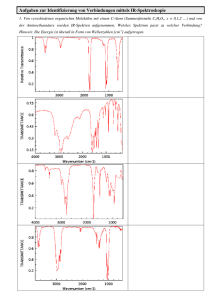
OP II Praktikum Gruppenpräparat: Versuch 1.3.1 3
Werbung

-1/23- OP II Praktikum Gruppenpräparat: Versuch 1.3.1 3- Diethylaminoanisol Gruppe π: Georg Hiltensperger Sebastian Völker Fabian Zieschang -2/23- Gliederung 1. Mustervorschrift 2. Problemstellung 3. Literaturrecherche 4. Reaktionsgleichung 5. Gefahrenpotential 6. Entsorgung 7. Mechanismus 8. Durchführung 8.1. Reaktionsdurchführungen mit Natriumamid 8.1.1.1 Charakterisierung Ansatz 1 8.1.1.2 Fehlerdiskussion Ansatz 1 8.1.2.1 Charakterisierung Ansatz 2 8.1.2.2 Fehlerdiskussion Ansatz 2 8.2 Reaktionsdurchführungen mit Kaliumtertbutanolat 9. Fazit 10. Anhang 8.2.1.1 Durchführung Ansatz 3 8.2.1.2 Charakterisierung Ansatz 3 8.2.1.3 Fehlerdiskussion Ansatz 3 8.2.2.1 Durchführung Ansatz 4 8.2.2.2 Charakterisierung Ansatz 4 8.2.2.3 Fehlerdiskussion Ansatz 4 -3/23- 1. Mustervorschrift Zum Einkondensieren von Ammoniak wird folgender Versuchsaufbau gewählt: Ein trockener 250 ml Dreihalskolben mit Magnetrührstab wird mit einem Gaseinleitungsaufsatz und einem Trockeneiskühler bestückt. Der dritte Schliff wird mit einem Stopfen verschlossen. Am Trockeneiskühler wird ein Blasenzähler angebracht, gefolgt von zwei Waschflaschen, wobei die zweite Flasche mit 10 %iger HCl-Lösung gefüllt wird. Zum Trocknen des Ammoniakgases wird zwischen der Ammoniakbombe und der Reaktionsapparatur eine Waschflasche, gefüllt mit KOH-Plätzchen, geschaltet. Die Apparatur wird mit trockenem Ammoniak gespült, indem kurzzeitig ein leichter Ammoniakstrom durch die Apparatur geleitet wird. Nun wird ein Dewar-Gefäß [1], mit einem Aceton-Trockeneis Gemisch gefüllt, zur Kühlung unter die Apparatur geschoben und mit einem Tieftemperaturthermometer versehen. Der Trockeneiskühler wird ebenfalls mit einem Aceton-Trockeneis Gemisch gefüllt [2]. Nach Einkondensieren von ca. 100 ml Ammoniak [3] wird der Gaseinleitungsaufsatz durch den Blasenzähler des Trockeneiskühlers ersetzt. Der Trockeneiskühler wird wiederum durch einen Rückflusskühler mit Stickstoffaufsatz ausgetauscht [4]. Unter Rühren werden zunächst 0,20g (8,70 mmol) Natrium und eine Spatelspitze Eisen-(III)nitrat-hexahydrat (Fe(NO3)3*6H2O) als Katalysator über den unbesetzten Schliff zugegeben. Anschließend werden weitere 0.86g (37,6 mmol) Natrium zugegeben [5] und 30 min gerührt [6]. Der Blasenzähler wird auf den Stickstoffaufsatz angebracht [7] und der freie Schliff mit einem Tropftrichter versehen. Das Kältebad wird entfernt, so dass überschüssiges Ammoniak verdampfen kann. Während der Ammoniak entweicht, werden 7.50g (0.11 mol; 10,6 ml) Diethylamin [8] langsam unter Stickstoffatmosphäre zugetropft. Nach vollständigem -4/23Entweichen des Ammoniaks (siehe Blasenzähler), wird die Schutzgas Blasenzähler Lösung für 10 min zum Sieden erhitzt, um auch letzte überschüssige Ammoniakreste zu entfernen. Nach Abkühlen auf Raumtemperatur, werden 2,10g (11,2 mmol; 1,38 ml) 2Bromanisol langsam unter Rühren und Stickstoffatmosphäre zugetropft. Anschließend lässt man die Reaktionsmischung für 21h bei 90°C unter Rückfluss erhitzten. Die Reaktion wird mittels DC-Kontrolle verfolgt [9]. Die noch heiße Reaktionsmischung wird in einen zur Hälfte mit Eis gefüllten 500 ml Scheidetrichter gegossen und 40 ml Diethylether zum Extrahieren zugegeben. Nach Phasentrennung wird die wässrige Phase mit Diethylether gewaschen (3 x 40 ml) und die vereinigten organischen Phasen über Kaliumcarbonat getrocknet. Das Lösungsmittel wird unter vermindertem Druck entfernt und der Rückstand mit einer Mikrodestille fraktionierend unter stark vermindertem Druck destilliert. 3-Diethylaminoanisol (gelbe Flüssigkeit) siedet bei 104°C/1mbar. Anmerkungen [1] Es ist darauf zu achten, dass das Dewar-Gefäß keinen Metallboden besitzt, da sonst das später notwendige Rühren mittels Magnetrührer nicht funktioniert. [2] Hierbei ist darauf zu achten, dass die Mischung nicht überläuft und evtl. über den Schliff in den trockenen Kolben gelangt. Deshalb wird Aceton vorgelegt und ganz langsam kleine Stückchen Trockeneis zugegeben bis der Trockeneiskühler gefüllt ist. [3] Die Füllhöhe von 100 ml kann zuvor mittels Klebeband markiert werden. [4] Beim Umbau darf das Aceton-Trockeneis-Kühlbad nicht entfernt werden, da sonst der kondensierte Ammoniak entweicht. [5] Natrium sollte in kleinen krustenfreien Stückchen zugegeben werden. [6] Durch Zugabe von Natrium erfolg eine Blaufärbung (solvatisierte Elektronen), die dann ins grau-metallische übergeht. Die vollständige Umsetzung erkennt man an einer letztendlich braunen Färbung der Lösung -5/23[7] Wenn der Blasenzähler nicht auf dem Rückflusskühler angebracht wird, entweicht das leicht flüchtige Diethylamin beim Verdampfen des Ammoniaks, sowie beim anschließenden Erhitzen unter Rückfluss, unverbraucht aus der Reaktionsmischung [8] Diethylamin wird vor Gebrauch 3 Stunden über KOH unter Rückfluss erhitzt und anschließend über Molekularsieb 3Å gelagert. [9] Als Laufmittel dient ein Gemisch aus n-Hexan/Ethylacetat = 100:1 2. Problemstellung Ursprünglich wurde dieser Versuch im Praktikum mit 50%iger NaNH2/Toluol Suspension durchgeführt, um die Gefahren von hochexplosivem reinem Natriumamid zu umgehen. Allerdings wurden auf diesem Wege, wenn überhaupt, nur sehr geringe Ausbeuten erhalten. Ziel dieses Gruppenpräparates war es, eine effiziente Versuchsdurchführung zur Erstellung von 3-Diethylaminoanisol zu erarbeiten. 3. Literaturrecherche Bei der Literaturrecherche wurde sich auf die Suche nach alternativen Darstellungsmöglichkeiten für 3-Diethylaminoanisol konzentriert. Dabei wurde darauf geachtet, dass diese sich einer ähnlichen Synthesestrategie (über die intermediäre Bildung eines Arins) bediente. Die Verwendung einer anderen Base als Natriumamid schien sinnvoll, auch in Anbetracht des davon ausgehenden Gefahrenpotentials. Die Literaturrecherche lieferte ein dem unseren ähnliches System. Hier wurde 2-Chloranisol verwendet. Als Base diente leichter handhabbares Kaliumtertbutanolat und die erhaltene Ausbeute lag bei 81%, was darauf schließen ließ, dass auch bei der Variation des Edukts noch gute oder zumindest ausreichende Ausbeuten erwartet werden konnten. -6/23- Verwendete Literatur - L. Gattermann, T. Wieland, Die Praxis des Organischen Chemikers, 43. Aufl., deGruyter-Verlag, Berlin, 1982,S. 108 und 116 (wie bei Pyridin beschrieben) (Lit. 1) - Internet: http://www.aist.go.jp/RIODB/SDBS/ IR-, NMREduktvergleichsspektren (Lit. 2) - M. Beller, C. Breindl, T. H. Riermeier, A. Tillack, Synthesis of 2,3Dihydroindoles,Indoles, and Anilines by Transition Metal-Free Amination of Aryl Chlorides, J. Org. Chem., 2001, 66, 1403-1412 (Lit. 3) - Vorschrift in Anlehnung an: J. F. Bunnet, T. K. Brotherton, J. Org. Chem., 1957, 22, 832-834 (Lit. 4) - Versuchsvorschrift für die in-situ Herstellung von Natriumamid aus dem Praktikum (Lit. 5) - M. Breuning, Seminarskript OP II, Kapitel 8 Metallorganik, 2006, S.8 (Lit. 6) - R. Brückner, Reaktionsmechanismen, Spektrum Verlag München, 3. Aufl., 2004, S. 254 ff. (Lit. 7) 4. Reaktionsgleichung OMe OMe Br Base (KOtBu/ NaNH2) HNEt2 NEt2 -7/23- 5. Gefahrenpotential Natriumamid C, N R 14-19-29-34-50 S 6.3-7/8-26-36/37/39-45-61 Diethylamin F, C R 11-20/21/22-35 S 3-16-26-29-36/37/39-45 2-Methoxybrombenzol --- --- Kaliumhydroxid C R 22-35 S 26-36/37/39-45 Diethylether F+, Xn R 12-19-22-66-67 S 9-16-29-33 Kaliumcarbonat Xi R 36/37/38 S 26 Ammoniak T, N R 10-23-34-50 S 9-16-26-36/37/39-45-61 Natrium F, C R 14/15-34 S 1/2-5.3-8-43.7-45 Kaliumtertbutanolat F, C R 11-14-22-35 S 8-16-26-36/37/39-43.3-45 Toluol F, Xn R 11-38-48/20-63-65-67 S 36/37-46-62 Calciumhydrid F R 15 S 7/8-24/25-43.6 Magnesiumsulfat -- R 11 S 9-16-29-33 -8/23- 6. Entsorgung Natriumamid: Aufgeschlämmt in einem trockenem, inertem Lösemittel, z.B. Tetrahydrofuran, tropfenweise mit 2-Propanol versetzten. Nachdem die Reaktion abgeklungen ist, vorsichtig Wasser zugeben. Nach Neutralisation zu den organischen, halogenfreien Lösemittelabfällen geben Diethylamin: organische, aminhaltige Abfälle 2-Methoxybrombenzol: organische, halogenhaltige Lösemittelabfälle Kaliumhydroxid: die Alkali- und Erdalkalihydroxide und -oxide können nach vorsichtiger Diethylether: Neutralisation in das Abwasser gegeben werden je nach Begleitstoffen als halogenfreie oder halogenhaltige Lösungsmittel entsorgen Kaliumcarbonat: in Wasser lösen und zu den wässrigen, basischen Lösemittelabfällen geben Ammoniak: Defekte Druckgasflaschen müssen durch eine Spezialfirma entsorgt werden Natrium: Kleine Reste vorsichtig in Propanol oder Ethanol geben, danach mit Wasser versetzen und neutralisieren. Das Gemisch wird im Sammelbehälter für neutrale wässrige Lösungen entsorgt. Kaliumtertbutanolat: halogenfreie, organische Lösemittelabfälle Calciumhydrid: in inertem Lösungsmittel (z.B. Toluol) aufschlämmen und vorsichtig Isopropanol zutropfen -9/23- 7. Mechanismus Base HNEt 2 OMe NEt 2 OMe + H-Base H Br Et Base N Et H OMe NEt2 Das Diethylamin wird von der Base deprotoniert. Die Base induziert eine Eliminierung, wobei ein Arin entsteht. Das Nucleophil greift das Arin in meta-Stellung an, da diese gegenüber der ortho-Stellung sowohl sterisch, als auch elektronisch bevorzugt ist. 8. Durchführung Es wurden sowohl mit NaNH2, als auch mit KOt-Bu als Base jeweils zwei Versuche durchgeführt. Auf die im Praktikum ursprünglich angewendete Methode mit einer NaNH2/Toluol – Suspension wurde verzichtet, da diese von der anderen Gruppe ohne nennenswerten Erfolg durchgeführt wurde. Allgemein gab es einen regen Austausch zwischen den Gruppen, um so die Arbeit effizienter zu gestalten. -10/23- 8.1. Reaktionsdurchführungen mit Natriumamid Es wurden zwei Versuche nach der gleichen Vorschrift durchgeführt. Da der erste Versuch wegen technischer Probleme nicht zum gewünschten Resultat führte, wurde erneut nach der gleichen Vorschrift gearbeitet. Aus diesem Grund ist die folgende Durchführungsbeschreibung inklusive aller Mengenangaben für beide Ansätze gültig. Durchführung In einem 250 ml Dreihalskolben mit Trockeneiskühler, Blasenzähler und Gaseinleitungsaufsatz wurden unter Kühlung [1] ca. 100 ml Ammoniak [2] einkondensiert. Anschließend tauschte man den Trockeneiskühler und Gaseinleitungsaufsatz durch einen Rückflusskühler mit Stickstoffaufsatz aus. Unter Stickstoffatmosphäre und Kühlung wurden nun 0,20g (8,70mmol) Natrium zugegeben [3]. Anschließend gab man eine Spatelspitze Eisen-(III)-nitrat-hexahydrat (Fe(NO3)3*6H2O) zu, gefolgt von 0,86g (37,6mmol) Natrium [4]. Nach 30min Rühren entfernte man das Kühlbad. Während der überschüssige Ammoniak entwich, wurden 7,50g (0,10mol; 10,6 ml) Diethylamin [5] langsam unter Stickstoffatmosphäre zugetropft [6]. Die Lösung wurde für 10 min unter Rückfluss zum Sieden erhitzt und nach Abkühlen auf Raumtemperatur 2,10g (11,2mmol; 1,38 ml) 2- Bromanisol zugetropft. Die Reaktionsmischung wurde für 21 h unter Rückfluss zum Sieden erhitzt (90°C) und die Reaktion mittels DC-Kontrolle verfolgt [7]. Die heiße Lösung goss man auf Eis und extrahierte sie mit 40 ml Diethylether. Die wässrige Phase wurde mit weiterem Diethylether gewaschen (3 x 40ml), die vereinigten organischen Phasen über Kaliumcarbonat getrocknet und das Lösungsmittel unter vermindertem Druck entfernt. Der Rückstand wurde anschließend unter vermindertem Druck fraktionierend destilliert. Anmerkungen [1] Zur Kühlung wird ein Dewar-Gefäß mit einer Aceton/Trockeneis Mischung verwendet [2] Vor dem Einkondensieren muss das Ammoniak-Gas getrocknet werden. Dies geschieht durch Zwischenschalten einer mit KOH-Plätzchen gefüllten Waschflasche -11/23[3] Durch Zugabe von Natrium zu flüssigem Ammoniak kommt es zu einer Blaufärbung der Lösung (solvatisierte Elektronen) [4] Es ist darauf zu achten, dass nur kleine und vor allem krustenfreie Natriumstücke zugegeben werden [5] Diethylamin wird vor der Verwendung 3 Stunden über KOH unter Rückfluss erhitzt und auf Molekularsieb 3Å destilliert [6] Ab diesem Reaktionsschritt ist es wichtig, dass Stickstoffaufsatz und Blasenzähler auf dem Rückflusskühler angebracht sind, da sonst das leicht flüchtige Diethylamin unverbraucht aus der Reaktionsmischung entweicht [7] Zur DC-Kontrolle wird ein Laufmittelgemisch aus n-Hexan/Ethylacetat = 100:1 gewählt 8.1.1.1. Charakterisierung Ansatz 1 Ausbeute: 0,37g (2,06mmol) = 18% Siedepunkt: 107-109°C/3mbar Brechungsindex nD20,6 = 1,5427 IR: ν~ = 3095 cm-1 (=C-H, aromatisch) ν~ = 2980 cm-1 (-C-H, alkylisch) ν~ = 1605 cm-1 (C=C, aromatisch) 1 H-NMR: (250MHz, CDCl3) δ = ppm: 7.25 (dd, 1H, 3J=8.23Hz, 3J=8.55Hz; Ar-H) 6.50-6.30 (m, 3H, Ar-H) 3.95 (s, 3H, O-CH3) 3.50 (q, 4H, 3J=7.0 Hz; N-CH2) 1.20 (t, 6H, 3J=7.0 Hz; N-CH2-CH3) -12/23- -13/23- 8.1.1.2. Fehlerdiskussion Ansatz 1 Die schlechte Ausbeute lässt sich dadurch erklären, dass über Nacht die Wasserkühlung des Rückflusskühlers ausfiel, da nachts anscheinend ein geringerer Wasserdruck auf der Wasserleitung liegt. Dies hatte zur Folge, dass auch die Wärmezufuhr nach wenigen Stunden gestoppt wurde. Eine DC-Kontrolle zeigte, dass das gewünschte Produkt (Rf-Wert: 0,06) nur geringfügig entstanden war und sich noch Edukt (Rf-Wert: 0,45) in der Lösung befand. Die Lösung wurde dennoch aufgearbeitet und das Produkt isoliert. Immerhin konnte so festgestellt werden, dass die eigentliche Reaktionszeit von 21h von großer Bedeutung für die Umsetzung ist. Aus dem NMR–Spektrum lässt sich schließen, dass das gewünschte Produkt teilweise synthetisiert wurde. Allerdings sind noch einige andere Peaks zu erkennen, die dem Edukt zugeordnet werden können. (s. Anhang) 8.1.2.1. Charakterisierung Ansatz 2 Ausbeute: 1,22g (6,81mmol) = 61% Siedepunkt: 104°C/1mbar Brechungsindex nD25 = 1,5420 IR: ν~ = 3095 cm-1 (=C-H, aromatisch) ν~ = 2980 cm-1 (-C-H, alkylisch) ν~ = 1605 cm-1 (C=C, aromatisch) 1 H-NMR: (250MHz, CDCl3) δ = ppm: 7.19 (dd, 1H, 3J=8.23Hz, 3J=8.55Hz; Ar-H) 6.40-6.20 (m, 3H, Ar-H) 3.95 (s, 3H, O-CH3) 3.40 (q, 4H, 3J=7.0 Hz; N-CH2) 1.30 (t, 6H, 3J=7.0 Hz; N-CH2-CH3) -14/23- -15/23- 8.1.2.2. Fehlerdiskussion Ansatz 2 Nachdem die Umsetzung des ersten Ansatzes nicht vollständig war, wurde diesmal darauf geachtet die Reaktionszeit von 21 h auch wirklich eingehalten wurde. Anhand der DCKontrolle konnte festgestellt werden, dass das 2-Bromanisol (Rf-Wert: 0,49) komplett zum 3Diethylaminoanisol (Rf-Wert: 0.07) umgesetzt wurde. Die Aufarbeitung mit anschließender fraktionierter Destillation konnte problemlos durchgeführt werden und es wurde diesmal eine Ausbeute von 61% erzielt. Das NMR- und IR- Spektrum lässt auf hohe Reinheit schließen, da vor allem im NMR– Spektrum ausschließlich die gewünschten Peaks zu sehen sind. Dies war die quantitativ beste Durchführung und ist für die Darstellung von 3Diethylaminoanisol sehr zu empfehlen. 8.2 Reaktionsdurchführung mit Kaliumtertbutanolat Die folgenden zwei Versuchsansätze wurden in Anlehnung an Lit 3 durchgeführt. Hierbei wird auf das explosive Natriumamid verzichtet und Kaliumtertbutanolat als Base verwendet. Es wird in der Literatur eine allgemeine Vorschrift für die Aminierung von Chlorbenzolen beschrieben, sowie eine spezifische Vorschrift zur Herstellung von 3-Diethylaminoanisol. Diese unterscheiden sich lediglich in den verwendeten Eduktverhältnissen. Ansatz 3 wurde nach der allgemeinen, Ansatz 4 nach der spezifischen Vorschrift durchgeführt. 8.2.1.1 Durchführung Ansatz 3 In einem 250 ml Dreihalskolben wurden unter Stickstoffatmosphäre [1] 3,20g (17,1mmol / 1 eq) 2-Methoxybrombenzol und 2.49g (34,0mmol / 2 eq) Diethylamin [2] in 60ml trockenem Toluol [3] gelöst. Nach Zugabe von 5,72g (50,1mmol / 3 eq) KOt-Bu wurde die Mischung unter ständigem Rühren unter Rückfluss zum Sieden auf 135°C erhitzt. Der Reaktionsverlauf -16/23wurde dünnschichtchromatographisch verfolgt [4]. Nach 5 Tagen lies man die Reaktionsmischung auf Raumtemperatur abkühlen und versetzte sie mit 60 ml Wasser. Die Phasen wurden getrennt und die wässrige Phase mit Dichlormethan extrahiert (3 x 60 ml) [5]. Die vereinigten organischen Phasen wurden über Magnesiumsulfat getrocknet und das Lösungsmittel unter vermindertem Druck entfernt. Der Rückstand wurde mittels Säulenfiltration (Hexan/Ethylacetat = 5:1) gereinigt und anschließend fraktionierend destilliert. Anmerkungen [1] Wichtig beim Versuchsaufbau ist, dass der Stickstoffaufsatz sowie der Blasenzähler auf dem Rückflusskühler angebracht sind, da sonst das leicht flüchtige Diethylamin aus der Reaktionsmischung entweicht. [2] Vor der Verwendung wird das Diethylamin über Kaliumhydroxid 3 Stunden unter Rückfluss erhitzt und anschließend auf Molekularsieb 3 Å destilliert. [3] Zum Trocknen von Toluol wird es zunächst 3 Stunden über Calciumhydrid unter Rückfluss erhitzt und anschließend auf Molekularsieb 3 Å destilliert. [4] Als Laufmittel für die DC-Kontrolle diente ein Gemisch aus Cyclohexan und Ethylacetat im Verhältnis 5:1. [5] Durch Zugabe von 40 ml Wasser zur organischen Phase kommt es zur Trübung, wodurch die Phasengrenze nicht mehr sichtbar ist. Durch Zugabe einer gesättigten NaCl-Lösung (Aussalzen) löst sich die Trübung auf und die Phasengrenze ist wieder sichtbar. 8.2.1.2. Charakterisierung Ansatz 3 Ausbeute: 1,31g (7,31mmol) = 43% (Lit.: 81%) Siedepunkt: 97°C/2mbar Brechungsindex nD20,6 = 1,5468 IR: ν~ = 3065 cm-1 (=C-H, aromatisch) ν~ = 2980 cm-1 (-C-H, alkylisch) ν~ = 1605 cm-1 (C=C, aromatisch) -17/23- 1 H-NMR: (250MHz, CDCl3) δ = ppm: 7.19 (dd, 1H; Ar-H) 6.50-6.30 (m, 3H; Ar-H) 3.87 (s, 3H; O-CH3) 3.39 (q, 4H; N-CH2) 1.23 (t, 6H; N-CH2-CH3) -18/23- 8.2.1.3. Fehlerdiskussion Ansatz 3 Sowohl das IR – Spektrum, als auch das NMR – Spektrum lassen darauf schließen, dass die gewünschte Substanz synthetisiert wurde. Die DC-Kontrolle bestätigt dieses (Rf-Wert des Produkts: 0,32), zeigt aber auch, dass immer noch Edukt vorhanden ist (Rf-Wert: 0,61). Allerdings sind trotz Säulenfiltration und fraktionierter Destillation noch Unreinheiten zu erkennen. Das NMR-Spektrum zeigt Peaks, welche auf das Edukt schließen lassen könnten. (s. Anlage Vergleichsspektrum Edukt) Die Literaturausbeute von 81% konnte allerdings nicht erreicht werden. -19/23- 8.2.2.1. Durchführung Ansatz 4 In einem 250 ml-Dreihalskolben mit Rückflusskühler, Blasenzähler und Stickstoffaufsatz [1] wurden unter Stickstoff 3.20g (17.1mmol, 1 eq) 2-Bromanisol, 1.25g (17.1mmol, 1 eq) Diethylamin [2] und 2.86g (25.5mmol, 1.5 eq) Kaliumtertbutanolat in 60 ml trockenem Toluol [3] gelöst. Die Reaktionsmischung wurde für 45 Stunden bei 135°C erhitzt und der Verlauf mittels DC-Kontrolle verfolgt [4]. Nach Abkühlen auf Raumtemperatur versetzte man die Reaktionsmischung mit 45 ml Wasser. Die organische Phase wurde abgetrennt und mit weiteren 40 ml Wasser gewaschen [5]. Anschließend wurden die vereinigten wässrigen Phasen mit Dichlormethan (6 x 20 ml) extrahiert. Nach Vereinigen der organischen Phasen trocknete man diese über Magnesiumsulfat und entfernte das Lösungsmittel unter vermindertem Druck. Das Rohprodukt wurde mittels Säulenchromatographie über Kieselgel gereinigt [6]. -20/23Anmerkungen [1] Wichtig beim Versuchsaufbau ist, dass der Stickstoffaufsatz sowie der Blasenzähler auf dem Rückflusskühler angebracht sind, da sonst das leicht flüchtige Diethylamin aus der Reaktionsmischung entweicht. [2] Vor der Verwendung wird das Diethylamin über Kaliumhydroxid 3 Stunden unter Rückfluss erhitzt und anschließend auf Molekularsieb 3 Å destilliert. [3] Zum Trocknen von Toluol wird es zunächst 3 Stunden über Calciumhydrid unter Rückfluss erhitzt und anschließend auf Molekularsieb 3 Å destilliert. [4] Als Laufmittel für die DC-Kontrolle diente ein Gemisch aus Cyclohexan und Ethylacetat im Verhältnis 5:1. [5] Durch Zugabe von 40 ml Wasser zur organischen Phase kommt es zur Trübung, wodurch die Phasengrenze nicht mehr sichtbar ist. Durch Zugabe einer gesättigten NaCl- Lösung (Aussalzen) löst sich die Trübung auf und die Phasengrenze ist wieder sichtbar. [6] Als Laufmittel wird ein Gemisch aus Cyclohexan und Ethylacetat im Verhältnis 5:1 verwendet. 8.2.2.2. Charakterisierung Ansatz 4 Ausbeute: 375 mg (2.09 mmol) = 12% (Lit.: 81%) Brechungsindex: nD20,6 = 1.5598 IR: ν~ =3400 cm-1 (-O-H) ν~ = 3060 cm-1 (=C-H, aromatisch) ν~ = 2920cm-1 (-C-H, alkylisch) ν~ = 1600 (C=C, aromatisch) 1 H-NMR: Das 1H-NMR-Spektrum ließ keine sinnvolle Auswertung zu. -21/23- -22/23- 8.2.2.3. Fehlerdiskussion Ansatz 4 Da die erste Reaktionsdurchführung nicht die gewünschte Ausbeute brachte, wurde die Umsetzung mit Kaliumtertbutanolat in einem weiteren Ansatz erneut durchgeführt. Diesmal wurde genaustens darauf geachtet, dass die Reaktionszeit von 45 Stunden eingehalten wurde. Auch diesmal zeigte eine DC- Kontrolle eine unvollständige Umsetzung des 2-Bromanisols (Rf-Wert: 0,65) an. Allerdings ist auch ein Produktspot (Rf-Wert: 0,62) zu erkennen. Sowohl das IR- als auch das NMR-Spektrum lassen vermuten, dass das gewünschte Produkt nur in äußerst geringem Maße entstanden ist. Fraglich ist allerdings, warum das 1H-NMR-Spektrum dermaßen unübersichtlich ist, da die DC-Kontrollen der Säulenchromatographie jeweils nur einen Produktspot zeigten. Auch das IR-Spektrum ließ keine schlüssige Charakterisierung zu, da es sowohl das Vorhandensein einer OH-Gruppe anzeigte als auch eine enorme Abweichung im Fingerprintbereich im Vergleich zum IR-Spektrum des Ansatzes 8.2.1. aufwies. Welche Ursache sich hinter diesem Sachverhalt verbirgt, bleibt leider unergründlich. 9. Fazit Aufgrund unserer Versuchsdurchführungen sind wir zu dem Ergebnis gekommen, dass die Verwendung von in-situ-erzeugtem Natriumamid die besten Ausbeuten an 3-Diethylaminoanisol liefern. Nach unserer Meinung ist es daher sinnvoll für den Praktikumsversuch diesen Weg der Darstellung zu gehen, obwohl er aufwendiger ist und ein gewisses Gefahrenpotential birgt. Die Versuchsvorschrift mit KOtBu als Base schien weniger aufwendig, war allerdings auch weniger erfolgreich, sowohl was die Reinheit, als auch was die Ausbeute betrifft. 10. Anhang Vergleichspektren des Edukts (2-Bromanisol) -23/23-