Gase. Abbildung 7 Substrate der DFPase Außer DFP gehören die in dieser Abbildung aufgelisteten Substrate alle zur Klasse der Nervenkampfstoffe .Es h d l i h b i ll S b Nervenkampfstoffe sind Inhibitoren der Acetylcholinesterase, die sich im synaptischen Spalt befindet. Die Übertragung eines ankommenden Nervenreizes von einem präsynaptischen Neuron zu einem postsynaptischen Neuron erfolgt durch Ausschüttung des Neurotransmitters Acetylcholin (ACh) aus dem Synapsenendknöpfchen mittels synaptischer Vesikel des präsynaptischen Axons und durch die Bindung von ACh an Rezeptoren der postsynaptischen Membran. Diese Bindung bewirkt die Liganden-abhängige Öffnung der Ionenkanäle des postsynaptischen Neurons, wodurch es zur Änderung des Membranpotentials kommt. Der Nervenreiz wurde über die Synapse weitergeleitet. Das Enzym Acetylcholinesterase spaltet nun ACh in Acetat und Cholin, sodass der Nervenimpuls ca. 1ms anhält. Diese Moleküle werden wieder vom Synapsenendknöpfchen phagocytiert. Wird die Acetylcholinesterase durch Substrate der DFPase blockiert wird ACh nicht mehr gespalten. Da es in dieser Form jedoch nicht zurück ins Synapsenendknöpfchen transportiert werden kann, bleibt es im synaptischen Spalt und bewirkt durch die dauerhafte Bindung an den Rezeptor der postsynaptischen Membran eine Dauerreizung des postsynaptischen Neurons. Diese Dauerreizung bewirkt den Verlust der Kontrolle über sämtliche motorischen Muskelfunktionen und führt letztlich zum Tod durch Atemversagen. Zum Herzstillstand kommt es nicht, da das Herz andere Neurotransmitter hat und somit nicht ausschließlich von der Aktivität der Acetylcholinesterase abhängig ist. Außerdem ist der Herzmuskel aufgrund einer doppelt so langen Refraktärphase (20ms anstatt 10ms im Skelettmuskel) nicht tetanisierbar (kann nicht krampfen), da die Kontraktion des Herzmuskels noch vor dem Ende der Refraktärphase beendet ist. Der Reaktionsmechanismus der DFPase Reaktion Das Enzym spaltet eine Reihe von Organophosphat-Verbindungen durch Hydrolyse. Der exakte Mechanismus ist noch nicht verstanden, jedoch sind einige Prinzipien der SubstratUmsetzung bereits deutlich geworden. Das für die katalytische Aktivität benötigte Calcium Ion (positiv geladen) der Bindungstasche koordiniert den an das Phosphoratom gebundene Sauerstoff (negativ geladen) des Substrats. Dadurch kommt es zur Polarisierung innerhalb des Substrats (die Elektronen werden zum Sauerstoff hin gezogen, wodurch das Phosphoratom partial positiv geladen wird). Nun kann ein Wassermolekül bzw. ein OH- Ion nukleophil an das Phosphat angreifen. Dieser Angriff resultiert in einem fünffach koordinierten Übergangszustand, aus dem letztlich die elektronegative Gruppe (meist Fluor) entlassen wird. Abbildung 8 Reaktionsgleichung der DFPase-Reaktion Diese Reaktionsgleichung zeigt die Hydrolyse von DFP. Auto Dock 4.0 Unser in silico Experiment wird mit dem Programm Auto Dock 4.0 durchgeführt. Im Folgenden soll die Arbeitsweise von diesem Programm genauer betrachtet werden. Berechnung der Bindungsenergie Das Programm liefert als Ergebnis eines Docking Experiments die Bindungsenergie ΔG, welche sich aus mehreren Komponenten zusammensetzt. Gemessen werden zunächst die Wechselwirkungen zwischen Rezeptor und Ligand (Van-der-Waals, H-Brücken und elektrostatische Wechselwirkungen). Bei der Bindung des Liganden an seinen Rezeptor, der in Lösung in seiner günstigsten Konformation vorliegt, kann eine Konformationsänderung auftreten, dessen ΔG ebenso berechnet wird. Außerdem werden die Anzahl der Torsionsfreiheitsgrade berücksichtigt. In Lösung haben alle n-atomigen Moleküle zusätzlich zu den 3n Freiheitsgraden (3 Translation, 2-3 Rotation, und Vibration; Bsp. CO2 hat 2 Scherschwingungen und 2 Streckschwingungen, da es nur 2 Rotationsfreiheitsgrade aufweist, H2O hat eine Scherschwingung weniger, weil es 3 Rotationsfreiheitsgrade aufweist) an jeder drehbaren Bindung einen Torsionsfreiheitsgrad, bei dem eine Atom-Gruppe im Molekül rotiert. Diese gehen bei der Bindung an den Rezeptor verloren, weshalb die Bindung entropisch ungünstig ist. Als vierte Komponente wird die Energie der Solvatisierung vorhergesagt. Hierzu wird ein thermodynamischer Kreisprozess zu Hilfe genommen. Es wird ΔG f ür die Bindung im Vakuum berechnet, außerdem für die Solvatisierung des gebundenen Rezeptor-Ligand Komplexes und der ungebundenen Komponenten. Die Gibbs Energie für die Bindung in Lösung erhält das Computerprogramm nun über die Subtraktion des ΔG Wertes f ür die Solvatisierung des Rezeptor-Ligand Komplexes von dem ΔG Wert f ür die Solvatisierung der ungebundenen Komponenten. Dies ist daher möglich, weil ΔG eine Zustandsgröße und daher vom Weg unabhängig ist. Dies sei in folgender Abbildung veranschaulicht. Abbildung 9 Thermodynamischer Kreisprozess der Solvatisierung Ein weiterer Term (unbound system energy) ist bei der aktuellsten Version von Auto-Dock hinzugekommen, dessen Funktion uns bisher unbekannt ist. Die zugrunde liegenden Gleichungen aller Berechnungen sind hochkomplex. Die Addition aller Terme ergibt die Mastergleichung. Das resultierende ΔG soll sehr genau der freien Enthalpie des Bindungsprozesses entsprechen. Im Folgenden ist die Mastergleichung gezeigt. Abbildung 10 Die Mastergleichung von Auto-Dock Gezeigt werden die einzelnen Terme für die Berechnung der freien Gibbs Energie für die Van-der-Waals WW, für HBrücken, für elektrostatische WW, für die Torsionsfreiheitsgrade und die Solvatation. Es fehlt der bei Auto Dock 4.0 neu hinzugekommene Term für die Das Ergebnis ist nur eine Näherung Das Ergebnis ist allerdings nicht exakt. Auto Dock berechnet zwar die Solvatisierung über den thermodynamischen Kreisprozess, berücksichtigt aber nicht einzelne Lösungsmittelmoleküle, weshalb es sich nur um eine Annäherung handelt. Außerdem wird angenommen, dass der Rezeptor starr ist, was ebenso nicht korrekt ist. Auch der Rezeptor kann seine Konformation leicht verändern und weist geringe, in wenigen Fällen sogar große Bewegungen auf, welche die Bindung beeinflussen könnten (wie in Abschnitt 4.8 gezeigt). Außerdem berechnet Auto Dock 4.0 die Interaktionsenergie zwischen Rezeptor und Ligand durch molekülmechanische Ansätze, die sich von den tatsächlich quantenmechanischen Vorgängen sehr unterscheiden können. Es kommt hinzu, dass Auto-Dock nicht alle Konformationen berücksichtigt, sondern nur stichprobenartig einige auswählt. Das gefundene Ergebnis ist demnach nur zu einer gewissen Wahrscheinlichkeit das tatsächliche ΔG Minimum. Darüber hinaus liegen in der Zelle noch eine große Bandbreite anderer Moleküle vor, die um die Wechselwirkung mit dem aktiven Zentrum (Ionen, andere Proteine) oder der Bindung an diesem aktiven Zentrum (strukturell ähnliche Moleküle, Inhibitoren) mit dem Liganden konkurrieren. Zudem berechnet Auto-Dock nur die Interaktions-Energie zwischen Liganden und Rezeptor in einer bestimmten Position des aktiven Zentrums. Die Wahrscheinlichkeit mit der der Ligand nun tatsächlich in dieses Zentrum gelangt bleibt unberücksichtigt. So kann ein rein hydrophober Ligand eine sehr große Wechselwirkung mit einem rein hydrophoben Zentrum erreichen. Wenn allerdings die gesamte Oberfläche des Proteins hydrophob ist, so ist die Wahrscheinlichkeit, dass der Ligand im aktiven Zentrum landet erheblich eingeschränkt. Hinzu kommt, dass innerhalb der in der .pdb Datei gespeicherten Struktur Fehler sein können, wie bereits in Abschnitt 2.1 diskutiert wurde. Dies kann ebenso zu verfälschten Einschätzungen über die Bindungsaffinität führen. Aufgrund all dieser Vernachlässigungen und Annäherungen sind spätere in vitro (ITC) und in vivo Experimente nicht wegzudenken. Die Funktion von Auto-Grid 4.0 Um die Berechnung der energetisch günstigsten Position des Liganden zu beschleunigen, wird von Auto Dock ein engmaschiges Gitter als Hilfsmittel verwendet. Die Größe des Gitters und der Abstand der Gitterpunkte werden im Programm festgelegt. Die Position des Gitters wird über das aktive Zentrum gelegt. Wenn das Unterprogramm Auto-Grid seine Rechnung beginnt, werden in jeden Gitterpunkt alle im Liganden vorhandenen Atomtypen in Form eines „Testatoms“ eingesetzt und die auf ihn wirkende Energie berechnet. Ebenso wird mit den „Testladungen“ zur Berechnung der elektrostatischen Wechselwirkungen verfahren. Es resultieren die Gitterenergien der einzelnen Gitterpunkte je Testatom bzw. Testladung. Im eigentlichen Docking werden diese Gitterenergien verwendet, um die Gesamtenergie der Rezeptor-Ligand-Wechselwirkung zu berechnen. Dabei können Atome auch zwischen den Gitterpunkten positioniert werden, wobei hier ein Mittelwert der Energien der umgebenden Gitterpunkte als Energiewert genommen wird. Der Suchalgorithmus während des Docking-Prozesses Während des Dockings werden die Bindungsgeometrien (Konformation: Bindungswinkel, Bindungsabstände, Torsionswinkel) und die Position des Liganden (Translation und Rotation) stichprobenartig und willkürlich variiert. Daher gilt, dass die gefundene Position des Liganden, welches ein ΔG Minimum für die Bindung aufweist nur zu einer bestimmten Wahrscheinlichkeit das tatsächliche Minimum ist. Zur Findung des Minimums werden zwei Methoden miteinander kombiniert: der GenotypAlgorithmus und der Phänotyp-Algorithmus bilden zusammen den hybriden Suchalgorithmus von Auto-Dock. Die folgende Abbildung soll diesen veranschaulichen. Abbildung 11 Schematische Darstellung des hybriden Suchalgorithmus’ von Auto Dock 4.0 Die untere Linie stellt den Genotyp dar, der die Suche an den Phänotyp weitergibt. Der Phänotyp Algorithmus startet die lokale Suche nach einem Minimum. Im Genotyp Zunächst wird im Genotyp-Algorithmus ein bestimmter Bezugspunkt im Gitter ausgewählt, der als „Parent“ bezeichnet wird. Alle Atome des Liganden richten sich nach diesem Bezugspunkt aus. Diese Ausrichtung resultiert in einer bestimmten Rezeptor-Ligand Interaktionsenergie. Nun wird diese Position beibehalten und der Phänotyp-Algorithmus aktiviert. Dadurch werden die Positionen der einzelnen Atome im Raum zueinander und somit die Bindungslängen und Winkel willkürlich verändert, so dass der zuvor gesetzte Bezugspunkt leicht verändert wird. Dadurch wird auch die zuvor berechnete Energie verändert. Ist die neu berechnete Energie kleiner als die vorherige werden die Atompositionen weiter in dieselbe Richtung verändert. Ist sie größer werden sie die Richtung der Positionsänderung variieren. Die Entfernungen, welche die Atome pro Schritt in diesem Algorithmus zurücklegen, werden von Schritt zu Schritt kleiner. Somit verändert sich die Interaktions-Energie auch immer geringfügiger. Der Phänotyp-Algorithmus läuft solange weiter, bis ein lokales Energie-Minimum gefunden wurde oder eine zuvor festgelegte Abbruchskondition vorliegt. Abbruchskondition kann zum Beispiel eine bestimmte Anzahl sein, mit der die Energie neu berechnet werden soll. Das lokale Minimum ist nun das Ergebnis des ersten Phänotyp Algorithmus. Dieses wird zwischengespeichert und an den Genotyp-Algorithmus weitergegeben, wodurch eine erste Child-Generation im Genotyp erzeugt wurde. Aufgrund der Tatsache, dass sich der Phänotyp direkt auf den Genotyp auswirkt, wird hier vom Lamark’schen hybriden Suchalgorithmus gesprochen. Der Genotyp kann seinen Bezugspunkt auch durch Mutation willkürlich verändern. Der Bezugspunkt der Child-Generation wird nun erneut an den Phänotyp Algorithmus weiter gegeben, um ein weiteres Minimum zu detektieren. Da die Wechselwirkung zwischen Molekülen von sehr vielen Faktoren abhängig ist, resultiert für einen durchschnittlichen Liganden ein 30-40 dimensionaler Graph, in der jede Dimension ein Minimum hat. Daher ist es wichtig mehrmals zurück in den Genotyp-Algorithmus zu gehen, um mehrere lokale Minima zu detektieren. Je öfter in den Genotyp-Algorithmus gewechselt wird, desto wahrscheinlicher ist es das globale Minimum zu finden. Dabei spielt natürlich auch die breite des Minimums eine Rolle. Ist das globale Minimum schmal, verringert sich die Wahrscheinlichkeit seiner Detektion, wie bei einem Blick auf die SchemaDarstellung deutlich wird. Die PDB, Corina und PyMol Die Struktur des Proteins wird online aus der PDB (Protein-Data-Bank: http://www.rcsb.org/pdb/home/home.do) herunter geladen. Die PDB lebt durch die Strukturaufklärung mittels Röntgenstrukturanalyse und den anderen genannten strukturaufklärenden Methoden (siehe Abschnitt 2.1). Fachleute sind sich uneinig, ob die .pdb Dateien wirklich verlässlich sind. Es handelt sich um die größte Datenbank der Welt. Dennoch ist ihre Validität aufgrund einer unterschiedlich großen Fehlerrate in den Strukturen umstritten. Im Gegensatz dazu verbessern sich die Auflösungen der in der PDB hinterlegten Dateien ständig, was die Fehlerrate senkt, Fehler jedoch nicht ausschließt. Aufgrund der Tatsache, dass wir den Liganden selbst entworfen haben, ist es sehr unwahrscheinlich, dass wir die Struktur in einer .pdb finden werden. Daher müssen wir für unsere Liganden die .pdb Datei selbst erstellen. Hierzu wird das Webprogramm Corina (http://www2.chemie.unierlangen.de/software/corina/free_struct.html) verwendet. Dabei wird die Struktur gezeichnet und anschließend in einen eindeutigen Smiley-Code übersetzt, welcher nach Bestätigung über einen Start Button in eine energieminimierte dreidimensionale Struktur übertragen und in einer .pdb Datei gespeichert wird, die nun herunterladen werden kann. Wenn das ΔG Minimum für die Rezeptor-Ligand-Bindung mittels Auto-Dock gefunden wurde, kann die zu diesem Minimum korrelierte Liganden-Struktur und die Struktur des fixen Rezeptors exportiert werden. Die exportierten Strukturen lassen sich nun mittels Pymol komfortabel betrachten. In diesem Programm lassen sich mittels Wizzard ebenso u.a. Bindungswinkel und Bindungslängen messen und andere elementare Operationen durchführen (siehe Material und Methoden).