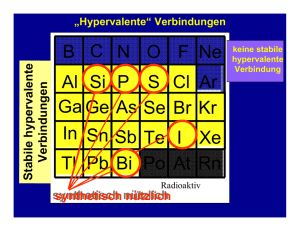
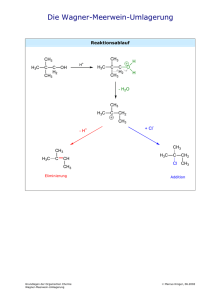
Neue Aspekte der Chemie des supersauren Mediums HF/GeF4 und
Werbung

Dissertation zur Erlangung des Doktorgrades der Fakultät für Chemie und Pharmazie der Ludwig-Maximilians-Universität München Neue Aspekte der Chemie des supersauren Mediums HF/GeF4 und Beiträge zur Protonierung sehr starker organischer Säuren THERESA SOLTNER aus Cottbus 2011 New aspects in superacid chemistry of HF/GeF4 and protonation of strong organic acids THERESA SOLTNER Cottbus 2011 Erklärung Diese Dissertation wurde im Sinne von § 13 Abs. 3 bzw. 4 der Promotionsordnung vom 29. Januar 1998 (in der Fassung der sechsten Änderungssatzung vom 16. August 2010) von Herrn Prof. Dr. ANDREAS KORNATH betreut. Ehrenwörtliche Versicherung Diese Dissertation wurde selbstständig, ohne unerlaubte Hilfe erarbeitet. München, 27.06.2011 …………………………………. Dissertation eingereicht am: 30.06.2011 1. Gutachter: Prof. Dr. Andreas Kornath 2. Gutachter: Prof. Dr. Thomas M. Klapötke Mündliche Prüfung am: 12.12.2011 meinen Eltern PETRA, ULRICH, KATHARINA, BRIGITTE, KARL-HEINZ, HILDEGARD, VICTOR, HELENE, GERTRUD, RAINER, KAORI, EMLYN, FLORIAN, NADINE, FABIAN, STEFAN, HUBERT, PIET, FLORIAN, ASUKA, CAN, BEN, GABI Inhaltsverzeichnis Inhaltsverzeichnis 1 Einleitung ......................................................................................................................... 1 2 Aufgabenstellung.............................................................................................................. 4 3 Ergebnisse und Diskussion............................................................................................... 5 3.1 4 5 Protonierung starker organischer Säuren ................................................................... 5 3.1.1 Trifluormethansulfonsäure ................................................................................. 8 3.1.2 Monofluoressigsäure ........................................................................................ 16 3.1.3 Difluoressigsäure .............................................................................................. 25 3.1.4 Trifluoressigsäure ............................................................................................. 33 3.1.5 Trichloressigsäure ............................................................................................ 41 3.2 Tricyanomethan (Cyanoform) ................................................................................. 49 3.3 Die strukturelle Vielfalt von Fluorogermanaten ...................................................... 56 3.3.1 Umsetzung von HF/GeF4 mit Nitroaromaten ................................................... 58 3.3.2 [(Me2OH)2]2+[Ge2F10]2−.................................................................................... 65 3.3.3 [(Me2OH)4]4+[Ge3F16]4−.................................................................................... 70 3.3.4 [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF ......................................................................... 74 3.3.5 [(Me2SH)3]3+[Ge4F19]3− ⋅ HF ............................................................................ 82 3.3.6 [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF ............................................................................ 90 3.3.7 [(H3O)2]2+[GeF6]2−............................................................................................ 94 3.3.8 Vergleich der Fluorogermanatanionen ............................................................. 97 Ausblick und Zusammenfassung.................................................................................. 101 4.1 Protonierung starker organischer Säuren ............................................................... 101 4.2 Tricyanomethan (Cyanoform) ............................................................................... 102 4.3 Die strukturelle Vielfalt von Fluorogermanaten .................................................... 103 Experimenteller Teil ..................................................................................................... 104 5.1 Apparaturen und Chemikalien ............................................................................... 104 I Inhaltsverzeichnis 5.1.1 Allgemeine Arbeitstechnik ............................................................................. 104 5.1.2 Chemikalien.................................................................................................... 104 5.2 Analytik und quantenchemische Rechnungen ....................................................... 106 5.2.1 Raman-Spektroskopie .................................................................................... 106 5.2.2 IR-Spektroskopie ............................................................................................ 106 5.2.3 Kernresonanzspektroskopie............................................................................ 107 5.2.4 Einkristall-Röntgenstrukturanalyse ................................................................ 108 5.2.5 Quantenchemische Rechnungen..................................................................... 109 5.3 Synthesen ............................................................................................................... 110 5.3.1 Darstellung von [CF3SO3H2]+[SbF6]− ............................................................ 110 5.3.2 Darstellung von [CH2FC(OH)2]+[AsF6]− ....................................................... 110 5.3.3 Darstellung von [CHF2C(OH)2]+[AsF6]− ....................................................... 111 5.3.4 Darstellung von [CF3C(OH)2]+[AsF6]− .......................................................... 111 5.3.5 Darstellung von [Cl3CC(OH)2]+[AsF6]− ......................................................... 112 5.3.6 Darstellung von HC(CN)3 .............................................................................. 112 5.3.7 Darstellung von [(C6H5NO2H)2]2+[Ge2F10]2− ................................................. 113 5.3.8 Darstellung von [(Me2OH)2]2+[Ge2F10]2− ....................................................... 113 5.3.9 Darstellung von [(Me2OH)4]4+[Ge3F16]4− ....................................................... 114 5.3.10 Darstellung von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF ............................................. 114 5.3.11 Darstellung von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF ................................................ 115 5.3.12 Darstellung von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF ............................................... 115 5.3.13 Darstellung von [(H3O)2]2+[GeF6]2− ............................................................... 116 5.3.14 Darstellung der deuterierten Verbindungen ................................................... 116 6 Röntgenstrukturanalysen .............................................................................................. 117 7 Verzeichnisse................................................................................................................ 130 7.1 Abkürzungs- und Einheitenverzeichnis ................................................................. 130 7.2 Abbildungsverzeichnis .......................................................................................... 131 II Inhaltsverzeichnis 7.3 Tabellenverzeichnis ............................................................................................... 134 7.4 Literaturverzeichnis ............................................................................................... 137 III Einleitung 1 Einleitung Säuren und Basen stellen seit Menschen Gedenken einen wichtigen Bestandteil unseres Lebens dar. Die keimtötende Wirkung der Essigsäure wurde zur Wundbehandlung eingesetzt und machte es römischen Legionären erst möglich, das bisweilen stark verkeimte Wasser zu trinken, indem sie es mit Essig mischten.[1] Jedoch brachte die Essigsäure nicht nur Vorteile. Durch den süßen Geschmack des Bleiacetats zu reichlichem Genuss verleitet, starben viele römische Bürger an einer Bleivergiftung.[2] Der Begriff „Säure“ ist ein weitläufiger Begriff in der Chemie, der in zahlreichen Veröffentlichungen diskutiert wurde. Im 18. Jahrhundert sah unter anderem der französische Chemiker Lavoisier den Sauerstoff als maßgebliche Ursache saurer Eigenschaften eines Stoffes, da das Auflösen von Nichtmetalloxiden in Wasser zu sauren Lösungen führt. Das Element erhielt auf diesem Wege seinen Namen Sauerstoff oder auch Oxygenium (lateinisch: „es gebärt Säure“). Ende des 19. Jahrhunderts wurde durch den späteren Nobelpreisträger Arrhenius eine zutreffendere Theorie aufgestellt, in der er behauptete, Säuren sind Wasserstoffverbindungen, die in wässriger Lösung H+-Ionen abgeben. Die Bildung von OH−-Ionen wurde durch Basen bewirkt. Die Definition von Arrhenius ist auf Wasser als Lösungsmittel beschränkt.[3] Nach Ergänzungen von Lowry und Brønsted aus dem Jahr 1923 werden Protonen von Säuren abgegeben und von Basen aufgenommen. Die Brønsted-Definition ist nicht mehr auf Wasser als Lösungsmittel beschränkt.[4-5] Mit Hilfe dieser Theorie ließen sich beispielsweise die sauren Eigenschaften der Salz-, Schwefel- oder Salpetersäure anschaulich erklären. Um aber das saure Verhalten einer Eisen(III)chlorid-Lösung verstehen zu können, bedurfte es einiger Erweiterungen dieses Konzeptes. In der umfassenden Theorie von Lewis aus dem Jahr 1923 bezeichnete dieser Säuren als Elektronenpaarakzeptoren und Basen als Elektronenpaardonatoren.[6] Das in den vierziger Jahren des 20. Jahrhunderts von Lux aufgestellte und von Flood weiterentwickelte Konzept übertrug den Säure-Base-Begriff auf protonenfreie Systeme, wie sie in anorganischen Schmelzen und Interhalogenen zu finden sind.[7-9] Durch diese Theorie waren nun auch basische Eigenschaften, beispielsweise von Calciumoxid, oder das saure Verhalten von Schwefeltrioxid in Wasser zu erklären, was wiederum mit den anfänglichen Erklärungsversuchen von Lavoisier im Einklang stand. Durch dieses Konzept war der Begriff „Säure“ weitestgehend definiert. 1 Einleitung Um verschiedene Säuren hinsichtlich ihrer Säurestärke vergleichen zu können, wurde als Skala der pH-Wert eingeführt, der sich gemäß (1.1) ausschließlich auf die Aktivität der in der Lösung vorhandenen Oxoniumionen bezieht. Der pH-Wert ist definitionsgemäß aufgrund der folgenden zwei Sachverhalte eingeschränkt. Zum einen ist die Löslichkeit einer Säure in Wasser durch ihr Löslichkeitsprodukt beschränkt, somit kann je nach Säure nur eine spezifische Konzentration erreicht werden. Zum anderen lassen sich starke Säuren mit einer hohen Protolyse-Gleichgewichtskonstante ab einem Protolysegrad α ≈ 1 hinsichtlich ihrer Stärke nicht mehr unterscheiden, da die dissoziierte Spezies bereits zu annähernd 100 % vorliegt. Daher kann eine Einordnung der Säurestärke bei sehr starken Säuren nur in einem Medium stattfinden, welches eine schwächere Base als Wasser darstellt. Zur Berechnung der Säurestärke wurde von Deyrup und Hammet 1932 die Hammet-Konstante H0 eingeführt, welche sich nach Gleichung (1.2) ergibt.[10] (1.2) Gleichgewichtskonstante KBH+ für die Reaktion: B + H+ BH+ Experimentell wird mittels UV/Vis-Spektroskopie der Protonierungsgrad [BH+]/[B] einer Indikatorbase B nach der Reaktion mit der zu bestimmenden Substanz ermittelt und somit auch die Säurestärke. Das Spektrum, der zur Aciditätsbestimmung verwendeten Indikatorbasen reicht von stärkeren Basen wie aromatischen Aminen bis hin zu schwächeren Basen wie Nitroverbindungen.[11] Aufgrund der Vielzahl verschiedener Indikatoren und verwendeter Lösungsmittel lässt sich kein allgemeiner Wert für Säurestärken finden, jedoch erwiesen sich die H0-Werte als brauchbare Richtlinien. Eine einheitliche pH-Skala ist dabei schon lange das Ziel einiger Forschungsgruppen, entweder auf einem theoretischen[12-13] oder einem experimentell vergleichenden Ansatz[14] basierend. Supersäuren werden als Säuren definiert, welche einen niedrigeren H0-Wert als 100%ige wasserfreie Schwefelsäure (H0 = –12) aufweisen.[15] Der Begriff Supersäure wurde bereits 1927 von Conant und Hall eingeführt, blieb jedoch zu dieser Zeit eher unbeachtet.[16] Erst als es Olah 1966 gelang, das Trimethylcarbeniumion nachzuweisen und kinetisch zu stabilisieren, wurde das Thema wieder aufgegriffen.[17-18] 2 Einleitung Eine von Olah häufig verwendete Supersäure war ein Gemisch aus Fluorsulfonsäure und Antimonpentafluorid, welches auch als magic acid bekannt ist. In den folgenden Jahrzehnten wurden weitere Systeme auf supersaure Eigenschaften untersucht, wobei sich Gemische aus starken Brønsted- und starken Lewis-Säuren als Systeme mit den niedrigsten H0-Werten herausstellten. Als stärkstes bisher untersuchtes System gilt HF/SbF5, mit welchem sogar die magic acid selbst protoniert und isoliert werden konnte.[19] 2 HF + SbF5 (1.3) [H2F]+ + [SbF6]- Die hohe Acidität wird dabei hauptsächlich durch die Stabilität des Anions bewirkt, wodurch das Gleichgeweicht der Reaktion auf (Gleichung (1.3)). 3 die rechte Seite geschoben wird Aufgabenstellung 2 Aufgabenstellung In dieser Arbeit werden unterschiedliche Verbindungen hinsichtlich ihres Reaktionsverhaltens unter supersauren Bedingungen untersucht. Ein Ziel dieser Arbeit ist die Protonierung starker organischer Säuren und deren vollständige Charakterisierung. Zu diesen Säuren zählen die Trifluormethansulfonsäure, welche zu den stärksten bekanntesten organischen Säuren zählt und selbst per Definition eine Supersäure ist, die Fluoressigsäuren und die Trichloressigsäure (Gleichung (2.1) und (2.2)). Die Salze sollen schwingungsspektroskopisch und strukturell charakterisiert werden. CF3SO3H + HF/MF5 [CF3SO3H2]+[MF6]- (2.1) RCO2H + HF/MF5 [RC(OH)2]+[MF6]- (2.2) R = CH2F, CHF2, CF3, CCl3 M = As, Sb Der zweite Teil der Arbeit beschäftigt sich mit der Fragestellung ob Tricyanomethan im Festkörper als Cyanoform oder als Dicyanoketenimin vorliegt (Abbildung 2.1). Da Umsetzungen mit HCl oder anderen protischen Säuren keine Ergebnisse lieferten,[20] sollten zur Klärung dieser Frage Tricyanomethanide mit HF umgesetzt werden. H NC Abbildung 2.1: NC CN CN C C NH NC Strukturen von Cyanoform (links) und Dicyanoketenimin (rechts). Der letzte Teil der Arbeit beschäftigt sich mit dem supersauren System HF/GeF4. Lange Zeit wurde angenommen, dass sich die Fluorogermanatanionen strukturell ähnlich verhalten wie die Anionen der sehr gut untersuchten Systeme HF/AsF5 und HF/SbF5. Es soll untersucht werden, ob die Fluorogermanatanionen eher als Monomere (oktaedrisches [GeF6]2– oder trigonal-bipyramidales [GeF5]–) vorliegen oder Tendenzen zur Polymerisation zeigen, wie es schon von Bartlett für zwei Strukturen beschrieben wurde.[21] 4 Ergebnisse und Diskussion 3 Ergebnisse und Diskussion 3.1 Protonierung starker organischer Säuren Zu den stärksten bisher bekannten organischen Säuren zählen die Trifluormethansulfonsäure (H0 = –14.1) und die Fluorsulfonsäure (H0 = –15.1).[11] Ihre Säurestärken sind vergleichbar mit der von Perchlorsäure (H0 = –13) und sie sind per Definition Supersäuren, da ihre H0Werte niedriger als der von konzentrierter Schwefelsäure sind (H0 = –12).[11,22] Eine Protonierung der Fluorsulfonsäure, wurde erst kürzlich im Arbeitskreis Kornath gezeigt, und das Produkt konnte als [FSO3H2]+[SbF6]– isoliert und charakterisiert werden.[19] Die Trifluormethansulfonsäure wurde 1954 entdeckt und ist aufgrund ihrer nichtoxidierenden Eigenschaften und ihrer hohen Stabilität gegenüber Hitze, Wasser und Alkohol seitdem Gegenstand intensiver Forschung.[23-24] Die Säure, ihre Salze und ihre organischen Derivate (bekannt als Triflate) sind Verbindungen, welche ein breites Anwendungsspektrum unter anderem in Polymerisationsreaktionen und der organischen Synthesechemie besitzen.[25-27] Beispielsweise gelang erst kürzlich Schulz et al. die Verwendung von [CF3SO3(SiMe3)2]+[B(C6F5)4]− als Sylilierungsreagenz.[28] Das Autoprotolysegleichgewicht der Trifluormethansulfonsäure lässt sich wie in Gleichung (3.1) beschreiben. (3.1) [CF3SO(OH)2]+ + [CF3SO3]- 2 CF3SO3H Das Gleichgewicht kann auf die rechte Seite verschoben werden, indem das Anion von starken Lewis-Säuren als Triflat gebunden wird. Hierbei erhöht sich die Konzentration der sauren Spezies gemäß dem Massenwirkungsgesetz. Die größte Säurestärke (H0 = –18.5) wurde bisher mit den Lewis-Säuren B(OSO2CF3)3 und SbF5 gemessen.[11] Aufgrund der begrenzten Löslichkeit von B(OSO2CF3)3 in Trifluormethansulfonsäure ist zu erwarten, dass F 2 F O F Sb O F F S F OH F F F F OH F Sb O F F S F 5 OH F F F + F F Sb O F F O S F OH F F (3.2) Ergebnisse und Diskussion eine noch größere Acidität von Trifluormethansulfonsäure-Systemen mit SbF5 erreicht werden kann. In Anlehnung an Gleichung (3.1) sollte sich bei hohen SbF5-Konzentrationen das Gleichgewicht gemäß Gleichung (3.2) einstellen.[11] Tatsächlich, haben Mootz et al. eine Reaktion von Trifluormethansulfonsäure und SbF5 beschrieben, in welcher ein Addukt gebildet wurde, welches kristallographisch nachgewiesen werden konnte (Abbildung 3.1).[29] Abbildung 3.1: Kristallstruktur von [CF3SO3H⋅SbF5].[29] Für seine Arbeiten auf dem Gebiet der Carbokationen erhielt Olah 1994 den Nobelpreis. Das Interesse an diesen Verbindungen ist dadurch begründet, dass viele Intermediate bei Reaktionsmechanismen der organischen Chemie Carbokationen sind.[30] Dihydroxycarbeniumionen sind eine bekannte und gut charakterisierte Substanzklasse. Bisher konnten einige Dihydroxycarbeniumsalze, unter anderem die protonierte Essigsäure und die protonierte Ameisensäure, isoliert und charakterisiert werden.[31-32] Durch Substitution der Wasserstoffatome der Methylgruppe durch Fluoratome nimmt der –I-Effekt der Methylgruppe mit steigender Anzahl an Fluoratomen zu und dadurch die Stabilisierung des Carbokations ab.[33] Olah gelang ein erster Nachweis der Mono-, Di- und Trifluormethyldihydroxycarbeniumionen in supersauren Lösungen von HSO3F/SbF5/SO2 mittels NMR-Spektroskopie. Er konnte jedoch keine strukturelle Aufklärung erreichen.[34] Eine Isolierung der Salze ermöglicht zusätzlich die Einkristall-Röntgenstrukturanalyse und die Durchführung schwingungsspektroskopischer Untersuchungen, um genauere Informationen über die Struktur der Salze zu erhalten. Das [CF3C(OH)2]+-Kation ist aufgrund seiner Ähnlichkeit zur Fluorameisensäure ein für die Forschung sehr interessantes Molekül, da diese bisher nicht in reiner Form isoliert werden konnte und die Elektronegativität der CF3-Gruppe ähnlich der des Fluoratoms ist.[35-36] 6 Ergebnisse und Diskussion Von Olah wurde der Zerfall von CF3CO2H in supersauren Lösungen zu CF4 untersucht, in welchem er sogar ein Dikation als Intermediat postulierte.[37] Die Trifluoressigsäure besitzt einen pKs-Wert von 0.23 und ist somit bereits 100.000 mal saurer als Essigsäure.[38] Untersuchungen der Di- und Trifluoressigsäure sowie der Trichloressigsäure als Hexafluoroantimonat-Salze wurden bereits von Seelbinder im Rahmen seiner Promotion im Arbeitskreis Minkwitz durchgeführt.[39] Es gelang aber bisher nicht die protonierte Monofluoressigsäure strukturell zu untersuchen. In dieser Arbeit konnte [CH2FC(OH)2]+[AsF6]− synthetisiert und charakterisiert werden. Zusätzlich wurden die Salze [CF3C(OH)2]+[AsF6]−, [CHF2C(OH)2]+[AsF6]− und [CCl3C(OH)2]+[AsF6]− untersucht, da die Qualität der Datensätze der Hexafluoroantimonate nicht zufrieden stellend waren.[39] Eine erneute Diskussion der schwingungsspektroskopischen Ergebnisse wird durchgeführt, da die bisherige Interpretation der Spektren zum Teil unschlüssig war. Aufgrund dessen wurden auch zusätzliche Isotopenstudien und theoretische Berechnungen durchgeführt. 7 Ergebnisse und Diskussion 3.1.1 Trifluormethansulfonsäure 3.1.1.1 Bildung von [CF3SO3H2]+[SbF6] − [CF3SO3H2]+[SbF6]− wurde quantitativ in drei Schritten nach folgenden Gleichungen synthetisiert. 2 HF + SbF5 -40 °C (CF3SO2)2O + HF CF3SO3H (3.3) [H2F]+[SbF6]-40 °C, [H2F]+[SbF6]- + [H2F]+[SbF6]- -50 °C CF3SO3H + CF3SO2F [CF3SO3H2]+[SbF6]- + HF (3.4) (3.5) Die deuterierte Verbindung wurde analog mit DF anstelle von HF hergestellt. Im ersten Schritt wurde die Supersäure hergestellt, um die höchstmögliche Konzentration von [H2F]+[SbF6]− in der HF-Lösung zu erhalten (Gleichung (3.3)). Im zweiten Schritt wurde Trifluormethansulfonsäureanhydrid, welches flüchtiger als CF3SO3H ist und deshalb leichter quantitativ zu kondensieren, zu der gefrorenen Supersäure kondensiert. Anschließend wurde das Reaktionsgemisch erwärmt und die Supersäure schmolz bei –40 °C, das Anhydrid wurde gespaltet und bildete CF3SO2F und CF3SO3H, wobei [H2F]+[SbF6]− als Katalysator diente (Gleichung (3.4)). CF3SO3H wurde dann, wie in Gleichung (3.5) beschrieben, protoniert. Nach langsamem Entfernen von HF und CF3SO2F bei –78 °C bildeten sich farblose Kristalle von [CF3SO3H2]+[SbF6]−, welche bis –50 °C beständig waren und sich für eine EinkristallRöntgenstrukturanalyse eigneten. Umsetzungen der Trifluormethansulfonsäure mit den supersauren Systemen HF/AsF5 und HF/GeF4 waren nicht erfolgreich. Wahrscheinlich ist die Säurestärke dieser Systeme nicht ausreichend, um CF3SO3H zu protonieren oder ein eventuell entstandenes Salz bei diesen Temperaturen nicht beständig. Die Bildung eines Addukts, wie bei Mootz et al. beschrieben, wurde in Gegenwart von HF nicht beobachtet.[29] 3.1.1.2 Röntgenstrukturanalyse [CF3SO3H2]+[SbF6]− kristallisiert in der triklinen Raumgruppe P–1 (Nr. 2) mit zwei Formeleinheiten in der Elementarzelle. Ausgewählte Bindungslängen und -winkel sind in 8 Ergebnisse und Diskussion Tabelle 3.1 aufgelistet. Die Struktur des [CF3SO3H2]+-Kations und seine − Wasserstoffbrückenbindungen zu den benachbarten Fluoratomen der [SbF6] -Anionen sind in Abbildung 3.2 dargestellt. Abbildung 3.3 zeigt einen Ausschnitt aus der Kristallpackung. Abbildung 3.2: Struktur von [CF3SO3H2]+ im Kristall mit Wasserstoffbrückenbindungen zu den Fluoratomen der benachbarten [SbF6]–-Anionen (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome, Symmetrieoperationen: a: x,y,z+1, b: x,y+1,z). Alle Atome einschließlich der Wasserstoffatome wurden mittels Differenz-Fourier-Synthese gefunden und frei verfeinert. Alle Nicht-Wasserstoffatome wurden anisotrop verfeinert. Das [CF3SO3H2]+ besitzt, wie erwartet, eine kurze S=O-Doppelbindung mit 140.5(2) pm, welche mit der S=O-Doppelbindung in CF3SO3H vergleichbar ist. Die zwei S–O-Abstände mit Längen von 148.3(2) und 150.5(2) pm sind für eine durchschnittliche formale Einfachbindung (d(S–O) = 170 pm) verhältnismäßig kurz. Der Grund hierfür ist die elektronenziehende CF3-Gruppe. Dieser kurze S–O-Abstand zeigt sich auch bei der unprotonierten Säure. Der C–S-Abstand von 185.5(2) pm ist gegenüber der unprotonierten Trifluormethansulfonsäure etwas verlängert (183.2(3) pm). Die C–F-Bindungslängen und -winkel liegen in den typischen Bereichen. Die Geometrie der CF3-Gruppe wird durch eine Protonierung nicht beeinflusst. Im Kristall ist jedes Kation zu den zwei benachbarten [SbF6]−-Anionen über zwei starke, fast lineare Wasserstoffbrückenbindungen mit O···F-Abständen von 242.6(2) und 252.5(2) pm verknüpft. Diese Verknüpfung führt zu ([CF3SO3H2]+[SbF6]–)n-Ketten entlang der [011]Achse. Die Symmetrie des [SbF6]−-Anions ist von einem idealen Oktaeder zu einer gestreckten quadratischen Bipyramide erniedrigt. Die Sb–F-Abstände in der äquatorialen Ebene sind annähernd gleich lang (von 185.1(2) bis 186.6(2) pm), während die transverknüpfenden Sb–F-Bindungslängen um rund 6 pm länger sind (190.6(2) und 192.7(2) pm). 9 Ergebnisse und Diskussion Tabelle 3.1: C(1)–S(1) S(1)–O(1) S(1)–O(2) S(1)–O(3) C(1)–F(1) C(1)–F(2) C(1)–F(3) O(1)–H(1) O(2)–H(2) O(1)···F(4a) O(2)···F(9b) Ausgewählte Bindungslängen [pm] und -winkel [°] für CF3SO3H, [CF3SO3H2]+[SbF6]–, das berechnete [CF3SO3H2]+ und die berechnete [CF3SO3H2(HF)2]+-Einheit + CF3SO3H [40] exp. [CF3SO3H2] [SbF6] exp. 183.5(4) 153.4(3) 142.7(3) 141.4(4) 131.6(5) 131.3(4) 131.3(5) 99(9) 185.5(2) 150.5(2) 148.3(2) 140.5(2) 130.5(2) 130.7(2) 132.0(2) 84(4) 73(3) 252.5(2) 242.6(2) – O(1)–S(1)–C(1) 101.2(2) 102.26(9) O(2)–S(1)–C(1) 105.2(2) 101.1(2) O(3)–S(1)–C(1) 107.4(2) 111.8(2) F(1)–C(1)–S(1) 109.2(3) 109.7(2) F(2)–C(1)–S(1) 109.5(3) 107.5(2) F(3)–C(1)–S(1) 110.2(3) 107.6(2) S(1)–O(1)–H(1) 115(5) 117(3) S(1)–O(2)–H(2) 114(2) O(1)–H(1)···F(4a) 173.09(4) O(2)–H(2)···F(9b) 162.84(4) [a] berechnet mit PBE1PBE/6-311G(3df,3pd); [b] berechnet Symmetrieoperationen: a: x,y,z+1; b: x,y+1,z. Abbildung 3.3: [CF3SO3H2] [a] C1, ber. 188.8 153.7 152.3 139.8 130.6 130.1 129.5 97.5 97.5 + [CF3SO3H2(HF)2] [b] C1, ber. + 187.3 151.2 151.2 140.5 130.4 130.4 130.7 100.5 100.5 254.1 254.1 103.8 105.3 113.2 106.7 106.2 107.1 114.6 117.3 mit 104.4 104.4 111.0 108.6 106.8 106.8 115.3 115.3 177.7 177.7 PBE1PBE/6-311G++(3df,3pd); Kristallpackung von [CF3SO3H2]+[SbF6]– (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). 10 Ergebnisse und Diskussion 3.1.1.3 Theoretische Berechnungen Die DFT-Berechnungen der Gasphasenstruktur der freien [CF3SO3A2]+-Kationen (A = H, D) wurden mit der Methode PBE1PBE und dem 6-311G(3df,3pd)-Basissatz durchgeführt.[41-43] Anschließend wurden die Schwingungsfrequenzen der relaxierten Strukturen sowie die IRund Raman-Intensitäten berechnet. Die berechnete Struktur des freien Kations stimmt zufriedenstellend mit den experimentellen Daten der Kristallstruktur überein (Tabelle 3.1), zeigt jedoch große Abweichungen Schwingungsfrequenzen zwischen (Tabelle 3.2). den Insbesondere berechneten die und gefundenen O–H-Streckschwingungen (νs = 3589 und νas = 3578 cm–1) werden bis zu 500 cm–1 überschätzt im Vergleich zu den experimentellen Daten (νs = 3076 cm–1). Der Grund hierfür sind die starken Wasserstoffbrückenbindungen, welche in der Kristallstruktur zu finden sind. Die Bildung von Wasserstoffbrückenbindungen führt im Allgemeinen zu einer Erniedrigung der korrespondierenden O–H-Streckschwingung. Zur Bestätigung dieser Annahme wurden zum freien [CF3SO3A2]+-Kation (A = H, D) zwei HF-Moleküle hinzugefügt und diese [CF3SO3A2(HF)2]+-Einheit mit der Methode PBE1PBE und dem 6-311++G(3df,3pd)-Basissatz berechnet (Abbildung 3.4).[41-44] Die berechnete Geometrie der [CF3SO3A2(HF)2]+-Einheit ist vergleichbar zu der in der Kristallstruktur gefundenen. Die Addition der zwei HF-Moleküle verändert die Geometrieparameter nur wenig, beeinflusst aber deutlich die Schwingungsfrequenzen. Insbesondere die O–H-Streckschwingungen des Kations (νs = 3059 und νas = 3006 cm–1) werden aufgrund der Bildung von S–O···H···F–H-Wasserstoffbrücken in einen Bereich, der gut mit den experimentellen Daten übereinstimmt, rotverschoben. Insgesamt stimmen die beobachteten und die berechneten Schwingungen der [CF3SO3A2(HF)2]+-Einheiten gut überein, obwohl die [CF3SO3A2(HF)2]+-Einheiten nur eine sehr vereinfachte Simulation der Festphase darstellen. Abbildung 3.4: Quantenchemisch berechnete Struktur der [CF3SO3H2(HF)2]+-Einheit. 11 Ergebnisse und Diskussion 3.1.1.4 Schwingungsspektren Die IR- und Raman-Spektren von [CF3SO3H2]+[SbF6]− und [CF3SO3D2]+[SbF6]− sind in Abbildung 3.5 und Abbildung 3.6 dargestellt. Die dazugehörigen Schwingungsfrequenzen sind in Tabelle 3.2 und Tabelle 3.3 zusammen mit den quantenchemisch berechneten Frequenzen von [CF3SO3A2(HF)2]+ (A = H, D) aufgelistet. Die Zuordnung der Schwingungsbanden erfolgt Trifluormethansulfonsäure und durch den Vergleich theoretisch mit dem berechneten Spektrum Schwingungen der der [CF3SO3A2(HF)2]+-Einheiten (A = H, D).[45-47] Das Kation besitzt C1-Symmetrie, weshalb 24 Grundschwingungen im Spektrum zu erwarten sind. Beide Spektren zeigen Schwingungsbanden für die CF3-Gruppe, die vergleichbar mit denen von CF3SO3H sind. Die νs(OH)-Bande tritt bei 3076 cm–1 und die νs(OD)-Bande bei 2162 cm–1 auf. Beide Frequenzen stimmen gut mit den berechneten Werten überein und sind im Einklang mit der Teller-Redlich-Regel (ν(OH)/ν(OD) = 1.42, theoretisch 1.41).[48] Die breite Form der Banden ist typisch für Streckschwingungen von O–H-Gruppen, die an Wasserstoffbrücken beteiligt sind.[49] Abbildung 3.5: IR- und Raman-Spektrum von [CF3SO3H2]+[SbF6]−. 12 Ergebnisse und Diskussion Abbildung 3.6: IR- und Raman-Spektrum von [CF3SO3D2]+[SbF6]−. Die Streckschwingung der S=O-Doppelbindung ist bei ca. 1400 cm–1 zu finden, was vergleichbar mit der S–O-Schwingung im [H3SO4]+ und [H2SO3F]+ ist.[19,49] Die zwei S–O-Streckschwingungen bei 1073 und 956 cm–1 können als Schwingungen zwischen dem Schwefelatom und den Hydroxygruppen beschrieben werden. Die C–S-Streckschwingung tritt bei der bemerkenswert niedrigen Wellenzahl von 267 cm–1 auf. Das ist noch niedriger als im CF3SO3H beobachtet (312 cm–1). Offensichtlich führt die Protonierung der Trifluormethansulfonsäure zu einer Schwächung der C–S-Bindung, was in Übereinstimmung mit den kristallographischen Ergebnissen ist.[46,50] Für ein [SbF6]−-Anion mit idealer oktaedrischer Geometrie werden zwei Banden im IRSpektrum und drei Banden im Raman-Spektrum mit gegenseitiger Auslöschung erwartet. Wie schon in der Kristallstruktur bestätigt ist das [SbF6]−-Anion mindestens bis zur D4hSymmetrie verzerrt, für welche fünf IR-aktive Schwingungen und fünf Schwingungen im Raman-Spektrum mit Alternativverbot erwartet werden. Diese erniedrigte Symmetrie erklärt die große Anzahl an Schwingungen im niedrigen Wellenzahlenbereich. 13 Ergebnisse und Diskussion Tabelle 3.2: Beobachtete Schwingungsfrequenzen [cm–1] von [CF3SO3H2]+[SbF6]− und CF3SO3H sowie berechnete Schwingungsfrequenzen von [CF3SO3H2(HF)2]+ und [CF3SO3H2]+ + [CF3SO3H2] [SbF6] IR − [CF3SO3H2(HF)2] Raman 3076 (w,br) 1392 (s) 1400 (22) 1261 (vs) 1239 (7) 1163 1073 989 956 777 (m) (m) (w) (s) (vw) 610 576 524 483 469 (m) (m) (w) (vw) (w) 1211 (10) 1162 (14) 957 (41) 779 (62) 607 567 525 483 461 338 321 267 (10) (20) (0.9) (11) (8) (34) (28) (15) C1, ber. + [a] [CF3SO3H2] C1, ber. + [b] 3059 (302/196) 3006 (3793/21) 1421 (205/10) 3589 (237/68) 3578 (453/33) 1439 (162/8) 1288 1288 1234 1212 1145 1029 1316 1286 1080 1012 1110 945 (229/0.2) (202/1) (50/3) (85/5) (178/5) (287/1) 891 758 215 203 576 550 552 443 432 335 310 262 157 141 47 (226/4) (80/13) (124/0.8) (106/0.4) (80/1) (1/1) (2/2) (11/0.9) (27/2) (4/2) (1/2) (3/6) (21/0.3) (8/1) (4/0.01) 959 778 757 714 593 560 558 490 471 329 325 275 173 171 37 (223/0.2) (198/1) (38/1) (121/2) (153/7) (311/2) (252/9) (64/10) (81/3) (14/0.1) (143/1) (5/2) (1/2) (25/2) (0.2/2) (3/2) (5/2) (8/5) (2/0.2) (7/0.03) (0.01/0.03) CF3SO3H Lit. Zuordnung [46] 3000 1400 1244 1232 1122 1152 930 775 499 620 570 478 340 312 νs(OH) νas(OH) ν(S=O) ν(S=O) νas(CF3) νas(CF3) δ(SOH) δ(SOH) νs(CF3) νas(S–O) νs(S–O) δ(CF3) δ(SOH) δ(SOH) δ(SO3) δ(CF3) δ(CF3) δ(SO3) δ(SO3) δ(SO3/CF3) δ(SO3/CF3) ν(C–S) δ(SO3/CF3) δ(SO3/CF3) τ 691 (s) 665 (100) − 592 (9) [SbF6] 384 (10) 299 (46) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.97 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 14 Ergebnisse und Diskussion Tabelle 3.3: Beobachtete Schwingungsfrequenzen [cm–1] von [CF3SO3D2]+[SbF6]− sowie berechnete Schwingungsfrequenzen von [CF3SO3D2(HF)2]+ und [CF3SO3D2]+ + – [CF3SO3D2] [SbF6] IR Raman 2162 (w,br) 1389 (m) 1260 (vs) 1167 1108 989 950 (m) (m) (vw) (s) 813 (w) 774 (w) 585 (s) 610 562 530 472 (m) (m) (m) (w) 2295 (10) 2333 (2) 1408 (15) 1252 (7.4) 1167 (15) 982 (10) 952 (25) 818 (9) 776 (42) 609 (7) 566 (14) 472 438 336 318 267 (7) (10) (24) (22) (13) [CF3SO3D2(HF)2] [a] C1, ber. + [CF3SO3D2] [b] C1, ber. + Zuordnung 2241 2192 1420 1288 1285 1144 1077 (152/95) (1977/10) (184/9) (205/1) (253/0.4) (148/6) (135/3) 2614 2605 1435 1315 1285 1106 998 (133/33) (260/15) (155/8) (233/0.3) (206/0.5) (181/4) (154/3) νs(OD) νas(OD) ν(S=O) νas(CF3) νas(CF3) νs(CF3) νas(S–O) 955 887 874 775 575 533 591 563 554 457 445 327 323 274 173 169 37 (238/10) (73/2) (104/0.2) (22/13) (48/2) (16/0.1) (170/2) (1/2) (6/1) (31/1) (6/1) (2/2) (6/2) (8/5) (2/0.2) (7/0.02) (0.01/0.03) 906 790 741 761 185 169 572 549 548 427 393 324 307 261 141 118 47 (179/7) (93/2) (108/4) (27/10) (13/0.1) (32/0.1) (93/1) (2/1) (2/1) (18/1) (23/2) (1/2) (0.5/2) (9/6) (80/0.5) (7/0.8) (4/0.04) νs(S–O) δ(SOD) δ(SOD) δ(CF3) δ(SOD) δ(SOD) δ(SO3) δ(CF3) δ(CF3) δ(SO3) δ(SO3) δ(SO3/CF3) δ(SO3/CF3) ν(C–S) δ(SO3/CF3) δ(SO3/CF3) τ 691 (vs) 664 (100) 585 (8) − [SbF6] 504 (8) 378 (9) 300 (42) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.97 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 15 Ergebnisse und Diskussion 3.1.2 Monofluoressigsäure 3.1.2.1 Bildung von [CH2FC(OH)2] +[AsF6] − [CH2FC(OH)2]+[AsF6]− wurde quantitativ in einem Schritt nach Gleichung (3.6) hergestellt. Die deuterierte Verbindung wurde analog mit DF anstelle von HF synthetisiert. -20 °C, HF CH2FCO2H + [H2F]+[AsF6]- [CH2FC(OH)2]+[AsF6]- + HF (3.6) -60 °C, HF 2 HF + AsF5 Monofluoressigsäure wurde in einem Reaktor vorgelegt und anschließend wurden im hohen Überschuss HF (150 : 1) und AsF5 (1.5 : 1) kondensiert. Als das Reaktionsgemisch langsam auf –60 °C aufgetaut wurde, bildete sich die Supersäure [H2F]+[AsF6]−. Bei weiterem Erwärmen auf –20 °C löste sich die Monofluoressigsäure in der supersauren Lösung und wurde protoniert. Nach langsamem Entfernen von HF und überschüssigem AsF5 bei –78 °C bildeten sich farblose Kristalle von [CH2FC(OH)2]+[AsF6]−, welche bis –20 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 3.1.2.2 Röntgenstrukturanalyse [CH2FC(OH)2]+[AsF6]− kristallisiert in der monoklinen Raumgruppe P21/c (Nr. 14) mit vier Formeleinheiten in der Elementarzelle. Ausgewählte Bindungslängen und -winkel des Edukts, von [CH2FC(OH)2]+[AsF6]− und der berechneten [CH2FC(OH)2(HF)2]+-Einheit sind in Tabelle 3.4 + aufgelistet. Abbildung 3.7 zeigt die asymmetrische Einheit von − [CH2FC(OH)2] [AsF6] und Abbildung 3.8 zeigt einen Ausschnitt aus der Kristallpackung. Alle Atome einschließlich der Wasserstoffatome wurden mittels Differenz-Fourier-Synthese gefunden und frei verfeinert. Alle Nicht-Wasserstoffatome wurden anisotrop verfeinert. Die Kristallstruktur der Monofluoressigsäure wurde 1972 von Kanters und Kroon gelöst.[51] 16 Ergebnisse und Diskussion Abbildung 3.7: Asymmetrische Einheit [CH2FC(OH)2]+[AsF6]− im Kristall (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). Das [CH2FC(OH)2]+ besitzt zwei gleich lange C–O-Bindungen (126.6(3) und 126.4(3) pm) mit Bindungsabständen, welche zwischen einer typischen Einfach- und einer Doppelbindung (d(C–O) = 143 pm und d(C=O) = 119 pm) liegen.[52] Die C–C-Bindung ist gegenüber der unprotonierten Säure leicht verlängert, wobei die Länge der C–F-Bindung und die Geometrie der CH2F-Gruppe von der Protonierung nicht beeinflusst werden. Abbildung 3.8: Kristallpackung von [CH2FC(OH)2]+[AsF6]– (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). 17 Ergebnisse und Diskussion Tabelle 3.4: Ausgewählte Bindungslängen [pm] und -winkel [°] für CH2FCO2H, [CH2FC(OH)2]+[AsF6]− und die berechnete [CH2FC(OH)2(HF)2]+-Einheit CH2FCO2H exp. C(2)–F(7) C(2)–H(3) C(2)–H(4) C(1)–C(2) C(1)–O(1) C(1)–O(2) O(1)–H(1) O(1)–H(2) H(1)···F(1) H(2)···F(2a) O(1)···F(1) O(2)···F(2a) + [CH2FC(OH)2] [AsF6] [51] − [CH2FC(OH)2(HF)2] exp. 137(1) 111(7) 103(7) 146(1) 131(1) 122(1) 100(7) 137.2(2) 93(2) 96(2) 148.1(3) 126.6(3) 126.4(3) 68(3) 63(2) 190(3) 202(2) 257.0(2) 261.1(2) C(1)–O(1)–H(1) 113(4) 114(2) C(1)–O(2)–H(2) 111(3) O(1)–C(1)–O(2) 123.9(5) 119.8(2) C(2)–C(1)–O(1) 112.3(5) 116.18(19) C(2)–C(1)–O(2) 123.8(5) 123.95(19) F(7)–C(2)–C(1) 111.4(5) 108.51(17) F(7)–C(2)–H(3) 109(4) 113.5(13) C(1)–C(2)–H(3) 103(4) 110.4(13) F(7)–C(2)–H(4) 106(4) 107.4(12) C(1)–C(2)–H(4) 108(4) 108.8(12) H(3)–C(2)–H(4) 119(6) 108.1(18) O(1)–H(1)···F(1) 167(3) O(2)–H(2)···F(2a) 159(3) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd); Symmetrieoperationen: a: –x,–y+1,–z+1. Abbildung 3.9: C1, ber. + [a] 134.1 109.8 109.8 149.8 127.2 125.7 98.7 99.6 114.7 114.7 119.8 118.4 121.9 110.8 110.7 108.1 110.7 108.1 108.2 Wasserstoffbrückenbindungen in [CH2FC(OH)2]+[AsF6]– (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome, Symmetrieoperationen: a: –x,–y+1,–z+1). 18 Ergebnisse und Diskussion Die zwei aciden Protonen der Hydroxygruppen sind strukturell nicht äquivalent. Das eine Proton befindet sich in cis-, das andere in trans-Position zur CH2F-Gruppe. Jedes Kation ist zu zwei benachbarten [AsF6]−-Anionen über zwei starke Wasserstoffbrückenbindungen mit O···F-Abständen von 257.0(2) und 261.1(2) pm und O–H···F-Winkeln von 167(3)° bzw. 159(3)° verknüpft. Diese Verknüpfung führt zu 4-gliedrigen Ringen aus 16 Atomen (Abbildung 3.9). Die Struktur des [AsF6]−-Anions ist verzerrt und weicht somit von der idealen Oktaedersymmetrie ab. Die As–F-Abstände, der an den Wasserstoffbrückenbindungen beteiligten Atome, sind leicht verlängert (173.23(13) und 174.02(12) pm), wobei die übrigen As–F-Abstände zwischen 168.83(13) und 171.38(11) pm liegen. Die Fluoratome des Anions, die an den Wasserstoffbrückenbindungen beteiligt sind, befinden sich in cis-Position zueinander. Der F(1)–As(1)–F(2)-Winkel weicht daher mit 88.58(7)°von den idealen 90° ab. Abbildung 3.10: Koordination zweier [AsF6]−-Anionen zur C(OH)2-Gruppe (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome, Symmetrieoperationen: a: 1–x,0.5+y,1.5+z; b: –x, 0.5+y,1.5+z). Die C(OH)2-Gruppe ist planar (Winkelsumme: 359.98°). Sie ist durch die Protonierung sehr elektronenarm und wird von oben und unten durch zwei Fluoratome der [AsF6]−-Anionen koordiniert (Abbildung 3.10). Die C···F-Abstände betragen 279.8 und 293.9 pm, was deutlich unter der Summe der Van-der-Waals-Radien von 320 pm liegt. Diese Koordination wird durch die negative Ladung der Fluoratome bedingt, welche in das leere pz-Orbital des Kohlenstoffs donieren. Beide Fluoratome stehen annähernd in einer Linie über dem Kohlenstoffatom. Diese Art der Wechselwirkung wurde bereits bei Christe in [(CH3)2CF]+[AsF6]− beschrieben.[53] 19 Ergebnisse und Diskussion 3.1.2.3 Theoretische Berechnungen Die DFT-Berechnungen der Gasphasenstrukturen der [CH2FC(OA)2(HF)2]+-Einheiten (A = H, D) wurden mit der Methode PBE1PBE und dem 6-311G++(3df,3pd)-Basissatz durchgeführt (Abbildung 3.11).[41-44] Anschließend wurden die Schwingungsfrequenzen der relaxierten Strukturen, sowie deren IR- und Raman-Intensitäten berechnet. Eine Berechnung der freien [CH2FC(OA)2]+-Kationen führte nur zu bedingt befriedigenden schwingungsspektroskopischen Ergebnissen, was bereits im Kapitel der Trifluormethansulfonsäure diskutiert wurde (siehe 3.1.1.4). Abbildung 3.11: Quantenchemisch berechnete Struktur der [CH2FC(OH)2(HF)2]+-Einheit. Die starken Wasserstoffbrückenbindungen, welche in der Kristallstruktur zu finden sind, werden durch die [CH2FC(OA)2(HF)2]+-Einheiten sehr gut wiedergegeben. Durch die Simulation von C–O–H···F–H-Wasserstoffbrückenbindungen werden die O–H-Deformationsschwingungen bedeutend besser berechnet (932 und 841 cm–1) und sind im Vergleich zu denen des berechneten freien Kation um ca. 200 cm–1 blau verschoben. Insgesamt stimmen die beobachteten [CH2FC(OA)2(HF)2]+-Einheiten und die berechneten zufriedenstellend überein, + Schwingungen der obwohl die [CH2FC(OA)2(HF)2] -Einheiten nur eine sehr vereinfachte Simulation der Festphase darstellen. 20 Ergebnisse und Diskussion 3.1.2.4 Schwingungsspektren Die IR- und Raman-Spektren von [CH2FC(OH)2]+[AsF6]− und [CH2FC(OD)2]+[AsF6]− sind in Abbildung 3.12 und Abbildung 3.13 wiedergegeben. Die dazugehörigen Schwingungsfrequenzen sind in Tabelle 3.5 und Tabelle 3.6, zusammen mit den quantenchemisch berechneten Frequenzen von [CH2FC(OA)2(HF)2]+ (A = H, D) aufgelistet. Das Kation besitzt C1 Symmetrie, weshalb 21 Grundschwingungen im Spektrum zu erwarten sind. Für die Zuordnung der Schwingungsbanden wird mit dem Spektrum der dimeren Monofluoressigsäure verglichen, da diese durch ihre Wasserstoffbrückenbindungen der Festphase stärker ähnelt als die monomere Form. Zusätzlich werden die theoretisch berechneten Schwingungen der [CH2FC(OA)2(HF)2]+-Einheiten (A = H, D) berücksichtigt.[54] Beide Spektren zeigen Frequenzen für die CH2F-Gruppe, die vergleichbar mit denen in CH2FCO2H sind. Die ν(OH) treten nur sehr schwach bei 3092 und 2995 cm–1 im IRSpektrum auf, man kann sie aber im Raman-Spektrum als breite Bande unter den CHValenzschwingungen vermuten. Die ν(OD) sind bei 2303 und 2274 cm–1 zu finden und stimmen gut Abbildung 3.12: mit den berechneten Werten überein. Die IR- und Raman-Spektrum von [CH2FC(OH)2]+[AsF6]–. 21 breite Form der Ergebnisse und Diskussion Linien ist typisch für Streckschwingungen Wasserstoffbrücken beteiligt sind. von O–H-Gruppen, welche an [49] Im Raman-Spektrum von CH2FCO2H sind zwei Streckschwingungen für die C(OH)2Gruppe bei 1662 und 1570 cm–1 zu finden. Im Vergleich zum Edukt zeigt sich hier sehr schön der Effekt der Protonierung, welcher die Streckschwingung der C=O-Doppelbindung durch Verlängerung dieser Bindung rotverschiebt und diejenige der C–O-Einfachbindung aufgrund einer Verkürzung dieser Bindung um über 100 cm–1 blauverschiebt. Das steht auch im Einklang mit den in der Kristallstruktur gefundenen Bindungslängen für die C(OH)2Gruppe. Die ν(C–C) ist schwach rotverschoben (ca. 40 cm–1) und bestätigt die Schwächung der Bindung. Die große Abweichung des Anions von der idealen Oktaedersymmetrie spiegelt sich, wie schon im kristallographischen Teil beschrieben, auch im Schwingungsspektrum wieder. Durch die erniedrigte Symmetrie, sind daher für das Anion in [CH2FC(OH)2]+[AsF6]− acht und für [CH2FC(OD)2]+[AsF6]− sieben Schwingungsbanden zu beobachten. Abbildung 3.13: IR- und Raman-Spektrum von [CH2FC(OD)2]+[AsF6]−. 22 Ergebnisse und Diskussion Tabelle 3.5: Beobachtete Schwingungsfrequenzen [cm–1] von [CH2FC(OH)2]+[AsF6]− und CH2FCO2H sowie berechnete Schwingungsfrequenzen von [CH2FC(OH)2(HF)2]+ und [CH2FC(OH)2]+ + [CH2FC(OH)2] [AsF6] IR − Raman 3092 (vw) 2995 (vw) [CH2FC(OH)2(HF)2] C1, ber. + [a] [CH2FC(OH)2] C1, ber. (764/157) (2054/65) (5/49) (33/117) [b] 3021 (13) 2977 (35) 3234 3192 3048 3000 1663 (m) 1633 (m) 1570 (m) 1662 (2) 1677 (325/0.4) 1667 (237/2) 1570 (8) 1576 (197/6) 1554 (179/4) 1547 1419 1360 1272 1244 1214 1183 1093 1023 927 889 822 791 635 1549 (2) 1420 (39) 1364 (6) 3669 3609 2998 2959 + (226/49) (296/61) (4/48) (36/122) CH2FCO2H Lit. Zuordnung [54] 3140 2957 2865 ν(OH) ν(OH) ν(CH2) ν(CH2) 2356 (w) 2054 (vw) 1729 (m) (m) (vw) (vw) (vw) (w) (m) (m) (vw) (vw) (m) (w) (w) (s) 497 (m) 1250 (30) 1194 (16) 1096 (8) 1023 (3) 893 (61) 639 (8) 507 (25) ν(C=O) ν(CO2) 1447 ν(CO2) ν(C−O) 1416 1355 1285 1237 (61/9) (2/1) (108/0.6) (2/3) 1408 1360 1174 1256 (47/10) (61/1) (83/1) (0.2/4) 1302 1219 1256 792 δ(CH2F) δ(CH2F) δ(COH)in plane δ(CH2F) 1198 1147 1016 924 892 886 (249/11) (161/1) (2/0.1) (5/0.01) (14/8) (191/0.02) 1161 1194 1009 744 865 607 (167/6) (102/3) (1/0.2) (76/1) (16/5) (207/1) 1076 770 899 931 δ(COH)in plane ν(CF) δ(CCO) δ(COH)out of plane ν(CC) δ(COH)out of plane 645 540 514 275 123 (6/1) (2/0.09) (23/3) (21/0.05) (4/2) 608 492 464 252 101 (43/1) (0.1/0.3) (13/3) (3/0.2) (4/0.4) 646 546 498 483 284 δ(CO2) δ(CO2/CH2F) δ(CO2/CH2F) δ(CO2/CH2F) τ 291 (3) 716 (41) 702 (14) 671 (100) 574 (25) − [AsF6] 548 (14) 403 (3) 391 (2) 372 (59) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.98 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 713 698 683 565 (s,sh) (vs,sh) (vs,br) (m) 23 Ergebnisse und Diskussion Tabelle 3.6: Beobachtete Schwingungsfrequenzen [cm–1] von [CH2FC(OD)2]+[AsF6]− sowie berechnete Schwingungsfrequenzen von CH2FC(OD)2(HF)2+ und CH2FC(OD)2+ + [CH2FC(OD)2] [AsF6] IR 3020 (vw) 2972 (vw) 2261 1702 1626 1532 1431 1411 1357 1326 1263 1192 1078 (w,br) (w) (w) (vw) (vw) (vw) (vw) (vw) (vw) (vw) (m) 1016 916 890 839 (w) (w) (m,sh) (m) − C1, ber. Raman [a] + CH2FC(OD)2 C1, ber. + Zuordnung [b] (21) (42) 3048 (5/49) 3000 (24/126) 2998 (5/48) 2959 (33/124) ν(CH2) ν(CH2) (14) (7) 2361 (403/67) 2331 (1090/32) 2674 (127/23) 2628 (176/28) ν(OD) ν(OD) 1647 (2) 1536 (11) 1666 (379/0.4) 1539 (107/7) 1649 (291/2) 1524 (110/5) ν(CO2) ν(CO2) 1416 (32) 1359 (8) 1412 (59/10) 1348 (18/1) 1404 (57/11) 1358 (52/1) δ(CH2F) δ(CH2F) 1250 1199 1098 1028 1020 920 890 845 709 1237 (2/3) 1150 (74/3) 1256 (0.4/4) 1193 (87/3) δ(CH2F) ν(CF) 1038 (65/3) 1015 (3/0.1) 1002 (47/5) 1008 (2/0.1) δ(COD)in plane δ(CCO) 3023 2975 2859 2303 2274 (25) (1) (8) (14) (1) (7) (3) (29) (9) 595 (5) 486 (m) CH2FC(OD)2(HF)2 497 (12) 911 842 689 649 602 520 502 266 122 (156/3) (16/7) (6/0.02) (95/0.001) (15/1) (0.06/0.06) (26/3) (20/0.04) (16/7) 883 812 449 620 439 565 433 246 100 (45/2) (26/3) (110/0.1) (35/0.1) (1/0.3) (40/2) (19/2) (3/0.2) (4/0.4) δ(COD)in plane ν(CC) δ(COD)out of plane δ(CO2) δ(COD)out of plane δ(CO2/CH2F) δ(CO2/CH2F) δ(CO2/CH2F) τ 280 (2) 717 (34) 673 (100) 572 (16) − 546 (6) [AsF6] 402 (3) 389 (2) 371 (43) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.98 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 729 (vs) 676 (s) 24 Ergebnisse und Diskussion 3.1.3 Difluoressigsäure 3.1.3.1 Bildung von [CHF2C(OH)2] +[AsF6] − [CHF2C(OH)2]+[AsF6]− wurde quantitativ in einem Schritt nach Gleichung (3.7) synthetisiert. Die deuterierte Verbindung wurde analog mit DF anstelle von HF hergestellt. CHF2CO2H + [H2F]+[AsF]6- -30 °C, HF [CHF2C(OH)2]+[AsF6]- + HF (3.7) -60 °C, HF 2 HF + AsF5 Difluoressigsäure wurde in einem Reaktor vorgelegt und anschließend wurden im großen Überschuss HF (150 : 1) und AsF5 (1.5 : 1) kondensiert. Als das Reaktionsgemisch langsam auf –60 °C aufgetaut wurde, bildete sich die Supersäure [H2F]+[AsF6]−. Beim weiteren Erwärmen auf –30 °C löste sich die Difluoressigsäure in der supersauren Lösung und wurde protoniert. Nach langsamem Entfernen von HF und des überschüssigen AsF5 bei –78 °C bildeten sich farblose Kristalle von [CHF2C(OH)2]+[AsF6]−, welche bis –30 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 3.1.3.2 Röntgenstrukturanalyse [CHF2C(OH)2]+[AsF6]− kristallisiert in der triklinen Raumgruppe P–1 (Nr. 2) mit zwei Formeleinheiten in der Elementarzelle. Ausgewählte Bindungslängen und -winkel des Edukts, des [CHF2C(OH)2]+[AsF6]−-Salzes und der berechneten [CHF2C(OH)2(HF)2]+ Einheit sind in Tabelle 3.7 aufgelistet. Abbildung 3.14 zeigt die asymmetrische Einheit von [CHF2C(OH)2]+[AsF6]− und Abbildung 3.15 zeigt einen Ausschnitt aus der Kristallpackung. Alle Atome einschließlich der Wasserstoffatome wurden mittels Differenz-Fourier-Synthese gefunden und frei verfeinert. Alle Nicht-Wasserstoffatome wurden anisotrop verfeinert. Von der Difluoressigsäure ist eine Gasphasenstruktur bekannt.[55] 25 Ergebnisse und Diskussion Abbildung 3.14: Asymmetrische Einheit [CHF2C(OH)2]+[AsF6]− im Kristall (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). Eine Diskussion der Struktur des Kations wurde bereits für [CHF2C(OH)2]+[SbF6]− durchgeführt. Da die Kationen beider Verbindungen annähernd identisch sind, wird hier darauf verzichtet.[39] Jedes Kation ist zu den zwei benachbarten [AsF6]−-Anionen über zwei starke Wasserstoffbrückenbindungen mit O···F-Abständen von 258.4(2) und 259.7(2) pm sowie mit O–H···F-Winkeln von 173(4)° bzw. 161(3)° verknüpft. Diese Verknüpfung führt zu ([CH2FC(OH)2]+[AsF6]−)n-Ketten entlang der [001]-Achse. Die Struktur des [AsF6]− ist verzerrt und weicht von der idealen Oktaedersymmetrie ab. Die As–F-Bindungslängen, der an den Wasserstoffbrückenbindungen beteiligten Atome, sind Abbildung 3.15: Kristallpackung von [CHF2C(OH)2]+[SbF6]− (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). 26 Ergebnisse und Diskussion um ca. 4 – 5 pm länger (174.42(14) und 176.17(13) pm) als die übrigen As–F-Bindungen (170.00(13) und 171.72(14) pm). Die Fluoratome, welche an den Wasserstoffbrückenbindungen beteiligt sind, befinden sich in cis-Position zueinander. Eine Koordination der Fluoratome der Anionen zum positiven Kohlenstoffzentrum der C(OH)2-Gruppe tritt auch hier auf (Abbildung 3.16). Tabelle 3.7: Ausgewählte Bindungslängen [pm] und -winkel [°] für CHF2CO2H, [CHF2C(OH)2]+[AsF6]− und die berechnete [CHF2C(OH)2(HF)2]+-Einheit CHF2CO2H exp. C(2)-F(7) C(2)-F(8) C(2)-H(3) C(1)-C(2) C(1)-O(1) C(1)-O(2) O(1)-H(1) O(1)-H(2) H(1)···F(1a) H(2)···F(2b) O(1)···F(1a) O(2)···F(2b) + [CHF2C(OH)2] [AsF6] [55] exp. 135.4(7) 135.4(7) 110.2 151.7(6) 134.5(9) 121.2(4) 96(2) 133.8(3) 134.2(3) 93(3) 152.6(3) 125.9(3) 126.0(3) 69(3) 69(3) 190(3) 194(3) 258.4(2) 259.7(2) − [CHF2C(OH)2(HF)2] C1, ber. + [a] 133.2 133.8 109.3 153.5 125.7 125.7 100 100 C(1)-O(1)-H(1) 107 113(3) 115.1 C(1)-O(2)-H(2) 114(2) 116.8 O(1)-C(1)-O(2) 121.3(2) 121.5 C(2)-C(1)-O(1) 110.6(10) 115.5(2) 115.7 C(2)-C(1)-O(2) 123.9(10) 123.2(2) 122.8 F(7)-C(2)-C(1) 108.7(7) 107.76(19) 106.8 F(8)-C(2)-C(1) 108.7(7) 109.0(2) 107.6 H(3)-C(2)-C(1) 109.5 111.0(15) 111.4 F(7)-C(2)-F(8) 108.6(6) 107.76(19) 109.7 F(7)-C(2)-H(3) 111.6(15) 110.8 F(8)-C(2)-H(3) 108.4(15) 110.4 O(1)–H(1)···F(1a) 173(4) O(2)–H(2)···F(2b) 161(3) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd); Symmetrieoperationen: a: –x,–y+1,–z+2; b: –x,–y+1,–z+1. 27 Ergebnisse und Diskussion Abbildung 3.16: Koordination zweier [AsF6]−-Anionen zur C(OH)2-Gruppe (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome, Symmetrieoperationen: a: x,-1+y,z). 3.1.3.3 Theoretische Berechnungen Die DFT-Berechnungen der Gasphasenstrukturen der [CHF2C(OA)2(HF)2]+-Einheiten (A = H, D) wurde mit der Methode PBE1PBE und dem 6-311G++(3df,3pd)-Basissatz durchgeführt (Abbildung 3.17).[41-44] Anschließend wurden die Schwingungsfrequenzen der relaxierten Strukturen, sowie die IR- und Raman-Intensitäten berechnet. Eine Berechnung der freien [CHF2C(OA)2]+-Kationen führte nur zu bedingt befriedigenden schwingungsspektroskopischen Ergebnissen (siehe 3.1.1.4). Die starken Wasserstoffbrückenbindungen, welche in der Kristallstruktur zu finden sind, werden durch die [CHF2C(OA)2(HF)2]+-Einheiten sehr gut wiedergegeben. Durch die Simulation von C–O···H···F–H-Wasserstoffbrückenbindungen werden tendenziell alle Frequenzen besser berechnet. Man erhält somit eine zufriedenstellende Übereinstimmung zwischen den beobachteten und den berechneten Schwingungen der [CHF2C(OA)2(HF)2]+-Einheiten, obwohl die [CHF2C(OA)2(HF)2]+-Einheiten nur eine sehr vereinfachte Simulation der Festphase darstellen. 28 Ergebnisse und Diskussion Abbildung 3.17: Quantenchemisch berechnete Struktur der [CHF2C(OH)2(HF)2]+-Einheit. 3.1.3.4 Schwingungsspektren Die IR- und Raman-Spektren von [CHF2C(OH)2]+[AsF6]− und [CHF2C(OD)2]+[AsF6]− sind in Abbildung 3.18 und Abbildung 3.19 dargestellt. Die dazugehörigen Schwingungsfrequenzen sind in Tabelle 3.8 und Tabelle 3.9, zusammen mit den quantenchemisch berechneten Frequenzen von [CHF2C(OA)2(HF)2]+ (A = H, D) aufgelistet. Das Kation besitzt C1-Symmetrie, weshalb 21 Grundschwingungen im Spektrum zu erwarten sind. Für die Zuordnung der Schwingungsbanden wird mit dem Spektrum der dimeren Difluoressigsäure verglichen, da diese durch ihre Wasserstoffbrückenbindungen der Festphase stärker ähnelt als die monomere Form. Zusätzlich werden die theoretisch berechneten Schwingungen der [CHF2C(OA)2(HF)2]+-Einheiten (A = H, D) berücksichtigt.[54] Die Zuordnung der beobachteten Banden und Intensitäten stimmt nur teilweise mit den in der Literatur veröffentlichten für [CHF2C(OH)2]+ überein.[39] Die Bande bei 927 cm–1 (IR) bzw. 930 cm–1 (Raman) wurde hier der C–C-Streckschwingung zugeordnet (Seelbinder: 833 cm–1), was mit den C–C-Schwingungsfrequenzen der protonierten Mono- und Trifluoressigsäure im Einklang steht. Das Anion ist verzerrt, es sind daher für das Anion von [CHF2C(OH)2]+[AsF6]− sechs und für das Anion von [CH2FC(OD)2]+[AsF6]− acht Schwingungsbanden im Spektrum zu beobachten. Das stimmt gut mit den geometrischen Parametern der Struktur überein. 29 Ergebnisse und Diskussion Tabelle 3.8: Beobachtete Schwingungsfrequenzen [cm–1] von [CHF2C(OH)2]+[AsF6]− und CHF2CO2H sowie berechnete Schwingungsfrequenzen von [CHF2C(OH)2(HF)2]+ und [CHF2C(OH)2]+ + [CHF2C(OH)2] [AsF6] IR − Raman 2995 (33) 2661 (1) [CHF2C(OH)2(HF)2] C1, ber. + [CHF2C(OH)2] [a] C1, ber. + [b] 3149 (796/168) 3098 (2393/60) 3055 (8/64) 3595 (225/79) 3551 (353/25) 3043 (7/66) 1700 (282/0.6) 1563 (165/4) CHF2CO2H Lit. 3180 2995 1739 1671 (m) 1563 (m) 1676 (2) 1564 (23) 1709 (340/0.7) 1573 (141/7) 1350 (m) 1356 1346 1249 1193 1151 1139 1346 1338 1294 1235 1158 1153 944 921 906 (25/1) (9/2) (91/0.3) (326/5) (188/3) (171/1) (10/0.2) (41/5) (165/0.4) 1345 1332 1202 1179 1175 1135 732 907 688 (25/2) (29/1) (63/5) (93/1) (174/5) (193/0.2) (112/2) (39/3) (169/2) 765 635 579 455 298 266 24 (14/4) (17/1) (4/2) (41/2) (13/0.6) (5/0.2) (3/0.05) 770 583 550 425 292 230 56 (12/2) (6/1) (29/0.8) (1/1) (27/0.6) (1/0.4) (2/0.6) 1255 (m) 1192 (m) 1137 (vs) 927 885 797 741 577 536 484 (m) (m) (m) (s) (m) (m) (w) (3) (15) (3) (15) (4) (25) 930 (29) 750 575 540 480 (43) (42) (9) (18) Zuordnung [54] 1455 1346 1122 1255 1217 1085 936 897 574 562 482 338 ν(OH) ν(OH) ν(CH) ν(C=O) ν(CO2) ν(CO2) ν(C–O) δ(CHF2) δ(CHF2) δ(COH)in plane δ(COH)in plane ν(CF2) ν(CF2) δ(COH)out of plane ν(CC) δ(COH)out of plane δ(CCO) δ(CHF2) δ(CO2) δ(CO2/CHF2) δ(CO2/CHF2) δ(CO2/CHF2) τ 278 (2) 275 705 (100) 671 (89) 414 (2) − [AsF6] 394 (2) 372 (59) 333 (13) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.98 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 704 (vs) 673 (vs) 30 Ergebnisse und Diskussion Tabelle 3.9: Beobachtete Schwingungsfrequenzen [cm–1] von [CHF2C(OD)2]+[AsF6]− sowie berechnete Schwingungsfrequenzen von CHF2C(OD)2(HF)2+ und CHF2C(OD)2+ + − [CHF2C(OD)2] [AsF6] IR Raman 2988 (vw) 2993 (28) 2859 (2) 2657 (3) 2264 (vw) 2306 (6) 2178 (vw) 2183 (5) 2024 (vw) 2030 (2) 1667 (m) 1672 (2) 1529 (w) 1532 (28) 1342 (m) 1346 (16) 1152 1135 1020 910 894 728 703 652 567 559 (m) (s) (m) (m) (m) (s) (vs) (s,sh) (m) (m) 472 (w) 1157 1138 1023 932 903 744 719 701 (8) (20) (20) (2) (11) (3) (6) (7) 571 559 521 471 (21) (3) (4) (10) [CHF2C(OD)2(HF)2] [a] C1, ber. 3055 (5/70) + [CHF2C(OD)2] [b] C1, ber. 3043 (7/67) + Zuordnung ν(CH) 2302 (419/75) 2264 (1255/30) 2618 (125/38) 2585 (204/11) ν(OD) ν(OD) 1698 1534 1342 1336 1155 1159 1039 935 893 755 682 659 1687 1528 1344 1327 1145 1184 1006 918 866 711 546 502 ν(CO2) ν(CO2) δ(CHF2) δ(CHF2) ν(CF2) ν(CF2) δ(COD)in plane δ(COD)in plane ν(CC) δ(CCO) δ(COD)out of plane δ(COD)out of plane (393/0.7) (50/8) (26/2) (29/2) (86/3) (167/3) (87/3) (232/2) (4/3) (27/3) (15/1) (83/0.2) (329/1) (78/5) (23/2) (33/1) (87/2) (140/2) (48/4) (124/1) (2/2) (29/4) (36/0.1) (75/0.4) 604 (14/1) 566 (11/1) 600 (25/1) 516 (40/1) δ(CHF2) δ(CO2) 449 289 261 24 410 279 223 55 δ(CO2/CHF2) δ(CO2/CH2F) δ(CO2/CH2F) τ (44/2) (12/0.6) (5/0.2) (3/0.05) (3/1) (25/1) (2/0.3) (2/1) 273 (1) 711 (43) 675 (vs) 680 (100) 601 (3) 539 (m) 543 (7) − [AsF6] 411 (3) 394 (2) 373 (38) 325 (12) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.98 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 31 Ergebnisse und Diskussion Abbildung 3.18: IR- und Raman-Spektrum von [CHF2C(OH)2]+[AsF6]−. Abbildung 3.19: IR- und Raman-Spektrum von [CHF2C(OD)2]+[AsF6]−. 32 Ergebnisse und Diskussion 3.1.4 Trifluoressigsäure 3.1.4.1 Bildung von [CF3C(OH)2] +[AsF6] − [CF3C(OH)2]+[AsF6]− wurde quantitativ nach folgenden Gleichungen synthetisiert. 2 HF + AsF5 -60 °C (CF3CO)2O + HF CF3CO2H (3.8) [H2F]+[AsF6]- -40 °C, [H2F]+[AsF6]-40 °C + [H2F]+[AsF6]- (3.9) CF3CO2H + CF3COF [CF3C(OH)2]+[AsF6]- + HF (3.10) Die deuterierte Verbindung wurde analog mit DF anstelle von HF hergestellt. Trifluoressigsäureanhydrid wurde in einem Reaktor vorgelegt und anschließend wurden im großen Überschuss HF (150 : 1) und AsF5 (1.5 : 1) kondensiert. Als das Reaktionsgemisch langsam auf –60 °C aufgetaut wurde, bildete sich zuerst die Supersäure [H2F]+[AsF6]− (Gleichung (3.8)), welche bei weiterer Erwärmung auf –40 °C das Anhydrid löst und mit diesem zu CF3CO2H und CF3COF reagiert (Gleichung (3.9)). CF3CO2H wurde dann, wie in Gleichung (3.10) beschrieben, protoniert. Nach langsamen Entfernen von HF, CF3COF und des überschüssigen AsF5 bei –78 °C bildeten sich farblose Kristalle von [CF3C(OH)2]+[AsF6]−, welche bis –40 °C beständig waren und sich für eine EinkristallRöntgenstrukturanalyse eigneten. 3.1.4.2 Röntgenstrukturanalyse [CF3C(OH)2]+[AsF6]− kristallisiert in der triklinen Raumgruppe P–1 (Nr. 2) mit zwei Formeleinheiten in der Elementarzelle. Ausgewählte Bindungslängen und -winkel des Edukts, des [CF3C(OH)2]+[AsF6]−-Salzes und der berechneten [CF3C(OH)2(HF)2]+-Einheit sind in Tabelle 3.10 aufgelistet. Abbildung 3.20 zeigt die asymmetrische Einheit von [CF3C(OH)2]+[AsF6]− und Abbildung 3.21 zeigt einen Ausschnitt aus der Kristallstruktur. Alle Atome einschließlich der Wasserstoffatome wurden mittels Differenz-Fourier-Synthese gefunden und frei verfeinert. Alle Nicht-Wasserstoffatome wurden anisotrop verfeinert. Die Kristallstruktur der Trifluoressigsäure wurde 1978 im Arbeitskreis von Andersen gelöst.[56] 33 Ergebnisse und Diskussion Abbildung 3.20: Asymmetrische Einheit von [CF3C(OH)2]+[AsF6]− im Kristall (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). Abbildung 3.21: Kristallpackung von [CF3C(OH)2]+[AsF6]− (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). Eine Diskussion der Struktur des Kations wurde bereits für [CF3C(OH)2]+[SbF6]− vorgenommen, daher wird hier darauf verzichtet, da die Kationen beider Verbindungen annähernd identisch sind.[39] Jedes Kation ist zu den zwei benachbarten [AsF6]−-Anionen über zwei starke Wasserstoffbrückenbindungen mit O···F-Abständen von 257.7(3) und 256.2(3) pm sowie O–H···F-Winkeln von 172(5)° bzw. 172(4)° verbunden. Diese Verknüpfung führt zu 4-gliedrigen Ringen aus 16 Atomen. 34 Ergebnisse und Diskussion Die Geometrie des [AsF6]−-Anions ist auch in dieser Struktur verzerrt und weicht von der idealen Oktaedersymmetrie ab. Die As–F-Bindungslängen, der an den Wasserstoffbrückenbindungen beteiligten Atome, sind um ca. 4 – 5 pm länger (174.61(18) und 175.32(18) pm) als die übrigen As–F-Bindungen (170.05(18) bis 171.24(17) pm). Die Fluoratome, welche an den Wasserstoffbrückenbindungen beteiligt sind, befinden sich in trans-Position zueinander. Die Winkel liegen bei annähernd 90°. Das Anion ist daher ein schwach gestrecktes Oktaeder mit D4h-Symmetrie. Eine Stabilisierung der elektronenarmen C(OH)2-Gruppe ist auch hier durch die Fluoratome der Anionen gegeben. In diesem Fall doniert aber nur ein Fluoratom (F(4a)) direkt in das pzOrbital des Carbeniumkohlenstoffs. Zwei Fluoratome des zweiten Anions (F(5b) und F(6b)) haben den kürzesten (Abbildung 3.22). Die Abstand zu C(OH)2-Gruppe den ist, Sauerstoffatomen wie bei den der C(OH)2-Gruppe anderen protonierten Fluoressigsäuren, ebenfalls planar (Winkelsumme: 359.9°). Tabelle 3.10: Ausgewählte Bindungslängen [pm] und -winkel [°] für CF3CCO2H, [CF3C(OH)2]+[AsF6]− und die berechnete [CF3CC(OH)2(HF)2]+ Einheit + CF3CO2H exp. C(2)–F(7) C(2)–F(8) C(2)–F(9) C(1)–C(2) C(1)–O(1) C(1)–O(2) O(1)–H(1) O(1)–H(2) H(1)···F(1) H(2)···F(2a) O(1)···F(1) O(2)···F(2a) [CF3C(OH)2] [AsF6] [56] − [CF3C(OH)2(HF)2] exp. 143.0 133.1 132.4 152.6 130.3 121.3 132.2(4) 131.3(4) 131.5(4) 154.6(5) 125.3(4) 124.1(4) 72(4) 64(4) 186(4) 192(4) 257.7(3) 256.2(3) C(1)–O(1)–H(1) 110.3 109(4) C(1)–O(2)–H(2) 121(4) O(1)–C(1)–O(2) 128.1 123.2(3) C(2)–C(1)–O(1) 111.5 113.6(3) C(2)–C(1)–O(2) 120.3 123.3(3) F(7)–C(2)–C(1) 110.8 108.9(3) F(8)–C(2)–C(1) 112.4 110.6(3) F(9)–C(2)–C(1) 108.9 108.7(3) F(7)–C(2)–F(8) 107.6 109.9(3) F(7)–C(2)–F(9) 108.4 109.3(3) F(8)–C(2)–F(9) 108.8 109.4(3) O(1)–H(1)···F(1) 172(5) O(2)–H(2)···F(2a) 172(4) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd); Symmetrieoperationen: a: –x+1,–y+1,–z. 35 C1, ber. 131.8 131.4 131.0 155.4 125.3 125.7 100.2 100.1 115.6 117.3 121.8 115.3 122.8 108.6 108.0 109.4 110.1 110.3 110.7 [a] + Ergebnisse und Diskussion Abbildung 3.22: Koordination zweier [AsF6]−-Anionen zur positiven C(OH)2-Gruppe (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome, Symmetrieoperationen: a: x,–1+y,z, b: –1+x,y,z). 3.1.4.3 Theoretische Berechnungen Die DFT-Berechnungen der Gasphasenstrukturen der [CF3C(OA)2(HF)2]+-Einheiten (A = H, D) wurde mit der Methode PBE1PBE und dem 6-311G++(3df,3pd)-Basissatz durchgeführt (Abbildung 3.23).[41-44] Anschließend wurden die Schwingungsfrequenzen der relaxierten Strukturen, sowie die IR- und Raman-Intensitäten berechnet. Eine Berechnung der freien [CF3C(OA)2]+-Kationen führte nur zu bedingt befriedigenden schwingungs- spektroskopischen Ergebnissen (siehe 3.1.1.4). Abbildung 3.23: Quantenchemisch berechnete Struktur der CF3C(OH)2(HF)2+ Einheit. 36 Ergebnisse und Diskussion Die starken Wasserstoffbrückenbindungen, welche in der Kristallstruktur zu finden sind, werden durch die [CF3C(OA)2(HF)2]+-Einheiten sehr gut wiedergegeben. Durch die Simulation von C–O···H···F–H-Wasserstoffbrücken wird die O–H-Streckschwingung bedeutend genauer berechnet (3087 cm–1) und weicht vom experimentell gefundenen Wert von 2781 cm–1 um 500 cm–1 weniger ab, als die berechnete Schwingung des freien Kations. Insgesamt stimmen die beobachteten und die berechneten Schwingungen der + [CF3C(OA)2(HF)2] -Einheiten gut überein. 3.1.4.4 Schwingungsspektren Die IR- und Raman-Spektren von [CF3C(OH)2]+[AsF6]− und [CF3C(OD)2]+[AsF6]− sind in Abbildung 3.24 und Abbildung 3.25 wiedergegeben. Die dazugehörigen Schwingungsfrequenzen sind in Tabelle 3.11 und Tabelle 3.12, zusammen mit den quantenchemisch berechneten Frequenzen von CF3C(OA)2(HF)2+ (A = H, D) aufgelistet. Das Kation besitzt C1-Symmetrie, weshalb 21 Grundschwingungen im Spektrum zu erwarten sind. Für die Zuordnung der Schwingungsbanden wird mit dem Spektrum der dimeren Trifluoressigsäure verglichen, da diese durch ihre Wasserstoffbrückenbindungen der Festphase stärker ähnelt als die monomere Form, und es werden die theoretisch berechneten Schwingungen der [CF3C(OA)2(HF)2]+-Einheiten (A = H, D) berücksichtigt.[54] Die Interpretation der Schwingungen des Kations in [CF3C(OH)2]+[AsF6]− weicht von den bereits diskutierten Schwingungen des Kations in [CF3C(OH)2]+[SbF6]− ab.[39] Anstatt der Carboxyl-Gruppe drei Streckschwingungen zuzuweisen, kann man hier davon ausgehen, dass die Schwingungen bei 1705 und 1570 cm–1 von der Carboxyl-Gruppe stammen und die Bande bei 1766 cm–1 wahrscheinlich eine Kombination der Schwingungen bei 1254 und 512 cm–1 ist (∑ = 1766 cm–1). Ebenfalls wurden die Deformationsschwingungen neu zugeordnet. 37 Ergebnisse und Diskussion Tabelle 3.11: Beobachtete Schwingungsfrequenzen [cm–1] von [CF3C(OH)2]+[AsF6]− und CF3CO2H sowie berechnete Schwingungsfrequenzen von [CF3C(OH)2(HF)2]+ und [CF3C(OH)2]+ + [CF3C(OH)2] [AsF6] IR − Raman [CF3C(OH)2(HF)2] C1, ber. + [a] [CF3C(OH)2] C1, ber. + [b] 2781 (4) 3134 (862/147) 3078 (2508/72) 3589 (139/85) 3572 (468/18) 1575 (7) 1714 (339/1) 1571 (120/7) 1706 (275/0.8) 1565 (123/5) CF3CO2H Lit. 962 449 675 ν(OH) ν(OH) ν(CF3)+δ(CF3)= 1254+512=1766 ν(C=O) ν(CO2) ν(CO2) ν(C−O) ν(CF3) δ(COH)in plane ν(CF3) ν(CF3) δ(COH)in plane δ(COH)out of plane δ(COH)out of plane ν(CC) δ(CCO) δ(CO2) 700 δ(CF3) 598 515 435 401 284 261 δ(CF3) δ(CF3) δ(CO2/CF3) δ(CO2/CF3) δ(CO2/CF3) τ 3180 1766 (vw) 1739 1705 (w,br) 1570 (m) 1270 (2) 1254 (s,sh) 1215 (vs,sh) 1189 (vs) 1229 (1) 1210 (1) 1193 (3) 819 (m) 823 (26) 667 633 599 535 512 450 598 (9) 538 (9) (m) (s) (m,sh) (m) (m) (m) 450 (7) 260 (3) 1276 1293 1256 1184 1217 944 907 827 767 700 (41/0.5) (161/2) (253/2) (202/0.7) (558/6) (10/0.03) (178/0.04) (6/10) (6/0.2) (17/0.7) 1295 1172 1277 1192 1149 711 674 811 793 645 597 (1/1) 510 452 244 198 177 20 (259/1) (82/2) (144/1) (343/3) (180/2) (47/0.9) (194/0.5) (22/7) (32/0.2) (63/1) 593 (2/0.7) (6/0.4) (42/2) (2/0.3) (23/0.01) (4/0.1) (0.3/0.1) 500 419 366 228 221 41 724 (vs) 691 (s) (0.5/0.3) (2/1) (16/2) (4/0.1) (0.003/0.6) (0.8/0.8) Zuordnung [54] 1456 1290 1207 1234 1162 886 691 (100) 427 (4) − [AsF6] 384 (21) 371 (11) 359 (6) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.98 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 38 Ergebnisse und Diskussion Abbildung 3.24: IR- und Raman-Spektrum von [CF3C(OH)2]+[AsF6]−. Abbildung 3.25: IR- und Raman-Spektrum von [CF3C(OD)2]+[AsF6]−. 39 Ergebnisse und Diskussion Tabelle 3.12: Beobachtete Schwingungsfrequenzen [cm–1] von [CF3C(OD)2]+[AsF6]− sowie berechnete Schwingungsfrequenzen von [CF3C(OD)2(HF)2]+ und [CF3C(OD)2]+ + [CF3C(OD)2] [AsF6] IR 2409 2349 2169 2006 1737 1691 1564 1535 1246 (vw) (vw) (vw) (vw) (vw) (m) (vw) (vw) (m) 1200 1012 919 797 782 677 635 604 588 536 (vs) (m) (w) (vw) (w) (m) (w) (w) (w) (vw) − [CF3C(OD)2(HF)2] Raman 2859 (1) C1, ber. + [a] [CF3C(OD)2] C1, ber. + Zuordnung [b] 2168 (10) 2007 (1) 2292 (448/67) 2247 (1325/36) 2615 (70/41) 2601 (277/9) ν(OD) ν(OD) 1691 1564 1538 1262 1224 1206 1019 921 802 1702 (392/0.8) 1692 (324/0.7) ν(CO2) (1) (1) (14) (2) (2) (1) (9) (5) (24) 649 (4) 593 (5) 540 (9) 443 (8) 258 (2) 1535 1291 1237 1182 1026 930 804 788 678 657 653 592 509 446 237 199 179 11 (30/9) (264/1) (262/2) (227/1) (101/3) (123/3) (13/8) (27/0.2) (5/0.1) (17/1) (75/0.05) (0.3/1) (4/0.5) (38/1) (3/0.3) (24/0.01) (3/0.1) (1/0.2) 1533 1295 1272 1169 993 886 792 779 587 579 554 515 471 413 350 220 216 40 (47/6) (251/1) (227/0.6) (231/0.6) (66/4) (60/1) (19/5) (21/0.1) (2/0.6) (61/2) (12/0.5) (89/0.03) (25/0.2) (2/1) (17/1) (5/0.1) (0.04/1) (0.7/0.7) ν(CO2) ν(CF3) ν(CF3) ν(CF3) δ(COD)in plane δ(COD)in plane ν(CC) δ(CCO) δ(COD)out of plane δ(CO2) δ(COD)out of plane δ(CF3) δ(CF3) δ(CF3) δ(CO2/CF3) δ(CO2/CF3) δ(CO2/CF3) τ 707 (vs) 691 (100) 423 (3) − 399 (2) [AsF6] 384 (16) 371 (9) 359 (6) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.98 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 40 Ergebnisse und Diskussion 3.1.5 Trichloressigsäure 3.1.5.1 Bildung von [CCl3C(OH)2] +[AsF6] − [CCl3C(OH)2]+[AsF6]− wurde quantitativ in einem Schritt nach Gleichung (3.11) hergestellt. Die deuterierte Verbindung wurde analog mit DF anstelle von HF hergestellt. CCl3CO2H + [H2F]+[AsF6]- -20 °C, HF [CCl3C(OH)2]+[AsF6]- + HF (3.11) -60 °C, HF 2 HF + AsF5 Trichloressigsäure wurde in einem Reaktor vorgelegt und anschließend wurden im großen Überschuss HF (150 : 1) und AsF5 (1.5 : 1) kondensiert. Als das Reaktionsgemisch langsam auf –60 °C aufgetaut wurde, bildete sich die Supersäure [H2F]+[AsF6]−. Beim weiteren Erwärmen auf –20 °C löste sich die Trichloressigsäure in der supersauren Lösung und wurde protoniert. Nach langsamem Entfernen von HF und des überschüssigen AsF5 bei –78 °C bildeten sich farblose Kristalle von [CCl3C(OH)2]+[AsF6]−, welche bis –20 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 3.1.5.2 Röntgenstrukturanalyse [CCl3C(OH)2]+[AsF6]− kristallisiert in der monoklinen Raumgruppe P21/c (Nr. 14) mit vier Formeleinheiten in der Elementarzelle. Ausgewählte Bindungslängen und -winkel des Edukts, des [CCl3C(OH)2]+[AsF6]−-Salzes und der berechneten CCl3C(OH)2(HF)2+-Einheit sind in Tabelle 3.13 aufgelistet. Abbildung 3.26 zeigt die asymmetrische Einheit von [CCl3C(OH)2]+[AsF6]− und Abbildung 3.27 zeigt einen Ausschnitt aus der Kristallstruktur. Alle Atome einschließlich der Wasserstoffatome wurden mittels Differenz-Fourier-Synthese gefunden und frei verfeinert. Alle Nicht-Wasserstoffatome wurden anisotrop verfeinert. Die Kristallstruktur der Trichloressigsäure wurde 1972 im Arbeitskreis von Jonsson gelöst.[57] 41 Ergebnisse und Diskussion Abbildung 3.26: Asymmetrische Einheit [CCl3C(OH)2]+[AsF6]− im Kristall (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). Abbildung 3.27: Kristallpackung von [CCl3C(OH)2]+[AsF6]− (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). Eine Diskussion der Struktur des Kations wurde bereits für [CCl3C(OH)2]+[SbF6]− vorgenommen, daher wird hier darauf verzichtet, da die Geometrieparameter der Kationen beider Salze fast identisch sind.[39] Die Struktur des [AsF6]−-Anions ist verzerrt und weicht von der idealen Oktaedersymmetrie ab. Die As–F-Bindungslängen, der an den Wasserstoffbrückenbindungen beteiligten Atome, sind um ca. 4 – 5 pm länger (174.10(12) und 175.88(12) pm) als die übrigen As–FBindungen (von 169.67(12) bis 171.24(13) pm). Die Fluoratome, die an den Wasserstoffbrückenbindungen beteiligt sind, befinden sich in trans-Position zueinander. Die F–As–F-Winkel liegen bei fast 90°. Das Anion ist daher ein leicht gestrecktes Oktaeder mit annähernder D4h-Symmetrie. 42 Ergebnisse und Diskussion Tabelle 3.13: Ausgewählte Bindungslängen [pm] und -winkel [°] für CCl3CO2H, [CCl3C(OH)2]+[AsF6]− und die berechnete CCl3C(OH)2(HF)2+ Einheit + CCl3CO2H exp. C(2)–Cl(1) C(2)–Cl(2) C(2)–Cl(3) C(1)–C(2) C(1)–O(1) C(1)–O(2) O(1)–H(1) O(1)–H(2) H(1)···F(1a) H(2)···F(2b) O(1)···F(1a) O(2)···F(2b) [CCl3C(OH)2] [AsF6] [57] exp. 173.9(4) 174.2(4) 174.5(4) 153.9(4) 129.0(4) 120.5(4) 100.9(11) 175.5(2) 175.2(2) 176.8(2) 153.1(3) 126.1(2) 126.3(2) 70(2) 83(2) 185(2) 177(3) 255.0(2) 255.9(2) − [CCl3C(OH)2(HF)2] C1, ber. + [a] 173.5 176.3 176.4 153.2 126.4 125.7 99.4 99.8 Cl(1)–C(2)–C(1) 109.7(3) 110.13(15) 111.7 Cl(2)–C(2)–C(1) 110.3(2) 111.00(15) 111.2 Cl(3)–C(2)–C(1) 106.9(2) 102.53(13) 105.4 Cl(1)–C(2)–Cl(2) 109.4(2) 110.71(11) 111.5 Cl(1)–C(2)–Cl(3) 109.4(2) 110.96(12) 111.2 Cl(2)–C(2)–Cl(3) 111.0(2) 111.25(12) 105.5 C(2)–C(1)–O(1) 112.9(3) 115.64(18) 118.1 C(2)–C(1)–O(2) 121.2(3) 123.47(19) 122.4 O(1)–C(1)–O(2) 125.9(3) 120.69(19) 119.6 C(1)–O(1)–H(1) 111.1(4) 116(2) 114.3 C(1)–O(2)–H(2) 117.4(18) 117.1 O(1)–H(1)···F(1a) 173(3) O(2)–H(2)···F(2b) 169(3) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd); Symmetrieoperationen: a: x+1,y–1,z; b: –x+1,y–1/2,–z+1/2. Abbildung 3.28: Wasserstoffbrückenbindungen in [CCl3C(OH)2]+[AsF6]− (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). 43 Ergebnisse und Diskussion Das Hexafluoroantimonat und das Hexafluoroarsenat unterscheiden sich jedoch in der Anordnung der Wasserstoffbrückenbindungen. Im Antimonat bilden sich ([CCl3C(OH)2]+[SbF6]–)n-Ketten entlang einer Achse.[39] Im [CCl3C(OH)2]+[AsF6]− hingegen bilden die Wasserstoffbrücken ein drei dimensionales Netzwerk (Abbildung 3.28). 3.1.5.3 Theoretische Berechnungen Die DFT-Berechnungen der Gasphasenstrukturen der [CCl3C(OA)2(HF)2]+-Einheiten (A = H, D) wurde mit der Methode PBE1PBE und dem 6-311G++(3df,3pd)-Basissatz durchgeführt (Abbildung 3.29).[41-44] Anschließend wurden die Schwingungsfrequenzen der relaxierten Strukturen, sowie deren IR- und Raman-Intensitäten berechnet. Eine Berechnung der freien [CF3F(OA)2]+-Kationen führte nur zu bedingt befriedigenden schwingungsspektroskopischen Ergebnissen (siehe 3.1.1.4). Die starken Wasserstoffbrückenbindungen, welche in der Kristallstruktur zu finden sind, werden durch [CCl3C(OA)2(HF)2]+-Einheiten sehr gut wiedergegeben. Durch die Simulation von C–O···H···F–H-Wasserstoffbrücken werden die Schwingungsfrequenzen bedeutend besser berechnet als bei den freien CF3F(OA)2+ Kationen. Die O–H-Streckschwingung weist zum Beispiel nur eine Abweichung von 50 cm–1 auf. Insgesamt stimmen die beobachteten und die berechneten Schwingungen der [CCl3C(OA)2(HF)2]+-Einheiten gut überein. Abbildung 3.29: Quantenchemisch berechnete Struktur der [CCl3C(OH)2(HF)2]+-Einheit. 44 Ergebnisse und Diskussion 3.1.5.4 Schwingungsspektren Die IR- und Raman-Spektren von [CCl3C(OH)2]+[AsF6]− und [CCl3C(OD)2]+[AsF6]− sind in Abbildung 3.30 und Abbildung 3.31 dargestellt. Die dazugehörigen Schwingungsfrequenzen sind in Tabelle 3.14 und Tabelle 3.15, zusammen mit den quantenchemisch berechneten Frequenzen von [CCl3C(OA)2(HF)2]+ (A = H, D) aufgelistet. Das Kation besitzt C1-Symmetrie, weshalb 21 Grundschwingungen im Spektrum zu erwarten sind. Für die Zuordnung der Schwingungsbanden werden das Spektrum der Trichloressigsäure und die theoretisch berechneten Schwingungen der CCl3C(OA)2(HF)2+ Einheiten (A = H, D) berücksichtigt.[58] Bei der Interpretation der Schwingungsspektren von [CCl3C(OH)2]+[AsF6]− gab es Unterschiede zu den bereits diskutierten Spektren von [CCl3C(OH)2]+[SbF6]−.[39] Es erfolgt daher eine komplett neue Zuordnung der Banden von 3181 bis 961 cm–1. Die νas(CCl3) sind bei 874 und 816 cm–1 und die νs(CCl3) ist bei 516 cm–1 zu beobachten. Weitere Banden bei niedrigeren Wellenzahlen können den Deformationsschwingungen des Kations und den Schwingungen des Anions zugeordnet werden. Die Symmetrie des Anions ist zu D4h erniedrigt und es weist daher sieben Schwingungen auf. Abbildung 3.30: IR- und Raman-Spektrum von [CCl3C(OH)2]+[AsF6]−. 45 Ergebnisse und Diskussion Abbildung 3.31: IR- und Raman-Spektrum von [CCl3C(OD)2]+[AsF6]−. 46 Ergebnisse und Diskussion Tabelle 3.14: Beobachtete Schwingungsfrequenzen [cm–1] von [CCl3C(OH)2]+[AsF6]− und CCl3CO2H sowie berechnete Schwingungsfrequenzen von [CCl3C(OH)2(HF)2]+ und [CCl3C(OH)2]+ + [CCl3C(OH)2] [AsF6] IR − Raman 3181 (vs,br) [CCl3C(OH)2(HF)2] C1, ber. + [a] [CCl3C(OH)2] C1, ber. + [b] 3230 (857/101) 3163 (2061/142) 3620 (323/88) 3392 (293/25) 1668 (233/1) 1536 (279/8) CCl3CO2H Lit. 3100 1694 1640 (w) 1513 (vw) 1648 (1) 1522 (11) 1675 (258/1) 1532 (180/10) 1255 (w) 1206 (w) 956 (vw) 1255 (4) 1206 (4) 959 (10) 1285 1220 961 937 870 855 799 691 685 (154/1) (340/8) (26/2) (95/0.1) (103/3) (158/2) (0.2/3) (14/6) (50/1) 1191 1174 951 757 860 613 822 619 728 (193/5) (103/7) (13/2) (2/0.8) (102/3) (43/2) (67/5) (60/5) (212/2) 459 454 309 278 278 225 189 2 (34/8) (5/7) (3/1) (9/1) (0.2/2) (2/1) (0.2/1) (10/0.06) 444 419 300 275 268 197 175 27 (6/11) (12/3) (20/1) (1/2) (1/2) (0.3/1) (1/1) (1/0.4) 871 (vw) 837 (vw) 725 (m,sh) 679 (m,sh) 614 (s,br) 874 832 816 723 677 (10) (4) (4) (62) (28) 518 455 446 361 (9) (21) (95) (11) Zuordnung [58] 1433 1260 961 852 837 686 456 433 400 327 ν(OH) ν(OH) ν(C=O) ν(CO2) ν(CO2) ν(C–O) δ(COH)in plane δ(COH)in plane ν(CC) δ(COH)out of plane ν(CCl3) δ(COH)out of plane ν(CCl3) δ(CO2) δ(CCO) ν(CCl3) δ(CO2/CCl3) δ(CO2/CCl3) δ(CO2/CCl3) δ(CCl3) δ(CCl3) δ(CCl3) τ 284 304 (17) 219 283 (65) 255 (7) 210 708 (m) 704 (30) 693 (13) 672 (m) 665 (100) − 561 (w) 559 (25) [AsF6] 397 (5) 389 (8) 370 (44) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.98 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 47 Ergebnisse und Diskussion Tabelle 3.15: Beobachtete Schwingungsfrequenzen [cm–1] von [CCl3C(OD)2]+[AsF6]− sowie berechnete Schwingungsfrequenzen von [CCl3C(OD)2(HF)2]+ und [CCl3C(OD)2]+ + [CCl3C(OD)2] [AsF6] IR 2120 2040 1748 1634 1518 1471 1258 1214 1033 928 889 867 850 − Raman (vw) (vw) (w) (m) (vw) (w) (vw) (vw) (w) (m) (m) (m) 822 (m) 729 (vs) 665 (s) 616 (m) 529 (m) [CCl3C(OD)2(HF)2] C1, ber. + [a] [CCl3C(OD)2] C1, ber. + Zuordnung [b] 2128 (4) 2049 (3) 2361 (442/46) 2309 (1093/69) 2636 (188/42) 2469 (160/12) 1642 (4) 1527 (10) 1482 (15) 1662 (293/1) 1658 (250/1) ν(OD) ν(OD) 928+822=1750 ν(CO2) 1484 (139/14) 1487 (196/11) ν(CO2) 1041 932 894 863 (11) (6) (11) (14) 1043 935 917 863 829 817 727 633 623 616 521 449 434 361 299 282 (9) (19) (31) (9) (11) (10) (7) (90) (10) (9) (17) (42) 815 734 639 638 608 456 443 305 278 273 223 187 2 (56/2) (209/3) (64/2) (46/3) (49/5) (116/1) (19/6) (2/1) (43/0.01) (40/11) (3/3) (3/1) (0.1/2) (10/1) (2/1) (0.2/1) (10/0.1) 1016 931 886 856 (25/3) (94/4) (33/0.5) (95/3) δ(COD)in plane ν(CC) δ(COD)in plane ν(CCl3) 821 716 559 549 468 433 407 292 273 264 194 171 26 (69/5) (84/0.8) (39/5) (31/3) (53/0.6) (5/9) (13/3) (20/1) (1/2) (3/2) (0.3/1) (2/1) (1/0.4) ν(CCl3) δ(CCO) δ(CO2) δ(COD)out of plane δ(COD)out of plane ν(CCl3) δ(CO2/CCl3) δ(CO2/CCl3) δ(CCl3) δ(CO2/CCl3) δ(CCl3) δ(CCl3) τ 255 (5) 704 (16) 692 (10) 667 (100) − 557 (m) 559 (15) [AsF6] 396 (8) 389 (5) 370 (35) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 0.98 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 705 (vs) 48 Ergebnisse und Diskussion 3.2 Tricyanomethan (Cyanoform) Die Synthese und die Eigenschaften von Cyanoform (HC(CN)3) beschäftigen Chemiker seit über einem Jahrhundert.[20,59-63] Bereits 1896 versuchte Schmidtmann diese Verbindung aus Schwefelsäure und Natriumtricyanomethanid zu synthetisieren.[59] Durch Zufügen von Ether erhielt er eine grünliche Flüssigkeit, die durch Einengen orangebis rotbraun-gefärbte Kristalle ergab. Er führte dies auf die Bildung eines Polymers von Tricyanomethan zurück. Hantzsch und Osswald konnten 1899 die Ergebnisse größtenteils bestätigen und schlugen Dicyanoketenimin als gleichwertig neben Cyanoform existierendes Tautomer vor (Abbildung 3.32Abbildung 4.2).[63] H NC NC CN CN Abbildung 3.32: C C NH + [H3O] NC NC Strukturen von Cyanoform, cyanomethanid[20]. Dicyanoketenimin CN - CN und Oxoniumtri- Diese Befunde konnten Fontaine und Cox hingegen nicht reproduzieren und versuchten die Darstellung über einen neuen Reaktionsweg, indem [64] Brommalonsäuredinitril bei –15 °C in Ether umsetzten. sie Kaliumcyanid und Während Cox und Fontaine angaben, bei Raumtemperatur stabile Kristalle erhalten zu haben, konnte Trofimenko 1963 diese Ergebnisse wiederum nicht bestätigen. Er erhielt einen kristallinen Feststoff, der bei Raumtemperatur polymerisierte.[61-62] Das Infrarotspektrum deutete seiner Meinung nach auf das Dicyanoketenimin hin. Dunitz wiederholte 2010 die oben beschriebenen Experimente und identifizierte (Abbildung 3.32). die erhaltenen Kristalle als Oxoniumtricyanomethanid [20] Die Umsetzung von Natriumtricyanomethanid mit Chlorwasserstoff in Tetrahydrofuran lieferte hingegen ausschließlich Additionsprodukte mit HCl. Somit gelang es bisher nicht, Tricyanomethan in reiner Form darzustellen.[20] Tricyanomethan ist mit einem pKs-Wert von –5.1, eine sehr starke Säure.[14,65] Leito et al. konnte diese Verbindung in sehr verdünnter Form untersuchen und deren Säurestärke bestimmen und damit auch den 1962 von Boyd bestimmten pKs-Wert bestätigen. 49 Ergebnisse und Diskussion In dieser Arbeit wurden zwei Wege beschritten, Tricyanomethan zu synthetisieren. Der eine Weg beschäftigte sich mit der Umsetzung von Alkali- und Erdalkali-Salzen des Tricyanomethans mit wasserfreier HF. Es bildete sich Tricyanomethan, welches NMR- und schwingungsspektroskopisch untersucht werden konnte. Bei Kristallisationsversuchen dieser Ansätze konnte zwar nicht ein für eine Einkristall-Röntgenstrukturanalyse geeigneter Kristall von Tricyanomethan gezüchtet werden, aber es gelang die Struktur des NaH4F5 aufzuklären. Bislang ist nur eine weitere Kristallstruktur dieses Anions bekannt und zwar das raumtemperaturbeständige KH4F5.[66] NaH4F5 bildet farblose Kristalle, welche bis –20 °C beständig sind (weitere Informationen befinden sich in Kapitel 6). Der andere Weg befasste sich mit der Synthese von Trimethylsilyltricyanomethan (TMS– TCM) und der anschließenden Umsetzung mit HF analog der Synthese der protonierten Blausäure.[67-68] Durch die Umsetzung mit HF würde Trimethylsilylfluorid entstehen, welches durch seinen sehr niedrigen Siedepunkt leicht entfernt werden könnte. Der große Vorteil dieser Synthese wäre die Isolierung von Tricyanomethan und die Möglichkeit dieses auch kristallographisch untersuchen zu können. Die Synthese von TMS–TCM analog zur Herstellung von Trimethylsilylcyanid (TMS–CN) gelang jedoch nicht.[68] Die beginnende Polymerisation des Produktes bei –40 °C und die Wahl des Lösungsmittels bereiten bei dieser Reaktion große Schwierigkeiten. Anstatt des erwünschten TMS–TCM konnten mehrmals Additionsprodukte mit dem Lösungsmittel beobachtet werden. 3.2.1.1 Bildung von HC(CN)3 Calciumtricyanomethanid, hergestellt nach Literaturvorschrift, wurde mit HF gemäß der folgenden Reaktionsgleichung (3.12) umgesetzt.[62,69-71] Ca(C(CN)3)2 + 4 HF -50 °C, HF 2 HC(CN)3 + Ca(HF2)2 (3.12) Das Calciumtricyanomethanid wurde in einem Reaktor vorgelegt und anschließend wurde in großem Überschuss HF kondensiert. Die Reaktion wurde für 10 min bei –50 °C durchgeführt, bis eine klare farblose Lösung vorlag. Nach langsamem Entfernen von HF bei –78 °C blieb ein farbloser Rückstand im Reaktor zurück. Das Gemisch ist bis –40 °C beständig und ändert die Farbe bei beginnender Zersetzung zu gelb und wird bei weiterer Erwärmung tiefrot. Das Gemisch besteht aus HC(CN)3 und Ca(HF2)2. Die deuterierte Verbindung wurde analog mit DF anstelle von HF hergestellt. 50 Ergebnisse und Diskussion 3.2.1.2 NMR-Spektren Von der Umsetzung von Ca(C(CN)3)2 mit HF wurden NMR-Spektren bei –45 °C und SO2 als Lösungsmittel gemessen. Es wurden 1H-, 19 F-NMR-Spektren aufgenommen. 13 Als C- (1H-gekoppelt und -entkoppelt), externer Standard 14 wurde N- und eine d6-Aceton/CFCl3/Nitromethan-Mischung (3 : 1 : 1) eingesetzt. 1 H-NMR-Spektrum In Abbildung 3.33 ist das 1H-NMR-Spektrum der Umsetzung abgebildet. Das Singulett bei 6.68 ppm gehört zum [HF2]– des Nebenprodukts Ca(HF2)2. Das Proton des Tricyanomethans ist bei 5.79 ppm zu beobachten. Die starke Verschiebung ins Tieffeld deckt sich mit der hohen Acidität des Protons (pKs = –5.1).[14,65] Die weiteren Signale gehören zu den eingesetzten Standards Nitromethan (4.41 ppm) und d6-Aceton (2.04 ppm). Abbildung 3.33: 1 H-NMR-Spektrum der Umsetzung von Ca(C(CN)3)2 und HF (* Signale der externen Standards: d6-Aceton und Nitromethan). 13 C-NMR-Spektren Von der Probe wurden zwei 13C-NMR-Spektren gemessen, 1H-gekoppelt und 1H-entkoppelt (Abbildung 3.34). Das Singulett bei 106.10 ppm ist den Kohlenstoffatomen der drei äquivalenten (C≡N)-Gruppen zuzuordnen. Dieses Signal ist um ca. 15 ppm gegenüber dem Tricyanomethanid-Ion ins Hochfeld verschoben (121.97 ppm).[72] Das Duplett im 1Hgekoppelten Spektrum bei 16.91 ppm ist ein Beweis dafür, dass das Proton an dem zentralen Kohlenstoff gebunden ist (1JCH = 147 Hz). Im C(CN)3− liegt dieses Signal bei 6.72 ppm.[72] 51 Ergebnisse und Diskussion Abbildung 3.34: 19 13 C-NMR-Spektrum von HC(CN)3 (oben: 1H-gekoppelt, unten: 1Hentkoppelt, * Signale der externen Standards: d6-Aceton, CFCl3 und Nitromethan). F-NMR-Spektrum Im 19 F-NMR-Spektrum beobachtet man ein Singulett, das dem Nebenprodukt Ca(HF2)2 zugeordnet werden kann. Eine mögliche Addition von HF an eine oder mehrere Cyanogruppen des HC(CN)3 kann ausgeschlossen werden. 14 N-NMR-Spektrum Im 14 N-NMR-Spektrum ist ein breites Singulett bei -127.1 ppm zu beobachten, das auf den Stickstoff der Cyanogruppen zurückzuführen ist. Bei C(CN)3− findet man diese Signal bei 124 ppm.[72] Eine Protonierung einer der Cyanogruppen ist daher unwahrscheinlich, da in diesem Fall ein weiteres Signal im Bereich von ungefähr –300 ppm zu erwarten wäre. 52 Ergebnisse und Diskussion 3.2.1.3 Theoretische Berechnungen Die DFT-Berechnungen der Gasphasenstrukturen des AC(CN)3 (A = H, D) und des Isomers C(CN)2CNH wurden mit der Methode PBE1PBE und dem 6-311G(3df,3pd)-Basissatz durchgeführt (Abbildung 3.35).[41-43] Anschließend wurden die Schwingungsfrequenzen der relaxierten Strukturen, sowie deren IR- und Raman-Intensitäten berechnet. Dicyanoketenimin hat eine um 5.7 kJ/mol höhere Energie als das Cyanoform. Das ist im Einklang mit anderen literaturbekannten Berechnungen auf DFT-Level.[73] Abbildung 3.35: links: Struktur von C(CN)3− in Ca(C(CN)3)2, mittig: berechnete Struktur von HC(CN)3, rechts: berechnete Struktur von C(CN)2CNH. 3.2.1.4 Schwingungsspektren Die IR- und Raman-Spektren von HC(CN)3 und das Raman-Spektrum von DC(CN)3 werden in Abbildung 3.36 wiedergegeben. Die dazugehörigen Schwingungsfrequenzen sind, zusammen mit den quantenchemisch berechneten Frequenzen von AC(CN)3 (A = H, D) und dem Edukt in Tabelle 3.16 aufgelistet. Das berechnete freie AC(CN)3 besitzt C3v-Symmetrie. Die experimentellen Befunde zeigen jedoch, dass die Entartung einiger Schwingungen aufgehoben ist und das AC(CN)3 im Festkörper somit eine niedrigere Symmetrie besitzt. Die Raman-Spektren von HC(CN)3 und DC(CN)3 belegen, dass das Tricyanomethan in der Cyanoform vorliegt. Das entstandene Nebenprodukt Ca(HF2)2 störte die NMR- und schwingungsspektroskopische Auswertung der Spektren nicht, da alle Schwingungen von Ca(HF2)2 Raman-inaktiv sind. Im IR dagegen überlagern die starken Schwingungsbanden des [HF2]– zum Teil die Banden von HC(CN)3. 53 Ergebnisse und Diskussion Tabelle 3.16: Beobachtete und berechnete Schwingungsfrequenzen [cm–1] von HC(CN)3 und DC(CN)3 IR 2915 (s) 2886 (m) 2310 (vw) 2212 (vw) 1349 1248 1025 1006 918 829 (w) (vw) (s) (m) (w) (s) 569 (vs) HC(CN)3 Raman C3v, ber. [a] 2885 (38) 2922 (14/57) 2287 (100) 2316 (0.1/100) 2259 (7) 2310 (2/40) 1259 (3) 1253 (5) 1022 (7) 825 (24) 575 (7) 567 (16) Raman DC(CN)3 [a] C3v, ber. 2286 (100) 2319 (13) 2126 (66) Ca(C(CN)3)2 exp. 2316 (0.1/100) 2310 (2/42) 2145 (4/30) 1232 (6/4) 1002 (32/2) 813 (4/4) 559 (0.1/2) 1116 861 849 806 570 561 (6) (10) (15) (27) (15) (7) 1098 (30/2) 831 (6/0.1) 794 (4/5) 559 (0.1/2) 556 (1/3) 556 (1/3) 345 (0/0) 345 (0/0) 347 (45) 345 (0.5/2) 339 (67) 336 (0.6/2) 160 (23/1) 159 (23/1) 126 (14/4) 126 (14/4) [a] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem skaliert; IR-Intensität in km/mol, Raman-Aktivität in %. 2244 2188 1265 1256 1045 Zuordnung ν(CH) ν(CH) νs(CN) νas(CN) ν(CD) δ(CCH) νas(CC) δ(CCD) δ(CCD) νs(CC) 683 δ(CCC) δ(C(CN)3) ω(C(CN)3) τ(C(CN)3) δ(C(CN)3) ω(C(CN)3) δ(C(CN)3) empirischen Faktor von 0.96 Die (C–H)-Valenzschwingung ist bei 2885 cm–1 zu beobachten. Durch die Deuterierung verschiebt sich die Schwingung nach 2126 cm–1, was zu einem Frequenzverhältnis ν(CH)/ν(CD) von 1.36 führt, welches gut mit dem theoretisch berechneten Wert von 1.41 übereinstimmt. Bei 2212 und 2310 cm–1 treten die ν(CN) als intensivste Schwingungen auf und sind im Vergleich zum Edukt um ca. 80 cm–1 blauverschoben. Die (C–C)-Valenzschwingung ist von 1265 cm–1 nach 1022 cm–1 um ca. 240 cm–1 rotverschoben. Das lässt auf eine Verkürzung der C≡N-Bindung und eine Schwächung der C–C-Bindung schließen. Durch das Proton im HC(CN)3 wird das freie Elektronenpaar des C(CN)3− gebunden, was die geometrischen Veränderungen sehr gut erklärt. Theoretische Berechnungen der Geometrie des Anions und des Tricyanomethans bestätigen diese Beobachtungen (Abbildung 3.35). 54 Ergebnisse und Diskussion Abbildung 3.36: Schwingungsspektren der Umsetzung von Ca(C(CN)3)2 mit HF: b) Raman-Spektrum, c) IR Spektrum; und von der Umsetzung von Ca(C(CN)3)2 mit DF: a) Raman-Spektrum (* Schwingungsbanden von Ca(HF2)2). 55 Ergebnisse und Diskussion 3.3 Die strukturelle Vielfalt von Fluorogermanaten Es sind nur wenige binäre Verbindungen bekannt, die ausschließlich Germanium und Fluor enthalten. Das GeF4, mit Ge–F-Abständen von 166.07(6) pm, wurde von Hoppe untersucht.[74] Dieses ist isotyp mit SiF4 und besteht aus nicht vernetzten Tetraedern. Das GeF2 wurde 1966 von Trotter et al. beschrieben.[75] Es sind noch zwei gemischt valente Strukturen der binären Germaniumfluoride bekannt. Zum einen das Ge5F12, in welchem [Ge4F6]2+-Schichten über [GeF6]2−-Oktaeder verbunden sind, und zum anderen das Ge7F16, in der [Ge6F10]2+-Schichten vorliegen, welche auch über [GeF6]2−-Oktaeder verbrückt sind.[76-77] Die Struktureinheit des [GeF6]2−-Oktaeders ist in der Literatur wohl bekannt. Bereits 1939 wurden die Kristallstrukturen von Fluorogermanaten in Form von [NH4]2+[GeF6]2− und K2+[GeF6]2− von Hoard und Vincent beschrieben.[78] Das monomere Hexafluorogermanatanion wurde dabei als schwach verzerrtes Oktaeder beschrieben. Das [GeF6]2−-Anion wurde schwingungsspektroskopisch von Griffiths im Jahr 1964 untersucht, wobei er die von Hoard und Vincent gefundene Struktur des Anions bestätigte.[79] Nur drei Jahre später stieß Clark bei einer Elementaranalyse einer Tetraphenylarsonium-Fluorogermantverbindung auf ein Ge-F-Verhältnis von 1 : 5, woraufhin er eine polymere Struktur des Anions vermutete.[80] Diese Vermutung wurde 1976 von Christe durch seine Arbeit zum Dioxygenylpentafluorogermanat [O2]+[GeF5]− gestützt.[81] Er fand ein deutlich komplexeres Ramanspektrum, als es bei einem regulären [GeF6]2−-Oktaeder zu erwarten gewesen wäre. Mallouk und Bartlett konnten 1984 mittels einer Röntgenstrukturanalyse von [XeF5]+[GeF5]− und [ClO2]+[GeF5]− die ersten polymeren Fluorogermanate bestätigen.[21] Bei beiden handelt es sich um eine Kettenstruktur, in der [GeF6]2−-Oktaeder über Ecken miteinander verknüpft sind. Bei [ClO2]+[GeF5]− liegt eine cis- und bei [XeF5]+[GeF5]− eine trans-Verknüpfung vor. Mallouk und Bartlett schließen aus ihren Ergebnissen, dass auch bei den [SF3]+-, [NO2]+und [NF4]+-Salzen polymere Strukturen vorhanden sein müssten. Es wurde viel darüber diskutiert, ob ein monomeres [GeF5]−-Anion analog zu [SiF5]− existiert. In [(C4H9)4N]+[GeF5]−, welches erstmalig von Wharf und Onyszchuk synthetisiert wurde, wird das Anion schwingungsspektroskopisch auf Basis einer D3h-Symmetrie durch den Vergleich mit anderen MX5-Verbindungen zugeordnet.[21,82] Ein eindeutiger Beweis, dass in dieser Struktur [GeF5]− als Monomer vorliegt, konnte aber nicht erbracht werden, da von dieser Struktur keine kristallographischen Daten, sondern nur Schwingungsspektren vorliegen. 56 Ergebnisse und Diskussion Eine Fünffach-Koordination eines Ge(IV)-Atoms kann aber in der Tat durch große Liganden erreicht werden und wurde von Brauer in Tetramethylammonium-difluor- tris(trifluormethyl)germanat gefunden.[83] Das Anion ist eine trigonale Bipyramide, in der die drei CF3-Gruppen in der Äquatorialebene liegen und die zwei Fluor-Atome die axialen Positionen einnehmen. Kondensierte MF6-Oktaeder, welche über Ecken miteinander verknüpft sind, sind gerade im Bereich der Supersäurechemie wohl bekannt. Bei den Fluoroarsenaten ist es zwar nur das [As2F11]−, aber bei den Fluoroantimonaten wurden die Anionen des Typs [SbxF5x+1]− (x = 1 − 4) mittels Röntgenstrukturanalyse charakterisiert. Doch können Oktaeder nicht nur linear miteinander verbunden werden, sondern auch Ringe bilden. Vierer-Ringe[84] binärer Pentafluoride der Form M4F20 sind z.B. von Tantal[85-86], Niob[86], Ruthenium[87-88], Platin[89] und Vanadium[90] bekannt. Eine dimere Einheit mit einer Kantenverknüpfung zweier Oktaeder war bisher hauptsächlich aus dem Bereich der binären Übergangsmetallhalogenide bekannt, wenn das Halogenid Chlor, Brom oder Iod ist. Beispiele hierfür wären Nb2Cl10, Ta2Br10 oder [Ti2Cl10]2−.[91] Nur sehr wenige binäre Metallfluoride sind dagegen kristallographisch untersucht worden. [Al2F10]4− und [Ti2F10]4− konnten 2000 sowie 2001 von Tang und Dadachov als Piperazinium-Salze synthetisiert werden (d(Al⋅⋅⋅Al) = 296.0(1) pm, d(Ti⋅⋅⋅Ti) = 280.7(1)).[9293] Becker konnte 1996 erstmalig das isoelektronische [Sb2F10]4− strukturell untersuchen, in welchem aber die [SbF5]2−-Untereinheiten nur sehr schwach aneinander gebunden sind.[94] Die Sb–F-Abstände zu den verbrückenden Fluoratomen betragen 199.2(2) und 296.7(2) pm, der Sb⋅⋅⋅Sb-Abstand 418.6(1) pm. 57 Ergebnisse und Diskussion 3.3.1 Umsetzung von HF/GeF4 mit Nitroaromaten Die Protonierung von Nitrobenzol (pKBH+ = –12.14) erfolgte primär, um die Säurestärke des Systems HF/GeF4 zu testen.[11] Analog zu dieser Reaktion wurde auch 2,4,6-Trinitrotoluol (TNT) umgesetzt, welches einen pKBH+-Wert von –15.60 hat.[11] Wie schon bei Götz beschrieben, ist die Protonierung von C6H5NO2 mit den Systemen HF/AsF5 und HF/SbF5 möglich.[95] Auch das System HF/GeF4 ist stark genug, um C6H5NO2 zu protonieren, es konnte aber kein Salz des protonierten TNT isoliert werden. Eine Protonierung von TNT gelang bisher nur mit dem stärksten bekannten supersauren System HF/SbF5.[96] 3.3.1.1 Umsetzung von C6H5NO2 mit HF/GeF4 [(C6H5NO2H)2]2+[Ge2F10]2− wurde quantitativ in einem Schritt nach Gleichung (3.13) synthetisiert. 2 C6H5NO2 + 2 [(H2F)2]2+[GeF6]2- -50 °C, HF [(C6H5NO2H)2]2+[Ge2F10]2- + 6 HF -60 °C, HF (3.13) 8 HF + 2 GeF4 Nitrobenzol wurde in einem Reaktor vorgelegt und anschließend wurden im hohen Überschuss HF (150 : 1) und GeF4 (2.0 : 1) kondensiert. Als das Reaktionsgemisch langsam auf –60 °C aufgetaut wurde, bildete sich die Supersäure [(H2F)2]2+[GeF6]2−. Bei weiterem Erwärmen auf –50 °C löste sich das Nitrobenzol in der supersauren Lösung und wurde protoniert. Nach langsamem Entfernen von HF bei –70 °C für 3 h und bei –50 °C für 12 h bildeten sich gelbe Kristalle von [(C6H5NO2H)2]2+[Ge2F10]2−, welche bis –25 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 3.3.1.2 Umsetzung von 2,4,6-Trinitrotoluol mit HF/GeF4 TNT wurde in einem Reaktor vorgelegt und anschließend wurden im hohen Überschuss HF (150 : 1) und GeF4 (2.0 : 1) dazu kondensiert. Das TNT löste sich bei –30 °C in der supersauren Lösung. Nach langsamem Entfernen der flüchtigen Verbindungen bei –70 °C 58 Ergebnisse und Diskussion für 16 h und bei –60 °C für 24 h blieb ein farbloser Feststoff im Reaktor zurück, der sich mittels Raman-Spektroskopie als das Edukt identifizieren ließ. Das System HF/GeF4 konnte TNT nicht protonieren. GeF4 wurde mit dem Lösungsmittel HF ebenfalls entfernt. 3.3.1.3 Röntgenstrukturanalyse von [(C6H5NO2H)2] 2+[Ge2F10] 2− [(C6H5NO2H)2]2+[Ge2F10]2− kristallisiert in der triklinen Raumgruppe P–1 (Nr. 2) mit zwei Formeleinheiten in der Elementarzelle. Ausgewählte Bindungslängen und -winkel von [(C6H5NO2H)2]2+[Ge2F10]2− sind zusammen mit den berechneten Strukturdaten der [Ge2F10]2−-Einheit in Tabelle 3.17 aufgelistet. Alle Atome, einschließlich der Wasserstoffatome am Sauerstoff, wurden mittels DifferenzFourier-Synthese gefunden und frei verfeinert. Die Wasserstoffatome des Benzolrings wurden berechnet. Alle Nicht-Wasserstoffatome wurden anisotrop verfeinert. Die asymmetrische Einheit von [(C6H5NO2H)2]2+[Ge2F10]2− besteht aus einem [C6H5NO2H]+-Kation und einem halben [Ge2F10]2−-Anion, welches ein Inversionszentrum auf der Mitte der gemeinsamen Kante besitzt (Abbildung 3.37 und Abbildung 3.38). Ein Ausschnitt aus der Kristallpackung ist in Abbildung 3.39 dargestellt. Abbildung 3.37: Struktur von [(C6H5NO2H)2]2+[Ge2F10]2−, Beschriftung der asymmetrischen Einheit (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome). Götz untersuchte Nitrobenzol schon mit den Systemen HF/AsF5 und HF/SbF5. Hierbei gelang die Isolierung von [(C6H5NO2)2H]+[AsF6]− und [(C6H5NO2)2H]+[SbF6]−. In beiden Fällen wurde das Nitrobenzol hemiprotoniert, d.h. zwei Nitrobenzolmoleküle teilen sich ein acides Proton.[95,97] Bei [(C6H5NO2H)2]2+[Ge2F10]2− liegt dagegen eine vollständige Protonierung vor. 59 Ergebnisse und Diskussion Tabelle 3.17: Ausgewählte Bindungslängen [pm] und -winkel 2+ 2− [(C6H5NO2H)2] [Ge2F10] und des berechneten [Ge2F10]2− 2+ [(C6H5NO2H)2] [Ge2F10] 2− [Ge2F10] 132.0(3) 75(4) 119.7(3) 142.8(3) 138.3(4) 140.3(4) 137.4(4) 139.7(4) 138.3(4) 138.6(4) N(1)–O(1)–H(1) O(2)–N(1)–O(1) O(2)–N(1)–C(1) O(1)–N(1)–C(1) C(5)–C(1)–C(2) C(5)–C(1)–N(1) C(2)–C(1)–N(1) C(3)–C(2)–C(1) C(2)–C(3)–C(6) C(6)–C(4)–C(5) C(1)–C(5)–C(4) C(4)–C(6)–C(3) 113(3) 121.5(2) 124.2(2) 114.3(2) 123.7(3) 119.9(2) 116.4(3) 117.4(3) 120.1(3) 120.2(3) 117.5(3) 121.2(3) von 2− D2h, ber. exp. O(1)–N(1) O(1)–H(1) O(2)–N(1) N(1)–C(1) C(1)–C(5) C(1)–C(2) C(2)–C(3) C(3)–C(6) C(4)–C(6) C(4)–C(5) [°] Ge(1)–F(4) Ge(1)–F(2) Ge(1)–F(3) Ge(1)–F(1) Ge(1)–F(5) Ge(1)–F(5)#1 Ge(1)–Ge(1)#1 172.47(17) 173.36(15) 173.76(16) 179.20(15) 192.01(16) 194.48(16) 308.11(5) 177.0 175.9 175.9 177.0 195.7 195.7 312.4 F(4)–Ge(1)–F(2) F(4)–Ge(1)–F(3) F(2)–Ge(1)–F(3) F(4)–Ge(1)–F(1) F(2)–Ge(1)–F(1) F(3)–Ge(1)–F(1) F(4)–Ge(1)–F(5) F(2)–Ge(1)–F(5) F(1)–Ge(1)–F(5) F(2)–Ge(1)–F(5)#1 F(3)–Ge(1)–F(5)#1 F(1)–Ge(1)–F(5)#1 F(5)–Ge(1)–F(5)#1 Ge(1)–F(5)–Ge(1)#1 95.18(8) 101.46(8) 92.92(8) 91.53(8) 171.80(7) 90.35(8) 92.44(8) 87.93(7) 87.08(7) 87.26(7) 91.73(8) 85.13(7) 74.27(8) 105.73(8) 91.8 100.7 91.8 91.8 174.5 91.8 92.6 87.8 87.8 87.8 92.6 87.8 74.1 105.9 [a] H(1)···F(1) 176(4) O(1)···F(1) 249.8(3) O(1)–H(1)···F(1) 173(4) [a] berechnet mit PBE1PBE/6-311G(3df,3pd); Symmetrieoperation: #1: –x,–y,–z. Abbildung 3.38: Struktur des [Ge2F10]2−-Anions (Symmetrieoperation: #1: –x,–y,–z). Die N=O-Doppelbindung ist mit einer Länge von 199.7(3) pm fast gleich lang wie bei der hemiprotonierten Spezies (120.6(3) pm). Die N−O-Einfachbindung ist mit einer Länge von 132.0(3) pm wie zu erwarten um ca. 4 pm verlängert. Ansonsten weist das Kation gegenüber der hemiprotonierten Spezies keine weiteren strukturellen Besonderheiten auf. 60 Ergebnisse und Diskussion Sehr interessant an dieser Verbindung ist jedoch das Anion. Das [Ge2F10]2−-Ion war bisher unbekannt. Es bildet sich nicht wie zuerst erwartet der [GeF6]2−-Oktaeder, sondern es liegen in dieser Struktur zwei kantenverknüpfte Oktaeder vor (Abbildung 3.38). In der Mitte dieses Anions befindet sich ein Inversionszentrum. Aufgrund der starken Spannungen im [Ge2F10]2− ist diese Einheit stark verzerrt. Die längsten Ge–F-Bindungsabstände sind mit 192.01(16) und 194.48(16) pm die Bindungen zu den verbrückenden Fluoratomen. Diese sind damit um über 20 pm länger als der kürzeste Ge–F-Bindungsabstand (172.47(17) pm) in dieser Struktur. Verlängert ist auch die Ge–F-Bindung des axialen Fluoratoms, welches an der Wasserstoffbrückenbindung zum [C6H5NO2H]+ beteiligt ist. Es liegt eine starke, fast lineare Wasserstoffbrücke mit einem O···F-Abstand von nur 249.8(3) pm vor. In dieser Struktur beträgt der Ge···Ge-Abstand 308.11(5) pm, was mit den Abständen der vergleichbaren Zentralatome von [AlF5]24− und [TiF5]24− im Einklang ist ((Al⋅⋅⋅Al) = 296.0(1) pm, Ti⋅⋅⋅Ti = 280.7(1)).[92-93] Abbildung 3.39: Kristallpackung von [(C6H5NO2H)2]2+[Ge2F10]2− mit interionischen Kontakten entlang der a-Achse (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome). Die F–Ge–F-Winkel des [Ge2F10]2− weichen ebenfalls deutlich von den idealen 90° eines Oktaeders ab. Die Verzerrung kommt durch das Herausrücken des Zentralatoms aus der 61 Ergebnisse und Diskussion Polyedermitte zustande. Die Abweichung ist mit 74.27(8)° am stärksten beim F(5)–Ge(1)–F(5)#1-Winkel, an dem die verbrückenden Fluoratome beteiligt sind. Die größte Aufweitung befindet sich auf der gegenüberliegenden Seite beim F(4)−Ge(1)−F(3)-Winkel mit 101.46(8)°. Die Verzerrung wird zusätzlich durch die Koordination von F(2) zum positiven Stickstoff der NO(OH)-Gruppe verstärkt (Abbildung 3.40). Diese Koordination wurde auch von Götz bei [(C6H5NO2)2H]+[MF6]− (M = As, Sb) beobachtet. Bei [(C6H5NO2H)2]2+[Ge2F10]2− bildet das Anion eine Wasserstoffbrückenbindung aus und koordiniert zusätzlich mit einem zweiten Fluoratom zum elektronenarmen Stickstoffatom der NO(OH)-Gruppe. Die zweite Koordination auf der Gegenseite erfolgt durch ein anderes [Ge2F10]2−-Anion. Durch diese Kontakte werden ([(C6H5NO2H)2]2+[Ge2F10]2−)n-Stränge entlang der b-Achse gebildet (Abbildung 3.39). Abbildung 3.40: [C6H5NO2H]+ mit interionischen Kontakten (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome, Symmetrieoperationen: #1: –x,–y,–z, #2: –x,1–y,–z). 3.3.1.4 Theoretische Berechnungen von [Ge2F10] 2− Die DFT-Berechnungen der Gasphasenstruktur von [Ge2F10]2− (D2h-Symmetrie) wurde mit der Methode PBE1PBE und dem 6-311G(3df,3pd)-Basissatz durchgeführt (Abbildung 3.41).[41-43] Anschließend wurden die Schwingungsfrequenzen der relaxierten Struktur, sowie die IR- und Raman-Intensitäten berechnet. Die geometrischen Parameter der optimierten Struktur sind in Tabelle 3.17 aufgelistet. Man kann erkennen, dass das berechnete Anion in sehr guter Übereinstimmung mit der Geometrie des Anions in der Kristallstruktur von [(C6H5NO2H)2]2+[Ge2F10]2− ist. 62 Ergebnisse und Diskussion Abbildung 3.41: Quantenchemisch berechnete Struktur von [Ge2F10]2−. 3.3.1.5 Schwingungsspektren Das Kation weist im Verhältnis zum Anion in dieser Verbindung eine sehr starke RamanAktivität auf. Daher wird das [Ge2F10]2−-Anion erst in Kapitel 3.2.2 schwingungsspektroskopisch näher betrachtet und diskutiert und hier hauptsächlich auf das Kation eingegangen. Abbildung 3.42: Raman-Spektrum von [(C6H5NO2H)2]2+[Ge2F10]2−. 63 Ergebnisse und Diskussion Tabelle 3.18: Beobachtete Schwingungsfrequenzen [cm–1] von [(C6H5NO2H)2]2+[Ge2F10]2− 2+ - [(C6H5NO2H)2] [Ge2F10] 2+ Zuordnung Raman 3138 (2) 3094 (5) 1644 (12) 1563 (71) 1459 (18) 1333 (1) 1322 (1) 1207 (60) 1170 (6) 1156 (100) 1105 (15) 1086 (6) 1022 (13) Raman-Aktivität in %. - [(C6H5NO2H)2] [Ge2F10] Zuordnung Raman ν(CH) ν(CH) ν(N=O) ν(CC) δ(CCH)in plane ν(CC) δ(CCH)in plane δ(CCH)in plane δ(CCH)in plane ν(CN) δ(CCH) δ(CCH) ν(N-O) 1004 828 801 670 651 533 279 698 608 489 360 333 (43) (1) (33) (2) (1) (3) (3) (5) (10) (2) (1) (2) δ(CCC) δ(CCH)out of plane δ(ONO) δ(CNO)out of plane δ(CCC)in plane δ(CNO)in plane δ(CCN)in plane [Ge2F10] 2− Das Raman-Spektrum von [(C6H5NO2H)2]2+[Ge2F10]2− ist in Abbildung 3.42 wiedergegeben. Die dazugehörigen Schwingungsfrequenzen sind in Tabelle 3.18 aufgelistet. Die (O–H)-Streckschwingung kann im Raman-Spektrum nicht beobachtet werden. Der Grund hierfür ist wahrscheinlich die starke O–H···F-Wasserstoffbrückenbindung, die in [(C6H5NO2H)2]2+[Ge2F10]2− zu finden ist und zu einer Schwächung der Aktivität führt. Die Spektren zeigen zwei sehr unterschiedliche NO-Streckschwingungen, die ν(N=O) ist bei 1644 cm–1 zu finden und die ν(N–O) bei 1022 cm–1. Im Vergleich zum unprotonierten Nitrobenzol ist die Schwingung der N=O-Doppelbindung daher blau- und die der N–OEinfachbindung stark rotverschoben (C6H5NO2: ν(N=O) = 1525 cm–1, ν(N–O) = 1346 cm–1), was ein guter Beweis für die erfolgreiche Protonierung des Nitrobenzols darstellt. Die restlichen beobachteten Linien und Aktivitäten stimmen sehr gut mit denen der hemiprotonierten Spezies überein.[95] Die Linien bei 698, 608, 489, 360 und 333 cm–1 werden dem Anion zugeordnet, auf welches in Kapitel 3.3.2.3 näher eingegangen wird. 64 Ergebnisse und Diskussion 3.3.2 [(Me2OH)2]2+[Ge2F10]2− 3.3.2.1 Bildung von [(Me2OH)2]2+[Ge2F10] 2− [(Me2OH)2]2+[Ge2F10]2− wurde quantitativ in einem Schritt nach Gleichung (3.14) synthetisiert. 2 Me2O + 2 [(H2F)2]2+[GeF6]2- -50 °C, HF [(Me2OH)2]2+[Ge2F10]2- + 6 HF -60 °C, HF (3.14) 8 HF + 2 GeF4 Dimethylether wurde in einem Reaktor vorgelegt und anschließend wurden im hohen Überschuss HF (150 : 1) und GeF4 (2.0/1.0/0.5 : 1) kondensiert. Als das Reaktionsgemisch langsam auf –60 °C aufgetaut wurde, bildete sich die Supersäure [(H2F)2]2+[GeF6]2−. Bei weiterem Erwärmen auf –50 °C löste sich der Dimethylether in der supersauren Lösung und wurde protoniert. Nach langsamem Entfernen von HF und, je nach Ansatz, überschüssigem GeF4 oder Me2O für 12 h bei –78 °C und für 8 h bei –50 °C bildeten sich farblose Kristalle von [(Me2OH)2]2+[Ge2F10]2−, welche bis –30 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 3.3.2.2 Röntgenstrukturanalyse von [(Me2OH)2] 2+[Ge2F10] 2− [(Me2OH)2]2+[Ge2F10]2− kristallisiert in der triklinen Raumgruppe P–1 (Nr. 2) mit einer Formeleinheit in der Elementarzelle. Ausgewählte Bindungslängen und -winkel von [(Me2OH)2]2+[Ge2F10]2− sowie der berechneten [Ge2F10]2−-Einheit, welche schon in Kapitel 3.3.1.4 beschrieben wurde, sind in Tabelle 3.19 aufgelistet, zusammen mit den berechneten Strukturdaten der [Ge2F10]2−-Einheit. Abbildung 3.43 zeigt die asymmetrische Einheit von [(Me2OH)2]2+[Ge2F10]2− und Abbildung 3.44 zeigt einen Ausschnitt aus der Kristallpackung. Alle Atome, einschließlich der Wasserstoffatome am Sauerstoff wurden mittels Differenz-Fourier-Synthese gefunden und frei verfeinert. Die Wasserstoffatome der Methylgruppen wurden berechnet. Alle NichtWasserstoffatome wurden anisotrop verfeinert. 65 Ergebnisse und Diskussion Abbildung 3.43: Struktur von [(Me2OH)2]2+[Ge2F10]2−, Beschriftung der asymmetrischen Einheit (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome). Tabelle 3.19: Ausgewählte Bindungslängen [pm] und -winkel [(Me2OH)2]2+[Ge2F10]2− und des berechneten [Ge2F10]2− 2+ [(Me2OH)2] [Ge2F10] 2− [Ge2F10] [°] 2− D2h, ber. exp. O(1)–C(2) 147.1(2) Ge(1)–F(2) 173.93(12) 177.0 O(1)–C(1) 147.8(2) Ge(1)–F(4) 174.26(12) 175.9 O(1)–H(1) 80(3) Ge(1)–F(3) 174.50(12) 175.9 Ge(1)–F(1) 179.18(12) 177.0 C(2)–O(1)–C(1) 114.51(16) Ge(1)–F(5) 191.28(12) 195.7 C(2)–O(1)–H(1) 118(2) Ge(1)–F(5)#1 195.15(12) 195.7 C(1)–O(1)–H(1) 112(2) Ge(1)–Ge(1)#1 306.58(5) 312.4 F(2)–Ge(1)–F(4) 92.47(6) 91.8 H(1)···F(1) 169(3) F(2)–Ge(1)–F(3) 93.11(6) 91.8 O(1)···F(1) 249.30(19) F(4)–Ge(1)–F(3) 174.30(6) 174.5 O(1)–H(1)···F(1) 175(4) F(2)–Ge(1)–F(1) 101.16(6) 100.7 F(4)–Ge(1)–F(1) 90.62(6) 91.8 F(3)–Ge(1)–F(1) 89.46(6) 91.8 F(2)–Ge(1)–F(5) 93.33(6) 92.6 F(4)–Ge(1)–F(5) 88.88(5) 87.8 F(3)–Ge(1)–F(5) 89.62(5) 87.8 F(4)–Ge(1)–F(5)#1 87.15(5) 87.8 F(3)–Ge(1)–F(5)#1 87.14(5) 87.8 F(1)–Ge(1)–F(5)#1 90.51(5) 92.6 F(5)–Ge(1)–F(5)#1 75.00(6) 74.1 Ge(1)–F(5)–Ge(1)#1 105.00(5) [a] berechnet mit PBE1PBE/6-311G(3df,3pd); Symmetrieoperationen: #1: –x,–y,–z. 66 105.9 [a] von Ergebnisse und Diskussion Die asymmetrische Einheit von [(Me2OH)2]2+[Ge2F10]2− besteht aus einem 2− Dimethyloxoniumkation und einem halben [Ge2F10] -Anion. Das [Me2OH]+-Kation ist pyramidal aufgebaut und ähnelt dem [Me2OH]+-Kation in [(Me2OH)2]2+[SbCl5]2−, welches von Jin et al. beschrieben wurde.[98] Das Anion besteht, wie beim [(C6H5NO2H)2]2+[Ge2F10]2−, aus zwei kantenverknüpften Oktaedern und besitzt auf der verknüpfenden Kante ein Inversionszentrum. Die Oktaeder sind stark verzerrt, da sich auch hier die Zentralatome nicht mehr in der Polyedermitte befinden. Der kleinste in dieser Struktur gefundene F–Ge–F-Winkel beträgt 75.00(6)° und der größte 101.16(6)°. Auch hier treten die längsten Ge–F-Bindungslängen mit 191.28(12) und 195.15(12) pm bei den verbrückenden Fluoratomen auf. Der kürzeste beobachtete Ge–F-Abstand beträgt nur 173.93(12) pm. Eine fast lineare Wasserstoffbrückenbindung mit einem O···F-Abstand von nur 249.8(3) pm liegt zwischen dem aciden Proton des [Me2OH]+ und einem äquatorialen Fluoratom des [Ge2F10]2− vor. In dieser Struktur beträgt der Ge–Ge-Abstand nur 306.58(5) pm und ist somit der kürzeste Ge–Ge-Abstand, der bisher für binäre oktaedrisch koordinierte Germaniumverbindungen gefunden wurde. Abbildung 3.44: Kristallstruktur von [(Me2OH)2]2+[Ge2F10]2− (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). 3.3.2.3 Schwingungsspektren 67 Ergebnisse und Diskussion Für die Zuordnung der Schwingungsfrequenzen des Dimethyloxoniumkations wurden ebenfalls Berechnungen einer [Me2OH(HF)]+-Einheit zu Hilfe genommen, sowie das Spektrum mit dem von Dimethylether verglichen.[99] Die v(CO2) verschieben sich durch die Protonierung auf 979 und 827 cm–1, also zu niedrigeren Wellenzahlen (Me2O: νas(CO2) = 1095 cm–1 und νs(CO2) = 920 cm–1), was durch die Schwächung der C–O-Bindung bedingt wird. Die Streckschwingungen der CH3-Gruppe verschieben sich leicht zu höheren Wellenzahlen, die Deformationsschwingungen bleiben durch die Protonierung nahezu unbeeinflusst. In [(C6H5NO2H)2]2+[Ge2F10]2− werden bei 670, 489, 360 und 333 cm–1 die Schwingungen des Anions beobachtet. Das ist in guter Übereinstimmung mit den gefundenen Werten bei [(Me2OH)2]2+[Ge2F10]2−. Die kleinen Abweichungen lassen sich durch die unterschiedliche Ausbildung der Wasserstoffbrücken erklären. Die beteiligten Fluoratome sind bei [(C6H5NO2H)2]2+[Ge2F10]2− in axialer und bei [(Me2OH)2]2+[Ge2F10]2− in äquatorialer Stellung. Abbildung 3.45: Raman-Spektrum von [(Me2OH)2]2+[Ge2F10]2−. Tabelle 3.20: Beobachtete Schwingungsfrequenzen [cm–1] von [(Me2OH)2]2+[Ge2F10]2− sowie berechnete Schwingungsfrequenzen von [Me2OH(HF)]+ und [Ge2F10]2− 68 Ergebnisse und Diskussion 2+ [(Me2OH)2] [Ge2F10] Raman 2− [Me2OH(HF)] + 2+ Zuordnung [(Me2OH)2] [Ge2F10] [a] C1, ber. 3279 (1267/79) 3157 (0/45) 3156 (1/22) 3141 (1/12) 3139 (1/51) 3030 (0.4/1) 3029 (4/245) Raman ν(OH) νas(CH3) νas(CH3) νas(CH3) νas(CH3) νs(CH3) νs(CH3) 2− [Ge2F10] 2- Zuordnung [b] D2h, ber. 739 (294/0) 720 (0/0) 684 (0/15) 667 (265/0) 666 (81/0) 654 (0/1) 608 (0/4) 606 (180/0) νs(Ge–Fe) νas(Ge–Fe) 684 (62) νs(Ge–Fe) νas(Ge–Fe) 3066 (17) νas(Ge–Fe) νas(Ge–Fe) 2992 (100) 621 (32) νs(Ge–Fe) 2873 (9) νas(Ge–Fe) 2834 (10) 554 (3) ν(Ge–F) 2654 (3) 500 (8) ν(Ge–Fμ) 508 (0/2) 2558 (4) 432 (115/0) ν(Ge–Fμ) δ(COH) 1517 (4) 1500 (48/2) ν(Ge–Fμ) 425 (77/0) δas(CH3) 1482 (36) 1467 (14/6) ν(Ge–Fμ) 407 (0/0) δas(CH3) 1458 (19) 1460 (28/1) 393 (5) δ(Fe–Ge–Fμ) 382 (43/0) δas(CH3) 1448 (9) 1449 (15/1) δ(Fe–Ge–Fe) 371 (0/0.5) δas(CH3) 1446 (8/2) 355 (5) δ(F–Ge–F) 349 (33/0) δas(CH3) 1435 (1/5) δ(Fe–Ge–Fe) 347 (0/1) δs(CH3) 1423 (6) 1430 (2/1) 339 (13) δ(Fe–Ge–Fe) 341 (0/1) ρ(CH3) 1260 (8) 1253 (2/2) 315 (248/0) δ(Fe–Ge–Fe) ρ(CH3) 1198 (23) 1187 (6/1) δ(Fe–Ge–Fe) 311 (0/0) ρ(CH3) 1146 (4) 1130 (4/1) δ(Fe–Ge–Fμ) 290 (17/0) ρ(CH3) 1123 (3) 1095 (1/0.3) 250 (0/0.04) δ(Fe–Ge–Fμ) νas(CO2) 957 (25) 979 (116/2) δ(Fe–Ge–Fe) 209 (0/0.3) δ(COH) 932 (77/1) 208 (0/0.02) δ(Fe–Ge–Fμ) νs(CO2) 836 (48) 827 (60/8) δ(Fe–Ge–Fe) 205 (1/0) δ(COC) 409 (6) 384 (103/1) δ(Gerüst) 203 (0/0.4) 190 (13/0.01) τ(CH3)i.p. δ(Gerüst) 179 (0/0.8) τ(CH3)o.o.p. 145 (1/0.1) 120 (0/0.04) δ(Gerüst) δ(Gerüst) 115 (0.3/0) δ(Gerüst) 94 (0.3/0) τ 92 (0/0) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 1.04 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %; Fe: endständiges Fluoratom; Fμ: verbrückendes Fluoratom. 3082 (48) 69 Ergebnisse und Diskussion 3.3.3 [(Me2OH)4]4+[Ge3F16]4− 3.3.3.1 Bildung von [(Me2OH)4]4+[Ge3F16] 4− Bei der Synthese von [(Me2OH)2]2+[Ge2F10]2− bildeten sich, nachdem der Reaktor 6 Tage in einem Dewargefäß mit Trockeneis stehen gelassen wurde, im oberen Bereich des Reaktors farblose Kristalle. Die Temperatur an der Kristallisationsstelle lag bei ca. –30 °C. Die Kristalle konnten mittels Einkristall-Röntgenstrukturanalyse untersucht und als [(Me2OH)4]4+[Ge3F16]4− identifiziert werden. Sie sind hydrolyseempfindlich und bis ca. –15 °C beständig. Wahrscheinlich schmolz bei einer kurzen Erwärmung des oberen Teils des Reaktors ein Teil der Kristalle von [(Me2OH)2]2+[Ge2F10]2− und kristallisierte anschließend als [(Me2OH)4]4+[Ge3F16]4− wieder aus. Es wurden somit 0.5 Äquivalente GeF4 pro [(Me2OH)2]2+[Ge2F10]2− aus dem Kristall entfernt. Da kein Überdruck im Reaktor vorlag, bildete sich wahrscheinlich im kälteren Teil des Reaktors [(H2F)2]2+[GeF6]2−, dessen Existenz aber nicht nachgewiesen werden konnte (Gleichung (3.15)). Eine gezielte Synthese dieser Verbindung gelang nicht. 2 [(Me2OH)2]2+[Ge2F10]2- -30 °C [(Me2OH)4]4+[Ge3F16]4- + GeF4 (3.15) 3.3.3.2 Röntgenstrukturanalyse von [(Me2OH)4] 4+[Ge3F16] 4− [(Me2OH)4]4+[Ge3F16]4− kristallisiert in der triklinen Raumgruppe P–1 (Nr. 2) mit einer Formeleinheit in der Elementarzelle. Das [Ge3F16]4−-Ion war bisher unbekannt. Ausgewählte Bindungslängen und -winkel von [(Me2OH)4]4+[Ge3F16]4− sind in Tabelle 3.21 aufgelistet, zusammen mit den berechneten Strukturdaten der [Ge3F16]4−-Einheit. Alle Atome, einschließlich der Wasserstoffatome am Sauerstoff, wurden mittels Differenz-FourierSynthese gefunden und frei verfeinert. Die Wasserstoffatome der Methylgruppen wurden berechnet. Alle Nicht-Wasserstoffatome wurden anisotrop verfeinert. Die asymmetrische Einheit von [(Me2OH)4]4+[Ge3F16]4− besteht aus zwei Dimethyloxoniumkationen und einem halben [Ge3F16]4−-Anion (Abbildung 3.46). Ein Ausschnitt aus der Kristallpackung ist in Abbildung 3.47 dargestellt. 70 Ergebnisse und Diskussion Das [Ge3F16]4− ist ein Trimer aus drei eckenverknüpften [GeF6]−-Oktaedern und besitzt ein Inversionszentrum auf Ge(2). Die Verknüpfung über das mittlere Oktaeder erfolgt in transPosition. Die Oktaeder sind in einer Zick-Zack-Kette angeordnet mit 157.08(9)° gegeneinander verkippt sowie um ca. 20° gegeneinander verdreht (staggered). Eine ähnliche Struktur des Anions findet man in [Br2]+[Sb3F16]−. Auch hier sind die Oktaeder gegeneinander verkippt (148(2)°) und staggered angeordnet (36(2)°).[100] Abbildung 3.46: Struktur von [(Me2OH)4]4+[Ge3F16]4− im Kristall, Beschriftung der asymmetrischen Einheit (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome). Abbildung 3.47: Kristallstruktur von [(Me2OH)4]4+[Ge3F16]4− (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). 71 Ergebnisse und Diskussion Je zwei der Fluoratome der endständigen Oktaeder bilden Wasserstoffbrücken zu den aciden Protonen des [Me2OH]+ aus. Die Ge–F-Abstände sind bei diesen Bindungen gegenüber den restlichen endständigen Ge–F-Bindungen um bis zu 9 pm verlängert. Die längsten Ge–FAbstände sind, wie zu erwarten, zum verbrückenden Fluoratom mit 190.10(14) und 193.20(14) pm zu finden. Die kürzeste beobachtete Ge–F-Bindungslänge liegt bei 174.06(14) pm. Tabelle 3.21: Ausgewählte Bindungslängen [pm] und -winkel [°] 4+ 4− 4− [(Me2OH)4] [Ge3F16] und von der berechneten [Ge3F16] -Einheit 4+ 4− [(Me2OH)4] [Ge3F16] exp. O(1)–C(2) 147.0(3) Ge(1)–F(4) 174.06(14) O(1)–C(1) 148.2(3) Ge(1)–F(2) 174.11(14) O(1)–H(1) 89(4) Ge(1)–F(3) 175.20(15) O(2)–C(3) 146.2(3) Ge(1)–F(6) 178.58(15) O(2)–C(4) 146.4(3) Ge(1)–F(1) 183.08(14) O(2)–H(2) 79(4) Ge(1)–F(5) 193.20(14) C(2)–O(1)–C(1) 113.6(2) Ge(2)–F(7) 174.06(14) Ge(2)–F(8) 175.15(15) C(2)–O(1)–H(1) 111.3(19) Ge(2)–F(5) 190.10(14) C(1)–O(1)–H(1) 112.6(19) C(3)–O(2)–C(4) 114.7(2) F(4)–Ge(1)–F(2) 95.08(7) C(3)–O(2)–H(2) 104(3) F(4)–Ge(1)–F(3) 95.01(7) C(4)–O(2)–H(2) 113(3) F(2)–Ge(1)–F(3) 93.12(7) F(4)–Ge(1)–F(6) 93.58(7) H(1)···F(6) 158(4) F(2)–Ge(1)–F(6) 91.84(7) O(1)···F(6) 246.8(3) F(3)–Ge(1)–F(6) 169.66(7) O(1)–H(1)···F(6) 175(3) F(4)–Ge(1)–F(1) 91.64(7) F(2)–Ge(1)–F(1) 173.10(7) H(2)···F(1) 165(4) F(3)–Ge(1)–F(1) 87.85(7) O(2)···F(1) 242.7(2) F(6)–Ge(1)–F(1) 86.15(7) O(2)–H(2)···F(1) 167(5) F(4)–Ge(1)–F(5) 176.11(7) F(2)–Ge(1)–F(5) 88.80(6) F(3)–Ge(1)–F(5) 84.98(7) F(6)–Ge(1)–F(5) 86.06(7) F(1)–Ge(1)–F(5) 84.48(6) F(7)–Ge(2)–F(8) 89.92(8) F(7)–Ge(2)–F(5)#1 91.04(7) F(8)–Ge(2)–F(5)#1 88.91(7) F(7)–Ge(2)–F(5) 88.96(7) F(8)–Ge(2)–F(5) 91.09(7) Ge(2)–F(5)–Ge(1) 157.08(9) [a] berechnet mit PBE1PBE/6-311G(3df,3pd); Symmetrieoperationen: #1: –x,–y+1,–z+1. 72 von 4− [Ge3F16] [a] C2h, ber. 182.6 178.9 178.9 178.9 178.9 199.8 176.6 176.6 200.1 91.3 91.3 90.0 91.3 90.0 90.0 91.3 177.3 90.0 90.0 180.0 88.7 88.6 88.7 88.6 90.0 90.0 90.0 90.0 90.0 179.9 Ergebnisse und Diskussion 3.3.3.3 Theoretische Berechnungen von [Ge3F16] 4− Die DFT-Berechnungen der Gasphasenstruktur von [Ge3F16]4− (C2h-Symmetrie) wurde mit der Methode PBE1PBE und dem 6-311G(3df,3pd)-Basissatz durchgeführt (Abbildung 3.48).[41-43] Anschließend wurden die Schwingungsfrequenzen der relaxierten Struktur sowie die IR- und Raman-Intensitäten berechnet. Die geometrischen Parameter sind in Tabelle 3.21 aufgelistet. Die Geometrie des berechneten Anions ist in akzeptabler Übereinstimmung mit der Struktur des Anions im Kristall. In der berechneten Struktur ist die Kette linear aufgebaut, wohingegen der Ge–F–Ge-Winkel im Kristall 157.08(9)° beträgt. Die Oktaeder sind jedoch genau wie im Kristall staggered angeordnet. Bei der Berechnung der Schwingungsfrequenzen fällt auf, dass die niedrigste Frequenz negativ ist. Dieser Wert ist mit –23 cm–1 aber relativ klein, daher kann davon ausgegangen werden, dass bei der Strukturoptimierung trotzdem ein energetisches Minimum und kein Übergangszustand gefunden wurde. Abbildung 3.48: Quantenchemisch berechnete Struktur von [Ge3F16]4−. 73 Ergebnisse und Diskussion 3.3.4 [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF 3.3.4.1 Bildung von [(Me2SH)3] 3+[Ge4F19] 3− ⋅ 2 HF [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF wurde quantitativ in einem Schritt nach Gleichung (3.16) synthetisiert. 3 Me2S + 3 [(H2F)2]2+[GeF6]2- -50 °C, HF [(Me2SH)3]3+[Ge4F19]3- 2 HF + 7 HF (3.16) -60 °C, HF 12 HF + 3 GeF4 Dimethylsulfid wurde in einem Reaktor vorgelegt und anschließend wurden im hohen Überschuss HF (150 : 1) und GeF4 (0.5 : 1) kondensiert. Als das Reaktionsgemisch langsam auf –60 °C aufgetaut wurde, bildete sich die Supersäure [(H2F)2]2+[GeF6]2−. Bei weiterem Erwärmen auf –50 °C löste sich das Dimethylsulfid in der supersauren Lösung und wurde protoniert. Nach langsamem Entfernen von HF bei –78 °C für 48 h und bei –60 °C für 10 h bildeten sich farblose Kristalle von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF, welche bis –30 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. Es ist davon auszugehen, dass unprotoniertes Dimethylsulfid, welches einen Schmelzpunkt von –98 °C besitzt, beim Entfernen des Fluorwasserstoffs ebenfalls entfernt wurde, da die Stöchiometrie des Produktes von der Stöchiometrie der Edukte deutlich abweicht. 3.3.4.2 Röntgenstrukturanalyse von [(Me2SH)3] 3+[Ge4F19] 3− ⋅ 2 HF [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF kristallisiert in der triklinen Raumgruppe P–1 (Nr. 2) mit einer Formeleinheit in der Elementarzelle. Ein [Ge4F19]3−-Ion mit dieser Struktur war bisher unbekannt. Ausgewählte Bindungslängen und -winkel von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF und den berechneten Strukturdaten der [Ge4F20]4−-Einheit sind in Tabelle 3.22 aufgelistet. Abbildung 3.49 und Abbildung 3.50 [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF. In zeigen die Abbildung 3.51 Strukturen wird ein der Ionen Ausschnitt aus von der Kristallstruktur dargestellt. Alle Atome, einschließlich der Wasserstoffatome am Schwefel und am Fluor, wurden mittels Differenz-Fourier-Synthese gefunden und frei verfeinert. Die Wasserstoffatome der Methylgruppen wurden berechnet. Alle Nicht-Wasserstoffatome wurden anisotrop verfeinert. 74 Ergebnisse und Diskussion Abbildung 3.49: Struktur von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF im Kristall (Wasserstoffatome wurden der Übersicht halber weggelassen; thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). In dieser Struktur liegen Inversionszentren zum einen in der Mitte des vierer-Rings[84], auf F(5) und zwischen den zwei Kohlenstoffatomen (C(3)) eines Dimethylsulfoniumkations. Eine Verdoppelung der Zelle entlang der a-Achse, um die Fehlordnung des protonierten Dimethylsulfid Moleküls aufzuheben, führte zu keinen zufriedenstellenden Ergebnissen. Es liegen zwei kristallographisch unabhängige Kationen vor, bei denen die S–H-Bindung aus der CSC-Ebene mit einem Winkel von 78.25° bzw. 82.46° herausstehen. Aufgrund der geringen Fähigkeit des Schwefels zur sp2-Hybridisierung ist das Proton stärker abgewinkelt als beim protonierten Dimethylether. Beide Kationen bilden je eine Wasserstoffbrückenbindung zum Anion aus. Bindungslängen und Bindungswinkel des Dimethylsulfoniumkations stehen mit den von Mootz und Deeg erhaltenen Werten bei Me2S ∙ 4 HCl und Me2S ∙ 5 HCl im Einklang.[101] Das Anion hat eine polymere Kettenstruktur. Darin bilden vier [GeF6]2−-Oktaeder durch cisFluor-Verknüpfungen einen vierer-Ring. Diese Ringe sind ebenfalls miteinander über ein weiteres Fluoratom verknüpft. Die dadurch entstehenden, zur a-Achse parallel verlaufenden Stränge, welche sich in eine Ebene anordnen und somit Schichten senkrecht zur b-Achse ausbilden. Dabei entsteht eine AB-Struktur, da die Bänder in jeder zweiten Schicht um eine halbe Einheit versetzt sind. 75 Ergebnisse und Diskussion Die Ge–F-Abstände des Anions lassen sich in drei Kategorien unterteilen. Endständige Fluoratome weisen, bedingt durch die starke Verzerrung, sehr kurze Ge–F-Abstände auf (171.07(16)−175.71(14) pm), was deutlich kürzer ist als die Ge–F-Abstände eines regulären [GeF6]2−-Oktaeders (d(Ge–F) = 177 – 178 pm).[102] Auffallend dabei ist, dass axiale Ge–F-Bindungen im Vergleich mit äquatorialen Ge–F-Bindungen die marginal kürzere Spezies darstellen. Bei der dreifach verknüpften Oktaedereinheit des Ge(1)-Atoms liegen die Ge–FBindungslängen von verbrückenden Fluoratomen zwischen 182.51(14) und 186.86(14) pm. Bei der zweifach verknüpften Oktaedereinheit des Ge(2)-Atoms sind bei den verbrückenden Fluoratomen die längsten Ge–F-Bindungslängen der Struktur zu beobachten (191.71(14) bis 195.08(14) pm). Innerhalb des vierer-Rings liegen alle Germaniumatome in einer Ebene. Um die Ringspannung zu erniedrigen und die Verknüpfung der Ringe untereinander zu ermöglichen, weisen zwei Fluoratome in den Ring hinein und zwei hinaus. Das führt zu F–Ge–F-Winkeln von 83.71(6)° und 88.41(7)° sowie Ge–F–Ge-Winkeln von 157.97(9)° und 206.53(9)° (Ring-Innenwinkel). Die Dimethylsulfoniumkationen sind zwischen den erwähnten anionischen Schichten eingelagert und bilden je eine starke, fast lineare Wasserstoffbrückenbindung zu den Fluoratomen des Anions aus (d(S···F) = 330.00(17) und 325.2(2) pm). Abbildung 3.50: Links: Struktur der Kationen in [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF, rechts: repetitive Einheit des Anions mit Winkeln [°] (blau) und Bindungslängen [pm] (schwarz) (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome). 76 Ergebnisse und Diskussion Tabelle 3.22: Ausgewählte Bindungslängen [pm] und -winkel [°] von 3+ 3− 4− [(Me2SH)3] [Ge4F19] ⋅ 2 HF und von der berechneten [Ge4F20] -Einheit 3+ 3− [(Me2SH)3] [Ge4F19] ⋅ 2 HF exp. S(1)–C(2) 177.9(3) Ge(1)–F(9) 171.07(16) S(1)–C(1) 178.4(3) Ge(1)–F(10) 171.67(15) S(1)–H(1) 110(3) Ge(1)–F(2) 172.73(14) Ge(1)–F(3) 182.51(14) S(2)–C(3) 169.0(3) Ge(1)–F(5) 186.82(3) Ge(1)–F(4) 186.86(14) S(2)–C(3)#3 181.6(3) S(2)–H(2) 133(5) Ge(2)–F(8) 171.37(15) Ge(2)–F(6) 172.67(15) F(11)–H(11) 74(4) Ge(2)–F(1) 173.40(14) Ge(2)–F(7) 175.71(14) C(2)–S(1)–C(1) 102.74(14) Ge(2)–F(4) 191.71(14) C(2)–S(1)–H(1) 93.7(15) Ge(2)–F(3)#1 195.08(14) C(1)–S(1)–H(1) 101.6(16) F(9)–Ge(1)–F(10) 172.45(8) C(3)–S(2)–C(3)#3 104.37(14) F(10)–Ge(1)–F(2) 93.10(8) C(3)–S(2)–H(2) 97(2) F(2)–Ge(1)–F(3) 95.20(7) C(3)#3–S(2)–H(2) 93(2) F(2)–Ge(1)–F(5) 90.71(5) F(3)–Ge(1)–F(4) 88.41(7) H(1)···F(1)#4 223(3) F(5)–Ge(1)–F(4) 85.68(4) S(1)···F(1)#4 330.00(17) F(8)–Ge(2)–F(6) 167.81(7) S(1)–H(1)···F(1)#4 163(2) F(1)–Ge(2)–F(7) 96.97(7) F(1)–Ge(2)–F(4) 88.97(7) H(2)···F(2)#5 193(5) F(7)–Ge(2)–F(3)#1 90.33(7) S(2)···F(2)#5 325.2(2) F(4)–Ge(2)–F(3)#1 83.71(6) S(2)–H(2)···F(2)#5 172(4) Ge(1)–F(3)–Ge(2)#1 153.47(9) Ge(1)–F(4)–Ge(2) 157.97(9) Ge(1)#2–F(5)–Ge(1) 180.000(15) [a] berechnet mit PBE1PBE/6-311G(3df,3pd); Symmetrieoperationen: #1 –x+1,–y,–z+1; #3 –x+2,–y+1,–z+1; #4 –x+1,–y+1,–z+1; #5 x+1,y+1,z. Abbildung 3.51: 4− [Ge4F20] [a] C2h, ber. 175.7 175.7 178.4 194.3 178.4 194.5 175.7 175.7 178.4 178.4 194.3 194.5 172.8 90.5 90.5 93.6 85.4 90.5 172.8 93.6 90.5 90.5 85.4 175.1 175.7 #2 –x,–y,–z+1; Kristallstruktur von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF (Wasserstoffatome wurden der Übersicht halber weggelassen; thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). 77 Ergebnisse und Diskussion 3.3.4.3 Theoretische Berechnungen von [Ge4F20] 4− Die DFT-Berechnungen der Gasphasenstruktur von [Ge4F20]4− (C2h-Symmetrie) wurde mit der Methode PBE1PBE und dem 6-311G(3df,3pd)-Basissatz durchgeführt (Abbildung 3.48).[41-43] Anschließend wurden die Schwingungsfrequenzen der relaxierten Struktur, sowie die IR- und Raman-Intensitäten berechnet. Die geometrischen Parameter des [Ge4F20]4−-Anions sind in Tabelle 3.22 den strukturellen Daten von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF gegenübergestellt. Die Geometrie des berechneten vierer-Rings ist in akzeptabler Übereinstimmung mit der Geometrie des Anions im Kristall. Unterschiede ergeben sich deshalb, da nur die isolierte Einheit berechnet werden konnte, im Kristall diese Ringe jedoch untereinander zu einer Kette verknüpft sind. Die unterschiedlichen Ge–F-Bindungslängen in Abhängigkeit vom Verknüpfungsgrad des zugehörigen Oktaeders, sowie die unterschiedlichen Ge–F-Abstände bei axialer oder äquatorialer Stellung der Fluoratome werden trotzdem gut wiedergegeben. Die Fluor- und Germaniumatome, welche den Ring bilden, liegen auch in der berechneten Struktur in einer Ebene. Typische Frequenzmuster im Spektrum, welche den Deformationsschwingungen des Rings zugeordnet werden können, finden sich auch im aufgenommenen Spektrum wieder. Abbildung 3.52: Quantenchemisch berechnete Struktur von [Ge4F20]4− mit Winkeln [°] (blau) und Bindungslängen [pm] (schwarz). 78 Ergebnisse und Diskussion 3.3.4.4 Schwingungsspektren Das erhaltene Raman-Spektrum stimmt mit den Ergebnissen der Röntgenstrukturanalyse überein (Abbildung 3.53). Die Zuordnung der Schwingungsfrequenzen erfolgte durch Vergleich mit vorhandenen Literaturdaten und quantenchemischen Berechnungen der Schwingungsfrequenzen von [Ge4F20]4− und [Me2SH(HF)]+.[21,81,103] Die beobachteten Linien und Aktivitäten stimmen gut mit den bereits Veröffentlichen für [Me2SH]+ überein.[103] Die vollständige Zuordnung ist in Tabelle 3.23 dargestellt. Im Bereich zwischen 3100 und 830 cm–1 befinden sich ausschließlich die Linien des [Me2SH]+-Kations. Insbesondere die (S–H)-Streckschwingung bei 2525 cm–1 ist gut zu beobachten und ein eindeutiger Nachweis für die erfolgreiche Protonierung des Dimethylsulfids. Die Linienform ist breit und lässt auf eine starke Assoziation der Salze im Feststoff schließen. Bei 784 cm–1 befindet sich die δ(SCH) und die zwei ν(C–S) sind bei 731 und 666 cm–1 zu beobachten. Die intensivste Bande im niederfrequenten Bereich bei 289 cm–1 wird der δ(CSC) des Kations zugeordnet. Abbildung 3.53: Raman-Spektrum von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF. 79 Ergebnisse und Diskussion Die Schwingungen des Anions weisen deutlich geringere Aktivitäten gegenüber denen des Kations auf und werden teilweise durch dessen intensivere Schwingungsbanden überlagert. Die ν(Ge–F) bei 677 cm–1, welche der Berechnung zufolge die größte Raman-Aktivität aufweist, wird teilweise durch die (C–S)-Streckschwingung des Kations (666 cm–1) überlagert. Eine weitere Linie bei 689 cm–1 wird ebenfalls zum Teil überlagert. Sie ist, bezogen auf die Berechnungen, nicht dem isolierten vierer-Ring zuzuordnen, sondern wird wahrscheinlich aufgrund der Symmetrieerniedrigung durch die Polymerstruktur hervorgerufen. Die Linien bei 677 und 621 cm–1 werden den Streckschwingungen des Rings zugeordnet. Die Deformationsschwingungen des Rings werden bei 363, 351 und 337 cm–1 beobachtet. Im Vergleich mit den berechneten Werten zeigt sich eine akzeptable Übereinstimmung, wenn auch nicht alle beobachteten Schwingungen den berechneten zugeordnet werden können. Die theoretischen Berechnungen sind nur eine Annäherung an die experimentell gefundenen Strukturen, da aufgrund des resultierenden Rechenaufwands weder die Wechselwirkungen im Kristall, noch die Polymerstruktur des Anions wiedergegeben werden können. Tabelle 3.23: Beobachtete Schwingungsfrequenzen [cm–1] von 3+ 3− [(Me2SH)3] [Ge4F19] ⋅ 2 HF und berechnete Schwingungsfrequenzen von [Me2SH(HF)]+ und [Ge4F20]4− 3+ [(Me2SH)3] [Ge4F19] ⋅ 2 HF Raman 3− [Me2SH(HF)] ber. + Zuordnung [a] 3+ [(Me2SH)3] [Ge4F19] ⋅ 2 HF Raman 3− [Ge4F20] 4− C2h, ber. Zuordnung [b] 3058 (56) 3136 (2/12) νas(CH3) 752 (543/0) νs(Ge–Fe) 3048 (100) 3134 (9/60) νas(CH3) 737 (0/0.04) νas(Ge–Fe) 2962 (78) 3120 (12/65) νas(CH3) 2821 (0) 3120 (5/31) νas(CH3) 689 (8) 3018 (11/0.4) νs(CH3) 677 (21) 3016 (1/284) νs(CH3) 2525 (76) 2579 (170/136) ν(SH) 1433 (16) 1432 (33/1) δas(CH3) 1419 (14) 730 (0.01/0) νas(Ge–Fe) ν(Ge–F) 682 (0/30) νs(Ge–Fe) 675 (2/0) νas(Ge–F) 675 (0.01/0.1) ν(Ge–F) 621 (4) 636 (0.1/0.5) ν(Ge–F) 1428 (12/7) δas(CH3) 633 (1022/0) νas(Ge–Fe) 1417 (0.03/8) δas(CH3) 618 (0/0) νas(Ge–Fe) 1415 (16/1) δas(CH3) 614 (0/0.02) νas(Ge–Fe) 1348 (2/0.1) δs(CH3) 609 (0/7) ν(Ge–Fe) 1325 (0.01/0.1) δs(CH3) 608 (200/0) ν(Ge–Fe) 600 (1) 1088 (5) 1097 (4/2) δ(CSH) 501 (0/0.1) ν(Ge–Fμ) 1075 (4) 1069 (10/2) ρ(CH3) 478 (206/0) ν(Ge–Fμ) 1052 (1) 1038 (8/1) 830 (4) ρ(CH3) 423 (0/0) ν(Ge–Fμ) 916 (3/0.1) ρ(CH3) 385 (0/1) ν(Ge–Fμ) 812 (1/2) ρ(CH3) 370 (0/0) ν(Ge–Fμ) 80 Ergebnisse und Diskussion Fortsetzung Tabelle 3.23 3+ [(Me2SH)3] [Ge4F19] ⋅ 2 HF Raman 3− [Me2SH(HF)] ber. + Zuordnung [a] Raman 784 (7) 768 (16/4) δ(CSH) 731 (17) 732 (0.2/5) νas(CS2) 666 (45) 661 (5/16) νs(CS2) 258 (57/2) δ(CSC) 198 (1/0.3) 174 (0.2/0.01) 289 (18) 3+ [(Me2SH)3] [Ge4F19] ⋅ 2 HF 3− [Ge4F20] 4− C2h, ber. Zuordnung [b] 363 (119/0) δ(Ring) 362 (0.6/0) δ(Fe–Ge–Fe) 354 (0/1) δ(F–Ge–F) 348 (0/0) δ(F–Ge–F) τ(CH3)i.p. 339 (6/0) δ(F–Ge–F) τ(CH3)o.o.p. 338 (0/0) δ(F–Ge–F) 337 (0/0) δ(F–Ge–F) 363 (2) 351 (1) δ(F–Ge–F) 337 (1) 319 (0/0.3) δ(F–Ge–F) 314 (0/0.02) δ(F–Ge–F) 312 (1156/0) δ(F–Ge–F) δ(F–Ge–F) 307 (2) 301 (0/0) δ(F–Ge–F) 282 (0/2) δ(F–Ge–F) 272 (1/0) δ(F–Ge–F) 232 (0/0.5) δ(F–Ge–F) 215 (0/0) δ(F–Ge–F) 209 (0/0.3) δ(F–Ge–F) 200 (0/0.01) δ(F–Ge–F) 199 (20/0) δ(F–Ge–F) 189 (0/0) δ(F–Ge–F) 158 (12/0) δ(F–Ge–F) 151 (0/0) δ(F–Ge–F) 141 (0/0) δ(F–Ge–F) 139 (0/2) δ(Gerüst) 137 (0/0.3) δ(Gerüst) 110 (0/0) δ(Gerüst) 92 (0.8/0) δ(Gerüst) 69 (0/0.2) δ(Gerüst) 44 (0/0) δ(Gerüst) 38 (0.2/0) δ(Gerüst) 31 (0/0) δ(Gerüst) 29 (0/0) δ(Gerüst) δ(Gerüst) 15 (0/0) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 1.05 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %; Fe: endständiges Fluoratom; Fμ: verbrückendes Fluoratom. 81 Ergebnisse und Diskussion 3.3.5 [(Me2SH)3]3+[Ge4F19]3− ⋅ HF 3.3.5.1 Bildung von [(Me2SH)3] 3+[Ge4F19] 3− ⋅ HF [(Me2SH)3]3+[Ge4F19]3− ⋅ HF wurde quantitativ in einem Schritt nach Gleichung (3.17) synthetisiert. 3 Me2S + 3 [(H2F)2]2+[GeF6]2- -50 °C, HF [(Me2SH)3]3+[Ge4F19]3- HF + 8 HF -60 °C, HF (3.17) 12 HF + 3 GeF4 Dimethylsulfid wurde in einem Reaktor vorgelegt und anschließend wurden im hohen Überschuss HF (150 : 1) und GeF4 (1 : 1) kondensiert. Als das Reaktionsgemisch langsam auf –60 °C aufgetaut wurde, bildete sich die Supersäure [(H2F)2]2+[GeF6]2−. Bei weiterem Erwärmen auf –50 °C löste sich das Dimethylsulfid in der supersauren Lösung und wurde protoniert. Nach langsamem Entfernen von HF für 16 h bei –78 °C und für 15 h bei –40 °C bildeten sich farblose Kristalle von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF, welche bis –20 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. Es ist davon auszugehen, dass unprotoniertes Dimethylsulfid, wie im Fall von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF, beim Entfernen des Fluorwasserstoffs ebenfalls entfernt wurde, da die Stöchiometrie des Produktes von der Stöchiometrie der Edukte leicht abweicht. 3.3.5.2 Röntgenstrukturanalyse von [(Me2SH)3] 3+[Ge4F19] 3− ⋅ HF [(Me2SH)3]3+[Ge4F19]3− ⋅ HF kristallisiert in der orthorhombischen Raumgruppe Pbca (Nr. 61) mit acht Formeleinheiten in der Elementarzelle. Das [Ge4F19]3−-Ion mit dieser Struktur war bisher unbekannt. Ausgewählte Bindungslängen und -winkel von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF sind in Tabelle 3.24 aufgelistet, zusammen mit den berechneten Strukturdaten der [Ge3F15]3−-Einheit. Abbildung 3.54 und Abbildung 3.55 zeigen die Strukturen der Ionen von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF und Abbildung 3.56 zeigt die 82 Ergebnisse und Diskussion Kristallpackung und die Elementarzelle entlang der a- bzw. der b-Achse. Alle Atome, einschließlich der Wasserstoffatome am Schwefel und am Fluor, wurden mittels DifferenzFourier-Synthese gefunden und frei verfeinert. Die Wasserstoffatome der Methylgruppen wurden berechnet. Alle Nicht-Wasserstoffatome wurden anisotrop verfeinert. Es liegen drei kristallographisch unabhängige Kationen vor (Abbildung 3.55), welche auch hier in guter Übereinstimmung mit der Literatur sind.[101] Zwei dieser Kationen sind über je eine Wasserstoffbrückenbindung zu einem Anion koordiniert. Das acide Proton des dritten Kations weist zwei schwächere Wasserstoffbrückenbindungen auf (Tabelle 3.25). Das Anion bildet eine polymere Struktur. Darin bilden drei [GeF6]2−-Oktaeder durch cisFluor-Verknüpfungen einen dreier-Ring[84]. Dieser Ring ist über einen weiteren cisverbrückenden [GeF6]2−-Oktaeder mit dem nächsten dreier-Ring verbunden. Die dadurch entstehenden Bänder verlaufen entlang der b-Achse und bilden Schichten in der [011]Ebene. Dabei entsteht eine AB-Schichtstruktur, in welcher die Bänder in jeder zweiten Schicht um eine halbe Einheit versetzt sind. Die Ge–F-Bindungslängen gleichen denen, der bereits diskutierten Struktur, aus Abschnitt 3.2.4. Auch hier weisen endständige Fluoratome die kürzesten Ge–F-Bindungslängen mit Werten zwischen 171.17(14) und 174.97(16) pm auf. Bei den verbrückenden Fluoratomen der dreifachverknüpfenden Oktaeder von Ge(1) und Ge(3) sind Ge–F-Abstände von 182.48(16) bis 188.63(16) pm zu beobachten. Die längsten interatomaren Ge–FBindungslängen befinden sich auch hier bei verbrückenden Fluoratomen an den zweifach verknüpfenden Oktaedern von Ge(2) und Ge(4) mit Ge–F-Abständen von 192.01(16) bis 197.09(17) pm. Die drei F–Ge–F-Winkel innerhalb des dreier-Rings liegen bei den dreifach-verbrückenden Oktaedern bei annähernd 90° (88.20(7)° und 89.54(7)°), bei dem zweifach verknüpfenden Oktaeder ist er mit 85.64(7)° deutlich kleiner. Der kleinste F–Ge–F-Winkel der gesamten Struktur befindet sich mit 80.32(7)° in der Oktaedereinheit des Ge(4), welcher die dreierRinge miteinander verbindet. Die Dimethylsulfoniumionen sind zwischen den anionischen Schichten eingelagert und über Wasserstoffbrückenbindungen zu den Fluoratomen des Anions koordiniert. 83 Ergebnisse und Diskussion Abbildung 3.54: Struktur von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF im Kristall (die Wasserstoffatome wurden der Übersicht halber weggelassen; thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome). Abbildung 3.55: Oben: Struktur der Kationen in [(Me2SH)3]3+[Ge4F19]3− ⋅ HF, unten: repetitive Einheit des Anions mit Winkeln [°] (blau) und Bindungslängen (schwarz) (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome). 84 Ergebnisse und Diskussion Tabelle 3.24: Ausgewählte Bindungslängen [pm] und -winkel 3+ 3− [(Me2SH)3] [Ge4F19] ⋅ HF und vom berechneten [Ge3F15]3− 3+ [(Me2SH)3] [Ge4F19] 3− [Ge3F15] ⋅ HF S(2)–C(4) S(2)–C(3) S(2)–H(2) S(3)–C(6) S(3)–C(5) S(3)–H(3) F(20)–H(20) C(1)–S(1)–C(2) C(1)–S(1)–H(1) C(2)–S(1)–H(1) C(4)–S(2)–C(3) C(4)–S(2)–H(2) C(3)–S(2)–H(2) C(6)–S(3)–C(5) C(6)–S(3)–H(3) C(5)–S(3)–H(3) [a] berechnet #2: –x+1/2,y+1/2,z. von 3− C3v, ber. exp. S(1)–C(1) S(1)–C(2) S(1)–H(1) [°] Ge(1)–F(15) 171.17(14) Ge(1)–F(14) 171.77(14) Ge(1)–F(12) 173.77(15) Ge(1)–F(13) 182.93(16) Ge(1)–F(11) 188.26(16) 177.7(3) 179.0(3) Ge(1)–F(5) 188.63(16) Ge(2)–F(9) 172.14(15) 110(3) Ge(2)–F(8) 172.29(15) Ge(2)–F(7) 172.86(17) 178.1(3) 178.6(3) Ge(2)–F(10) 174.97(16) Ge(2)–F(11) 192.01(16) 112(3) Ge(2)–F(6) 197.09(17) Ge(3)–F(2) 171.53(15) 68(5) Ge(3)–F(1) 172.24(15) Ge(3)–F(3) 174.18(17) 101.67(17) Ge(3)–F(4) 182.48(16) 100.6(14) Ge(3)–F(6) 183.58(16) 100.3(14) Ge(3)–F(5) 188.39(16) Ge(4)–F(16) 170.95(15) 102.82(17) Ge(4)–F(17) 173.10(14) 95.3(16) Ge(4)–F(18) 173.43(17) 97.4(16) Ge(4)–F(19) 174.09(17) Ge(4)–F(4) 196.35(16) 103.00(16) Ge(4)–F(13)#1 196.64(16) 99.3(15) F(15)–Ge(1)–F(14) 172.70(8) 98.2(14) F(12)–Ge(1)–F(13) 95.91(7) F(13)–Ge(1)–F(11) 86.51(7) F(12)–Ge(1)–F(5) 89.37(7) F(11)–Ge(1)–F(5) 88.20(7) F(9)–Ge(2)–F(8) 166.82(9) F(7)–Ge(2)–F(10) 97.79(8) F(7)–Ge(2)–F(11) 89.67(8) F(10)–Ge(2)–F(6) 86.91(7) F(11)–Ge(2)–F(6) 85.64(7) F(2)–Ge(3)–F(1) 172.70(8) F(3)–Ge(3)–F(4) 92.31(8) F(3)–Ge(3)–F(6) 91.64(8) F(4)–Ge(3)–F(5) 86.51(7) F(6)–Ge(3)–F(5) 89.54(7) F(16)–Ge(4)–F(17) 166.94(8) F(18)–Ge(4)–F(19) 99.31(9) F(19)–Ge(4)–F(4) 89.65(8) F(18)–Ge(4)–F(13)#1 90.62(8) F(4)–Ge(4)–F(13)#1 80.32(7) Ge(3)–F(4)–Ge(4) 154.31(9) Ge(3)–F(5)–Ge(1) 150.81(9) Ge(3)–F(6)–Ge(2) 151.96(9) Ge(1)–F(11)–Ge(2) 153.56(10) Ge(1)–F(13)–Ge(4)#2 147.76(9) mit PBE1PBE/6-311G(3df,3pd); Symmetrieoperationen: 177.1(3) 177.9(3) 124(3) 85 #1: [a] 176.0 176.0 178.5 176.0 176.0 178.5 178.5 193.7 193.9 176.0 176.0 178.5 193.7 193.9 173.3 95.2 89.7 89.7 85.4 173.3 95.2 89.7 89.7 84.4 173.3 95.2 89.7 89.7 84.5 154.5 154.5 154.6 –x+1/2,y–1/2,z; Ergebnisse und Diskussion Tabelle 3.25: Wasserstoffbrückenbindungen von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF (Bindungslängen [pm] und -winkel [°]) D–H···A d(D–H) d(H···A) S(1)–H(1)···F(10)#2 124(3) 212(3) S(2)–H(2)···F(10)#2 110(3) 241(3) S(2)–H(2)···F(18)#1 110(3) 253(3) S(3)–H(3)···F(19) 112(3) 227(3) F(20)–H(20)···F(3)#1 68(5) 196(5) F(20)–H(20)···F(7) 68(5) 239(5) Symmetrieoperationen: #1: –x+1/2,y+1/2,z; #2: –x+1/2,–y,z+1/2. Abbildung 3.56: d(D···A) <(DHA) 330.45(18) 335.32(18) 347.77(18) 327.34(18) 258.6(3) 283.4(3) 160(2) 143(2) 143(2) 148(2) 154(6) 125(6) Kristallstruktur von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF, links: entlang der bAchse, rechts: entlang der c-Achse (Wasserstoffatome wurden der Übersicht halber weggelassen; thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome). 3.3.5.3 Theoretische Berechnungen von [Ge3F15] 3− Die DFT-Berechnungen der Gasphasenstruktur von [Ge3F15]3− (C3v-Symmetrie) wurde mit der Methode PBE1PBE und dem 6-311G(3df,3pd)-Basissatz durchgeführt (Abbildung 3.57).[41-43] Anschließend wurden die Schwingungsfrequenzen der relaxierten Struktur sowie die IR- und Raman-Intensitäten berechnet. Die geometrischen Parameter sind in Tabelle 3.24 den experimentellen Daten gegenüber gestellt. Man erkennt, dass das berechnete Anion in akzeptabler Übereinstimmung mit der Struktur des Anions im Kristall ist. Unterschiede ergeben sich, wie schon in Abschnitt 3.3.4.3 erwähnt, weil nur eine isolierte Ringeinheit berechnet werden konnte, im Kristall diese Ringe aber über ein weiteres Oktaeder zu einer Kette verknüpft sind. Die unterschiedlichen Ge–F-Bindungslängen in Abhängigkeit vom Verknüpfungsgrad des 86 Ergebnisse und Diskussion zugehörigen Oktaeders, sowie die unterschiedlichen Ge–F-Abstände bei axialer oder äquatorialer Stellung der Fluoratome werden dennoch gut wiedergegeben. Die Fluor- und Germaniumatome, welche den Ring bilden liegen auch in der berechneten Struktur in einer Ebene. Abbildung 3.57: Quantenchemisch berechnete Struktur von [Ge3F15]3− mit Winkeln [°] (blau) und Bindungslängen [pm] (schwarz). 3.3.5.4 Schwingungsspektren Das erhaltene Raman-Spektrum ist mit den Ergebnissen der Röntgenstrukturanalyse im Einklang (Abbildung 3.58). Die Zuordnung der Schwingungsfrequenzen erfolgte durch Vergleich mit vorhandenen Literaturdaten und quantenchemischen Berechnungen der Schwingungsfrequenzen von [Ge3F15]3− und [Me2SH(HF)]+.[21,81,103] Die beobachteten Linien und Intensitäten stimmen gut mit den bereits für [Me2SH]+ veröffentlichen Daten überein.[103] Die vollständige Zuordnung ist in Tabelle 3.26 aufgelistet. Im Bereich zwischen 3100 und 830 cm–1 befinden sich auch in dieser Verbindung ausschließlich die Schwingungen des [Me2SH]+-Kations. Die breite ν(S–H), bei 2526 cm–1, ist ein eindeutiger Nachweis für die erfolgreiche Protonierung von Dimethylsulfid. Bei den Schwingungen für das Anion unterscheidet sich das Spektrum von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF in zwei Bereichen (siehe Kapitel 3.3.4.4). Zum einen ist eine neue Schwingungsbande bei 573 cm–1 zu beobachten. Nach Bartlett liegen Streckschwingungen einer cis-verknüpften Fluorogermanat-Kette im Bereich von 560–600 cm–1.[21] Eine solche Zuordnung der Bande bei 573 cm–1 würde mit 87 Ergebnisse und Diskussion den Ergebnissen der Einkristall-Röntgenstrukturanalyse im Einklang stehen, da sich in der gefundenen Struktur ein cis-verknüpfendes [GeF6]2−-Oktaeder befindet, welcher in der in Kapitel 3.3.4 diskutierten Struktur nicht vorkommt. Zum Anderen tritt eine Veränderung des Frequenzmusters im Bereich um 350 cm–1 auf. Im Schwingungsspektrum von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF (vierer-Ring) waren bei 363, 352 und 337 cm–1 drei relativ schwache Schwingungen zu beobachten. In diesem Spektrum ist nur noch eine breitere und intensivere Bande bei 337 cm–1 vorhanden. Die Schwingung bei 675 cm–1 ist eine Überlagerung der symmetrischen νs(Ge–F) des Anions und der δ(CSC) des Kations. Hierfür spricht auch die relativ große Aktivität der Linie. Beim Vergleich vom berechneten mit dem gemessenen Spektrum des [Ge3F15]3−-Anions müssen größere Abweichungen in Kauf genommen werden. Eine Berechnung der Polymerstruktur ist zu rechenintensiv, daher erfolgte nur die Berechnung des dreier-Rings. Die intensivsten Schwingungen des [Ge3F15]3−-Anions, die Streckschwingungen, stimmen trotzdem gut mit den experimentellen Werten überein. Abbildung 3.58: Raman-Spektrum von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF. 88 Ergebnisse und Diskussion Tabelle 3.26: Beobachtete Schwingungsfrequenzen [cm–1] von [Me3SH]3+[Ge4F19]3− ⋅ HF und berechnete Schwingungsfrequenzen von [Me2SH(HF)]+ und [Ge3F15]3− 3+ 3− [(Me2SH)3] [Ge4F19] ⋅ HF Raman [Me2SH(HF)] C1, ber. + Zuordnung 3+ 3− [(Me2SH)3] [Ge4F19] ⋅ HF [a] Raman 3− C3v, ber. Zuordnung [b] νs(Ge–Fe) νas(Ge–Fe) νs(Ge–Fe) 675* (75) 3050 (55) νas(Ge–Fe) ν(Ge–F) 641 (3) νas(Ge–Fe) 632 (628/0) 2956 (100) νas(Ge–Fe) 622 (0/0) 2526 (84) 605 (294/0.02) ν(Ge–Fe) 1427 (34) ν(Ge–Fe) 605 ν(Ge–Fe) 598 (0/5) ν(Ge–F) 573 (6) 497 (1) 488 (142/0.02) ν(Ge–Fμ) ν(Ge–Fμ) 445 (0/0) δ(Ring) 374 (0/0.08) 1093 (4) δ(Fμ–Ge–Fμ) 370 (73/1) 1075 (7) δ(Fe–Ge–Fμ) 363 (80/0) 1052 (3) δ(Fe–Ge–Fe) 830 (5) 346 (0/1) δ(Ring) 337 (4) 341 (0/1) 808 (2) δ(Fe–Ge–Fe) 338 (21/0) 774 (6) δ(Fe–Ge–Fe) 335 (0/0.1) 734 (18) 675* (75) 315 (652/0.2) δ(Fe–Ge–Fe) δ(Fe–Ge–Fe) 299 (0/0) 290 (14) δ(F–Ge–F) 267 (0/0.1) δ(F–Ge–F) 272 (4) 251 (0.3/0.2) δ(F–Ge–F) 211 (0/0) δ(F–Ge–F) 210 (5/0.3) δ(F–Ge–F) 210 (0/0.3) δ(F–Ge–F) 172 (5/0.3) δ(Gerüst) 164 (0/1) δ(Gerüst) 142 (0/0) δ(Gerüst) 122 (5/0.3) δ(Gerüst) 109 (0/0) δ(Gerüst) 109 (2/0.3) δ(Gerüst) 46 (0.1/0) δ(Gerüst) 35 (0/0) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 1.03 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %; * Linie wird dem Anion und dem Kation zugeordnet; Fe: endständiges Fluoratom; Fμ: verbrückendes Fluoratom. 3136 3134 3120 3120 3018 3016 2579 1432 1428 1417 1415 1348 1325 1097 1069 1038 916 812 768 732 661 258 198 174 (2/12) (9/60) (12/65) (5/31) (11/0.4) (1/284) (170/136) (33/1) (12/7) (0/8) (16/1) (2/0) (0/0) (4/2) (10/2) (8/1) (3/0) (1/2) (16/4) (0/5) (5/16) (57/2) (1/0.3) (0.2/0.01) νas(CH3) νas(CH3) νas(CH3) νas(CH3) νs(CH3) νs(CH3) ν(SH) δas(CH3) δas(CH3) δas(CH3) δas(CH3) δs(CH3) δs(CH3) δ(CSH) ρ(CH3) ρ(CH3) ρ(CH3) ρ(CH3) δ(CSH) νas(CS2) νs(CS2) δ(CSC) τ(CH3)i.p. τ(CH3)o.o.p. [Ge3F15] 737 719 671 665 . 89 (418/0) (0/0.02) (0/23) (2/0.02) Ergebnisse und Diskussion [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF 3.3.6 3.3.6.1 Bildung von [(Me2SH)2] 2+[GeF6] 2− ⋅ 2 HF [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF wurde quantitativ in einem Schritt nach Gleichung (3.18) synthetisiert. -50 °C, HF 2 Me2S + [(H2F)2]2+[GeF6]2- [(Me2SH)2]2+[GeF6]2- 2 HF (3.18) -60 °C, HF 4 HF + GeF4 Dimethylsulfid wurde in einem Reaktor vorgelegt und anschließend wurden im hohen Überschuss HF (150 : 1) und GeF4 (2.0 : 1) kondensiert. Als das Reaktionsgemisch langsam auf –60 °C aufgetaut wurde, bildete sich die Supersäure [(H2F)2]2+[GeF6]2−. Bei weiterem Erwärmen auf –50 °C löste sich das Dimethylsulfid in der supersauren Lösung und wurde protoniert. Nach langsamem Entfernen von HF und überschüssigem GeF4 für 30 h bei –78 °C und für 13 h bei –40 °C bildeten sich farblose Kristalle von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF, welche bis –40 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 3.3.6.2 Röntgenstrukturanalyse von [(Me2SH)2] 2+[GeF6]2− ⋅ 2 HF [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF kristallisiert in der orthorhombischen Raumgruppe Fdd2 (Nr. 43) mit acht Formeleinheiten in der Elementarzelle. Ausgewählte Bindungslängen und -winkel von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF sind in Tabelle 3.27 aufgelistet, zusammen mit den berechneten Strukturdaten des [GeF6]2−-Oktaeders. Alle Atome, einschließlich der Wasserstoffatome am Sauerstoff, wurden mittels Differenz-Fourier-Synthese gefunden und frei verfeinert. Die Wasserstoffatome der Methylgruppen wurden berechnet. Alle NichtWasserstoffatome wurden anisotrop verfeinert. Die asymmetrische Einheit von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF besteht aus einem Dimethylsulfoniumkation, einem Lösungsmittel HF und einem halben [GeF6]2−-Anion (Abbildung 3.59). Das [GeF6]2−-Anion hat eines eine zweizählige Drehachse entlang F(4)–Ge(1)–F(1). 90 Ergebnisse und Diskussion Das Anion ist ein leicht verzerrtes [GeF6]2−-Oktaeder, welcher zwei Wasserstoffbrückenbindungen ausbildet. Die Ge–F-Bindungslängen der Fluoratome, welche die Wasserstoffbrücken ausbilden, sind um 3 − 4 pm gegenüber den anderen Ge–FAbständen verlängert. Die Winkel des Oktaeders liegen alle bei annähernd 90°. Abbildung 3.59: Struktur von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF im Kristall (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für NichtWasserstoffatome, Symmetrieoperationen: #1: –x,1–y,z). Tabelle 3.27: Ausgewählte Bindungslängen [pm] und -winkel [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF und vom berechneten [GeF6]2− 2+ [(Me2SH)2] [GeF6] 2− [GeF6] ⋅ 2 HF 179.35(13) 179.60(14) 119.2(15) F(5)–H(5) 59(3) C(2)–S(1)–C(1) C(2)–S(1)–H(1) C(1)–S(1)–H(1) 102.70(6) 97.5(7) 100.1(7) S(1)···F(1) H(1)···F(1) S(1)–H(1)···F(1) 324.01(7) 205.0(16) 175.9(12) Ge(1)–F(3) Ge(1)–F(4) Ge(1)–F(1) Ge(1)–F(2) 176.92(6) 177.52(14) 179.55(14) 182.49(6) 181.9 181.9 181.9 181.9 F(3)–Ge(1)–F(3)#1 F(3)–Ge(1)–F(4) F(3)–Ge(1)–F(1) F(3)–Ge(1)–F(2) F(4)–Ge(1)–F(2) F(1)–Ge(1)–F(2) F(3)–Ge(1)–F(2)#1 F(2)–Ge(1)–F(2)#1 178.19(6) 90.90(3) 89.10(3) 89.86(3) 89.93(3) 90.07(3) 90.15(3) 179.86(6) 180 90 90 90 90 90 90 180 F(5)···F(2)#2 246.18(12) H(5)···F(2)#2 190(3) F(5)–H(5)···F(2)#2 163(4) [a] berechnet mit PBE1PBE/6-311G(3df,3pd); Symmetrieoperationen: #1: –x,–y+1,z; #2: x,y,z+1. 91 von 2− Oh, ber. exp. S(1)–C(2) S(1)–C(1) S(1)–H(1) [°] [a] Ergebnisse und Diskussion 3.3.6.3 Schwingungsspektren Das erhaltene Raman-Spektrum steht mit den Ergebnissen der Röntgenstrukturanalyse im Einklang (Abbildung 3.58). Die Zuordnung des Spektrums ist in Tabelle 3.28 aufgelistet. Im Bereich zwischen 3100 und 830 cm–1 befinden sich auch hier ausschließlich die Schwingungslinien des [Me2SH]+-Kations. Die Abweichung des Anions von der idealen Oktaedersymmetrie spiegelt sich auch im Schwingungsspektrum wieder. Durch die erniedrigte Geometrie, sind für das Anion vier Linien zu beobachten. Die Streckschwingungen des Anions sind bei 629 und 495 cm–1 zu beobachten und eine Deformationsschwingung bei 333 cm–1. Tabelle 3.28: Beobachtete Schwingungsfrequenzen [cm–1] von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF und berechnete Schwingungsfrequenzen von [Me2SH(HF)]+ und [GeF6]2− 2+ [(Me2SH)2] [GeF6] ⋅ 2 HF Raman 3061 (11) 3050 (25) 3041 (14) 2965 2957 2538 2462 2412 1449 1438 1425 (43) (85) (48) (77) (92) (29) (14) (24) 2− [Me2SH(HF)] + Zuordnung [a] C1, ber. 3136 (2/12) 3134 (9/60) 3120 (12/65) 3120 (5/31) 3018 (11/0.4) 3016 (1/284) 2579 (170/136) 2+ [(Me2SH)2] [GeF6] ⋅ 2 HF Raman νas(CH3) νas(CH3) νas(CH3) νas(CH3) νs(CH3) νs(CH3) 629 (20) 495 (3) 333 (7) 2− [GeF6] 2− Zuordnung [b] Oh, ber. 621 (263/0) 616 (0/10) 483 (0/1) 370 (150/0) 326 (0/2) 207 (0/0) νas(Ge–F) νs(Ge–F) νas(Ge–F) δ(F–Ge–F) δ(F–Ge–F) δ(F–Ge–F) ν(SH) δas(CH3) 1432 (33/1) δas(CH3) 1428 (12/7) δas(CH3) 1417 (0.03/8) δas(CH3) 1415 (16/1) δs(CH3) 1348 (2/0.1) δs(CH3) 1325 (0.01/0.07) δ(CSH) 1097 (11) 1097 (4/2) ρ(CH3) 1086 (13) 1069 (10/2) ρ(CH3) 1038 (8/1) ρ(CH3) 843 (8) 916 (3/0.1) ρ(CH3) 805 (12) 812 (1/2) δ(CSH) 731 (35) 768 (16/4) νas(CS2) 732 (0.2/5) νs(CS2) 666 (100) 661 (5/16) δ(CSC) 289 (31) 258 (57/2) τ(CH3)i.p. 198 (0.8/0.3) τ(CH3)o.o.p. 174 (0.2/0.01) [a] berechnet mit PBE1PBE/6-311G++(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor von 0.98 skaliert; [b] berechnet mit PBE1PBE/6-311G(3df,3pd), die Frequenzen wurden mit einem empirischen Faktor 4 –1 von 1.05 skaliert; IR-Intensität in km/mol, Raman-Aktivität in Å µ bzw. in %. 92 Ergebnisse und Diskussion Abbildung 3.60: Raman-Spektrum von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF. 93 Ergebnisse und Diskussion 3.3.7 [(H3O)2]2+[GeF6]2− 3.3.7.1 Bildung von [(H3O)2] 2+[GeF6] 2− Bei der Umsetzung von Nitrobenzol mit HF/GeF4 wurde bei einer Synthese ein Gemisch aus gelben und farblosen Kristallen erhalten. Durch eine Einkristall-Röntgenstrukturanalyse konnten die farblosen Kristalle als [(H3O)2]2+[GeF6]2− identifiziert werden. Die Kristalle sind hydrolyseempfindlich und bis ca. –5 °C beständig. Wahrscheinlich wurde bei einem Arbeitsschritt Wasser in den Reaktor eingeschleppt. 3.3.7.2 Röntgenstrukturanalyse von [(H3O)2] 2+[GeF6] 2− [(H3O)2]2+[GeF6]2− kristallisiert in der monoklinen Raumgruppe P21/c (Nr. 14) mit zwei Formeleinheiten in der Elementarzelle. Ausgewählte Bindungslängen und -winkel von [(H3O)2]2+[GeF6]2− sind in Tabelle 3.29 aufgelistet, zusammen mit den berechneten Strukturdaten des [GeF6]2−-Oktaeders. Alle Atome, einschließlich der Wasserstoffatome, wurden mittels Differenz-Fourier-Synthese gefunden und frei verfeinert. Alle NichtWasserstoffatome wurden anisotrop verfeinert. Die asymmetrische Einheit von [(H3O)2]2+[GeF6]2− besteht aus einem Oxoniumkation und einem halben [GeF6]2−-Anion (Abbildung 3.61). Ein Ausschnitt aus der Kristallpackung ist in Abbildung 3.62 dargestellt. Das Anion ist ein [GeF6]2−-Oktaeder mit annähernder Oh-Symmetrie, welches ein Inversionszentrum auf Ge(1) besitzt. Diese hohe Symmetrie wird erreicht, weil jedes Fluoratom eine Abbildung 3.61: ähnliche Umgebung hat und eine Wasserstoffbrückenbindung Struktur von [(H3O)2]2+[GeF6]2− im Kristall, Beschriftung der asymmetrischen Einheit (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome). 94 Ergebnisse und Diskussion Tabelle 3.29: Ausgewählte Bindungslängen [pm] und -winkel [°] von [(H3O)2]2+[GeF6]2− und des berechneten [GeF6]2− 2+ O(1)–H(2) O(1)–H(1) O(1)–H(3) 101(4) 87(3) 77(4) H(2)–O(1)–H(1) H(2)–O(1)–H(3) H(1)–O(1)–H(3) 114(3) 109(3) 110(3) 2− 2− [(H3O)2] [GeF6] exp. Ge(1)–F(1) Ge(1)–F(2) Ge(1)–F(3) 179.65(13) 179.02(14) 178.42(14) F(2)–Ge(1)–F(1) F(3)–Ge(1)–F(1) F(3)–Ge(1)–F(2) F(2)#1–Ge(1)–F(1) F(3)–Ge(1)–F(2)#1 F(3)#1–Ge(1)–F(1) [a] berechnet mit PBE1PBE/6-311G(3df,3pd). Abbildung 3.62: 89.53(6) 89.78(7) 90.73(6) 90.47(6) 89.27(6) 90.22(7) [GeF6] [a] Oh, ber. 181.9 181.9 181.9 90 90 90 90 90 90 Kristallstruktur von [(H3O)2]2+[GeF6]2− mit interionischen Kontakten (thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome). zu einem Kation ausbildet. Die Ge–F-Bindungslängen sind gegenüber den Literaturbekannten Daten für [GeF6]2−-Oktaeder um ca. 10 pm verlängert, was durch Ausbildung von starken Wasserstoffbrückenbindungen hervorgerufen wird.[78] Die Winkel des Oktaeders liegen alle bei annähernd 90°. Die Wasserstoffbrückenbindungen sind in Abbildung 3.63 dargestellt und in Tabelle 3.30 aufgelistet. Durch diese Bindungen entsteht ein dreidimensionales Netzwerk. Das Kation ist gewinkelt und weist eine Winkelsumme der H–O–H-Winkel von 333° auf. 95 Ergebnisse und Diskussion Tabelle 3.30: Wasserstoffbrückenbindungen von [(H3O)2]2+[GeF6]2− (Bindungslängen [pm] und -winkel [°]) D–H···A d(D–H) d(H···A) O(1)–H(1)···F(1) 87(3) 165(3) O(1)–H(2)···F(2)#1 101(4) 149(4) O(1)–H(3)···F(3)#2 077(4) 179(4) Symmetrieoperationen: #1: x,–y+1/2,z–1/2; #2 x+1,y,z. Abbildung 3.63: d(D···A) 252.2(3) 250.3(3) 255.4(3) <(DHA) 173(3) 174(3) 169(4) [H3O]+-Kation mit Wasserstoffbrückenbindungen (schwarz: Bindungslängen [pm], blau: Bindungswinkel, thermische Auslenkungsellipsoide mit 50 % Aufenthaltswahrscheinlichkeit für Nicht-Wasserstoffatome, Symmetrieoperationen: #1: x,1/2–y,-1/2+z, #2: 1+x,y,z). 96 Ergebnisse und Diskussion 3.3.8 Vergleich der Fluorogermanatanionen Die bisher bekannten Strukturen der binären Fluorogermanten waren der [GeF6]2−-Oktaeder und die cis- und trans-eckenverknüpften polymeren ([GeF5]−)n-Ketten.[21,104-105] Die Strukturen des [Ge2F10]2−-Dimers, des [Ge3F16]4−-Trimers sowie die zwei polymeren Ringstrukturen von [Ge4F19]3− waren bisher nicht bekannt (Abbildung 3.64 und Abbildung 3.65). Abbildung 3.64: Isolierte Fluorogermanate: links: [GeF6]2−-Oktaeder in [(H3O)2]2+[GeF6]2−; mittig: [Ge2F10]2−-Dimer in [(Me2OH)2]2+[Ge2F10]2−; rechts: [Ge3F16]4−Trimer in [(Me2OH)4]4+[Ge3F16]4−. Durch die starke Verzerrung der Oktaeder in den Strukturen kann man sowohl extrem kurze als auch sehr lange Ge–F-Bindungen beobachten. Der kürzeste Ge–F-Abstand der bisher beschriebenen Anionen ist mit 170.95(15) pm im dreier-Ring zu finden. In der Struktur des [Ge2F10]2−-Anions ist sowohl der kleinste bisher gefundene F–Ge–F-Winkel für oktaedrisch koordiniertes Ge(IV) mit nur 74.27(8)° zu beobachten, sowie der größte Winkel mit 105.73(8)°. Betrachtet man die Ge-F-Stöchiometrien der verschiedenen Anionen, so stellt man fest, dass alle ein ähnliches oder sogar identisches Verhältnis besitzen. Eine Übersicht liefert hierfür Tabelle 3.31. Drei der verschiedenen Anionen haben exakt die gleiche Ge-FStöchiometrie von 1 : 5 wie das [GeF5]−-Monomer, und drei weitere sind im Verhältnis doch sehr ähnlich. Tabelle 3.31: Vergleich der Ge:F-Stöchiometrien der Fluorogermanatanionen Anion Verhältnis Ge : F 2− Anion Verhältnis Ge : F − [GeF6] -Oktaeder 1:6 cis-μ-([GeF5] )n 1:5 [Ge2F10] -Dimer 1:5 trans-μ-([GeF5] )n 1:5 [Ge3F16] -Trimer 1:5.33 2− 4− − 3− ([Ge4F19] )n (vierer-Ringe) 1:4.75 3− ([Ge4F19] )n (dreier-Ringe) 1:4.75 97 Ergebnisse und Diskussion Abbildung 3.65: Polymere Fluorogermanate: oben/links: cis-μ-([GeF5]−)n in + −[21] − [XeF5] [GeF5] ; oben/rechts: trans-μ-([GeF5] )n in [XeF5]+[GeF5]−[21]; mittig: ([Ge4F19]3−)n in [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF; unten: ([Ge4F19]3−)n in [(Me2SH)3]3+[Ge4F19]3− ⋅ HF. Bisher war eine Vorhersage der Struktur unbekannter Fluorogermanatanionen nur anhand von schwingungsspektroskopischen Experimenten so gut wie unmöglich. Mit den in dieser Arbeit gewonnenen Daten sind nun grundlegende Strukturmerkmale der Anionen zumindest mit hoher Wahrscheinlichkeit zu identifizieren (Abbildung 3.66). Eine Auflistung der Raman-Schwingungen der bisher gefundenen Fluorogermanatanionen ist in Tabelle 3.32 wiedergegeben. Tabelle 3.32: Vergleich der Ramanfrequenzen der Fluorogermanatanionen trans-μcis-μ− [21] − [21] ([GeF5] )n ([GeF5] )n 713 700 657 654 603 526 590 518 559 463 399 337 303 381 339 331 2− [GeF6] 2− [Ge2F10] 629 495 455 684 621 554 500 333 393 355 339 98 3− 3− ([Ge4F19] )n ([Ge4F19] )n (dreier-Ringe) (vierer-Ringe) 675 689 641 677 573 621 497 600 337 272 363 351 337 307 Zuordnung ν(Ge–F) δ(F–Ge–F) Ergebnisse und Diskussion Trotzdem ist zu bedenken, dass eine Unterschiedliche kristallographische Umgebung des Anions durchaus zu Abweichungen führen kann, wie im Fall des [Ge2F10]2−-Ions gezeigt werden konnte. Die zwei Raman-Spektren des [Ge2F10]2−-Dimers sind in Abbildung 3.67 vergrößert dargestellt. Das Muster der Linien bleibt zwar erhalten, aber man erkennt eine systematische Abweichung, gerade im Bereich der Streckschwingungen. Durch die unterschiedlich ausgebildeten Wasserstoffbrückenbindungen tritt bei den asymmetrischen und symmetrischen Streckschwingungen der Verbindungen eine Umkehr der Intensitätsverhältnisse auf. Abbildung 3.66: Raman-Spektren von: a) [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF; 3+ 3− b) [(Me2SH)3] [Ge4F19] ⋅ HF; c) [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF; d) [(Me2OH)2]2+[Ge2F10]2−; (* Schwingung des Kations). 99 Ergebnisse und Diskussion Abbildung 3.67: Raman-Spektren der Anionen von [(Me2OH)2]2+[Ge2F10]2− (schwarz) und [(C6H5NO2H)2]2+[Ge2F10]2− (blau) (* Schwingungen des Kations). Die schwingungsspektroskopische Zuordnung für das [GeF5]− in [Bu4N]+[GeF5]− erfolgte damals auf Grundlage der D3h-Symmetrie, da das Schwingungsmuster, den bis dato gefunden Anionen nicht glich.[21,82] Stellt man aber die Anionenschwingungen von [Bu4N]+[GeF5]− denen des Dimers in [(Me2OH)2]2+[Ge2F10]2− gegenüber, so kann es durchaus sein, dass auch in [Bu4N]+[GeF5]− zwei kantenverknüpfte Oktaeder vorliegen (Tabelle 3.33). Tabelle 3.33: Gegenüberstellung der Schwingungen von [GeF5]− in [Bu4N]+[GeF5]− und [(Me2OH)2]2+[Ge2F10]2− − − [GeF5] in + [Bu4N] [GeF5] [GeF5] in −[21] 2+ [(Me2OH)2] [Ge2F10] 690 684 665 654 621 520 554 500 345 355 337 339 317 ~117 100 2− Ausblick und Zusammenfassung 4 Ausblick und Zusammenfassung 4.1 Protonierung starker organischer Säuren Das [CF3SO3H2]+-Kation konnte zum ersten Mal synthetisiert und charakterisiert werden. Die Darstellung erfolgte durch die Reaktion von (CF3SO2)2O mit einer supersauren Lösung von HF/SbF5, bei welcher sich in situ CF3SO3H bildet, das anschließend protoniert wird. Das entstandene [CF3SO3H2]+[SbF6]−-Salz wurde mittels Schwingungsspektroskopie und Einkristall-Röntgenstrukturanalyse charakterisiert. Die Bildung von [CF3SO3H2]+[SbF6]– kann als das Ergebnis einer Konkurrenzreaktion von CF3SO3H/SbF5 und HF/SbF5 gesehen werden und zeigt somit, dass CF3SO3H/SbF5 eine schwächere konjugierte Brønsted-LewisSupersäure ist, als das HF/SbF5-System. Ebenfalls gelang die Darstellung der Reihe der Fluormethyldihydroxycarbeniumionen und des Trichlordihydroxycarbeniumions als Hexafluoroarsenat-Salze. Alle Salze konnten isoliert und schwingungsspektroskopisch, sowie mittels Einkristall-Röntgenstrukturanalyse charakterisiert werden. Im Festkörper dieser Verbindungen werden starke O–H···F-Wasserstoffbrückenbindungen zwischen den Kationen und den Fluoratomen der benachbarten Anionen beobachtet. Daher beschreiben Berechnungen der freien [CF3SO3A2]+- und [RC(OA)2]+-Kationen (A = H, D; R = CH2F, CHF2, CF3, CCl3) die experimentellen Schwingungsfrequenzen nur ungenügend. Um den Einfluss [CF3SO3H2(HF)2]+-, der Wasserstoffbrückenbindungen bzw. [RC(OA)2(HF)2]+-Einheiten Wasserstoffbrückenbindungen (O–H···F) zwischen den zu berücksichtigen, berechnet, wurden welche Hydroxygruppen und zwei den Fluoratomen der HF-Moleküle besitzen. Dieses einfache Modell führte zu guten Übereinstimmungen zwischen den berechneten und Schwingungsfrequenzen, sowie der Geometrie der Kationen. 101 experimentell gefundenen Ausblick und Zusammenfassung 4.2 Tricyanomethan (Cyanoform) Tricyanomethan konnte erstmalig als Feststoff untersucht werden. Die Synthese gelang durch die Umsetzung von Calciumtricyanomethanid mit wasserfreier HF (Gleichung (4.1)). Ca(C(CN)3)2 + 4 HF -50 °C, HF 2 HC(CN)3 + Ca(HF2)2 (4.1) Durch NMR- und Schwingungsspektroskopie, unterstützt durch quantenchemische Berechnungen konnte belegt werden, dass Tricyanomethan im Feststoff als Cyanoform vorliegt (Abbildung 4.2). Bereits bei –40 °C setzt eine Polymerisierung des Produkts ein, wobei diese durch eine mit steigender Temperatur immer intensiver werdende Rotfärbung einhergeht. Abbildung 4.1: Berechnete Struktur von Cyanoform. Calciumtricyanomethanid ist nicht das einzige untersuchte Edukt für die Darstellung von Tricyanomethan. Es konnten auch das Natrium-, Magnesium- und Bleitricyanomethanid erfolgreich umgesetzt werden. Die entstehenden Metallhydrogenfluoride überlagerten jedoch im IR-Spektrum stark die Banden des Tricyanomethans. Wenn es künftig gelingen sollte Cyanoform in reiner Form zu isolieren, sollte es möglich sein, dieses auch strukturell untersuchen zu können. Interessant wäre dann auch, ob sich Cyanoform mit den bekannten supersauren Systemen HF/MF5 (M = As, Sb) protonieren ließe. 102 Ausblick und Zusammenfassung 4.3 Die strukturelle Vielfalt von Fluorogermanaten Bei Untersuchungen der Säurestärke des Systems HF/GeF4 stellte sich heraus, dass das [GeF6]2−-Anion stark zur Polymerisation neigt. Erstmalig konnten Ringstrukturen binärer Germaniumfluoride synthetisiert werden. Die gleichzeitige spektroskopische Untersuchung dieser Anionen ermöglicht einen Ansatz für eine spektroskopische Bibliothek für Fluorogermanatanionen. Es wurden vier neue strukturell unterschiedliche Anionen gefunden. Dabei wurden erstmals Ringstrukturen von Fluorogermanaten beschrieben. Die Strukturen des [Ge2F10]2−-Dimers, des [Ge3F16]4−-Trimer sowie die zwei polymeren Ringstrukturen von [Ge4F19]3− waren bisher nicht bekannt (Abbildung 4.2). Abbildung 4.2: Oben: Isolierte Fluorogermanate: links: [Ge2F10]2−-Dimer in 2+ 2− [(Me2OH)2] [Ge2F10] ; rechts: [Ge3F16]4−-Trimer in 4+ 4− [(Me2OH)4] [Ge3F16] ; unten: polymere Fluorogermanate: links: ([Ge4F19]3−)n in [(Me2SH)3]3+[Ge4F19]3− ⋅ HF, rechts: ([Ge4F19]3−)n in [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF. Ein Zusammenhang zwischen der Stöchiometrie der eingesetzten Edukte und der resultierenden Struktur des Anions konnte nicht festgestellt werden. Die Ausbildung der verschiedenen Strukturen wird wahrscheinlich über die Kristallisationstemperatur und die Größe der Kationen bestimmt. Weitere Untersuchungen in dieser Richtung lassen auf neue Strukturen hoffen. 103 Experimenteller Teil 5 Experimenteller Teil 5.1 Apparaturen und Chemikalien 5.1.1 Allgemeine Arbeitstechnik Reaktionen mit hydrolyse- oder oxidationsempfindlichen Reagenzien bzw. Produkten wurden soweit nicht anders angegeben mittels Standard-Schlenck-Technik durchgeführt. Als Schutzgas diente Stickstoff (5.0, Fa. Messner Griesheim), welcher zusätzlich über Orangegel und Sicapent® geleitet wurde, um Restspuren von Wasser zu entfernen. Kondensierbare, Glas nicht angreifende Verbindungen, wurden in Hochvakuumapparaturen und Gefäßen aus Duranglas mit fettfreien Hähnen (Fa. Young) gehandhabt. Reaktionen mit Fluor oder Fluorwasserstoff wurden an einer Vakuum-Linie aus elektropoliertem Edelstahl durchgeführt. Als Reaktoren dienten Röhrchen aus FEP (Perfluorethylenpropylen-Copolymer, øaußen = 3/8‘‘; øinnen = ca. 6 mm), welche auf einer Seite verschlossen sind und auf der anderen Seite ein Nadelventil aus Edelstahl (Swagelok®) besitzen. Vor jeder Reaktion wurden die Edelstahllinie und die Reaktoren mit Fluor chemisch getrocknet. Das verwendete Fluor wurde anschließend über eine Säule mit Sodalime® geleitet und auf diese Weise vernichtet. 5.1.2 Chemikalien Die verwendeten Chemikalien wurden in den üblichen Qualitäten puriss., p.a. und purum bezogen. Herkunft und ggf. Aufreinigung der verwendeten Chemikalien sind in Tabelle 5.1 aufgelistet. Die Reinigung und Trocknung organischer Lösungsmittel erfolgte nach Standardmethoden. DF DF wurde aus der Umsetzung von trockenem CaF2 und D2SO4 gewonnen, unter Vakuum destilliert und anschließend mit F2 für zwei Wochen in einem Edelstahlzylinder getrocknet. D2SO4 wurde durch Umsetzung von D2O mit SO3 erhalten (SO3 aus H2SO4, 65%, Merck). 104 Experimenteller Teil Tabelle 5.1: Verwendete Chemikalien Bezeichnung Antimon(V)-fluorid Arsen(V)-fluorid Brom Calciumchlorid Chlortrimethylsilan Deuteriumfluorid Difluoressigsäure 1,2-Dimethoxyethan Dimethylether Dimethylmercaptan Fluorwasserstoff Germanium(IV)-fluorid Kaliumbromid Kaliumcyanid Malonsäuredinitril Monofluoressigsäure Nitrobenzol Silbernitrat Trichloressigsäure Trifluoressigsäureanhydrid Trifluorsulfonsäureanhydrid chem. Formel SbF5 AsF5 Br2 CaCl2 SiCl(CH3)3 DF CHF2COOH C4H10O2 Me2O Me2S HF GeF4 KBr KCN CH2(CN)2 CH2FCOOH C6H5NO2 AgNO3 CCl3COOH (CF3CO)2O (CF3SO2)2O Reinheit Herkunft 98% ABCR Solvay Acros Fluka >99% Merck Arbeitskreis 98% ABCR Fluka Linde Alfa Aesar Linde Arbeitskreis Fluka Fluka VWR 98% ABCR VWR VWR >99% Sigma-Aldrich 99.5% ABCR 99% ABCR 105 Reinigung dreifach destilliert Trocknung mit F2 Umkondensation Trocknung mit F2 Experimenteller Teil 5.2 Analytik und quantenchemische Rechnungen 5.2.1 Raman-Spektroskopie Die Raman-Messungen wurden in einer Tieftemperatur-Schutzgasküvette (Abbildung 5.1) unter Schutzgas (Stickstoff) und unter Kühlung mit flüssigem Stickstoff (–196 °C) durchgeführt. Für die Messung wurde die Küvette evakuiert. Die Küvette war mit einem Young®-Hahn verschlossen. Nach der Messung wurden die Proben mit Wasser hydrolysiert. Die Messungen wurden an dem MultiRAM FTRamanspektrometer der Fa. Bruker mit eine Nd:YAG Laser bei einer Wellenlänge von λ = 1064 nm durchgeführt. Die Spektren wurden mit der OPUS-6.5-Software ausgewertet.[106] Falls nötig wurde eine Grundlinienkorrektur angewendet. Die Spektren werden ab 250 cm–1 dargestellt. Abbildung 5.1: 5.2.2 Tieftemperatur-Schutzgasküvette. IR-Spektroskopie Die IR-Spektren wurden in einer speziell angefertigten Tieftemperatur-IR-Zelle aus Aluminium mit kühlbarem CsBr-Einkristallfenster aufgenommen (Abbildung 5.2).[107] Das Einkristallfenster wurde durch eine Kupferwendel mit flüssigem Stickstoff gekühlt. Die Substanzen wurden unter Schutzgas auf das gekühlte Fenster aufgebracht und verrieben. Die 106 Experimenteller Teil Messungen wurden an einem Vertex-80V-FT-IR-Spektrometer der Fa. Bruker mit evakuierbarer Probenkammer durchgeführt. Die Spektren wurden mit der OPUS-6.5Software ausgewertet.[106] Falls nötig wurde eine Grundlinienkorrektur angewendet. Die Spektren werden ab 450 cm–1 dargestellt. Abbildung 5.2: 5.2.3 Tieftemperatur-IR-Zelle. Kernresonanzspektroskopie Die NMR-Spektren wurden an einem Eclipse 400+ der Firma JEOL aufgenommen. Die Messfrequenzen des NMR-Spektrometer sind bei 1 H-NMR 399.782 MHz, 13 C-NMR 100.535 MHz, 19F-NMR 376.171 MHz, und 14N-NMR 28.918 MHz. Für die NMR-Experimente wurden die Synthesen der zu messenden Substanzen in FEPReaktoren (øaußen = 4 mm) durchgeführt und die Menge auf 0.25 mmol reduziert. In die Reaktoren wurde nach der Synthese SO2 kondensiert, welches als Lösungsmittel diente. Anschließend wurden die Reaktoren abgeschmolzen und in ein Standard-NMR-Rohr aus Glas (øinnen = 4 mm) eingesetzt. Als externer Standard konnte zwischen Glaswandung und FEP-Reaktor noch ein Gemisch aus einem deuteriertem Lösungsmittel und einer Referenzsubstanz gegeben werden. Die Angabe der chemischen Verschiebung δ erfolgt in ppm bezogen auf die eingesetzten Standardverbindungen. Die eingesetzten Standards Referenzverschiebungen sind in Tabelle 5.2 angegeben. 107 mit den entsprechenden Experimenteller Teil Tabelle 5.2: Deuterierte Lösungsmittel und Standards für die NMR-Spektroskopie Isotop 1 H 13 C 19 F 14 N Standard 6 d -Aceton 6 d -Aceton CFCl3 CH3NO2 Verschiebung 2.05 28.84 / 206.26 0.00 0.00 Die Messtemperatur ist explizit zusammen mit den jeweiligen Daten angegeben. Kopplungskonstanten nJ über n Bindungen werden in Hertz angegeben. Zur Beschreibung der Spektren und der darin auftretenden Signalmultiplizität werden die üblichen Abkürzungen verwendet: s (Singulett), d (Duplett), t (Triplett), q (Quartett). Verbreiterte Signale sind mit br (broad bzw. breit) gekennzeichnet. Die Auswertung und Darstellung der NMR-Spektren erfolgte mit dem Programm DELTA 4.3.6 der Firma Jeol.[108] 5.2.4 Einkristall-Röntgenstrukturanalyse Einkristall-Röntgenstrukturanalysen wurden an einem Oxford Diffraction XCalibur3 Diffraktometer mit einem Sapphire CCD-Detektor durchgeführt. Die Messungen erfolgten mit Mo-Kα Strahlung der Wellenlänge λ = 0.71073 Å. Die Einkristalle wurden im gekühlten Schutzgasstrom (N2) an einem Polarisations-Stereomikroskop überprüft und mit Hilfe eines Tieftemperatur-PFPE-Öls auf einem Polyimid-Loop (Hampton Research) fixiert, welcher auf biszu –60 °C gekühlt werden konnte. Die Datensätze wurden mit den Programmpaketen des Diffraktometers reduziert. Die Strukturen wurden mittels direkter Methoden gelöst (SHELXS-97) und mit Diffrenz-Fourier Rechnung mit vollständiger Matrix nach der Methode der kleinsten Fehlerquadrate gegen Fo2Fc2 verfeinert (SHELXL-97).[109-110] Die Überprüfung der Raumgruppe wurde mit dem Programm PLATON durchgeführt.[111] Die Abbildung der Molekülstrukturen erfolgte mit dem Programm MERCURY 2.3 und die der Polyeder erfolgte mit DIAMOND 3.2f, aus den entsprechenden cif-Dateien.[112-113] Die ergänzenden kristallographischen Daten einiger Verbindungen (Röntgenstrukturanalysen −> CCDC-Nummer) sind in der Datenbank des Camebridge Crystallographic Data Centre hinterlegt und können dort kostenfrei unter http://www.ccdc.cam.ac.uk/data_request/cif abgerufen werden. 108 Experimenteller Teil 5.2.5 Quantenchemische Rechnungen Theoretische Rechnungen wurden mit dem Programmpaket Gaussian03 durchgeführt.[114] Die jeweils angewendeten Methoden und Basissätze sind in den jeweiligen Kapiteln angegeben. Soweit nicht anders erwähnt wurden die Strukturoptimierungen mit dem GDIISAlgorithmus und die Kationen zusätzlich mit dem Konvergenzkriterium tight berechnet.[115] 109 Experimenteller Teil 5.3 Synthesen 5.3.1 Darstellung von [CF3SO3H2]+[SbF6]− F O F S F OH F F OH F Sb F F F SbF5 (1.00 mmol, 0.22 g) wurde in einen gekühlten FEP-Reaktor (–196 °C) kondensiert. Anschließend wurde HF (ca. 150 : 1) im Überschuss kondensiert und das Gemisch auf –40 °C erwärmt, um die Supersäure zu bilden. Der Reaktor wurde wieder auf –196 °C gekühlt und (CF3SO2)2O (0.28 g, 1.00 mmol) wurde unter Stickstoff zur gefrorenen Lösung gegeben. Das Reaktionsgemisch wurde für 10 min auf –40 °C erwärmt, bis eine homogene, farblose und klare Lösung vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF und CF3SO2F wurden im dynamischen Vakuum bei –78 °C über Nacht entfernt. Man erhielt in quantitativer Ausbeute farblose, hydrolyseempfindliche Kristalle, welche bis –50 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 5.3.2 Darstellung von [CH2FC(OH)2]+[AsF6]− H F OH H F OH F F As F F F CH2FCO2H (0.08 g, 1.00 mmol) wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und im Überschuss HF (ca. 150 : 1) und anschließend AsF5 (0.25 g, 1.50 mmol) kondensiert. Das Gemisch wurde für 10 min auf –20 °C erwärmt, bis eine homogene, farblose Phase vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF und AsF5 wurden im dynamischen Vakuum für 15 h bei –78 °C und für 7h bei –25 °C entfernt. Man erhielt in quantitativer Ausbeute farblose, hydrolyseempfindliche Kristalle, welche bis –20 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 110 Experimenteller Teil 5.3.3 Darstellung von [CHF2C(OH)2]+[AsF6]− H F OH F F OH F F As F F F CHF2CO2H (0.08 g, 1.00 mmol) wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und im Überschuss HF (ca. 150 : 1) und anschließend AsF5 (0.25 g, 1.50 mmol) kondensiert. Das Gemisch wurde für 10 min auf –30 °C erwärmt, bis eine homogene, farblose Phase vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF und AsF5 wurden im dynamischen Vakuum für 16 h bei –78 °C entfernt. Man erhielt in quantitativer Ausbeute farblose, hydrolyseempfindliche Kristalle, welche bis –30 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 5.3.4 Darstellung von [CF3C(OH)2]+[AsF6]− F F OH F F OH F F As F F F (CF3CO)2O (0.21 g, 1.00 mmol) wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und HF (ca. 150 : 1) im Überschuss und anschließend AsF5 (0.25 g, 1.50 mmol) kondensiert. Das Gemisch wurde für 15 min auf –40 °C erwärmt, bis eine homogene farblose Phase vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF, CF3COF und AsF5 wurden im dynamischen Vakuum für 5 h bei –78 °C und für 2 h bei –60 °C entfernt. Man erhielt in quantitativer Ausbeute farblose, hydrolyseempfindliche Kristalle, welche bis –40 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 111 Experimenteller Teil 5.3.5 Darstellung von [Cl3CC(OH)2]+[AsF6]− OH Cl Cl Cl F OH F F As F F F Cl3CCO2H (0.16 g, 1.00 mmol) wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und im Überschuss HF (ca. 150 : 1) und anschließend AsF5 (0.26 g, 1.50 mmol) kondensiert. Das Gemisch wurde für 5 min auf –50 °C erwärmt, bis eine homogene, farblose Phase vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF und AsF5 wurden im dynamischen Vakuum bei –78 °C über Nacht entfernt. Man erhielt in quantitativer Ausbeute farblose, hydrolyseempfindliche Kristalle, welche bis –20 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 5.3.6 Darstellung von HC(CN)3 H NC CN CN Ca(C(CN)3)2, hergestellt nach Literaturvorschrift, wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und HF (ca. 150 : 1) im Überschuss kondensiert.[62,69-71] Das Gemisch wurde für 10 min auf –50 °C erwärmt, bis eine homogene, farblose Phase vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF wurde im dynamischen Vakuum bei –78 °C über Nacht entfernt. Es blieb ein farbloser Rückstand im Reaktor zurück, welcher eine Mischung aus HC(CN)3 und Ca(HF2)2 war. Das Gemisch war bis –40 °C beständig, veränderte bei bei beginnender Zersetzung die Farbe nach gelb und wurde bei weiterer Erwärmung tiefrot. 112 Experimenteller Teil 5.3.7 Darstellung von [(C6H5NO2H)2]2+[Ge2F10]2− O N F OH F F Ge 2 F F F F Ge F 2 F F C6H5NO2 (0.13 g, 1.00 mmol) wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und im Überschuss HF (ca. 150 : 1) und anschließend GeF4 (0.30 g, 2.00 mmol) kondensiert. Das Gemisch wurde für 5 min auf –50 °C erwärmt, bis eine homogene, gelbe und klare Lösung vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF wurde im dynamischen Vakuum für 3 h bei –70 °C und für 12 h bei –50 °C entfernt. Man erhielt in quantitativer Ausbeute gelbe, hydrolyseempfindliche Kristalle, welche bis –25 °C beständig waren und sich für eine Einkristall- Röntgenstrukturanalyse eigneten. 5.3.8 Darstellung von [(Me2OH)2]2+[Ge2F10]2− Me O H Me F F 2 F Ge F F F F Ge F 2 F F Me2O (0.13 g, 1.00 mmol) wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und im Überschuss HF (ca. 150 : 1) und anschließend GeF4 (0.30 g, 2.00 mmol / 0.15 g, 1.00 mmol / 0.07 g, 0.50 mmol) kondensiert. Das Gemisch wurde für jeweils 10 min auf –50 °C erwärmt, bis eine homogene, farblose klare Lösung vorlag und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF und GeF4 wurde im dynamischen Vakuum für 12 h bei –78 °C und für 8 h bei –50 °C entfernt. Man erhielt in quantitativer Ausbeute farblose, hydrolyseempfindliche Kristalle, welche bis –30 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 113 Experimenteller Teil 5.3.9 Darstellung von [(Me2OH)4]4+[Ge3F16]4− Me O H Me 4 F F F F F F F F Ge Ge Ge F F F F F F F F 4 Bei der Synthese von [(Me2OH)2]2+[Ge2F10]2− bildeten sich, wenn der Reaktor 6 Tage in einem Dewar mit Trockeneis stehen gelassen wurde, im oberen Bereich des Reaktors farblose Kristalle. Die Temperatur an der Kristallisationsstelle lag bei ca. –30 °C. Die Kristalle konnten mittels 4+ 4− Einkristall-Röntgenstrukturanalyse untersucht und als [(Me2OH)4] [Ge3F16] identifiziert werden. Sie waren hydrolyseempfindlich und bis ca. – 15 °C beständig. 5.3.10 Darstellung von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF F Me Me F F 3 Ge F F F F F F Ge Ge F F F F F F Ge F F F F S H 3 2 HF Me2S (0.124 g, 2.00 mmol) wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und HF (ca. 150 : 1) im Überschuss und anschließend GeF4 (0.15 g, 1.00 mmol) kondensiert. Das Gemisch wurde für 10 min auf –50 °C erwärmt, bis eine homogene, farblose klare Lösung vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF und Me2S wurden im dynamischen Vakuum für 48 h bei –78 °C und für 10 h bei –60 °C entfernt. Man erhielt in quantitativer Ausbeute farblose, hydrolyseempfindliche Kristalle, welche bis –30 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 114 Experimenteller Teil 5.3.11 Darstellung von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF F F Me Me F S H Ge F F F Ge F F F 3 F F F F Ge F 3 F F Ge F F F HF F Me2S (0.062 g, 1.00 mmol) wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und im Überschuss HF (ca. 150 : 1) und anschließend GeF4 (0.15 g, 1.00 mmol) kondensiert. Das Gemisch wurde für 10 min auf –50 °C erwärmt, bis eine homogene farblose klare Lösung vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF und Me2S wurden im dynamischen Vakuum für 16 h bei –78 °C und für 15 h bei –40 °C entfernt. Man erhielt in quantitativer Ausbeute farblose hydrolyseempfindliche Kristalle, welche bis –20 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 5.3.12 Darstellung von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF Me Me F S H F F Ge 2 F 2 F F 2 HF Me2S (0.062 g, 1.00 mmol) wurde in einem FEP-Reaktor vorgelegt. Dann wurde dieser auf –196 °C gekühlt und im Überschuss HF (ca. 150 : 1) und anschließend GeF4 (0.30 g, 2.00 mmol) kondensiert. Das Gemisch wurde für 10 min auf –50 °C erwärmt, bis eine homogene, farblose und klare Lösung vorlag, und dann mit Trockeneis auf –78 °C gekühlt. Das überschüssige HF und GeF4 wurden im dynamischen Vakuum für 30 h bei –78 °C und für 13 h bei –40 °C entfernt. Man erhielt in quantitativer Ausbeute farblose hydrolyseempfindliche Kristalle, welche bis –40 °C beständig waren und sich für eine Einkristall-Röntgenstrukturanalyse eigneten. 115 Experimenteller Teil 5.3.13 Darstellung von [(H3O)2]2+[GeF6]2− H O H H F F 2 F Ge F 2 F F Bei einer Umsetzung von Nitromethan mit HF/GeF4 wurden neben den gelben Kristallen von [(C6H5NO2H)2]2+[Ge2F10]2− farblose Kristalle entdeckt. Die farblosen Kristalle konnten mittels Einkristall-Röntgenstrukturanalyse untersucht und als [(H3O)2]2+[GeF6]2−identifiziert werden. 5.3.14 Darstellung der deuterierten Verbindungen Die Synthese der deuterierten Verbindungen erfolgte, soweit nicht anders beschrieben, analog zu den Synthesen der protonierten Verbindungen, mit dem einzigen Unterschied, dass DF anstatt HF verwendet wurde. 116 Röntgenstrukturanalysen 6 Röntgenstrukturanalysen Tabelle 6.1: Strukturdaten von [CF3SO3H2]+[SbF6]− Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus Kristallsystem Raumgruppe a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung CH2F9O3SSb 386.836 0.4 x 0.3 x 0.2 farbloser Block triklin P−1 7.0929(5) 7.6346(5) 8.8215(6) 71.884(6) 72.488(6) 84.509(6) −8 ≤ h ≤ 8, −9 ≤ k ≤ 9, −10 ≤ l ≤ 10 432.97(5) 2 2.9673(3) 3.569 360 4.31 − 25.99 99.4 100 3364 1693 0.0135 0.0144 / 0.0342 0.0164 / 0.0345 1693 / 0 / 144 1.011 −0.528 / 0.602 multiscan 0.257 / 0.490 Oxford XCalibur SapphireCCD MoKα SHELXS-97 Verfeinerung Messcode CCDC SHELXL-97 xa005 782908 117 Röntgenstrukturanalysen Tabelle 6.2: Strukturdaten von [CH2FC(OH)2]+[AsF6]− Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus C2H4AsF7O2 267.962 0.46 x 0.16 x 0.09 farbloser Block Kristallsystem Raumgruppe a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC monoklin P21/c 8.1734(3) 9.7654(3) 11.4889(4) 90 129.158(2) 90 −10 ≤ h ≤ 10, −12 ≤ k ≤ 11, −12 ≤ l ≤ 14 711.05(4) 4 2.50316(14) 4.878 512 4.76−26.50 99.3 140 4882 1459 0.0229 0.0191 / 0.0428 0.0256 / 0.0436 1431 / 0 /117 0.980 0.496 / 0.421 multiscan 0.077 / 0.391 Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa002 − 118 Röntgenstrukturanalysen Tabelle 6.3: Strukturdaten von [CHF2C(OH)2]+[AsF6]− Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus Kristallsystem Raumgruppe C2H3AsF8O2 285.953 0.25 x 0.21 x 0.19 farbloser Block triklin P−1 a [Å] 6.1889(8) b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z 7.3562(8) 8.0695(8) 91.244(9) 96.293(9) 90.768(10) −7 ≤ h ≤ 7, −9 ≤ k ≤ 6, −9 ≤ l ≤ 6 365.03(7) 2 ρcalc [g/cm3] 2.6017(5) μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe 4.779 272 4.29 − 25.99 99.40 100 2339 1421 Rint 0.0181 R1 / wR2 [I>2σ>(I)] 0.0214 / 0.045 R1 / wR2 [alle Reflexe] 0.0251 / 0.0456 Daten / Restraints / Parameter GooF 1421 / 0 / 130 0.968 min. / max. ρe [e/Å3] −0.459 / 0.325 Absorptionskorrektur multiscan Tmin / Tmax 0.375 / 0.411 Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa066 − 119 Röntgenstrukturanalysen Tabelle 6.4: Strukturdaten von [CF3C(OH)2]+[AsF6]− Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus Kristallsystem Raumgruppe C2H2AsF9O2 303.943 0.27 x 0.20 x 0.16 farbloser Block triklin P−1 a [Å] 5.6941(4) b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z 6.6096(5) 10.3171(8) 86.946(6) 89.227(6) 79.312(7) −6 ≤ h ≤ 7, −8 ≤ k ≤ 7, −12 ≤ l ≤ 11 381.01(5) 2 ρcalc [g/cm3] 2.6494(3) μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe 4.605 288 4.35 – 26.00 99.30 100 2454 1487 Rint 0.0203 R1 / wR2 [I>2σ>(I)] 0.0274 / 0.0625 R1 / wR2 [alle Reflexe] 0.0336 / 0.0636 Daten / Restraints / Parameter GooF 1487 / 0 / 135 0.983 min. / max. ρe [e/Å3] −0.409 / 0.901 Absorptionskorrektur analytisch Tmin / Tmax 0.440 / 0.567 Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa063 − 120 Röntgenstrukturanalysen Tabelle 6.5: Strukturdaten von [CCl3C(OH)2]+[AsF6]− Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus Kristallsystem C2H2AsCl3F6O2 353.306 0.35 x 0.22 x 0.18 farbloser Block monoklin Raumgruppe P21/c a [Å] 5.4531(2) b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z 8.9550(3) 19.3461(8) 90° 92.442(4)° 90° −7 ≤ h<=7, −11 ≤ k ≤ 11, −25 ≤ l ≤ 14 943.86(6) 4 ρcalc [g/cm3] 2.48633(16) −1 μ [mm ] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe 4.514 672 4.22 − 28.00 99.40 100 5459 2267 Rint 0.0239 R1 / wR2 [I>2σ>(I)] 0.0212 / 0.0397 R1 / wR2 [alle Reflexe] 0.0312 / 0.0407 Daten / Restraints / Parameter GooF 2267 / 0 / 135 0.868 min. / max. ρe [e/Å3] −0.324 / 0.475 Absorptionskorrektur analytisch Tmin / Tmax 0.361 / 0.521 Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa064 − 121 Röntgenstrukturanalysen Tabelle 6.6: Strukturdaten von [(C6H5NO2H)2]2+[Ge2F10]2− Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus C6H6F5GeNO2 291.72 0.3 x 0.3 x 0.15 gelber Block Kristallsystem Raumgruppe a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC triklin P−1 7.4719(9) 7.4969(9) 9.0481(11) 65.896(12) 79.398(10) 86.675(10) −9 ≤ h ≤ 8, −9 ≤ k ≤ 9, −11 ≤ l ≤ 10 454.67(10) 2 2.1309(5) 3.429 284 4.35 − 25.99 98.90 100 2989 1766 0.0185 0.0275 / 0.0702 0.0326 / 0.0717 1766 / 0 / 140 0.991 −0.656 / 1.277 multiscan 0.403 / 0.598 Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa049 − 122 Röntgenstrukturanalysen Tabelle 6.7: Strukturdaten von [(Me2OH)2]2+[Ge2F10]2− Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus C4H14F10Ge2O2 429.360 0.3 x 0.3 x 0.1 farbloser Block Kristallsystem Raumgruppe a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC triklin P−1 5.8272(5) 7.8348(7) 7.8935(8) 62.147(10) 80.850(8) 75.678(8) −7 ≤ h ≤ 8, −11 ≤ k ≤ 10, −10 ≤ l ≤ 11 308.33(5) 1 2.3124(4) 4.993 208 4.17 − 30.08 99.6 100 3185 1798 0.0250 0.0242 / 0.0473 0.0304 / 0.0483 1798 / 0 / 88 0.940 −0.589 / 0.531 multiscan 0.467 / 0.607 Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa014 − 123 Röntgenstrukturanalysen Tabelle 6.8: Strukturdaten von [(Me2OH)4]4+[Ge3F16]4− Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus C8H28F16Ge3O4 710.110 0.2 x 0.05 x 0.05 farbloses Prisma Kristallsystem Raumgruppe a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC triklin P−1 6.7681(3) 8.8979(7) 9.7025(7) 85.661(6) 81.253(5) 75.484(5) −8 ≤ h ≤ 8, −10 ≤ k ≤ 10, −11 ≤ l ≤ 11 558.64(6) 1 2.1108(2) 4.154 348 4.25 − 25.99 99.4 123 3819 2181 0.0212 0.0233 / 0.0498 0.0305 / 0.0507 2181 / 0 / 154 0.942 −0.605 / 0.371 multiscan 0.608 / 0.812 Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa015 − 124 Röntgenstrukturanalysen Tabelle 6.9: Strukturdaten von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF Summenformel M [g/mol] C6H23F21Ge4S3 880.850 Kristallgröße [mm3] Farbe, Habitus Kristallsystem Raumgruppe a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] 0.41 x 0.27 x 0.27 farbloser Block triklin Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC 2370 / 0 / 171 0.982 −0.356 / 0.434 multiscan 0.158 / 0.244 Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa035 − P−1 8.3534(6) 8.8428(6) 8.9804(6) 104.223(6) 104.710(6) 96.945(6) −10 ≤ h ≤ 10, −10 ≤ k ≤ 10, −11 ≤ l ≤ 11 609.95(7) 1 2.3981(3) 5.292 424 4.76 – 26.00 99.3 123 7490 2370 0.0281 0.0211 / 0.0485 0.0282 / 0.0498 125 Röntgenstrukturanalysen Tabelle 6.10: Strukturdaten von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus Kristallsystem C6H22F20Ge4S3 860.840 0.4 x 0.3 x 0.2 farbloses Prisma triklin Raumgruppe a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC P−1 13.0897(4) 17.2701(5) 21.4416(6) 90 90 90 −13 ≤ h ≤ 15, −11 ≤ k ≤ 20, −25 ≤ l ≤ 14 4847.1(2) 8 2.35933(10) 5.319 3312 4.27 – 25.00 99.3 123 12258 4242 0.0288 0.0214 / 0.0359 0.0392 / 0.0371 4242 / 0 / 326 0.801 −0.508 / 0.400 multiscan 0.216 / 0.345 Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa038 − 126 Röntgenstrukturanalysen Tabelle 6.11: Strukturdaten von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus Kristallsystem Raumgruppe a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint C4H16F8GeS2 352.900 0.3 x 0.2 x 0.1 farbloser Block orthorhombisch Fdd2 17.6926(9) 22.0465(12) 6.3959(3) 90 90 90 −24 ≤ h ≤ 24, −30 ≤ k ≤ 30, −8 ≤ l ≤ 8 2494.8(2) 8 1.87914(15) 2.858 1408 4.36 − 29.99 99.7 100 7911 1808 0.0225 R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC 0.0123 / 0.0267 0.0134 / 0.0268 1808 / 1 / 80 0.975 −0.133 / 0.278 multiscan 0.536 / 0.751 Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa040 − 127 Röntgenstrukturanalysen Tabelle 6.12: Strukturdaten von [(H3O)2]2+[GeF6]2− Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus Kristallsystem Raumgruppe F6GeH6O2 224.650 0.17 x 0.09 x 0.08 farbloses Plättchen monoklin P21/c a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC 5.2756(5) 9.2306(10) 5.8885(7) 90 95.227(9) 90 −5 ≤ h ≤ 6, −6 ≤ k ≤ 1, −7 ≤ l ≤ 7 285.56(5) 2 2.6127(5) 5.433 216 4.42 − 26.83 98.4 123 1058 607 0.0181 0.0211 / 0.0387 0.0298 / 0.0396 607 / 0 / 55 0.891 −0.417 / 0.388 multiscan 0.556 / 0.657 Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa048 − 128 Röntgenstrukturanalysen Tabelle 6.13: Strukturdaten von NaH4F5 Summenformel M [g/mol] Kristallgröße [mm3] Farbe, Habitus F5H4Na 122.014 0.37 x 0.07 x 0.04 farbloses Plättchen Kristallsystem Raumgruppe a [Å] b [Å] c [Å] α [°] β [°] γ [°] Index Bereich V [Å3] Z ρcalc [g/cm3] μ [mm−1] F(000) θ-Bereich [°] Completeness to θ [%] Temperatur [K] Gesammelte Reflexe Unabhängige Reflexe Rint R1 / wR2 [I>2σ>(I)] R1 / wR2 [alle Reflexe] Daten / Restraints / Parameter GooF min. / max. ρe [e/Å3] Absorptionskorrektur Tmin / Tmax Diffraktometer Strahlung Lösung Verfeinerung Messcode CCDC orthorhombisch I41/a 6.0284(3) 6.0284(3) 11.6317(15) 90 90 90 −7 ≤ h ≤ 7, −7 ≤ k ≤ 7, −11 ≤ l ≤ 14 422.71(6) 4 1.9173(3) 0.356 240 5.93 − 28 98.1 100 1375 254 0.0211 0.0181 / 0.0396 0.0255 / 0.0405 254 / 0 /18 0.987 −0.12 / 0.153 analytisch 0.966 / 0.985 Oxford XCalibur SapphireCCD MoKα SHELXS-97 SHELXL-97 xa044 − 129 Verzeichnisse 7 Verzeichnisse 7.1 Abkürzungs- und Einheitenverzeichnis a Å Å4µ–1 as ber. br bzw. ca. d δ et al. exp. Fe Fμ GooF h i.p. IR J m Me μ NMR ν o.o.p. Ph ppm q RT s sh TNT vs vw w Z λ ρ τ ω Winkel (angle) Ångström (10‐10 m) Raman Aktivität; μ: induziertes Dipolmoment antisymmetrisch berechnet broad (IR) beziehungsweise circa Abstand, Dublett (NMR) Deformationsschwingung (IR und Raman); chemische Verschiebung (NMR) et alii experimentell Fluoratom in äquatorialer Stellung verbrückendes Fluoratom Goodness of fit Stunde in phase Infrarot Kopplungskonstante medium (IR) Methyl verbrückend Nuclear Magnetic Resonance Streckschwingung out of phase Phenyl parts per million Quartett Raumtemperatur strong (IR); Singulett (NMR); symmetrisch (Schwingungsspektroskopie) shoulder 2,4,6-Trinitrotuluol very strong (IR) very weak (IR) weak (IR) Anzahl der Formeleinheiten in der Zelle Wellenlänge rocking-Schwingung (IR und Raman), Dichte Torsionsschwingung Schaukelschwingung (wagging) 130 Verzeichnisse 7.2 Abbildungsverzeichnis Abbildung 2.1: Strukturen von Cyanoform (links) und Dicyanoketenimin (rechts). ............ 4 Abbildung 3.1: Kristallstruktur von [CF3SO3H⋅SbF5].[29] ...................................................... 6 Abbildung 3.2: Struktur von [CF3SO3H2]+ im Kristall mit Wasserstoffbrückenbindungen zu den Fluoratomen der benachbarten [SbF6]–-Anionen .................................. 9 Abbildung 3.3: Kristallpackung von [CF3SO3H2]+[SbF6]– ................................................. 10 Abbildung 3.4: Quantenchemisch berechnete Struktur der [CF3SO3H2(HF)2]+-Einheit. .... 11 Abbildung 3.5: IR- und Raman-Spektrum von [CF3SO3H2]+[SbF6]−. ................................. 12 Abbildung 3.6: IR- und Raman-Spektrum von [CF3SO3D2]+[SbF6]−. ................................. 13 Abbildung 3.7: Asymmetrische Einheit [CH2FC(OH)2]+[AsF6]− im Kristall ..................... 17 Abbildung 3.8: Kristallpackung von [CH2FC(OH)2]+[AsF6]– ............................................ 17 Abbildung 3.9: Wasserstoffbrückenbindungen in [CH2FC(OH)2]+[AsF6]– ........................ 18 Abbildung 3.10: Koordination zweier [AsF6]−-Anionen zur C(OH)2-Gruppe ..................... 19 Abbildung 3.11: Quantenchemisch berechnete Struktur der [CH2FC(OH)2(HF)2]+-Einheit. 20 Abbildung 3.12: IR- und Raman-Spektrum von [CH2FC(OH)2]+[AsF6]–. ............................ 21 Abbildung 3.13: IR- und Raman-Spektrum von [CH2FC(OD)2]+[AsF6]−. ............................ 22 Abbildung 3.14: Asymmetrische Einheit [CHF2C(OH)2]+[AsF6]− im Kristall ...................... 26 Abbildung 3.15: Kristallpackung von [CHF2C(OH)2]+[SbF6]− ............................................ 26 Abbildung 3.16: Koordination zweier [AsF6]−-Anionen zur C(OH)2-Gruppe ...................... 28 Abbildung 3.17: Quantenchemisch berechnete Struktur der [CHF2C(OH)2(HF)2]+-Einheit. 29 Abbildung 3.18: IR- und Raman-Spektrum von [CHF2C(OH)2]+[AsF6]−. ............................ 32 Abbildung 3.19: IR- und Raman-Spektrum von [CHF2C(OD)2]+[AsF6]−. ............................ 32 Abbildung 3.20: Asymmetrische Einheit von [CF3C(OH)2]+[AsF6]− im Kristall .................. 34 Abbildung 3.21: Kristallpackung von [CF3C(OH)2]+[AsF6]− ............................................... 34 Abbildung 3.22: Koordination zweier [AsF6]−-Anionen zur positiven C(OH)2-Gruppe ...... 36 Abbildung 3.23: Quantenchemisch berechnete Struktur der CF3C(OH)2(HF)2+ Einheit....... 36 Abbildung 3.24: IR- und Raman-Spektrum von [CF3C(OH)2]+[AsF6]−. ............................... 39 Abbildung 3.25: IR- und Raman-Spektrum von [CF3C(OD)2]+[AsF6]−. ............................... 39 Abbildung 3.26: Asymmetrische Einheit [CCl3C(OH)2]+[AsF6]− im Kristall ...................... 42 Abbildung 3.27: Kristallpackung von [CCl3C(OH)2]+[AsF6]− .............................................. 42 Abbildung 3.28: Wasserstoffbrückenbindungen in [CCl3C(OH)2]+[AsF6]− ......................... 43 Abbildung 3.29: Quantenchemisch berechnete Struktur der [CCl3C(OH)2(HF)2]+-Einheit. . 44 Abbildung 3.30: IR- und Raman-Spektrum von [CCl3C(OH)2]+[AsF6]−. ............................. 45 Abbildung 3.31: IR- und Raman-Spektrum von [CCl3C(OD)2]+[AsF6]−. ............................. 46 131 Verzeichnisse Abbildung 3.32: Strukturen von Cyanoform, Dicyanoketenimin und Oxoniumtricyanomethanid[20]. ....................................................................................... 49 Abbildung 3.33: 1H-NMR-Spektrum der Umsetzung von Ca(C(CN)3)2 und HF ................. 51 Abbildung 3.34: 13 C-NMR-Spektrum von HC(CN)3 ............................................................ 52 Abbildung 3.35: links: Struktur von C(CN)3− in Ca(C(CN)3)2, mittig: berechnete Struktur von HC(CN)3, rechts: berechnete Struktur von C(CN)2CNH. .................... 53 Abbildung 3.36: Schwingungsspektren der Umsetzung von Ca(C(CN)3)2 mit HF: b) RamanSpektrum, c) IR Spektrum; und von der Umsetzung von Ca(C(CN)3)2 mit DF: a) Raman-Spektrum (* Schwingungsbanden von Ca(HF2)2). ............. 55 Abbildung 3.37: Struktur von [(C6H5NO2H)2]2+[Ge2F10]2−. .................................................. 59 Abbildung 3.38: Struktur des [Ge2F10]2−-Anions .................................................................. 60 Abbildung 3.39: Kristallpackung von [(C6H5NO2H)2]2+[Ge2F10]2− mit interionischen Kontakten entlang der a-Achse .................................................................. 61 Abbildung 3.40: [C6H5NO2H]+ mit interionischen Kontakten .............................................. 62 Abbildung 3.41: Quantenchemisch berechnete Struktur von [Ge2F10]2−. .............................. 63 Abbildung 3.42: Raman-Spektrum von [(C6H5NO2H)2]2+[Ge2F10]2−. ................................... 63 Abbildung 3.43: Struktur von [(Me2OH)2]2+[Ge2F10]2− ......................................................... 66 Abbildung 3.44: Kristallstruktur von [(Me2OH)2]2+[Ge2F10]2− ............................................. 67 Abbildung 3.45: Raman-Spektrum von [(Me2OH)2]2+[Ge2F10]2−. ......................................... 68 Abbildung 3.46: Struktur von [(Me2OH)4]4+[Ge3F16]4− im Kristall ....................................... 71 Abbildung 3.47: Kristallstruktur von [(Me2OH)4]4+[Ge3F16]4− ............................................. 71 Abbildung 3.48: Quantenchemisch berechnete Struktur von [Ge3F16]4−. .............................. 73 Abbildung 3.49: Struktur von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF im Kristall ........................... 75 Abbildung 3.50: Links: Struktur der Kationen in [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF, rechts: repetitive Einheit des Anions mit Winkeln [°] (blau) und Bindungslängen [pm] (schwarz) ........................................................................................... 76 Abbildung 3.51: Kristallstruktur von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF .................................. 77 Abbildung 3.52: Quantenchemisch berechnete Struktur von [Ge4F20]4− mit Winkeln [°] (blau) und Bindungslängen [pm] (schwarz)................................................ 78 Abbildung 3.53: Raman-Spektrum von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF. ............................... 79 Abbildung 3.54: Struktur von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF im Kristall .............................. 84 Abbildung 3.55: Oben: Struktur der Kationen in [(Me2SH)3]3+[Ge4F19]3− ⋅ HF, unten: repetitive Einheit des Anions mit Winkeln [°] (blau) und Bindungslängen (schwarz) .................................................................................................... 84 Abbildung 3.56: Kristallstruktur von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF, links: entlang der bAchse, rechts: entlang der c-Achse ............................................................ 86 Abbildung 3.57: Quantenchemisch berechnete Struktur von [Ge3F15]3− mit Winkeln [°] (blau) und Bindungslängen [pm] (schwarz)................................................ 87 Abbildung 3.58: Raman-Spektrum von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF. .................................. 88 132 Verzeichnisse Abbildung 3.59: Struktur von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF im Kristall .............................. 91 Abbildung 3.60: Raman-Spektrum von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF. ................................. 93 Abbildung 3.61: Struktur von [(H3O)2]2+[GeF6]2− im Kristall ............................................... 94 Abbildung 3.62: Kristallstruktur von [(H3O)2]2+[GeF6]2− mit interionischen Kontakten ...... 95 Abbildung 3.63: [H3O]+-Kation mit Wasserstoffbrückenbindungen .................................... 96 Abbildung 3.64: Isolierte Fluorogermanate: links: [GeF6]2−-Oktaeder in [(H3O)2]2+[GeF6]2−; mittig: [Ge2F10]2−-Dimer in [(Me2OH)2]2+[Ge2F10]2−; rechts: [Ge3F16]4−Trimer in [(Me2OH)4]4+[Ge3F16]4−. ............................................................. 97 Abbildung 3.65: Polymere Fluorogermanate: oben/links: cis-μ-([GeF5]−)n in [XeF5]+[GeF5]−[21]; oben/rechts: trans-μ-([GeF5]−)n in [XeF5]+[GeF5]−[21]; mittig: ([Ge4F19]3−)n in [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF; unten: ([Ge4F19]3−)n in [(Me2SH)3]3+[Ge4F19]3− ⋅ HF. .................................................................. 98 Abbildung 3.66: Raman-Spektren von: a) [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF; b) [(Me2SH)3]3+[Ge4F19]3− ⋅ HF; c) [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF; d) [(Me2OH)2]2+[Ge2F10]2−; (* Schwingung des Kations). .............................. 99 Abbildung 3.67: Raman-Spektren der Anionen von [(Me2OH)2]2+[Ge2F10]2− (schwarz) und [(C6H5NO2H)2]2+[Ge2F10]2− (blau) ........................................................... 100 Abbildung 4.1: Berechnete Struktur von Cyanoform. ....................................................... 102 Abbildung 4.2: Oben: Isolierte Fluorogermanate: links: [Ge2F10]2−-Dimer in [(Me2OH)2]2+[Ge2F10]2−; rechts: [Ge3F16]4−-Trimer in [(Me2OH)4]4+[Ge3F16]4−; unten: polymere Fluorogermanate: links: ([Ge4F19]3−)n in [(Me2SH)3]3+[Ge4F19]3− ⋅ HF, rechts: ([Ge4F19]3−)n in [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF. ................................................................. 103 Abbildung 5.1: Tieftemperatur-Schutzgasküvette. ............................................................ 106 Abbildung 5.2: Tieftemperatur-IR-Zelle. ........................................................................... 107 133 Verzeichnisse 7.3 Tabellenverzeichnis Tabelle 3.1: Ausgewählte Bindungslängen [pm] und -winkel [°] für CF3SO3H, [CF3SO3H2]+[SbF6]–, das berechnete [CF3SO3H2]+ und die berechnete [CF3SO3H2(HF)2]+-Einheit .............................................................................. 10 Tabelle 3.2: Beobachtete Schwingungsfrequenzen [cm–1] von [CF3SO3H2]+[SbF6]− und CF3SO3H sowie berechnete Schwingungsfrequenzen von [CF3SO3H2(HF)2]+ und [CF3SO3H2]+ ............................................................................................. 14 Tabelle 3.3: Beobachtete Schwingungsfrequenzen [cm–1] von [CF3SO3D2]+[SbF6]− sowie berechnete Schwingungsfrequenzen von [CF3SO3D2(HF)2]+ und [CF3SO3D2]+ ......................................................................................................................... 15 Tabelle 3.4: Ausgewählte Bindungslängen [pm] und -winkel [°] für CH2FCO2H, [CH2FC(OH)2]+[AsF6]− und die berechnete [CH2FC(OH)2(HF)2]+-Einheit.... 18 Tabelle 3.5: Beobachtete Schwingungsfrequenzen [cm–1] von [CH2FC(OH)2]+[AsF6]− und CH2FCO2H sowie berechnete Schwingungsfrequenzen von [CH2FC(OH)2(HF)2]+ und [CH2FC(OH)2]+ ..................................................... 23 Tabelle 3.6: Beobachtete Schwingungsfrequenzen [cm–1] von [CH2FC(OD)2]+[AsF6]− sowie berechnete Schwingungsfrequenzen von CH2FC(OD)2(HF)2+ und CH2FC(OD)2+ .................................................................................................. 24 Tabelle 3.7: Ausgewählte Bindungslängen [pm] und -winkel [°] für CHF2CO2H, [CHF2C(OH)2]+[AsF6]− und die berechnete [CHF2C(OH)2(HF)2]+-Einheit.... 27 Tabelle 3.8: Beobachtete Schwingungsfrequenzen [cm–1] von [CHF2C(OH)2]+[AsF6]− und CHF2CO2H sowie berechnete Schwingungsfrequenzen von [CHF2C(OH)2(HF)2]+ und [CHF2C(OH)2]+ ..................................................... 30 Tabelle 3.9: Beobachtete Schwingungsfrequenzen [cm–1] von [CHF2C(OD)2]+[AsF6]− sowie berechnete Schwingungsfrequenzen von CHF2C(OD)2(HF)2+ und CHF2C(OD)2+ .................................................................................................. 31 Tabelle 3.10: Ausgewählte Bindungslängen [pm] und -winkel [°] für CF3CCO2H, [CF3C(OH)2]+[AsF6]− und die berechnete [CF3CC(OH)2(HF)2]+ Einheit ....... 35 Tabelle 3.11: Beobachtete Schwingungsfrequenzen [cm–1] von [CF3C(OH)2]+[AsF6]− und CF3CO2H sowie berechnete Schwingungsfrequenzen von [CF3C(OH)2(HF)2]+ und [CF3C(OH)2]+ ........................................................................................... 38 Tabelle 3.12: Beobachtete Schwingungsfrequenzen [cm–1] von [CF3C(OD)2]+[AsF6]− sowie berechnete Schwingungsfrequenzen von [CF3C(OD)2(HF)2]+ und [CF3C(OD)2]+ .................................................................................................. 40 Tabelle 3.13: Ausgewählte Bindungslängen [pm] und -winkel [°] für CCl3CO2H, [CCl3C(OH)2]+[AsF6]− und die berechnete CCl3C(OH)2(HF)2+ Einheit ......... 43 Tabelle 3.14: Beobachtete Schwingungsfrequenzen [cm–1] von [CCl3C(OH)2]+[AsF6]− und CCl3CO2H sowie berechnete Schwingungsfrequenzen von [CCl3C(OH)2(HF)2]+ und [CCl3C(OH)2]+ ....................................................... 47 Tabelle 3.15: Beobachtete Schwingungsfrequenzen [cm–1] von [CCl3C(OD)2]+[AsF6]− sowie berechnete Schwingungsfrequenzen von [CCl3C(OD)2(HF)2]+ und [CCl3C(OD)2]+ ................................................................................................. 48 134 Verzeichnisse Tabelle 3.16: Beobachtete und berechnete Schwingungsfrequenzen [cm–1] von HC(CN)3 und DC(CN)3 ................................................................................................... 54 Tabelle 3.17: Ausgewählte Bindungslängen [pm] und -winkel [°] von [(C6H5NO2H)2]2+[Ge2F10]2− und des berechneten [Ge2F10]2− .......................... 60 Tabelle 3.18: Beobachtete Schwingungsfrequenzen [cm–1] von [(C6H5NO2H)2]2+[Ge2F10]2− ......................................................................................................................... 64 Tabelle 3.19: Ausgewählte Bindungslängen [pm] und -winkel [°] von [(Me2OH)2]2+[Ge2F10]2− und des berechneten [Ge2F10]2− ................................ 66 Tabelle 3.20: Beobachtete Schwingungsfrequenzen [cm–1] von [(Me2OH)2]2+[Ge2F10]2− sowie berechnete Schwingungsfrequenzen von [Me2OH(HF)]+ und [Ge2F10]2− ......................................................................................................................... 68 Tabelle 3.21: Ausgewählte Bindungslängen [pm] und -winkel [°] von [(Me2OH)4]4+[Ge3F16]4− und von der berechneten [Ge3F16]4−-Einheit ............ 72 Tabelle 3.22: Ausgewählte Bindungslängen [pm] und -winkel [°] von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF und von der berechneten [Ge4F20]4−-Einheit .. 77 Tabelle 3.23: Beobachtete Schwingungsfrequenzen [cm–1] von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF und berechnete Schwingungsfrequenzen von [Me2SH(HF)]+ und [Ge4F20]4− ......................................................................... 80 Tabelle 3.24: Ausgewählte Bindungslängen [pm] und -winkel [°] von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF und vom berechneten [Ge3F15]3− ....................... 85 Tabelle 3.25: Wasserstoffbrückenbindungen von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF (Bindungslängen [pm] und -winkel [°]) ........................................................................... 86 Tabelle 3.26: Beobachtete Schwingungsfrequenzen [cm–1] von [Me3SH]3+[Ge4F19]3− ⋅ HF und berechnete Schwingungsfrequenzen von [Me2SH(HF)]+ und [Ge3F15]3−. 89 Tabelle 3.27: Ausgewählte Bindungslängen [pm] und -winkel [°] von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF und vom berechneten [GeF6]2− ......................... 91 Tabelle 3.28: Beobachtete Schwingungsfrequenzen [cm–1] von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF und berechnete Schwingungsfrequenzen von [Me2SH(HF)]+ und [GeF6]2− ... 92 Tabelle 3.29: Ausgewählte Bindungslängen [pm] und -winkel [°] von [(H3O)2]2+[GeF6]2− und des berechneten [GeF6]2− .......................................................................... 95 Tabelle 3.30: Wasserstoffbrückenbindungen von [(H3O)2]2+[GeF6]2− (Bindungslängen [pm] und -winkel [°])................................................................................................ 96 Tabelle 3.31: Vergleich der Ge:F-Stöchiometrien der Fluorogermanatanionen ................... 97 Tabelle 3.32: Vergleich der Ramanfrequenzen der Fluorogermanatanionen ........................ 98 Tabelle 3.33: Gegenüberstellung der Schwingungen von [GeF5]− in [Bu4N]+[GeF5]− und [(Me2OH)2]2+[Ge2F10]2− ................................................................................. 100 Tabelle 5.1: Verwendete Chemikalien............................................................................... 105 Tabelle 5.2: Deuterierte Lösungsmittel und Standards für die NMR-Spektroskopie ........ 108 Tabelle 6.1: Strukturdaten von [CF3SO3H2]+[SbF6]−......................................................... 117 Tabelle 6.2: Strukturdaten von [CH2FC(OH)2]+[AsF6]−.................................................... 118 Tabelle 6.3: Strukturdaten von [CHF2C(OH)2]+[AsF6]−.................................................... 119 135 Verzeichnisse Tabelle 6.4: Strukturdaten von [CF3C(OH)2]+[AsF6]−....................................................... 120 Tabelle 6.5: Strukturdaten von [CCl3C(OH)2]+[AsF6]− ..................................................... 121 Tabelle 6.6: Strukturdaten von [(C6H5NO2H)2]2+[Ge2F10]2− ............................................. 122 Tabelle 6.7: Strukturdaten von [(Me2OH)2]2+[Ge2F10]2− ................................................... 123 Tabelle 6.8: Strukturdaten von [(Me2OH)4]4+[Ge3F16]4− ................................................... 124 Tabelle 6.9: Strukturdaten von [(Me2SH)3]3+[Ge4F19]3− ⋅ 2 HF ......................................... 125 Tabelle 6.10: Strukturdaten von [(Me2SH)3]3+[Ge4F19]3− ⋅ HF ............................................ 126 Tabelle 6.11: Strukturdaten von [(Me2SH)2]2+[GeF6]2− ⋅ 2 HF ........................................... 127 Tabelle 6.12: Strukturdaten von [(H3O)2]2+[GeF6]2− ........................................................... 128 Tabelle 6.13: Strukturdaten von NaH4F5 ............................................................................. 129 136 Verzeichnisse 7.4 Literaturverzeichnis [1] Z. Sir Cope, Med. Hist. 1958, 2, 163. [2] S. C. Gilfillan, J. Occup. Environ. Med. 1965, 7, 53. [3] S. Arrhenius, Z. Phys. Chem. 1887, 1, 631. [4] J. N. Brönstedt, Rec. Trav. Chim. Pay-Bas 1923, 42, 718. [5] T. M. Lowry, Chem. Ind. 1923, 42, 42. [6] G. N. Lewis, Valence and the Structure of Molecules, The chemical catalogue Co., New York, 1923. [7] H. Flood, Acta Chem. Scand. 1947, 1, 781. [8] H. Flood, Acta Chem. Scand. 1947, 1, 592. [9] H. Lux, Z. Elektrochem. Angew. Phys. Chem. 1939, 45, 303. [10] L. P. Hammet, A. J. Deyrup, J. Am. Chem. Soc. 1932, 54, 2721. [11] G. A. Olah, G. K. Prakash, J. Sommer, A. Molnar, Superacid Chemistry, 2nd ed., John Wiley & Sons: New Jersey, 2009. [12] I. A. Koppel, P. Burk, I. Koppel, I. Leito, T. Sonoda, M. Mishima, J. Am. Chem. Soc. 2000, 122, 5114. [13] D. Himmel, S. K. Goll, I. Leito, I. Krossing, Angew. Chem. 2010, 122, 7037; Angew. Chem., Int. Ed. 2010, 49, 6885. [14] A. Kutt, T. Rodima, J. Saame, E. Raamat, V. Maemets, I. Kaljurand, I. A. Koppel, R. Y. Garlyauskayte, Y. L. Yagupolskii, L. M. Yagupolskii, E. Bernhardt, H. Willner, I. Leito, J. Org. Chem. 2010, 76, 391. [15] R. J. Gillespie, T. E. Peel, E. A. Robinson, J. Am. Chem. Soc. 1971, 93, 5083. [16] N. F. Hall, J. B. Conant, J. Am. Chem. Soc. 1927, 49, 3047. [17] G. A. Olah, E. Namanworth, J. Am. Chem. Soc. 1966, 88, 5327. [18] A. Commeyras, G. A. Olah, J. Am. Chem. Soc. 1969, 91, 2929. [19] R. Seelbinder, N. R. Götz, J. Weber, R. Minkwitz, A. J. Kornath, Chem.-Eur. J. 2010, 16, 1026. [20] D. Šišak, L. B. McCusker, A. Buckl, G. Wuitschik, Y.-L. Wu, W. B. Schweizer, J. D. Dunitz, Chem.-Eur. J. 2010, 16, 7224. [21] T. E. Mallouk, B. Desbat, N. Bartlett, Inorg. Chem. 1984, 23, 3160. [22] R. J. Gillespie, Acc. Chem. Res. 1968, 1, 202. [23] R. N. Haszeldine, J. M. Kidd, J. Chem. Soc. 1954, 4228. [24] A. Senning, Chem. Rev. 1965, 65, 385. 137 Verzeichnisse [25] R. D. Howells, J. D. Mc Cown, Chem. Rev. 1977, 77, 69. [26] J. B. Hendrickson, D. D. Sternbach, K. W. Bair, Acc. Chem. Res. 1977, 10, 306. [27] V. W. Cicha, A. Kornath, R. J. McKinney, V. N. M. Rao, J. S. Trasher, A. Waterfeld, U.S. 5,773,637, E. I. du Pont de Nemours and Company, U.S., 1998, p. 6. [28] A. Schulz, J. Thomas, A. Villinger, Chem. Commun. 2010, 46, 3696. [29] D. Mootz, K. Bartmann, Z. Naturforsch. B 1991, 46, 1659. [30] G. A. Olah, Angew. Chem. 1995, 107, 1519; Angew. Chem., Int. Ed. 1995, 34, 1393. [31] R. Minkwitz, T. Hertel, Z. Naturforsch. 1997, 52b, 1283. [32] R. Minkwitz, S. Schneider, M. Seifert, H. Hartl, Z. Anorg. Allg. Chem. 1996, 622, 1404. [33] T. T. Tidwell, Angew. Chem. 1984, 96, 16; Angew. Chem., Int. Ed. 1984, 23, 20. [34] G. A. Olah, A. Germain, H. C. Lin, J. Am. Chem. Soc. 1975, 97, 5481. [35] G. A. Olah, A. Burrichter, T. Mathew, Y. D. Vankar, G. Rasul, G. K. S. Prakash, Angew. Chem. 1997, 109, 1958; Angew. Chem., Int. Ed. 1997, 36, 1875. [36] J. J. Lagowski, Q. Rev. Chem. Soc. 1959, 13, 233. [37] G. K. S. Prakash, G. Rasul, A. Burrichter, K. K. Laali, G. A. Olah, J. Org. Chem. 1996, 61, 9253. [38] J. F. J. Dippy, S. R. C. Hughes, A. Rozanski, J. Chem. Soc 1959, 2492. [39] R. Seelbinder, Dissertation 2004, Universität Dortmund. [40] K. Bartmann, D. Mootz, Acta Crystallogr. C 1990, 46, 319. [41] J. P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 1996, 77, 3865. [42] A. D. McLean, G. S. Chandler, J. Chem. Phys. 1980, 72, 5639. [43] R. Krishnan, J. S. Binkley, R. Seeger, J. A. Pople, J. Chem. Phys. 1980, 72, 650. [44] T. Clark, J. Chandrasekhar, G. W. Spitznagel, P. v. R. Schleyer, J. Comp. Chem. 1983, 4, 294. [45] S. P. Gejji, K. Hermansson, J. Lindgren, J. Phys. Chem. 1993, 97, 6986. [46] Y. Katsuhara, R. M. Hammaker, D. D. DesMarteau, Inorg. Chem. 1980, 19, 607. [47] E. S. Stoyanov, K.-C. Kim, C. A. Reed, J. Phys. Chem. A 2004, 108, 9310. [48] J. Weidlein, U. Müller, K. Dehnicke, Schwingungsspektroskopie, Thieme, Stuttgart; 2. neubearb. Auflage, 1988. [49] R. Minkwitz, R. Seelbinder, R. Schöbel, Angew. Chem. 2002, 114, 119; Angew. Chem., Int. Ed. 2002, 41, 111. [50] G. Schultz, I. Hargittai, R. Seip, Z. Naturforsch. A 1981, 36, 917. [51] J. A. Kanters, J. Kroon, Acta Crystallogr. 1972, B28, 1946. 138 Verzeichnisse [52] A. F. Holleman, E. Wiberg, 101. Auflage, Berlin (Germany), 1995, p. Anhang V. [53] K. O. Christe, X. Zhang, R. Bau, J. Hegge, G. A. Olah, G. K. S. Prakash, J. A. Sheehy, J. Am. Chem. Soc. 1999, 122, 481. [54] J. R. Barceló, C. Otero, Spectrochim. Acta 1962, 18, 1231. [55] J. M. J. M. Bijen, J. L. Derissen, J. Mol. Struct. 1975, 27, 233. [56] I. Nahringbauer, J.-O. Lundgren, E. K. Andersen, Acta Crystallogr. 1979, B35, 508. [57] P.-G. Jonsson, W. C. Hamilton, J. Chem. Phys. 1972, 56, 4433. [58] R. Fausto, J. J. C. Teixeira-Dias, J. Mol. Struct. 1986, 144, 241. [59] H. Schmidtmann, Ber. Dtsch. Chem. Ges. 1896, 29, 1168. [60] B. Bak, C. Bjorkman, J. Mol. Struct. 1975, 25, 131. [61] S. Trofimenko, J. Org. Chem. 1963, 28, 217. [62] S. Trofimenko, E. L. Little, H. F. Mower, J. Org. Chem. 1962, 27, 433. [63] A. Hantzsch, G. Osswald, Ber. Dtsch. Chem. Ges. 1899, 32, 641. [64] A. F. E. Cox, Bull. Soc. Chim. Fr. 1954, 948. [65] R. H. Boyd, J. Phys. Chem. 1963, 67, 737. [66] B. A. Coyle, L. W. Schroeder, J. A. Ibers, J. Solid State Chem. 1970, 1, 386. [67] T. Soltner, N. R. Götz, A. Kornath, Eur. J. Inorg. Chem. 2011, 20, 3076. [68] M. T. Reetz, I. Chatziiosifidis, Synthesis 1982, 330. [69] R. A. Carboni, Org. Synth. 1959, 39, 64. [70] J. Konnert, D. Britton, Inorg. Chem. 1966, 5, 1193. [71] K. E. Bessler, C. C. Gatto, L. L. Romualdo, J. A. Ellena, M. J. d. A. Sales, Z. Naturforsch. 2008, 63b, 285. [72] S. V. Garbuz, V. V. Skopenko, V. D. Khavryuchenko, N. N. Gerasimchuk, Theor. Exp. Chem. 1989, 25, 83. [73] H. Brand, J. F. Liebman, A. Schulz, Eur. J. Org. Chem. 2008, 2008, 4665. [74] J. Köhler, A. Simon, R. Hoppe, J. Less Common Met. 1988, 137, 333. [75] J. Trotter, M. Akhtar, N. Barlett, J. Chem. Soc. A 1966, 1966, 30. [76] J. C. Taylor, P. W. Wilson, J. Am. Chem. Soc. 1973, 95, 1834. [77] J. Koehler, J.-H. Chang, Z. Anorg. Allg. Chem. 1997, 623, 596. [78] J. L. Hoard, W. B. Vincent, J. Am. Chem. Soc. 1939, 61, 2849. [79] J. E. Griffiths, D. E. Irish, Inorg. Chem. 1964, 3, 1134. [80] H. C. Clark, Chem. Commun. (London), , 1967, 14. [81] K. O. Christe, R. D. Wilson, I. B. Goldberg, Inorg. Chem. 1976, 15, 1271. [82] I. Wharf, M. Onyszchuk, Can. J. Chem. 1970, 48, 2250. 139 Verzeichnisse [83] D. J. Brauer, J. Wilke, R. Eujen, J. Organomet. Chem. 1986, 316, 261. [84] Benennung nach Liebau (F. Liebau, Structural Chemistry of Silicates, Springer, Berlin, 1985). [85] S. A. Brewer, A. K. Brisdon, J. Fawcett, P. J. Holliman, J. H. Holloway, E. G. Hope, D. R. Russell, Z. Anorg. Allg. Chem. 2006, 632, 325. [86] A. J. Edwards, J. Chem. Soc. (Resumed) 1964, 3714. [87] W. J. Casteel, A. P. Wilkinson, H. Borrmann, R. E. Serfass, N. Bartlett, Inorg. Chem. 1992, 31, 3124. [88] J. H. Holloway, R. D. Peacock, R. W. H. Small, J. Chem. Soc. (Resumed) 1964, 644. [89] B. G. Mueller, M. Serafin, Eur. J. Solid State Inorg. Chem. 1992, 29, 625. [90] A. J. Edwards, G. Jones, J. Chem. Soc. A 1969, 958. [91] U. Müller, Anorganische Strukturchemie, Teubner Studienbücher Chemie, Stuttgart, 1991. [92] L. Q. Tang, M. S. Dadachov, X. D. Zou, Z. Kristallogr. - New Cryst. Struct. 2001, 216, 389. [93] M. S. Dadachov, L. Q. Tang, X. D. Zou, Z. Kristallogr. - New Cryst. Struct. 2000, 215, 605. [94] I. K. Becker, R. Mattes, Z. Anorg. Allg. Chem. 1996, 622, 105. [95] N. R. Götz, Dissertation 2011; Ludwig-Maximilians Universität, München. [96] J. Axhausen, A. Kornath, to be published. [97] N. R. Götz, A. Kornath, to be published. [98] W.-J. Feng, Z. Shu, X.-J. Ma, Z.-M. Jin, Acta Crystallogr. E 2007, 63, m2411. [99] I. W. Levin, R. A. R. Pearce, J. R. C. Spiker, J. Chem. Phys. 1978, 68, 3471. [100] A. J. Edwards, G. R. Jones, J. Chem. Soc. A 1971, 1971, 2318. [101] D. Mootz, A. Deeg, Z. Anorg. Allg. Chem. 1992, 615, 109. [102] J. L. Hoard, W. B. Vincent, J. Am. Chem. Soc. 1939, 61, 2849. [103] R. Minkwitz, V. Gerhard, T. Norkat, Z. Naturforsch., B: Chem. Sci. 1989, 44, 1337; Z. Naturforsch., B: Chem. Sci. [104] T. E. Mallouk, G. L. Rosenthal, G. Mueller, R. Brusasco, N. Bartlett, Inorg. Chem. 1984, 23, 3167. [105] A. J. Edwards, K. O. Christe, J. Chem. Soc., Dalton Trans. 1976, 175. [106] OPUS Version 6.5, Bruker, Ettlingen, 2008. [107] L. Bayersdorfer, R. Minkwitz, J. Jander, Z. Anorg. Allg. Chem. 1972, 392, 137. [108] DELTA Version 4.3.6, JEOL Inc., Peabody, USA, 2000. 140 Verzeichnisse [109] G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, Universität Göttingen: Göttingen, Germany, 1997. [110] G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen: Göttingen, Germany, 1997. [111] L. A. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University: Utrecht, The Netherlands, 1999. [112] MERCURY 2.3 (Build RC4), Copyright CCDC 2001-2009. [113] K. Brandenburg, DIAMOND 3.2f, Crystal Impact GbR, Bonn, Germany. [114] G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. V. Jr., K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, J. A. Pople, Revision B.03 ed., Gaussian Inc., Pittsburgh (PA, USA), 2003, Pittsburgh (PA, USA), 2003. [115] P. Császár, P. Pulay, J. Mol. Struct. 1984, 114, 31. 141 Lebenslauf geb. am 30.10.1979 in Cottbus Nationalität: deutsch AUSBILDUNG Promotion im Bereich der Anorganischen Chemie, Arbeitskreis Prof. Dr. A. Kornath „Neue Aspekte der Chemie des supersauren Mediums HF/GeF4 und Beiträge zur Protonierung sehr starker organischer Säuren“ Ludwig-Maximilians Universität München seit 10/07 Betriebswirt (IWW) Institut für Wirtschaftswissenschaften (IWW), FernUniversität Hagen 09/09 – 03/11 Master of Science, Chemie Ludwig-Maximilians Universität München 09/04 – 11/06 Bachelor of Science, Chemie/Biochemie Ludwig-Maximilians Universität München 10/01 – 09/04 BERUFLICHE ERFAHRUNGEN ABCR GmbH & Co. KG, Karlsruhe Freiberuflicher wissenschaftlicher Berater • Synthese von Feinchemikalien • Informationsbeschaffung in den Bereichen Silicium- und Fluorchemie 08/07 – 03/11 Wacker Chemie AG (Consortium für elektrochemische Industrie), München Mitarbeiter im Forschungsbereich Organische Synthese • Durchführung von Hochdruckreaktionen inklusive Apparate-technischer Aspekte (Planung, Aufbau, Wartung) • Charakterisierung der Reaktionsprodukte mit verschiedenen Methoden • Synthese von Reagenzien und Katalysatoren 02/07 – 08/07 STUDIENBEGLEITENDE TÄTIGKEITEN Fachschaftsinitiative Chemie, LMU, München gewählter studentischer Vertreter 10/02 – 11/06 Ludwig-Maximilians-Universität, München studentische Hilfskraft in der Arbeitsgruppe von PD. Dr. H. Huppertz 03/05 – 09/06 Fachbereichsrat der Fakultät Chemie u. Pharmazie, LMU, München gewählter studentischer Vertreter 10/03 – 10/05 Forschungspraktikum in Organischer Chemie, Arbeitskreis Prof. Dr. P. van Leeuwen Universität von Amsterdam, Niederlande 08/05 – 10/05 SCHULBILDUNG Comenius-Gymnasium Deggendorf, Abitur 09/90 – 07/99 PUBLIKATIONSLISTE ZUSCHRIFTEN • The Protonation of Mono-, Di-, and Trifluoroacetic Acid T. Soltner, A. Kornath in preperation • The Structural Diversity of Fluorogermanates T. Soltner, A. Kornath in preperation • The Protonation of CF3SO3H: Preperation and Characterization of Trifluoromethanedihydroxyoxosulfonium Hexafluoroantimonate [CF3SO3H2]+[SbF6]T. Soltner, N. R. Goetz, A. Kornath Eur. J. Inorg. Chem, in press, DOI: 10.1002/ejic.201001339 • Spiking of Hydrocarbon Fuels with Silanes-based Combustion Enhancers B. Hidding, M. Fikri, M. Bozkurt, C. Schulz, T. Soltner, A. Kornath, M. Pfitzner, A. Adamczyk, L. Broadbelt, H. Ellerbrock, D. Simone, C. Bruno ISTS Special Issue: Selected papers from the 27th International Symposium on Space Technology and Science, 2010, 8, 39. • High-Precision Catalysts: Regioselective Hydroformylation of Internal Alkenes by Encapsulated Rhodium Complexes M. Kuil, T. Soltner, Piet W. N. M. van Leeuwen, Joost N. H. Reek J. Am. Chem. Soc. 2006, 128, 11344. • δ-La(BO2)3 (= δ-LaB3O6): A New High-Pressure Modification of Lanthanum metaOxoborate G. Heymann, T. Soltner, H. Huppertz Solid State Sci. 2006, 8, 821. • The High-Pressure Modification of CePtSn – Synthesis, Structure, and Magnetic Properties J. Riecken, G. Heymann, T. Soltner, R.-D. Hoffmann, H. Huppertz, D. Johrendt, R. Pöttgen Z. Naturforsch. 2005, 60b, 821. KONFERENZBEITRÄGE • Die Strukturelle Vielfalt von Fluorgermanaten T. Soltner 14. Deutscher Fluortag, Dorfweil (D), 27.–29. September 2010, Vortrag. • The Protonation of CF3SO3H: Preperation and Characterization of Trifluoromathanedihydroxyoxosulfonium Hexafluoroantimonate [CF3SO3H2]+[SbF6]T. Soltner, A. Kornath 16th European Symposium on Fluorine Chemistry, Ljubljana (SLO), 18.–23. July 2010, Poster. • Protonation of Mono-, Di-, and Trifluoroacetic Acid T. Soltner, A. Kornath 19th International Symposium on Fluorine Chemistry, Wyoming (USA), 23.–28. August 2009, Poster.


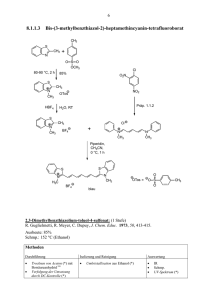


![6.3.1 1-Oxa-spiro[2.5]octan - Institut für Organische Chemie](http://s1.studylibde.com/store/data/001356875_1-96e669e5c88ad586db9f9f199d424d05-300x300.png)